
Nitric oxide pros and cons: The role of l-arginine, a nitric oxide precursor, and idebenone, a...
8
Brain Research Bulletin 83 (2010) 49–56 Contents lists available at ScienceDirect Brain Research Bulletin journal homepage: www.elsevier.com/locate/brainresbull Research report Nitric oxide pros and cons: The role of l-arginine, a nitric oxide precursor, and idebenone, a coenzyme-Q analogue in ameliorating cerebral hypoxia in rat Nayira A. Abdel Baky a,∗ , Zeenat F. Zaidi b , Amal J. Fatani a , Mohamed M. Sayed-Ahmed a , Hazar Yaqub a a Pharmacology Department, Faculty of Pharmacy, King Saud University, Riyadh, Saudi Arabia b Anatomy Department, Faculty of Medicine, King Saud University, Riyadh, Saudi Arabia article info Article history: Received 18 April 2010 Received in revised form 6 July 2010 Accepted 7 July 2010 Available online 15 July 2010 Keywords: Nitric oxide Hypoxia ROS l-Arginine Idebenone ATP Antioxidant abstract Evidence exists that nitric oxide (NO) may mediate both protective and pathological responses dur- ing brain hypoxia (HP). Reactive oxygen species have also been implicated in the pathophysiological response of the brain tissues to HP. Therefore, this study investigated whether a NO precursor, l-arginine (l-arg), a free radical scavenger, idebenone (ID), and their combination would reduce neurological injury resulting from hemic hypoxia (HP) in rats. Adult male Wistar albino rats were injected with sodium nitrite (60 mg/kg, s.c.) to establish hemic hypoxia. ID (100 mg kg −1 , i.p.) and/or l-arg (100 mg kg −1 , i.p.) were administrated 24 and 1 h prior to sodium nitrite intoxication, respectively. Hypoxia signifi- cantly decreased hemoglobin concentration, while significantly increased serum lactate dehydrogenase (LDH), creatine phosphokinase (CPK), total nitrate/nitrite, sialic, and uric acids concentrations. More- over, brain lipid peroxides were significantly enhanced, while reduced glutathione, l-ascorbic acids, adenosine triphosphate (ATP) contents, and the activities of catalase and superoxide dismutase, were significantly reduced in the brain tissue. Pretreatment with either ID or l-arg altered the majority of the above-mentioned biochemical changes in hypoxic rats. Additionally, the combination of these two agents significantly reduced injury marker enzyme activities as well as serum sialic, and uric acids level (P > 0.05 vs. control). Moreover, this combination exerted a synergistic antioxidant effect by blocking the induction of lipid peroxidation, preserving brain energy (ATP) content, and greatly reducing the hypoxic alterations in brain enzymatic and non-enzymatic antioxidants. Histopathological examination of the brain tissue supported these biochemical findings. This study showed that ID and l-arg were capable of reducing neurological injury following HP in rat, and support the idea of the usefulness of l-arg and ID as prophylaxis from hypoxic brain injury. © 2010 Elsevier Inc. All rights reserved. 1. Introduction Cerebral hypoxia–ischemia is predisposing factor for the inci- dence of cerebral infarction that represents the third leading cause of death and a major cause of long-term disability [55]. The pathogenesis of cerebral hypoxia has been associated with a time- dependent cascade of molecular events including: rapid depletion of intracellular adenosine triphosphate (ATP) stores, anaerobic gly- colysis, activation of calcium-stimulated enzymes, mitochondrial dysfunction, and formation of reactive oxygen species (ROS) that contribute to the oxidative brain damage [2,19,73]. One of the experimental models used to simulate human cerebral hypoxia syndrome is the administration of sodium nitrite in high dose ∗ Corresponding author at: Department of Pharmacology, Faculty of Pharmacy, King Saud University, Riyadh 11495, P.O. Box 22452, Saudi Arabia. Tel.: +966 1 2913728; fax: +966 1 2913728. E-mail addresses: [email protected], [email protected] (N.A.A. Baky). to induce hemic hypoxia that finally result in neuronal damage [41,42]. Formation of methemoglobin, a substance that interferes with the ability of blood cells to carry oxygen, is the most recog- nizable and detectable sign of nitrite toxicity in humans [21]. In addition, high dose of nitrite salts generate excessive free radicals that finally aggravate cellular brain injury [60,12]. In the last years, the involvement of nitric oxide (NO) in control of the physiological responses to brain hypoxia has been sug- gested, however its role in the mechanisms of hypoxic/ischemic brain injury has been debatable [11,37,66]. Part of the problem has been that the complex biological characteristics of NO, suggest- ing that this agent can be either detrimental or beneficial to the injured brain. The NO produced by the neuronal and inducible iso- forms of NO synthetase (NOS) can be neurotoxic during hypoxia [35,56]. In contrast, NO produced by the endothelial isoform of NOS is beneficial [36]. Endothelial-derived NO may limit neuronal damage through its powerful vasodilating effects on vascular beds [93]. In the brain itself NO may has neuroprotective role through scavenging of reactive oxygen species [52,67,90], and by its anti- inflammatory effect [5]. Consequently, administration of NO has 0361-9230/$ – see front matter © 2010 Elsevier Inc. All rights reserved. doi:10.1016/j.brainresbull.2010.07.004
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Nitric oxide pros and cons: The role of l-arginine, a nitric oxide precursor, and idebenone, a...

R
Ni
Na
b
a
ARRAA
KNHRlIAA
1
dopdocdces
KT
(
0d
Brain Research Bulletin 83 (2010) 49–56
Contents lists available at ScienceDirect
Brain Research Bulletin
journa l homepage: www.e lsev ier .com/ locate /bra inresbul l
esearch report
itric oxide pros and cons: The role of l-arginine, a nitric oxide precursor, anddebenone, a coenzyme-Q analogue in ameliorating cerebral hypoxia in rat
ayira A. Abdel Bakya,∗, Zeenat F. Zaidib, Amal J. Fatania, Mohamed M. Sayed-Ahmeda, Hazar Yaquba
Pharmacology Department, Faculty of Pharmacy, King Saud University, Riyadh, Saudi ArabiaAnatomy Department, Faculty of Medicine, King Saud University, Riyadh, Saudi Arabia
r t i c l e i n f o
rticle history:eceived 18 April 2010eceived in revised form 6 July 2010ccepted 7 July 2010vailable online 15 July 2010
eywords:itric oxideypoxiaOS-ArgininedebenoneTPntioxidant
a b s t r a c t
Evidence exists that nitric oxide (NO) may mediate both protective and pathological responses dur-ing brain hypoxia (HP). Reactive oxygen species have also been implicated in the pathophysiologicalresponse of the brain tissues to HP. Therefore, this study investigated whether a NO precursor, l-arginine(l-arg), a free radical scavenger, idebenone (ID), and their combination would reduce neurological injuryresulting from hemic hypoxia (HP) in rats. Adult male Wistar albino rats were injected with sodiumnitrite (60 mg/kg, s.c.) to establish hemic hypoxia. ID (100 mg kg−1, i.p.) and/or l-arg (100 mg kg−1,i.p.) were administrated 24 and 1 h prior to sodium nitrite intoxication, respectively. Hypoxia signifi-cantly decreased hemoglobin concentration, while significantly increased serum lactate dehydrogenase(LDH), creatine phosphokinase (CPK), total nitrate/nitrite, sialic, and uric acids concentrations. More-over, brain lipid peroxides were significantly enhanced, while reduced glutathione, l-ascorbic acids,adenosine triphosphate (ATP) contents, and the activities of catalase and superoxide dismutase, weresignificantly reduced in the brain tissue. Pretreatment with either ID or l-arg altered the majority ofthe above-mentioned biochemical changes in hypoxic rats. Additionally, the combination of these two
agents significantly reduced injury marker enzyme activities as well as serum sialic, and uric acids level(P > 0.05 vs. control). Moreover, this combination exerted a synergistic antioxidant effect by blocking theinduction of lipid peroxidation, preserving brain energy (ATP) content, and greatly reducing the hypoxicalterations in brain enzymatic and non-enzymatic antioxidants. Histopathological examination of thebrain tissue supported these biochemical findings. This study showed that ID and l-arg were capable ofury foxic b
reducing neurological injas prophylaxis from hypo
. Introduction
Cerebral hypoxia–ischemia is predisposing factor for the inci-ence of cerebral infarction that represents the third leading causef death and a major cause of long-term disability [55]. Theathogenesis of cerebral hypoxia has been associated with a time-ependent cascade of molecular events including: rapid depletionf intracellular adenosine triphosphate (ATP) stores, anaerobic gly-olysis, activation of calcium-stimulated enzymes, mitochondrial
ysfunction, and formation of reactive oxygen species (ROS) thatontribute to the oxidative brain damage [2,19,73]. One of thexperimental models used to simulate human cerebral hypoxiayndrome is the administration of sodium nitrite in high dose∗ Corresponding author at: Department of Pharmacology, Faculty of Pharmacy,ing Saud University, Riyadh 11495, P.O. Box 22452, Saudi Arabia.el.: +966 1 2913728; fax: +966 1 2913728.
