
A Dinucleotide Deletion in CD24 Confers Protection against Autoimmune Diseases
Upload
independentCategory
view
1download
0
NLRP3 deletion protects from hyperoxia-induced acute lung injury
Jutaro Fukumoto,1 Itsuko Fukumoto,1 Prasanna Tamarapu Parthasarathy,1 Ruan Cox,1 Bao Huynh,1
Gurukumar Kollongod Ramanathan,1 Rajan Babu Venugopal,1 Diane S. Allen-Gipson,2
Richard F. Lockey,1 and Narasaiah Kolliputi11Division of Allergy and Immunology, Department of Internal Medicine, Morsani College of Medicine, University of SouthFlorida, Tampa, Florida; and 2Department of Pharmaceutical Sciences, College of Pharmacy, Morsani College of Medicine,University of South Florida, Tampa, Florida
Submitted 29 March 2013; accepted in final form 29 April 2013
Fukumoto J, Fukumoto I, Parthasarathy PT, Cox R, Huynh B,Ramanathan GK, Venugopal RB, Allen-Gipson DS, Lockey RF,Kolliputi N. NLRP3 deletion protects from hyperoxia-induced acutelung injury. Am J Physiol Cell Physiol 305: C182–C189, 2013. Firstpublished May 1, 2013; doi:10.1152/ajpcell.00086.2013.—Inspirationof a high concentration of oxygen, a therapy for acute lung injury(ALI), could unexpectedly lead to reactive oxygen species (ROS)production and hyperoxia-induced acute lung injury (HALI). Nucleo-tide-binding domain and leucine-rich repeat PYD-containing protein 3(NLRP3) senses the ROS, triggering inflammasome activation andinterleukin-1� (IL-1�) production and secretion. However, the role ofNLRP3 inflammasome in HALI is unclear. The main aim of this studyis to determine the effect of NLRP3 gene deletion on inflammatoryresponse and lung epithelial cell death. Wild-type (WT) and NLRP3�/�
mice were exposed to 100% O2 for 48–72 h. Bronchoalveolar lavagefluid and lung tissues were examined for proinflammatory cytokineproduction and lung inflammation. Hyperoxia-induced lung patholog-ical score was suppressed in NLRP3�/� mice compared with WTmice. Hyperoxia-induced recruitment of inflammatory cells and ele-vation of IL-1�, TNF�, macrophage inflammatory protein-2, andmonocyte chemoattractant protein-1 were attenuated in NLRP3�/�
mice. NLRP3 deletion decreased lung epithelial cell death andcaspase-3 levels and a suppressed NF-�B levels compared with WTcontrols. Taken together, this research demonstrates for the first timethat NLRP3-deficient mice have suppressed inflammatory responseand blunted lung epithelial cell apoptosis to HALI.
hyperoxia; inflammation; injury; lung; reactive oxygen species
ACUTE LUNG INJURY (ALI) is characterized by severe alveolardamage resulting from an acute inflammatory response thatleads to proinflammatory cytokine production, neutrophil,macrophage infiltration, and edema. The most severe form ofALI is acute respiratory distress syndrome (ARDS), which is amajor cause for admission to critical care units. Hyperoxiatherapy is a necessary part of treatment for patients with acuteand chronic cardiovascular and pulmonary diseases. However,prolonged exposure to hyperoxia could deteriorate ALI (19,47). Currently, there are several animal models available tostudy the mechanism of ALI. The hyperoxia-induced acutelung injury (HALI) animal model became widely used to studyhuman ALI after Cochrane et al. (7) revealed the increase ofoxidants in the lungs of patients with ARDS. It is now wellestablished that there are clinically relevant similarities be-tween the animal model of HALI and human lung injury (26).However, the molecular mechanisms that initiate and amplify
the lung inflammation in response to inhaled oxygen are notwell understood.
IL-1� is one of the most potent early cytokines found in ALIpatients, and it induces the production of other cytokines (12).The proinflammatory cytokine IL-1� is also known to be oneof the most biologically important inflammatory mediators inthe air space of patients with early ALI (35). Interestingly,IL-1� can also act as an important activator and prosurvivalcytokine for neutrophils (37). However, mechanisms that ini-tiate IL-1� processing in ALI are not clearly defined.
Martinon et al. (25) first reported in 2002 that caspase-1-mediated processing of IL-1� is mediated by the nucleotide-binding domain and leucine-rich repeat PYD-containing pro-tein 3 (NLRP3) inflammasome. The NLRP3 inflammasome isa multiprotein complex, which contains NLRP3, the caspaserecruitment domain containing protein Cardinal, apoptosis-associated speck-like protein (ASC), and caspase-1 (33). NLRP3inflammasome is implicated in sensing stress caused by reac-tive oxygen species (9, 32). Recently we showed that hyper-oxia induces inflammasome activation (21, 22). However,whether inhibition or deletion of NLRP3 inflammasome iscritical to confer protection against HALI has not been studiedyet. Since the HALI model is thoroughly characterized in termsof reactive oxygen species involvement, assessing the effect ofNLRP3 deletion on HALI will provide important informationabout how NLRP3 plays a role in HALI and might result innovel therapeutic strategies to treat ALI. In this study for thefirst time we used NLRP3-deficient mice to identify the role ofinflammasomes in hyperoxia-induced lung injury.
MATERIALS AND METHODS
Mice. The University of South Florida Institutional Animal Careand Use Committee (IACUC) approved all animal procedures.NLRP3 knockout mice (gift from Dr. Vishva Dixit, Genentech, SanFrancisco, CA) and C57BL/6J mice (Harlan Laboratories, Indianap-olis, IN) were used to conduct in vivo experiments. NLRP3 �/� micewere derived from C57BL/6J strain.
Mice, aged 7–9 wk, were used in all experiments. Mice were placedin cages in a chamber (75�50�50 cm) and exposed to 100% O2 for48 or 72 h. The oxygen concentration in the chamber was monitoredand regulated with the proOx P100 sensor (BioSpherix). Mice wereeuthanized 48 or 72 h after exposure to 100% O2 for sample collec-tion.
