
Nitric oxide mediates neurologic injury after hypothermic circulatory arrest
9
1999;67:65-71 Ann Thorac Surg Lowenstein, Mary E. Blue, Michael V. Johnston and William A. Baumgartner Elaine E. Tseng, Malcolm V. Brock, Mary S. Lange, Juan C. Troncoso, Charles J. Nitric oxide mediates neurologic injury after hypothermic circulatory arrest http://ats.ctsnetjournals.org/cgi/content/full/67/1/65 on the World Wide Web at: The online version of this article, along with updated information and services, is located Print ISSN: 0003-4975; eISSN: 1552-6259. Southern Thoracic Surgical Association. Copyright © 1999 by The Society of Thoracic Surgeons. is the official journal of The Society of Thoracic Surgeons and the The Annals of Thoracic Surgery by on May 30, 2013 ats.ctsnetjournals.org Downloaded from
Transcript of Nitric oxide mediates neurologic injury after hypothermic circulatory arrest

1999;67:65-71 Ann Thorac SurgLowenstein, Mary E. Blue, Michael V. Johnston and William A. Baumgartner
Elaine E. Tseng, Malcolm V. Brock, Mary S. Lange, Juan C. Troncoso, Charles J. Nitric oxide mediates neurologic injury after hypothermic circulatory arrest
http://ats.ctsnetjournals.org/cgi/content/full/67/1/65on the World Wide Web at:
The online version of this article, along with updated information and services, is located
Print ISSN: 0003-4975; eISSN: 1552-6259. Southern Thoracic Surgical Association. Copyright © 1999 by The Society of Thoracic Surgeons.
is the official journal of The Society of Thoracic Surgeons and theThe Annals of Thoracic Surgery
by on May 30, 2013 ats.ctsnetjournals.orgDownloaded from

Nitric Oxide Mediates Neurologic Injury AfterHypothermic Circulatory ArrestElaine E. Tseng, MD, Malcolm V. Brock, MD, Mary S. Lange, MA,Juan C. Troncoso, MD, Charles J. Lowenstein, MD, Mary E. Blue, PhD,Michael V. Johnston, MD, and William A. Baumgartner, MDDivision of Cardiac Surgery, Johns Hopkins Medical Institutions and Kennedy-Krieger Research Institute, Baltimore, Maryland
Background. Prolonged hypothermic circulatory arrest(HCA) causes neurologic injury. However, the mecha-nism of this injury is unknown. We hypothesized thatHCA causes nitric oxide production to result in neuronalnecrosis. This study was undertaken to determinewhether the neuronal nitric oxide synthase inhibitor17477AR reduces necrosis after HCA.
Methods. Thirty-two dogs underwent 2 hours of HCAat 18°C. Nitric oxide synthase catalytic assay and intrace-rebral microdialysis for nitric oxide production wereperformed in acute nonsurvival experiments (n 5 16).Sixteen animals survived for 72 hours after HCA: Group1 (n 5 9) was treated with 17477AR (Astra Arcus), andgroup 2 (n 5 7) received vehicle only. Animals werescored from 0 (normal) to 500 (coma) for neurologicfunction and from 0 (normal) to 100 (severe) for neuronalnecrosis.
Results. Administration of 17477AR reduced nitric ox-ide production in the striatum by 94% (HCA alone),3.65 6 2.42 mmol/L; HCA and 17477AR, 0.20 6 0.14mmol/L citrulline). Dogs treated with 17477AR after HCAhad superior neurologic function (62.22 6 29.82 for group1 versus 141.86 6 61.53 for group 2, p 5 0.019) andsignificantly reduced neuronal necrosis (9.33 6 4.67 forgroup 1 versus 38.14 6 2.23 for group 2, p < 0.00001)compared with untreated HCA dogs.
Conclusions. Our results provide evidence that neuro-nal nitric oxide synthase mediates neuronal necrosis afterHCA and plays a significant role in HCA-induced neu-rotoxicity. Pharmacologic strategies to inhibit neuronalnitric oxide synthase after the ischemic period of HCAmay be clinically beneficial.
(Ann Thorac Surg 1999;67:65–71)© 1999 by The Society of Thoracic Surgeons
Hypothermic circulatory arrest (HCA) enables abloodless operative field, unobstructed by vascular
clamps and cannulas. It facilitates repair of aortic archand thoracic aortic lesions in addition to complex con-genital heart lesions [1–3]. However, the brain is exquis-itely sensitive to ischemia and prohibits lengthy periodsof arrest. Morbidity and mortality increase substantiallywhen the duration of HCA exceeds 45 to 60 minutes [4].Impaired intellectual development, choreoathetosis, andlearning and memory deficits are characteristically de-layed neurologic sequelae after prolonged HCA [5–7].
Previous studies in our laboratory have shown thatglutamate excitotoxicity plays a role in HCA-inducedneurologic damage [8–10]. Recent evidence suggests thatnitric oxide (NO) mediates glutamate excitotoxicity [11].In addition, we previously showed that HCA causesinduction of neuronal nitric oxide synthase (nNOS) andthat inhibition of nNOS reduces apoptosis, one form ofneuronal cell death after HCA [12, 13]. In the presentstudy, we hypothesized that HCA results in increasedNO production and that NO mediates ischemic necrosis.
