
Carboxylic Acid Derivatives: Nucleophilic Acyl Substitution ...

BASIC SCIENCE
Impact on Replicative Fitness of the G48ESubstitution in the Protease of HIV-1
An In Vitro and In Silico Evaluation
Jean-Marie Zimmer, MSc,* Francxois Roman, MD, PhD,* Christine Lambert, MSc,*
Abel Jonckheer, MSc,† Ana Vazquez,‡ Jean-Marc Plesseria, BSc,* Jean-Yves Servais, MSc,*
Kris Covens, MSc,§ Jan Weber, PhD,‡ Kristel Van Laethem, PhD,§ Jean-Claude Schmit, MD, PhD,*
Anne-Mieke Vandamme, PhD,§ Miguel E. Quinones-Mateu, PhD,‡ and Marc De Maeyer, PhD†
Summary: We observed an unusual glycine-to-glutamate sub-
stitution at protease (PR) residue position 48 (G48E) in an African
patient infected with a subtype A1 HIV-1 strain failing a saquinavir-
containing regimen. Phenotypic analysis of protease inhibitor (PI)
susceptibility showed that the G48E site-directed mutant, when
introduced into an NL4-3 HIV-1 PR backbone, was slightly resistant
to SQV (2-fold when compared with the wild-type virus). In addition,
the G48E and G48E/V82A site-directed mutants were associated with
a decrease in fitness, whereas a reversion to the wild type at position
48 was observed in vitro. Growth competition experiments using
a novel growth competition assay based on enhanced green
fluorescent protein– or Discosoma spp. red fluorescent protein–
expressing viruses showed that the replicative fitness of the G48E
virus was reduced to 55% compared with the parental NL4-3 virus.
Synthesizing all possible site-directed mutants found in the patient
strain is too time-consuming; therefore, a molecular dynamics (MD)
simulation approach was used to understand why this mutation
survived despite its fitness cost. These simulations documented that
the G48E mutant interacted with PI resistance mutations (M46I, I54V,
Q58E, and L63P) and with natural polymorphisms specific to subtype
A1 (E35D, M36I, and R57K) that were present in the patient’s virus.
We hypothesize that the polymorphisms contained in the PR flap
regions of the patient’s virus may compensate for the presence of
G48E, possibly by restoring the flexibility of the PR flaps. In
summary, our results demonstrate that the G48E substitution, when
introduced in the context of an HIV-1 subtype B strain, is highly
unstable and gives rise to viruses with a poor replicative fitness
in vitro. We also showed that when confronted with too many
mutations to evaluate in vitro, MD simulations are helpful to draft
hypotheses on how polymorphisms can interact with resistance
mutations to stabilize their potential fitness cost.
Key Words: HIV-1, protease, replicative capacity, subtypes
(J Acquir Immune Defic Syndr 2008;48:255–262)
The HIV-1 protease (PR) enzyme cleaves the Gag andGag-Pol polyproteins to produce the viral enzymes and struc-tural proteins necessary for the release of mature virionparticles.1 It is composed of 2 noncovalently associatedsequence and structurally identical monomers and contains anactive site, which includes an Asp-Thr-Gly motif at residuepositions 25 to 27.1 Protease inhibitor (PI) resistance muta-tions have been described in the substrate cleft of the enzymethat impair the binding between the inhibitor and the mutantPR.2–4 Some major primary mutations are seen frequently inpatients failing highly active antiretroviral therapy (HAART),including 1 or more PIs.5 In addition, secondary mutations inthe PR gene and in Gag-Pol cleavage site regions are oftenselected, which seem to play a major role in the recovery ofviral replicative fitness after the selection of primary muta-tions.6–11 Rare amino acid substitutions can also occur, and theirrole as natural polymorphic variants or as minor mutationsrequired for resistance to emerge in vivo is a subject ofinvestigation.12
We observed an unusual glycine-to-glutamate substi-tution at PR residue position 48 (G48E) in sequential isolatesfrom an African patient infected with a subtype A1 HIV-1strain. This amino acid substitution was observed after saquin-avir (SQV) had been introduced in the treatment regimen ofthe patient. Resistance to SQV is usually associated witha glycine-to-valine substitution at position 48 (G48V), locatedon the tip of the PR flap, a region that extends into the substratecleft. G48V frequently follows the development of a valine-to-alanine change at position 82 (V82A). Other mutations at posi-tion 48 are extremely rare, particularly in untreated patients.13
In this study, we performed a longitudinal analysis ofthe complete PR and Gag-Pol cleavage site regions throughoutthe course of infection of the patient. We showed the impactof G48E on viral replicative fitness and PI susceptibility on
Received for publication September 3, 2007; accepted March 17, 2008.From the *Retrovirology Laboratory, Centre de Recherche Publique-Sante,
Luxembourg, Luxembourg; †Laboratory for Biomolecular Modelling andBioMacS, Department of Chemistry, Leuven, Belgium; ‡Section ofVirology, Department of Molecular Genetics, Lerner Research Institute,Cleveland Clinic Foundation, Cleveland, OH; and §Rega Institute forMedical Research, Katholieke Universiteit Leuven, Leuven, Belgium.
Jean-Marie Zimmer and Francxois Roman contributed equally to this study andshould both be considered first authors.
Supported by the Fondation Recherche sur le SIDA and the Centre deRecherche Publique-Sante of Luxembourg.
Correspondence to: Francxois Roman, MD, PhD, Retrovirology Laboratory,Centre de Recherche Public-Sante, 84, Rue Val Fleuri, L-1256Luxembourg (e-mail: [email protected]).
Copyright � 2008 by Lippincott Williams & Wilkins
J Acquir Immune Defic Syndr � Volume 48, Number 3, July 1, 2008 255
Copyright © 2008 Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.

the basis of in vitro phenotypic evaluation. Finally, using amolecular dynamics (MD) simulation approach, we investi-gated whether G48E might have an impact on HIV-1replicative capacity and how several polymorphisms containedin the PR flap regions of the patient’s virus might compensatethe fitness cost of G48E.
