
Gene profiling reveals unknown enhancing and suppressive actions of glucocorticoids on immune cells
Upload
independentCategory
view
0download
0
Inhibition of human T cell leukemia virus type 2replication by the suppressive action of class IItransactivator and nuclear factor YGiovanna Tosi*, Elisabetta Pilotti†, Lorenzo Mortara*, Andrea De Lerma Barbaro*, Claudio Casoli†,and Roberto S. Accolla*‡
*Department of Clinical and Biological Sciences, University of Insubria, 21100 Varese, Italy; and †Department of Clinical Medicine, Nephrology,and Health Sciences, University of Parma, 43100 Parma, Italy
Edited by Robert C. Gallo, University of Maryland, Baltimore, MD, and approved July 6, 2006 (received for review February 25, 2006)
The master regulator of MHC-II gene transcription, class II trans-activator (CIITA), acts as a potent inhibitor of human T cell leukemiavirus type 2 (HTLV-2) replication by blocking the activity of the viralTax-2 transactivator. Here, we show that this inhibitory effecttakes place at the nuclear level and maps to the N-terminal 1–321region of CIITA, where we identified a minimal domain, frompositions 64–144, that is strictly required to suppress Tax-2 func-tion. Furthermore, we show that Tax-2 specifically cooperates withcAMP response element binding protein-binding protein (CBP) andp300, but not with p300�CBP-associated factor, to enhance tran-scription from the viral promoter. This finding represents a uniquedifference with respect to Tax-1, which uses all three coactivatorsto transactivate the human T cell leukemia virus type 1 LTR. Directsequestering of CBP or p300 is not the primary mechanism by whichCIITA causes suppression of Tax-2. Interestingly, we found that thetranscription factor nuclear factor Y, which interacts with CIITA toincrease transcription of MHC-II genes, exerts a negative regula-tory action on the Tax-2-mediated HTLV-2 LTR transactivation.Thus, CIITA may inhibit Tax-2 function, at least in part, throughnuclear factor Y. These findings demonstrate the dual defensiverole of CIITA against pathogens: it increases the antigen-presentingfunction for viral determinants and suppresses HTLV-2 replicationin infected cells.
MHC class II � viral replication � Tax-2
Human T cell leukemia virus type 1 (HTLV-1) and human T cellleukemia virus type 2 (HTLV-2) are related retroviruses with
similar genomic organization and common modes of transmissionbut different disease manifestations (1–3).
HTLV-1 is the etiologic agent of adult T cell leukemia�lymphoma and tropical spastic paraparesis�HTLV-1-associatedmyelopathy (4–7).
Although originally isolated from a case of atypical hairy T cellleukemia (8), HTLV-2 has not been epidemiologically linked tolymphoproliferative disorders (9). Few reports have described anassociation with tropical spastic paraparesis�HTLV-1-associatedmyelopathy-‘‘like’’ cases (10, 11).
HTLV-1 and HTLV-2 show a preferential tropism for CD4� andCD8� T cells, respectively, but they can also infect other popula-tions, including monocytes and B cells (12–16). Both viruses encodehomologous transcription activators, respectively designated asTax-1 and Tax-2, that are important mediators of viral pathogenesisand essential for immortalization of T lymphocytes (17, 18).
Tax-1 activates transcription of the HTLV-1 viral genome byinteracting with the cAMP response element binding protein-binding protein (CBP)�ATF family of transcription factors, whichbind to the viral LTR (19, 20). This interaction stabilizes thecomplex and facilitates the recruitment of general transcriptionfactors and coactivators, such as the histone acetyltransferases(HATs), CBP, p300, and p300�CBP-associated factor (PCAF),resulting in the enhancement of transcription (21–26). Tax-1 alsoderegulates the expression of a variety of cellular genes and
signaling pathways involved in cell cycle progression, cell growth,DNA repair, and apoptosis. This deregulation is believed to con-tribute to the initiation of leukemogenesis and maintenance of themalignant phenotype in adult T cell leukemia�lymphoma (27–29).
Tax-2 has �75% amino acid sequence homology with Tax-1, andalthough its mechanism of action is assumed to be similar to that ofTax-1, very little is known about the cellular factors interacting withand�or used by Tax-2 to mediate its biological functions. Moreover,several reports have shown that Tax-1 and Tax-2 have distinctbiological properties (30–37).
We reported that Tax-2 transactivation of the HTLV-2 LTR isstrongly inhibited by the host transcription factor class II transac-tivator (CIITA). As a consequence, susceptible T and B human cellsdo not support HTLV-2 replication when expressing CIITA (38).Similarly, CIITA targets the viral transactivator Tat to inhibit thereplication of the HIV-1 virus (39, 40).
The AIR-1 locus-encoded CIITA is the master regulator of theexpression of MHC-II genes (41–43) that play a key role in thehomeostasis of the immune system. MHC-II-encoded moleculespresent peptides to the antigen receptor of CD4� T cells, whoseactivation is required to trigger and modulate both humoral andcellular immune responses (44). CIITA is a non-DNA-bindingtranscriptional integrator recruited to MHC-II promoters via mul-tiple interactions with transcription factors bound to DNA, includ-ing the regulatory factor X and the nuclear factor Y (NF-Y)complexes (45–50).
CIITA interacts with CBP, p300, PCAF, and the cyclin T1subunit of the positive transcription elongation factor b to enhanceMHC-II gene transcription (51–54).
The positive transcription elongation factor b is also used by Tatto promote the elongation of HIV-1 viral transcripts (55), and wehave shown that sequestration of cyclin T1 is the major mechanismby which CIITA blocks the transactivating function of Tat (40). Butthe molecular basis of the CIITA-mediated inhibition of Tax-2 isstill elusive.
To further understand how CIITA affects Tax-2, in the presentstudy we investigated: (i) whether the block of Tax-2 functionmediated by CIITA takes place in the cytoplasm or the nucleus; (ii)the minimal region of CIITA retaining the ability to block Tax-2transactivation; (iii) whether cellular factors used by CIITA toactivate MHC-II gene transcription are also used by Tax-2 totransactivate the viral promoter; and (iv) if so, whether CIITAtargets one or more of these factors to inhibit Tax-2 function.