Our purpose was to determine whether nNOS inhibitionreduces neuronal necrosis in a canine model of HCA.
Material and Methods
PreparationOur canine model of hypothermic circulatory arrest hasbeen described elsewhere [12, 13]. Thirty-two heart-worm-negative conditioned male hound dogs (weight, 20to 27 kg; age, 7 to 12 months) were used. Anesthesticinduction with sodium thiopental (3.0 mg/kg intrave-nously) followed by endotracheal intubation was per-formed. Animals were maintained with halothane (0.5%to 2.0%) anesthesia and 100% oxygen.
Bilateral tympanic membrane, nasopharyngeal, andrectal temperature probes were placed. Tympanic mem-brane temperature closely correlates with brain temper-ature. Electrocardiographic monitoring was used. ASwan-Ganz catheter was placed percutaneously throughthe left external jugular vein, and a left femoral arterycatheter was placed for blood pressure and arterial bloodgas determinations.
Cardiopulmonary Bypass and HypothermicCirculatory ArrestThe cardiopulmonary bypass (CPB) circuit consisted of amembrane oxygenator (Cobe Laboratories, Inc, Lake-
Presented at the Forty-fourth Annual Meeting of the Southern ThoracicSurgical Association, Naples, FL, Nov 6–8, 1997.
Address reprint requests to Dr Baumgartner, Division of Cardiac Surgery,Johns Hopkins Hospital, Blalock 618, 600 N Wolfe St, Baltimore,MD 21287 (e-mail: [email protected]).
© 1999 by The Society of Thoracic Surgeons 0003-4975/99/$20.00Published by Elsevier Science Inc PII S0003-4975(98)01363-0
by on May 30, 2013 ats.ctsnetjournals.orgDownloaded from

wood, CO), roller pump (Sarns Inc, Ann Arbor, MI), and40-mm in-line arterial filter. The circuit was primed with1.5 L of lactated Ringer’s solution with 50 mEq of sodiumbicarbonate and 10 mEq of potassium chloride. Aftersystemic heparinization (300 U/kg intravenously), dogswere cannulated for closed chest CPB. An arterial can-nula (12F to 14F) was placed in the descending aorta fromthe right femoral artery, and venous cannulas (18F to 20F)were advanced to the level of the right atrium throughthe right external jugular and femoral veins.
Cardiopulmonary bypass was instituted, and the dogswere cooled by surface (ice bags around the head and acooling blanket) and core (CPB) cooling to a tympanicmembrane temperature of 18°C, at which time the arte-rial pump was turned off, and venous blood was drainedby gravity into the reservoir. During CPB, mean arterialpressure was maintained at 50 to 60 mm Hg with pumpflows of 80 to 100 mL/kg and was reduced to 60 mL/kgwhen the temperature was less than 32°C. Arterial bloodgas levels were controlled using the alpha-stat strategy.
Circulatory arrest was maintained for 2 hours at 18°C,followed by reinstitution of CPB and rewarming. Sodiumbicarbonate (50 mEq) and furosemide (20 mg) weregiven. At normothermia (37°C), dogs were weaned fromCPB and decannulated. The right femoral artery and veinwere ligated. Protamine was administered. Wounds wereirrigated and closed.
Postoperatively, 16 dogs were monitored for 8 to 20hours after HCA and were sacrificed for acute experi-ments. Ventilation was continued, and anesthesia wasmaintained with intravenous fentanyl (10 to 20 mg/kg)and midazolam (1 mg) as needed. The remaining 16animals survived for 72 hours and were weaned from theventilator at 20 hours. The electrocardiogram, arterialblood pressure, Swan-Ganz catheter variables, and uri-nary output, as well as arterial blood gas, hemoglobin,and glucose levels were monitored in the intensive careunit. Animals were sacrificed fully anesthetized andperfused with either ice-cold saline or 4% paraformalde-hyde. Five normal dogs that did not undergo HCA werealso sacrificed, and their brains were harvested for nor-mal control values.
17477AR ProtocolExperimental dogs (group 1, n 5 9) received the selectivenNOS inhibitor 17477AR (Astra Arcus USA, Rochester,NY) as a 1.5-mg/kg intravenous infusion for 30 minutes(0.5 mg/mL in lactated Ringer’s solution titrated to pH4.0) after HCA at decannulation and every 12 hours for 24hours postoperatively. The half-life of 17477AR’s abilityto inhibit nitric oxide synthase (NOS) activity was ap-proximately 12 hours, according to Astra Arcus. The HCAcontrol animals (group 2, n 5 7) received vehicle only.
