
INFECTIVITY AND HOST SPECIFICITY OF T. DANILEWSKYI STRAIN FCC-1
Upload
independentCategory
view
0download
0
IC
MV*U
BmrptorehtMpthgwpxRasaiCeCigdCIf
Pin5cdit
GASTROENTEROLOGY 2003;125:1203–1218
nfectivity Enhanced, Cyclooxygenase-2 Promoter-Basedonditionally Replicative Adenovirus for Pancreatic Cancer
ASATO YAMAMOTO,*,‡,§,¶,� JULIA DAVYDOVA,*,‡,§,¶,� MINGHUI WANG,*,‡,§,¶,� GENE P. SIEGAL,§,#,�
ICTOR KRASNYKH,*,‡,§,¶,� SELWYN M. VICKERS,¶ and DAVID T. CURIEL*,‡,§,¶,�
Division of Human Gene Therapy, Departments of ‡Medicine, §Pathology, ¶Surgery, and #Cell Biology, and �the Gene Therapy Center,niversity of Alabama at Birmingham, Birmingham, Alabama
cdriptaosiw
sahT
ackground & Aims: Pancreatic cancer is one of theost aggressive human malignancies. Conditionally
eplicative adenoviruses (CRAds) have shown someromise in the treatment of cancers. However, to date,heir application for pancreatic cancer has met severalbstacles: one is lack of a good control element toegulate replication, and the other is relatively low ad-noviral infectivity. Thus, we constructed infectivity en-anced cyclooxygenase (COX)-2 promoter-based CRAdso develop a safe and effective therapeutic modality.ethods: The CRAds were designed to achieve COX-2romoter-controlled E1 expression for regulated replica-ion (COX-2 CRAds). The infectivity-enhanced CRAds alsoave an RGD-4C motif in the adenoviral fiber-knob re-ion. The selectivity and efficacy of these constructsere analyzed with cell lines in vitro. The in vivo thera-eutic effect and viral replication were analyzed with aenograft model. Pathology of the major organs and E1NA levels in the liver were also studied after systemicdministration. Results: The COX-2 CRAds showed aelective cytocidal effect in vitro in COX-2–positive cellsnd killed most of the pancreatic cancer cells. In vivo,ntratumoral administration of the infectivity-enhancedOX-2 CRAds (109 particles) showed a strong antitumorffect comparable to wild-type virus, whereas the COX-2RAds without infectivity enhancement showed a lim-
ted effect. Viral replication was confirmed in the xeno-raft tumors. Systemic administration did not cause anyetectable toxicity; the E1 RNA level in the liver afterOX-2 CRAd administration was minimal. Conclusions:
nfectivity-enhanced COX-2 CRAd is a promising agentor the treatment of pancreatic cancer.
ancreatic cancer is one of the most aggressive anddevastating human malignancies. Its aggressiveness
s represented by a tightly matched number of estimatedew cancer cases and actual cancer deaths,1 and the-year survival rate is dismal (�2%).2 In addition, pan-reatic cancer ranks as the fourth leading cause of cancereath and the eighth most frequent type of solid tumorn North America.1 So far, neither early detection norreatment of advanced disease is possible. Ninety-one
ercent of lesions are unresectable at the time of diag-osis, resulting in an average survival time of 4–5onths.3 Gemcitabine is currently the most effective
hemotherapeutic agent for pancreatic cancer, showing aetter 1-year survival rate when compared with 5-flu-rouracil.4 However, even after the introduction of gem-itabine, the most advanced combination chemotherapyesults in a median survival of �1 year.5 This clearlyndicates that development of new modalities is needed,nd gene therapy represents one such strategy.
Adenoviral vectors (Ads) have been used in a largeumber of gene therapy approaches due to their capacityo accomplish effective in vivo gene delivery.6 However,he numbers of cancer gene therapy clinical trials per-ormed to date have fallen short with respect to initialxpectations of demonstrable therapeutic outcomes. Ofote, these trials have shown limited tumor transductionrequencies and have also suggested that this limitationay represent a fundamental barrier to realizing the
enefits of cancer gene therapy.7–10 Notably in the fieldf gastrointestinal cancers (especially in pancreatic can-ers), the transduction efficiency of Ads is suboptimalue to the limited expression of the primary adenoviraleceptor (Coxsackie-adenovirus receptor [CAR]).11 Thensufficient infectivity is a critical issue for clinical ap-lication because it leads to a higher administrative doseo achieve the requisite level of gene transfer for a ther-peutic outcome. At a high dose, there is increased riskf a vector-induced innate immune reaction and toxicide effects,12–17 which have resulted in a lethal systemicnflammatory response syndrome in the case of a patientith an ornithine transcarbamylase deficiency.18
Abbreviations used in this paper: Ad, adenoviral vector; CAR, Cox-ackie-adenovirus receptor; CMV, cytomegalovirus; CRAd, condition-lly replicative adenovirus; COX, cyclooxygenase; GAPDH, glyceralde-yde-3-phosphate dehydrogenase; PCR, polymerase chain reaction;SP, tumor-specific promoter; vp, viral particles.
© 2003 by the American Gastroenterological Association0016-5085/03/$30.00
doi:10.1053/S0016-5085(03)01196-X
pnmcbocria
nttf
enfmbo
We have endeavored to overcome this problem byrational engineering of Ad agents. The most widely usedvectors have been rendered replication incompetent, andeffective tumor transduction has been limited. In con-trast to conventional nonreplicative vectors, with a ther-apeutic end point dependent on the transgene expression,conditionally replicative adenoviruses (CRAds) have beendesigned to replicate selectively in tumor cells. Theprogeny virus can then infect neighboring cells andspread multiplicatively to eventually cause oncolysis ofthe whole tumor.19,20 Initially, several CRAds with amutation or deletion in the E1 region were developed(e.g., dl1520/ONYX-015, Ad�24)21,22 and used in clin-ical trials. Also, tumor-specific promoters (TSPs) havebeen applied to achieve a tumor-selective replication ofthe adenoviral agents. Such an outcome has been accom-plished by restricting the expression of key adenoviralgenes with a TSP.6,23–26 For cancer of the pancreas, anumber of candidate TSPs have been proposed,27–33 butnone of the described promoters has exhibited the levelsof activity and specificity required to develop CRAds foreradication of pancreatic cancer.
In the field of gastrointestinal cancers, cyclooxygenase(COX)-2 is frequently expressed in neoplastic tissues andplays an essential role in many aspects of cancer progres-sion both directly and indirectly.34–36 Especially in thefield of pancreatic cancer, the differential expression be-tween cancerous and normal pancreatic tissues, alongwith the in vitro antitumor effect of COX-2 inhibitors,have led to the recognition of the importance of COX-2as a potential target of anticancer therapy.37–42 We havebeen studying promoter-based targeting in adenoviralgene therapy and have established the COX-2 promoteras a promoter with an optimal activity profile for gas-trointestinal cancers.43,44 We have reported that theCOX-2 promoter in an adenoviral construct shows auniquely beneficial “tumor-ON/liver-OFF” profile forpancreatic cancers.45 These data suggested that theCOX-2 promoter is promising as a control element forthe construction of pancreatic cancer CRAds. In thisstudy, we constructed such CRAds by controlling E1gene expression with the COX-2 promoter.
In the context of infectivity enhancement via theaugmentation of viral binding, we have reported thatAds with the RGD-4C motif configured in the HI loopof the fiber-knob region showed superior infectivity incells showing low CAR expression compared with vec-tors with wild-type Ad5 fiber. This enhancement ismainly mediated by the binding of the RGD motif ontointegrins that are frequently overexpressed on the surfaceof the target cancer cells.46 Furthermore, our previous
studies have indicated that low CAR expression andintegrin overexpression is typically observed in pancre-atic cancers.11 When the RGD modification was appliedto another CRAd (�24),22 the viral replication and cy-tocidal effect was dramatically enhanced.47 These studiesclearly indicate that the RGD-4C motif-based infectivityenhancement should augment the therapeutic potentialof the COX-2 CRAds in pancreatic cancer.
In this study, we constructed CRAds controlled by theCOX-2 promoter and also enhanced its infectivity byRGD modification of the fiber protein. We evaluatedthese CRAds in vitro and in vivo to assess their potentialtherapeutic utility. These studies establish that an infec-tivity-enhanced, COX-2 promoter-driven CRAd is apromising agent for the therapy of pancreatic cancer.
Materials and MethodsCells and Animals
The MIA PaCa-2, Capan-1, Hs 766T, and PANC-1pancreatic cancer cell lines (CRL-1420, HTB-79, HTB-134,and CRL-1469; American Type Culture Collection, Manassas,VA) were grown in Dulbecco’s modified Eagle medium (Me-diatech, Herndon, VA) with 10% fetal calf serum (HyClone,Logan, UT). The A549 (COX-2–positive lung cancer cell line,CCL-185; American Type Culture Collection), MKN45(COX-2–positive gastric cancer cell line, JCRB0254; JapaneseCollection of Research Bioresources, Tokyo, Japan), andBT474 (COX-2–negative breast cancer cell line, HTB-20;American Type Culture Collection) cells were maintained inRPMI 1640 (Mediatech) containing 10% fetal calf serum. Themedium of BT474 cells was supplemented with bovine insulin(0.01 mg/mL; Life Technologies, Rockville, MD). The 911cells (a kind gift from Dr. Van Der Eb, Leiden University, TheNetherlands)48 were maintained in Dulbecco’s modified Eaglemedium containing 10% fetal calf serum. Each medium wasalso supplemented with penicillin (100 IU/mL) and strepto-mycin (100 �g/mL). Cells were incubated in a 37°C and 5%CO2 environment under humidified conditions.
Female C57BL/6 mice (Charles River, Wilmington, MA)and female athymic NCr-nu nude mice (Frederick CancerResearch, Frederick, MD) (6–8 weeks of age) were used in thein vivo experiments. All animals received humane care basedon the guidelines set by the American Veterinary Association.All of the experimental protocols involving live animals werereviewed and approved by the institutional Animal Care andUse Committee of the University of Alabama at Birmingham.
