
Septal-lateral annular cinching abolishes acute ischemic mitral regurgitation

Aging Cell
(2009)
8
, pp2–17 Doi: 10.1111/j.1474-9726.2008.00441.x
2
© 2009 The AuthorsJournal compilation © Blackwell Publishing Ltd/Anatomical Society of Great Britain and Ireland 2009
Blackwell Publishing Ltd
MSH2 deficiency abolishes the anticancer and pro-aging activity of short telomeres
Paula Martinez,
1
Irene Siegl-Cachedenier,
1
Juana M. Flores
2
and Maria A. Blasco
1
1
Telomeres and Telomerase Group, Molecular Oncology Program, Spanish National Cancer Centre, 28029 Madrid, Spain
2
Animal Surgery and Medicine Department, Facultad de Veterinaria, Universidad Complutense de Madrid, 28040 Madrid, Spain
Summary
Mutations in the mismatch repair (MMR) pathway occurin human colorectal cancers with microsatellite instability.Mounting evidence suggests that cell-cycle arrest inresponse to a number of cellular stresses, includingtelomere shortening, is a potent anticancer barrier. Thetelomerase-deficient mouse model illustrates the anticancereffect of cell-cycle arrest provoked by short telomeres.Here, we describe a role for the MMR protein, MSH2, insignaling cell-cycle arrest in a p21/p53-dependent mannerin response to short telomeres in the context of telomerase-deficient mice. In particular, progressively shorter telomeresat successive generations of
MSH2
–/–
Terc
–/–-
mice did notsuppress cancer in these mice, indicating that MSH2deficiency abolishes the tumor suppressor activity of shorttelomeres. Interestingly, MSH2 deficiency preventeddegenerative pathologies in the gastrointestinal tractof
MSH2
–/–
Terc
–/–
mice concomitant with a rescue of pro-liferative defects. The abolishment of the anticancer andpro-aging effects of short telomeres provoked by MSH2abrogation was independent of changes in telomerelength. These results highlight a role for MSH2 in theorganismal response to dysfunctional telomeres, which inturn may be important in the pathobiology of humancancers bearing mutations in the MMR pathway.Key words: aging; cancer; mouse models; MMR; telomeres.
Introduction
The DNA mismatch repair (MMR) pathway maintains genomic
integrity by correcting DNA replication errors and preventing
recombination between divergent DNA sequences (reviewed in
Kunkel & Erie, 2005; Jiricny, 2006). At least three protein
heterodimers are involved in the initiation step of the eukaryotic
MMR process: MSH2/MSH6 (MutS
α
), MSH2/MSH3 (MutS
β
) and
MLH1/PMS2 (MutL
α
). Muts
α
binds both to base–base mismatches
and short insertion/deletion loops, while MutS
β
can only
recognize insertion/deletion loops containing up to 16 extra
nucleotides in one strand (Kunkel & Erie, 2005). MutL
α
possesses an intrinsic endonuclease activity, activated upon
mismatch recognition, that introduces incisions in the discon-
tinuous strand of the heteroduplex DNA, thereby generating
entry sites so exonucleases can digest unwound DNA (Kadyrov
et
al
., 2006). The resulting gap is filled by DNA polymerase III;
ligation of the remaining nick completes repair (Jiricny, 2006).
Furthermore,
in vitro
reconstitution of the MMR system showed
a requirement for EXO1 for both the 5
′
–3
′
and 3
′
–5
′
mismatch
corrections, suggesting that EXO1 is a downstream player of
the MMR pathway (Genschel
et
al
., 2000; Jiricny, 2006). In
support of this, mice deficient in EXO1 exhibited defects in the
MMR of DNA (Wei
et
al
., 2003).
Mutations in a subset of MMR genes, including
MSH2
,
MSH3
,
MSH6
,
MLH1
and
PMS2
, associate with human hereditary
non-polyposis colon cancer, stressing the crucial role of MMR
in the maintenance of genome integrity (Jiricny, 2006). Mice
deficient in different MMR genes, as well as in EXO1, show
an increase in both the spontaneous mutation rates and
susceptibility to develop cancer; the MSH2 knockout mouse having
the most severe cancer-prone phenotype (Baker
et
al
., 1995; de
Wind
et
al
., 1995; Prolla
et
al
., 1998; Wei
et
al
., 2003). A role
for MMR proteins in signaling DNA damage by direct involvement
of the p53 pathway has also been proposed (Duckett
et
al
., 1999;
Peters
et
al
., 2003; Luo
et
al
., 2004), which in turn could affect
tumorigenesis. Additionally, MMR factors activate ataxia tel-
angiectasia mutated (ATM) signaling in the S-phase checkpoint
response to DNA damage (Brown
et
al
., 2003).
Telomeres are protective structures at the ends of chromo-
somes that consist of tandem TTAGGG repeats bound to a
protein complex known as shelterin (Chan & Blackburn, 2002;
de Lange, 2005). Critical telomere shortening and loss of function
of telomere-binding proteins result in loss of telomere protection,
end-to-end chromosome fusions, and cell-cycle arrest or
apoptosis (van Steensel
et
al
., 1998; Goytisolo & Blasco, 2002;
de Lange, 2005). As a consequence, critical telomere shortening
in the context of the telomerase-deficient mouse model leads
to premature aging pathologies and reduced lifespan (Lee
et
al
.,
1998; Herrera
et
al
., 1999; Blasco, 2005). Telomere shortening is
also envisioned as a potent tumor suppressor mechanism (Gonzalez-
Suarez
et
al
., 2000; Blasco & Hahn, 2003; Blasco, 2005). Mice
lacking telomerase activity are resistant to cancer (Greenberg
et
al
., 1999; Gonzalez-Suarez
et
al
., 2000), with the only exception
of p53-deficient and TRF2-overexpressing genetic backgrounds
(Chin
et
al
., 1999; Artandi
et
al
., 2000; Blanco
et
al
., 2007).
Correspondence
Maria A. Blasco, Telomeres and Telomerase Group, Molecular Oncology
Program, Spanish National Cancer Centre, 28029 Madrid, Spain.
Tel.: +34 9 1732 8031; fax: +34 9 1732 8028; e-mail: [email protected]
Paula Martinez and Irene Siegl-Cachedenier contributed equally to this work.
Accepted for publication
29 September 2008
MSH2 and signaling of short telomeres, P. Martinez
et al.
© 2009 The AuthorsJournal compilation © Blackwell Publishing Ltd/Anatomical Society of Great Britain and Ireland 2009
3
Telomerase activation, detected in a vast majority of human
cancers (Shay & Wright, 2006), is able to extend the lifespan of
cells in culture by maintaining telomeres (Bodnar
et
al
., 1998).
We have recently shown that PMS2 deficiency rescues
organismal survival and proliferative defects in telomerase-
deficient mice by attenuating p21-dependent cell cycle arrest
in response to short telomeres (Siegl-Cachedenier
et
al
., 2007).
These observations go in line with recent work from other
laboratories indicating that short telomeres provoke organismal
aging through a p21-dependent induction of cellular senescence
(Choudhury
et
al
., 2007; Feldser & Greider, 2007). Similarly,
previous findings in yeast also indicated that MMR deficiency
increases the survival of telomerase-deficient cells (Rizki &
Lundblad, 2001). MMR deficiency may rescue proliferative
defects in telomerase-deficient cells, this proposed mechanism
involves the activation of recombination-based telomere
elongation mechanisms associated with MMR deficiency the
so-called alternative lengthening of telomeres (ALT) (Rizki &
Lundblad, 2001; Bechter
et
al
., 2004). However, previous
observations from the study of PMS2- and MSH2-deficient mice
suggest that MMR defects do not lead to an increased telomere
length or telomere recombination (Campbell
et
al
., 2006; Siegl-
Cachedenier
et
al
., 2007).
Here, we demonstrate that, in the context of a telomerase-
deficient mouse background, MSH2 abrogation does not impact
on telomere length homeostasis. The work described here
strongly supports instead our previously proposed notion that
MMR pathway plays an important role in the cellular response
to telomere dysfunction by signaling a p21/p53-dependent cell-
cycle arrest in cells with critically short telomeres (Siegl-Cachedenier
et
al
., 2007). MSH2 deficiency was sufficient to abolish the
tumor suppressor effect of short telomeres, in marked contrast
to PMS2-deficiency, arguing that MSH2 is a more potent blocker
of the cell cycle in response to short telomeres than PMS2.
MSH2 deficiency is the third genetic scenario described to date
in which short telomeres fail to work as tumor suppressors,
along with p53 deficiency and TRF2 overexpression (Chin
et
al
.,
1999; Blanco
et
al
., 2007).