MATERIALS AND METHODS
Population Nucleotide Sequencing of thePatient’s Virus
Viral RNA was extracted from plasma samples of thepatient and directly sequenced on an automated ABIPrism3130 (Applied Biosystems, Foster city, CA). For sequencingthe PR and the gag and pol cleavage site regions, we usedViroseq technology (Abbott N.V. Diagnostics, Louvain-la-Neuve, Belgium) and BigDye Terminator v3.1 chemistry (PEApplied Biosystems, Foster City, CA) according to previouslydescribed protocols.14,15 Deduced amino acid sequences werecompared with the subtype B HXB2 reference sequence. Anadditional comparison was made with a subtype A1 referencesequence for the Gag-Pol cleavage site sequences.
Site-Directed MutagenesisG48E, V82A, and G48E/V82A mutations were inserted
into a plasmid encoding the polymerase gene of NL4 to 3(pUC19-NL4 to 3, a gift from the Rega Institute, KatholiekeUniversiteit Leuven, Leuven, Belgium) by site-directedmutagenesis using the following primers: 5#-gatggaaaccaaaaatgatagagggaattgg-3# (sense primer, positions 2374 to 2404 ofthe HXB2 genome) and 5#-actttgataaaacctccaattccctctatca-3#(antisense primer, HXB2 positions 2419 to 2389) for G48Eand 5#-gtaggacctacacctgccaacataattggaagaaatctg-3# (sense primer,HXB2 positions 2481 to 2519) and 5#-cagatttcttccaattatgttggcaggtgtaggtcctac-3# (antisense primer, HXB2 positions 2519to 2481) for V82A. V82A mutant was created, because V82Ais a common mutation and we were interested to investigate itsfitness effects in the settings of our experiment. Mutagenesisproducts were transformed to One-shot Escherichia colielectrocompetent cells (Invitrogen, Merelbeke, Belgium) afterdigestion of the parental plasmid DNA with DpnI. Afterovernight incubation on Luria-Burtani (LB) agar plates, cloneswere cultured in 5 mL of LB medium and plasmid DNA wasextracted using the QIAquick Minispin extraction kit(QIAGEN, KJ Venlo, The Netherlands). The presence ofdesired mutations was screened for by sequencing plasmids onan automated ABIPrism3100, using BigDye Terminator cycle-sequencing chemistry and RVP5 (sense, 5#-gggaagatctggccttcctacaaggg-3#, HXB2 positions 2092 to 2117) and RVP3(antisense, 5#-ggcaaatactggagtattgtatgg-3#, HXB2 positions2712 to 2735) as sequencing primers.
Phenotypic Analysis of PI Susceptibility andReplicative Fitness
After detection of clones incorporating the desiredmutation, a polymerase chain reaction (PCR) assay wasperformed on mutant plasmid DNA using RVP5 and RVP3 toamplify 620–base pair (bp) PR fragments. PR fragments werethen recombined into a proviral plasmid deleted of the PR
gene, as described previously.16 Recombinant viruses wereused to infect MT4 cells. After detection of cytopathic effects(CPEs) in culture, viral supernatants of G48E, V82A, andG48E/V82A mutants were titrated and analyzed by directsequencing. Phenotypic resistance tests (ie, inhibitory con-centrations [IC50] values, defined as the concentration of druginhibiting 50% of HIV-1 replication in MT4 cells) wereperformed using a standard 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) procedure as described.17
In addition, for every mutant, pol recombinant viruseswere generated by cotransfection of PCR-amplified pol gene(carrying G48E, V82A, or G48E/V82A mutations) witha pol-deleted proviral plasmid tagged with the enhancedgreen fluorescent protein (EGFP) or Discosoma spp. redfluorescent protein (DsRed2) as described.18 Viral productionwas monitored every 3 to 4 days for CPEs, and supernatantwas analyzed using an in-house reverse transcriptase (RT)assay. Culture supernatants were harvested, cleared by low-speed centrifugation, filtered using a 0.45-mm sterile vacuumfiltration system, and aliquoted. Tissue culture dose for50% infectivity (TCID50) was determined as describedpreviously.19
Viral replicative fitness was individually evaluated usingviral growth kinetics in MT-4 cells and by growth competitionexperiments in peripheral blood mononuclear cells (PBMCs)against wild-type NL4-3 strain. To estimate growth kinetics3 3 104 MT-4 cells were infected separately in 96-well platesin triplicate with each virus at a multiplicity of infection (MOI)of 0.01 IU/cell in 200 mL of RPMI 1640 and incubated for2 hours at 37�C in 5% CO2. Infected cells were then washed 3times with phosphate-buffered saline (PBS) and cultured ingrowth medium. RT activity in the supernatant was measuredon day 2 after infection and every day thereafter. RT activitywas also used for evaluating susceptibility of the G48E, V82A,or G48E/V82A mutants to PIs.
In growth competition experiments, EGFP- or DsRed2-tagged viruses competed with awild-type NL4-3 strain in a 1:1initial proportion using a MOI of 0.01 IU/cell. One milliliter ofthese virus mixtures was incubated with 1 3 106 PBMCs for2 hours at 37�C, 5% CO2. Cells were subsequently washed3 times with 13 PBS and then resuspended in culture medium(1 3 106 cells/mL). Dual infections were monitored dailyusing a Leica DMIRB inverted, upright, wide-field fluo-rescence microscope (Heidelberg, Germany). After the detec-tion of green and/or red fluorescent cells, aliquots of HIV-infected cultures were homogenized and diluted in 1 3 PBSuntil reaching a single-cell monolayer in a 96-well plate. Eachwell was individually analyzed, and 4 representative picturesper well were obtained using a MicroMax 5-MHz charge-coupled device (CCD) digital camera (Princeton Instru-ments, Trenton, NJ). Images were processed with Image-Pro Plus 5.0 software (Media Cybernetics, Silver Springs,MD), allowing the quantification of green and red cells in eachcompetition by measuring their area and mean intensity. Todetermine HIV-1 replicative fitness, the final proportion ofthe 2 viruses produced in each growth competition exper-iment was quantified by fluorescence microscopy after nor-malizing to viral production in the HIV-1 monoinfections asdescribed elsewhere.18
256 q 2008 Lippincott Williams & Wilkins
Zimmer et al J Acquir Immune Defic Syndr � Volume 48, Number 3, July 1, 2008
Copyright © 2008 Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.