Vector Construction and StructureConfirmation
The CRAd genomes were constructed via homologousrecombination in Escherichia coli49,50 (Figure 1). The COX-2CRAds contain the nucleotide 1–358 sequence of Ad5 as aleft-end structure (containing left inverted terminal repeat and
1204 YAMAMOTO ET AL. GASTROENTEROLOGY Vol. 125, No. 4

viral packaging signal) and the COX-2 promoter-controlledE1 expression cassette in place of the original E1 region of theAd genome. Except for RGD-4C insertion into the Ad fiber-knob region, other parts (corresponding to Ad5 nucleotides3511 and later) are identical to adenovirus type 5.46,51 Specif-ically, we constructed the shuttle vectors for COX-2 CRAdgeneration using pShuttleGL3Bcox-2L, a shuttle vector forCOX-2 promoter-driven luciferase expression vector AdCox2LLuc, as a starting material. This plasmid contains the �1432/�59 region of the COX-2 promoter derived from phPES2(Drs. Inoue and Tanabe at the National Cardiovascular CenterResearch Institute, Suita, Japan52,53) and a simian virus 40polyadenylation signal (see Yamamoto et al.43 for details). ThisCOX-2 promoter has shown high selectivity in an Ad config-uration.43 First, to restore the adenoviral protein IX promoter,the fragment nucleotides 3511–3924 containing intact proteinIX promoter was amplified by polymerase chain reaction(PCR) using a high-fidelity polymerase (LA Taq; PanVera/TaKaRa, Madison, WI) with primers 3511-Hc-S (5� gcgtcga-cagatcttgtactgaaatgtgtgggcgtggctt 3�) and 3511-Hc-AS (5�gtgccaaaagagccgtcaactt 3�) and treated with SalI and HincII.Then, the SalI–HincII region of the pShuttleGL3Bcox-2L wasreplaced with the new fragment. Next, the KpnI site at the 5�end of the COX-2 promoter was converted to a SalI site byusing a SalI linker for later construction of vectors with the E1expression cassette in reverse orientation. The E1 region (Ad5nucleotides 556–3512) was amplified by PCR using a high-fidelity polymerase (LA Taq) with primers E1-Hd-S (5�cccaagcttgaaaatgagacatattatctggcacgga 3�) and E1-Xba-AS (5�gctctagaacctcaatctgtatcttcatcgctaga 3�). After confirmation ofthe sequence, the 3� side 1/3 of E1 gene (HindIII–XbaI frag-ment, from nucleotide 2805 to the 3� end) was cloned into theshuttle vector in place of the luciferase gene (HindIII–XbaI),and then the rest of the E1 gene (HindIII–HindIII fragment,from the 5� end to nucleotide 2805) was cloned into theHindIII site of the resultant vector, resulting in a pShuttlecox-2L E1 F. The vector with the E1 expression cassette in theopposite direction was constructed by cleavage and religationwith 2 SalI sites at both ends of the E1 expression cassette
(pShuttle cox-2L E1 R). After cleavage with PmeI, the shuttlevectors were recombined with Ad5 DNA and ClaI linearizedpVK50346 to generate the CRAd genomes with wild-typefiber and RGD-modified fiber, respectively. The resultant plas-mids encoding COX-2 CRAds were linearized with PacI andtransfected into 911 cells using Superfect (Qiagen, Valencia,CA).
The structures of the CRAds were analyzed for severalcritical vector components by PCR. The virus (5.0 � 108 viralparticles [vp]) was processed with a blood DNA kit (Qiagen),and 1/50 of the DNA solution was analyzed by PCR forvarious regions with Taq polymerase (Qiagen). Thermal cy-cling conditions were as follows: initial denaturation, 5 min-utes at 95°C; 25 cycles of 30 seconds at 95°C, 30 seconds at55°C, and 1 minute at 72°C; and a final extension for 10minutes at 72°C. The primers used for each region were asfollows: forward direction CRAd (392 base pairs) (CRA-dITR[S] 5� GATAATGAGGGGGTGGAGTTTGTG 3� andCRAdcoxL[AS] 5� CAGTTATGTCCTAAGTCCTTAGCAT-TACA 3�), wild-type Ad5 (485 base pairs) (CRAdITR[S] andCRAdWt[AS] 5� GAAAACTCTACTCGCTGGCACTCA 3�),COX-2 promoter (405 base pairs) (COX2p check [S] 5�CCCATCCAAGGCGATCAGTC 3� and COX2p check [AS]5� GACGTGCTCCTGACGCTCACTGCAA 3�), E1a (338base pairs) (E1a [S] 5� GAGACATATTATCTGCCACG-GAGG 3� and [AS] 5� TTGGCATAGAAACCGGACCCA-AGG 3�), and fiber-knob region (wild type, 247 base pairs;RGD, 274 base pairs) (fiber up 5� CAAACGCTGTTGGATT-TATG 3� and fiber down 5� GTGTAAGAGGATGTGGC-AAAT 3�).
Wild-type Ad5 (Ad5) and its RGD-modified version(Ad5wtRGD, generated from pVK50346) were used as ubiq-uitous replication control vectors, whereas AdCMVLuc (E1-deleted nonreplicative luciferase expression vector)43 and itsRGD-modified version (RGDCMVLuc) were used as nonrep-licative controls in the CRAd analysis.
For infectivity-enhancement analysis, a cytomegalovirus(CMV) promoter-driven luciferase expression vector with theRGD modification (Ad5lucRGD)46 and its counterpart withwild-type fiber (Ad5Luc1) were used. These 2 vectors areidentical except for the HI loop of the fiber-knob region.
The viruses were propagated in the adenovirus packagingcell line 911 and purified by double CsCl density gradientcentrifugation, followed by dialysis against phosphate-bufferedsaline with 10% glycerol. The vectors were titrated by plaqueassay, and vp number was determined spectrophotometricallybased on absorbance at a wavelength of 260 nm.54 The vectorswere stored at �80°C until use.
Analysis of COX-2 RNA Status
The COX-2 RNA status of cell lines was analyzed byreverse-transcription PCR as described previously.43 Briefly,total RNA was extracted from semiconfluent cell culturesusing the RNeasy Mini RNA Extraction Kit (Qiagen) andanalyzed for COX-2 and glyceraldehyde-3-phosphate dehydro-genase (GAPDH) RNA with the GeneAMP RNA PCR Kit
Figure 1. Structure of CRAds. The vectors were constructed based onhuman adenovirus type 5 sequences. After restoration of the pIXpromoter region, a COX-2 promoter-driven E1 expression cassettewas inserted in both orientations, respectively (CRAdcox2F andCRAdcox2R). The infectivity-enhanced versions of COX-2 CRAds werealso generated (RGD CRAdcox2F and RGD CRAdcox2R). The E3 regionwas maintained in all vectors.
October 2003 ENHANCED COX-2 CRAD FOR PANCREATIC CANCER 1205

(Perkin-Elmer, Branchburg, NJ). Total RNA (500 ng) wasreverse transcribed with oligo(dT) primer and murine leuke-mia virus reverse transcriptase and amplified by PCR with 50nmol/L primers using a cycling program as described previ-ously.43 This reverse-transcription analysis has been confirmedto be semiquantitative.43
Analysis of Infectivity
The infectivity of the vectors with wild-type fiber andRGD-modified fiber was analyzed and compared by usingCMV promoter-driven luciferase expression vectors with the 2types of fibers. One day after plating 50,000 cells/well on a24-well plate, cells were infected with Ad5lucRGD andAd5Luc1 using 2 different amounts of virus per cell (500 and5000 vp/cell) in Dulbecco’s modified Eagle medium with 5%fetal calf serum (infection medium). Two hours later, theinfection medium was replaced with the appropriate completemedium. After 48 hours of cultivation, the cells were lysedwith Cell Culture Lysis Buffer (Promega, Madison, WI) andthe resultant lysates were analyzed with the Luciferase AssaySystem (Promega). The protein concentration was determinedwith the DC protein assay (Bio-Rad, Hercules, CA). All ex-periments were performed in triplicate.
In Vitro Analysis of Cytocidal Effect
The in vitro cytocidal effect of the COX-2 CRAds wasanalyzed by determining the viability of the cells with crystalviolet staining after infection. One day after 25,000 cells/wellwere plated on a 12-well plate, cells were infected at 0.01vp/cell with CRAds in infection medium. Two hours later, theinfection medium was replaced with the appropriate completemedium. After 10 days of cultivation, the cells were fixed with10% buffered formalin for 10 minutes and stained with 1%crystal violet in 70% ethanol for 20 minutes, followed bywashing 3 times with tap water and air drying.
Analysis of Viral Replication and Regulation
To analyze the viral replication and regulation ofCRAd selectivity, 2.5 � 104 A549 and BT474 cells wereinoculated onto 12-well plates and infected with 10 vp/cell ofeach vector the next day. Three days later, supernatant andcells were separately recovered for analysis. Encapsidated viralDNA in the supernatant was isolated as follows. The mediumwas recovered from the culture dish and spun in a microcen-trifuge at 3000 rpm for 5 minutes, and 360 �L of supernatantwas recovered. Then, 40 �L of 10� deoxyribonuclease buffer(60 mmol/L MgCl2 , 400 mmol/L Tris-Cl [pH 7.5], 20mmol/L CaCl2) and 10 U of deoxyribonuclease I (Roche Mo-lecular Biochemicals, Indianapolis, IN) were added, followedby incubation at 37°C for 1 hour. A total of 20 �L of 0.5mol/L ethylenediaminetetraacetic acid, 20 �L of 10% sodiumdodecyl sulfate, and 10 �L of proteinase K were added to themix, and the samples were then incubated at 52°C for 2 hours.After phenol-chloroform extraction, the DNA was ethanolprecipitated together with 10 �g of glycogen. After rinsingwith 70% ethanol and air drying, the DNA was dissolved with
100 �L of 10 mmol/L Tris-Cl, pH 7.4. For viral DNAisolation from the cells, after washing with phosphate-bufferedsaline twice, the cells were scraped from the plate and thesuspension was split in two. One half was directly processedwith a QIAamp Blood DNA Mini Kit (Qiagen) for totalcellular DNA, and the other half was processed by a spermine-HCl method for encapsidated viral DNA isolation.55 Briefly,the cells were resuspended with 50 �L of 100 mmol/L Tris-Cl(pH 9.0) and then lysed with 50 �L of DOC lysis buffer (0.4%sodium deoxycholate, 0.1 mol/L Tris-Cl [pH 9.0], 20% eth-anol) followed by incubation with 1 �L of 500 mmol/Lspermine on ice for 10 minutes. After centrifugation at 14,000rpm at 4°C for 4 minutes, the supernatant was recovered andprocessed with a QIAamp Blood DNA Mini Kit.
