
Adsorption of Cd,Zn-metallothionein on covered Hg electrodes and its voltammetric determination
Upload
independentCategory
view
0download
0
Metallothionein Reduces Central NervousSystem Inflammation, Neurodegeneration,and Cell Death Following Kainic Acid-Induced Epileptic Seizures
Milena Penkowa,1 Sergi Florit,2 Mercedes Giralt,2 Albert Quintana,2
Amalia Molinero,2 Javier Carrasco,2 and Juan Hidalgo2*1Department of Medical Anatomy, The Panum Institute, University of Copenhagen, Copenhagen, Denmark2Institute of Neurosciences and Department of Cellular Biology, Physiology and Immunology,Autonomous University of Barcelona, Barcelona, Spain
We examined metallothionein (MT)-induced neuropro-tection during kainic acid (KA)-induced excitotoxicity bystudying transgenic mice with MT-I overexpression(TgMT mice). KA induces epileptic seizures and hip-pocampal excitotoxicity, followed by inflammation anddelayed brain damage. We show for the first time thateven though TgMT mice were more susceptible to KA,the cerebral MT-I overexpression decreases the hip-pocampal inflammation and delayed neuronal degener-ation and cell death as measured 3 days after KA admin-istration. Hence, the proinflammatory responses ofmicroglia/macrophages and lymphocytes and their ex-pression of interleukin (IL)-1, IL-6, IL-12, tumor necrosisfactor-� and matrix metalloproteinases (MMP-3, MMP-9)were significantly reduced in hippocampi of TgMT micerelative to wild-type mice. Also by 3 days after KA, theTgMT mice showed significantly less delayed damage,such as oxidative stress (formation of nitrotyrosine,malondialdehyde, and 8-oxoguanine), neurodegenera-tion (neuronal accumulation of abnormal proteins), andapoptotic cell death (judged by TUNEL and activatedcaspase-3). This reduced bystander damage in TgMTmice could be due to antiinflammatory and antioxidantactions of MT-I but also to direct MT-I effects on theneurons, in that significant extracellular MT presencewas detected. Furthermore, MT-I overexpression stimu-lated astroglia and increased immunostaining of antiin-flammatory IL-10, growth factors, and neurotrophins(basic fibroblastic growth factor, transforming growthfactor-�, nerve growth factor, brain-derived neurotrophicfactor, glial-derived neurotrophic factor) in hippocampus.Accordingly, MT-I has different functions that likely con-tribute to the increased neuron survival and improvedCNS condition of TgMT mice. The data presented hereadd new insight into MT-induced neuroprotection andindicate that MT-I therapy could be used against neuro-logical disorders. © 2004 Wiley-Liss, Inc.
Key words: excitotoxicity; inflammation; neuroprotec-tion; apoptosis
Kainic acid (KA) is a glutamate receptor agonist thatinduces significant excitotoxicity in the hippocampus,which results in epileptic seizures as well as neuronal celldeath mainly in the hippocampal CA1 and CA3 subfields(Faherty et al., 1999; Carrasco et al., 2000; Kim et al.,2003). The KA-induced damage is followed by inflamma-tory responses, cytokine expression, formation of reactiveoxygen species (ROS), and oxidative stress in the hip-pocampal area (Carrasco et al., 2000; Chung and Han,2003; Lehtimaki et al., 2003), so intervening in theseprocesses might be a useful therapeutic approach. To this
Contract grant sponsor: The Lundbeck Foundation; Contract grant spon-sor: The Danish Medical Research Council; Contract grant sponsor: Kath-rine og Vigo Skovgaards Fond; Contract grant sponsor: Hørslev-fonden;Contract grant sponsor: Novo Nordisk Fonden; Contract grant sponsor:Toyota Fonden; Contract grant sponsor: Direktør Ib Henriksens Fond;Contract grant sponsor: The Danish Medical Association Research Fund—The Wacherhausens Legat; Contract grant sponsor: Grosserer Johan Quen-tin og Hustrus Legat; Contract grant sponsor: Dagmar Marshalls Fond;Contract grant sponsor: Dir. Jacob Madsen’s Fond; Contract grant sponsor:Eva og Henry Frænkels Mindefond; Contract grant sponsor: Sclerose-foreningen; Contract grant sponsor: Warwara Larsens Fond; Contract grantsponsor: Fru Lily Benthine Lunds Fond; Contract grant sponsor: RagnhildIbsens Legat for Medicinsk Forskning; Contract grant sponsor: Fonden tilLægevidenskabens Fremme; Contract grant sponsor: Haensch’s Fond; Con-tract grant sponsor: Fonden af 17.12.1981; Contract grant sponsor: Johannog Hanne Weimanns Legat; Contract grant sponsor: Vald. Foersom ogHustrus Fond; Contract grant sponsor: Ministerio de Ciencia y Tecnologıaand Feder; Contract grant number: SAF2002-01268; Contract grant spon-sor: Direccio General de Recerca; Contract grant number: 2001SGR00203.
*Correspondence to: Dr. Juan Hidalgo, Departamento de Biologıa Celular,Fisiologıa e Inmunologıa, Unidad de Fisiologıa Animal, Facultad de Cien-cias, Universidad Autonoma de Barcelona, Bellaterra, Barcelona, Spain08193. E-mail: [email protected]
Received 16 September 2004; Revised 1 November 2004; Accepted2 November 2004
Published online 21 December 2004 in Wiley InterScience (www.interscience.wiley.com). DOI: 10.1002/jnr.20387
Journal of Neuroscience Research 79:522–534 (2005)
© 2004 Wiley-Liss, Inc.

end, accumulating studies have shown that CNS cell deathas seen during a range of neuropathological conditions canbe ameliorated by neurotrophins and neuroprotective fac-tors (Alberch et al., 2004; Tabakman et al., 2004; Villo-slada and Genain, 2004), among which metallothionein-Iand -II (MT-I � II) may have a therapeutic potential(Penkowa and Hidalgo, 2000, 2003; Giralt et al., 2002b;Penkowa et al., 2002; Trendelenburg et al., 2002; Chunget al., 2003; Carmel et al., 2004).
MT-I � II are low-molecular-weight, cysteine-richproteins that induce neuroprotection during various neu-ropathological conditions and brain disorders (for reviewsee Hidalgo et al., 2001, 2002; Coyle et al., 2002; Chungand West, 2004). MT-I � II appear to have different rolesin the central nervous system (CNS), where they cansignificantly reduce 1) inflammatory responses, includingexpression of proinflammatory cytokines; 2) oxidativestress and formation of reactive oxygen species (ROS; freeradicals); and 3) apoptotic cell death and progression ofneural damage. During excitotoxicity, free radical forma-tion is increased, leading to oxidative injury in the hip-pocampus (Carrasco et al., 2000; Ueda et al., 2002; Chungand Han, 2003; Rego and Oliveira, 2003), which con-tributes to neuronal cell death (Bishop and Cashman,2003; Bossy-Wetzel et al., 2003; Mattson, 2003; Rego andOliveira, 2003). Thus, our previous findings in MT-I �II-deficient mice demonstrating increased oxidative stress,neuron death, and epileptic seizures following KA stronglysuggested a neuroprotective role of these proteins in thismodel of excitotoxicity (Carrasco et al., 2000). This isimportant considering that, in fact, excitotoxicity-inducedneuropathology might be involved not only in epilepsybut also in other acute neurological diseases, such as strokeand neurotrauma, as well as in chronic neurodegeneration,for instance, during Alzheimer’s, Parkinson’s, and Hun-tington’s diseases and amyotrophic lateral sclerosis (ALS;for review see Mattson, 2003; Rego and Oliveira, 2003).It is likely that increasing the MT-I and/or -II levelsduring excitotoxicity could reduce the cerebral damageand improve neuron survival, but this has not been stud-ied. By using transgenic mice overexpressing MT-I(TgMT mice), we hereby show for the first time thatendogenous MT-I overexpression sensitizes mice to KA-induced seizures, but that, despite this, it inhibits thedelayed damage (measured 3 days after KA administration)in the CA3 hippocampal area, whereby MT-I and/or -IIcould be a therapeutic target for counteracting neurolog-ical diseases.
