
A new p38 MAP kinase-regulated transcriptional coactivator that stimulates p53-dependent apoptosis

ISSN: 1524-4563 Copyright © 2007 American Heart Association. All rights reserved. Print ISSN: 0194-911X. Online
72514Hypertension is published by the American Heart Association. 7272 Greenville Avenue, Dallas, TX
DOI: 10.1161/01.HYP.0000256831.33459.ea published online Jan 15, 2007; Hypertension
Friedrich C. Luft and Dominik N. Muller Anette Fiebeler, Jeffrey B. Madwed, Alexander Schirdewan, Hermann Haller,
Gapeljuk, Maren Wellner, Silke Meiners, Petra Gratze, Nidal Al-Saadi, Sandra Feldt, Joon-Keun Park, Robert Fischer, Ralf Dechend, Erdenechimeg Shagdarsuren, Andrej
II-Induced Target Organ Damagep38 Mitogen-Activated Protein Kinase Inhibition Ameliorates Angiotensin
http://hyper.ahajournals.orglocated on the World Wide Web at:
The online version of this article, along with updated information and services, is
http://www.lww.com/static/html/reprints.htmlReprints: Information about reprints can be found online at
[email protected], Baltimore, MD 21202-2436. Phone 410-5280-4050. Fax: 410-528-8550. Email: Permissions: Permissions & Rights Desk, Lippincott Williams & Wilkins, 351 West Camden
http://hyper.ahajournals.org/subsriptions/Subscriptions: Information about subscribing to Hypertension is online at
at University of Kentucky--Lexington on January 26, 2007 hyper.ahajournals.orgDownloaded from

p38 Mitogen-Activated Protein Kinase InhibitionAmeliorates Angiotensin II–Induced Target Organ Damage
Joon-Keun Park, Robert Fischer, Ralf Dechend, Erdenechimeg Shagdarsuren, Andrej Gapeljuk,Maren Wellner, Silke Meiners, Petra Gratze, Nidal Al-Saadi, Sandra Feldt, Anette Fiebeler,
Jeffrey B. Madwed, Alexander Schirdewan, Hermann Haller, Friedrich C. Luft, Dominik N. Muller
Abstract—We investigated whether or not p38 mitogen-activated protein kinase inhibition ameliorates angiotensinII–induced target organ damage. We used double transgenic rats harboring both human renin and angiotensinogen genes(dTGRs). dTGR, with or without p38 inhibitor (BIRB796; 30 mg/kg per day in the diet), and nontransgenicSprague–Dawley rats were studied in 2 protocols. In protocol 1 (week 7), systolic blood pressure of untreated dTGRswas 204�4 mm Hg, but partially reduced after BIRB796 treatment (166�7 mm Hg), whereas Sprague–Dawley ratswere normotensive. The cardiac hypertrophy index was unchanged in untreated and BIRB796-treated dTGRs. The�-myosin heavy chain expression of BIRB796-treated hearts was significantly lower in BIRB796 compared withdTGRs, indicating a delayed switch to the fetal isoform. BIRB796 treatment significantly reduced cardiac fibrosis,connective tissue growth factor, tumor necrosis factor-�, interleukin-6, and macrophage infiltration. Albuminuria wasnot reduced in BIRB796-treated dTGRs. Tubular and glomerular damage with tumor necrosis factor-� expression wasunaltered, although serum creatinine and cystatin C were normalized. Renal macrophage infiltration, fibrosis, and vesseldamage were reduced. In protocol 2 (week 8), we focused on mortality and arrhythmogenic electrical remodeling.Mortality of untreated dTGRs was 100% but was reduced to 10% in the BIRB796 group. Cardiac magnetic fieldmapping showed prolongation of depolarization and repolarization in untreated dTGRs compared with Sprague–Dawleyrats with a partial reduction by BIRB796. Programmed electrical stimulation elicited ventricular tachycardias in 81% ofuntreated dTGRs but only in 48% of BIRB796-treated dTGRs. In conclusion, BIRB796 improved survival, target organdamage, and arrhythmogenic potential in angiotensin II–induced target organ damage. (Hypertension. 2007;49:1-9.)
Key Words: angiotensin II � p38 � electrical remodeling � cardiac and renal damage
Mitogen-activated protein kinase (MAPK) p38 is a4-isoform (p38� p38�, p38�, and p38�) serine/threonine
kinase originally isolated from lipopolysaccharide-stimulatedmonocytes.1 p38 kinase pathway activation results in phosphor-ylation of transcription factors affecting cell division, apoptosis,and invasiveness of inflammatory cells.2–4 p38� is involved inthe biosynthesis of tumor necrosis factor (TNF)-�, interleukin(IL)-1�, and other important inflammatory cytokines that repre-sent a convergence point for many proinflammatory signalingprocesses.5 p38�/� inhibitors were developed as a potentialtreatment for inflammatory diseases.6 However, p38 is alsoactivated by catecholamines, angiotensin (Ang) II, endothelin-1,hypoxia, and vascular wall stress.7,8 Ang II activates p38 invarious cell types, including cardiomyocytes, vascular smoothmuscle cells, mesangial cells, and monocytes.9–12 p38 MAPKand extracellular signal regulated kinase (ERK) 1/2 activationdepend on the epidermal growth factor receptor.13 p38 MAPK,
ERK, and tyrosine kinase signaling also play a role in patho-genesis of cardiovascular disease.8,14–16 p38 MAPK inhibitionmay, therefore, be of interest for cardiovascular diseases, includ-ing electrical remodeling. We tested this notion in an AngII–dependent double transgenic rat (dTGR) model harboring thehuman angiotensinogen and renin genes.17 The rats develophypertension, cardiac hypertrophy, proteinuria, and renal failureand die abruptly between age 7 to 8 weeks.18,19 Inflammationand innate and acquired immunity are important in the dTGRmodel.19,20 Others,21,22 as well as our laboratories,18,19,23–26
showed earlier that strategies directed at nuclear factor �B,TNF-�, corticosteroids, aldosterone, antioxidants, and antilym-phocyte treatments all ameliorate target organ damage thatprimarily involves the heart, kidneys, and vessels.18 We inves-tigated MAPK p38 signaling in our model by using a novel p38MAPK-� and -� inhibitor (BIRB796).27 Because transgenic ratsdie suddenly, we also entertained the possibility of an arrhyth-
Received August 19, 2006; first decision September 5, 2006; revision accepted December 19, 2006.From the Medical Faculty of the Charite (R.F., R.D., A.G., M.W., S.M., N.A.-S., S.F., A.F., A.S., F.C.L., D.N.M.), Franz Volhard Clinic, HELIOS
Klinikum, Berlin, Germany; Max Delbruck Center for Molecular Medicine (P.G., F.C.L., D.N.M.), Berlin-Buch, Germany; Institute for MolecularCardiovascular Research (E.S.), RWTH Aachen University, University Hospital Aachen, Aachen, Germany; the Medical School of Hannover (J.-K.P.,H.H.), Hannover, Germany; and Boehringer-Ingelheim Pharmaceuticals (J.B.M.), Ridgefield, Conn.
