
Novel Inhibitors of Alkaline Phosphatase Suppress Vascular Smooth Muscle Cell Calcification
11
Novel Inhibitors of Alkaline Phosphatase Suppress Vascular Smooth Muscle Cell Calcification Sonoko Narisawa, 1 Dympna Harmey, 1 Manisha C Yadav, 1 W Charles O’Neill, 2 Marc F Hoylaerts, 3 and Jose Luis Millán 1 ABSTRACT: We report three novel inhibitors of the physiological pyrophosphatase activity of alkaline phosphatase and show that these compounds are capable of reducing calcification in two models of vascular calcification (i.e., they suppress in vitro calcification by cultured Enpp1 -/- VSMCs and they inhibit the increased pyrophosphatase activity in a rat aortic model). Introduction: Genetic ablation of tissue-nonspecific alkaline phosphatase (TNALP) leads to accumulation of the calcification inhibitor inorganic pyrophosphate (PP i ). TNALP deficiency ameliorates the hypermineral- ization phenotype in Enpp1 -/- and ank/ank mice, two models of osteoarthritis and soft tissue calcification. We surmised that the pharmacological inhibition of TNALP pyrophosphatase activity could be used to prevent/ suppress vascular calcification. Materials and Methods: Comprehensive chemical libraries were screened to identify novel drug-like com- pounds that could inhibit TNALP pyrophosphatase function at physiological pH. We used these novel com- pounds to block calcification by cultured vascular smooth muscle cells (VSMCs) and to inhibit the upregulated pyrophosphatase activity in a rat aortic calcification model. Results: Using VSMC cultures, we determined that Enpp1 -/- and ank/ank VSMCs express higher TNALP levels and enhanced in vitro calcification compared with wildtype cells. By high-throughput screening, three novel compounds, 5361418, 5923412, and 5804079, were identified that inhibit TNALP pyrophosphatase function through an uncompetitive mechanism, with high affinity and specificity when measured at both pH 9.8 and 7.5. These compounds were shown to reduce the calcification by Enpp1 -/- VSMCs. Furthermore, using an ex vivo rat whole aorta PP i hydrolysis assay, we showed that pyrophosphatase activity was inhibited by all three lead compounds, with compound 5804079 being the most potent at pH 7.5. Conclusions: We conclude that TNALP is a druggable target for the treatment and/or prevention of ectopic calcification. The lead compounds identified in this study will serve as scaffolds for medicinal chemistry efforts to develop drugs for the treatment of soft tissue calcification. J Bone Miner Res 2007;22:1700–1710. Published online on July 16, 2007; doi: 10.1359/JBMR.070714 Key words: vascular calcification, computer modeling, docking, chemical library screening, pyrophosphatase, druggable target INTRODUCTION V ASCULAR CALCIFICATION REFERS to the deposition of hydroxyapatite in cardiovascular tissues such as arter- ies and heart valves and is a significant risk factor in the pathogenesis of cardiovascular disease, being associated with myocardial infarction and coronary death. (1) Vascular disease is also common in diabetes, obesity, aging, and renal failure, where it is responsible for much of the morbidity and mortality in end-stage renal disease. (2) Two types of calcification are recognized: the first occurs primarily in the form of intimal calcification, usually associated with athero- sclerosis, and the second, also known as Mönckeberg’s scle- rosis, is defined as calcification restricted to the arterial media. (3) A key regulator of mineralization, both in bone and vessels, is extracellular pyrophosphate (ePP i ). (4) This compound is a potent inhibitor of hydroxyapatite formation at concentrations normally found in plasma (5) and prevents calcification of rat aortas in culture. (6) Conversely, ePP i depletion promotes spontaneous arterial calcifica- tion. (7) Mice lacking ecto-nucleotide pyrophosphatase/ phosphodiesterases-1 (NPP1, a.k.a PC-1), a major genera- tor of ePP i , spontaneously develop articular cartilage, perispinal, and medial aortic calcification at a young age. (8) These NPP1 knockout mice (Enpp1 -/- ) share phenotypic features with a human disease, idiopathic infantile arterial calcification. (9,10) Interestingly, another mouse model of de- pressed ePP i levels, this time caused by defective transport The authors state that they have no conflicts of interest. 1 The Burnham Institute, La Jolla, California, USA; 2 Renal Division, Emory University, Atlanta, Georgia, USA; 3 Center for Molecular and Vascular Biology, University of Leuven, Leuven, Belgium. JOURNAL OF BONE AND MINERAL RESEARCH Volume 22, Number 11, 2007 Published online on July 16, 2007; doi: 10.1359/JBMR.070714 © 2007 American Society for Bone and Mineral Research 1700
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Novel Inhibitors of Alkaline Phosphatase Suppress Vascular Smooth Muscle Cell Calcification

Novel Inhibitors of Alkaline Phosphatase Suppress Vascular SmoothMuscle Cell Calcification
Sonoko Narisawa,1 Dympna Harmey,1 Manisha C Yadav,1 W Charles O’Neill,2 Marc F Hoylaerts,3 andJose Luis Millán1
ABSTRACT: We report three novel inhibitors of the physiological pyrophosphatase activity of alkalinephosphatase and show that these compounds are capable of reducing calcification in two models of vascularcalcification (i.e., they suppress in vitro calcification by cultured Enpp1−/− VSMCs and they inhibit theincreased pyrophosphatase activity in a rat aortic model).
Introduction: Genetic ablation of tissue-nonspecific alkaline phosphatase (TNALP) leads to accumulation ofthe calcification inhibitor inorganic pyrophosphate (PPi). TNALP deficiency ameliorates the hypermineral-ization phenotype in Enpp1−/− and ank/ank mice, two models of osteoarthritis and soft tissue calcification. Wesurmised that the pharmacological inhibition of TNALP pyrophosphatase activity could be used to prevent/suppress vascular calcification.Materials and Methods: Comprehensive chemical libraries were screened to identify novel drug-like com-pounds that could inhibit TNALP pyrophosphatase function at physiological pH. We used these novel com-pounds to block calcification by cultured vascular smooth muscle cells (VSMCs) and to inhibit the upregulatedpyrophosphatase activity in a rat aortic calcification model.Results: Using VSMC cultures, we determined that Enpp1−/− and ank/ank VSMCs express higher TNALPlevels and enhanced in vitro calcification compared with wildtype cells. By high-throughput screening, threenovel compounds, 5361418, 5923412, and 5804079, were identified that inhibit TNALP pyrophosphatasefunction through an uncompetitive mechanism, with high affinity and specificity when measured at both pH9.8 and 7.5. These compounds were shown to reduce the calcification by Enpp1−/− VSMCs. Furthermore, usingan ex vivo rat whole aorta PPi hydrolysis assay, we showed that pyrophosphatase activity was inhibited by allthree lead compounds, with compound 5804079 being the most potent at pH 7.5.Conclusions: We conclude that TNALP is a druggable target for the treatment and/or prevention of ectopiccalcification. The lead compounds identified in this study will serve as scaffolds for medicinal chemistry effortsto develop drugs for the treatment of soft tissue calcification.J Bone Miner Res 2007;22:1700–1710. Published online on July 16, 2007; doi: 10.1359/JBMR.070714
Key words: vascular calcification, computer modeling, docking, chemical library screening, pyrophosphatase,druggable target
INTRODUCTION
VASCULAR CALCIFICATION REFERS to the deposition ofhydroxyapatite in cardiovascular tissues such as arter-
ies and heart valves and is a significant risk factor in thepathogenesis of cardiovascular disease, being associatedwith myocardial infarction and coronary death.(1) Vasculardisease is also common in diabetes, obesity, aging, and renalfailure, where it is responsible for much of the morbidityand mortality in end-stage renal disease.(2) Two types ofcalcification are recognized: the first occurs primarily in theform of intimal calcification, usually associated with athero-sclerosis, and the second, also known as Mönckeberg’s scle-
rosis, is defined as calcification restricted to the arterialmedia.(3) A key regulator of mineralization, both in boneand vessels, is extracellular pyrophosphate (ePPi).(4) Thiscompound is a potent inhibitor of hydroxyapatite formationat concentrations normally found in plasma(5) and preventscalcification of rat aortas in culture.(6) Conversely,ePPi depletion promotes spontaneous arterial calcifica-tion.(7) Mice lacking ecto-nucleotide pyrophosphatase/phosphodiesterases-1 (NPP1, a.k.a PC-1), a major genera-tor of ePPi, spontaneously develop articular cartilage,perispinal, and medial aortic calcification at a young age.(8)
These NPP1 knockout mice (Enpp1−/−) share phenotypicfeatures with a human disease, idiopathic infantile arterialcalcification.(9,10) Interestingly, another mouse model of de-pressed ePPi levels, this time caused by defective transportThe authors state that they have no conflicts of interest.