Intracerebral MicrodialysisIntracerebral microdialysis was performed in 16 nonsur-vival experiments animals that subsequently underwentHCA. Eight dogs were used to measure inhibition of NOproduction by 17477AR; the remaining 8 served as theHCA control group. Animals were positioned in a Kopf
stereotactic device. The right side of the skull was ex-posed, and burr holes were placed 3 mm caudal to thecoronal suture and 8 mm from the midsagittal axis. Thedura was opened, and microdialysis probes (CMA 10/4;Acton, MA) were stereotactically placed to a depth of20 mm into the corpus striatum. Confirmation of properplacement was determined at sacrifice by brain cuttingand visualization of the probe tract. Tissue was allowedto equilibrate after probe placement for 180 minutes.Warmed artificial cerebrospinal fluid (mmol/L concen-tration: sodium chloride, 131.8, sodium bicarbonate, 24.6,calcium chloride, 2.0, potassium chloride, 3.0, magnesiumchloride, 0.65, urea, 6.7, and dextrose, 3.7) was filteredand continuously bubbled with 95% nitrogen and 5%carbon dioxide until oxygen and carbon dioxide tensionswere similar to those of normal brain cerebrospinal fluid.Artificial cerebrospinal fluid was infused through theinflow cannula at 1 mL/min and serially collected every 30minutes. Effluent was assayed by high-performance liq-uid chromatography with electrochemical detection forextracellular amino acid concentrations [14, 15]. Citrul-line concentration was measured and used as a marker ofNO production. All concentrations were not corrected forprobe efficiency.
Nitric Oxide Synthase AssayCortical tissues were collected, homogenized, and as-sayed for NOS activity, as described by Bredt and Snyder[16]. This assay measured the amount of carbon-14 (14C)arginine converted to 14C-citrulline by calcium-dependent NOS.
Clinical Neurologic Injury EvaluationSurviving dogs were neurologically assessed every 12hours for 72 hours after HCA, according to a species-specific behavior scale developed and validated for dogsat the International Resuscitation Research Center, Uni-versity of Pittsburgh [17]. Five components of neurologicfunction were evaluated, including level of conscious-ness, breathing pattern, cranial nerve function, motorand sensory function, and behavior; each was scoredfrom 0 (normal) to 100 (severe injury), for a total of 0(normal function) to 500 (brain death).
Histopathologic AnalysisHistopathologic analysis was performed in all survivingdogs at 72-hours after HCA. The right hemisphere ofeach brain was post-fixed in 10% formalin and embeddedin paraffin, and 8-mm sections were stained with hema-toxylin and eosin or cresyl violet. The left hemispherewas sectioned coronally, frozen on dry ice, and stored at285°C for NOS assay.
Hematoxylin and eosin–stained sections of 25 anatomicregions were examined in blinded manner by a neuro-pathologist (J.T.) for neuronal necrosis. Each populationof neurons was scored as follows: normal 5 0; fewischemic neurons 5 1; moderate ischemic neurons 5 2;infarct with loss of neurons 5 3; and hemorrhagic in-farct 5 4. Neuronal populations were then divided intosix anatomically related regions for comparison between
66 TSENG ET AL Ann Thorac SurgNO MEDIATES NEUROLOGIC INJURY AFTER HCA 1999;67:65–71
by on May 30, 2013 ats.ctsnetjournals.orgDownloaded from

experimental groups of dogs. Total histopathologic scorefor each brain was the sum of all scores for each of 25anatomic regions, ranging from 0 (normal) to 100 (severeinjury).
Statistical AnalysesResults are expressed as mean 6 standard deviation ofthe population. Comparisons between groups were madeby analysis of variance for repeated measures or Stu-dent’s t test where appropriate.
Animal CareAll experimental protocols were preapproved by theAnimal Care and Use Committee of the Johns HopkinsMedical Institutions. Animals received humane care incompliance with the “Guide for the Care and Use ofLaboratory Animals” published by the National Insti-tutes of Health (NIH Publications No. 85-23, revised1985).
Results
Intracerebral MicrodialysisIn the brain, NOS converts oxygen and l-arginine to NOand citrulline in a 1:1 ratio. Because the only othersynthetic enzyme for citrulline— ornithine transcar-bamylase in the urea cycle—is present in the liver but notin the brain, citrulline concentration can be used as amarker of NO production. We used microdialysis tomeasure in vivo formation of extracellular citrulline inthe basal ganglia as a function of time. In dogs undergo-ing HCA alone, peak citrulline concentration rose signif-icantly (p , 0.05) above baseline during reperfusion CPBand 2 to 8 hours after HCA (Fig 1). Treatment with17477AR at 2 hours after HCA resulted in a significantlydecreased citrulline concentration and therefore a re-duced NO production 2 to 8 hours after HCA ( p , 0.05)(Fig 1). Administration of 17477AR reduced peak extra-cellular citrulline to 0.20 6 0.14 mmol/L compared with
3.65 6 2.42 mmol/L for HCA alone and 1.01 6 1.06 mmol/Lat baseline. Mean arterial pressure was not affected byadministration of 17477AR.
