
mTOR-Independent Translational Control of the Extrinsic Cell Death Pathway by RalA
13
MOLECULAR AND CELLULAR BIOLOGY, Oct. 2006, p. 7345–7357 Vol. 26, No. 20 0270-7306/06/$08.000 doi:10.1128/MCB.00126-06 Copyright © 2006, American Society for Microbiology. All Rights Reserved. mTOR-Independent Translational Control of the Extrinsic Cell Death Pathway by RalA † Amith Panner, 1,2,3 Jean L. Nakamura, 3 Andrew T. Parsa, 1,2,3 Pablo Rodriguez-Viciana, 3 Mitchel S. Berger, 1,2,3 David Stokoe, 1,2,3 and Russell O. Pieper 1,2,3 * Department of Neurological Surgery, 1 The Brain Tumor Research Center, 2 and The UCSF Comprehensive Cancer Center, 3 University of California—San Francisco, 2340 Sutter St., San Francisco, California 94115-0875 Received 20 January 2006/Returned for modification 10 March 2006/Accepted 27 July 2006 Oncogenic potential is associated with translational regulation, and the prevailing view is that oncogenes use mTOR-dependent pathways to up-regulate the synthesis of proteins critical for transformation. In this study, we show that RalA, a key mediator of Ras transformation, is also linked to the translational machinery. At least part of this linkage, however, is independent of mTOR and acts through RalBP1 to suppress cdc42-mediated activation of S6 kinase and the translation of the antiapoptotic protein FLIP S . This action, rather than contributing to transformation, opens a latent tumor-suppressive mechanism that can be activated by tumor necrosis factor-related apoptosis-inducing ligand. These results show that the translational machinery is linked to tumor suppression as well as cell-proliferative pathways and that the reestablishment of cell death pathways by activation of the Ral oncogenic program provides a means for selective therapeutic targeting of Ral-driven malignancies. Controlled proliferation depends on accurate and timely regulation of cellular protein levels and activity. To accommo- date this need, cells have evolved a variety of mechanisms to control gene expression, the most proximal of which regulate translation. The process of translation is highly regulated, be- ginning at the rate-limiting step of initiation (36). For capped mRNAs, the binding of eukaryotic initiating factor 4E (eIF- 4E) to the mRNA cap structure facilitates the formation of the eIF-4F complex, the melting of secondary structures near the 5 cap, and the ribosomal recruitment/loading of the mRNA onto polyribosome complexes (polysomes) (13, 14). For un- capped mRNAs with 5 terminal oligopyrimidine sequences, the initiation process may be facilitated by S6K1-mediated phosphorylation of S6, a component of the 40S ribosome (10, 12–14), although recent evidence suggests that translational regulation of 5 terminal oligopyrimidine mRNAs occurs even in the absence of S6K1 (43). The activities of both eIF-4E and S6K are closely regulated by a variety of factors, the most intensely studied being mTOR. mTOR phosphorylates S6K1, allowing the recruitment of PDK-1, which in turn activates S6K1 by Thr-229 phosphorylation (14). mTOR also phosphor- ylates the three isoforms of 4E-BP, inhibiting the ability of these proteins to compete with eIF-4G for eIF-4E binding and block translation initiation (11). Given the importance of mTOR in regulating translation, it is not surprising that mTOR and the translational process itself are co-opted by transform- ing oncogenes. Oncogenic activation of the Ras pathway, for example, stimulates translation in at least two ways: phospha- tidylinositol 3-kinase (PI3K), via the extensively described Akt- mTOR pathway, stimulates translation by activating eIF-4E and S6K (14), while in some circumstances, the Raf-extracel- lular signal-regulated kinase pathway stimulates translation via MNK-mediated phosphorylation of eIF-4E (2, 34, 45). The ability of multiple oncogenic pathways to stimulate translation as well as the observation that the mTOR pathway is hyperac- tive in many tumors, has led to the suggestions that enhanced protein synthesis provides a growth advantage to tumors and that regulators of translation, in particular mTOR, may be promising therapeutic targets (17). In addition to stimulating translation, however, growth-pro- moting oncogenic pathways also paradoxically activate latent tumor suppressor pathways that leave tumors vulnerable to cell death. Raf activation, in addition to activating eIF-4E, also induces the expression of p16/ARF, indirectly activating Rb and/or p53, which in turn influences the expression/activity of controllers of senescence, including p21 and PML (24, 40). The activation of the Ras/Raf/extracellular signal-regulated kinase pathway has also been shown (perhaps by myc stabilization) to transcriptionally up-regulate the death receptors DR4 and DR5 and stimulate caspase-8 recruitment to the death-induced signaling complex (DISC), all of which sensitize cells to apop- tosis induced by the death ligand TRAIL (29, 37, 46, 47). Enhanced translation mediated by oncogenic activation is therefore countered by an “oncogenic checkpoint” which, when activated, sensitizes cells to death signals in their envi- ronments and limits tumor growth. While the Raf and PI3K pathways have been linked to both control of translation and activation of innate tumor-suppres- sive mechanisms, relatively little is known about a third re- cently identified effector of Ras oncogenesis, Ral. The Ral proteins (RalA and RalB) are small GTPases whose activity is controlled by the RalGEF family of proteins, of which at least three members (RalGDS, Rgl, and Rlf) are known (48). Ac- tivated Ras binds RalGDS via a Ras-binding domain and re- cruits it to the membrane, where it stimulates an exchange of * Corresponding author. Mailing address: UCSF Cancer Center, 2340 Sutter St., Rm N219, San Francisco, CA 94115-0875. Phone: (415) 502-7132. Fax: (415) 502-6779. E-mail: [email protected]. † Supplemental material for this article may be found at http://mcb .asm.org/. Published ahead of print on 7 August 2006. 7345 on April 2, 2016 by guest http://mcb.asm.org/ Downloaded from
Transcript of mTOR-Independent Translational Control of the Extrinsic Cell Death Pathway by RalA

MOLECULAR AND CELLULAR BIOLOGY, Oct. 2006, p. 7345–7357 Vol. 26, No. 200270-7306/06/$08.00�0 doi:10.1128/MCB.00126-06Copyright © 2006, American Society for Microbiology. All Rights Reserved.
mTOR-Independent Translational Control of the Extrinsic Cell DeathPathway by RalA�†
Amith Panner,1,2,3 Jean L. Nakamura,3 Andrew T. Parsa,1,2,3 Pablo Rodriguez-Viciana,3Mitchel S. Berger,1,2,3 David Stokoe,1,2,3 and Russell O. Pieper1,2,3*
Department of Neurological Surgery,1 The Brain Tumor Research Center,2 and The UCSF Comprehensive Cancer Center,3
University of California—San Francisco, 2340 Sutter St., San Francisco, California 94115-0875
Received 20 January 2006/Returned for modification 10 March 2006/Accepted 27 July 2006
Oncogenic potential is associated with translational regulation, and the prevailing view is that oncogenes usemTOR-dependent pathways to up-regulate the synthesis of proteins critical for transformation. In this study,we show that RalA, a key mediator of Ras transformation, is also linked to the translational machinery. At leastpart of this linkage, however, is independent of mTOR and acts through RalBP1 to suppress cdc42-mediatedactivation of S6 kinase and the translation of the antiapoptotic protein FLIPS. This action, rather thancontributing to transformation, opens a latent tumor-suppressive mechanism that can be activated by tumornecrosis factor-related apoptosis-inducing ligand. These results show that the translational machinery islinked to tumor suppression as well as cell-proliferative pathways and that the reestablishment of cell deathpathways by activation of the Ral oncogenic program provides a means for selective therapeutic targeting ofRal-driven malignancies.
Controlled proliferation depends on accurate and timelyregulation of cellular protein levels and activity. To accommo-date this need, cells have evolved a variety of mechanisms tocontrol gene expression, the most proximal of which regulatetranslation. The process of translation is highly regulated, be-ginning at the rate-limiting step of initiation (36). For cappedmRNAs, the binding of eukaryotic initiating factor 4E (eIF-4E) to the mRNA cap structure facilitates the formation of theeIF-4F complex, the melting of secondary structures near the5� cap, and the ribosomal recruitment/loading of the mRNAonto polyribosome complexes (polysomes) (13, 14). For un-capped mRNAs with 5� terminal oligopyrimidine sequences,the initiation process may be facilitated by S6K1-mediatedphosphorylation of S6, a component of the 40S ribosome (10,12–14), although recent evidence suggests that translationalregulation of 5� terminal oligopyrimidine mRNAs occurs evenin the absence of S6K1 (43). The activities of both eIF-4E andS6K are closely regulated by a variety of factors, the mostintensely studied being mTOR. mTOR phosphorylates S6K1,allowing the recruitment of PDK-1, which in turn activatesS6K1 by Thr-229 phosphorylation (14). mTOR also phosphor-ylates the three isoforms of 4E-BP, inhibiting the ability ofthese proteins to compete with eIF-4G for eIF-4E binding andblock translation initiation (11). Given the importance ofmTOR in regulating translation, it is not surprising that mTORand the translational process itself are co-opted by transform-ing oncogenes. Oncogenic activation of the Ras pathway, forexample, stimulates translation in at least two ways: phospha-tidylinositol 3-kinase (PI3K), via the extensively described Akt-
mTOR pathway, stimulates translation by activating eIF-4Eand S6K (14), while in some circumstances, the Raf-extracel-lular signal-regulated kinase pathway stimulates translation viaMNK-mediated phosphorylation of eIF-4E (2, 34, 45). Theability of multiple oncogenic pathways to stimulate translationas well as the observation that the mTOR pathway is hyperac-tive in many tumors, has led to the suggestions that enhancedprotein synthesis provides a growth advantage to tumors andthat regulators of translation, in particular mTOR, may bepromising therapeutic targets (17).
In addition to stimulating translation, however, growth-pro-moting oncogenic pathways also paradoxically activate latenttumor suppressor pathways that leave tumors vulnerable to celldeath. Raf activation, in addition to activating eIF-4E, alsoinduces the expression of p16/ARF, indirectly activating Rband/or p53, which in turn influences the expression/activity ofcontrollers of senescence, including p21 and PML (24, 40). Theactivation of the Ras/Raf/extracellular signal-regulated kinasepathway has also been shown (perhaps by myc stabilization) totranscriptionally up-regulate the death receptors DR4 andDR5 and stimulate caspase-8 recruitment to the death-inducedsignaling complex (DISC), all of which sensitize cells to apop-tosis induced by the death ligand TRAIL (29, 37, 46, 47).Enhanced translation mediated by oncogenic activation istherefore countered by an “oncogenic checkpoint” which,when activated, sensitizes cells to death signals in their envi-ronments and limits tumor growth.
