
Induction of heme oxygenase-1 after hyperosmotic opening of the blood-brain barrier
11
Ž . Brain Research 780 1998 108–118 Research report Induction of heme oxygenase-1 after hyperosmotic opening of the blood-brain barrier Jeremy D. Richmon a,c , Kazumasa Fukuda a,c , Nino Maida a,c , Motoki Sato a,c , Marcelle Bergeron b,d , Frank R. Sharp b,d , S. Scott Panter a,d , L.J. Noble a,c, ) a Department of Neurosurgery, UniÕersity of California, San Francisco, CA, USA b Department of Neurology, UniÕersity of California, San Francisco, CA, USA c San Francisco General Hospital, San Francisco, CA, USA d Veterans Affairs Medical Center, San Francisco, CA, USA Accepted 21 October 1997 Abstract Ž . The induction of the stress protein heme oxygenase-1 HO-1 was studied in the rat brain after intracarotid administration of Ž . hyperosmolar mannitol. HO-1 was immunolocalized in fixed sections of brain 24 h to 7 days after injection. Immunoglobulin G IgG was immunolocalized in adjacent sections to demonstrate areas of breakdown of the blood–brain barrier. Induction of HO-1 was also evaluated by Western immunoblots, performed at 24 h after the insult. Immunofluorescent double labelling with monoclonal antibodies to HO-1 and either glial fibrillary acidic protein or the complement C3bi receptor was used to determine if gliarmacrophages expressed HO-1. There was pronounced, widespread induction of HO-1 in the ipsilateral hemisphere and cerebellum by 24 h both by immunocytochemistry and by Western blots. This induction was markedly attenuated at later times. HO-1 was induced in astrocytes and microgliarmacrophages in the ipsilateral hemisphere. In addition, the protein was induced in Bergmann glia and scattered microgliarmacrophages in the cerebellum. The mechanism of induction of HO-1 in glia after opening of the blood–brain barrier could include exposure to heme proteins, denatured proteins and other plasma constituents known to induce HO-1. This glial induction may reflect a protective response of these cells. q 1998 Elsevier Science B.V. Keywords: Astrocyte; Bergmann glia; Heat shock protein; Macrophage; Mannitol; Microglia 1. Introduction Heme oxygenase is a microsomal enzyme that oxida- tively cleaves heme and produces biliverdin, carbon w x monoxide and iron 24 . There are two isozymes of heme oxygenase, designated HO-1 and HO-2. HO-2 is the con- stitutive form and is found in neurons throughout the brain w x Ž . 50 . HO-1 is a stress protein HSP32 that is expressed in neurons in the hilus of the dentate gyrus, hypothalamus, w x cerebellum and brainstem 50 and can be induced in the brain by a variety of pathologic events insults including w x w x ischemia 35,44 , UVA radiation 49 , concussive brain w x w x injury 12,13 , bacterial toxins 23 , glutathione depletion w x w x 11 and subarachnoid haemorrhage 26,27 . H0-1 is regu- lated by a number of elements in the promoter region of ) Corresponding author. Department of Neurosurgery, 521 Parnassus Avenue, Room C224, San Francisco, CA 94143-0520, USA. Fax: q1 Ž . 415 476-5634. w x the gene. These include a heat shock element 18,19,33 , wx wx an AP-1 site 1 , a NFkB site 3 and a metalrheme w x binding site 8,29 . We have hypothesized that hyperosmotic opening of the blood–brain barrier results in the induction of HO-1. The blood–brain barrier is located at the level of the microvas- w x cular endothelium 16,17 . Under normal conditions the blood–brain barrier selectively regulates transport of molecules across this interface. Breakdown of the barrier disturbs the microenvironment of the brain and exposes cells to plasma constituents. There are several ways that the disturbed microenviron- ment could induce HO-1. Extravasated heme proteins could directly induce HO-1 via the heme binding site on the promoter region of the gene. Alternatively, circulating elements in the blood, such as bradykinin, amino acids, or other circulating peptides, may injure brain cells, thereby stimulating HO-1 transcription. Finally, denatured plasma proteins may activate heat shock factors that stimulate the heat shock element on the HO-1 gene. 0006-8993r98r$19.00 q 1998 Elsevier Science B.V. All rights reserved.
