
Natural selection of adaptive mutations in non-structural genes increases trans-encapsidation of...
13
Natural selection of adaptive mutations in non- structural genes increases trans-encapsidation of hepatitis C virus replicons lacking envelope protein genes Carole Fournier, 1,2 Franc ¸ ois Helle, 2 Ve ´ ronique Descamps, 1,2 Virginie Morel, 1,2 Catherine Franc ¸ ois, 1,2 Sarah Dedeurwaerder, 2,3 Czeslaw Wychowski, 4 Gilles Duverlie 1,2 and Sandrine Castelain 1,2 Correspondence Sandrine Castelain [email protected] Received 13 November 2012 Accepted 24 December 2012 1 Virology Department, Amiens University Hospital, South Hospital, Amiens, France 2 EA4294, Jules Verne University of Picardy, Amiens, France 3 Laboratory of Cancer Epigenetics, Universite ´ Libre de Bruxelles, Faculty of Medicine, Brussels, Belgium 4 INSERM U1019, CNRS UMR 8204, Center for Infection and Immunity of Lille, Institut de Biologie de Lille, Lille, France A trans-packaging system for hepatitis C virus (HCV) replicons lacking envelope glycoproteins was developed. The replicons were efficiently encapsidated into infectious particles after expression in trans of homologous HCV envelope proteins under the control of an adenoviral vector. Interestingly, expression in trans of core or core, p7 and NS2 with envelope proteins did not enhance trans-encapsidation. Expression of heterologous envelope proteins, in the presence or absence of heterologous core, p7 and NS2, did not rescue single-round infectious particle production. To increase the titre of homologous, single-round infectious particles in our system, successive cycles of trans-encapsidation and infection were performed. Four cycles resulted in a 100-fold increase in the yield of particles. Sequence analysis revealed a total of 16 potential adaptive mutations in two independent experiments. Except for a core mutation in one experiment, all the mutations were located in non-structural regions mainly in NS5A (four in domain III and two near the junction with the NS5B gene). Reverse genetics studies suggested that D2437A and S2443T adaptive mutations, which are located at the NS5A-B cleavage site did not affect viral replication, but enhanced the single-round infectious particles assembly only in trans- encapsidation model. In conclusion, our trans-encapsidation system enables the production of HCV single-round infectious particles. This system is adaptable and can positively select variants. The adapted variants promote trans-encapsidation and should constitute a valuable tool in the development of replicon-based HCV vaccines. INTRODUCTION Hepatitis C virus (HCV) is an important human pathogen; worldwide, over 170 million people are chronically infected. Indeed, 75–80 % of HCV-infected patients develop chronic infection, which may lead to cirrhosis and, ultimately, hepatocellular carcinoma. HCV is a positive-strand RNA virus from the genus Hepacivirus in the family Flaviviridae (Choo et al., 1991). Its RNA genome is 9600 nt long and features two untranslated regions at the 59 and 39 ends (59 UTR and 39 UTR) (Kato et al. , 1990). The genome encodes a large polyprotein (with approximately 3010 aa) that is co- and post-translationally cleaved by cellular and viral proteases to form structural proteins (core, E1 and E2) and non- structural proteins (p7, NS2, NS3, NS4A, NS4B, NS5A and NS5B) (Tang & Grise ´, 2009). Following the discovery of HCV in 1989, the investigation of viral replication and pathogenesis has notably been hampered by the lack of an appropriate viral culture system (Gottwein & Bukh, 2008). Recently, full-genome replicons capable of producing infectious virus particles in cell culture (HCVcc) have been developed. These include the genotype 2a JFH1 strain and the derived inter- and intragenotypic chimeras (Gottwein et al., 2009; Pietschmann et al., 2006; Wakita et al., 2005). Viral particle release from some of these cell culture systems was initially quite low. However, infectivity titres were improved by successive infections of Supplementary figures are available with the online version of this paper. Journal of General Virology (2013), 94, 996–1008 DOI 10.1099/vir.0.049676-0 996 049676 G 2013 SGM Printed in Great Britain
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Natural selection of adaptive mutations in non-structural genes increases trans-encapsidation of...

Natural selection of adaptive mutations in non-structural genes increases trans-encapsidation ofhepatitis C virus replicons lacking envelope proteingenes
Carole Fournier,1,2 Francois Helle,2 Veronique Descamps,1,2
Virginie Morel,1,2 Catherine Francois,1,2 Sarah Dedeurwaerder,2,3
Czeslaw Wychowski,4 Gilles Duverlie1,2 and Sandrine Castelain1,2
Correspondence
Sandrine Castelain
Received 13 November 2012
Accepted 24 December 2012
1Virology Department, Amiens University Hospital, South Hospital, Amiens, France
2EA4294, Jules Verne University of Picardy, Amiens, France
3Laboratory of Cancer Epigenetics, Universite Libre de Bruxelles, Faculty of Medicine, Brussels,Belgium
4INSERM U1019, CNRS UMR 8204, Center for Infection and Immunity of Lille, Institut de Biologiede Lille, Lille, France
A trans-packaging system for hepatitis C virus (HCV) replicons lacking envelope glycoproteins
was developed. The replicons were efficiently encapsidated into infectious particles after
expression in trans of homologous HCV envelope proteins under the control of an adenoviral
vector. Interestingly, expression in trans of core or core, p7 and NS2 with envelope proteins did
not enhance trans-encapsidation. Expression of heterologous envelope proteins, in the presence
or absence of heterologous core, p7 and NS2, did not rescue single-round infectious particle
production. To increase the titre of homologous, single-round infectious particles in our system,
successive cycles of trans-encapsidation and infection were performed. Four cycles resulted in a
100-fold increase in the yield of particles. Sequence analysis revealed a total of 16 potential
adaptive mutations in two independent experiments. Except for a core mutation in one experiment,
all the mutations were located in non-structural regions mainly in NS5A (four in domain III and two
near the junction with the NS5B gene). Reverse genetics studies suggested that D2437A and
S2443T adaptive mutations, which are located at the NS5A-B cleavage site did not affect viral
replication, but enhanced the single-round infectious particles assembly only in trans-
encapsidation model. In conclusion, our trans-encapsidation system enables the production of
HCV single-round infectious particles. This system is adaptable and can positively select variants.
The adapted variants promote trans-encapsidation and should constitute a valuable tool in the
development of replicon-based HCV vaccines.
INTRODUCTION
Hepatitis C virus (HCV) is an important human pathogen;worldwide, over 170 million people are chronically infected.Indeed, 75–80 % of HCV-infected patients develop chronicinfection, which may lead to cirrhosis and, ultimately,hepatocellular carcinoma. HCV is a positive-strand RNAvirus from the genus Hepacivirus in the family Flaviviridae(Choo et al., 1991). Its RNA genome is 9600 nt long andfeatures two untranslated regions at the 59 and 39 ends (59
UTR and 39 UTR) (Kato et al., 1990). The genome encodes alarge polyprotein (with approximately 3010 aa) that is co-and post-translationally cleaved by cellular and viral proteases
to form structural proteins (core, E1 and E2) and non-structural proteins (p7, NS2, NS3, NS4A, NS4B, NS5A andNS5B) (Tang & Grise, 2009).
Following the discovery of HCV in 1989, the investigationof viral replication and pathogenesis has notably beenhampered by the lack of an appropriate viral culture system(Gottwein & Bukh, 2008). Recently, full-genome repliconscapable of producing infectious virus particles in cell culture(HCVcc) have been developed. These include the genotype2a JFH1 strain and the derived inter- and intragenotypicchimeras (Gottwein et al., 2009; Pietschmann et al., 2006;Wakita et al., 2005). Viral particle release from some of thesecell culture systems was initially quite low. However,infectivity titres were improved by successive infections ofSupplementary figures are available with the online version of this paper.
Journal of General Virology (2013), 94, 996–1008 DOI 10.1099/vir.0.049676-0
996 049676 G 2013 SGM Printed in Great Britain

naıve cells to obtain culture-adapted variants (Delgrangeet al., 2007; Kato et al., 2007; Kaul et al., 2007; Russell et al.,2008).