The first 3 authors contributed equally to this work.Correspondence to Dominik N. Muller, Max-Delbruck Center, Wiltberg Strasse 50, 13125 Berlin, Germany. E-mail [email protected]© 2007 American Heart Association, Inc.
Hypertension is available at http://www.hypertensionaha.org DOI: 10.1161/01.HYP.0000256831.33459.ea
1

mogenic death in the animals and investigated electricalremodeling.
MethodsExperimental DesignWe studied male transgenic dTGRs (RCC Ltd) and age-matchednontransgenic Sprague–Dawley (SD) rats (MDC).17–20,23,25,26 Localauthorities approved the studies, and American Physiological Soci-ety guidelines for animal care were followed. We performed 2different protocols. In protocol 2, untreated dTGR (n�15),dTGR�BIRB796 (30 mg/kg per day in the diet for 3 weeks; n�11),and SD (n�8 each group) rats were analyzed. The structure ofBIRB796 and kinase selectivity is shown in Figure IA (availableonline at http://hyper.ahajournals.org). Systolic blood pressure wasmeasured weekly by tail cuff. Twenty-four–hour urine samples werecollected in metabolic cages from weeks 5 to 7. Serum was collectedat week 7. Serum creatinine and cystatin C were measured by clinicalroutine assays. Urinary rat albumin was determined by enzyme-linked immunosorbent assay (CellTrend). The aim of protocol 2 wasto focus on electrophysiological alterations and mortality. UntreateddTGR (n�10), dTGR�BIRB796 (n�10), and SD (n�10) rats werestudied up to week 8. Cardiac magnetic field mapping (CMFM) wasperformed at week 7 under isoflurane anesthesia. Echocardiographywas performed as described earlier.18
Cardiac Magnetic Field Mapping and InVivo ElectrophysiologyMagnetic fields of the rat heart were recorded over the anterior chest.Details are given in the online supplement. Programmed electricalstimulation was performed to test for the inducibility of ventriculararrhythmias. Details are given in the online supplement.
ImmunohistochemistryIce-cold acetone-fixed cryosections (6 �m) were stained by immu-nofluorescence or alkaline phosphatase-anti-alkaline phosphatasetechniques as described earlier.19,20 Details are given in theonline supplement.
Quantitative TaqMan RT-PCRRNA isolation and TaqMan RT-PCR were performed as describedearlier.23 We analyzed left ventricular tissue for �-myosin heavychain, connective tissue growth factor, and IL-6. Each sample was intriplicate. For quantification, the target sequences were normalized inrelation to the 36B4 product. Biotez synthesized the primers. Thesequences are available on request.
StatisticsData are presented as mean�SEM. Statistically significant differ-ences in mean values were tested by ANOVA, and blood pressureand albuminuria by repeated-measures ANOVA and the Scheffe ttest. A value of P�0.05 was considered statistically significant. Thedata were analyzed using Statview statistical software.
Resultsp38 MAPK Inhibition Reduces Blood PressureWithout Reduction of Cardiac Hypertrophy butAffected Fetal �-Myosin Heavy Chain Expressionand Proliferation of CardiomyocytesSystolic blood pressure increased progressively in untreateddTGRs from 155�3 mm Hg in week 5 to 204�4 mm Hg inweek 7. The BIRB796 treatment slightly reduced bloodpressure (166�7 mm Hg at week 7; P�0.05; Figure 1A),whereas SD rats were normotensive (123�3 mm Hg). De-spite the reduction in blood pressure, untreated and BIRB796-treated dTGRs had similar heart weight (data not shown) and
cardiac hypertrophy indices (heart-to-tibia ratio), which weresignificantly higher compared with nontransgenic SD rats(310�6 versus 307�6 versus 206�5 mg/cm, respectively;P�0.05; Figure 1B). Echocardiography confirmed the resultsof cardiac hypertrophy. Total wall thickness (expressed assum of septum�left ventricular posterior wall) of dTGRs was3.3�0.1 mm, 3.8�0.1 mm for BIRB796, and 1.8�0.05 mmfor SD rats (Figure 1C). In contrast to untreated dTGRs,BIRB796-treated hearts showed a significantly lower expres-sion of the fetal �-myosin heavy chain isoform indicating adifferent pattern of cardiac hypertrophy (Figure 1D). We nextanalyzed cardiomyocyte proliferation. The number of Ki-67–positive cells in the heart (Figure 1E) was significantly higherin untreated dTGRs compared with SD rats. BIRB796 treat-ment resulted in a further significant increase of proliferatingcardiomyocytes and fibroblasts. Localization of Ki-67 posi-tive nuclei is shown in Figure II.