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: HTLV-1, human T cell leukemia virus type 1; HTLV-2, human T cell leukemiavirus type 2; CIITA, class II transactivator; HAT, histone acetyltransferase; CBP, cAMPresponse element binding protein-binding protein; PCAF, p300�CBP-associated factor;NF-Y, nuclear factor Y.
‡To whom correspondence should be addressed. E-mail: [email protected].
© 2006 by The National Academy of Sciences of the USA
www.pnas.org�cgi�doi�10.1073�pnas.0601589103 PNAS � August 22, 2006 � vol. 103 � no. 34 � 12861–12866
MED
ICA
LSC
IEN
CES

We found that the CIITA 1–321 N-terminal region, with anexclusive nuclear distribution, inhibits Tax-2 function and viralreplication. We identified CBP and p300 as crucial factors for theTax-2-directed LTR transactivation. However, they are not in-volved in CIITA-mediated inhibition of Tax-2. Instead the tran-scription factor NF-Y was found to inhibit Tax-2 transactivatingfunction.
We present a model to account for the involvement of NF-Y inthe negative control of the viral replication exerted by CIITA.
ResultsThe N-Terminal Region of CIITA Inhibits HTLV-2 Replication in SusceptibleB Cells. We previously demonstrated that the N-terminal regionof CIITA mediates the inhibition of HTLV-2 LTR transactiva-tion directed by Tax-2 (38).
To determine whether this inhibition interferes with the life cycleof HTLV-2, we analyzed the capacity of human target cells tosupport viral replication when expressing different fragments ofCIITA. CIITA-defective RJ2.2.5 B cells were stably transfectedwith chimeric constructs expressing a fusion protein between theGFP and the Flag-tagged full-length CIITA (residues 1–1130) orthe deletion mutants 1–321 or 322-1130. Transfectants were sortedon the basis of the green fluorescence and tested for the expressionof MHC-II molecules. As shown in Fig. 1, FACS analysis revealedthat GFP did not affect transcriptional activity of CIITA becausetransfection of the GFPfCIITA1-1130 chimera resulted in cellsurface expression of MHC-II-DR. As expected, the C-terminaland N-terminal truncated forms of CIITA failed to activate class IIgene expression.
To further characterize RJ2.2.5 transfectants, we analyzed thesubcellular distribution of the various GFPfCIITA molecules byconfocal microscopy (Fig. 1, confocal microscopy). In control cellstransfected with unmodified GFP a diffuse fluorescent pattern wasobserved throughout the cells. GFPfCIITA1-1130 displayed dualcytoplasmic and nuclear localization, but was clearly excluded fromnucleoli. GFPfCIITA1-321 predominantly accumulated into thenucleus, but not in the nucleoli. In contrast, GFPfCIITA322-1130showed a strictly cytoplasmic localization. Of note, the observedlocalization of the various GFPfCIITA chimeras reflects the rela-
tive subcellular distribution of the corresponding untagged CIITAmolecules (data not shown).
The RJ2.2.5 transfectants were then infected with HTLV-2. Theinfection efficiency was comparable for all cell lines as assessed byreal-time PCR of the tax-2b gene 2 days after infection (Fig. 1,tax-2b). Viral replication was extremely different in the varioustransfectants as indicated by the proviral load values at day 15 afterinfection. RJ2.2.5 cells supported viral replication when expressingthe GFP protein alone or the C-terminal 322-1130 region. Incontrast, RJ2.2.5 cells expressing the N-terminal 1–321 fragment orthe full-length CIITA did not allow viral replication as demon-strated by their very low proviral load at day 15 (178 and 192 versus13,680 in the RJ�GFP cells, respectively). This distinct behavior wasnot caused by different expression levels of the various GFPfCIITAchimeras because they were equally expressed in the transfectants(Fig. 1, �Flag IP, �CIITA WB).
These findings demonstrate that the constitutive expression atphysiological levels of the N-terminal 1–321 fragment of CIITAmay effectively inhibit viral replication in natural cellular targets ofHTLV-2 infection.
A Minimal Sequence of 80 Amino Acids in the N-Terminal 1–321 Regionof CIITA Is Required to Inhibit the Tax-2-Mediated HTLV-2 LTR Transac-tivation. To identify the minimal sequence of CIITA that inhibitsTax-2 activity we performed a functional mapping of CIITA. Agene reporter assay was carried out in COS cells cotransfectedwith a HTLV-2 LTR-Luciferase construct, an expression vectorfor Tax-2, and increasing amounts of the plasmids coding foreither CIITA full-length or CIITA deletion mutants. Westernblot analyses with anti-Flag antibody showed that all recombi-nant CIITA proteins were equivalently expressed in the cells(Fig. 2A). We assessed the capacity of the various CIITAfragments to inhibit Tax-2-mediated HTLV-2 LTR transactiva-
Fig. 1. The N-terminal 1–321 fragment of CIITA inhibits HTLV-2 viral repli-cation in B cells. RJ2.2.5 B cells stably expressing GFP alone or GFP fused toFlag-tagged CIITAs (fC1-1130, fC1-321, or fC322-1130) were infected withHTLV-2 virus. tax-2b values express the HTLV-2 proviral load, as assessed atdays 2 and 15 after infection by real-time PCR of tax-2b sequences. GFP andMHC-II DR expression was monitored by FACS analysis. The log fluorescenceintensity is reported in the abscissa as arbitrary units. The subcellular localiza-tion of the exogenous GFPfCIITA proteins was monitored by confocal fluo-rescence microscopy. The expression levels of the GFPfCIITA chimeras wereassessed by anti-Flag immunoprecipitation (IP) and anti-CIITA immunoblot-ting (WB).
Fig. 2. Mapping of the minimal CIITA region required for inhibition of Tax-2activity. (A) COS cells were transiently cotransfected with fixed amounts (0.2�g) of pLTRII-Luc and pcTax-2b vectors and increasing amounts (0.2–0.8 �g) ofvectors coding for Flag-tagged CIITA WT or the indicated deletion mutants.Lower amounts of plasmid DNA (0.1–0.4 �g) were transfected for fCIITA1-321.Results are presented as percentage of luciferase activity relative to 100%activation by Tax-2 (black bars). Bar 1 (hatched) represents the activity of thepcDNA3 vector. CIITA did not affect basal promoter activity (data not shown).Expression levels of recombinant fCIITA proteins were evaluated by anti-FlagWestern blot (WB) analyses of the cell extracts. (B) Schematic representationof the results of the gene reporter assays illustrated in A. CIITA proteins usedfor the mapping are shown along with their capacity to inhibit Tax-2 function(�). The endpoints of each CIITA form are indicated. At the top is a diagramof CIITA with its domains: AD, activation domain; P�S�T, proline�serine�threonine-rich domain; GBD, GTP-binding domain; LRR, leucine-rich repeats.The black boxes represent the minimal domain from positions 64–144 that isnecessary to block the transactivation capacity of Tax-2.
