
Translational and morphological effects of signalling alcohols ...
Upload
independentCategory
view
1download
0
A
astwaTfHtCC©
K
1
addntT([
0d
Cell Calcium 40 (2006) 553–560
Mitochondrial calcium signalling and cell death: Approaches forassessing the role of mitochondrial Ca2+ uptake in apoptosis
Gyorgy Hajnoczky ∗, Gyorgy Csordas, Sudipto Das, Cecilia Garcia-Perez,Masao Saotome, Soumya Sinha Roy, Muqing Yi
Department of Pathology, Anatomy and Cell Biology, Thomas Jefferson University,Philadelphia, PA 19107, USA
Received 1 August 2006; accepted 23 August 2006
bstract
Local Ca2+ transfer between adjoining domains of the sarcoendoplasmic reticulum (ER/SR) and mitochondria allows ER/SR Ca2+ release toctivate mitochondrial Ca2+ uptake and to evoke a matrix [Ca2+] ([Ca2+]m) rise. [Ca2+]m exerts control on several steps of energy metabolism toynchronize ATP generation with cell function. However, calcium signal propagation to the mitochondria may also ignite a cell death programhrough opening of the permeability transition pore (PTP). This occurs when the Ca2+ release from the ER/SR is enhanced or is coincidentith sensitization of the PTP. Recent studies have shown that several pro-apoptotic factors, including members of the Bcl-2 family proteins
nd reactive oxygen species (ROS) regulate the Ca2+ sensitivity of both the Ca2+ release channels in the ER and the PTP in the mitochondria.o test the relevance of the mitochondrial Ca2+ accumulation in various apoptotic paradigms, methods are available for buffering of [Ca2+],or dissipation of the driving force of the mitochondrial Ca2+ uptake and for inhibition of the mitochondrial Ca2+ transport mechanisms.owever, in intact cells, the efficacy and the specificity of these approaches have to be established. Here we discuss mechanisms that recruit
he mitochondrial calcium signal to a pro-apoptotic cascade and the approaches available for assessment of the relevance of the mitochondriala2+ handling in apoptosis. We also present a systematic evaluation of the effect of ruthenium red and Ru360, two inhibitors of mitochondriala2+ uptake on cytosolic [Ca2+] and [Ca2+]m in intact cultured cells.2006 Elsevier Ltd. All rights reserved.
2+ ; VDAC
lpCcime
eywords: Calcium; Ca ; IP3 receptor; Ryanodine receptor; Mitochondria
. Mitochondrial Ca2+ transport mechanisms
The pathways of the mitochondrial Ca2+ import and exportre illustrated in Fig. 1 Ca2+ traverses the outer mitochon-rial membrane (OMM) primarily through the voltage depen-ent anion-selective channel (VDAC) [1–3]. The molecularature of the proteins mediating the Ca2+ transport across
he inner mitochondrial membrane (IMM) remains unknown.he protein mediating Ca2+ uptake is referred as the uniporterUP) and has been identified as a Ca2+ selective ion channel4]. The UP passes Ca2+ along the electrochemical gradient
∗ Corresponding author. Tel.: +1 215 503 1427; fax: +1 215 923 2218.E-mail address: [email protected] (G. Hajnoczky).
dtetPpdr
143-4160/$ – see front matter © 2006 Elsevier Ltd. All rights reserved.oi:10.1016/j.ceca.2006.08.016
; Ruthenium red; Ru360
argely due to the highly negative mitochondrial membraneotential, �Ψm of 180 mV. Both the VDAC and the UP showa2+-dependent activation that is relevant for the homeostaticontrol of cytoplasmic [Ca2+] ([Ca2+]c) [1,3,4]. Ca2+ enter-ng the mitochondrial matrix stimulates the Ca2+ sensitiveitochondrial dehydrogenases (CSMDH) to increase the H+
xtrusion that is important for both the maintenance of theriving force for the Ca2+ uptake and for the ATP produc-ion. Ca2+ export across the IMM is mediated by the Ca2+
xchangers (Na+/Ca2+ and H+/Ca2+) and under some condi-ions (see below), the matrix Ca2+ induces formation of the
TP that traverses both the IMM and OMM and allows freeassage of Ca2+, other ions and small molecules. Detailediscussion of the mitochondrial Ca2+ transport is available inecent comprehensive reviews [5–8].
554 G. Hajnoczky et al. / Cell Calci
Fig. 1. Mechanisms of the mitochondrial Ca2+ transport. Abbreviations:Ctc
2m
ovdbetCiDiR
otloeg(am
torgaCP[tNaitaiaorptbiaa
Fms
SMDH, Ca2+ sensitive mitochondrial dehydrogenase; PTP, permeabilityransition pore; UP, uniporter; VDAC, voltage dependent anion selectivehannel.