MATERIALS AND METHODS
Animals
Homozygous breeding pairs of TgMT mice in a C57BL/6 genetic background (Palmiter et al., 1993) were purchasedfrpm The Jackson Laboratory [C57BL/6J-Tg(Mt1)174Bri/J].These mice carry 56 copies of a minimally mutated Mt1 (Mt1*)gene; as controls, C57BL/6 mice were used. The animal exper-iments were carried out in accordance with the proper NationalCommittees of Animal Research as well as the National Insti-
tutes of Health guidelines and the European CommunitiesCouncil Directive of 24 November 1986. All efforts were madeto minimize animal suffering and to reduce the number ofanimals used.
Experimental Procedures
To induce epileptic seizures, mice were injected i.p.,based on previous results (Penkowa et al., 2001), with 10 mg/kgkainic acid (KA; Sigma Aldrich, St. Louis, MO; code K-0250),which is a glutamate receptor agonist with excitotoxic effects. Inthe C57BL/6 genetic background, this KA dosage causes sig-nificant seizures activity in most of the mice, with low mortality.Saline i.p. injection was used as control treatment. Twenty-fiveTgMT mice were injected with either KA (n � 20) or saline(n � 5), whereas 28 wild-type mice received either KA (n � 22)or saline (n � 6). All mice were 3–4-month-old males. AfterKA injection, the behavior of each mouse was recorded onvideo tape for 120 min for further analysis, which was carriedout in 11 wild-type and 15 TgMT mice. The time of latencyand the number and duration of limb clonus and tonic-clonicconvulsions were quantitated as an index of the total seizureactivity. Others seizure-associated behaviors were also measured(see Table I).
Three days after KA or saline injection, the mice weredeeply anesthetized and fixed by vascular perfusion, after whichthe brain tissue was processed as described earlier (Penkowa etal., 2000, 2003). The brains were cut into 3-�m coronal sec-tions, which were processed as described previously (Penkowaet al., 2000, 2003). We focused our histopathological analysis onthe hippocampal CA3 area.
Histochemistry
Stainings for H&E were done according to standard proce-dures. Stainings with tomato lectin from Lycopersicon esculentum(Sigma; code L9389) were used to identify microglia/macrophagesas previously described (Penkowa et al., 2003). Lectin staining alsodemonstrates blood vessels.
Immunohistochemistry
Sections were incubated overnight at 4°C with one of thefollowing primary antibodies: rabbit anti-GFAP 1:250 (Dako-patts, Copenhagen, Denmark; code Z 334); rat anti-MOMA-11:20 (Serotec, Bicester, United Kingdom; code MCA-947;monocyte-macrophage marker-1; a marker of peripherally de-rived, activated macrophages); mouse anti-CD3 1:50 (Serotec;code MCA-772; as a marker for all T lymphocytes); rabbitanti-MT-1 � 2 1:500 (Gasull et al., 1993; Penkowa et al.,2002); mouse antiinterleukin-1� (IL-1�) 1:50 (Biogenesis; code5375-4329); rat anti-IL-6 1:50 (Harlan Seralab, Indianapolis,IN; code MAS584); mouse anti-IL-10 1:50 (Biosource; codeARC9104); rat anti-IL-12 1:50 (Biosource; code AMC0122);rabbit antitumor necrosis factor (TNF-�) 1:100 (Biosource;code AMC 3012); rabbit antibasic fibroblastic growth factor(bFGF) 1:100 (Santa Cruz Biotechnology, Snata Cruz, CA;code sc-79); rabbit antitransforming growth factor-� (TGF-�) 1:200 (Santa Cruz Biotechnology; code sc-146); mouseantibrain-derived neurotrophic factor (BDNF) 1:50 (OncogeneResearch Products, Germany; code GF35L); mouse antiglial-derived neurotrophic factor (GDNF) 1:50 (Oncogene Research
Metallothionein Has Neuroprotective Actions 523

Products, Germany; code GF38L); rabbit antinerve growth fac-tor (NGF) 1:50 (Chemicon, United Kingdom; code AB927);mouse antimatrix metalloproteinase-3 (MMP-3) 1:100 (Neo-markers; code MS-810-P1ABX); mouse anti-MMP-9 1:100(Neomarkers; code MS-820-P1ABX); mouse anti-8-oxo-guanine 1:100 (Chemicon; code MAB-3560; marking a freeradical-induced base modification in the DNA); rabbit antini-trotyrosine (NITT) 1:100 (Alpha Diagnostics Int.; code NITT12-A; marker for peroxynitrite-induced nitration of tyrosine
residues); rabbit antimalondialdehyde (MDA) 1:100 (Alpha Di-agnostics Int.; code M0DA 11-S; a marker for byproducts offatty acid peroxidation); mouse anti-Cu/Zn-superoxide dis-mutase (Cu/Zn-SOD) 1:50 (Sigma-Aldrich; code S2147); sheepanticatalase 1:50 (The Binding Site, United Kingdom; codePC136); rabbit antineurofibrillary tangles 1:200 (Chemicon;code AB1518; marking all neurofibrillary tangles/neurodegen-eration); goat antiamyloid precursor protein (APP; the mutatedAPP form) 1:50 (Chemicon; code AB5342); and rabbit anti-caspase-3 (activated/cleaved caspase-3) 1:50 (Cell SignalingTechnology, Bevrly, MA; code 9661; a marker for apoptoticsignaling).
When a mouse-derived primary antibody was used forimmunohistochemistry, the sections were preincubated withBlocking Solutions A � B from the HistoMouse-SP Kit(Zymed, South San Francisico, CA; code 95-9544), to quenchendogenous mouse IgG. The primary antibodies were detectedusing biotinylated anti-mouse IgG 1:200 (Sigma; code B8774),or biotinylated anti-mouse IgM (� chain specific) 1:50 (JacksonImmunoresearch, West Grove, PA; code 115-065-020), or bi-otinylated anti-rabbit IgG 1:400 (Sigma; code B3275), or bio-tinylated anti-rat IgG 1:1,500 (Amersham, Amersham, UnitedKingdom; code RPN 1005), or biotinylated anti-goat/sheepIgG 1:20 (Amersham; code RPN 1025), followed by stre-ptavidin-biotin-peroxidase complex (StreptABComplex/HRP;Dakopatts; code K377) prepared at the manufacturer’s recom-mended dilution. These secondary and tertiary steps in theimmunoreaction were performed for 30 min at room temper-ature. Afterward, sections were incubated with biotinylated
Fig. 1. H&E stainings of hippocampal CA3 area. As expected, the morphology of the CA3 area of theTgMT mice (B) was comparable to that of wild-type mice (A) after saline treatment. Three days afterKA injection, wild-type mice (C) showed severe disorganization of the CA3 neuronal layers and cellloss, effects dramatically decreased in the TgMT mice (D). Scale bars � 56 �m.
