
Kinetic analysis of the phosphorylation-dependent interactions of synapsin I with rat brain synaptic...
15
Journal of Physiology (1997), 504.3, pp. 501-515 Kinetic analysis of the phosphorylation-dependent interactions of synapsin I with rat brain synaptic vesicles Giovanni Stefani *, Franco Onofri *, Flavia Valtorta t, Paola Vaccaro *, Paul Greengard t and Fabio Benfenati *§ *Department of Experimental Medicine, University of Roma Tor Vergata, 00133 Roma and Department of Biomedical Sciences, Section of Physiology, University of Modena, 41100 Modena, tDIBIT, S.Raffaele Scientific Institute, CNR Centre of Cellular and Molecular Pharmacology, Department of Medical Pharmacology, University of Milano, 20100 Milano, Italy and t Laboratory of Molecular and Cellular Neuroscience, The Rockefeller University, New York, NY 10021-6399, USA 1. Synapsin I, a major synaptic vesicle (SV)-associated phosphoprotein, is involved in the regulation of neurotransmitter release and synapse formation. By binding to both phospholipid and protein components of SV with high affinity and in a phosphorylation- dependent fashion, synapsin I is believed to cluster SV and to attach them to the actin-based cytoskeleton of the nerve terminal. 2. In the present study we have investigated the kinetic aspects of synapsin I-SV interactions and the mechanisms of their modulation by ionic strength and site-specific phosphorylation, using fluorescence resonance energy transfer between suitable fluorophores linked to synapsin I and to the membrane bilayer. 3. The binding of synapsin I to the phospholipid and protein components of SV has fast kinetics: mean time constants ranged between 1 and 4 s for association and 9 and 11 s for ionic strength-induced dissociation at 20 'C. The interaction with the phospholipid component consists predominantly of a hydrophobic binding with the core of the membrane which may account for the membrane stabilizing effect of synapsin I. 4. Phosphorylation of synapsin I by either SV-associated or purified exogenous Ca2+/calmodulin-dependent protein kinase II (CaMPKII) inhibited the association rate and the binding to SV at steady state by acting on the ionic strength-sensitive component of the binding. When dephosphorylated synapsin I was previously bound to SV, exposure of SV to Ca2+/calmodulin in the presence of ATP triggered a prompt dissociation of synapsin I with a time constant similar to that of ionic strength-induced dissociation. 5. In conclusion, the reversible interactions between synapsin I and SV are highly regulated by site-specific phosphorylation and have kinetics of the same order of magnitude as the kinetics of SV recycling determined in mammalian neurons under comparable temperature conditions. These findings are consistent with the hypothesis that synapsin I associates with, and dissociates from, SV during the exo-endocytotic cycle. The on-vesicle phosphorylation of synapsin I by the SV-associated CaMPKII, and the subsequent dissociation of the protein from the vesicle membrane, though not involved in mediating exocytosis of primed vesicles evoked by a single stimulus, may represent a prompt and efficient mechanism for the modulation of neurotransmitter release and presynaptic plasticity. The synapsins (synapsin I and II) are a family of synaptic 1990; Greengard, Valtorta, Czernik & Benfenati, 1993 for vesicle (SV)-associated phosphoproteins that participate in review). Synapsin I, more abundant than II and the most the regulation of neurotransmitter release and synapse thoroughly studied of the synapsins, is phosphorylated on formation (see De Camilli, Benfenati, Valtorta & Greengard, distinct sites by multiple protein kinases both in vitro and § To whom correspondence should be addressed at the Department of Biomedical Sciences, Section of Physiology, Via Campi 287, I-41100 Modena, Italy. 7005 501 ) by guest on July 15, 2011 jp.physoc.org Downloaded from J Physiol (
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Kinetic analysis of the phosphorylation-dependent interactions of synapsin I with rat brain synaptic...

Journal of Physiology (1997), 504.3, pp. 501-515
Kinetic analysis of the phosphorylation-dependentinteractions of synapsin I with rat brain synaptic vesicles
Giovanni Stefani *, Franco Onofri *, Flavia Valtorta t, Paola Vaccaro *,Paul Greengard t and Fabio Benfenati *§
*Department of Experimental Medicine, University of Roma Tor Vergata, 00133 Romaand Department of Biomedical Sciences, Section of Physiology, University of Modena,41100 Modena, tDIBIT, S.Raffaele Scientific Institute, CNR Centre of Cellular and
Molecular Pharmacology, Department of Medical Pharmacology, University of Milano,20100 Milano, Italy and t Laboratory of Molecular and Cellular Neuroscience,
The Rockefeller University, New York, NY 10021-6399, USA
1. Synapsin I, a major synaptic vesicle (SV)-associated phosphoprotein, is involved in theregulation of neurotransmitter release and synapse formation. By binding to bothphospholipid and protein components of SV with high affinity and in a phosphorylation-dependent fashion, synapsin I is believed to cluster SV and to attach them to the actin-basedcytoskeleton of the nerve terminal.
2. In the present study we have investigated the kinetic aspects of synapsin I-SV interactionsand the mechanisms of their modulation by ionic strength and site-specific phosphorylation,using fluorescence resonance energy transfer between suitable fluorophores linked tosynapsin I and to the membrane bilayer.
3. The binding of synapsin I to the phospholipid and protein components of SV has fastkinetics: mean time constants ranged between 1 and 4 s for association and 9 and 11 s forionic strength-induced dissociation at 20 'C. The interaction with the phospholipidcomponent consists predominantly of a hydrophobic binding with the core of the membranewhich may account for the membrane stabilizing effect of synapsin I.
4. Phosphorylation of synapsin I by either SV-associated or purified exogenousCa2+/calmodulin-dependent protein kinase II (CaMPKII) inhibited the association rate andthe binding to SV at steady state by acting on the ionic strength-sensitive component of thebinding. When dephosphorylated synapsin I was previously bound to SV, exposure of SV toCa2+/calmodulin in the presence of ATP triggered a prompt dissociation of synapsin I witha time constant similar to that of ionic strength-induced dissociation.
5. In conclusion, the reversible interactions between synapsin I and SV are highly regulated bysite-specific phosphorylation and have kinetics of the same order of magnitude as thekinetics of SV recycling determined in mammalian neurons under comparable temperatureconditions. These findings are consistent with the hypothesis that synapsin I associates with,and dissociates from, SV during the exo-endocytotic cycle. The on-vesicle phosphorylationof synapsin I by the SV-associated CaMPKII, and the subsequent dissociation of the proteinfrom the vesicle membrane, though not involved in mediating exocytosis of primed vesiclesevoked by a single stimulus, may represent a prompt and efficient mechanism for themodulation of neurotransmitter release and presynaptic plasticity.
The synapsins (synapsin I and II) are a family of synaptic 1990; Greengard, Valtorta, Czernik & Benfenati, 1993 forvesicle (SV)-associated phosphoproteins that participate in review). Synapsin I, more abundant than II and the mostthe regulation of neurotransmitter release and synapse thoroughly studied of the synapsins, is phosphorylated onformation (see De Camilli, Benfenati, Valtorta & Greengard, distinct sites by multiple protein kinases both in vitro and
§To whom correspondence should be addressed at the Department of Biomedical Sciences,Section of Physiology, Via Campi 287, I-41100 Modena, Italy.
7005 501
) by guest on July 15, 2011jp.physoc.orgDownloaded from J Physiol (

0. Stefani and others
in vivo. Both synapsins are phosphorylated at a single site(site 1) on the NH2-terminal region (head) by cAMP-dependent protein kinase (PKA) and Ca2+/calmodulin-dependent protein kinase I (CaMPKI). In addition,synapsin I is the best known substrate for Ca2+/calmodulin-dependent protein kinase II (CaMPKII) whichphosphorylates two serine residues (sites 2 and 3) in theelongated, basic and proline-rich COOH-terminal region(tail). Recently synapsin I has been reported to bephosphorylated at three serine residues (sites 4, 5 and 6) bya mitogen-activated protein kinase (MAPK) (Jovanovic et al.1996).
Synapsin I binds to the cytoplasmic side of the SV membraneand interacts with both actin filaments and monomers(Greengard et al. 1993). Site-specific phosphorylationmodulates these interactions selectively. Phosphorylation byCaMPKII greatly reduces the binding of synapsin I to SVby inducing a 5-fold decrease in binding affinity atequilibrium (Schiebler, Jahn, Doucet, Rothlein & Greengard,1986; Benfenati, Biihler, Jahn & Greengard, 1989a;Benfenati, Valtorta, Rubenstein, Gorelick, Greengard &Czernik, 1992b), while phosphorylation by PKA or MAPKis without effect (Schiebler et at. 1986; Jovanovic et al. 1996).The interactions of synapsin I with actin filments and actinare dramatically inhibited or virtually abolished byCaMPKII phosphorylation, but in this case phosphorylationby MAPK and PKA is also effective although to a smallerextent (Bahler & Greengard, 1987; Valtorta, Greengard,Fesce, Chieregatti & Benfenati, 1992; Jovanovic et al. 1996).
The ability to interact simultaneously with vesicles andactin allows synapsin I to reversibly cross-link SV to theactin-based cytoskeleton in a phosphorylation-dependentmanner (Benfenati, Valtorta, Chieregatti & Greengard,1992a; Ceccaldi, Grohovaz, Benfenati, Chieregatti,Greengard & Valtorta, 1995). In addition, synapsin I is ableto aggregate vesicles in clusters and to stabilize theirphospholipid membrane into a stable bilayer structure(Benfenati, Valtorta, Rossi, Onofri, Sihra & Greengard,1993). These biological properties of synapsin I account, atleast in part, for the functional consequences of alteredexpression of the protein in vivo. Thus, dephosphorylatedsynapsin I injected in high doses into nerve terminalsinhibits neurotransmitter release, while the CaMPKII-phosphorylated form is ineffective (Llinas, Gruner,Sugimori, McGuinness & Greengard, 1991). Injection ofanti-synapsin antibodies into lamprey reticulospinal axonscauses the disappearance of synaptic vesicle clusters and animpaired release after high-frequency stimulation(Pieribone, Shupliakov, Brodin, Hilfiker-Rothenfluh, Czernik& Greengard, 1995). Synapsin I-deficient mice obtained bytargeted gene disruption exhibit changes in the number anddistribution of SV, decreased maximal neurotransmitterrelease and altered presynaptic plasticity (Li et al. 1995;Chin, Li, Ferreira, Kosik & Greengard, 1995; Rosahl et al.1995; Takei et al. 1995; Ryan, Li, Chin, Greengard &Smith, 1996). Precocious expression of synapsin I in
Xenopus embryos induces an accelerated structural andfunctional maturation of neuromuscular synapses withincreased number and clustering of SV within developingnerve terminals (Lu, Greengard & Poo, 1992; Valtorta,Iezzi, Benfenati, Lu, Poo & Greengard, 1995).
