
Induction of the Upstream Regulatory Region of Human Papillomavirus Type 31 by Dexamethasone Is...
9
JOURNAL OF VIROLOGY, Oct. 2003, p. 10975–10983 Vol. 77, No. 20 0022-538X/03/$08.000 DOI: 10.1128/JVI.77.20.10975–10983.2003 Copyright © 2003, American Society for Microbiology. All Rights Reserved. Induction of the Upstream Regulatory Region of Human Papillomavirus Type 31 by Dexamethasone Is Differentiation Dependent Jennifer L. Bromberg-White,† Ellora Sen, Samina Alam, Jason M. Bodily, and Craig Meyers* Department of Microbiology and Immunology, The Pennsylvania State University College of Medicine, Hershey, Pennsylvania 17033 Received 2 December 2002/Accepted 10 July 2003 Glucocorticoids have been shown to play a role in the transforming abilities of human papillomaviruses (HPVs), and glucocorticoid response elements (GREs) have been identified in the upstream regulatory regions (URRs) of various HPV types. These findings have made glucocorticoids potential therapeutic targets for HPV infection. We have previously shown that the URR of HPV type 31 (HPV31) is insensitive to induction by the synthetic glucocorticoid dexamethasone (dex) in monolayer culture, despite the identification of three potential GREs in the 5 region of the URR. Due to the fact that the HPV life cycle is intimately linked to the differentiation of the host tissue, we chose to determine whether the URR of HPV31 was inducible by dex under differentiating conditions. Upon suspension of cells in a semisolid medium of methylcellulose, we found that the URR of HPV31 was inducible by dex. The three GREs appear to play roles as independent repressors of this inducibility. By 5 deletion analysis, the element(s) responsible for this induction was localized to nucleotides (nt) 7238 to 7557. Furthermore, we found that the region between nt 7883 and 7900 appears to act as a repressor of dex inducibility. These findings indicate that epithelial differentiation has a profound effect on the action of dex on the URR of HPV31, suggesting that glucocorticoids play an important role in the differentiation-dependent life cycle of HPV. Human papillomavirus (HPV) comprises a group of over 100 types of small, double-stranded DNA viruses that infect differentiating squamous epithelium. HPV infection is thought to occur primarily through microabrasions in the epithelium, upon which HPV DNA is maintained in the basal cells of the epithelium as extrachromosomal episomes. Viral early-gene expression is controlled by promoter and enhancer elements located in the upstream regulatory region (URR) of the ge- nome (21). Early viral genes E1 and E2 regulate viral DNA replication, which occurs along with cellular DNA replication in the mitotically active basal cells (27, 45). Expression of the late viral genes occurs as HPV-infected cells migrate up through the epithelium and undergo a complex program of differentiation (7, 17, 35). The life cycle of HPV is tightly linked to the differentiation of the epithelium it infects (41). In undifferentiated keratino- cytes, viral DNA is maintained at low copy numbers and viral gene expression is limited (9). Upon epithelial differentiation, viral DNA is amplified to over 1,000 copies per cell and an increase in viral gene expression is observed. While the major- ity of early viral genes are expressed in both undifferentiated and differentiated epithelial cells, expression of the late genes occurs only in the terminally differentiated layers of the epi- thelium (23). These differentiation-dependent events occur in preparation for virion morphogenesis. Late-gene expression is controlled by a differentiation-dependent promoter; in HPV31, it is p742 (22), and in HPV16, it is p670 (20). Both p742 pro- moter activity and late-gene expression have been shown to be upregulated in differentiating organotypic (raft) cultures com- pared with what occurs in monolayers of the CIN612-9E (9E) cell line (35, 36). This cell line is a clonal isolate of a cervical intraepithelial neoplasia grade I lesion that maintains episomal copies of HPV type 31b (HPV31b) (7) and has been shown to produce infectious virus in raft cultures (32). Cellular transcription factors have been shown to modulate the activity of the early viral promoter by binding to cis ele- ments in the URR. Changes in the expressions of viral genes upon epithelial differentiation can be explained in part by differences in the expressions and activities of these cellular factors. Numerous studies have demonstrated that changes in viral gene expression upon differentiation are strongly influ- enced by the levels and activities of cellular transcription fac- tors and that this influence is mediated by the binding, or lack thereof, to the viral URR (1). The dependence of a productive HPV infection on epithelial differentiation has caused difficulties in the study of the com- plete HPV life cycle in vitro. Advances in the organotypic (raft) culture system have allowed for the growth of differentiating epithelium in vitro and have made it possible to provide all of the factors necessary for the complete viral life cycle (10, 32, 33, 35). However, preparation of raft cultures can be time consuming, requiring up to 12 days in culture prior to analysis of late viral events, which can limit the rapid analysis of mul- tiple constructs. Suspension of epithelial cells in a semisolid medium of methylcellulose has been shown to allow for the * Corresponding author. Mailing address: Department of Microbi- ology and Immunology, The Pennsylvania State University College of Medicine, Hershey, PA 17033. Phone: (717) 531-6240. Fax: (717) 531- 4600. E-mail: [email protected]. † Present address: Laboratory of Tumor Metastasis and Angiogen- esis, The Van Andel Research Institute, Grand Rapids, MI 49503. 10975 on February 18, 2016 by guest http://jvi.asm.org/ Downloaded from
-
Upload
independent -
Category
Documents
-
view
8 -
download
0
Transcript of Induction of the Upstream Regulatory Region of Human Papillomavirus Type 31 by Dexamethasone Is...

JOURNAL OF VIROLOGY, Oct. 2003, p. 10975–10983 Vol. 77, No. 200022-538X/03/$08.00�0 DOI: 10.1128/JVI.77.20.10975–10983.2003Copyright © 2003, American Society for Microbiology. All Rights Reserved.
Induction of the Upstream Regulatory Region of HumanPapillomavirus Type 31 by Dexamethasone Is
Differentiation DependentJennifer L. Bromberg-White,† Ellora Sen, Samina Alam, Jason M. Bodily,
and Craig Meyers*Department of Microbiology and Immunology, The Pennsylvania State University
College of Medicine, Hershey, Pennsylvania 17033
Received 2 December 2002/Accepted 10 July 2003
Glucocorticoids have been shown to play a role in the transforming abilities of human papillomaviruses(HPVs), and glucocorticoid response elements (GREs) have been identified in the upstream regulatory regions(URRs) of various HPV types. These findings have made glucocorticoids potential therapeutic targets for HPVinfection. We have previously shown that the URR of HPV type 31 (HPV31) is insensitive to induction by thesynthetic glucocorticoid dexamethasone (dex) in monolayer culture, despite the identification of three potentialGREs in the 5� region of the URR. Due to the fact that the HPV life cycle is intimately linked to thedifferentiation of the host tissue, we chose to determine whether the URR of HPV31 was inducible by dex underdifferentiating conditions. Upon suspension of cells in a semisolid medium of methylcellulose, we found thatthe URR of HPV31 was inducible by dex. The three GREs appear to play roles as independent repressors ofthis inducibility. By 5� deletion analysis, the element(s) responsible for this induction was localized tonucleotides (nt) 7238 to 7557. Furthermore, we found that the region between nt 7883 and 7900 appears to actas a repressor of dex inducibility. These findings indicate that epithelial differentiation has a profound effecton the action of dex on the URR of HPV31, suggesting that glucocorticoids play an important role in thedifferentiation-dependent life cycle of HPV.
Human papillomavirus (HPV) comprises a group of over100 types of small, double-stranded DNA viruses that infectdifferentiating squamous epithelium. HPV infection is thoughtto occur primarily through microabrasions in the epithelium,upon which HPV DNA is maintained in the basal cells of theepithelium as extrachromosomal episomes. Viral early-geneexpression is controlled by promoter and enhancer elementslocated in the upstream regulatory region (URR) of the ge-nome (21). Early viral genes E1 and E2 regulate viral DNAreplication, which occurs along with cellular DNA replicationin the mitotically active basal cells (27, 45). Expression of thelate viral genes occurs as HPV-infected cells migrate upthrough the epithelium and undergo a complex program ofdifferentiation (7, 17, 35).
The life cycle of HPV is tightly linked to the differentiationof the epithelium it infects (41). In undifferentiated keratino-cytes, viral DNA is maintained at low copy numbers and viralgene expression is limited (9). Upon epithelial differentiation,viral DNA is amplified to over 1,000 copies per cell and anincrease in viral gene expression is observed. While the major-ity of early viral genes are expressed in both undifferentiatedand differentiated epithelial cells, expression of the late genesoccurs only in the terminally differentiated layers of the epi-thelium (23). These differentiation-dependent events occur in
preparation for virion morphogenesis. Late-gene expression iscontrolled by a differentiation-dependent promoter; in HPV31, itis p742 (22), and in HPV16, it is p670 (20). Both p742 pro-moter activity and late-gene expression have been shown to beupregulated in differentiating organotypic (raft) cultures com-pared with what occurs in monolayers of the CIN612-9E (9E)cell line (35, 36). This cell line is a clonal isolate of a cervicalintraepithelial neoplasia grade I lesion that maintains episomalcopies of HPV type 31b (HPV31b) (7) and has been shown toproduce infectious virus in raft cultures (32).