While the Raf and PI3K pathways have been linked to bothcontrol of translation and activation of innate tumor-suppres-sive mechanisms, relatively little is known about a third re-cently identified effector of Ras oncogenesis, Ral. The Ralproteins (RalA and RalB) are small GTPases whose activity iscontrolled by the RalGEF family of proteins, of which at leastthree members (RalGDS, Rgl, and Rlf) are known (48). Ac-tivated Ras binds RalGDS via a Ras-binding domain and re-cruits it to the membrane, where it stimulates an exchange of
* Corresponding author. Mailing address: UCSF Cancer Center,2340 Sutter St., Rm N219, San Francisco, CA 94115-0875. Phone:(415) 502-7132. Fax: (415) 502-6779. E-mail: [email protected].
† Supplemental material for this article may be found at http://mcb.asm.org/.
� Published ahead of print on 7 August 2006.
7345
on April 2, 2016 by guest
http://mcb.asm
.org/D
ownloaded from
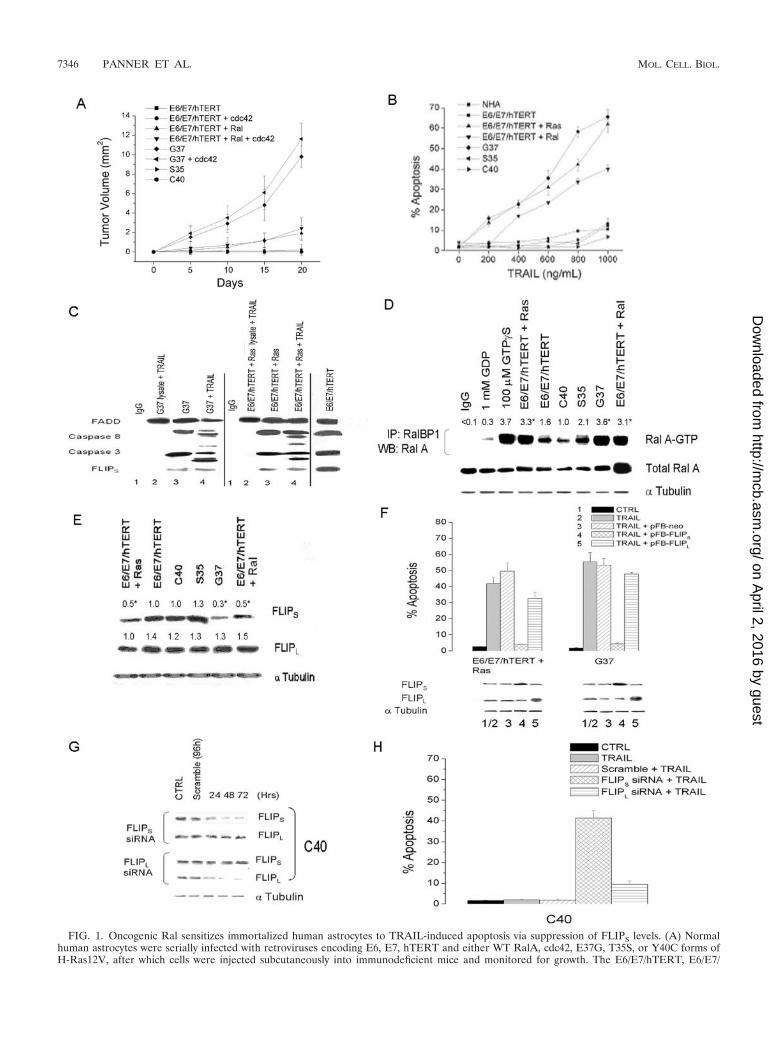
FIG. 1. Oncogenic Ral sensitizes immortalized human astrocytes to TRAIL-induced apoptosis via suppression of FLIPS levels. (A) Normalhuman astrocytes were serially infected with retroviruses encoding E6, E7, hTERT and either WT RalA, cdc42, E37G, T35S, or Y40C forms ofH-Ras12V, after which cells were injected subcutaneously into immunodeficient mice and monitored for growth. The E6/E7/hTERT, E6/E7/
7346 PANNER ET AL. MOL. CELL. BIOL.
on April 2, 2016 by guest
http://mcb.asm
.org/D
ownloaded from

GDP for GTP on both forms of Ral (22, 28, 35). Activated ralmolecules in turn interact with a myriad of Ral effectors, in-cluding RalBP1 (a cdc42 GTPase-activating protein) (4, 21),subunits of the exocyst complex (Sec5 and Exo84) that directvesicles to the plasma membrane (28), the transcription factorZonab (38), and the actin-binding protein filamin (30). WhileRalA/RalB activation plays a role in membrane trafficking,actin organization, and gene expression, the expression of aRas point effector mutant selective for Ral GDS activation(H-Ras12V E37G) also transforms immortalized human fibro-blasts, embryonic kidney epithelial cells, and astrocytes (18).Consistent with this idea, RalGDS was shown to be requiredfor tumor formation in a mouse model of skin carcinogenesis(16); this requirement is perhaps related to Ral’s ability tostimulate the Jun N-terminal protein kinase/stress-activatedprotein kinase pathway and suppress apoptosis and enhancecell survival or alternatively related to interactions betweenRal and cytoskeletal reorganizing proteins, such as RalBP1,Rac1, and cdc42. The relative contributions of RalA and RalBto cellular transformation remain uncertain, although the pro-teins clearly differ in function (for example, RalA binds moreeffectively to the exocyst components than RalB does) (41).The suppression of RalA blocks Ras- and Ral-mediated trans-formation of cultured fibroblasts (23), while the suppression ofRalB leads to apoptosis (6), suggesting that both RalA andRalB may play key roles. While these studies firmly establishthe Ral pathway as a key contributor to Ras-mediated tumor-igenesis in mouse and humans, they leave open the question ofwhether the Ral pathway, like other oncogenic pathways, islinked to the translational machinery and innate tumor-sup-pressive mechanisms. We address these possibilities in thepresent study and show that the RalA pathway is linked to boththe translational control and the tumor-suppressive pathways.Unlike other oncogenes, however, the ability of RalA to reg-ulate translation is independent of its transforming ability butdirectly responsible for its ability to sensitize glioma cells to theactivation of the extrinsic death pathway by TRAIL. Theseresults define an mTOR-independent link between RalA and
translation and identify a means by which Ral-regulated ma-lignancies may be attacked therapeutically.
MATERIALS AND METHODS
Cell culture and drug treatment. Immortalized or Ras-transformed humanastrocytes were generated and cultured as described previously (42). Humanxenografted glioblastoma multiforme (GBM) cells were obtained from theUCSF Brain Tumor Research Center Tissue Bank and cultured as previouslydescribed (31). Human recombinant TRAIL and rapamycin were purchasedfrom Sigma and Cell Signaling Technology, respectively, and dissolved in di-methyl sulfoxide.
Analysis of tumorigenesis. Cells (1 � 107) were resuspended in a 1:1 mixtureof phosphate-buffered saline-Matrigel and injected subcutaneously into theflanks of rag1�/� immunodeficient mice (C57BL6/129Sv), after which tumorvolumes were determined at regular intervals as described previously (23).
Immunoblot analysis and analysis of apoptosis by flow cytometry. Cell lysateswere prepared and carried through Western blot analysis as previously described(31) by using antibody against alpha tubulin, p70 S6K1, or phospho-S6K1 (Thr-389) (all mouse polyclonal antibodies; Santa Cruz Biotechnology), phopho-S6(rabbit polyclonal antibody; Santa Cruz Biotechnology), FLIPL and FLIPS (goatmonoclonal antibody; Santa Cruz Biotechnology), FADD (mouse monoclonalantibody; Abcam), RalBP1 (rabbit monoclonal antibody; Cell Signaling Tech-nology), and cdc42, caspase 3, and caspase 8 (rabbit polyclonal antibodies; CellSignaling Technology). Bound antibody was detected with mouse anti-goat im-munoglobulin G (IgG), goat anti-rabbit IgG, or goat anti-mouse IgG (Santa CruzBiotechnology) by using ECL Western blotting detection reagents (AmershamPharmacia Biotech, Inc.). The expression of alpha tubulin was used to verifyequal loading in all studies. The extent of apoptosis in cultures (attached andfloating cells) was determined by fluorescence-activated cell sorter analysis(sub-G1 DNA content), with measurements verified by annexin V-propidiumiodide staining as previously described (5).
Retroviral infection, transfection of plasmids, and siRNA. Retroviral con-structs encoding FLIPS or FLIPL were provided by L. Bin (3). An expressionvector encoding hemagglutinin-tagged wild-type (WT) p70 S6K1 (pRK7/HA-S6K1) was kindly provided by Robert Abraham (Burnham Institute, San Diego,CA). Constructs encoding Ras point effector mutants (H-Ras12V E37G, T35S,or Y40C) or WT RalA were provided by Pablo Rodriguez-Viciana (UCSFCancer Center, San Francisco, CA), while the WT cdc42 and constitutively active(Q61L) and kinase-dead (T17N) cdc42 constructs were provided by DavidStokoe (UCSF Cancer Center San Francisco, CA). Constructs encoding WTRalBP1, RalBP1�65-80, or RalBP1�154-219 were provided by Sanjay Awasthi(University of Texas—Arlington) (51), while a construct encoding RalBP1 withan N-terminal deletion of the GAP domain (RalBP1�GAP) was provided byPablo Rodriguez-Viciana (9). Pools of productively infected cells (obtained byselection with 1 �g/ml neomycin, 7 days, or 9 �g/ml puromycin, 7 days) (38) were
hTERT�cdc42, T35S, and Y40C cells did not form tumors, and as such, the data points for these groups are compressed along the x axis. Errorbars indicate standard deviations. (B) Select cells in panel A were exposed to TRAIL (0 to 1,000 ng/ml, 24 h), stained with propidium iodide, andanalyzed by flow cytometry for the percentage of cells having a �2N DNA content (apoptotic cells). Error bars indicate standard deviations. (C)Expression of DISC components (FADD, caspase-8, caspase-3, and FLIPS) was assessed by Western blot analysis from lysates of TRAIL-sensitiveG37 and E6/E7/hTERT/Ras cells (lanes 3) or TRAIL resistant E6/E7/hTERT cells and from anti-FADD antibody DISC immunoprecipitates fromTRAIL-exposed (800 ng/ml) cells (lanes 4) or TRAIL-exposed cell lysates (lanes 2). Lanes 1, negative control using IgG antibody rather thanFADD antibody. (D) TRAIL-resistant/nontransformed (E6/E7/hTERT, C40, and S35) or TRAIL-sensitive/transformed (E6/E7/hTERT �Ras,E6/E7/hTERT/Ral, and G37) cells were lysed, preincubated with vehicle, GDP (negative control to inactivate RalA-GTP), or a nonhydrolyzableform of GTP (GTP�S, positive control to activate RalA-GTP), and incubated with agarose-conjugated RalBP1, a substrate of the activated (GTPbound) form of RalA, or an agarose-conjugated nonspecific substrate (normal rabbit IgG). Substrate-bound (activated) RalA-GTP was theneluted, and levels were assessed by Western blotting (WB) using a RalA antibody and compared to levels of total RalA and alpha tubulin in lysatesprior to incubation with RalBP1. *, P � 0.05. IP, immunoprecipitate. (E) Western blot analysis of FLIPS, FLIPL, and alpha tubulin (loadingcontrol) expression in TRAIL-resistant/nontransformed (E6/E7/hTERT, C40, and S35) or TRAIL-sensitive/transformed (E6/E7/hTERT�Ras,E6/E7/hTERT/Ral, and G37) cells. *, P � 0.05. (F) TRAIL-sensitive cells were sham infected (CTRL) or stably infected with blank (pFB-Neo),FLIPS- or FLIPL-encoding constructs. Cells were then either analyzed for FLIPS, FLIPL, and alpha tubulin (loading control) expression by Westernblotting (bottom panel), or were exposed to TRAIL (800 ng/ml, 24 h) and analyzed for the percentage of apoptotic cells (top panel). Error barsindicate standard deviations. (G) TRAIL-resistant C40 cells were sham transfected (CTRL) or transiently transfected with either siRNA targetingFLIPS or FLIPL, or a nonspecific scrambled siRNA and analyzed for levels of FLIPS, FLIPL, or alpha tubulin protein 0 to 72 h after siRNAaddition. Alpha tubulin expression shown is derived from FLIPS siRNA cells and is representative of both cell lines used. (H) Cells created in panelG were exposed to TRAIL (800 ng/ml, 24 h) beginning 48 h after the addition of siRNA and analyzed for the percentage of apoptotic cells. Errorbars indicate standard errors of the means.
