
Laboratory Study of Simulated Atmospheric Transformations of Chromium in Ultrafine Combustion...
12
Aerosol Science and Technology, 40:545–556, 2006 Copyright c American Association for Aerosol Research ISSN: 0278-6826 print / 1521-7388 online DOI: 10.1080/02786820600714353 Laboratory Study of Simulated Atmospheric Transformations of Chromium in Ultrafine Combustion Aerosol Particles Michelle Werner, 1 Peter Nico, 2 Bing Guo, 3 Ian Kennedy, 3 and Cort Anastasio 1 1 Department of Land, Air & Water Resources, University of California, Davis, California, USA 2 Earth Sciences Division, Lawrence Berkeley National Laboratory, One Cyclotron Road, Berkeley, California, USA 3 Department of Mechanical and Aeronautical Engineering, University of California, Davis, California, USA While atmospheric particles can have adverse health effects, the reasons for this toxicity are largely unclear. One possible rea- son is that the particles can contain toxic metals such as chromium. Chromium exists in the environment in two major oxidation states: III, which is an essential nutrient, and VI, which is highly toxic and carcinogenic. Currently little is known about the speciation of chromium in airborne particles or how this speciation is altered by atmospheric reactions. To investigate the potential impacts of atmospheric aging on the speciation and toxicity of chromium- containing particles, we collected chromium and chromium-iron combustion ultrafine particles on Teflon filters and exposed the particles to a combination of light, ozone, water vapor, and, in Received 2 September 2005; accepted 23 March 2006. We thank Dr. Matthew Newville of the GSECARS at the Advanced Photon Source (APS), Argonne National Laboratory, for his assis- tance. The majority to the X-ray data presented above were collected at GeoSoilEnviroCARS (Sector 13), which is supported by the Na- tional Science Foundation—Earth Sciences (EAR-0217473), Depart- ment of Energy—Geosciences (DE-FG02-94ER14466) and the State of Illinois. Use of the APS was supported by the U.S. Department of En- ergy under Contract No. W-31-109-Eng-38. We would also like to thank Dr. Matthew Marcus of beamline 10.3.2 of the Advanced Light Source (Lawrence Berkeley National Laboratory), which is supported by the US Department of Energy (DE-AC02-05CH11231). Portions of this research were also carried out at the Stanford Synchrotron Radiation Laboratory, operated by Stanford University on behalf of the U.S. De- partment of Energy. Finally, we thank David Paige (Paige Instruments) for constructing the ozone generator, John Newberg and Mike Jimenez- Cruz for building the ozone dilution and delivery system, and Michelle Gras of the UC Davis Interdisciplinary Center for Plasma Mass Spec- trometry for conducting the ICP-MS analyses. This work was supported by grant number 5 P42 ES04699 from the National Institute of Envi- ronmental Health Sciences (NIEHS) of the NIH, and by a fellowship from the UC Davis NEAT-IGERT program to Michelle Werner (NSF IGERT Grant # 9972741). Partial support was also provided by the U.S. Department of Energy under contract number DE-AC02-05CH11231. The contents of this paper are solely the responsibility of the authors and do not necessarily represent the official views of the NIEHS, NIH. Address correspondence to Cort Anastasio, One Shields Avenue, Department of Land, Air & Water Resources, University of California, Davis, CA 95616. E-mail: [email protected] some cases, basic or acidic conditions. After the aging process, the aged and not-aged samples were analyzed for Cr oxidation state using X-ray Absorption Near Edge Spectroscopy (XANES). We found that the aging process reduced Cr(VI) by as much as 20% in chromium particles that had high initial Cr(VI)/Cr(total) ra- tios. This reduction of Cr(VI) to Cr(III) appears to be due to reac- tions primarily with light and hydroperoxyl radical (HO 2 ) in the chamber. Particles that had low initial Cr(VI)/Cr(total) ratios ex- perienced no significant change in Cr oxidation states after aging. Compared to particles containing only Cr, the addition of Fe to the flame decreased the Cr(VI)/Cr(total) ratio in fresh Cr-Fe particles by ∼60%. Aging of these Cr-Fe particles had no additional effects on the Cr(VI)/Cr(total) ratio. INTRODUCTION Understanding the concentrations and composition of partic- ulate matter (PM) in the atmosphere has become a focus for re- search because fine particles can be inhaled deeply into the lungs and cause a variety of negative health affects, such as bronchial irritation, reduced lung function, respiratory disease, cancer, and mortality (Pope et al. 1995; Schlesinger 2000). On a mass basis, ultrafine PM (particles with aerodynamic equivalent diameters ≤0.1 µm) can have more adverse health effects than larger par- ticles of the same composition (Ferin et al. 1990; Oberdorster et al. 1992). Ferin et al. propose that this is because the smaller particles can penetrate the interstitial space of the lungs and because the large number of particles being deposited can over- whelm lung defenses, such as removal by alveolar macrophages. While ultrafine particles can be present at very high num- ber concentrations, they make up only a small portion of the total particle mass in the atmosphere (Anastasio and Martin 2001). Though their high number concentrations might influence the toxicity of ultrafine particles, their composition, specifically their transition metal content, can also influence toxicity. For example, it has been observed that the transition metal compo- nent of particles is associated with acute lung injury (Dreher et al. 1997). The initial composition of “fresh” ultrafine parti- cles is determined by the PM sources, which include fossil fuel 545
Transcript of Laboratory Study of Simulated Atmospheric Transformations of Chromium in Ultrafine Combustion...

Aerosol Science and Technology, 40:545–556, 2006Copyright c© American Association for Aerosol ResearchISSN: 0278-6826 print / 1521-7388 onlineDOI: 10.1080/02786820600714353
Laboratory Study of Simulated Atmospheric Transformationsof Chromium in Ultrafine Combustion Aerosol Particles
Michelle Werner,1 Peter Nico,2 Bing Guo,3 Ian Kennedy,3 and Cort Anastasio1
1Department of Land, Air & Water Resources, University of California, Davis, California, USA2Earth Sciences Division, Lawrence Berkeley National Laboratory, One Cyclotron Road,Berkeley, California, USA3Department of Mechanical and Aeronautical Engineering, University of California, Davis,California, USA
While atmospheric particles can have adverse health effects,the reasons for this toxicity are largely unclear. One possible rea-son is that the particles can contain toxic metals such as chromium.Chromium exists in the environment in two major oxidation states:III, which is an essential nutrient, and VI, which is highly toxicand carcinogenic. Currently little is known about the speciation ofchromium in airborne particles or how this speciation is alteredby atmospheric reactions. To investigate the potential impacts ofatmospheric aging on the speciation and toxicity of chromium-containing particles, we collected chromium and chromium-ironcombustion ultrafine particles on Teflon filters and exposed theparticles to a combination of light, ozone, water vapor, and, in
Received 2 September 2005; accepted 23 March 2006.We thank Dr. Matthew Newville of the GSECARS at the Advanced
Photon Source (APS), Argonne National Laboratory, for his assis-tance. The majority to the X-ray data presented above were collectedat GeoSoilEnviroCARS (Sector 13), which is supported by the Na-tional Science Foundation—Earth Sciences (EAR-0217473), Depart-ment of Energy—Geosciences (DE-FG02-94ER14466) and the Stateof Illinois. Use of the APS was supported by the U.S. Department of En-ergy under Contract No. W-31-109-Eng-38. We would also like to thankDr. Matthew Marcus of beamline 10.3.2 of the Advanced Light Source(Lawrence Berkeley National Laboratory), which is supported by theUS Department of Energy (DE-AC02-05CH11231). Portions of thisresearch were also carried out at the Stanford Synchrotron RadiationLaboratory, operated by Stanford University on behalf of the U.S. De-partment of Energy. Finally, we thank David Paige (Paige Instruments)for constructing the ozone generator, John Newberg and Mike Jimenez-Cruz for building the ozone dilution and delivery system, and MichelleGras of the UC Davis Interdisciplinary Center for Plasma Mass Spec-trometry for conducting the ICP-MS analyses. This work was supportedby grant number 5 P42 ES04699 from the National Institute of Envi-ronmental Health Sciences (NIEHS) of the NIH, and by a fellowshipfrom the UC Davis NEAT-IGERT program to Michelle Werner (NSFIGERT Grant # 9972741). Partial support was also provided by the U.S.Department of Energy under contract number DE-AC02-05CH11231.The contents of this paper are solely the responsibility of the authorsand do not necessarily represent the official views of the NIEHS, NIH.
Address correspondence to Cort Anastasio, One Shields Avenue,Department of Land, Air & Water Resources, University of California,Davis, CA 95616. E-mail: [email protected]
some cases, basic or acidic conditions. After the aging process, theaged and not-aged samples were analyzed for Cr oxidation stateusing X-ray Absorption Near Edge Spectroscopy (XANES). Wefound that the aging process reduced Cr(VI) by as much as 20%in chromium particles that had high initial Cr(VI)/Cr(total) ra-tios. This reduction of Cr(VI) to Cr(III) appears to be due to reac-tions primarily with light and hydroperoxyl radical (HO2) in thechamber. Particles that had low initial Cr(VI)/Cr(total) ratios ex-perienced no significant change in Cr oxidation states after aging.Compared to particles containing only Cr, the addition of Fe to theflame decreased the Cr(VI)/Cr(total) ratio in fresh Cr-Fe particlesby ∼60%. Aging of these Cr-Fe particles had no additional effectson the Cr(VI)/Cr(total) ratio.