Molecular Dynamics SimulationAll MD simulations were carried out using the
GROMACS software package (University of Groningen,Groningen, the Netherlands).20 The simulations were performedin the presence of explicit water and counter ions. Periodicboundaries conditions were applied in all directions. Theapplied force field was the GROMOS96 force field. Initializa-tion of the MD simulation included an energy minimization of500 steps and a 20-picosecond MD simulation with harmonicpositional restraints on the solute. The final production MDsimulations were run for 2 nanoseconds at 300 K, keepingpressure constant at a reference of 1 bar. All calculations weredone on an Apple X-serve cluster with 12 nodes.
RESULTS
Evolutional Characteristics of the PR andGag-Pol Cleavage Site Regions of theSubtype A1 Patient’s Virus
Variation of the PR and Gag-Pol cleavage site regions isshown Table 1. Natural PR non-B polymorphisms (L10I, I13V,E35D, M36I, R57K, H69K, and L89M) were present atbaseline and throughout the course of infection. The PR regionexhibited increasing variability over time. Although severalmutations were attributable to PI-based regimen pressure(M46I, I54V, Q58E, I62V, L63P, V82F, and L90M), otherpolymorphisms were classically not related to resistance to PI(K20I, E35N, K55Q, Q61E, and L89I). The G48E mutationwas first detected 3 months after the introduction of SQV in thepatient’s treatment regimen and persisted after removal of SQV(see Table 1). At baseline, Gag-Pol cleavage site regions werecomparable to a subtype A1 reference sequence. Severalmutations were later observed, among which were an A-to-Vsubstitution at residue P2 of the conserved NC/p1-NC/TFPcleavage site before the development of G48E in the patient’svirus and a P-to-L change at residue P5# of the p1/p6 cleavagesite after the emergence of G48E.
Replication and Drug Susceptibility of VirusesCarrying the G48E, V82A, and G48E/V82AMutation in the PR Gene
Replication characteristics of each site-directed mutantusing the standard MTT assay are summarized in Table 2.After 8 days of viral growth, CPEs were observed for theV82A mutant. The valine-to-alanine substitution at residueposition 82 was confirmed after sequencing of the viral super-natant. The virus was titrated at 2795 TCID50/mL. The G48Emutant was cultured for 7 days until CPEs were observed. Thetiter was determined at 2795 TCID50/mL. Direct sequencing ofthe viral supernatant revealed the appearance of a G/E mix atposition 48, however, and the presence of a second mutation,K20M. Three G48E/V82A double-mutant viruses, namely,G48E/V82A-1, G48E/V82A-2, and G48E/V82A-3, were grownin vitro for 36, 46, and 60 days, respectively, until CPEs wereobserved. For all mutants, the V82A mutation was conservedat the time of titration. The titer of G48E/V82A-1 wascalculated at 559 TCID50/mL. Similar to the G48E mutant,direct sequencing of the viral supernatant revealed a G/E mix
at position 48, which was confirmed after an additional 5-dayvirus culture in vitro. Furthermore, a mixed viral populationwas observed at residue positions 20 and 34 (20K/M and34E/K, respectively). The titer of G48E/V82A-2 was 559TCID50/mL, and the sequence analysis revealed the presenceof mixed viral populations at residue positions 20 (K/M), 48(G/E), and 71 (A/V). G48E/V82A-3 exhibited a slower growthin vitro and a lower titer of 22 TCID50/mL. Interestingly, theG48E mutation was predominant in the viral supernatant andwas associated with the emergence of an E21K mutation.When G48E/V82A-3 was grown for 15 additional days,however, the virus exhibited progressive loss of the E atposition 48, in parallel with a higher titer in culture (559TCID50/mL).
Finally, the susceptibility of V82A and G48E mutants tonelfinavir (NFV), lopinavir (LPV), ritonavir (RTV), indinavir(IDV), amprenavir (APV), and SQV was determined (Fig. 1).As described elsewhere in this article, the G48E mutant viruswas characterized by impaired replication as compared withthe NL4-3 virus control (ie, low RT activity in the absence ofPI) and exhibited slightly decreased susceptibility to SQV(2-fold when compared with the wild-type virus). The G48Esubstitution showed no effect on the susceptibility to other PIsin vitro, however.
Replicative FitnessWe evaluated the replicative fitness of viruses carrying
G48E, V82A, or G48E/V82A mutations in the PR gene usinga novel assay based on recombinant viruses expressing theEGFP or the DsRed2 protein in an HIV-1 NL4-3 backbone.18
EGFP- and DsRed2-tagged viruses with G48E and V82Amutations were generated. Attempts to create recombinantvirus with G48E/V82A mutations resulted in virus carryingonly the V82A mutation. Replicative fitness was initiallyevaluated using a viral growth kinetics assay (Fig. 2A). Resultsshowed that the G48E mutant virus replicated with reducedkinetics compared with the wild-type NL4-3 strain and V82Amutant virus (at day 3 and day 4; P , 0.001, t test).
To confirm the effect of the G48E substitution on HIVreplicative fitness, we performed growth competition experi-ments in PBMCs using 4 fluorescent viruses: G48E-EGFPversus NL4-3–DsRed2 and G48E-DsRed2 versus NL4-3–EGFP, respectively. The mean replicative fitness of the G48Emutant virus (1.146 0.67 [mean6 SD], n = 6) was reduced to55% compared with the wild-type NL4 to 3 (2.07 6 1.07; n =6; P = 0.041, Mann-Whitney rank sum test; see Fig. 2B).
Molecular Dynamics Simulation of theG48E Mutant
Root-mean-square deviation (RMSD) plots were ob-tained for the PR sequences of residues 35 to 63 (cor-responding to the PR flap) of (1) the wild-type NL4-3reference strain (based on the protein data bank (PDB) code:5HVP, in blue in Fig. 3), (2) the site-directed mutant G48Emutant in the context of the wild type (in red in Fig. 3), and (3)the subtype A1 clinical isolate of the patient (in green in Fig.3). RMSD plots (represented on the y axis) are an indication ofthe flexibility of the PR flap, on the tip of which the 48 residue
q 2008 Lippincott Williams & Wilkins 257
J Acquir Immune Defic Syndr � Volume 48, Number 3, July 1, 2008 G48E Protease Substitution Impact on HIV-1 Fitness
Copyright © 2008 Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.