The binding of synapsin I to SV and its regulation byphosphorylation are likely to play a crucial role in thebiological activity of synapsin I. The binding results frommultiple interactions of distinct sites of synapsin I withprotein and phospholipid components of SV. The highlyhydrophobic head region of synapsin I binds with highaffinity to acidic phospholipids enriched in the cytoplasmicleaflet of the vesicles and penetrates the hydrophobic core ofthe vesicle membrane (phospholipid site; Benfenati,Greengard, Brunner & Bahler, 1989b; Benfenati et al.1989a). On the other hand the basic and elongated tailregion specifically binds the regulatory domain of a SV-associated form of CaMPKII and its binding is virtuallyabolished by CaMPKII phosphorylation of synapsin I(protein site; Benfenati et al. 1989b; Benfenati et al. 1992a).The synaptic vesicle-associated CaMPKII seems to be themajor enzyme responsible for phosphorylation of synapsin Iwithin nerve terminals and for the regulation of itsassociation with the vesicle membrane (Benfenati, Onofri,Czernik & Valtorta, 1996).
Although the interactions between synapsin I and SV havebeen investigated in detail, all data have been obtained atequilibrium by using perturbing and time-consumingmethods to separate vesicle-bound from free synapsin I,such as high-speed centrifugation or gel filtration (Schiebleret al. 1986). Kinetic information, which could indicatewhether the association and dissociation reactions occur on atime scale compatible with a fast regulation of presynapticfunction, has not been available so far. Recently, thedevelopment of extracellular fluorescent tracers which arecaptured by SV upon endocytosis and released uponsubsequent exocytosis of loaded SV has allowed thedetermination of the dynamics of SV recycling in livingneurons. In both central and peripheral synapses, the half-time of SV endocytosis was found to be about 20 s at roomtemperature, while the minimum time required for a newlyendocytosed SV to become available again for exocytosisranged between 15 and 45 s, with a minimum overallduration of a complete exo-endocytotic cycle ofapproximately 1 min (Ryan, Reuter, Wendland, Schweizer,Tsien & Smith, 1993; Ryan & Smith, 1995; Betz & Wu,1995; De Camilli & Takei, 1996).
In this paper, we have used non-perturbing techniques suchas fluorescence resonance energy transfer (FRET) toinvestigate the topology and kinetics of the interactionsbetween synapsin I and SV, as well as their sensitivity tosite-specific phosphorylation of synapsin I. The resultsdemonstrate that the kinetics of association and dissociationat both the phospholipid and the protein sites occur withhalf-times of a few seconds at 20 °C, that the penetrationinto the bilayer core accounts for the majority of the
502 J Physiol.504.3
) by guest on July 15, 2011jp.physoc.orgDownloaded from J Physiol (

Kinetics of synapsin-synaptic vesicle binding
phospholipid interaction and finally, that activation of theSV-associated CaMPKII induces prompt dissociation ofsynapsin I from the vesicle membrane.
METHODSMaterials[y-32P]ATP (specific activity > 3000 Ci mmol-'), [14C]phosphatidyl-choline (0-1 Ci mmolF1) and "25I-labelled goat anti-rabbit IgGs(90 1sCi #sgl) were from Amersham (Milan, Italy). Phosphatidyl-choline (PC), phosphatidylethanolamine (PE), phosphatidylserine(PS), phosphatidylinositol (PI) and N-(lissamine rhodamine Bsulphonyl)-L-,8-phosphatidylethanolamine (Rh-PE) were fromAvanti Polar Lipids (Alabaster, AL, USA). Fluorescein-5-maleimide(FMA), fluorescein-6-isothiocyanate (FITC), 3-palmitoyl-2-(1-pyrenedecanoyl)-L-a-phosphatidylcholine (10-py-PC) and N-(1-pyrenesulphonyl)dipalmitoyl-L-fl-phosphatidylethanolamine (N-py-PE) were from Molecular Probes (Eugene, OR, USA). Molecularweight markers, ATP, octyl glucoside and 2-nitro-5-thio-cyanobenzoic acid were from Sigma, Sephacryl S-200 was fromPharmacia (Milan, Italy), hydroxylapatite was from BioRad (Milan,Italy), carboxymethyl cellulose (Cm-52) was from Whatman (Milan,Italy), nitrocellulose membranes (pore size, 0'2 /sm) were fromSchleicher and Schuell (Milan, Italy) and calmodulin (CaM) wasfrom Boehringer Mannheim (Milan, Italy). CaMPKII was purifiedfrom rat forebrain as previously described (McGuinness, Lai &Greengard, 1985) and stored in 5 mm Tris-HCl/5 mM /,-mercapto-ethanol/50% glycerol, pH 7'4 at a protein concentration of0'4-0'8 mg ml-'. The catalytic subunit of cAMP-dependent proteinkinase (PKA) was purified from calf heart as described (Kaczmareket al. 1980). Protein kinase activity was assessed using purifiedsynapsin I as substrate. Phosphorylation state-specific antibodiesdirected against site 1 and sites 2-3 of synapsin I (G-257 andRU-20, respectively) were gifts of Dr Andrew J. Czernik (TheRockefeller University, New York). All other chemicals were ofreagent grade and were obtained from standard commercialsuppliers.
Purification and fluorescent labelling of synapsin I and ofsynapsin I COOH-terminal fragmentSynapsin I was purified from bovine forebrain as described bySchiebler et al. (1986) and modified as described by Biihler &Greengard (1987) and stored in 200 mm NaCl, 25 mm Tris-HCl,pH 8'0 at -80 °C. The COOH-terminal fragment (molecular mass,39-40 and 35-36 kDa for the a and b isoforms, respectively) wasgenerated by cysteine-specific cleavage with 2-nitro-5-thiocyano-benzoic acid and purified to homogeneity as previously described(Biihler, Benfenati, Valtorta, Czernik & Greengard, 1989).
Synapsin I was labelled with either FMA, which reacts withsulphydryl groups, or FITC, which reacts with amino groups. TheCOOH-terminal fragment, lacking cysteine residues, was labelledonly with FITC. For FMA labelling, synapsin I (18 /M 54jFMsulphydryl groups) was extensively dialysed against 200 mm NaCl,25 mm Tris-HCl, pH 8'0 under N2-saturated atmosphere tocompletely remove reducing agents. FMA dissolved in dimethyl-sulphoxide was then added to synapsin I to a final concentration of0'5 mM and incubated for 12 h at 4 °C in the dark. The reaction wasquenched by adding 2-mercaptoethanol (1 mM), followed bycentrifugation to sediment the undissolved reagent and extensivedialysis against 200 mm NaCl/25 mm Tris-HCl, pH 7'4 to removeunbound probe.
For FITC labelling, synapsin I or its COOH-terminal fragment(12 FM) was extensively dialysed against 50 mm sodium borate/
100 mM NaCl, pH 8'4. FITC was then added to a finalconcentration of 0'5 mm and incubation was carried out for 12 h at4 °C in the dark with gentle mixing. At the end of the incubationperiod, 100 mm Tris-HCl, pH 7'4 was added to stop the reactionand the unbound FITC was removed as described above.
The amount of FITC or FMA bound to synapsin I was calculatedfrom the absorption spectrum of the derivatized protein, assuminga molar extinction coefficient of 80000 for protein-boundfluorescein (Pick & Karlish, 1980). The obtained labellingstoichiometries were 0 75-0 9 mol fluorescein (mol synapsin I)-' forFMA labelling and 0'85-1'07 mol fluorescein (mol synapsin I)-' or(mol COOH-terminal fragment)-1 for FITC labelling.
Phosphorylation of synapsin IFluorescein-labelled synapsin I was stoichiometricallyphosphorylated in vitro on site 1 by the catalytic subunit of PKAor on sites 2-3 by purified CaMPKII as described by Schiebler et al.(1986). Phosphorylation stoichiometry was 1'9-2'2 and 08-1 molphosphate (mol synapsin I)-' for CaMPKII and PKAphosphorylation, respectively. Fluoresceinated synapsin I orsynapsin I COOH-terminal fragments were also phosphorylated byPKA or CaMPKII in the cuvettes in the presence of 1-5 mM MgCl2/50 FM ATP and of either 0'5 mM EGTA (PKA phosphorylation) or1 mM CaCl2/5jug ml' CaM (for CaMPKII phosphorylation). Thephosphorylation state of synapsin I (or of its fragments) wasevaluated at the end of the incubation period by quantitativeimmunoblotting with phosphorylation state-specific antibodies.
Preparation and fluorescent labelling of phospholipidvesiclesPure PC liposomes or liposomes mimicking the phospholipidcomposition of SV (mixed liposomes; 43 PC, 32 PE, 19 PS and 6%PI) were prepared using the detergent solubilization/removalprocedure (Benfenati et al. 1989b). All liposomes containedcholesterol (10% w/w of total lipid). For the preparation offluorescently labelled liposomes, fluorescent phospholipids (either10-py-PC, N-py-PE or Rh-PE) were added prior to the solubilizationprocedure (1-2% w/w of total phospholipids).
Purification and fluorescent labelling of synaptic vesiclesSynaptic vesicles were purified through the step of controlled-poreglass (CPG) chromatography (Huttner, Schiebler, Greengard &De Camilli, 1983) from the forebrain of rats. Briefly, fifteenSprague-Dawley rats (Harlan Nossan, Milan, Italy), weighing150-200 g and housed under standard temperature and humiditywith a 12 h light-dark cycle, were deeply anaesthetized with C02,killed by decapitation, and the brains rapidly removed from theskull and processed. The procedure conformed with the guidelinesof the Italian National Research Council and the Italian HealthMinistry and was approved by the local University Animal CareCommittee.