INTRODUCTIONUnderstanding the concentrations and composition of partic-
ulate matter (PM) in the atmosphere has become a focus for re-search because fine particles can be inhaled deeply into the lungsand cause a variety of negative health affects, such as bronchialirritation, reduced lung function, respiratory disease, cancer, andmortality (Pope et al. 1995; Schlesinger 2000). On a mass basis,ultrafine PM (particles with aerodynamic equivalent diameters≤0.1 µm) can have more adverse health effects than larger par-ticles of the same composition (Ferin et al. 1990; Oberdorsteret al. 1992). Ferin et al. propose that this is because the smallerparticles can penetrate the interstitial space of the lungs andbecause the large number of particles being deposited can over-whelm lung defenses, such as removal by alveolar macrophages.
While ultrafine particles can be present at very high num-ber concentrations, they make up only a small portion of thetotal particle mass in the atmosphere (Anastasio and Martin2001). Though their high number concentrations might influencethe toxicity of ultrafine particles, their composition, specificallytheir transition metal content, can also influence toxicity. Forexample, it has been observed that the transition metal compo-nent of particles is associated with acute lung injury (Dreheret al. 1997). The initial composition of “fresh” ultrafine parti-cles is determined by the PM sources, which include fossil fuel
545

546 M. WERNER ET AL.
combustion, incineration, and other human activities (Anastasioand Martin 2001). In addition, the composition of ultrafinePM will be altered by atmospheric aging involving oxidation-reduction reactions as well as the condensation of secondaryspecies such as organics, nitrate, and sulfate (Finlayson-Pitts andPitts 2000). Because ultrafine particles can have relatively longatmospheric lifetimes (between 1–10 days for particles between10–100 nm (Brasseur et al. 1999); they can be transported overlong distances and undergo extensive atmospheric reactions.
Chromium is a transition metal of particular interest in PMbecause its toxicity varies based on oxidation state. Chromiumexists in two major oxidation states: +3, which is an essen-tial nutrient, and +6, which is highly toxic and carcinogenic(Cieslak-Golonka 1996). It has been proposed that Cr(VI) istoxic because it can penetrate cells, be reduced to Cr(III), andthen generate reactive oxygen species, such as hydroxyl radical,that can cause DNA damage (Cohen et al. 1993). The highestexposures to Cr(VI)-containing particulate matter likely occursto workers during welding, chrome plating, spray painting, andchrome pigment productions (Cohen et al. 1993). However, astudy in Los Angeles showed that ambient fine particles can alsocontain significant amounts of chromium and that the amount ofCr (relative to the total mass of metals) is greater in the ultrafineparticles than that in the fine particles (Hughes et al. 1998). Inthat study chromium accounted for up to ∼10% of the transi-tion metal mass in ultrafine particles (diameters <0.097 µm).Chromium is also among the top five most abundant metalsin particle emissions from diesel fuel combustion (Wang et al.2003).
Sampling of emissions of particles from specific applications,such as welding fumes and fly ash from coal burning (Minniet al. 1984; Shoji et al. 2002), have shown that Cr(VI) accountsfor approximately 40–90% of the total PM mass in metal arcwelding fumes and 0–30% in fly ash (depending on the originof the coal). However, very few studies have examined howthe speciation of chromium, and therefore its toxicity, changesduring atmospheric transport and aging.
Computer modeling results suggest that under typical atmo-spheric conditions Cr(VI) is reduced to Cr(III) by the reduc-tants As(III), Fe(II), V(II), V(III), VO2+, and SO2 (Seigneur andConstantinou 1995). However, this model did not include reac-tions of chromium species with atmospheric oxidants such asozone (O3), hydrogen peroxide (H2O2), hydroxyl radical (OH),or hydroperoxyl radical (HO2). These reactive species are re-sponsible for the oxidation of most trace pollutants in the at-mosphere (Finlayson-Pitts and Pitts 2000) and it is possible thatthey also affect the speciation of Cr on atmospheric particles.Zhang and Bartlett (1999), for example, have found that aqueousCr(III) is oxidized to Cr(VI) in the presence of light under acidicconditions, and hypothesized that this oxidation is mediated byOH generated during the photolysis of Fe(OH)2+. Further stud-ies showed that the presence of organic acids, such as acetate,could have significant impacts on the light- and Fe(III)-inducedoxidation of Cr(III) (Zhang 2000; Zhang and Bartlett 1999).
There has also been only one previous study of particulatechromium transformation during simulated atmospheric reac-tions, however, they did not include illumination during theirexperiments (Grohse et al. 1988). In this study, particles weregenerated using an aerosol nebulizer containing an aqueous so-lution of Cr(VI), dried, and collected on Teflon or PVC filters.These filters were then exposed to pollutants such as HNO3,transition metals, organic acids, and ozone via a nebulizer in aplexiglass or aluminum chamber. From their chamber studiesthey concluded that: (1) under acidic conditions Cr(VI) reactswith oxidizable species, such as unsaturated hydrocarbons andtransition metals, to form Cr(III); (2) there is an overall reduc-tion of Cr(VI) in the reaction chamber, as well as in separatesolution studies; (3) at ambient Cr levels (typically 20–100 ng)particulate Cr(VI) has an average half-life of ∼13 hours, thoughthe range is broad; (4) the role of relative humidity on Cr chem-istry is unclear; and (5) there is no indication of Cr(III) oxidationto Cr(VI). Cr(VI) in the experiments of Grohse et al. (1988) wasdetermined by digestion of the filter followed by column sepa-ration with ion chromatography detection.
An alternative analytical method to determine Cr oxidationstates is X-ray Absorption Near Edge Spectroscopy (XANES),which is non-destructive and requires no sample extraction. An-other advantage of using XANES to measure Cr oxidation state isthat the octahedrally coordinated Cr(VI) has a distinct pre-edgepeak in the Cr spectrum that is nearly independent of the pres-ence of the tetrahedrally coordinated Cr(III). Although XANEShas not been previously used to determine metal oxidation statesin atmospheric particles, it has been used to quantify Cr(VI) incoal (Huggins et al. 1999), on rice husks (Hu et al. 2004), andon treated wood (Nico et al. 2004).
The overall goal of this project was to determine how atmo-spheric aging affects the oxidation state of Cr in combustion-generated ultrafine particles in order to understand how atmo-spheric reactions might alter the chromium-associated toxicityof PM. In this work, we generate particles containing chromium(or a mixture of chromium and iron) in a diffusion flame, agethe collected PM in a Teflon-coated reaction chamber with light,ozone, and water vapor, and determine Cr oxidation states usingXANES on pairs of aged and not-aged portions of our samples.
EXPERIMENTAL
Sample PreparationChromium and chromium-iron aerosols were generated by
a hydrogen diffusion flame that was stabilized on a nozzle lo-cated on the axis of a wind tunnel; the tunnel working sectionwas 300 mm by 300 mm with a working length of ∼1.5 m.Filtered, particle-free air flowed through the wind tunnel toform a uniform laminar flow in the working section. Air ve-locity in the wind tunnel was kept constant for all experi-ments. Chromium hexacarbonyl (Cr(CO)6) and iron pentacar-bonyl (Fe(CO)5), organometallic compounds with high satura-tion vapor pressures and moderate decomposition temperatures,

Cr TRANSFORMATIONS IN ULTRAFINE COMBUSTION PM 547
were used as the precursors for chromium and iron. To deliverchromium to the flame, a slow stream of fuel gas (H2) flowedthrough a Cr(CO)6 cartridge (built from a commercial 47-mm fil-ter holder) to saturate the flow with Cr(CO)6 vapor. In the caseof iron, liquid Fe(CO)5 was contained in an air-tight bubbler,which was slowly purged with a separate stream of H2 gas toproduce saturated Fe(CO)5 vapors. The metal-laden H2 streamswere then mixed with clean fuel gas and/or the diluent gas beforethey were delivered to the flame. The concentrations of Cr(CO)6
and Fe(CO)5 in the fuel mixture were estimated from the satura-tion vapor pressures of the precursors and the mixing ratio of thegases. Ambient temperature was kept constant at 23 ± 0.5◦C,minimizing fluctuations in the precursor vapor pressures. Wediluted the H2 fuel gas with Ar to vary the flame temperature.
A sampling probe was positioned 520 mm above the nozzleon-axis, where the temperature and concentrations were so lowthat no significant chemical reaction and aerosol developmentwas expected to take place. The post-flame aerosols were drawnthrough the sampling probe by vacuum and the particles werecollected onto 47 mm Zefluor filters with nominal pore size of0.2 µm (Pall Gelman Laboratory). Based on analysis by SMPSparticles were typically between 10–100 nm in size. More detailson the apparatus for synthesizing the Cr and Cr-Fe aerosols canbe found elsewhere (Guo and Kennedy 2004).
Aging ProcedureThe atmospheric aging system is illustrated in Figure 1.
Particle-free air was generated using a 737-R Aadco Pure AirGenerator or introduced from a zero air cylinder (99.8% purity).