TABLE 1. PR and Gag-Pol Cleavage Site Sequence Characteristics of the Subtype A1 HIV-1 Clinical Isolate Over Time*
Sampling Date Treatment VL† 48 Other PR Mutations‡
March 23, 1995 AZT/ddI ,10,000 G L10I, I13V, E35D, M36I, R41K, R57K, H69K, L89M
October 25, 1995 AZT/ddC 39,900 G L10I, I13V, E35D, M36I, R41K, R57K, H69K, L89M
January 30, 1996 AZT/ddC 152,800 G L10I, I13V, E35D, M36I, R41K, R57K, H69K, L89M
April 4, 1997 AZT/ddC/IDV 45,340 G L10I, I13V, E35D, M36I, R41K, M46I, I54V, R57K,H69K, V82F, L89M
August 20, 1998 AZT/ddC/IDV 18,954 G L10I, I13V, E35D, M36I, M46I, I54V, R57K, Q58E, H69K, V82F
November 17, 1998 3TC/d4T/SQV 18,060 G L10I, I13V, M36I, M46I, I54V, Q58E, H69K, V82F
February 1, 1999 3TC/d4T/SQV 17,605 G/E L10I, I13V, K20I, M36I, M46I, I54V, Q58E, H69K, V82F
May 21, 1999 3TC/d4T/SQV 10,663 G/E L10I, I13V, K20I, M36I, M46I, I54V, R57K, Q58E, H69K, V82F
November 4, 1999 ddI/EFV/SQV/HU 3617 E L10I, I13V, K20I, E35D, M36I, M46I, I54V, R57K, Q58E,L63P, H69K, V82F, L89I
January 18, 2000 ddI/EFV/SQV/RTV/HU 7501 G/E L10I, I13V, K20I, E35D, M36I, M46I, I54V, R57K, Q58E, L63P,H69K, V82F, L89I
April 13, 2000 ddI/d4T/EFV/SQV/RTV 4607 E L10I, I13V, K20I, E35N, M36I, M46I, I54V, K55Q, R57K, Q58E,Q61E, H69K, V82F, L89I
April 11, 2001 ddI/ABC/EFV 2680 E L10I, I13V, K20I, E35N, M36I, M46I, I54V, K55Q, R57K,Q58E, Q61E, L63P, H69K, V82F, L89I, L90M
October 1, 2001 ddI/ABC/NVP 1176 E L10I, I13V, K20I, M36I, M46I, I54V, K55Q, R57K, Q58E,Q61E, L63P, H69K, V82F, L89I, L90M
June 19, 2002 AZT/3TC/ABC/LPV 3398 E L10I, I13V, K20I, E35N, M36I, M46I, I54V, K55Q, R57K,Q58E, Q61E, L63P, H69K, V82F, L89I, L90M
January 22, 2003 AZT/3TC/ddI/ABC/LPV 3567 E L10I, I13V, K20I, E35N, M36I, M46I, I54V, K55Q, R57K,Q58E, Q61E, L63P, H69K, V82F, L89I, L90M
June, 19, 2003 AZT/3TC/ddI/ABC/LPV 3623 E L10I, I13V, K20I, E35N, M36I, M46I, I54V, K55Q, R57K,Q58E, Q61E, I62V, L63P, H69K, V82F, L89I, L90M
September 23, 2003 AZT/3TC/ddI/SQV/RTV 2580 E L10I, I13V, K20I, E35N, M36I, M46I, I54V, K55Q, R57K, Q58E,Q61E, I62V, L63P, H69K, V82F, L89I, L90M
February 5, 2004 ddI/NVP/LPV 6521 E L10I, I13V, K20I, E35N, M36I, M46I, I54V, K55Q, R57K, Q58E,Q61E, I62V, L63P, H69K, V82F, L89I, L90M
May 4, 2004 ddI/NVP/LPV 5526 E L10I, I13V, K20I, E35N, M36I, M46I, I54V, K55Q, R57K, Q58E,Q61E, I62V, L63P, H69K, V82F, L89I, L90M
November 16, 2004 ddI/NVP/LPV 11,183 E L10I, I13V, K20I, E35N, M36I, M46I, I54V, K55Q, R57K, Q58E,Q61E, I62V, L63P, H69K, V82F, L89I, L90M
Sampling Date p2/NC NC/p1 p1/p6 TFP/p6 p6/PR§
March 23, 1995 TNIMM-MQRGN ERQAN-FLGKI RPGNF-PQSRP ENLAF-QQGEA PPFSF-PQITL
October 25, 1995 TNIMM-MQRGN ERQAN-FLGKI RPGNF-PQSRP ENLAF-QQQGEA PPFSF-PQITL
January 30, 1996 TNIMM-MQRGN ERQAN-FLGKI RPGNF-PQSRP ENLAF-QQQGEA PTFSFPQITL
April 4, 1997 TNIMM-MQRGN ERQAN-FLGKI RPGNF-PQSRP ENLAF-QQQGEA PTFSF-PQITL
August 20, 1998 TNIMM-MQRGN ERQVN-FLGKI RPGNF-PQSRP ENLAF-q/pQQGEA PTFSF-PQITL
November 17, 1998 TNIMM-MQRGN ERQVN-FLGKI RPGNF-PQSRP ENLAF-PQQGEA PTFSF-PQITL
February 1, 1999 TNIMM-MQRGN ERQVN-FLGKI RPGNF-PQSRP ENLAF-q/pQQGEA PTFSF-PQITL
May 21, 1999 TNIMM-MQRGN ERQVN-FLGKI RPGNF-PQSRP ENLAF-PQQGEA PTFs/nF-PQITL
November 4, 1999 TNIMM-MQRGN ERQVN-FLGKI RPGNF-PQSRP ENLAF-PQQGEA PTFSF-PQITL
January 18, 2000 TNIMM-MQRGN ERQVN-FLGKI RPGNF-PQSRP ENLAF-PQQGEA PTFSF-PQITL
April 13, 2000 TNIMM-MQRGN ERQVN-FLGKI RPGNF-PQSRP ENLAF-q/pQQGEA PTFNF-PQITL
April 11, 2001 TNIMM-MQRGN ERQVN-FLGKI RPGNF-PQSRP ENLAF-q/pQQGEA PTFNF-PQITL
October 1, 2001 TNIMM-MQRGN ERQVN-FLGKI RPGNF-PQSRP ENLAF-q/pQQGEA PTFNF-PQITL
June 19, 2002 TNIMM-MQRGN ERQVN-FLGKI RPGNF-PQSRP ENLAF-QQQGEA PTFNF-PQITL
January 22, 2003 TNIm/lM-MQRGN ERQVN-FLGKI RPGNF-PQSRP ENLAF-QQQGEA Pp/tFNF-PQITL