Although the flaviviridae genome is translated in cis, it hasbeen shown for some viruses that some proteins could becomplemented in trans. In particular, it was reported thatenvelope glycoprotein Erns in classical swine fever virus(a pestivirus) can be complemented in trans by an SK6 cel-lular cell line that constitutively expresses this protein(Widjojoatmodjo et al., 2000). Pigs vaccinated with thesedefective viral particles were protected against a lethalchallenge with the virulent Brescia strain. This observationopened up new perspectives in the development ofmodified, live-attenuated vaccines. The first trans-comple-mentation studies in HCV used NS5A to rescue lethalmutations in replicons (Appel et al., 2005; Tong &Malcolm, 2006). Following the development of HCVcc,several groups have used trans-complementation to rescuelethal mutations or deletions in core, p7, NS2, NS4B andNS5A proteins (Appel et al., 2008; Brohm et al., 2009;Jirasko et al., 2008; Jones et al., 2009; Miyanari et al., 2007).Furthermore, trans-encapsidation of HCV subgenomicreplicon RNA or replicons lacking envelope-encodinggenes has been achieved with viral structure proteins(Adair et al., 2009; Ishii et al., 2008; Steinmann et al., 2008).
In the present study, we developed and optimized a trans-encapsidation system based on HCV envelope glycopro-teins for the production of single-round infectiousparticles. To complement replicons lacking E1E2 glyco-proteins, we used an adenoviral system to transducehomologous or heterologous envelope proteins expressedalone or in combination with core or core, p7 and NS2. Wethen increased the yield of viral particles in this trans-encapsidation system by applying successive cycles of trans-encapsidation and infection. Our data agree with previoustrans-encapsidation studies of structural proteins and alsodemonstrate that the trans-encapsidation system is adapt-able in cell culture, as replicons or full-length genomes.Adaptation of this trans-encapsidation system enabled usto obtain high titres of single-round infectious particlesand should be a useful tool in vaccine development.
RESULTS
Production of single-round infectious viralparticles
In order to establish whether HCV JFH1 replicons lackingthe E1E2 genes can be complemented in trans, two repliconswere used (JFH1-LucDE1E2 and JFH1-PuroDE1E2, expres-sing Renilla luciferase and puromycin acetyltransferaseproteins, respectively) (Fig. 1a). Expression of NS3 proteinby JFH1-LucDE1E2 and JFH1-PuroDE1E2 replicons wasconfirmed by indirect immunofluorescence (Fig. 1b).
We also built an efficient system for envelope proteinexpression in trans. Previous work has shown that
adenoviral systems can efficiently express proteins inhepatoma cell lines (Huh-7 cells, in particular) (Dimitrovaet al., 2003; Eyre et al., 2009). Hence, cDNA encoding the 21carboxy-terminal residues of core, E1 and E2 from the JFH1strain was used to generate adenovirus constructs expressinggenotype 2a E1E2 (Ad-E1E2-2a). Indirect immunofluores-cence (Fig. 1c) and Western blot analysis (Fig. 2b) confirmedhigh levels of E1E2 expression after the transduction of Huh-7 cells with Ad-E1E2-2a.
Subsequently, Huh-7 cells were electroporated with JFH1-LucDE1E2 RNA and transduced 24 h later with Ad-E1E2-2aor, as a negative control, empty adenovirus (Ad-control).Non-transduced, full-length JFH1-Luc virus (JFH1-Luc-WT) was used as a positive control. Seventy-two hours post-transduction, supernatants were harvested and infectiousvirus release was measured, after infection of naıve Huh-7cells, by quantifying luciferase activity, intracellular HCVRNA and f.f.u. (Fig. 1d, e, f, respectively).
According to the luciferase activity results (Fig. 1d),infectious particle production by JFH1-LucDE1E2 trans-complemented with Ad-E1E2-2a was similar to that seenwith JFH1-Luc-WT. Similar results were obtained afterquantification of HCV RNA in infected cells using primersin the NS3 region to avoid quantification of the RNAexpressed from adenoviruses (Fig. 1e). Surprisingly,whereas measure of luciferase activities suggested that theproduction of trans-complemented particles was similar tothat of authentic virions we observed a 2 log10 differencewhen measuring infectious titres (Fig. 1f). This may be dueto specificity differences between the two assays. Asexpected, no infectious viral particles were detected aftertrans-encapsidation with Ad-control.
We also worked with Huh-7 cells stably replicating a JFH1-PuroDE1E2 genome. We hypothesized that trans-encapsida-tion would be more efficient under these conditions sincethe cells replicating DE1E2 replicon were selected bypuromycin addition and thus all cells transduced containedthe replicon. As demonstrated by quantification of intracel-lular HCV RNA and f.f.u. in cells inoculated with thesupernatant from transduced cells, JFH1-PuroDE1E2 couldalso be complemented by expression of E1E2 in trans toproduce single-round infectious particles. However, theyield of trans-complemented infectious particles was 3 log10
lower than for the JFH1-Puro-WT (Fig. 1f). Likewise,intracellular HCV RNA quantification was 1.6 log10 lowerthan for the JFH1-Puro-WT (5.32 log10 IU mg21 and3.76 log10 IU mg21 RNA for JFH1-Puro-WT and JFH1-PuroDE1E2 transduced with Ad-E1E2-2a, respectively; Fig.1e). Our data show that single-round infectious particles canbe produced by complementing DE1E2 replicons in transwith envelope proteins expressed from Ad-E1E2-2a.
To confirm that infection by trans-complemented particlesobtained from transduced JFH1-PuroDE1E2 stable cells,depends on the presence of E1E2 at the virion surface, weinfected naıve Huh-7 cells with the supernatant fromtransduced cells in the presence of an E2-specific neutralizing
Adaptive mutations enhance trans-encapsidation
http://vir.sgmjournals.org 997

(a)
(b)
(c)
(d)
1,E+02
1,E+03
1,E+04
1,E+05
1,E+06
RLU
E2 E2 E2
Ad-E1E2-2a E1 E2
CMV
JFH1-PuroΔE1E2
JFH1-LucΔE1E2
JFH1-Puro-WT
JFH1-Puro-WT
JFH1-GND
Ad-E1E2-2aAd-control
Ad-E1E2-2a NTAd-control
JFH1-PuroΔE1E2 JFH1-LucΔE1E2
JFH1-LucΔE1E2
JFH1-Luc-WT
ΔC1
ΔC1
ΔC2
5’
5’
3’Puro p7 NS2 NS3 NS4A NS4B NS5A NS5BC
E1 E2
217 567 2AUbi
2AUbi
3’ NS2 NS3 NS4A NS4B NS5A NS5BCLuc p7
E1 E2
217 567
NS3 NS3 NS3 NS3
C. Fournier and others
998 Journal of General Virology 94

mAb. As shown in Fig. 1(g), the presence of 30 mg ml21 ofanti-E2 mAb decreased the infectious titre by 88.4 %. We didnot detect any focus formation at a mAb concentration of60 mg ml21. The fact that an E2-specific antibody inhibitsthe infection of single-round infectious particles stronglysuggests that the particles’ entry into Huh-7 cells is mediatedby glycoproteins.