FIG. 1. Schematic of the aging chamber and experimental set up. All tubing is PTFE while fittings are either stainless steel or Teflon.
This air was passed through a charcoal trap, a 47 mm Teflo fil-ter (2.0 µm pore; Pall Gelman Labs), then two glass impingersfilled with Milli-Q water (≥18.2 M� cm) to bring the relativehumidity of the air to approximately 100%. Ozone was gener-ated by passing oxygen (medical grade, Puritan Bennett; 0.25sLpm) through a GE 021 quartz tube (7′′ long, 5 mm ID, 7 mmOD) that ran parallel to an ozone-generating Hg lamp (P/N 81-1127-1; BHK). The amount of ozone produced in the ozonatorwas varied by moving an adjustable steel pipe to cover differentamounts of the UV lamp. Prior to each experiment, ozone mix-ing ratios in the gas stream from the ozonator were measuredusing a Dasibi 1003-PC analyzer after mixing with 1.75 sLpmof purified, humidified air to obtain sufficient flow. During ex-periments the humidified air flow was adjusted to 0.75 sLpmand was combined with the ozone stream to make a total flowinto the chamber of 1.00 sLpm. The final ozone mixing ratio inthe aging chamber was ∼1 ppmv.
The aging chamber is a Teflon-coated, air-tight cylinder witha diameter of 14 cm, height of 6.5 cm, and internal volume of∼1000 cm3. The air-tight Teflon lid contains a circular quartzwindow with a diameter of 7 cm so that samples can be irradi-ated during aging. The inlet and exhaust ports on the chamberare located 2.5 cm from the base of the reaction chamber and op-posite one another. A charcoal trap was attached to the exhaustand vented appropriately. Samples in the chamber are held on aTeflon stand (6 cm height and 12.5 cm diameter base) with a 47mm dia recessed edge in order to center the filter.
The illumination system (Spectral Energy Corp., model LH151N) contains a 1000 W xenon bulb (Osram Sylvania Inc.,model XBO 1000W/HS OFR) powered by an Oriel Spectral
548 M. WERNER ET AL.
Physics power supply (model 69920) run at an average currentof 43 A and voltage of 22.5 V. We use a dichroic cold mirrorin the lamp housing to transmit wavelengths between 300–500nm to the sample but remove more energetic UV as well as IRwavelengths to reduce heating of the sample and chamber.
Before each aging experiment, sampled filters were cut into8 equal pie-shaped wedges using a custom-made cutting guide.This guide was a 2-inch high, 47 mm diameter polyvinylchloridecylinder with a 1/8 pie wedge removed (as a cutting template)and with a 2-mm high rim on the bottom to be able to holddown the perimeter of the filter without disturbing the sampledparticles. For each sample, four pieces of filter were placed in aPetri dish and stored in the dark in an airtight, N2-purged box(Pelican, model #1050, with a Swagelock fitting and septumadded for purging). The other four pieces of filter were placedin the aging chamber described above.
Samples were aged in the chamber for 4–24 hours. They wereexposed to simulated sunlight, ∼75% relative humidity, and 0.8–1.0 ppm ozone. In several experiments we aged samples withoutozone or illumination. In a few cases, we exposed samples toacidic or basic vapors prior to aging by placing the filters inair-tight jars on a platform above either concentrated H2SO4 orNH4OH, respectively, for 24 h. After aging, both aged and notaged pieces of sample were placed in Petri dishes and storedin an air-tight, nitrogen-purged box in a cool, dark place untilanalysis.
Sample Analysis by XANESBulk X-Ray Absorption Near Edge Spectroscopy (XANES)
analysis was performed at three beamlines: (1) the majority ofour data was taken at GSECARS beamline 13-BM at the Ad-vanced Photon Source (APS) of Argonne National Laboratoryusing a 16 element Ge detector, a Si (111) monochromator, abeam size of ∼1.5 × 6.0 mm, and a relatively constant beamcurrent of ∼100 mA; (2) beamline 4–3 at Stanford SynchrotronRadiation Laboratory (SSRL) using a Lytle type ionization de-tector, a Si (220) monochromator, a beam size of ∼20 × 4 mmand a beam current ranging from ∼50–100 mA; and (3) Beam-line 10.3.2 at the Advanced Light Source (ALS) of LawrenceBerkeley National Lab using a seven element Ge detector, aSi(111) monochromator, a beam size of ∼16 × 7 µm, and abeam current between 200–400 mA.
Pie-shaped wedges of sample filters were mounted withKapton tape onto slide mounts then placed in the beam. Sam-ples were run in fluorescence mode at a 45
◦angle to the incident
beam; the fluorescent intensity was normalized to the incidentX-ray intensity (Io), which was measured using in-line ionizationchambers. The energy scan was typically 5990–6150 eV in 0.5eV steps to encompass the K-edge of Cr. Because the concentra-tions of Cr in our samples were relatively high, only one or twoscans were taken of each filter. The monochromator energy wascalibrated to a suite of chromium standards, with the Cr(VI)pre-edge peak being assigned a value of 5994.5 eV. Figure 2
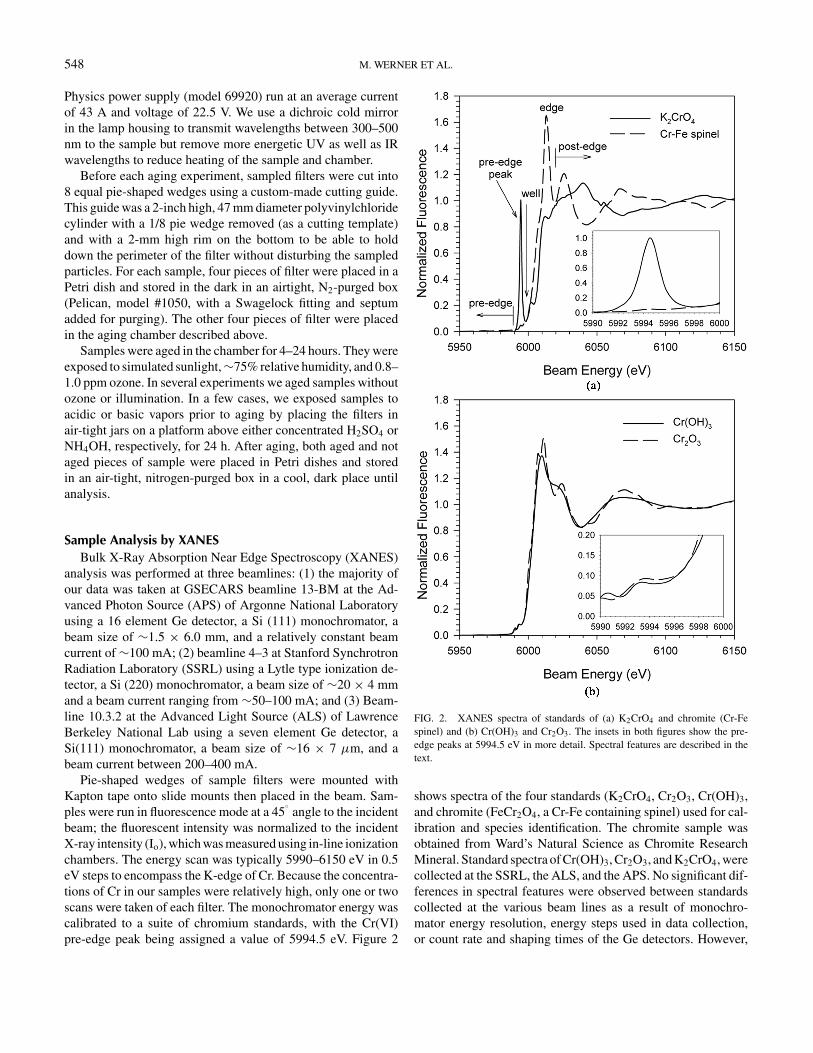
FIG. 2. XANES spectra of standards of (a) K2CrO4 and chromite (Cr-Fespinel) and (b) Cr(OH)3 and Cr2O3. The insets in both figures show the pre-edge peaks at 5994.5 eV in more detail. Spectral features are described in thetext.
shows spectra of the four standards (K2CrO4, Cr2O3, Cr(OH)3,and chromite (FeCr2O4, a Cr-Fe containing spinel) used for cal-ibration and species identification. The chromite sample wasobtained from Ward’s Natural Science as Chromite ResearchMineral. Standard spectra of Cr(OH)3, Cr2O3, and K2CrO4, werecollected at the SSRL, the ALS, and the APS. No significant dif-ferences in spectral features were observed between standardscollected at the various beam lines as a result of monochro-mator energy resolution, energy steps used in data collection,or count rate and shaping times of the Ge detectors. However,
Cr TRANSFORMATIONS IN ULTRAFINE COMBUSTION PM 549
some standard spectra did appear to be somewhat over absorbed.The spectra in Figure 2 are the best representatives obtained foreach standard species independent of where they were collected.The chromite spectra were collected solely at the APS becauseit was added to the set of standards after we observed it in oursample data. As shown in Figure 2a, the “pre-edge” of the spec-trum, which includes the pre-edge peak refers to the normalizedfluorescence below ∼6000 eV, the “edge” is the region between6000–6020 (depending on the coordination chemistry of the Cratoms), and the “post-edge” region is between approximately6020–6150 eV.