Results
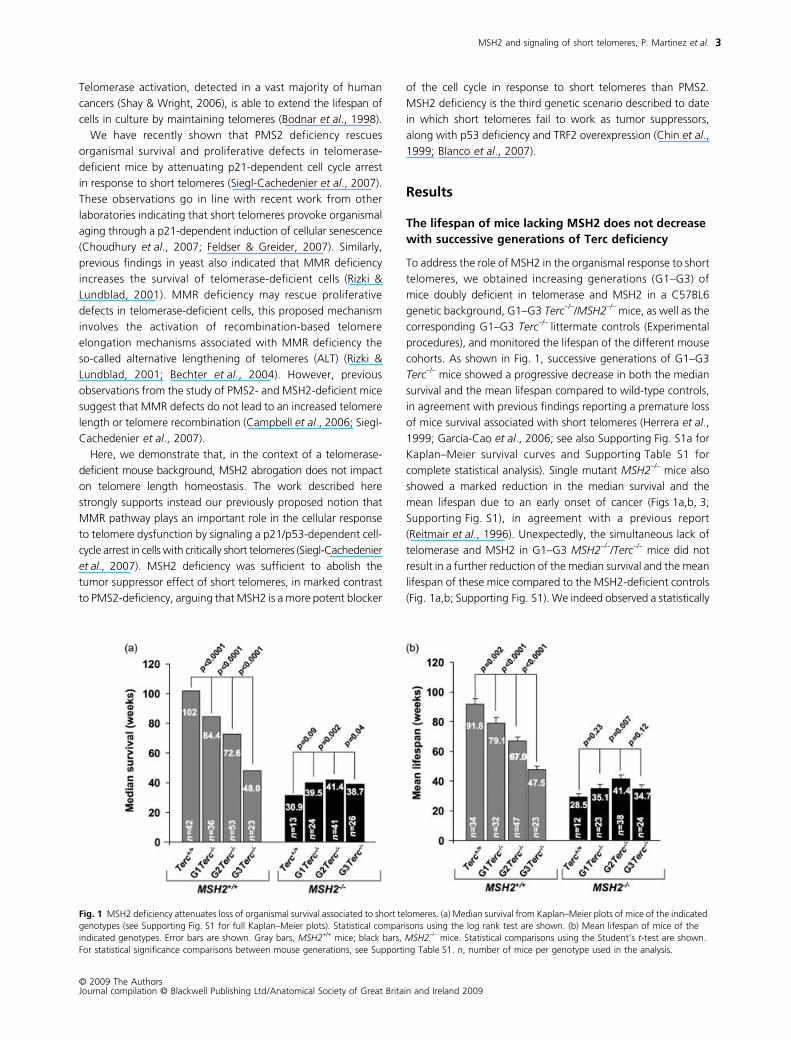
The lifespan of mice lacking MSH2 does not decrease with successive generations of Terc deficiency
To address the role of MSH2 in the organismal response to short
telomeres, we obtained increasing generations (G1–G3) of
mice doubly deficient in telomerase and MSH2 in a C57BL6
genetic background, G1–G3
Terc
–/–
/MSH2
–/–
mice, as well as the
corresponding G1–G3
Terc
–/–
littermate controls (Experimental
procedures), and monitored the lifespan of the different mouse
cohorts. As shown in Fig. 1, successive generations of G1–G3
Terc
–/–
mice showed a progressive decrease in both the median
survival and the mean lifespan compared to wild-type controls,
in agreement with previous findings reporting a premature loss
of mice survival associated with short telomeres (Herrera
et
al
.,
1999; García-Cao
et
al
., 2006; see also Supporting Fig. S1a for
Kaplan–Meier survival curves and Supporting Table S1 for
complete statistical analysis). Single mutant
MSH2
–/–
mice also
showed a marked reduction in the median survival and the
mean lifespan due to an early onset of cancer (Figs 1a,b, 3;
Supporting Fig. S1), in agreement with a previous report
(Reitmair
et
al
., 1996). Unexpectedly, the simultaneous lack of
telomerase and MSH2 in G1–G3
MSH2
–/–
/Terc
–/–
mice did not
result in a further reduction of the median survival and the mean
lifespan of these mice compared to the MSH2-deficient controls
(Fig. 1a,b; Supporting Fig. S1). We indeed observed a statistically
Fig. 1 MSH2 deficiency attenuates loss of organismal survival associated to short telomeres. (a) Median survival from Kaplan–Meier plots of mice of the indicated genotypes (see Supporting Fig. S1 for full Kaplan–Meier plots). Statistical comparisons using the log rank test are shown. (b) Mean lifespan of mice of the indicated genotypes. Error bars are shown. Gray bars, MSH2+/+ mice; black bars, MSH2–/– mice. Statistical comparisons using the Student’s t-test are shown. For statistical significance comparisons between mouse generations, see Supporting Table S1. n, number of mice per genotype used in the analysis.

MSH2 and signaling of short telomeres, P. Martinez
et al.
© 2009 The AuthorsJournal compilation © Blackwell Publishing Ltd/Anatomical Society of Great Britain and Ireland 2009
4
significant increase in the median survival when comparing
G2 and G3
MSH2
–/–
/Terc
–/–
mice to the MSH2-deficient
Terc
+/+
controls (Fig. 1a,b; Supporting Fig. S1; Supporting Table S1 for
complete statistical significance analysis). These results suggest
that telomerase deficiency partially ameliorates the MSH2-
associated loss of organismal survival.
MSH2 abrogation rescues aging pathologies associated to short telomeres
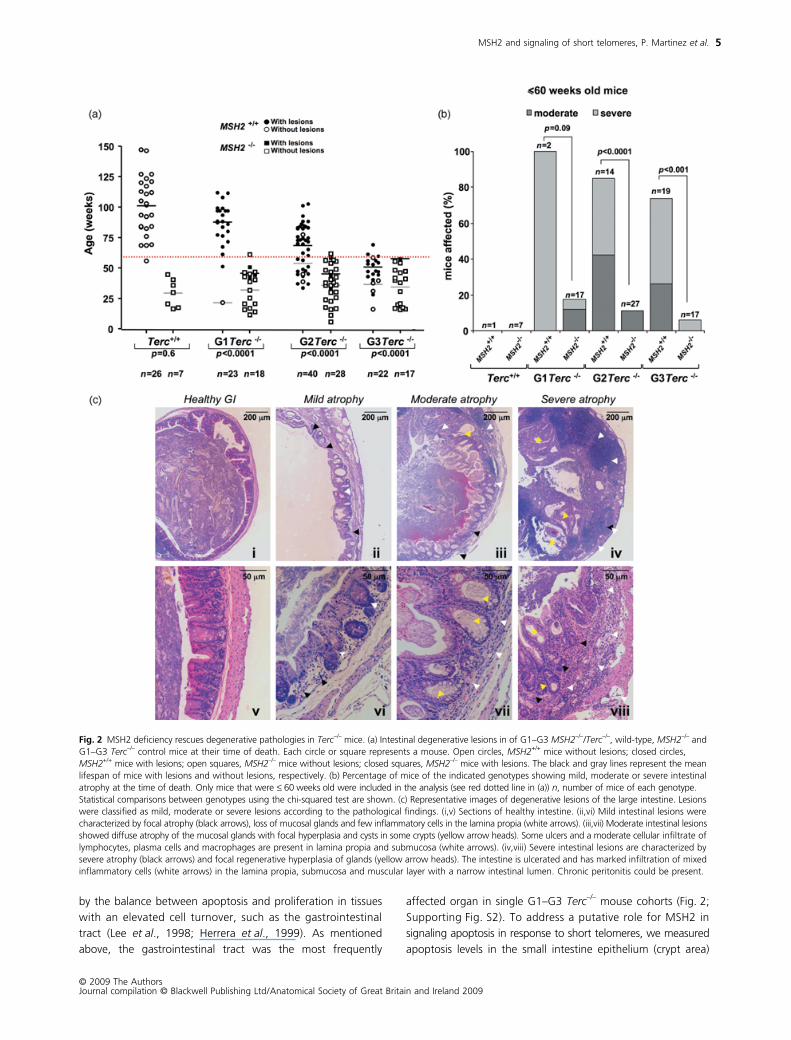
We performed next a thorough histopathological analysis to
determine the cause of death of the different mouse cohorts
(Experimental procedures). The most frequent and life-
threatening degenerative lesions associated to short telomeres in
G1–G3 Terc−/– mice are the atrophies of the intestinal epithelia;
they contribute to the early mortality of these mice (Herrera
et al., 1999; García-Cao et al., 2006; Siegl-Cachedenier et al.,2007). As illustrated in Fig. 2a, the age of onset of these lesions
progressively anticipates with increasing Terc–/– generations (see
also Fig. 2b,c for a comparison between mouse cohorts of a
similar age range at their time of death, as well as for some
representative images of the lesions). These lesions, however,
were largely absent in both wild-type and single MSH2-deficient
mice at the time of their death (Fig. 2a–c). Strikingly, the per-
centage of G1–G3 MSH2–/–/Terc–/– mice showing intestinal
degenerative lesions was dramatically reduced compared to
their G1–G3 Terc–/– controls (p < 0.001 for all comparisons)
(Fig. 2a–c). Furthermore, when we considered the whole set of
degenerative lesions in the different organs of these mice,
we observed a similar effect (Supporting Fig. S2). When we
analyzed mouse cohorts that were ≤ 60 weeks old at the time
of their death, we obtained similar results, ruling out an age-
dependent effect in the protection from degenerative
pathologies associated to MSH2 deficiency (Fig. 2b).
Indeed, very few animals deficient in MSH2 display intestinal
atrophy regardless of Terc–/– generation status (G1, G2 or G3),
while late-generation Terc–/– exhibit an early onset and high
incidence of intestinal atrophy (Fig. 2b). Finally, the rescue of
degenerative pathologies in the G1–G3 MSH2–/–/Terc–/– mice
correlated with the observed attenuation of the low body
weight phenotype in mice with dysfunctional telomeres (data
not shown). Altogether, these results indicate that MSH2
deficiency rescues degenerative pathologies associated with
increasing generations of telomerase-deficient mice.
MSH2 abrogation largely abolishes the tumor suppressor activity of short telomeres
Premature death of MSH2-deficient mice was associated to an
early onset of malignant tumors (Fig. 3a), in agreement with the
findings of previous reports (Reitmair et al., 1995; de Wind
et al., 1995). In particular, MSH2–/– mice presented a very high
incidence of lymphoma, which affected 67% of the mouse
cohort compared to less than 40% of wild-type controls bearing
this tumor at the time of their death. To rule out an age-
dependent effect, we analyzed tumor incidence in mouse
cohorts that were ≤ 60 weeks old at the time of their death
(Fig. 3b). In particular, MSH2–/– mice die of tumors and the same
trend is observed in different generations MSH2–/–Terc–/– mice
within the same age range (Fig. 3b). In marked contrast, single
Terc-deficient mice displayed a progressively decreased cancer
incidence with increasing mice generations (Fig. 3a,b), which is
in agreement with the tumor suppressor activity of short
telomeres and telomerase deficiency when in a p53-proficient
genetic background (reviewed in Gonzalez-Suarez et al.,2000; Blasco & Hahn, 2003). Indeed, when we compared
age-matched mice from 60 to 100 weeks old, we observed that
50% of wild-type mice (n = 10) developed cancer whereas none
of the G2 MSH2+/+Terc–/– mice (n = 24) showed malignant
tumors at the time of their death. Strikingly, the tumor suppressor
activity of short telomeres was completely abolished in successive
generations of Terc–/–/MSH2–/– mice, with 82% of G3 Terc–/–/MSH2–/– mice succumbing to malignant tumors compared to
less than 20% of G3 Terc–/– controls (Fig. 3b). Again, the age
at the time of death of these two mouse cohorts was very similar
(Figs 1a,b and 3a), ruling out an age-dependent effect on cancer
incidence in these mice. The fact that a higher percentage of
G3 Terc–/–/MSH2–/– mice developed tumors compared to wild-type
animals (82% and 67%, respectively) suggests that telomere
dysfunction may cooperate with Msh2 deficiency to induce
tumor formation (Fig. 3b). In summary, mice harboring a MSH2deletion showed a high incidence of cancer regardless of the
Terc genotype, indicating that the tumor suppressor effect of
telomerase deficiency and short telomeres was fully blocked by
the MSH2 null mutation.