E-mail addresses: [email protected], [email protected]. Baky).
361-9230/$ – see front matter © 2010 Elsevier Inc. All rights reserved.oi:10.1016/j.brainresbull.2010.07.004
llowing HP in rat, and support the idea of the usefulness of l-arg and IDrain injury.
© 2010 Elsevier Inc. All rights reserved.
to induce hemic hypoxia that finally result in neuronal damage[41,42]. Formation of methemoglobin, a substance that interfereswith the ability of blood cells to carry oxygen, is the most recog-nizable and detectable sign of nitrite toxicity in humans [21]. Inaddition, high dose of nitrite salts generate excessive free radicalsthat finally aggravate cellular brain injury [60,12].
In the last years, the involvement of nitric oxide (NO) in controlof the physiological responses to brain hypoxia has been sug-gested, however its role in the mechanisms of hypoxic/ischemicbrain injury has been debatable [11,37,66]. Part of the problem hasbeen that the complex biological characteristics of NO, suggest-ing that this agent can be either detrimental or beneficial to theinjured brain. The NO produced by the neuronal and inducible iso-forms of NO synthetase (NOS) can be neurotoxic during hypoxia[35,56]. In contrast, NO produced by the endothelial isoform of
NOS is beneficial [36]. Endothelial-derived NO may limit neuronaldamage through its powerful vasodilating effects on vascular beds[93]. In the brain itself NO may has neuroprotective role throughscavenging of reactive oxygen species [52,67,90], and by its anti-inflammatory effect [5]. Consequently, administration of NO has
5 search
bhaahtN
imTctbii[tbetIi
obonipirn
2
2
pw
2
1Ksa(bTC
2
riI(pIl
tfdfwctH
0 N.A.A. Baky et al. / Brain Re
een considered to be a possible candidate treatment for acuteypoxia–ischemia. One of the useful sources of NO for study innimal models is l-arginine (l-arg). It has been reported that l-rg supplementation enhances endothelial NO production both inumans [34,53] and in experimental animals [8]. In addition, sys-emic as well as central administration of l-arg is able to enhanceO production in rat brain [70,91].
It has been suggested that ROS are generated after hypoxicnsult [29,89], and may constitute the basis to explain the enhance-
ent of brain injury occurring hours after the onset of hypoxia.hus, antioxidants and/or free radical scavengers capable of easilyrossing the brain barrier were postulated to provide protec-ion in experimental hypoxia models [28,75]. Idebenone (ID), aenzoquinone derivative, is a compound with protective capabil-
ty against cerebrovascular disorders in vivo, including cerebralschemia [61] as well as hypertension-induced vascular lesions92]. It appears to act both as a radical scavenger and as an electronrapping agent. The antioxidant properties of ID were determinedy the inhibition of lipid peroxidation through its free radical scav-nger activity [80]. The protective effects of ID are also attributedo improvement in cerebral energy metabolism [6,7]. Furthermore,D penetrates the brain rapidly and ameliorates neurological deficitn rats with experimental cerebral ischemia [83,58].
Since time is of narrow therapeutical window that can reverser interfere with the progression of neuronal damage after cere-ral hypoxia, the search for effective agents capable of preventingr reducing the progression of neuronal injury in patients at risk ofeurodegenerative diseases further supports the research. Accord-
ng to this purpose, it has been decided to investigate whetherharmacologic increase of NO using l-arginine, and a free rad-
cal scavenger, idebenone, would ameliorate neurological injuryesulting from experimental model of cerebral hypoxia (sodiumitrite-induced hypoxia) in rats.
. Materials and methods
.1. Drugs and chemicals
l-Arginine, idebenone, and sodium nitrite used in this study were analyticallyure product of Sigma–Aldrich Chemical Co., St. Louis, MO, USA. All other chemicalsere of the highest analytical grade.
.2. Experimental animals
All experiments were carried out with male Wistar albino rats weighing60–180 g, obtained from Experimental Animal Care Center, College of Pharmacy,ing Saud University, Riyadh, Saudi Arabia. The animals were housed in stainlessteel cages (5 animals/cage). Animals were acclimated with free access to tap waternd standard pellet diet (Purina Chow) in a facility with controlled temperature22–24 ◦C) and humidity (50–60%), on a 12-h light/12-h dark cycle, for at least 1 weekefore the experiments. All animals were fasted for 3 h prior to drugs administration.he protocol of this study has been approved by the Research Ethics Committee ofollege of Pharmacy, King Saud University, Riyadh, kingdom of Saudi Arabia (KSA).
.3. Experimental protocol
The animals were randomized into five groups, each group consisted of 10ats—(I) Normal group: normal animals treated with saline; (II) Hypoxic (HP) group:n which rats treated with sodium nitrite (60 mg/kg, s.c) to induce hypoxia [24]; (III)D/HP group: in which rats were injected with (100 mg/kg, i.p) of ID 1 h prior to HP;IV) l-arg/HP group: animals were injected with l-arg (100 mg/kg, i.p) 24 h then 1 hrior to HP; and (V) l-arg + ID/HP group: rats were treated with the same doses of
D and l-arg prior to sodium nitrite as previously mentioned. The doses of ID and-arg were determined based on prior studies [26,18].
After 1 h of sodium nitrite injection, all rats were sacrificed by cervical decapi-ation, blood was collected, plasma and serum were separated and stored at −80 ◦Cor further various biochemical estimations. The brain tissue was separated imme-
iately from each animal, washed with ice-chilled physiological saline and used forurther biochemical estimations. A known weight of the brain tissue from each groupas homogenized in normal saline or the appropriate buffer as indicated in the pro-edures of measurement of each parameter. The homogenate was centrifuged andhe supernatant was used for the estimation of various biochemical parameters.istological assessment was carried out at the end of the experiment.
Bulletin 83 (2010) 49–56
2.4. Biochemical analysis
2.4.1. Determination of blood hemoglobin and serum biochemical parametersHemoglobin concentration was determined colorimetrically using Drabkin’s
reagent according to the method of Kjeldsberg [44]. Serum biochemical parame-ters including the activity of either LDH or CPK were measured using commerciallyavailable kits (Bio-Mèrieux-RCS, Lyon, France). In addition, serum sialic acid concen-tration was assessed using periodate-resorcinol method described by Jourdian et al.[40], while serum uric acid was determined according to the method of Slaunwhiteet al. [74]. Total serum nitrate/nitrite level was estimated according to the methoddescribed by Green et al. [25] using Griess reagent.
2.4.2. Determination of lipid peroxides level in brain tissueThe degree of lipid peroxidation in brain tissues was determined by measuring
thiobarbituric acid reactive substances (TBARS) in the supernatant tissue from brainhomogenate [84]. The absorbance was measured spectrophotometrically at 532 nmand quantified as nanomoles of malondialdehyde (MDA)/g wet tissue.
2.4.3. Determination of non-enzymatic antioxidants concentrations in braintissue
Brain tissue levels of acid-soluble thiols, mainly reduced glutathione (GSH), weredetermined colorimetrically at 412 nm [17]. Homogenates were precipitated withtrichloroacetic acid, and after centrifugation, supernatants were used for the estima-tion of protein thiols (Protein-SH) expressed as �mol/gm wet tissue. l-Ascorbic acid(vitamin C) concentration was determined using Folin-Ciocalteu reagent, and thecolor reaction was measured spectrophotometrically at 760 nm. Vitamin C contentwas expressed as �g ascorbic acid/g wet tissue [45].