. Induction of cell death as a consequence ofitochondrial Ca2+ uptake
It has been known for several decades that sequestrationf vast amounts of Ca2+ in the mitochondria occurs underarious pathophysiological conditions and contributes to theemise of the cells [9]. In these paradigms, the loss of thealance between plasma membrane Ca2+ influx and Ca2+
xport leads to a sustained elevation in [Ca2+]c from 100 nMo ≥1 �M, inducing a progressive increase in mitochondriala2+ uptake. When large quantities of Ca2+ are accumulated
n the mitochondrial matrix, Ca2+ interacts with cyclophilinto induce opening of the PTP [10]. Furthermore, the rise
n [Ca2+]m stimulates the generation of factors, includingOS and free fatty acids, which also promote the opening
“db(
ig. 2. Ca2+-induced mitochondrial membrane permeabilization schemes illustratinobilization (B and C) induced mitochondrial membrane permeabilization. Pro-s
hown in purple. Abbreviations used: AA, arachidonic acid; ROS, reactive oxygen
um 40 (2006) 553–560
f the PTP [11,12]. Opening of the PTP causes dissipation ofhe �Ψm and release of Ca2+. If the cytoplasmic Ca2+ over-oad persists, the PTP stays open and allows accumulationf solutes in the mitochondrial matrix. This in turn, leads toxpansion of the matrix space and to rupture of the OMM,iving rise to release of the intermembrane space content [13]Fig. 2A). Finally, impairment of the mitochondrial functionnd activation of cytoplasmic mechanisms by the releaseditochondrial factors leads to execution of the cells.In the late 90 s, studies of the propagation of the IP3 recep-
or (IP3R)-and ryanodine receptor (RyR)-mediated [Ca2+]cscillations to the mitochondria provided the unexpectedesult that physiological [Ca2+]m transients can also trig-er mitochondrial membrane permeabilization, leading topoptotic cell death [14–16]. This pathway also depends ona2+-induced activation of the PTP. However, opening of theTP in this case, required the coincidental detection of theCa2+]m oscillations and pro-apoptotic stimuli that loweredhe threshold for the Ca2+-induced PTP opening (Fig. 2B).either the [Ca2+]m oscillations nor the sensitizing factor
lone was sufficient to initiate these events. The PTP open-ng was followed by the release of pro-apoptotic factors fromhe intermembrane space to the cytoplasm. The exact mech-nism of the OMM permeabilization remains elusive. Anmportant attribute of this path is the absence of a prolongednd massive [Ca2+]c load. Therefore closure of the PTP mayccur, preventing large scale swelling and allowing functionalecovery of the mitochondria. Mitochondrial ATP productionrovides ATP for caspase activation enhancing the fidelity ofhe execution of the apoptotic program. Opening of the PTPy Ca2+ is effectively modulated by several factors, includ-ng ROS, pH, �Ψm [5] the level of which is affected by
variety of metabolic intermediates, signalling moleculesnd drugs. Therefore, it is not surprising that the role of a
coincidence detection” or a “two hit” mechanism of Ca2+-ependent mitochondrial membrane permeabilization haseen implicated in a broad range of cell death and tissue injurye.g. [17]).g possible mechanisms for the cellular Ca2+ overload (A) and ER/SR Ca2+
urvival mechanisms are depicted in green and pro-death mechanisms arespecies; cyto c, cytochrome c.

ll Calci
hmafcCDcethbCfItdbtbCpElcTaoTaBItaotf
obmEmppcB(budttwm
bsoeammbtdaC[emasdoftTlmt
3m
cfpupaFeebbt
3
abbnt
G. Hajnoczky et al. / Ce
Recent studies of the ER-mitochondrial communicationave also provided evidence for regulation of the IP3R-ediated Ca2+ release by both pro-and anti-apoptotic mech-
nisms (Fig. 2C). Pro-apoptotic factors have been reported toacilitate the ER Ca2+ mobilization and in turn, to increase thealcium signal propagation to the mitochondria, resulting ina2+-dependent mitochondrial membrane permeabilization.ue to a complex, local interaction between IP3Rs and mito-
hondrial Ca2+ uptake [18–20], regulation of ER Ca2+ releasevokes more extensive changes in the [Ca2+]m signal than inhe [Ca2+]c signal [21]. Boehning and co-workers showedigh affinity binding of cytochrome c to the IP3Rs, whichlocks the Ca2+-dependent inhibition of the IP3-induceda2+ efflux [22]. By this mechanism, cytochrome c released
rom the mitochondria at the onset of apoptosis recruits theP3-linked Ca2+ mobilization from the ER and Ca2+ loadingo the mitochondria to enhance and propagate the mitochon-rial membrane permeabilization. A similar mechanism maye supported by the ROS that are produced and released fromhe mitochondria to the cytosol during mitochondrial mem-rane permeabilization [23] and promote the IP3R-mediateda2+ release [24]. The studies of Scorrano and co-workersrovided evidence that the presence of Bax and Bak in theR membrane is required to optimize the storage and mobi-
ization of Ca2+ and showed that this effect is the criticalontribution of Bax/Bak in several models of apoptosis [25].he effect of Bax/Bak may be due to suppression of the inter-ction between the IP3R and the anti-apoptotic Bcl-2 [26,27]r Bcl-xL [28], which controls the Ca2+ leakage from the ER.he recruitment of Bax to the ER and the ER Ca2+ release arelso regulated by Bik, a BH3-only Bcl-2 family protein[29].cl-2/Bcl-xL seems to serve both as a direct effector of the
P3R [26,28] and as a regulator of the phosphorylation ofhe type 1 IP3R [27]. Thus the effect of Bcl-2/Bcl-xL on thectivation of the IP3R by IP3 [26,28] may reflect more thanne mechanism of the interaction or interaction with morehan one IP3R isotype and may be modulated by numerousactors that target the IP3R.