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Induction of heme oxygenase-1 after hyperosmotic opening of the blood-brain barrier

Ž .Brain Research 780 1998 108–118
Research report
Induction of heme oxygenase-1 after hyperosmotic opening of theblood-brain barrier
Jeremy D. Richmon a,c, Kazumasa Fukuda a,c, Nino Maida a,c, Motoki Sato a,c, Marcelle Bergeron b,d,Frank R. Sharp b,d, S. Scott Panter a,d, L.J. Noble a,c,)
a Department of Neurosurgery, UniÕersity of California, San Francisco, CA, USAb Department of Neurology, UniÕersity of California, San Francisco, CA, USA
c San Francisco General Hospital, San Francisco, CA, USAd Veterans Affairs Medical Center, San Francisco, CA, USA
Accepted 21 October 1997
Abstract
Ž .The induction of the stress protein heme oxygenase-1 HO-1 was studied in the rat brain after intracarotid administration ofŽ .hyperosmolar mannitol. HO-1 was immunolocalized in fixed sections of brain 24 h to 7 days after injection. Immunoglobulin G IgG
was immunolocalized in adjacent sections to demonstrate areas of breakdown of the blood–brain barrier. Induction of HO-1 was alsoevaluated by Western immunoblots, performed at 24 h after the insult. Immunofluorescent double labelling with monoclonal antibodies toHO-1 and either glial fibrillary acidic protein or the complement C3bi receptor was used to determine if gliarmacrophages expressedHO-1. There was pronounced, widespread induction of HO-1 in the ipsilateral hemisphere and cerebellum by 24 h both byimmunocytochemistry and by Western blots. This induction was markedly attenuated at later times. HO-1 was induced in astrocytes andmicrogliarmacrophages in the ipsilateral hemisphere. In addition, the protein was induced in Bergmann glia and scatteredmicrogliarmacrophages in the cerebellum. The mechanism of induction of HO-1 in glia after opening of the blood–brain barrier couldinclude exposure to heme proteins, denatured proteins and other plasma constituents known to induce HO-1. This glial induction mayreflect a protective response of these cells. q 1998 Elsevier Science B.V.
Keywords: Astrocyte; Bergmann glia; Heat shock protein; Macrophage; Mannitol; Microglia
1. Introduction
Heme oxygenase is a microsomal enzyme that oxida-tively cleaves heme and produces biliverdin, carbon
w xmonoxide and iron 24 . There are two isozymes of hemeoxygenase, designated HO-1 and HO-2. HO-2 is the con-stitutive form and is found in neurons throughout the brainw x Ž .50 . HO-1 is a stress protein HSP32 that is expressed inneurons in the hilus of the dentate gyrus, hypothalamus,
w xcerebellum and brainstem 50 and can be induced in thebrain by a variety of pathologic events insults including
w x w xischemia 35,44 , UVA radiation 49 , concussive brainw x w xinjury 12,13 , bacterial toxins 23 , glutathione depletion
w x w x11 and subarachnoid haemorrhage 26,27 . H0-1 is regu-lated by a number of elements in the promoter region of
) Corresponding author. Department of Neurosurgery, 521 ParnassusAvenue, Room C224, San Francisco, CA 94143-0520, USA. Fax: q1Ž .415 476-5634.
w xthe gene. These include a heat shock element 18,19,33 ,w x w xan AP-1 site 1 , a NFkB site 3 and a metalrheme
w xbinding site 8,29 .We have hypothesized that hyperosmotic opening of the
blood–brain barrier results in the induction of HO-1. Theblood–brain barrier is located at the level of the microvas-
w xcular endothelium 16,17 . Under normal conditions theblood–brain barrier selectively regulates transport ofmolecules across this interface. Breakdown of the barrierdisturbs the microenvironment of the brain and exposescells to plasma constituents.
There are several ways that the disturbed microenviron-ment could induce HO-1. Extravasated heme proteins coulddirectly induce HO-1 via the heme binding site on thepromoter region of the gene. Alternatively, circulatingelements in the blood, such as bradykinin, amino acids, orother circulating peptides, may injure brain cells, therebystimulating HO-1 transcription. Finally, denatured plasmaproteins may activate heat shock factors that stimulate theheat shock element on the HO-1 gene.
0006-8993r98r$19.00 q 1998 Elsevier Science B.V. All rights reserved.Ž .PII S0006-8993 97 01314-0

( )J.D. Richmon et al.rBrain Research 780 1998 108–118 109
2. Materials and methods
2.1. Experimental model
2.1.1. Surgical proceduresAdult male Sprague–Dawley rats were anaesthetized
Ž .with 4% chloral hydrate 10 mlrkg, intraperitoneally . Acatheter was inserted into the right femoral artery tomonitor blood pressure. The right external and internalcarotid arteries were exposed and the pterygopalatine arteryand the distal part of the external carotid artery wereligated. The occipital and superior thyroid arteries were
Ž .ligated. Polyethylene PE 50 tubing connected to a hep-arinized syringe was inserted into the external carotidartery and advanced to the carotid bifurcation. MannitolŽ .1.6 M D-Mannitol, Sigma, St. Louis, MO dissolved in0.9% saline was warmed to 378C, filtered through a 0.22
Ž .nitrocellulose filter Micron Separation and injected intothe internal carotid artery at a rate of 1 mlrmin for 1 min
Žusing a Harvard syringe infusion pump Harvard Appara-. Ž .tus, South Natick, MA . Body temperature 37.0–37.58C
was maintained with a heating pad prior to surgery andafter mannitol infusion. Animals were euthanized at 24 hŽ . Ž . Ž . Ž .ns5 , 2 days ns4 , 3 days ns4 , and 7 days ns3after surgery. Animals were reanaesthetized and perfusedthrough the ascending aorta with 500 ml of 4% para-
Ž .formaldehyde PFA in 0.1 M phosphate buffer, pH 7.4Ž .PB . Coronal sections, 50 um in thickness, were cutthrough the forebrain and cerebellum using a vibratome.