12862 � www.pnas.org�cgi�doi�10.1073�pnas.0601589103 Tosi et al.
tion. Full-length CIITA significantly reduced the effect of Tax-2on the HTLV-2 promoter, and the 1–321 CIITA fragmentexerted an even more potent inhibition (Fig. 2 A, compare bars3–5 and 6–8 with bar 2), supporting the results of the infectionexperiments in RJ2.2.5-transfected cells (Fig. 1). On the con-trary, the C-terminal 322-1130 and 201-1130 fragments of CIITAdid not affect Tax-2-mediated transactivation (Fig. 2 A, comparebars 18–20 and 21–23 with bar 2), indicating that the first 200 aaof CIITA are necessary to block Tax-2 function. Interestingly,additional mutants with further deletions of the 1–200 region(fCIITA26-200, fCIITA26-144, and fCIITA64-200) still retainedthe ability to inhibit Tax-2. Quantitation of luciferase activityshowed up to a 20-fold reduction in Tax-2 transactivation at thehighest doses of these CIITA fragments (Fig. 2 A, compare bars11, 14, and 17 with bar 2).
To rule out a toxic effect of the N-terminal CIITA fragments onthe transcription machinery during the transient transfection assay,CIITA1-321 or CIITA64-200 were cotransfected in Cos cells withGal4–VP16 hybrid plasmid and pG5bCAT reporter plasmid (56).We found that the overexpression of both CIITA1-321 andCIITA64-200 did not inhibit the transcriptional activity of theGal4–VP16 chimera on the Gal4 responsive promoter of thepG5bCAT construct (data not shown).
We conclude that a short stretch of 80 aa encompassing positions64–144 of the activation domain of CIITA (Fig. 2B, black boxes) isrequired for the functional and specific inhibition of Tax-2.
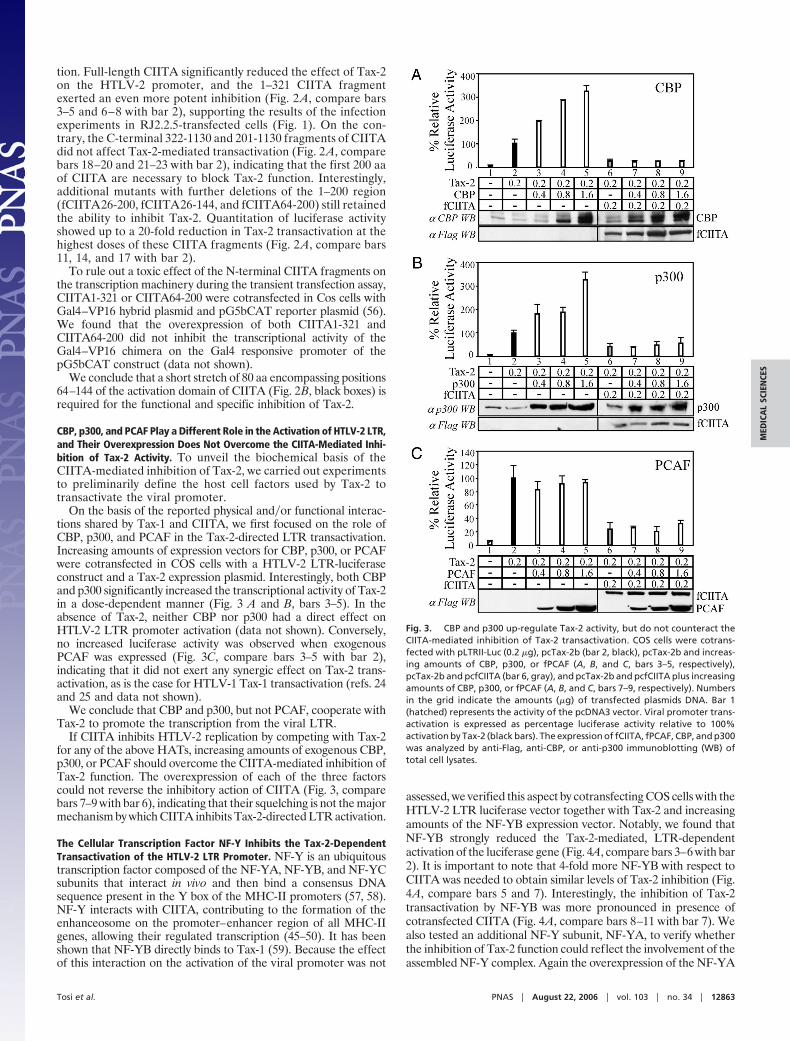
CBP, p300, and PCAF Play a Different Role in the Activation of HTLV-2 LTR,and Their Overexpression Does Not Overcome the CIITA-Mediated Inhi-bition of Tax-2 Activity. To unveil the biochemical basis of theCIITA-mediated inhibition of Tax-2, we carried out experimentsto preliminarily define the host cell factors used by Tax-2 totransactivate the viral promoter.
On the basis of the reported physical and�or functional interac-tions shared by Tax-1 and CIITA, we first focused on the role ofCBP, p300, and PCAF in the Tax-2-directed LTR transactivation.Increasing amounts of expression vectors for CBP, p300, or PCAFwere cotransfected in COS cells with a HTLV-2 LTR-luciferaseconstruct and a Tax-2 expression plasmid. Interestingly, both CBPand p300 significantly increased the transcriptional activity of Tax-2in a dose-dependent manner (Fig. 3 A and B, bars 3–5). In theabsence of Tax-2, neither CBP nor p300 had a direct effect onHTLV-2 LTR promoter activation (data not shown). Conversely,no increased luciferase activity was observed when exogenousPCAF was expressed (Fig. 3C, compare bars 3–5 with bar 2),indicating that it did not exert any synergic effect on Tax-2 trans-activation, as is the case for HTLV-1 Tax-1 transactivation (refs. 24and 25 and data not shown).