In the sequence from the opening of the IP3Rs to thepening of the PTP, the calcium signal propagation maye affected by several factors, including the activity of theitochondrial Ca2+ uptake sites, the organization of theR-mitochondrial interface, and the morphology of theitochondria. tBid, a pro-apoptotic BH3 only Bcl-2 family
rotein induces permeabilization of the OMM to promote theropagation of the IP3R-mediated [Ca2+]c signal to the mito-hondria [30]. Overexpression of Mcl-1, an anti-apoptoticcl-2 family protein [31] or PPARgamma-coactivator-1alpha
PGC-1�) that triggers mitochondrial biogenesis [32] haseen shown to lower the efficacy of the mitochondrial Ca2+
ptake, the IP3R-mediated [Ca2+]m signal and the Ca2+-ependent apoptosis. Neutralization or down-regulation of
hese factors may favor to an increase in the apoptotic poten-ial of the [Ca2+]m signal. Regarding the role of the interface,e observed that the ER-mitochondrial association becomesore extensive in the beginning of apoptosis inducedtapb
um 40 (2006) 553–560 555
y tunicamycin or serum starvation. Furthermore, whentrengthening of the associations was induced by expressionf a synthetic bi-functional ER-mitochondrial crosslinker,nhanced Ca2+ transfer from ER to the mitochondriand sensitization to the Ca2+-dependent mitochondrialembrane permeabilization were noted [49]. The ER-itochondrial interface may be supported by direct links
etween the organelles or by anchorage of the organelles tohe cytoskeletal framework. Both the ER and mitochondriaisplay dynamic positioning along the cytoskeletal tracksnd through the control of the organellar motility the locala2+ signalling between the organelles is also modulated
33,34]. The fusion and fission dynamics of the mitochondriamerges as an important factor for the organization of the ER-itochondrial interface as well as for the distribution of Ca2+
ccumulated to the mitochondrial matrix. Szabadkai et al.howed that Drp1-mediated fragmentation of the mitochon-ria inhibited the spreading of the [Ca2+]m signal along therganelles [35]. These authors also found that mitochondrialragmentation suppressed the ceramide-induced apoptosishat is mediated through Ca2+ delivery to the mitochondria.hus evidence is on the rise that some determinants of the
ocal Ca2+ transfer between ER and mitochondria and of theitochondrial morphology may also serve as checkpoints for
he calcium signal to support either cell survival or cell death.
. Methods for evaluating the relevance of theitochondrial handling in the induction of apoptosis
The emerging evidence for the calcium signal-driven mito-hondrial membrane permeabilization underscores the needor methods to test the dependence of various apoptosisaradigms on mitochondrial Ca2+ uptake. The commonlysed strategies include (1) buffering of [Ca2+]c, (2) dissi-ation of the driving force of the mitochondrial Ca2+ uptakend (3) inhibition of the mitochondrial uptake sites (Fig. 3).or each strategy, fairly straightforward protocols have beenstablished in suspensions of isolated mitochondria. How-ver, in intact cells, the barrier formed by the plasma mem-rane, the complex cellular response to mitochondrial inhi-ition and the extra-mitochondrial targets of the drugs limithe utility of these protocols.
.1. Buffering of [Ca2+]c
Introduction of Ca2+ chelators into the cytoplasm providesmeans to attenuate the [Ca2+]c signal. BAPTA or EGTA cane loaded to the cells as acetoxymethylesters that are cleavedy intracellular esterases to release the free acid form. Alter-atively, BAPTA or EGTA free acids can be introduced tohe cells by microinjection. To assess the effect of the chela-
or on the [Ca2+]c fluorescence measurement of [Ca2+]c bycytoplasmic probe (e.g. fura2, fluo4 or cytoplasm targetedericam) can be used. It is important to note that effectiveuffering of bulk [Ca2+]c does not necessarily result in pre-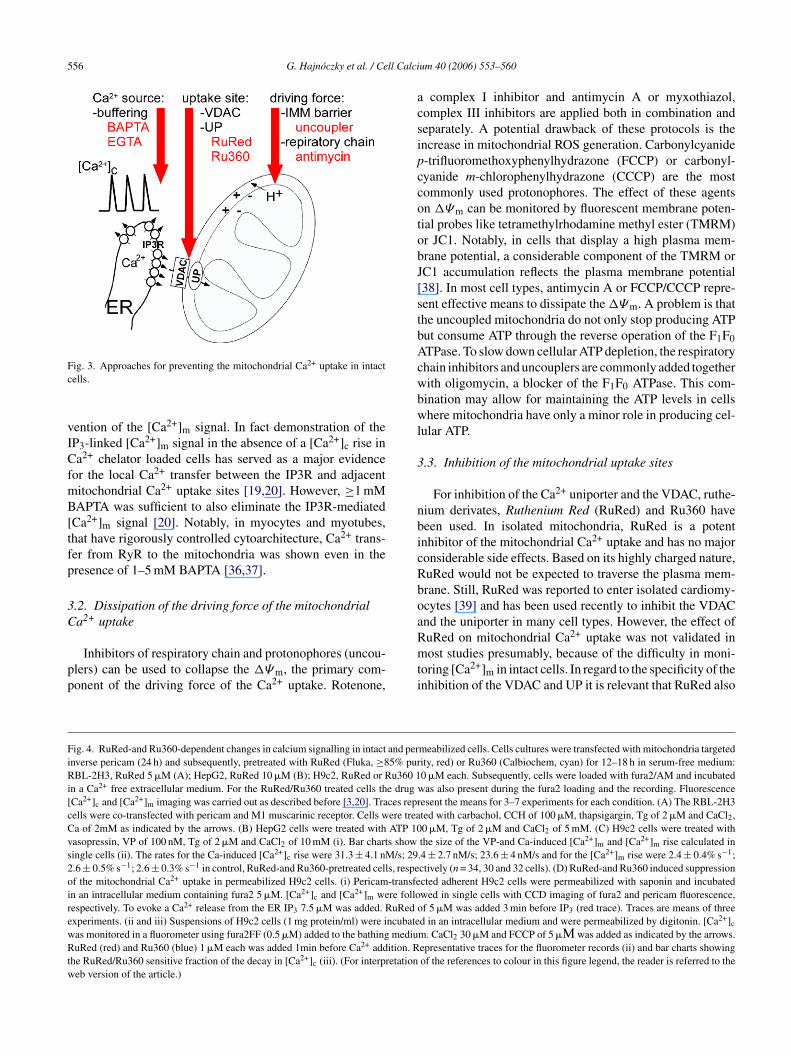
556 G. Hajnoczky et al. / Cell Calci
Fc
vICfmB[tfp
3C
pp
acsipccotobJ[stbAcwbwl
3
nbicRboa
FiRi[cCvs2oirewRtw
ig. 3. Approaches for preventing the mitochondrial Ca2+ uptake in intactells.