2.1.2. Surgical controlsŽ .Vehicle-control animals ns2 were infused with saline
at 378C at a rate of 1 mlrmin for 1 min. Sham-operatedŽ .animals ns2 underwent the same surgical procedure
including cannulations without infusion. These surgicalcontrols were euthanized at 24 h post surgery.
2.2. Immunocytochemistry
2.2.1. Immunocytochemical localization of IgG and HO-1Alternate sections were immunostained for IgG, to de-
fine patterns of barrier breakdown, and HO-1. After beingŽ .rinsed in 0.05 M phosphate-buffered saline, pH 7.4 PBS ,Ž .sections were incubated in 1.0% hydrogen peroxide H O2 2
in PBS for 10 min to quench any endogenous peroxidaticactivity, and then rinsed again in PBS. To immunolocalizeIgG, sections were incubated at room temperature as fol-
Ž .lows: a 2.0% rabbit serumr0.2% Triton-X-100r0.1%Ž . Ž .bovine serum albumin RSrTXrBSA , 5 min; b 10%
rabbit serumr0.2% Triton-X-100r0.1% bovine serum al-Ž . Ž .bumin 10% RSrTXrBSA , 20 min; c biotinylated rab-
Žbit anti-rat IgG 1:1000 in RSrTXrBSA, Vector Labora-.tories, Burlingame, CA , 2 h.
The HO-1 antibody was originally prepared by Trakshelw xet al. 47 using purified rat liver HO-1 as the immunogen.
The antibody produces a single band on Western im-munoblots. To immunolocalize HO-1, sections were incu-
Ž .bated after peroxidase block as follows: a 2% goatserumr0.2% Triton-Xr0.1% bovine serum albuminŽ . Ž .GSrTXrBSA , 5 min; b 10% goat serumr0.2% Triton-
Ž .Xr0.1% bovine serum albumin 10% GSrTXrBSA , 1 h;Ž . Žc antiheme oxygenase-1 polyclonal antibody 1:10,000 in
.GSrTXrBSA; StressGen, Victoria, BC, Canada , mini-Ž . Ž .mum 36 h at 48C; d PBS, 3=5 min; e biotinylated goat
Ž .anti-rabbit IgG 1:200 in GSrTXrBSA; Vector Labs. , 1h.
All sections were rinsed in PBS and incubated inŽ .avidin–biotin horseradish peroxidase HRP complex
Ž .ABC, 1:100 in PBS, Vector Labs for 30 min. Aftersections were rinsed in PBS, the reaction product wasvisualized using 0.05% 3,3-diaminobenzidine tetrachlorideas the chromogen in the presence of 0.02% H O . The2 2
immunostained sections were rinsed in PB, mounted ongelatinized slides, dehydrated in graded alcohols, cleared
Ž .in Hemo-De Fisher Scientific, Pittsburg, PA , and pre-pared for brightfield microscopy.
2.2.2. Immunofluorescent labelling of gliaŽ .Animals ns3 were euthanized at 24 h after manni-
tol-infusion and prepared for immunofluorescent doublelabelling using antibodies to HO-1 and either glial fibril-lary acidic protein for astrocytes or complement C3bireceptor for microgliarmacrophages.
After being rinsed in PBS, sections were incubated atroom temperature, unless otherwise specified, in each of
Ž .the following solutions for the time indicated: aŽ . Ž .GSrTXrBSA, 5 min; b 10% GSrTXrBSA, 1 h; c
Žantiheme oxygenase-1 polyclonal antibody 1:10,000 in. ŽGSrTXrBSA and either anti-GFAP 1:3000 in
. ŽGSrTXrBSA, ICN, Costa Mesa, CA or OX-42 1:3000.in GSrTXrBSA, Serotec, Kidlington Oxford, England ,
Ž . Ž .36–48 h at 48C; d PBS, 3=5 min; e 2% sheepserumr0.2% Triton-Xr0.1% bovine serum albuminŽ . Ž .SSrTXrBSA , 5 min; f 10% sheep serumr0.2% Tri-
Ž .ton-Xr0.1% bovine serum albumin 10% SSrTXrBSA ,Ž . Ž20 min; g Rhodamine conjugated anti-rabbit IgG 1:100
.in SSrTXrBSA, Boehringer-Mannheim, Indianapolis, INŽand biotinylated sheep anti-mouse IgG 1:200 in
. Ž . Ž .SSrTXrBSA, Amersham ; h PBS, 5 min; i 0.1 MŽ .sodium bicarbonate buffer saline, pH 8.5 BBS , 2=5
Ž .min; and j avidin-conjugated fluorescein isothiocyanateŽ .FITC, 1:100 in BBS, Vector Labs , 2 h. Sections werethen washed in PBS, mounted on slides, and prepared forfluorescent microscopy.