TABLE I. Response of TgMT and Wild-Type Mice to KainicAcid Administration*
Wild type TgMT P
Mortality 0/22 3/20Animals convulsing 9/11 13/15Latency time (min) 51.4 � 8.1 34.1 � 4.6 0.058Number of convulsions 9.4 � 2.6 33.4 � 14.8 0.14Time of convulsion (sec) 14.0 � 3.2 118.5 � 42.9 0.025Animals scratching 1/9 7/13Unilateral clonic-tonic 4/9 10/13Bilateral clonic-tonic 9/9 13/13Full clonic-tonic 0/9 3/13Unable to stand 0/9 4/13Animals jumping 0/9 3/13Animals grooming 9/9 11/13
*Mice were injected intraperitoneally with kainic acid at 10 mg/kg bodyweight. Latency time, number of convulsions, and convulsion time aremean � SEM (WT, n � 9; TgMT, n � 13).
524 Penkowa et al.

tyramide and streptavidin-peroxidase complex (tyramide signalamplification, TSA indirect; NEN, Life Science Products, Bos-ton, MA; code NEL700A) prepared following the manufac-turer’s recommendations. The immunoreaction was visualizedby using 0.015% H2O2 in DAB/TBS for 10 min at roomtemperature.
To evaluate the extent of nonspecific binding in theimmunohistochemical experiments, control sections were incu-bated in normal goat or donkey serum or in the absence ofprimary antibody, secondary antibody, the StreptABComplex/HRP, or the TSA indirect. A further control was to preabsorbthe primary antibodies with the corresponding antigenic pro-teins (such as IL-1�, IL-6, TNF-�, bFGF), as described previ-ously (Penkowa et al., 2003). Results were considered only ifthese controls were negative.
In Situ Detection of DNA Fragmentation (TUNEL)
TUNEL was performed as previously described (Carrascoet al., 2000; Penkowa et al., 2003).
Cell Counts
For most of the variables studied, cell counts were carriedout in the CA3 hippocampal area in five mice per group and ina blinded manner, which allowed statistical analysis. Five miceper group were used, and, from each, one 0.5-mm2 microscopicfield was examined in matched CA3 areas of 3-�m brain sec-tions. The cell counts were made in a blinded and randomizedmanner by the same independent attending person, who wasblinded to the animal identity, genotype, and KA vs. salineinjection. To this end, positively stained cells, defined as cellswith staining of the soma, or in the case of TUNEL andcaspase-3 cells with nuclear staining, were counted.
Statistical Analysis
The results for behavioral traits induced by KA wereanalyzed by Student’s t-test or by the Mann-Whitney U-testdepending on the homogeneity of variances. The results ofcellular counts were evaluated by two-way ANOVA, with strainand KA injection as the main factors.
RESULTS
SeizuresThe seizures-related behaviors were recorded for
2 hr after KA administration; results are shown in Table I.As expected, the convulsive activity elicited by 10 mg/kgKA was not dramatic. The results together strongly suggestthat the TgMT mice are more susceptible to KA thanwild-type mice: Mortality was observed only in them, andthe latency time was less and the number and time ofconvulsions greater in the TgMT mice. A more detailedinspection of the behavioral traits elicited by KA alsosuggested a higher susceptibility of the TgMT mice, es-pecially regarding the number of animals scratching,showing unilateral or full clonic-tonic convulsions, orjumping. Finally, KA treatment caused a transient, smallbody weight loss (0.66 � 0.14 and 0.52 � 0.13 g for wildtype and TgMT, respectively, at 24 hr), although it wasnot statistically significant (P � 0.109).
H&E StainingsThe general morphology of the brain was analyzed
by using H&E stainings. A general inspection of the dam-age caused by KA (after 3 days) demonstrated that thehippocampus was the most affected brain area, in line withprevious results (Carrasco et al., 2000; Penkowa et al.,2001), so we have focused our analysis on the hippocampal
Fig. 2. MT-I � II immunoreactivity in CA3 of KA injected wild-type(A) and TgMT (B) mice. The MT-I � II immunoreactivity is in-creased 3 days after KA-induced seizures, especially in reactive astro-cytes. The number of cells positive for MT-I � II was higher in TgMTmice, which also showed significant MT-I � II immunostaining in theextracellular hippocampal tissue, something that was not observed inthe wild-type mice. Specificity of the immunostaining was fully dem-onstrated by the complete lack of staining in KA-injected MT-I � IIdeficient mice (C). Scale bars � 32 �m.
Metallothionein Has Neuroprotective Actions 525

area and especially on the CA3 subfield. As expected, nodifferences between strains were observed in the CA3 areaof saline-injected animals (Fig. 1A,B). After KA injection,the wild-type mice showed a severe disorganization of theneuronal layers of Ammon’s horn and neuron loss subfieldCA3 (Fig. 1C), an effect dramatically decreased in theTgMT mice (Fig. 1D).
MT-I � II ExpressionAs expected (Giralt et al., 2002b; Penkowa et al.,
2002, 2003; Molinero et al., 2003), low levels of MT-I �II immunostaining were observed throughout the CNS,including the hippocampus, which were increased in theTgMT mice. As previously shown (Carrasco et al., 2000;Penkowa et al., 2001; Kim et al., 2003), KA-inducedseizures increased significantly MT-I � II immunohisto-chemistry in the hippocampal formation 3 days later,which reflects increased gene up-regulation (Carrasco etal., 2000). As could be expected, MT-I � II immunore-activity was much higher in TgMT mice (Fig. 2B) than inwild-type mice (Fig. 2A, Table II). In both genotypes,increased MT-I � II immunostaining was observed inreactive astroglia mainly, but also in the activatedmicroglia/macrophages, perivascular cells, and vessel en-dothelium (Fig. 2). A remarkable feature observed in theKA-injected TgMT mice was a significant extracellularMT-I � II immunoreactivity, especially around hypertro-phic reactive astroglia, which did not occur (or was notdetectable) in wild-type mice (Fig. 2). Specificity ofthe antibody used is fully demonstrated by the absence ofimmunostaining in KA-injected Mt-1 � 2-deficient mice(Carrasco et al., 2000; Fig. 2C).