Expression of C-E1E2 or C-E1E2-P7-NS2, ratherthan E1E2 alone, does not enhance theproduction of single-round infectious particles
Although previous results had shown that replicons andenvelope proteins were efficiently expressed, viral particleproduction for JFH1-DE1E2 replicons complemented withAd-E1E2-2a was relatively low. Hence, we decided to lookat whether the expression of other viral proteins in transcould influence the yield of single-round infectiousparticles. Additional adenoviruses expressing C-E1E2(Ad-59-E2-2a) or C-E1E2-P7-NS2 (Ad-59-NS2-2a) fromJFH1 cDNA were produced (Fig. 2a). After transduction ofnaıve Huh-7 cells with Ad-59-E2-2a or Ad-59-NS2-2a, highlevels of E2 expression were detected by Western blotanalysis (Fig. 2b). Huh-7 cells containing JFH1-LucDE1E2or JFH1-PuroDE1E2 replicons were transduced with theseadenoviruses or Ad-control. Seventy-two hours later,supernatants were harvested and single-round infectiousparticle production was measured, by quantifying lucifer-ase activity, intracellular HCV RNA and f.f.u., afterinfection of naıve Huh-7 cells. As shown in Fig. 2(c), theproduction of single-round infectious particles, as meas-ured by the viral titre, was not improved by the expressionof core-E2 or core-NS2, rather than E1E2 alone (1.81, 1.30and 2.22 log10 f.f.u. ml21, respectively, after trans-encapsi-dation of the JFH1-LucDE1E2 replicon; 1.37, 1.13 and
(g)
1,E+00
No antibody Anti-E2-30 μg ml–1 Anti-E2-60 μg ml–1
1,E+01
1,E+02
f.f.u. m
l–1
JFH1-PuroΔE1E2
(e)
1,E+01
1,E+02
1,E+03
1,E+04
1,E+05
1,E+06
HC
V IU
per μg
to
tal R
NA
(f)
1,E+00
1,E+01
1,E+02
1,E+03
1,E+04
1,E+05
f.f.u. m
l–1
JFH1-LucΔE1E2
JFH1-PuroΔE1E2
JFH1-Luc-WT
JFH1-Puro-WT
JFH1-LucΔE1E2
JFH1-PuroΔE1E2JFH1-Luc-WTJFH1-Puro-WT
Ad-E1E2-2a NTAd-control
Ad-E1E2-2a NTAd-control
Fig. 1. Trans-encapsidation of the envelope-gene-deleted JFH1replicons by envelope proteins enables HCV virion production. (a)Schematic representation of the two JFH1 replicons and theadenoviral constructions expressing genotype 2a E1E2 proteins. Inthe replicons, 350 codons within the E1- and E2-encoding regionwere deleted (positions 217–567). The puromycin acetyltransfer-ase and luciferase genes are indicated by Puro and Luc,respectively. 2AUbi represents the 17-residue fragment of theself-cleaving foot-and-mouth disease virus 2A protein (2A) and theubiquitin monomer (Ubi), which were fused to the C terminus of theluciferase or puromycin acetyltransferase gene. CMV indicated thepromoter of cytomegalovirus, DC1 contained the first 19 aaresidues of core and DC2 contained the C-terminal residues of
core (positions 171–191). (b) Detection of NS3 expression byindirect immunofluorescence 72 h after the electroporation ofreplicons JFH1-PuroDE1E2, JFH1-LucDE1E2, JFH1-Puro-WTand JFH1-GND into Huh-7 cells. (c) Detection of E2 expressionby indirect immunofluorescence 48 h after transduction of Huh-7cells with Ad-E1E2-2a or Ad-control and infection with JFH1-Puro-WT. (d, e, f) Huh-7 cells containing JFH1-PuroDE1E2 orJFH1-LucDE1E2 replicons were transduced with adenoviruscontrol or Ad-E1E2-2a (m.o.i.510). Supernatants were harvested72 h later and placed in contact with naıve Huh-7 cells for 4 h. In acontrol experiment, naıve Huh-7 cells were infected with JFH1-Puro-WT or JFH1-Luc-WT. Luciferase (d), intracellular RNA (e)and f.f.u. (f) assays were performed 72 h post-infection. Datarepresent the mean±SD of three independent experiments. NT,Non-transduced. (g) Supernatant from JFH1-PuroDE1E2 repli-cating cells transduced with Ad-E1E2-2a was incubated for 2 h inthe absence or presence (30 or 60 mg ml”1) of anti-E2 mAb 3/11.Next, mixtures were placed in contact with naıve Huh-7 cells for4 h f.f.u. assays were performed 72 h post-infection. Datarepresent the mean±SD of three independent experiments.
Adaptive mutations enhance trans-encapsidation
http://vir.sgmjournals.org 999

1.48 log10 f.f.u. ml21 after trans-encapsidation of theJFH1-PuroDE1E2 replicon). These results were con-firmed with intracellular HCV RNA assays after trans-encapsidation of JFH1-LucDE1E2 (4.11, 3.40 and4.12 log10 IU mg21 RNA with Ad-59-E2-2a, Ad-59-NS2-2a and Ad-E1E2-2a, respectively) or JFH1-PuroDE1E2
(3.94, 3.74 and 3.76 log10 IU mg21 RNA with Ad-59-E2-2a, Ad-59-NS2-2a and Ad-E1E2-2a, respectively, Fig S1a,available in JGV Online). Similarly, we did not find anincrease in yield when measuring luciferase activity incells infected with trans-complemented particles pro-duced with Ad-59-E2-2a or Ad-59-NS2-2a, relative to Ad-E1E2-2a (Fig S1b). Thus, our results suggest that theconcomitant expression of core or core, p7, NS2 withE1E2 envelope proteins does not facilitate the trans-encapsidation of DE1E2 replicons.
Trans-encapsidation with heterologous envelopeproteins does not product single-round infectiousviruses
We continued our investigation of the mechanism of trans-encapsidation of JFH1-DE1E2 replicons by determiningwhether single-round infectious particle production couldbe restored by the expression of heterologous envelopeproteins. To this end, we produced additional adeno-viruses: Ad-E1E2-1a, Ad-E1E2-1b, Ad-E1E2-3a and Ad-E1E2-4 expressing envelope proteins from genotype 1a, 1b,3a and 4 (the UKN1A2-1, UKN1B2-16, UKN3A-1.28 andUKN4-21.16 strains, respectively) and Ad-59-NS2-1bexpressing C-E1E2-P7-NS2 from genotype 1b (the Lexstrain). Importantly, these envelope proteins are known tobe functional, since they enable the production ofinfectious retroviral pseudoparticles (Lavillette et al.,2005). Expression of E2 by the different constructs wasmonitored by Western blot analysis; high expression levelswere detected (Fig. 3a). Huh-7 cells containing JFH1-LucDE1E2 or JFH1-PuroDE1E2 replicons were transducedwith these adenoviruses or Ad-control and the productionof trans-complemented infectious particles was assayed asdescribed above. Luciferase activity values obtained afterinoculation with the supernatant from JFH1-LucDE1E2replicating cells transduced with Ad-E1E2-1a, Ad-E1E2-1b,Ad-E1E2-3a and Ad-E1E2-4 were close to those obtainedwith JFH1-LucDE1E2 replicating cells transduced with Ad-control (Fig S2b). Similar results were obtained aftermeasuring HCV RNA levels in infected cells (Fig S2a). Onthe basis of an f.f.u. assay, we did not observe anyproduction of single-round infectious particles after trans-encapsidation of JFH1-LucDE1E2 or JFH1-PuroDE1E2(Fig. 3b). This set of results suggests that heterologousenvelope proteins cannot trans-complement JFH1-DE1E2replicons. Luciferase activity obtained after trans-encapsi-dation tests with Ad-59-NS2-1b was similar to thebackground level for the assay (Fig S2b). Furthermore,results obtained after intracellular HCV RNA quantifica-tion were also similar to control values (Fig S2a). Nosingle-round infectious particles were found in the super-natants, as shown by the f.f.u. assay results for JFH1-LucDE1E2 and JFH1-PuroDE1E2 (Fig. 3b). Thus, expres-sion in trans of genotype 1b core, p7 and NS2 with E1E2does not enable the production of single-round infectiousparticles.
(a)
(b)
(c)
1,E+00
1,E+01
1,E+02
1,E+03
1,E+04
1,E+05
Ad-control Ad-E1E2-2a Ad-5'-E2-2a Ad-5'-NS2-2a NT
f.f.u. m
l–-1
Ad- 5’- NS2-2a 5’ UTR
C E1 E2 p7 NS2
CMV
Ad- 5’- E2-2a
CMV
5’ UTRC E1 E2
Ad-
E1E2
2a
E2
β-Actin
Ad-
5’-E2
2a
Ad-
5’-NS2
2a
JFH1-Puro
WT
Ad
control
JFH1-LucΔE1E2
JFH1-PuroΔE1E2
JFH1-Luc-WT
JFH1-Puro-WT
Fig. 2. Expression of C-E1E2 or C-E1E2-P7-NS2 instead ofE1E2 proteins does not enhance trans-encapsidation. (a)Schematic representation of adenoviral constructions expressinggenotype 2a C-E1E2 or C-E1E2-P7-NS2. (b) Detection of E2expression by Western blot analysis after infection of Huh-7 cellswith Ad-E1E2-2a, Ad-59-NS2-2a, Ad-59-E2-2a, Ad-control orJFH1-Puro-WT. (c) Huh-7 cells containing JFH1-PuroDE1E2 orJFH1-LucDE1E2 replicons were transduced with the aforemen-tioned adenoviruses (m.o.i.510). Supernatants were harvested72 h later and placed in contact with naıve Huh-7 cells for 4 h. Asa control, naıve Huh-7 cells were infected with JFH1-Puro-WT orJFH1-Luc-WT. f.f.u. (c) assays were performed 72 h post-infection. Data represent the mean±SD of three independentexperiments. NT, Non-transduced.
C. Fournier and others
1000 Journal of General Virology 94

Successive cycles of trans-encapsidation andinfection with JFH1-PuroDE1E2 improve theproduction of trans-encapsidated infectiousparticles
It has been shown that cell culture-adaptive mutations,especially in non-structural proteins, enhance HCVccproduction and are selected after successive infections.Hence, we decided to perform successive infections withtrans-complemented particles. Cells infected with trans-complemented particles produced from JFH1-PuroDE1E2(the P0 population) were selected by puromycin addition,in order to obtain a population containing the JFH1-PuroDE1E2 replicon (designated P1). These trans-encapsi-dation, infection and selection steps were repeated a furtherthree times to obtain additional JFH1-PuroDE1E2 cellpopulations (designated P2, P3 and P4). We performedtwo independent experiments (series 1 and 2). Virion
production was measured in supernatants collected aftertransduction with Ad-E1E2-2a by assaying core release. Asshown in Fig. 4(a), we observed a gradual increase in corerelease with each cycle. We observed a 1 log10 increase incore release after trans-encapsidation by the P4 JFH1-PuroDE1E2 cell population, relative to the P0 population.