BIRB796 Reduces p38 MAPK and ERK 1/2Phosphorylation, Fibrosis, Cytokine Expression,and Macrophage Infiltration in the HeartWe investigated whether or not BIRB796 treatment affectedMAPK phosphorylation in vivo. Untreated dTGRs show in-creased p-p38 MAPK and p-ERK 1/2 expression, which wassignificantly reduced by BIRB796 (Figure 2A). Untreated dT-GRs showed marked signs of fibrosis. Collagen I was expressedperivascularly around damaged dTGR vessels. The BIRB796-treated rats and SD rats showed a similar expression (Figure 2A).Cardiac fibronectin immunoreactivity was observed predomi-nantly in the interstitium and perivascularly (data not shown).BIRB796 treatment significantly reduced TNF-� (Figure 2A),connective tissue growth factor, and IL-6 treatment (Figure 2B).We next analyzed cardiac connective tissue growth factor(Figure 2C) and IL-6 (Figure 2D) gene expression. Both geneswere significantly reduced in the left ventricle after BIRB796treatment. Cardiac macrophage and monocyte infiltration(ED-1� cells) was increased in untreated dTGRs, significantlyreduced by BIRB796, but still elevated compared with SD rats(Figure IIIA).
p38 MAPK Inhibition Does Not AffectAlbuminuria but Normalizes Serum Creatinineand Cystatin C and ReducesMacrophage InfiltrationSurprisingly, BIRB796 treatment did not ameliorate albumin-uria compared with untreated dTGRs (30�5 versus 33�5mg/day, respectively; Figure 3A). Both groups showed asignificantly increased urinary albumin excretion comparedwith SD (0.1�0.02 mg/day). In contrast, only untreateddTGRs had an elevated serum creatinine (Figure 3B) andaltered cystatin C (Figure 3C), whereas BIRB796 treatmentnormalized creatinine and cystatin C to SD levels. Monocyte/macrophage infiltration in the kidney was significantly in-creased in untreated dTGRs, reduced by BIRB796, but stillelevated compared with SD (Figure IIIB). The effect ofBIRB796 on p38 MAPK in the kidney is shown in FigureIVA-C.
2 Hypertension March 2007

p38 MAPK Inhibition Reduces Renal Interstitialbut Not Glomerular Fibrosis and PreventedVascular but Not Tubular andGlomerular DamageRenal histology (Masson trichrome staining) revealed thatuntreated dTGRs had increased perivascular, interstitial, andglomerular matrix deposition; vessel with hypertrophied me-dia; and damaged tubules and glomeruli (Figure 3D). Tubuleswere often filled with protein in BIRB796-treated rats evenmore frequently compared with untreated dTGRs (Figure3D). Chronic BIRB796 treatment resulted in a significantlyreduced interstitial matrix formation and a reduction of vesseldamage (Figure 3D). In contrast, tubules and glomeruli werestill damaged (Figure 3D). Interstitial fibronectin expressionwas significantly reduced by BIRB796 (Figure 3E), whereasglomeruli from BIRB796-treated rats showed an unchangedcollagen III expression compared with dTGRs (Figure 3F).Renal TNF-� immunoreactivity was increased in damagedtubules, glomeruli, and vessels of untreated dTGRs. However,BIRB796 reduced vascular TNF-� (see Figure 3D, inset) buthad no effect on glomerular and tubular expression (Figure 3G).
p38 MAPK Inhibition ImprovesElectrophysiological AlterationsWe next focused on electrophysiological alterations andmortality by analyzing CMFM recordings. Untreated dTGRs
had a significantly prolonged QRS duration and QT intervalcompared with SD controls. BIRB796 treatment resulted in apartial but significant reversal of this pattern (Table I). Wealso analyzed parameters reflecting the inhomogeneity ofdepolarization (mean inhomogeneity index for CMFM distri-bution during QRS interval) and repolarization (peak of theT-wave [TPeak]-end of the T-wave [TEnd] interval and TPeak-Dispersion), which were significantly increased in untreateddTGRs compared with SD controls and partially restored byBIRB796 (Table I). Indicators of regional/spatial differencesin the process of repolarization were significantly increasedin dTGRs compared with SD controls (Table I). Inhomoge-neity of repolarization was improved by BIRB796 treatment(Table I).
CMFM distribution for QRS and ST-T wave is shown inFigure VA and VB. Untreated dTGRs show an increase in fieldstrength and duration of the QRS compared with controls.BIRB796 treatment ameliorated these changes (Figure VA).ST-T wave maps visualize prolongation of repolarization inuntreated dTGRs and were partially ameliorated by BIRB796treatment. These results were confirmed by a reorientation of thecardiac magnetic field in the end of the T wave (Table I).
“Butterfly” plots are CMFM waveform composites fromall of the animals per group (Figure VIA). Inserted magnifi-cation show that the dispersion of the T wave peak was
Figure 1. A, BIRB796 reduced blood pressure only slightly at week 7. B and C, Untreated and BIRB796-treated dTGRs both showedincreased cardiac hypertrophy (heart weight/tibia ratio) and total wall thickness (expressed as septum�left ventricular wall diameter)compared with SD. D, BIRB796 treatment reduced the fetal �-myosin heavy chain isoform. E, Untreated dTGR hearts have moreKi-67� cells compared with SD, whereas BIRB796 treatment further increased it by 20% compared with untreated dTGRs. BIRB796results are mean�SEM. *P�0.05 vs untreated dTGR, #P�0.05 vs dTGR�BIRB796.