The viral DNA isolated by the aforementioned methods wasanalyzed by real-time PCR analysis to determine the adenovi-ral DNA copy number at the UAB Gene Therapy CenterCorrelative Labs.56 Briefly, 1 �L of extracted DNA sample wasadded to 9 �L per reaction of a master mix containing 1�Universal PCR Master Mix (Applied Biosystems, Foster City,CA), 100 nmol/L forward primer, 100 nmol/L reverse primer,1 nmol/L probe, and 0.025% bovine serum albumin in eachreaction capillary. For the standard curve to quantify the E4copy numbers, E4 template DNA with known copy number(108, 106, 104, and 102/�L) was also analyzed. All PCR reac-tions were performed using the LightCycler System (RocheMolecular Biochemicals) as described by the manufacturer.Thermal cycling conditions were as follows: initial denatur-ation, 10 minutes at 95°C, and 40 cycles of 15 seconds at95°C and 1 minute at 60°C. Data were analyzed with theLightCycler software. The primers were designed to detecta 68 – base pair region in E4: forward primer (5� GGAGT-GCGCCGAGACAAC 3�, nucleotides 34,007–34,024),reverse primer (5� ACTACGTCCGGCGTTCCAT 3�, nu-cleotides 34,074 –34,056), and 6-FAM–labeled probe(6FAM-TGGCATGACACTACGACCAACACGATCT-TAMRA, nucleotides 34,054 –34,027).
To compare the promoter property in COX-2 CRAds withdifferent expression-cassette orientations, E1 RNA levels wereanalyzed in cells infected with CRAdcox2F and CRAdcox2R.A549, a COX-2–positive control cell line, was infected withthe aforementioned viruses (500 vp/cell), and the total RNAwas isolated with an RNAeasy Mini Kit (Qiagen) after 24hours of incubation. The RNA samples were analyzed withTaqMan EZ reverse-transcription PCR kit (Applied Biosys-tems). Briefly, 1 �L of RNA sample was added to 9 �L ofPCR mixture (1� TaqMan EZ buffer; 3 mmol/L ofMn[CH3COOH]2; 300 �mol/L of deoxyadenosine triphos-phate, deoxycytidine triphosphate, and deoxyguanosinetriphosphate; 600 �mol/L of deoxyuridine triphosphate; 100nmol/L of forward and reverse primers and probe; 0.1 U/�L ofrTth DNA polymerase; and 0.025% bovine serum albumin inribonuclease-free water) into each reaction capillary. Knownamounts of control E1a RNA standards (108, 106, 104, and102/�L) were also analyzed to obtain a standard curve toquantify the E1A copy numbers in the unknown samples.
1206 YAMAMOTO ET AL. GASTROENTEROLOGY Vol. 125, No. 4

Reverse-transcription PCR reactions were performed usingthe LightCycler System. Thermal cycling conditions includedreverse transcription (2 minutes at 50°C, 30 minutes at 60°C,and 5 minutes at 95°C) and 40 cycles of 20 seconds at 94°Cand 1 minute at 62°C. Data were analyzed using the Light-Cycler software. The control E1a RNA was prepared by invitro transcription (MAXIscript In Vitro Transcription Kit;Ambion, Austin, TX) with T7 polymerase from a cloned E1atemplate as described by the manufacturer. TaqMan primersand probe designed for the E1a gene were as follows: forwardprimer, 5� AACCAGTTGCCGTGAGAGTTG 3�; reverseprimer, 5�CTCGTTAAGCAAGTCCTCGATACAT 3�; 6-FAM–labeled probe, 5� CACAGCCTGGCGACGCCCA-TAMRA 3�.
In Vivo Antitumor Effect
To analyze the antitumor effect of the CRAds in an invivo model, 2 � 107 Hs766T cells were inoculated subcuta-neously into the flank of female athymic NCr-nu nude mice,and a single dose (109 vp) of CRAds or control viruses wasinjected into the tumors after they reached a maximum diam-eter of 6–8 mm. Hs766T cells were selected because this cellline produced the most consistent subcutaneous tumors amongthe cells tested. The condition of the mice was monitoreddaily, and the tumor diameter was measured twice a week withcalibers. The tumor volume was calculated using the followingformula: Tumor Volume � Width2 � Length/2. The micewere killed 32 days after the viral injection in accordance withthe institutional approved animal experimental protocol due tothe volume of the tumors in the control group.
In Vivo CRAd Replication
To investigate the in vivo replication of the CRAds,intratumoral Ad DNA copy number was quantified at varioustime points, and the expression of adenoviral hexon protein inthe tumor was analyzed by immunostaining. Hs766T subcu-taneous xenografts were established in nude mice by thepreviously mentioned procedure, and intratumoral injectionsof COX-2 CRAds and control vectors (1010 vp) were deliveredto established nodules. The mice were killed on days 1, 5, 10,and 15, and the tumor specimens were cut in half at the center;the first half was quickly frozen with dry ice and kept at�80°C for DNA isolation, and the second half was fixed withbuffered formaldehyde for immunostaining. For DNA quan-titation, the tumor specimens were processed with a DNeasyTissue Kit (Qiagen). Each DNA sample (20 ng) was analyzedby real-time quantitative PCR analysis for adenovirus E4 copynumber using the aforementioned procedure at the UAB GeneTherapy Center Correlative Labs. The result is shown as ad-enoviral copy number per microgram DNA. Immunostainingof adenoviral hexon protein was performed as described previ-ously.57 Briefly, after fixation (buffered formaldehyde) andcryoprotection (15% sucrose, 0.1 mol/L phosphate buffer, pH7.2), the specimens were frozen and cut into 15-�m slices. Theslices were embedded onto silane-coated slides and stainedwith anti-hexon goat polyclonal antibody (primary) and Alexa
Fluor 488–labeled donkey anti-goat immunoglobulin G (sec-ondary) and counterstained with Hoechst 33342.57
In Vivo Toxicity Study
To investigate the potential toxicity of the COX-2CRAds following systemic administration, each group ofCRAds and control viruses was injected into the tail veins offemale C57BL/6 mice (Charles River). The mice were carefullyobserved for 5 days and killed under approved protocols. Forhistopathologic analysis, the major organs were fixed with10% buffered formalin, paraffin-embedded, and cut at 4-�mthickness, followed by deparaffinization and staining withH&E under standard conditions. To analyze the E1 RNA level,total RNA from the liver was extracted. The liver tissue wasfrozen on dry ice, avoiding contamination, and stored at�80°C until assayed. On the day of analysis, tissues wereground into a fine powder with a pestle and mortar in anethanol/dry ice bath and total RNA was extracted with anRNAeasy Mini Kit (Qiagen). After adjusting the concentra-tion to 100 ng/�L, 1 �L of each RNA sample was analyzedwith the same E1a RNA quantitative PCR procedure as pre-viously described.
Statistics
The data were analyzed with Smirnov’s extreme valuetest, and Student t test was used to compare different groupsof data as indicated. A 2-tailed P value of �0.05 was consid-ered significant.
ResultsAttributes of COX-2 CRAds
The vectors shown in Figure 1 were constructedand amplified using 911 cells. The yields were between0.5 and 1.5 � 1012 vp (per 20 of 15-cm dishes), com-parable to the yields of ordinary E1-deleted vectorsgrown in 911 cells. The vp/plaque-forming unit ratioswere in the range of 24–100. The vector structure wasconfirmed by PCR of the viral DNA. All vectors lackedthe wild-type left-end sequence (Figure 2A ). This indi-cated the absence of the wild-type vector as a result ofundesired homologous recombination during vectorpropagation. These vectors possessed the E1a gene andthe COX-2 promoter sequence as part of the E1 expres-sion cassette (Figure 2B and C ). The 392–base pairfragment was amplified from CRAdcox2F andRGDCRAdcox2F with the sense primer correspondingto the left inverted terminal-repeat sequence and theantisense primer corresponding to the COX-2 promotersequence (Figure 2D). In the analysis of the fiber region,the vectors with wild-type Ad5 fiber (CRAdcox2F andCRAdcox2R) gave a signal at 247 base pairs, whereas thevectors with the RGD-modified fiber (RGDCRAdcox2Fand RGDCRAdcox2R) yielded a band at 274 base pairs
October 2003 ENHANCED COX-2 CRAD FOR PANCREATIC CANCER 1207

(Figure 2E ), representing the 9–amino acid insertion inthe HI loop. These data confirmed the structural accu-racy of the CRAd constructs.
COX-2 RNA Status of the Pancreatic CancerCell Lines
The COX-2 RNA status of the cell lines used forthis experiment was analyzed by reverse-transcriptionPCR (Figure 3). Because the primers were designed tohave 3 introns between them, the signal detected at 723base pairs represented a complementary DNA sequencethat was reverse transcribed from RNA. The BT474breast cancer cell line (COX-2–negative control) wasappropriately negative for COX-2 RNA, whereas theMKN45 gastric cancer cell line was strongly positive.The pancreatic cancer cell lines Hs766T and Capan-1were positive for COX-2 RNA, whereas MiaPaca-2 andPanc-1 were negative for COX-2 messenger RNA. TheGAPDH RNA levels analyzed by reverse-transcriptionPCR (Figure 3B) were the same among all the cellstested, serving as a control for RNA isolation and thereverse-transcription PCR procedure.
Enhancement of the Adenoviral Infectivityin Pancreatic Cancer Cells by Incorporationof RGD-4C
The infectivity enhancement was analyzed usingCMV promoter-driven luciferase expression vectors. Thevectors Ad5Luc1 and Ad5lucRGD have the same struc-ture except that the fiber of the latter vector has a9–amino acid insertion in the HI loop corresponding tothe RGD modification. The luciferase activities withAd5lucRGD were 2.2-fold to 13.6-fold higher than thatof Ad5Luc1 in 4 of 4 human pancreatic cancer cell lines(n � 3, P � 0.005). Moreover, the enhancements in eachcell line were consistent regardless of the viral dose oninfection (50 and 500 vp/cell) (Figure 4). This indicatedthat the RGD modification confers infectivity enhance-ment in a wide range of pancreatic cancer cells.
In Vitro Oncolytic Potency of COX-2 CRAds
To analyze the oncolytic potency in conjunctionwith viral replication, we infected each cell line with alow dose of the CRAds (0.01 vp/cell) and analyzed thecells by crystal violet staining at day 10 (Figure 5). Inthis experiment, the result shows the presence of viralreplication and spread because the toxicity based on viralprotein expression (e.g., E1a) does not cause a detectablecytocidal effect at this low inoculation dose. In A549cells (COX-2 positive), all 4 CRAds (CRAdcox2F,CRAdcox2R, RGDCRAdcox2F, and RGDCRAdcox2R)showed a strong cytocidal effect comparable to wild-type virus (Ad5) and its RGD-modified version(Ad5wtRGD), whereas the nonreplicative luciferaseexpression vectors did not show any cytocidal effect.None of these COX-2 CRAds indicated a detectablecytocidal effect in COX-2–negative BT474 cells. Inpancreatic cancer cells, CRAds with the left-to-right
Figure 3. COX-2 RNA profile of the pancreatic cancer cells. The RNAof the cell lines used in these experiments was analyzed by reversetranscription followed by PCR. (A) The signal for COX-2 RNA wasdetected at the position of 723 base pairs with COX-2 sense (5�ggtctggtgcctggtctgatgatg 3�) and COX-2 antisense (5� gtcctttcaag-gagaatggtgc 3�) primers. (B) GAPDH RNA was detected with GAPDHsense (5� caactacatggtttacatgttccaa 3�) and GAPDH antisense (5�gccagtggactccacgacgt 3�) primers. Lane 1, BT474; lane 2, MKN45;lane 3, Hs766T; lane 4, Panc-1; lane 5, Capan-1; lane 6, MIA PaCa-2;lane 7, H2O.
