
Multidimensional potential energy surface determination by modified Shepard interpolation for a...
10
Multi-dimensional potential energy surface determination by modified Shepard interpolation for a molecule–surface reaction: H 2 + Pt(1 1 1) C. Crespos a , M.A. Collins b , E. Pijper a , G.J. Kroes a, * a Leiden Institute of Chemistry, Gorlaeus Laboratories, Leiden University, P.O. Box 9502, 2300 RA Leiden, The Netherlands b Research School of Chemistry, Australian National University, ACT 0200, Australia Received 20 March 2003; in final form 4 June 2003 Published online: 9 July 2003 Abstract A modified Shepard interpolation method, developed for constructing potential energy surfaces (PESs) for gas phase reactions, has been adapted to generate PESs for molecule–surface reactions, and applied to the dissociative chemi- sorption of H 2 on Pt(1 1 1). To provide a test of the method, the input data were taken from an existing PES. Reaction probabilities computed using classical and quantum dynamics on the new PES are in excellent agreement with results for the old PES, the construction of which required twice as many points. This shows that the modified Shepard in- terpolation method can be used efficiently to build PESs which yield accurate dynamics results for molecule–surface reactions. Ó 2003 Elsevier B.V. All rights reserved. 1. Introduction A key step in theoretical studies of chemical re- actions dynamics is the determination of the po- tential energy surface (PES). Many fitting and interpolation methods have been proposed to con- struct continuous representations of PESs using ab initio calculated points. Exact dynamics calcula- tions of, for instance, initial-state selected reaction probabilities require the use of a sufficiently accu- rate PES, defined on a large area of configuration space. Although the development of low-dimen- sional models [1–3] to understand and interpret the dynamics is very important, the validation of elec- tronic structure methods through comparisons with experiment often requires accurate high-dimen- sional simulations [4–7]. The study of gas–surface reactions implies many degrees of freedom and the development of an efficient scheme to produce ac- curate high-dimensional PESs is a crucial task. In recent years, significant advances have been made in the evaluation of the energy of a molecule interacting with a metal surface. In particular, density functional theory (DFT) [8,9] within the generalized gradient approximation (GGA) [10,11] is a convenient tool to compute molecule–surface Chemical Physics Letters 376 (2003) 566–575 www.elsevier.com/locate/cplett * Corresponding author. Fax: +31-71-527-4488. E-mail address: [email protected] (G.J. Kroes). 0009-2614/$ - see front matter Ó 2003 Elsevier B.V. All rights reserved. doi:10.1016/S0009-2614(03)01033-9
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Multidimensional potential energy surface determination by modified Shepard interpolation for a...

Chemical Physics Letters 376 (2003) 566–575
www.elsevier.com/locate/cplett
Multi-dimensional potential energy surface determinationby modified Shepard interpolation for amolecule–surface reaction: H2 + Pt(1 1 1)
C. Crespos a, M.A. Collins b, E. Pijper a, G.J. Kroes a,*
a Leiden Institute of Chemistry, Gorlaeus Laboratories, Leiden University, P.O. Box 9502, 2300 RA Leiden, The Netherlandsb Research School of Chemistry, Australian National University, ACT 0200, Australia
Received 20 March 2003; in final form 4 June 2003
Published online: 9 July 2003
Abstract
A modified Shepard interpolation method, developed for constructing potential energy surfaces (PESs) for gas phase
reactions, has been adapted to generate PESs for molecule–surface reactions, and applied to the dissociative chemi-
sorption of H2 on Pt(1 1 1). To provide a test of the method, the input data were taken from an existing PES. Reaction
probabilities computed using classical and quantum dynamics on the new PES are in excellent agreement with results
for the old PES, the construction of which required twice as many points. This shows that the modified Shepard in-
terpolation method can be used efficiently to build PESs which yield accurate dynamics results for molecule–surface
reactions.
� 2003 Elsevier B.V. All rights reserved.
1. Introduction
A key step in theoretical studies of chemical re-
actions dynamics is the determination of the po-
tential energy surface (PES). Many fitting andinterpolation methods have been proposed to con-
struct continuous representations of PESs using ab
initio calculated points. Exact dynamics calcula-
tions of, for instance, initial-state selected reaction
probabilities require the use of a sufficiently accu-
rate PES, defined on a large area of configuration
* Corresponding author. Fax: +31-71-527-4488.
E-mail address: [email protected] (G.J. Kroes).
0009-2614/$ - see front matter � 2003 Elsevier B.V. All rights reserv
doi:10.1016/S0009-2614(03)01033-9
space. Although the development of low-dimen-
sional models [1–3] to understand and interpret the
dynamics is very important, the validation of elec-
tronic structure methods through comparisons with
experiment often requires accurate high-dimen-sional simulations [4–7]. The study of gas–surface
reactions implies many degrees of freedom and the
development of an efficient scheme to produce ac-
curate high-dimensional PESs is a crucial task.
In recent years, significant advances have been
made in the evaluation of the energy of a molecule
interacting with a metal surface. In particular,
density functional theory (DFT) [8,9] within thegeneralized gradient approximation (GGA) [10,11]
is a convenient tool to compute molecule–surface
ed.