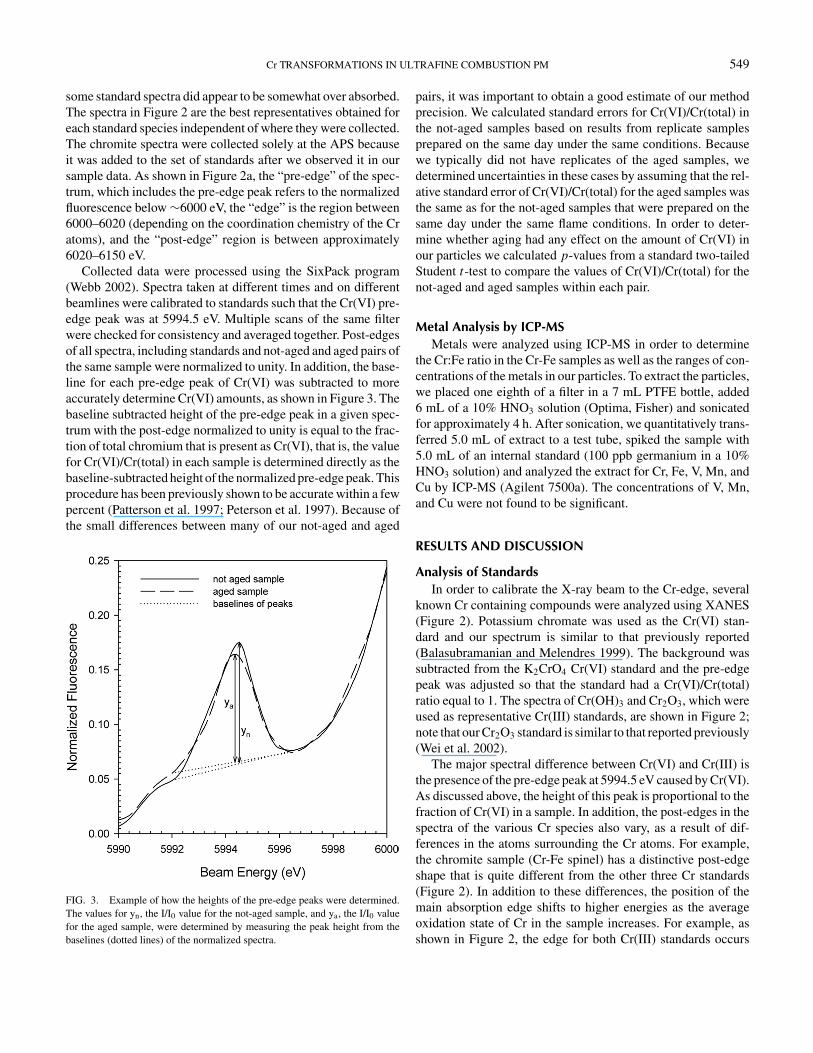
Collected data were processed using the SixPack program(Webb 2002). Spectra taken at different times and on differentbeamlines were calibrated to standards such that the Cr(VI) pre-edge peak was at 5994.5 eV. Multiple scans of the same filterwere checked for consistency and averaged together. Post-edgesof all spectra, including standards and not-aged and aged pairs ofthe same sample were normalized to unity. In addition, the base-line for each pre-edge peak of Cr(VI) was subtracted to moreaccurately determine Cr(VI) amounts, as shown in Figure 3. Thebaseline subtracted height of the pre-edge peak in a given spec-trum with the post-edge normalized to unity is equal to the frac-tion of total chromium that is present as Cr(VI), that is, the valuefor Cr(VI)/Cr(total) in each sample is determined directly as thebaseline-subtracted height of the normalized pre-edge peak. Thisprocedure has been previously shown to be accurate within a fewpercent (Patterson et al. 1997; Peterson et al. 1997). Because ofthe small differences between many of our not-aged and aged
FIG. 3. Example of how the heights of the pre-edge peaks were determined.The values for yn, the I/I0 value for the not-aged sample, and ya, the I/I0 valuefor the aged sample, were determined by measuring the peak height from thebaselines (dotted lines) of the normalized spectra.
pairs, it was important to obtain a good estimate of our methodprecision. We calculated standard errors for Cr(VI)/Cr(total) inthe not-aged samples based on results from replicate samplesprepared on the same day under the same conditions. Becausewe typically did not have replicates of the aged samples, wedetermined uncertainties in these cases by assuming that the rel-ative standard error of Cr(VI)/Cr(total) for the aged samples wasthe same as for the not-aged samples that were prepared on thesame day under the same flame conditions. In order to deter-mine whether aging had any effect on the amount of Cr(VI) inour particles we calculated p-values from a standard two-tailedStudent t-test to compare the values of Cr(VI)/Cr(total) for thenot-aged and aged samples within each pair.
Metal Analysis by ICP-MSMetals were analyzed using ICP-MS in order to determine
the Cr:Fe ratio in the Cr-Fe samples as well as the ranges of con-centrations of the metals in our particles. To extract the particles,we placed one eighth of a filter in a 7 mL PTFE bottle, added6 mL of a 10% HNO3 solution (Optima, Fisher) and sonicatedfor approximately 4 h. After sonication, we quantitatively trans-ferred 5.0 mL of extract to a test tube, spiked the sample with5.0 mL of an internal standard (100 ppb germanium in a 10%HNO3 solution) and analyzed the extract for Cr, Fe, V, Mn, andCu by ICP-MS (Agilent 7500a). The concentrations of V, Mn,and Cu were not found to be significant.
RESULTS AND DISCUSSION
Analysis of StandardsIn order to calibrate the X-ray beam to the Cr-edge, several
known Cr containing compounds were analyzed using XANES(Figure 2). Potassium chromate was used as the Cr(VI) stan-dard and our spectrum is similar to that previously reported(Balasubramanian and Melendres 1999). The background wassubtracted from the K2CrO4 Cr(VI) standard and the pre-edgepeak was adjusted so that the standard had a Cr(VI)/Cr(total)ratio equal to 1. The spectra of Cr(OH)3 and Cr2O3, which wereused as representative Cr(III) standards, are shown in Figure 2;note that our Cr2O3 standard is similar to that reported previously(Wei et al. 2002).
The major spectral difference between Cr(VI) and Cr(III) isthe presence of the pre-edge peak at 5994.5 eV caused by Cr(VI).As discussed above, the height of this peak is proportional to thefraction of Cr(VI) in a sample. In addition, the post-edges in thespectra of the various Cr species also vary, as a result of dif-ferences in the atoms surrounding the Cr atoms. For example,the chromite sample (Cr-Fe spinel) has a distinctive post-edgeshape that is quite different from the other three Cr standards(Figure 2). In addition to these differences, the position of themain absorption edge shifts to higher energies as the averageoxidation state of Cr in the sample increases. For example, asshown in Figure 2, the edge for both Cr(III) standards occurs

550 M. WERNER ET AL.
between 5995–6005 eV, while the edge for K2CrO4 occurs be-tween 6005 and 6015 eV.
Effects of Flame Temperature and Compositionon the Cr(VI)/Cr(total) Ratio
The first variable we explored was the effect of flame tem-perature on the Cr(VI)/Cr(total) ratio in the particles. The twodominant fuel conditions we used were a hotter, 100% H2 flamewith a measured temperature of approximately 2480 K and acooler, 33% H2/67% Ar flame with a calculated temperature ofapproximately 1840 K (Reynolds 1986). We also prepared a se-ries of samples using a cooler 33% C3H8/67% Ar flame, whichproduced particles containing soot as well as metal. Based onICP-MS results from a subset of samples, the amount of Cr col-lected on each filter was typically 10–30 µg for particles from a33% H2 flame and 140–340 µg per filter for particles from the100% H2 flame.
As shown in Figure 4, the fuel composition, and thus thetemperature of the flame, has a large effect on the oxidation stateof the chromium in the resulting particles. The inset of Figure 4shows that particles from the 100% H2 flame seeded with Cr havea baseline-subtracted pre-edge peak height (i.e., Cr(VI)/Cr(total)ratio) of 0.43, indicating that 43% of the chromium is present asCr(VI). In contrast, the sample from the cooler 33% H2 flameseeded with Cr has a Cr(VI)/Cr(total) ratio of 0.051, almost anorder of magnitude less than that from the 100% H2 sample.Samples from a propane flame, by far the coolest of three flameconditions, had no detectable pre-edge peak, indicating there islittle, if any, Cr(VI) in these samples (Figure 4).
Figure 4 also shows that as the Cr(VI)/Cr(total) ratio in-creases, the edge is shifted to higher beam energies, consistent
FIG. 4. XANES spectra of particles collected from flames fed with 100% H2,33% H2, and 33% C3H8 and containing Cr(CO)6 or both Cr(CO)6 and Fe(CO)5.The laboratory where each sample was analyzed is listed in parentheses.
with the theoretically expected trend observed with our standards(Figure 2). The edge for the 100% H2 Cr only sample begins atapproximately 6000 eV, which coincides with the more tightlybound electrons around the Cr(VI) atoms in K2CrO4. In addi-tion, the post-edge oscillations of Cr particles from the 100%H2 flame resemble those of K2CrO4 (Figure 2a), while the post-edge oscillations of both the 33% H2 and C3H8 flames resemblethose of Cr2O3 (Figure 2b).