We conclude that CBP and p300, but not PCAF, cooperate withTax-2 to promote the transcription from the viral LTR.
If CIITA inhibits HTLV-2 replication by competing with Tax-2for any of the above HATs, increasing amounts of exogenous CBP,p300, or PCAF should overcome the CIITA-mediated inhibition ofTax-2 function. The overexpression of each of the three factorscould not reverse the inhibitory action of CIITA (Fig. 3, comparebars 7–9 with bar 6), indicating that their squelching is not the majormechanism by which CIITA inhibits Tax-2-directed LTR activation.
The Cellular Transcription Factor NF-Y Inhibits the Tax-2-DependentTransactivation of the HTLV-2 LTR Promoter. NF-Y is an ubiquitoustranscription factor composed of the NF-YA, NF-YB, and NF-YCsubunits that interact in vivo and then bind a consensus DNAsequence present in the Y box of the MHC-II promoters (57, 58).NF-Y interacts with CIITA, contributing to the formation of theenhanceosome on the promoter–enhancer region of all MHC-IIgenes, allowing their regulated transcription (45–50). It has beenshown that NF-YB directly binds to Tax-1 (59). Because the effectof this interaction on the activation of the viral promoter was not
assessed, we verified this aspect by cotransfecting COS cells with theHTLV-2 LTR luciferase vector together with Tax-2 and increasingamounts of the NF-YB expression vector. Notably, we found thatNF-YB strongly reduced the Tax-2-mediated, LTR-dependentactivation of the luciferase gene (Fig. 4A, compare bars 3–6 with bar2). It is important to note that 4-fold more NF-YB with respect toCIITA was needed to obtain similar levels of Tax-2 inhibition (Fig.4A, compare bars 5 and 7). Interestingly, the inhibition of Tax-2transactivation by NF-YB was more pronounced in presence ofcotransfected CIITA (Fig. 4A, compare bars 8–11 with bar 7). Wealso tested an additional NF-Y subunit, NF-YA, to verify whetherthe inhibition of Tax-2 function could reflect the involvement of theassembled NF-Y complex. Again the overexpression of the NF-YA
Fig. 3. CBP and p300 up-regulate Tax-2 activity, but do not counteract theCIITA-mediated inhibition of Tax-2 transactivation. COS cells were cotrans-fected with pLTRII-Luc (0.2 �g), pcTax-2b (bar 2, black), pcTax-2b and increas-ing amounts of CBP, p300, or fPCAF (A, B, and C, bars 3–5, respectively),pcTax-2b and pcfCIITA (bar 6, gray), and pcTax-2b and pcfCIITA plus increasingamounts of CBP, p300, or fPCAF (A, B, and C, bars 7–9, respectively). Numbersin the grid indicate the amounts (�g) of transfected plasmids DNA. Bar 1(hatched) represents the activity of the pcDNA3 vector. Viral promoter trans-activation is expressed as percentage luciferase activity relative to 100%activation by Tax-2 (black bars). The expression of fCIITA, fPCAF, CBP, and p300was analyzed by anti-Flag, anti-CBP, or anti-p300 immunoblotting (WB) oftotal cell lysates.
Tosi et al. PNAS � August 22, 2006 � vol. 103 � no. 34 � 12863
MED
ICA
LSC
IEN
CES

subunit led to a dose-dependent reduction of luciferase activity(Fig. 4B, compare bars 3–6 with bar 2), but the effect was lessdramatic compared with NF-YB.
These data show that the NF-Y complex, rather than a singleNF-Y subunit, is most likely responsible for the observed inhibitoryeffect on Tax-2-mediated LTR transactivation. The data alsoindicate that in the presence of CIITA the inhibitory effect of NF-Yis enhanced.
DiscussionThe findings reported here clearly establish that host factorsinvolved in the regulation of MHC-II gene expression, namelyCIITA and NF-Y, can negatively regulate HTLV-2 replication byblocking the function of Tax-2 viral transactivator. Moreover, ourresults demonstrate that Tax-2 differs from Tax-1 in the selectiveuse of HATs for the activation of the viral LTR.
CIITA is a transcription factor with both nuclear and cytoplasmiclocalization. This dual subcellular distribution is the net result ofmultiple nuclear import and nuclear export signals (53, 60–65).Because Tax-2 activates the viral promoter in the nucleus, it is likelythat CIITA inhibits Tax-2 at the nuclear level. Nevertheless, it wasrecently shown that Tax-2 is a shuttling protein with predominantcytoplasmic accumulation (66). Thus, the possibility existed thatCIITA interferes with Tax-2 in the cytoplasm.
To address this issue we analyzed and correlated the subcellulardistribution of several CIITA fragments with their capacity toinhibit HTLV-2 replication. We show that, beside CIITA wild type,
the N-terminal 1–321 fragment, which exhibits a restricted nuclearlocalization, is able to inhibit viral replication. Similarly, we foundthat the minimal 64–200 fragment of CIITA that still suppressesTax-2 transactivation of viral LTR, is mostly, if not exclusively,localized in the nucleus (data not shown). In contrast, CIITA322-1130, which displays cytoplasmic localization, does not affect viralreplication. These results demonstrate a strict correlation betweennuclear localization of CIITA and its capacity to inhibit Tax-2transactivation. This conclusion is corroborated by our previousfinding that BLS2 cells support HTLV-2 replication (38). Thesecells express a mutant form of CIITA that retains all of thefunctional domains of the molecule, but shows an exclusive cyto-plasmic localization because of a short in-frame deletion thatremoves a nuclear localization motif (60).
The region 1–321 of CIITA is critical not only for the inhibitionof HTLV-2 replication but also for the transcription of MHC-IIgenes. It includes the transcriptional activation domain and, frompositions 253–321, the dimerization domain (56). The N terminusof CIITA also interacts with CBP, p300, PCAF, and subunits of theregulatory factor X and NF-Y complexes (48, 50–53). Our mappingexperiments define the boundary of the CIITA fragment that isminimally required to suppress Tax-2 transactivation to residuesfrom positions 64–144 (Fig. 2). This sequence does not overlap withthe dimerization domain, implying that CIITA self-association isnot a prerequisite for Tax-2 inhibition.