In agreement with these results, we also observed a gradualincrease in intracellular HCV RNA after inoculation ofnaıve Huh-7 cells with the supernatants of the transducedpopulations (Fig. 4b). Most importantly, we observed a2 log10 increase when comparing viral titres for P4 and P0(Fig. 4c). This set of results demonstrates that cell cultureadaptation of trans-encapsidation systems can improve theyield of single-round infectious particles.
Identification of conserved adaptive mutationsselected during successive trans-encapsidation
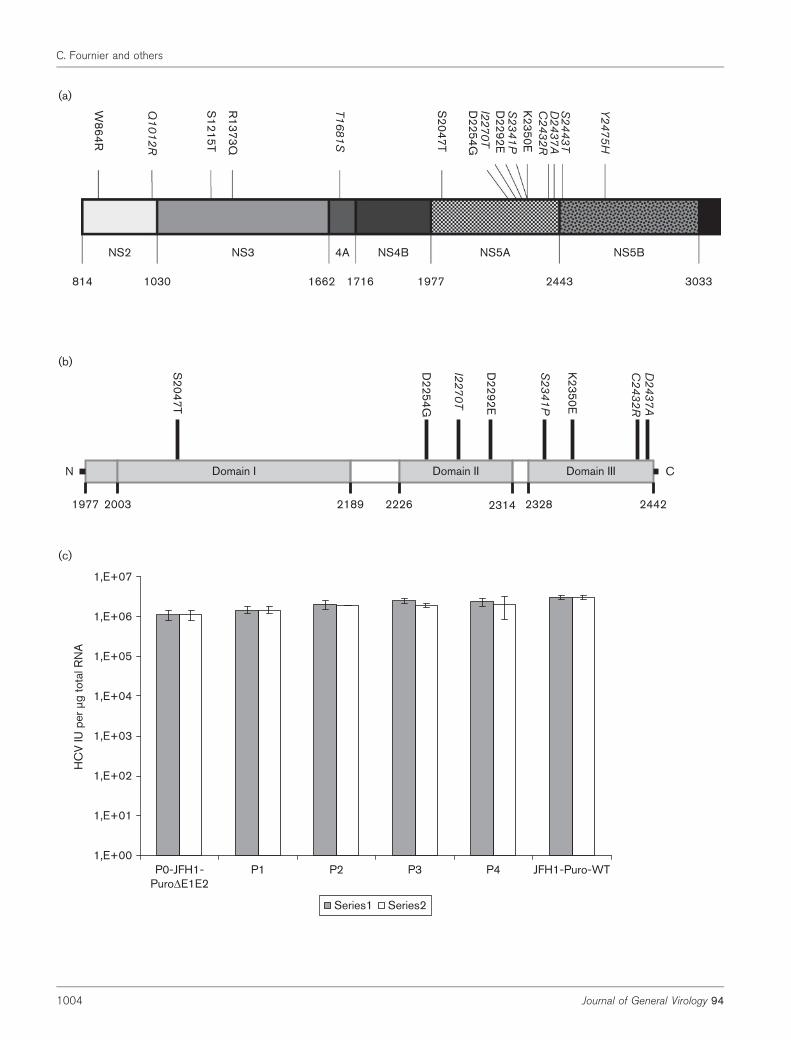
As is seen with JFH1-derived full-length genomes, wereasoned that the higher yields of trans-complementedparticles might be due to the selection of adaptive mutations.We therefore sequenced JFH1-PuroDE1E2 replicons frompopulations P0, P1, P2, P3 and P4 in series 1 and 2. Theresults for series 1 revealed seven conserved mutations in P2(D21G in core, Q1012R in NS2, T1681S in NS4A as well asI2270T, S2341P, C2432R and D2437A in NS5A) and twoadditional mutations in P4 (S2443T, Y2475H in NS5B). Forseries 2, we found seven different, conserved mutations in P3(W864R in NS2, S1215T and R1373Q in NS3, as well asS2047T, D2254G, D2292E and K2350E in NS5A). Hence,with the exception of D21G in core, all the selected mutationswere located in non-structural genes (Fig. 5a). It isnoteworthy that eight of the mutations were in NS5A (fourin domain III and two near the junction with the NS5B gene;Fig. 5b); this finding suggests that NS5A has an essential rolein the trans-encapsidation mechanism.
To characterize these mutations, we first assessed theirimpact on HCV RNA replication by measuring intracellularHCV RNA in the P0–P4 populations of JFH1-PuroDE1E2replicons. As shown in Fig. 5(c), intracellular RNA levels inP0–P4 were similar and did not differ greatly from the valuefound with JFH1-Puro-WT. This suggests that thesemutations do not affect genomic replication.
D2437A and S2443T mutations improved theproduction of trans-encapsidated virions
Various mutations that enhance infectious virus produc-tion were described close to the C terminus of NS5A(Fig. 6a). In contrast, mutations close to the N terminus ofNS5B were not known (Han et al., 2009; Jiang & Luo, 2012;Kang et al., 2009; Kaul et al., 2007; Russell et al., 2008;Scheel et al., 2008; Takeda et al., 2012).
In order to examine the role of the two mutations near theNS5A-B cleavage site on HCV RNA replication andparticles production, the D2437A and S2443T mutations
(a)
(b)
1,E+05
1,E+04
JFH1-LucΔE1E2
JFH1-PuroΔE1E2
JFH1-Luc-WT
JFH1-Puro-WT
1,E+03
1,E+02
1,E+01
Ad
control
Ad
E1E2-2a
Ad
E1E2-1a
Ad
E1E2-1b
Ad
E1E2-3a
Ad
E1E2-4
Ad
5’-NS2-1b
NT
f.f.u. m
l–1
1,E+00
E2
β-Actin
Ad-
E1E2-
1a
Ad-
E1E2-
1b
Ad-
E1E2-
3a
Ad-
E1E2-
4
Ad-
5’-
NS2-
1b
JFH1-
Puro-
WT
Ad
control
Fig. 3. The expression of heterologous envelope proteins does nottrans-complement JFH1DE1E2 replicons. (a) Detection of E2expression by Western blot analysis after transduction of Huh-7cells with Ad-E1E2-1a, Ad-E1E2-1b, Ad-E1E2-3a, Ad-E1E2-4,Ad-59-NS2-1b or Ad-control. (b) Huh-7 cells containing JFH1-PuroDE1E2 or JFH1-LucDE1E2 replicons were transduced withthe aforementioned adenoviruses (m.o.i.510). Supernatants wereharvested 72 h later and placed in contact with naıve Huh-7 cellsfor 4 h. As a control, naıve Huh-7 cells were infected with JFH1-Puro-WT or JFH1-Luc-WT. f.f.u. (b) assays were performed 72 hpost-infection. Data represent the mean±SD of three independentexperiments. NT, Non-transduced.