2.4.4. Determination of brain enzymatic antioxidant activitiesSuperoxide dismutase (SOD) activity was assayed spectrophotometrically as
described by Marklund [51]. This method is based on the capacity of SOD in inhibit-ing autoxidation of pyrogallol. The color reaction was measured at 420 nm. One unitof enzyme was defined as the amount of enzyme required to inhibit the rate of pyro-gallol autoxidation by 50%. The enzymatic activity was expressed as units (U)/mgprotein. Catalase (CAT) activity was assayed by the method of Aebi [1]. The princi-ple of the method was based on the determination of the rate constant (k) of theH2O2 decomposition rate at 240 nm. The H2O2 added to the assessment medium isdecomposed by the catalyzing activity of CAT enzyme, and reduction in absorbanceat 240 nm is monitored with as spectrophotometer. The reduction in absorbanceis direct proportional to the enzyme activity. The results were expressed as k/mgprotein.
2.4.5. Determination of adenosine triphosphate (ATP) content in brain tissueATP content in brain tissues was estimated using HPLC technique according
to the method described by Volonté et al. [87]. In brief, brain tissue was homoge-nized with 0.4 M perchloric acid (20% homogenate), and then homogenates wereprecipitated with 0.8 ml 0.2 M potassium hydroxide and centrifuged at 3000 rpm at0 ◦C for 10 min. An aliquot (20 �l) of the filtered supernatant was injected into theHPLC system. The chromatographic separation of ATP was performed using a C18reversed-phase column, at flow rate 1 ml/min. The mobile phase was composed of215 mM potassium dihydrogen phosphate, 2.3 mM tetrabutylammonium hydrogensulfate, 4% acetonitrile, 0.4% potassium hydroxide (1 M). The peak elution was fol-lowed at 220 nm. Standard solutions of ATP (0.4 mg/ml) in 0.4 M perchloric acidwere prepared in the same way as described for the sample preparation. The ATPconcentration is expressed as nmol/g wet tissue.
2.4.6. Determination of tissue protein contentProtein was determined according to Lowry et al. [50] using Bovine Serum Albu-
min (BSA) as standard, at 660 nm.
2.5. Histopathologial examination
The brains were removed and placed overnight in fixative containing 10% for-malin. Cerebellum were paraffin-embedded for hematoxylin and eosin (H and E)staining and cut at 5 �m in the coronal plane. Fifty Purkinje cells along the line ofcells randomly chosen portions of each section were counted at a magnification of20×. The reduction in cerebellar Purkinje cells damage was used as a parameter toexamine the effects of ID and/or l-arg on experimentally induced brain hypoxia. Thecriteria for neuronal damage were: (1) Cell swelling: which is represented by cellswith visible intact nucleus, and with an increased cytoplasm/nucleus ratio of 20%or more compared to adjacent cell; (2) Autolytic cells: cells with no nucleus and nodistinct cellular morphology; (3) Darkened shrunken cell: cells filled with dark stainand have a darkened outline. Cells in a total of five sections were counted to computethe ratio of damaged cells to intact neurons, and calculated as a percentage.
2.6. Statistical analysis
The experimental data were statistically analyzed using one-way analysis ofvariance (ANOVA) followed by Bonferroni test for multiple comparisons. Data wereexpressed as mean ± S.D. Differences were considered significant at P ≤ 0.05.
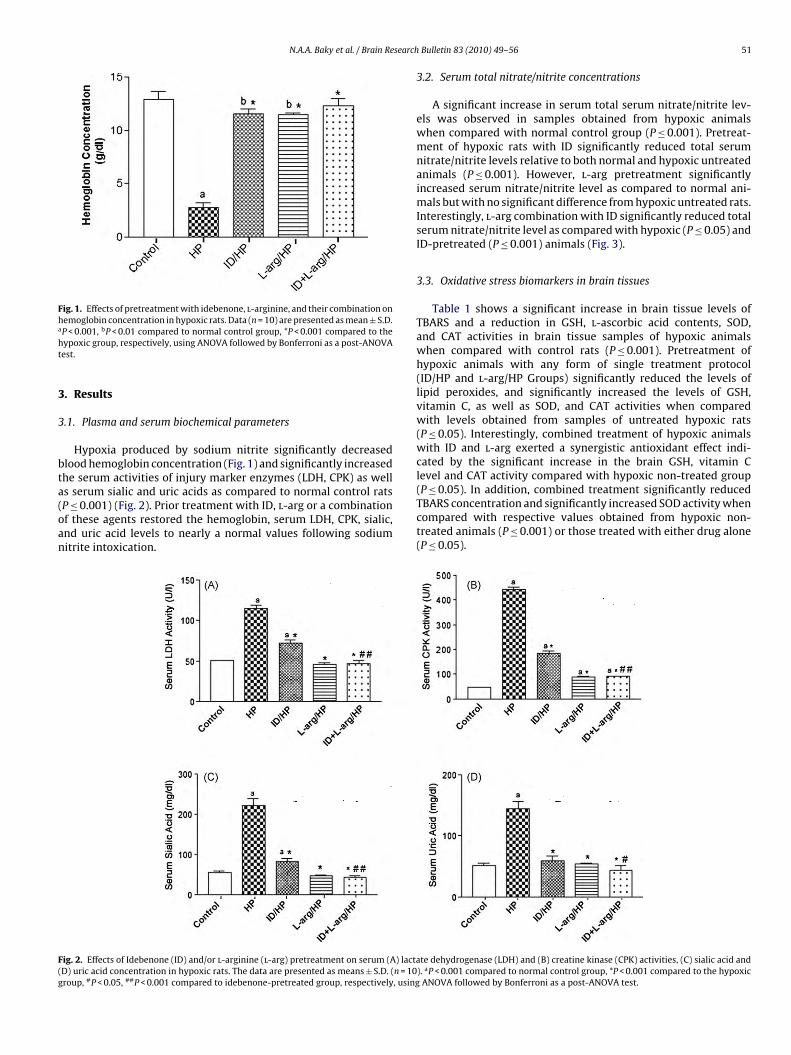
N.A.A. Baky et al. / Brain Research
Fig. 1. Effects of pretreatment with idebenone, l-arginine, and their combination onhemoglobin concentration in hypoxic rats. Data (n = 10) are presented as mean ± S.D.a
ht
3
3
bta(oan
(P ≤ 0.05). In addition, combined treatment significantly reduced
F(g
P < 0.001, bP < 0.01 compared to normal control group, *P < 0.001 compared to theypoxic group, respectively, using ANOVA followed by Bonferroni as a post-ANOVAest.
. Results
.1. Plasma and serum biochemical parameters
Hypoxia produced by sodium nitrite significantly decreasedlood hemoglobin concentration (Fig. 1) and significantly increasedhe serum activities of injury marker enzymes (LDH, CPK) as wells serum sialic and uric acids as compared to normal control rats
P ≤ 0.001) (Fig. 2). Prior treatment with ID, l-arg or a combinationf these agents restored the hemoglobin, serum LDH, CPK, sialic,nd uric acid levels to nearly a normal values following sodiumitrite intoxication.ig. 2. Effects of Idebenone (ID) and/or l-arginine (l-arg) pretreatment on serum (A) lactaD) uric acid concentration in hypoxic rats. The data are presented as means ± S.D. (n = 10roup, #P < 0.05, ##P < 0.001 compared to idebenone-pretreated group, respectively, using
Bulletin 83 (2010) 49–56 51
3.2. Serum total nitrate/nitrite concentrations
A significant increase in serum total serum nitrate/nitrite lev-els was observed in samples obtained from hypoxic animalswhen compared with normal control group (P ≤ 0.001). Pretreat-ment of hypoxic rats with ID significantly reduced total serumnitrate/nitrite levels relative to both normal and hypoxic untreatedanimals (P ≤ 0.001). However, l-arg pretreatment significantlyincreased serum nitrate/nitrite level as compared to normal ani-mals but with no significant difference from hypoxic untreated rats.Interestingly, l-arg combination with ID significantly reduced totalserum nitrate/nitrite level as compared with hypoxic (P ≤ 0.05) andID-pretreated (P ≤ 0.001) animals (Fig. 3).