Reactive AstrogliosisGFAP-positive astrocytes were observed throughout
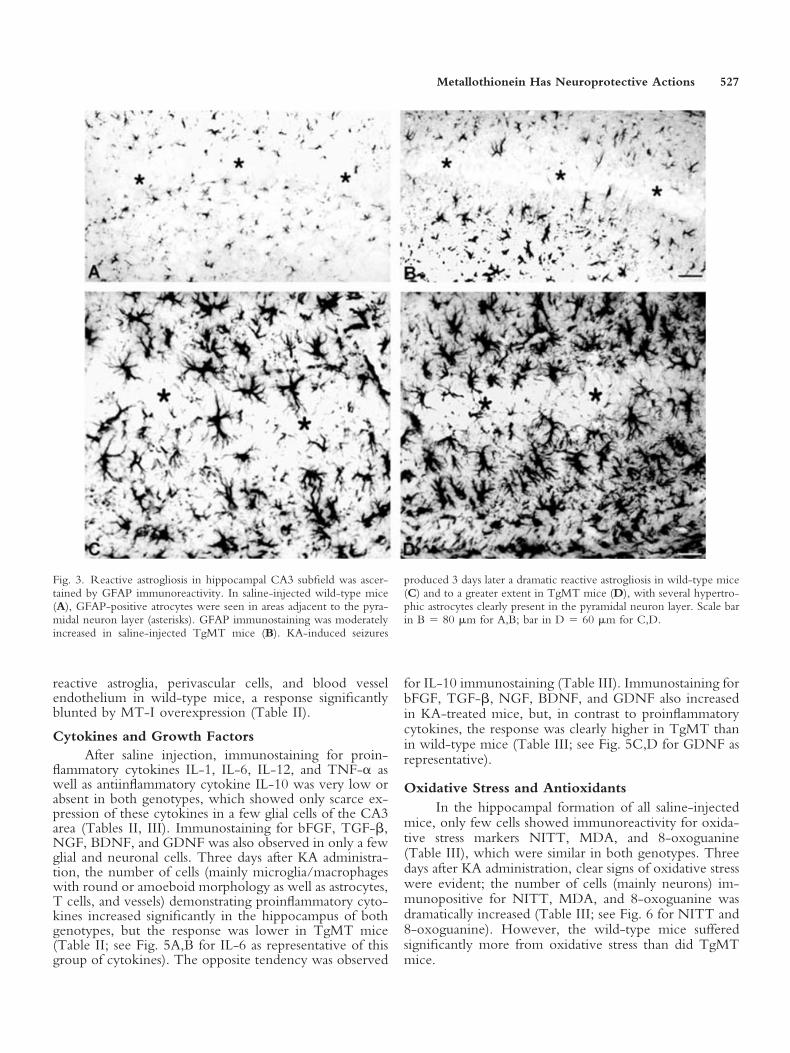
the hippocampal formation in areas adjacent to the pyra-midal neuron layer in saline-injected wild-type mice(Fig. 3A); a mild but clear astrogliosis was observed in
TgMT mice (Fig. 3B, Table II). After KA administration,reactive astrogliosis occurred in hippocampus of both ge-notypes as determined 3 days later, with the most pro-nounced astrogliosis occurring in the CA3 subfield, wherereactive astrocytes were infiltrating the pyramidal layers;astrogliosis was clearly higher in TgMT (Fig. 3D) than inwild-type mice (Fig. 3C, Table II).
Microglia/MacrophagesIn saline-injected mice, lectin staining was seen only
in some hippocampal blood vessels and in ramified micro-glia, indicative of resting cells, in both wild-type (Fig. 4A)and TgMT (Fig. 4B) mice. Three days after KA adminis-tration, wild-type mice showed many activated, round,or amoeboid microglia/macrophages in the CA3 area(Fig. 4C), a response dramatically blunted in TgMT mice(Fig. 4D, Table II). Results with MOMA-1 immunore-activity, which is observed only in peripheral monocytes/macrophages, indicate that few macrophages appeared tobe blood-derived peripheral cells (Table II).
LymphocytesIn all saline-injected mice, the brain tissue, including
the CA3 hippocampal area, was basically devoid of CD3-positive lymphocytes (Table II). Three days after KA-induced seizures, some CD3-positive T cells were seen inthe CA3 subfield in wild-type mice, an effect almosttotally blunted in TgMT mice (Table II).
MMP ExpressionMatrix metalloproteinases MMP-3 and MMP-9 are
endopeptidases mediating breakdown of the blood–brainbarrier (BBB) and invasion of activated, blood-derivedleukocytes into the brain (Leppert et al., 2001; Avolio etal., 2003), and, as expected, they were not detected insaline-injected mice (Table II). Three days after KA ad-ministration, MMP-3 and MMP-9 immunostainings in-creased in activated microglia/macrophages, lymphocytes,
TABLE II. Effect of MT-I Overexpression on KA-Induced Inflammation in the CA3 Subfield of theHippocampus (Positive Cells/0.5 mm2)†
Wild-type saline TgMT saline Wild-type KA TgMT KA
MT I � II 6.00 � 1.12 26.20 � 5.23* 58.80 � 4.52** 148.60 � 11.44*,**,***GFAP 8.20 � 1.39 12.20 � 1.85* 63.20 � 3.58** 125.60 � 8.35*,**,***Lectin 8.20 � 1.78 5.80 � 1.29* 75.40 � 9.33** 40.40 � 6.17*,**,***MOMA 0 � 0 0 � 0* 8.00 � 1.77** 1.00 � 0.71*,**,***CD-3 0 � 0 0 � 0* 7.20 � 1.68** 0.60 � 0.45*,**,***MMP-3 0 � 0 0 � 0* 28.00 � 4.40** 11.40 � 2.20*,**,***MMP-9 0 � 0 0 � 0* 22.80 � 3.61** 7.60 � 1.79*,**,***IL-1 1.40 � 0.57 1.20 � 0.42* 81.80 � 3.90** 20.60 � 3.10*,**,***IL-6 0.60 � 0.45 0.40 � 0.28* 83.80 � 3.80** 19.00 � 3.48*,**,***IL-12 0 � 0 0 � 0* 83.20 � 5.52** 22.60 � 2.89*,**,***TNF-� 0 � 0 0 � 0* 80.40 � 4.22** 16.40 � 3.23*,**,***†Mice were killed 3 days after saline or KA treatment. Results (mean � SEM; n � 5) were analyzed via two-wayANOVA with strain and treatment as main factors.*Effect of MT-I overexpression significant (P � 0.05).**Effect of KA administration significant (P � 0.05).***Interaction between both factors significant (P � 0.05).
526 Penkowa et al.
reactive astroglia, perivascular cells, and blood vesselendothelium in wild-type mice, a response significantlyblunted by MT-I overexpression (Table II).
Cytokines and Growth FactorsAfter saline injection, immunostaining for proin-
flammatory cytokines IL-1, IL-6, IL-12, and TNF-� aswell as antiinflammatory cytokine IL-10 was very low orabsent in both genotypes, which showed only scarce ex-pression of these cytokines in a few glial cells of the CA3area (Tables II, III). Immunostaining for bFGF, TGF-�,NGF, BDNF, and GDNF was also observed in only a fewglial and neuronal cells. Three days after KA administra-tion, the number of cells (mainly microglia/macrophageswith round or amoeboid morphology as well as astrocytes,T cells, and vessels) demonstrating proinflammatory cyto-kines increased significantly in the hippocampus of bothgenotypes, but the response was lower in TgMT mice(Table II; see Fig. 5A,B for IL-6 as representative of thisgroup of cytokines). The opposite tendency was observed
for IL-10 immunostaining (Table III). Immunostaining forbFGF, TGF-�, NGF, BDNF, and GDNF also increasedin KA-treated mice, but, in contrast to proinflammatorycytokines, the response was clearly higher in TgMT thanin wild-type mice (Table III; see Fig. 5C,D for GDNF asrepresentative).
Oxidative Stress and AntioxidantsIn the hippocampal formation of all saline-injected
mice, only few cells showed immunoreactivity for oxida-tive stress markers NITT, MDA, and 8-oxoguanine(Table III), which were similar in both genotypes. Threedays after KA administration, clear signs of oxidative stresswere evident; the number of cells (mainly neurons) im-munopositive for NITT, MDA, and 8-oxoguanine wasdramatically increased (Table III; see Fig. 6 for NITT and8-oxoguanine). However, the wild-type mice sufferedsignificantly more from oxidative stress than did TgMTmice.
Fig. 3. Reactive astrogliosis in hippocampal CA3 subfield was ascer-tained by GFAP immunoreactivity. In saline-injected wild-type mice(A), GFAP-positive atrocytes were seen in areas adjacent to the pyra-midal neuron layer (asterisks). GFAP immunostaining was moderatelyincreased in saline-injected TgMT mice (B). KA-induced seizures
produced 3 days later a dramatic reactive astrogliosis in wild-type mice(C) and to a greater extent in TgMT mice (D), with several hypertro-phic astrocytes clearly present in the pyramidal neuron layer. Scale barin B � 80 �m for A,B; bar in D � 60 �m for C,D.