Lung perfusion and tissue collection. Mice were anesthetized by anintraperitoneal injection of ketamine/xylazine mixture. Postmortem,the abdominal cavity was opened, and the inferior vena cava (IVC)was severed for blood removal. Following thoracotomy, the IVC wasclamped just above the diaphragm and then the lungs were perfusedthrough the right ventricle. The left lower lobe of the lung wasremoved and fixed in 10% buffered formalin for standard histological
Address for reprint requests and other correspondence: N. Kolliputi, Divi-sion of Allergy and Immunology, Dept. of Internal Medicine, Morsani Collegeof Medicine, Univ. of South Florida, 12901 Bruce B. Downs Blvd., Tampa, FL33612 (e-mail:[email protected]).
Am J Physiol Cell Physiol 305: C182–C189, 2013.First published May 1, 2013; doi:10.1152/ajpcell.00086.2013.
0363-6143/13 Copyright © 2013 the American Physiological Society http://www.ajpcell.orgC182

processing and paraffin embedding (FFPE). The remaining portion ofthe lungs was cut into several pieces and stored in an ultralowtemperature freezer.
Bronchoalveolar lavage analysis. Blood was removed from theIVC, as described previously, followed by a small transverse incisionin the skin of the ventral neck. The trachea was exposed, a catheterwas inserted, and a whole lung lavage was performed by using sterilePBS. Bronchoalveolar lavage (BAL) fluid, (2–3 ml), was centrifugedat 400 g for 10 min at 4°C. Cell pellets were resuspended in 1 ml ofice-cold sterile PBS. The total number of cells in the cell suspensionwas counted using a disposable hemocytometer. Aliquots, 200–400�l, of cell suspension were centrifuged onto glass slides at 800 rpmfor 3 min in a cytocentrifuge (Shandon Cytospin 2, Pittsburgh, PA).The cytospined cells were stained with Diff-Quik stain set (AndwinScientific, Schaumburg, IL), and a differential white blood cell countwas performed (200 cells).
ELISA. The levels of IL-1�, monocyte chemoattractant protein-1(MCP-1), macrophage inflammatory protein-2 (MIP-2; eBioscience,San Diego, CA), TNF-� (RayBiotech, Norcross, GA), and IL-6 (BDBiosciences, San Diego, CA) in BAL fluid were measured usingcommercial ELISA kits as per the manufacturer’s instructions.
Histology and evaluation of lung injury. FFPE lung tissue sectionswere stained with hematoxylin and eosin, and the extent of lung injurywas evaluated. Evaluation of the lung tissue pathological severity wasperformed in a blinded manner following the methods describedpreviously (17, 45) by independently scoring four parameters: alveo-lar congestion, hemorrhage, aggregation of neutrophil or leukocyteinfiltration, and thickness of the alveolar wall. 0: no lung abnormal-ities; 1: lesions involving �25% of the lung; 2: lesions involving25–50% of the lung; 3: lesions involving 50–75% of the lung; and 4:lesions involving 75% or more of the lung. The total histopathologicalscore was expressed as the sum of the scores for all parameters.
Terminal deoxynucleotidyl transferase dUTP-mediated nick-endlabeling staining on lung tissue sections. Terminal deoxynucleotidyltransferase dUTP-mediated nick-end labeling (TUNEL) staining wasperformed on FFPE lung tissue sections using the in situ apoptosisdetection kit (TaKaRa Biomedicals, Tokyo, Japan) according to themanufacturer’s instructions with some modifications. Briefly, the lungtissue sections were deparaffinized and then incubated with 20 �g/mlproteinase K for 40 min at 37°C to unmask antigen binding sites. Afterbeing blocked with 3% hydrogen peroxide in PBS, the sections wereincubated with TdT enzyme and FITC-labeled dUTP for 90 min at37°C. Subsequently, the sections were incubated with anti-FITChorseradish peroxidase conjugate for 60 min at 37°C. Colorimetricdetection of apoptotic cells was performed using 3,3=-diaminobenzi-dine reagent.
Lung homogenization. Lung tissue samples were thawed on ice andhomogenized using a PowerGen 500 homogenizer (Fisher Scientific,Pittsburgh, PA). For protein extraction, lysis buffer (20 mM Tris·HCl,pH 7.4, 150 mM NaCl, and 0.5% Triton X-100) was supplementedwith protease and phosphatase inhibitors (Sigma-Aldrich, St. Louis,MO). After two freeze-thaw cycles, the lung homogenates werecentrifuged at 15,000 g for 10 min at 4°C. Supernatants were collectedand stored at �80°C until use.
Western blotting. Supernatants from lung homogenates were boiledin Laemmli 4� SDS sample buffer (Boston BioProducts, Worcester,MA), and equal amounts of protein were subjected to SDS-PAGE.Proteins were then transferred onto polyvinylidene difluoride mem-branes. The membranes were blocked in Tris-buffered saline (20 mMTris·HCl at pH 7.5 and 150 mM NaCl) with 0.1% Tween 20 (TBS-T)containing 5% skim milk and then incubated with anti-NF-�B, anti-phosphor-NF-�B antibody, anti-cleaved casp-3 antibody (Cell Signal-ing Technology, Beverly, MA), or anti-caspase-1 p10 antibody (SantaCruz Biotechnology, Dallas, TX). The membranes were washed withTBS-T and incubated with horseradish peroxidase-conjugated second-ary antibody for 30 min at room temperature. Following TBS-T
washes, the proteins were visualized using Pierce ECL Westernblotting substrate (Thermo Fisher Scientific, Hudson, NH).
Statistical analysis. Statistical analysis was performed with Graph-Pad Prism 5 (GraphPad Software, San Diego, CA). Comparison ofcontinuous variables between two groups was performed using theStudent’s t-test, while total histopathological scores were comparedusing the Mann-Whitney U test. All tests were two-tailed, and valuesof P � 0.05 were considered significant.