VOL. 26, 2006 RalA CONTROL OF CELL DEATH 7347
on April 2, 2016 by guest
http://mcb.asm
.org/D
ownloaded from

used for further analysis. In cells expressing multiple constructs, all retroviralinfections and selections were done serially. For short interfering RNA (siRNA)studies, 200 nM FLIPS-, FLIPL- (33) or RalBP1-targeted siRNA (Ambion;identification no. 18693) or 1 �M p70 S6 kinase SMARTpool siRNA or scramblesiRNA (Dharmacon, Lafayette, CO) was transfected into cells and protein levelswere analyzed 1 to 4 days later.
Analysis of DISC components. For the analysis of components of the assem-bled DISC, cells were incubated with TRAIL (800 ng/ml, 24 h) and lysed, afterwhich the DISC was immunoprecipitated using an anti-FADD antibody andlevels of components were assessed by Western blot analysis. Immunoprecipita-tions carried out using a nonspecific normal mouse IgG antibody were includedas negative controls, as were analyses of cells to which TRAIL (800 ng/ml) wasadded following lysis.
Analysis of cdc42 GTP levels. Cells were lysed, preincubated with vehicle,GDP (negative control to inactivate cdc42-GTP), or a nonhydrolyzable form ofGTP (GTP�S, positive control to activate cdc42-GTP), and incubated with anagarose-conjugated p21-binding domain (PBD) of PAK-1, a substrate of theactivated (GTP bound) form of cdc42, or an agarose-conjugated nonspecificsubstrate (normal rabbit IgG). Substrate-bound (activated) cdc42 was theneluted, and levels were assessed by Western blot analysis by using a cdc42antibody. Levels of total cdc42 were also determined in lysates prior to incuba-tion with PAK-1 PBD.
Sucrose density gradient fractionation and RNA isolation/analysis. The frac-tionation of cells by sucrose density gradient centrifugation was performed aspreviously described (20). The gradient was divided into 48 fractions, each ofwhich was analyzed for absorbance at 260 nm then pooled into a total of 12fractions. RNA from each fraction or from total cell lysates was spiked with 0.5�g exogenous Drosophila ribosomal protein L3 (RPL3) mRNA (Ambion) (tocontrol for losses of mRNA during purification) before purification using TRIzolreagent (Invitrogen; QIAGEN). Northern blot analyses were carried out usingprobes generated by reverse transcription-PCR.
RESULTS
Ral pathway activation coordinately transforms immortal-ized human astrocytes and opens a latent tumor-suppressivemechanism activated by TRAIL. Previous studies showed thatthe expression of a Ras effector mutant selective for the Ralpathway (H-Ras12V E37G) or of activated RalA itself wassufficient to transform immortalized human fibroblasts, embry-onic kidney epithelial cells, and astrocytes (18, 23). Consistentwith these results, the expression of the Ral-selective G37mutant (but not of T35S or Y40C mutants selective for Raf orPI3K activation, respectively) was sufficient in our studies toallow immortalized human astrocytes (E6/E7/hTERT) to growin soft agar (not shown) and form tumors following injectioninto immunodeficient animals (Fig. 1A). Similar albeit smallereffects were noted in immortalized astrocytes retrovirally in-fected with a construct encoding WT RalA itself (Fig. 1A).Because Ras-mediated transformation sensitizes fibroblasts tothe extrinsic cell death activator TRAIL (29), we also exam-ined TRAIL sensitivity in the various Ras-pathway-modulatedastrocytes. As shown in Fig. 1B, the expression of only con-structs that led to cellular transformation (H-Ras12V, G37,and WT RalA) sensitized immortalized astrocytes to TRAIL-induced apoptosis. This sensitization was not the result ofchanges in DR4/DR5 receptor expression as is noted followingmyc transformation of fibroblasts (see Fig. S1 in the supple-mental material) (39, 46), nor was it associated with TRAIL-induced formation of a FADD antibody immunoprecipitableDISC complex, which was noted in both TRAIL-resistant E6/E7/hTERT cells and TRAIL-sensitive Ras or G37 cells (Fig.1C). Rather, TRAIL sensitization was associated with the con-version of caspase-3 and caspase-8 from uncleaved formsfound in whole-cell lysates (Fig. 1C, lanes 3) to cleaved/acti-
vated forms in the DISC (lanes 4 versus lanes 3) exclusively inTRAIL-sensitive cells.
RalA activation is linked to TRAIL sensitivity via the sup-pression of FLIPS levels. FLIP, a protein that exists in long(55-kDa, FLIPL) and short (28-kDa, FLIPS) forms, is a mod-ulator of DISC-associated caspase cleavage and TRAIL sensi-tivity (19), and the suppression of FLIPS levels sensitizes cellsto TRAIL-induced apoptosis (31). To determine whetherFLIP expression was associated with RalA activation or playeda role in Ral-mediated enhancement of TRAIL sensitivity, wefirst compared the expression of activated RalA and FLIP inRas- or Ral-transformed TRAIL-sensitive cells to that inTRAIL-resistant T35S or Y40C mutant-expressing cells. Whilelevels of total RalA were essentially constant across all celllines (except the E6/E7/hTERT cells manipulated to overex-press Ral A), RalA-GTP levels were significantly higher inRas- or Ral-transformed TRAIL-sensitive cells (E6/E7/hTERT/Ras, G37, or RalA) than those in the TRAIL-resistantcell lines (E6/E7/hTERT, C40, and S35) (Fig. 1D). Consistentwith the idea that RalA-mediated increases in TRAIL sensi-tivity might be associated with altered FLIP expression, FLIPS
levels (but not FLIPL levels) were significantly lower in theTRAIL-sensitive cells (E6/E7/hTERT/Ras, G37, or RalA) withhigh RalA activation than those in the TRAIL-resistant celllines (E6/E7/hTERT, C40, and S35) with low RalA activation(Fig. 1E). To further assess the importance of both forms ofFLIP in controlling TRAIL resistance, TRAIL-sensitive Ras-or Ral-transformed cells were stably infected with a blankretrovirus or with retroviral constructs encoding FLIPS orFLIPL and assessed for levels of FLIPS/FLIPL protein andTRAIL sensitivity. Cells expressing the FLIPL-encoding con-struct exhibited a twofold increase in FLIPL expression butunderwent TRAIL-induced apoptosis to the same extent as didcontrol cells that received a blank construct (Fig. 1F, bars 5versus bars 2 and 3). Cells expressing the FLIPS-encodingconstruct exhibited twofold increases in FLIPS levels but ex-hibited significantly less TRAIL-induced apoptosis than didcells that received an empty construct (Fig. 1F, bars 4 versusbars 3). In converse experiments, TRAIL-resistant cells ex-pressing the Y40C Ras mutant selective for PI3K (C40 cells)were transfected with siRNA targeting FLIPS or FLIPL, afterwhich TRAIL-induced apoptosis was assessed. While bothsiRNAs selectively decreased the expressions of their targets(Fig. 1G), only FLIPS-targeted siRNA significantly increasedTRAIL-induced apoptosis (Fig. 1H). These results show thatthe activation of the RalA pathway suppresses levels of FLIPS,that the suppression of FLIPS levels sensitizes cells to TRAIL-mediated apoptosis, and that FLIPS links the Ral oncogenicpathway to the tumor-suppressive mechanism activated byTRAIL.
RalA activation is linked to FLIPS and the extrinsic celldeath pathway via RalBP1 and cdc42. The means by whichRalA is linked to the modulation of FLIPS levels have not beenexamined, although a variety of downstream targets of Ralhave been suggested. To define the pathway coupling RalA toapoptotic sensitization, we first examined the role of the Raltarget RalBP1. TRAIL-sensitive Ras- or Ral-transformed cellswere incubated with siRNA targeting RalBP1, after which theeffects of RalBP1 suppression on FLIPS expression andTRAIL sensitivity were monitored. E6/E7/hTERT/Ras, G37,