Of note, these results are in marked contrast to those
previously described for G1–G3 Terc–/–/PMS2–/– mice, which lack
a different MMR component: the PMS2 protein (Siegl-Cachedenier
et al., 2007). In particular, progressive telomere shortening in
G1–G3 Terc–/–/PMS2–/– mice effectively protected against PMS2-
induced cancer. This marked difference in cancer behavior
between PMS2- and MSH2-deficient mice suggests that MSH2
deletion has a stronger tumorigenic effect than PMS2 deficiency
(see Discussion). Finally, the fact that short telomeres cannot
suppress the cancer-prone phenotype associated with MSH2
deficiency explains that the median lifespan and survival of
MSH2–/–/Terc–/– mice are not prolonged compared to Terc–/–
cohorts despite their marked reduction in degenerative
pathologies. Therefore, while MSH2 null mutation could
rescue the premature aging phenotypes of Terc–/– mice,
lack of telomerase activity, however, could not exert its
well-known tumor suppressor effect in the context of MSH2
deficiency.
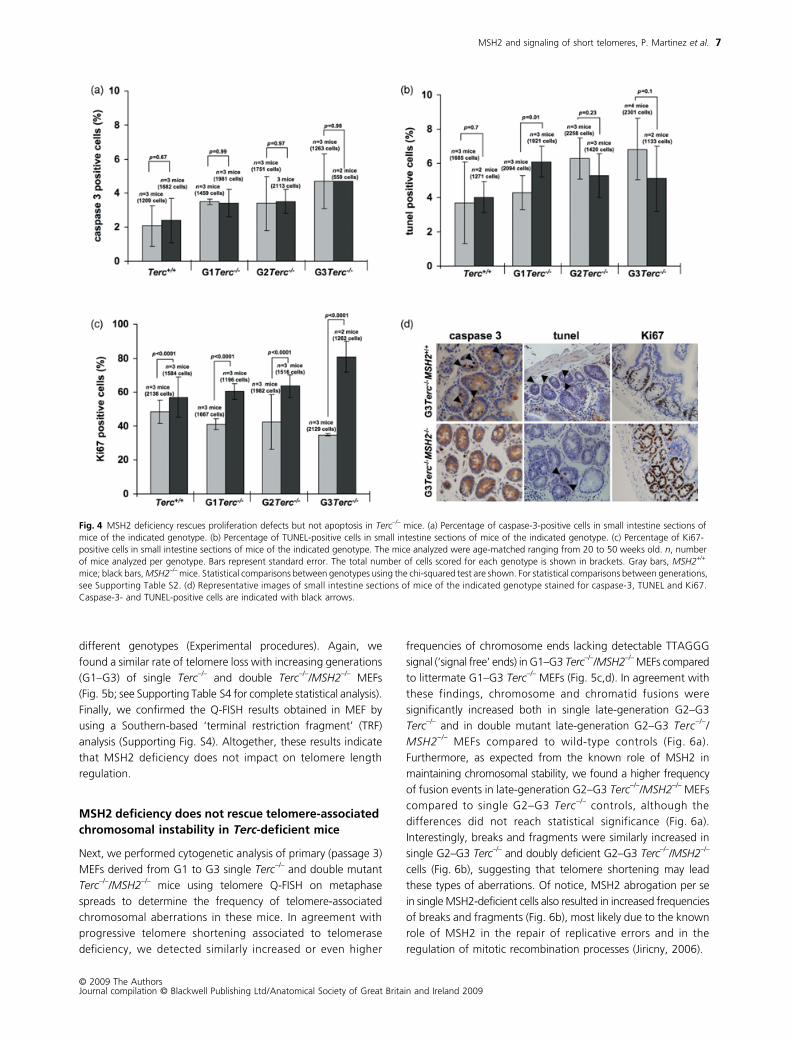
MSH2 abrogation rescues cell-cycle arrest but not apoptosis provoked by short telomeres
Decreased survival of late-generation Terc-deficient mice has
been previously shown to be the consequence of a decreased
tissue renewal capacity, which in turn is thought to be determined
MSH2 and signaling of short telomeres, P. Martinez et al.
© 2009 The AuthorsJournal compilation © Blackwell Publishing Ltd/Anatomical Society of Great Britain and Ireland 2009
5
by the balance between apoptosis and proliferation in tissues
with an elevated cell turnover, such as the gastrointestinal
tract (Lee et al., 1998; Herrera et al., 1999). As mentioned
above, the gastrointestinal tract was the most frequently
affected organ in single G1–G3 Terc–/– mouse cohorts (Fig. 2;
Supporting Fig. S2). To address a putative role for MSH2 in
signaling apoptosis in response to short telomeres, we measured
apoptosis levels in the small intestine epithelium (crypt area)
Fig. 2 MSH2 deficiency rescues degenerative pathologies in Terc–/– mice. (a) Intestinal degenerative lesions in of G1–G3 MSH2–/–/Terc–/–, wild-type, MSH2–/– and G1–G3 Terc–/– control mice at their time of death. Each circle or square represents a mouse. Open circles, MSH2+/+ mice without lesions; closed circles, MSH2+/+ mice with lesions; open squares, MSH2–/– mice without lesions; closed squares, MSH2–/– mice with lesions. The black and gray lines represent the mean lifespan of mice with lesions and without lesions, respectively. (b) Percentage of mice of the indicated genotypes showing mild, moderate or severe intestinal atrophy at the time of death. Only mice that were ≤ 60 weeks old were included in the analysis (see red dotted line in (a)) n, number of mice of each genotype. Statistical comparisons between genotypes using the chi-squared test are shown. (c) Representative images of degenerative lesions of the large intestine. Lesions were classified as mild, moderate or severe lesions according to the pathological findings. (i,v) Sections of healthy intestine. (ii,vi) Mild intestinal lesions were characterized by focal atrophy (black arrows), loss of mucosal glands and few inflammatory cells in the lamina propia (white arrows). (iii,vii) Moderate intestinal lesions showed diffuse atrophy of the mucosal glands with focal hyperplasia and cysts in some crypts (yellow arrow heads). Some ulcers and a moderate cellular infiltrate of lymphocytes, plasma cells and macrophages are present in lamina propia and submucosa (white arrows). (iv,viii) Severe intestinal lesions are characterized by severe atrophy (black arrows) and focal regenerative hyperplasia of glands (yellow arrow heads). The intestine is ulcerated and has marked infiltration of mixed inflammatory cells (white arrows) in the lamina propia, submucosa and muscular layer with a narrow intestinal lumen. Chronic peritonitis could be present.

MSH2 and signaling of short telomeres, P. Martinez et al.
© 2009 The AuthorsJournal compilation © Blackwell Publishing Ltd/Anatomical Society of Great Britain and Ireland 2009
6
using two independent techniques: caspase-3 immunohisto-
chemistry and TUNEL staining (Fig. 4a,b,d) (Experimental
procedures). As expected, G1–G3 Terc-deficient mice showed
increased apoptosis in the gastrointestinal tract compared to
wild-type controls (Fig. 4a,b,d; see Supporting Table S2 for
complete statistical significance analysis). Interestingly, the
frequency of apoptotic cells was indistinguishable between G1–
G3 Terc–/–/MSH2–/– mice and their corresponding single G1–G2
Terc–/– controls (Fig. 4a,b,d; see Supporting Table S2 for complete
statistical significance analysis), arguing that the rescue of
degenerative pathologies associated to MSH2 abrogation is not
due to decreased apoptosis in Terc–/–/MSH2-–/– mice compared
to the single Terc–/– counterparts.
We determined next the proliferative index of the gastro-
intestinal tract epithelium in mice of the different genotypes
(crypt area) by performing immunohistochemistry with the Ki67
mitotic marker (Experimental procedures). Successive generations
of telomerase-deficient mice presented a lower percentage
of proliferating cells in the gastrointestinal tract compared
to wild-type controls, in agreement with their higher incidence
of intestinal degenerative pathologies (Fig. 4c,d; see Supporting
Table S2 for complete statistical significance analysis). Single
MSH2-deficient mice showed significantly higher proliferation
rates compared to wild-type mice (Fig. 4c,d; see Supporting
Table S2 for complete statistical significance analysis).
Importantly, MSH2 abrogation in G1–G3 Terc–/–/MSH2–/– mice
significantly rescued proliferative defects in the gastrointestinal
tract epithelium of these mice compared to single G1–G3 Terc-
deficient controls (p ≤ 0.0001 for all comparisons; Fig 4c,d;
see Supporting Table S2 for complete statistical significance
analysis). This was particularly striking in the case of G3 Terc–/–/MSH2–/– mice, where 80% of the gastrointestinal tract epithelium
cells were positive for Ki67 compared to less than 40% in the
single G3 Terc–/– controls. Altogether, these results indicate that
MSH2 deficiency resumes proliferation of cells from Terc–/–/MSH2–/– mice, resulting in a rescue of degenerative pathologies
in these mice.
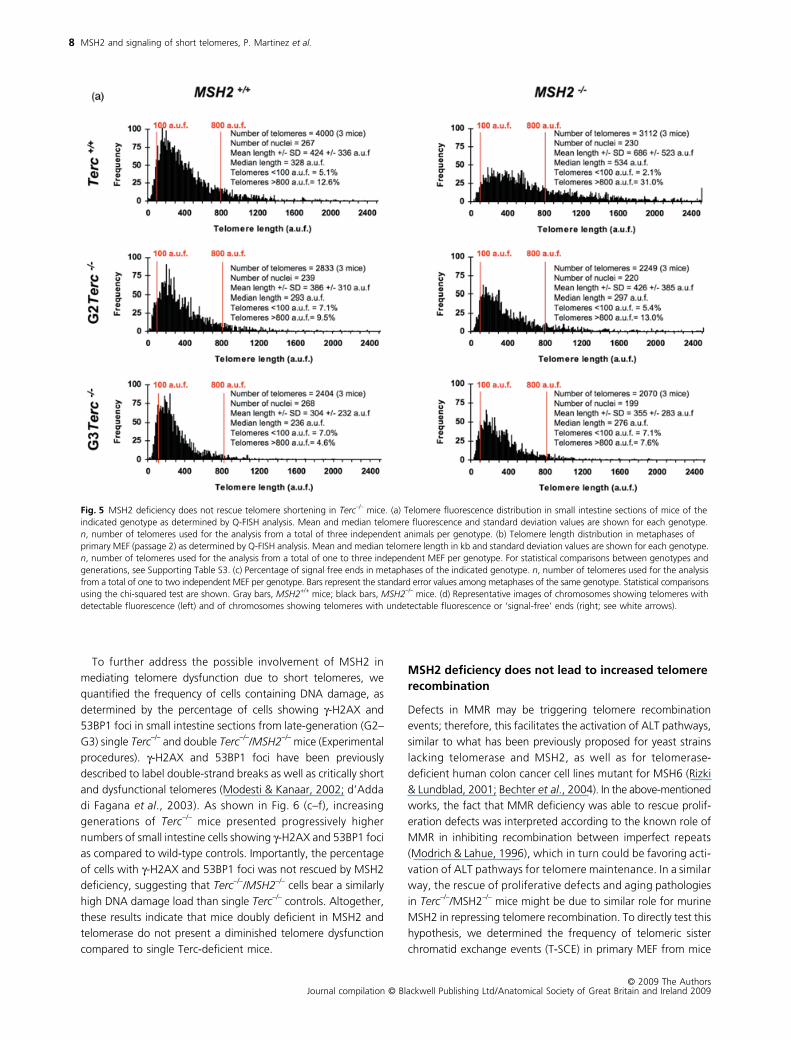
MSH2 deficiency does not prevent telomere shortening in Terc–/– mice
Given the well-known role of MMR as a repressor of recombi-
nation processes (Modrich, 1994; Jiricny, 2006), we next addressed
whether MSH2 deficiency could be rescuing telomere length and
telomere capping defects associated to Terc deficiency by increasing
recombination between telomeric sequences. To address this,
we first measured telomere length directly on small intestine
sections using quantitative fluorescence in situ hybridization
(Q-FISH; Experimental procedures). Single mutant Terc–/– and
double mutant Terc–/–/MSH2–/– mice showed similar shortening
of telomeres and frequency of critically short telomeres associated
to successive mouse generations (G2–G3) in the gastrointestinal
tract epithelium (Fig. 5a; see Supporting Fig. S3 and Table S3 for
statistical analysis), indicating that the rescue of gastrointestinal
tract degenerative pathologies in G1–G3 Terc–/–/MSH2–/– mice
is not associated to a rescue of short telomeres in these mice.