Immediately after elution from the column,SV (10 ug protein ml-') were diluted with an equal volume of 0'4 MNaCl and pelleted after 2 h incubation at 0 'C. The latter procedurequantitatively removes endogenous synapsin I from the vesiclemembrane (Huttner et al. 1983; Schiebler et al. 1986). Aftercentrifugation, the vesicles were resuspended in 0'3 M glycine/5 mMHepes-NaOH, pH 7'4 (glycine buffer) at a protein concentration of1'5-2 mg ml-'. Synapsin I-depleted SV were fluorescently labelledupon incubation with non-pelletable liposomes containingfluorescent phospholipids.
Unilamellar, non-pelletable liposomes containing 50%w/w PC and50% w/w Rh-PE were vesiculized under N2 in a sonicating waterbath for 15 min. Liposomes were subjected to high-speed
J. Phy8iol.504.3 503
) by guest on July 15, 2011jp.physoc.orgDownloaded from J Physiol (

0. Stefani and others
centrifugation at 300 000 g for 20 min, repeated at least 5 times.The resulting non-pelletable liposomes were incubated with SV at37 °C for 30 min with occasional mixing and the vesicles were
recovered by centrifugation at 300 000 g for 20 min and extensivelywashed with glycine buffer to remove the non-incorporatedphospholipids (Benfenati et al. 1989a).
Fluorometric binding assays
The binding was studied by evaluating FRET between a donorfluorophore linked to synapsin I and an acceptor fluorophore linkedto either liposomes or SV. Incubations were carried out in 2 mlquartz cuvettes at 20 °C under constant stirring using a Perkin-Elmer LS50 spectrofluorometer equipped with a water-jacketedcuvette holder and a circulating water bath. Time-drive recordingswere obtained with sampling times of 0 5 s. Excitation andemission slits were set at 5 and 10 nm, respectively. Specific FRETwas analysed between the following couples: (a) synapsin Itryptophan residues (donor) and pyrene-labelled liposomes(acceptor), by exciting tryptophan at 296 nm wavelength andmeasuring either tryptophan emission quenching at 330 nm or
pyrene-fluorescence enhancement at 380 nm; (b) fluorescein linkedto either synapsin I or synapsin I COOH-terminal fragment (donor)and rhodamine-labelled liposomes or SV (acceptor), by excitingfluorescein at 490 nm and measuring fluorescein emissionquenching at 520 nm. The specificity of the FRET was verified byrepeating the experiments using unlabelled vesicles under variousionic strength conditions. The specific FRET was calculated fromthe difference in fluorescence levels between samples run in thepresence of unlabelled and labelled vesicles, respectively. In orderto overcome the slight fluorescein bleaching apparent duringsuccessive fluorometric runs, all experiments were run in parallelwith control samples in which fluorescein was subjected to the sameextent of irradiation. Binding assays were repeated at least fivetimes on different days and with different liposome and SVpreparations. Results were reproducible with mean variabilitieswithin the same experimental session and among variousexperimental sessions, lower than 5% and 10%, respectively.
Data analysis and curve fittingThe association and dissociation reactions were analysed by fittingthe time-resolved specific FRET values to a mono- or multi-exponential function as detailed below:
f(t) = E [a (1 - exp(-t/t))].for the association reaction, where f(t) is specific FRET at time t,a is the plateau value reached at equilibrium and Tr is the timenecessary to reach 63% of the binding in equilibrium conditions(= association time constant, Ton) and n is the number ofexponential components; and
f(t) = E [aexp(-t/T)], + b
for the dissociation reaction, where f(t) is specific FRET at time t,T is the time necessary to reach 37% of the binding in equilibriumconditions ( = dissociation time constant, Toff) a is the plateauvalue reached in equilibrium conditions, b is the lower plateau valueafter dissociation and n is the number of decay components. Theparameters of the equations yielding the best possible fit to theexperimental points were calculated using the least-squares fittingroutine of the computer program Sigmaplot 4.0 (Jandel). Results ofthe fittings obtained with various models were then analysed bymeans of the 'runs' test and the F test (Munson & Rodbard, 1980)in order to evaluate the best-fitting model.
Starting from the association and dissociation time constants, thedissociation rate constant, the observed association rate constant
and the association rate constant (koff , kolb and kor, respectively)were calculated, according to the following equations:
koff = 1/T7off,kobs = /7on,
kon = (kobs -koff)/L,where koff and kobS are in s-5, ko1 is in s-' M-1 and L is the ligandconcentration used. The dissociation constant under equilibriumconditions (Kd) was then calculated from the k00 and koff values asfollows:
Kd = koff/kon-Miscellaneous techniquesProtein determinations were performed using the BioRad or BCAprotein assays with bovine serum albumin as a standard or spectro-photometrically, using the reported extinction coefficients.SDS-polyacrylamide gel electrophoresis was performed accordingto Laemmli (1970). Proteins in the gels were electrophoreticallytransferred to nitrocellulose membranes (Towbin, Staehelin &Gordon, 1979). Immunolabelling of nitrocellulose sheets wasperformed using affinity-purified polyclonal antibodies, followed by'251-labelled anti-rabbit IgG overlay. Autoradiography wasperformed using Kodak Bio-Max films. Statistical analysis wasperformed using one-way ANOVA followed by multiplecomparison tests.
RESULTSInteractions with phospholipid vesiclesSynapsin I was previously demonstrated to interact withliposomes sharing the same phospholipid composition asnative SV (Benfenati et al. 1989b). We investigated theseinteractions by studying the specific transfer of energybetween fluorophores linked to synapsin I and fluorophoresincorporated into the phospholipid bilayer. The intrinsicfluorescence of synapsin I is contributed to by fourtryptophan residues (trp126, trp227, trp335 and trp356). Theextrinsic fluorescence was obtained by covalently bindingfluorescein to cysteine residues (cys223, Cys360 and cys370), allof which are located in the hydrophobic C domain (Siidhof etal. 1989). In both cases, fluorophores were localized withinthe NH2-terminal head region which is entirely responsiblefor the interaction with the phospholipid site. The intrinsicand extrinsic fluorescence of synapsin I were matched withthe presence of pyrene- and lissamine rhodamine-labelledphospholipids incorporated into the vesicle bilayer,respectively.
The binding of fluoresceinated synapsin I to liposomescontaining acidic phospholipids and bearing PE labelledwith rhodamine on the polar head group was associatedwith an intense and specific quenching of fluoresceinemission and by a concomitant enhancement of rhodamineemission (Fig. 1A). The larger quenching in fluoresceinemission with respect to the increase in rhodamine emissionis likely to be due to inner filtering and not to eitherfluorescein bleaching or to the turbidity of the vesiclesuspension. The addition of rhodamine-labelled PC liposomeswhich do not interact with synapsin I (Benfenati et al.1989b) did not bring about any noticeable quenching of the
504 J Phy8iol.504.3
) by guest on July 15, 2011jp.physoc.orgDownloaded from J Physiol (

Kinetics of synapsin-synaptic vesicle binding
fluorescein-synapsin I fluorescence. FRET was markedlyinhibited by increasing the ionic strength to 200 mm NaCl.The time-resolved analysis of the changes in FRETindicated that the association reaction is very fast andreaches a plateau within a few seconds (Fig. IB). Once thebinding plateau had been reached, bringing the ionicstrength to 200 mm NaCl promoted a slower dissociationreaction which involved about 50% of bound synapsin I.Association and dissociation curves were best fittedaccording to a mono-exponential model, yielding a kon of0 045 + 0 007 108 s-' M-l and a koff of 0-103 + 0-013 s-'(Table 1). These values allowed a calculation of a kinetic Kdof 24-7 + 4-2 nm, similar to that determined in equilibriumbinding experiments (13 + 5 nM; Schiebler et al. 1986).
It was previously shown that surface binding of synapsin Ito the negatively charged head groups of acidicphospholipids is accompanied by penetration of hydrophobicdomains of synapsin I into the membrane core (Benfenati etal. 1989b). By using phospholipid vesicles containingphospholipids labelled with pyrene alternatively on thesurface head groups or on the end of the aliphatic chains and
A
00C0)(0)0)0
ii
jL
50
40
30
10
o
Control.+PC-LS
*+Mix-HS
by analysing the FRET between synapsin I tryptophanresidues and pyrene, we intended to clarify on a quantitativebasis which one of the surface and hydrophobic interactionsaccounts for synapsin I binding to phospholipid vesicles.
As shown in Fig. 2A, when tryptophan was excited at296 nm, the occurrence of FRET between synapsin I andphospholipid-linked pyrene was demonstrated by theconcomitant quenching of tryptophan and enhancement ofpyrene fluorescence emission, which were of approximatelythe same extent. This effect was present only whenphospholipid vesicles included acidic phospholipids and wasnegligible when pure PC vesicles were used. A significantFRET was present both when phospholipids were labelledon the polar head and when they were labelled on thealiphatic chains.
A quantitative analysis performed by increasing theconcentrations of the acceptor (phospholipid vesicles) at aconstant concentration of the donor (synapsin I) revealedthat the hydrophobic interaction predominates over thesurface interaction. The analysis of the specific FRET,calculated for tryptophan quenching as the difference
B
00C00
0032-
520 560 600Wavelength (nm)
200 300Time (s)
500
Figure 1. Association of synapsin I with phospholipid vesicles containing acidic phospholipids isfast and sensitive to ionic strengthA, control, steady-state emission spectrum of fluoresceinated synapsin I (50 nM) alone. Curves markedMix-LS and Mix-HS represent the spectra of a mixture of fluoresceinated synapsin I with liposomescontaining acidic phospholipids (100 /sg phospholipid, 2% labelled with rhodamine) incubated at 20 °C for10 min under low (Mix-LS) or high (Mix-HS) salt conditions, respectively. PC-LS represents the spectrumof a mixture of fluoresceinated synapsin I with phosphatidylcholine liposomes (100 jug phospholipid, 2%labelled with rhodamine) incubated at 20 °C for 10 min under low salt conditions. Spectra were obtained byexciting the samples at 490 nm and recording the fluorescence emission as a function of wavelength.B, time course of the fluorescence emission of fluoresceinated synapsin I (50 nM) following addition attime 30 s of either PC or Mix liposomes under low salt (LS) or high salt (HS) conditions (upper traces). Thespecific FRET (lower trace) was calculated as the difference in fluorescence levels between samples rununder LS conditions in the presence of PC and Mix liposomes. Dissociation was induced by increasing theionic strength to 200 mm NaCl (arrow). Association and dissociation curves between synapsin I and Mixliposomes under low salt conditions were best fitted according to a mono-exponential model. Here and inother figures, a.u. stands for arbitrary units.