ention of the [Ca2+]m signal. In fact demonstration of theP3-linked [Ca2+]m signal in the absence of a [Ca2+]c rise ina2+ chelator loaded cells has served as a major evidence
or the local Ca2+ transfer between the IP3R and adjacentitochondrial Ca2+ uptake sites [19,20]. However, ≥1 mMAPTA was sufficient to also eliminate the IP3R-mediated
Ca2+]m signal [20]. Notably, in myocytes and myotubes,hat have rigorously controlled cytoarchitecture, Ca2+ trans-er from RyR to the mitochondria was shown even in theresence of 1–5 mM BAPTA [36,37].
.2. Dissipation of the driving force of the mitochondriala2+ uptake
Inhibitors of respiratory chain and protonophores (uncou-lers) can be used to collapse the �Ψm, the primary com-onent of the driving force of the Ca2+ uptake. Rotenone,
Rmti
ig. 4. RuRed-and Ru360-dependent changes in calcium signalling in intact and pernverse pericam (24 h) and subsequently, pretreated with RuRed (Fluka, ≥85% purBL-2H3, RuRed 5 �M (A); HepG2, RuRed 10 �M (B); H9c2, RuRed or Ru360 1
n a Ca2+ free extracellular medium. For the RuRed/Ru360 treated cells the drugCa2+]c and [Ca2+]m imaging was carried out as described before [3,20]. Traces reprells were co-transfected with pericam and M1 muscarinic receptor. Cells were treaa of 2mM as indicated by the arrows. (B) HepG2 cells were treated with ATP 10asopressin, VP of 100 nM, Tg of 2 �M and CaCl2 of 10 mM (i). Bar charts showingle cells (ii). The rates for the Ca-induced [Ca2+]c rise were 31.3 ± 4.1 nM/s; 29.6 ± 0.5% s−1; 2.6 ± 0.3% s−1 in control, RuRed-and Ru360-pretreated cells, respef the mitochondrial Ca2+ uptake in permeabilized H9c2 cells. (i) Pericam-transfen an intracellular medium containing fura2 5 �M. [Ca2+]c and [Ca2+]m were folloespectively. To evoke a Ca2+ release from the ER IP3 7.5 �M was added. RuRed oxperiments. (ii and iii) Suspensions of H9c2 cells (1 mg protein/ml) were incubateas monitored in a fluorometer using fura2FF (0.5 �M) added to the bathing mediuuRed (red) and Ru360 (blue) 1 �M each was added 1min before Ca2+ addition. R
he RuRed/Ru360 sensitive fraction of the decay in [Ca2+]c (iii). (For interpretationeb version of the article.)
um 40 (2006) 553–560
complex I inhibitor and antimycin A or myxothiazol,omplex III inhibitors are applied both in combination andeparately. A potential drawback of these protocols is thencrease in mitochondrial ROS generation. Carbonylcyanide-trifluoromethoxyphenylhydrazone (FCCP) or carbonyl-yanide m-chlorophenylhydrazone (CCCP) are the mostommonly used protonophores. The effect of these agentsn �Ψm can be monitored by fluorescent membrane poten-ial probes like tetramethylrhodamine methyl ester (TMRM)r JC1. Notably, in cells that display a high plasma mem-rane potential, a considerable component of the TMRM orC1 accumulation reflects the plasma membrane potential38]. In most cell types, antimycin A or FCCP/CCCP repre-ent effective means to dissipate the �Ψm. A problem is thathe uncoupled mitochondria do not only stop producing ATPut consume ATP through the reverse operation of the F1F0TPase. To slow down cellular ATP depletion, the respiratoryhain inhibitors and uncouplers are commonly added togetherith oligomycin, a blocker of the F1F0 ATPase. This com-ination may allow for maintaining the ATP levels in cellshere mitochondria have only a minor role in producing cel-
ular ATP.