Cellular transcription factors have been shown to modulatethe activity of the early viral promoter by binding to cis ele-ments in the URR. Changes in the expressions of viral genesupon epithelial differentiation can be explained in part bydifferences in the expressions and activities of these cellularfactors. Numerous studies have demonstrated that changes inviral gene expression upon differentiation are strongly influ-enced by the levels and activities of cellular transcription fac-tors and that this influence is mediated by the binding, or lackthereof, to the viral URR (1).
The dependence of a productive HPV infection on epithelialdifferentiation has caused difficulties in the study of the com-plete HPV life cycle in vitro. Advances in the organotypic (raft)culture system have allowed for the growth of differentiatingepithelium in vitro and have made it possible to provide all ofthe factors necessary for the complete viral life cycle (10, 32,33, 35). However, preparation of raft cultures can be timeconsuming, requiring up to 12 days in culture prior to analysisof late viral events, which can limit the rapid analysis of mul-tiple constructs. Suspension of epithelial cells in a semisolidmedium of methylcellulose has been shown to allow for the
* Corresponding author. Mailing address: Department of Microbi-ology and Immunology, The Pennsylvania State University College ofMedicine, Hershey, PA 17033. Phone: (717) 531-6240. Fax: (717) 531-4600. E-mail: [email protected].
† Present address: Laboratory of Tumor Metastasis and Angiogen-esis, The Van Andel Research Institute, Grand Rapids, MI 49503.
10975
on February 18, 2016 by guest
http://jvi.asm.org/
Dow
nloaded from

rapid induction of differentiation in only 48 h (16, 24). HPV31-positive epithelial cells have been shown to express the differ-entiation markers involucrin and transglutaminase after sus-pension in methylcellulose as well as to express transcriptsfrom p742, although no capsid proteins have been detected inthis system (38). Therefore, suspension in methylcellulose is aconvenient system for use in a quick analysis of the effect ofdifferentiation on a variety of life cycle events, even though itcannot facilitate the final steps of the viral life cycle.
In addition to the other transcription factors mentioned,glucocorticoids have been shown to affect viral early-gene ex-pression and promoter activity by binding to glucocorticoidresponse elements (GREs) in the URR (12, 14, 19, 30). Brom-berg-White and Meyers have previously shown that the URRof HPV31 is not inducible by the synthetic glucocorticoid dexa-methasone (dex) in monolayer culture (10). In the presentstudy, we demonstrate that suspension of the HPV31b-positive9E cell line in the semisolid medium methylcellulose results ina dex-inducible activation of a luciferase promoter constructdriven by the URR of HPV31 that was not seen in monolayerculture. The three GREs we identified by sequence analysisand their ability to bind the glucocorticoid receptor (GR) inthe URR of HPV31 do not appear to be responsible for thisinducibility. However, loss of all three GREs resulted in anincrease in the induction, suggesting that each of the threeGREs acts as a repressor of glucocorticoid action. By 5� dele-tion analysis, we localized the element(s) responsible for thedex inducibility of the URR of HPV31 to nucleotides (nt) 7238to 7557. Furthermore, replacement of wild-type sequences be-tween nt 7883 and 7900 by linker scanning mutagenesis re-sulted in an increased dex inducibility. Taken together, theseresults show that differentiation has a profound effect on theaction of dex on the URR of HPV31, suggesting that glucocor-ticoids may play an important role in the differentiation-de-pendent life cycle of HPV.
MATERIALS AND METHODS
Cell culture. The 9E cell line is a clonal isolate derived from a cervicalintraepithelial neoplasia grade I lesion that maintains 50 episomal copies ofHPV31b per cell (7). Primary human foreskin keratinocyte (HFK) cultures werederived from newborn foreskin via trypsin digestion at 37°C (38). 9E cells weregrown in E medium in the presence of mitomycin-C-treated J2 fibroblasts aspreviously described (7, 31, 32). HFK cells were maintained in KGM-2 (Bio-Whittaker, Walkersville, Md.). Deficient E medium was used to culture 9E cellsfor transfection experiments as described previously (10).
Plasmid constructs. The full-length URR of HPV31 or HPV18 was clonedinto the pGL2-Basic luciferase reporter vector as described previously (10, 40).All other constructs were created as described previously (10, 40).
Transfection and luciferase assay. For monolayer studies, 9E cells were trans-fected as previously described (10). HFK cells were seeded at a density of 2 � 105
cells per 35-mm-diameter dish in KGM-2. Twelve hours later, cells were trans-fected by using Lipofectamine PLUS (Invitrogen-Life Technologies, Carlsbad,Calif.). Each well was transfected in KGM-2 with 1 �g of experimental construct,6 �l of PLUS reagent, and 4 �l of Lipofectamine for 3 h. The transfectionsolution was replaced with fresh KGM-2 that contained either 0.1% ethyl alcohol(vehicle) or 10�6 M dex. After transfection, monolayer cells were incubated for48 h at 37°C in an atmosphere of 5% CO2. For differentiation studies, 9E or HFKcells were seeded at a density of 8 � 105 cells per 100-mm-diameter dish. 9E cellswere first seeded in deficient E medium (which contains no hydrocortisone orphenol red and is supplemented with dextran-coated, charcoal-stripped serum)for 3 h, at which point the medium was switched to KGM-2. HFK cells wereseeded and maintained in KGM-2 for the duration of the transfection. At 12 h(9E) or 16 h (HFK) postseeding, cells were transfected in KGM-2 with 4 �g ofexperimental construct, 20 �l of PLUS reagent, and 30 �l of Lipofectamine per
100-mm-diameter dish. After a 3-h incubation, cells were trypsinized, pelleted,and resuspended in 0.5 ml of deficient E medium. Cells were then suspended in15 ml of 1.6% methylcellulose and treated with vehicle or 10�6 M dex. Methyl-cellulose was prepared exactly as described previously, using Deficient E medium(38, 40). After a 48-h incubation, cells in methylcellulose were recovered by threerounds of washes in phosphate-buffered saline. Luciferase lysates of all transfec-tions were prepared using passive lysis buffer (Promega Corp., Madison, Wis.).Luciferase assays were performed by using the Luciferase Assay System (Pro-mega) with a Turner Designs TD 20/20 luminometer as recommended by themanufacturer. For methylcellulose experiments, luciferase lysates were normal-ized to total protein by the Bradford assay by using a protein assay dye reagent(Bio-Rad Laboratories, Hercules, Calif.).
Nuclear-extract preparation and EMSA. Nuclear extracts from 9E cells wereprepared as described previously (26). Briefly, 2 � 106 cells were suspended in400 �l of buffer A (10 mM HEPES [pH 7.9], 10 mM KCl, 0.1 mM EDTA, 0.1mM EGTA, 1 mM dithiothreitol, 0.5 mM phenylmethylsulfonyl fluoride). Thecells were allowed to swell on ice for 20 min, after which 25 �l of IGEPALCA-630 (Sigma) was added, followed by vigorous mixing for 20 s. Followingcentrifugation for 30 s, the pellet was recovered and resuspended in buffer B (20mM HEPES [pH 7.9], 400 mM KCl, 1 mM EDTA, 0.5 mM phenylmethylsulfonylfluoride), shaken at 4°C for 30 min, and centrifuged for 5 min at 4°C. Thesupernatants were recovered, and protein concentration was measured by usinga BC protein assay kit (Pierce, Rockford, Ill.). Blunt-ended, double-strandedoligonucleotides were end labeled using [�32-P]ATP and T4 polynucleotide ki-nase as previously described (10). Competition electrophoretic mobility shiftassays (EMSA) were performed by incubating 10 �g of nuclear extract with 2 �gof poly(dI-dC) (Pharmacia, Kalamazoo, Mich.) in the presence or absence of a100-fold excess of unlabeled homologous oligonucleotide or oligonucleotidecorresponding to the consensus GRE (GREc) sequence for 20 min at roomtemperature in a 20-�l reaction volume containing 5% glycerol, 150 mM KCl, 0.5mM EDTA, and 0.5 mM dithiothreitol. Following incubation, 0.75 ng of 32P-labeled oligonucleotide probe was added to the reaction, followed by incubationat room temperature for an additional 30 min. The DNA-protein complexeswere separated from free probe by electrophoresis on a 6% Tris-glycine-EDTApolyacrylamide gel (acrylamide to bisacrylamide, 37.5:1) at 200 V at 4°C. Gelswere dried at 80°C for 2 h and subjected to autoradiography.