We also explored how the presence of Fe affects the oxida-tion state of Cr in ultrafine combustion particles. Iron is a com-mon constituent in combustion particles as well as in ambientaerosols (Puxbaum et al. 2004; Wang et al. 2003). For example,ratios of Cr:Fe in ambient particulate matter collected in andaround Vienna range from 1:2 to 1:10 in urban and rural set-tings respectively, depending on the types of industry in the area(Puxbaum et al. 2004). The Cr:Fe ratios in our samples, whichwere determined by ICP-MS, were between 1:2 to 1:3, with atypical iron concentration of 190–460 µg per filter.
As shown in Figure 4, this sample from a 100% H2 flameseeded with both Cr and Fe has a Cr(VI)/Cr(total) ratio of 0.12,compared to a value of 0.43 in the particles from the 100%H2 flame that contain Cr but no Fe. This indicates that addingFe decreases, but does not eliminate, the presence of Cr(VI) incombustion particles. Note that the edge of the Cr-Fe sample alsolies between the edges for the Cr-only particles created from the100% H2 and 33% H2 flames, consistent with the intermediateCr(VI)/Cr(total) ratio in the presence of iron.
Interestingly, in addition to decreasing the Cr(VI)/Cr(total)ratio, the presence of Fe in the flame appears to lead to the forma-tion of a new solid phase material. As seen in Figure 4, except forthe presence of a small amount of Cr(VI), the edge and post-edgesections of the spectra of the Cr/Fe samples strongly resemblethat of the Cr-Fe mixed phase (chromite) standard spectra shownin Figure 2. The similarity between these spectra implies that themajor form of Cr in these mixed phase samples is that of a Cr-Fespinel with a structure very similar to natural chromite.
Effects of Atmospheric Aging on Cr Only SamplesThough it is important to understand the composition of par-
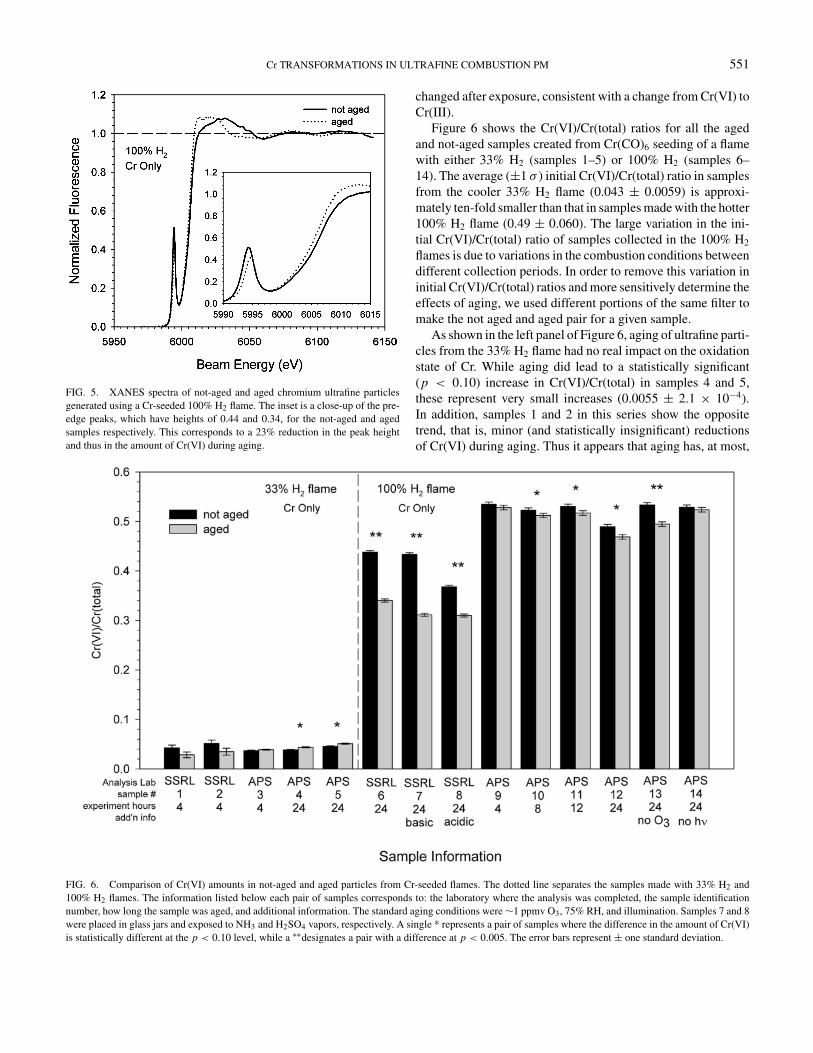
ticles from combustion processes, it is equally important toknow what happens to the composition during particle trans-port. We performed simulated aging experiments on particlescontaining only Cr as well as those containing both Cr andFe in order to see how atmospheric chemistry affects the ox-idation state of Cr. Figure 5 shows one example of the ef-fects of simulated atmospheric aging on Cr speciation in par-ticles created from a 100% H2 flame seeded with Cr(CO)6.Prior to aging, the Cr(VI)/Cr(total) ratio is 0.44, while after 24hours of aging (with ozone, humidified air, and illumination) theCr(VI)/Cr(total) ratio decreased to 0.34, a ∼23% reduction in theinitial amount of Cr(VI). The post-edge oscillations in the agedsample are also somewhat different from the not-aged sample,indicating that the bonding environment around the Cr atoms has
Cr TRANSFORMATIONS IN ULTRAFINE COMBUSTION PM 551
FIG. 5. XANES spectra of not-aged and aged chromium ultrafine particlesgenerated using a Cr-seeded 100% H2 flame. The inset is a close-up of the pre-edge peaks, which have heights of 0.44 and 0.34, for the not-aged and agedsamples respectively. This corresponds to a 23% reduction in the peak heightand thus in the amount of Cr(VI) during aging.
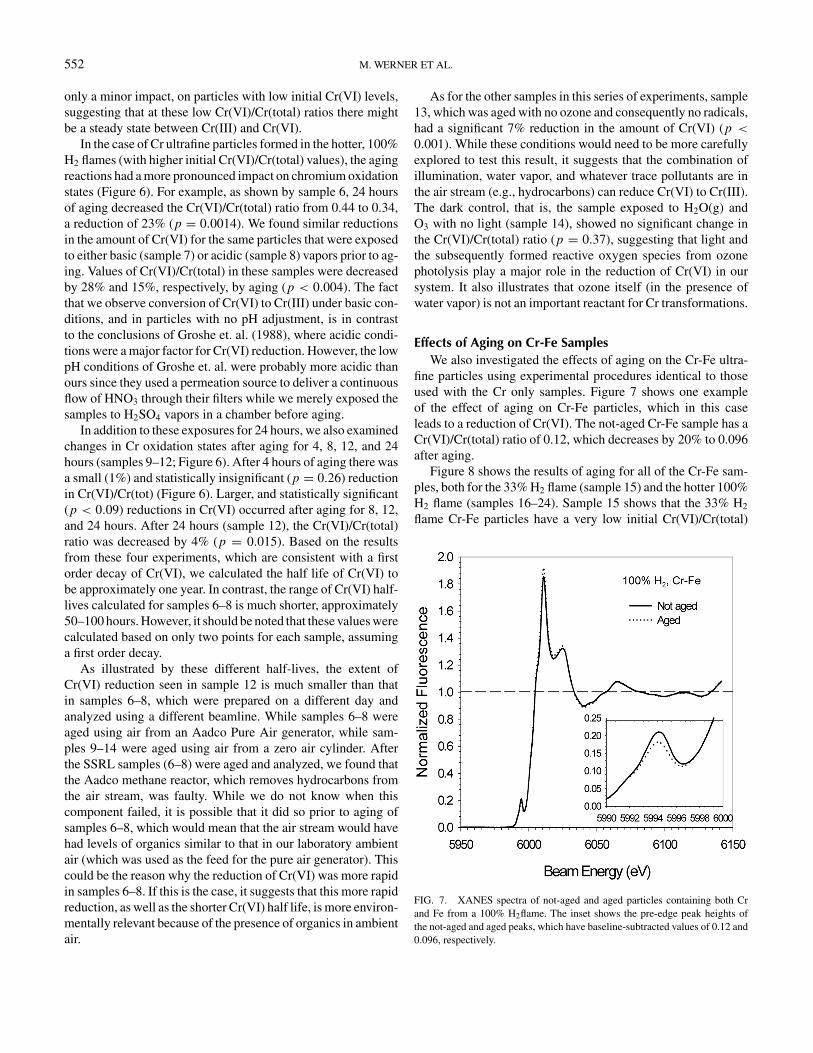
FIG. 6. Comparison of Cr(VI) amounts in not-aged and aged particles from Cr-seeded flames. The dotted line separates the samples made with 33% H2 and100% H2 flames. The information listed below each pair of samples corresponds to: the laboratory where the analysis was completed, the sample identificationnumber, how long the sample was aged, and additional information. The standard aging conditions were ∼1 ppmv O3, 75% RH, and illumination. Samples 7 and 8were placed in glass jars and exposed to NH3 and H2SO4 vapors, respectively. A single * represents a pair of samples where the difference in the amount of Cr(VI)is statistically different at the p < 0.10 level, while a ∗∗designates a pair with a difference at p < 0.005. The error bars represent ± one standard deviation.
changed after exposure, consistent with a change from Cr(VI) toCr(III).