.3. Inhibition of the mitochondrial uptake sites
For inhibition of the Ca2+ uniporter and the VDAC, ruthe-ium derivates, Ruthenium Red (RuRed) and Ru360 haveeen used. In isolated mitochondria, RuRed is a potentnhibitor of the mitochondrial Ca2+ uptake and has no majoronsiderable side effects. Based on its highly charged nature,uRed would not be expected to traverse the plasma mem-rane. Still, RuRed was reported to enter isolated cardiomy-cytes [39] and has been used recently to inhibit the VDACnd the uniporter in many cell types. However, the effect of
uRed on mitochondrial Ca2+ uptake was not validated inost studies presumably, because of the difficulty in moni-oring [Ca2+]m in intact cells. In regard to the specificity of thenhibition of the VDAC and UP it is relevant that RuRed also
meabilized cells. Cells cultures were transfected with mitochondria targetedity, red) or Ru360 (Calbiochem, cyan) for 12–18 h in serum-free medium:0 �M each. Subsequently, cells were loaded with fura2/AM and incubated
was also present during the fura2 loading and the recording. Fluorescenceesent the means for 3–7 experiments for each condition. (A) The RBL-2H3ted with carbachol, CCH of 100 �M, thapsigargin, Tg of 2 �M and CaCl2,0 �M, Tg of 2 �M and CaCl2 of 5 mM. (C) H9c2 cells were treated withthe size of the VP-and Ca-induced [Ca2+]m and [Ca2+]m rise calculated in.4 ± 2.7 nM/s; 23.6 ± 4 nM/s and for the [Ca2+]m rise were 2.4 ± 0.4% s−1;ctively (n = 34, 30 and 32 cells). (D) RuRed-and Ru360 induced suppressioncted adherent H9c2 cells were permeabilized with saponin and incubatedwed in single cells with CCD imaging of fura2 and pericam fluorescence,f 5 �M was added 3 min before IP3 (red trace). Traces are means of three
d in an intracellular medium and were permeabilized by digitonin. [Ca2+]c
m. CaCl2 30 �M and FCCP of 5 �М was added as indicated by the arrows.epresentative traces for the fluorometer records (ii) and bar charts showingof the references to colour in this figure legend, the reader is referred to the

ll Calci
i(
csc
mc
G. Hajnoczky et al. / Ce
nhibits plasma membrane and intracellular cation channelse.g. TASK-3 [40], TRPV [41] and RyR [42], respectively).
To evaluate the utility of RuRed in inhibition of mito-hondrial Ca2+ uptake in intact cells, we recorded [Ca2+]mimultaneously with [Ca2+]c in 3 cell lines, RBL-2H3 rat mastells, HepG2 human hepatoma cells and H9c2 rat cardiac
wmut
um 40 (2006) 553–560 557
yoblast cells using a mitochondrial matrix-targeted peri-am construct [43] and cytosolic fura2 (Fig. 4). The cells
ere pretreated 12–18 h with RuRed of 5 or 10 �M, a supra-aximal dose for the inhibition of the mitochondrial Ca2+ptake in permeabilized cells or in isolated mitochondria. Inhe single cell [Ca2+] imaging experiments, the cells were first

5 ll Calci
iuCa2aaCwce([taftsptR
ciamaHwiafcotif[tiala[csTmlaiI[tsu
adtttHteTnHCtui
docDIotdcpriNclcti
4
dtcbmtfimgClc
58 G. Hajnoczky et al. / Ce
ncubated in a Ca2+ free medium and were sequentially stim-lated by an IP3-linked agonist and thapsigargin to mobilizea2+ from the ER. Then, extracellular Ca2+ was added back tollow the Ca2+ entry (basal + store-depletion induced). RBL-H3 cells expressing the M1 muscarinic receptor displayedrobust carbachol-induced [Ca2+]c transient that was associ-ted with a sharp and large increase in [Ca2+]m (Fig. 4A). Thea2+ entry elicited a substantial increase in [Ca2+]c, whichas followed by a further rise in [Ca2+]m. RuRed did not
ause any change in either the [Ca2+]c or [Ca2+]m signalvoked by the intracellular Ca2+ mobilization or Ca2+ entryFig. 4A red traces). In HepG2 cells, the agonist-inducedCa2+]c and [Ca2+]m increase was relatively small, whereashe Ca2+ entry resulted in a large elevation in both [Ca2+]cnd [Ca2+]m (Fig. 4B). Similar to the RBL-2H3 cells, RuRedailed to suppress either the [Ca2+]c or the [Ca2+]m signal inhe HepG2 cells (Fig. 4B, red traces). Previously, RuRed washown to inhibit IP3 and the Ca2+-induced [Ca2+]m signal inermeabilized cells [20]. Therefore, the lack of an effect inhe intact cells seems to indicate that RuRed did not enter theBL-2H3 or the HepG2 cells.