3.3. Oxidative stress biomarkers in brain tissues
Table 1 shows a significant increase in brain tissue levels ofTBARS and a reduction in GSH, l-ascorbic acid contents, SOD,and CAT activities in brain tissue samples of hypoxic animalswhen compared with control rats (P ≤ 0.001). Pretreatment ofhypoxic animals with any form of single treatment protocol(ID/HP and l-arg/HP Groups) significantly reduced the levels oflipid peroxides, and significantly increased the levels of GSH,vitamin C, as well as SOD, and CAT activities when comparedwith levels obtained from samples of untreated hypoxic rats(P ≤ 0.05). Interestingly, combined treatment of hypoxic animalswith ID and l-arg exerted a synergistic antioxidant effect indi-cated by the significant increase in the brain GSH, vitamin Clevel and CAT activity compared with hypoxic non-treated group
TBARS concentration and significantly increased SOD activity whencompared with respective values obtained from hypoxic non-treated animals (P ≤ 0.001) or those treated with either drug alone(P ≤ 0.05).
te dehydrogenase (LDH) and (B) creatine kinase (CPK) activities, (C) sialic acid and). aP < 0.001 compared to normal control group, *P < 0.001 compared to the hypoxic
ANOVA followed by Bonferroni as a post-ANOVA test.

52 N.A.A. Baky et al. / Brain Research Bulletin 83 (2010) 49–56
Table 1Effects of pretreatment with idebenone and/or l-arginine, and their combination on lipid peroxidation, non-enzymatic, and enzymatic antioxidants of hypoxic brain tissues.
Group MDA (nmol/g tissue) Vitamin C (�g/g tissue) GSH ((mol/g tissue) SOD (U/mg protein) CAT (k/mg protein)
Control 106.09 ± 2.32 19.84 ± 1.86 2.032 ± 1.03 0.925 ± 0.03 2.111 ± 0.21HP 182.55 ± 12.94a 8.19 ± 0.40a 1.576 ± 0.06a 0.171 ± 0.05a 0.367 ± 0.10a
ID/HP 128.62 ± 8.33ab 14.30 ± 1.02ab 1.938 ± 0.11b 0.558 ± 0.14ab 1.073 ± 0.26ab
l-arg/HP 114.28 ± 13.64b 11.76 ± 0.56ab 1.713 ± 0.05ab 0.721 ± 0.09ab 1.613 ± 0.33ab
ID + l-arg/HP 89.51 ± 5.46bcd 14.48 ± 0.56abd
Values are means of 10 ± S.D. (a), (b), (c), and (d) indicate significant change from contrrespectively, at P < 0.05 using ANOVA followed by Bonferoni as a post-ANOVA test.
Fig. 3. Effects of pretreatment with idebenone, l-arginine, and their combi-nation on serum total nitrate/nitrite concentrations in hypoxic rats. Data arep a
*ir
3
rbtIAsnw
Fompr
resented as mean ± S.D. (n = 10). P < 0.001 compared to normal control group,P < 0.05,**P < 0.001 compared to the hypoxic group, #P < 0.001 compared todeenone-pretreated group, $P < 0.001 compared to l-arginine-pretreated group,espectively, using ANOVA followed by Bonferroni as a post-ANOVA test.
.4. Adenosine triphosphate (ATP) content in brain tissue
The effects of ID and l-arg on ATP level in brain tissues of hypoxicats are shown in Fig. 4. Hypoxia resulted in a significant decease inrain ATP level (383.3 ± 14.5 nmol/g tissue vs. 822.01 ± 1.04 nmol/gissue in normal control group, P < 0.001). Administration of eitherD or l-arg prior to induction of hypoxia significantly elevated brain
TP level (574.6 ± 3.25 nmol/g tissue and 479.05±7.85 nmol/g tis-ue, respectively, P < 0.001) as compared to the values of hypoxicon-treated group. Interestingly, pretreatment of hypoxic animalsith ID and l-arg produced significant increase in brain ATP levelsig. 4. Effects of pretreatment with idebenone, l-arginine, and their combinationn adenosine triphosphate level in hypoxic brain tissues. Data are presented asean ± S.D. (n = 10). aP < 0.001 compared to normal control group, *P < 0.001 com-
ared to the hypoxic group, $P < 0.001 compared to l-arginine-pretreated group,espectively, using ANOVA followed by Bonferroni as a post-ANOVA test.
1.711 ± 0.03abc 0.910 ± 0.04bcd 1.669 ± 0.32bc
ol, sodium nitrite-treated, idebenone-, and l-arginine-pretreated hypoxic groups,
(583.8 ± 18.2 nmol/g tissue, P < 0.001) as compared to l-arg pre-treated and hypoxic non-treated groups, respectively.
3.5. Histopathological examination of brain tissues
Histopathological examination of brain tissues revealed thatthere was no marked difference in the general appearance andthickness of the cerebellar cortex in all groups. However, the Purk-inje cells showed marked autolytic changes in all of the treatedgroups (Fig. 5B–F). Extensive damage was noted in HP group, inwhich almost all the neurons were affected. Most of the cells werelarge, swollen, their arrangement as a single layer was also dis-turbed (Fig. 5B and C). ID pretreated rats showed little inhibition inthe number of damaged purkinje cells, while l-arg/HP and ID + l-arg/HP pretreated groups showed marked reduction in the numberof damaged Purkinje cells compared to HP group. The percent ofdamaged cell in different groups are shown in Table 2.
4. Discussion
Cerebellar Purkinje cells represent a group of neurons highlyvulnerable to ischemic and hypoxic stress [88]. Short-termischemia/hypoxia exposure elicits dose-dependent cerebellarPurkinje and fastigial neuron damage [64]. Cerebellar structures,are preferentially targeted during chronic hypoxic exposure [9,10].Rat cerebellar slice exposed to hypoxia elicited by a 30 min expo-sure to artificial cerebrospinal fluid continuously gassed with 95%N2:5% CO2 induced a characteristic type of toxicity of Purkinjecells in which these cells exhibited marked rounded appearancewith cytoplasmic darkening, nuclear condensation and cytoplas-mic vacuolization [4]. This is consistent with the hypoxic structuralmodifications caused by sodium nitrite treatment in the currentstudy as shown in Fig. 4B and C. Pretreatment of hypoxic ratswith combination of ID and l-arg nearly prevented the damagingeffect of nitrite intoxication that was manifested microscopicallyby extensive improvements in the histological features of Purk-inje cells as well as marked reduction in the number of damagedPurkinje cells of pretreated rats (Fig. 4F, and Table 2).
Nitrite in toxic doses has been reported to induce hemichypoxia. The underlying mechanism of such effect is mainly dueto increased methemoglobin formation [21,23]. The significantdecrease in hemoglobin concentration after sodium nitrite treat-ment in the current study was previously reported by Kohan et al.[46]. In this study, l-arg- or ID-pretreatment significantly increasedhemoglobin concentration in hypoxic rats. More interestingly, theircombination resulted in a nearly complete reversal of sodiumnitrite-induced anemia. These findings are in line with the result ofTarumoto et al. [82] in which l-arg administration to anemic elderlypatients suffering from renal disease, resulted in an increases intheir hemoglobin levels and improvement in their renal function.
Nitrite methemoglobinemia is a known to cause for free-radicalgeneration [46], as nitrite can stimulate oxidation of ferrous ionsin oxyhemoglobin to form methemoglobin as well as various ROS[23,63]. The brain is particularly susceptible to hypoxic oxida-tive injury because of its abundant lipid content, and its limited

N.A.A. Baky et al. / Brain Research Bulletin 83 (2010) 49–56 53
Fig. 5. Light microscopic photographs representing Purkinje cells of brain tissue stained with H&E (original magnification 20×). (A) A single row of normal Purkinje cells(black arrows), which are pyramidal cells arranged in a single row between the outer molecular layer and the inner granular layers of the cerebellar cortex; (B and C) differents k arroa of norw r, G =t
arpetitadCTwdaabclobs
TE
tages of neuronal damage due to HP; normal (short black arrow), swollen (long blacnd the regular arrangement in a single layer is also disturbed; (D–F) a single rowith idebenone, l-arginine, and their combination, respectively. M = molecular laye
his figure legend, the reader is referred to the web version of the article.)
ntioxidant defense systems [30]. These ROS can initiate a wideange of toxic oxidative reactions, including an increase in lipideroxidation, and an impairment of both enzymatic and non-nzymatic antioxidant defense systems of brain tissues [31,14]. Inhe present study, acute nitrite intoxication caused a significantncrease in brain MDA content, a hallmark of oxidative modificationo membrane lipids, a significant decrease in brain non-enzymaticntioxidants (GSH and l-ascorbic acid) as well as a significantecrease in the activities of brain antioxidant enzymes (SOD andAT) which support the possibility of increased ROS production.he current results are in agreement with other previous reports inhich sodium nitrite treatment significantly increased lipid peroxi-ation and produced significant decrease in SOD and CAT activitiess well as depleted intracellular GSH contents of the brain, heartnd liver of rats [24,63,68]. Brain lipid peroxidation was inhibitedy ID and l-arg pretreatment, and the same combination signifi-antly altered the hypoxic-induced changes in brain GSH content.