Figure 2. Confirmation of the CRAd structure by PCR. To confirmstructural accuracy, the viral DNAs were analyzed using sets of prim-ers corresponding to several important regions of the virus. (A) De-tection of wild-type Ad5. The primers that recognize wild-type Ad5left-end sequence did not amplify any fragments from any of the 4COX-2 CRAds. (B) Detection of the E1a sequence. All 4 COX-2 CRAdshave the E1a sequence because they possess COX-2 promoter-drivenE1 expression cassettes. (C) Detection of the COX-2 promoter se-quence. All 4 CRAds contain the COX-2 promoter sequence to driveE1 expression. (D) Direction of the E1 expression cassette. A setof the primers recognizing the COX-2 promoter, placed in a left-to-right direction, amplified the sequence only in CRAdcox2F andRGDCRAdcox2F. (E) Fiber structure. The primers designed to distin-guish the presence of an RGD motif in the HI-loop region amplified a247–base pair fragment from CRAdcox2F and CRAdcox2R, whereasthey amplified a 274–base pair fragment containing a 9–aminoacid insertion from RGDCRAdcox2F and RGDCRAdcox2R. Lane 1,CRAdcox2F; lane 2, RGDCRAdcox2F; lane 3, CRAdcox2R; lane 4,RGDCRAdcox2R.
1208 YAMAMOTO ET AL. GASTROENTEROLOGY Vol. 125, No. 4

direction E1 expression cassettes (CRAdcox2F andRGDCRAdcox2F) exhibited a cytocidal effect in 4 of4 cell lines, whereas the CRAds with the right-to-leftdirection E1 expression cassette (CRAdcox2R andRGDCRAdcox2R) indicated a cytocidal effect in 2 of4 cell lines. In the context of infectivity enhancement,the infectivity-enhanced vectors showed more cyto-cidal effect in pancreatic cancer cells in general. How-ever, under this in vitro condition, the magnitudes ofthe enhancement and the time points that showed thelargest difference were variable depending on each celland CRAd. Microscopically, the cells undergoing cy-tocidal effect showed the typical characteristic of anadenoviral cytopathic effect (data not shown).
Regulation of the COX-2 CRAd Replication
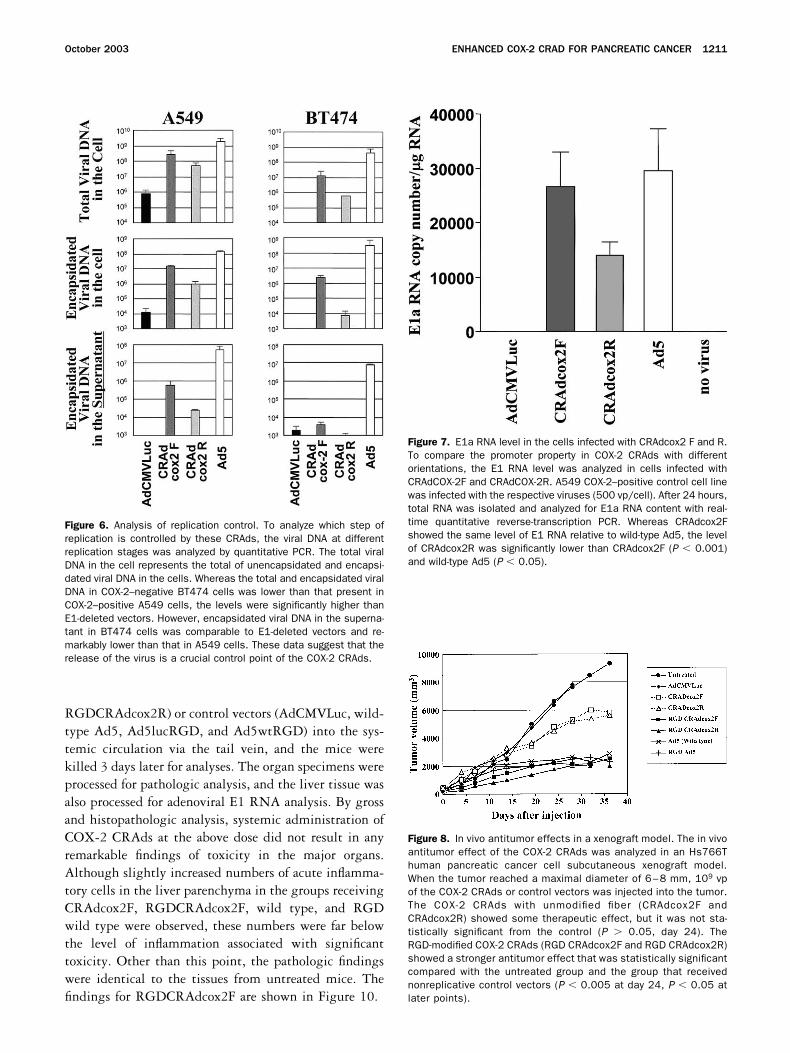
To determine which step(s) plays a major role inthe replication control of the COX-2 CRAds, we quan-tified the viral DNA in 3 different stages of viral repli-cation. The first was the total cellular DNA representingvirion encapsidated and unencapsidated DNA in thecells, the second was the encapsidated DNA in the cells,and the third was the encapsidated DNA in the culturemedium (Figure 6). The total and encapsidated viral
DNA levels after CRAd infection were lower than that ofwild-type controls in both COX-2–positive and –nega-tive cells (P � 0.05), and those in COX-2–negative cellswere lower than those in COX-2–positive cells. Thistendency was more remarkable in CRAdcox-2R thanCRAdcox-2F. However, these copy numbers were stillsignificantly higher than nonreplicating AdCMVLuc(P � 0.05). The encapsidated CRAd DNA (CRAdcox2Fand R) in the supernatant in BT474 cells was remarkablylower than that in A549 cells (P � 0.02) and was as lowas an E1-deleted control virus (P 0.1). These datasuggest that COX-2 promoter-controlled E1 expressionaffects multiple steps of viral replication and that therelease of the virus was one of the crucial control pointsin this CRAd type. The same experiment with RGD-modified vectors showed the same tendency.
To analyze the effect of the orientation of E1 expres-sion cassette on COX-2 promoter activity, A549 COX-2–positive control cells were infected with CRAdcox2Fand R vectors. After 24 hours, the total RNA wasisolated from the cells and the E1 RNA level was ana-lyzed (Figure 7). Whereas CRAdcox2F showed the samelevel of E1 RNA compared with wild-type Ad5 (P �0.32), the RNA level of CRAdcox2R was significantlylower than CRAdcox2F (P � 0.001) and wild-type Ad5(P � 0.05). This result indicates that COX-2 promoter-driven E1 expression is lower when the cassette is placedin a right-to-left direction. We also tested the promoteractivity by using COX-2 promoter-driven luciferase ex-pression vectors with both orientations, but no differencewas observed between the 2 directions in these E1-deleted vectors (data not shown).
In Vivo Antitumor Effect of COX-2 CRAds
The antitumor effect was analyzed in vivo usingHs766T pancreatic cancer subcutaneous xenografts. Af-ter establishment of the tumor (6–8 mm), the CRAds(109 vp) were injected into the tumors. This dose is morethan 2 orders of magnitude lower than the dose we havebeen using for suicide gene therapy with nonreplicativevectors. Compared with the untreated group (day 24,n � 8, 6596 3319) and the group that received anonreplicative luciferase expression vector (day 24, n �6, 6369 4182), the groups with CRAdcox2F (day 24,n � 6, 4750 4144) and CRAdcox2R (day 24, n � 6,4510 2279) showed tumor growth suppression butthe effect was not statistically significant (P 0.05).However, on day 24, RGDCRAdcox2F (day 24, n � 8,2215 1666) and RGDCRAdcox2R (day 24, n � 6,1739 1280) showed stronger tumor growth suppres-sion comparable to that of the wild-type virus, withstatistical significance when compared with the un-
Figure 4. Infectivity enhancement by RGD modification in pancreaticcancer cells. The infectivity enhancement was analyzed using CMVpromoter-driven luciferase expression vectors with and without RGDmodification. Two days after infection at 50 and 500 vp/cell, the cellswere analyzed for luciferase expression. The result is shown as apercentage infectivity of that of Ad5Luc1 used as the control. In allpancreatic cancer cell lines tested, RGD-modified vectors showed astatistically significant (n � 3, P � 0.005) increase in transduction(2.2-fold to 13.6-fold). This was seen consistently regardless of thevp/cell used for the experiment.
October 2003 ENHANCED COX-2 CRAD FOR PANCREATIC CANCER 1209

treated group (day 24, P � 0.005) and the group withthe nonreplicative control (day 24, P � 0.025). Thisdifference was also observed at later time points (P �0.05) (Figure 8). This experiment was repeated 3 timesand showed the same tendency. Thus, whereas non-RGDCOX-2 CRAds possess unremarkable in vivo therapeuticeffect, the oncolytic potential of RGD-modified COX-2CRAds was statistically significant. Some tumors thatreceived COX-2 CRAds (regardless of RGD or non-RGD) experienced tumor necrosis and subsequent tumorregression, whereas the tumors of the untreated groupand the group with the nonreplicative control showedconsistent growth without any tumor necrosis.
In Vivo Replication of CRAds
In situ replication is the most important functionof CRAds. We tested the in situ replication of COX-2CRAds in the Hs766T xenograft model. COX-2 CRAds(CRAdcox2F and RGDCRAdcox2F) and control vectors(AdCMVLuc and Ad5) were injected intratumorally(1010 vp), and the tumors were analyzed for viral copynumber (Figure 9A ) and hexon immunostaining (Figure9B–E). Ten days after administration, intratumoral viral
copy number of all replicative vectors (except nonrepli-cative AdCMVLuc) increased by 2–3 orders of magni-tude (P � 0.02). Although the increase of the copynumber of COX-2 CRAds was slower than wild-typeAd5, the copy numbers of all 3 replicative virusesreached a plateau in 10–15 days.
To further show CRAd replication in the tumor, westained the tumor specimens on day 10 with anti-hexonantibody. Although there were no stained cells in thetumor with AdCMVLuc (Figure 9B), tumors withCRAdcox2F, RGDCRAdcox2F, and Ad5 contained cellsexpressing the hexon protein (Figure 9C–E, respectively).In the tumor with RGDcox2F (Figure 9D), there wastumor necrosis in the center and hexon staining was mostintense in areas adjacent to the tumor necrosis.