2.2.3. Immunocytochemical controlsImmunocytochemical controls, completed on adjacent
sections of brain, consisted of the same reaction procedurein the absence of primary antibody.

( )J.D. Richmon et al.rBrain Research 780 1998 108–118110
2.3. Western blotting
Brains were quickly removed from anaesthetized ani-Ž .mals Ns2 at 24 h after administration of mannitol. The
brains were quickly frozen on dry ice and stored at y708C,w xuntil analyzed by Western immunoblotting 6 . The apical
Ž .surface ;2 mm of the frontoparietal cortex from ipsilat-eral and contralateral hemispheres was dissected out on ice
Žand placed in Laemmli solubilizing buffer 2.5% sodiumdodecyl sulfate, 10% glycerol, 62.5 mmolrl Tris–HCl, pH
.6.8, 5% 2-mercaptoethanol and boiled for 10 min. Equal
Ž .amounts 55 mg of protein per sample were separated on12% sodium dodecyl sulfate polyacrylamide gels with4.5% stacking gel. After electrotransfer onto a nitrocellu-
Žlose membrane 0.2 mm pore size, Schleicher and Schuell,.Keene, NH , immobilized proteins were stained with Pon-
ceau solution to verify equal protein loading and homoge-nous transfer. After a brief rinse in deionized water, themembranes were incubated overnight at 48C, in sodium
Ž .phosphate buffer PB, 0.1 M, pH 7.4 , containing 5%nonfat dry milk, 1% BSA and 0.1% Tween-20, rinsed
Ž .briefly in PB 0.1 M containing 1% BSA and 0.1%
Ž . Ž .Fig. 1. Immunolocalization of IgG A,B and HO-1 C,D in adjacent sections of the contralateral and ipsilateral cortices 24 h after hyperosmotic insult.Ž . Ž . Ž .Neither IgG A nor HO-1 C is immunolocalized in the contralateral cortex. In contrast, pronounced barrier breakdown B, arrows corresponds to
Ž .prominent induction of HO-1 D, arrows in the ipsilateral outer cortical layers. Scale bars0.25 mm.
( )J.D. Richmon et al.rBrain Research 780 1998 108–118 111
Tween-20, then incubated for 1.5 h with a 1:3325 dilutionŽof rabbit polyclonal anti-rat HO-1 antibody StressGen,
.Victoria, BC, Canada . After three washes, membraneswere incubated with a 1:2500 dilution of anti-rabbit Ig-
Žhorseradish peroxidase antibody Amersham, Arlington.Heights, IL for 1 h. Finally, the membranes were washed
three times and the bound antibody was visualized with theECL chemiluminescence system according to the manufac-
Ž .turer’s protocol Amersham . A computer-based imagingŽ .system Alpha Innotec, San Leandro, CA was used to
measure the relative optical density of HO-1 protein onŽ .Western immunoblots 32 kDa bands in both the ipsilat-
eral and contralateral hemispheres after subtracting back-ground. Data were expressed as the percentage change inthe ipsilateral cortex and hippocampus as compared to thecontralateral cortex and hippocampus in each animal.
3. Results
3.1. Physiological parameters
Ž .The mean arterial blood pressure MABP prior toŽ .mannitol infusion was 65 "16 S.D. mmHg in the anaes-
Ž .thetized animal. An increase in MABP to 76 "21 S.D.mmHg occurred by 30 s after infusion of mannitol andreturned to preinfusion levels thereafter.
3.2. Time course for induction of HO-1
3.2.1. Immunocytochemical controlsNo immunostaining was noted in sections of mannitol-
infused, vehicle-control, or sham-operated brains that wereprocessed without primary antibody.