-arg is an important nutrient for critically ill patients, and mostf its pharmacological actions are attributed to NO. One possi-le mechanism to explain the observed cellular protection in thistudy is through NO termination of the chain reaction of lipidable 2ffects of pretreatment with idebenone, l-arginine, and their combination on different hi
Group Swollen cells Autolysed cells
Control 26 16HP 187 347ID/HP 43 171l-arg/HP 29 88ID+l-arg/HP 31 79
w), darkly stained (long red arrows), and autolysed (short red arrow) Purkinje cells,mal (black arrows) and darkly stained (red arrow) Purkinje cells of rat pretreatedgranular layer, WM = white matter. (For interpretation of the references to color in
peroxidation by scavenging peroxyl radicals [54]. NO has beenalso reported to prevent both Cu2+ induced lipid peroxidation inlow-density lipoproteins [69] and radical-induced accumulationof lipid hydroperoxides in phosphatidylcholine liposomes [33].Moreover, Utepbergenov et al. [85] reported that NO offered pro-tection from hypoxia/reperfusion-induced increase in blood-brainbarrier permeability and lipid peroxidation through scavengingROS. l-arg itself has been reported to protect tissues and organsagainst ischemia–reperfusion injury as evidenced from the signifi-cant increase in level of total antioxidant, and the decrease in levelof MDA in rats [47]. Moreover, l-arg significantly reduced MDAlevel and increased GSH content in the heart tissue of exhaustivelyexercised rats [49]. On the other hand, ID has been reported to pre-serve non-protein thiols, and inhibit lipid peroxidation in rat brainduring post-cardiac arrest reperfusion [26]. Cardoso et al. [6] alsodemonstrated that ID treatment prevented the increase in lipidperoxidation and the depletion in GSH content of synaptosomes
exposed to ascorbate/iron oxidative stress. All of these findings aug-ment the current results in which the combination of both agentssignificantly decreased brain damage by synergistically inhibitingmembrane lipid peroxidation and increasing cellular level of GSH.stopathological features of hypoxic brain cells.
Dark shrunken cells Damaged cell %
23 1345 95.818 66.454 34.247 31.4

5 search
aptitipwLdHitacoIstL[
oRtnadsetpst
saiarsriheaoAema[
rbswiIse
iqw
4 N.A.A. Baky et al. / Brain Re
ROS not only caused peroxidation of the polyunsaturated fattycids in the neuronal membrane structure, but also affected theroteins localized within the membrane, therefore altering thehree dimensional structure of the membrane, and finally disrupt-ng its selective permeability characteristics [43]. This will lead tohe consequent release of injury marker enzymes whose releases correlated with changes in plasma membrane integrity and/orermeability. These are in harmony with the results of the presentork in which hypoxic rats showed significant increase in serum
DH and CPK activities. Such increase offers another supporting evi-ence of hypoxic oxidative damage due to sodium nitrite treatment.owever, the level of these markers were significantly reduced
n ID and/or l-arg pretreated hypoxic rats, which coincides withhe improvement in histological architecture of Purkinje cells inll treated groups (Fig. 4D–F), with maximal improvement in theombination group. These findings are in line with the resultsf Muscoli et al. [57] who reported that liposomally entrappedD significantly reduced ethanol-induced injury of astrocytes ashown by the decrease of ethanol-induced LDH release. In addi-ion, l-arg supplementation was also reported to decrease plasmaDH and CPK activities following acute exhaustive exercise in rats49].
Ascorbic acid has been reported as strong reducing againstxidants, thus affording considerable protection to the cells [86].educed level of ascorbic acid in hypoxic brain tissue suggestshe participation of this non-enzymatic antioxidant defense in theitrite-evoked toxic in brain of rats. This observed decrease inscorbic acid level, that associated with increased lipid peroxi-ation, clearly characterize hypoxic oxidative stress induced byodium nitrite. The present data is in agreement with the knowl-dge that normal physiological levels of ascorbate in the brainissue cannot be maintained during hypoxia [76]. Both ID and l-argretreatment significantly increased ascorbic acid level in brain tis-ue as compared to hypoxic rats which strength the possibility ofheir synergistic antioxidant effect.
Reduction in the activities of antiperoxidative enzymes inodium nitrite intoxicated rats is most probably an adaptive mech-nism to counteract lipid peroxidation of brain tissue. As thisncreased generation of free radicals mostly exceeded the collectivebility of free radical scavenging enzymes to quench these radicals,esulting in oxidative brain damage. ID and/or l-arg pretreatmentignificantly elevated brain SOD and CAT activities in hypoxicats. This indicates that both agents have free radicals scaveng-ng activity allow them to attenuate the nitrite-induced oxidativeypoxic stress. In harmony with this, ID has been shown to scav-nge a variety of radical species including peroxyl, peroxy nitritend organic hydroperoxides, as well as reducing non-respiratoryxygen consumption associated with lipid peroxidation [32,79].dditionally, ID also may indirectly reduce oxidative brain stress bylevating nerve growth factor [81,62] which acutely blocks ROS for-ation in the brain [65,16]. l-arg has a protective role against ROS
ttack due to its direct chemical interaction with superoxide anions48].
The current significant elevation in serum sialic acid of hypoxicats is in line with data of Baev et al. [3], in which elevated lipid-ound sialic acid content was previously reported in the earliesttages of sodium nitrite-induced hypoxia in rats. Serum sialic acidas also reported to be significantly increased in oxidative stress-
nduced tissue damage [72]. Pretreatment of hypoxic rat with eitherD and/or l-arg significantly ameliorated this increase in serumialic acid level, which further supports their powerful antioxidant
ffect.Another important finding in this study was the significantncrease in serum nitrite/nitrate level in hypoxic rats, with conse-uent increase in brain MDA concentration. Serum nitrite/nitrateas significantly decreased upon pretreatment of hypoxic animals
Bulletin 83 (2010) 49–56
with ID or ID and l-arg combination but not with l-arg pretreat-ment. However, the increase in NO level in l-arg pretreated groupwas accompanied with a significant decrease in MDA concentra-tion, and an increase in GSH content of hypoxic brain. Obviously,rats in hypoxic group had significant increases in free radicals gen-eration after hypoxia with resultant damaging effect on the braintissue. Data of l-arg pretreated group suggests that pharmaco-logic increase in NO levels did not exacerbate the increase in freeradical formation. In fact, high level of NO/or l-arg itself in l-argpretreated group may be protective, probably due to their abilityto scavenging of free radicals as well as inhibiting xanthenes oxi-dase (XO) enzyme [54,20]. The present results are in agreementwith the study of Lin et al. [49], in which NO was significantlyincreased in rats treated with l-arg after exhaustive exercise, anincrease accompanied by a significant decrease in MDA level andXO activity in myocardial tissue. On the other hand, ID treatmentsuppressed leukocyte-enhanced cold ischemia/reperfusion injuryof liver endothelium through almost complete suppression of theendothelial constitutive NOS mRNA expression after reperfusion[71]. Moreover, ID treatment reduced NO generation and apoptosisin hepatocytes treated with toxic bile salt glycochenodeoxycholate[27].