1The Burnham Institute, La Jolla, California, USA; 2Renal Division, Emory University, Atlanta, Georgia, USA; 3Center for Molecularand Vascular Biology, University of Leuven, Leuven, Belgium.
JOURNAL OF BONE AND MINERAL RESEARCHVolume 22, Number 11, 2007Published online on July 16, 2007; doi: 10.1359/JBMR.070714© 2007 American Society for Bone and Mineral Research
1700
JO702077 1700 1710 November

function of the transmembrane protein ANK (ank/ank mu-tant mice), also develops soft tissue calcification, includingvascular calcification.(7,11,12)
Alkaline phosphatases (ALPs; E.C.3.1.3.1) are dimericenzymes present in most organisms.(13) They catalyze thehydrolysis of phosphomonoesters with release of inorganicphosphate (Pi) and alcohol. In humans, three of the fourisozymes are tissue-specific, i.e., the intestinal (IALP), pla-cental (PLALP), and germ cell (GCLP) APs, whereas thefourth ALP is tissue-nonspecific (TNALP) and is expressedin bone, liver, and kidney. Recent studies have providedcompelling proof that a major role for TNALP in bonetissue is to hydrolyze ePPi to avoid accumulation of thismineralization inhibitor, thus ensuring normal bone miner-alization.(14–16) Normalization of ePPi levels in NPP1 nulland ANK-deficient mice improves their calcification abnor-malities.(14,15) Crossbreeding either the Enpp1−/− or theank/ank mice to mice deficient in TNALP (Akp2−/−) micenormalizes ePPi levels and induces a secondary upregula-tion of osteopontin (OPN) levels, another calcification in-hibitor.(16,17) Importantly, these studies have indicated thatTNALP may be a useful therapeutic target for the treat-ment of diseases such as ankylosis and osteoarthritis, butalso arterial calcification. The presence of TNALP-enriched matrix vesicles (MVs) in human atheroscleroticlesions suggests an active role in the promotion of the ac-companying vascular calcification.(18–22) Increased expres-sion of TNALP accelerates calcification by bovine vascularsmooth muscle cells (VSMCs)(23) and macrophages mayinduce a calcifying phenotype in human VSMCs by activat-ing TNALP in the presence of IFN� and 1,25(OH)2D3.(24)
Calcification of rat aorta in culture and of human valveinterstitial cells has been shown to be dependent onTNALP activity.(6,25) Thus, there is ample evidence war-ranting exploration of the therapeutic potential of TNALPinhibition at sites of arterial calcification to increase localconcentrations of ePPi, which is expected to antagonize thedeposition of hydroxyapatite. For these reasons, in thisstudy, we have undertaken the screening of large, compre-hensive small, drug-like, molecule libraries to identify andcharacterize novel potent TNALP inhibitors that might beuseful for the treatment of vascular calcification. We pres-ently describe three new compounds, one of which acts as apotent uncompetitive TNALP inhibitor, capable of inhibit-ing TNALP’s pyrophosphatase activity at physiological pH,thereby suppressing calcification in primary cultures ofVSMCs as well as in aortic explants.
MATERIALS AND METHODS
Reagents
All routine chemicals were of analytical grade fromSigma (St Louis, MO, USA), unless otherwise indicated.
Expression and preparation of test enzymes
Expression plasmids containing a secreted epitope-tagged TNALP, PLALP, and IALP were transfected intoCOS-1 cells for transient expression using a standard elec-troporation method. Medium was replaced to Opti-MEM
24 h later, and the serum free media containing secretedproteins were collected 60 h after electroporation. Condi-tioned medium was dialyzed against TBS containing 1 mMMgCl2 and 20 �M ZnCl2 (to remove phosphate) and fil-tered with a 2-mm cellulose acetate filter. The TNALPstock solution was obtained from the dialyzed conditionedmedium. The PLALP and IALP solutions were producedin the same way. To compare the percentage of inhibition,dilutions of PLALP- and IALP-conditioned media wereadjusted to obtain the same value of activity as TNALP-conditioned medium without inhibitor.
High-throughput screening
The TNALP stock solution was diluted 120-fold, and 12�l of diluted TNALP solution was dispensed into 96-wellmicrotiter plates with half area bottom (Costar, Corning,NY, USA) by an auto dispenser (Matrix, Hudson, NH,USA). The maximum volume in each well was 190 �l (welldepth, 10.54 mm; well bottom area, 0.1586 cm2). The librarycompounds were dissolved in 100% dimethylsulfoxide(DMSO) in the master plates, and our working plates con-tained 10% DMSO, giving 1% DMSO in the final enzy-matic reaction. Whereas 10% DMSO inhibits TNALP byabout 30%, the final 1% DMSO concentration does notaffect TNALP activity. A robotic liquid handler, BiomekFX (Beckman Coulter, Fullerton, CA, USA) dispensed 2.5�l of each compound (dissolved in 10% DMSO) from thelibrary plates. Plates were incubated at room temperaturefor at least 1 h to allow TNALP to interact with each com-pound before addition of 10.5 �l substrate solution (1.19mM pNPP). After 30-min incubation, A405 nm was mea-sured with a microtiter plate reader, Analyst HT (Molecu-lar Devices, Sunnyvale, CA, USA). Both the enzyme(TNALP) and substrate (pNPP) solution were made in di-ethanolamine (DEA) buffers; the final reaction consists of1 M DEA-HCl buffer, pH 9.8, containing 1 mM MgCl2 and20 �M ZnCl2. The concentration of TNALP and pNPP(final 0.5 mM) were adjusted to obtain A405 nm ∼0.4, whilemaintaining good sensitivity to the known inhibitors le-vamisole and phosphate, used as positive controls. Km ob-tained with a 1/120 dilution of TNALP and a fixed incuba-tion period of 30 min was 0.58 ± 0.081 mM.
Enzyme kinetic experiments
To determine the inhibition selectivity for inhibitor can-didates, human TNALP, PLALP, or IALP was added tomicrotiter plates followed by addition of the substratepNPP (0.5 mM), and activity was measured in 1 M DEA-HCl buffer, pH 9.8, or in 1 M Tris-HCl buffer, pH 7.5,(26)
containing 1 mM MgCl2 and 20 �M ZnCl2, in the presenceof potential inhibitors (0–30 �M). TNALP, PLALP, andIALP activities were adjusted to an approximate �A405 nm,equivalent to 1, measured after 30 min. Residual ALP ac-tivity in the presence of inhibitors was expressed as per-centage of the control activity. To study the mechanism ofinhibition, double reciprocal plots of enzyme activity (ex-pressed as mA405 nm min−1) versus substrate concentrationwere constructed in the presence of various concentrationsof added inhibitors (0–30 �M). The y-axis intercepts of the
TNALP INHIBITION AND VASCULAR CALCIFICATION 1701

1/v versus 1/[S] plots were plotted versus [I] to graphicallyextract Ki values as the x-intercept in this plot. The numeri-cal values from y- and x-intercepts were derived using linearregression analysis, using software Prism 3.02 (GraphPadSoftware). These analyses were performed, using pNPP asa substrate in 1 M DEA-HCl buffer, pH 9.8, as well as in 1M Tris-HCl buffer, pH 7.5, to determine Ki at optimal andphysiological pH, respectively. The TNALP concentrationin those experiments was chosen to generate a �A405 nm,equivalent to 0.3, measured after 1 h, to allow a linear in-crease of A405 nm with time, for the lowest substrate con-centration tested ([pNPP] � 100 �M). Inhibitors were fur-ther tested and sorted based on their kinetic properties atpH 7.4 using PPi, the relevant natural substrate ofTNALP.(27) In this part of the study, pyrophosphate so-dium salt (99% ACS reagent; Sigma-Aldrich, St Louis, MO,USA) was used as a substrate. Amounts of released phos-phate were measured using the Biomol Green Reagent(Biomol Research Laboratories, Plymouth Meeting, PA,USA). Finally, to document the potency of selected inhibi-tors in physiological media, TNALP inhibition by com-pound 5804079 (0–30 �M) was studied at pH 7.4, duringcatalysis of 0.1 mM pNPP, in the presence of increasingconcentrations of Na2HPO4 (0–10 mM) and pyrophosphate(0–40 mM).