Metallothionein Has Neuroprotective Actions 527

Fig. 4. Microglia/macrophages were judged by using lectin histochemistry. In saline-injected mice, lectinstainings showed only some vessels and resting microglia in both wild-type (A) and TgMT (B) mice. Threedays after KA-induced seizures, many activated, round microglia/macrophages were observed in the CA3area of wild-type mice (C), a response significantly blunted in TgMT mice (D). Scale bars � 33 �m.
TABLE III. Effect of MT-I Overexpression on KA-Induced Growth Factors, Neurotropins andAnti-oxidants as well as KA-Involved Oxidative Stress, Degeneration and Apoptosis in the CA3Subfield of the Hippocampus†
Wild-type saline TgMT saline Wild-type KA TgMT KA
IL-10 0 � 0 0 � 0 26.00 � 2.85** 34.80 � 5.51**bFGF 4.60 � 1.61 4.40 � 2.02* 41.00 � 5.42** 59.20 � 5.46*,**,***BDNF 1.80 � 0.65 2.20 � 0.90* 31.80 � 4.71** 64.20 � 9.03*,**,***NGF 2.20 � 0.74 2.80 � 1.30* 28.00 � 4.68** 46.40 � 6.93*,**,***GDNF 3.60 � 1.35 4.80 � 1.20* 34.20 � 4.04** 54.40 � 4.86*,**,***Cu/Zn-SOD 5.80 � 0.96 4.40 � 1.61 55.80 � 5.40** 49.80 � 6.70**Catalase 7.40 � 1.61 6.20 � 1.20 28.60 � 3.60** 24.60 � 2.78**NITT 1.40 � 0.57 0.60 � 0.28* 38.60 � 2.71** 14.20 � 2.44*,**,***MDA 1.00 � 0.36 0.80 � 0.65* 38.00 � 3.41** 13.80 � 2.22*,**,***8-Oxoguanine 1.40 � 0.76 1.00 � 0.61* 43.20 � 2.97** 14.20 � 2.25*,**,***Neurofibrilary tangles 0 � 0 0 � 0* 15.00 � 2.48** 7.40 � 1.35*,**,***APP 0 � 0 0 � 0* 18.60 � 4.62** 4.60 � 1.89*,**,***TUNEL 1.00 � 0.36 0.40 � 0.28* 41.20 � 5.17** 15.80 � 2.22*,**,***Caspase-3 2.20 � 0.65 1.20 � 0.42* 41.20 � 5.07** 14.80 � 2.16*,**,***†Mice were killed 3 days after saline or KA treatment. Results (mean � SEM; n � 5) were analyzed via two-wayANOVA with strain and treatment as main factors.*Effects of MT-I overexpression significant (P � 0.05).**Effect of KA administration significant (P � 0.05).***Interaction between both factors significant (P � 0.05).
528 Penkowa et al.

During the KA-induced oxidative injury, all miceincreased the number of cells demonstrating other anti-oxidants than MT-I � II such as Cu/Zn-SOD and cata-lase (Table III), which may counteract ROS and oxidativestress in the brain tissue (Nagano et al., 2001; Takuma etal., 2004). However, both strains showed comparable lev-els of Cu/Zn-SOD and catalase both after saline and KA(Table III). In wild-type mice, SOD and catalase wereseen in astroglia, microglia, and macrophages, whereas, inTgMT mice, these antioxidants were seen almost only inastroglia (not shown).
Neurodegeneration and ApoptosisTo examine neurodegeneration and delayed cell
death (apoptosis), we have immunostained for neuronalaggregation of neurofibrillary tangles and APP and forcleaved caspase-3 as well as TUNEL, respectively. Neuro-fibrillary tangles and APP were generally not detectable inthe brain tissue of all saline-injected mice (Table III).Three days after KA injection, neurofibrillary tangles andAPP appeared in all the mice, mainly in pyramidal neurons
of the CA3 area, but MT-I-overexpressing mice showeda dramatic reduction compared with wild-type mice(Table III, Fig. 7A–D). Signs of apoptosis were barelydetectable in saline-injected mice, as expected (Table III,Fig. 8A,B). In contrast, 3 days after KA, many CA3pyramidal neurons were positive for TUNEL and immu-nopositive for caspase-3, TgMT mice (Fig. 8D) showing adramatic reduction in apoptosis compared with wild-typemice (Fig. 8C; see also Table III). Signs of neurodegen-eration and apoptosis were also observed in other hip-pocampal areas (not shown).
DISCUSSIONThe present study shows for the first time the neuro-
pathological responses to KA-induced excitotoxicity intransgenic mice overexpressing MT-I. These mice weremore susceptible to KA-induced seizures, results in prin-ciple surprising in that MT-I � II KO mice are also moresusceptible (Carrasco et al., 2000). Zinc has long beensuggested to be related to seizure-induced neuronal dam-age, although much remains to be understood (Lee et al.,
Fig. 5. Expression of IL-6 and GDNF in hippocampal subfield CA3 3 days after KA administration.In wild-type mice (A), IL-6 was increased in microglia/macrophages, glial cells, T cells, and somevessels. In TgMT mice (B), IL-6 expression was reduced and appeared only in some glial cells and invessels. GDNF was increased in both wild-type (C) and to a greater extent in TgMT (D) mice. Scalebar in B � 40 �m for A,B; bar in D � 50 �m for C,D.
Metallothionein Has Neuroprotective Actions 529

Fig. 6. Oxidative stress markers NITT (A,B) and 8-oxoguanine (C,D) in CA3 3 days after KAinjection. In wild-type mice, many pyramidal neurons demonstrated NITT (A) and8-oxoguanine (C) immunoreactivities. KA-induced oxidative stress was dramatically decreased inTgMT mice, in which both NITT (B) and 8-oxoguanine (D) immunostainings were clearlyreduced. Scale bars � 54 �m.
Fig. 7. Neurodegeneration in hippocampal subfield CA3 3 days after KA treatment as judged byneuronal accumulation of neurofibrillary tangles (A,B) and APP (C,D). KA-induced immunoreac-tivities for neurofibrillary tangles and APP in CA3 neurons were higher in wild-type (A,C) than inTgMT (B,D) mice. Scale bars � 55 �m.
530 Penkowa et al.

2000), and MT-I and -II are major zinc-binding proteins:For peripheral tissues, it is clear that MT-I overexpressionincreases zinc accumulation and provides a biologicallyimportant labile pool of zinc (Dalton et al., 1996; Davis etal., 1998), and the same is expected to happen in the brain.Recent results obtained in cultured astrocytes, whereMT-II was overexpressed by means of an adenoviral vec-tor, further support a zinc-buffering role of MT as well asZn-MT being a releasable pool of this essential metal(Malaiyandi et al., 2004). The present results would sug-gest that both MT deficiency and MT excess are coupledto the induction of seizures, which deserves further atten-tion. Alterations in membrane-associated zinc transporters,glutamatergic and GABAergics receptors, and voltage-gated ion channels as a consequence of changes in MTlevels are among a number of factors that could be under-lying these paradoxical results (for an excellent review seeSensi and Jeng, 2004).