J Physiol.504.3 505
) by guest on July 15, 2011jp.physoc.orgDownloaded from J Physiol (

5. Stefani and others
between the changes in fluorescence in the presence of purePC liposomes and those with mixed liposomes, made itpossible to deduce distinct curves of saturation for thesurface and the hydrophobic interactions. Although thesurface interaction exhibited an apparently higher affinity,the hydrophobic interaction reached a plateau which wasabout three-fold higher than that of the surface interaction,accounting for the majority of binding at saturation(Fig. 2B and C).
Interactions with synaptic vesiclesIn order not to alter the structure and activity of vesicle-associated proteins, purified SV depleted of endogenoussynapsin I were labelled by phospholipid exchange uponincubation of SV at 37 °C with non-sedimentable,unilamellar liposomes containing 50% rhodaminated PE.In addition to the FMA-labelled synapsin I used for theliposome experiments, we also used synapsin I labelled onprimary amino groups with FITC, bearing the fluorophore
A
150 b
~~100Cd
co
0 . V~oFL 50 1 /
350 400
Wavelength (nm)
preferentially concentrated in the COOH-terminal region.The purified COOH-terminal fragment of synapsin I,originated from cysteine-specific cleavage, was also labelledwith FITC. Previous studies have shown that labellingsynapsin I or its COOH-terminal fragment on primaryamino groups induces only a slight change in isoelectricpoint and does not significantly alter the interaction withSV, as evaluated by equilibrium binding assays (Benfenati etal. 1992b and 1996; F. Benfenati and F. Valtorta,unpublished observations).
When either form of fluoresceinated synapsin I wasincubated with rhodaminated SV and fluorescein wasexcited at 490 nm, a noticeable FRET between fluoresceinand rhodamine was observed (Fig. 3A). No significantdifferences in the extent of the FRET were observedbetween synapsin I labelled by FMA or by FITC (notshown), confirming that the association of synapsin I withSV involves multiple sites of the protein within both the
B
-100
0
25
0
a.)
c -2040.
c 0 20 40 60 8025
20-
U-o 15 -
10_CD.1
20 40 60
PL concentration (mg 1-1)
Figure 2. The hydrophobic interaction with the core of the vesicle membrane accounts for themajority of synapsin I bindingA, spectrum a represents the algebraic sum of the spectra recorded from separate samples containing eitherunlabelled synapsin 1 (50 nM) or pyrene-labelled mixed liposomes (100 sg phospholipid containing 2% ofeither 10-py-PC or N-py-PE) alone. Spectrum b was recorded from a mixture of unlabelled synapsin I withpyrene-labelled liposomes incubated at 20 °C for 10 min. Spectra were obtained by exciting tryptophan at296 nm and recording the fluorescence as a function of wavelength. B and C, the association betweensynapsin I and the phospholipid membrane was quantitatively analysed- by incubating synapsin I withincreasing concentrations of either pure PC (triangles) or mixed phospholipid (circles) liposomes containing2% of pyrene-labelled phospholipid with the fluorophore either exposed at the membrane surface (opensymbols) or buried into the membrane core (closed symbols). FRET was evaluated by exciting the donor at296 nm and recording the quenching of tryptophan emission at 330 nm. B refers to the changes influorescence at steady state expressed as a percentage of the control sample (unquenched tryptophan). Cshows the specific FRET, calculated as the difference between the changes in fluorescence in the presence ofmixed liposomes and those with pure PC liposomes containing the same type of pyrene-labelledphospholipid.
J Phy8iol. 504.3506
) by guest on July 15, 2011jp.physoc.orgDownloaded from J Physiol (

J Phy8iol. 504.3 Kinetics of synapsin-synaptic vesicle binding
Table 1. Kinetic parameters for the binding of synapsin I to synaptic vesicles
Ligand Dp-syn I Dp-syn I IP-syn I 2,3P-syn I TailReceptor Mix PIs SV SV SV SV
Equilibrium binding assays (4 °C, 40 mm NaCl equivalent)Kd(nM) 13 + 5 10 + 6 16 + 6 52 + 13 55 +10
Association/dissociation kinetics (20 °C, 40 mm NaCl equivalent)ron (s) 3-15 + 0-31 * 1P74 + 0-32 2-78 + 0-28n 3-26 + 0.42* 3-29 + 0.33*Toff-NaC1 (s) 10 3 + 12ns 9.89 + 1P94 12-08 + 2 83ns 12-53 + 3-36'skoff NaCl (s ) 04103 + 0-013 04123 + 0-028 0 094 + 0-021 0-127 + 0'022kon (S- SM ') 4-56 + 0 79 10-66 + 1P83 5f56 + 0 57 - 198 + 0-13Kd (nM) 24-7 + 4-2 11P9 +1P8 16-9 + 2-81 65-8 + 12-3Toff-phospho (s) 7 63 + 051. n.d.koff phosphO (s1 04132 + 0.010 n.d.
Values are indicated as means + S.E.M. (n = 5-7). All kinetic values have been calculated for eachindividual curve as described in Methods and averaged. When the best fitting model was bi-exponential,only the rTn and the Toff of the fast component (accounting for about 90% of the binding at steady state)have been considered. Equilibrium binding data were obtained from Schiebler et al. 1986 and Benfenati etal. 1989a, b. Mix Pls, mixed phospholipid vesicles; SV, synaptic vesicles; Dp-syn I, dephosphorylatedsynapsin I; 1P-syn I, PKA-phosphorylated synapsin I (site 1); 2,3P-syn I, CaMPKII-phosphorylatedsynapsin I (sites 2 and 3); Tail, dephosphorylated COOH-terminal fragment of synapsin I; Toff-NaC1'koff-NaCl, ionic strength-induced dissociation; Toff-phosphog koffphosphO dissociation induced by CaMPKIIphosphorylation; n.d., not determined. Changes in Ton and Toff values were statistically analysed using one-way ANOVA followed by Dunnet's multiple comparison test, with Dp-syn I binding to SV as the controlgroup; ns, not significant; * P < 0 05.
B200
150
LI-
8 100.C
CD
I . .,'; r-*.*f --:;3i
520 560 600Wavelength (nm)
50 100 IFree synapsin I (nM)
Figure 3. The association of fluorescein-labelled synapsin I with rhodamine-labelled synapticvesicles can be evaluated by specific FRETA, steady-state spectra of samples containing fluoresceinated synapsin I (Fl-syn I, 50 nM) incubated at20 °C for tO min in the absence or presence of either unlabelled SV (20 jug protein ml-', dotted line) or
rhodamine-PE labelled SV (Rh-SV, 20 jug protein ml-', continuous line). Spectra were obtained by excitingthe samples at 490 nm and recording the fluorescence as a function of wavelength. The spectra resultingfrom the analysis of equal amounts of either unlabelled or Rh-labelled SV are also reported. B, thequenching in fluorescein emission was calculated at various concentrations of fluoresceinated synapsin I(20-200 nM) in the presence of Rh-labelled SV (20 jug protein ml-'; total binding, 0) or of a correspondingamount of unlabelled SV (non-specific binding, *). The resulting specific binding (A) isotherm describes a
high-affinity and saturable interaction.
507
50A
co
00C0C)0
0
150
0 1-
d,
11.0
11
) by guest on July 15, 2011jp.physoc.orgDownloaded from J Physiol (
5. Stefani and others
NH2-terminal and COOH-terminal regions. The quenchingof fluorescein was not due to sample turbidity, since it wastotally absent when labelled synapsin I was incubated withan equal amount of unlabelled SV. Furthermore, theobserved FRET seemed to be specific for SV, as theincubation of fluoresceinated synapsin I with rhodaminatedbovine serum albumin induced only a minor decrease influorescein emission (Ceccaldi, Benfenati, Chieregatti,Greengard & Valtorta, 1993; F. Benfenati and F. Valtorta,unpublished observations). The association of synapsin Iwith SV, as evaluated by FRET, appeared to be saturable(Fig. 3B). The analysis of the isotherms of specific bindingat steady state calculated as the difference betweenfluorescein quenching obtained in the presence ofrhodamine-labelled and of unlabelled vesicles revealed asteady-state Kd value in the low nanomolar range as thatdetermined with conventional binding techniques,indicating that the covalent modification of synapsin I didnot alter its ability to interact with SV.