Adaptive mutations enhance trans-encapsidation
http://vir.sgmjournals.org 1001

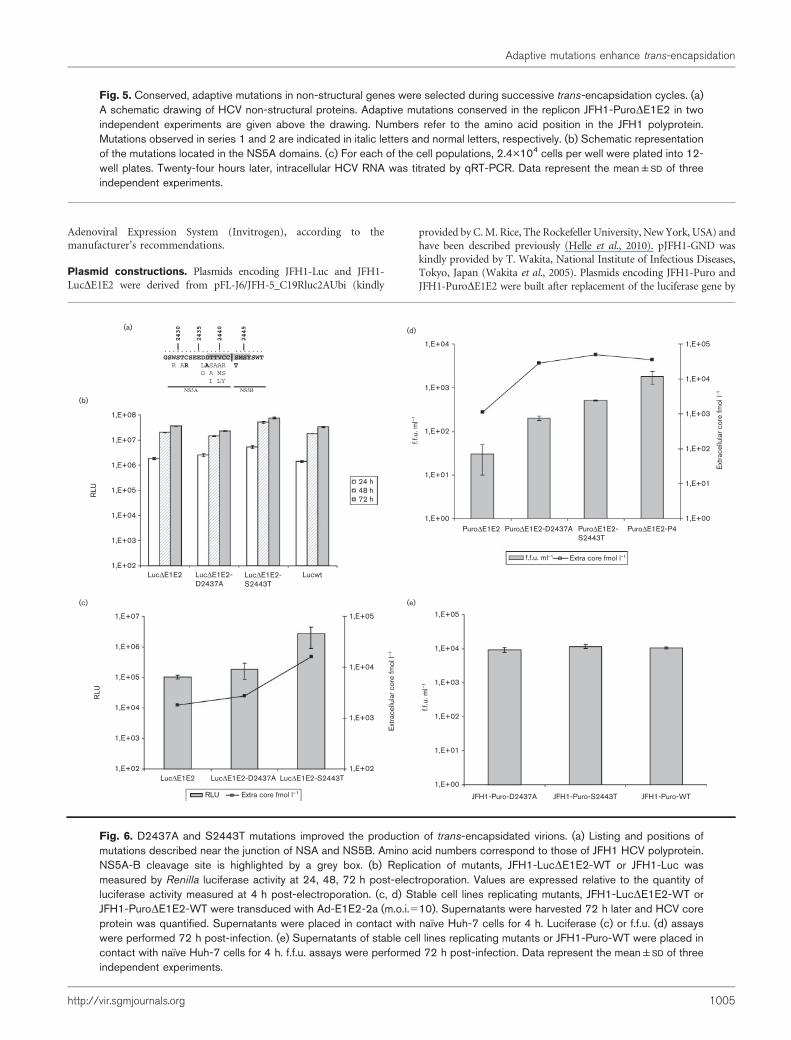
were independently introduced into JFH1-LucDE1E2 byreverse genetics. The luciferase activities of the D2437A orS2443T mutants were comparable to those of JFH1-LucDE1E2-WT and JFH1-Luc, showing that the mutationsdo not affect RNA replication (Fig. 6b). Single-roundparticle production was then determined in the cell culturesupernatant after Ad-E1E2-2a transduction. The infectiv-ities of JFH1-LucDE1E2-D2437A and JFH1-LucDE1E2-S2443T were, respectively, 1.8- and 26-fold higher thanJFH1-LucDE1E2-WT (Fig. 6c). In the same context, theD2437A and S2443T mutations increased core releaseabout 1.5- and 10.5-fold, respectively, as compared withthe JFH1-LucDE1E2-WT (Fig. 6c). To confirm our resultsthe D2437A and S2443T mutations were independentlyintroduced into pJFH1-PuroDE1E2. Stable cells linesreplicating D2437A or S2443T mutants were producedand transduced with Ad-E1E2-2a, f.f.u. assays andmeasurement of core release were performed on super-natants. As previously observed, a 6.5- and 17-fold increaseof infectivity was obtained with D2437A and S2443Tmutants, respectively, relative to the JFH1-PuroDE1E2-WT. In the same context, the D2437A and S2443Tmutations increased core release about 26- and 45-fold,respectively, as compared with the JFH1-PuroDE1E2-WT(Fig. 6d).
Finally, the mutations were introduced into pJFH1-Puro-WT and stable cells lines replicating JFH1-Puro-D2437A andJFH1-Puro-S2443T mutants were produced. Interestingly,the virus titres obtained were comparable to those of JFH1-Puro-WT, thus demonstrating that the D2437A and S2443Tmutations have no impact on infectious virus production ofJFH1-Puro-WT in cis (Fig. 6e).
DISCUSSION
Trans-encapsidation of HCV genomes lacking at least theE1E2 encoding region has recently been reported by severalgroups, with expression of the corresponding proteins byhelper viruses, transient or stable transfections or baculovirustransduction (Adair et al., 2009; Ishii et al., 2008; Steinmannet al., 2008). Moreover, it has been suggested that this type ofdefective genome can be found in the blood of infectedpatients and be trans-encapsidated in vivo into infectiousparticles by helper WT viruses (Pacini et al., 2009; Sugiyamaet al., 2009). Here, we built, characterized and optimized asystem for producing single-round infectious particles bytrans-encapsidation of JFH1-DE1E2 replicons with homo-logous envelope proteins produced by adenoviral vectors.Importantly, in agreement with previous reports (Adair et al.,2009; Ishii et al., 2008; Steinmann et al., 2008), wedemonstrated that these particles, like HCVcc particles, canbe neutralized by an anti-E2 neutralizing antibody and aretherefore likely to enter target cells in an envelope protein-dependent manner. We observed that production ofinfectious viral particles after trans-encapsidation of eitherJFH1-LucDE1E2 or JFH1-PuroDE1E2 replicons was 100-fold less efficient than with a full-length genome.
(a)
1,E+00
1,E+01
1,E+02
1,E+03
1,E+04
1,E+05
P0-JFH1-PuroΔE1E2
P2
Ext
rac
ellu
lar
core
fm
ol l
–1
Series1 Series2
(b)
1,E+02
1,E+03
1,E+04
1,E+05
1,E+06
P0-JFH1-PuroΔE1E2
P1
HC
V I
U p
er
µg
tota
l R
NA
Series1 Series2
JFH1-Puro-WTP3 P4
JFH1-Puro-WTP3 P4P2
(c)
1,E+00
1,E+01
1,E+02
1,E+03
1,E+04
1,E+05
P0-JFH1-PuroΔE1E2
P2
f.f.u. m
l–1
Series1 Series2
P3 P4 JFH1-Puro-WT
Fig. 4. Viral production is enhanced by successive trans-
encapsidations of JFH1-PuroDE1E2 by homologous envelopeproteins. (a) Huh-7 cells stably expressing JFH1-PuroDE1E2 weretransduced with Ad-E1E2-2a (m.o.i.510). Supernatants wereharvested 72 h later and placed in contact with naıve Huh-7 cellsfor 4 h. Puromycin was added 72 h post-infection. A newpopulation (P1) stably expressing JFH1-PuroDE1E2 was obtained.Trans-encapsidation, infection and selection were repeated threemore times to obtain P2, P3 and P4. Two independent experimentswere performed (series 1 and 2). These two series of fourpopulations were transduced with Ad-E1E2-2a (m.o.i.510).Supernatants were harvested 72 h later and HCV core proteinwas quantified. (b, c) Supernatants were placed in contact withnaıve Huh-7 cells for 4 h. Intracellular RNA (b) and f.f.u. (c) assayswere performed 72 h post-infection. Data represent the mean±SD
of three independent experiments.
C. Fournier and others
1002 Journal of General Virology 94

Surprisingly, the use of JFH1-PuroDE1E2 replicon did notenhance trans-encapsidation whereas, contrary to JFH1-LucDE1E2, all cells transduced with Ad-E1E2-2a containedthe replicon. This finding could be explained by the differentstructure of the packaged genomes that could be encapsi-dated with different efficiency.
Interestingly, we observed that the concomitant expressionof core or core, p7, NS2 with E1E2 did not facilitate themechanism of trans-encapsidation. Indeed, transduction ofcells replicating DE1E2 replicons with Ad-59-E2-2a or Ad-59-NS2-2a resulted in lower levels of trans-encapsidationthan with Ad-E1E2-2a, despite the likely presence of higherlevels of core or core, p7, NS2 expression, respectively. Onepossible explanation for this observation is that theprocessing of the polyproteins expressed by Ad-59-E2-2aand Ad-59-NS2-2a constructs into functional E1E2 is lowerthan with Ad-E1E2-2a. Also, Adair et al. observed thatexpression of NS2 in cis with replicons greatly enhancedreplicon transmission to naıve recipient cells, whencompared with the provision of NS2 in trans (Adair et al.,2009). Moreover, Pacini et al. observed that the best resultswere obtained after complementation of a DE1E2 repliconwith core, E1, E2 and p7 expressed in trans (Pacini et al.,2009). The latter authors highlighted significant cross-talkbetween core and NS2, which should apparently beexpressed by two separate constructs for efficient trans-encapsidation. However, we observed that trans-encapsida-tion of DE1E2 replicons with either Ad-E1E2-2a or Ad-59-NS2-2a was achievable. One could also imagine that theamounts of core, p7 and NS2 expressed in cis and in transwere far higher than that of NS5A, which plays a key role inHCV assembly and in our trans-encapsidation system (seebelow).