Minimal Toxicity of COX-2 CRAds inRodents
Although human adenoviruses do not replicate inmice, in vivo toxicologic studies were performed in miceto obtain preliminary data on toxicity due to systemicvector administration. We injected the CRAds (109 vp)(CRAdcox2F, CRAdcox2R, RGDCRAdcox2F, and
Figure 5. In vitro analysis of cytocidal effect. The COX-2 CRAds were analyzed for their cytocidal effect. The cells were infected at 0.01 vp/celland cultivated for 10 days. Cytotoxicity was assessed by crystal violet staining. The virus used in each well is indicated in the left lower panel.E1-deleted vectors (AdCMVLuc and RGDCMVLuc: nonreplicative) did not kill any cells. The wild-type viruses (Ad5 and Ad5wtRGD) killed all cells.Both COX-2 CRAds killed COX-2–positive control cells (A549), whereas neither killed COX-2–negative control cells (BT474). In pancreatic cancercells, the CRAds with the left-to-right expression cassette (CRAdcox2F and RGDCRAdcox2F) showed cytocidal effect in 4 of 4 cells, whereas thosewith the cassette in the opposite orientation (CRAdcox2R and RGDCRAdcox2R) killed only 2 of 4 cells. In the context of infectivity enhancement,RGD CRAds showed a slightly better cytocidal effect that the non-RGD counterparts.
1210 YAMAMOTO ET AL. GASTROENTEROLOGY Vol. 125, No. 4
RGDCRAdcox2R) or control vectors (AdCMVLuc, wild-type Ad5, Ad5lucRGD, and Ad5wtRGD) into the sys-temic circulation via the tail vein, and the mice werekilled 3 days later for analyses. The organ specimens wereprocessed for pathologic analysis, and the liver tissue wasalso processed for adenoviral E1 RNA analysis. By grossand histopathologic analysis, systemic administration ofCOX-2 CRAds at the above dose did not result in anyremarkable findings of toxicity in the major organs.Although slightly increased numbers of acute inflamma-tory cells in the liver parenchyma in the groups receivingCRAdcox2F, RGDCRAdcox2F, wild type, and RGDwild type were observed, these numbers were far belowthe level of inflammation associated with significanttoxicity. Other than this point, the pathologic findingswere identical to the tissues from untreated mice. Thefindings for RGDCRAdcox2F are shown in Figure 10.
Figure 7. E1a RNA level in the cells infected with CRAdcox2 F and R.To compare the promoter property in COX-2 CRAds with differentorientations, the E1 RNA level was analyzed in cells infected withCRAdCOX-2F and CRAdCOX-2R. A549 COX-2–positive control cell linewas infected with the respective viruses (500 vp/cell). After 24 hours,total RNA was isolated and analyzed for E1a RNA content with real-time quantitative reverse-transcription PCR. Whereas CRAdcox2Fshowed the same level of E1 RNA relative to wild-type Ad5, the levelof CRAdcox2R was significantly lower than CRAdcox2F (P � 0.001)and wild-type Ad5 (P � 0.05).
Figure 8. In vivo antitumor effects in a xenograft model. The in vivoantitumor effect of the COX-2 CRAds was analyzed in an Hs766Thuman pancreatic cancer cell subcutaneous xenograft model.When the tumor reached a maximal diameter of 6–8 mm, 109 vpof the COX-2 CRAds or control vectors was injected into the tumor.The COX-2 CRAds with unmodified fiber (CRAdcox2F andCRAdcox2R) showed some therapeutic effect, but it was not sta-tistically significant from the control (P 0.05, day 24). TheRGD-modified COX-2 CRAds (RGD CRAdcox2F and RGD CRAdcox2R)showed a stronger antitumor effect that was statistically significantcompared with the untreated group and the group that receivednonreplicative control vectors (P � 0.005 at day 24, P � 0.05 atlater points).
Figure 6. Analysis of replication control. To analyze which step ofreplication is controlled by these CRAds, the viral DNA at differentreplication stages was analyzed by quantitative PCR. The total viralDNA in the cell represents the total of unencapsidated and encapsi-dated viral DNA in the cells. Whereas the total and encapsidated viralDNA in COX-2–negative BT474 cells was lower than that present inCOX-2–positive A549 cells, the levels were significantly higher thanE1-deleted vectors. However, encapsidated viral DNA in the superna-tant in BT474 cells was comparable to E1-deleted vectors and re-markably lower than that in A549 cells. These data suggest that therelease of the virus is a crucial control point of the COX-2 CRAds.
October 2003 ENHANCED COX-2 CRAD FOR PANCREATIC CANCER 1211

Next, we analyzed the E1 RNA level in the liver toobtain key information regarding replication specificityof the COX-2 CRAds. Compared with the wild-typecontrols (wild-type and Ad5wtRGD), the E1 RNA levelsin the livers of COX-2 CRAd-treated animals, where theE1 gene is under the control of the COX-2 promoter,were much lower and near background level, regardlessof the fiber modification (Figure 11). Because the selec-tivity of the COX-2 CRAds is based on the selectivity ofE1 expression, this provides encouraging critical infor-mation with respect to the in vivo replication selectivityof the COX-2 CRAds.
DiscussionThe prognosis of pancreatic cancer is devastating,
showing minimal long-term survival.2,58 This fact has
motivated many efforts in the development of novelcancer therapeutic strategies. For almost 5 decades, var-ious replicative viruses have been tried in the context ofcancer therapy.59–66 Recently, several conditionally rep-licative viral agents (adenovirus and herpes simplex vi-rus), designed with the advanced molecular and biolog-ical understanding of the viruses, have been tested inpancreatic cancer.61,67,68 In the field of adenovirus,dl1520/ONYX-015 virus showed good safety data in aphase I clinical trial. However, the therapeutic efficacy inpancreatic cancer was not as noteworthy as that seen inhead-neck cancers.69,70 Thus, the further development ofCRAds designed specifically for pancreatic cancer isneeded.
In this study, we designed CRAds based on COX-2promoter selectivity43,45 and enhanced the infectivity by
Figure 9. In vivo replication of COX-2 CRAds. To investigate the in vivo replication of the CRAds, the intratumoral Ad DNA copy number wasquantified while expression of adenoviral hexon protein was analyzed by immunostaining. After intratumoral injection of COX-2 CRAds and controlvectors (1010 vp) into Hs766T xenografts, the mice were killed on days 1, 5, 10, and 15. (A) Adenoviral DNA copy number. The DNA samplesisolated from tumor specimens were analyzed by real-time quantitative PCR analysis for adenovirus E4 copy number, and the result is shown asadenoviral copy number per microgram DNA. Ten days after administration, intratumoral viral copy number of replicative vectors (exceptnonreplicative AdCMVLuc) increased 2–3 orders of magnitude (P � 0.02) and reached a plateau by 10–15 days. (B–E) Hexon immunostaining.Immunostaining of adenoviral hexon protein was performed as described previously.57 Briefly, after fixation and cryoprotection, the specimenswere frozen and cut into 15-�m slices and stained with anti-hexon goat polyclonal antibody (primary) and Alexa Fluor 488–labeled donkeyanti-goat immunoglobulin G (secondary) and counterstained with Hoechst 33342. While there were no stained cells in (B) the tumor withAdCMVLuc, tumors with (C) CRAdcox2F, (D) RGDCRAdcox2F, and (E) Ad5 showed cells expressing hexon. (D) In the tumor with RGDcox2F, thereis necrosis in the center (*) and hexon staining is most intense adjacent to the necrosis area. The outer edge of the tumor is shown with arrowsin B–D. The edge is outside the field of view in E.
1212 YAMAMOTO ET AL. GASTROENTEROLOGY Vol. 125, No. 4

incorporating an RGD-4C motif in the HI loop of thefiber-knob region with the expectation of increased effi-cacy.11,46,47,51 During vector amplification, the COX-2CRAds produced consistent replication regardless of theexistence of the RGD modification, and the yield wascomparable to regular �E1 vectors. Also, the resultantviruses maintained their correct genomic structures andwere free from unconditionally replicative recombinantscontaining the wild-type E1 control region (Figure 2).These data indicate that these vectors fulfill the requiredfeatures for clinical grade vector production.
We have reported that the COX-2 promoter in anadenovirus vector delivers strong activity in all of theestablished and near primary pancreatic cancer cell linesthat we have tested.45 Interestingly, the reverse-tran-scription PCR analysis of COX-2 RNA in the pancreaticcancer cell lines showed that COX-2 RNA is negative in2 cell lines (Figure 3, MiaPaca-2 and Panc-1). Becausethe COX-2 promoter was confirmed to be active in these
cells (data not shown), there is a discrepancy between theCOX-2 RNA status and the activity of an extrinsicallydelivered COX-2 promoter. In pancreatic cancer cells,structural rearrangements in chromosome 1 are one ofthe most frequently observed phenomena,71 and theCOX-2 gene and its promoter are in the 1q25.2-3 regionof human chromosome 1 (see D28235 by Tanabe, DDBJ/EMBL/GenBank). The functional loss of the COX-2gene or its promoter, a result of its deletion or thealteration of adjacent regions, could lead to the lack ofCOX-2 expression in these cells but would not necessar-ily influence the positive control signal of the COX-2promoter. Thus, this might explain the discrepancy be-tween the COX-2 RNA status and the COX-2 promoteractivity profile in some cells.
In the context of infectivity enhancement, we reeval-uated infectivity enhancement using a new set of vectorswith and without the RGD-4C motif in the HI-loopregion. In these CMV promoter-driven luciferase expres-
Figure 10. Microscopic analysis of major organs after systemic CRAd administration. Major organs were analyzed histopathologically aftersystemic administration of CRAds. Three days after injection of the CRAds (109 vp) (CRAdcox2F, CRAdcox2R, RGDCRAdcox2F, orRGDCRAdcox2R) and control vectors (AdCMVLuc, wild-type Ad5, RGDAdCMVLuc, and Ad5wtRGD), the major organs were analyzed histologically.Although a slight increase in inflammatory cells was noted in the parenchyma of the liver for the groups that received CRAdcox2F, RGDCRAdcox2F,wild type, and Ad5wtRGD, this amount was far below the level of significant hepatotoxicity. Other than this feature, the histologic findings wereidentical among all groups examined. Importantly, there were no remarkable findings of toxicity in any of the major organs. The findings withRGDCRAdcox2F, which indicated the most promising result in the therapeutic experiments, are shown in Figure 9 (A, liver; B, lung; C, spleen; D,kidney).