RESULTS
HPV31 URR is inducible by dex in methylcellulose. It haspreviously been shown that the URR of HPV31 was insensitiveto induction by dex in monolayer 9E culture (10) comparedwith the URR of HPV18, which has been shown previously tocontain a functional GRE (12, 14, 30). Because the life cycle ofHPV is intimately linked to the differentiation of the epithe-lium it infects, we were interested in determining whether theURR of HPV31 was inducible by dex in a differentiating sys-tem. Therefore, we chose to analyze the induction of transcrip-tional activity of the URR of HPV31 by dex in a semisolidmedium, methylcellulose. Suspension in methylcellulose pro-vides a simplified system for differentiation of epithelial cells ina shorter time frame than that with the organotypic (raft) cul-ture system (16, 24). While the growth of raft cultures is nec-essary in order to obtain the complete viral life cycle, includinginfectious-virus production, cells suspended in methylcelluloseare able to undergo a variety of differentiation-dependent lateevents of the viral life cycle, including viral-genome amplifica-tion and late-gene transcript expression (38). To determine ifcells suspended in methylcellulose are in fact undergoing dif-ferentiation, differentiation-specific cell markers are com-monly analyzed (15, 32, 38, 40). We have previously shown thatthe 9E cells are able to differentiate when suspended in meth-ylcellulose, as evidenced by an increase in the expression of thedifferentiation markers K10 and involucrin (40).
We wanted to determine whether suspension of these cellsin methylcellulose would alter the dex inducibility of the wild-
10976 BROMBERG-WHITE ET AL. J. VIROL.
on February 18, 2016 by guest
http://jvi.asm.org/
Dow
nloaded from
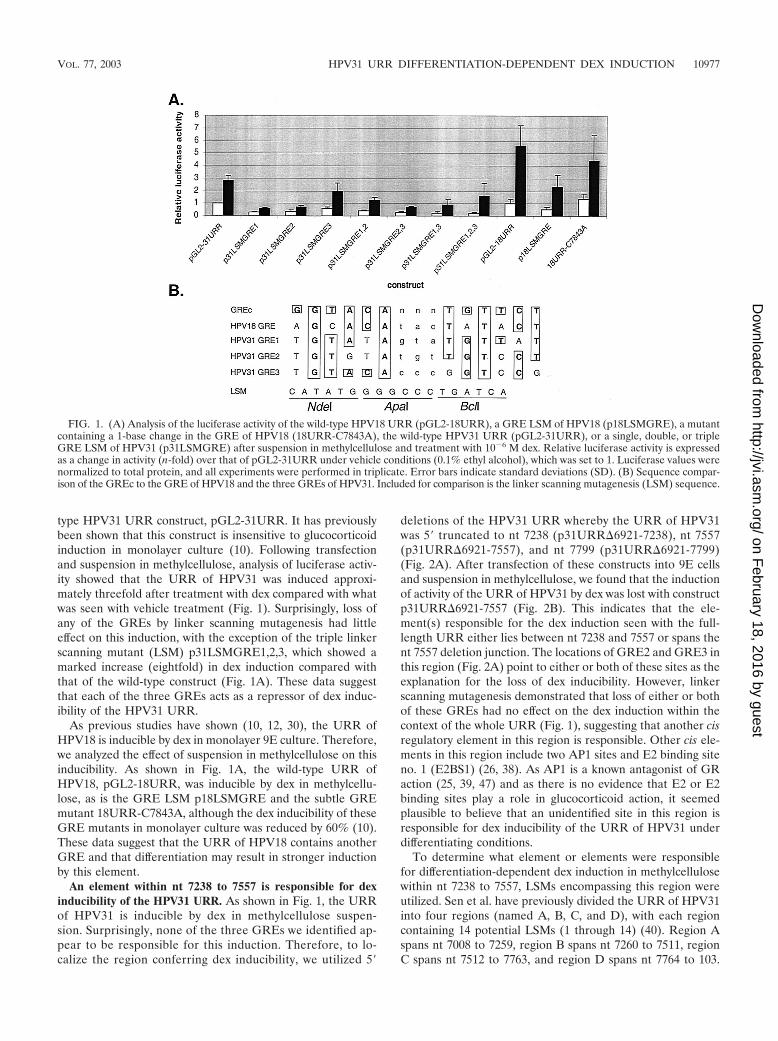
type HPV31 URR construct, pGL2-31URR. It has previouslybeen shown that this construct is insensitive to glucocorticoidinduction in monolayer culture (10). Following transfectionand suspension in methylcellulose, analysis of luciferase activ-ity showed that the URR of HPV31 was induced approxi-mately threefold after treatment with dex compared with whatwas seen with vehicle treatment (Fig. 1). Surprisingly, loss ofany of the GREs by linker scanning mutagenesis had littleeffect on this induction, with the exception of the triple linkerscanning mutant (LSM) p31LSMGRE1,2,3, which showed amarked increase (eightfold) in dex induction compared withthat of the wild-type construct (Fig. 1A). These data suggestthat each of the three GREs acts as a repressor of dex induc-ibility of the HPV31 URR.
As previous studies have shown (10, 12, 30), the URR ofHPV18 is inducible by dex in monolayer 9E culture. Therefore,we analyzed the effect of suspension in methylcellulose on thisinducibility. As shown in Fig. 1A, the wild-type URR ofHPV18, pGL2-18URR, was inducible by dex in methylcellu-lose, as is the GRE LSM p18LSMGRE and the subtle GREmutant 18URR-C7843A, although the dex inducibility of theseGRE mutants in monolayer culture was reduced by 60% (10).These data suggest that the URR of HPV18 contains anotherGRE and that differentiation may result in stronger inductionby this element.
An element within nt 7238 to 7557 is responsible for dexinducibility of the HPV31 URR. As shown in Fig. 1, the URRof HPV31 is inducible by dex in methylcellulose suspen-sion. Surprisingly, none of the three GREs we identified ap-pear to be responsible for this induction. Therefore, to lo-calize the region conferring dex inducibility, we utilized 5�
deletions of the HPV31 URR whereby the URR of HPV31was 5� truncated to nt 7238 (p31URR�6921-7238), nt 7557(p31URR�6921-7557), and nt 7799 (p31URR�6921-7799)(Fig. 2A). After transfection of these constructs into 9E cellsand suspension in methylcellulose, we found that the inductionof activity of the URR of HPV31 by dex was lost with constructp31URR�6921-7557 (Fig. 2B). This indicates that the ele-ment(s) responsible for the dex induction seen with the full-length URR either lies between nt 7238 and 7557 or spans thent 7557 deletion junction. The locations of GRE2 and GRE3 inthis region (Fig. 2A) point to either or both of these sites as theexplanation for the loss of dex inducibility. However, linkerscanning mutagenesis demonstrated that loss of either or bothof these GREs had no effect on the dex induction within thecontext of the whole URR (Fig. 1), suggesting that another cisregulatory element in this region is responsible. Other cis ele-ments in this region include two AP1 sites and E2 binding siteno. 1 (E2BS1) (26, 38). As AP1 is a known antagonist of GRaction (25, 39, 47) and as there is no evidence that E2 or E2binding sites play a role in glucocorticoid action, it seemedplausible to believe that an unidentified site in this region isresponsible for dex inducibility of the URR of HPV31 underdifferentiating conditions.
To determine what element or elements were responsiblefor differentiation-dependent dex induction in methylcellulosewithin nt 7238 to 7557, LSMs encompassing this region wereutilized. Sen et al. have previously divided the URR of HPV31into four regions (named A, B, C, and D), with each regioncontaining 14 potential LSMs (1 through 14) (40). Region Aspans nt 7008 to 7259, region B spans nt 7260 to 7511, regionC spans nt 7512 to 7763, and region D spans nt 7764 to 103.
FIG. 1. (A) Analysis of the luciferase activity of the wild-type HPV18 URR (pGL2-18URR), a GRE LSM of HPV18 (p18LSMGRE), a mutantcontaining a 1-base change in the GRE of HPV18 (18URR-C7843A), the wild-type HPV31 URR (pGL2-31URR), or a single, double, or tripleGRE LSM of HPV31 (p31LSMGRE) after suspension in methylcellulose and treatment with 10�6 M dex. Relative luciferase activity is expressedas a change in activity (n-fold) over that of pGL2-31URR under vehicle conditions (0.1% ethyl alcohol), which was set to 1. Luciferase values werenormalized to total protein, and all experiments were performed in triplicate. Error bars indicate standard deviations (SD). (B) Sequence compar-ison of the GREc to the GRE of HPV18 and the three GREs of HPV31. Included for comparison is the linker scanning mutagenesis (LSM) sequence.