To investigate whether heterologous trans-encapsidationwas possible, we generated adenoviral constructs expres-sing E1E2 from strains UKN1A2-1, UKN1B2-16, UKN3A-1.28 and UKN4-21.16 (genotypes 1a, 1b, 3a and 4,respectively). We observed that these envelope proteinsdid not enable the production of infectious viral particlesfrom DE1E2 replicons. These data confirm the resultsobtained by Li et al., who demonstrated that envelopeproteins from genotype 2a strains JFH1 and J6 couldrescue DE1E2 replicons, whereas H77 and Con1 (geno-types 1a and 1b, respectively) envelope proteins could not(Li et al., 2011). In contrast, Steinmann et al. reported thatan NS3-NS5B JFH1 replicon can be rescued by a helper virusencoding Con1-derived genome segments fused with JFH1at a junction within the NS2 coding region (Steinmann et al.,2008). Overall, these results suggest that, as for inter-genotypic chimeras (Morel et al., 2011), trans-encapsidationis possible only when core, E1, E2, p7 and NS2 are from thesame genotype. Surprisingly, we did not observe trans-encapsidation after transduction with Ad-59-NS2-1b. Thiscould mean that the co-expression of heterologous andhomologous core, p7 and NS2 proteins may perturb theassembly process.
Similarly to other groups, our infectious titres of trans-
complemented viral particles were relatively low. In the
literature, several strategies have been used to increase
single-round particle infectious titres: (i) subcloning of a
packaging cell line stably expressing the structural proteins
(Steinmann et al., 2008), (ii) improvement of the
transduction efficiency (Adair et al., 2009) and (iii)
introduction of adaptive mutations (Adair et al., 2009; Li
et al., 2011). In our present study, we took advantage of the
ability to select cells infected with trans-complemented
particles through expression of the puromycin resistance
gene. This allowed us to perform successive cycles of trans-
encapsidation and select adaptive mutations that improve
the trans-encapsidation efficiency. After four cycles, we
observed that several adaptive mutations had been selected
and that single-round particle infectious titres were ten-
fold higher. It is tempting to speculate that additional
cycles could select for other mutations and further increase
infectious titres still. It is noteworthy that the majority of
selected mutations appeared in NS5A; our results suggest
that the mutations increase the efficiency of HCV assembly
or release. These findings are in agreement with recent
work illustrating the key role of NS5A in HCV assembly
(Appel et al., 2008; Tellinghuisen et al., 2008) and suggest
that NS5A also plays a key role in the trans-encapsidation
mechanism. In particular, several mutations were located
in NS5A’s domain III, which is known to interact with core
protein to promote virion assembly (Masaki et al., 2008).
Thus, some of the mutations selected in our study could
facilitate the interaction of NS5A with core and enhance
the process of trans-encapsidation. Therefore, the two
novel mutations D2437A and S2443T identified, which are
located at the P6 and P1’ positions, respectively, of the
NS5A-B cleavage site were investigated. Various mutations
that apparently enhanced the JFH1 production ability, have
already be described into the NS5A-B cleavage site, as the
reported T2438I (Han et al., 2009) and V2440L (Kaul et al.,
2007) mutations. Similarly, we determined that these
mutations enhanced infectious particles production by
trans-encapsidation, but surprisingly no similar effects were
obtained when introduced into the JFH1 genome. These
results suggest that the two mutations are specifically
implicated into trans- but not cis-encapsidation mechanism.
In summary, we have established a trans-encapsidationsystem that can be easily adapted to produce high titres ofsingle-round infectious particles. This achievement greatlybroadens the scope of previous systems and opens up newperspectives for studying HCV assembly and developingmodified, live-attenuated vaccines.
METHODS
Cells and viruses. Huh-7 human hepatoma and 293A humanembryo kidney cells were grown in Dulbecco’s modified minimalessential medium (DMEM) supplemented with 10 % FBS.Recombinant adenoviruses were generated by using the ViraPower
Adaptive mutations enhance trans-encapsidation
http://vir.sgmjournals.org 1003
(a)
NS2 NS3 4A NS4B NS5A NS5B
303324431977171616621030
W8
64
R
Q1012R
S1
21
5T
R1
37
3Q
S2
04
7T
D2
25
4G
D2
29
2E
I2270T
S2341P
C2432R
D2437A
T1681S
K2
35
0E
814
S2443T
Y2475H
(b)
(c)
1,E+00
1,E+01
1,E+02
1,E+03
1,E+04
1,E+05
1,E+06
1,E+07
P0-JFH1-
PuroΔE1E2
P1 P2 P3 P4 JFH1-Puro-WT
HC
V IU
per
µg
tota
l R
NA
Series1 Series2
Domain I Domain II Domain IIIN C
1977 2003 2189 2226 2314 2328 2442
S2
04
7T
D2
25
4G
I2270T
D2
29
2E
S2341P
K2
35
0E
C2432R
D2437A
C. Fournier and others
1004 Journal of General Virology 94
Adenoviral Expression System (Invitrogen), according to themanufacturer’s recommendations.
Plasmid constructions. Plasmids encoding JFH1-Luc and JFH1-
LucDE1E2 were derived from pFL-J6/JFH-5_C19Rluc2AUbi (kindly
provided by C. M. Rice, The Rockefeller University, New York, USA) and
have been described previously (Helle et al., 2010). pJFH1-GND was
kindly provided by T. Wakita, National Institute of Infectious Diseases,
Tokyo, Japan (Wakita et al., 2005). Plasmids encoding JFH1-Puro and
JFH1-PuroDE1E2 were built after replacement of the luciferase gene by
(a)
................ .......GSWSTCSEEDDTTVCC SMSYSWT
R AR LASAAR TG A MSI LY
(b)
1,E+02
1,E+03
1,E+04
1,E+05
1,E+06
1,E+07
1,E+08
RLU
24 h48 h72 h
(c)
1,E+02
1,E+03
1,E+04
1,E+05
1,E+06
1,E+07
LucDE1E2 LucDE1E2-D2437A LucDE1E2-S2443T
LucDE1E2-
D2437A
LucDE1E2 LucDE1E2-
S2443T
Lucwt
RLU
1,E+02
1,E+03
1,E+04
1,E+05
Ext
racellu
lar
co
re f
mo
l l–
1
RLU Extra core fmol l–1
NS5A NS5B
2430
2445
2440
2435 (d)
1,E+00
1,E+01
1,E+02
1,E+03
1,E+04
PuroΔE1E2 PuroΔE1E2-D2437A PuroΔE1E2-
S2443T
PuroΔE1E2-P4
f.f.u. m
l–1
1,E+00
1,E+01
1,E+02
1,E+03
1,E+04
1,E+05
Ext
racellu
lar
core
fm
ol l–
1
f.f.u. ml–1 Extra core fmol l–1
(e)
1,E+00
1,E+01
1,E+02
1,E+03
1,E+04
1,E+05
JFH1-Puro-D2437A JFH1-Puro-S2443T JFH1-Puro-WT
f.f.u. m
l–1
Fig. 6. D2437A and S2443T mutations improved the production of trans-encapsidated virions. (a) Listing and positions ofmutations described near the junction of NSA and NS5B. Amino acid numbers correspond to those of JFH1 HCV polyprotein.NS5A-B cleavage site is highlighted by a grey box. (b) Replication of mutants, JFH1-LucDE1E2-WT or JFH1-Luc wasmeasured by Renilla luciferase activity at 24, 48, 72 h post-electroporation. Values are expressed relative to the quantity ofluciferase activity measured at 4 h post-electroporation. (c, d) Stable cell lines replicating mutants, JFH1-LucDE1E2-WT orJFH1-PuroDE1E2-WT were transduced with Ad-E1E2-2a (m.o.i.510). Supernatants were harvested 72 h later and HCV coreprotein was quantified. Supernatants were placed in contact with naıve Huh-7 cells for 4 h. Luciferase (c) or f.f.u. (d) assayswere performed 72 h post-infection. (e) Supernatants of stable cell lines replicating mutants or JFH1-Puro-WT were placed incontact with naıve Huh-7 cells for 4 h. f.f.u. assays were performed 72 h post-infection. Data represent the mean±SD of threeindependent experiments.
Fig. 5. Conserved, adaptive mutations in non-structural genes were selected during successive trans-encapsidation cycles. (a)A schematic drawing of HCV non-structural proteins. Adaptive mutations conserved in the replicon JFH1-PuroDE1E2 in twoindependent experiments are given above the drawing. Numbers refer to the amino acid position in the JFH1 polyprotein.Mutations observed in series 1 and 2 are indicated in italic letters and normal letters, respectively. (b) Schematic representationof the mutations located in the NS5A domains. (c) For each of the cell populations, 2.4�104 cells per well were plated into 12-well plates. Twenty-four hours later, intracellular HCV RNA was titrated by qRT-PCR. Data represent the mean±SD of threeindependent experiments.
Adaptive mutations enhance trans-encapsidation
http://vir.sgmjournals.org 1005

the puromycin acetyltransferase gene in pJFH1-Luc and pJFH1-
LucDE1E2, respectively. D2437A and S2443T mutations which are
located at the P6 and P1’ positions, respectively, of the NS5A-B cleavage
site were introduced into the plasmids by using the QuikChange II XL
site-directed mutagenesis kit, according to the manufacturer’s recom-
mendations (Agilent Technologies).