RESULTS
Effect of hyperoxia and NLRP3 deletion on lung histology.Hematoxylin-eosin-stained lung tissue sections obtained fromwild-type (WT) and NLRP3�/� mice exposed to 100% O2 for48 h were compared to determine if NLRP3 deletion amelio-rates oxygen toxicity. Hyperoxia-exposed WT mice show dif-fuse pathological changes characterized by alveolar congestionand inflammatory cell infiltration into the air space. However,these changes are suppressed in NLRP3�/� mice (Fig. 1A).Evaluation of pathological severity of the lung, as performed byindependently scoring four parameters, shows a significant de-crease in total histopathological score in NLRP3�/� comparedwith WT mice (Fig. 1B). These results suggest that NLRP3�/�
mice are resistant to hyperoxia-induced ALI.Hyperoxia-induced recruitment of inflammatory cells and
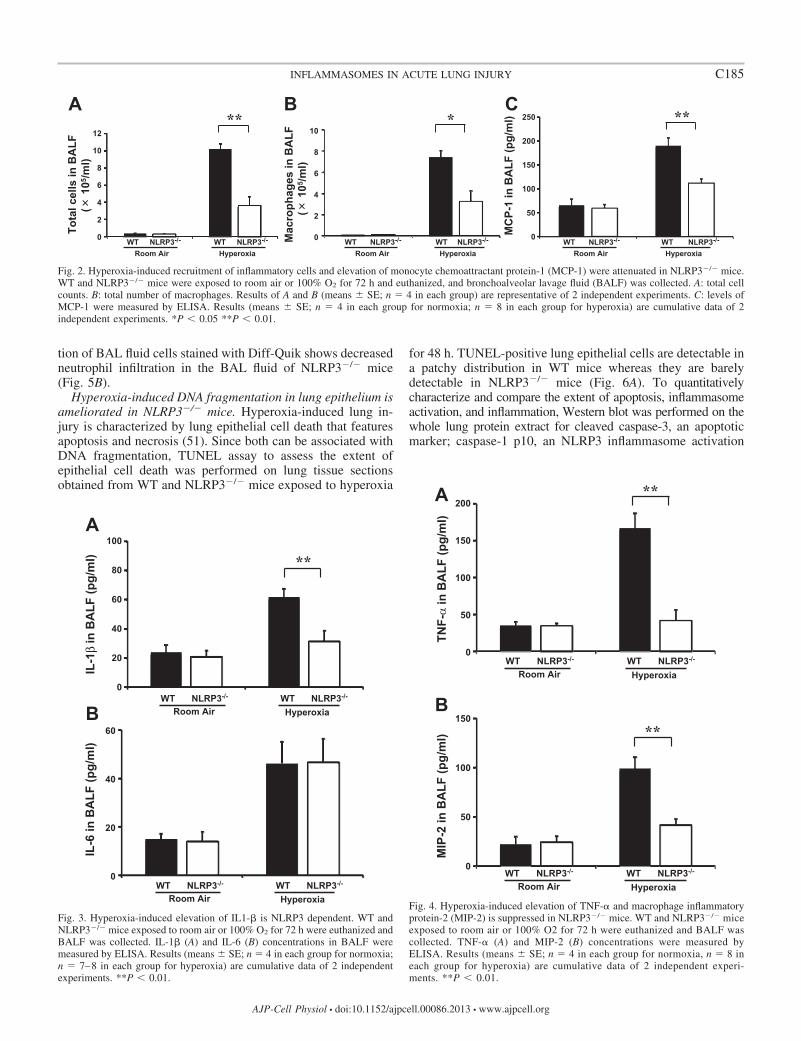
elevation of MCP-1 is attenuated in NLRP3�/� mice. Quanti-fication of total number of cells recovered from BAL fluid isuseful to evaluate alveolitis in hyperoxia-induced murine mod-els (38). Results show that the total number of cells in BAL fluidobtained from NLRP3�/� mice exposed to 100% O2 is signifi-cantly decreased compared with their WT controls (Fig. 2A).Macrophages are the most abundant immune cells in lungparenchyma (5). Activation of these alveolar macrophages inlungs is regulated by different cytokines and inflammatorymediators (13, 23). The number of macrophages present inBAL fluid was assessed by performing total and differentialcell counts. The results show that total number of macrophagesin BAL fluid in NLRP3�/� mice exposed to 100% O2 for 72 his significantly lower compared with WT mice (Fig. 2B).MCP-1 is known to be a potent chemoattractant mainly actingon monocytes/macrophages (18). MCP-1 concentrations inBAL fluid obtained from WT and NLRP3�/� mice exposed to100% O2 for 72 h were measured to determine whethersuppressed macrophage recruitment in NLRP3�/� mice isrelated to a decrease in MCP-1 levels in BAL fluid. The resultsshow that hyperoxia-induced elevated MCP-1 levels in WTmice (means � SE; 189.9 � 16.4 pg/ml) is significantlysuppressed in NLRP3�/� mice (111.6 � 8.3 pg/ml), consistentwith decreased macrophage infiltration (Fig. 2C). These resultssuggest that hyperoxia-induced immune cell infiltration andMCP-1 secretion in lung are, at least in part, under the controlof the NLRP3 inflammasome activation.
Hyperoxia-induced elevation of IL-1� is NLRP3 dependent.IL-1� is one of the most potent cytokines in the lungs of earlyALI patients (12, 50). Although inflammasome-mediatedIL-1� processing is involved in a variety of inflammatorydiseases, its significance in HALI has not been studied. WTand NLRP3�/� mice were exposed to hyperoxia for 72 h, andIL-1� levels in BAL fluid was measured by ELISA to deter-mine whether IL-1� processing in HALI occurs throughNLRP3 inflammasome activation. Results show that hyperoxiainduces elevated IL-1� levels in WT mice (61.5 � 5.8 pg/ml).