Park et al Angiotensin II and p38 3

increased in dTGRs compared with controls. BIRB796 treat-ment ameliorated the “butterfly” pattern (Figure VIA) andsignificantly reduced the T wave dispersion (Figure VIB).Untreated dTGRs showed a prolongation of repolarization by15 ms and a flattening of the peak of T wave, which waspartially corrected by BIRB796 (Figure VIC).
p38 MAPK Inhibition Reduces Arrhythmias byProgrammed Electrical StimulationWe next analyzed whether untreated dTGRs are prone toventricular arrhythmia induction and whether BIRB796 treat-ment affects arrhythmia induction. Programmed electricalstimulation showed a high nonsustained and sustained ven-tricular tachycardia induction rate in untreated dTGRs (81%,Figure 4A). In SD controls, the same protocol never initiatedarrhythmias. The arrhythmia induction was significantly re-duced in BIRB796-treated dTGRs (48%). In addition, themean cycle lengths of induced ventricular tachycardias weresignificantly shorter in untreated compared with BIRB796-treated dTGRs (Figure 4B). Representative recordings of per-formed electrophysiological studies are shown (Figure 4C).
p38 MAPK Inhibition Reduces MortalityFinally, chronic BIRB796 treatment significantly reducedmortality (Figure 5). Although mortality in untreated dTGRswas 100%, only 1 (10%) of 10 BIRB796-treated dTGRs diedat the last day of the study (day 56). SD controls had nomortality by week 8.
DiscussionThree major findings of the present study are that chronic p38MAPK inhibition reduced mortality in Ang II–induced end-organ damage, that p38 MAPK inhibition showed a highlycell type–specific action leading to protection of the heart andvessels, and that renal tubular and glomerular damage wereless ameliorated, although glomular filtration rate wasimproved. Furthermore, p38 MAPK inhibition improvedelectrophysiological alterations and reduced arrhythmiasby programmed electrical stimulation.
We found that p38 MAPK inhibition with BIRB796reduced cardiac fibrosis, connective tissue growth factor,TNF-�, and IL-6 expression, as well as vascular damage andmacrophage infiltration. Although BIRB796 treatment didnot alter the heart weight/tibia ratio and total wall thickness,
Figure 2. A and B, BIRB796 reduced p38 MAPK and ERK 1/2 phosphorylation, perivascular collagen I, CTGF deposition, and TNF-�.TNF-� and IL-6 were expressed in the media of untreated dTGRs. BIRB796 reduced the expression toward the SD control level.Semiquantitative scorings are given as insets. C and D, Cardiac CTGF and IL-6 mRNA expression confirmed the results. Results aremean�SEM. *P�0.05 vs untreated dTGR, #P�0.05 vs dTGR�BIRB796.
4 Hypertension March 2007
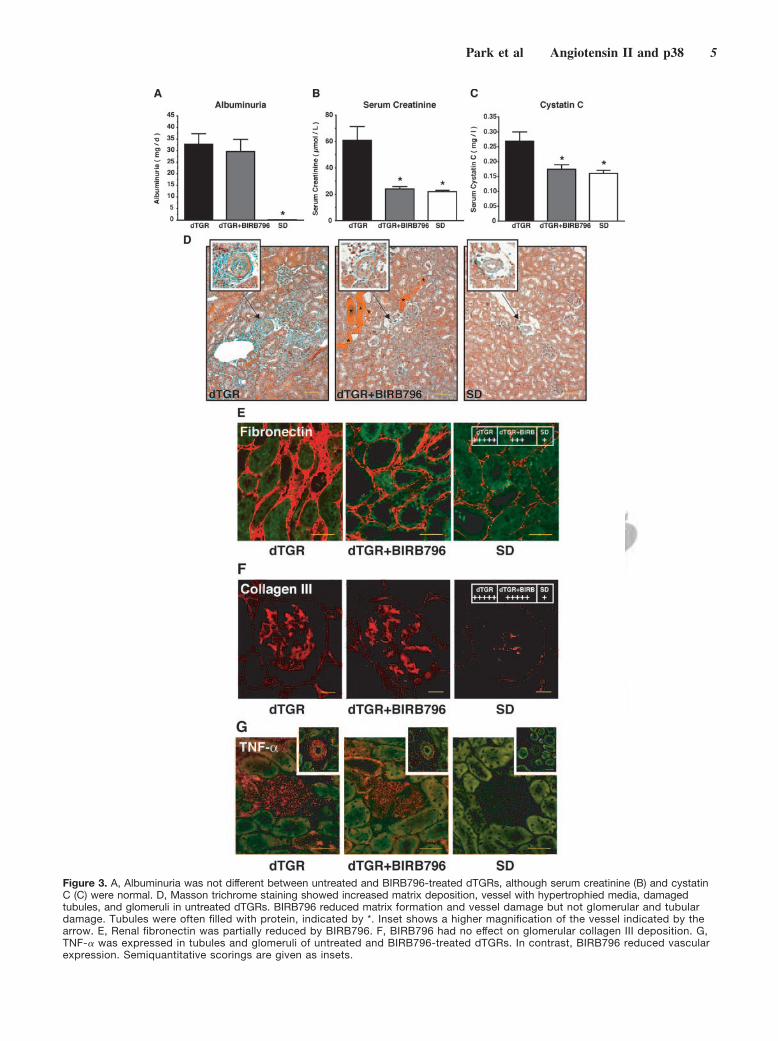
Figure 3. A, Albuminuria was not different between untreated and BIRB796-treated dTGRs, although serum creatinine (B) and cystatinC (C) were normal. D, Masson trichrome staining showed increased matrix deposition, vessel with hypertrophied media, damagedtubules, and glomeruli in untreated dTGRs. BIRB796 reduced matrix formation and vessel damage but not glomerular and tubulardamage. Tubules were often filled with protein, indicated by *. Inset shows a higher magnification of the vessel indicated by thearrow. E, Renal fibronectin was partially reduced by BIRB796. F, BIRB796 had no effect on glomerular collagen III deposition. G,TNF-� was expressed in tubules and glomeruli of untreated and BIRB796-treated dTGRs. In contrast, BIRB796 reduced vascularexpression. Semiquantitative scorings are given as insets.