Computer docking
Compound docking was performed using the Flexx pro-gram, part of the Sybyl package from Trios, as before.(28)
Formal charges were used for protein and compound at-oms. Heteroatoms (phosphate, zinc, and magnesium) wereconsidered as part of the pocket while docking.
Maintenance of Enpp1−/− and ank/ank mice
The generation and characterization of Enpp1−/− micehas been described earlier.(29) Mice carrying the ank muta-tion were purchased from The Jackson Laboratory. To de-termine genotypes, genomic DNA was isolated from tailsand analyzed using PCR protocols.(12)
Tissue preparation and morphological analysis
Whole mount skeletal preparations were prepared by re-moval of skin and viscera of mice followed by a 1-wk im-mersion in 100% ethanol, followed by 100% acetone.Samples were transferred to a 100% ethanol solution con-taining 0.01% Alizarin Red S, 0.015% Alcian Blue 8GX,and 0.5% acetic acid for 3 wk. Samples were destained with1% (vol/vol) KOH/50% glycerol solution. Cleared sampleswere stored in 100% glycerol.(12)
Isolation and culture of primary calvarialosteoblasts and VSMCs
Mouse calvarial cells were isolated from 3-day-old WTmice through sequential collagenase digestion, as previ-ously described.(12,15) Vascular smooth muscle cells(VSMCs) were isolated from explants using a collagenasedigestion method, and the smooth muscle phenotype wasconfirmed by RT-PCR analysis for smooth muscle �-actin.One mouse aorta provided on average 5 × 105 cells. These
cells were cultured (in triplicate) at a density of 3 × 104
cells/cm2 using �-MEM supplemented with �-glycerophos-phate (10 mM) and 50 �g/ml ascorbic acid for 3 wk. Mediawas renewed every third day and inhibitors were freshlyprepared and added each time. To quantify calcium depos-ited in these cultures, either the o-cresolphthalein complex-one method(16) or the standard Alizarin Red method(15)
was used.
Reverse transcription and quantitativereal-time PCR
Total RNA was extracted from the osteoblast and VSMCpellets and 2 �g of RNA used for reverse transcriptionusing the Superscript kit (Invitrogen, Carlsbad, CA, USA).OPN mRNA was quantified by real-time PCR using dual-labeled hydrolysis probes (FAM-TAMRA). The sequencesfor mouse OPN and 18S primers and probes were asfollows: OPN forward 5�-TGAGGTCAAAGTCTAG-GAGTTTCC-3�, OPN reverse 5�-TTAGACTCACCG-CTCTTCATGTG-3�, OPN Probe 5�-TTCTGATGAA-CAGTATCCTG-3�, 18S forward 5�-CGGCTACCA-CATCCAAGGAA-3�, 18S reverse 5�-GCTGGAATTAC-CGCGGCT-3� and 18S Probe 5�-TGCTGGCACCA-GACTTGCCCTC-3�. For quantitative real-time PCR, 2 �lof the cDNA and the reaction mixture used 12.5 �l of plati-num qPCR UDG supermix (Invitrogen). The reaction wasperformed in a 96-well plate on a Stratagene MX2000P realtime machine (Stratagene, La Jolla, CA, USA). The reac-tion was run at an initial temperature of 95°C for 10 minand at 95°C for 30 s, 55°C for 1 min, followed by 72°C for30 s for 45 cycles. Ct values were determined by theMX2000P Software according to the optimization of thebaseline. For computing the relative amount of OPN in thesamples, the average Ct for 18S was subtracted from that ofOPN to give changes in Cts (�Ct). Relative units (log2
�Cts) were calculated and used as a measure of OPN ex-pression.
PPi hydrolysis by whole aortas ex vivo
Sprague-Dawley rats were killed, and aortas perfusedwith Hanks salt solution to remove blood. The aortas wereremoved and, after the adventitia was dissected away, werecut into rings ∼2 mm in length. Four rings were placed in 1ml of DMEM without serum containing the compounds tobe tested. After 90 min at 37°C, sodium PPi (final concen-tration, 1 �M) and [32P]PPi (final concentration, 1 �Curie/ml) were added, and six samples were removed over 4 h.(6)
Pi was separated from PPi by adding 800 �l of 0.028 Mammonium molybdate in 0.75 M H2SO4 to the samples andextracting with 1600 �l of isobutanol and petroleum ether(4:1). 32P was counted in the organic phase by Cerenkovradiation. Hydrolysis of PPi was linear over 4 h, and the ratewas determined by linear regression.
RESULTS
Aortic calcification in Enpp1−/− and ank/ank miceis associated with elevations in TNALP expression
Given the coordinated function of NPP1 and ANK inestablishing extracellular PPi concentrations and the simi-
NARISAWA ET AL.1702
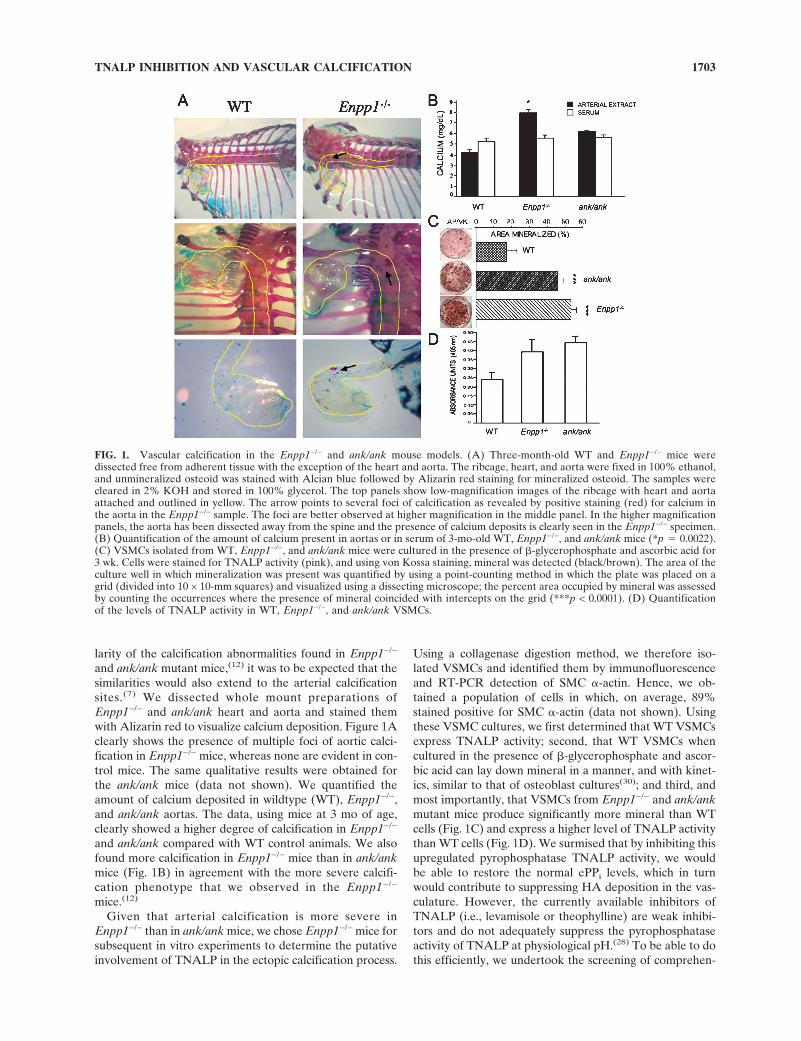
larity of the calcification abnormalities found in Enpp1−/−
and ank/ank mutant mice,(12) it was to be expected that thesimilarities would also extend to the arterial calcificationsites.(7) We dissected whole mount preparations ofEnpp1−/− and ank/ank heart and aorta and stained themwith Alizarin red to visualize calcium deposition. Figure 1Aclearly shows the presence of multiple foci of aortic calci-fication in Enpp1−/− mice, whereas none are evident in con-trol mice. The same qualitative results were obtained forthe ank/ank mice (data not shown). We quantified theamount of calcium deposited in wildtype (WT), Enpp1−/−,and ank/ank aortas. The data, using mice at 3 mo of age,clearly showed a higher degree of calcification in Enpp1−/−
and ank/ank compared with WT control animals. We alsofound more calcification in Enpp1−/− mice than in ank/ankmice (Fig. 1B) in agreement with the more severe calcifi-cation phenotype that we observed in the Enpp1−/−
mice.(12)
Given that arterial calcification is more severe inEnpp1−/− than in ank/ank mice, we chose Enpp1−/− mice forsubsequent in vitro experiments to determine the putativeinvolvement of TNALP in the ectopic calcification process.