Figure 6 shows the Cr(VI)/Cr(total) ratios for all the agedand not-aged samples created from Cr(CO)6 seeding of a flamewith either 33% H2 (samples 1–5) or 100% H2 (samples 6–14). The average (±1 σ ) initial Cr(VI)/Cr(total) ratio in samplesfrom the cooler 33% H2 flame (0.043 ± 0.0059) is approxi-mately ten-fold smaller than that in samples made with the hotter100% H2 flame (0.49 ± 0.060). The large variation in the ini-tial Cr(VI)/Cr(total) ratio of samples collected in the 100% H2
flames is due to variations in the combustion conditions betweendifferent collection periods. In order to remove this variation ininitial Cr(VI)/Cr(total) ratios and more sensitively determine theeffects of aging, we used different portions of the same filter tomake the not aged and aged pair for a given sample.
As shown in the left panel of Figure 6, aging of ultrafine parti-cles from the 33% H2 flame had no real impact on the oxidationstate of Cr. While aging did lead to a statistically significant(p < 0.10) increase in Cr(VI)/Cr(total) in samples 4 and 5,these represent very small increases (0.0055 ± 2.1 × 10−4).In addition, samples 1 and 2 in this series show the oppositetrend, that is, minor (and statistically insignificant) reductionsof Cr(VI) during aging. Thus it appears that aging has, at most,
552 M. WERNER ET AL.
only a minor impact, on particles with low initial Cr(VI) levels,suggesting that at these low Cr(VI)/Cr(total) ratios there mightbe a steady state between Cr(III) and Cr(VI).
In the case of Cr ultrafine particles formed in the hotter, 100%H2 flames (with higher initial Cr(VI)/Cr(total) values), the agingreactions had a more pronounced impact on chromium oxidationstates (Figure 6). For example, as shown by sample 6, 24 hoursof aging decreased the Cr(VI)/Cr(total) ratio from 0.44 to 0.34,a reduction of 23% (p = 0.0014). We found similar reductionsin the amount of Cr(VI) for the same particles that were exposedto either basic (sample 7) or acidic (sample 8) vapors prior to ag-ing. Values of Cr(VI)/Cr(total) in these samples were decreasedby 28% and 15%, respectively, by aging (p < 0.004). The factthat we observe conversion of Cr(VI) to Cr(III) under basic con-ditions, and in particles with no pH adjustment, is in contrastto the conclusions of Groshe et. al. (1988), where acidic condi-tions were a major factor for Cr(VI) reduction. However, the lowpH conditions of Groshe et. al. were probably more acidic thanours since they used a permeation source to deliver a continuousflow of HNO3 through their filters while we merely exposed thesamples to H2SO4 vapors in a chamber before aging.
In addition to these exposures for 24 hours, we also examinedchanges in Cr oxidation states after aging for 4, 8, 12, and 24hours (samples 9–12; Figure 6). After 4 hours of aging there wasa small (1%) and statistically insignificant (p = 0.26) reductionin Cr(VI)/Cr(tot) (Figure 6). Larger, and statistically significant(p < 0.09) reductions in Cr(VI) occurred after aging for 8, 12,and 24 hours. After 24 hours (sample 12), the Cr(VI)/Cr(total)ratio was decreased by 4% (p = 0.015). Based on the resultsfrom these four experiments, which are consistent with a firstorder decay of Cr(VI), we calculated the half life of Cr(VI) tobe approximately one year. In contrast, the range of Cr(VI) half-lives calculated for samples 6–8 is much shorter, approximately50–100 hours. However, it should be noted that these values werecalculated based on only two points for each sample, assuminga first order decay.
As illustrated by these different half-lives, the extent ofCr(VI) reduction seen in sample 12 is much smaller than thatin samples 6–8, which were prepared on a different day andanalyzed using a different beamline. While samples 6–8 wereaged using air from an Aadco Pure Air generator, while sam-ples 9–14 were aged using air from a zero air cylinder. Afterthe SSRL samples (6–8) were aged and analyzed, we found thatthe Aadco methane reactor, which removes hydrocarbons fromthe air stream, was faulty. While we do not know when thiscomponent failed, it is possible that it did so prior to aging ofsamples 6–8, which would mean that the air stream would havehad levels of organics similar to that in our laboratory ambientair (which was used as the feed for the pure air generator). Thiscould be the reason why the reduction of Cr(VI) was more rapidin samples 6–8. If this is the case, it suggests that this more rapidreduction, as well as the shorter Cr(VI) half life, is more environ-mentally relevant because of the presence of organics in ambientair.
As for the other samples in this series of experiments, sample13, which was aged with no ozone and consequently no radicals,had a significant 7% reduction in the amount of Cr(VI) (p <
0.001). While these conditions would need to be more carefullyexplored to test this result, it suggests that the combination ofillumination, water vapor, and whatever trace pollutants are inthe air stream (e.g., hydrocarbons) can reduce Cr(VI) to Cr(III).The dark control, that is, the sample exposed to H2O(g) andO3 with no light (sample 14), showed no significant change inthe Cr(VI)/Cr(total) ratio (p = 0.37), suggesting that light andthe subsequently formed reactive oxygen species from ozonephotolysis play a major role in the reduction of Cr(VI) in oursystem. It also illustrates that ozone itself (in the presence ofwater vapor) is not an important reactant for Cr transformations.
Effects of Aging on Cr-Fe SamplesWe also investigated the effects of aging on the Cr-Fe ultra-
fine particles using experimental procedures identical to thoseused with the Cr only samples. Figure 7 shows one exampleof the effect of aging on Cr-Fe particles, which in this caseleads to a reduction of Cr(VI). The not-aged Cr-Fe sample has aCr(VI)/Cr(total) ratio of 0.12, which decreases by 20% to 0.096after aging.
Figure 8 shows the results of aging for all of the Cr-Fe sam-ples, both for the 33% H2 flame (sample 15) and the hotter 100%H2 flame (samples 16–24). Sample 15 shows that the 33% H2
flame Cr-Fe particles have a very low initial Cr(VI)/Cr(total)
FIG. 7. XANES spectra of not-aged and aged particles containing both Crand Fe from a 100% H2flame. The inset shows the pre-edge peak heights ofthe not-aged and aged peaks, which have baseline-subtracted values of 0.12 and0.096, respectively.
Cr TRANSFORMATIONS IN ULTRAFINE COMBUSTION PM 553
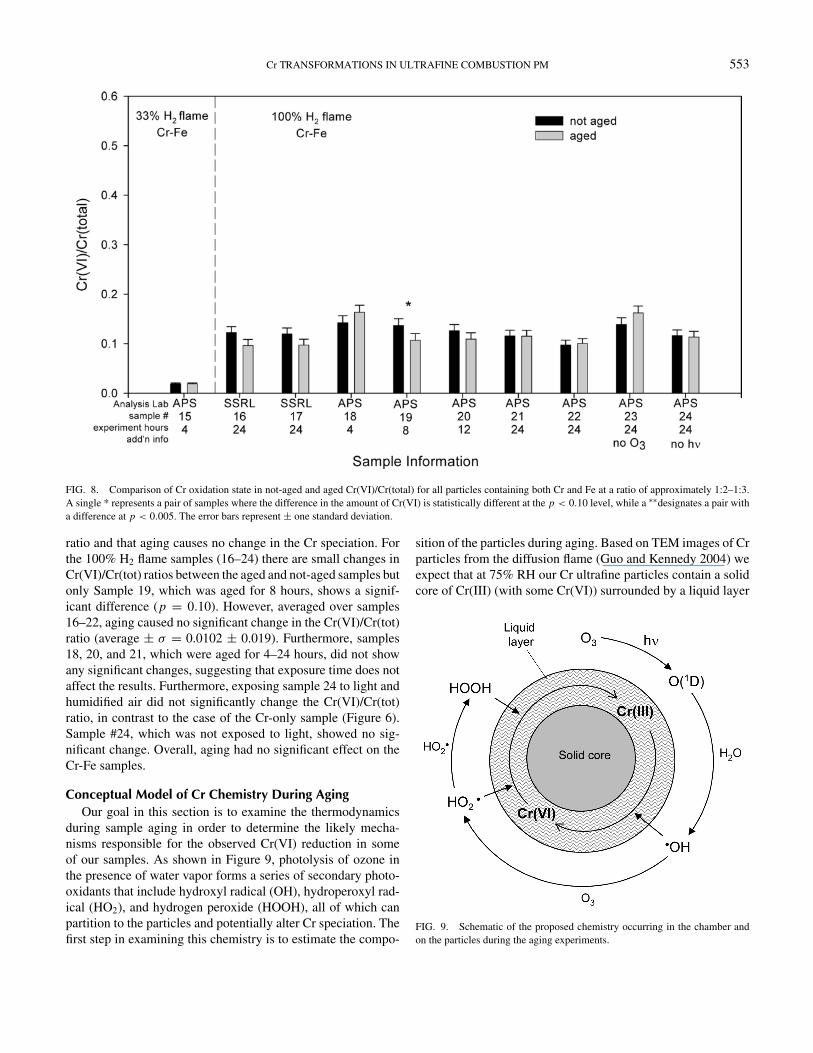
FIG. 8. Comparison of Cr oxidation state in not-aged and aged Cr(VI)/Cr(total) for all particles containing both Cr and Fe at a ratio of approximately 1:2–1:3.A single * represents a pair of samples where the difference in the amount of Cr(VI) is statistically different at the p < 0.10 level, while a ∗∗designates a pair witha difference at p < 0.005. The error bars represent ± one standard deviation.