June, 19, 2003 TNILM-MQRGN ERQVN-FLGKI RPGNF-PQSRp/l ENLAF-q/pQQGEA PTFNF-PQITL
September 23, 2003 TNILM-MQRGN ERQVN-FLGKI RPGNF-PQSRp/l ENLAF-q/pQQGEA PTFNF-PQITL
February 5, 2004 TNILM-MQRGN ERQVN-FLGKI RPGNF-PQSRp/l ENLAF-QQQGEA PTFNF-PQITL
May 4, 2004 TNILM-MQRGN ERQVN-FLGKI RPGNF-PQSRp/l ENLAF-QQQGEA PTFNF-PQITL
November 16, 2004 TNILM-MQRGN ERQVN-FLGKI RPGNF-PQSRp/l ENLAF-QQQGEA PTFNF-PQITL
*Emerging mutations are shown in bold.†In copies per milliliter of plasma.‡Only predominant substitutions are shown.§All other cleavage sites are invariant compared with the subtype A1 reference sequence.ABC indicates abacavir; AZT, zidovudine; ddC, zalcitabine; ddI, didanosine; d4T, stavudine; EFV, efavirenz; HU, hydroxyurea; 3TC, lamivudine; VL, viral load.
258 q 2008 Lippincott Williams & Wilkins
Zimmer et al J Acquir Immune Defic Syndr � Volume 48, Number 3, July 1, 2008
Copyright © 2008 Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.

is located. Over 2-nanosecond simulations, the wild type ismuch more flexible, compared with the G48E mutant, when
site-directed in an NL4-3 backbone. The patient isolate,corresponding to a sample from April 11, 1999 (see Table 1 for
complete amino acid composition), displays an intermediateflexibility (see Fig. 3).
DISCUSSIONWe have demonstrated that G48E, when introduced in
the context of a subtype B HIV-1 strain, is associated withmoderate resistance to SQV but is also highly unstable, givingrise to viruses with diminished replicative fitness in vitro.Mixed viral subpopulations at position 48 appeared after 7
TABLE 2. Replication Characteristics of G48E, V82A, and G48E/V82A Recombinant Viruses
Virus CPE (d) Titer (TCID50/mL) AA at Position 48 AA at Position 82 Other PR Mutations
Parental* 7 4180 G V None
V82A 8 2795 G A None
G48E 7 2795 G/E V K20M
G48E/V82A-1 36 559 G/E A 20K/M, 34E/K
G48E/V82A-2 46 559 G/E A 20K/M, 71A/V
G48E/V82A-3† 60 22 E A E21K
*The parental virus corresponds to the NL4-3 pol gene inserted in the pHIVdeltaPROBstEII clone.†When grown for 15 additional days, the virus exhibited a G/E mix at residue position 48.
FIGURE 1. Drug susceptibility anal-ysis of V82A and G48E mutants.Mutant viruses and the NL4-3 con-trol virus were grown in the presenceof increasing concentrations of 6 dif-ferent PIs (NFV, LPV, RTV, IDV, APV,and SQV). Viral replication was de-termined 7 days after infection usingan in-house RT assay. cpm indicatescounts per minute.
q 2008 Lippincott Williams & Wilkins 259
J Acquir Immune Defic Syndr � Volume 48, Number 3, July 1, 2008 G48E Protease Substitution Impact on HIV-1 Fitness
Copyright © 2008 Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.
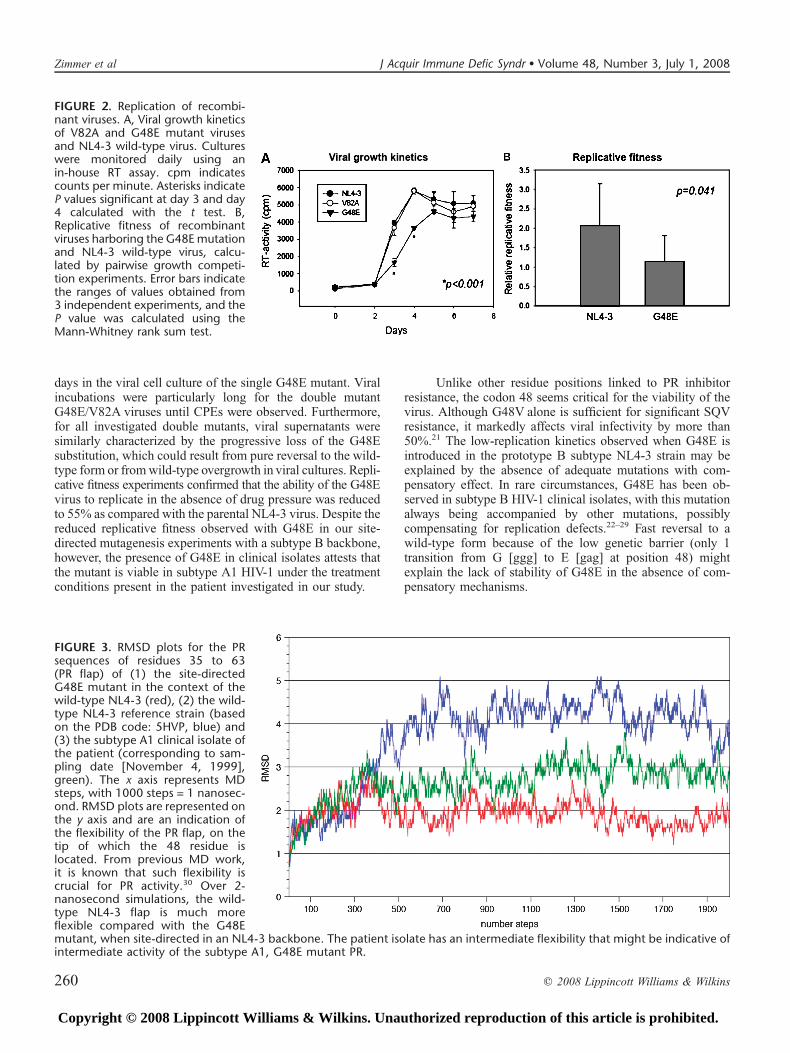
days in the viral cell culture of the single G48E mutant. Viralincubations were particularly long for the double mutantG48E/V82A viruses until CPEs were observed. Furthermore,for all investigated double mutants, viral supernatants weresimilarly characterized by the progressive loss of the G48Esubstitution, which could result from pure reversal to the wild-type form or fromwild-type overgrowth in viral cultures. Repli-cative fitness experiments confirmed that the ability of the G48Evirus to replicate in the absence of drug pressure was reducedto 55% as compared with the parental NL4-3 virus. Despite thereduced replicative fitness observed with G48E in our site-directed mutagenesis experiments with a subtype B backbone,however, the presence of G48E in clinical isolates attests thatthe mutant is viable in subtype A1 HIV-1 under the treatmentconditions present in the patient investigated in our study.