In searching for the intimate molecular basis of Tax-2 inhibitionby CIITA we focused on CBP, p300, PCAF, and the NF-Y complex,which are known to interact with both Tax-1 and CIITA. Ourstudies demonstrate that CBP and p300, but not PCAF, enhanceTax-2-directed LTR transactivation. The fact that PCAF, p300, andCBP all are essential for optimal transactivation by Tax-1 (23–25)substantiates the existence of important differences betweenHTLV-2 Tax-2 and HTLV-1 Tax-1. The different requirement forPCAF between the two viral transactivators also implies that Tax-1,but not Tax-2, might influence nuclear PCAF-containing com-plexes, potentially contributing to the pleiotropic deregulated ex-pression of cellular genes during leukemogenesis. Importantly,Tax-1 mutants, which interact poorly with PCAF, exhibit an im-paired transactivation capacity and are defective for transformation(24, 67). The theme of differential usage of coactivators with HATactivity between Tax-2 and Tax-1 is not new. A recent work byMeertens et al. (68) has shown that whereas Tax-1 can use CBP orp300 for inhibiting p53 Tax-2 uses only CBP. In addition, it has beenshown that Tax-1 transforms rat fibroblasts and inhibits p53 func-tion more efficiently than Tax-2 (30, 31). These observationssuggest that a selective use of HATs in different transcriptionalpathways could be responsible, at least in part, for the higheroncogenic potential of Tax-1 with respect to Tax-2.
We previously demonstrated that CIITA inhibits HIV-1 repli-cation by competing with the Tat transactivator for cyclin T1 (40).This finding prompted us to determine whether squelching of CBP,p300, or PCAF could account for the observed inhibition of Tax-2by CIITA. These HATs were attractive candidates in light of thefact that their squelching seems to be a common mechanism bywhich CIITA mediates gene suppression (69–72). We found thatdirect sequestration of the above cofactors was not the primarymechanism by which CIITA causes suppression of Tax-2 transac-tivating function.
Interestingly, we found that overexpression of NF-Y significantlyinhibited Tax-2-directed HTLV-2 LTR transactivation. A transcrip-tionally competent NF-Y complex is expressed in COS and RJ2.2.5cells. Thus, apparently, physiologic levels of NF-Y do not preventeither the Tax-2-mediated LTR-driven gene expression in COScells or the replication of HTLV-2 virus in RJ2.2.5 cells (Figs. 1 and2). On the contrary, CIITA constitutively expressed at physiologiclevels in Raji B cells (38) or RJ2.2.5 cells after stable transfection(Fig. 1 and ref. 38) dramatically suppresses HTLV-2 productiveinfection. Cotransfection of NF-Y and suboptimal amounts of
Fig. 4. Overexpression of B and A subunits of the NF-Y complex repressesTax-2 activity. Shown are the results of Luciferase gene reporter assays per-formed in COS cells cotransfected with 0.2 �g of pLTRII-Luc vector, and theindicated amounts (�g) of plasmid DNA are listed on the left of the grids. Bar1 (hatched), bar 2 (black), and bar 7 (gray) indicate the luciferase activitiesinduced by pcDNA3 vector, Tax-2, and Tax-2 plus CIITA, respectively. Bars 3–5indicate luciferase activity induced by Tax-2 in the presence of increasingamounts of NF-YB (A) or NF-YA (B). Bars 8–11 indicate the luciferase activityinduced by Tax-2 in the presence of CIITA and increasing amount of NF-YB (A)or NF-YA (B). The data are set relative to 100% activation of the LTRII-Lucreporter by Tax-2. The expression of fCIITA, mNF-YA, and mNF-YB was ana-lyzed by immunoblotting (WB) of the cell extracts with anti-Flag and anti-Mycantibodies.
12864 � www.pnas.org�cgi�doi�10.1073�pnas.0601589103 Tosi et al.

CIITA in COS cells resulted in even stronger NF-Y-mediatedinhibition of Tax-2 transactivation capacity, which suggests that theNF-Y complex requires CIITA to contribute its inhibitory activityon Tax-2.
On the basis of these findings we propose a molecular model forthe CIITA-mediated inhibition of Tax-2 activity via the binding ofthe CIITA–NF-Y complex to Tax-2 (Fig. 5). NF-Y expressed incells at physiologic levels does not efficiently bind Tax-2, which isrecruited to the LTR promoter to activate transcription (Fig. 5A).On the contrary, the overexpression of NF-Y can facilitate theinteraction with Tax-2, preventing its recruitment to the viralpromoter (Fig. 5B). When expressed, CIITA interacts with theNF-Y complex; this interaction changes the conformation of NF-Y,increasing its binding affinity for Tax-2. The Tax-2–NF-Y complexis not recruited to the LTR and does not activate transcription (Fig.5C). Alternatively, the binding of NF-Y to Tax-2 could still permitits recruitment to the LTR, but not its transcriptional activity.Following our model, the inhibition of Tax-2 by CIITA is not causedby the squelching of a transcriptional positive coactivator, butinstead by the recruitment of a cellular factor, NF-Y, with a negativeregulatory action on Tax-2.
The results of this study may contribute to therapeutic strategiesaimed at counteracting retroviral infections through the control ofCIITA expression and�or the selective use of CIITA fragments.
MethodsPlasmids. pcfCIITA1-1130, pcf1-321, and pcf322-1130 vectors havebeen described (56). All Flag-tagged CIITA deletion mutants weregenerated from pcfCIITA1-1130 by PCR.