ratio and that aging causes no change in the Cr speciation. Forthe 100% H2 flame samples (16–24) there are small changes inCr(VI)/Cr(tot) ratios between the aged and not-aged samples butonly Sample 19, which was aged for 8 hours, shows a signif-icant difference (p = 0.10). However, averaged over samples16–22, aging caused no significant change in the Cr(VI)/Cr(tot)ratio (average ± σ = 0.0102 ± 0.019). Furthermore, samples18, 20, and 21, which were aged for 4–24 hours, did not showany significant changes, suggesting that exposure time does notaffect the results. Furthermore, exposing sample 24 to light andhumidified air did not significantly change the Cr(VI)/Cr(tot)ratio, in contrast to the case of the Cr-only sample (Figure 6).Sample #24, which was not exposed to light, showed no sig-nificant change. Overall, aging had no significant effect on theCr-Fe samples.
Conceptual Model of Cr Chemistry During AgingOur goal in this section is to examine the thermodynamics
during sample aging in order to determine the likely mecha-nisms responsible for the observed Cr(VI) reduction in someof our samples. As shown in Figure 9, photolysis of ozone inthe presence of water vapor forms a series of secondary photo-oxidants that include hydroxyl radical (OH), hydroperoxyl rad-ical (HO2), and hydrogen peroxide (HOOH), all of which canpartition to the particles and potentially alter Cr speciation. Thefirst step in examining this chemistry is to estimate the compo-
sition of the particles during aging. Based on TEM images of Crparticles from the diffusion flame (Guo and Kennedy 2004) weexpect that at 75% RH our Cr ultrafine particles contain a solidcore of Cr(III) (with some Cr(VI)) surrounded by a liquid layer
FIG. 9. Schematic of the proposed chemistry occurring in the chamber andon the particles during the aging experiments.

554 M. WERNER ET AL.
containing primarily Cr(VI). We estimate that this liquid layerhas a pH of ∼5 based on rough estimates made using pH paperon very slightly moistened PM samples. From the pKa values forH2CrO4 and HCrO−
4 (0.75 and 6.49, respectively (Perrin 1982);most Cr(VI) in the liquid layer will be present as HCrO−
4 . Assum-ing that Cr(III) solubility is controlled by equilibrium with solidCr(OH)3, we estimate that the liquid layer also contains 6 µMCr(H2O)3+
6 and 90 µM CrOH(H2O)2+5 . Finally, based on esti-
mated concentrations of the gas phase oxidants and their Henry’slaw constants (Finlayson-Pitts and Pitts 2000) we estimate thatthe concentrations of oxidants in the particle liquid layer are:OH (1 × 10−12 M), HO2 (8 × 10−9 M), H2O2 (8 × 10−5 M),O3 (1 × 10−8 M), HO3 (assumed to be 1 × 10−15 M), and O2
(2.6 × 10−4 M). Although only approximations, these oxidantand chromium concentrations are used to adjust standard state�G0 values to more realistic thermodynamic values, �G, asdiscussed below.
The five main photo-oxidants in our system vary significantlyin strength from OH (the strongest oxidant) to O2 (the weakest)as can be seen from their standard state reduction half-reactions(Wardman 1989):
OH + H+ + e− → H2O �G0 = −263 kJ/mol [R1]
HO2 + H+ + e− → HOOH �G0 = −140 kJ/mol [R2]
O3 + H+ + e− → HO3 �G0 = −73.7 kJ/mol [R3]
HOOH+H+ + e− → H2O + OH �G0 = −67.5 kJ/mol
[R4]
H+ + O2 + e− → HO2 �G0 = 41.5 kJ/mol [R5]
Similarly, the chromium species have different stabilities asshown by the standard state reduction half-reactions of bichro-mate to form different Cr(III) species (Woods and Garre1987):
3e− + 4H+ + HCrO−4 (aq)
→ 1
2Cr2O3(s) + 2
1
2H2O �G0 = −734.0 kJ/mol [R6]
3e− + 7H+ + HCrO−4 (aq)
→ Cr3+(aq) + 4H2O �G0 = −388.2 kJ/mol [R7]
3e− + 7H+ + HCrO−4 (aq)
→ Cr(OH)3(s) + H2O �G0 = −363.2 kJ/mol [R8]
3e− + H2O + HCrO−4 (aq)
→ Cr(OH)(H2O)2+5 (aq) �G0 = 829.2 kJ/mol [R9]
By combining the oxidant couples (half-reactions (R1–R5)with the chromium half-reactions (R6–R9), and adjusting thestandard state values using the assumed reaction concentrationsabove, we can estimate whether a given reaction should occurspontaneously in our model system (i.e., �G (or �G0) < 0).As a first step at understanding the chemistry, we focus hereon the two main Cr(III) species, Cr2O3(s) and Cr(OH)3(s). Forexample, the combination of OH (R1) with Cr2O3 (R6) is non-
spontaneous:
3OH + 1
2Cr2O3(s) →
HCrO−4 + 1
2H2O + H+�G = 20 kJ/mol [R10]
Indeed, solid Cr2O3 does not react spontaneously with any ofthe oxidants to form Cr(VI), indicating that Cr2O3 in our ag-ing chamber is essentially inert to oxidation. The situation issimilar, although more complicated, for solid Cr(OH)3. Whileoxidation of Cr(OH)3 by OH and HO2 (R11 and R12) is ther-modynamically favored, oxidation by the other photo-oxidantsis not spontaneous (R13–R15).
Cr(OH)3(s) + 3OH → H+ + HCrO−4 (aq) + 2H2O
�G = −250 kJ/mol [R11]
Cr(OH)3(s) + H2O + 3HO2 → H+ + HCrO−4 (aq)
+3HOOH �G = −22.1 kJ/mol [R12]
Cr(OH)3(s) + H2O + 3O3 → H+ + HCrO−4 (aq) + 3HO3
�G = 14 kJ/mol [R13]
Cr(OH)3(s) + 3HOOH → H+ + HCrO−4 (aq)
+2H2O + 3OH �G = 24 kJ/mol [R14]
Cr(OH)3(s) + H2O + 3O2 → H+ + HCrO−4 (aq) + 3HO2
�G = 377 kJ/mol [R15]
In fact, these calculations show that HO2 can actually serveto reduce Cr(VI) to Cr(III), with a very strong thermodynamicdriving force (i.e., �G = −377 kJ/mol for the reverse of R15).This potential role of HO2 as a reductant is interesting becausesimple kinetic photochemical modeling of our aging chamberconditions indicates that the gas phase concentration of HO2is approximately 20 times higher than that of OH. When theseobservations are combined with the known kinetic stability ofCr(III) species (Nico and Zasoski 2000), it seems reasonablethat we observed no significant oxidation of Cr(III) species, butrather saw Cr(VI) reduction. Based on the abundance of HO2
and our thermodynamic calculations, this reduction might havebeen due to HO2, but it is unclear whether this reaction is rapidenough to account for our observations. Overall, the low solubil-ity and kinetic stability of the Cr(III) compounds strongly limitstheir tendency to be oxidized. On the other hand, HCrO−
4 is moreavailable for reaction because of its solubility and it is gener-ally thermodynamically poised for reduction. It should also bementioned that since a number of the �G values are close tozero (within ± 50 kJ/mol) there is a possibility that a pseudo-equilibrium condition could exist between Cr(OH)3 and HCrO−
4under atmospheric conditions. Similarly, since both oxidationand reduction reactions are thermodynamically allowed underthe current experimental conditions, it is possible that a steadystate between Cr(III) and Cr(IV) may exist under some circum-stances. The presence of a such a steady state could explain the

Cr TRANSFORMATIONS IN ULTRAFINE COMBUSTION PM 555
observation that the distribution of Cr oxidation states remainsunchanged during aging of particles with low initial Cr(VI), pos-sibly because the rates of Cr(III) oxidation by OH and Cr(VI) re-duction by HO2 are approximately equal under these conditions.