Sequences encoding the 21 carboxy-terminal residues of core with E1-
E2 (aa 171–750), 59 UTR-E2 or 59 UTR-NS2 from the genotype 2a
JFH1 strain (kindly provided by T. Wakita) were amplified by PCR,
cloned into the pENTR Directional TOPO vector and transferred into
pAd/CMv/v5-DEST by gateway recombination. The same strategy
was used to construct pAd/CMv/v5-DEST encoding E1E2 from
genotype 1a, 1b, 3a and 4 (UKN1A2-1, UKN1B2-16, UKN3A-1.28
and UKN4-21.16 strains, respectively, kindly provided by J. K. Ball,
University of Nottingham, Nottingham, UK) or core to NS2 from
genotype 1b (Lex strain, GenBank accession no. JN120912). The
primer sequences are available on request. These constructs were used
to generate adenovirus according to the manufacturer’s recommen-
dations (Invitrogen).
In vitro transcription, electroporation and establishment of
stable cell lines. HCV RNA were produced by in vitro transcription
of JFH1-Puro-WT, JFH1-PuroDE1E2, JFH1-Luc-WT and JFH1-
LucDE1E2 genomes and delivered into Huh-7 cells by electropora-
tion, as described previously (Helle et al., 2010). Electroporated cells
were seeded at a density of 106 cells per 25 cm2 flask. For the
establishment of JFH1-PuroDE1E2 cell populations, puromycin
(Invitrogen) was added to DMEM supplemented with 10 % FBS to
give a final concentration of 5 mg ml21.
Trans-encapsidation experiments. One million cells containing
JFH1-PuroDE1E2 or JFH1-LucDE1E2 replicon were seeded into a
25 cm2 flask. On the following day, cells were exposed to the
adenovirus (m.o.i. of 10) and incubated overnight. Supernatants were
recovered 72 h later, centrifuged (1000 g for 5 min) and filtered
through a 0.45 mm syringe filter.
Indirect immunofluorescence. Cells were fixed with paraformalde-
hyde for 10 min at room temperature, permeabilized with 0.5 %
Triton X-100 and washed three times with PBS. Next, the cells were
incubated with primary antibodies [rat anti-E2 mAb 3/11, kindly
provided by J. A. McKeating, or anti-NS3 (Virogen)] for 1 h at room
temperature, washed with PBS and then incubated with secondary
antibodies [rhodamine red-X-conjugated affiniPure donkey anti-rat
IgG (Jackson ImmunoResearch) or FITC-conjugated anti-mouse IgG
(Rockland)] for 30–45 min at room temperature.
Western blot analysis. Cells were lysed with PBS and 1 % Triton X-
100. Protein content of pre-cleared cell lysates was determined by the
BCA method as recommended by the manufacturer (Sigma), using
BSA as a standard. Total proteins were separated by SDS-PAGE,
transferred to nitrocellulose membranes (Hybond-ECL; Amersham)
and revealed with a specific mAb followed by anti-rat IgG conjugated
to peroxidize (Jackson Immunoresearch). The immune complexes
were visualized by enhanced chemiluminescence detection (ECL;
Amersham) as recommended by the manufacturer.
HCV core protein quantification. HCV core antigen secreted into
the supernatant was quantified by a fully automated chemilumin-
escent microparticle immunoassay (Architect HCVAg; Abbott),
according to the manufacturer’s instructions.
HCV RNA quantification. Huh-7 cells (1.26105) per well in a 12-well
plate were inoculated for 4 h with 1 ml of trans-encapsidation
supernatant. Cells were incubated overnight and washed with
DMEM. Seventy-two hours later, total RNA in cell lysates was extracted
using the Qiagen RNeasy kit. cDNA was synthesized using the High
Capacity cDNA Reverse Transcription kit (Applied BioSystems) and
titrated in a quantitative, real-time RT-PCR assay (qRT-PCR) on an
ABI Prism 7900 (Applied Biosystems) using primers within the NS3
region (sequences available on request). Values were normalized
against the b-actin-specific signal. For replication analysis, stable cell
lines P0, P1, P2, P3, P4 were plated in 12-well plates, 72 h post-plating
total RNA was extracted and titrated as previously described.
Measurement of luciferase activity. Cells were inoculated as
described for HCV RNA quantification. After 72 h, cell monolayers
were lysed by the addition of 200 ml Renilla Luciferase Assay Lysis
Buffer (Promega). Luminescence was measured according to the
manufacturer’s instruction on a Centro XS3 LB960 luminometer
(Berthold Technologies). For replication analysis, cells were electro-
porated with various mutants or wild-type RNAs, then were plated in
24-well plates and lysed with 100 ml 16 Renilla lysis buffer (Promega)
at 4, 24, 48 and 72 h post-electroporation. Fifty microlitres of cell
lysate was used to determine the luciferase activity. Values were
normalized relative to those at 4 h.
HCV infectivity quantification. Naıve Huh-7 cells were inoculated
with 300 ml trans-encapsidation supernatant. Forty-eight hours after
inoculation, the infected cells were fixed with paraformaldehyde,
permeabilized with 0.5 % Triton X-100, immunostained with anti-
NS3 antibody (Virogen) and counted for f.f.u.
Neutralization assay. Neutralization assays were performed by pre-
incubating trans-encapsidation supernatants with the 3/11 anti-E2
mAb for 2 h at 37 uC (Helle et al., 2010). The infection efficiency was
measured 72 h post-infection, as described above.
Sequencing of adaptive mutations. We determined the sequence
of JFH1-PuroDE1E2 replicons in the different Huh-7 cell populations
(P0, P1, P2, P3 and P4) by directly sequencing overlapping PCR
fragments spanning the entire ORF. The purified PCR products were
directly sequenced with the BigDye Terminator v1.1 kit (Applied
Biosystems). Sequences were read on an ABI Prism 310 genetic
analyser (Applied Biosystems) and its related Sequence Navigator
software. The sequences of primers used for the RT-PCR, PCR and
sequencing reactions are available on request.
ACKNOWLEDGEMENTS
We thank T. Wakita, C. M. Rice, J. K. Ball and J. A. McKeating for
providing us with reagents. We are grateful to A. Baron for her expert
technical assistance. This work was funded by the Conseil Regional de
Picardie (Hepanovir, grant no. AAP08-12) and the Universite de
Picardie–Jules Verne.
REFERENCES
Adair, R., Patel, A. H., Corless, L., Griffin, S., Rowlands, D. J. &McCormick, C. J. (2009). Expression of hepatitis C virus (HCV)
structural proteins in trans facilitates encapsidation and transmission
of HCV subgenomic RNA. J Gen Virol 90, 833–842.
Appel, N., Herian, U. & Bartenschlager, R. (2005). Efficient rescue of
hepatitis C virus RNA replication by trans-complementation with
nonstructural protein 5A. J Virol 79, 896–909.
Appel, N., Zayas, M., Miller, S., Krijnse-Locker, J., Schaller, T., Friebe,P., Kallis, S., Engel, U. & Bartenschlager, R. (2008). Essential role of
domain III of nonstructural protein 5A for hepatitis C virus infectious
particle assembly. PLoS Pathog 4, e1000035.
C. Fournier and others
1006 Journal of General Virology 94

Brohm, C., Steinmann, E., Friesland, M., Lorenz, I. C., Patel, A.,Penin, F., Bartenschlager, R. & Pietschmann, T. (2009).Characterization of determinants important for hepatitis C virus p7
function in morphogenesis by using trans-complementation. J Virol
83, 11682–11693.
Choo, Q. L., Richman, K. H., Han, J. H., Berger, K., Lee, C., Dong, C.,Gallegos, C., Coit, D., Medina-Selby, R. & Barr, P. J. (1991). Geneticorganization and diversity of the hepatitis C virus. Proc Natl Acad Sci
U S A 88, 2451–2455.
Delgrange, D., Pillez, A., Castelain, S., Cocquerel, L., Rouille, Y.,Dubuisson, J., Wakita, T., Duverlie, G. & Wychowski, C. (2007).Robust production of infectious viral particles in Huh-7 cells by
introducing mutations in hepatitis C virus structural proteins. J GenVirol 88, 2495–2503.
Dimitrova, M., Imbert, I., Kieny, M. P. & Schuster, C. (2003). Protein-protein interactions between hepatitis C virus nonstructural proteins.
J Virol 77, 5401–5414.