C183INFLAMMASOMES IN ACUTE LUNG INJURY
AJP-Cell Physiol • doi:10.1152/ajpcell.00086.2013 • www.ajpcell.org

However, this elevation was significantly suppressed inNLRP3�/� mice (31.2 � 7.3 pg/ml; Fig. 3A). Additionally,levels of IL-6, another major proinflammatory cytokine inHALI were measured. Results show that hyperoxia-inducedIL-6 elevation in BAL fluid seen in WT mice (46.1 � 9.0pg/ml) is not altered in NLRP3�/� mice (46.6 � 9.8 pg/ml;Fig. 3B). These results suggest that IL-1� secretion in HALI isNLRP3 dependent.
Hyperoxia-induced expression of TNF-� and MIP-2 is sup-pressed in NLRP3�/� mice. TNF-� is the most widely studiedproinflammatory cytokine member of the TNF super familyand is recognized as a mediator of the pulmonary inflammatoryresponse (30). TNF-� concentrations in BAL fluid obtainedfrom WT and NLRP3�/� mice exposed to 100% O2 for 72 hwere measured. Results show that hyperoxia-induced TNF-�elevation in BAL fluid is significantly suppressed in NLRP3�/�
mice (41.0 � 14.9 pg/ml) compared with WT mice (166.4 �20.3 pg/ml; Fig. 4A). MIP-2, a functional homologue of human
IL-8, is known to be chemotactic for neutrophils in ALI (41,46). MIP-2 levels in BAL fluid obtained from mice exposed to100% O2 for 72 h were measured to check whether protection ofNLRP3�/� mice is accompanied by modulation of MIP-2. Resultsshow that MIP-2 levels in hyperoxia-induced WT mice (98.7 � 11.9pg/ml) were significantly higher compared with NLRP3�/� mice(41.5 � 5.9 pg/ml; Fig. 4B).
Hyperoxia-induced neutrophil accumulation is suppressedin NLRP3�/� mice. Neutrophil infiltration into the pulmonaryinterstitium and alveoli is one of the key features of ALI.Suppression of neutrophils infiltration reduces hemorrhage- orendotoxemia-induced lung injury (1). The number of neutro-phils in BAL fluid in mice exposed to 100% O2 for 72 h wasassessed to determine whether protection of NLRP3�/� miceagainst HALI is related to suppression of neutrophil infiltra-tion. There is a significantly lower number of neutrophils inBAL fluid in hyperoxia-exposed NLRP3�/� mice comparedwith WT mice (Fig. 5A). Consistently, microscopic observa-
NLRP3-/-WT
A
B
HyperoxiaRoom air
WT
NLRP3-/-
His
topa
thol
ogic
al s
core
*
12
10
8
6
4
2
0
Fig. 1. Hyperoxia-exposed Nucleotide-binding domain and leucine-rich repeat PYD-containing protein 3-deficient (NLRP3�/�) mice show attenuated lunginjury. wild-type (WT) and NLRP3�/� mice were exposed to room air or 100% O2 for 48 h. Mice were euthanized and lung tissues were collected.A: representative hematoxylin-eosin (H&E)-stained lung tissue sections. Original magnifications: �40 (insets, �100). B: semiquantitative lung injury analysisby evaluation of H&E-stained lung tissue sections. Total histopathological score was expressed as summed scores for 4 independent parameters: alveolarcongestion, hemorrhage, aggregation of neutrophil or leukocyte infiltration, and thickness of the alveolar wall. Results (means � SE; n 8 in each group) arecumulative data of 2 independent experiments. *P � 0.05.
C184 INFLAMMASOMES IN ACUTE LUNG INJURY
AJP-Cell Physiol • doi:10.1152/ajpcell.00086.2013 • www.ajpcell.org
tion of BAL fluid cells stained with Diff-Quik shows decreasedneutrophil infiltration in the BAL fluid of NLRP3�/� mice(Fig. 5B).
Hyperoxia-induced DNA fragmentation in lung epithelium isameliorated in NLRP3�/� mice. Hyperoxia-induced lung in-jury is characterized by lung epithelial cell death that featuresapoptosis and necrosis (51). Since both can be associated withDNA fragmentation, TUNEL assay to assess the extent ofepithelial cell death was performed on lung tissue sectionsobtained from WT and NLRP3�/� mice exposed to hyperoxia
for 48 h. TUNEL-positive lung epithelial cells are detectable ina patchy distribution in WT mice whereas they are barelydetectable in NLRP3�/� mice (Fig. 6A). To quantitativelycharacterize and compare the extent of apoptosis, inflammasomeactivation, and inflammation, Western blot was performed on thewhole lung protein extract for cleaved caspase-3, an apoptoticmarker; caspase-1 p10, an NLRP3 inflammasome activation
A B C
Tota
l cel
ls in
BA
LF(
105 /m
l)
Room AirWT NLRP3-/-
HyperoxiaWT NLRP3-/-
**
Room AirWT NLRP3-/-
HyperoxiaWT NLRP3-/-M
acro
phag
es in
BA
LF(
105 /m
l)
*
MC
P-1
in B
ALF
(pg/
ml)
Room AirWT NLRP3-/-
HyperoxiaWT NLRP3-/-
**12
10
8
6
4
2
0
10
8
6
4
2
0
250
200
150
100
50
0
Fig. 2. Hyperoxia-induced recruitment of inflammatory cells and elevation of monocyte chemoattractant protein-1 (MCP-1) were attenuated in NLRP3�/� mice.WT and NLRP3�/� mice were exposed to room air or 100% O2 for 72 h and euthanized, and bronchoalveolar lavage fluid (BALF) was collected. A: total cellcounts. B: total number of macrophages. Results of A and B (means � SE; n 4 in each group) are representative of 2 independent experiments. C: levels ofMCP-1 were measured by ELISA. Results (means � SE; n 4 in each group for normoxia; n 8 in each group for hyperoxia) are cumulative data of 2independent experiments. *P � 0.05 **P � 0.01.