To obtain more robust and extensive data of telomere length
frequencies on individual chromosomes, we next determined
telomere length by Q-FISH on metaphase spreads from primary
(passage 3) littermate mouse embryonic fibroblasts (MEF) of the
Fig. 3 MSH2 abrogation abolishes the tumor suppression effect of Terc deficiency. (a) Incidence of malignant tumors (lymphoma and carcinoma) in increasing generations of MSH2–/–/Terc–/–, wild-type, MSH2–/– and G1–G3 Terc–/– control mice at their time of death. Open circles, MSH2+/+ mice without tumors; closed circles, MSH2+/+ mice with tumors; open squares, MSH2–/– mice without tumors; closed squares, MSH2–/– mice with tumors. The black and gray lines represent the mean lifespan of mice with and without tumors, respectively. (b) Percentage of mice of the indicated genotype showing lymphoma and carcinoma tumors at their time of death. Only mice that were ≤ 60 weeks old were included in the analysis (see red dotted line in part a). n, number of mice of each genotype. Statistical comparisons between genotypes using the chi-squared test are shown.
MSH2 and signaling of short telomeres, P. Martinez et al.
© 2009 The AuthorsJournal compilation © Blackwell Publishing Ltd/Anatomical Society of Great Britain and Ireland 2009
7
different genotypes (Experimental procedures). Again, we
found a similar rate of telomere loss with increasing generations
(G1–G3) of single Terc–/– and double Terc–/–/MSH2–/– MEFs
(Fig. 5b; see Supporting Table S4 for complete statistical analysis).
Finally, we confirmed the Q-FISH results obtained in MEF by
using a Southern-based ‘terminal restriction fragment’ (TRF)
analysis (Supporting Fig. S4). Altogether, these results indicate
that MSH2 deficiency does not impact on telomere length
regulation.
MSH2 deficiency does not rescue telomere-associated chromosomal instability in Terc-deficient mice
Next, we performed cytogenetic analysis of primary (passage 3)
MEFs derived from G1 to G3 single Terc–/– and double mutant
Terc–/–/MSH2–/– mice using telomere Q-FISH on metaphase
spreads to determine the frequency of telomere-associated
chromosomal aberrations in these mice. In agreement with
progressive telomere shortening associated to telomerase
deficiency, we detected similarly increased or even higher
frequencies of chromosome ends lacking detectable TTAGGG
signal (‘signal free’ ends) in G1–G3 Terc–/–/MSH2–/– MEFs compared
to littermate G1–G3 Terc–/– MEFs (Fig. 5c,d). In agreement with
these findings, chromosome and chromatid fusions were
significantly increased both in single late-generation G2–G3
Terc–/– and in double mutant late-generation G2–G3 Terc–/–/MSH2–/– MEFs compared to wild-type controls (Fig. 6a).
Furthermore, as expected from the known role of MSH2 in
maintaining chromosomal stability, we found a higher frequency
of fusion events in late-generation G2–G3 Terc–/–/MSH2–/– MEFs
compared to single G2–G3 Terc–/– controls, although the
differences did not reach statistical significance (Fig. 6a).
Interestingly, breaks and fragments were similarly increased in
single G2–G3 Terc–/– and doubly deficient G2–G3 Terc–/–/MSH2–/–
cells (Fig. 6b), suggesting that telomere shortening may lead
these types of aberrations. Of notice, MSH2 abrogation per se
in single MSH2-deficient cells also resulted in increased frequencies
of breaks and fragments (Fig. 6b), most likely due to the known
role of MSH2 in the repair of replicative errors and in the
regulation of mitotic recombination processes (Jiricny, 2006).
Fig. 4 MSH2 deficiency rescues proliferation defects but not apoptosis in Terc−/– mice. (a) Percentage of caspase-3-positive cells in small intestine sections of mice of the indicated genotype. (b) Percentage of TUNEL-positive cells in small intestine sections of mice of the indicated genotype. (c) Percentage of Ki67-positive cells in small intestine sections of mice of the indicated genotype. The mice analyzed were age-matched ranging from 20 to 50 weeks old. n, number of mice analyzed per genotype. Bars represent standard error. The total number of cells scored for each genotype is shown in brackets. Gray bars, MSH2+/+ mice; black bars, MSH2–/– mice. Statistical comparisons between genotypes using the chi-squared test are shown. For statistical comparisons between generations, see Supporting Table S2. (d) Representative images of small intestine sections of mice of the indicated genotype stained for caspase-3, TUNEL and Ki67. Caspase-3- and TUNEL-positive cells are indicated with black arrows.
MSH2 and signaling of short telomeres, P. Martinez et al.
© 2009 The AuthorsJournal compilation © Blackwell Publishing Ltd/Anatomical Society of Great Britain and Ireland 2009
8
To further address the possible involvement of MSH2 in
mediating telomere dysfunction due to short telomeres, we
quantified the frequency of cells containing DNA damage, as
determined by the percentage of cells showing γ-H2AX and
53BP1 foci in small intestine sections from late-generation (G2–
G3) single Terc–/– and double Terc–/–/MSH2–/– mice (Experimental
procedures). γ-H2AX and 53BP1 foci have been previously
described to label double-strand breaks as well as critically short
and dysfunctional telomeres (Modesti & Kanaar, 2002; d’Adda
di Fagana et al., 2003). As shown in Fig. 6 (c–f), increasing
generations of Terc–/– mice presented progressively higher
numbers of small intestine cells showing γ-H2AX and 53BP1 foci
as compared to wild-type controls. Importantly, the percentage
of cells with γ-H2AX and 53BP1 foci was not rescued by MSH2
deficiency, suggesting that Terc–/–/MSH2–/– cells bear a similarly
high DNA damage load than single Terc–/– controls. Altogether,
these results indicate that mice doubly deficient in MSH2 and
telomerase do not present a diminished telomere dysfunction
compared to single Terc-deficient mice.
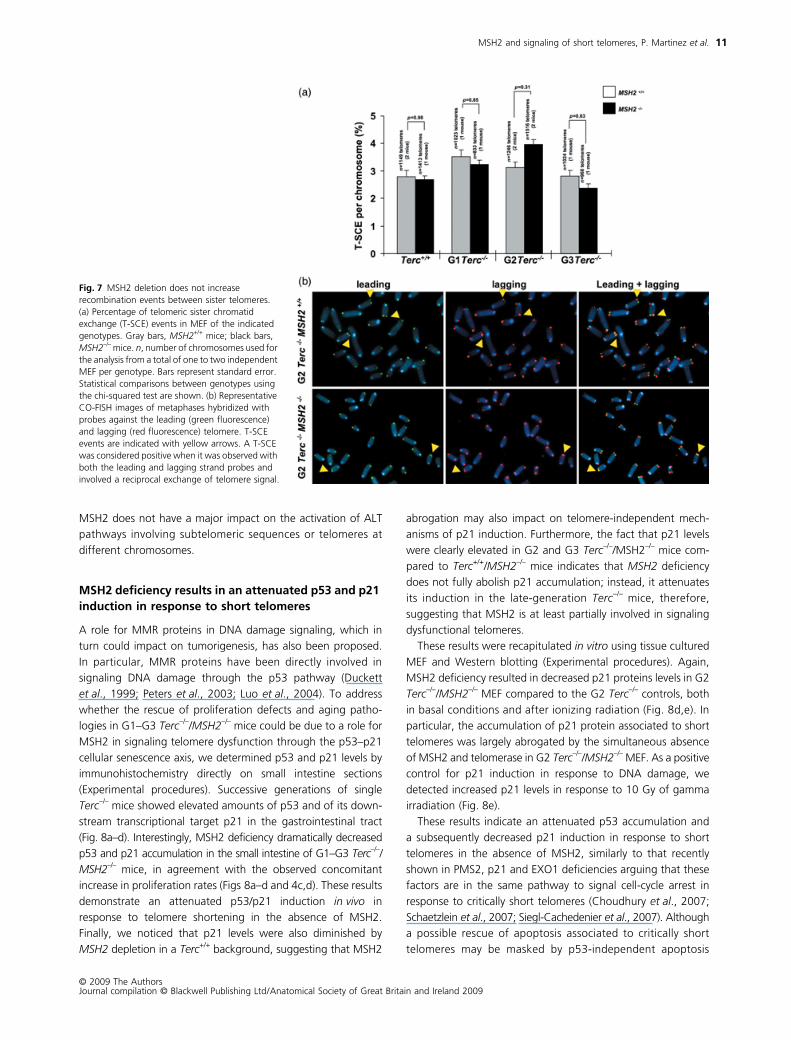
MSH2 deficiency does not lead to increased telomere recombination
Defects in MMR may be triggering telomere recombination
events; therefore, this facilitates the activation of ALT pathways,
similar to what has been previously proposed for yeast strains
lacking telomerase and MSH2, as well as for telomerase-
deficient human colon cancer cell lines mutant for MSH6 (Rizki
& Lundblad, 2001; Bechter et al., 2004). In the above-mentioned
works, the fact that MMR deficiency was able to rescue prolif-
eration defects was interpreted according to the known role of
MMR in inhibiting recombination between imperfect repeats
(Modrich & Lahue, 1996), which in turn could be favoring acti-
vation of ALT pathways for telomere maintenance. In a similar
way, the rescue of proliferative defects and aging pathologies
in Terc–/–/MSH2–/– mice might be due to similar role for murine
MSH2 in repressing telomere recombination. To directly test this
hypothesis, we determined the frequency of telomeric sister
chromatid exchange events (T-SCE) in primary MEF from mice
Fig. 5 MSH2 deficiency does not rescue telomere shortening in Terc–/– mice. (a) Telomere fluorescence distribution in small intestine sections of mice of the indicated genotype as determined by Q-FISH analysis. Mean and median telomere fluorescence and standard deviation values are shown for each genotype. n, number of telomeres used for the analysis from a total of three independent animals per genotype. (b) Telomere length distribution in metaphases of primary MEF (passage 2) as determined by Q-FISH analysis. Mean and median telomere length in kb and standard deviation values are shown for each genotype. n, number of telomeres used for the analysis from a total of one to three independent MEF per genotype. For statistical comparisons between genotypes and generations, see Supporting Table S3. (c) Percentage of signal free ends in metaphases of the indicated genotype. n, number of telomeres used for the analysis from a total of one to two independent MEF per genotype. Bars represent the standard error values among metaphases of the same genotype. Statistical comparisons using the chi-squared test are shown. Gray bars, MSH2+/+ mice; black bars, MSH2–/– mice. (d) Representative images of chromosomes showing telomeres with detectable fluorescence (left) and of chromosomes showing telomeres with undetectable fluorescence or ‘signal-free’ ends (right; see white arrows).