ENVIRONMENTAL IMPLICATIONSAND CONCLUSIONS
Our goal was to examine how atmospheric aging affectsCr speciation on ultrafine particulate matter from combustionsources. We simulated a simple atmosphere with humidified air,ozone, and light to determine the fate of Cr on ultrafines gener-ated from a laboratory flame. Our results show that the amountof Cr(VI) present in the particles (prior to aging) decreases withdecreasing flame temperature and that the addition of Fe to thefuel also reduces the amount of Cr(VI). Our aging experimentsshow that after 24 hours of aging there is up to a 20% reduc-tion in the Cr(VI)/Cr(total) ratio for particles produced from thehottest flame (100% H2). The half life for Cr(VI) in these ex-periments was typically 50–100 hours. Groshe and co-workersreported an average half-life of 13 ± 5.8 hours for experimentsthat included organics. However, the half life of Cr(VI) in theirexperiments also ranged as high as ∼200 hours under conditionswhere there were no organics (Grohse et al. 1988). Our observedreduction of ultra fine Cr(VI) to Cr(III) during simulated at-mospheric aging is supported by thermodynamic calculations,which suggest that Cr(VI) reduction is more important thanCr(III) oxidation during aging. This net reduction of Cr(VI) isconsistent with previous model predictions (Seigneur and Con-stantinou 1995), although our mechanism of reduction is verydifferent.
The fact that Cr(VI) is generally reduced, both in our ex-periments as well as those of Grohse and co-workers (1988),suggests that the Cr-associated toxicity in particulate matterdecreases over time. However, there are a number of impor-tant caveats to this statement. First, we find that particles whereCr(VI)/Cr(total) is relatively low (≤15%), the Cr(VI) is not sig-nificantly reduced by aging (e.g., in our Cr PM from the 33% H2
flame or in the Cr-Fe particles from the 100% H2 flame). Grohseand co-workers observed a wide range in the reduction and halflife of Cr(VI) depending on the experimental conditions underwhich their particles were aged (Grohse et al. 1988). Second,both our particles and aging conditions are simple comparedto ambient PM and atmospheric conditions, and thus our ex-periments could be missing significant reactions that affect Croxidation states in the atmosphere. Finally, even if Cr(VI) isreduced to Cr(III) on a timescale of tens of hours in the atmo-sphere, there can still be significant exposure for populationsthat are relatively near Cr(VI) emission sources.
REFERENCESAnastasio, C. A., and Martin, S. T. (2001). Atmospheric Nanoparticles, Reviews
in Minerology and Geochem. 44:293–349.
Balasubramanian, M., and Melendres, C. A. (1999). An X-Ray Absorption Near-Edge Spectroscopy Study of the Oxidation State of Chromium in Electrode-posited Oxide Films, Electrochimica Acta 44:2941–2945.
Brasseur, G. P., Orlando, J. J., and Tyndall, G. S. (1999). Atmospheric Chemistryand Global Change, Oxford University Press, New York.
Cieslak-Golonka, M. (1996). Toxic and Mutagenic Effects of Chromium(VI).A Review, Polyhedron. 15:3667–3689.
Cohen, M. D., Kargacin, B., Klein, C. B., and Costa, M. (1993). Mechanismsof Chromium Carcinogenicity and Toxicity, Crit. Rev. Toxicol. 23:255–281.
Dreher, K. L. Jaskot, R. H., Lehmann, J. R., Richards, J. H., McGee, J. K., Ghio,A. J., and Costa, D. L. (1997). Soluble Transition Metals Mediate Residual OilFly Ash Induced Acute Lung Injury, J. Toxicol. Environ. Health 50:285–305.
Ferin, J., Oberdorster, G., Penney, D. P., Soderholm, S. C., Gelein, R., and Piper,H. C. (1990). Increased Pulmonary Toxicity of Ultrafine Particles. 1. ParticleClearance, Translocation, Morphology, J. Aerosol Science 21:381–384.
Finlayson-Pitts, B. J., and Pitts, J. N. (2000). Chemistry of the Upper and LowerAtmospere, Academic Press, San Diego.
Grohse, P. M., Gutknect, W. F., Hodson, L., and Wilson, B. M. (1988). The Fateof Hexavalent Chromium in the Atmosphere, CARB contract No. A6-096-32,Research Triangle Institute.
Guo, B., and Kennedy, I. M. (2004). The Speciation and Morphology ofChromium Oxide Nanoparticles in a Diffusion Flame, Aerosol Sci. and Tech-nol. 38:424–436.
Hu, M. J., Wei, Y. L., Yang, Y. W., and Lee, J. F. (2004). X-Ray AbsorptionSpectroscopy Study of Chromium Recovered from Cr(VI)-Containing Waterwith Rice Husk, J. Phy. Cond. Mat. 16:S3473–S3478.
Huggins, F. E., Najih, M., and Huffman, G. P. (1999). Direct Speciation ofChromium in Coal Combustion by-Products by X-Ray Absorption Fine-Structure Spectroscopy, Fuel 78:233–242.
Hughes, L. S., Cass, G. R., Gone, J., Ames, M., and Olmez, I. (1998). Physicaland Chemical Characterization of Atmospheric Ultrafine Particles in the LosAngeles Area, Environ. Sci. Technol. 32:1153–1161.
Minni, E., Gustafsson, T. E., Koponen, M., and Kalliomaki, P. L. (1984). AStudy of the Chemical-Structure of Particles in the Welding Fumes of Mildand Stainless-Steel, J. Aerosol Sci. 15:57–68.
Nico, P. S., Fendorf, S. E., Lowney, Y. W., Holm, S. E., and Ruby, M. V. (2004).Chemical Structure of Arsenic and Chromium in CCA-Treated Wood: Impli-cations of Environmental Weathering, Environ. Sci. Technol. 38:5253–5260.
Nico, P. S., and Zasoski, R. J. (2000). Importance of Mn(II) Availability ofCr(III) Oxidation on Birnessite, Environ. Sci. Technol. 38:5253–5260.
Oberdorster, G., Finkelstein, J., Ferin, J., and Cox, C. (1992). Particle InducedLung Injury—Studies with Particles of High and Low Intrinsic Toxicity andof Different Particle Sizes, Faseb Journal 6:A2048–A2048.
Patterson, R. R., Fendorf, S., and Fendorf, M. (1997). Reduction of Hexava-lent Chromium by Amorphous Iron Sulfide, Environ. Sci. Technol. 31:2039–2044.
Peterson, M. L., Brown, G. E., Parks, G. A., and Stein, C. L. (1997) DifferentialRedox and Sorption of Cr(III/VI) on Natural Silicate and Oxide Minerals:EXAFS and XANES results, Geochimica et Cosmochimica Acta 61:3399–3412.
Perrin, D. D. (1982), Ionization Constants of Inorganic Acids and Bases inAqueous Solution, Pergamon, Oxford.
Pope, C. A., Dockery, D. W., and Schwartz, J. (1995). Review of EpidemiologicalEvidence of Health-Effects of Particulate Air-Pollution, Inhal. Toxicol. 7:1–18.
Puxbaum, H., Gomiscek, B., Kalina, M., Bauer, H., Salam, A., Stopper, S.,Preining, O., and Hauck, H. (2004). A Dual Site Study of PM2.5 and PM10Aerosol Chemistry in the Larger Region of Vienna, Austria, Atmos. Environ.38:3949–3958.
Reynolds, W. C. (1986). The Element Potential Method for Chemical Equi-librium Analysis: Implementation in the Interactive Program STANJAB,Stanford University.
Schlesinger, R. B. (2000). Properties of Ambient Pm Responsible for HumanHealth Effects: Coherence between Epidemiology and Toxicology, Inhal. Tox-icol. 12:23–25.

556 M. WERNER ET AL.
Seigneur, C., and Constantinou, E. (1995). Chemical Kinetic Mechanism forAtmospheric Chromium, Environ. Sci. Technol. 29:222–231.
Shoji, T., Huggins, F. E., Huffman, G. P., Linak, W. P., and Miller, C. A. (2002).XAFS Spectroscopy Analysis of Selected Elements in Fine Particulate MatterDerived from Coal Combustion, Energy & Fuels 16:325–329.
Wang, Y. F., Huang, K. L., Li, C. T., Mi, H. H., Luo, J.-H., and Tsai, P.-J. (2003).Emissions of Fuel Metals Content from a Diesel Vehicle Engine, Atmos.Environ. 37:4637–4643.
Wardman, P. (1989). Reduction Potentials of One-Electron Couples InvolvingFree Radicals in Aqueous Solution, J. Phy. Chem. Ref. Data 18:1637–1755.
Webb, S., Sixpack, version 0.52, 2002.
Wei, Y. L., Chiu, S. Y., Tsai, H. N., Yang, Y. W., and Lee, J.-F. (2002). ThermalStabilization of Chromium (VI) in Kaolin, Environ. Sci. Technol. 36:4633–4641.
Woods, T. L., and Garrel, R. M. (1987) Thermodyrnamic Values at Low Tem-peratures for Natural Inorganic Materials: An Uncritical Summary. OxfordUniversity Press, Inc., New York.
Zhang, H. (2000). Light and Iron(III)-Induced Oxidation of Chromium(III) inthe Presence of Organic Acids and Manganese(II) in Simulated AtmosphericWater, Atmos. Environ. 34:1633–1640.
Zhang, H., and Bartlett, R. J. (1999). Light-Induced Oxidation of AqueousChromium(III) in the Presence of Iron(III), Environ. Sci. Technol. 33:588–594.




