Using a collagenase digestion method, we therefore iso-lated VSMCs and identified them by immunofluorescenceand RT-PCR detection of SMC �-actin. Hence, we ob-tained a population of cells in which, on average, 89%stained positive for SMC �-actin (data not shown). Usingthese VSMC cultures, we first determined that WT VSMCsexpress TNALP activity; second, that WT VSMCs whencultured in the presence of �-glycerophosphate and ascor-bic acid can lay down mineral in a manner, and with kinet-ics, similar to that of osteoblast cultures(30); and third, andmost importantly, that VSMCs from Enpp1−/− and ank/ankmutant mice produce significantly more mineral than WTcells (Fig. 1C) and express a higher level of TNALP activitythan WT cells (Fig. 1D). We surmised that by inhibiting thisupregulated pyrophosphatase TNALP activity, we wouldbe able to restore the normal ePPi levels, which in turnwould contribute to suppressing HA deposition in the vas-culature. However, the currently available inhibitors ofTNALP (i.e., levamisole or theophylline) are weak inhibi-tors and do not adequately suppress the pyrophosphataseactivity of TNALP at physiological pH.(28) To be able to dothis efficiently, we undertook the screening of comprehen-
FIG. 1. Vascular calcification in the Enpp1−/− and ank/ank mouse models. (A) Three-month-old WT and Enpp1−/− mice weredissected free from adherent tissue with the exception of the heart and aorta. The ribcage, heart, and aorta were fixed in 100% ethanol,and unmineralized osteoid was stained with Alcian blue followed by Alizarin red staining for mineralized osteoid. The samples werecleared in 2% KOH and stored in 100% glycerol. The top panels show low-magnification images of the ribcage with heart and aortaattached and outlined in yellow. The arrow points to several foci of calcification as revealed by positive staining (red) for calcium inthe aorta in the Enpp1−/− sample. The foci are better observed at higher magnification in the middle panel. In the higher magnificationpanels, the aorta has been dissected away from the spine and the presence of calcium deposits is clearly seen in the Enpp1−/− specimen.(B) Quantification of the amount of calcium present in aortas or in serum of 3-mo-old WT, Enpp1−/−, and ank/ank mice (*p � 0.0022).(C) VSMCs isolated from WT, Enpp1−/−, and ank/ank mice were cultured in the presence of �-glycerophosphate and ascorbic acid for3 wk. Cells were stained for TNALP activity (pink), and using von Kossa staining, mineral was detected (black/brown). The area of theculture well in which mineralization was present was quantified by using a point-counting method in which the plate was placed on agrid (divided into 10 × 10-mm squares) and visualized using a dissecting microscope; the percent area occupied by mineral was assessedby counting the occurrences where the presence of mineral coincided with intercepts on the grid (***p < 0.0001). (D) Quantificationof the levels of TNALP activity in WT, Enpp1−/−, and ank/ank VSMCs.
TNALP INHIBITION AND VASCULAR CALCIFICATION 1703
Fig 1 live 4/C

sive chemical libraries to identify and characterize novellead compounds that could enable the development of po-tent drug-like inhibitor of TNALP’s physiological pyro-phosphatase function.
High-throughput screening
To identify such novel small molecule inhibitors ofTNALP activity, we optimized an assay to screen chemicallibraries containing 53,280 compounds. These included (1)the Spectrum Collection (from MicroSource, www.msdis-covery.com), containing 2000 compounds (25 plates, 80compounds/plate); about one half of the collection containsknown bioactive agents, permitting the evaluation of hun-dreds of marketed drugs and biochemical standards;the other half of the collection includes pure natural prod-ucts and their derivatives; (2) the LOPAC1280 Collection(www.sigmaaldrich.com/Area_of_Interest/Chemistry/D r u g _ D i s c o v e r y / A s s a y _ D e v _ a n d _ S c r e e n i n g /C o m p o u n d _ L i b r a r i e s / V a l i d a t i o n _ L i b r a r i e s /Lopac1280home.html), containing 1280 pharmacologicallyactive compounds; this library contains effector moleculesfor all major target classes and all of the compounds in thiscollection are available for powder resupply from SIGMA;and (3) the Chembridge DIVERSet Collection (fromChembridge, www.chembridge.com) that contains 50,000diverse, predesigned compounds (625 plates, 80 compoundsper plate); this collection was selected by a “rational” ap-proach based on 3D pharmacophore analysis to cover thebroadest part of biologically relevant pharmacophore di-versity space.
Screening the chemical libraries was based on a 96-wellplate assay using 0.5 mM pNPP as substrate. We used 30�M of the uncompetitive inhibitor levamisole and 300 �Mof the competitive inhibitor Pi in each individual assay plateas positive controls. The concentration of the chemical li-brary compounds in the reaction mixture was ∼10 �M.Even though some of the compounds in the libraries absorbat 405 nm, monitoring p-nitrophenol production proved tobe more sensitive in our hands than other methods, detect-ing liberated Pi through alternative colorimetric assays,such as the Biomol green reagent that absorbs at 620 nm.Instead, any compound with inherent A405 nm absorptionwas retested manually in the laboratory, at various concen-trations, to complement the single point assay of the roboticstation. After each daily run of assay, we manually tested allthose compounds that had shown >20% inhibition. A totalof 11 hits with reproducible inhibition were retested, andfinally, four compounds were identified as effectiveTNALP inhibitors: one was levamisole, a well-known albeitweak ALP inhibitor, contained within the 2000 SpectrumCollection of known drugs and presently used as a positivecontrol during our screening. The other three representednovel structures as shown in Fig. 2. The physicochemicalproperty of these compounds is summarized in Table 1. Allthree compounds conform to Lipinski’s rule of 5 (http://www.acdlabs.com/products/phys_chem_lab/logp/ruleof5.html): they have a molecular weight of <500, have<5 H-bond donors; have <5 H-bond acceptors; have <10
rotational bonds, and have an octanol/water repartition co-efficient (LogP) � 5. Their nitrogen content ranges from 3to 7 N atoms per inhibitor (Fig. 2).
Kinetic properties of the inhibitors
None of the three identified novel TNALP inhibitorsseemed to inhibit, either at pH 9.8 or at physiological pH,other relevant human ALPs, such as PLALP or IALP,which share 50% and 52% sequence identity with TNALP,respectively. Figure 3 shows the inhibition of TNALP,PLALP, and IALP for increasing concentrations (0–30�M) of the inhibitors at physiological pH. Furthermore,none of the inhibitors had any effect on PHOSPHO1, anovel phosphatase proposed to be involved in the initiationof MV-mediated calcification.(31) The double reciprocalplots of 1/v versus 1/[S], for various inhibitor concentra-tions, showed parallel lines for all three inhibitors, indicat-ing that each TNALP inhibitor acts in an uncompetitivemanner, both at pH 9.8 and at physiological pH. Figure 4Ashows the Lineweaver-Burk plot of 1/v versus 1/[S] for com-pound 5804079 at pH 9.8 and pH 7.5. The Eadie-Hofsteeplot at pH 9.8 confirms that the mechanism is uncompeti-
FIG. 2. Chemical structure of the three lead compounds thatinhibit TNALP activity, identified through high-throughput li-brary screening.