Nitric Oxide Synthase AssayNitric oxide synthase assay measures tissue NOS activityby the amount of 14C-citrulline produced from 14C-arginine over time. There are three forms of NOS; induc-ible NOS is calcium independent, whereas endothelialand nNOS are calcium dependent. Nitric oxide synthaseassay in the presence of calcium was performed in thedorsolateral neocortex. Nitric oxide synthase activity wassignificantly reduced in dogs treated with 17477AR at 72hours after HCA compared with NOS activity in un-treated HCA dogs at 72 hours (group 1 versus group 2:0.497 6 0.217 versus 4.138 6 0.064 3 10212 moles 14C-citrulline formed per milligram of protein per minute,p , 0.05) or untreated control dogs (group 1 versusuntreated control dogs: 0.497 6 0.217 versus 4.552 60.62 3 10212 moles 14C-citrulline formed per milligram ofprotein per minute, p 5 0.0157) (Fig 2). The nNOSinhibitor 17477AR had a long half-life of approximately12 hours and when administered 24 hours after HCA,had lingering effects at 72 hours. Untreated control dogswere normal dogs sacrificed without CPB or HCA.
Physiologic MonitoringCooling times during CPB for both groups ranged from25 to 30 minutes. Tympanic membrane temperatureswere similar for both groups throughout cooling, arrest,rewarming, and recovery phases (Fig 3). No statisticallysignificant differences were found between the twogroups to explain the cerebroprotection of 17477AR.There were no significant differences in esophageal andrectal temperatures between the two groups throughoutthe experiment. Mean arterial pressure was similar inboth groups throughout the experiment, notably evenwith 17477AR administration, as was cardiac output.There were no significant differences in arterial bloodgases between the two groups, with similar pH andcarbon dioxide tension. Glucose and hemoglobin levels
Fig 1. In vivo measurement of NOS activity as citrulline concentra-tion (mmol/L) over time. Peak levels of citrulline were plotted overthree time periods: HCA, reperfusion CPB 2 to 8 hours after HCA.(* 5 p , 0.05, 2 to 8 hours versus baseline. ** 5 p , 0.05,17477AR versus untreated HCA control [Ctl] group.)
Fig 2. Nitric oxide synthase (NOS) activity (pmol 14C-citrulline z
mg protein21 z min21) in the dorsolateral neocortex in untreatedcontrol dogs, HCA dogs at 72 hours, and 17477AR-treated dogs at72 hours after HCA.*p , 0.05 compared with untreated HCA dogs.
67Ann Thorac Surg TSENG ET AL1999;67:65–71 NO MEDIATES NEUROLOGIC INJURY AFTER HCA
by on May 30, 2013 ats.ctsnetjournals.orgDownloaded from
were monitored, and both groups exhibited similar hy-perglycemia and hemodilution.
Neurologic OutcomeAt each 12-hour neurologic evaluation after HCA, dogstreated with the nNOS inhibitor 17477AR had signifi-cantly better neurologic deficit scores than untreatedHCA dogs (Fig 4). Final neurologic scores were 62.22 629.82 for the group treated with 17477AR and 141.86 661.53 for the untreated HCA group ( p 5 0.019).
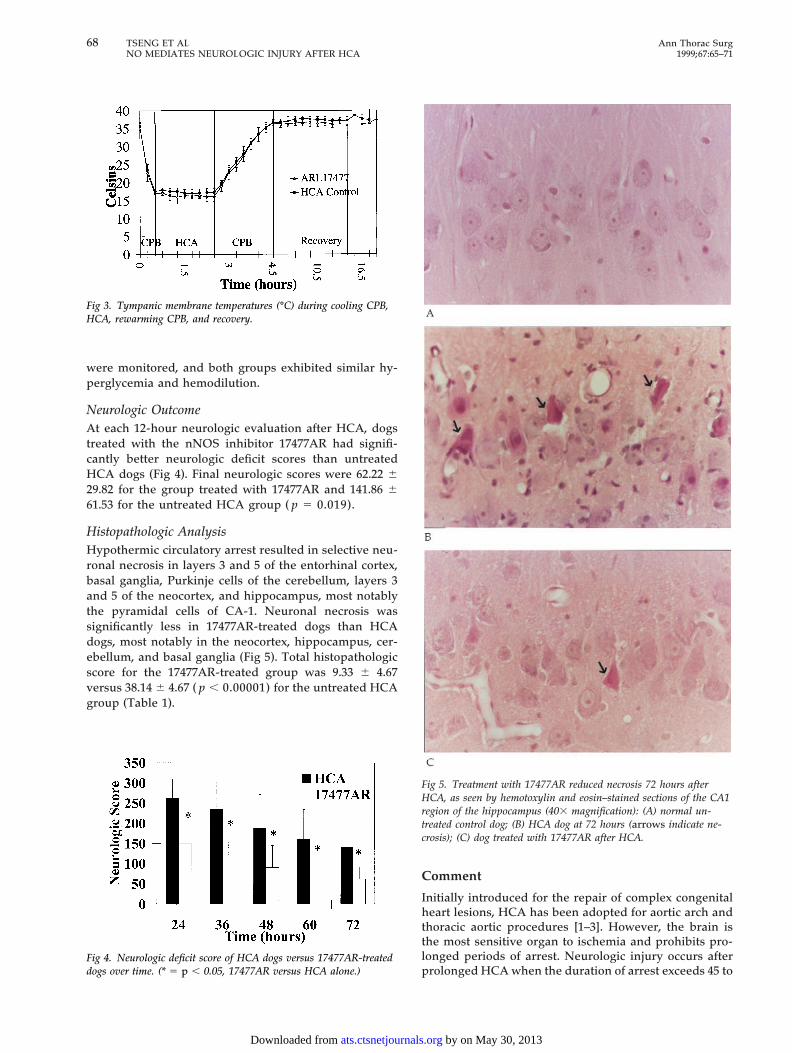
Histopathologic AnalysisHypothermic circulatory arrest resulted in selective neu-ronal necrosis in layers 3 and 5 of the entorhinal cortex,basal ganglia, Purkinje cells of the cerebellum, layers 3and 5 of the neocortex, and hippocampus, most notablythe pyramidal cells of CA-1. Neuronal necrosis wassignificantly less in 17477AR-treated dogs than HCAdogs, most notably in the neocortex, hippocampus, cer-ebellum, and basal ganglia (Fig 5). Total histopathologicscore for the 17477AR-treated group was 9.33 6 4.67versus 38.14 6 4.67 ( p , 0.00001) for the untreated HCAgroup (Table 1).