C. Crespos et al. / Chemical Physics Letters 376 (2003) 566–575 567
interactions involving many degrees of freedom.
Thus, comparable advances in interpolation and
fitting techniques are highly desirable.
The first PES models used in gas–surface dy-
namics simulations were derived from the general
London–Eyring–Polanyi–Sato (LEPS) analyticalform fitted on ab initio data [12–14]. Other authors
have expanded the potential of a diatomic mole-
cule interacting with a surface in functions which
are adapted to the symmetry associated with the
surface unit cell [15,16]. A new idea has been to
first remove the greater part of the corrugation
and the anisotropy from the molecule–surface in-
teraction, by taking sums of atom–surface inter-actions (for the atoms constituting the molecule)
as a zero-order expression for the potential, and
then using symmetry-adapted functions to inter-
polate the remainder of the interaction. This
method has been called the corrugation reducing
procedure (CRP) [17]. This new interpolation
scheme [17] has proved its efficiency in many
studies of diatomic molecules reacting or scatter-ing on metal surfaces, for instance, in six-dimen-
sional quantum and classical calculations on the
reaction of H2 on Pt(1 1 1) [4]. However, it is not
easy to extend this method to problems involving
more than six degrees of freedom.
A major goal in theoretical surface science is to
be able to study the dynamics of a polyatomic
molecule reacting on a surface. The achievement ofthis goal requires the availability of accurate, glo-
bal high-dimensional PESs for such systems. As a
step towards this goal, we have therefore tested
and adapted a method for generating PESs based
on modified Shepard interpolation, that has pre-
viously been applied to gas phase reactions [18–
22]. This method does not require the knowledge
of an a priori analytical form of the PES, and usesclassical trajectory simulations to provide an iter-
ative scheme for successively improving the PES.
The PES is given by an interpolation of local
Taylor expansions, and an iterative procedure
places new points only in regions of configuration
space that are important for dynamics. So instead
of computing a regular grid of ab initio points and
performing interpolation on it, the method focuseson �dynamically interesting� parts of the PES. One
of the great advantages of this method is its
economy in terms of the number of ab initio points
needed and, as a consequence, its economy in CPU
time. For example, only about 200 ab initio points
were needed for the reaction OH +H2 !H2O + H
[21].
The main goals of this work are: (i) to pro-duce a modified version of this interpolation
method that allows the construction of PESs for
gas–surface reactions and (ii) to test the method
by applying it to the system H2 + Pt(1 1 1). The
diffraction and dissociation processes of H2 on
Pt(1 1 1) surface have already been studied [4],
using an accurate representation of the PES [23]
based on the corrugation reducing procedure[17]. The latter PES (designated as �CRP-PES� for
corrugation reducing procedure PES) has been
used here to generate a new PES via the modified
Shepard interpolation method (named �MS-PES�for modified Shepard PES). Thus, the CRP-PES
plays the role of an ab initio calculation code
and gives for any geometry a value of the energy
and the first derivatives, considered as the exactones. The efficiency and the accuracy of the
modified Shepard interpolation method is tested
by comparing the results of dynamics simulations
on both the CRP-PES and the MS-PES (which
should be identical if the interpolation were
exact).
Section 2 gives a brief overview of the modified
Shepard interpolation method as revised for mol-ecule–surface interactions, and describes the way
we have prepared the MS-PES from the CRP-PES.
In Section 3, a discussion of the accuracy of the
interpolation method is provided by comparing
reaction probabilities obtained from classical and
quantum dynamics calculations that were ob-
tained with both PESs.
2. Method
In this section, the method for iteratively de-
veloping an interpolated PES, by modified Shep-
ard interpolation, is briefly presented. The
complete methodology will be detailed elsewhere
[24]. Here, the method will be applied, for the firsttime, to the system of a diatomic molecule reacting
on a surface.

568 C. Crespos et al. / Chemical Physics Letters 376 (2003) 566–575
2.1. Interpolated PES
The interpolated PES is given by a weighted
series of Taylor expansions centered at ab initio
data points, sampled throughout the configurationspace of the system. This sample of data points is
non-uniform and describes the regions of the
configuration space that are most important for
reaction with the highest accuracy. This sample of
points will be called the �PES data set� in the fol-
lowing. The method to determine the location of
these ab initio data points will be detailed below.
The H2 +Pt(1 1 1) system is described using in-verse inter-atomic distances, Q ¼ f1=R1; 1=R2; . . . ;1=RNðN�1Þ=2g with inter-atomic distances Ri and Nbeing the number of atoms. The model and the
system of coordinates to be used in the interpola-
tion method is depicted in Fig. 1. The impinging
molecule is a diatomic and the frozen surface is
represented by three atoms which are kept fixed
(these three atoms, defining the surface unit cellvectors, are required to represent the motion of the
molecule center of mass parallel to the surface).