It is very remarkable that, despite being more vul-nerable to KA as far as seizures is concerned, the TgMTmice were resistant to KA-induced excitotoxic damage inthe hippocampus. Hence, 3 days after KA-induced sei-zures, more viable neurons and less oxidative stress andneurodegeneration were observed in the TgMT mice.These neuroprotective roles of MT-I are in agreementwith previous studies of mice with MT-I overexpressionor that are deficient in MT-I � II. Thus, after focal CNSischemia, transgenic TgMT mice show reduced cerebraledema, infarct size, neurodegeneration, and damage and,accordingly, better sensory and motor performance (Van
Lookeren Campagne et al., 1999). In contrast, mice withMT-I � II deficiency develop about threefold larger ce-rebral infarcts and poorer neurological outcome (Tren-delenburg et al., 2002). Moreover, MT-I and -II are likelyinvolved in the protective phenomenon ischemic precon-ditioning, in which a short episode of cerebral ischemiacan provide ischemic tolerance and protection againstsuccessive ischemia and infarcts (Carmel et al., 2004).Moreover, in transgenic mice with neurotrauma, MT-Ioverexpression protects against oxidative stress and apo-ptosis and enhances tissue repair, neuronal regrowth, andrevascularization (Giralt et al., 2002b; Penkowa et al.,2003). Hence, transgenic MT-I overexpression could in-hibit peroxynitrite-induced cell injury, fatty acid peroxi-dation, and activation of caspase-3, which likely are im-portant in the prevention of oxidative stress and cell death.These actions were also seen in the present study afterKA-induced excitotoxicity. However, we further showthat MT-I overexpression can also inhibit ROS-inducedbase modifications in the genome, as judged by 8-oxoguanine levels. Protecting the genome from ROS-induced DNA modifications adds to the beneficial actionsof MT-I in CNS disorders, insofar as especially an oxida-tive attack on the DNA leading to 8-oxoguanine accu-mulation is crucial for the development of human neuro-degenerative diseases, such as Alzheimer’s disease (Diaz-Arrastia and Baskin, 2001; Iida et al., 2002) and ALS(Kikuchi et al., 2002). Indeed, MT-I and -II have protec-tive roles against development of familial ALS, in thatMT-I � II deficiency advanced ALS clinically and in-
Fig. 8. TUNEL-positive cells in hippocampus CA3 following saline (A,B) and KA (C,D) injection.Saline-treated wild-type (A) and TgMT (B) genotypes showed generally no TUNEL-positive cells.Three days after KA, wild-type mice (C) showed many apoptotic neurons in comparison with TgMT(D) mice. Scale bars � 64 �m.
Metallothionein Has Neuroprotective Actions 531

creased the morbidity and mortality relative to controls(Nagano et al., 2001; Puttaparthi et al., 2002). Thesestudies indicate that the worsening of ALS in MT-I �II-deficient mice may be due to a lack of MT-I � IIinhibition of ROS-induced neuronal toxicity.
The MT-induced inhibition of brain cell death hasbeen shown comprehensively by using MT-I-over-expressing mice or MT-I � II-deficient mice as well as bytreating animals with exogenous MT-II during variousneuropathological conditions (Masters et al., 1994; VanLookeren Campagne et al., 1999; Carrasco et al., 2000;Penkowa and Hidalgo, 2000, 2001, 2003; Penkowa et al.,2000, 2002, 2003; Trendelenburg et al., 2002; Hidalgo etal., 2001, 2002; Giralt et al., 2002a,b; Puttaparthi et al.,2002; Køhler et al., 2003; Molinero et al., 2003; Chung etal., 2003; Chung and West, 2004; Xie et al., 2004). In thepresent study, an antiinflammatory effect of MT-I wasevident in hippocampus, in that microglia/macrophageactivation; T lymphocytes recruitment; and IL-1, IL-6,IL-12, and TNF-� up-regulation 3 days after KA admin-istration were efficiently inhibited. Such effects are likelyprotective, insofar as ongoing inflammatory responses inthe brain are recognized as key contributors to variousneuropathological conditions and diseases (for review seeWersinger and Sidhu, 2002; Allan and Rothwell, 2003;Villoslada and Genain, 2004). Moreover, proinflammatorycytokines are heavily involved in CNS degeneration andcell death (Martino et al., 2000; Wang et al., 2002; Tabak-man et al., 2004). In addition, we show here that trans-genic MT-I overexpression stimulates astroglia and in-creases antiinflammatory cytokine IL-10 and the growthfactors/neurotrophins bFGF, TGF-�, NGF, BDNF, andGDNF. Astroglia deliver metabolic and trophic support tothe neurons and provide the CNS with key antioxidantdefenses (Faulkner et al., 2004; Petrova et al., 2004;Takuma et al., 2004), so the MT-I-induced stimulation ofastrocytes likely contributes to the observed reduction inKA-induced excitotoxicity and delayed neurodegenera-tion as seen in the TgMT mice.
The observation that MT-I and -II inhibit IL-1,IL-6, and TNF-� while increasing bFGF, TGF-�, andNGF is in agreement with data obtained in TgMT miceafter cortical trauma, glial toxicity, and IL-6-induced in-flammation (Penkowa et al., 2002, 2003; Molinero et al.,2003) and in MT-I � II-deficient mice (Penkowa et al.,1999, 2000; Giralt et al., 2002a) as well as data from studiesof exogenous MT-II treatment during experimental au-toimmune encephalomyelitis (EAE), brain injury, and glialtoxicity (Penkowa et al., 1999, 2002; Penkowa andHidalgo, 2001, 2003; Giralt et al., 2002b). According tothese studies, MT-I and -II inhibit both the mRNA andthe protein levels of IL-1, IL-6, and TNF-� as shown inMT-I � II-deficient mice (Penkowa et al., 2000). How-ever, the present KA study is the first to show that trans-genic MT-I overexpression also inhibits the proinflamma-tory cytokine IL-12 and stimulates the antiinflammatorycytokine IL-10 and the neurotrophins BDNF and GDNF.The neurotrophins induce intracellular signaling cascades
through the Trk tyrosine kinase receptors, whereby theyprovide survival and repair of neurons and restore neuro-logical functions (Allan and Rothwell, 2003; Tabakman etal., 2004; Villoslada and Genain, 2004). Thereby, theMT-induced shift in the balance between pro- and anti-inflammatory molecules in the CNS could explain thereduced neuronal degeneration and cell death of TgMTmice after the KA-induced injury.
However, from this in vivo model of excitotoxicity,it is rather difficult to determine whether the neuropro-tective effects of MT-I overexpression are induced by theMT antiinflammatory effects or whether the MT-I hasdirect actions on the neurons. This is intriguing in thatMT-I and -II are almost never observed inside neuronsbut are observed primarily in nonneuronal cells, so it hasappeared somewhat unlikely that MT-I and -II wouldhave direct neuroactive properties. However, independentgroups showed that in fact MT-I and -II act directly onneurons, as judged from primary neuron cultures devoidof immune and glial cells, which received exogenousMT-II treatment (Chung et al., 2003; Køhler et al., 2003).This would call for an MT-I � II secretory process, and,although the mechanism is unknown, it must exist, be-cause circulating MT is a normal feature (for a recentreview see Chung and West, 2004). MT-I and -II releasein the brain, from astrocytes, microglia/macrophages,and/or other cells, has normally been inferred rather thandemonstrated, presumably at least in part because of lack ofadequate experimental approaches and/or lack of sensitiv-ity for MT measurement. For instance, MT-I and -II arereadily detectable in primary cultures of neurons whenmeasured by radioimmunoassay (Hidalgo et al., 1994).Interestingly, MT-I � II can be detected in the culturemedia of both astrocyte and neuron primary cultures aswell as in cerebrospinal fluid (unpublished results). In thisstudy, we could observe a clear extracellular MT-I � IIimmunostaining, mainly surrounding reactive astrocytes,which is suggestive of secretion of these proteins by thesecells. This phenomenon was observed only in TgMTmice, but, at this point, we speculate that this has to domore with the higher endogenous MT content than witha specific effect of the strain. Once in the extracellularspace, and undoubtedly favored by their low molecularweight, MT-I and -II could difuse and act on neighboringcells, such as neurons. It is also conceivable that extracel-lular MT-I � II could act on microglia/macrophages (forfurther discussion see Penkowa and Hidalgo, 2000, 2001).