VOL. 77, 2003 HPV31 URR DIFFERENTIATION-DEPENDENT DEX INDUCTION 10977
on February 18, 2016 by guest
http://jvi.asm.org/
Dow
nloaded from
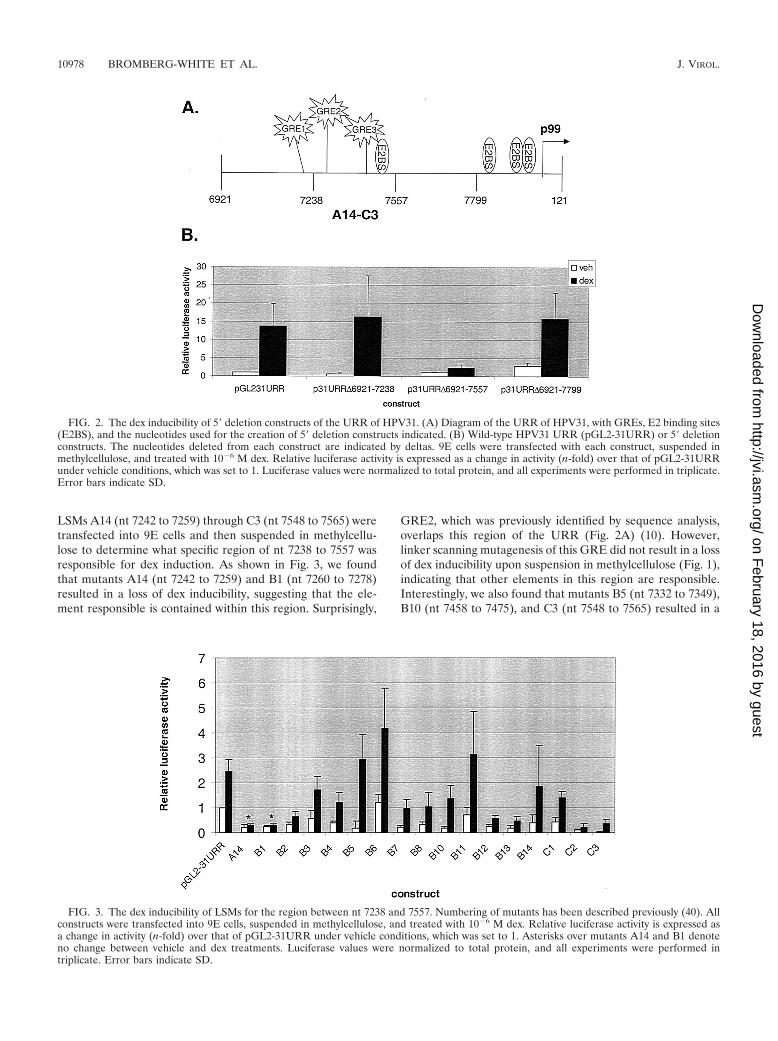
LSMs A14 (nt 7242 to 7259) through C3 (nt 7548 to 7565) weretransfected into 9E cells and then suspended in methylcellu-lose to determine what specific region of nt 7238 to 7557 wasresponsible for dex induction. As shown in Fig. 3, we foundthat mutants A14 (nt 7242 to 7259) and B1 (nt 7260 to 7278)resulted in a loss of dex inducibility, suggesting that the ele-ment responsible is contained within this region. Surprisingly,
GRE2, which was previously identified by sequence analysis,overlaps this region of the URR (Fig. 2A) (10). However,linker scanning mutagenesis of this GRE did not result in a lossof dex inducibility upon suspension in methylcellulose (Fig. 1),indicating that other elements in this region are responsible.Interestingly, we also found that mutants B5 (nt 7332 to 7349),B10 (nt 7458 to 7475), and C3 (nt 7548 to 7565) resulted in a
FIG. 2. The dex inducibility of 5� deletion constructs of the URR of HPV31. (A) Diagram of the URR of HPV31, with GREs, E2 binding sites(E2BS), and the nucleotides used for the creation of 5� deletion constructs indicated. (B) Wild-type HPV31 URR (pGL2-31URR) or 5� deletionconstructs. The nucleotides deleted from each construct are indicated by deltas. 9E cells were transfected with each construct, suspended inmethylcellulose, and treated with 10�6 M dex. Relative luciferase activity is expressed as a change in activity (n-fold) over that of pGL2-31URRunder vehicle conditions, which was set to 1. Luciferase values were normalized to total protein, and all experiments were performed in triplicate.Error bars indicate SD.
FIG. 3. The dex inducibility of LSMs for the region between nt 7238 and 7557. Numbering of mutants has been described previously (40). Allconstructs were transfected into 9E cells, suspended in methylcellulose, and treated with 10�6 M dex. Relative luciferase activity is expressed asa change in activity (n-fold) over that of pGL2-31URR under vehicle conditions, which was set to 1. Asterisks over mutants A14 and B1 denoteno change between vehicle and dex treatments. Luciferase values were normalized to total protein, and all experiments were performed intriplicate. Error bars indicate SD.
10978 BROMBERG-WHITE ET AL. J. VIROL.
on February 18, 2016 by guest
http://jvi.asm.org/
Dow
nloaded from
strong upregulation of dex induction of the URR of HPV31(Fig. 3), suggesting the presence of repressors of dex inductionin this region.
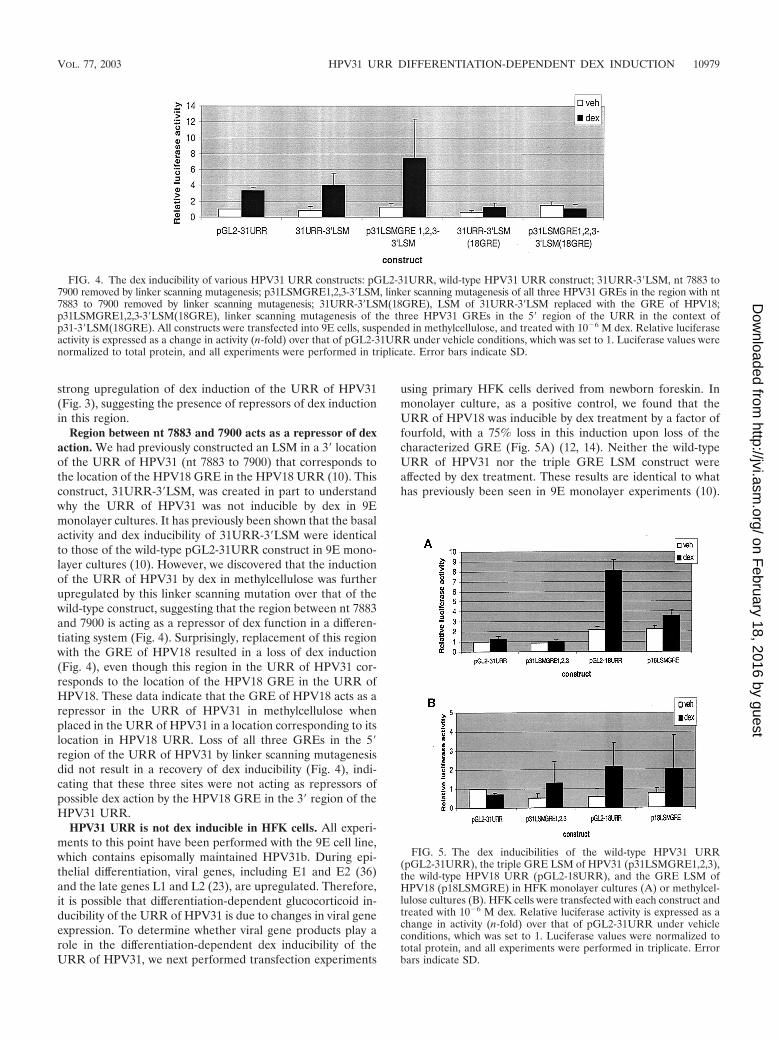
Region between nt 7883 and 7900 acts as a repressor of dexaction. We had previously constructed an LSM in a 3� locationof the URR of HPV31 (nt 7883 to 7900) that corresponds tothe location of the HPV18 GRE in the HPV18 URR (10). Thisconstruct, 31URR-3�LSM, was created in part to understandwhy the URR of HPV31 was not inducible by dex in 9Emonolayer cultures. It has previously been shown that the basalactivity and dex inducibility of 31URR-3�LSM were identicalto those of the wild-type pGL2-31URR construct in 9E mono-layer cultures (10). However, we discovered that the inductionof the URR of HPV31 by dex in methylcellulose was furtherupregulated by this linker scanning mutation over that of thewild-type construct, suggesting that the region between nt 7883and 7900 is acting as a repressor of dex function in a differen-tiating system (Fig. 4). Surprisingly, replacement of this regionwith the GRE of HPV18 resulted in a loss of dex induction(Fig. 4), even though this region in the URR of HPV31 cor-responds to the location of the HPV18 GRE in the URR ofHPV18. These data indicate that the GRE of HPV18 acts as arepressor in the URR of HPV31 in methylcellulose whenplaced in the URR of HPV31 in a location corresponding to itslocation in HPV18 URR. Loss of all three GREs in the 5�region of the URR of HPV31 by linker scanning mutagenesisdid not result in a recovery of dex inducibility (Fig. 4), indi-cating that these three sites were not acting as repressors ofpossible dex action by the HPV18 GRE in the 3� region of theHPV31 URR.