Thus N ¼ 5 atoms are required to fully represent
the problem. The inter-atomic distances are kept
fixed for the surface atoms (the three Pt–Pt inter-
atomic distances). Only six coordinates are needed
to describe the reaction of a diatomic molecule
Fig. 1. Representation of the H2 +Pt(1 1 1) system, used in the
modified Shepard interpolation scheme.
with a frozen surface, so that any given geometry
nðiÞ ¼ n½QðiÞ is expressed in terms of 6 linear
combinations n ¼ ðn1; . . . . . . :n6Þ of the ½NðN � 1Þ=2 � 3 ¼ 7 remaining inter-atomic distances (one
of them being redundant).
The potential energy V , at a given configurationn, in the vicinity of an ab initio data point nðiÞ, can
be expanded as a second-order Taylor series, TiðnÞ:
TiðnÞ ¼ V ½nðiÞ þX6
k¼1
½nk � nkðiÞoVonk
����n¼nðiÞ
þ 1
2!
X6
k¼1
X6
j¼1
½nk � nkðiÞ
� ½nj � njðiÞo2V
onkonj
����n¼nðiÞ
þ � � � ð1Þ
An accurate evaluation of V can be obtained by a
modified Shepard interpolation [25,26], where the
potential energy is obtained as a weighted average
of the different Taylor series Ti (i ¼ 1; . . . ;Ndata)
calculated from each data point of our configura-
tion space sample (the number of data points in
the PES data set being Ndata) and their symmetry
equivalent points:
V ðnÞ ¼X
g2G
XNdata
i¼1
wg�iðnÞTg�iðnÞ; ð2Þ
where G denotes the symmetry group of the system
and g � i express the fact that the ith data point is
transformed by the group element g (in the origi-
nal study of the H2 + Pt(1 1 1) system, the differ-
ence between the fcc and hcp sites is neglected and
the symmetry group of the surface unit cell is C6v
[23]). The sum over the group elements in Eq. (2)
ensures that the PES has the correct symmetry
with respect to the permutation of indistinguish-
able particles. The symmetry of the PES with re-
spect to translation on the surface is ensured by
translating (in X and Y ) the center of mass of the
impinging molecule to the primary unit cell before
the PES of Eq. (2) is evaluated. The weight wg�i ofeach Taylor series in Eq. (2) is evaluated in a
similar manner to that used previously [22,27].
Further technical details concerning the weights
and other aspects of the PES interpolation method
will be presented elsewhere [24].

C. Crespos et al. / Chemical Physics Letters 376 (2003) 566–575 569
2.2. Iterative development of the PES
In order to build and optimize the PES data set,
an iterative scheme has been developed [18]. The
idea is to start with only a few calculated pointsand compute classical trajectories using this initial
version of the PES sample. The initial guess con-
sists of calculated data points mainly located close
to some hypothetic reaction pathways, assuming a
good accuracy in these regions is required. The
configurations visited by each of the classical tra-
jectories are stored in files and a choice of relevant
data points to be added to the initial sample ismade according to two different criteria, with the
aim of computing accurate observables (reaction
probabilities). For this purpose, one argument
suggests that the best location for a new data point
would be in the region most frequently visited by
the classical trajectories (�h-weight� criterion). A
second argument suggests that the accuracy of the
PES could be improved if a new data point isadded in the region where the interpolated poten-
tial is expected to be least accurate (�variance
sampling� criterion). The least accurate regions are
the ones for which the variance of the weighted
average in Eq. (2) is largest.
The distribution of data points chosen to be
added to the PES data set is driven by the con-
figurations visited by the classical trajectories. Theselection criteria for growing the sample of data
points ensure an efficient improvement of the PES
and a convergence of observables like reaction
probabilities. Indeed, the observation of such
quantities gives us a criterion to control the gain in
accuracy during the growing process of the PES
data set. In this study we have chosen to check the
convergence of the initial-state selected moleculardissociation probability as the size of the data set
increases. Many past studies show a good con-
vergence of reaction probability with the growth of
the PES data set for gas phase reactions [21,28–
33].
In summary, an initial PES data set is grown by
iteratively adding points corresponding to config-
uration space regions where accurate determina-tion of the potential is required. The growth of the
PES is stopped when a computed observable (in
our case the reaction probability) is considered as
converged, within a given tolerance. The key point
of the modified Shepard interpolation method is
that classical trajectories are used to scan the
configuration space of the system and locate the
dynamically important regions.
2.3. H2 +Pt(1 1 1) system as a benchmark
The aim of the present study is to test the re-
vised interpolation method for the case of gas–
surface reactions. We have modified the original
code to be able to treat the translational symmetry
of the problem (motion of H2 center of mass
parallel to the surface).The diffraction and dissociation processes of H2
on Pt(1 1 1) surface have already been extensively
studied [4], using an accurate representation of the
PES based on the corrugation reducing procedure
[17]. We use this accurate PES (CRP-PES) to
produce data points required in the modified
Shepard interpolation method. At the end of the
interpolated PES (MS-PES) growing process, weare left with two versions of the H2 + Pt(1 1 1)
potential, one made from the other. Quantum and
classical dynamics simulations are then carried out
on both PESs to test the efficiency and accuracy of
the modified Shepard interpolation method.