7348 PANNER ET AL. MOL. CELL. BIOL.
on April 2, 2016 by guest
http://mcb.asm
.org/D
ownloaded from
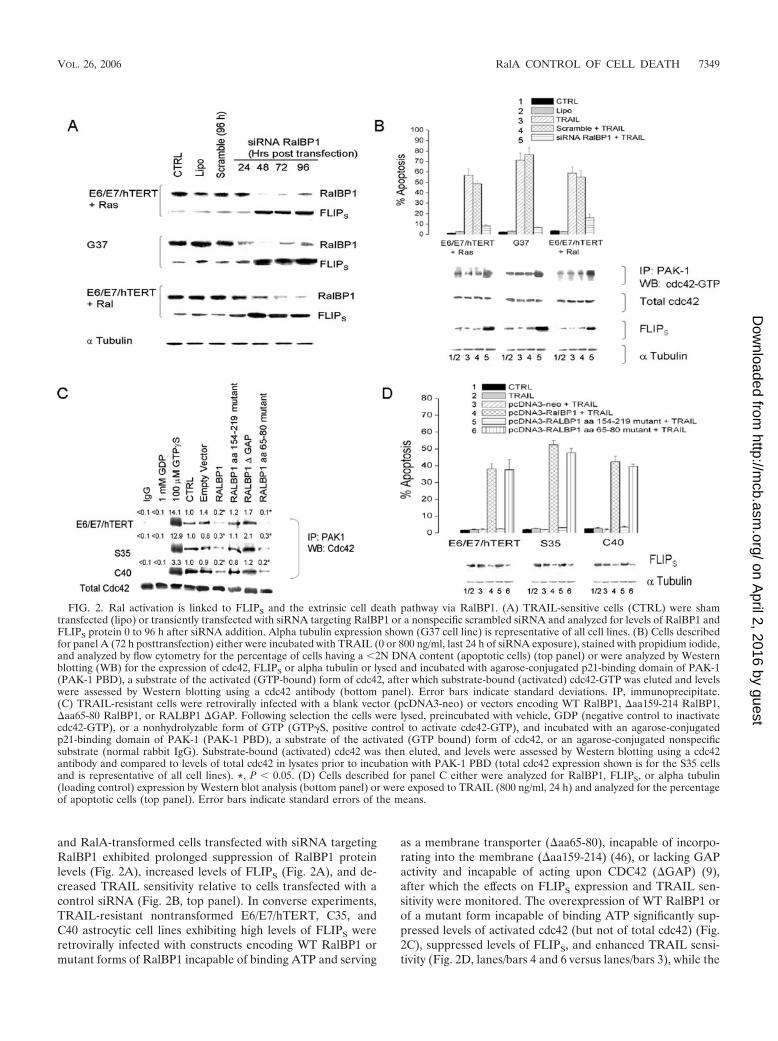
and RalA-transformed cells transfected with siRNA targetingRalBP1 exhibited prolonged suppression of RalBP1 proteinlevels (Fig. 2A), increased levels of FLIPS (Fig. 2A), and de-creased TRAIL sensitivity relative to cells transfected with acontrol siRNA (Fig. 2B, top panel). In converse experiments,TRAIL-resistant nontransformed E6/E7/hTERT, C35, andC40 astrocytic cell lines exhibiting high levels of FLIPS wereretrovirally infected with constructs encoding WT RalBP1 ormutant forms of RalBP1 incapable of binding ATP and serving
as a membrane transporter (�aa65-80), incapable of incorpo-rating into the membrane (�aa159-214) (46), or lacking GAPactivity and incapable of acting upon CDC42 (�GAP) (9),after which the effects on FLIPS expression and TRAIL sen-sitivity were monitored. The overexpression of WT RalBP1 orof a mutant form incapable of binding ATP significantly sup-pressed levels of activated cdc42 (but not of total cdc42) (Fig.2C), suppressed levels of FLIPS, and enhanced TRAIL sensi-tivity (Fig. 2D, lanes/bars 4 and 6 versus lanes/bars 3), while the
FIG. 2. Ral activation is linked to FLIPS and the extrinsic cell death pathway via RalBP1. (A) TRAIL-sensitive cells (CTRL) were shamtransfected (lipo) or transiently transfected with siRNA targeting RalBP1 or a nonspecific scrambled siRNA and analyzed for levels of RalBP1 andFLIPS protein 0 to 96 h after siRNA addition. Alpha tubulin expression shown (G37 cell line) is representative of all cell lines. (B) Cells describedfor panel A (72 h posttransfection) either were incubated with TRAIL (0 or 800 ng/ml, last 24 h of siRNA exposure), stained with propidium iodide,and analyzed by flow cytometry for the percentage of cells having a �2N DNA content (apoptotic cells) (top panel) or were analyzed by Westernblotting (WB) for the expression of cdc42, FLIPS or alpha tubulin or lysed and incubated with agarose-conjugated p21-binding domain of PAK-1(PAK-1 PBD), a substrate of the activated (GTP-bound) form of cdc42, after which substrate-bound (activated) cdc42-GTP was eluted and levelswere assessed by Western blotting using a cdc42 antibody (bottom panel). Error bars indicate standard deviations. IP, immunoprecipitate.(C) TRAIL-resistant cells were retrovirally infected with a blank vector (pcDNA3-neo) or vectors encoding WT RalBP1, �aa159-214 RalBP1,�aa65-80 RalBP1, or RALBP1 �GAP. Following selection the cells were lysed, preincubated with vehicle, GDP (negative control to inactivatecdc42-GTP), or a nonhydrolyzable form of GTP (GTP�S, positive control to activate cdc42-GTP), and incubated with an agarose-conjugatedp21-binding domain of PAK-1 (PAK-1 PBD), a substrate of the activated (GTP bound) form of cdc42, or an agarose-conjugated nonspecificsubstrate (normal rabbit IgG). Substrate-bound (activated) cdc42 was then eluted, and levels were assessed by Western blotting using a cdc42antibody and compared to levels of total cdc42 in lysates prior to incubation with PAK-1 PBD (total cdc42 expression shown is for the S35 cellsand is representative of all cell lines). *, P � 0.05. (D) Cells described for panel C either were analyzed for RalBP1, FLIPS, or alpha tubulin(loading control) expression by Western blot analysis (bottom panel) or were exposed to TRAIL (800 ng/ml, 24 h) and analyzed for the percentageof apoptotic cells (top panel). Error bars indicate standard errors of the means.
VOL. 26, 2006 RalA CONTROL OF CELL DEATH 7349
on April 2, 2016 by guest
http://mcb.asm
.org/D
ownloaded from
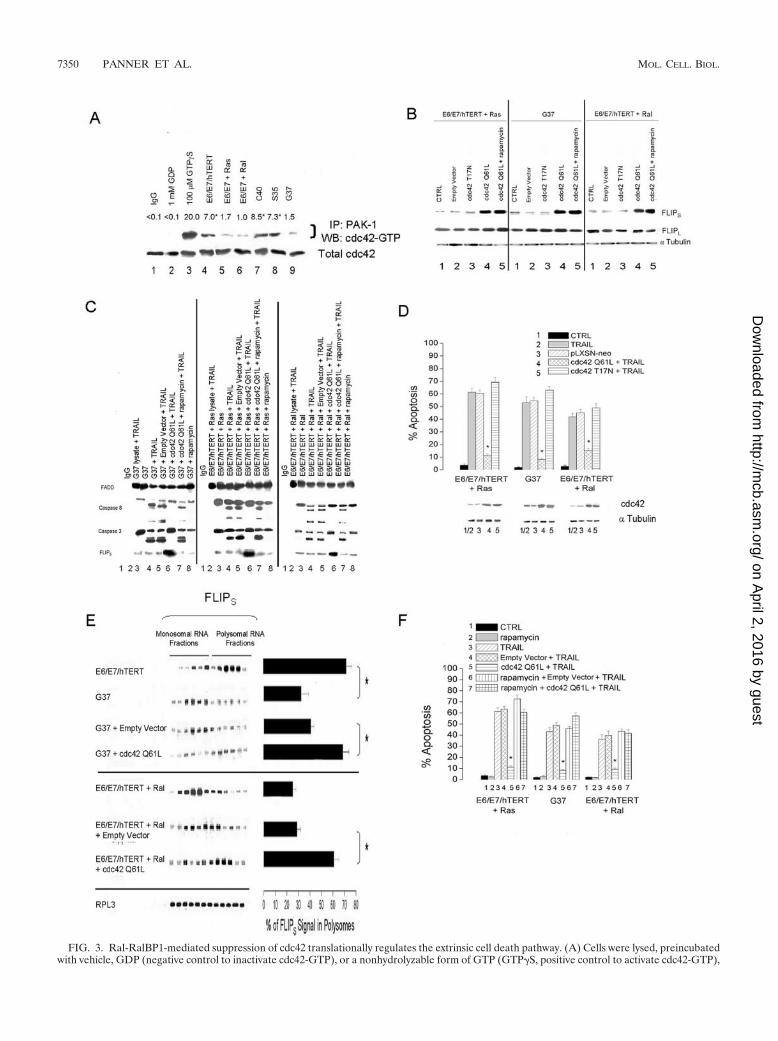
FIG. 3. Ral-RalBP1-mediated suppression of cdc42 translationally regulates the extrinsic cell death pathway. (A) Cells were lysed, preincubatedwith vehicle, GDP (negative control to inactivate cdc42-GTP), or a nonhydrolyzable form of GTP (GTP�S, positive control to activate cdc42-GTP),
7350 PANNER ET AL. MOL. CELL. BIOL.
on April 2, 2016 by guest
http://mcb.asm
.org/D
ownloaded from

overexpression of the RalBP1 mutants incapable of incorpo-rating into the membrane (�aa159-214) or lacking GAP activ-ity (�GAP) did not alter levels of active cdc42, FLIPS, orTRAIL sensitivity relative to vector controls (Fig. 2C and D,lanes/bars 5 versus lanes/bars 3). These results show thatRalBP1 serves in a membrane-bound, GAP-dependent man-ner to link RalA activation to the suppression of cdc42 activa-tion, the suppression of FLIPS expression, and enhanced sen-sitivity to TRAIL-induced apoptosis.
To further examine the potential link between RalBP1,cdc42, and FLIPS expression, we first compared levels of acti-vated cdc42 (cdc42-GTP) among Ras-modulated cell lines. Asin Fig. 2C, cell lysates were preincubated with vehicle, GDP (anegative control to deplete cdc42-GTP), or a nonhydrolyzableform of GTP (positive control to enhance cdc42-GTP levels),followed by incubation with an agarose-conjugated substrate ofthe activated (GTP bound) form of cdc42 (PAK-1 PBD). Lev-els of eluted cdc42-GTP and total cdc42 were then assessed byWestern blot analysis. TRAIL-sensitive Ras- or Ral-trans-formed cells with low levels of FLIPS (Fig. 1E) and low levelsof activated RalA (Fig. 1D) also had low levels of active cdc42(Fig. 3A, lanes 5, 6, and 9), while TRAIL-resistant E6/E7/hTERT, S35, and C40 cells with high levels of FLIPS andactivated RalA had higher levels of cdc42-GTP (lanes 4, 7, and8, respectively). The Ras- or Ral-transformed cells transfectedwith siRNA targeting RalBP1 that exhibited significantly in-creased levels of FLIPS and increased TRAIL resistance (Fig.2B) also exhibited increased levels of activated cdc42 (Fig. 2B,bottom panel, lanes/bars 5 versus lanes/bars 4), while the E6/E7/hTERT, C35, and C40 cells retrovirally infected with WTor mutant RalBP1 constructs that exhibited suppressed levelsof FLIPS, decreased levels of activated cdc42, and increasedTRAIL sensitivity (Fig. 2C and D, lanes 4 and 6) also exhibitedsuppressed levels of activated cdc42 (Fig. 2C).
To more directly determine whether cdc42 activity modu-lated FLIPS levels and TRAIL response, TRAIL-sensitiveRas- or Ral-transformed cells with high levels of active RalAand low levels of cdc42-GTP and FLIPS (E6/E7/hTERT/Ras,G37, and E6/E7/hTERT/RalA) were retrovirally infected witha blank retroviral construct (pLXSN-neo) or a construct en-coding either constitutively active or dominant-negative cdc42(Q61L and T17N, respectively) (1), after which stably express-ing cells were selected and effects on FLIPS levels and TRAIL
sensitivity were assessed. Ras- or Ral-transformed cells in-fected with the active cdc42 construct had increased levels ofFLIPS (but not FLIPL) relative to those of controls (Fig. 3B,lanes 4 versus lanes 2), and the DISC complex immunopre-cipitated from these cells following exposure to TRAIL hadincreased levels of FLIPS and decreased levels of cleavedcaspase-8/caspase-3 (Fig. 3C, compare lanes 6 to lanes 4 and5). The increased FLIPS expression and decreased DISCcaspase cleavage was also accompanied by changes in TRAILsensitivity, with both the Ras- and Ral-transformed cells ex-hibiting decreased TRAIL-induced apoptosis relative to thatof control cells or cells expressing the dominant-negative(T17N) cdc42 construct (Fig. 3D, bars 4 versus bars 3 or 5,respectively). These results show that cdc42 activity is sup-pressed by Ras/Ral/RalBP1 activation and that decreases incdc42 activity lead to decreased levels of FLIPS and enhancedTRAIL sensitivity.