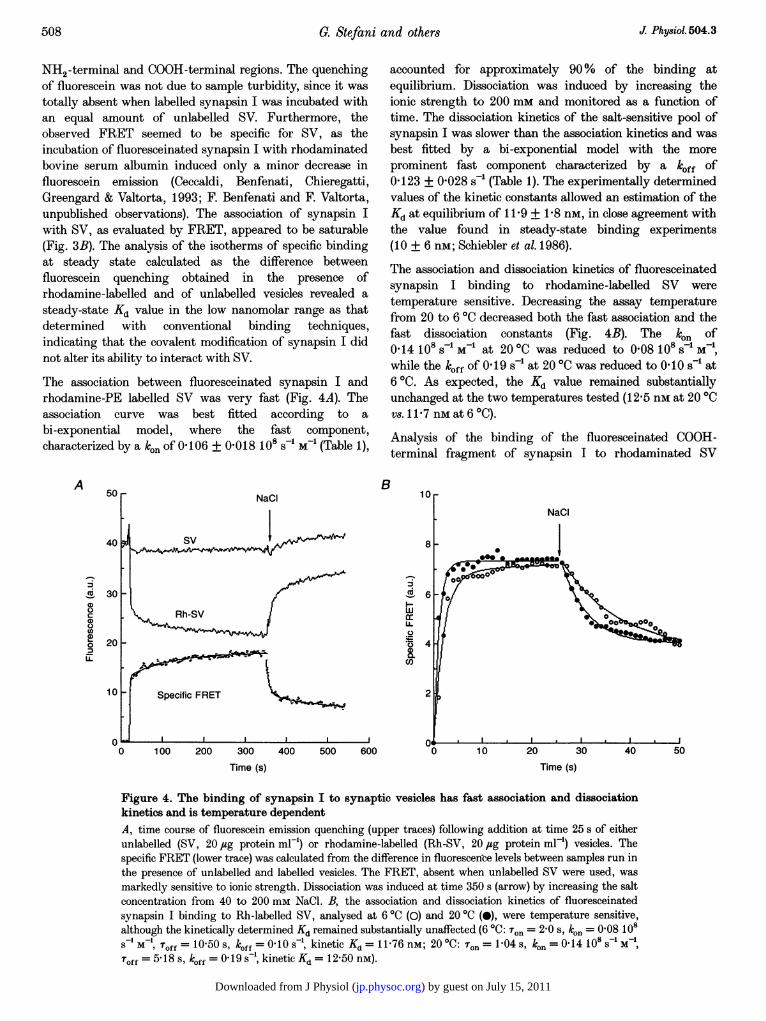
The association between fluoresceinated synapsin I andrhodamine-PE labelled SV was very fast (Fig. 4A). Theassociation curve was best fitted according to abi-exponential model, where the fast component,characterized by a ken of 0 106 + 0-018 108 s-1 M-1 (Table 1),
A 50
40
cX 300
a)00na)O 20U-
10
0
B
accounted for approximately 90% of the binding atequilibrium. Dissociation was induced by increasing theionic strength to 200 mm and monitored as a function oftime. The dissociation kinetics of the salt-sensitive pool ofsynapsin I was slower than the association kinetics and wasbest fitted by a bi-exponential model with the moreprominent fast component characterized by a klff of0 123 + 0-028 s-' (Table 1). The experimentally determinedvalues of the kinetic constants allowed an estimation of theKd at equilibrium of 11-9 + 1P8 nm, in close agreement withthe value found in steady-state binding experiments(10 + 6 nM; Schiebler et al. 1986).
The association and dissociation kinetics of fluoresceinatedsynapsin I binding to rhodamine-labelled SV weretemperature sensitive. Decreasing the assay temperaturefrom 20 to 60C decreased both the fast association and thefast dissociation constants (Fig. 4B). The kln of0 14 10 s-1 M-1 at 200C was reduced to 0-08 108 s-1 M-1,while the k0ff of 0 19 s' at 200C was reduced to 0I10 s-' at60C. As expected, the Kd value remained substantiallyunchanged at the two temperatures tested (12-5 nm at 200Cvs.1 1 7 nMat 6 °C).
Analysis of the binding of the fluoresceinated COOH-terminal fragment of synapsin I to rhodaminated SV
I
I-w
IL)C.)
CO
0 100 200 300 400Time (s)
500 600 30Time (s)
Figure 4. The binding of synapsin I to synaptic vesicles has fast association and dissociationkinetics and is temperature dependentA, time course of fluorescein emission quenching (upper traces) following addition at time 25 s of eitherunlabelled (SV, 20 jug protein ml-') or rhodamine-labelled (Rh-SV, 20 jug protein ml') vesicles. Thespecific FRET (lower trace) was calculated from the difference in fluorescence levels between samples run inthe presence of unlabelled and labelled vesicles. The FRET, absent when unlabelled SV were used, wasmarkedly sensitive to ionic strength. Dissociation was induced at time 350 s (arrow) by increasing the saltconcentration from 40 to 200 mm NaCl. B, the association and dissociation kinetics of fluoresceinatedsynapsin I binding to Rh-labelled SV, analysed at 6 °C (0) and 20 °C (0), were temperature sensitive,although the kinetically determined Kd remained substantially unaffected (6 °C: Ton = 2-0 s, ko. = 0-08 108S M1, Toff = 10-50 s, k0ff = 0 10 s , kinetic Kd = 11-76 nM; 20 °C: T.. = 1-04 s, kon = 0-14 108s MToff = 5 18 s, koff = 0 19 s- , kinetic Kd = 12-50 nM).
508 J: Physiol.504.3
) by guest on July 15, 2011jp.physoc.orgDownloaded from J Physiol (

Kinetics of synapsin-synaptic vesicle binding
revealed a binding stoichiometrically comparable with thatof holosynapsin I. However, at variance with holosynapsin I,the association reaction was slower and the dissociationinduced by increasing the ionic strength virtually complete(Fig. 5A). As in the case of synapsin I, both association anddissociation curves could be effectively interpolated using abi-exponential model which yielded a kon value of0.019 + 0.001 108 s-' M-1 and a koff value of 0 127 +0-022 s-' for the fast component, with a kinetically calculatedKd of 65 + 12 nm, close to that previously described inequilibrium binding studies (55 + 10 nM; Benfenati et al.1989a; Table 1).
Effects of site-specific phosphorylation of synapsin IThe effects of site-specific phosphorylation on the kinetics ofthe interaction between synapsin I and SV were analysed byincubating rhodaminated SV with fluoresceinatedsynapsin I in dephosphorylated form or stoichiometricallyphosphorylated in vitro by either PKA or CaMPKII. Theanalysis of the specific quenching of fluorescein emission dueto FRET at steady state (Fig. 6A and B) revealed thatstoichiometric phosphorylation by CaMPKII, but not byPKA, significantly decreased the extent of energy transferbetween synapsin I and SV. The ability of the SV-associated CaMPKII in modulating the association between
synapsin I and SV was evaluated by reacting fluorescein-labelled synapsin I and rhodamine PE-labelled SV underbasal (EGTA, no ATP/Mg2+) or phosphorylation(Ca2+/CaM + ATP/Mg2+) conditions (Fig. 6C). Activation ofthe SV-associated CaMPKII by Ca2+/CaM in the presenceof ATP/Mg2+ was sufficient to significantly decrease thespecific FRET to a level which could not be further modifiedby the subsequent addition of the purified soluble enzyme.The decrease in binding caused by phosphorylation wasquantitatively similar to that induced by increasing theionic strength and the two effects were not additive.Stoichiometric phosphorylation by CaMPKII also sloweddown the association of synapsin I to SV (son = 3-26 +0-42 s with respect to 1P74 + 0X32 s of the dephosphorylatedform; Table 1), although the absolute values of koff and koncould not be calculated in the absence of a significant ionicstrength-induced dissociation. The fact that the activationof the kinase in the absence of phosphorylation (by theaddition of Ca2+/CaM in the absence of ATP/Mg2+) wasunable to affect the binding (not shown) further supportedthe key role of phosphorylation in changing the bindingactivity of synapsin I. While the phosphorylation-inducedinhibition of synapsin I binding was not complete,phosphorylation of the COOH-terminal fragment of
B
Cda)0
C,)a)0
LL
0 100 200 300 400 500Time (s)
520 540
Wavelength (nm)
Figure 5. The association of the COOH-terminal fragment of synapsin I with synaptic vesicles issensitive to ionic strength and phosphorylationA, dephosphorylated or CaMPKII phosphorylated synapsin I COOH-terminal fragment labelled withfluorescein (100 nM) was incubated at 20 °C and 40 mm NaCl with rhodamine-labelled vesicles(20 jug protein ml-') and the specific fluorescein quenching was followed as a function of time. Nosignificant quenching of fluorescein was observed when unlabelled SV were used (not shown). Dissociationwas induced at time 300 s (arrow) by increasing the salt concentration from 40 to 200 mm NaCl. B, thespecific quenching of fluorescein emission of synapsin I COOH-terminal fragment was monitored at steadystate as a function of wavelength 5 min after the following sequential additions: 20 ,ug ml-' rhodamine-labelled SV (Rh-SV), 1 mM MgCl2/50 /uM ATP/1 mm CaCl2/50Jug ml-' calmodulin (Rh-SV/CaM),0-25 /tg ml-' purified CaMPKII (Rh-SV/CaM/PK) and 200 mm NaCl. The displayed spectra result fromthe subtraction of the contribution of rhodamine (Rh-SV alone) to the fluorescein region.
A100
80
a)a)C)CO)a)0
I-L
40
0560
J Physiol.504.3 509
) by guest on July 15, 2011jp.physoc.orgDownloaded from J Physiol (

G. Stefani and others
synapsin I by either the SV-associated or exogenously addedCaMPKII virtually abolished its binding to SV (Fig. 5B). Asobserved for synapsin I, activation of the SV-associatedCaMPKII was sufficient to achieve a maximal effect. Astatistical evaluation of the effects of site-specificphosphorylation showed a highly significant inhibition ofthe ionic strength-sensitive component of FRET afterCaMPKII phosphorylation of synapsin I and its COOH-terminal fragment (79 + 4 and 97 + 0f8 %, respectively)and no significant effects after PKA phosphorylation ofsynapsin I (Fig. 7).
An important issue in evaluating the physiologicalimportance of synapsin I phosphorylation is whether itoccurs on a sufficiently short time-scale to be able toparticipate in the regulation of the fast process of neuro-transmitter release. However, all previous data wereobtained under steady-state conditions and no clearinformation on the time course of the phosphorylation-induced dissociation of synapsin I from the vesicles wasavailable. Taking advantage of the time resolution of the
A
co
U-
0
co_
201
15
10
B
FRET technique, fluoresceinated dephospho-synapsin I wasbound to rhodamine-labelled SV in the presence ofMg2+/ATP and after the binding had reached a stableplateau, either Ca2+/CaM (to activate the endogenousCaMPKII; Fig. 8A), or the catalytic subunit of PKA(Fig. 8B) was added. Phosphorylation by PKA wasrelatively ineffective, but activation of the endogenousCaMPKII induced a prompt dissociation of synapsin I fromSV. Fitting the dissociation induced by the on-vesiclephosphorylation of synapsin I with a bi-exponential modelyielded a koff of the fast component of 0-132 + 0-010 s-i,closely similar to that observed after increase in the ionicstrength of the medium (0-123 + 0-028 s-1; see Table 1). Asmentioned above, the subsequent addition of salt did notfurther significantly modify the binding. Interestingly, thefluorescence plateau reached after the dissociation ofsynapsin I induced by activation of the endogenousCaMPKII exactly overlapped the plateau reached byfluoresceinated synapsin I which had been stoichiometricallyphosphorylated in vitro by purified CaMPKII (not shown).