MSH2 and signaling of short telomeres, P. Martinez et al.
© 2009 The AuthorsJournal compilation © Blackwell Publishing Ltd/Anatomical Society of Great Britain and Ireland 2009
9
of the different genotypes using two-color chromosome orien-
tation FISH (CO-FISH) (Bailey et al., 2004; Gonzalo et al., 2006).
The strand-specific nature of the CO-FISH yields typically two
telomeric signals of each color (red: lagging; green: leading) per
chromosome in the absence of recombination events. A sister
chromatid exchange within telomeric DNA (T-SCE) leads to the
mixture of red and green fluorescent signals. We counted as
positive T-SCE events only those that were detected with both
the leading and the lagging telomere probes. T-SCE frequencies
in the double mutant Terc–/–/MSH2–/– mice were not significantly
different from those of the single Terc–/– controls (Fig. 7a,b),
suggesting that an increased telomere recombination between
sister telomeres is unlikely to be responsible for the higher pro-
liferation capacity observed in these mice. These results are also
in agreement with the fact that Terc–/–/MSH2–/– mice did not
show either a lower incidence of chromosomes lacking telomere
Fig. 5 Continued

MSH2 and signaling of short telomeres, P. Martinez et al.
© 2009 The AuthorsJournal compilation © Blackwell Publishing Ltd/Anatomical Society of Great Britain and Ireland 2009
10
signals or a significant rescue of telomere-associated chromo-
somal aberrations (Figs 5d,e and 6). However, we cannot rule
out subtle differences because in these analyses we examined
only one to two mice per genotype and generation. It should
also be pointed out that CO-FISH allows for the detection of
recombination events only between sister telomeres but not
between sister subtelomeric repeats or between telomeres
located at different chromosomes. The fact that telomere length
and the frequency of H2AX and 53BP1-positive cells is not
significantly rescued by MSH2 deficiency argues, however, that
Fig. 6 Telomere dysfunction and chromosomal instability in MSH2/Terc-deficient cells. (a) Frequency of fusions events and (b) breaks and fragments events in primary MEF metaphases of the indicated genotypes. End-to-end fusion events lacking (– TTAGGG) or not (+ TTAGGG) telomeric signals at the fusion point are indicated. n, number of metaphases used for the analysis from a total of two to four independent MEF per genotype. Bars represent standard error. Statistical comparisons using the chi-squared test are shown. (c) Percentage of γ-H2AX- and (d) 53BP1-positive cells in small intestine sections of mice of the indicated genotype. n, number of mice analyzed per genotype. The total number of cells scored is indicated. Bars represent standard error. Statistical comparisons using the chi-squared test are shown. Gray bars, MSH2+/+ mice; black bars, MSH2−/– mice. For statistical comparisons between generations, see Supporting Table S2. (e) Representative images of γ-H2AX- and (f) 53BP1-positive cells (upper panel) and of the DAPI combined-images (lower panel) in small intestine sections of G3Terc–/–MSH2+/+ and G3Terc–/–MSH2–/–mice.
MSH2 and signaling of short telomeres, P. Martinez et al.
© 2009 The AuthorsJournal compilation © Blackwell Publishing Ltd/Anatomical Society of Great Britain and Ireland 2009
11
MSH2 does not have a major impact on the activation of ALT
pathways involving subtelomeric sequences or telomeres at
different chromosomes.
MSH2 deficiency results in an attenuated p53 and p21 induction in response to short telomeres
A role for MMR proteins in DNA damage signaling, which in
turn could impact on tumorigenesis, has also been proposed.
In particular, MMR proteins have been directly involved in
signaling DNA damage through the p53 pathway (Duckett
et al., 1999; Peters et al., 2003; Luo et al., 2004). To address
whether the rescue of proliferation defects and aging patho-
logies in G1–G3 Terc–/–/MSH2–/– mice could be due to a role for
MSH2 in signaling telomere dysfunction through the p53–p21
cellular senescence axis, we determined p53 and p21 levels by
immunohistochemistry directly on small intestine sections
(Experimental procedures). Successive generations of single
Terc–/– mice showed elevated amounts of p53 and of its down-
stream transcriptional target p21 in the gastrointestinal tract
(Fig. 8a–d). Interestingly, MSH2 deficiency dramatically decreased
p53 and p21 accumulation in the small intestine of G1–G3 Terc–/–/MSH2–/– mice, in agreement with the observed concomitant
increase in proliferation rates (Figs 8a–d and 4c,d). These results
demonstrate an attenuated p53/p21 induction in vivo in
response to telomere shortening in the absence of MSH2.
Finally, we noticed that p21 levels were also diminished by
MSH2 depletion in a Terc+/+ background, suggesting that MSH2
abrogation may also impact on telomere-independent mech-
anisms of p21 induction. Furthermore, the fact that p21 levels
were clearly elevated in G2 and G3 Terc–/–/MSH2–/– mice com-
pared to Terc+/+/MSH2–/– mice indicates that MSH2 deficiency
does not fully abolish p21 accumulation; instead, it attenuates
its induction in the late-generation Terc–/– mice, therefore,
suggesting that MSH2 is at least partially involved in signaling
dysfunctional telomeres.
These results were recapitulated in vitro using tissue cultured
MEF and Western blotting (Experimental procedures). Again,
MSH2 deficiency resulted in decreased p21 proteins levels in G2
Terc–/–/MSH2–/– MEF compared to the G2 Terc–/– controls, both
in basal conditions and after ionizing radiation (Fig. 8d,e). In
particular, the accumulation of p21 protein associated to short
telomeres was largely abrogated by the simultaneous absence
of MSH2 and telomerase in G2 Terc–/–/MSH2–/– MEF. As a positive
control for p21 induction in response to DNA damage, we
detected increased p21 levels in response to 10 Gy of gamma
irradiation (Fig. 8e).
These results indicate an attenuated p53 accumulation and
a subsequently decreased p21 induction in response to short
telomeres in the absence of MSH2, similarly to that recently
shown in PMS2, p21 and EXO1 deficiencies arguing that these
factors are in the same pathway to signal cell-cycle arrest in
response to critically short telomeres (Choudhury et al., 2007;
Schaetzlein et al., 2007; Siegl-Cachedenier et al., 2007). Although
a possible rescue of apoptosis associated to critically short
telomeres may be masked by p53-independent apoptosis
Fig. 7 MSH2 deletion does not increase recombination events between sister telomeres. (a) Percentage of telomeric sister chromatid exchange (T-SCE) events in MEF of the indicated genotypes. Gray bars, MSH2+/+ mice; black bars, MSH2–/– mice. n, number of chromosomes used for the analysis from a total of one to two independent MEF per genotype. Bars represent standard error. Statistical comparisons between genotypes using the chi-squared test are shown. (b) Representative CO-FISH images of metaphases hybridized with probes against the leading (green fluorescence) and lagging (red fluorescence) telomere. T-SCE events are indicated with yellow arrows. A T-SCE was considered positive when it was observed with both the leading and lagging strand probes and involved a reciprocal exchange of telomere signal.

MSH2 and signaling of short telomeres, P. Martinez et al.
© 2009 The AuthorsJournal compilation © Blackwell Publishing Ltd/Anatomical Society of Great Britain and Ireland 2009
12
associated to severe chromosomal instability in MSH2/Terc
doubly deficient mice, previous findings using p21/Terc and
PMS2/Terc doubly deficient mice also showed that, at least in
the gastrointestinal tract, cell-cycle arrest is predominant over
apoptosis in eliciting age-related pathologies and decreasing
survival in telomerase-deficient mice (Choudhury et al., 2007;
Schaetzlein et al., 2007; Siegl-Cachedenier et al., 2007). We do
not rule out, however, that a different scenario could be observed
in other tissues.
Discussion
The data presented here strongly support a role for the MSH2
MMR gene in mediating cell-cycle arrest in response to short
and dysfunctional telomeres in the context of the organism,
which in turn impacts on mouse aging. In particular, MSH2
deficiency is able to prevent degenerative pathologies in
telomerase-deficient mice by attenuating a p53/p21-dependent
cell-cycle arrest in response to critically short telomeres. A similar
role has been recently proposed for the PMS2 MMR factor
(Siegl-Cachedenier et al., 2007). Furthermore, these findings go
in line with recent reports showing that abrogation of additional
factors, which are also related to the MMR signaling pathway
(namely, EXO1 and p21), are also able to significantly delay
aging pathologies in telomerase-deficient mice by rescuing
proliferative defects associated to short telomeres (Choudhury
et al., 2007; Schaetzlein et al., 2007). In particular, mice doubly
deficient in Terc and p21 show a rescue of organismal survival
and a rescue of proliferative defects but not of apoptosis, which
is independent of telomere length (Choudhury et al., 2007).