In the following, we will use the system of co-
ordinates depicted in Fig. 2a to discuss the results
of the classical and quantum dynamics calcula-tions. This system of coordinates is the one com-
monly used to discuss diatomic molecules reacting
on surfaces. The motion of the H2 center of mass is
represented by the set of coordinates (X , Y , Z), the
orientation of the molecular axis by the angles (h,
/) and the stretching of the molecular bond by the
coordinate r. To aid the discussion in Section 3,
some two-dimensional cuts through the CRP-po-tential [23] are shown in Fig. 3. Fig. 3a shows the
dependence of the potential on r and Z, for a ge-
ometry in which the molecule is located at the
threefold hollow site, the molecular axis being
parallel to the surface. Fig. 3a shows where the
reactants valley, the barrier region, and the prod-
ucts valley are approximately located for the sys-
tem considered here. Fig. 3b shows how thereaction barrier height varies across the surface,
for the molecule lying parallel to the surface (the
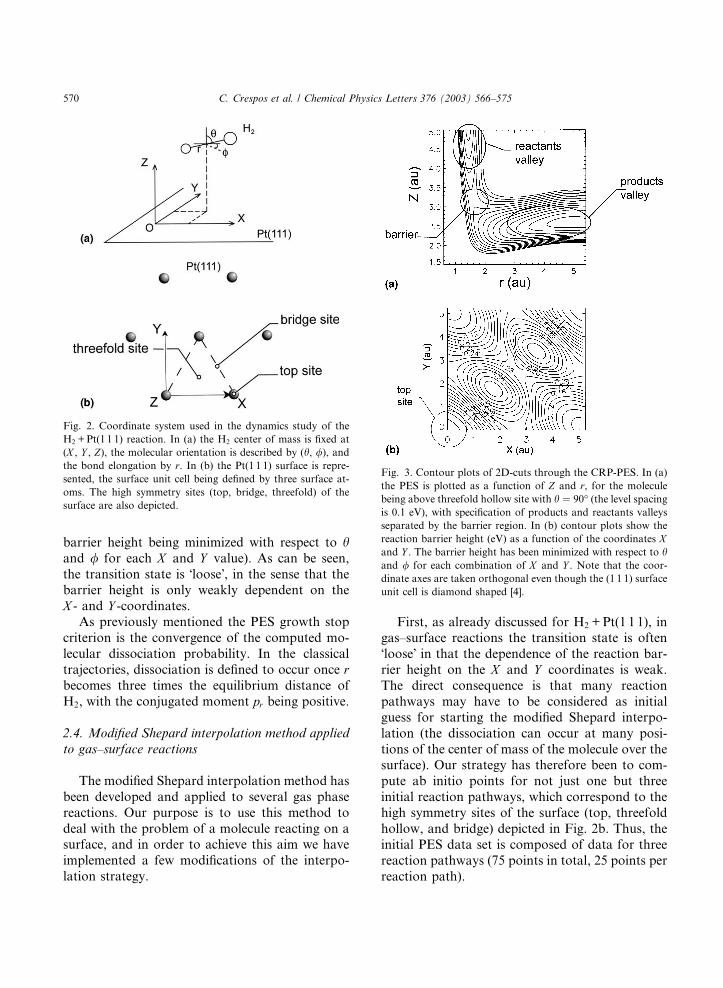
Fig. 2. Coordinate system used in the dynamics study of the
H2 +Pt(1 1 1) reaction. In (a) the H2 center of mass is fixed at
(X , Y , Z), the molecular orientation is described by (h, /), and
the bond elongation by r. In (b) the Pt(1 1 1) surface is repre-
sented, the surface unit cell being defined by three surface at-
oms. The high symmetry sites (top, bridge, threefold) of the
surface are also depicted.
Fig. 3. Contour plots of 2D-cuts through the CRP-PES. In (a)
the PES is plotted as a function of Z and r, for the molecule
being above threefold hollow site with h ¼ 90� (the level spacing
is 0.1 eV), with specification of products and reactants valleys
separated by the barrier region. In (b) contour plots show the
reaction barrier height (eV) as a function of the coordinates Xand Y . The barrier height has been minimized with respect to hand / for each combination of X and Y . Note that the coor-
dinate axes are taken orthogonal even though the (1 1 1) surface
unit cell is diamond shaped [4].
570 C. Crespos et al. / Chemical Physics Letters 376 (2003) 566–575
barrier height being minimized with respect to hand / for each X and Y value). As can be seen,
the transition state is �loose�, in the sense that thebarrier height is only weakly dependent on the
X - and Y -coordinates.
As previously mentioned the PES growth stop
criterion is the convergence of the computed mo-
lecular dissociation probability. In the classical
trajectories, dissociation is defined to occur once rbecomes three times the equilibrium distance of
H2, with the conjugated moment pr being positive.
2.4. Modified Shepard interpolation method applied
to gas–surface reactions
The modified Shepard interpolation method has
been developed and applied to several gas phase
reactions. Our purpose is to use this method to
deal with the problem of a molecule reacting on asurface, and in order to achieve this aim we have
implemented a few modifications of the interpo-
lation strategy.
First, as already discussed for H2 + Pt(1 1 1), ingas–surface reactions the transition state is often
�loose� in that the dependence of the reaction bar-
rier height on the X and Y coordinates is weak.