Acute reduction in cerebral oxygen delivery known to lead tothe breakdown of neuronal energy metabolism [39,15]. Addition-ally, high concentrations of ROS, resulting from impaired oxidativephosphorylation or electron leakage, eventually leads to ATP deple-tion [77]. This is consistent with results of this study in whichATP level was markedly reduced in hypoxic brain tissue. However,ID and/or l-arg pretreatment ameliorated the depleting effect ofnitrite-induced hypoxia on brain ATP content, suggesting that theirprotective effect may be mediated through improving the cere-bral energy metabolism. Moreover, ID has been proved to inhibitreduction in brain ATP content in experimental cerebral ischemiaand stroke-prone spontaneously hypertensive rats [58,59]. It seemsthat, ID improve cerebral mitochondrial oxidative metabolism inpatients with mitochondrial encephalopathy [38]. In addition, itsrole in scavenging ROS and inhibiting lipid peroxidation could bean important factor in improving mitochondrial respiratory chain.Studies have shown that under cellular low oxygen conditions IDprevents the free radical damaging effect and maintains relativelynormal cell ATP levels [22].
Hypoxia compromises membrane functions as well as cellularenzymes and may result in conversion of xanthine dehydrogenaseto XO, which oxidizes hypoxanthine in an NADPH-dependent man-ner, producing xanthine and uric acid, and catalyzing the reductionof molecular oxygen to form both O2
− and hydrogen peroxide [78].Therefore, serum uric acid level has been postulated as an impor-tant marker in oxidative stress [13]. The present data support thesefacts as the decrease in brain ATP content in hypoxic rats wasaccompanied with significant increase in brain MDA content andserum uric acid concentrations. l-arg supplementation has beenproven to significantly reduce the increased in cardiac XO activity,MDA levels, and serum uric acid caused by exhaustive exercise inrats [49]. This is in harmony with the present result in which eitherl-arg alone or in combination with ID significantly reduced serumuric acid level in hypoxic rats. Hence, the protective role of l-argand ID could probably be due to their property of scavenging freeradicals, in addition to the inhibitory effect of l-arg on XO.
The present results suggest that l-arg and/or ID are likelyresponsible for brain tissue protection in hypoxic rats through theiraction on NO generation, reduction as well as scavenging of ROS,
maintenance of normal enzymatic and non-enzymatic antioxidantsystems, and improvement of energy production. Therefore, thecurrent finding may have important implications in the develop-ment of therapeutic strategies aimed at manipulating l-arg and IDsupplementation for prophylaxis from hypoxic brain injury.
search
A
Cw
R
[
[[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
N.A.A. Baky et al. / Brain Re
cknowledgment
The authors gratefully acknowledge the Saudi Basic Industriesorporation (SABIC)-Riyadh-KSA for the financial support of thisork in the form of Research Fellowship.
eferences
[1] H. Aebi, Catalase, in: H.U. Bergmeyer (Ed.), Methods of Enzymatic Analysis, 2nded., Verlag Chemie, Weinheim, Germany, 1974, pp. 673–685.
[2] S.F. Amerisco, S. Sahai, Mechanisms of ischemia in situ vascular occlusivedisease, in: K.M.A. Welch, L.R. Caplan, D.J. Reis, B.K. Siesjö (Eds.), Primer onCerebrovascular Disease, Academic Press, San Diego, 1997, pp. 279–285.
[3] V.I. Baev, I.V. Vasil’eva, N.N. Nalivaeva, Rat brain gangliosides during hypoxia,Bull. Exp. Biol. Med. 118 (1) (1994) 698–700.
[4] P. Barenberg, H. Strahlendorf, J. Strahlendorf, Hypoxia induces an excitotoxic-type of dark cell degeneration in cerebellar Purkinje neurons, Neurosci. Res. 40(2001) 245–254.
[5] P.M.W. Bath, D.G. Hassall, A.M. Gladwin, R.M.J. Palmer, J.F. Martin, Nitricoxide and prostacyclin. Divergence of inhibitory effects on monocyte chemo-taxis and adhesion to endothelium in vitro, Arterioscler. Thromb. 11 (1991)254–260.
[6] S.M. Cardoso, C. Pereira, C.R. Oliveira, The protective effect of vitamin E,idebenone and reduced glutathione on free radical mediated injury in rat brainsynaptosomes, Biochem. Biophys. Res. Commun. 246 (1998) 703–710.
[7] S.M. Cardoso, C. Pereira, C.R. Oliveira, Mitochondrial function is differentiallyaffected upon oxidative stress, FRBM 26 (1999) 3–13.
[8] M.R. Cernadas, A. López-Farré, A. Riesco, M.J. Gallego, G. Espinosa, E. Digiuni,L. Hernando, S. Casado, C. Caramelo, Renal and systemic effects of aminoacidsadministered separately: comparison between l-arginine and non-nitric oxidedonor aminoacids, J. Pharmacol. Exp. Ther. 263 (1992) 1023–1029.
[9] J. Cervos-Navarro, N.H. Diemer, Selective vulnerability in brain hypoxia, Crit.Rev. Neurobiol. 6 (1991) 149–182.
10] J. Cervos-Navarro, S. Sampaolo, G. Hamdorf, Brain changes in experimentalchronic hypoxia, Exp. Pathol. (Jena) 42 (1991) 205–212.
11] D.W. Choi, Proc. Natl. Acad. Sci. U.S.A. 90 (1993) 9741–9743.12] C.K. Chow, C.B. Hong, Dietary vitamin E and selenium and toxicity of nitrite and
nitrate, Toxicology 180 (2002) 195–207.13] C.C. Chuang, S.C. Shiesh, C.H. Chi, Y.F. Tu, L.I. Hor, C.C. Shieh, M.F. Chen, Serum
total antioxidant capacity reflects severity of illness in patients with severesepsis, Crit. Care 10 (2006) 1–7.
14] P.M. Clarkson, H.S. Thompson, Antioxidants: what role do they play in physicalactivity and health? Am. J. Clin. Nutr. 72 (2000) 637S–646S.
15] M. Delivoria-Papadopoulos, O.P. Mishra, Mechanisms of prenatal cerebralinjury in fetus and newborn, Ann. N.Y. Acad. Sci. 900 (2000) 159–168.
16] L.L. Dugan, D.J. Creedon, E.M. Johnson Jr., D.M. Holtzman, Rapid suppression offree radical formation by nerve growth factor involves the mitogen-activatedprotein kinase pathway, Proc. Natl. Acad. Sci. U.S.A. 94 (1997) 4086–4091.
17] G.L. Ellman, Tissue sulfhydryl groups, Arch. Biochem. Biophys. 74 (1959)214–226.
18] M.A. El-Missiry, A.I. Othman, M.A. Amer, l-arginine ameliorates oxidative stressin alloxan-induced experimental diabetes mellitus, J. Appl. Toxicol. 24 (2004)93–97.
19] M. Endres, D. Biniszkiewicz, R.W. Sobol, Harás Ch, M. Ahmadi, J. Katchanovb, P.Mergenthaler, U. Dirnagl, S.H. Wilson, A. Meisel, R. Jaenisch, Increased postis-chemic brain injury in mice deficient in uracil-DNA glycosylase, J. Clin. Invest.113 (2004) 1711–1721.
20] M. Fukahori, K. Ichimori, H. Ishida, H. Nakagawa, H. Okino, Nitric oxidereversibly suppresses xanthine oxidase activity, Free Radic. Res. 21 (1994)203–212.
21] J. Ger, H. Kao, T.S. Shih, J.F. Deng, Fatal toxic methemoglobinemia due to occu-pational exposure to methyl nitrite, Clin. Med. J. 57 (1996) S78.
22] V. Geromel, N. Darin, D. Chretien, B. Paule, P. Benit, P. DeLonlay, A. Rotig, A.Munnich, P. Rustin, Coenzyme Q10 and idebenone in the therapy of respira-tory chain diseases: rationale and comparative benefits, Mol. Genet. Metab. 77(2002) 21–30.
23] M.T. Gladwin, J.H. Crawford, R.P. Patel, The biochemistry of nitric oxide, nitrite,and hemoglobin: role in blood flow regulation, Free Rad. Biol. Med. 36 (2004)707–717.
24] O. Gonchar, I. Mankovskaya, E. Klyuchko, Role of complex nucleosides in thereversal of oxidative stress and metabolic disorders induced by acute nitritepoisoning, IJP 38 (2006) 414–418.
25] L. Green, D. Wagner, J. Glogowski, P. Skipper, J. Wishnok, S. Tannenbaum, Anal-ysis of nitrate, nitrite, and [15N] nitrate in biological fluids, Anal. Biochem. 126(1982) 131–138.