A
B
IL-6
in B
ALF
(pg/
ml)
IL-1
β in
BA
LF (p
g/m
l)
Room AirWT NLRP3-/-
HyperoxiaWT NLRP3-/-
**
Room AirWT NLRP3-/-
HyperoxiaWT NLRP3-/-
100
80
60
40
20
0
60
40
20
0
Fig. 3. Hyperoxia-induced elevation of IL1-� is NLRP3 dependent. WT andNLRP3�/� mice exposed to room air or 100% O2 for 72 h were euthanized andBALF was collected. IL-1� (A) and IL-6 (B) concentrations in BALF weremeasured by ELISA. Results (means � SE; n 4 in each group for normoxia;n 7–8 in each group for hyperoxia) are cumulative data of 2 independentexperiments. **P � 0.01.
TNF-
α in
BA
LF (p
g/m
l)M
IP-2
in B
ALF
(pg/
ml)
A
B
Room AirWT NLRP3-/-
HyperoxiaWT NLRP3-/-
Room AirWT NLRP3-/-
HyperoxiaWT NLRP3-/-
**
**
200
150
100
50
0
150
100
50
0
Fig. 4. Hyperoxia-induced elevation of TNF-� and macrophage inflammatoryprotein-2 (MIP-2) is suppressed in NLRP3�/� mice. WT and NLRP3�/� miceexposed to room air or 100% O2 for 72 h were euthanized and BALF wascollected. TNF-� (A) and MIP-2 (B) concentrations were measured byELISA. Results (means � SE; n 4 in each group for normoxia, n 8 ineach group for hyperoxia) are cumulative data of 2 independent experi-ments. **P � 0.01.
C185INFLAMMASOMES IN ACUTE LUNG INJURY
AJP-Cell Physiol • doi:10.1152/ajpcell.00086.2013 • www.ajpcell.org

marker; and NF-�B, phospho-NF-�B, markers for inflamma-tory activation. Consistent with the results from TUNEL stain-ing, the expression of cleaved caspase-3 in the lung after 48-hexposure to hyperoxia was decreased in NLRP3�/� micecompared with WT controls. Likewise, the expression ofcaspase-1 p10, NF-�B, and phospho-NF-�B was decreased inNLRP3�/� mice compared with WT controls (Fig. 6B). Ourresults showed that NLRP3 deletion decreased lung epithelial celldeath, caspase-3 levels and suppressed NF-�B pathway comparedwith WT controls. These results suggest that hyperoxia-inducedepithelial cell death is ameliorated in NLRP3-deficient mice.
DISCUSSION
The NLRP3 inflammasome is a multiprotein complex re-sponsible for IL-1� processing in various inflammatory dis-eases (28, 50). In this study to determine whether inflam-masome activation is critical in hyperoxia-induced lung injurywe used NLRP3 homozygous knockout mice. IL-1� inducesthe production of other proinflammatory cytokines such asTNF-� and IL-8 (a human homologue of MIP-2; Refs. 4, 6, 20,43, 49). IL-1� also causes surfactant abnormalities (16). Ele-vated TNF-� concentrations are associated with morbidity andmortality in patients with ARDS (2).
Our results show that hyperoxia-induced TNF-� and IL-1�elevation in BAL fluid is significantly suppressed in NLRP3�/�
mice compared with WT controls, suggesting that hyperoxia-induced TNF-� elevation in lung is NLRP3 dependent. Previ-ously, IL-1� was reported to induce IL-6 elevation (40). However,in the present study, there is no significant difference in IL-6 levelin BAL fluid between hyperoxic WT and NLRP3�/� mice, suggest-ing that hyperoxia-induced IL-6 elevation is NLRP3 independent.
Macrophages are the most abundant immune cells in lungparenchyma, and different cytokines and inflammatory medi-ators activate these cells (5, 13, 23, 38). Recruitment ofmacrophages or monocytes to inflammatory sites in HALI ismainly controlled by MCP-1 (34), and a significant correlationexists between the BAL fluid MCP-1 level and severity ofARDS (39). The accumulation of macrophages in the lungs asseen in HALI should reflect enhanced recruitment of macro-phages and/or their resistance to apoptosis. To investigate therole of NLRP3 inflammasome in macrophage accumulation inHALI, we assessed the number of macrophages and levels ofMCP-1 in BAL fluid obtained from WT and NLRP3�/� miceexposed to hyperoxia. NLRP3�/� mice showed a significantdecrease in the number of total cells and macrophages in BALfluid under hyperoxic condition compared with WT mice. Thelevels of MCP-1 in BAL fluid were also significantly decreasedin NLRP3�/� mice under hyperoxic conditions compared withWT mice. Neutrophil infiltration in the lung is a key event thatcorrelates well with the severity of ALI/ARDS (14). Theelimination of neutrophils markedly decreases the severity ofALI in experimental models (1). MIP-2 and IL-8 (a humanhomologue of MIP-2) are well-known neutrophil-attractingCXC chemokines and were reported to be elevated in ALI/ARDS (3, 29, 38, 42). In terms of the relationship between theinflammasome and neutrophil recruitment, it was recentlyreported that neutrophil recruitment to the thermally injuredlocal site of the liver surface was blunted in NLRP3- orASC-deficient mice compared with WT controls (27). Simi-larly, we show that the accumulation of neutrophils seen inHALI was diminished in NLRP3�/� mice. This suppression ofneutrophil infiltration may be due to complete deletion of
A
Neu
trop
hils
in B
ALF
(x 1
04 (/m
l)
BRoom Air
WT NLRP3-/-
HyperoxiaWT NLRP3-/-
**40
30
20
10
0
Fig. 5. Hyperoxia-induced neutrophil recruit-ment was decreased in NLRP3�/� mice. WTand NLRP3�/� mice exposed to room air or100% O2 were euthanized and BALF wascollected. A: means � SE of total number ofneutrophils (n 4 in each group) from 1 ofthe 2 independent experiments is shown.B: mixture of equal amount of BALF from 4different mice per each group was cytospinedand stained with Diff-Quik. Infiltrating neu-trophils are indicated by arrows. Magnifica-tion: �200. **P � 0.01.