Ral-RalBP1-mediated suppression of cdc42 translationallyregulates the extrinsic cell death pathway. Because FLIPS pro-tein levels can be controlled by the distribution of the FLIPS
mRNA between translating polyribosomes and nontranslatingmonosomes (31), we used sucrose gradient density centrifuga-tion in combination with Northern blot analysis to assess theability of cdc42 to control ribosomal distribution of FLIPS
mRNA and Ral-enhanced TRAIL sensitivity. Lysates fromTRAIL-resistant E6/E7/hTERT cells, TRAIL-sensitive Ral-transformed cells (G37 or Ral), or the same cells madeTRAIL-resistant by the introduction of a construct encodingconstitutively active cdc42 were layered on a continuous 5 to70% sucrose gradient and centrifuged. Fractions were thencollected and analyzed for ribosomal distribution (see Fig. S2in the supplemental material), spiked with Drosophila RPL3RNA (as an internal control), and analyzed for FLIPS, FLIPL,and RPL3 RNA levels. While FLIPL mRNA associated withboth polysomal and monosomal fractions in a pattern that wasindependent of TRAIL sensitivity (not shown), FLIPS mRNAwas selectively associated with the nontranslating monosomesin TRAIL-sensitive G37- or Ral-transformed cells but with thetranslating polysomes in TRAIL-resistant parental E6/E7/hTERT cells (Fig. 3E). Furthermore, the overexpression ofconstitutively active cdc42 in the G37- or Ral-transformed cellsshifted the distribution of FLIPS mRNA from the monosomalfraction to the polysomal fraction relative to control cells (Fig.
and incubated with an agarose-conjugated p21-binding domain of PAK-1 (PAK-1 PBD), a substrate of the activated (GTP bound) form of cdc42,or an agarose-conjugated nonspecific substrate (normal rabbit IgG). Substrate-bound (activated) cdc42 was then eluted, and levels were assessedby Western blotting (WB) using a cdc42 antibody and compared to levels of total cdc42 in lysates prior to incubation with PAK-1 PBD. IP,immunoprecipitate. *, P � 0.05. (B) TRAIL-sensitive E6/E7/hTERT�Ras, G37, and E6/E7/hTERT/Ral cells (CTRL) were infected with an emptyvector or with constructs encoding constitutively active or dominant-negative cdc42 (Q61L and T17N, respectively) and after selection wereincubated with rapamycin (0 or 100 nM, 30 min) and analyzed for FLIPS, FLIPL, and alpha tubulin expression by Western blotting. (C) Cellsdescribed for panel B were analyzed for expression of DISC components as described in the legend for Fig. 1C. (D) Cells described for panel Bwere analyzed for expression of cdc42 (and alpha tubulin as a loading control) by Western blot analysis (bottom panel) or exposed to TRAIL (800ng/ml, 24 h) and analyzed for the percentage of apoptotic cells (top panel). Error bars indicate standard deviations. *, P � 0.05. (E) E6/E7/hTERT,E6/E7/hTERT � Ral, G37, G37�empty vector, and G37�cdc42Q61L cells were lysed and subjected to sucrose density gradient centrifugation,with subsequent RNA from fractions containing unassembled ribosomal subunits (fractions 1 to 6) or assembled polyribosomes (fractions 7 to 12)spiked with Drosophila RPL3 RNA (to normalize for equal isolation/loading) and analyzed for FLIPS and RPL3 content by Northern blotting (leftpanel). The percentage of the FLIPS signal localized to the polysomal fractions is displayed in the right panel. *, P � 0.05. The RPL3 expressionshown is for the G37 cells and is representative of all cell lines. Error bars indicate standard deviations. (F) Cells described for panel B wereexposed to rapamycin (0 or 100 nM, 30 min) and then TRAIL (800 ng/ml, 24 h) and analyzed for the percentage of apoptotic cells. Error barsindicate standard errors of the means. *, P � 0.05.
VOL. 26, 2006 RalA CONTROL OF CELL DEATH 7351
on April 2, 2016 by guest
http://mcb.asm
.org/D
ownloaded from
FIG
.4.
Ral
-Ral
BP1
supp
ress
ion
ofcd
c42
tran
slat
iona
llyre
gula
tes
the
extr
insi
cce
llde
ath
path
way
via
S6K
.(A
)W
este
rnbl
otan
alys
isof
phos
pho-
S6K
(act
ivat
ed),
phos
pho-
S6,t
otal
S6K
,an
dal
pha
tubu
lin(l
oadi
ngco
ntro
l)ex
pres
sion
inT
RA
IL-r
esis
tant
/non
tran
sfor
med
(E6/
E7/
hTE
RT
,C40
,and
S35)
orT
RA
IL-s
ensi
tive/
tran
sfor
med
(E6/
E7/
hTE
RT
�R
as,E
6/E
7/hT
ER
T/R
al,
and
G37
)ce
lls.(
B)
TR
AIL
-sen
sitiv
ece
lls(E
6/E
7/hT
ER
T�
Ras
,G37
,and
E6/
E7/
hTE
RT
�R
al)
that
wer
esh
amtr
ansf
ecte
d(l
ipo)
ortr
ansi
ently
tran
sfec
ted
with
siR
NA
targ
etin
gR
alB
P1or
ano
nspe
cific
scra
mbl
edsi
RN
A(s
cram
ble)
(72
h),w
ere
incu
bate
dw
ithra
pam
ycin
(0or
100
nM,l
ast
24h
orsi
RN
Ain
cuba
tion)
and
anal
yzed
for
leve
lsof
Ral
BP1
,pho
spho
-S6K
(act
ivat
ed),
phos
pho-
S6,t
otal
S6K
,and
alph
atu
bulin
(loa
ding
cont
rol)
prot
ein.
Alp
hatu
bulin
expr
essi
onsh
own
isfo
rth
eG
37ce
llsan
dis
repr
esen
tativ
eof
allc
elll
ines
.(C
)E
6/E
7/hT
ER
T/R
asce
llsw
ere
sham
tran
sfec
ted,
tran
sien
tlytr
ansf
ecte
dw
ithei
ther
siR
NA
targ
etin
gS6
K1
ora
nons
peci
ficsc
ram
bled
siR
NA
(96
h),o
rsta
bly
tran
sfec
ted
with
anem
pty
vect
orco
ntro
l(pR
K7-
neo
orL
XSN
-neo
)
7352 PANNER ET AL. MOL. CELL. BIOL.
on April 2, 2016 by guest
http://mcb.asm
.org/D
ownloaded from

3E), consistent with the increased levels of FLIPS proteinnoted in these cells. These results suggest that Ral/RalBP1activation suppresses cdc42 activity, which in turn controlsFLIPS mRNA distribution and translation, and by doing socontrols FLIPS protein levels and ultimately TRAIL sensitivity.
Ral-mediated control of FLIPS expression is mTOR inde-pendent. Because mTOR is a target of Ras and can controlribosomal distribution of FLIPS mRNA (31), we examined thepossibility that mTOR was involved in the Ras/Ral/RalBP1/cdc42 pathway that suppresses FLIPS mRNA translation andenhances extrinsic apoptosis. For these studies, E6/E7/hTERT/Ras, G37, and Ral cells overexpressing constitutively activecdc42 (and subsequently exhibiting high levels of FLIPS
mRNA in translating polysomal fractions, high levels of FLIPS,lack of TRAIL-induced caspase cleavage in the DISC, andhigh TRAIL resistance) were incubated with the mTOR inhib-itor rapamycin, after which the effects on Ral-related signaltransduction were assessed. Although rapamycin exposure hadno effect on cdc42-driven FLIPS protein overexpression (Fig.3B, lanes 5 versus lanes 4), it altered events in the DISC ofRas-or Ral-transformed cells, decreasing levels of FLIPS in theDISC and increasing cleavage of caspase-8/caspase-3 (Fig. 3C,lanes 7 versus lanes 6), while reversing cdc42-mediated TRAILsensitivity (Fig. 3F, compare bars 7 to bars 5 and bars 3). Theseresults show that while mTOR does not alter the ability of theRal/RalBP1/cdc42 pathway to control FLIPS levels, it maymodulate the ability of the FLIPS protein, once created, tolocalize to the DISC complex and to regulate the apoptoticprocess.