TABLE 1. STRUCTURAL PROPERTIES OF PRESENTLY IDENTIFIED
NOVEL TNALP INHIBITORS
Compound ID5361418 5923412 5804079
Formula C16H13N3OS C12H14N4O2S C19H17N7SMolecular
weight 295.365 278.335 375.458LogP 3.18 0.26 5.34Rot. B 5 7 6Hdon 1 2 2Hacc 3 4 5
Chemical names: 1,2-diphenyl-2-(1H-1,2,4-triazol-5-ylthio)athanone(5361418), N-(2-phenoxyethyl)-2-(4H-1,2,4-triazol-3-ylthio)acetamide(5923412), 6-[(1-methyl-1H-imidazol-2-yl)thio]-N,N’-diphenyl-1,3,5-triazine-2,4-diamine (5804079).
LogP, octanol/water repartition coefficient; Rot. B, number of rotationalbonds; Hdon, number of hydrogen donors; Hacc, number of hydrogenacceptors.
NARISAWA ET AL.1704
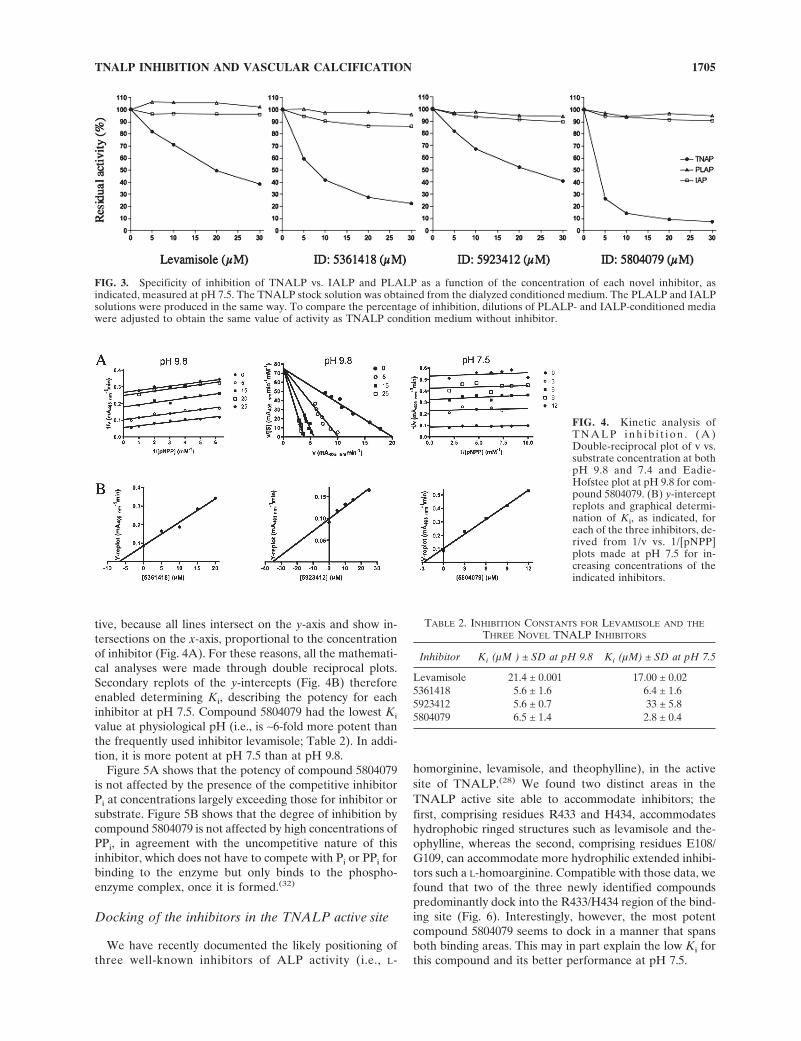
tive, because all lines intersect on the y-axis and show in-tersections on the x-axis, proportional to the concentrationof inhibitor (Fig. 4A). For these reasons, all the mathemati-cal analyses were made through double reciprocal plots.Secondary replots of the y-intercepts (Fig. 4B) thereforeenabled determining Ki, describing the potency for eachinhibitor at pH 7.5. Compound 5804079 had the lowest Ki
value at physiological pH (i.e., is ∼6-fold more potent thanthe frequently used inhibitor levamisole; Table 2). In addi-tion, it is more potent at pH 7.5 than at pH 9.8.
Figure 5A shows that the potency of compound 5804079is not affected by the presence of the competitive inhibitorPi at concentrations largely exceeding those for inhibitor orsubstrate. Figure 5B shows that the degree of inhibition bycompound 5804079 is not affected by high concentrations ofPPi, in agreement with the uncompetitive nature of thisinhibitor, which does not have to compete with Pi or PPi forbinding to the enzyme but only binds to the phospho-enzyme complex, once it is formed.(32)
Docking of the inhibitors in the TNALP active site
We have recently documented the likely positioning ofthree well-known inhibitors of ALP activity (i.e., L-
homorginine, levamisole, and theophylline), in the activesite of TNALP.(28) We found two distinct areas in theTNALP active site able to accommodate inhibitors; thefirst, comprising residues R433 and H434, accommodateshydrophobic ringed structures such as levamisole and the-ophylline, whereas the second, comprising residues E108/G109, can accommodate more hydrophilic extended inhibi-tors such a L-homoarginine. Compatible with those data, wefound that two of the three newly identified compoundspredominantly dock into the R433/H434 region of the bind-ing site (Fig. 6). Interestingly, however, the most potentcompound 5804079 seems to dock in a manner that spansboth binding areas. This may in part explain the low Ki forthis compound and its better performance at pH 7.5.
FIG. 3. Specificity of inhibition of TNALP vs. IALP and PLALP as a function of the concentration of each novel inhibitor, asindicated, measured at pH 7.5. The TNALP stock solution was obtained from the dialyzed conditioned medium. The PLALP and IALPsolutions were produced in the same way. To compare the percentage of inhibition, dilutions of PLALP- and IALP-conditioned mediawere adjusted to obtain the same value of activity as TNALP condition medium without inhibitor.
FIG. 4. Kinetic analysis ofT N A L P i n h i b i t i o n . ( A )Double-reciprocal plot of v vs.substrate concentration at bothpH 9.8 and 7.4 and Eadie-Hofstee plot at pH 9.8 for com-pound 5804079. (B) y-interceptreplots and graphical determi-nation of Ki, as indicated, foreach of the three inhibitors, de-rived from 1/v vs. 1/[pNPP]plots made at pH 7.5 for in-creasing concentrations of theindicated inhibitors.