Park et al Angiotensin II and p38 5

p38 MAPK inhibition reduced �-myosin heavy chain expres-sion, suggesting a less pathological and more physiologicalhypertrophy in BIRB796-treated dTGR hearts. p38 MAPK-inhibited hearts also showed an improved electrical remod-eling and a decreased susceptibility to ventricular tachycardiainduction. Although BIRB796 treatment did not reduce albu-minuria, serum creatinine did not increase in BIRB796-
treated dTGRs. Visible glomerular and tubular damage waspresent BIRB796-treated dTGRs. Nevertheless, p38 MAPKinhibition reduced renal matrix formation and macrophageinfiltration, as well as vascular damage. We believe that mostof the effects are mediated via p38 �/� MAPK. Nevertheless,we cannot completely exclude the role of c-Jun NH2-terminalkinase (JNK)-2�2. In vitro, BIRB796 inhibits JNK-2�2 withan IC50 of 2 nM, whereas BIRB796 has no effect on JNK-1.Nevertheless, it could well be that, in vivo, our chronicBIRB796 treatment resulted in drug levels sufficient toinhibit JNK-2�2.
We also found that blood pressure was partially reducedcompared with untreated dTGRs. Nonetheless, hypertensionpersisted at a level of �165 mm Hg. We do not believe thatblood pressure reduction alone was a major cause for theimprovement of target organ damage.19,25 Several pointssuggest a blood pressure–independent effect. First, despite ablood pressure decrease of �40 mm Hg, cardiac mass/hyper-trophy was similar in untreated and BIRB796-treated dTGRs,but the nature of the cardiac phenotype was different. Ingeneral, cardiac mass/hypertrophy clearly depends on after-load. Therefore, we could well imagine that a reduction of40 mm Hg afterload might have facilitated to reverse the
Figure 4. A, Programmed electrical stimulation revealed a reduced susceptibility to ventricular arrhythmias in BIRB796-treated dTGRs com-pared with untreated dTGRs. No arrhythmias could be induced in SD controls. The percentage indicates the number of performed stimula-tion protocols with reproducible induced ventricular arrhythmias. B, BIRB796-treated dTGRs had induced ventricular tachycardias with longermean cycle length (slower heart rate) compared with untreated dTGRs. In SD rats, no tachycardias could be detected (n.d. indicates notdetected). Results are mean�SEM. *P�0.05 vs untreated dTGR. C, representative results of programmed electrical stimulation. One ECG isrecorded simultaneously with endocardial left ventricular (LV) electrograms and indicators of delivered electrical stimuli (Stim). UntreateddTGRs had faster ventricular tachycardias compared with BIRB796-treated dTGRs. In SD controls, electrical stimuli did not elicit reproduciblearrhythmias.
Figure 5. All untreated dTGRs died up to week 8. BIRB796treatment reduced mortality to 10%.
6 Hypertension March 2007

pathophysiological hypertrophy in untreated dTGRs to aphysiological hypertrophy observed in BIRB796-treated dT-GRs. Nonetheless, load, per se, cannot be the sole reason.BIRB796-treated dTGRs were still hypertensive but showedalmost normalized matrix and cytokine expression, as well asreduced cell infiltration. We did not include a parallel bloodpressure control group (eg, hydralazine treatment) in thisstudy. However, earlier we performed several studies thatseparated Ang II–related from blood pressure–related effectsin our model.25 We treated dTGRs treated with triple therapy(hydralazine, hydrochlorothiazide, and reserpine). This treat-ment merely delayed the course of the disease by 1 week. Inother experiments, we treated dTGRs with dexamethasone.Dexamethasone-treated dTGRs were severely hypertensive(blood pressure levels �200 mm Hg). Nevertheless, dexa-methasone reduced mortality to 0 with significantly improvedtarget organ damage.19
In an earlier study, we found that untreated dTGRs exhib-ited an �/�-myosin heavy chain switch to the fetal isoform, aswe confirm here.28 In that study, atrial natriuretic peptideprecursor, a marker of cardiac hypertrophy, genes of the cellcycle (cyclin D3, cyclin D2, and cyclin D), heat shock proteins(HSP70 and HSP27), and genes of the MAPK pathway, namely,heparin-binding epidermal growth factor (epidermal growthfactor-like) growth factor, p38 MAPK, c-fos, junB, fra, andc-myc, were all significantly increased in the left ventricles ofterminally ill dTGRs. Interestingly, heat shock proteins them-selves activate the p38 pathway. These findings are relevant tothe present results, because BIRB796 treatment amelioratedtarget organ damage and reduced mortality significantly.Whether the heart can grow by multiplication of myocytes hasbeen controversial over the decades. Recent data indicate that theadult heart has a subpopulation of myocytes that is not termi-nally differentiated. These myocytes evidently reentered the cellcycle and underwent nuclear mitotic division. Beltrami et al29
provided convincing proof of myocyte replication in the failinghuman heart and showed that this form of cell growth couldcompensate for exhaustion of myocyte hypertrophy. Recently,Engel et al30 demonstrated that p38 MAPK inhibition promotescardiomyocyte cytokinesis. Our data show that untreated hyper-trophied dTGR hearts have a higher number of Ki-67–positivecardiomyocytes compared with nontransgenic hearts. Interest-ingly, BIRP796 treatment further increased myocyte prolifera-tion by 20%. It is tempting to speculate that increased cardio-myocyte proliferation might have contributed to tissue repair inour model.