C186 INFLAMMASOMES IN ACUTE LUNG INJURY
AJP-Cell Physiol • doi:10.1152/ajpcell.00086.2013 • www.ajpcell.org

NLRP3 in all cell types. However, cell-specific knockoutstudies are further warranted in the future.
Both IL-1� and MIP-2 are also decreased in BAL fluidobtained from NLRP3�/� mice compared with WT controls.These results suggest that neutrophil accumulation in HALI isNLRP3 dependent through suppression of MIP-2 (as a neutro-phil chemoattractant). Enhanced and sustained neutrophil ac-cumulation in the lung would further facilitate the secretion ofabundant proinflammatory cytokines or elastase from neutro-phils. Several lines of evidence support the idea that oxidantstress directly injures lung epithelium (11, 24, 36, 44). InHALI, it is well known that inhaled oxygen toxicity inducesoxidative damage and DNA fragmentation, an apoptoticmarker, in lung epithelium (15, 48). Our results show thatDNA fragmentation and cleaved caspase-3 expression aredecreased in NLRP3�/� mice compared with WT controls.Although we also found decreased NF-�B activity in whole lungsobtained from NLRP3�/� mice compared with WT controls,additional studies are required to confirm how NF-�B pathway isdirectly involved in NLRP3-mediated HALI protection.
Taken together, this research demonstrates for the first timethat NLRP3-deficient mice have suppressed inflammatory re-sponse to HALI. Based on our current results, we believe thata cytokine burst that follows cytokine-dependent inflammationand epithelial cell apoptosis in HALI is dependent on theNLRP3 inflammasome. The cytokine burst will facilitate epi-thelial permeability, epithelial barrier dysfunction, and epithe-lial cell death (8, 10, 31), key features of ALI.
From a clinical perspective, targeting inflammasome by apharmacological approach that disrupts inflammasome activa-tion may provide a novel therapeutic strategy for the protectionof lung tissue from cellular injury during acute or chronicillness associated with ALI syndromes. However, one majorlimitation of our study is using a single insult (hyperoxia) as atrigger to study the inflammasome. In many clinical conditions,hyperoxia is currently being used to treat critically ill adultswho may already have vigorous inflammasome activation.Further, it is also totally possible that inflammasome-indepen-dent mechanisms of transcription and release of IL-1� andIL-18 may coexist and represent a fundamental considerationfor future therapeutics. Future investigation is needed to gainmechanistic insights into the regulation of NLRP3 inflammasomein ALI. Understanding the mechanisms of NLRP3-mediatedHALI may lead to the development of clinical interventions forthe protection of patients requiring supplemental oxygen.
ACKNOWLEDGMENTS
We thank Dr. Brenda Flam for critical reading of the manuscript.
GRANTS
N. Kolliputi was funded by the American Heart Association NationalScientist Development Grant 09SDG2260957 and National Heart, Lung, andBlood Institute Grant R01-HL-105932 and the Joy McCann CulverhouseEndowment to the Division of Allergy and Immunology.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
WT NLRP3-/-A
Casp-1 p10
GAPDH
Cleaved Casp-3
NFκB
Phospho-NFκB
BWT NLRP3-/-
Fig. 6. Hyperoxia-induced lung epithelialcell death is attenuated in NLRP3�/� mice.A: WT and NLRP3�/� mice (n 8 in eachgroup) exposed to 100% O2 for 48 h wereeuthanized and lung tissues were collected.Lung tissue sections were stained using ter-minal deoxynucleotidyl transferase dUTP-mediated nick-end labeling (TUNEL) assay.Shown are representative images of 2 inde-pendent experiments. TUNEL-positive apop-totic cells are observed in lung epithelium inWT mice, but they are hardly detectable inNLRP3�/� mice. Magnification: �200. B: West-ern blot analysis of NF-�B, phosphor-NF-�B,caspase-1 p10, and cleaved caspase-3 in wholelung lysates after 48-h hyperoxic exposure.Results are representative of 2 independentexperiments.
C187INFLAMMASOMES IN ACUTE LUNG INJURY
AJP-Cell Physiol • doi:10.1152/ajpcell.00086.2013 • www.ajpcell.org

AUTHOR CONTRIBUTIONS
Author contributions: J.F., I.F., P.T.P., R.C., and B.H. performed experi-ments; J.F. analyzed data; J.F. prepared figures; J.F. drafted manuscript; P.T.P.,D.S.A.-G., R.F.L., and N.K. edited and revised manuscript; G.K.R., R.B.V.,and N.K. interpreted results of experiments; N.K. approved final version ofmanuscript.
REFERENCES
1. Abraham E, Carmody A, Shenkar R, Arcaroli J. Neutrophils as earlyimmunologic effectors in hemorrhage- or endotoxemia-induced acute lunginjury. Am J Physiol Lung Cell Mol Physiol 279: L1137–L1145, 2000.
2. Agouridakis P, Kyriakou D, Alexandrakis MG, Prekates A, PerisinakisK, Karkavitsas N, Bouros D. The predictive role of serum and bronchoal-veolar lavage cytokines and adhesion molecules for acute respiratory distresssyndrome development and outcome. Respir Res 3: 25, 2002.
3. Ayala A, Chung CS, Lomas JL, Song GY, Doughty LA, Gregory SH,Cioffi WG, LeBlanc BW, Reichner J, Simms HH, Grutkoski PS.Shock-induced neutrophil mediated priming for acute lung injury in mice:divergent effects of TLR-4 and TLR-4/FasL deficiency. Am J Pathol 161:2283–2294, 2002.
4. Bethea JR, Chung IY, Sparacio SM, Gillespie GY, Benveniste EN.Interleukin-1 beta induction of tumor necrosis factor-alpha gene expres-sion in human astroglioma cells. J Neuroimmunol 36: 179–191, 1992.