HPV31 URR is not dex inducible in HFK cells. All experi-ments to this point have been performed with the 9E cell line,which contains episomally maintained HPV31b. During epi-thelial differentiation, viral genes, including E1 and E2 (36)and the late genes L1 and L2 (23), are upregulated. Therefore,it is possible that differentiation-dependent glucocorticoid in-ducibility of the URR of HPV31 is due to changes in viral geneexpression. To determine whether viral gene products play arole in the differentiation-dependent dex inducibility of theURR of HPV31, we next performed transfection experiments
using primary HFK cells derived from newborn foreskin. Inmonolayer culture, as a positive control, we found that theURR of HPV18 was inducible by dex treatment by a factor offourfold, with a 75% loss in this induction upon loss of thecharacterized GRE (Fig. 5A) (12, 14). Neither the wild-typeURR of HPV31 nor the triple GRE LSM construct wereaffected by dex treatment. These results are identical to whathas previously been seen in 9E monolayer experiments (10).
FIG. 4. The dex inducibility of various HPV31 URR constructs: pGL2-31URR, wild-type HPV31 URR construct; 31URR-3�LSM, nt 7883 to7900 removed by linker scanning mutagenesis; p31LSMGRE1,2,3-3�LSM, linker scanning mutagenesis of all three HPV31 GREs in the region with nt7883 to 7900 removed by linker scanning mutagenesis; 31URR-3�LSM(18GRE), LSM of 31URR-3�LSM replaced with the GRE of HPV18;p31LSMGRE1,2,3-3�LSM(18GRE), linker scanning mutagenesis of the three HPV31 GREs in the 5� region of the URR in the context ofp31-3�LSM(18GRE). All constructs were transfected into 9E cells, suspended in methylcellulose, and treated with 10�6 M dex. Relative luciferaseactivity is expressed as a change in activity (n-fold) over that of pGL2-31URR under vehicle conditions, which was set to 1. Luciferase values werenormalized to total protein, and all experiments were performed in triplicate. Error bars indicate SD.
FIG. 5. The dex inducibilities of the wild-type HPV31 URR(pGL2-31URR), the triple GRE LSM of HPV31 (p31LSMGRE1,2,3),the wild-type HPV18 URR (pGL2-18URR), and the GRE LSM ofHPV18 (p18LSMGRE) in HFK monolayer cultures (A) or methylcel-lulose cultures (B). HFK cells were transfected with each construct andtreated with 10�6 M dex. Relative luciferase activity is expressed as achange in activity (n-fold) over that of pGL2-31URR under vehicleconditions, which was set to 1. Luciferase values were normalized tototal protein, and all experiments were performed in triplicate. Errorbars indicate SD.
VOL. 77, 2003 HPV31 URR DIFFERENTIATION-DEPENDENT DEX INDUCTION 10979
on February 18, 2016 by guest
http://jvi.asm.org/
Dow
nloaded from
Upon suspension of transfected cell in methylcellulose, wefound that the URR of HPV18 was only weakly inducible bydex and that the URR of HPV31 was not inducible at all (Fig.5B). This is in direct contrast to what was seen with 9E cells,where the URR of HPV18 and the URR of HPV31 wereinducible by dex (Fig. 1 and 2). Furthermore, we also notedthat the triple LSM GRE construct of HPV31 and the LSMGRE construct of HPV18 were not strongly inducible by dex inHFK methylcellulose cultures (Fig. 5B).
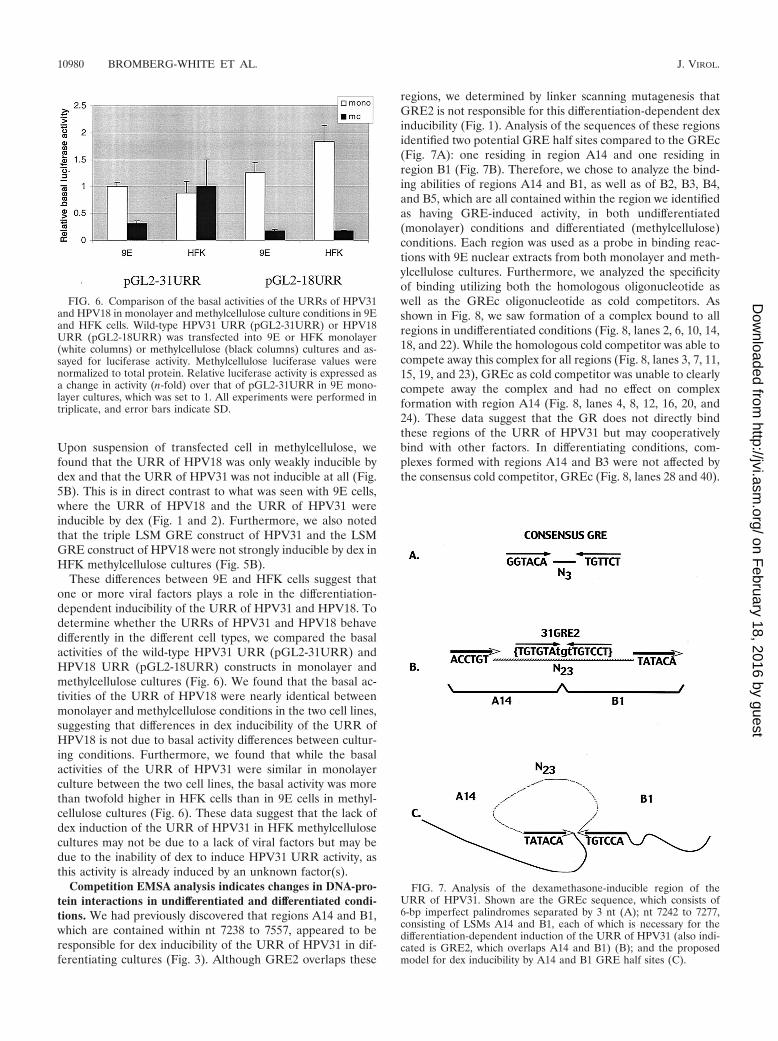
These differences between 9E and HFK cells suggest thatone or more viral factors plays a role in the differentiation-dependent inducibility of the URR of HPV31 and HPV18. Todetermine whether the URRs of HPV31 and HPV18 behavedifferently in the different cell types, we compared the basalactivities of the wild-type HPV31 URR (pGL2-31URR) andHPV18 URR (pGL2-18URR) constructs in monolayer andmethylcellulose cultures (Fig. 6). We found that the basal ac-tivities of the URR of HPV18 were nearly identical betweenmonolayer and methylcellulose conditions in the two cell lines,suggesting that differences in dex inducibility of the URR ofHPV18 is not due to basal activity differences between cultur-ing conditions. Furthermore, we found that while the basalactivities of the URR of HPV31 were similar in monolayerculture between the two cell lines, the basal activity was morethan twofold higher in HFK cells than in 9E cells in methyl-cellulose cultures (Fig. 6). These data suggest that the lack ofdex induction of the URR of HPV31 in HFK methylcellulosecultures may not be due to a lack of viral factors but may bedue to the inability of dex to induce HPV31 URR activity, asthis activity is already induced by an unknown factor(s).
Competition EMSA analysis indicates changes in DNA-pro-tein interactions in undifferentiated and differentiated condi-tions. We had previously discovered that regions A14 and B1,which are contained within nt 7238 to 7557, appeared to beresponsible for dex inducibility of the URR of HPV31 in dif-ferentiating cultures (Fig. 3). Although GRE2 overlaps these
regions, we determined by linker scanning mutagenesis thatGRE2 is not responsible for this differentiation-dependent dexinducibility (Fig. 1). Analysis of the sequences of these regionsidentified two potential GRE half sites compared to the GREc(Fig. 7A): one residing in region A14 and one residing inregion B1 (Fig. 7B). Therefore, we chose to analyze the bind-ing abilities of regions A14 and B1, as well as of B2, B3, B4,and B5, which are all contained within the region we identifiedas having GRE-induced activity, in both undifferentiated(monolayer) conditions and differentiated (methylcellulose)conditions. Each region was used as a probe in binding reac-tions with 9E nuclear extracts from both monolayer and meth-ylcellulose cultures. Furthermore, we analyzed the specificityof binding utilizing both the homologous oligonucleotide aswell as the GREc oligonucleotide as cold competitors. Asshown in Fig. 8, we saw formation of a complex bound to allregions in undifferentiated conditions (Fig. 8, lanes 2, 6, 10, 14,18, and 22). While the homologous cold competitor was able tocompete away this complex for all regions (Fig. 8, lanes 3, 7, 11,15, 19, and 23), GREc as cold competitor was unable to clearlycompete away the complex and had no effect on complexformation with region A14 (Fig. 8, lanes 4, 8, 12, 16, 20, and24). These data suggest that the GR does not directly bindthese regions of the URR of HPV31 but may cooperativelybind with other factors. In differentiating conditions, com-plexes formed with regions A14 and B3 were not affected bythe consensus cold competitor, GREc (Fig. 8, lanes 28 and 40).