The p38 MAPK module is activated by a plethora ofother MAPK kinases in response to numerous physical andchemical stresses, including Ang II, aldosterone, variouscytokines, oxidative stress, hypoxia, and ischemia.1,31–34
Recently, we found that blockade of aldosterone synthaseameliorates fibrosis and inflammation in our model.23
Possibly, aldosterone-p38 signaling might have contributedto the pathogenesis of organ damage in our dTGR model.Interestingly, various in vivo studies in transgenic rats over-expressing the mouse ren-2 gene, another model of AngII–induced target organ damage, demonstrated that not onlyp38 MAPK but also ERK 1/2 and JNK signaling are involvedin the pathogenesis.8,14,16 In our model, we found that
BIRB796 also reduced cardiac ERK 1/2 phosphorylation. Webelieve that this result might have been an indirect phenom-enon because of improved cardiac function with reduced cellinfiltration and cytokine production. Altogether the findingsin both transgenic models suggest that MAPK signaling playsan important role in Ang II–induced target organ damage.
Innate and acquired immunity also plays an important rolein our dTGR model. Untreated dTGRs show elevated TNF-�,IL-6, and C-reactive protein levels.18–20,26 The p38� kinaseinhibitor BIRB796 blocks the biosynthesis of TNF-�,C-reactive protein, IL-1�, and other important inflammatorycytokines in humans.35 In vivo, the proinflammatory situationis not only determined by cytokine production and release ofimmune cells but also by other cell types, like smooth musclecells, tubular cells, and glomerular cells. BIRB796 treatmentled to an organ and cell type–specific reduction of inflamma-tory actions. Although BIRB796 reduced IL-6 and TNF-�expression in the heart and macrophage infiltration in heartand kidney, the drug only reduced vascular but not tubularand glomerular TNF-� expression in the kidney. Shao et alreported that Ang II–infused rats showed an increase in theT-helper 1 cytokine interferon-�.36 Rincon et al37 reportedthat the p38 MAPK pathway is necessary for interferon-�production by T-helper 1 effector cells without affecting theT-helper 2 response. P38 is also activated in macrophages,neutrophils, and T cells, where it participates in respiratoryburst activity, chemotaxis, granular exocytosis, adherence,and apoptosis. P38 also mediates immune responses bystabilizing specific cellular mRNAs involved in these pro-cesses. The precise functions of p38 are only now beginningto emerge; however, p38 has been shown to phosphorylateseveral cellular targets, including cytosolic phospholipaseA2, the microtubule-associated protein Tau, and the tran-scription factors ATF1 and -2, MEF2A, Sap-1, Elk-1, nuclearfactor �B, Ets-1, and p53. There are numerous p38 targetsthat could have influenced the course of target organ damagein our model. Nuclear factor �B is a particularly attractivetarget that received our attention in earlier studies.18,26 Wehave not explored the pathways involved. However, otherinvestigators have drawn attention to interactions among AngII, reactive oxygen species, and p38 MAPK31,32,34 that couldvery well be implicated here. In vitro, but also in vivo, AngII also promotes p38 MAPK activation via the epidermalgrowth factor receptor and platelet-derived growth factorreceptor �.14,16
Among our major focal points was electrical remodeling.The term “remodeling” is generally used morphologically.Wijffels et al38 defined the term electrical remodeling as theoccurrence of cardiac arrhythmias because of acquiredchanges in cardiac structure or function. Increased fibrosis isan important factor that affects electrical conduction leadingto an increased susceptibility to arrhythmias.39 Inflammationalso promotes electrical remodeling.40 The morphologicalalterations in the heart of our dTGR rats have been addressedin an earlier study.28 A part of our hypothesis in the currentstudy involved the cause of death in these animals. We havenot addressed that issue directly. Continuous ECG telemetryin a large number of animals would be necessary. Neverthe-less, we have introduced a preliminary approach to address
Park et al Angiotensin II and p38 7

this issue. Our presumption requires verification; however,the results presented here suggest that the electrical remod-eling process can be influenced by an anti-inflammatoryintervention mediated by a signaling molecule, namely, p38.
PerspectivesOur findings underscore the importance of inflammatory andelectrophysiological pathways in target organ damage. Viewson p38 have invariably considered anti-inflammatory appli-cations. Cardiovascular disease is not generally considered tobe an inflammatory or immune disorder. Our model and thedata presented here support the therapeutic perspective ofanti-inflammatory and antiarrhythmic approaches to cardio-vascular disease.
AcknowledgmentsWe thank Petra Berkefeld, Astrid Schiche, Jutta Meisel, May-BrittKohler, Gabriele N�diaye, Mathilde Schmidt, and Jana Czychi fortheir excellent technical assistance.
Sources of FundingThe studies were supported by grants-in-aid from the NGFN2(Nationales Genom Netzwerk KGCV1 01GS0416) and the EuropeanUnion (EuReGene). D.N.M. is a Helmholtz fellow. The DeutscheForschungsgemeinschaft supported D.N.M. and F.C.L. Boehringer-Ingelheim Pharmaceuticals, USA, supported the study and suppliedthe p38 inhibitor.
DisclosuresF.C.L. has served as an advisor for Boehringer-Ingelheim Pharma-ceuticals. The remaining authors report no conflicts.
References1. Han J, Lee JD, Bibbs L, Ulevitch RJ. A MAP kinase targeted by
endotoxin and hyperosmolarity in mammalian cells. Science. 1994;265:808–811.
2. Raingeaud J, Whitmarsh AJ, Barrett T, Derijard B, Davis RJ. MKK3- andMKK6-regulated gene expression is mediated by the p38 mitogen-acti-vated protein kinase signal transduction pathway. Mol Cell Biol. 1996;16:1247–1255.
3. Takenaka K, Moriguchi T, Nishida E. Activation of the protein kinasep38 in the spindle assembly checkpoint and mitotic arrest. Science.1998;280:599–602.
4. Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposingeffects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331.