Similarly, Terc–/–/EXO1–/– double mutant mice display an improved
organ maintenance and longer lifespan compared to single
Terc–/– mice (Schaetzlein et al., 2007). As mentioned above,
EXO1 is a downstream player of the MMR pathway through its
Fig. 8 MSH2 is involved in signaling telomere dysfunction. Percentage of (a) p53- and (b) p21-positive cells in small intestine sections of mice of the indicated genotypes. The mice analyzed were age-matched ranging from 20 to 50 weeks old. n, number of mice analyzed per genotype. Bars represent standard error. The total number of cells scored for each genotype is shown in brackets. Gray bars, MSH2+/+ mice; black bars, MSH2–/– mice. Statistical comparisons between genotypes using the chi-squared test are shown. For statistical comparisons between generations, see Supporting Table S2. (c) Representative images of small intestine sections of mice of the indicated genotype stained for p53 and p21. Note the higher amount of p21- and p53-positive cells in G3 Terc–/– single mutant mice compared to G3 Terc–/–/MSH2–/– mice. Intestine sections of p53- and p21-deficient mice were used as negative controls. (d) Quantification of p21 protein levels in MEF (passage 2) of the indicated genotypes before (–) and 18 h after (+) ionizing radiation after normalization to actin levels. n, number of independent MEF per genotype used for the analysis. (e) Representative Western blot images of p21 protein levels in primary MEF. Actin was used as a loading control.

MSH2 and signaling of short telomeres, P. Martinez et al.
© 2009 The AuthorsJournal compilation © Blackwell Publishing Ltd/Anatomical Society of Great Britain and Ireland 2009
13
exonuclease activity (Genschel et al., 2000; Jiricny, 2006; Kadyrov
et al., 2006). Furthermore, EXO1 deletion has been recently shown
to impair the upstream induction of DNA damage responses by
reducing single stranded DNA formation and the recruitment
of replication protein A (RPA) and ATR at DNA breaks. In this
regard, a similar rescue of ATR foci was not observed by MSH2
deletion in successive generations of Terc-deficient mice
(Supporting Fig. S5), arguing that rescue of ATR foci in late-
generation Terc-deficient mice by EXO1 abrogation (Schaetzlein
et al., 2007) may be related to a role of this enzyme in additional
DNA repairs pathways where MMR genes are not involved. In
summary, these results suggest a role for the MMR pathway in
mediating the cellular and organismal response to short telo-
meres, thereby impinging on cancer and aging.
Based on these findings, we would like to propose a specu-
lative model by which MMR may be signaling cellular arrest in
response to critical telomere shortening. It is conceivable that
when telomeres reach a critically short length, annealing of the
single DNA G-strand overhang to its complementary 3′–5′strand during T-loop formation (de Lange, 2005) may be forced
to take place at the vicinity of subtelomeric regions where
degenerated telomeric repeats are found. This would lead to
generation of heteroduplex DNA containing mismatches (i.e.
TTAGGG repeats would anneal to degenerated subtelomeric
repeats), which could then be signaled through components of
the MMR pathway, such as MSH2 and PMS2, as well as through
downstream MMR effectors, such as p21 (Choudhury et al.,2007; Siegl-Cachedenier et al., 2007; this paper). Activation of
the MMR pathway will then induce a DNA damage response
that imposes a p53/p21-dependent cell cycle arrest in the
gastrointestinal tract epithelium in the case of telomerase-
deficient mice. In telomerase-null mice simultaneously deficient
for MMR components (PMS2, MSH2) or for its downstream
effectors (p53, p21), this DNA damage response is attenuated
resulting in normal cell proliferation rates and maintenance of
tissue fitness. At the same time, rescue of cell-cycle arrest may
lead to increased cancer incidence depending on the oncogenic
activity of the MMR mutation. In this regard, MSH2 deletion has
been previously shown to be more potent in inducing tumori-
genesis than PMS2, EXO1 or p21 deletion (Baker et al., 1995;
de Wind et al., 1995; Prolla et al., 1998; Wei et al., 2003), in
agreement with the fact that MSH2 deficiency but not p21,
EXO1 or PMS2 deficiency abolishes the tumor suppressor
activity of short telomeres (Choudhury et al., 2007; Schaetzlein
et al., 2007; Siegl-Cachedenier, 2007; this paper). The tumor
prone phenotype associated to MSH2 deficiency explains that
the survival of MSH2–/–/Terc–/– mice is not prolonged compared
to single Terc–/– mouse counterpart, even though degenerative
pathologies in these mice are greatly prevented. These results
suggest that telomerase inhibitors would not be effective in the
treatment of tumors bearing MSH2 mutations. This situation is
similar to that previously described for Terc–/–/p53–/– mice (Chin
et al., 1999). In both cases, MSH2 and p53 deficiency leads to
rapid and robust tumor development (all mice die with tumors
within the first 50 weeks of life), which is not impaired by
telomerase deficiency and the presence of critically short
telomeres. Indeed, telomere dysfunction accelerates p53-induced
carcinogenesis by promoting chromosomal instability (Artandi
et al., 2000). Similarly, mutations in MSH2 are by far the most
aggressive, leading to tumorigenesis, compared to other MMR
components, in agreement with the fact that MSH2–/– animals
lack all mismatch recognition functions and have a higher
chromosomal instability (de Wind et al., 1995). Of notice,
together with p53 and MSH2 deletions, TRF2 overexpression is
the third genetic scenario, which abolishes the tumor suppressor
activity of short telomeres (Blanco et al., 2007).
Finally, although to the best of our knowledge no direct physical
interaction between MMR components and telomeric DNA has
been reported to date, the telomere-interacting protein WRN
has been described to interact with MMR components. In particular,
when MutSα and MutSβ encounter DNA heteroduplexes,
they recruit the WRN DNA helicase to disrupt the joined DNA
molecule (Saydam et al., 2007). Interestingly, WRN interacts
with TRF2 and can be detected at telomeres (reviewed in
Opresko et al., 2002; Crabbe et al., 2004; Opresko et al., 2004;
de Lange, 2005). In addition, human MutSα has been shown
to bind efficiently to G-quadruplex DNA (Larson et al., 2005),
a DNA structure that is also proposed to form at telomeric
repeats.
In conclusion, the results shown here strongly support an
important role of the MMR pathway in the upstream signaling
events in response to dysfunctional short telomeres and indicate
that at least at the gastrointestinal tract, cell-cycle arrest is
predominant over apoptosis in eliciting age-related pathologies
and decreasing survival in telomerase-deficient mice.
Experimental procedures
Generation of mice and primary mouse cells
To generate MSH2–/–/Terc–/– mice, MSH2–/– mice (Reitmair et al.,1995) were crossed with Terc–/– animals (Blasco et al., 1997) and
the resulting double heterozygous breeding pairs were used to
generate G1 of single Terc–/– and of double Terc–/–/MSH2–/–.
Mouse colonies of successive generations, G2 and G3, were
generated by intercrossing G1 and G2 MSH2+/–/Terc–/– pairs,
respectively. Genotyping was performed as described elsewhere
(Reitmair et al., 1995; Blasco et al., 1997). Mice were maintained
at the Spanish National Cancer Centre under specific-pathogen-
free conditions in accordance with the recommendations of the
Federation of European Laboratory Animal Science Associations.
Primary MEFs were prepared from day 13.5 embryos, as
previously described (Blasco et al., 1997).
Telomere length analysis by Q-FISH
Exponentially growing primary MEFs were incubated with
0.1 μg mL–1 colcemide (Gibco, Paisley, UK) for 4 h at 37 °C and
then fixed in methanol : acetic acid (3 : 1). Q-FISH was performed
as described (Herrera et al., 1999; Samper et al., 2000). To correct

MSH2 and signaling of short telomeres, P. Martinez et al.
© 2009 The AuthorsJournal compilation © Blackwell Publishing Ltd/Anatomical Society of Great Britain and Ireland 2009
14
for lamp intensity and alignment, images from fluorospheres
(fluorescent beads; Molecular Probes, Carlsbad, CA, USA) were
analyzed using the TFL-Telo software (gift from Dr Peter
Lansdorp). Telomere fluorescence values were extrapolated
from the telomere fluorescence of lymphoma cell lines LY-R (R
cells) and LY-S (S cells) with known telomere lengths of 80 and
10 kb, respectively. There was a linear correlation (r2 = 0.999)
between the fluorescence intensity of the R and S telomeres.
We recorded the images using a Cohu CCD camera (San Diego,
CA, USA) on a fluorescence microscope (DMRb, Leica, Wetzlar,
Germany). A mercury vapour lamp (CS 100 W-2; Philips,
Amsterdam, the Netherlands) was used as a source. We captured
the images using the Leica Q-FISH software in a linear acquisi-
tion mode to prevent oversaturation of fluorescence intensity.
We used the TFL-Telo software to quantify the fluorescence
intensity of telomeres from at least 10 metaphases for each data
point (Zijlmans et al., 1997).
For Q-FISH analysis on small intestine sections, de-paraffinated
sections were hybridized with a PNA-telomeric probe and
telomere fluorescence was determined as previously described
(Herrera et al., 1999; Gonzalez-Suarez et al., 2000). More
than 200 nuclei from each genotype were captured at
×100 magnification using a Leica CTR MIC microscope and a
Cohu High Performance CCD camera. Telomere fluorescence
was integrated using spot Integrated Optical Density analysis
in the TFL-TELO program (kindly provided by Dr Landsdorp,
Vancouver, BC, Canada) (Zijlmans et al., 1997).
TRF-based telomere length analysis of MEF
Primary MEF of the indicated genotypes were included in
agarose plugs following instructions provided by the manufacturer
(Bio-Rad, Munich, Germany) and TRF analysis was performed
as previously described (Blasco et al., 1997).
Cytogenetic analysis using telomere Q-FISH on metaphases
For analysis of chromosomal aberrations, at least 50 metaphases
per genotype were analyzed by superimposing the telomere
image on the 4′,6-diamidino-2-phenylindole (DAPI) image using
the TFL-telo software.
Telomere recombination measurements using CO-FISH
Exponentially growing primary MEFs were subcultured in the
presence of 5′-bromo-2′-deoxyuridine (Sigma, St Louis, MO,
USA) at a final concentration of 1 × 10–5 M, and then allowed
to replicate their DNA once at 37 °C for 24 h. Colcemide was
added at a concentration of 1 μg mL–1 during the last 4 h. Cells
were then recovered and metaphases prepared as described
(Samper et al., 2000). CO-FISH was performed as described
(Bailey et al., 2004; Gonzalo et al., 2006) using first a (TTAGGG)7probe labeled with Cy3 and then a second (CCCTAA)7 probe
labeled with Rhodamine Green (Applied Biosystems, Bedford,
MA, USA). Metaphase spreads were captured on a Leitz Leica
DMRB fluorescence microscope.