FIG. 6. Comparison of the basal activities of the URRs of HPV31and HPV18 in monolayer and methylcellulose culture conditions in 9Eand HFK cells. Wild-type HPV31 URR (pGL2-31URR) or HPV18URR (pGL2-18URR) was transfected into 9E or HFK monolayer(white columns) or methylcellulose (black columns) cultures and as-sayed for luciferase activity. Methylcellulose luciferase values werenormalized to total protein. Relative luciferase activity is expressed asa change in activity (n-fold) over that of pGL2-31URR in 9E mono-layer cultures, which was set to 1. All experiments were performed intriplicate, and error bars indicate SD.
FIG. 7. Analysis of the dexamethasone-inducible region of theURR of HPV31. Shown are the GREc sequence, which consists of6-bp imperfect palindromes separated by 3 nt (A); nt 7242 to 7277,consisting of LSMs A14 and B1, each of which is necessary for thedifferentiation-dependent induction of the URR of HPV31 (also indi-cated is GRE2, which overlaps A14 and B1) (B); and the proposedmodel for dex inducibility by A14 and B1 GRE half sites (C).
10980 BROMBERG-WHITE ET AL. J. VIROL.
on February 18, 2016 by guest
http://jvi.asm.org/
Dow
nloaded from
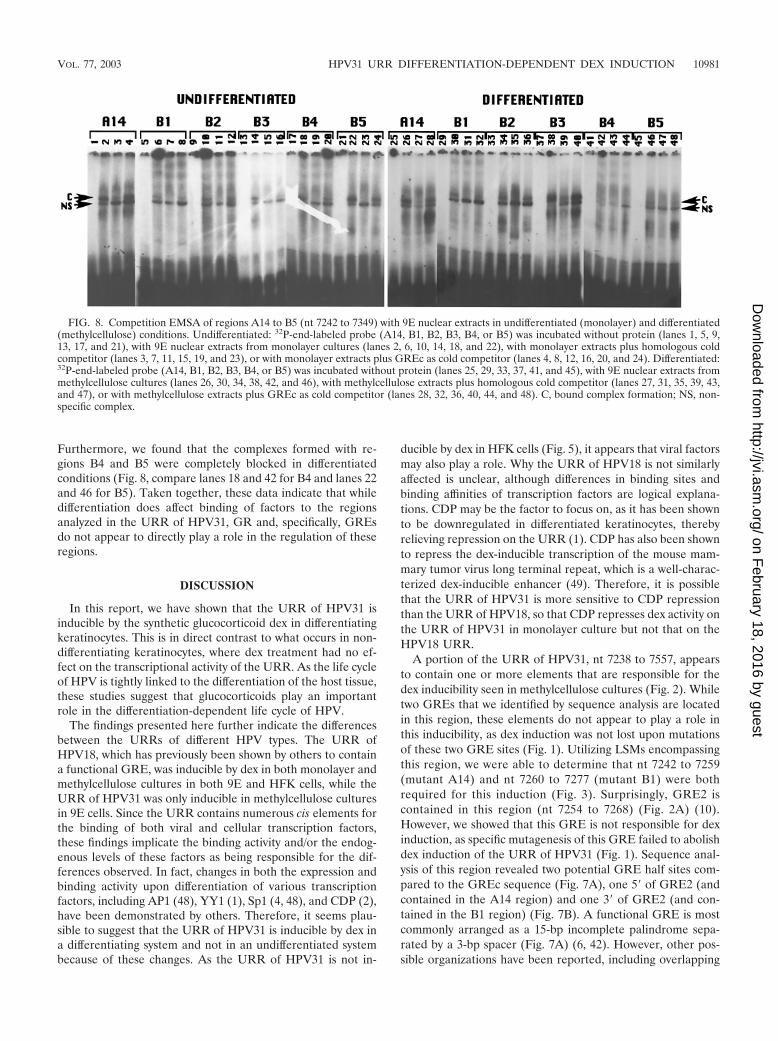
Furthermore, we found that the complexes formed with re-gions B4 and B5 were completely blocked in differentiatedconditions (Fig. 8, compare lanes 18 and 42 for B4 and lanes 22and 46 for B5). Taken together, these data indicate that whiledifferentiation does affect binding of factors to the regionsanalyzed in the URR of HPV31, GR and, specifically, GREsdo not appear to directly play a role in the regulation of theseregions.
DISCUSSION
In this report, we have shown that the URR of HPV31 isinducible by the synthetic glucocorticoid dex in differentiatingkeratinocytes. This is in direct contrast to what occurs in non-differentiating keratinocytes, where dex treatment had no ef-fect on the transcriptional activity of the URR. As the life cycleof HPV is tightly linked to the differentiation of the host tissue,these studies suggest that glucocorticoids play an importantrole in the differentiation-dependent life cycle of HPV.
The findings presented here further indicate the differencesbetween the URRs of different HPV types. The URR ofHPV18, which has previously been shown by others to containa functional GRE, was inducible by dex in both monolayer andmethylcellulose cultures in both 9E and HFK cells, while theURR of HPV31 was only inducible in methylcellulose culturesin 9E cells. Since the URR contains numerous cis elements forthe binding of both viral and cellular transcription factors,these findings implicate the binding activity and/or the endog-enous levels of these factors as being responsible for the dif-ferences observed. In fact, changes in both the expression andbinding activity upon differentiation of various transcriptionfactors, including AP1 (48), YY1 (1), Sp1 (4, 48), and CDP (2),have been demonstrated by others. Therefore, it seems plau-sible to suggest that the URR of HPV31 is inducible by dex ina differentiating system and not in an undifferentiated systembecause of these changes. As the URR of HPV31 is not in-
ducible by dex in HFK cells (Fig. 5), it appears that viral factorsmay also play a role. Why the URR of HPV18 is not similarlyaffected is unclear, although differences in binding sites andbinding affinities of transcription factors are logical explana-tions. CDP may be the factor to focus on, as it has been shownto be downregulated in differentiated keratinocytes, therebyrelieving repression on the URR (1). CDP has also been shownto repress the dex-inducible transcription of the mouse mam-mary tumor virus long terminal repeat, which is a well-charac-terized dex-inducible enhancer (49). Therefore, it is possiblethat the URR of HPV31 is more sensitive to CDP repressionthan the URR of HPV18, so that CDP represses dex activity onthe URR of HPV31 in monolayer culture but not that on theHPV18 URR.
A portion of the URR of HPV31, nt 7238 to 7557, appearsto contain one or more elements that are responsible for thedex inducibility seen in methylcellulose cultures (Fig. 2). Whiletwo GREs that we identified by sequence analysis are locatedin this region, these elements do not appear to play a role inthis inducibility, as dex induction was not lost upon mutationsof these two GRE sites (Fig. 1). Utilizing LSMs encompassingthis region, we were able to determine that nt 7242 to 7259(mutant A14) and nt 7260 to 7277 (mutant B1) were bothrequired for this induction (Fig. 3). Surprisingly, GRE2 iscontained in this region (nt 7254 to 7268) (Fig. 2A) (10).However, we showed that this GRE is not responsible for dexinduction, as specific mutagenesis of this GRE failed to abolishdex induction of the URR of HPV31 (Fig. 1). Sequence anal-ysis of this region revealed two potential GRE half sites com-pared to the GREc sequence (Fig. 7A), one 5� of GRE2 (andcontained in the A14 region) and one 3� of GRE2 (and con-tained in the B1 region) (Fig. 7B). A functional GRE is mostcommonly arranged as a 15-bp incomplete palindrome sepa-rated by a 3-bp spacer (Fig. 7A) (6, 42). However, other pos-sible organizations have been reported, including overlapping
FIG. 8. Competition EMSA of regions A14 to B5 (nt 7242 to 7349) with 9E nuclear extracts in undifferentiated (monolayer) and differentiated(methylcellulose) conditions. Undifferentiated: 32P-end-labeled probe (A14, B1, B2, B3, B4, or B5) was incubated without protein (lanes 1, 5, 9,13, 17, and 21), with 9E nuclear extracts from monolayer cultures (lanes 2, 6, 10, 14, 18, and 22), with monolayer extracts plus homologous coldcompetitor (lanes 3, 7, 11, 15, 19, and 23), or with monolayer extracts plus GREc as cold competitor (lanes 4, 8, 12, 16, 20, and 24). Differentiated:32P-end-labeled probe (A14, B1, B2, B3, B4, or B5) was incubated without protein (lanes 25, 29, 33, 37, 41, and 45), with 9E nuclear extracts frommethylcellulose cultures (lanes 26, 30, 34, 38, 42, and 46), with methylcellulose extracts plus homologous cold competitor (lanes 27, 31, 35, 39, 43,and 47), or with methylcellulose extracts plus GREc as cold competitor (lanes 28, 32, 36, 40, 44, and 48). C, bound complex formation; NS, non-specific complex.