Unlike other residue positions linked to PR inhibitorresistance, the codon 48 seems critical for the viability of thevirus. Although G48V alone is sufficient for significant SQVresistance, it markedly affects viral infectivity by more than50%.21 The low-replication kinetics observed when G48E isintroduced in the prototype B subtype NL4-3 strain may beexplained by the absence of adequate mutations with com-pensatory effect. In rare circumstances, G48E has been ob-served in subtype B HIV-1 clinical isolates, with this mutationalways being accompanied by other mutations, possiblycompensating for replication defects.22–29 Fast reversal to awild-type form because of the low genetic barrier (only 1transition from G [ggg] to E [gag] at position 48) mightexplain the lack of stability of G48E in the absence of com-pensatory mechanisms.
FIGURE 2. Replication of recombi-nant viruses. A, Viral growth kineticsof V82A and G48E mutant virusesand NL4-3 wild-type virus. Cultureswere monitored daily using anin-house RT assay. cpm indicatescounts per minute. Asterisks indicateP values significant at day 3 and day4 calculated with the t test. B,Replicative fitness of recombinantviruses harboring the G48E mutationand NL4-3 wild-type virus, calcu-lated by pairwise growth competi-tion experiments. Error bars indicatethe ranges of values obtained from3 independent experiments, and theP value was calculated using theMann-Whitney rank sum test.
FIGURE 3. RMSD plots for the PRsequences of residues 35 to 63(PR flap) of (1) the site-directedG48E mutant in the context of thewild-type NL4-3 (red), (2) the wild-type NL4-3 reference strain (basedon the PDB code: 5HVP, blue) and(3) the subtype A1 clinical isolate ofthe patient (corresponding to sam-pling date [November 4, 1999],green). The x axis represents MDsteps, with 1000 steps = 1 nanosec-ond. RMSD plots are represented onthe y axis and are an indication ofthe flexibility of the PR flap, on thetip of which the 48 residue islocated. From previous MD work,it is known that such flexibility iscrucial for PR activity.30 Over 2-nanosecond simulations, the wild-type NL4-3 flap is much moreflexible compared with the G48Emutant, when site-directed in an NL4-3 backbone. The patient isolate has an intermediate flexibility that might be indicative ofintermediate activity of the subtype A1, G48E mutant PR.
260 q 2008 Lippincott Williams & Wilkins
Zimmer et al J Acquir Immune Defic Syndr � Volume 48, Number 3, July 1, 2008
Copyright © 2008 Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.

The restoration of the flexibility of the PR flaps is one ofthose mechanisms. We were interested in exploring whetherpolymorphisms that were present in the PR of the subtype A1patient’s virus could contribute to such a compensatorymechanism, when added to a single G48E mutation. To avoidmultiple syntheses of site-directed mutants and time-consum-ing viral culture experiments, however, we used an in silicoapproach for testing such an effect. The MD simulationssuggest that the single G48E mutation drastically alters suchflexibility when introduced in a wild-type subtype B back-bone. Previous MD work had already indicated that PR flapmovements were central to the function of the enzyme, andhence to the viability of the virus. The tip of those flaps(including PR residues 48 to 52) must curl in and bury thehydrophobic tip, Ile-50, against the inside wall of the activesite so as to open enough space for PR substrates.30 Using MDsimulations, we observed that the loss of PR flap flexibilitycaused by the G48E mutation was partially restored whenpolymorphisms present in the patient’s virus (namely, E35D,M36I, M46I, I54V, R57K, Q58E, and L63P) were added to theinitial G48E mutant sequence. Both mutations selected in thecontext of PI utilization regardless of subtype (eg, M46I, I54V,Q58E, L63P) or natural polymorphisms specific to subtype A1(in this case, E35D, M36I, and R57K) might contribute tothis compensating effect. Although their respective contribu-tion was difficult to evaluate in the settings of our study, it isinteresting to note that E35D, a natural subtype A1 poly-morphism, has already been identified as a critical componentof a gag-pol substitution complex responsible for improvingthe fitness of NFV-resistant isolates.31 Similarly, M36I isa polymorphism that naturally occurs in 12% of subtype Bviruses and more than 80% of non-B viruses and has beenshown to increase the replication rate of HIV-1 in vitro.32
Other PR substitutions that were not investigated in MDsimulation but that were present in the patient’s virus mightalso have modified the fitness of the G48E mutant. In thisrespect, L10I can naturally occur in the context of non-Bsubtype (which was the case in our study patient) and is knownto compensate for the loss of replication capacity conferred byG48V and V82A in vitro.21 In the context of drug pressure,particularly SQV, which selects for mutations at residueposition 48, K20I provides a significant replication benefitin vitro.32 Although not present at baseline, K20I emergedsimultaneous to G48E in the patient’s virus (see Table 1).Variation at residue position 20 is more common in subtypenon-B versus subtype B under PI pressure,33 which suggestsa non-B–specific compensatory pathway for fixing G48E inthe patient.