Histopathology and immunohistochemistry
Small intestine sections were fixed in 10% buffered formalin,
embedded in paraffin wax and sectioned at 2–3 μm. Immuno-
histochemistry was performed on de-paraffinated intestine
sections processed with 10 mM sodium citrate (pH 6.5) and
cooked under pressure for 2 min. Slides were washed in water,
then in buffer TBS Tween 20 0.5%, blocked with peroxidase,
washed with TBS Tween 20 0.5% again and blocked with fetal
bovine serum followed by another wash. Then the slides were
incubated with the primary antibodies: rabbit monoclonal to
Ki-67 antibody (prediluted, SP6, Master Diagnostica, Madrid,
Spain), rabbit polyclonal to active caspase-3 at 1 : 200 (R&D
Systems, Minneapolis, MN, USA), rabbit polyclonal to p53 at
1 : 200 (CM5, Novocastra, Newcastle upon Tyne, UK) or goat
polyclonal to p21 at 1 : 250 (C-19-G, Santa Cruz Biotechnology,
Santa Cruz, CA, USA). Slides were then incubated with second-
ary antibodies conjugated with peroxidase from Dako (Glostrup,
Denamark), goat anti-rabbit (1 : 50) in the case of Ki-67, active
caspase-3 and p53 and rabbit anti-goat in the case of p21.
For signal development DAB (Dako) was used as a substrate.
Sections were lightly counterstained with haematoxylin and
analyzed by light microscopy. Apoptosis was also determined
by in situ hybridization TUNEL kit according to the manufacturer’s
instruction (ApopTag, Chemicon, Temecula, CA, USA).
Phosphorylated H2AX foci were detected using a mouse
monoclonal anti-phospho-histone H2AX antibody (1 : 500;
Upstate Biotechnology, Lake Placid, NY, USA). 53BP1 foci were
detected using a rabbit polyclonal anti-53BP1 (1 : 100; Novus
Biologicals, Littleton, CO, USA). ATR foci were detected using a
mouse monoclonal ant ATR (1 : 100; Cell Signalling Technology,
Danvers, MA, USA). After incubation with Cy3-goat anti-mouse
or 488 Alexa-goat anti-rabbit antibody (1 : 400; Jackson Immuno-
Research Laboratories, West Grove, PA, USA), for 30 min at
room temperature, slides were mounted in Vectashield with
DAPI. Images were obtained using a fluorescence microscope
(Leica DMRB).
The intestinal histopathology was analyzed in sections of
duodenum, cecum and colon. Five fields at ×20 were evaluated
by a pathologist in a blind manner. The length of the intestinal
wall, number and distribution of the glands, inflammation,
ulcerations, and compensatory glandular hyperplasia were
evaluated. Intestinal lesions were classified as mild, moderate
or severe lesions according to the pathological findings. Mild
intestinal lesions were characterized by focal atrophy, loss of
mucosal glands and few inflammatory cells in the lamina propia.
Moderate intestinal lesions showed diffuse atrophy of the
mucosal glands with focal hyperplasia and cysts in some crypts.
Some ulcers and a moderate cellular infiltrate of lymphocytes,
plasma cells and macrophages are present in lamina propia and
submucosa. Severe intestinal lesions are characterized by severe
atrophy and focal regenerative hyperplasia of glands. The intestine

MSH2 and signaling of short telomeres, P. Martinez et al.
© 2009 The AuthorsJournal compilation © Blackwell Publishing Ltd/Anatomical Society of Great Britain and Ireland 2009
15
is ulcerated and has marked infiltration of mixed inflammatory
cells in the lamina propia, submucosa and muscular layer with
a narrow intestinal lumen. Chronic peritonitis could be present.
Western blots
Whole-cell extracts were prepared from non-irradiated and
irradiated primary MEF as described (Blanco et al., 2007).
Protein concentration was determined using the Bio-Rad
DC Protein Assay (Bio-Rad). Fifty micrograms of each extract
were separated in 4–20% gradient sodium dodecyl sulfate–
polyacrylamide gels by electrophoresis. After transfer, the
membranes were incubated with an anti-p21 C-19 poly-
clonal antibody (1 : 500) (Santa Cruz Biotechnology) and
anti-β-actin monoclonal (1 : 10.000) (Sigma). Antibody
binding was detected after incubation with a secondary
antibody coupled to horseradish peroxidase using enhanced
chemiluminescence.
γγγγ-Irradiation of primary MEF
A total of 3 × 105 MEFs were seeded in 60-mm plates and
incubated overnight at 37 °C. Then, MEFs were treated with
10 Gy by using a 137Cs source (MARK 1–30 Irradiator; Shepherd
& Associates, San Fernando, CA, USA) at a rate of 2.11 Gy min–1.
After removing the medium, cells were washed and fresh
medium was added. To analyze the effect of γ-irradiation on
protein expression, cells were incubated for 18 h at 37 °C and
then recovered and frozen for further analysis by Western blot.
Statistical analysis
A log rank test was used to calculate statistical differences in
median survival of the different mouse cohorts. A Student’s
t-test was used to calculate the statistical significance of the
observed differences in mean lifespan, The Wilcoxon–Mann–
Whitney rank sum test was used for statistical comparisons of
the mean telomere length in MEFs and small intestine sections.
A chi-squared test was used to calculate statistical significance
in pathologies, signal-free ends, chromosomal aberrations,
T-SCE, γH2AX, 53BP1, apoptosis, proliferation, as well as p21
and p53 expression.
Acknowledgments
We thank R. Serrano and E. Santos for mouse care and
genotyping. P.M. is a ‘Ramon y Cajal’ senior scientist. I.S.-C. is
a Predoctoral Fellow from the Spanish Ministry of Education and
Culture (MEC). M. A. Blasco’s laboratory is funded by the MEC
(SAF2005-00277, GEN2001-4856-C13-08), by the Regional
Government of Madrid (GR/SAL/0597/e2004), European Union
(Telosens-CT-2002-00217, Intact LSHC-CT-2003-506803,
Zincage FOOD-CT-2003-506850, RISC-RAD FI6R-CT-e2003-
508842, Mol Cancer Med Lshc-CT-2004-502943) and the
Spanish Association Against Cancer.
References
d’Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, VonZglinicki T, Saretzki G, Carter NP, Jackson SP (2003) A DNA damagecheckpoint response in telomere-initiated senescence. Nature 426,194–198.
Artandi SE, Chang S, Lee SL, Alson S, Gottlieb GJ, Chin L, DePinho RA(2000) Telomere dysfunction promotes non-reciprocal translocationsand epithelial cancers in mice. Nature 406, 641–645.
Bailey SM, Brenneman MA, Goodwin EH (2004) Frequent recombinationin telomeric DNA may extend the proliferative life of telomerase-negative cells. Nucleic Acids Res. 32, 3743–3751.
Baker SM, Bronner CE, Zhang L, Plug AW, Robatzek M, Warren G, ElliottEA, Yu J, Ashley T, Arnheim N, Flavell RA, Liskay RM (1995) Male micedefective in the DNA mismatch repair gene PMS2 exhibit abnormalchromosome synapsis in meiosis. Cell 82, 309–319.
Bechter OE, Zou Y, Walker W, Wright WE, Shay JW (2004) Telomericrecombination in mismatch repair deficient human colon cancer cellsafter telomerase inhibition. Cancer Res. 64, 3444–3451.
Blanco R, Muñoz P, Flores JM, Klatt P, Blasco MA (2007) Telomeraseabrogation dramatically accelerates TRF2-induced epithelial carcinogenesis.Genes Dev. 21, 206–220.
Blasco MA (2005) Telomeres and human disease: ageing, cancer andbeyond. Nat. Rev. Genet. 6, 611–622.
Blasco MA, Hahn WC (2003) Evolving views of telomerase and cancer.Trends Cell Biol. 13, 289–294.
Blasco MA, Lee HW, Hande MP, Samper E, Lansdorp PM, DePinho RA,Greider CW (1997) Telomere shortening and tumor formation bymouse cells lacking telomerase RNA. Cell 91, 25–34.
Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, HarleyCB, Shay JW, Lichtsteiner S, Wright WE (1998) Extension of life-spanby introduction of telomerase into normal human cells. Science 279,349–352.
Brown KD, Rathi A, Kamath R, Beardsley DI, Zhan Q, Mannino JL,Baskaran R (2003) The mismatch repair system is required for S-phasecheckpoint activation. Nat. Genet. 33, 80–84.
Campbell MR, Wang Y, Andrew SE, Liu Y (2006) Msh2 deficiency leadsto chromosomal abnormalities, centrosome amplification, andtelomere capping defect. Oncogene 25, 2531–2536.
Chan SW, Blackburn EH (2002) New ways not to make ends meet:telomerase, DNA damage proteins and heterochromatin. Oncogene21, 553–563.
Chin L, Artandi SE, Shen Q, Tam A, Lee SL, Gottlieb GJ, Greider CW,DePinho RA (1999) p53 deficiency rescues the adverse effects oftelomere loss and cooperates with telomere dysfunction to acceleratecarcinogenesis. Cell 97, 527–538.
Choudhury AR, Ju Z, Djojosubroto MW, Schienke A, Lechel A, Schaetzlein S,Jiang H, Stepczynska A, Wang C, Buer J, Lee HW, von Zglinicki T,Ganser A, Schirmacher P, Nakauchi H, Rudolph KL (2007) Cdkn1adeletion improves stem cell function and lifespan of mice withdysfunctional telomeres without accelerating cancer formation. Nat.Genet. 39, 99–105.
Crabbe L, Verdun RE, Haggblom CI, Karlseder J (2004) Defectivetelomere lagging strand synthesis in cells lacking WRN helicaseactivity. Science 306, 1951–1953.
Duckett DR, Bronstein SM, Taya Y, Modrich P (1999) hMutSα- andhMutLα-dependent phosphorylation of p53 in response to DNAmethylator damage. Proc. Natl Acad. Sci. USA 96, 12384–12388.
Feldser DM, Greider CW (2007) Short telomeres limit tumor progressionin vivo by inducing senescence. Cancer Cell. 11, 461–469.
García-Cao I, García-Cao M, Tomás-Loba A, Martín-Caballero J, FloresJM, Klatt P, Blasco MA, Serrano M (2006) Increased p53 activity doesnot accelerate telomere-driven aging. EMBO Rep. 7, 546–552.

MSH2 and signaling of short telomeres, P. Martinez et al.
© 2009 The AuthorsJournal compilation © Blackwell Publishing Ltd/Anatomical Society of Great Britain and Ireland 2009
16
Genschel J, Curth U, Urbanke C (2000) Interaction of E. coli single-strandedDNA binding protein (SSB) with exonuclease I. The carboxy-terminusof SSB is the recognition site for the nuclease. Biol. Chem. 381, 183–192.
Gonzalez-Suarez E, Samper E, Flores JM, Blasco MA (2000) Telo-merase-deficient mice with short telomeres are resistant to skintumorigenesis. Nat. Genet. 26, 114–117.