The direct consequence is that many reaction
pathways may have to be considered as initial
guess for starting the modified Shepard interpo-
lation (the dissociation can occur at many posi-
tions of the center of mass of the molecule over thesurface). Our strategy has therefore been to com-
pute ab initio points for not just one but three
initial reaction pathways, which correspond to the
high symmetry sites of the surface (top, threefold
hollow, and bridge) depicted in Fig. 2b. Thus, the
initial PES data set is composed of data for three
reaction pathways (75 points in total, 25 points per
reaction path).

C. Crespos et al. / Chemical Physics Letters 376 (2003) 566–575 571
Second, previous studies in dynamics of gas–
surface reactions exhibit quite a large complexity of
mechanisms leading to dissociation. In some cases,
like H2 reacting on Pd(1 1 1) [34], the molecular
dissociation mechanisms are different at low and
high value of the translational energy of the H2
molecule (collision energy), and the low energy
mechanism is sensitive to small structural features
of the PES. As a consequence, itmay be necessary to
grow the potential at different values of the trans-
lational energy of the impinging molecule. Indeed,
the scan of the PES could be dependent of the choice
of the collision energy at which the trajectories are
performed. In order to obtain a good level of ac-curacy for the whole range of energies implied by
our dynamics study, we have decided, in this work,
to grow the PES data set at three different collision
energies simultaneously. For example, in the case of
H2 + Pt(1 1 1), the dissociation dynamics is studied
for collision energies between E ¼ 0:05 and 0.5 eV.
Therefore, we have decided to grow the PES at
E ¼ 0:1, 0.3 and 0.5 eV simultaneously.From a more technical point of view, the inter-
polation of the PES, in the approximation of frozen
surface, is performed by using fixed surface atoms
and periodic boundary conditions. In order to re-
produce the symmetry of the potential with respect
to the motion of the molecular center of mass par-
allel to the surface, we have chosen to use the pri-
mary unit cell of the surface (the vectors ofwhich aredefined by three atoms) as a periodic box. Appro-
priate constraints on the surface atoms have been
used to keep them in fixed positions during the
classical trajectories calculations. Additional details
concerning, for instance, the need of three surface
atoms will be presented elsewhere [24]. Keeping all
the surface atoms fixed is not a restriction imposed
by the MS-method and we could in principle allow afew surface atoms to move, in order to build a PES
enabling a study of the energy exchange between the
molecule and the surface atoms. However, this was
not the goal of our research.
3. Results and discussion
We present here the results of the interpolation
and results of quantum and classical dynamics
calculations performed using the CRP-PES and
the MS-PES. We focus on initial-state selected
reaction probabilities, where the initial rovibra-
tional state of the impinging molecules is fixed at
(v ¼ 0, j ¼ 0), v being the vibrational and j the
rotational angular momentum quantum number.
3.1. Sampling of the configuration space
One very important aspect of the interpolation
method is that it avoids the calculation of a uni-
form grid of data points over configuration space
but rather focuses on computing points in the re-
gions relevant for the dynamics. The modifiedShepard interpolation method should locate the
so-called dynamically important region of the PES
and in that way give essential information on
where the reaction takes place.
In Fig. 4, different representations of the PES
data set are plotted. First, 2D representations in
(Z, r) are depicted for two different molecular
translational energies at which the PES was grown,E ¼ 0:1 eV (Fig. 4a) and E ¼ 0:5 eV (Fig. 4b).
Thus, the regions visited by the classical trajecto-
ries, relevant in terms of dynamics, are repre-
sented. As the figures show, the dynamically
relevant region is located in the region of high Zvalues and r values close to the H2 equilibrium
bond distance. This means that the reactivity is
mainly determined by the region corresponding tothe approach towards the surface and the region of
the barrier, as expected (see also Fig. 3a). For
obtaining initial-state selected reaction probabili-
ties, the accuracy of the potential beyond the dis-
sociation barrier (products valley) is not as
important as the accuracy of the potential in the
incoming part (reactants valley and barrier re-
gion). It is interesting to note that the sampledregion is slightly different for the lower and higher
value of the energy during the growing process.