pcfCIITA210-1130 was made with primers F201 (5�-GGGGG-GAATTCGCCACCATGGACTACAAGGACGACGACG-ACAAGCGCCTGGAGAAAACCGACCAG-3�) and 1130R (5�-CGATGCTCTAGATCATCTCAGGCTGATCCGTGA-3�).PCR product was ligated into EcoRI–XbaI-digested pcDNA3vector (Invitrogen, Milan, Italy). pcfCIITA26-200 and pcfCI-ITA26-144 were made by using the common primer F26(5�-GGGGGGAATTCGCCACCATGGACTACAAGGACGA-CGACGACAAGGCAGAGTTGGGGCCCCTAGAAGGTGG-CTACC-3�) and the 200R (5�-GGGGGCTCGAGTCACATCTG-GCCGGAGGCTGGCTCCTGG-3�) or 144R (5�-GGGGCTC-GAGTCATTTCTGACTTTTCTGCCCAACTTCTG-3�) reverseprimers, respectively, and ligation into EcoRI–XhoI-cleavedpcDNA3.1(�) vector. Primers used to make pcfCIITA64-200 were:F64 (5�-GGGGGGAATTCGCCACCATGGACTACAAGGAC-GACGACGACAAGGCACTCTACTCAGAACCCGACACAG-ACACCATC-3�) and the 200R primer. PCR product was ligatedinto EcoRI–XhoI-cleaved pcDNA3.1(�). All constructs were se-quenced. pLTRII-Luc vector containing the HTLV-2 LTR pro-
moter linked to the firefly Luciferase gene, pcTax-2b, pcRmNF-YA, pcmNF-YB, pCMVCBP, and pCMV�p300 have beendescribed (38, 49, 52, 40). To obtain the pcfPCAF vector, fPCAFcDNA was excised from the pcxPCAF plasmid (73) by EcoRI–KpnI digestion and inserted into EcoRI–KpnI-cleavedpcDNA3.1(�) vector.
pGFPfCIITA1-1130, pGFPfCIITA1-321, and pGFPfCIITA322-1130 constructs were generated by excision of the correspondingCIITA cDNA from the pcDNA3 vector by digestion with EcoRI,EcoRI–XhoI, and EcoRI–XbaI, respectively. The resulting frag-ments were ligated into pEGFP-C2 vector (Clontech, Palo Alto,CA) digested with EcoRI, EcoRI–SalI, and EcoRI–XbaI, respec-tively, to produce GFP chimeras in which the GFP was fusedin-frame to the N terminus of Flag-tagged CIITA proteins. TheRenilla luciferase reporter vector phRL-CMV was purchased fromPromega (Milan, Italy).
Transient Transfections, Luciferase Assay, and Western Blotting. COScells grown in DMEM supplemented with 10% FCS and 5 mML-glutamine in 35-mm-diameter plates were transfected with vari-able amounts of plasmids DNA as indicated for each gene reporterassay and 5 ng of phRL-CMV, using Lipofectamine (Invitrogen).Empty pcDNA3 vector was used as a stuffer DNA in all experi-ments. Cell extracts were prepared 24 h posttransfection andassayed for luciferase activity by using the Dual Luciferase ReporterAssay System (Promega) according to the manufacturer’s instruc-tions. The values of three independent experiments performed intriplicate were calculated as mean luciferase�renilla ratio � stan-dard error and expressed as percentages relative to luciferaseactivity produced by Tax-2 (100%). Cell lysates were analyzed forthe expression of recombinant proteins by SDS�PAGE on 4–12%polyacrylamide gels and Western blotting as described (40). Thefollowing antibodies were used: anti-Flag M2 (Sigma, Milan, Italy)to detect fCIITA proteins and fPCAF; anti-Myc 9E10 (Santa CruzBiotechnology, Santa Cruz, CA) to detect myc-tagged NF-YA andNF-YB; and anti-CBP (A22) and anti-p300 (N15) (Santa CruzBiotechnology) to detect CBP and p300, respectively. HRP-conjugated anti-mouse Ig or anti-rabbit Ig secondary antibodies(Amersham Pharmacia, Milan, Italy) were used. Blots were devel-oped by chemiluminescence assay (ECL, Amersham Pharmacia).
Stable Transfections, FACS Analysis, and Immunoprecipitations. TheMHC-II-negative RJ2.2.5 human B cells (4 � 106), lacking theexpression of the AIR-1 locus product CIITA (41), were transfectedby electroporation with 5 �g of pGFPfCIITA1-1130, pGFPfCI-ITA1-321, pGFPfCIITA322-1130, or pEGFP-C2 vectors by usingthe GenePulser II apparatus (Bio-Rad, Richmond, CA) at 320V�350 �F. Transfected cells were selected and cultivated with 1mg�ml neomycin (GIBCO�BRL, Milan, Italy). GFP� cells werepurified by fluorescence-activated cell sorting and analyzed for thecell surface expression of MHC-II molecules with a phycoerythrin-conjugated anti-HLA-DR antibody (L243) (Becton Dickinson,Milan, Italy) by cytofluorometry.
The lysate obtained from 70 � 106 cells was assayed for thepresence of GFPfCIITA recombinant proteins by immunopre-cipitation with anti-FlagM2-agarose beads (Sigma) followed byimmunoblotting with rabbit sera raised against residues 911–980(40) or residues 255–321 of CIITA and HRP-conjugated anti-rabbitIg secondary antibody (Amersham Pharmacia).
GFP Analyses. Stably transfected RJ2.2.5 cells, suspended in PBS,were mounted on microscope slides with coverslips and immedi-ately viewed with a confocal laser microscope (FV500, Olympus,Hamburg, Germany), at room temperature, �60 magnification,1.40 numerical aperture of the objective lenses, and Fluoviewacquisition software.
Fig. 5. A possible model for the CIITA-mediated inhibition of Tax-2 tran-scriptional activity. (A) Physiologic amounts of NF-Y are insufficient to affectthe activity of Tax-2 on the HTLV-2 LTR. (B) Overexpression of NF-Y results inNF-Y�Tax-2 interaction and inhibition of transcription from the LTR. (C) Whenpresent, CIITA interacts with NF-Y and increases NF-Y binding affinity forTax-2, resulting in a lack of HTLV-2 LTR transactivation.
Tosi et al. PNAS � August 22, 2006 � vol. 103 � no. 34 � 12865
MED
ICA
LSC
IEN
CES

Viral Infection and Assessment of HTLV-2 Proviral Load. Stably trans-fected RJ2.2.5 cells were cocultured with a double share of HTLV-2strain Gu-infected BJAB cells and assessed for tax-2b sequences per105 cells by Taqman real-time PCR as described (38).