TABLE 2. INHIBITION CONSTANTS FOR LEVAMISOLE AND THE
THREE NOVEL TNALP INHIBITORS
Inhibitor Ki (µM ) ± SD at pH 9.8 Ki (µM) ± SD at pH 7.5
Levamisole 21.4 ± 0.001 17.00 ± 0.025361418 5.6 ± 1.6 6.4 ± 1.65923412 5.6 ± 0.7 33 ± 5.85804079 6.5 ± 1.4 2.8 ± 0.4
TNALP INHIBITION AND VASCULAR CALCIFICATION 1705

TNALP inhibition abrogates calcification andpyrophosphatase activity of VSMCs
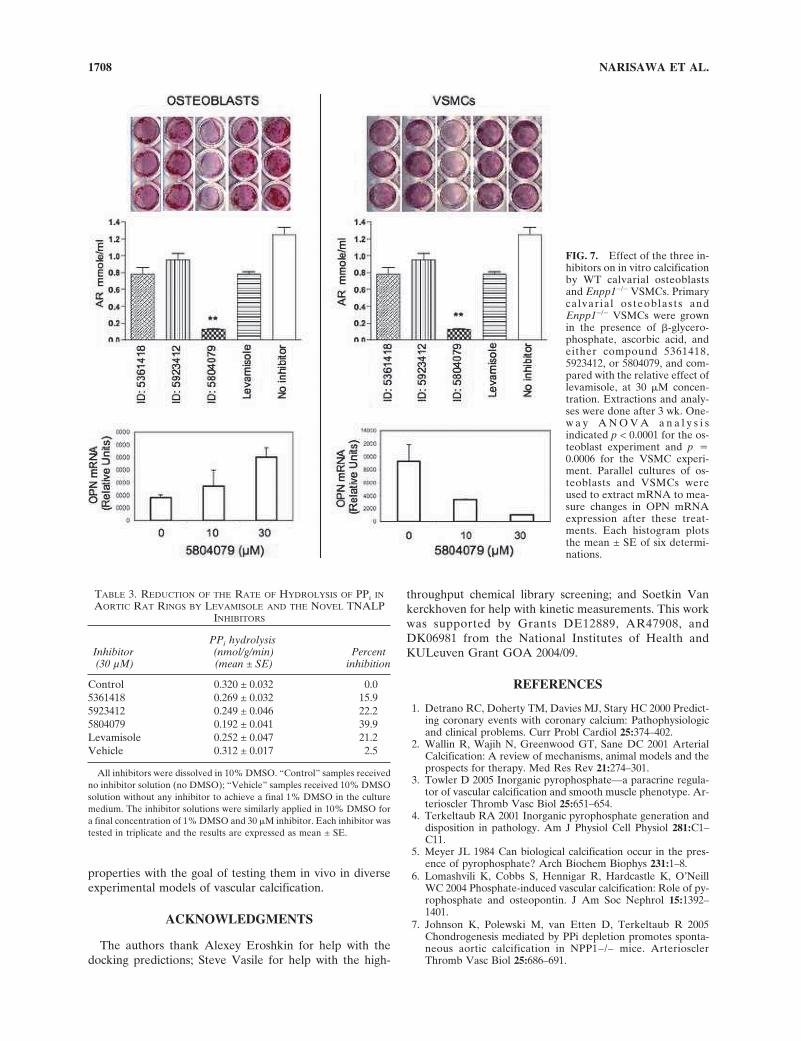
To validate the inhibitory potential of all three inhibitorson in vitro calcification, we tested the ability of all threenovel compounds, using levamisole as control, to inhibitTNALP activity in primary calvarial osteoblast cultures andin Enpp1−/− VSMCs. As shown in Fig. 7, all four com-pounds inhibited mineralization to some degree in bothculture systems, with compound 5804079 being the mosteffective in both in vitro assays. As expected,(12,17) the in-hibitor treatment induced OPN mRNA expression in pri-mary calvarial osteoblast cultures but had the opposite ef-fect on OPN expression in Enpp1−/− VSMC cultures.
To measure the degree of pyrophosphatase inhibition bythe new TNALP inhibitors, we used an ex vivo organ cul-ture system in whole aortas. For this analysis, rat ratherthan mouse aortas were selected, because they are largerand easier to dissect. This analysis also showed that com-pound 5804079 was the most effective in suppressing en-dogenous pyrophosphatase activity at the site of vascularcalcification (Table 3) at the maximal concentration of 30�M (chosen for all these highly aromatic inhibitors to avoidsolubility problems), i.e., 40% inhibition. Given that ap-proximately one half of the pyrophosphatase activity found
in aortic tissue is attributable to TNALP (WC O’Neill, un-published data), this ex vivo assay indicates that compound5804079 is able to pharmacologically ablate ∼80% ofTNALP’s pyrophosphatase activity in the aortic rings.
DISCUSSION
According to the World Health Organization (WHO) anestimated 17 million people die every year of cardiovasculardiseases, particularly heart attacks and strokes. Vascularcalcification is a major cause of cardiovascular morbidityand mortality. It is commonly found in atherosclerotic le-sions and has been associated with increased accumulationof macrophages in the chronically inflamed atherogenicvessel wall,(33) but also with enhanced vascular TNALPactivity. At the cellular level, calcifying vascular cells havethe potential to undergo osteoblastic differentiation andmineralization,(34–36) and it has been recently shown thatthe adult artery wall also contains mesenchymal progenitorcells with myogenic and chondrogenic potential.(37) Matrixvesicles (MVs), the membrane limited chondroblast- andosteoblast-derived structures in which the process of min-eralization is initiated,(38,39) have also been documented incalcified atherosclerotic lesions.(22,40,41)
These findings document that murine VSMCs produceTNALP in culture in sufficiently large amounts to play arole in the hydrolysis of PPi, which acts as an inhibitor ofmineralization. Accordingly, the elevated levels of TNALPin Enpp1−/− and ank/ank VSMCs, compounded with theinherent deficient production/transport of ePPi in theseNPP1- and ANK-deficient cells, respectively, provide thebasis to explain excessive arterial calcification in these ani-mal models. We tested whether inhibition of vascularTNALP through potent new inhibitors can raise PPi con-centrations to levels capable of blocking ectopic vascularcalcification in these genetic models of medial calcification.Whereas a number of ALP inhibitors have been describedin the literature, none of them are entirely specific forTNALP, and some of the best, such as levamisole and the-ophylline, have effects in vivo that are unrelated to ALPfunction. Levamisole is an FDA-approved drug used in pa-tients as an adjuvant treatment for colon cancer(42,43) andthe drug has also been used as an anti-helmintic in ani-mals.(44–46) Theophylline is a bronchodilator approved forthe treatment of asthma. Thus, there is a need to developspecific TNALP inhibitors with drug-like properties suit-able for further development toward in vivo therapeuticsfor vascular calcification. We presently report how the useof high-throughput library screening has identified threenovel lead compounds able to inhibit TNALP with highaffinity and specificity. We validated the usefulness of thesecompounds kinetically and for their ability to prevent cal-cification in primary cultures of osteoblasts and Enpp1−/−
VSMCs and through their inhibition of TNALP’s pyro-phosphatase activity in whole rat aortas ex vivo.
The presently selected inhibitors are very aromatic mol-ecules; all three inhibit TNALP through an uncompetitivemechanism, both at pH 9.8 and at physiological pH. Thesmall differences in Ki measured at pH 9.8 and 7.5, for two
FIG. 5. Residual TNALP activity at pH 7.5, measured in thepresence of 0.1 mM pNPP as a function of the concentration ofcompound 5804079, in the absence (�) or presence (●) of 10 mMPi (A) or 40 mM PPi (B).
NARISAWA ET AL.1706

of the three inhibitors, imply that inhibitor positioning isnot ionic, in accordance with the high number of aromaticnitrogen atoms in their backbone structure. The most po-tent inhibitor (i.e., compound 5804079) is even more potentat pH 7.5 and can be modeled in the active site pocket ofTNALP in a manner that spans the large enzyme pocket.The uncompetitive nature of enzyme neutralization has theadditional advantage that inhibition of TNALP can beachieved by micromolar concentrations of inhibitor, evenon a background of millimolar concentrations of Pi and PPi.However, we expect that future medicinal chemistry effortson these compounds will enable us to optimize their chemi-cal structure to increase solubility and improve affinity evenfurther, to enable in vivo trials.
When compound 5804079 was used to treat calvarial os-teoblasts cultures, we observed the expected increase inOPN mRNA expression that is secondary to the increase inePPi concentrations resulting from inhibiting TNALP’s py-rophosphatase activity. This is entirely in agreement withour previous data, indicating that the expression of thesetwo potent calcification inhibitors is strictly correlated inskeletal tissue.(12,16,17) However, this TNALP inhibitor hadthe opposite effect on Enpp1−/− VSMCs. The reasons forthis apparent misregulation are not clear at this moment,but given that these are genetically abnormal cells wherethe production of ePPi is affected, it is possible that ePPi
and OPN, which normally act as counter-regulatory mecha-nisms balancing each others concentrations in skeletalcells,(17) may not be operating normally in these knockoutcells, which already have reduced levels of OPN.(7) Never-
theless, when added to Enpp1−/− VSMCs, compound5804079 potently reduced the cell-mediated calcificationprocess.