Gonzalo S, Jaco I, Fraga MF, Chen T, Li E, Esteller M, Blasco MA (2006)DNA methyltransferases control telomere length and telomererecombination in mammalian cells. Nat. Cell Biol. 8, 416–424.
Goytisolo FA, Blasco MA (2002) Many ways to telomere dysfunction:in vivo studies using mouse models. Oncogene 21, 584–591.
Greenberg RA, Chin L, Femino A, Lee KH, Gottlieb GJ, Singer RH,Greider CW, DePinho RA (1999) Short dysfunctional telomeres impairtumorigenesis in the INK4aΔ2/3 cancer-prone mouse. Cell 97, 515–525.
Herrera E, Samper E, Martin-Caballero J, Flores JM, Lee HW, Blasco MA(1999) Disease states associated with telomerase deficiency appearearlier in mice with short telomeres. EMBO J. 18, 2950–2960.
Jiricny J (2006) The multifaceted mismatch-repair system. Nat. Rev. Mol.Cell Biol. 7, 335–346.
Kadyrov FA, Dzantiev L, Constantin N, Modrich P (2006) Endonucleolyticfunction of MutLα in human mismatch repair. Cell 126, 297–308.
Kunkel TA, Erie DA (2005) DNA mismatch repair. Annu. Rev. Biochem.74, 681–710.
de Lange T (2005) Shelterin: the protein complex that shapes andsafeguards human telomeres. Genes Dev. 19, 2100–2110.
Larson ED, Duquette ML, Cummings WJ, Streiff RJ, Maizels N (2005)MutSα binds to and promotes synapsis of transcriptionally activatedimmunoglobulin switch regions. Curr. Biol. 15, 470–474.
Lee HW, Blasco MA, Gottlieb GJ, Horner JW, Greider CW, DePinho RA(1998) Essential role of mouse telomerase in highly proliferativeorgans. Nature 392, 569–574.
Luo Y, Lin FT, Lin WC (2004) ATM-mediated stabilization of hMutL DNAmismatch repair proteins augments p53 activation during DNAdamage. Mol. Cell. Biol. 24, 6430–6444.
Modesti M, Kanaar R (2002) DNA repair: spot (light) s on chromatin.Curr. Biol. 11, 229–232.
Modrich P (1994) Mismatch repair, genetic stability, and cancer. Science266, 1959–1960.
Modrich P, Lahue R (1996) Mismatch repair in replication fidelity, geneticrecombination, and cancer biology. Annu. Rev. Biochem. 65, 101–133.
Opresko PL, von Kobbe C, Laine JP, Harrigan J, Hickson ID, Bohr VA(2002) Telomere-binding protein TRF2 binds to and stimulates theWerner and Bloom syndrome helicases. J. Biol. Chem. 277, 41110–41119.
Opresko PL, Otterlei M, Graakjaer J, Bruheim P, Dawut L, Kolvraa S,May A, Seidman MM, Bohr VA (2004) The Werner syndrome helicaseand exonuclease cooperate to resolve telomeric D loops in a mannerregulated by TRF1 and TRF2. Mol. Cell 14, 763–774.
Peters AC, Young LC, Maeda T, Tron VA, Andrew SE (2003) MammalianDNA mismatch repair protects cells from UVB-induced DNA damageby facilitating apoptosis and p53 activation. DNA Repair 2, 427–435.
Prolla TA, Baker SM, Harris AC, Tsao JL, Yao X, Bronner CE, Zheng B,Gordon M, Reneker J, Arnheim N, Shibata D, Bradley A, Liskay RM(1998) Tumour susceptibility and spontaneous mutation in micedeficient in Mlh1, Pms1 and Pms2 DNA mismatch repair. Nat. Genet.18, 276–279.
Reitmair AH, Schmits R, Ewel A, Bapat B, Redston M, Mitri A,Waterhouse P, Mittrucker HW, Wakeham A, Liu B, Thomason A,Griesser H, Gallinger S, Ballhausen WG, Fishel R, Mak TW (1995)MSH2 deficient mice are viable and susceptible to lymphoid tumours.Nat. Genet. 11, 64–70.
Reitmair AH, Redston M, Cai JC, Chuang TC, Bjerknes M, Cheng H, Hay K,Gallinger S, Bapat B, Mak TW (1996) Spontaneous intestinal carcinomasand skin neoplasms in Msh2-deficient mice. Cancer Res. 56, 3842–3849.
Rizki A, Lundblad V (2001) Defects in mismatch repair promote telomerase-independent proliferation. Nature 411, 713–716.
Samper E, Goytisolo FA, Slijepcevic P, van Buul PP, Blasco MA (2000)Mammalian Ku86 protein prevents telomeric fusions independentlyof the length of TTAGGG repeats and the G-strand overhang. EMBORep. 1, 244–252.
Saydam N, Kanagaraj R, Dietschy T, Garcia PL, Pena-Diaz J, Shevelev I,Stagljar I, Janscak P (2007) Physical and functional interactionsbetween Werner syndrome helicase and mismatch-repair initiationfactors. Nucleic Acids Res. 35, 5706–5716.
Schaetzlein S, Kodandaramireddy NR, Ju Z, Lechel A, Stepczynska A, Lilli DR,Clark AB, Rudolph C, Kuhnel F, Wei K, Schlegelberger B, Schirmacher P,Kunkel TA, Greenberg RA, Edelmann, W, Rudolph KL (2007) Exo-nuclease-1 deletion impairs DNA damage signaling and prolongslifespan of telomere-dysfunctional mice. Cell 130, 863–877.
Shay JW, Wright WE (2006) Telomerase therapeutics for cancer:challenges and new directions. Nat. Rev. Drug Discov. 5, 577–584.
Siegl-Cachedenier I, Munoz P, Flores JM, Klatt P, Blasco MA (2007)Deficient mismatch repair improves organismal fitness and survival ofmice with dysfunctional telomeres. Genes Dev. 21, 2234–2247.
van Steensel B, Smogorzewska A, de Lange T (1998) TRF2 protectshuman telomeres from end-to-end fusions. Cell 92, 401–413.
Wei K, Clark AB, Wong E, Kane MF, Mazur DJ, Parris T, Kolas NK, RussellR, Hou H Jr, Kneitz B, Yang G, Kunkel TA, Kolodner RD, Cohen PE,Edelmann W (2003) Inactivation of Exonuclease 1 in mice results inDNA mismatch repair defects, increased cancer susceptibility, andmale and female sterility. Genes Dev. 17, 603–614.
de Wind N, Dekker M, Berns A, Radman M, te Riele H (1995) Inactivationof the mouse Msh2 gene results in mismatch repair deficiency,methylation tolerance, hyperrecombination, and predisposition tocancer. Cell 82, 321–330.
Zijlmans JM, Martens UM, Poon SS, Raap AK, Tanke HJ, Ward RK,Lansdorp PM (1997) Telomeres in the mouse have large inter-chromosomal variations in the number of T2AG3 repeats. Proc. NatlAcad. Sci. USA 94, 7423–7428.
Supporting Information
Additional Supporting Information may be found in the online
version of this article:
Fig. S1 Kaplan–Meier survival curves of the different mouse
cohorts. (A) MSH2+/+/Terc+/+, G1MSH2+/+/Terc–/–, G2MSH2+/+/Terc–/– and G3MSH2+/+/Terc–/–; (B) MSH2–/–/Terc+/+, G1MSH2–/–/Terc–/–, G2MSH2–/–/Terc–/–- and G3MSH2–/–/Terc–/–. n, number of
mice of each genotype included in the analysis. Statistical
comparisons among genotypes using the log rank test are
shown. For statistical comparisons between generations, see
Supporting Table S1.
Fig. S2 Histopathological analysis of mice of the different
genotypes at their time of death. Percentage of mice of the
indicated genotype showing degenerative pathologies in the
indicated organs at time of death. Intestinal lesions include atrophy,
hyperplasia, cysts, hemorrhage, edema and lymphoid infiltrations

MSH2 and signaling of short telomeres, P. Martinez et al.
© 2009 The AuthorsJournal compilation © Blackwell Publishing Ltd/Anatomical Society of Great Britain and Ireland 2009
17
(see also Fig. 2). Kidney lesions include atrophy, tubular
degeneration, amyloidosis, glomerulonephritis, hydronephrosis,
intersticial nephritis, cysts and lymphoid infiltrations. Spleen
lesions include atrophy, amyloidosis, lymphoid and myeloid
hyperplasia. Liver lesions include vacuolar degeneration, necrosis,
cyst and lymphoid infiltration. Testis, seminal vesicle, ovary and
uterus compose the reproductive tract group. The reproductive
tract lesions include atrophy, cyst, hyperplasia of Leydig cells,
inflammation of seminal vesicles and cystic endometrial hyper-
plasia. Heart lesions include congestion, ventricular dilatation
and myocardiopathy. Lung lesions include congestion,
hemorrhage, alveolar hyperplasia and lymphoid infiltration.
Statistical comparisons between genotypes using the chi-squared
test are shown.
Fig. S3 Representative combined images of telomere flu-
orescence and DAPI staining on small intestine sections of mice
of the indicated genotype as determined by Q-FISH.
Fig. S4 Telomere length as determined by TRF in MEF of the
indicated genotypes. No significant differences in TRF fragment
size were detected between single Terc–/– and double Terc–/–
MSH2–/– mice of the indicated mouse generations.
Fig. S5 (a) Percentage of ATR-positive cells in small intestine
sections of mice of the indicated genotypes. n, number of mice
analyzed per genotype. The total number of cells scored is
indicated. Bars represent standard error. Statistical comparisons
using the chi-squared test are shown. Gray bars, MSH2+/+ mice;
black bars, MSH2–/– mice. For statistical comparisons between
generations, see Supporting Table S2. (b) Representative images of
ATR-positive cells (upper panel) and of the DAPI combined-
images (lower panel) in small intestine sections of G3Terc–/–
MSH2+/+ and G3Terc–/– MSH2–/– mice.
Table S1 Statistical significance in median survival and mean
lifespan analysis by log rank and Student’s t-test, respectively
Table S2 Comparisons between generations. Statistical
significance in immunohistochemistry and in situ hybridization
determinations by chi-squared test
Table S3 Statistical significance in Q-FISH analysis in small
intestine sections
Table S4 Statistical significance in Q-FISH analysis in MEF
metaphases
Please note: Wiley-Blackwell are not responsible for the content
or functionality of any supporting materials supplied by the
authors. Any queries (other than missing material) should be
directed to the corresponding author for the article.
Copyright © 2022 FDOKUMEN



















![[Regulation of telomeres length: making the telomeres accessible?]](https://static.fdokumen.com/doc/165x107/633f1028d121719806096682/regulation-of-telomeres-length-making-the-telomeres-accessible.jpg)