We thank A. Parodi for technical assistance; L. Chiossone for help withconfocal microscopy; and Drs. B. M. Peterlin (University of California,
San Francisco, CA), K. Ozato (National Institutes of Health, Bethesda,MD), J. Ting (University of North Carolina, Chapel Hill, NC), and M.Giacca (International Center for Genetic Engineering and Biotechnol-ogy, Triste, Italy) for providing reagents. This work was supported byIstituto Superiore di Sanita National Research Project on AIDS Grants40D.01 (to R.S.A.) and 40F.24 (to C.C.), a grant from Fondi di Ateneoper la Ricerca 2005 (to R.S.A. and G.T.), and Cariparma BankingFoundation Grant 2004.0190.
1. Gallo, R. C. (2005) Retrovirology 2, 17–23.2. Manns, A. & Blattner, W. A. (1991) Transfusion 31, 67–75.3. Franchini, G. (1995) Blood 86, 3619–3639.4. Poiesz, B. J., Ruscetti, F. W., Gazdar, A. F., Bunn, P. A., Minna, J. D. & Gallo,
R. C. (1980) Proc. Natl. Acad. Sci. USA 77, 7415–7419.5. Yoshida, M., Miyoshi, I. & Hinuma, Y. (1982) Proc. Natl. Acad. Sci. USA 79,
2031–2035.6. Gessain, A., Barin, F., Vernant, J. C., Gout, O., Maurs, L., Calender, A. & de
The, G. (1985) Lancet 2, 407–410.7. Osame, M., Usuku, K., Izumu, S., Ijichi, N., Amitani, H., Igata, A., Matsumoto,
M. & Tara, M. (1986) Lancet 1, 1031–1032.8. Kalyanaraman, V. S., Sarngadharan, M. G., Robert-Guroff, M., Miyoshi, I.,
Golde, D. & Gallo, R. C. (1982) Science 218, 571–573.9. Hjelle, B., Mills, R., Swenson, S., Mertz, G., Key, C. & Allen, S. (1991) J. Infect.
Dis. 163, 435–440.10. Lehky, T. J., Flerlage, N., Katz, D., Houff, S., Hall, W. H., Ishii, K., Monken,
C., Dhib-Jalbut, S., McFarland, H. F. & Jacobson, S. (1996) Ann. Neurol. 40,714–723.
11. Roucoux, D. F. & Murphy, E. L. (2004) AIDS Rev. 6, 144–154.12. Ijichi, S., Ramundo, M. B., Takahashi, H. & Hall, W. W. (1992) J. Exp. Med.
176, 293–296.13. Hoffman, P. M., Dhib-Jalbut, S., Mikovits, J. A., Robbins, D. S., Wolf, A. L.,
Bergey, G. K., Lohrey, N. C., Weislow, O. S. & Ruscetti, F. W. (1992) Proc. Natl.Acad. Sci. USA 89, 11784–11788.
14. Lal, R. B., Owen, S. M., Rudolph, D. L., Dawson, C. & Prince, H. (1995)Virology 210, 441–447.
15. Casoli, C., Cimarelli, A. & Bertazzoni, U. (1995) Virology 206, 1126–1128.16. Wang, T. G., Ye, J., Lairmore, M. D. & Green, P. L. (2000) AIDS Res. Hum.
Retroviruses 16, 1661–1668.17. Grassmann, R. C., Dengler, C., Muller-Fleckenstein, I., Fleckenstein, B.,
McGuire, M. C., Dokhelar, J. G., Sodrosky, J. G. & Haseltine, W. A. (1989)Proc. Natl. Acad. Sci. USA 86, 3351–3355.
18. Ross, T. M., Narayan, M., Fang, Z.-Y., Minella, A. C. & Green, P. L. (2000)J. Virol. 74, 2655–2662.
19. Giam, C. Z. & Xu, Y. L. (1989) J. Biol. Chem. 264, 15236–15241.20. Goren, I., Semmes, O. J., Jeang, K. T. & Moelling, K. (1995) J. Virol. 69,
5806–5811.21. Kwok, R. P., Laurance, M. E., Lundblad, J. R., Goldman, P. S., Shih, H.,
Connor, L. M., Marriott, S. J. & Goodman, R. H. (1996) Nature 380, 642–646.22. Kashanchi, F., Duvall, J. F., Kwok, R. P. S., Lundblad, J., Goodman, R. &
Brady, J. N. (1998) J. Biol. Chem. 51, 34646–34652.23. Harrod, R., Tang, Y., Nicot, C., Lu, H. S., Vassilev, A., Nakatani, Y. & Giam,
C.-Z. (1998) Mol. Cell. Biol. 18, 5052–5061.24. Jiang, H., Lu, H., Schiltz, R. L., Pise-Masison, C. A., Ogryzko, V. V., Nakatani,
Y. & Brady, J. N. (1999) Mol. Cell. Biol. 19, 8136–8145.25. Harrod, R., Kuo, Y.-L., Tang, Y., Yao, Y., Vassilev, A., Nakatani, Y. & Giam,
C.-Z. (2000) J. Biol. Chem. 16, 11852–11857.26. Georges, S. A., Giebler, H. A., Cole, P. A., Luger, K., Laybourn, P. J. & Nyborg,
J. K. (2003) Mol. Cell. Biol. 23, 3392–3404.27. Yoshida, M. (2005) Oncogene 24, 5931–5937.28. Kashanchi, F. & Brady, J. N. (2005) Oncogene 24, 5938–5951.29. Hall, W. W. & Fujii, M. (2005) Oncogene 24, 5965–5975.30. Endo, K., Hirata, A., Iwai, K., Sakurai, M., Fukushi, M., Oie, M., Higuchi, M.,
Hall, W. W., Gejyo, F. & Fujii, M. (2002) J. Virol. 76, 2648–2653.31. Mahieux, R., Pise-Masison, C. A., Lambert, P. F., Nicot, C., De Marchis, L.,
Gessain, A., Green, P., Hall, W. & Brady, J. N. (2000) J. Virol. 74, 6866–6874.32. Semmes, O. J., Majone, F., Cantemir, C., Turchetto, L., Hjelle, B. & Jeang,
K. T. (1996) Virology 217, 373–379.33. Tripp, A., Liu, Y., Sieburg, M., Montalbano, J., Wrzesinski, S. & Feuer, G.
(2003) J. Virol. 77, 12152–12164.34. Tanaka, Y., Hayashi, M., Takagi, S. & Yoshie, O. (1996) J. Virol. 70, 8508–8517.35. Sieburg, M., Tripp, A., Ma, J.-W. & Feuer, G. (2004) J. Virol. 78, 10399–10409.36. Niinuma, A., Higuchi, M., Takahashi, M., Oie, M., Tanaka, Y., Gejyo, F.,
Tanaka, N., Sugamura, K., Xie, L., Green, P. L., et al. (2005) J. Virol. 79,11925–11934.