5. Lee JC, Laydon JT, McDonnell PC, Gallagher TF, Kumar S, Green D,McNulty D, Blumenthal MJ, Heys JR, Landvatter SW, Strickler JE,McLaughlin MM, Siemens IR, Fisher SM, Livi GP, White JR, Adams JL,Young PR. A protein kinase involved in the regulation of inflammatorycytokine biosynthesis. Nature. 1994;372:739–746.
6. Dominguez C, Powers DA, Tamayo N. p38 MAP kinase inhibitors: manyare made, but few are chosen. Curr Opin Drug Discov Devel. 2005;8:421–430.
7. Behr TM, Nerurkar SS, Nelson AH, Coatney RW, Woods TN, SulpizioA, Chandra S, Brooks DP, Kumar S, Lee JC, Ohlstein EH, AngermannCE, Adams JL, Sisko J, Sackner-Bernstein JD, Willette RN. Hypertensiveend-organ damage and premature mortality are p38 mitogen-activatedprotein kinase-dependent in a rat model of cardiac hypertrophy anddysfunction. Circulation. 2001;104:1292–1298.
8. de Borst MH, Navis G, de Boer RA, Huitema S, Vis LM, van Gilst WH,van Goor H. Specific MAP-kinase blockade protects against renaldamage in homozygous TGR(mRen2)27 rats. Lab Invest. 2003;83:1761–1770.
9. Naito T, Masaki T, Nikolic-Paterson DJ, Tanji C, Yorioka N, Kohno N.Angiotensin II induces thrombospondin-1 production in human mesangialcells via p38 MAPK and JNK: a mechanism for activation of latentTGF-beta1. Am J Physiol Renal Physiol. 2004;286:F278–F287.
10. Nishida M, Tanabe S, Maruyama Y, Mangmool S, Urayama K,Nagamatsu Y, Takagahara S, Turner JH, Kozasa T, Kobayashi H, Sato Y,Kawanishi T, Inoue R, Nagao T, Kurose H. G alpha 12/13- and reactiveoxygen species-dependent activation of c-Jun NH2-terminal kinase andp38 mitogen-activated protein kinase by angiotensin receptor stimulationin rat neonatal cardiomyocytes. J Biol Chem. 2005;280:18434–18441.
11. Touyz RM, He G, El Mabrouk M, Schiffrin EL. p38 Map kinase regulatesvascular smooth muscle cell collagen synthesis by angiotensin II in SHRbut not in WKY. Hypertension. 2001;37:574–580.
12. Wei C, Cardarelli MG, Downing SW, McLaughlin JS. The effect ofangiotensin II on mitogen-activated protein kinase in human cardiomyo-cytes. J Renin Angiotensin Aldosterone Syst. 2000;1:379–384.
13. Eguchi S, Dempsey PJ, Frank GD, Motley ED, Inagami T. Activationof MAPKs by angiotensin II in vascular smooth muscle cells.Metalloprotease-dependent EGF receptor activation is required for acti-vation of ERK and p38 MAPK but not for JNK. J Biol Chem. 16.2001;276:7957–7962.
14. de Boer RA, Pokharel S, Flesch M, van Kampen DA, Suurmeijer AJ,Boomsma F, van Gilst WH, van Veldhuisen DJ, Pinto YM. Extracellularsignal regulated kinase and SMAD signaling both mediate the angiotensinII driven progression towards overt heart failure in homozygousTGR(mRen2)27. J Mol Med. 2004;82:678 – 687.
15. Pandya N, Santani D, Jain S. Role of mitogen-activated protein (MAP)kinases in cardiovascular diseases. Cardiovasc Drug Rev. 2005;23:247–254.
16. Schellings MW, Baumann M, van Leeuwen RE, Duisters RF, Janssen SH,Schroen B, Peutz-Kootstra CJ, Heymans S, Pinto YM. Imatinib attenuatesend-organ damage in hypertensive homozygous TGR(mRen2)27 rats.Hypertension. 2006;47:467–474.
17. Ganten D, Wagner J, Zeh K, Bader M, Michel JB, Paul M, ZimmermannF, Ruf P, Hilgenfeldt U, Ganten U, Kaling M, Bachmann S, Fukamizu A,Mullins J, Murakami K. Species specificity of renin kinetics in transgenicrats harboring the human renin and angiotensinogen genes. Proc NatlAcad Sci U S A. 1992;89:7806–7810.
18. Muller DN, Heissmeyer V, Dechend R, Hampich F, Park JK, Fiebeler A,Shagdarsuren E, Theuer J, Elger M, Pilz B, Breu V, Schroer K, Ganten D,Dietz R, Haller H, Scheidereit C, Luft FC. Aspirin inhibits NF-kappaBand protects from angiotensin II-induced organ damage. FASEB J. 2001;15:1822–1824.
19. Muller DN, Shagdarsuren E, Park JK, Dechend R, Mervaala E, HampichF, Fiebeler A, Ju X, Finckenberg P, Theuer J, Viedt C, Kreuzer J,Heidecke H, Haller H, Zenke M, Luft FC. Immunosuppressive treatmentprotects against angiotensin II-induced renal damage. Am J Pathol. 2002;161:1679–1693.
20. Shagdarsuren E, Wellner M, Braesen JH, Park JK, Fiebeler A, Henke N,Dechend R, Gratze P, Luft FC, Muller DN. Complement activation inangiotensin II-induced organ damage. Circ Res. 2005;97:716–724.
21. Ruiz-Ortega M, Ruperez M, Esteban V, Rodriguez-Vita J, Sanchez-LopezE, Carvajal G, Egido J. Angiotensin II: a key factor in the inflammatoryand fibrotic response in kidney diseases. Nephrol Dial Transplant. 2006;21:16–20.