The possibility that treatment with TNALP inhibitorsmay have secondary deleterious effects on skeletal tissuesneeds to be considered. TNALP activity is highest in skel-etal tissues undergoing growth spurts (children and adoles-cents) or during the repair of fractures or in cases of osteo-blastic metastasis. An adult individual only has steady-statelevels of TNALP activity needed for bone homeostasis.Given that the amount of TNALP expressed by VSMCs atthe site of arterial calcification is upregulated comparedwith the steady-state levels of TNALP in osteoblasts, onewould expect VSMC activity to be selectively sensitive toinhibitor treatment. However, in the event that TNALPinhibitor treatments would lead to adverse side effects inbone or other tissues and that this toxicity could not bemanipulated by dosage or route of administration, targeteddelivery strategies can be contemplated to achieve a highconcentration of TNALP inhibitors only at the site of vas-cular calcification.
In conclusion, this study showed that potent inhibitors ofTNALP pyrophosphatase activity can be obtained by sys-tematic chemical library screening efforts. These com-pounds have the potential to be used to treat and/or preventectopic soft tissue calcification. The lead compounds iden-tified in this paper will serve as the foundation for thoroughstructure–activity relationship studies to optimize thesenovel inhibitors for optimal solubility and pharmacokinetic
FIG. 6. Computer docking ofthe three lead compounds intothe active site of the modeledstructure of TNALP. The topleft panel shows a detail of allthe active site residues in-volved in stabilization of in-hibitors as previously deter-mined.(28) The other threepanels shows a close up view ofeach novel inhibitor docked inthe active site environment.
TNALP INHIBITION AND VASCULAR CALCIFICATION 1707
Fig 6 live 4/C
properties with the goal of testing them in vivo in diverseexperimental models of vascular calcification.
ACKNOWLEDGMENTS
The authors thank Alexey Eroshkin for help with thedocking predictions; Steve Vasile for help with the high-
throughput chemical library screening; and Soetkin Vankerckhoven for help with kinetic measurements. This workwas supported by Grants DE12889, AR47908, andDK06981 from the National Institutes of Health andKULeuven Grant GOA 2004/09.
REFERENCES
1. Detrano RC, Doherty TM, Davies MJ, Stary HC 2000 Predict-ing coronary events with coronary calcium: Pathophysiologicand clinical problems. Curr Probl Cardiol 25:374–402.
2. Wallin R, Wajih N, Greenwood GT, Sane DC 2001 ArterialCalcification: A review of mechanisms, animal models and theprospects for therapy. Med Res Rev 21:274–301.
3. Towler D 2005 Inorganic pyrophosphate—a paracrine regula-tor of vascular calcification and smooth muscle phenotype. Ar-terioscler Thromb Vasc Biol 25:651–654.
4. Terkeltaub RA 2001 Inorganic pyrophosphate generation anddisposition in pathology. Am J Physiol Cell Physiol 281:C1–C11.
5. Meyer JL 1984 Can biological calcification occur in the pres-ence of pyrophosphate? Arch Biochem Biophys 231:1–8.
6. Lomashvili K, Cobbs S, Hennigar R, Hardcastle K, O’NeillWC 2004 Phosphate-induced vascular calcification: Role of py-rophosphate and osteopontin. J Am Soc Nephrol 15:1392–1401.
7. Johnson K, Polewski M, van Etten D, Terkeltaub R 2005Chondrogenesis mediated by PPi depletion promotes sponta-neous aortic calcification in NPP1−/− mice. ArteriosclerThromb Vasc Biol 25:686–691.
FIG. 7. Effect of the three in-hibitors on in vitro calcificationby WT calvarial osteoblastsand Enpp1−/− VSMCs. Primarycalvarial osteoblasts andEnpp1−/− VSMCs were grownin the presence of �-glycero-phosphate, ascorbic acid, andeither compound 5361418,5923412, or 5804079, and com-pared with the relative effect oflevamisole, at 30 �M concen-tration. Extractions and analy-ses were done after 3 wk. One-w a y A N O V A a n a l y s i sindicated p < 0.0001 for the os-teoblast experiment and p �0.0006 for the VSMC experi-ment. Parallel cultures of os-teoblasts and VSMCs wereused to extract mRNA to mea-sure changes in OPN mRNAexpression after these treat-ments. Each histogram plotsthe mean ± SE of six determi-nations.
TABLE 3. REDUCTION OF THE RATE OF HYDROLYSIS OF PPI IN
AORTIC RAT RINGS BY LEVAMISOLE AND THE NOVEL TNALPINHIBITORS
Inhibitor(30 µM)
PPi hydrolysis(nmol/g/min)(mean ± SE)
Percentinhibition
Control 0.320 ± 0.032 0.05361418 0.269 ± 0.032 15.95923412 0.249 ± 0.046 22.25804079 0.192 ± 0.041 39.9Levamisole 0.252 ± 0.047 21.2Vehicle 0.312 ± 0.017 2.5
All inhibitors were dissolved in 10% DMSO. “Control” samples receivedno inhibitor solution (no DMSO); “Vehicle” samples received 10% DMSOsolution without any inhibitor to achieve a final 1% DMSO in the culturemedium. The inhibitor solutions were similarly applied in 10% DMSO fora final concentration of 1% DMSO and 30 �M inhibitor. Each inhibitor wastested in triplicate and the results are expressed as mean ± SE.
NARISAWA ET AL.1708
Fig 7 live 4/C

8. Okawa A, Nakamura I, Goto S, Moriya H, Nakamura Y,Ikegawa S 1998 Mutation in Npps in a mouse model of ossifi-cation of the posterior longitudinal ligament of the spine. NatGenet 19:271–273.
9. Rutsch F, Vaingankar S, Johnson K, Goldfine I, Maddux B,Schauerte P, Kalhoff H, Sano K, Boisvert WA, Superti-FurgaA, Terkeltaub R 2001 PC-1 nucleoside triphosphate pyrophos-phohydrolase deficiency in idiopathic infantile arterial calcifi-cation. Am J Pathol 158:543–554.
10. Rutsch F, Ruf N, Vaingankar S, Toliat MR, Suk A, Hohne W,Schauer G, Lehmann M, Roscioli T, Schnabel D, Epplen JT,Knisely A, Superti-Furga A, McGill J, Filippone M, SinaikoAR, Vallance H, Hinrichs B, Smith W, Ferre M, Terkeltaub R,Nurnberg P 2003 Mutations in Enpp1 are associated with ‘id-iopathic’ infantile arterial calcification. Nat Genet 34:379–381.
11. Ho AM, Johnson MD, Kingsley DM 2000 Role of the mouseank gene in control of tissue calcification and arthritis. Science289:265–270.
12. Harmey D, Hessle L, Narisawa S, Johnson K, Terkeltaub R,Millán JL 2004 Concerted regulation of inorganic pyrophos-phate and osteopontin by Akp2, Enpp1 and Ank. An inte-grated model of the pathogenesis of mineralization disorders.Am J Pathol 164:1199–1209.
13. Millán JL 2006 Mammalian Alkaline Phosphatases. From Bi-ology to Applications in Medicine and Biotechnology. Wiley-VCH Verlag, Weinheim, Germany.
14. Johnson KA, Hessle L, Wennberg C, Mauro S, Narisawa S,Goding J, Sano K, Millán JL, Terkeltaub R 2000 Tissue-nonspecific alkaline phosphatase ( TNALP) and plasma cellmembrane glycoprotein-1 (PC-1) act as selective and mutualantagonists of mineralizing activity by murine osteoblasts. AmJ Phys Regulat Integrat Physiol 279:R1365–R1377.
15. Hessle L, Johnson KA, Anderson HC, Narisawa S, Sali A,Goding JW, Terkeltaub R, Millán JL 2002 Tissue-nonspecificalkaline phosphatase and plasma cell membrane glycopro-tein-1 are central antagonistic regulators of bone mineraliza-tion. Proc Natl Acad Sci USA 99:9445–9449.