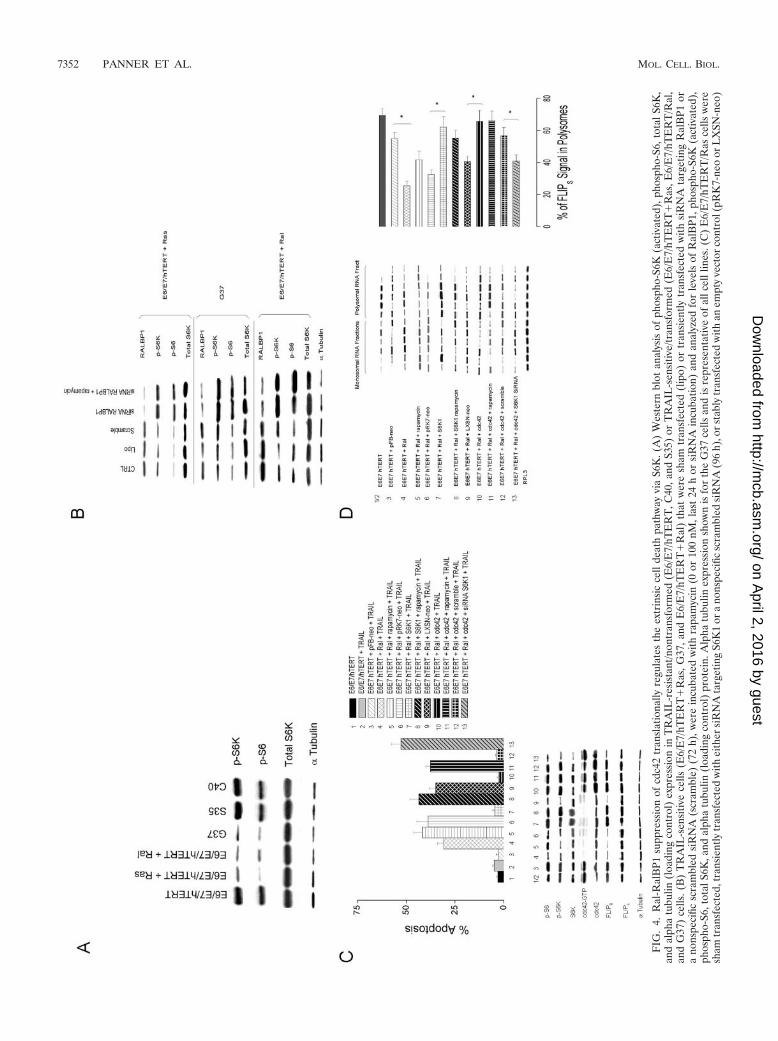
S6K1 links cdc42 activation to enhanced FLIPS translation.Because cdc42 activation increases FLIPS translation and con-fers TRAIL resistance, we considered the possible involvementof S6K1, a translational regulator and known downstream tar-get of cdc42. Initial Western blot analysis showed that TRAIL-sensitive cells (E6/E7/hTERT/Ras, G37, and E6/E7/hTERT/Ral) with high levels of activated RalA and low levels ofactivated cdc42 and FLIPS also had lower levels of pS6K andpS6 than did TRAIL-resistant cells (E6/E7/hTERT, S35, andC40) (Fig. 4A). The introduction of an siRNA targetingRalBP1 also suppressed RalBP1 levels (relative to sham-trans-fected cells or cells transfected with a scrambled siRNA) whilesimultaneously increasing levels of pS6K and pS6 in threedifferent TRAIL-sensitive cell lines with high levels of RalAactivation (Fig. 4B). Furthermore, the ability of siRNA target-ing RalBP1 to enhance pS6K and pS6 levels was independentof mTOR as the incubation of the above-described cells withrapamycin had no effect on the ability of the RalBP1 siRNA toenhance pS6K and pS6 levels (Fig. 4B). We therefore per-formed further studies in which Ral-transformed cells wereretrovirally infected with a construct encoding either constitu-tively active cdc42 or wild-type p70 S6K1. Levels of S6K1,phosphorylated (active) S6K1, phosphorylated S6, cdc42, ac-tive cdc42-GTP, FLIPS, and FLIPL were then assessed in un-treated or rapamycin-treated cells, as was the ribosomal distri-bution of the FLIPS mRNA. The same cells were alsoincubated with either TRAIL or rapamycin followed byTRAIL, after which the extent of apoptosis was assessed. Cellsthat received the cdc42 construct (Fig. 4C, bottom panel, lane8) exhibited increased levels of cdc42, cdc42-GTP, pS6K, pS6,and FLIPS relative to those of the blank vector E6/E7/hTERT/or
aco
nstr
uct
enco
ding
S6K
1or
cdc4
2.C
ells
wer
eth
enan
alyz
edby
Wes
tern
blot
ting
for
leve
lsof
pS6K
1,pS
6,S6
K1,
cdc4
2-G
TP,
cdc4
2,F
LIP
S,F
LIP
L,a
ndal
pha
tubu
lin(l
oadi
ngco
ntro
l)(b
otto
mpa
nel)
,and
anal
yzed
for
the
perc
enta
geof
apop
totic
cells
afte
rex
posu
reto
rapa
myc
in(0
or10
0nM
,30
min
)an
dT
RA
IL(8
00ng
/ml,
24h)
(top
pane
l).E
rror
bars
indi
cate
stan
dard
devi
atio
ns.(
D)
Cel
lsus
edin
pane
lCw
ere
lyse
d,se
para
ted
into
mon
osom
alan
dpo
lyso
mal
frac
tions
,and
spik
edw
ithD
roso
phila
RPL
3R
NA
,aft
erw
hich
RN
Aw
asis
olat
edan
dan
alyz
edfo
rF
LIP
San
dR
PL3
RN
Ale
vels
byN
orth
ern
blot
anal
ysis
(lef
tpa
nel)
.The
perc
enta
geof
the
FL
IPS
sign
allo
caliz
edto
the
poly
som
alfr
actio
nsis
disp
laye
din
the
righ
tpa
nel.
*,P
�0.
05.T
heR
PL3
expr
essi
onsh
own
isfo
rth
eE
6/E
7/hT
ER
Tce
llsan
dis
repr
esen
tativ
eof
allc
elll
ines
.Err
orba
rsin
dica
test
anda
rder
rors
ofth
em
eans
.
VOL. 26, 2006 RalA CONTROL OF CELL DEATH 7353
on April 2, 2016 by guest
http://mcb.asm
.org/D
ownloaded from
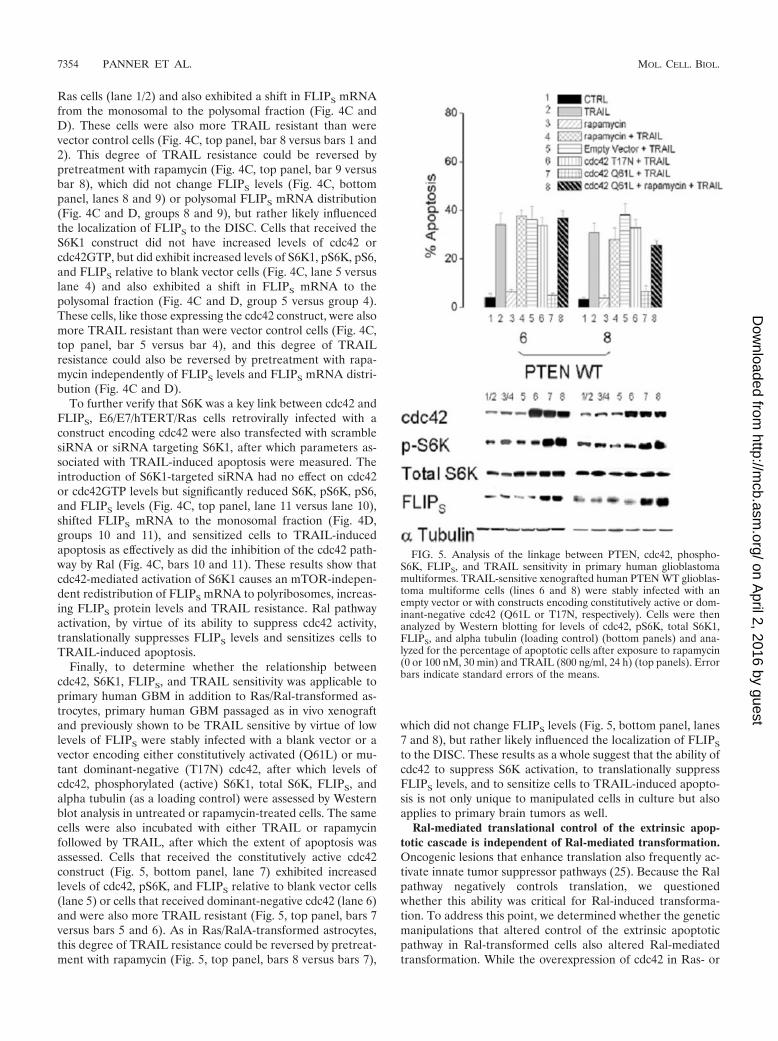
Ras cells (lane 1/2) and also exhibited a shift in FLIPS mRNAfrom the monosomal to the polysomal fraction (Fig. 4C andD). These cells were also more TRAIL resistant than werevector control cells (Fig. 4C, top panel, bar 8 versus bars 1 and2). This degree of TRAIL resistance could be reversed bypretreatment with rapamycin (Fig. 4C, top panel, bar 9 versusbar 8), which did not change FLIPS levels (Fig. 4C, bottompanel, lanes 8 and 9) or polysomal FLIPS mRNA distribution(Fig. 4C and D, groups 8 and 9), but rather likely influencedthe localization of FLIPS to the DISC. Cells that received theS6K1 construct did not have increased levels of cdc42 orcdc42GTP, but did exhibit increased levels of S6K1, pS6K, pS6,and FLIPS relative to blank vector cells (Fig. 4C, lane 5 versuslane 4) and also exhibited a shift in FLIPS mRNA to thepolysomal fraction (Fig. 4C and D, group 5 versus group 4).These cells, like those expressing the cdc42 construct, were alsomore TRAIL resistant than were vector control cells (Fig. 4C,top panel, bar 5 versus bar 4), and this degree of TRAILresistance could also be reversed by pretreatment with rapa-mycin independently of FLIPS levels and FLIPS mRNA distri-bution (Fig. 4C and D).
To further verify that S6K was a key link between cdc42 andFLIPS, E6/E7/hTERT/Ras cells retrovirally infected with aconstruct encoding cdc42 were also transfected with scramblesiRNA or siRNA targeting S6K1, after which parameters as-sociated with TRAIL-induced apoptosis were measured. Theintroduction of S6K1-targeted siRNA had no effect on cdc42or cdc42GTP levels but significantly reduced S6K, pS6K, pS6,and FLIPS levels (Fig. 4C, top panel, lane 11 versus lane 10),shifted FLIPS mRNA to the monosomal fraction (Fig. 4D,groups 10 and 11), and sensitized cells to TRAIL-inducedapoptosis as effectively as did the inhibition of the cdc42 path-way by Ral (Fig. 4C, bars 10 and 11). These results show thatcdc42-mediated activation of S6K1 causes an mTOR-indepen-dent redistribution of FLIPS mRNA to polyribosomes, increas-ing FLIPS protein levels and TRAIL resistance. Ral pathwayactivation, by virtue of its ability to suppress cdc42 activity,translationally suppresses FLIPS levels and sensitizes cells toTRAIL-induced apoptosis.
Finally, to determine whether the relationship betweencdc42, S6K1, FLIPS, and TRAIL sensitivity was applicable toprimary human GBM in addition to Ras/Ral-transformed as-trocytes, primary human GBM passaged as in vivo xenograftand previously shown to be TRAIL sensitive by virtue of lowlevels of FLIPS were stably infected with a blank vector or avector encoding either constitutively activated (Q61L) or mu-tant dominant-negative (T17N) cdc42, after which levels ofcdc42, phosphorylated (active) S6K1, total S6K, FLIPS, andalpha tubulin (as a loading control) were assessed by Westernblot analysis in untreated or rapamycin-treated cells. The samecells were also incubated with either TRAIL or rapamycinfollowed by TRAIL, after which the extent of apoptosis wasassessed. Cells that received the constitutively active cdc42construct (Fig. 5, bottom panel, lane 7) exhibited increasedlevels of cdc42, pS6K, and FLIPS relative to blank vector cells(lane 5) or cells that received dominant-negative cdc42 (lane 6)and were also more TRAIL resistant (Fig. 5, top panel, bars 7versus bars 5 and 6). As in Ras/RalA-transformed astrocytes,this degree of TRAIL resistance could be reversed by pretreat-ment with rapamycin (Fig. 5, top panel, bars 8 versus bars 7),
which did not change FLIPS levels (Fig. 5, bottom panel, lanes7 and 8), but rather likely influenced the localization of FLIPS
to the DISC. These results as a whole suggest that the ability ofcdc42 to suppress S6K activation, to translationally suppressFLIPS levels, and to sensitize cells to TRAIL-induced apopto-sis is not only unique to manipulated cells in culture but alsoapplies to primary brain tumors as well.