26] P. Grieb, M.S. Ryba, G.S. Debicki, W. Gordon-Krajcer, S. Januszewski, S.J. Chra-pusta, Changes in oxidative stress in the rat brain during post-cardiac arrestreperfusion, and the effect of treatment with the free radical scavenger
idebenone, Resuscitation 39 (1998) 107–113.27] E. Gumpricht, R. Dahl, B. Yerushalmi, M.W. Devereaux, R.J. Sokol, Nitric oxideameliorates hydrophobic bile acid-induced apoptosis in isolated rat hepato-cytes by non-mitochondrial pathways, J. Biol. Chem. 277 (2002) 25823–25830.
28] E.D. Hall, P.K. Andrus, S.L. Smith, J.A. Oostveen, H.M. Scherch, B.S. Lutzke, T.J.Raub, G.A. Sawada, J.R. Palmer, L.S. Banitt, J.S. Tustin, K.L. Belonga, D.E. Ayer,
[
[
Bulletin 83 (2010) 49–56 55
G.L. Bundy, Neuroprotective efficacy of microvascularly-localized versus brain-penetrating antioxidants, Acta Neurochir. (Wien) 66 (Suppl.) (1996) 107–113.
29] B. Halliwell, Reactive oxygen species and the central nervous system, J. Neu-rochem. 59 (1992) 1609–1623.
30] B. Halliwell, J.M.C. Gutteridge, Oxygen radicals and the nervous system, TrendsNeurosci. 8 (1985) 22–26.
31] B. Halliwell, J.M.C. Gutteridge, Free Radicals in Biology and Medicine, 3rd ed.,Oxford University Press, Oxford, 1999.
32] J.T. Hancock, Superoxide, hydrogen peroxide and nitric oxide as signalingmolecules: their production and role in disease, Br. J. Med. Sci. 54 (1997) 38–46.
33] K. Hayashi, N. Noguchi, E. Niki, FEBS Lett. 370 (1995) 37–40.34] K. Hishikawa, T. Nakaki, M. Tsuda, H. Esumi, H. Ohshima, H. Suzuki, T. Saruta, R.
Kato, Effect of systemic l-arginine administration on hemodynamic and nitricoxide release in man, Jpn. Heart J. 33 (1992) 41–48.
35] Z. Huang, P.L. Huang, N. Panahian, T. Dalkara, M.C. Fishman, M.A. Moskowitz,Effects of cerebral ischemia in mice deficient in neuronal nitric oxide synthase,Science 265 (1994) 1883–1885.
36] Z. Huang, P.L. Huang, J. Ma, W. Meng, C. Ayata, M.C. Fishman, M.A. Moskowitz,Enlarged infarcts in endothelial nitric oxide synthase knockout mice are atten-uated by nitro-l-arginine, J. Cereb. Blood Flow Metab. 16 (1996) 981–987.
37] C. Iadecola, D.A. Pelligrino, M.A. Moskowitz, N.A. Lassen, Nitric oxide synthaseinhibition and cerebrovascular regulation, J. Cereb. Blood Flow Metab. 14 (2)(1994) 175–192.
38] Y. Ikejiri, E. Mori, K. Ishii, K. Nishimoto, M. Yasuda, M. Sasaki, Idebenoneimproves cerebral mitochondrial oxidative metabolism in a patient withMELAS, Neurology 47 (1996) 583–585.
39] A. Jensen, Y. Garnier, J. Middelanis, R. Berger, Perinatal brain damage frompathophysiology to prevention, Eur. J. Obstet. Gynecol. Reprod. Biol. 110 (2003)70–79.
40] G.W. Jourdian, L. Dean, S.A. Roseman, Periodate-resorcinol method for thequantitative estimation of free sialic acids and the glycosides, J. Biol. Chem.246 (1971) 430–435.
41] S. Jyoti, S. Sushma, S. Shashi, T. Anjana, Protective effect of tulsi (Ocimum sanc-tum) on lipid peroxidation in stress induced by anemic hypoxia in rabbits, Ind.J. Physiol. Pharmacol. 47 (1) (2003) 115–119.
42] S. Jyoti, S. Satendra, S. Sushma, T. Anjana, S. Shashi, Antistressor activity of Oci-mum sanctum (Tulsi) against experimentally induced oxidative stress in rabbits,Methods Find Exp. Clin. Pharmacol. 29 (6) (2007) 411–416.
43] S. Kahraman, A.T. Demiryurek, Propofol is a peroxynitrite scavenger, Anesth.Analg. 84 (1997) 1127–1129.
44] C.R. Kjeldsberg, Principles of hematologic examination, in: G.R. Lee, T.C. Bittell,J. Foerster, J.W. Athens, J.N. Lukens (Eds.), Wintrobe’s Clinical Hematology, vol.1, Lea & Febiger, Philadelphia, London, 1993, pp. 7–37.
45] T. Kleszczewski, E. Kleszczewska, Flow injection spectrophotometric determi-nation of l-ascorbic acid in biological matters, J. Pharm. Biomed. Anal. 29 (2002)755–759.
46] M.C. Kohn, R.L. Melnick, F. Ye, C.J. Portier, Pharmacokinetics of sodium nitrite-induced methemoglobinemia in the rat, Drug Metab. Dispos. 30 (2002)676–683.
47] H. Krauss, A. Jablecka, P. Sosnowski, P. Bogdanski, Influence of l-arginineon the nitric oxide concentration and level of oxidative stress duringischemia–reperfusion injury in a rat model, Int. J. Clin. Pharmacol. Ther. 47(2009) 533–538.
48] A. Lass, A. Suessenbacher, G. Wolkart, B. Mayer, F. Brunner, Functional andanalytical evidence for scavenging of oxygen radicals by l-arginine, Mol. Phar-macol. 61 (2002) 1081–1088.
49] W. Lin, S. Yang, S. Tsai, C. Huang, N. Lee, l-Arginine attenuates xanthine oxidaseand myeloperoxidase activities in hearts of rats during exhaustive exercise, Br.J. Nutr. 95 (2006) 67–75.
50] O.H. Lowry, M.J. Roesborough, A.L. Farr, R.J. Randall, Protein measurement withFolin-Phenol reagent, J. Biol. Chem. 193 (1951) 265–275.
51] S.L. Marklund, Pyrogallol antioxidation, in: R.A. Greenwald (Ed.), Handbook ofMethods for Oxygen Radical Research, CRC Press, Boca Raton, FL, USA, 1985,pp. 243–247.
52] R.B. Mason, R.M. Pluta, S. Walbridge, D.A. Wink, E.H. OldWeld, R.J. Boock,Production of reactive oxygen species after reperfusion in vitro and in vivo:protective effect of nitric oxide, J. Neurosurg. 93 (2000) 99–107.
53] S. Mehta, D.J. Stewart, R.D. Levy, The hypotensive effect of l-arginine isassociated with increased expired nitric oxide in humans, Chest 109 (1996)1550–1555.
54] A.M. Miles, D.S. Bohle, P.A. Glassbrenner, B. Hansert, D.A. Wink, M.B. Grisham,J. Biol. Chem. 271 (1996) 40–47.
55] J.P. Mohr, L.R. Caplan, J.W. Melski, The Harvard cooperative stroke registry: aprospective registry, Neurology 28 (1978) 754–762.
56] M.A. Moro, A. Cardenas, O. Hurtado, J.C. Leza, I. Lizasoain, Role of nitric oxideafter brain ischaemia, Cell Calcium 36 (2004) 265–275.
57] C. Muscoli, M. Fresta, V. Cardile, M. Palumbo, M. Renis, G. Puglisi, D. Paolino, S.Nistico, D. Rotiroti, V. Mollace, Ethanol-induced injury in rat primary corticalastrocytes involves oxidative stress: effect of idebenone, Neurosci. Lett. 329(2002) 21–24.
58] A. Nagaoka, M. Suno, M. Shibota, M. Kakihana, Effects of idebenone on neuro-logical deficits, local cerebral blood flow, and energy metabolism in rats withexperimental cerebral ischemia, Arch Gerontol. Geriatr. 8 (1989) 193–202.
59] A. Nagaoka, M. Kakihana, K. Fujiwara, Effects of idebenone on neurologi-cal deficits following cerebrovascular lesions in stroke-prone spontaneouslyhypertensive rats, Arch Gerontol. Geriatr. 8 (1989) 203–212.