37. Feuer, G. & Green, P. L. (2005) Oncogene 24, 5996–6004.
38. Casoli, C., De Lerma Barbaro, A., Pilotti, E., Bertazzoni, U., Tosi, G. &Accolla, R. S. (2004) Blood 103, 995–1001.
39. Accolla, R. S., De Lerma Barbaro, A., Mazza, S., Casoli, C., De Maria, A. &Tosi, G. (2001) Trends Immunol. 22, 560–563.
40. Accolla, R. S., Mazza, S., De Lerma Barbaro, A., De Maria, A. & Tosi, G.(2002) Eur. J. Immunol. 32, 2783–2791.
41. Accolla, R. S., Jottrand-Bellomo, M., Scarpellino, L., Maffei, A., Carra, G. &Guardiola, J. (1986) J. Exp. Med. 164, 369–374.
42. Steimle, V., Otten, L. A., Zufferey, M. & Mach, B. (1993) Cell 75, 135–146.43. Reith, W., LeibundGut-Landmann, S. & Waldburger, J. M. (2005) Nat. Rev. 5,
703–806.44. Germain, R. N. (1994) Cell 76, 287–299.45. Caretti, G., Cocchiarella, F., Sidoli, C., Villard, J., Peretti, M., Reith, W. &
Mantovani, R. (2000) J. Mol. Biol. 302, 539–552.46. De Sandro, A. M., Nagarajan, U. M. & Boss, J. M. (2000) Mol. Cell. Biol. 20,
6587–6599.47. Masternak, K., Muhlethaler-Mottet, A., Villard, J., Zufferey, M., Steimle, V.
& Reith, W. (2000) Genes Dev. 14, 1156–1166.48. Zhu, X.-S., Linhoff, M. W., Li, G., Chin, K. & Ting, J. P.-Y. (2000) Mol. Cell.
Biol. 20, 6051–6064.49. Jabrane-Ferrat, N., Nekrep, N., Tosi, G., Esserman, L. & Peterlin, B. M. (2002)
Mol. Cell. Biol. 22, 5616–5625.50. Jabrane-Ferrat, N., Nekrep, N., Tosi, G., Esserman, L. & Peterlin, B. M. (2003)
Int. Immunol. 15, 467–475.51. Kretsovali, A., Agalioti, T., Spilianakis, C., Tzortzakaki, E., Merika, M. &
Papamatheakis, J. (1998) Mol. Cell. Biol. 18, 6777–6783.52. Fontes, J. D., Kanazawa, S., Jean, D. & Peterlin, B. M. (1999) Mol. Cell. Biol.
19, 941–947.53. Spilianakis, C., Papamatheakis, J. & Kretsovali, A. (2000) Mol. Cell. Biol. 20,
8489–8498.54. Kanazawa, S., Takashi, O. & Peterlin, B. M. (2000) Immunity 12, 61–70.55. Wei, P., Garber, M. E., Fang, S. M., Fisher, W. H. & Jones, K. A. (1998) Cell
92, 451–462.56. Tosi, G., Jabrane-Ferrat, N. & Peterlin, B. M. (2002) EMBO J. 21, 5467–5476.57. Dorn, A., Bollekens, J., Staub, A., Benoist, C. & Mathis, D. (1987) Cell 50,
863–872.58. Mantovani, R. (1999) Gene 239, 15–27.59. Pise-Masison, C. A., Dittmer, J., Clemens, K. E. & Brady, J. N. (1997) Mol. Cell.
Biol. 17, 1236–1243.60. Cressman, D. E., Chin, K.-C., Taxman, D. J. & Ting, J. P.-Y. (1999) Immunity
10, 163–171.61. Harton, J. A., Cressman, D. E., Chin, K.-C., Der, C. J. & Ting, J. P.-Y. (1999)
Science 285, 1402–1405.62. Hake, S. B., Masternak, K., Kammerbauer, C., Janzen, C., Reith, W. & Steimle,
V. (2000) Mol. Cell. Biol. 20, 7716–7725.63. Cressman, D. E., O’Connor, W. J., Greer, S. F., Zhu, X.-S. & Ting, J. P.-Y.
(2001) J. Immunol. 167, 3626–3634.64. Kretsovali, A., Spilianakis, C., Dimakopoulos, A., Makatounakis, T. & Pa-
pamatheakis, J. (2001) J. Biol. Chem. 276, 32191–32197.65. Raval, A., Weissman, J. D., Howcroft, T. K. & Singer, D. S. (2003) J. Immunol.
170, 922–930.66. Meertens, L., Chevalier, S., Weil, R., Gessain, A. & Mahieux, R. (2004) J. Biol.
Chem. 279, 43307–43320.67. Smith, M. R. & Green, W. C. (1991) J. Clin. Invest. 88, 1038–1042.68. Meertens, L., Pise-Masison, C., Quere, N., Brady, J., Gessain, A. & Mahieux,
R. (2004) Oncogene 23, 5447–5458.69. Sisk, T. J., Gourley, T., Roys, S. & Chang, C.-H. (2000) J. Immunol. 165,
2511–2517.70. Zhu, X.-S. & Ting, J. P.-Y. (2001) Mol. Cell. Biol. 21, 7078–7088.71. Nozell, S., Ma, Z., Wilson, C., Shah, R. & Benveniste, E. N. (2004) J. Biol.
Chem. 279, 38577–38589.72. Yee, C. S., Yao, Y., Li, P., Klemsz, M. J., Blum, J. S. & Chang, C. H. (2004)
J. Immunol. 172, 5528–5534.73. Blanco, J. C., Minucci, S., Lu, J., Yang, X. J., Walker, K. K., Chen, H., Evans,
R. M., Nakatani, Y. & Ozato, K. (1998) Genes Dev. 12, 1638–1651.
12866 � www.pnas.org�cgi�doi�10.1073�pnas.0601589103 Tosi et al.
Copyright © 2022 FDOKUMEN




