A more complex picture emerged in the H9c2 cells. RuRedaused attenuation of the agonist-induced [Ca2+]c signal,ndicating an effect of the drug on the cascade from thegonist receptor (V1 vasopressin receptor) to the ER Ca2+
obilization (Fig. 4Ci, red versus black). Accordingly, thegonist-induced [Ca2+]m signal was also suppressed in the9c2 cells. The Ca2+ entry resulted in a [Ca2+]c signal thatas followed by a rapid and large [Ca2+]m elevation both
n the control and RuRed-treated H9c2 cells. RuRed did notffect the rate or the extent of the [Ca2+]m rise. Thus RuRedailed to inhibit the mitochondrial Ca2+ uptake in the H9c2ells. Ru360 has been reported as a more potent inhibitorf the mitochondrial Ca2+ uptake than RuRed, which lackshe inhibitory effect of RuRed on the RyR and enters rapidlyntact cells [44]. Exposure of the H9c2 cells to Ru360 (10 �Mor 12–24 h) led to some attenuation of the agonist-inducedCa2+]c and [Ca2+]m response and no change occurred inhe Ca2+ entry dependent [Ca2+]c and [Ca2+]m rise. Since thendividual H9c2 cells show some heterogeneity in the [Ca2+]cnd [Ca2+]m and a modest drug-induced change in the under-ying Ca2+ fluxes may remain unnoticed in the cell populationverage, the size and the rate for the agonist and Ca2+-inducedCa2+]c and [Ca2+]m signal was also quantitated in singleells (Fig. 4Cii). However, the single cell analysis did nothow any effects of RuRed or Ru360 on the [Ca2+]m rise.hus, similar to RuRed, Ru360 also failed to inhibit theitochondrial Ca2+ uptake in intact H9c2 cells. In permeabi-
ized H9c2 cells (Fig. 4D), the IP3-induced Ca2+ mobilizationppeared as a sharp [Ca2+]c increase closely followed by anncrease in [Ca2+]m (Fig. 4Di). When RuRed was present theP3-induced [Ca2+]c rise was somewhat suppressed but the
Ca2+]m increase was almost abolished (Fig. 4Di red). Fur-hermore, when Ca2+ was added to permeabilized H9c2 celluspensions the mitochondrial Ca2+ uptake led to a rapid andncoupler-sensitive decay in [Ca2+]c, which was practicallyopca
um 40 (2006) 553–560
bolished by both RuRed and Ru360 (Fig. 4Dii,iii). Theseata confirmed that RuRed or Ru360 is a potent inhibitor ofhe mitochondrial Ca2+ uptake in permeabilized H9c2. Takenogether the results, neither RuRed nor Ru360 interferes withhe [Ca2+]c signal propagation to the mitochondria in intact9c2 cells, presumably due to the limited penetration across
he plasma membrane. Furthermore, both RuRed and Ru360xert an inhibition in the agonist-induced Ca2+ mobilization.he main conclusion is that the extracellular application ofeither RuRed nor Ru360 can be used in intact RBL-2H3,epG2 or H9c2 cells for evaluating the role of mitochondriala2+ uptake in cell death. For broader implications, it seems
o be necessary to validate the effect on mitochondrial Ca2+
ptake when RuRed or Ru360 is intended to be used in anyntact cells.
Complementary approaches to test the role of mitochon-rial Ca2+ in cell death also use inhibitors of the PTPpening. Two commonly used cell-permeable inhibitors areyclosporin A and bongkrekic acid, which bind to cyclophilin
and to the adenine nucleotide translocase, respectively.mportantly, cyclosporin A does not abolish the openingf the pore, it only causes an increase in the Ca2+ reten-ion capacity of the mitochondria. The broad extramitochon-rial effects of cyclosporin A, including the inhibition ofalcineurin and the impairments of the mitochondrial ATProduction evoked by bongkrekic acid represent concernsegarding the prolonged treatment of intact cells. However,n the absence of molecular approaches, cyclosporin A (or the-methyl-4-valine-cyclosporin that inhibits the PTP without
alcineurin inactivation [45]) and bongkrekic acid particu-arly, in combination with approaches to suppress the mito-hondrial Ca2+ uptake, have been proven practical for testinghe relevance of the mitochondrial calcium PTP pathway innduction of cell death.
. Conclusions
Decoding of calcium signals in the mitochondria to celleath inducing mechanisms depends on coincidental detec-ion of a [Ca2+]m rise and pro-apoptotic stimuli. In the cal-ium signal propagation from the ER to the mitochondria,asically every step offers an opportunity for other signallingechanisms to alter the spatial and temporal properties of
he [Ca2+]m signal. Furthermore, a variety of pro-apoptoticactors converge on the PTP to control its Ca2+ sensitiv-ty. To sort out the pathophysiological relevance of these
echanisms the mitochondrial Ca2+ uptake has to be tar-eted. The drugs available for the inhibition of mitochondriala2+ uptake have limitations and may evoke complex cellu-
ar responses. At least, the monitoring of [Ca2+]m in intactells has become feasible by the recent addition of a series
f luminescent or green fluorescent protein-based fluorescentrobes and fluorophores.[43,46–48]. Evaluation of the effi-acy and specificity of the available protocols may provide andequate solution until the molecular structure of the mito-
ll Calci
ci
A
a
R
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
G. Hajnoczky et al. / Ce
hondrial Ca2+ transporters will be defined and new potentnhibitors will be synthetized.
cknowledgments
This work was supported by grants from the NIH to G.H.nd an AHA Scientist Development Grant 0435236N to G.C.
eferences
[1] D. Gincel, H. Zaid, V. Shoshan-Barmatz, Calcium binding and translo-cation by the voltage-dependent anion channel: a possible regula-tory mechanism in mitochondrial function, Biochem. J. 358 (2001)147–155.
[2] E. Rapizzi, P. Pinton, G. Szabadkai, M.R. Wieckowski, G. Vandecas-teele, G. Baird, R.A. Tuft, K.E. Fogarty, R. Rizzuto, Recombinantexpression of the voltage-dependent anion channel enhances the trans-fer of Ca2+ microdomains to mitochondria, J. Cell. Biol. 159 (2002)613–624.
[3] G. Bathori, G. Csordas, C. Garcia-Perez, E. Davies, G. Hajnoczky,Ca2+-dependent control of the permeability properties of the mito-chondrial outer membrane and VDAC, J. Biol. Chem. 281 (2006)17347–17358.
[4] Y. Kirichok, G. Krapivinsky, D.E. Clapham, The mitochondrial calciumuniporter is a highly selective ion channel, Nature 427 (2004) 360–364.
[5] P. Bernardi, Mitochondrial transport of cations: channels, exchangers,and permeability transition, Physiol. Rev. 79 (1999) 1127–1155.
[6] D.R. Pfeiffer, T.E. Gunter, R. Eliseev, K.M. Broekemeier, K.K. Gunter,Release of Ca2+ from mitochondria via the saturable mechanisms andthe permeability transition, IUBMB Life 52 (2001) 205–212.
[7] E. Carafoli, Historical review: mitochondria and calcium: ups anddowns of an unusual relationship, Trends Biochem. Sci. 28 (2003)175–181.