Comment
Initially introduced for the repair of complex congenitalheart lesions, HCA has been adopted for aortic arch andthoracic aortic procedures [1–3]. However, the brain isthe most sensitive organ to ischemia and prohibits pro-longed periods of arrest. Neurologic injury occurs afterprolonged HCA when the duration of arrest exceeds 45 to
Fig 3. Tympanic membrane temperatures (°C) during cooling CPB,HCA, rewarming CPB, and recovery.
Fig 4. Neurologic deficit score of HCA dogs versus 17477AR-treateddogs over time. (* 5 p , 0.05, 17477AR versus HCA alone.)
Fig 5. Treatment with 17477AR reduced necrosis 72 hours afterHCA, as seen by hemotoxylin and eosin–stained sections of the CA1region of the hippocampus (403 magnification): (A) normal un-treated control dog; (B) HCA dog at 72 hours (arrows indicate ne-crosis); (C) dog treated with 17477AR after HCA.
68 TSENG ET AL Ann Thorac SurgNO MEDIATES NEUROLOGIC INJURY AFTER HCA 1999;67:65–71
by on May 30, 2013 ats.ctsnetjournals.orgDownloaded from

60 minutes [4]. Clinical neurologic sequelae include sei-zures, choreoathetosis, learning and memory deficits,and impaired intellectual development, which occurs inup to 12% of children and 15% of adults [4–7].
Neuronal cell death causes HCA-induced neurologicinjury. Selective neuronal necrosis is the hallmark ofneuropathologic injury after HCA. Much evidence nowsupports the role of glutamate excitotoxicity in mediatinghypoxic and ischemic neuronal necrosis [18]. Hypoxiaand ischemia result in overaccumulation of the excitatoryamino acid glutamate. Extracellular glutamate stimulatesglutamate receptors, including N-methyl-d-aspartate,a-amino-3-hydroxy-5-methyl-4-isoxazolepropionate(AMPA), and kainate receptors, to result in a rise inintracellular calcium [18]. Calcium then activates phos-pholipases, protein kinases, proteases, and NOS andtriggers a cascade of cellular events that leads to lipidperoxidation, DNA degradation, and eventually neuronalcell death. We previously showed that glutamate recep-tor antagonists, including N-methyl-d-aspartate anda-AMPA receptor antagonists, reduced HCA-inducedneuronal necrosis [8–10].
Recently, NO has been implicated in mediating gluta-mate excitotoxicity [11, 19–21]. In transgenic mice miss-ing the neuronal NOS gene, cerebral infarcts from mid-dle cerebral artery occlusion were significantly smaller insize than control mice [21]. Activation of NOS by calciumentering through the N-methyl-d-aspartate receptorchannels results in increased NO, which may be neuro-protective or neurodestructive, depending on its redoxstate [19]. Nitrosonium ion may be neuroprotective byreacting with sulfhydryl groups on the N-methyl-d-aspartate receptor, whereas NO may be neurodestructiveby reacting with superoxide anion to form peroxynitrite.Peroxynitrite causes lipid peroxidation and oxidation ofsulphydryls [19]. In addition, NO can damage DNA bybase deamination and activate poly(adenosine 59-diphosphoribose) polymerase (PARP) to repair damaged
DNA. Activation of PARP may then lead to progressivecell depletion of adenosine triphosphate and death [20].
Nitric oxide has beneficial effects in other tissues, suchas the heart, during ischemia and reperfusion, because ofits endothelial-relaxing factor effect of vasodilatation.This aspect may also be beneficial during reperfu-sion injury in the brain to increase cerebral bloodflow; however, it is counteracted by the neurotoxic effectsof NO, hydroxyl radical, nitrogen dioxide, andperoxynitrite.