The genetic make-up of Gag-Pol cleavage site regionsalso contributes to rescue the fitness of HIV-1 PR mutants. Infunctional analyses of the HIV-1 PR, Moody and colleagues34
demonstrated that a synthetic G48E mutant had altered sub-strate specificity and that such G48E phenotype could bereversed by changing the P3# aspartic acid of the RT/RTp66cleavage site to glycine or asparagine. Although such specificchange was not identified in our patient’s virus, other sub-stitutions were observed in cleavage site regions, before orafter the emergence of G48E. An A-to-V substitution at res-idue P2 of the conserved NC/p1-NC/TFP cleavage site
(A431V) appeared after G48E had emerged and, as alreadyillustrated in previous studies,9–11 likely compensated for thepartially defective PR in the patient’s virus. A P-to-L change atresidue P5# of the p1/p6 cleavage site (P453L) was observedafter the emergence of G48E. P453L, which is commonlyselected with the I50V PR mutation under APV pressure,improves the catalytic activity of PR.35 Other substitutionswere observed in less well-conserved cleavage site regions (eg,p2/NC, TFP/p6, p6/PR); however, their role in restoring fitnessis currently unknown. Whether the natural variability of gag-pol cleavage site regions in subtype A1 favors the emergenceof A431V and P453L (or other less well-characterized poly-morphisms) during treatment remains to be established.Nevertheless, non-B subtypes are naturally more variable atNC/p1-NC/TFP and p1/p6 cleavage sites than subtype B HIV-1,36 and one could expect that such intersubtype difference ingag-pol cleavage sites might affect fitness more easily overtime. In addition, a practical implication of such intersubtypedifference may be that PR-recombinant virus assays that do notinclude cleavage sites in the recombination process are notoptimal for evaluating drug sensitivities of non-B virusesin vitro. In particular, most gag cleavage sites, including allamino acids N-terminal to and P1# and P2# amino acids of theNC/p1 cleavage site as well as cleavage sites of the RT andintegrase genes were not represented in the recombinant virusused for the MTT assay in our study.
In conclusion, we showed that G48E severely impairsthe replicative fitness of subtype B HIV-1 in vitro, althoughbeing viable in a clinical subtype A1 context. Several non-B–specific compensatory mechanisms could account for thefitness recovery observed in the patient’s virus investigated inour study. Those mechanisms, linked to the natural or drug-induced variability in the PR and Gag-Pol cleavage sites,might ultimately have an impact on the PI-based treatmentresponse of patients infected with non-B viruses. Addressingthis issue has now become a crucial task if one considers thatHIV-1 subtype B viruses are responsible for just more than12% of the global HIV pandemic37 and that the number ofpersons with non-B viruses starting therapy are likely toincrease dramatically in the coming years. In this respect, ourfindings illustrate that when confronted with too manymutations to evaluate in vitro, MD simulations are helpfulfor elucidating how polymorphisms can compensate for thepotential fitness cost of resistance mutations.
REFERENCES1. Erickson JW, Burt SK. Structural mechanisms of HIV drug resistance.
Annu Rev Pharmacol Toxicol. 1996;36:545–571.2. Velazquez-Campoy A, Muzammil S, Ohtaka H, et al. Structural and
thermodynamic basis for resistance to HIV-1 protease inhibition:implications for inhibitor design. Curr Drug Targets Infect Disord.2003;3:311–328.
3. Kagan RM, Shenderovich MD, Heseltine PNR, et al. Structural analysis ofan HIV-1 protease I47A mutant resistant to the protease inhibitorlopinavir. Protein Sci. 2005;14:1870–1878.
4. King NM, Prabu-Jeyabalan M, Nalivaika EA, et al. Structural andthermodynamic basis for the binding of TMC114, a next-generationhuman immunodeficiency virus type 1 protease inhibitor. J Virol. 2004;78:12012–12021.
5. Shafer RW. Genotypic testing for human immunodeficiency virus type 1drug resistance. Clin Microbiol Rev. 2002;15:247–277.
q 2008 Lippincott Williams & Wilkins 261
J Acquir Immune Defic Syndr � Volume 48, Number 3, July 1, 2008 G48E Protease Substitution Impact on HIV-1 Fitness
Copyright © 2008 Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.

6. Berkhout B. HIV-1 evolution under pressure of protease inhibitors:climbing the stairs of fitness. J Biomed Sci. 1999;6:298–305.
7. Nijhuis M, Deeks S, Boucher C. Implications of antiretroviral resistanceon viral fitness. Curr Opin Infect Dis. 2001;14:23–28.
8. Quinones-Mateu ME, Aerts EJ. HIV-1 fitness: implications for drugresistance, disease progression and global epidemic evolution. In: KuikenC, Foley B, Hahn B, et al, eds. HIV Sequence Compendium 2001. LosAlamos, NM: Los Alamos National Laboratory/Theoretical Biology andBiophysics; 2001:134–170.
9. Doyon L, Croteau G, Thibeault D, et al. Second locus involved in humanimmunodeficiency virus type 1 resistance to protease inhibitors. J Virol.1996;70:3763–3769.
10. Feher A, Weber IT, Bagossi P, et al. Effect of sequence polymorphism anddrug resistance on two HIV-1 Gag processing sites. Eur J Biochem. 2002;269:4114–4120.
11. Zhang YM, Imamichi H, Imamichi T, et al. Drug resistance duringindinavir therapy is caused by mutations in the protease gene and in itsGag substrate cleavage sites. J Virol. 1997;71:6662–6670.
12. Svicher V, Ceccherini-Silberstein F, Erba F, et al. Novel humanimmunodeficiency virus type 1 protease mutations potentially involvedin resistance to protease inhibitors. Antimicrob Agents Chemother. 2005;49:2015–2025.
13. Stanford University HIV Drug Resistance Database. September 1, 2007.Available at: http://hivdb.stanford.edu/cgi-bin/PRMut.cgi.