[8] D.G. Nicholls, S. Chalmers, The integration of mitochondrial calciumtransport and storage, J. Bioenerg. Biomembr. 36 (2004) 277–281.
[9] C. Krieger, M.R. Duchen, Mitochondria Ca2+ and neurodegenerativedisease, Eur. J. Pharmacol. 447 (2002) 177–188.
10] E. Basso, L. Fante, J. Fowlkes, V. Petronilli, M.A. Forte, P. Bernardi,Properties of the permeability transition pore in mitochondria devoidof Cyclophilin D, J. Biol. Chem. 280 (2005) 18558–18561.
11] A.A. Starkov, C. Chinopoulos, G. Fiskum, Mitochondrial calcium andoxidative stress as mediators of ischemic brain injury, Cell Calcium 36(2004) 257–264.
12] L. Scorrano, D. Penzo, V. Petronilli, F. Pagano, P. Bernardi, Arachidonicacid causes cell death through the mitochondrial permeability transitionImplications for tumor necrosis factor-alpha aopototic signalling, J.Biol. Chem. 276 (2001) 12035–12040.
13] D.R. Green, G. Kroemer, The pathophysiology of mitochondrial celldeath, Science 305 (2004) 626–629.
14] G. Szalai, R. Krishnamurthy, G. Hajnoczky, Apoptosis driven by IP(3)-linked mitochondrial calcium signals, EMBO J. 18 (1999) 6349–6361.
15] P. Pinton, D. Ferrari, E. Rapizzi, F. Di Virgilio, T. Pozzan, R. Rizzuto,The Ca2+ concentration of the endoplasmic reticulum is a key deter-minant of ceramide-induced apoptosis: significance for the molecularmechanism of Bcl-2 action, EMBO J. 20 (2001) 2690–2701.
16] P. Pacher, G. Hajnoczky, Propagation of the apoptotic signal by mito-chondrial waves, EMBO J. 20 (2001) 4107–4121.
17] J. Jacobson, M.R. Duchen, Mitochondrial oxidative stress and cell death
in astrocytes—requirement for stored Ca2+ and sustained opening ofthe permeability transition pore, J. Cell. Sci. 115 (2002) 1175–1188.18] R. Rizzuto, M. Brini, M. Murgia, T. Pozzan, Microdomains with highCa2+ close to IP3-sensitive channels that are sensed by neighboringmitochondria, Science 262 (1993) 744–747.
[
um 40 (2006) 553–560 559
19] R. Rizzuto, P. Pinton, W. Carrington, F.S. Fay, K.E. Fogarty, L.M.Lifshitz, R.A. Tuft, T. Pozzan, Close contacts with the endoplasmicreticulum as determinants of mitochondrial Ca2+ responses, Science280 (1998) 1763–1766.
20] G. Csordas, A.P. Thomas, G. Hajnoczky, Quasi-synaptic calcium sig-nal transmission between endoplasmic reticulum and mitochondria,EMBO J. 18 (1999) 96–108.
21] X. Lin, P. Varnai, G. Csordas, A. Balla, T. Nagai, A. Miyawaki, T. Balla,G. Hajnoczky, Control of calcium signal propagation to the mitochon-dria by inositol 1,4,5-trisphosphate-binding proteins, J. Biol. Chem.280 (2005) 12820–12832.
22] D. Boehning, R.L. Patterson, L. Sedaghat, N.O. Glebova, T. Kurosaki,S.H. Snyder, Cytochrome c binds to inositol (1,4,5) trisphosphate recep-tors, amplifying calcium-dependent apoptosis, Nat. Cell Biol. 5 (2003)1051–1061.
23] D.B. Zorov, C.R. Filburn, L.O. Klotz, J.L. Zweier, S.J. Sollott, Reac-tive oxygen species (ROS)-induced ROS release: a new phenomenonaccompanying induction of the mitochondrial permeability transitionin cardiac myocytes, J. Exp. Med. 192 (2000) 1001–1014.
24] Y. Zheng, X. Shen, H2O2 directly activates inositol 1,4,5-trisphosphatereceptors in endothelial cells, Redox Rep. 10 (2005) 29–36.
25] L. Scorrano, S.A. Oakes, J.T. Opferman, E.H. Cheng, M.D. Sorcinelli,T. Pozzan, S.J. Korsmeyer, BAX and BAK regulation of endoplas-mic reticulum Ca2+: a control point for apoptosis, Science 300 (2003)135–139.
26] R. Chen, I. Valencia, F. Zhong, K.S. McColl, H.L. Roderick, M.D.Bootman, M.J. Berridge, S.J. Conway, A.B. Holmes, G.A. Mignery,P. Velez, C.W. Distelhorst, Bcl-2 functionally interacts with inositol1,4,5-trisphosphate receptors to regulate calcium release from the ERin response to inositol 1,4,5-trisphosphate, J. Cell. Biol. 166 (2004)193–203.
27] S.A. Oakes, L. Scorrano, J.T. Opferman, M.C. Bassik, M. Nishino, T.Pozzan, S.J. Korsmeyer, Proapoptotic BAX and BAK regulate the type1 inositol trisphosphate receptor and calcium leak from the endoplasmicreticulum, Proc. Natl. Acad. Sci. U.S.A. 102 (2005) 105–110.
28] C. White, C. Li, J. Yang, N.B. Petrenko, M. Madesh, C.B. Thomp-son, J.K. Foskett, The endoplasmic reticulum gateway to apoptosis byBcl-X(L) modulation of the InsP3R, Nat. Cell Biol. 7 (2005) 1021–1028.