C
560 500 520 540 560 500 520 540 560Wavelength (nm)
Figure 6. The association with synaptic vesicles is modulated by site-specific phosphorylation ofsynapsin IA and B, fluorescein-labelled synapsin I (50 nM) in dephosphorylated form (Dp-syn I) or stoichiometricallyphosphorylated (P-syn I) in vitro either by the catalytic subunit of PKA (A) or by CaMPKII (B) wasincubated at 20 °C for 10 min with rhodamine-labelled SV (20 ug protein ml-). The specific quenching offluorescein emission of synapsin I was monitored at steady state as a function of wavelength by excitingthe samples at 490 nm. C, fluorescein-labelled dephosphorylated synapsin I (50 nM) was incubated withrhodamine-labelled SV (20 jug protein ml-') at 20 °C for 10 min and the specific quenching of fluoresceinemission was monitored 5 min after the sequential additions of -5 mm MgCl2/50 /SM ATP/1 mMCaCl2/5 jug ml-' calmodulin (ATP/CaM) and 0'25 jug ml-' purified CaMPKII. The contribution ofrhodamine was subtracted from the steady-state spectra of fluorescein and the specific FRET wascalculated, as a function of wavelength, from the difference between the emission quenching of fluoresceinobserved in the presence of unlabelled and rhodamine-labelled SV. Phosphorylation of synapsin I by theendogenous synaptic vesicle CaMPKII yielded an inhibition comparable with that obtained afterstoichiometric phosphorylation of synapsin I by purified exogenous CaMPKII (B).
J Phy8iol. 504.3510
) by guest on July 15, 2011jp.physoc.orgDownloaded from J Physiol (

J Physiol.504.3 Kinetics of synapsin-synaptic vesicle binding
2
00)Ic
0Qr-
C')
m
CL0a
100
* _
1 P-syn I 2,3P-syn I 2,3P-tail
Figure 7. Effects of site-specific phosphorylation on the association of synapsin I or of itsCOOH-terminal fragment with synaptic vesicles at steady stateThe effects of phosphorylation of synapsin I by PKA at site 1 (IP-syn I) or by CaMPKII at sites 2,3(2,3P-syn I) or of its COOH-terminal fragment by CaMPKII (2,3P-tail) on the ionic strength-sensitivecomponent of the binding was evaluated by expressing the FRET detected at steady state withphosphorylated proteins as percentage of the FRET between dephosphorylated synapsin I (or itsdephosphorylated COOH-terminal fragment) and synaptic vesicles. Data represent means + S.E.M. of sixexperiments. * P < 0-01, Dunnet's multiple comparison test.
B
40rATP + CaM
NaCI
Ico
cn
CO,
Dp-syn I
0 200 400Time (s)
600 800Time (s)
Figure 8. The on-vesicle phosphorylation of synapsin I by vesicular CaMPKII is effective inpromoting its dissociation from the synaptic vesicle membraneRhodamine-PE labelled or unlabelled SV (20 jug protein ml-') were added at time 100 s to fluorescein-labelled, dephosphorylated synapsin I (Dp-syn I, 50 nM) at 20 'C and the specific fluorescein quenching(calculated as the difference in fluorescence between the samples run in the presence of unlabelled andlabelled vesicles) was followed as a function of time. At time 300 s, either 1 mm CaCl2 and 5 ,g ml-'calmodulin (ATP + CaM) to activate endogenous CaMPKII (A) or the catalytic subunit of PKA (CS, B)were added to the sample. Ionic strength-dependent dissociation was induced at time 700 s by bringing theNaCl concentration from 40 to 200 mm.
511
A40 r-
30
20
I-
wErU-._
cO
101-
) by guest on July 15, 2011jp.physoc.orgDownloaded from J Physiol (

5. Stefani and others
DISCUSSIONSynapsin I was previously demonstrated to interact withhigh affinity with both the phospholipid and proteincomponents of SV. The interaction with the phospholipidbilayer is selective for acidic phospholipids and the surfacebinding of synapsin I to the negatively chargedphospholipid head groups is accompanied by the penetrationof hydrophobic domains of synapsin I into the membranecore (Benfenati et al. 1989a,b). In addition, the binding toSV was found to be sensitive to phosphorylation ofsynapsin I by CaMPKII but not by PKA or MAPK(Schiebler et al. 1986; Jovanovic et al. 1996). In contrast, thebinding to phospholipid vesicles was not affected byphosphorylation (Benfenati et al. 1989b). The identificationof a SV-associated form of CaMPKII as a binding site forthe COOH-terminal region of synapsin I (Benfenati et al.1992b) strengthened the potential importance of themodulation by phosphorylation. However, no informationwas available thus far on the kinetics of the association anddissociation reactions which could contribute to evaluatingthe physiological significance of the interaction of synapsin Iwith SV and its modulation by phosphorylation. Inaddition, although the hydrophobic interaction with themembrane bilayer may play a crucial role in the clusteringand membrane stabilization of SV (Benfenati et al. 1993), itwas never investigated quantitatively due to the variabilityof cross-linking/labelling efficiency in hydrophobic labellingexperiments (Benfenati et al. 1989b).
In this paper we have characterized the association ofsynapsin I with the synaptic vesicle membrane at themolecular level by using the time-resolved and non-perturbing FRET technique and found that: (1) theinteraction of synapsin I with acidic phospholipids(phospholipid site) has fast association and dissociationkinetics. (2) The interaction with the hydrophobic core of themembrane appears to be saturable and accounts for themajority of the binding to the phospholipid site. (3) Thebinding of holosynapsin I to intact SV has faster kineticsthan that of its NH2-terminal region binding to liposomeswith the same phospholipid composition of SV or of itsCOOH-terminal region binding to SV. (4) Phosphorylationby exogenous or endogenous CaMPKII markedly reducesthe interactions between synapsin I and SV by acting on theionic strength-sensitive component of the binding, whilephosphorylation by PKA has no effect on these interactions.(5) The on-vesicle phosphorylation of synapsin I by the SV-associated CaMPKII promotes a very rapid dissociationfrom SV, by acting on the ionic strength-sensitivecomponent of the binding.
The experimental data have been obtained undertemperature and medium conditions different from thosefound in vivo owing to technical constraints. However,previous studies suggest that the results can be extrapolatedto the in vivo conditions. Most of the physiological studiesaimed at determining the properties of synaptictransmission and plasticity and the rates of exo-endocytosis
in the mammalian central nervous system are performed attemperatures below 30 °C (see e.g. Ryan et al. 1993, 1996).In addition, a large body of experimental evidenceconcerning the function of synapsin I has been obtained inexperiments in which mammalian synapsin I was injectedinto nerve terminals from non-mammalian species, namelysquid, goldfish, lamprey or Xenopus laevis at roomtemperature (Hackett, Cochran, Greenfield, Brosius & Ueda,1990; Llinas et al. 1991; Lu et al. 1992; Valtorta et al. 1995;Pieribone et al. 1995). Moreover, the experiments on thetemperature dependence of the kinetics ofassociation-dissociation suggest that temperature affectsthe rates of association and dissociation, but leaves theaffinity of the binding unchanged. Previous studies (Huttneret al. 1983; Schiebler et al. 1986) have demonstrated that thebinding of synapsin I to synaptic vesicles is sensitive to theoverall ionic strength, but not to the type of cations andanions present and that binding buffers containing intra-cellular ion species are comparable with buffers containingan equivalent amount of NaCl. The ionic strength-sensitivity of the synapsin I-vesicle interaction in vitrodoes not argue against a physiological relevance of suchinteraction. Although the intracellular fluid is iso-osmoticwith the extracellular 'milieu', potassium proteinatesconstitute a very effective buffer which is only partlydissociated. Moreover, the estimated concentrations ofsynapsin I and vesicle binding sites in nerve terminals aremuch higher than those used in vitro (micromolar versusnanomolar; De Camilli et al. 1990), thus achieving avirtually maximal saturation of binding also at high ionicstrength. This is confirmed by the observations that innerve terminals virtually all dephosphorylated synapsin I isassociated with the vesicle membrane (De Camilli, Harris,Huttner & Greengard, 1983, Schiebler et al. 1986; TorriTarelli, Bossi, Fesce, Greengard & Valtorta, 1992).
The analysis of FRET between synapsin I and eitherphospholipid vesicles with the same phospholipidcomposition of SV or rat brain SV was found to be anefficient and specific method for evaluating thesynapsin I-vesicle interactions both qualitatively andquantitatively. The technique, widely used for theassessment of protein-protein interactions, is based on thespecific transfer of energy between a donor fluorophore,which is directly excited by the source, and an acceptorfluorophore having an excitation spectrum partially ortotally overlapping with the emission spectrum of the donor(for review, see Stryer, 1978). FRET occurs only when donorand acceptor fluorophores are brought into strict contact andcan be detected by the concomitant quenching of the donoremission and increase in the acceptor emission. Thismethodology appeared well-suited for the analysis of thesynapsin I-vesicle interactions. It allowed a time resolutionwhich was impossible to achieve with conventional bindingassays requiring high-speed sedimentation of SV, andprovided information on the topology of the interactionsusing site-specific labelling of synapsin I and of the vesiclemembrane.
512 J: Physiol.504.3
) by guest on July 15, 2011jp.physoc.orgDownloaded from J Physiol (

Kinetics of synapsin-synaptic vesicle binding
The interactions of synapsin I with phospholipid vesicleshave been studied using two couples of fluorophores. In onecase, the endogenous fluorescence of tryptophan residuespresent in the head region of synapsin I was challengedwith pyrenylated phospholipids in which the pyrene moietywas covalently linked either to the acyl chain or to the polarhead exposed at the surface of the membrane. In the secondcase, the exogenous fluorescence of synapsin I labelled oncysteine residues by fluorescein maleimide was challengedwith phospholipid vesicles incorporating rhodaminatedphospholipids. In both cases, the synapsin I donor fluoro-phore (either tryptophan or fluorescein) was located withinthe hydrophobic domains of the head region, i.e. in domainsdirectly responsible for the interaction with the'phospholipid site' (Benfenati et al. 1989a, b).