5 search
[
[
[
[
[
[
[[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
6 N.A.A. Baky et al. / Brain Re
60] S.R. Naik, V.W. Pilgaonkar, V.S. Panda, Evaluation of antioxidant activity ofGinkgo biloba phytosomes in rat brain, Phytother. Res. 20 (2006) 1013–1016.
61] A. Nakaoga, A. Shino, M. Kakihana, H. Iwatsuka, Inhibitory effect of idebenone(CV-2619), a novel compound, on vascular lesions in hypertensive rats, Jpn. J.Pharmacol. 36 (1984) 291–299.
62] A. Nitta, T. Hasegawa, T. Nabeshima, Oral administration of idebenone, a stimu-lator of NGF synthesis recovers reduced NGF content in aged rat brain, Neurosci.Lett. 163 (1993) 219–222.
63] S. Padmaja, G.L. Squadrito, W.A. Pryor, Inactivation of glutathione peroxidaseby peroxynitrite, Arch. Biochem. Biophys. 349 (1998) 1–6.
64] E. Pae, P. Chien, R.M. Harper, Intermittent hypoxia damages cerebellar cortexand deep nuclei, Neurosci. Lett. 375 (2005) 123–128.
65] Z. Pan, R. Perez-Polo, Role of nerve growth factor in oxidant homeostasis: glu-tathione metabolism, J. Neurochem. 61 (1993) 1713–1721.
66] D.A. Pelligrino, J. Neurosurg. Anesthesiol. 5 (1993) 221–231.67] R.M. Pluta, R. Rak, D.A. Wink, J.J. Woodward, A. Khaldi, E.H. OldWeld, J.C. Wat-
son, Effects of nitric oxide on reactive oxygen species production and infarctionsize after brain reperfusion injury, Neurosurgery 48 (2001) 884–892.
68] N.B. Poberezkina, O.V. Zadorina, P.I. Andriushchenko, Iu.V. Khmelevski, Therole of peroxidation processes and antioxidant protection in nitrite-inducedhypoxia and its correction with vitamins, Ukr. Biokhim. Zh. 64n (1992) 64–70.
69] H. Rubbo, S. Parthasarathy, S. Barnes, M. Kirk, B. Kalyanaraman, B.A. Freeman,Arch. Biochem. Biophys. 324 (1994) 15–25.
70] M. Salter, C. Duffy, J. Garthwaite, P.J.L.M. Strijbos, Ex vivo measurement of braintissue nitrite and nitrate accurately reflects nitric oxide synthase activity invivo, J. Neurochem. 66 (1996) 1683–1690.
71] E. Schutz, E. Wiland, A. Hensel, P. Niedmann, A. Dreiss, V.W. Armstrong, P.Schuff-werner, M. Oellerich, Suppression of leukocyte-enhanced cold ischemiareperfusion injury of liver endothelium with the benzoquinone antioxidantidebenone, Clin. Biochem. 30 (1997) 619–624.
72] K. Seyrek, E. Yaylak, H. Aksit, Serum sialic acid, malondialdehyde, retinol, zincand copper concentrations in dairy cows with Lameness, Bull. Vet. Inst. Pulawy52 (2008) 281–284.
73] N.R. Sims, M.F. Anderson, Mitochondrial contributions to tissue damage instroke, Neurochem. Int. 40 (2002) 511–526.
74] W.D. Slaunwhite, L.A. Pachla, D.C. Wenke, P.T. Kissinger, Colorimetric, enzy-matic, and liquid-chromatographic methods for serum uric acid, Clin. Chem.21 (1975) 1427–1429.
75] M. Soehle, A. Heimann, O. Kempski, Postischemic application of lipid perox-idation inhibitor U-101033E reduces neuronal damage after global cerebral
ischemia in rats, Stroke 29 (1998) 1240–1247.76] J.H. Song, S.H. Shin, G.M. Ross, Oxidative stress by ascorbate causes neuronaldamage in an in vitro system, Brain Res. 895 (2001) 66–72.
77] V. Srinivasan, S.R. Pandi-Perumal, G.J.M. Maestroni, A.I. Esquifino, R. Hardeland,D.P. Cardinali, Role of melatonin in neurodegenerative diseases, Neurotox. Res.7 (2005) 293–318.
[
[
Bulletin 83 (2010) 49–56
78] K.L. Stokes, D.N. Granger, Role of nitric oxide in ischemia reperfusion injury, in:L.J. Ignarro (Ed.), Nitric oxide: Biology and Pathobiology, Academic Press, NY,2000, pp. 633–647.
79] Y. Sugiyama, T. Fujita, Stimulation of the respiratory and phosphorylating activ-ities in rat brain mitochondria by idebenone (CV-2619), a new agent improvingcerebral metabolism, FEBS Lett. 184 (1985) 48–51.
80] M. Suno, Z. Terashita, A. Nagaoka, Inhibition of platelet aggregation byidebenone and the mechanism of inhibition, Arch. Gerontol. Geriatr. 8 (1989)313–321.
81] R. Takeuchi, K. Murase, Y. Furukawa, S. Furukawa, K. Hayashi, Stimulation ofnerve growth factor synthesis/secretion by 1,4-benzoquinone and its deriva-tives in cultured mouse astroglial cells, FEBS Lett. 261 (1990) 63–66.
82] T. Tarumoto, S. Imagawa, M. Kobayashi, A. Hirayama, K. Ozawa, T. Nagasawa,l-Arginine administration reverses anemia associated with renal disease, Int.J. Hematol. 86 (2007) 126–129.
83] H. Torii, K. Yoshida, T. Kobayashi, T. Tsukamoto, S. Tanayama, Disposition ofidebenone (CV-2619), a new cerebral metabolism improving agent, in rats anddogs, J. Pharmacobio. Dyn. 8 (1985) 457–467.
84] M. Uchiyama, M. Mihara, Determination of malondialdehyde precursor in tis-sues by thiobarbituric acid treatment, Anal. Biochem. 86 (1978) 271–278.
85] D.I. Utepbergenov, K. Mertsch, A. Sporbert, K. Tenz, M. Paul, R.F.Haselo, I.E. Blasig, Nitric oxide protects blood-brain barrier in vitro fromhypoxia/reoxygenation-mediated injury, FEBS Lett. 424 (1998) 197–201.
86] A. Van der Vliet, C.E. Cross, Oxidants, nitrosants, and the lung, Am. J. Med. 109(2000) 398–421.
87] M.G. Volonté, G. Yulna, P. Quiroga, A.E. Consolini, Development of an HPLCmethod for determination of metabolic compounds in myocardial tissue, J.Pharm. Biomed. Anal. 35 (2004) 647–653.
88] J.P. Welsh, G. Yuen, D.G. Placantonakis, T.Q. Vu, F. Haiss, E. O’Hearn, M.E. Mol-liver, S.A. Aicher, Why do Purkinje cells die so easily after global brain ischemia?Aldolase C, EAAT4, and the cerebellar contribution to post hypoxic myoclonus,Adv. Neurol. 89 (2002) 331–359.
89] J.X. Wilson, Antioxidant defense of the brain: a role for astrocytes, Can. J. Physiol.Pharmacol. 75 (1997) 1149–1163.
90] D.A. Wink, J.A. Cook, R. Pacelli, J. Liebmann, M.C. Krishna, J.B. Mitchell, Nitricoxide (NO) protects against cellular damage by reactive oxygen species, Toxicol.Lett. 82–83 (1995) 221–226.
91] K. Yamada, T. Nabeshima, Simultaneous measurement of nitrite and nitratelevels as indices of nitric oxide release in the cerebellum of conscious rats, J.Neurochem. 68 (1997) 1234–1243.
92] N. Yamazaki, Y. Take, A. Nagaoka, Y. Nagawa, Beneficial effect of idebenone(CV-2619) on cerebral ischemia-induced amnesia in rats, Jpn. J. Pharmacol. 36(1984) 349–356.
93] F. Zhang, C. Iadecola, Reduction of focal cerebral ischemic damage by delayedtreatment with nitric oxide donors, J. Cereb. Blood Flow Metab. 14 (1994)574–580.




