29] J.P. Mathai, M. Germain, G.C. Shore, BH3-only BIK regulatesBAX,BAK-dependent release of Ca2+ from endoplasmic reticulumstores and mitochondrial apoptosis during stress-induced cell death,J. Biol. Chem. 280 (2005) 23829–23836.
30] G. Csordas, M. Madesh, B. Antonsson, G. Hajnoczky, tcBid promotesCa(2+) signal propagation to the mitochondria: control of Ca(2+) perme-ation through the outer mitochondrial membrane, EMBO J. 21 (2002)2198–2206.
31] N. Minagawa, E.A. Kruglov, J.A. Dranoff, M.E. Robert, G.J. Gores,M.H. Nathanson, The anti-apoptotic protein Mcl-1 inhibits mitochon-drial Ca2+ signals, J. Biol. Chem. 280 (2005) 33637–33644.
32] K. Bianchi, G. Vandecasteele, C. Carli, A. Romagnoli, G. Szabadkai, R.Rizzuto, Regulation of Ca2+ signalling and Ca2+-mediated cell deathby the transcriptional coactivator PGC-1alpha, Cell Death Differ. 13(2006) 586–596.
33] M. Yi, D. Weaver, G. Hajnoczky, Control of mitochondrial motility anddistribution by the calcium signal: a homeostatic circuit, J. Cell. Biol.167 (2004) 661–672.
34] D. Brough, M.J. Schell, R.F. Irvine, Agonist-induced regulation ofmitochondrial and endoplasmic reticulum motility, Biochem. J. 392(2005) 291–297.
35] G. Szabadkai, A.M. Simoni, M. Chami, M.R. Wieckowski, R.J. Youle,R. Rizzuto, Drp-1-dependent division of the mitochondrial network
blocks intraorganellar Ca2+ waves and protects against Ca2+-mediatedapoptosis, Mol. Cell 16 (2004) 59–68.36] V.K. Sharma, V. Ramesh, C. Franzini-Armstrong, S.S. Sheu, Transportof Ca2+ from sarcoplasmic reticulum to mitochondria in rat ventricularmyocytes, J. Bioenerg. Biomembr. 32 (2000) 97–104.

5 ll Calci
[
[
[
[
[
[
[
[
[
[
[
[fluorescent proteins, Cell Calcium 38 (2005) 213–222.
60 G. Hajnoczky et al. / Ce
37] V.M. Shkryl, N. Shirokova, Transfer and tunneling of Ca2+ from sar-coplasmic reticulum to mitochondria in skeletal muscle, J. Biol. Chem.281 (2006) 1547–1554.
38] D.G. Nicholls, Simultaneous monitoring of ionophore- and inhibitor-mediated plasma and mitochondrial membrane potential changes incultured neurons, J. Biol. Chem. 281 (2006) 14864–14874.
39] J.G. McCormack, P.J. England, Ruthenium Red inhibits the activationof pyruvate dehydrogenase caused by positive inotropic agents in theperfused rat heart, Biochem. J. 214 (1983) 581–585.
40] G. Czirjak, P. Enyedi, Formation of functional heterodimers betweenthe TASK-1 and TASK-3 two-pore domain potassium channel subunits,J. Biol. Chem. 277 (2002) 5426–5432.
41] R. Amann, C.A. Maggi, Ruthenium red as a capsaicin antagonist, LifeSci. 49 (1991) 849–856.
42] L. Hymel, H. Schindler, M. Inui, S. Fleischer, Reconstitution of purified
cardiac muscle calcium release channel (ryanodine receptor) in planarbilayers, Biochem. Biophys. Res. Commun. 152 (1988) 308–314.43] T. Nagai, A. Sawano, E.S. Park, A. Miyawaki, Circularly permutedgreen fluorescent proteins engineered to sense Ca2+, Proc. Natl. Acad.Sci. U.S.A. 98 (2001) 3197–3202.
[
um 40 (2006) 553–560
44] M.A. Matlib, Z. Zhou, S. Knight, S. Ahmed, K.M. Choi, J. Krause-Bauer, R. Phillips, R. Altschuld, Y. Katsube, N. Sperelakis, D.M.Bers, Oxygen-bridged dinuclear ruthenium amine complex specificallyinhibits Ca2+ uptake into mitochondria in vitro and in situ in single car-diac myocytes, J. Biol. Chem. 273 (1998) 10223–10231.
45] P.C. Waldmeier, J.J. Feldtrauer, T. Qian, J.J. Lemasters, Inhibitionof the mitochondrial permeability transition by the nonimmunosup-pressive cyclosporin derivative NIM811, Mol. Pharmacol. 62 (2002)22–29.
46] R. Rudolf, M. Mongillo, R. Rizzuto, T. Pozzan, Looking forward toseeing calcium, Nat. Rev. Mol. Cell. Biol. 4 (2003) 579–586.
47] O. Gerasimenko, A. Tepikin, How to measure Ca2+ in cellularorganelles? Cell Calcium 38 (2005) 201–211.
48] N. Demaurex, Calcium measurements in organelles with Ca2+-sensitive
49] G. Csordas, C. Renken, P. Varnai, L. Walter, D. Weaver, K.F. Buttle, T.Balla, C.A. Mannella, G. Hajnoczky, Structural and functional featuresand significance of the physical linkage between ER and mitochondria,J. Cell. Biol. 174 (7) (2006) 915–921.
Copyright © 2022 FDOKUMEN




