Ral-mediated translational control of the extrinsic apop-totic cascade is independent of Ral-mediated transformation.Oncogenic lesions that enhance translation also frequently ac-tivate innate tumor suppressor pathways (25). Because the Ralpathway negatively controls translation, we questionedwhether this ability was critical for Ral-induced transforma-tion. To address this point, we determined whether the geneticmanipulations that altered control of the extrinsic apoptoticpathway in Ral-transformed cells also altered Ral-mediatedtransformation. While the overexpression of cdc42 in Ras- or
FIG. 5. Analysis of the linkage between PTEN, cdc42, phospho-S6K, FLIPS, and TRAIL sensitivity in primary human glioblastomamultiformes. TRAIL-sensitive xenografted human PTEN WT glioblas-toma multiforme cells (lines 6 and 8) were stably infected with anempty vector or with constructs encoding constitutively active or dom-inant-negative cdc42 (Q61L or T17N, respectively). Cells were thenanalyzed by Western blotting for levels of cdc42, pS6K, total S6K1,FLIPS, and alpha tubulin (loading control) (bottom panels) and ana-lyzed for the percentage of apoptotic cells after exposure to rapamycin(0 or 100 nM, 30 min) and TRAIL (800 ng/ml, 24 h) (top panels). Errorbars indicate standard errors of the means.
7354 PANNER ET AL. MOL. CELL. BIOL.
on April 2, 2016 by guest
http://mcb.asm
.org/D
ownloaded from

Ral-transformed (G37 and RalA) cells shifted FLIPS mRNAribosomal distribution, translationally up-regulated FLIPS lev-els, and increased TRAIL resistance, cdc42 expression had noeffect on the ability of the transformed cells to grow in soft agar(not shown) or in immunodeficient hosts (Fig. 1A). Theseresults suggest that the Ral pathway is linked to the control oftranslation but that this linkage neither is critical for nor in-terferes with Ral-mediated transformation.
DISCUSSION
Although oncogenic pathways frequently co-opt the transla-tional machinery for their own benefit, they also paradoxicallyopen tumor-suppressive mechanisms that can lead to their owndemises. The present study shows that the RalA oncogenicpathway fits this mold, with a few novel twists. Like otheroncogenic pathways, RalA activation hijacks the translationalmachinery. This translational co-option, however, is not thedriving force behind the transformation process, but rather, bysuppressing the translation of the extrinsic apoptotic pathwayinhibitor FLIPS, is itself the key that opens a pathway to celldeath.
The present work links the RalA pathway to control of theextrinsic apoptotic pathway activated by TRAIL. The mitogen-activated protein kinase pathway, but not the Ral or PI3Kpathways, has been previously linked to the stabilization ofmyc, increased DR5 expression, increased caspase activation,and enhanced TRAIL-induced apoptosis in fibroblasts andHEK cells (29, 37, 46, 47). In the present study, there was noindication of RalA-induced DR5 up-regulation or myc stabili-zation (unpublished data), although RalA but not a Raf-selec-tive T35S Ras effector mutant did increase caspase activation.These differences may be a consequence of different TRAILexposure conditions used or alternatively may reflect tissue-specific differences in the control of the extrinsic apoptoticpathway and subtle differences in the balance between themultiple factors that set the apoptotic threshold. The findingthat Ras, Raf, Ral, and myc all suppress TRAIL-inducedcaspase activation in multiple cell lines under multiple condi-tions (29, 37, 39, 46, 47), however, suggests that the regulationof DISC function, including that by FLIP up-regulation, maybe a common means by which oncogenic pathways regulate theextrinsic apoptotic pathway. While FLIPS regulation can beaccomplished transcriptionally, as has been previously re-ported for myc (37), the ability of the Ral pathway to do sotranslationally represents a novel means of controlling the ex-trinsic cell death pathway.
The means by which the RalA pathway is linked to thetranslational machinery is novel, as is the manner in which thisoncogenic pathway impacts translation. The present workshows that RalA is linked to the control of FLIPS translationnot via mTOR, the most commonly identified and extensivelystudied regulator of translation, but instead via a secondGTPase, cdc42, and by the ability of cdc42 to regulate theactivity of the mTOR target S6K1. Our observation that cdc42activates S6K1 is consistent with previous reports that suggestthat cdc42 coimmunoprecipitates with and activates S6K1 (7,8). How a GTPase such as cdc42 activates S6K1, however,remains unclear, although previous studies using isoprenyla-tion-deficient cdc42 mutants suggest that membrane targeting
of cdc42 is critical for S6K1 activation (7). Membrane local-ization of the cdc42-S6K complex may allow other membrane-associated proteins such as PDK1 to activate S6K1, although itis clear that at least part of the ability of cdc42 to activate S6K1is distinct from that of PDK1 (26). The observation that bothRalBP1 (this study) and cdc42 (32) function in a membrane-associated manner is consistent with the idea that RalBP1,cdc42, and S6K may be brought into intimate contact by RalAactivation in the cell membrane, after which S6K1 activationcan occur (15, 22, 27, 28).
The Ral-mediated control of translation noted in the presentstudy is also unusual in that input from the RalA oncogenicpathway negatively impacts translation, at least with regard toFLIPS mRNA. While the full range of Ral-mediated transla-tional regulation remains to be determined, it seems unlikelythat Ral pathway activation negatively regulates the translationof all mRNAs, particularly in light of recent studies showingthat even complete disruption of PDK, an activator of Akt-mTOR signaling, suppresses the translation of only a subset ofmRNAs and enhances the polysomal association of others(44). Rather, the Ral-mediated suppression of S6K1 activitymay suppress the translation of only a subset of structurallyrelated mRNAs. If this is the case, the examination of thedifferential sensitivity of the closely related FLIPS and FLIPL
mRNAs to Ral-mediated translational regulation may proveinformative. Alternatively, RalA may be linked to the transla-tional apparatus by both an mTOR-independent pathway thatsuppresses the translation of FLIPS and an mTOR-dependentpathway that does not regulate FLIPS mRNA translation butdoes regulate the translation of other mRNAs critical for Ral-mediated transformation. It’s worth noting that RalA appearsto control not only FLIPS translation but also FLIPS stability,as RalA pathway activation significantly decreases the half-lifeof the FLIPS protein in a manner that is reversible by theoverexpression of S6K1 (unpublished observations). RalA maytherefore have multiple inputs into translational and posttrans-lational regulation and multiple means of controlling the levelsof proteins critical for the translation process.
It is finally worth noting that the present studies also suggestthat mTOR contributes in a unique manner to the control ofthe extrinsic apoptotic pathway by enhancing FLIPS localiza-tion to the DISC. Although mTOR has not been reported to beinvolved in protein trafficking or to interact with FLIPS, FLIPS
is extensively posttranslationally modified in ways that couldpotentially be altered by mTOR (49, 50). Furthermore, be-cause mTOR plays a critical role in Akt-mediated enhance-ment of FLIPS translation (31), the ability of mTOR to facil-itate proper FLIPS localization may ensure proper shutdown ofthe extrinsic apoptotic pathway in tumors with activated Aktpathways. The ability of the Ral pathway to bypass this shut-down and to reopen the extrinsic cell death pathway indepen-dently of mTOR and even in the face of high Akt levels (un-published data) suggests that therapies designed to blockmTOR and the oncogenic stimulation of translation should becompatible and perhaps synergistic with approaches designedto activate the extrinsic pathway opened by RalA. It may alsobe possible that part of the effect of mTOR inhibition itself isthe unmasking of apoptotic pathways stimulated by RalA, par-ticularly if the Ral pathway suppresses the expression/transla-tion of a broad set of antiapoptotic proteins.
VOL. 26, 2006 RalA CONTROL OF CELL DEATH 7355
on April 2, 2016 by guest
http://mcb.asm
.org/D
ownloaded from

The present studies define several unique properties of theRal oncogenic pathway. RalA activation is linked to the trans-lational machinery in a novel, mTOR-independent, negativemanner. Furthermore, unlike other oncogenic pathways, RalAuses this link not to drive transformation but to sensitize cellsto cell death. While the exact means by which RalA can coop-erate with other Ras effectors to bring about transformationremain to be fully defined, the present studies provide a basisfor understanding the linkage between Ral pathway activationand translation and a mechanistic understanding of how theextrinsic apoptotic pathway could be manipulated for thera-peutic benefit in the multiple malignancies in which Ras andRalA activation play key roles.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health AwardsRO1 CA94989, RO1 CA115638, and P50 CA97257 to R.O.P.
REFERENCES
1. Ahmed, I., Y. Calle, S. Iwashita, and A. Nur-E-Kamal. 2006. Role of Cdc42in neurite outgrowth of PC12 cells and cerebellar granule neurons. Mol. Cell.Biochem. 281:17–25.
2. Allan, L. A., N. Morrice, S. Brady, G. Magee, S. Pathuk, and P. R. Clarke.2003. Inhibition of caspase-9 through phosphorylation at Thr 125 by ERKMAPK. Nat. Cell Biol. 5:647–654.
3. Bin, L., X. Li, L.-G. Xu, and H.-B. Shu. 2002. The short splice form ofCasper/c-FLIP is a major cellular inhibitor of TRAIL-induced apoptosis.FEBS Lett. 510:37–40.
4. Cantor, S. B., T. Urnao, and L. A. Feig. 1995. Identification and character-ization of Ral-binding protein 1, a potential downstream target of RalGTPases. Mol. Cell. Biol. 15:4578–4584.
5. Chang, G. H.-F., N. M. Barbaro, and R. O. Pieper. 2000. Phosphatidylserine-dependent phagocytosis of apoptotic glioma cells by normal human micro-glia, astrocytes, and glioma cells. Neuro-Oncol. 2:174–183.
6. Chien, Y., and M. A. White. 2003. RAL GTPases and linchpin modulators ofhuman tumor cell proliferation and survival. EMBO Rep. 4:800–806.
7. Chou, M. M., and J. Blenis. 1996. The 70 kDa S6 kinase complexes with andis activated by the Rho family G proteins Cdc42 and Rac1. Cell 85:573–583.
8. Coghlan, M. P., M. M. Chou, and C. L. Carpenter. 2000. Atypical proteinkinases C and -� associate with the GTP-binding protein Cdc42 and medi-ate stress fiber loss. Mol. Cell. Biol. 20:2880–2889.
9. DeRuiter, N. D., B. M. T. Burgering, and J. L. Bos. 2001. Regulation of theForkhead transcription factor AFX by Ral-dependent phosphorylation ofthreonines 447 and 451. Mol. Cell. Biol. 21:8225–8235.
10. Fingar, D. C., C. J. Richardson, A. R. Tee, L. Cheatham, C. Tsou, and J.Blenis. 2004. mTOR controls cell cycle progression through its cell growtheffectors S6K1 and 4E-BP1/eukaryotic translation initiation factor 4E. Mol.Cell. Biol. 24:200–216.