When the molecules approach the surface with a
relatively low energy (E ¼ 0:1 eV), the dynamics is
sensitive to the structure of the potential in the
incoming region, and the proportion of reflected
back trajectories is important. For the high energy
case (E ¼ 0:5 eV) the region of the barrier(2:5 a:u: < Z < 4 a.u.; r � 1:5 a.u.) and the region
behind it, are more extensively visited because
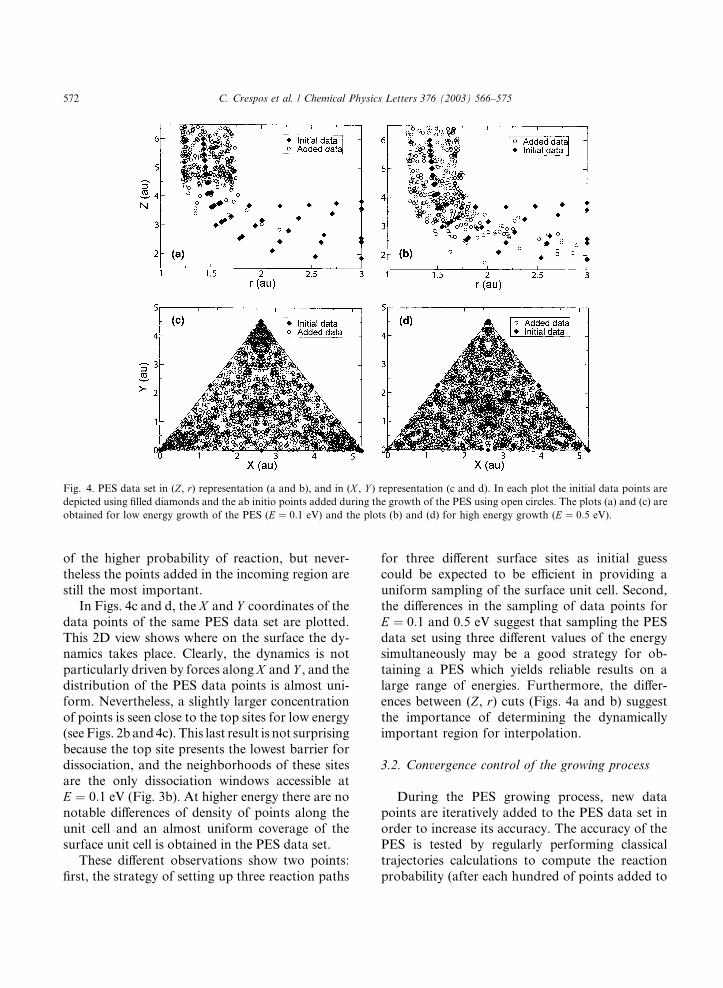
Fig. 4. PES data set in (Z, r) representation (a and b), and in (X , Y ) representation (c and d). In each plot the initial data points are
depicted using filled diamonds and the ab initio points added during the growth of the PES using open circles. The plots (a) and (c) are
obtained for low energy growth of the PES (E ¼ 0:1 eV) and the plots (b) and (d) for high energy growth (E ¼ 0:5 eV).
572 C. Crespos et al. / Chemical Physics Letters 376 (2003) 566–575
of the higher probability of reaction, but never-
theless the points added in the incoming region are
still the most important.
In Figs. 4c and d, the X and Y coordinates of the
data points of the same PES data set are plotted.
This 2D view shows where on the surface the dy-namics takes place. Clearly, the dynamics is not
particularly driven by forces along X and Y , and the
distribution of the PES data points is almost uni-
form. Nevertheless, a slightly larger concentration
of points is seen close to the top sites for low energy
(see Figs. 2b and 4c). This last result is not surprising
because the top site presents the lowest barrier for
dissociation, and the neighborhoods of these sitesare the only dissociation windows accessible at
E ¼ 0:1 eV (Fig. 3b). At higher energy there are no
notable differences of density of points along the
unit cell and an almost uniform coverage of the
surface unit cell is obtained in the PES data set.
These different observations show two points:
first, the strategy of setting up three reaction paths
for three different surface sites as initial guess
could be expected to be efficient in providing a
uniform sampling of the surface unit cell. Second,
the differences in the sampling of data points for
E ¼ 0:1 and 0.5 eV suggest that sampling the PES
data set using three different values of the energysimultaneously may be a good strategy for ob-
taining a PES which yields reliable results on a
large range of energies. Furthermore, the differ-
ences between (Z, r) cuts (Figs. 4a and b) suggest
the importance of determining the dynamically
important region for interpolation.
3.2. Convergence control of the growing process
During the PES growing process, new data
points are iteratively added to the PES data set in
order to increase its accuracy. The accuracy of the
PES is tested by regularly performing classical
trajectories calculations to compute the reaction
probability (after each hundred of points added to
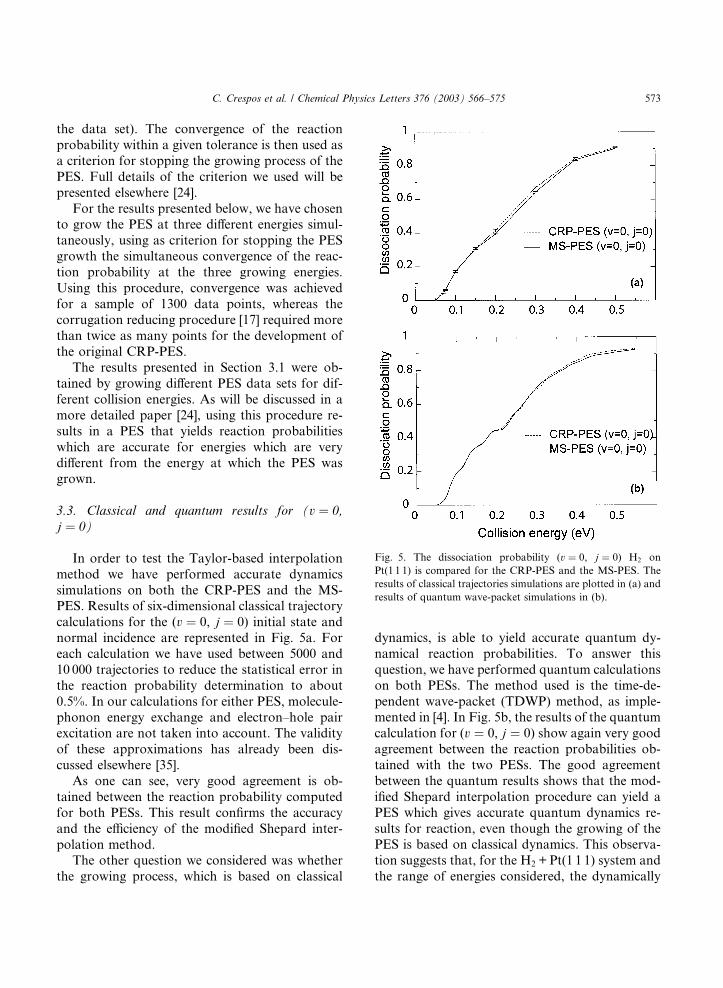
Fig. 5. The dissociation probability (v ¼ 0, j ¼ 0) H2 on
Pt(1 1 1) is compared for the CRP-PES and the MS-PES. The
results of classical trajectories simulations are plotted in (a) and
results of quantum wave-packet simulations in (b).