VOL. 77, 2003 HPV31 URR DIFFERENTIATION-DEPENDENT DEX INDUCTION 10981
on February 18, 2016 by guest
http://jvi.asm.org/
Dow
nloaded from

GREs (18), directly repeating half sites (13), and variable spac-ing of half sites (44). Furthermore, studies have shown that therotational positioning of a GRE in a nucleosome is critical forits accessibility (28) and that GR binding to the GRE inducesDNA bending (37). We have found that DNA bending of theURR of HPV31 in the region containing A14 and B1 couldresult in the creation of a complete GRE similar to the GREc(Fig. 7C).
We were surprised to find that loss of nt 7883 to 7900 bylinker scanning mutagenesis led to an increased induction ofthe URR of HPV31 by dex in differentiating cells (Fig. 4). Thisregion in the URR of HPV31 corresponds to the location ofthe GRE in the URR of HPV18. Sequence analysis of thisregion identified a weakly consensus GRE (7 of 12 nt consen-sus) as well as an overlapping, perfectly consensus GRE halfsite (nt 7898 to 7903). Whether GR can bind any of these sitesis unknown. We also identified a potential STAT5 binding sitein this region of the URR. STATs (signal transducers andactivators of transcription) are a family of transcription factorsthat are activated by cytokines such as interleukin-2 (8), inter-leukin-6 (43), and alpha interferon (29). This activation canresult in either positive or negative regulation of glucocorti-coid-induced transcription, the outcome of which is dependenton promoter context and cell type (8). It is possible thatSTAT5 may bind this region of the URR and repress dexactivity. The context of the URR in this region appears to beimportant, as placement of the HPV18 GRE in this locationresults in a repression of dex induction (Fig. 4). This result wasnot expected, as nt 7883 to 7900 in the URR of HPV31 cor-respond to the native location of the HPV18 GRE in the URRof HPV18. The fact that the HPV18 GRE acts as a positiveregulator of dex inducibility in the URR of HPV18 and anegative regulator in the URR of HPV31 suggests differencesin the overall context of the URRs of these two viral types.
The URR of HPV31 was not inducible by dex in HFK cellseither in monolayer or methylcellulose cultures (Fig. 5). It ispossible that viral gene products are necessary for this induc-tion, as the URR is inducible in 9E methylcellulose cultures,which contain endogenous HPV31b and express early viralgenes. E7 has been shown to act as a transcriptional activator(50) and can complex with AP1 factors such as c-jun, Jun B,and Jun D and can transactivate c-jun-induced transcription(3). As AP1 is a well-known antagonist of GR action, it seemedunlikely that E7 could cause induction of dex action. The viralprotein E2 could be important in glucocorticoid-dependentactivation of the URR of HPV31, as an E2 binding site islocated in the region of the URR responsible for dex induc-ibility (Fig. 2A).
Analysis of protein binding to regions A14 and B1, as well asregions B2, B3, B4, and B5, indicated that the GR did notdirectly bind these regions, as the GREc sequence did notclearly compete away the bound complex (Fig. 8), suggestingthat GR either (i) cooperatively binds these regions, resultingin the strengthening of binding of a second transcription factor,or (ii) strengthens the binding of another factor but does notitself bind these regions. Studies have shown that inductionof the mouse mammary tumor virus MMTV promoter by glu-cocorticoids requires functional synergism between GR andnuclear factor 1, with simultaneous occupancy of the promoterbinding sites for these factors (5). In addition, hormonal in-
duction of the MMTV promoter requires binding of othertranscription factors to adjacent promoter sequences in addi-tion to the binding of hormone receptors to their binding sites(11). Others have shown cooperative transactivation of genessuch as �-casein by STAT5, CCAAT/enhancer binding protein� (C/EBP�), and GR, with this cooperativity lost in the ab-sence of GR (46), as well as transcriptional cooperativity of thewhey acidic protein by NF1, STAT5, and GR (34). The presentstudy suggests that regions in the URR of HPV31 within nt7238 to 7557, especially regions A14 and B1, are involved inGR-mediated cooperative transactivation, as loss of these re-gions by linker scanning mutagenesis results in loss of glu-cocorticoid-dependent activation (Fig. 3), without any directinvolvement of GR binding to these regions (Fig. 8).
In conclusion, the URR of HPV31 has been shown to beinducible by the glucocorticoid dex in a differentiating system.From these findings, it is clear that there is a complex interplayof negative and positive factors on the URR of HPV. Further-more, we have shown that glucocorticoid-dependent activationof the URR of HPV is a complex process and is not the samebetween different HPV types.
ACKNOWLEDGMENTS
We thank David Spector and the Meyers laboratory for helpfuldiscussions.
This work was supported by NIH training grants CA60395-06(J.L.B.-W.) and CA79006 and PA-HEALTH 98-07-17 (C.M.).
REFERENCES
1. Ai, W., J. Marahari, and A. Roman. 2000. Yin Yang 1 negatively regulatesthe differentiation-specific E1 promoter of human papillomavirus type 6.J. Virol. 74:5198–5205.
2. Ai, W., E. Toussaint, and A. Roman. 1999. CCAAT displacement proteinbinds to and negatively regulates human papillomavirus type 6 E6, E7, andE1 promoters. J. Virol. 73:4220–4229.
3. Antinore, M. J., M. J. Birrer, D. Patel, L. Nader, and D. J. McCance. 1996.The human papillomavirus type 16 E7 gene product interacts with andtrans-activates the AP1 family of transcription factors. EMBO J. 15:1950–1960.
4. Apt, D., R. Watts, G. Suske, and H. Bernard. 1996. High Sp1/Sp3 ratios inepithelial cells during epithelial differentiation and cellular transformationcorrelate with the activation of the HPV-16 promoter. Virology 224:281–291.
5. Aumais, J. P., H. S. Lee, C. DeGannes, J. Horsford, and J. H. White. 1996.Function of directly repeated half-sites as response elements for steroidhormone receptors. J. Biol. Chem. 271:12568–12577.
6. Becker, P. B., B. Gloss, W. Schmid, U. Strahle, and G. Schutz. 1986. In vivoprotein-DNA interactions in a glucocorticoid response element require thepresence of the hormone. Nature 324:686–688.
7. Bedell, M., J. Hudson, T. Golub, M. Turyk, M. Hosken, G. Wilbanks, and L.Laimins. 1991. Amplification of human papillomavirus genomes in vitro isdependent on epithelial differentiation. J. Virol. 65:2254–2260.
8. Biola, A., P. Lefebvre, M. Perrin-Wolff, M. Sturm, J. Bertoglio, and M.Pallardy. 2001. Interleukin-2 inhibits glucocorticoid receptor transcriptionalactivity through a mechanism involving STAT5 (signal transducer and acti-vator of transcription 5) but not AP-1. Mol. Endocrinol. 15:1062–1076.
9. Broker, T., and M. Botchan. 1986. Papillomaviruses: retrospectives andprospectives. Cancer Cells 4:17–36.
10. Bromberg-White, J. L., and C. Meyers. 2002. The upstream regulatory re-gion of human papillomavirus type 31 is insensitive to glucocorticoid induc-tion. J. Virol. 76:9702–9715.
11. Bruggemeier, U., M. Kalff, S. Franke, C. Scheidereit, and M. Beato. 1991.Ubiquitous transcription factor OTF-1 mediates induction of the MMTVpromoter through synergistic interaction with hormone receptors. Cell 64:565–572.
12. Butz, K., and F. Hoppe-Seyler. 1993. Transcriptional control of human pap-illomavirus (HPV) oncogene expression: composition of the HPV type 18upstream regulatory region. J. Virol. 67:6476–6486.
13. Chan, G. C., P. Hess, T. Meenakshi, J. Carlstedt-Duke, J. A. Gustafsson, andF. Payvar. 1991. Delayed secondary glucocorticoid response elements. Un-usual nucleotide motifs specify glucocorticoid receptor binding to tran-scribed regions of 2u-globulin DNA. J. Biol. Chem. 266:22634–22644.
14. Chan, W., G. Klock, and H. Bernard. 1989. Progesterone and glucocorticoid
10982 BROMBERG-WHITE ET AL. J. VIROL.
on February 18, 2016 by guest
http://jvi.asm.org/
Dow
nloaded from

response elements occur in the long control region of several human papil-lomaviruses involved in anogenital neoplasia. J. Virol. 63:3261–3269.
15. Eckert, R., J. Crish, and N. Robinson. 1997. The epidermal keratinocyte asa model for the study of gene regulation and cell differentiation. Physiol.Rev. 77:397–424.
16. Flores, E. R., and P. F. Lambert. 1997. Evidence for a switch in the mode ofhuman papillomavirus type 16 DNA replication during the viral life cycle.J. Virol. 71:7167–7179.
17. Frattini, M. G., H. B. Lim, and L. A. Laimins. 1996. In vitro synthesis ofoncogenic human papillomaviruses requires episomal genomes for differen-tiation-dependent late expression. Proc. Natl. Acad. Sci. USA 93:3062–3067.