22. Savoia C, Schiffrin EL. Inflammation in hypertension. Curr OpinNephrol Hypertens. 2006;15:152–158.
23. Fiebeler A, Nussberger J, Shagdarsuren E, Rong S, Hilfenhaus G, Al-SaadiN, Dechend R, Wellner M, Meiners S, Maser-Gluth C, Jeng AY, Webb RL,Luft FC, Muller DN. Aldosterone synthase inhibitor ameliorates angiotensinII-induced organ damage. Circulation. 2005;111:3087–3094.
24. Mervaala E, Muller DN, Park JK, Dechend R, Schmidt F, Fiebeler A,Bieringer M, Breu V, Ganten D, Haller H, Luft FC. Cyclosporin Aprotects against angiotensin II-induced end-organ damage in doubletransgenic rats harboring human renin and angiotensinogen genes. Hyper-tension. 2000;35:360–366.
25. Mervaala E, Muller DN, Schmidt F, Park JK, Gross V, Bader M, Breu V,Ganten D, Haller H, Luft FC. Blood pressure-independent effects in rats withhuman renin and angiotensinogen genes. Hypertension. 2000;35:587–594.
26. Muller DN, Dechend R, Mervaala EM, Park JK, Schmidt F, Fiebeler A,Theuer J, Breu V, Ganten D, Haller H, Luft FC. NF-kappaB inhibitionameliorates angiotensin II-induced inflammatory damage in rats. Hyper-tension. 2000;35:193–201.
27. Regan J, Capolino A, Cirillo PF, Gilmore T, Graham AG, Hickey E, KroeRR, Madwed J, Moriak M, Nelson R, Pargellis CA, Swinamer A, Tor-cellini C, Tsang M, Moss N. Structure-activity relationships of thep38alpha MAP kinase inhibitor 1-(5-tert-butyl-2-p-tolyl-2H-pyrazol-3-yl)-3-[4-(2-morpholin-4-yl-ethoxy)n aph- thalen-1-yl]urea (BIRB 796).J Med Chem. 2003;46:4676–4686.
8 Hypertension March 2007

28. Wellner M, Dechend R, Park JK, Shagdarsuren E, Al-Saadi N, Kirsch T,Gratze P, Schneider W, Meiners S, Fiebeler A, Haller H, Luft FC, MullerDN. Cardiac gene expression profile in rats with terminal heart failure andcachexia. Physiol Genomics. 2005;20:256–267.
29. Beltrami AP, Urbanek K, Kajstura J, Yan SM, Finato N, Bussani R,Nadal-Ginard B, Silvestri F, Leri A, Beltrami CA, Anversa P. Evidencethat human cardiac myocytes divide after myocardial infarction. N EnglJ Med. 2001;344:1750–1757.
30. Engel FB, Schebesta M, Duong MT, Lu G, Ren S, Madwed JB, Jiang H,Wang Y, Keating MT. p38 MAP kinase inhibition enables proliferation ofadult mammalian cardiomyocytes. Genes Dev. 2005;19:1175–1187.
31. Callera GE, Touyz RM, Tostes RC, Yogi A, He Y, Malkinson S, SchiffrinEL. Aldosterone activates vascular p38MAP kinase and NADPH oxidasevia c-Src. Hypertension. 2005;45:773–779.
32. Chan SH, Hsu KS, Huang CC, Wang LL, Ou CC, Chan JY. NADPHoxidase-derived superoxide anion mediates angiotensin II-inducedpressor effect via activation of p38 mitogen-activated protein kinase inthe rostral ventrolateral medulla. Circ Res. 2005;97:772–780.
33. Schieven GL. The biology of p38 kinase: a central role in inflammation.Curr Top Med Chem. 2005;5:921–928.
34. Touyz RM, Yao G, Quinn MT, Pagano PJ, Schiffrin EL. p47phox asso-ciates with the cytoskeleton through cortactin in human vascular smoothmuscle cells: role in NAD(P)H oxidase regulation by angiotensin II.Arterioscler Thromb Vasc Biol. 2005;25:512–518.
35. Branger J, van den Blink B, Weijer S, Madwed J, Bos CL, Gupta A, YongCL, Polmar SH, Olszyna DP, Hack CE, van Deventer SJ, PeppelenboschMP, van der Poll T. Anti-inflammatory effects of a p38 mitogen-activatedprotein kinase inhibitor during human endotoxemia. J Immunol. 2002;168:4070–4077.
36. Shao J, Nangaku M, Miyata T, Inagi R, Yamada K, Kurokawa K, FujitaT. Imbalance of T-cell subsets in angiotensin II-infused hypertensive ratswith kidney injury. Hypertension. 2003;42:31–38.
37. Rincon M, Enslen H, Raingeaud J, Recht M, Zapton T, Su MS, Penix LA,Davis RJ, Flavell RA. Interferon-gamma expression by Th1 effector Tcells mediated by the p38 MAP kinase signaling pathway. EMBO J.1998;17:2817–2829.
38. Wijffels MC, Kirchhof CJ, Dorland R, Allessie MA. Atrial fibrillationbegets atrial fibrillation. A study in awake chronically instrumented goats.Circulation. 1995;92:1954–1968.
39. Verheule S, Sato T, Everett Tt, Engle SK, Otten D, Rubart-von derLohe M, Nakajima HO, Nakajima H, Field LJ, Olgin JE. Increasedvulnerability to atrial fibrillation in transgenic mice with selectiveatrial fibrosis caused by overexpression of TGF-beta1. Circ Res.2004;94:1458 –1465.
40. Shiroshita-Takeshita A, Brundel BJ, Lavoie J, Nattel S. Prednisoneprevents atrial fibrillation promotion by atrial tachycardia remodeling indogs. Cardiovasc Res. 2006;69:865–875.
Park et al Angiotensin II and p38 9
Copyright © 2022 FDOKUMEN




