14. Van Laethem K, Schrooten Y, Dedecker S, et al. A genotypic assay for theamplification and sequencing of gag and protease from diverse humanimmunodeficiency virus type 1 group M subtypes. J Virol Methods. 2006;132:181–186.
15. Snoeck J, Riva C, Steegen K, et al. Optimization of genotypicassay applicable to all human immunodeficiency virus type 1 prote-ase and reverse transcriptase subtypes. J Virol Methods. 2005;128:47–53.
16. Maschera B, Furfine E, Blair ED. Analysis of resistance to humanimmunodeficiency virus type 1 protease inhibitors by using matchedbacterial expression and proviral infection vectors. J Virol. 1995;69:5431–5436.
17. Vandamme AM, Witvrouw M, Pannecouque C, et al. Evaluating clinicalisolates for their phenotypic and genotypic resistance against anti-HIVdrugs. In: Kinchington D, Schinazi RF, eds. Methods in MolecularMedicine, vol. 24. Antiviral Methods and Protocols. Totowa, NJ: HumanaPress Inc., 2000:223–258.
18. Weber J, Weberova J, Carobene M, et al. Use of a novel assaybased on intact recombinant viruses expressing green (EGFP) or red(DsRed2) fluorescent proteins to examine the contribution of pol and envgenes to overall HIV-1 replicative fitness. J Virol Methods. 2006;136:102–117.
19. Quinones-Mateu ME, Ball SC, Marozsan AJ, et al. A dual infection/competition assay shows a correlation between ex vivo humanimmunodeficiency virus type 1 fitness and disease progression. J Virol.2000;74:9222–9233.
20. Van Der Spoel D, Lindhal E, Hess B, et al. GROMACS: fast, flexible, andfree. J Comput Chem. 2005;26:1701–1718.
21. Mammano F, Trouplin V, Zennou V, et al. Retracing the evolutionarypathways of human immunodeficiency virus type 1 resistance to proteaseinhibitors: virus fitness in the absence and in the presence of drug. J Virol.2000;74:8524–8531.
22. Baxter JD, Shapiro JM, Boucher CA, et al. Genotypic changes in humanimmunodeficiency virus type 1 protease associated with reducedsusceptibility and virologic response to the protease inhibitor tipranavir.J Virol. 2006;80:10794–10801.
23. Janini LM, Pieniazek D, Peralta JM, et al. Identification of single and dualinfections with distinct subtypes of human immunodeficiency virus type 1by using restriction fragment length polymorphism analysis. Virus Genes.1996;13:69–81.
24. Kantor R, Fessel WJ, Zolopa AR, et al. Evolution of primary proteaseinhibitor resistance mutations during protease inhibitor salvage therapy.Antimicrob Agents Chemother. 2002;46:1086–1092.
25. Monno L, Saracino A, Scudeller L, et al. HIV-1 phenotypic susceptibilityto lopinavir (LPV) and genotypic analysis in LPV/r-naive subjects withprior protease inhibitor experience. J Acquir Immune Defic Syndr. 2003;33:439–447.
26. Rhee SY, Fessel WJ, Zolopa AR, et al. HIV-1 protease and reverse-transcriptase mutations: correlations with antiretroviral therapy in subtypeB isolates and implications for drug-resistance surveillance. J Infect Dis.2005;192:456–465.
27. Rousseau CM, Birditt BA, McKay AR, et al. Large-scale amplification,cloning and sequencing of near full-length HIV-1 subtype C genomes.J Virol Methods. 2006;136:118–125.
28. Shulman NS, Zolopa AR, Passaro DJ, et al. Efavirenz- and adefovirdipivoxil-based salvage therapy in highly treatment-experienced patients:clinical and genotypic predictors of virologic response. J Acquir ImmuneDefic Syndr. 2000;23:221–226.
29. Wu TD, Schiffer CA, Gonzales MJ, et al. Mutation patterns and structuralcorrelates in human immunodeficiency virus type 1 protease followingdifferent protease inhibitor treatments. J Virol. 2003;77:4836–4847.
30. Scott WRP, Schiffer CA. Curling of flap tips in HIV-1 protease asa mechanism for substrate entry and tolerance of drug resistance.Structure. 2000;8:1259–1265.
31. Matsuoka-Aizawa S, Sato H, Hachiya A, et al. Isolation and molecularcharacterization of a nelfinavir (NFV)-resistant human immunodeficiencyvirus type 1 that exhibits NFV-dependent enhancement of replication.J Virol. 2003;77:318–327.
32. Holguin A, Sune C, Hamy F, et al. Natural polymorphisms in the proteasegene modulate the replicative capacity of non-B HIV-1 variants in theabsence of drug pressure. J Clin Virol. 2006;36:264–271.
33. Gonzales MJ, Machekano RM, Shafer RW. Human immunodeficiencyvirus type 1 reverse-transcriptase and protease subtypes: classification,amino acid mutation patterns, and prevalence in a northern Californiaclinic-based population. J Infect Dis. 2001;184:998–1006.
34. Moody MD, Pettit SC, Shao W, et al. A side chain at position 48 of thehuman immunodeficiency virus type-1 protease flap provides anadditional specificity determinant. Virology. 1995;207:475–485.
35. Maguire MF, Guinea R, Griffin P, et al. Changes in human immuno-deficiency virus type 1 gag at positions L449 and P453 are linked to I50Vprotease mutants in vivo and cause reduction of sensitivity to amprenavirand improved viral fitness in vitro. J Virol. 2002;76:7398–7406.
36. de Oliveira T, Engelbrecht S, Janse van Rensburg E, et al. Variability athuman immunodeficiency virus type 1 subtype C protease cleavage sites:an indication of viral fitness? J Virol. 2003;77:9422–9430.
37. Osmanov S, Patton C, Walker N, et al. Estimated global distribution andregional spread of HIV-1 genetic subtypes in the year 2000. J AcquirImmune Defic Syndr. 2002;29:184–190.
262 q 2008 Lippincott Williams & Wilkins
Zimmer et al J Acquir Immune Defic Syndr � Volume 48, Number 3, July 1, 2008
Copyright © 2008 Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.
Copyright © 2022 FDOKUMEN




