October 2003 ENHANCED COX-2 CRAD FOR PANCREATIC CANCER 1213

sion vectors, the genetic structure is identical except forthe HI loop. Additionally, infection with Ads (with andwithout RGD modification) did not affect the CMVpromoter activity (data not shown). Thus, luciferase ac-tivity obtained from this experiment truly represents theinfectivity of each vector. As expected from the fact thatthe pancreatic cell lines used in this experiment expressminimal CAR but much integrin(s) (either �v�3 or�v�5 integrin),11,46 the RGD-modified vectors showedmore infectivity than the vectors with the wild-typefiber. In fact, in HepG2 (hepatocellular carcinoma) cellline, which does not express �v�3 or �v�5 integrin, theinfectivity enhancement with the RGD-modified vectorwas minimal (data not shown). This suggests the infec-tivity enhancement with RGD modification correlateswith �v�3 and �v�5 integrin expression.
The COX-2 CRAds showed an in vitro cytocidal effectselectively in COX-2–positive cells (Figure 4, BT474and A549), and the same cytocidal selectivity dependenton cellular COX-2 promoter status has been observed inmany other cell lines (unpublished data, October 2001 toJune 2003). Because COX-2 CRAds lack cytocidal effectin COX-2–negative cells (BT474), which shows highadenoviral infectivity, the oncolytic effect clearly de-pends on the COX-2 promoter status. In pancreaticcancer cell lines, the COX-2 CRAds with E1 expressioncassettes in the left-to-right direction (F) showed stron-ger cytocidal effect than those with the reverse-directioncassettes (R). This difference is believed to be due to the
difference in promoter strength because the COX-2 pro-moter placed in the left-to-right direction shows moreactivity than when it is placed in a right-to-left directionin these CRAds (Figure 7). Interestingly, this differencewas not observed in COX-2 promoter-driven luciferaseexpression vectors that lack the E1 region (data notshown). The adenoviral E1a protein plays a major role inthe activation of E2F, which can bind to its bindingregions present in the adenoviral enhancer/packagingsignal region in the left end of the Ad genome andactivate transcription.72 This may explain why the left-to-right cassette where the Ad enhancer/packaging signalregion exists upstream of the extrinsic COX-2 promotercan provide stronger promoter activity in E1-expressingvectors (CRAds). However, this is not the case for E1-deleted vectors, which do not express E1a. When COX-2promoter-controlled CRAds were combined with infec-tivity enhancement, RGD-modified CRAds clearlyshowed a greater cytocidal effect than the unmodifiedversions, although complete cytopathic effect of the en-tire cell culture in some wells makes the detection of thedifferential effect difficult.
To further characterize the mechanism of the selectivecytocidal effect, we quantified the viral DNA at differentstages of viral replication. Interestingly, total and encap-sidated viral DNA levels in the cells were significantlylower in both CRAds than that in wild-type adenovirus.This might be due to the replacement of the adenoviralE1 promoter region with the specific but constitutiveCOX-2 promoter, therefore disputing the expressiontiming of viral genes and possibly affecting viral repli-cation.72,73 However, total and encapsidated viral DNAlevels in COX-2–negative cells were still significantlyhigher than E1-deleted vector. Therefore, the clear-cutselectivity of COX-2 CRAds observed in Figure 5 cannotbe fully explained by control of DNA replication andviral packaging levels. On the other hand, the encapsi-dated viral DNA in the supernatant of COX-2–negativecells was remarkably lower than that in COX-2–positivecells and comparable to that of the E1-deleted vector(Figure 6). These data strongly support the COX-2CRAd selectivity observed in Figure 5. Taken together,these data suggest that the replication of the CRAdsbased on the promoter-controlled E1 expression is con-trolled at multiple points of viral replication and thatviral release is one of the crucial control points. TheRGD-modified vectors showed the same tendency. Thisresult agrees with the report that some degree of repli-cation of the DNA and packaging does take place in thecell with no (or minimal) E1 expression.74 Conversely,the release of the encapsidated virus may require a higher
Figure 11. E1 RNA level in the liver after systemic CRAd administra-tion. Five days after systemic CRAd administration of 109 vp, RNA wasisolated from the liver and analyzed by quantitative reverse-transcrip-tion PCR for the E1a gene. The result is indicated as E1a RNA copynumber per 1 �g total RNA. The E1a RNA level in COX-2 CRAds wasminimal compared with that of the wild-type Ad5 and was close to thebackground level.
1214 YAMAMOTO ET AL. GASTROENTEROLOGY Vol. 125, No. 4

amount of E1 expression in the cells. Because the spreadof the cytocidal effect requires the effective release of theprogeny viruses and their infection of the surroundingcells,20 it is reasonable to conclude that the in vitrocytocidal effect was observed only in COX-2–positivecells (Figure 5).
In the subcutaneous xenograft model of pancreaticcancer, the COX-2 CRAds showed a therapeutic effectafter intratumoral injection of 109 vp per tumor. Inprevious suicide gene therapy experiments with Adsexpressing HSV-TK, the minimum dose resulting in aclear therapeutic effect in the gastrointestinal cancersubcutaneous xenograft model with low CAR expressionwas approximately 109 plaque-forming units (approxi-mately 1011 vp) per site.43,75 Thus, the COX-2 CRAdsshowed an antitumor effect with a 2-order lower dose.This is clearly beneficial because the innate immuneresponse after high-dose administration can be avoided orminimized by reducing the administration dose.12–14 Inthe xenograft experiment, RGD-modified CRAdsshowed a much stronger antitumor effect than the fiber-unmodified CRAds. The benefit of the infectivity en-hancement was bigger than that seen in the in vitroexperiment. In clinical trials with replicative adenovi-ruses, local viral replication was minimally detected.70
This suggests that in vivo circumstances are much morestringent for adenoviral replication than in vitro. Thus,in this situation, infectivity enhancement of the CRAdsmay give a clear advantage to achieve a replication-basedcytocidal effect.
The analysis of CRAd replication in the tumor iscrucial to understand the in vivo functionality of theCOX-2 CRAds. We injected the COX-2 CRAds witheither wild-type or RGD fibers into the subcutaneousnodules of Hs766T pancreatic cancer cells and analyzedviral DNA copy number and hexon expression in thetumors. The DNA copy number of CRAds (CRAdcox2Fand RGD CRAdcox2F) and wild-type Ad5 increased 2–3orders of magnitude over time, whereas E1-deleted Ad-CMVLuc did not show any increase (Figure 9A ). Al-though the in situ replication of COX-2 CRAds wasslightly slower than Ad5, the DNA level of COX-2CRAds reached that of wild-type Ad5 by 10 days. Thetumor specimens at day 10 were stained with anti-hexonantibodies. As shown in Figure 9B–E, hexon-expressingcells were detected in the tumors with CRAdcox2F,RGDCRAdcox2F, and Ad5, whereas the tumor withAdCMVLuc indicated no hexon-expressing cells. Hexonstaining has been used in clinical trials76 to prove theexistence and replication of the adenovirus because thehexon gene is under control of the adenoviral major late
promoter, which is active late in the replication cycle.72
Expression of the gene under the major late promoterwould indicate that the virus had reached a late stage ofreplication. Thus, the hexon staining in a tumor infectedwith CRAds provides strong evidence of viral replicationin the tumor. The fact that hexon expression was mostintense in regions adjacent to tumor necrosis may repre-sent the active process of viral replication-based cytocidaleffect. These results provide strong evidence of intratu-moral replication of COX-2 CRAds.
So far, there is no practical rodent model to directlyanalyze adenoviral replication and replication-based tox-icity because human adenoviruses do not replicate inrodents in general.77 Although the toxicity study in micedoes not represent the actual toxicity based on viralreplication, regulatory agencies require these results aspart of a preclinical toxicologic study. After tail veininjection of 109 vp of CRAds, no toxicity-related eventwas observed in the major internal organs. These resultsindicate that COX-2 CRAds, including the RGD ver-sion, do not cause severe organ damage under nonrepli-cative conditions. Ad5 is reported to replicate in cottonrats.12,78 Because there is no other small animal modelpermissive for Ad replication, this system is attractive asa model for preclinical toxicologic studies although themechanism enabling human Ad5 replication in theseanimals has not been clarified. We are in the process ofsetting up a preclinical toxicologic study with the Na-tional Cancer Institute/Rapid Access to Intervention De-velopment Program to assess these reagents in this modelsystem.
In our CRAds, the E1 RNA level is a key determiningfactor of the replication because their replication selec-tivity depends on the selective expression of the E1 genemediated by the COX-2 promoter. Thus, analysis of theE1 RNA after administration into mice provides us withkey information about in vivo replication potency andtoxicity as a consequence. In this experimental system,most of the intravenously administered adenovirus local-izes to the liver, and the liver is the primary target ofadenoviral toxicity. After systemic injection of 109 vp ofthe CRAds, the E1 RNA level in the liver was close tobackground. This indicates that COX-2 CRAds main-tain replication control functionality in vivo, a key find-ing that predicates the relative nontoxicity of our CRAdagents.
In the present study, we have established a novelconditionally replicative adenovirus using the COX-2promoter to control its replication and shown that theincorporation of an RGD-4C motif into these CRAds canenhance the antitumor efficacy, especially in vivo. The
October 2003 ENHANCED COX-2 CRAD FOR PANCREATIC CANCER 1215

COX-2 CRAds presented a good selectivity profile invitro and in vivo. Thus, these CRAds have the potentialto be a clinically usable agent for pancreatic cancer. Also,the COX-2 CRAds are promising not only for localinjection but also for transcatheter intra-arterial admin-istration to target intrahepatic lesions, where a vector-derived cytocidal effect in the liver can cause safetyproblems79 that are avoided with a COX-2 controlledvector.43 To make gene therapy not only feasible but alsoclinically useful, strategies to ensure both key safety andefficacy end points are required. In this regard, the RGDCOX-2 CRAds, which take advantage of the COX-2promoter selectivity and RGD modification– enhancedinfectivity, are useful in achieving enhanced but selectivereplication to reduce nonspecific adverse effects and havewidespread application for many kinds of COX-2–posi-tive cancers.34,38,80–82
References1. Jemal A, Thomas A, Murray T, Thun M. Cancer statistics, 2002.
CA Cancer J Clin 2002;52:23–47.2. Rosewicz S, Wiedenmann B. Pancreatic carcinoma. Lancet 1997;
349:485–489.3. Sener SF, Fremgen A, Menck HR, Winchester DP. Pancreatic
cancer: a report of treatment and survival trends for 100,313patients diagnosed from 1985–1995, using the National CancerDatabase. J Am Coll Surg 1999;189:1–7.