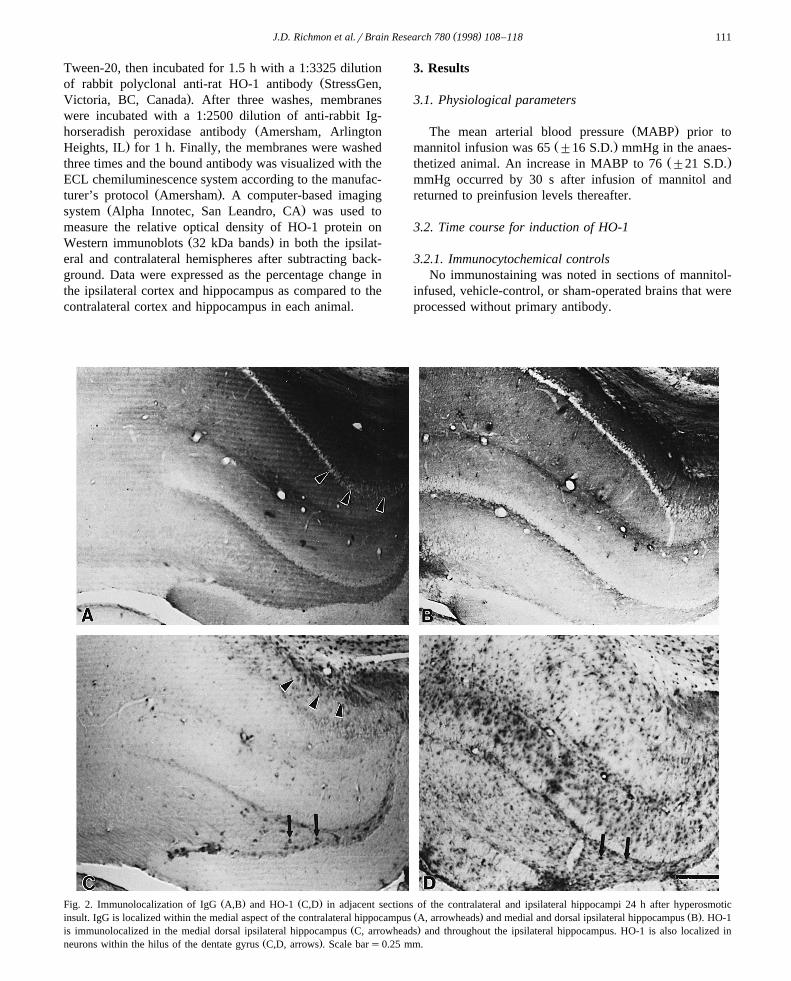
Ž . Ž .Fig. 2. Immunolocalization of IgG A,B and HO-1 C,D in adjacent sections of the contralateral and ipsilateral hippocampi 24 h after hyperosmoticŽ . Ž .insult. IgG is localized within the medial aspect of the contralateral hippocampus A, arrowheads and medial and dorsal ipsilateral hippocampus B . HO-1
Ž .is immunolocalized in the medial dorsal ipsilateral hippocampus C, arrowheads and throughout the ipsilateral hippocampus. HO-1 is also localized inŽ .neurons within the hilus of the dentate gyrus C,D, arrows . Scale bars0.25 mm.
( )J.D. Richmon et al.rBrain Research 780 1998 108–118112
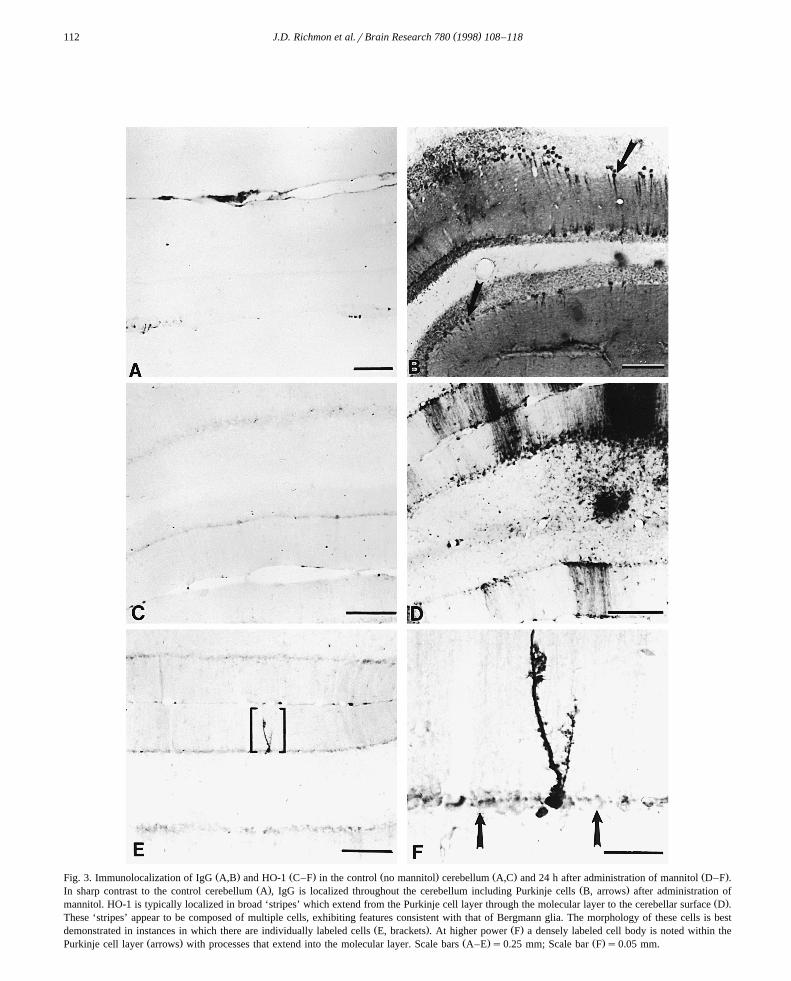
Ž . Ž . Ž . Ž . Ž .Fig. 3. Immunolocalization of IgG A,B and HO-1 C–F in the control no mannitol cerebellum A,C and 24 h after administration of mannitol D–F .Ž . Ž .In sharp contrast to the control cerebellum A , IgG is localized throughout the cerebellum including Purkinje cells B, arrows after administration of
Ž .mannitol. HO-1 is typically localized in broad ‘stripes’ which extend from the Purkinje cell layer through the molecular layer to the cerebellar surface D .These ‘stripes’ appear to be composed of multiple cells, exhibiting features consistent with that of Bergmann glia. The morphology of these cells is best
Ž . Ž .demonstrated in instances in which there are individually labeled cells E, brackets . At higher power F a densely labeled cell body is noted within theŽ . Ž . Ž .Purkinje cell layer arrows with processes that extend into the molecular layer. Scale bars A–E s0.25 mm; Scale bar F s0.05 mm.