Synapsin I had a very fast kinetics of association to vesiclescontaining acidic phospholipids exhibiting a single,virtually homogeneous, association component. Consistentwith the marked decrease in binding affinity at equilibrium,increasing the ionic strength promoted a markeddissociation of synapsin I. Interestingly, the majority of thesynapsin I bound to phospholipid vesicles at equilibriumwas involved in an interaction with the hydrophobic core ofthe membrane. Despite the ionic strength sensitivity, thisfinding confirms that the basic character of the protein,contributed by the COOH-terminal region, is not adeterminant of the binding to the phospholipid site. Rather,the interaction with the polar heads of acidic phospholipids,likely to be mediated by exposed charged residues withinthe hydrophobic head domain (which might explain thesensitivity of the binding to the ionic strength), mayrepresent a first and transient step in binding in whichsynapsin I approaches the membrane, followed by a morestable and low free-energy interaction of the hydrophobicdomains with the core of the membrane. Such an event maybe responsible for the stabilization of the membranestructure in the stable lamellar phase (Benfenati et al. 1993),similar to that described for colicins, which are pore-forming bacterial toxins (Geli, Koorengevel, Demel,Lazdunski & Killian, 1992). The insertion of synapsin I intothe bilayer may reduce the tendency of the vesiclemembrane to form concave surfaces or to experiencephospholipid packing defects. The occurrence of suchdefects in SV is facilitated by their high intrinsic curvatureand by their relative enrichment in non-bilayer structure-preferring phospholipids such as PE (Benfenati et al. 1993).
The interaction of synapsin I with SV was investigated bylabelling the protein with fluorescein covalently boundeither to the head cysteine residues or to primary aminogroups (i.e. lysine residues) preferentially distributed in theCOOH-terminal region. The association with SV was veryrapid and appeared to consist of fast and slow components,the former accounting for the majority of binding. Asimilarly fast, but not complete, dissociation was induced byincreasing the ionic strength. Both reactions weretemperature sensitive. The analysis of the association and
dissociation rates gave estimates of the dissociation constantin close agreement with the value obtained from equilibriumbinding assays. This agreement confirms that the covalentmodification of synapsin I did not alter its ability to interactwith SV.
Phosphorylation by CaMPKII, but not by PKA or MAPK,was demonstrated to effectively inhibit the binding ofsynapsin I to SV, by inducing a 5-fold shift in the Kd valueat steady state (Schiebler et al. 1986; Jovanovic et al. 1996).This modulation is believed to be responsible for thedissociation of synapsin I from the vesicles during high-frequency electrical stimulation or sustained depolarizationof nerve terminals (Sihra, Wang, Gorelick & Greengard,1989; Torri Tarelli et al. 1992) and for the release of SV fromthe cytoskeletal constraint as visualized by videomicroscopy(Ceccaldi et al. 1995). Phosphorylation by CaMPKII mayrepresent a very efficient way to modulate rapidly thefunction of synapsin I, as an enzyme-substrate complexbetween CaMPKII and synapsin I exists on the synapticvesicle surface (Benfenati et al. 1992b). In agreement withprevious observations carried out under equilibriumconditions (Schiebler et al. 1986; Benfenati et al. 1989a), theanalysis of FRET between fluoresceinated synapsin I andrhodaminated SV showed that phosphorylation of synapsinI by CaMPKII inhibits the binding of synapsin I by actingon the ionic strength-sensitive component of the binding.When synapsin I was stoichiometrically phosphorylated onthe CaMPKII sites, both the extent of binding at steadystate and the association constant were markedly decreased.The extent of inhibition of SV binding was comparablewhen synapsin I was previously phosphorylated by purifiedCaMPKII or endogenously phosphorylated by the SV-associated CaMPKII.
The effects of phosphorylation and ionic strength were ofsimilar extent but not additive, suggesting thatphosphorylation acts predominantly on the ionic strength-sensitive component of the binding. The effect ofphosphorylation may be the consequence of a majorstructural change. Thus, time-resolved fluorescenceanisotropy revealed a relatively large change in Stokesradius induced by CaMPKII phosphorylation (Benfenati,Neyroz, Bahler, Masotti, Greengard, 1990) and attributableto a folding of the elongated tail region over the head region.The available evidence suggests that the incorporation oftwo strongly negative phosphate groups in the basic COOH-terminal region removes the contribution of this region tothe overall binding of synapsin I. While the purelyelectrostatic binding of the COOH-terminal fragment istotally antagonized by either salt or CaMPKIIphosphorylation, the binding of holosynapsin I to SV is notcompletely abolished. The fact that the dissociation ofsynapsin I secondary to the loss in the binding of its COOH-terminal region is only partial may be accounted for by thepersistence of the hydrophobic interaction with SVphospholipids which does not appear to be greatly affectedby phosphorylation (Benfenati et al. 1989b).
J Physiol.504.3 513
) by guest on July 15, 2011jp.physoc.orgDownloaded from J Physiol (

5. Stefani and others
The addition of Ca2W/CaM in the presence of ATP/Mg2+ tothe sample containing SV-bound synapsin I was sufficientto promote a rapid dissociation of the protein from thevesicle membrane, with a time constant of less than 10 s at20 'C. In this time interval, a series of reactions have tooccur sequentially, namely activation of vesicle-associatedCaMPKII by binding of Ca2W/CaM to the regulatorydomain of the kinase, autophosphorylation of the kinase,phosphorylation of the COOH-terminal region of synapsin Ibound to the kinase, phosphorylation-dependentconformational change of synapsin I and dissociation of theprotein from the vesicle membrane. It has previously beenshown that either activation or autophosphorylation of theendogenous CaMPKII is not sufficient to inhibit synapsin Ibinding, but that phosphorylation of sites 2,3 is necessaryto bring about dissociation (Benfenati et al. 1996). Since thetime constant of association of dephosphorylated synapsin Iis extremely fast, the dissociation of synapsin I from thevesicle membrane after CaMPKII phosphorylation may befollowed by an even faster re-association of the protein upondephosphorylation, indicating that the phosphorylation-dephosphorylation cycles are strictly linked with cyclicchanges in the state of association between synapsin I andSv.
These results are consistent with evidence obtained inmammalian synaptosomes and frog neuromuscular junctionindicating that, following depolarization-inducedphosphorylation, synapsin I dissociates from the vesiclemembrane (Torri Tarelli et al. 1992; Sihra et al. 1989). Theseinteractions are also likely to have physiological relevance inthe more complex environment of the nerve terminal, sincethe association time constants between synapsin I and SVare not affected by the presence of F-actin, the majorcomponent of the nerve terminal cytomatrix (Ceccaldi et al.1995; F. Benfenati & F. Valtorta, unpublished observations).
The recent development of endocytic probes has allowed thedetermination of the dynamics of synaptic vesicle recyclingin mammalian neurons. The minimum time required for anewly internalized vesicle to be reprimed for exocytosisranges between 15 and 45 s in cultured rat hippocampalneurons at 24 'C, with a minimum duration of a completeexo-endocytotic cycle of approximately 1 min (Ryan et al.1993; Ryan & Smith, 1995). Thus, although the synapsin I-synaptic vesicle association-dissociation reactions may notbe involved in the exocytosis of primed vesicles evoked by asingle action potential, they are likely to play a physiologicalrole in regulating SV trafficking between the reserve andthe releasable pools, as well as in forms of presynapticplasticity involving recruitment of SV for exocytosis.
In conclusion, the data presented here indicate thatreversible interactions between synapsin I and SV occur andare markedly regulated by phosphorylation in a timedomain which is compatible with a function of synapsin I inthe modulation of the exo-endocytotic cycle of SV and inthe processes of presynaptic plasticity.
BAHLER, M., BENFENATI, F., VALTORTA, F., CZERNIK, A. J. &GREENGARD, P. (1989). Characterization of synapsin I fragmentsgenerated by cysteine-specific cleavage. A study of their interactionswith F-actin. Journal of Cell Biology 108, 1841-1849.
BAHLER, M. & GREENGARD, P. (1987). Synapsin I bundles F-actin in aphosphorylation-dependent manner. Nature 326, 704-707.
BENFENATI, F., BAHLER, M., JAHN, R. & GREENGARD, P. (1989a).Interactions of synapsin I with small synaptic vesicles: Distinct sitesin synapsin I bind to vesicle phospholipids and vesicle proteins.Journal of Cell Biology 108, 1863-1872.
BENFENATI, F., GREENGARD, P., BRUNNER, J. & BAHLER, M. (1989b).Electrostatic and hydrophobic interactions of synapsin I andsynapsin I fragments with phospholipid bilayers. Journal of CellBiology 108, 1851-1862.
BENFENATI, F., NEYROZ, P., BAHLER, M., MASOTTI, L. &GREENGARD, P. (1990). Time resolved fluorescence study of theneuron specific phosphoprotein synapsin I. Evidence forphosphorylation-dependent conformational changes. Journal ofBiological Chemistry 265, 12584-12595.
BENFENATI, F., ONOFRI, F., CZERNIK, A. J. & VALTORTA, F. (1996).Biochemical and functional characterization of the synaptic vesicle-associated form of Ca2+/calmodulin-dependent protein kinase II.MolecularBrain Research 40, 297-309.
BENFENATI, F., VALTORTA, F., CHIEREGATTI, E. & GREENGARD, P.(1992a). Interaction of free and synaptic vesicle-bound synapsin Iwith F-actin. Neuron 8, 377-386.
BENFENATI, F., VALTORTA, F., Rossi, M. C., ONOFRI, F., SIHRA, T. &GREENGARD, P. (1993). Interactions of synapsin I withphospholipids: possible role in synaptic vesicle clustering and in themaintenance of bilayer structures. Journal of Cell Biology 123,1845-1855.
BENFENATI, F., VALTORTA, F., RUBENSTEIN, J. R., GORELICK, F. S.,GREENGARD, P. & CZERNIK, A. J. (1992b). Synaptic vesicle-associated Ca2+/calmodulin-dependent protein kinase II is abinding protein for synapsin I. Nature 359, 417-420.