Eyre, N. S., Phillips, R. J., Bowden, S., Yip, E., Dewar, B., Locarnini,S. A. & Beard, M. R. (2009). Hepatitis B virus and hepatitis C virus
interaction in Huh-7 cells. J Hepatol 51, 446–457.
Gottwein, J. M. & Bukh, J. (2008). Cutting the gordian knot-
development and biological relevance of hepatitis C virus cell culture
systems. Adv Virus Res 71, 51–133.
Gottwein, J. M., Scheel, T. K., Jensen, T. B., Lademann, J. B., Prentoe,J. C., Knudsen, M. L., Hoegh, A. M. & Bukh, J. (2009). Development
and characterization of hepatitis C virus genotype 1-7 cell culturesystems: role of CD81 and scavenger receptor class B type I and effect
of antiviral drugs. Hepatology 49, 364–377.
Han, Q., Xu, C., Wu, C., Zhu, W., Yang, R. & Chen, X. (2009).Compensatory mutations in NS3 and NS5A proteins enhance the
virus production capability of hepatitis C reporter virus. Virus Res145, 63–73.
Helle, F., Vieyres, G., Elkrief, L., Popescu, C. I., Wychowski, C.,Descamps, V., Castelain, S., Roingeard, P., Duverlie, G. &Dubuisson, J. (2010). Role of N-linked glycans in the functions of
hepatitis C virus envelope proteins incorporated into infectious
virions. J Virol 84, 11905–11915.
Ishii, K., Murakami, K., Hmwe, S. S., Zhang, B., Li, J., Shirakura, M.,Morikawa, K., Suzuki, R., Miyamura, T. & other authors (2008).Trans-encapsidation of hepatitis C virus subgenomic replicon RNA
with viral structure proteins. Biochem Biophys Res Commun 371, 446–
450.
Jiang, J. & Luo, G. (2012). Cell culture-adaptive mutations promote
viral protein-protein interactions and morphogenesis of infectious
hepatitis C virus. J Virol 86, 8987–8997.
Jirasko, V., Montserret, R., Appel, N., Janvier, A., Eustachi, L., Brohm,C., Steinmann, E., Pietschmann, T., Penin, F. & Bartenschlager, R.(2008). Structural and functional characterization of nonstructural
protein 2 for its role in hepatitis C virus assembly. J Biol Chem 283,
28546–28562.
Jones, D. M., Patel, A. H., Targett-Adams, P. & McLauchlan, J. (2009).The hepatitis C virus NS4B protein can trans-complement viral RNA
replication and modulates production of infectious virus. J Virol 83,2163–2177.
Kang, J. I., Kim, J. P., Wakita, T. & Ahn, B. Y. (2009). Cell culture-
adaptive mutations in the NS5B gene of hepatitis C virus withdelayed replication and reduced cytotoxicity. Virus Res 144, 107–
116.
Kato, N., Hijikata, M., Ootsuyama, Y., Nakagawa, M., Ohkoshi, S.,Sugimura, T. & Shimotohno, K. (1990). Molecular cloning of the
human hepatitis C virus genome from Japanese patients with non-A,non-B hepatitis. Proc Natl Acad Sci U S A 87, 9524–9528.
Kato, T., Matsumura, T., Heller, T., Saito, S., Sapp, R. K., Murthy, K.,Wakita, T. & Liang, T. J. (2007). Production of infectious hepatitis Cvirus of various genotypes in cell cultures. J Virol 81, 4405–4411.
Kaul, A., Woerz, I., Meuleman, P., Leroux-Roels, G. &Bartenschlager, R. (2007). Cell culture adaptation of hepatitis C
virus and in vivo viability of an adapted variant. J Virol 81, 13168–
13179.
Lavillette, D., Tarr, A. W., Voisset, C., Donot, P., Bartosch, B., Bain, C.,Patel, A. H., Dubuisson, J., Ball, J. K. & Cosset, F. L. (2005).Characterization of host-range and cell entry properties of the majorgenotypes and subtypes of hepatitis C virus. Hepatology 41, 265–
274.
Li, R., Qin, Y., He, Y., Tao, W., Zhang, N., Tsai, C., Zhou, P. & Zhong, J.(2011). Production of hepatitis C virus lacking the envelope-encoding
genes for single-cycle infection by providing homologous envelopeproteins or vesicular stomatitis virus glycoproteins in trans. J Virol 85,
2138–2147.
Masaki, T., Suzuki, R., Murakami, K., Aizaki, H., Ishii, K., Murayama,A., Date, T., Matsuura, Y., Miyamura, T. & other authors (2008).Interaction of hepatitis C virus nonstructural protein 5A with core
protein is critical for the production of infectious virus particles.J Virol 82, 7964–7976.
Miyanari, Y., Atsuzawa, K., Usuda, N., Watashi, K., Hishiki, T., Zayas,M., Bartenschlager, R., Wakita, T., Hijikata, M. & Shimotohno, K.(2007). The lipid droplet is an important organelle for hepatitis C
virus production. Nat Cell Biol 9, 1089–1097.
Morel, V., Fournier, C., Francois, C., Brochot, E., Helle, F., Duverlie,G. & Castelain, S. (2011). Genetic recombination of the hepatitis C
virus: clinical implications. J Viral Hepat 18, 77–83.
Pacini, L., Graziani, R., Bartholomew, L., De Francesco, R. &Paonessa, G. (2009). Naturally occurring hepatitis C virus sub-genomic deletion mutants replicate efficiently in Huh-7 cells and are
trans-packaged in vitro to generate infectious defective particles.
J Virol 83, 9079–9093.
Pietschmann, T., Kaul, A., Koutsoudakis, G., Shavinskaya, A., Kallis,S., Steinmann, E., Abid, K., Negro, F., Dreux, M. & other authors(2006). Construction and characterization of infectious intragenoty-pic and intergenotypic hepatitis C virus chimeras. Proc Natl Acad Sci
U S A 103, 7408–7413.
Russell, R. S., Meunier, J. C., Takikawa, S., Faulk, K., Engle, R. E.,Bukh, J., Purcell, R. H. & Emerson, S. U. (2008). Advantages of a
single-cycle production assay to study cell culture-adaptive mutations
of hepatitis C virus. Proc Natl Acad Sci U S A 105, 4370–4375.
Scheel, T. K., Gottwein, J. M., Jensen, T. B., Prentoe, J. C., Hoegh,A. M., Alter, H. J., Eugen-Olsen, J. & Bukh, J. (2008). Development ofJFH1-based cell culture systems for hepatitis C virus genotype 4a and
evidence for cross-genotype neutralization. Proc Natl Acad Sci U S A
105, 997–1002.
Steinmann, E., Brohm, C., Kallis, S., Bartenschlager, R. &Pietschmann, T. (2008). Efficient trans-encapsidation of hepatitis C
virus RNAs into infectious virus-like particles. J Virol 82, 7034–7046.
Sugiyama, K., Suzuki, K., Nakazawa, T., Funami, K., Hishiki, T.,Ogawa, K., Saito, S., Shimotohno, K. W., Suzuki, T. & other authors(2009). Genetic analysis of hepatitis C virus with defective genome
and its infectivity in vitro. J Virol 83, 6922–6928.
Takeda, M., Ikeda, M., Ariumi, Y., Wakita, T. & Kato, N. (2012).Development of hepatitis C virus production reporter-assay
systems using two different hepatoma cell lines. J Gen Virol 93,1422–1431.
Tang, H. & Grise, H. (2009). Cellular and molecular biology of HCVinfection and hepatitis. Clin Sci (Lond) 117, 49–65.
Adaptive mutations enhance trans-encapsidation
http://vir.sgmjournals.org 1007

Tellinghuisen, T. L., Foss, K. L. & Treadaway, J. (2008). Regulation ofhepatitis C virion production via phosphorylation of the NS5Aprotein. PLoS Pathog 4, e1000032.
Tong, X. & Malcolm, B. A. (2006). Trans-complementation of HCVreplication by non-structural protein 5A. Virus Res 115, 122–130.
Wakita, T., Pietschmann, T., Kato, T., Date, T., Miyamoto, M., Zhao, Z.,Murthy, K., Habermann, A., Krausslich, H. G. & other authors (2005).
Production of infectious hepatitis C virus in tissue culture from a
cloned viral genome. Nat Med 11, 791–796.
Widjojoatmodjo, M. N., van Gennip, H. G., Bouma, A., van Rijn, P. A. &
Moormann, R. J. (2000). Classical swine fever virus Erns deletion
mutants: trans-complementation and potential use as nontransmis-
sible, modified, live-attenuated marker vaccines. J Virol 74, 2973–
2980.
C. Fournier and others
1008 Journal of General Virology 94




