4. Burris HA 3rd, Moore MJ, Andersen J, Green MR, Rothenberg ML,Modiano MR, Cripps MC, Portenoy RK, Storniolo AM, Tarassoff P,Nelson R, Dorr FA, Stephens CD, Von Hoff DD. Improvements insurvival and clinical benefit with gemcitabine as first-line therapyfor patients with advanced pancreas cancer: a randomized trial.J Clin Oncol 1997;15:2403–2413.
5. Gunzburg WH, Salmons B. Novel clinical strategies for the treat-ment of pancreatic carcinoma. Trends Mol Med 2001;7:30–37.
6. Hallenbeck PL, Chang YN, Hay C, Golightly D, Stewart D, Lin J,Phipps S, Chiang YL. A novel tumor-specific replication-restrictedadenoviral vector for gene therapy of hepatocellular carcinoma.Hum Gene Ther 1999;10:1721–1733.
7. Schmidt-Wolf G, Schmidt-Wolf IG. Human cancer and gene ther-apy. Ann Hematol 1994;69:273–279.
8. Herrmann F. Cancer gene therapy: principles, problems, andperspectives. J Mol Med 1995;73:157–163.
9. Seemayer TA, Cavenee WK. Molecular mechanisms of oncogen-esis. Lab Invest 1989;60:585–599.
10. Roth JA, Cristiano RJ. Gene therapy for cancer: what have wedone and where are we going? J Natl Cancer Inst 1997;89:21–39.
11. Wesseling JG, Bosma PJ, Krasnykh V, Kashentseva EA, BlackwellJL, Reynolds PN, Li H, Parameshwar M, Vickers SM, Jaffee EM,Huibregtse K, Curiel DT, Dmitriev I. Improved gene transfer effi-ciency to primary and established human pancreatic carcinomatarget cells via epidermal growth factor receptor and integrin-targeted adenoviral vectors. Gene Ther 2001;8:969–976.
12. Prince GA, Porter DD, Jenson AB, Horswood RL, Chanock RM,Ginsberg HS. Pathogenesis of adenovirus type 5 pneumonia incotton rats (Sigmodon hispidus). J Virol 1993;67:101–111.
13. Ginsberg HS, Prince GA. The molecular basis of adenoviruspathogenesis. Infect Agents Dis 1994;3:1–8.
14. Lieber A, He CY, Meuse L, Schowalter D, Kirillova I, Winther B,Kay MA. The role of Kupffer cell activation and viral gene expres-
sion in early liver toxicity after infusion of recombinant adenovirusvectors. J Virol 1997;71:8798–8807.
15. Nunes FA, Furth EE, Wilson JM, Raper SE. Gene transfer into theliver of nonhuman primates with E1-deleted recombinant adeno-viral vectors: safety of readministration. Hum Gene Ther 1999;10:2515–2526.
16. Schnell MA, Zhang Y, Tazelaar J, Gao GP, Yu QC, Qian R, Chen SJ,Varnavski AN, LeClair C, Raper SE, Wilson JM. Activation ofinnate immunity in nonhuman primates following intraportal ad-ministration of adenoviral vectors. Mol Ther 2001;3:708–722.
17. Raper SE, Haskal ZJ, Ye X, Pugh C, Furth EE, Gao GP, Wilson JM.Selective gene transfer into the liver of non-human primates withE1-deleted, E2A-defective, or E1-E4 deleted recombinant adeno-viruses. Hum Gene Ther 1998;9:671–679.
18. Raper SE, Yudkoff M, Chirmule N, Gao GP, Nunes F, Haskal ZJ,Furth EE, Propert KJ, Robinson MB, Magosin S, Simoes H,Speicher L, Hughes J, Tazelaar J, Wivel NA, Wilson JM, BatshawML. A pilot study of in vivo liver-directed gene transfer with anadenoviral vector in partial ornithine transcarbamylase defi-ciency. Hum Gene Ther 2002;13:163–175.
19. Curiel DT. The development of conditionally replicative adenovi-ruses for cancer therapy. Clin Cancer Res 2000;6:3395–3399.
20. Alemany R, Balague C, Curiel DT. Replicative adenoviruses forcancer therapy. Nat Biotechnol 2000;18:723–727.
21. Bischoff JR, Kirn DH, Williams A, Heise C, Horn S, Muna M, Ng L,Nye JA, Sampson-Johannes A, Fattaey A, McCormick F. An ade-novirus mutant that replicates selectively in p53-deficient humantumor cells. Science 1996;274:373–376.
22. Fueyo J, Gomez-Manzano C, Alemany R, Lee PS, McDonnell TJ,Mitlianga P, Shi YX, Levin VA, Yung WK, Kyritsis AP. A mutantoncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene 2000;19:2–12.
23. Yu DC, Sakamoto GT, Henderson DR. Identification of the tran-scriptional regulatory sequences of human kallikrein 2 and theiruse in the construction of calydon virus 764, an attenuatedreplication competent adenovirus for prostate cancer therapy.Cancer Res 1999;59:1498–1504.
24. Rodriguez R, Schuur ER, Lim HY, Henderson GA, Simons JW,Henderson DR. Prostate attenuated replication competent ade-novirus (ARCA) CN706: a selective cytotoxic for prostate-specificantigen-positive prostate cancer cells. Cancer Res 1997;57:2559–2563.
25. Rothmann T, Hengstermann A, Whitaker NJ, Scheffner M, zurHausen H. Replication of ONYX-015, a potential anticancer ade-novirus, is independent of p53 status in tumor cells. J Virol1998;72:9470–9478.
26. Goodrum FD, Ornelles DA. p53 status does not determine out-come of E1B 55-kilodalton mutant adenovirus lytic infection.J Virol 1998;72:9479–9490.
27. Ohashi M, Kanai F, Tanaka T, Lan KH, Shiratori Y, Komatsu Y,Kawabe T, Yoshida H, Hamada H, Omata M. In vivo adenovirus-mediated prodrug gene therapy for carcinoembryonic antigen-producing pancreatic cancer. Jpn J Cancer Res 1998;89:457–462.
28. Frazier ML. Gene expression in pancreatic adenocarcinoma. AnnN Y Acad Sci 1999;880:1–4.
29. Takahashi T, Namiki Y, Ohno T. Induction of the suicide HSV-TKgene by activation of the Egr-1 promoter with radioisotopes. HumGene Ther 1997;8:827–833.
30. Yan DH, Marin MC, Hung MC. Differential expression of the neuoncogene in mouse liver and pancreatic cell lines. BiochemBiophys Res Commun 1992;186:363–370.
31. DiMaio JM, Clary BM, Via DF, Coveney E, Pappas TN, Lyerly HK.Directed enzyme pro-drug gene therapy for pancreatic cancer invivo. Surgery 1994;116:205–213.
32. Sikora K, Harris J, Hurst H, Lemoine N. Therapeutic strategies
1216 YAMAMOTO ET AL. GASTROENTEROLOGY Vol. 125, No. 4

using c-erbB-2 promoter-controlled drug activation. Ann N Y AcadSci 1994;716:115–124; discussion 124–125, 140–143.
33. Kovarik A, Peat N, Wilson D, Gendler SJ, Taylor-Papadimitriou J.Analysis of the tissue-specific promoter of the MUC1 gene. J BiolChem 1993;268:9917–9926.
34. Williams C, Shattuck-Brandt RL, DuBois RN. The role of COX-2 inintestinal cancer. Ann N Y Acad Sci 1999;889:72–83.
35. Tsujii M, Kawano S, Tsuji S, Sawaoka H, Hori M, DuBois RN.Cyclooxygenase regulates angiogenesis induced by colon cancercells [published erratum appears in Cell 1998 Jul 24;94(2):following 271]. Cell 1998;93:705–716.
36. Tsujii M, DuBois RN. Alterations in cellular adhesion and apopto-sis in epithelial cells overexpressing prostaglandin endoperoxidesynthase 2. Cell 1995;83:493–501.
37. Ding XZ, Tong WG, Adrian TE. Cyclooxygenases and lipoxygen-ases as potential targets for treatment of pancreatic cancer.Pancreatology 2001;1:291–299.
38. Molina MA, Sitja-Arnau M, Lemoine MG, Frazier ML, Sinicrope FA.Increased cyclooxygenase-2 expression in human pancreatic car-cinomas and cell lines: growth inhibition by nonsteroidal anti-inflammatory drugs. Cancer Res 1999;59:4356–4362.
39. Yip-Schneider MT, Barnard DS, Billings SD, Cheng L, Heilman DK,Lin A, Marshall SJ, Crowell PL, Marshall MS, Sweeney CJ. Cyclo-oxygenase-2 expression in human pancreatic adenocarcinomas.Carcinogenesis 2000;21:139–146.
40. Tucker ON, Dannenberg AJ, Yang EK, Zhang F, Teng L, Daly JM,Soslow RA, Masferrer JL, Woerner BM, Koki AT, Fahey TJ 3rd.Cyclooxygenase-2 expression is up-regulated in human pancre-atic cancer. Cancer Res 1999;59:987–990.
41. Niijima M, Yamaguchi T, Ishihara T, Hara T, Kato K, Kondo F,Saisho H. Immunohistochemical analysis and in situ hybridiza-tion of cyclooxygenase-2 expression in intraductal papillary-muci-nous tumors of the pancreas. Cancer 2002;94:1565–1573.
42. Okami J, Yamamoto H, Fujiwara Y, Tsujie M, Kondo M, Noura S,Oshima S, Nagano H, Dono K, Umeshita K, Ishikawa O, Sakon M,Matsuura N, Nakamori S, Monden M. Overexpression of cycloox-ygenase-2 in carcinoma of the pancreas. Clin Cancer Res 1999;5:2018–2024.
43. Yamamoto M, Alemany R, Adachi Y, Grizzle WE, Curiel DT. Char-acterization of the cyclooxygenase-2 promoter in an adenoviralvector and its application for the mitigation of toxicity in suicidegene therapy of gastrointestinal cancers. Mol Ther 2001;3:385–394.
44. Casado E, Gomez-Navarro J, Yamamoto M, Adachi Y, CoolidgeCJ, Arafat WO, Barker SD, Wang MH, Mahasreshti PJ, HemminkiA, Gonzalez-Baron M, Barnes MN, Pustilnik TB, Siegal GP, AlvarezRD, Curiel DT. Strategies to accomplish targeted expression oftransgenes in ovarian cancer for molecular therapeutic applica-tions. Clin Cancer Res 2001;7:2496–2504.
45. Wesseling JG, Yamamoto M, Adachi Y, Bosma PJ, van Wijland M,Blackwell JL, Li H, Reynolds PN, Dmitriev I, Vickers SM,Huibregtse K, Curiel DT. Midkine and cyclooxygenase-2 promot-ers are promising for adenoviral vector gene delivery of pancre-atic carcinoma. Cancer Gene Ther 2001;8:990–996.