5. Byers DE, Holtzman MJ. Alternatively activated macrophages andairway disease. Chest 140: 768–774, 2011.
6. Chaudhary LR, Avioli LV. Regulation of interleukin-8 gene expression byinterleukin-1beta, osteotropic hormones, and protein kinase inhibitors innormal human bone marrow stromal cells. J Biol Chem 271: 16591–16596,1996.
7. Cochrane CG, Spragg R, Revak SD. Pathogenesis of the adult respira-tory distress syndrome. Evidence of oxidant activity in bronchoalveolarlavage fluid. J Clin Invest 71: 754–761, 1983.
8. Coyne CB, Vanhook MK, Gambling TM, Carson JL, Boucher RC,Johnson LG. Regulation of airway tight junctions by proinflammatorycytokines. Mol Biol Cell 13: 3218–3234, 2002.
9. Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, TschoppJ. Innate immune activation through Nalp3 inflammasome sensing ofasbestos and silica. Science 320: 674–677, 2008.
10. Finigan JH, Faress JA, Wilkinson E, Mishra RS, Nethery DE, WylerD, Shatat M, Ware LB, Matthay MA, Mason R, Silver RF, Kern JA.Neuregulin-1-human epidermal receptor-2 signaling is a central regulatorof pulmonary epithelial permeability and acute lung injury. J Biol Chem286: 10660–10670, 2011.
11. Freeman BA, Panus PC, Matalon S, Buckley BJ, Baker RR. Oxidantinjury to the alveolar epithelium: biochemical and pharmacologic studies.Res Rep Health Eff Inst 54: 1–30; discussion 31–39, 1993.
12. Ganter MT, Roux J, Miyazawa B, Howard M, Frank JA, Su G, Shep-pard D, Violette SM, Weinreb PH, Horan GS, Matthay MA, Pittet JF.Interleukin-1beta causes acute lung injury via alphavbeta5 and alphavbeta6integrin-dependent mechanisms. Circ Res 102: 804–812, 2008.
13. Gordon S, Martinez FO. Alternative activation of macrophages: mech-anism and functions. Immunity 32: 593–604, 2010.
14. Grommes J, Soehnlein O. Contribution of neutrophils to acute lunginjury. Mol Med 17: 293–307, 2011.
15. He CH, Waxman AB, Lee CG, Link H, Rabach ME, Ma B, Chen Q,Zhu Z, Zhong M, Nakayama K, Nakayama KI, Homer R, Elias JA.Bcl-2-related protein A1 is an endogenous and cytokine-stimulated medi-ator of cytoprotection in hyperoxic acute lung injury. J Clin Invest 115:1039–1048, 2005.
16. Hybertson BM, Lee YM, Cho HG, Cho OJ, Repine JE. Alveolar typeII cell abnormalities and peroxide formation in lungs of rats given IL-1intratracheally. Inflammation 24: 289–303, 2000.
17. Jain D, Atochina-Vasserman EN, Tomer Y, Kadire H, Beers MF.Surfactant protein D protects against acute hyperoxic lung injury. Am JRespir Crit Care Med 178: 805–813, 2008.
18. Jiang Y, Beller DI, Frendl G, Graves DT. Monocyte chemoattractantprotein-1 regulates adhesion molecule expression and cytokine productionin human monocytes. J Immunol 148: 2423–2428, 1992.
19. Kallet RH, Matthay MA. Hyperoxic acute lung injury. Respir Care 58:123–141, 2013.
20. Knofler M, Kiss H, Mosl B, Egarter C, Husslein P. Interleukin-1stimulates tumor necrosis factor-alpha (TNF-alpha) release from cytotro-
phoblastic BeWo cells independently of induction of the TNF-alphamRNA. FEBS Lett 405: 213–218, 1997.
21. Kolliputi N, Galam L, Parthasarathy PT, Tipparaju SM, Lockey RF.NALP-3 inflammasome silencing attenuates ceramide-induced transepi-thelial permeability. J Cell Physiol 227: 3310–3316, 2012.
22. Kolliputi N, Shaik RS, Waxman AB. The inflammasome mediates hyper-oxia-induced alveolar cell permeability. J Immunol 184: 5819–5826, 2010.
23. Lei D, Lancaster JR Jr, Joshi MS, Nelson S, Stoltz D, Bagby GJ,Odom G, Shellito JE, Kolls JK. Activation of alveolar macrophages andlung host defenses using transfer of the interferon-gamma gene. Am JPhysiol Lung Cell Mol Physiol 272: L852–L859, 1997.
24. Martin WJ, 2nd Gadek JE, Hunninghake GW, Crystal RG. Oxidantinjury of lung parenchymal cells. J Clin Invest 68: 1277–1288, 1981.
25. Martinon F, Burns K, Tschopp J. The inflammasome: a molecularplatform triggering activation of inflammatory caspases and processing ofproIL-beta. Mol Cell 10: 417–426, 2002.
26. Matute-Bello G, Frevert CW, Martin TR. Animal models of acute lunginjury. Am J Physiol Lung Cell Mol Physiol 295: L379–L399, 2008.
27. McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, WaterhouseCC, Beck PL, Muruve DA, Kubes P. Intravascular danger signals guideneutrophils to sites of sterile inflammation. Science 330: 362–366, 2010.
28. Menu P, Vince JE. The NLRP3 inflammasome in health and disease: thegood, the bad and the ugly. Clin Exp Immunol 166: 1–15, 2011.
29. Miller EJ, Cohen AB, Nagao S, Griffith D, Maunder RJ, Martin TR,Weiner-Kronish JP, Sticherling M, Christophers E, Matthay MA.Elevated levels of NAP-1/interleukin-8 are present in the airspaces ofpatients with the adult respiratory distress syndrome and are associatedwith increased mortality. Am Rev Respir Dis 146: 427–432, 1992.