( )J.D. Richmon et al.rBrain Research 780 1998 108–118 113
3.2.2. Immunocytochemical localization of IgGThere was little extravasation of IgG in the sham-oper-
ated group or vehicle-control groups. When present, IgGwas infrequently localized in small patches that encircledblood vessels.
At 24 h after infusion of mannitol, IgG was localizedthroughout the ipsilateral cerebral cortex, basal ganglia,hippocampus, thalamus, contralateral parasagittal cortex
Ž .and cerebellum Figs. 1 and 2 . The intensity and distribu-tion of immunostaining was maximal at 24 h after infusionof mannitol. It was markedly diminished by 3 days in allareas of the brain including the cerebellum.
In general, IgG was diffusely distributed throughout theipsilateral hemisphere. In addition, the protein was local-ized in small numbers of randomly distributed neurons andglia within the ipsilateral hemisphere.
3.2.3. Immunocytochemical localization of HO-1In sham-operated and vehicle control brains, HO-1 was
primarily localized in certain populations of neurons in thebrainstem and the hilus of the dentate gyrus of the hip-pocampus.
There was a widespread pattern of induction of HO-1 inglia in the ipsilateral hemisphere, including the cerebral
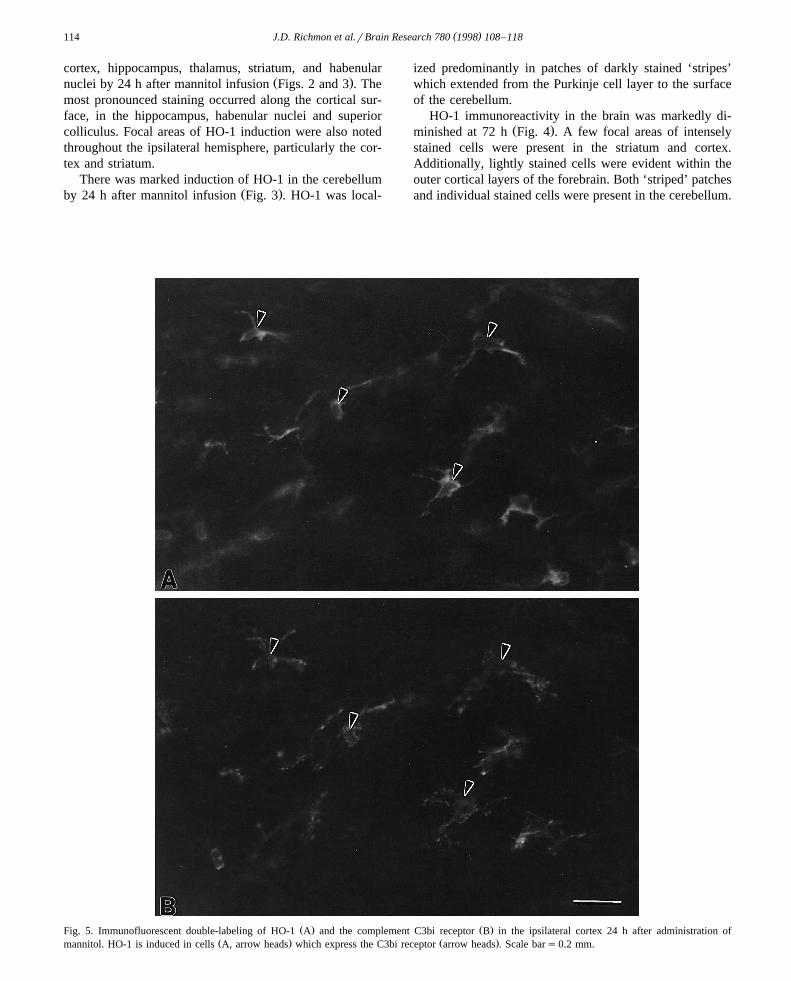
Ž . Ž . Ž .Fig. 4. Immunolocalization of HO-1 in the sham operated animal A and at 24 B and 48 C h after administration of mannitol. HO-1 appears to bemaximally induced in the ipsilateral cortex at 24 h. Although induction of HO-1 occurs throughout most of the cortex, it is most prominent in the outer
Ž .cortical layers arrow heads . Scale bars0.3 mm.
( )J.D. Richmon et al.rBrain Research 780 1998 108–118114
cortex, hippocampus, thalamus, striatum, and habenularŽ .nuclei by 24 h after mannitol infusion Figs. 2 and 3 . The
most pronounced staining occurred along the cortical sur-face, in the hippocampus, habenular nuclei and superiorcolliculus. Focal areas of HO-1 induction were also notedthroughout the ipsilateral hemisphere, particularly the cor-tex and striatum.