46. Dmitriev I, Krasnykh V, Miller CR, Wang M, Kashentseva E,Mikheeva G, Belousova N, Curiel DT. An adenovirus vector withgenetically modified fibers demonstrates expanded tropism viautilization of a coxsackievirus and adenovirus receptor-indepen-dent cell entry mechanism. J Virol 1998;72:9706–9713.
47. Suzuki K, Fueyo J, Krasnykh V, Reynolds PN, Curiel DT, AlemanyR. A conditionally replicative adenovirus with enhanced infectivityshows improved oncolytic potency. Clin Cancer Res 2001;7:120–126.
48. Fallaux FJ, Kranenburg O, Cramer SJ, Houweling A, Van OrmondtH, Hoeben RC, Van Der Eb AJ. Characterization of 911: a newhelper cell line for the titration and propagation of early region1-deleted adenoviral vectors. Hum Gene Ther 1996;7:215–222.
49. He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. Asimplified system for generating recombinant adenoviruses. ProcNatl Acad Sci U S A 1998;95:2509–2514.
50. Chartier C, Degryse E, Gantzer M, Dieterle A, Pavirani A, MehtaliM. Efficient generation of recombinant adenovirus vectors byhomologous recombination in Escherichia coli. J Virol 1996;70:4805–4810.
51. Krasnykh V, Dmitriev I, Mikheeva G, Miller CR, Belousova N,Curiel DT. Characterization of an adenovirus vector containing aheterologous peptide epitope in the HI loop of the fiber knob.J Virol 1998;72:1844–1852.
52. Inoue H, Yokoyama C, Hara S, Tone Y, Tanabe T. Transcriptionalregulation of human prostaglandin-endoperoxide synthase-2gene by lipopolysaccharide and phorbol ester in vascular endo-thelial cells. Involvement of both nuclear factor for interleukin-6expression site and cAMP response element. J Biol Chem 1995;270:24965–24971.
53. Inoue H, Nanayama T, Hara S, Yokoyama C, Tanabe T. The cyclicAMP response element plays an essential role in the expressionof the human prostaglandin-endoperoxide synthase 2 gene indifferentiated U937 monocytic cells. FEBS Lett 1994;350:51–54.
54. Maizel JV Jr, White DO, Scharff MD. The polypeptides of adeno-virus. I. Evidence for multiple protein components in the virionand a comparison of types 2, 7A, and 12. Virology 1968;36:115–125.
55. Hardy S, Kitamura M, Harris-Stansil T, Dai Y, Phipps ML. Con-struction of adenovirus vectors through Cre-lox recombination.J Virol 1997;71:1842–1849.
56. Adachi Y, Reynolds PN, Yamamoto M, Wang M, Takayama K,Matsubara S, Muramatsu T, Curiel DT. A midkine promoter-basedconditionally replicative adenovirus for treatment of pediatricsolid tumors and bone marrow tumor purging. Cancer Res 2001;61:7882–7888.
57. Suzuki K, Alemany R, Yamamoto M, Curiel DT. The presence ofthe adenovirus E3 region improves the oncolytic potency of con-ditionally replicative adenoviruses. Clin Cancer Res 2002;8:3348–3359.
58. Abbruzzese J, Evans D, Rich T. Principles and practice of oncol-ogy. Philadelphia: Lippincott-Raven, 1997.
59. Sinkovics J, Horvath J. New developments in the virus therapy ofcancer: a historical review. Intervirology 1993;36:193–214.
60. Smith R, Huebner R, Rowe W, Schatten W, Thomas L. Studies onthe use of viruses in the treatment of carcinoma of the cervix.Cancer 1956;9:1211–1218.
61. Bischoff JR, Kirn DH, Williams A, Heise C, Horn S, Muna M, Ng L,Nye JA, Sampson-Johannes A, Fattaey A, McCormick F. An ade-novirus mutant that replicates selectively in p53-deficient humantumor cells. Science 1996;274:373–376.
62. Southam CM, Moore AE. Clinical studies of viruses as antineo-plastic agents, with particular reference to Egypt 101 virus. Can-cer 1952;5:1025–1034.
63. Southam CM, Moore AE. Induced virus infection in man by Egyptisolated of west Nile virus. J Trop Med Hyg 1954;3:19–50.
64. Chambers R, Gillespie GY, Soroceanu L, Andreansky S, Chatter-jee S, Chou J, Roizman B, Whitley RJ. Comparison of geneticallyengineered herpes simplex viruses for the treatment of braintumors in a scid mouse model of human malignant glioma. ProcNatl Acad Sci U S A 1995;92:1411–1415.
65. Martuza RL, Malick A, Markert JM, Ruffner KL, Coen DM. Exper-imental therapy of human glioma by means of a geneticallyengineered virus mutant. Science 1991;252:854–856.
66. Telerman A, Tuynder M, Dupressoir T, Robaye B, Sigaux F,Shaulian E, Oren M, Rommelaere J, Amson R. A model for tumorsuppression using H-1 parvovirus. Proc Natl Acad Sci U S A1993;90:8702–8706.
67. McAuliffe PF, Jarnagin WR, Johnson P, Delman KA, Federoff H,
October 2003 ENHANCED COX-2 CRAD FOR PANCREATIC CANCER 1217

Fong Y. Effective treatment of pancreatic tumors with two mul-timutated herpes simplex oncolytic viruses. J Gastrointest Surg2000;4:580–588.
68. Lee JH, Federoff HJ, Schoeniger LO. G207, modified herpessimplex virus type 1, kills human pancreatic cancer cells in vitro.J Gastrointest Surg 1999;3:127–131; discussion 132–133.
69. Kirn D. Clinical research results with dl1520 (Onyx-015), a repli-cation-selective adenovirus for the treatment of cancer: whathave we learned? Gene Ther 2001;8:89–98.
70. Mulvihill S, Warren R, Venook A, Adler A, Randlev B, Heise C, KirnD. Safety and feasibility of injection with an E1B-55 kDa gene-deleted, replication-selective adenovirus (ONYX-015) into primarycarcinomas of the pancreas: a phase I trial. Gene Ther 2001;8:308–315.
71. Gorunova L, Hoglund M, Andren-Sandberg A, Dawiskiba S, Jin Y,Mitelman F, Johansson B. Cytogenetic analysis of pancreaticcarcinomas: intratumor heterogeneity and nonrandom pattern ofchromosome aberrations. Genes Chromosomes Cancer 1998;23:81–99.
72. Shenk T. Adenoviridae: the viruses and their replication. In: FieldsBN KD, Howley PM, eds. Virology. Volume 2. Philadelphia: Lip-pincott-Raven, 1996:2111–2148.
73. Kirch HC, Putzer B, Schwabe G, Gnauck HK, Schulte HolthausenH. Regulation of adenovirus 12 E1A transcription: E2F and ATFmotifs in the E1A promoter bind nuclear protein complexes in-cluding E2F1, DP-1, cyclin A and/or RB and mediate transcrip-tional (auto)activation. Cell Mol Biol Res 1993;39:705–716.
74. Steinwaerder DS, Carlson CA, Lieber A. DNA replication of first-generation adenovirus vectors in tumor cells. Hum Gene Ther2000;11:1933–1948.
75. Adachi Y, Raynolds PN, Yamamoto M, Grizzle WE, Overturf K,Matsubara S, Muramatsu T, Curiel DT. Midkine promoter-basedadenovirus vector gene delivery for pediatric solid tumors. CancerRes 2000;60:4305–4310.
76. DeWeese TL, van der Poel H, Li S, Mikhak B, Drew R, GoemannM, Hamper U, DeJong R, Detorie N, Rodriguez R, Haulk T, De-Marzo AM, Piantadosi S, Yu DC, Chen Y, Henderson DR, CarducciMA, Nelson WG, Simons JW. A phase I trial of CV706, a replica-tion-competent, PSA selective oncolytic adenovirus, for the treat-
ment of locally recurrent prostate cancer following radiation ther-apy. Cancer Res 2001;61:7464–7472.
77. Hay JG. “Man’s best friend”: a new model system for cancertherapeutics? Mol Ther 2003;7:144–145.
78. Pacini DL, Dubovi EJ, Clyde WA Jr. A new animal model for humanrespiratory tract disease due to adenovirus. J Infect Dis 1984;150:92–97.
79. Bilbao R, Gerolami R, Bralet MP, Qian C, Tran PL, Tennant B,Prieto J, Brechot C. Transduction efficacy, antitumoral effect, andtoxicity of adenovirus-mediated herpes simplex virus thymidinekinase/ganciclovir therapy of hepatocellular carcinoma: thewoodchuck animal model. Cancer Gene Ther 2000;7:657–662.
80. Wolff H, Saukkonen K, Anttila S, Karjalainen A, Vainio H, Risti-maki A. Expression of cyclooxygenase-2 in human lung carci-noma. Cancer Res 1998;58:4997–5001.
81. Shirvani VN, Ouatu-Lascar R, Kaur BS, Omary MB, Triadafilopou-los G. Cyclooxygenase 2 expression in Barrett’s esophagus andadenocarcinoma: ex vivo induction by bile salts and acid expo-sure. Gastroenterology 2000;118:487–496.
82. Yoshimura R, Sano H, Masuda C, Kawamura M, Tsubouchi Y,Chargui J, Yoshimura N, Hla T, Wada S. Expression of cyclooxy-genase-2 in prostate carcinoma. Cancer 2000;89:589–596.
Received October 7, 2002. Accepted July 10, 2003.Address requests for reprints to: Masato Yamamoto, M.D., Ph.D.,
Division of Human Gene Therapy, University of Alabama at Birming-ham, BMR2-408, 901 19th Street South, Birmingham, Alabama35294-2172. e-mail: [email protected]; fax: (205) 975-8565.
Supported in part by National Institutes of Health grants R01CA94084 (to D.T.C.), R01 DK63615 (to M.Y.), R01 HL67962 (toD.T.C.), P50 CA89019 (to D.T.C.), R01 CA86881 (to D.T.C.), R01 CA93796 (to G.P.S.), U19 PR15015 (to D.T.C.), Department of Defensegrant DAMD17-03-1-0104 (to M.Y.), an AVON Breast Cancer Researchand Care Program grant (to M.Y.), the Lustgarten Foundation Compet-itive Awards (to D.T.C.), and a UA-HSF grant (to G.P.S.).
The authors thank Drs. H. Inoue and T. Tanabe for providing aplasmid phPES2 containing the COX-2 promoter and Drs. RamonAlemany, Yasuo Adachi, Cristina Balague, Kaori Suzuki, Peter Nagi,and Long Le for helpful discussions.
1218 YAMAMOTO ET AL. GASTROENTEROLOGY Vol. 125, No. 4
Copyright © 2022 FDOKUMEN




