As a whole, the data presented here and in theliterature indicate that MT-I � II can induce neuropro-tection and neurological recovery following neuropatho-logical disorders in various anatomical regions. MT-I � IImay induce these functions by impeding the inflammatoryresponses but also by stimulating and influencing the neu-rons directly. However, the present study demonstratesforemost that endogenous MT-I overproduction inducessignificant antiinflammatory, antioxidative, and antiapop-totic effects in the hippocampal formation following KA-induced seizures and excitotoxicity. Moreover, we hereby
532 Penkowa et al.

add new insights into some of the MT-I-induced actionsin the CNS. Since excitotoxicity likely is implicated in thedevelopment of other major diseases, such as Alzheimer’s,Parkinson’s, and Huntington’s diseases; ALS; stroke; andtraumatic injuries (Mattson, 2003; Rego and Oliveira,2003), the data presented here might become clinicallyimportant, insofar as our results suggest that MT-I and/orMT-II could become future therapeutic approaches forpreventing excitotoxicity and counteracting neurologicaldiseases.
ACKNOWLEDGMENTSWe thank Hanne Hadberg, Ha Nguyen, Pernille S.
Froh, Tanja Hadberg Nielsen, and Grazyna Hahn forexcellent assistance. We also thank Jordi Camats.
REFERENCESAlberch J, Perez-Navarro E, Canals JM. 2004. Neurotrophic factors in
Huntington’s disease. Prog Brain Res 146:195–229.Allan SM, Rothwell NJ. 2003. Inflammation in central nervous system
injury. Philos Trans R Soc Lond B Biol Sci 358:1669–1677.Avolio C, Giuliani F, Liuzzi GM, Ruggieri M, Paolicelli D, Riccio P,
Livrea P, Trojano M. 2003. Adhesion molecules and matrix metallopro-teinases in multiple sclerosis: effects induced by interferon-beta. Brain ResBull 61:357–364.
Bishop A, Cashman NR. 2003. Induced adaptive resistance to oxidativestress in the CNS: a discussion on possible mechanisms and their thera-peutic potential. Curr Drug Metab 4:171–184.
Bossy-Wetzel E, Barsoum MJ, Godzik A, Schwarzenbacher R, Lipton SA.2003. Mitochondrial fission in apoptosis, neurodegeneration and aging.Curr Opin Cell Biol 15:706–716.
Carmel JB, Kakinohana O, Mestril R, Young W, Marsala M, Hart RP.2004. Mediators of ischemic preconditioning identified by microarrayanalysis of rat spinal cord. Exp Neurol 185:81–96.
Carrasco J, Penkowa M, Hadberg H, Molinero A, Hidalgo J. 2000. En-hanced seizures and hippocampal neurodegeneration following kainicacid induced seizures in metallothionein-1 � 2 deficient mice. EurJ Neurosci 12:2311–2322.
Chung RS, West AK. 2004. A role for extracellular metallothioneins inCNS injury and repair. Neuroscience 123:595–599.
Chung RS, Vickers JC, Chuah MI, West AK. 2003. Metallothionein-IIApromotes initial neurite elongation and postinjury reactive neurite growthand facilitates healing after focal cortical brain injury. J Neurosci 23:3336–3342.
Chung SY, Han SH. 2003. Melatonin attenuates kainic acid-inducedhippocampal neurodegeneration and oxidative stress through microglialinhibition. J Pineal Res 34:95–102.
Coyle P, Philcox JC, Carey LC, Rofe AM. 2002. Metallothionein: themultipurpose protein. Cell Mol Life Sci 59:627–647.
Dalton T, Fu K, Palmiter RD, Andrews GK. 1996. Transgenic mice thatoverexpress metallothionein-I resist dietary zinc deficiency. J Nutr 126:825–833.
Davis SR, McMahon RJ, Cousins RJ. 1998. Metallothionein knockout andtransgenic mice exhibit altered intestinal processing of zinc with uniformzinc-dependent zinc transporter-1 expression. J Nutr 128:825–831.
Diaz-Arrastia R, Baskin F. 2001. New biochemical markers in Alzheimerdisease. Arch Neurol 58:354–356.
Faherty CJ, Xanthoudakis S, Smeyne RJ. 1999. Caspase-3-dependentneuronal death in the hippocampus following kainic acid treatment. BrainRes Mol Brain Res 70:159–163.
Faulkner JR, Herrmann JE, Woo MJ, Tansey KE, Doan NB, SofroniewMV. 2004. Reactive astrocytes protect tissue and preserve function afterspinal cord injury. J Neurosci 24:2143–2155.
Gasull T, Rebollo DV, Romero B, Hidalgo J. 1993. Development of acompetitive double antibody radioimmunoassay for rat metallothionein.J Immunoassay 14:209–225.
Giralt M, Penkowa M, Hernandez J, Molinero A, Carrasco J, Lago N,Camats J, Campbell IL, Hidalgo J. 2002a. Metallothionein-1 � 2 defi-ciency increases brain pathology in transgenic mice with astrocyte-targeted expression of interleukin 6. Neurobiol Dis 9:319–338.
Giralt M, Penkowa M, Lago N, Camats J, Hernandez J, Molinero A,Hidalgo J. 2002b. Metallothionein-1 � 2 protect the CNS after a focalbrain injury. Exp Neurol 173:114–128.
Hidalgo J, Garcia A, Oliva AM, Giralt M, Gasull T, Gonzalez B, Milner-owicz H, Wood A, Bremner I. 1994. Effect of zinc, copper and glu-cocorticoids on metallothionein levels of cultured neurons and astrocytesfrom rat brain. Chem-Biol Interact 93:197–219.
Hidalgo J, Aschner M, Zatta P, Vasak M. 2001. Roles of the metallothio-nein family of proteins in the central nervous system. Brain Res Bull55:133–145.
Hidalgo J, Penkowa M, Giralt M, Carrasco J, Molinero A. 2002. Metal-lothionein expression and oxidative stress in the brain. Methods Enzymol348:238–249.
Iida T, Furuta A, Nishioka K, Nakabeppu Y, Iwaki T. 2002. Expression of8-oxoguanine DNA glycosylase is reduced and associated with neurofibrillarytangles in Alzheimer’s disease brain. Acta Neuropathol 103:20–25.
Kikuchi H, Furuta A, Nishioka K, Suzuki SO, Nakabeppu Y, Iwaki T.2002. Impairment of mitochondrial DNA repair enzymes against accu-mulation of 8-oxo-guanine in the spinal motor neurons of amyotrophiclateral sclerosis. Acta Neuropathol 103:408–414.
Kim D, Kim EH, Kim C, Sun W, Kim HJ, Uhm CS, Park SH, Kim H.2003. Differential regulation of metallothionein-I, -II, and -III mRNAexpression in the rat brain following kainic acid treatment. Neuroreport14:679–682.
Køhler LB, Berezin V, Bock E, Penkowa M. 2003. The role of metallo-thionein II in neuronal differentiation and survival. Brain Res 992:128–136.
Lee JY, Cole TB, Palmiter RD, Koh JY. 2000. Accumulation of zinc indegenerating hippocampal neurons of ZnT3-null mice after seizures:evidence against synaptic vesicle origin. J Neurosci 20:RC79.