11. Fingar, D. C., and J. Blenis. 2004. Target of rapamycin (TOR): an integratorof nutrient and growth factor signals and coordinator of cell growth and cellcycle progression. Oncogene 23:3151–3171.
12. Fingar, D. C., S. Salama, C. Tsou, E. Harlow, and J. Blenis. 2002. Mamma-lian cell size is controlled by mTOR and its downstream targets S6K1 and4E-BP1/eIF4E. Genes Dev. 16:1472–1487.
13. Gingras, A. C., S. P. Gygi, B. Raught, R. D. Polakiewicz, R. T. Abraham,M. F. Hoekstra, R. Abersold, and N. Sonenberg. 1999. Regulation of 4E-BP1phosphorylation: a novel two-step mechanism. Genes Dev. 13:1422–1437.
14. Gingras, A. C., B. Raught, and N. Sonenberg. 2001. Regulation of translationinitiation by FRAP/mTOR. Genes Dev. 15:807–826.
15. Goi, T., G. Rusanescu, T. Urano, and L. A. Feig. 1999. Ral-specific guaninenucleotide exchange factor activity opposes other Ras effectors in PC12 cellsby inhibiting neurite outgrowth. Mol. Cell. Biol. 19:1731–1741.
16. Gonzalez-Garcia, A., C. A. Pritchard, H. F. Paterson, G. Mavria, G. Stamp,and C. J. Marshall. 2005. RalGDS is required for tumor formation in amodel of skin carcinogenesis. Cancer Cell 7:219–226.
17. Guertin, D. A., and D. M. Sabatini. 2005. An expanding role for mTOR incancer. Trends Mol. Med. 11:353–361.
18. Hamad, N. M., J. H. Elconin, A. E. Karnoub, W. Bao, J. N. Rich, R. T.Abraham, C. J. Der, and C. M. Counter. 2002. Distinct requirements for Rasoncogenesis in human versus mouse cells. Genes Dev. 16:2045–2057.
19. Irmler, M., M. Thome, M. Hahne, P. Schneider, K. Hofmann, V. Steiner,J.-L. Bodmer, M. Schroter, K. Burns, C. Mattman, D. Rimoldi, L. E. French,and J. Tschopp. 1997. Inhibition of death receptor signals by FLIP. Nature388:190–195.
20. Johannes, G., and P. Sarnow. 1998. Cap-independent polysomal association
of natural mRNAs encoding c-myc, BiP, and eIF4G conferred by internalribosome entry sites. RNA 4:1500–1513.
21. Julien-Flores, V., O. Dorseuil, F. Romero, F. Letourneur, S. Saragosti, R.Berger, A. Tavitian, G. Gacon, and J. H. Camonis. 1995. Bridging RalGTPase to Rho pathway. RLIP76, a Ral effector with CDC42/Rac GTPase-activating protein activity. J. Biol. Chem. 270:22473–22477.
22. Kishida, S., S. Koyama, K. Matsubara, M. Kishida, Y. Matsuura, and A.Kikuchi. 1997. Colocalization of Ras and Ral on the membrane is requiredfor Ras-dependent Ral activation through RalGDP dissociation stimulator.Oncogene 15:2899–2907.
23. Lim, K.-H., A. T., Baines, J. J. Fiordalisi, M. Shipitsin, L. A. Feig, A. C. Cox,C. J. Der, and C. M. Counter. 2005. Activation of RalA is critical forRas-induced tumorigenesis of human cells. Cancer Cell 7:533–545.
24. Lowe, S. W., and C. J. Sherr. 2003. Tumour suppression by Ink4a-Arf:progress and puzzles. Curr. Opin. Genet. Dev. 13:77–83.
25. Lowe, S. W., E. Cepero, and G. Evan. 2004. Intrinsic tumour suppression.Nature 432:307–315.
26. Martin, K. A., S. S. Schalm, C. Richardson, A. Romanelli, K. L. Keon, andJ. Blenis. 2001. Regulation of ribosomal S6 kinase 2 by effectors of thephosphoinositide 3-kinase pathway. J. Biol. Chem. 276:7884–7891.
27. Matsubara, K., S. Kishida, Y. Matsuura, H. Kitayama, M. Noda, and A.Kikuchi. 1999. Plasma membrane recruitment of RalGDS is critical forRas-dependent Ral activation. Oncogene 18:1303–1312.
28. Moskalenko, S., C. Tong, C. Rosse, G. Mirey, E. Formstecher, L. Daviet, J.Camonis, and M. A. White. 2003. Ral GTPases regulate exocyst assemblythrough dual subunit interactions. J. Biol. Chem. 278:51743–51748.
29. Nesterov, A., M. Nikrad, T. Johnson, and A. S. Kraft. 2004. Oncogenic Rassensitizes normal human cells to tumor necrosis factor--related apoptosis-inducing ligand-induced apoptosis. Cancer Res. 64:3922–3927.
30. Ohta, Y., N. Suzuki, S. Nakamura, J. H. Hartwig, and T. P. Stossel. 1999.The small GTPase RalA targets filamin to induce filopodia. Proc. Natl. Acad.Sci. USA 96:2122–2128.
31. Panner, A., M. S. Berger, and R. O. Pieper. 2005. mTOR controls FLIPStranslation and TRAIL sensitivity in glioblastoma multiforme cells. Mol.Cell. Biol. 25:8809–8823.
32. Parsons, M. J., Monypenny, S. M. Ameer-Beg, T. H. Millard, L. M. Ma-chesky, M. Peter, M. D. Keppler, G. Schiavo, R. Watson, J. Chernoff, D.Zicha, B. Vojnovic, and T. Ng. 2005. Spatially distinct binding of Cdc42 toPAK1 and N-WASP in breast carcinoma cells. Mol. Cell. Biol. 25:1680–1695.
33. Perez, D., and E. White. 2003. E1A sensitizes cells to tumor necrosis factoralpha by downregulating c-FLIPS. J. Virol. 77:2651–2662.
34. Polunovsky, V. A., A. C. Gingras, N. Sonenberg, M. Peterson, A. Tan, J. B.Rubins, J. C. Manivel, and P. B. Bitterman. 2000. Translational control ofthe antiapoptotic function of Ras. J. Biol. Chem. 275:24776–24780.
35. Ponting, C. P., and D. R. Benjamin. 1996. A novel family of Ras-bindingdomains. Trends Biochem. Sci. 21:422–425.
36. Raught, B., and A. C. Gingras. 1999. eIF4E activity is regulated at multiplelevels. Int. J. Biochem. Cell. Biol. 31:43–57.
37. Ricci, M. S., Z. Jin, M. Dews, D. Yu, A. Thomas-Tikhonenko, D. T. Dicker,and W. S. El-Deiry. 2004. Direct repression of FLIP expression by c-myc isa major determinant of TRAIL sensitivity. Mol. Cell. Biol. 24:8541–8555.
38. Rodriguez-Viciana, P., and F. McCormick. 2005. RalGDS comes of age.Cancer Cell 7:205–206.
39. Rottmann, S., Y. Wang, M. Nasoff, Q. L. Ceveraux, and K. C. Quon. 2005. ATRAIL receptor-dependent synthetic lethal relationship between MYC ac-tivation and GSK3�/FBW7 loss of function. Proc. Natl. Acad. Sci. USA102:15195–15200.
40. Shay, J. W., and I. B. Roninson. 2004. Hallmarks of senescence in carcino-genesis and cancer therapy. Oncogene 23:2919–2933.
41. Shipitsin, M., and L. A. Feig. 2004. RalA but not RalB enhances polarizeddelivery of membrane proteins to the basolateral surface of epithelial cells.Mol. Cell. Biol. 24:5746–5756.
42. Sonoda, Y., T. Ozawa, Y. Hirose, K. D. Aldape, M. McMahon, M. S. Berger,and R. O. Pieper. 2001. Formation of intracranial tumors by geneticallymodified human astrocytes defines four pathways critical in the developmentof human anaplastic astrocytoma. Cancer Res. 61:4956–4960.
43. Stolovich, M., H. Tang, E. Hornstein, G. Levy, R. Cohen, S. S. Bae, M. J.Birnbaum, and O. Meyuhas. 2002. Transduction of growth or mitogenicsignals into translational activation of TOP mRNAs is fully reliant on thephosphatidylinositol 3-kinase-mediated pathway but requires neither S6K1nor rpS6 phosphorylation. Mol. Cell. Biol. 22:8101–8113.
44. Tominaga, Y., T. Tanguney, M. Kolesnichenko, B. Bilanges, and D. S.Stokoe. 2005. Translational deregulation in PDK-1�/� embryonic stem cells.Mol. Cell. Biol. 25:8465–8475.
45. Ueda, T., R. Watanabe-Fukunaga, H. Fukuyama, S. Nagata, and R. Fuku-naga. 2004. Mnk2 and Mnk1 are essential for constitutive and induciblephosphorylation of eukaryotic initiation factor 4E but not for cell growth ordevelopment. Mol. Cell. Biol. 24:6539–6549.
46. Wang, Y., I. H. Engels, D. A. Knee, M. Nasoff, Q. L. Deveraux, and K. C.Quan. 2005. Synthetic lethal targeting of MYC by activation of the DR5death receptor pathway. Cancer Cell 5:501–511.
47. Wang, Y., K. C. Quon, and D. A. Knee. 2005. RAS, MYC, and sensitivity to
7356 PANNER ET AL. MOL. CELL. BIOL.
on April 2, 2016 by guest
http://mcb.asm
.org/D
ownloaded from

tumor necrosis factor--related apoptosis-inducing ligand-induced apopto-sis. Cancer Res. 65:1615–1617.
48. Wolthius, R. M., and J. L. Bos. 1999. Ras caught in another affair: theexchange factors for Ral. Curr. Opin. Genet. Dev. 9:112–117.
49. Xiao, C., B. F. Yang, L. Li, J. H. Song, H. Schulman, and C. Hao. 2005.Inhibition of CaMKII-mediated c-FLIP expression sensitizes malignantmelanoma cells to TRAIL-induced apoptosis. Exp. Cell Res. 304:244–255.
50. Xiao, C., B. F. Yang, N. Asadi, F. Beguinot, and C. Hao. 2002. Tumornecrosis factor-related apoptosis-inducing ligand-induced death-inducingsignaling complex and its modulation by c-FLIP and PED/PEA-15 in gliomacells. J. Biol. Chem. 277:25020–25025.
51. Yadav, S., S. S. Singhal, J. Singhal, D. Wickramarachi, E. Knutson, B. A.Albrecht, Y. C. Awasthi, and S. Awasthi. 2004. Identification of membrane-anchoring domains of RLIP76 using deletion mutant analyses. Biochemistry43:16243–16253.
VOL. 26, 2006 RalA CONTROL OF CELL DEATH 7357
on April 2, 2016 by guest
http://mcb.asm
.org/D
ownloaded from




