We previously showed that nNOS was induced afterHCA and that nNOS inhibition reduced one form ofneuronal cell death (apoptosis) after HCA [12, 13]. In thepresent study, we hypothesized that HCA causes NOproduction and that NO or its metabolites, or both, causeneuronal necrosis. We examined citrulline production asa marker for NO production over time. Citrulline pro-duction increased significantly over baseline duringreperfusion CPB and at 2 to 8 hours after HCA. Weadministered 17477AR after reperfusion CPB and foundpeak citrulline production significantly reduced by 94%compared with HCA alone. Administration of 17477ARreduced basal NO production by 80%.
In addition, calcium-dependent NOS activity in neo-cortical tissues was determined. There are three forms ofNOS: calcium-independent inducible NOS, calcium-dependent endothelial NOS, and nNOS. Previously, weshowed that calcium-independent NOS activity was neg-ligible compared with calcium-dependent NOS activity[12]. In addition, we also demonstrated by immunohisto-chemical analysis that only nNOS, not endothelial orinducible NOS, was induced by HCA [12]. Because cal-cium-dependent NOS activity predominated, and17477AR did not inhibit endothelial NOS in vivo, as seenby its lack of elevation of mean arterial pressure, nNOSwas most likely responsible for the increases in NO seenafter HCA. In the present study, calcium-dependentNOS activity 72 hours after HCA was unchanged fromthat in normal dogs at baseline; however, 17477AR treat-ment significantly reduced nNOS activity compared withnNOS activity in untreated control and HCA dogs at 72hours.
Microdialysis experiments determined increases inNOS activity in the caudate over time, specifically duringand early after HCA, but was not measured as late as 72hours. Nitric oxide synthase assay revealed no changes inNOS activity in the neocortex at fixed time points ofsacrifice at 8, 20, and 72 hours after HCA. The lack ofincrease in NOS activity by NOS assay as opposed tohigh-performance liquid chromatographic assay mostlikely reflects two causes. Differences may be explainedby different intracerebral locations where activity wasmeasured and by the ability of one technique to measureactivity over time as opposed to the fixed time points,when the activity returned to baseline.
Anatomically, HCA demonstrated selective vulnerabil-ity of neurons to necrosis. Necrosis occurred in layers 3and 5 of the neocortex and entorhinal cortex, the pyra-midal cells of CA-1 hippocampus, dentate gyrus of thehippocampus, Purkinje cells of the cerebellum, and the
Table 1. Histopathologic Score for Each Regiona
Anatomic Locationb17477AR(group 1)
HCAControl
(group 2) p Value
Neocortex 3.3 6 2.4 11.3 6 2.7 0.001Hippocampus 2 6 2.4 8.6 6 2.4 0.001Deep central white matter 0 0.3 6 0.7 0.35Basal ganglia 1.9 6 1.0 5.4 6 1.9 0.01Brain stem 0.6 6 1.6 2.9 6 2.5 0.08Cerebellum 1.6 6 1.9 9.7 6 2.0 0.00001
Total histopathologic score 9.3 6 4.7 38.1 6 2.2 0.00001
a Data presented are mean 6 standard deviation. b Neocortex in-cludes frontal, parietal, temporal, occipital, insular, and cingulate regions(worst score is 24); hippocampus includes hippocampus, dentate gyrus,entorhinal cortex, and amygdala (worst score is 16); deep central whitematter includes white matter, anterior commissure, and corpus callosum(worst score is 12); basal ganglia includes globus pallidus, putamen,caudate, and thalamus (worst score is 16); brain stem includes midbrain,pons, substantia nigra, periaqueductal gray matter, and medulla (worstscore is 20); cerebellum includes Purkinje cells, granule cells, dentatenucleus (worst score is 12); total injury score for all 25 regions is 100.
69Ann Thorac Surg TSENG ET AL1999;67:65–71 NO MEDIATES NEUROLOGIC INJURY AFTER HCA
by on May 30, 2013 ats.ctsnetjournals.orgDownloaded from

basal ganglia. We demonstrated that NO mediated ne-crosis. Using the selective nNOS inhibitor 17477AR, weshowed that nNOS inhibition significantly reduced neu-ronal necrosis when given after HCA. Reduction inextracellular citrulline observed in the 17477AR-treatedgroup supported the hypothesis that the drug actedthrough inhibition of nNOS activity. Reduction in necro-sis was most notable in the cerebellum, hippocampus,neocortex, and basal ganglia.
Clinically, no adverse effects of 17477AR administra-tion were seen. Dogs treated with 17477AR demonstratedsuperior neurologic function with regard to level ofconsciousness, breathing pattern, cranial nerve function,motor and sensory function, and behavior comparedwith dogs that underwent HCA alone.