There was marked induction of HO-1 in the cerebellumŽ .by 24 h after mannitol infusion Fig. 3 . HO-1 was local-
ized predominantly in patches of darkly stained ‘stripes’which extended from the Purkinje cell layer to the surfaceof the cerebellum.
HO-1 immunoreactivity in the brain was markedly di-Ž .minished at 72 h Fig. 4 . A few focal areas of intensely
stained cells were present in the striatum and cortex.Additionally, lightly stained cells were evident within theouter cortical layers of the forebrain. Both ‘striped’ patchesand individual stained cells were present in the cerebellum.
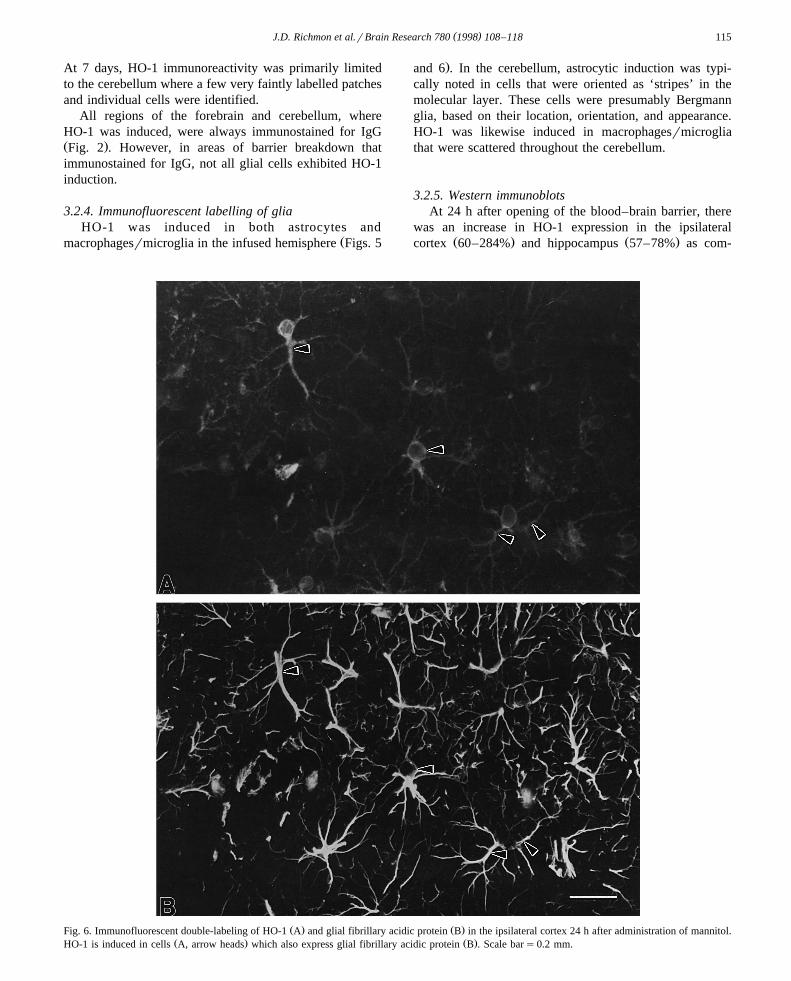
Ž . Ž .Fig. 5. Immunofluorescent double-labeling of HO-1 A and the complement C3bi receptor B in the ipsilateral cortex 24 h after administration ofŽ . Ž .mannitol. HO-1 is induced in cells A, arrow heads which express the C3bi receptor arrow heads . Scale bars0.2 mm.
( )J.D. Richmon et al.rBrain Research 780 1998 108–118 115
At 7 days, HO-1 immunoreactivity was primarily limitedto the cerebellum where a few very faintly labelled patchesand individual cells were identified.
All regions of the forebrain and cerebellum, whereHO-1 was induced, were always immunostained for IgGŽ .Fig. 2 . However, in areas of barrier breakdown thatimmunostained for IgG, not all glial cells exhibited HO-1induction.
3.2.4. Immunofluorescent labelling of gliaHO-1 was induced in both astrocytes and
Žmacrophagesrmicroglia in the infused hemisphere Figs. 5
.and 6 . In the cerebellum, astrocytic induction was typi-cally noted in cells that were oriented as ‘stripes’ in themolecular layer. These cells were presumably Bergmannglia, based on their location, orientation, and appearance.HO-1 was likewise induced in macrophagesrmicrogliathat were scattered throughout the cerebellum.
3.2.5. Western immunoblotsAt 24 h after opening of the blood–brain barrier, there
was an increase in HO-1 expression in the ipsilateralŽ . Ž .cortex 60–284% and hippocampus 57–78% as com-
Ž . Ž .Fig. 6. Immunofluorescent double-labeling of HO-1 A and glial fibrillary acidic protein B in the ipsilateral cortex 24 h after administration of mannitol.Ž . Ž .HO-1 is induced in cells A, arrow heads which also express glial fibrillary acidic protein B . Scale bars0.2 mm.