BETZ, W. J. & Wu, L.-G. (1995). Kinetics of synaptic vesicle recycling.Current Biology 5, 1096-1101.
CECCALDI, P. E., BENFENATI, F., CHIEREGATTI, E., GREENGARD, P. &VALTORTA, F. (1993). Rapid binding of synapsin I to F- and G-actin.A study using fluorescence resonance energy transfer. FEBS Letters329, 301-305.
CECCALDI, P. E., GROHOVAZ, F., BENFENATI, F., CHIEREGATTI, E.,GREENGARD, P. & VALTORTA, F. (1995). Dephosphorylatedsynapsin I anchors synaptic vesicles to actin cytoskeleton: ananalysis by videomicroscopy. Journal of Cell Biology 128, 905-912.
CHIN, L.-S., Li, L., FERREIRA, A., KoSIK, K. S. & GREENGARD, P.(1995). Impairment of axonal development and of synaptogenesis inhippocampal neurons of synapsin I-deficient mice. Proceedings ofthe National Academy of Sciences of the USA 92, 9230-9234.
DE CAMILLI, P., BENFENATI, F., VALTORTA, F. & GREENGARD, P.(1990). The synapsins. Annual Review of Cell Biology 6, 433-460.
DE CAMILLI, P., HARRIS, S. M., HUTTNER, W. B. & GREENGARD, P.(1983). Synapsin I (Protein I), a nerve terminal-specificphosphoprotein: II. Its specific association with synaptic vesiclesdemonstrated by immunocytochemistry in agarose-embeddedsynaptosomes. Journal of Cell Biology 96, 1355-1373.
DE CAMILLI, P. & TAKEI, K. (1996) Molecular mechanisms in synapticvesicle endocytosis and recycling. Neuron 16, 481-486.
GILI, V., KOORENGEVEL, M. C., DEMEL, R. A., LAZDUNSKI, C. &KILLIAN, J. A. (1992). Acidic interaction of the colicin A pore-forming domain with model membranes of Escherichia coli lipidsresults in a large perturbation of acyl chain order and stabilizationof the bilayer. Biochemistry 31, 11089-11094.
J Phy8iol. 504.3514
) by guest on July 15, 2011jp.physoc.orgDownloaded from J Physiol (

GxREENGARD, P., VALTORTA, F., CZERNIK, A. J. & BENFENATI, F.
(1993). Synaptic vesicle phosphoproteins and regulation of synaptic
function. Science 259, 780-785.HACKETT, J. T., COCHRAN, S. L., GREENFIELD, J., BROSIUS, D. C. &UEDA, T. (1990). Synapsin I injected presynaptically into Goldfish
Mauthner axons reduces quantal synaptic transmission. Journal of
Neurophysiology 63, 701-706.
HUTTNER, W. B., SCHIEBLER, W., GREENGARD, P. & DE CAMILLI, P.
(1983). Synapsin I (Protein I), a nerve terminal-specificphosphoprotein. III. Its association with synaptic vesicles studied in
a highly purified synaptic vesicle preparation. Journal of CellBiology 96, 1374-1388.
JOVANOVIC, J. N., BENFENATI, F., Siow, Y. L., SIHRA, T. S.,SANGHERA, J. S., PELECH, S. L., GREENGARD, P. & CZERNICK, A. J.(1996). Neurotrophins stimulate phosphorylation of synapsin I by
MAP kinase and regulate synapsin I-actin interactions. Proceedings
of the National Academy of Sciences of the USA 93, 3679-3683.KACZMAREK, L. K., JENNINGS, K. R., STRUMWASSER, F., NAIRN,A. C., WALTER, U., WILSON, F. D. & GREENGARD, P. (1980).Microinjections of catalytic subunit of cyclic AMP-dependent
protein kinase enhances calcium action potentials of bag cell
neurons in cell culture. Proceedings of the National Academy ofSciences of the USA 77, 7487-7491.
LAEMMLI, U. K. (1970). Cleavage of structural proteins during theassembly of the head of bacteriophage T4. Nature 227, 680-685.
Li, L., CHIN, L.-S., SHUPLIAKOV, O., BRODIN, L., SIHRA, T. S.,HVALBY, 0., JENSEN, V., ZHENG, D., MCNAMARA, J. O.,GREENGARD, P. & ANDERSEN, P. (1995). Impairment of synapticvesicle clustering and of synaptic transmission and increased seizurepropensity, in synapsin I-deficient mice. Proceedings of the NationalAcademy of Sciences of the USA 92, 9235-9239.
LLINAS, R., GRUNER, J. A., SUGIMORI, M., MCGUINNESS, T. L. &GREENGARD, P. (1991). Regulation by synapsin I and
Ca2+/calmodulin-dependent protein kinase II of transmitter release
in squid giant synapse. Journal of Physiology 436, 257-282.Lu, B., GREENGARD, P. & Poo, M.-M. (1992). Exogenous synapsin Ipromotes functional maturation of developing neuromuscular
synapses. Neuron 8, 521-529.
MCGUINNESS, T. L., LAI, Y. & GREENGARD, P. (1985).Ca2+/calmodulin-dependent protein kinase II. Isozymic forms from
rat forebrain and cerebellum. Journal of Biological Chemistry 260,1696-1704.
MUNSON, P. J. & RODBARD, D. (1980). LIGAND: A versatilecomputerized approach for characterization of ligand-binding
systems. Analytical Biochemistry 107, 220-239.
PICK, U. & KARLISH, S. J. (1980). Indications for an oligomericstructure and for conformational changes in sarcoplasmatic
reticulum Ca2+-ATP-ase labelled selectively with fluorescein.
Biochimica et Biophysica Acta 626, 255-261.
PIERIBONE, V. A., SHUPLIAKOV, O., BRODIN, L., HILFIKER-ROTHENFLUH, S., CZERNIK, A. J. & GREENGARD, P. (1995). Distinctpools of synaptic vesicles in neurotransmitter release. Nature 375,
493-497.
ROSAHL, T. W., SPILLANE, D., MISSLER, M., HERZ, J., SELING, 0. K.,WOLFF, J. R., HAMMER, R. E., MALENKA, R. C. & SUDHOF, T. C.(1995). Essential functions of synapsin I and II in synaptic vesicle
regulation. Nature 375, 488-493.
RYAN, T. A., Li, L., CHIN, L.-S., GREENGARD, P. & SMITH, S. J. (1996).Synaptic vesicle recycling in synapsin I knock-out mice. Journal of
Cell Biology 134, 1219-1227.
RYAN, T. A., REUTER, H., WENDLAND V. B., SCHWEIZER, E., TsIEN,R. W. & SMITH, S. J. (1993). The kinetics of synaptic vesicle
recycling measured at single presynaptic boutons. Neuron 11,
713-724.
515
RYAN, T. A. & SMITH, S. J. (1995). Vesicle pool mobilization duringaction potential firing at hippocampal synapses. Neuron 14,983-989.
SCHIEBLER, W., JAHN, R., DOUCET, J. P., ROTHLEIN, J. &
GREENGARD, P. (1986). Characterization of synapsin I binding tosmall synaptic vesicles. Journal of Biological Chemistry 261,8383-8390.
SIHRA, T. S., WANG, J. K. T., GORELICK, F. S. & GREENGARD, P.
(1989). Translocation of synapsin I in response to depolarization ofisolated nerve terminals. Proceedings of the National Academy ofSciences of the USA 6, 8108-8112.
STRYER, L. (1978). Fluorescence energy transfer as a spectroscopicruler. Annual Review of Biochemistry 47, 819-846.
SUDHoF, T. C., CZERNIK, A. J., KAo, H.-T., TAKEI, K., JOHNSTON,P. A., HoRIUCHI, A., KANAZIR, S. D., WAGNER, M. A., PERIN, M. S.,DE CAMILLI, P. & GREENGARD, P. (1989). Synapsins: Mosaics ofshared and individual domains in a family of synaptic vesiclephosphoproteins. Science 245, 1474-1480.
TAKEI, Y., HARADA, A., TAKEDA, S., KOBAYASHI, K., TERADA, S.,NODA, T., TAKAHASHI, T. & HIROKAWA, N. (1995). Synapsin Ideficiency results in structural changes in the presynaptic terminalsin the murine nervous system. Journal of Cell Biology 131,1789-1800.
TORRI TARELLI, F., Bossi, M., FESCE, R., GREENGARD, P. &VALTORTA, F. (1992). Synapsin I partially dissociates from synapticvesicles during exocytosis induced by electrical stimulation. Neuron9, 1143-1153.
ToWBIN, L., STAEHELIN, T. & GORDON, J. (1979). Electrophoretictransfer of proteins from poliacrylamide gels to nitrocellulosesheets: procedures and some applications. Proceedings of theNational Academy of Sciences of the USA 76, 4350-4354.
VALTORTA, F., GREENGARD, P, FESCE, R., CHIEREGATTI, F. &BENFENATI, F. (1992). Effects of the neuronal phosphoproteinsynapsin I on actin polymerization. I. Evidence for aphosphorylation-dependent nucleating effect. Journal of BiologicalChemistry 267, 11281-11288.
VALTORTA, F., IEZZI, N., BENFENATI, F., Lu, B., Poo, M-M. &GREENGARD, P. (1995). Accelerated structural maturation inducedby synapsin I at developing neuromuscular synapses of Xenopuslaevis. European Journal of Neuroscience 7, 261-270.
AcknowledgementsWe are indebted with Dr A. J. Czernik (The Rockefeller University,New York) for the anti-synapsin peptide antibodies and for criticalreading of the manuscript. This work was supported by grantsfrom Consiglio Nazionale delle Ricerche and Telethon (no. 753 toF.B. and no. 581 to F.V.) and US Public Health Service (MH 39327to P.G.). G.S. and F.O. are recipients of fellowships from theUniversity of Modena (Italy) and from the Anna Villa RusconiFoundation (Varese, Italy), respectively.
Author's email addressF. Benfenati: [email protected]
Received 6 June 1997; accepted 16 July 1997.
J Physiol. 504.3 Kinetics of synapsin-synaptic vesicle binding
) by guest on July 15, 2011jp.physoc.orgDownloaded from J Physiol (




