Lehtimaki KA, Peltola J, Koskikallio E, Keranen T, Honkaniemi J. 2003.Expression of cytokines and cytokine receptors in the rat brain after kainicacid-induced seizures. Brain Res Mol Brain Res 110:253–260.
Leppert D, Lindberg RL, Kappos L, Leib SL. 2001. Matrix metallopro-teinases: multifunctional effectors of inflammation in multiple sclerosisand bacterial meningitis. Brain Res Rev 36:249–257.
Malaiyandi LM, Dineley KE, Reynolds IJ. 2004. Divergent consequencesarise from metallothionein overexpression in astrocytes: zinc buffering andoxidant-induced zinc release. Glia 45:346–353.
Martino G, Furlan R, Brambilla E, Bergami A, Ruffini F, Gironi M,Poliani PL, Grimaldi LM, Comi G. 2000. Cytokines and immunity inmultiple sclerosis: the dual signal hypothesis. J Neuroimmunol 109:3–9.
Masters BA, Kelly EJ, Quaife CJ, Brinster RL, Palmiter RD. 1994. Tar-geted disruption of metallothionein I and II genes increases sensitivity tocadmium. Proc Natl Acad Sci U S A 91:584–588.
Mattson MP. 2003. Excitotoxic and excitoprotective mechanisms: abun-dant targets for the prevention and treatment of neurodegenerative dis-orders. Neuromol Med 3:65–94.
Molinero A, Penkowa M, Hernandez J, Camats J, Giralt M, Lago N,Carrasco J, Campbell IL, Hidalgo J. 2003. Metallothionein-I overexpres-sion decreases brain pathology in transgenic mice with astrocyte-targetedexpression of interleukin 6. J Neuropathol Exp Neurol 62:315–328.
Nagano S, Satoh M, Sumi H, Fujimura H, Tohyama C, Yanagihara T,Sakoda S. 2001. Reduction of metallothioneins promotes the diseaseexpression of familial amyotrophic lateral sclerosis mice in a dose-dependent manner. Eur J Neurosci 13:1363–1370.
Metallothionein Has Neuroprotective Actions 533

Palmiter RD, Sandgren EP, Koeller DM, Brinster RL. 1993. Distal regu-latory elements from the mouse metallothionein locus stimulate geneexpression in transgenic mice. Mol Cell Biol 13:5266–5275.
Penkowa M, Hidalgo J. 2000. Metallothionein I � II expression and theirrole in experimental autoimmune encephalomyelitis. Glia 32:247–263.
Penkowa M, Hidalgo J. 2001. Metallothionein treatment reduces proinflam-matory cytokines IL-6 and TNF-� and apoptotic cell death during experi-mental autoimmune encephalomyelitis (EAE). Exp Neurol 170:1–14.
Penkowa M, Hidalgo J. 2003. Treatment with metallothionein preventsdemyelination and axonal damage and increases oligodendrocyte precur-sors and tissue repair during experimental autoimmune encephalomyelitis.J Neurosci Res 72:574–586.
Penkowa M, Giralt M, Moos T, Thomsen PS, Hernandez J, Hidalgo J.1999. Impaired inflammatory response to glial cell death in geneticallymetallothionein-I � II deficient mice. Exp Neurol 156:149–164.
Penkowa M, Carrasco J, Giralt M, Molinero A, Hernandez J, Campbell IL,Hidalgo J. 2000. Altered central nervous system cytokine-growth factorexpression profiles and angiogenesis in metallothionein-I � II deficientmice. J Cereb Blood Flow Metab 20:1174–1189.
Penkowa M, Molinero A, Carrasco J, Hidalgo J. 2001. IL-6 deficiencyreduces the brain inflammatory response and increases oxidative stress andneurodegeneration after kainic acid-induced seizures. Neuroscience 102:805–818.
Penkowa M, Giralt M, Camats J, Hidalgo J. 2002. Metallothionein 1 � 2protect the CNS during neuroglial degeneration induced by 6-aminonicotinamide. J Comp Neurol 444:174–189.
Penkowa M, Camats J, Giralt M, Molinero A, Hernandez J, Carrasco J,Campbell IL, Hidalgo J. 2003. Metallothionein-I overexpression altersbrain inflammation and stimulates brain repair in transgenic mice withastrocyte targeted interleukin-6 expression. Glia 42:287–306.
Petrova PS, Raibekas A, Pevsner J, Vigo N, Anafi M, Moore MK, PeaireA, Shridhar V, Smith DI, Kelly J, Durocher Y, Commissiong JW. 2004.Discovering novel phenotype-selective neurotrophic factors to treat neu-rodegenerative diseases. Prog Brain Res 146:168–183.
Puttaparthi K, Gitomer WL, Krishnan U, Son M, Rajendran B, Elliott JL.2002. Disease progression in a transgenic model of familial amyotrophiclateral sclerosis is dependent on both neuronal and nonneuronal zincbinding proteins. J Neurosci 22:8790–8796.
Rego AC, Oliveira CR. 2003. Mitochondrial dysfunction and reactiveoxygen species in excitotoxicity and apoptosis: implications for thepathogenesis of neurodegenerative diseases. Neurochem Res 28:1563–1574.
Sensi SL, Jeng J-M. 2004. Rethinking the excitotoxic ionic milieu: theemerging role of Zn2� in ischemic neuronal injury. Curr Mol Med4:83–107.
Tabakman R, Lecht S, Sephanova S, Arien-Zakay H, Lazarovici P. 2004.Interactions between the cells of the immune and nervous system: neu-rotrophins as neuroprotection mediators in CNS injury. Prog Brain Res146:387–401.
Takuma K, Baba A, Matsuda T. 2004. Astrocyte apoptosis: implications forneuroprotection. Prog Neurobiol 72:111–127.
Trendelenburg G, Prass K, Priller J, Kapinya K, Polley A, Muselmann C,Ruscher K, Kannbley U, Schmitt AO, Castell S, Wiegand F, Meisel A,Rosenthal A, Dirnagl U. 2002. Serial analysis of gene expression identifiesmetallothionein-II as major neuroprotective gene in mouse focal cerebralischemia. J Neurosci 22:5879–5888.
Ueda Y, Yokoyama H, Nakajima A, Tokumaru J, Doi T, Mitsuyama Y.2002. Glutamate excess and free radical formation during and followingkainic acid-induced status epilepticus. Exp Brain Res 147:219–226.
Van Lookeren Campagne M, Thibodeaux H, Van Bruggen N, Caims B,Gerlai R, Palmer JT, Williams SP, Lowe D. 1999. Evidence for aprotective role of metallothionein-1 in focal cerebral ischemia. Proc NatlAcad Sci U S A 96:12870–12875.
Villoslada P, Genain CP. 2004. Role of nerve growth factor and othertrophic factors in brain inflammation. Prog Brain Res 146:403–414.
Wang J, Asensio VC, Campbell IL. 2002. Cytokines and chemokines asmediators of protection and injury in the central nervous system assessedin transgenic mice. Curr Top Microbiol Immunol 265:23–48.
Wersinger C, Sidhu A. 2002. Inflammation and Parkinson’s disease. CurrDrug Targets Inflamm Allerg 1:221–242.
Xie T, Tong L, McCann UD, Yuan J, Becker KG, Mechan AO, CheadleC, Donovan DM, Ricaurte GA. 2004. Identification and characterizationof metallothionein-1 and -2 gene expression in the context of (�/–)3,4-methylenedioxymethamphetamine-induced toxicity to brain dopaminer-gic neurons. J Neurosci 24:7043–7050.
534 Penkowa et al.
Copyright © 2022 FDOKUMEN




