Notably, reduction in neurologic injury was demon-strated despite administration after the ischemic periodof arrest. A number of reasons may explain this outcome.First, NO production increased significantly duringreperfusion CPB and recovery. Unlike glutamate, whichis released immediately during ischemia, NO productionis further downstream in the cascade. In cell cultures, thisdownstream effect may still be manifested as in conjunc-tion with glutamate release. However, NO productionrequires oxygen and in the present in vivo system,prolonged HCA is essentially an oxygen deprivationmodel. Although some NO production is likely to occurduring ischemia, the bulk of it occurred during reperfu-sion and thereafter, suggesting a beneficial effect ofpost-HCA administration. Second, nNOS is also locatedalong arterioles and is believed to play a role in regula-tion of cerebral blood flow; notably, inhibition of nNOSmay reduce cerebral blood flow. Post-HCA CPB reper-fuses the brain, reintroducing oxygen and nutrients;reduction of cerebral blood flow during this initial reper-fusion period may be harmful to neurons in the ischemicpenumbra. The beneficial effects of nNOS inhibition maydepend on a careful balance between cerebral blood flowand cellular toxic NO production. For these reasons, wechose to inhibit nNOS after reperfusion CPB. BecauseNO production continued in a delayed fashion afterHCA, we administered 17477AR for up to 24 hourspostoperatively.
Of note, inhibition of nNOS reduced neurologic injury;however, the relative contribution of reduction of NOversus hydroxyl radical, nitrogen dioxide, and peroxyni-trite production could not be determined in the presentstudy. Inhibition of nNOS presumably prevented furtherdownstream toxic effects of NO and its metabolites.Ultimately, the cause of direct tissue injury at the cellularlevel was not determined in the present study but is mostlikely due to peroxynitrite, formed by reaction of NOwith superoxide anion. However, we did demonstratethat inhibition of nNOS successfully blocked the neuro-toxic downstream cascade and reduced neuronalnecrosis.
We previously showed that both apoptosis and necro-sis were associated with neurologic injury after HCA [13].Hypothermic circulatory arrest results in problems withdeclarative memory. Declarative memory involves pro-
jections from the neocortex to perirhinal and parahip-pocampal cortices and then to the entorhinal cortex. Theentorhinal cortex, a major source of glutamate pathwaysto the hippocampus, subsequently projects to the dentategyrus, CA-3, and CA-1. Both necrosis and apoptosisoccurred in the hippocampus, entorhinal cortex, andneocortex [13]. Either or both processes could explainimpaired intellectual development and the learning andmemory deficits seen after HCA. Hypothermic circula-tory arrest also causes choreoathetosis. Necrosis predom-inated in the basal ganglia and cerebellum, which couldresult in uninhibited extrapyramidal activity, leading tochoreoathetoid movements. We previously showed thatapoptosis is reduced by nNOS inhibition. In the presentstudy, we demonstrated reduction of neuronal necrosisby 17477AR. Inhibition of nNOS significantly reducedboth forms of neuronal cell death—apoptosis and necro-sis—and clinically resulted in improved neurologic func-tion. Clinical safety of nNOS will require further evalu-ation; however, it may be beneficial in reducingneurologic complications even when given after HCA.
This work was supported by The Dana and Albert BroccoliCenter for Aortic Diseases and grant 2RO1NS31238-05 from theNational Institutes of Health. Elaine Tseng was supported by theNina Braunwald Research Fellowship Award from the ThoracicSurgery Foundation for Research and Education. We thankMelissa Haggerty, Jeffrey Brawn, Chieh A. Lee, and ChristopherKwon for technical assistance. We also thank David Reif andAstra Arcus, USA for providing 17477AR and its pharmacoki-netic data.
References
1. Niazi SA, Lewis FJ. Profound hypothermia: report of a case.Ann Surg 1957;147:264–6.
2. Coselli JS, Buket S, Djukanovic B. Aortic arch operation:current treatment and results. Ann Thorac Surg 1995;59:19–27.
3. Kouchoukos NT, Daily BB, Rokkas CK, Murphy SF, Bauer S,Abboud N. Hypothermic bypass and circulatory arrest foroperations on the descending thoracic and thoracoabdomi-nal aorta. Ann Thorac Surg 1995;60:67–77.
4. Kirklin JW, Barratt-Boyes BG. Hypothermia, circulatory ar-rest, and cardiopulmonary bypass. In: Kirklin JW, Barratt-Boyes BG, eds. Cardiac surgery. New York: Churchill Liv-ingstone, 1993:66–73.
5. Newburger JW, Jonas RA, Wernovsky G, et al. A comparisonof the perioperative neurologic effects of hypothermic circu-latory arest versus low-flow cardiopulmonary bypass ininfant heart surgery. N Engl J Med 1993;329:1057–64.
6. Bellinger DC, Jonas RA, Rappaport LA, et al. Developmentaland neurologic status of children after heart surgery withhypothermic circulatory arrest or low-flow cardiopulmonarybypass. N Engl J Med 1995;332:549–55.
7. Davis EA, Gillinov AM, Cameron DE, Reitz BA. Hypother-mic circulatory arrest as a surgical adjunct: a 5-year experi-ence with 60 adult patients. Ann Thorac Surg 1992;53:402–7.
8. Redmond JM, Gillinov AM, Blue ME, et al. The monosialo-ganglioside, GM1, reduces neurologic injury associated withhypothermic circulatory arrest. Surgery 1993;114:324–33.
9. Redmond JM, Gillinov AM, Zehr KJ, et al. Glutamate exci-totoxicity: a mechanism of neurologic injury associated withhypothermic circulatory arrest. J Thorac Cardiovasc Surg1994;107:776–87.