C. Crespos et al. / Chemical Physics Letters 376 (2003) 566–575 573
the data set). The convergence of the reaction
probability within a given tolerance is then used as
a criterion for stopping the growing process of the
PES. Full details of the criterion we used will be
presented elsewhere [24].
For the results presented below, we have chosento grow the PES at three different energies simul-
taneously, using as criterion for stopping the PES
growth the simultaneous convergence of the reac-
tion probability at the three growing energies.
Using this procedure, convergence was achieved
for a sample of 1300 data points, whereas the
corrugation reducing procedure [17] required more
than twice as many points for the development ofthe original CRP-PES.
The results presented in Section 3.1 were ob-
tained by growing different PES data sets for dif-
ferent collision energies. As will be discussed in a
more detailed paper [24], using this procedure re-
sults in a PES that yields reaction probabilities
which are accurate for energies which are very
different from the energy at which the PES wasgrown.
3.3. Classical and quantum results for (v ¼ 0,
j ¼ 0)
In order to test the Taylor-based interpolation
method we have performed accurate dynamics
simulations on both the CRP-PES and the MS-PES. Results of six-dimensional classical trajectory
calculations for the (v ¼ 0, j ¼ 0) initial state and
normal incidence are represented in Fig. 5a. For
each calculation we have used between 5000 and
10 000 trajectories to reduce the statistical error in
the reaction probability determination to about
0.5%. In our calculations for either PES, molecule-
phonon energy exchange and electron–hole pairexcitation are not taken into account. The validity
of these approximations has already been dis-
cussed elsewhere [35].
As one can see, very good agreement is ob-
tained between the reaction probability computed
for both PESs. This result confirms the accuracy
and the efficiency of the modified Shepard inter-
polation method.The other question we considered was whether
the growing process, which is based on classical
dynamics, is able to yield accurate quantum dy-
namical reaction probabilities. To answer this
question, we have performed quantum calculations
on both PESs. The method used is the time-de-
pendent wave-packet (TDWP) method, as imple-mented in [4]. In Fig. 5b, the results of the quantum
calculation for (v ¼ 0, j ¼ 0) show again very good
agreement between the reaction probabilities ob-
tained with the two PESs. The good agreement
between the quantum results shows that the mod-
ified Shepard interpolation procedure can yield a
PES which gives accurate quantum dynamics re-
sults for reaction, even though the growing of thePES is based on classical dynamics. This observa-
tion suggests that, for the H2 + Pt(1 1 1) system and
the range of energies considered, the dynamically

574 C. Crespos et al. / Chemical Physics Letters 376 (2003) 566–575
relevant regions for reaction in the quantum regime
do not really differ from the regions relevant for the
classical reaction dynamics.
4. Summary
The modified Shepard interpolation method,
previously developed for the construction of high-
dimensional potential energy surfaces for gas
phase reactions, has been adapted to molecule–
surface reactions and applied to the H2 +Pt(1 1 1)
system. Modifications, implemented to treat mol-
ecule–surface reactions, include the use of periodicboundary conditions for motion along the surface
and the freezing of the atoms representing the
surface. Furthermore, the initial data set used to
start the growing process contained data for three
reaction paths rather than one, the three reaction
paths describing dissociation over three different
surface sites. This strategy was adopted to pro-
mote a uniform sampling of the potential along themetal surface, because the dependence of the re-
action barrier height on the coordinates for mo-
tion along the surface is weak. Finally, a new
element has been that the PES was grown for three
different collision energies simultaneously, to allow
accurate reaction probabilities to be extracted
from its use for a large range of collision energies.
The accuracy and the efficiency of the modifiedShepard interpolation method was tested by ap-
plying the method to an initially known accurate
potential energy surface, thereby avoiding the
problem of errors related to ab initio calculations.