18. Garlatti, M., M. Daheshia, E. Slater, J. Bouguet, J. Hanoune, M. Beato, andR. Barouki. 1994. A functional glucocorticoid-responsive unit composed oftwo overlapping inactive receptor-binding sites: evidence for formation of areceptor tetramer. Mol. Cell. Biol. 14:8007–8017.
19. Gloss, B., H. U. Bernard, K. Seedorf, and G. Klock. 1987. The upstreamregulatory region of the human papilloma virus-16 contains an E2 protein-independent enhancer which is specific for cervical carcinoma cells andregulated by glucocorticoid hormones. EMBO J. 6:3735–3743.
20. Grassmann, K., B. Rapp, H. Mascjek, K. Petry, and T. Ifner. 1996. Identi-fication of a differentiation-inducible promoter in the E7 open reading frameof human papillomavirus type 16 (HPV-16) in raft cultures of a new cell linecontaining high copy numbers of episomal HPV-16 DNA. J. Virol. 70:2339–2349.
21. Howley, P. 1996. Papillomavirinae: the viruses and their replication, p. 2045–2076. In B. N. Fields, D. M. Knipe, and P. M. Howley (ed.), Fields virology,3rd ed., vol. 2. Lippincott-Raven Publishers, Philadelphia, Pa.
22. Hummel, M., J. B. Hudson, and L. A. Laimins. 1992. Differentiation-inducedand constitutive transcription of human papillomavirus type 31b in cell linescontaining viral episomes. J. Virol. 66:6070–6080.
23. Hummel, M., H. Lim, and L. Laimins. 1995. Human papillomavirus type 31blate gene expression is regulated through protein kinase C-mediated changesin RNA processing. J. Virol. 69:3381–3388.
24. Jeon, S., B. L. Allen-Hoffmann, and P. F. Lambert. 1995. Integration ofhuman papillomavirus type 16 into the human genome correlates with aselective growth advantage of cells. J. Virol. 69:2989–2997.
25. Jonat, C., H. Rahmsdorf, K. Park, A. Cato, S. Gebel, H. Ponta, and P.Herrlich. 1990. Antitumor promotion and antiinflammation: down-modula-tion of AP1 (Fos/Jun) activity by glucocorticoid hormone. Cell 62:1189–1204.
26. Kyo, S., A. Tam, and L. A. Laimins. 1995. Transcriptional activity of humanpapillomavirus type 31b enhancer is regulated through synergistic interactionof AP1 with two novel cellular factors. Virology 211:184–197.
27. Lambert, P. F. 1991. Papillomavirus DNA replication. J. Virol. 65:3417–3420.
28. Li, Q., and O. Wrange. 1995. Accessibility of a glucocorticoid responseelement in a nucleosome depends on its rotational positioning. Mol. Cell.Biol. 15:4375–4384.
29. Li, S., S. Labrecque, M. C. Gauzzi, A. R. Cuddihy, A. H. Wong, S. Pellegrini,G. J. Matlashewski, and A. E. Koromilas. 1999. The human papilloma virus(HPV)-18 E6 oncoprotein physically associates with Tyk2 and impairs Jak-STAT activation by interferon-. Oncogene 18:5727–5737.
30. Medina-Martinez, O., N. Morales-Peza, M. Yaniv, A. Garcia-Carranca, andF. Thierry. 1996. A single element mediates glucocorticoid hormone re-sponse of HPV18 with no functional interactions with AP1 or hbrm. Virology217:392–396.
31. Meyers, C. 1996. Organotypic (raft) epithelial tissue culture system for thedifferentiation-dependent replication of papillomavirus. Methods Cell Sci.18:1–10.
32. Meyers, C., M. Frattini, J. Hudson, and L. Laimins. 1992. Biosynthesis ofhuman papillomavirus from a continuous cell line upon epithelial differen-tiation. Science 257:971–973.
33. Meyers, C., T. Mayer, and M. Ozbun. 1997. Synthesis of infectious human
papillomavirus type 18 in differentiating epithelium transfected with viralDNA. J. Virol. 71:7381–7386.
34. Mukhopadhyay, S. S., S. L. Wyszomierski, R. M. Gronostajski, and J. M.Rosen. 2001. Differential interactions of specific nuclear factor I isoformswith the glucocorticoid receptor and STAT5 in the cooperative regulation ofWAP gene transcription. Mol. Cell. Biol. 21:6859–6869.
35. Ozbun, M., and C. Meyers. 1997. Characterization of late gene transcriptsexpressed during vegetative replication of human papillomavirus type 31b.J. Virol. 71:5161–5172.
36. Ozbun, M., and C. Meyers. 1998. Temporal usage of multiple promotersduring the life cycle of human papillomavirus type 31b. J. Virol. 72:2715–2722.
37. Petz, L. N., A. M. Nardulli, J. Kim, K. B. Horwitz, L. P. Freedman, and D. J.Shapiro. 1997. DNA bending is induced by binding of the glucocorticoidreceptor DNA binding domain and progesterone receptors to their responseelement. J. Steroid Biochem. Mol. Biol. 60:31–41.
38. Ruesch, M., F. Stubenrauch, and L. Laimins. 1998. Activation of papillo-mavirus late gene transcription and genome amplification upon differentia-tion in semisolid medium is coincident with expression of involucrin andtransglutaminase but not keratin-10. J. Virol. 72:5016–5024.
39. Schule, R., P. Rangarajan, S. Kliewer, L. J. Ransone, J. Bolado, N. Yang,I. M. Verma, and R. M. Evans. 1990. Functional antagonism between onco-protein c-Jun and the glucocorticoid receptor. Cell 62:1217–1226.
40. Sen, E., J. L. Bromberg-White, and C. Meyers. 2002. Genetic analysis of cisregulatory elements within the 5� region of the human papillomavirus type 31upstream regulatory region during different stages of the viral life cycle.J. Virol. 76:4798–4809.
41. Stoler, M. H., S. M. Wolinsky, A. Whitbeck, T. R. Broker, and L. T. Chow.1989. Differentiation-linked human papillomavirus types 6 and 11 transcrip-tion in genital condylomata revealed by in situ hybridization with message-specific RNA probes. Virology 172:331–340.
42. Strahle, U., G. Klock, and G. Schutz. 1987. A DNA sequence of 15 base pairsis sufficient to mediate both glucocorticoid and progesterone induction ofgene expression. Proc. Natl. Acad. Sci. USA 84:7871–7875.
43. Takeda, T., H. Kurachi, T. Yamamoto, Y. Nishio, Y. Nakatsuji, K. Mori-shige, A. Miyake, and Y. Murata. 1998. Crosstalk between the interleukin-6(IL-6)-JAK-STAT and the glucocorticoid-nuclear receptor pathway: syner-gistic activation of IL-6 response element by IL-6 and glucocorticoid. J.Endocrinol. 159:323–330.
44. Truss, M., and M. Beato. 1993. Steroid hormone receptors: interaction withdeoxyribonucleic acid and transcription factors. Endocr. Rev. 14:459–479.
45. Ustav, M., and A. Stenlund. 1991. Transient replication of BPV-1 requirestwo viral polypeptides encoded by the E1 and E2 open reading frames.EMBO J. 10:449–457.
46. Wyszomierski, S. L., and J. M. Rosen. 2001. Cooperative effects of STAT5(signal transducer and activator of transcription 5) and C/EBP� (CCAAT/enhancer-binding protein-�) on beta-casein gene transcription are mediatedby the glucocorticoid receptor. Mol. Endocrinol. 15:228–240.
47. Yang-Yen, H. F., J. C. Chambard, Y. L. Sun, T. Smeal, T. J. Schmidt, J.Drouin, and M. Karin. 1990. Transcriptional interference between c-Jun andthe glucocorticoid receptor: mutual inhibition of DNA binding due to directprotein-protein interaction. Cell 62:1205–1215.
48. Zhao, W., L. T. Chow, and T. R. Broker. 1997. Transcription activities ofhuman papillomavirus type 11 E6 promoter-proximal elements in raft andsubmerged cultures of foreskin keratinocytes. J. Virol. 71:8832–8840.
49. Zhu, Q., and J. P. Dudley. 2002. CDP binding to multiple sites in the mousemammary tumor virus long terminal repeat suppresses basal and glucocor-ticoid-induced transcription. J. Virol. 76:2168–2179.
50. Zwerschke, W., S. Joswig, and P. Jansen-Durr. 1996. Identification of do-mains required for transcriptional activation and protein dimerization in thehuman papillomavirus type-16 E7 protein. Oncogene 12:213–220.
VOL. 77, 2003 HPV31 URR DIFFERENTIATION-DEPENDENT DEX INDUCTION 10983
on February 18, 2016 by guest
http://jvi.asm.org/
Dow
nloaded from




