10. Redmond JM, Zehr KJ, Blue ME, et al. AMPA glutamate
70 TSENG ET AL Ann Thorac SurgNO MEDIATES NEUROLOGIC INJURY AFTER HCA 1999;67:65–71
by on May 30, 2013 ats.ctsnetjournals.orgDownloaded from

receptor antagonism reduces neurologic injury after hypo-thermic circulatory arrest. Ann Thorac Surg 1995;59:579–84.
11. Dawson TM, Snyder SH. Gases as biological messengers:nitric oxide and carbon monoxide in the brain. J Neurosci1994;14:5147–59.
12. Brock MV, Blue ME, Lowenstein CJ, et al. Induction ofneuronal nitric oxide after hypothermic circulatory arrest.Ann Thorac Surg 1996;62:1313–20.
13. Tseng EE, Brock MV, Lange MS, et al. Neuronal nitric oxidesynthase inhibition reduces neuronal apoptosis after hypo-thermic circulatory arrest. Ann Thorac Surg 1997;64:1639–47.
14. Carlberg M. Assay of neuronal nitric oxide synthase by HPLCdetermination of citrulline. J Neurosci Methods 1994;52:165–7.
15. Donzanti BA, Yamamoto BK. An improved and rapidHPLC-EC method for the isocratic separation of amino acidneurotransmitters from brain tissue and microdialysis per-fusates. Life Sci 1988;43:913–22.
16. Bredt DS, Snyder SH. Isolation of nitric oxide synthetase, acalmodulin-requiring enzyme. Proc Natl Acad Sci USA 1990;87:682–5.
17. Tisherman SA, Safar P, Radovsky A, et al. Profound hypo-thermia (,10°C) compared with deep hypothermia (15°C)improves neurologic outcome in dogs after two hours circu-latory arrest to enable resuscitative surgery. J Trauma 1991;31:1–11.
18. Lipton SA, Rosenberg PA. Excitatory amino acids as a finalcommon pathway for neurologic disorders. N Engl J Med1994;330:613–22.
19. Lipton SA, Choi YB, Pan ZH, et al. A redox-based mecha-nism for the neuroprotective and neurodestructive effects ofnitric oxide and related nitrosos-compounds. Nature 1993;364:626–31.
20. Zhang J, Dawon VL, Dawson TM, Snyder SH. Nitric oxideactivation of poly (ADP)-ribose synthetase in neurotoxicity.Science 1994;263:687–89.
21. Huang A, Huang PL, Panahian N, Dalkara R, FishmanMC, Moskowitz MA. Effects of cerebral ischemia in micedeficient in neuronal nitric oxide synthase. Science 1994;265:1883–5.
DISCUSSION
DR JOHN H. CALHOON (San Antonio, TX): Both papers werereally great. And in Dr Tseng’s case, I had a chance to review hermanuscript, which was equally as outstanding as herpresentation.
One question for her, if I could. Is the drug used to block NOSin your study, this 17477AR, safe in humans? Do you have anyplans to look at a trial for humans?
DR TSENG: There has not been any safety data on that drug inhumans. We examined its effects qualitatively in dogs and didnot find any adverse effects. However, we do not have plans fora clinical trial in humans yet.
DR JAKOB VINTEN-JOHANSEN (Atlanta, GA): In the brain,NO may reach very high levels because there is really no drainmechanism; for example, through combination with the hemo-globin moiety to form methemoglobin. Do you think that reallythe culprit could be peroxynitrite, which may also reach highlevels?
DR TSENG: We do believe that peroxynitrite is probably themechanism of injury. We have not directly measured levels ofperoxynitrite, but presumably the reaction of NO and superox-ide to form peroxynitrite results in lipid peroxidation anddegradation.
71Ann Thorac Surg TSENG ET AL1999;67:65–71 NO MEDIATES NEUROLOGIC INJURY AFTER HCA
by on May 30, 2013 ats.ctsnetjournals.orgDownloaded from

1999;67:65-71 Ann Thorac SurgLowenstein, Mary E. Blue, Michael V. Johnston and William A. Baumgartner
Elaine E. Tseng, Malcolm V. Brock, Mary S. Lange, Juan C. Troncoso, Charles J. Nitric oxide mediates neurologic injury after hypothermic circulatory arrest
& ServicesUpdated Information
http://ats.ctsnetjournals.org/cgi/content/full/67/1/65including high-resolution figures, can be found at:
References http://ats.ctsnetjournals.org/cgi/content/full/67/1/65#BIBL
This article cites 20 articles, 11 of which you can access for free at:
Citations http://ats.ctsnetjournals.org/cgi/content/full/67/1/65#otherarticles
This article has been cited by 8 HighWire-hosted articles:
Permissions & Licensing
[email protected]: orhttp://www.us.elsevierhealth.com/Licensing/permissions.jsp
in its entirety should be submitted to: Requests about reproducing this article in parts (figures, tables) or
Reprints [email protected]
For information about ordering reprints, please email:
by on May 30, 2013 ats.ctsnetjournals.orgDownloaded from




