In a comparison with results obtained for the
original PES, classical and quantum dynamics
calculations showed that the grown PES yields
accurate reaction probabilities for the collisionenergy range 0.05–0.5 eV. This result was achieved
using a data set which consists of a number of
points (1300) that was less than half the amount of
(ab initio) points required to construct the original
PES, which was constructed using a method (the
corrugation reducing procedure) that employs in-
terpolation of a uniform grid of points.
The results show that the modified Shepardinterpolation method, which does not require a
priori knowledge concerning potentially useful
analytical fitting forms, can be efficiently used to
construct PESs for molecule–surface reactions
from which accurate reaction probabilities can be
derived. Generalization of the method to reactions
of polyatomic molecules with surfaces is straight-
forward and we expect that the method can beeasily applied to, for instance, the dissociative
chemisorption of methane on metal surfaces.
References
[1] S. Holloway, M. Kay, G.R. Darling, Faraday Discuss. 105
(1996) 209.
[2] A. Gross, J. Chem. Phys. 110 (1999) 8696.
[3] G.R. Darling, Z.S. Wang, S. Holloway, Phys. Chem.
Chem. Phys. 2 (2000) 911.
[4] E. Pijper, G.J. Kroes, R.A. Olsen, E.J. Baerends, J. Chem.
Phys. 117 (2002) 5885.
[5] A. Gross, S. Wilke, M. Scheffler, Phys. Rev. Lett. 75 (1995)
2718.
[6] G.J. Kroes, E.J. Baerends, R.C. Mowrey, Phys. Rev. Lett.
78 (1997) 3583.
[7] J. Dai, J.C. Light, J. Chem. Phys. 107 (1997) 1676.
[8] P. Hohenberg, W. Kohn, Phys. Rev. 136 (1964) B864.
[9] W. Kohn, L.J. Sham, Phys. Rev. 140 (1965) A1133.
[10] A.D. Becke, Phys. Rev. A 38 (1988) 3098.
[11] J.P. Perdew, Phys. Rev. B 33 (1986) 8822.
[12] J. McCreery, G. Wolken Jr., J. Chem. Phys. 63 (1975)
2340.
[13] A. Forni, G. Wiesenekker, E.J. Baerends, G.F. Tantardini,
J. Phys. Condens. Matter 7 (1995) 7195.
[14] J. Dai, J.Z.H. Zhang, J. Chem. Phys. 102 (1995) 6280.
[15] A. Gross, M. Scheffler, Phys. Rev. B 57 (1998) 2493.
[16] G. Wiesenekker, G.J. Kroes, E.J. Baerends, R.C. Mowrey,
J. Chem. Phys. 103 (1995) 5168.
[17] H.F. Busnengo, A. Salin, W. Dong, J. Chem. Phys. 112
(2000) 7641.
[18] J. Ischtwan, M.A. Collins, J. Chem. Phys. 100 (1994) 8080.
[19] M.J.T. Jordan, K.C. Thompson, M.A. Collins, J. Chem.
Phys. 103 (1995) 9669.
[20] M.J.T. Jordan, M.A. Collins, J. Chem. Phys. 104 (1996)
4600.
[21] K.C. Thompson, M.A. Collins, J. Chem. Soc. Faraday
Trans. 93 (1997) 871.
[22] R.P.A. Bettens, M.A. Collins, J. Chem. Phys. 111 (1999)
816.
[23] R.A. Olsen, H.F. Busnengo, A. Salin, M.F. Somers, G.J.
Kroes, E.J. Baerends, J. Chem. Phys. 116 (2002) 3841.
[24] C. Crespos, M.A. Collins, E. Pijper, G.J. Kroes, J. Chem.
Phys., in preparation.
[25] R. Farwig, Math. Comput. 46 (1986) 577.
[26] R. Farwig, in: J.C. Mason, M.G. Cox (Eds.), Algorithms
for Approximation, Clarendon Press, Oxford, 1987, p. 194.
[27] M.A. Collins, Theor. Chem. Acc. 108 (2002) 313.

C. Crespos et al. / Chemical Physics Letters 376 (2003) 566–575 575
[28] M.A. Collins, D.H. Zhang, J. Chem. Phys. 111 (1999) 9924.
[29] R.P.A. Bettens, M.A. Collins, M.J.T. Jordan, D.H. Zhang,
J. Chem. Phys. 112 (2000) 10162.
[30] A.J. Chalk, S. Petrie, L. Radom, M.A. Collins, J. Chem.
Phys. 112 (2000) 6625.
[31] R.O. Fuller, R.P.A. Bettens, M.A. Collins, J. Chem. Phys.
114 (2001) 10711.
[32] K. Song, M.A. Collins, Chem. Phys. Lett. 335 (2001)
481.
[33] M. Yang, D.H. Zhang, M.A. Collins, S.Y. Lee, J. Chem.
Phys. 115 (2001) 174.
[34] C. Crespos, H.F. Busnengo, W. Dong, A. Salin, J. Chem.
Phys. 114 (2001) 10954.
[35] G.J. Kroes, Prog. Surf. Sci. 60 (1999) 1.




