16. Johnson K, Goding J, Van Etten D, Sali A, Hu SI, Farley D,Krug H, Hessle L, Millán JL, Terkeltaub R 2003 Linked defi-ciencies in extracellular PP(i) and osteopontin mediate patho-logic calcification associated with defective PC-1 and ANK ex-pression. J Bone Miner Res 18:994–1004.
17. Harmey D, Johnson KA, Zelken J, Camacho NP, HoylaertsMF, Noda M, Terkeltaub R, Millán JL 2006 Elevated osteo-pontin levels contribute to the hypophosphatasia phenotype inAkp2−/− mice. J Bone Miner Res 21:1377–1386.
18. Tanimura A, McGregor DH, Anderson HC 1986 Calcificationin atherosclerosis. I. Human studies. J Exp Pathol 2:261–273.
19. Tanimura A, McGregor DH, Anderson HC 1986 Calcificationin atherosclerosis. II. Animal studies. J Exp Pathol 2:275–297.
20. Hui M, Li SQ, Holmyard D, Cheng P 1997 Stable transfectionof nonosteogenic cell lines with tissue nonspecific alkalinephosphatase enhances mineral deposition both in the presenceand absence of beta-glycerophosphate: Possible role for alka-line phosphatase in pathological mineralization. Calcif TissueInt 60:467–472.
21. Hui M, Tenenbaum HC 1998 New face of an old enzyme:Alkaline phosphatase may contribute to human tissue aging byinducing tissue hardening and calcification. Anat Rec 253:91–94.
22. Hsu HH, Camacho NP 1999 Isolation of calcifiable vesiclesfrom human atherosclerotic aortas. Atherosclerosis 143:353–362.
23. Shioi A, Nishizawa Y, Jono S, Koyama H, Hosoi M, Morii H1995 Beta-glycerophosphate accelerates calcification in cul-tured bovine vascular smooth muscle cells. ArteriosclerThromb Vasc Biol 15:2003–2009.
24. Shioi A, Katagi M, Okuno Y, Mori K, Jono S, Koyama H,Nishizawa Y 2002 Induction of bone-type alkaline phosphatasein human vascular smooth muscle cells: Roles of tumor necro-sis factor-alpha and oncostatin M derived from macrophages.Circ Res 91:9–16.
25. Mathieu P, Voisine P, Pepin A, Shetty R, Savard N, Dagenais
F 2005 Calcification of human valve interstitial cells is depen-dent on alkaline phosphatase activity. J Heart Valve Dis14:353–357.
26. Hoylaerts MF, Ding L, Narisawa S, Van Kerckhoven S, MillánJL 2006 Mammalian alkaline phosphatase catalysis requiresactive site structure stabilization via the N-terminal amino acidmicroenvironment. Biochemistry 45:9756–9766.
27. Di Mauro S, Manes T, Hessle H, Kozlenkov A, Pizauro JM,Hoylaerts MF, Millán JL 2002 Kinetic characterization of hy-pophosphatasia mutations with physiological substrates. JBone Miner Res 17:1383–1391.
28. Kozlenkov A, Hoylaerts MF, Ny T, LeDu MH, Millán JL 2004Residues determining the binding specificity of uncompetitiveinhibitors to tissue-nonspecific alkaline phosphatase. J BoneMiner Res 19:1862–1872.
29. Sali A, Favaloro JM, Terkeltaub R, Goding JW 1999 Germlinedeletion of the nucleoside triphosphate pyrophosphohydrolase(NTPPPH) plasma cell membrane glycoprotein-1 (PC-1) pro-duces abnormal calcification of periarticular tissues. In: Van-duffel L, Lemmems R (eds.) Ecto-ATPases and Related Ec-toenzymes. Shaker Publishing, Maastrich, The Netherlands,pp. 267–282.
30. Wennberg C, Hessle L, Lundberg P, Mauro S, Narisawa S,Lerner UH, Millán JL 2000 Functional characterization of os-teoblasts and osteoclasts from alkaline phosphatase knockoutmice. J Bone Miner Res 15:1879–1888.
31. Roberts S, Narisawa S, Harmey D, Millán J, Farquharson C2007 Functional involvement of PHOSPHO1 in matrix vesicle-mediated skeletal calcification. J Bone Miner Res 22:617–627.
32. Hoylaerts MF, Manes T, Millán JL 1992 Molecular mechanismof uncompetitive inhibition of human placental and germ cellalkaline phosphatase. Biochem J 286:23–30.
33. Tintut Y, Patel J, Territo M, Saini T, Parhami F, Demer LL2002 Monocyte/macrophage regulation of vascular calcifica-tion. Circulation 105:650–655.
34. Watson KE, Bostrom K, Ravindranath R, Lam T, Norton B,Demer LL 1994 TGF-beta 1 and 25-hydroxycholesterol stimu-late osteoblast-like vascular cells to calcify. J Clin Invest93:2106–2113.
35. Wada T, McKee MD, Steitz S, Giachelli CM 1999 Calcificationof vascular smooth muscle cell cultures: Inhibition by osteo-pontin. Circ Res 84:166–178.
36. Steitz SA, Speer MY, Curinga G, Yang HY, Haynes P, Ae-bersold R, Schinke T, Karsenty G, Giachelli CM 2001 Smoothmuscle cell phenotypic transition associated with calcification:Upregulation of Cbfa1 and downregulation of smooth musclelineage markers. Circ Res 89:1147–1154.
37. Tintut Y, Alfonso Z, Saini T, Radcliff K, Watson K, BostromK, Demer LL 2003 Multilineage potential of cells from theartery wall. Circulation 108:2505–2510.
38. Hsu HH, Anderson HC 1978 Calcification of isolated matrixvesicles and reconstituted vesicles from fetal bovine cartilage.Proc Natl Acad Sci USA 75:3805–3808.
39. Ali SY, Sajdera SW, Anderson HC 1970 Isolation and charac-terization of calcifying matrix vesicles from epiphyseal carti-lage. Proc Natl Acad Sci USA 67:1513–1520.
40. Kim KM 1976 Calcification of matrix vesicles in human aorticvalve and aortic media. Fed Proc 35:156–162.
41. Hsu HHT, Camacho NP, Sun F, Tawik O, Aono H 2000 Iso-lation of calcifiable vesicles from aortas of rabbits fed with highcholesterol diets. Atherosclerosis 153:337–348.
42. Witte RS, Cnaan A, Mansour EG, Barylak E, Harris JE, Sch-utt AJ 2001 Comparison of 5-fluorouracil alone, 5-flourouracilwith levamisole, and 5-flourouracil with hepatic irradiation inthe treatment of patients with residual, nonmeasurable, intra-abdominal metastasis after undergoing resection for colorectalcarcinoma. Cancer 91:1020–1028.
43. Taal BG, van Tinteren H, Zoetmulder FA, NACCP group2001 Adjuvant 5FU plus levamisole in colonic or rectal cancer:
TNALP INHIBITION AND VASCULAR CALCIFICATION 1709

Improved survival in stage II and III. Br J Cancer 85:1437–1443.
44. Powe TA, Powers RD 1985 Periorchitis after tetramisole treat-ment in bulls implaned with Setaria labiatopapillos. J Am VetMed Assoc 186:588–589.
45. Bhopale GM, Bhatnagar BS 1984 Serum protein profile ofmice during infection of Ancylostoma caninum and after theadministration of tetramisole and levamisole. J Hyg EpidemiolMicrobiol Immunol 28:455–459.
46. Eisenberg E, Shklar G 1977 Levamisole and hamster pouchcarcinogenesis. Oral Surg Oral Med Oral Path 43:562–571.
Address reprint requests to:José Luis Millán, PhD
Burnham Institute for Medical Research10901 North Torrey Pines Road
La Jolla, CA 92037, USAE-mail: [email protected]
Received in original form February 5, 2007; revised form June 22,2007; accepted July 11, 2007.
NARISAWA ET AL.1710




