30. Mukhopadhyay S, Hoidal JR, Mukherjee TK. Role of TNFalpha inpulmonary pathophysiology. Respir Res 7: 125, 2006.
31. Nakamura M, Matute-Bello G, Liles WC, Hayashi S, Kajikawa O, LinSM, Frevert CW, Martin TR. Differential response of human lung epithelialcells to fas-induced apoptosis. Am J Pathol 164: 1949–1958, 2004.
32. O’Neill Immunology LA. How frustration leads to inflammation. Science320: 619–620, 2008.
33. Ogura Y, Sutterwala FS, Flavell RA. The inflammasome: first line of theimmune response to cell stress. Cell 126: 659–662, 2006.
34. Okuma T, Terasaki Y, Sakashita N, Kaikita K, Kobayashi H, Haya-saki T, Kuziel WA, Baba H, Takeya M. MCP-1/CCR2 signallingpathway regulates hyperoxia-induced acute lung injury via nitric oxideproduction. Int J Exp Pathol 87: 475–483, 2006.
35. Olman MA, White KE, Ware LB, Simmons WL, Benveniste EN, ZhuS, Pugin J, Matthay MA. Pulmonary edema fluid from patients with earlylung injury stimulates fibroblast proliferation through IL-1 beta-inducedIL-6 expression. J Immunol 172: 2668–2677, 2004.
36. Piotrowski WJ, Marczak J, Kurmanowska Z, Gorski P. Concentrationof TBA-reactive substances in type II pneumocytes exposed to oxidativestress. Arch Immunol Ther Exp (Warsz) 52: 435–440, 2004.
37. Prince LR, Allen L, Jones EC, Hellewell PG, Dower SK, Whyte MK,Sabroe I. The role of interleukin-1beta in direct and toll-like receptor4-mediated neutrophil activation and survival. Am J Pathol 165: 1819–1826, 2004.
38. Reddy NM, Kleeberger SR, Kensler TW, Yamamoto M, Hassoun PM,Reddy SP. Disruption of Nrf2 impairs the resolution of hyperoxia-inducedacute lung injury and inflammation in mice. J Immunol 182: 7264–7271,2009.
39. Rosseau S, Hammerl P, Maus U, Walmrath HD, Schutte H, Grim-minger F, Seeger W, Lohmeyer J. Phenotypic characterization of alve-olar monocyte recruitment in acute respiratory distress syndrome. Am JPhysiol Lung Cell Mol Physiol 279: L25–L35, 2000.
40. Cahill CM, Rogers JT. Interleukin (IL) 1beta induction of IL-6 ismediated by a novel phosphatidylinositol 3-kinase-dependent AKT/Ikap-paB kinase alpha pathway targeting activator protein-1. J Biol Chem 283:25900–25912, 2008.
41. Schmal H, Shanley TP, Jones ML, Friedl HP, Ward PA. Role formacrophage inflammatory protein-2 in lipopolysaccharide-induced lunginjury in rats. J Immunol 156: 1963–1972, 1996.
42. Shanley TP, Davidson BA, Nader ND, Bless N, Vasi N, Ward PA,Johnson KJ, Knight PR. Role of macrophage inflammatory protein-2 inaspiration-induced lung injury. Crit Care Med 28: 2437–2444, 2000.
43. Sica A, Matsushima K, Van Damme J, Wang JM, Polentarutti N,Dejana E, Colotta F, Mantovani A. IL-1 transcriptionally activates theneutrophil chemotactic factor/IL-8 gene in endothelial cells. Immunology69: 548–553, 1990.
C188 INFLAMMASOMES IN ACUTE LUNG INJURY
AJP-Cell Physiol • doi:10.1152/ajpcell.00086.2013 • www.ajpcell.org

44. Simon LM, Raffin TA, Douglas WH, Theodore J, Robin ED. Effects ofhigh oxygen exposure on bioenergetics in isolated type II pneumocytes. JAppl Physiol 47: 98–103, 1979.
45. Sue RD, Belperio JA, Burdick MD, Murray LA, Xue YY, Dy MC,Kwon JJ, Keane MP, Strieter RM. CXCR2 is critical to hyperoxia-induced lung injury. J Immunol 172: 3860–3868, 2004.
46. Tsujimoto H, Ono S, Mochizuki H, Aosasa S, Majima T, Ueno C,Matsumoto A. Role of macrophage inflammatory protein 2 in acute lunginjury in murine peritonitis. J Surg Res 103: 61–67, 2002.
47. Ward NS, Waxman AB, Homer RJ, Mantell LL, Einarsson O, Du Y,Elias JA. Interleukin-6-induced protection in hyperoxic acute lung injury.Am J Respir Cell Mol Biol 22: 535–542, 2000.
48. Waxman AB, Einarsson O, Seres T, Knickelbein RG, Warshaw JB,Johnston R, Homer RJ, Elias JA. Targeted lung expression of interleu-kin-11 enhances murine tolerance of 100% oxygen and diminishes hyper-oxia-induced DNA fragmentation. J Clin Invest 101: 1970–1982, 1998.
49. Weber A, Wasiliew P, Kracht M. Interleukin-1 (IL1) pathway. SciSignal 3: cm1, 2010.
50. Xiang M, Shi X, Li Y, Xu J, Yin L, Xiao G, Scott MJ, Billiar TR,Wilson MA, Fan J. Hemorrhagic shock activation of NLRP3 inflam-masome in lung endothelial cells. J Immunol 187: 4809–4817, 2011.
51. Zaher TE, Miller EJ, Morrow DM, Javdan M, Mantell LL. Hyperoxia-induced signal transduction pathways in pulmonary epithelial cells. FreeRadic Biol Med 42: 897–908, 2007.
C189INFLAMMASOMES IN ACUTE LUNG INJURY
AJP-Cell Physiol • doi:10.1152/ajpcell.00086.2013 • www.ajpcell.org
Copyright © 2022 FDOKUMEN




