( )J.D. Richmon et al.rBrain Research 780 1998 108–118116
Fig. 7. Western immunoblot analysis of adult rat brain HO-1 proteinlevels, 24 h after unilateral opening of the blood-brain barrier by manni-
Ž .tol. Whole tissue extracts of ipsilateral hippocampus Lanes 1 and 2 andŽ .cerebral cortex Lanes 3, 4, and 5 were prepared as described in Section
Ž .2. Equal protein samples 55 mg were analyzed by sodium dodecylsulfate polyacrylamide gel electrophoresis and immunoblotting usingrabbit anti-rat heme oxygenase-1 antibody. Lanes 1 and 3: contralateralhemisphere. Lanes 2 and 4: ipsilateral hemisphere. Lane 5: control rat
Ž .brain no mannitol . Molecular weight markers in kilodaltons are indi-cated on the left. There is a 57% increase in the ipsilateral hippocampusand a 284% increase in the ipsilateral cortex as compared to the contralat-eral hippocampus and cortex, respectively.
pared to the contralateral cortex and hippocampus, respec-Ž .tively Fig. 7 .
4. Discussion
4.1. Hyperosmotic opening of the blood–brain barrier
We have previously demonstrated that the blood–brainw xbarrier breaks down after traumatic brain injury 45 . The
possible contribution of barrier breakdown to producingtissue injury after traumatic brain injury is not understoodbecause abnormal permeability is coincident with the pri-mary insult and is sustained during the acute period whena variety of other secondary pathologic events evolve. Inthis report, the cellular response to hyperosmotic openingof the blood–brain barrier was evaluated. The advantage ofthis model is that it generates a characteristic transient
w xbarrier breakdown 37 in the absence of any traumaticmechanical perturbation. Barrier opening is typically great-est immediately after infusion of mannitol and is slowly
w xreconstituted thereafter 31 with re-establishment of bar-w xrier integrity by at least 6 h after the insult 15,37 . We
have used this model to begin to explore the cellularresponse to breakdown of the blood–brain barrier.
4.2. Induction of stress proteins after hyperosmotic insult
We have previously demonstrated that there is inductionof the stress protein HSP70 after hyperosmotic infusionsw x46 . It is not surprising that both HSP70 and HO-1 areinduced in the brain after hyperosmotic insult. Both theHSP70 and HO-1 genes contain a heat shock element inthe promoter region of the gene. It is known that denaturedproteins in cells activate heat shock factors that stimulate
w xheat shock transcription 2,36 . There are several scenarioswhereby proteins could become denatured after hyperos-motic insult. First, proteins may become denatured as aconsequence of cell injury. Such cell injury might occur in
response to exposure to amino acids such as glutamate andglycine which when present at high concentrations may be
w xtoxic to brain cells 40 . Second, cells may take up ex-travasated plasma proteins which in turn denature. Thispossibility is supported in part by this study as well as in a
w xprevious study 39 , in which we have demonstrated thatneurons and glia take up extravasated proteins.
We have also shown that there is focal rather thanwidespread induction of HSP70 after hyperosmotic insultw x39 . Thus, it seems unlikely that widespread induction ofHO-1 reflects a heat shock response, since the HSP70 andHO-1 distributions were so different. The HO-1 gene is
w xregulated not only by a heat shock element 18,19,33 butw x w xalso by an AP-1 site 1,2 , a NFkB site 3 and aw xmetalrheme binding site 8,29 . Collectively, these latter
elements render the gene responsive to AP1 stimulation byw ximmediate early gene family members 1 , hypoxia and
w x w xoxidative stress 20–22 and heme 10,29 .We can only speculate on the regulation of HO-1 after
hyperosmotic insult. Heme is a potent inducer of HO-1w x41,47 . This is exemplified in recent studies by Matz et al.w x26,27 in which intracisternal administration of blood oroxyhemoglobin resulted in widespread induction of HO-1.
ŽAfter hyperosmotic insult, extravasated hemoproteins e.g.,. Žhemoglobin andror cellular hemoproteins e.g., cy-
.tochromes, catalase, tryptophan oxygenase may denatureand release their heme moiety into the extracellular spacewhere they are taken up into glia which stimulate HO-1induction. In this study, widespread induction of HO-1 inmicroglia may therefore reflect the increased demand tosequester and metabolize the heme moiety of denaturedhemoproteins.
4.3. Glial induction of HO-1
HO-1 was consistently induced in glia in this study.HO-1 is induced in glia in response to a variety ofpathological conditions including experimental subarach-
w x w xnoid haemorrhage 26,27 , hyperthermia 10 , ischemiaw x w x14,32,35,43,44 , glutathione depletion 11 and brain in-
w xjury 7,12,13 . The generalized pattern of induction mayreflect the unique relationship of glia to microvesselsandror a defined role of these cells in responding toextravasated plasma proteins. It is noteworthy that certainpopulations of astrocytes and microglia maintain a veryintimate association with the vasculature. These cells couldrepresent the first ‘line of defense’ against extravasatedplasma proteins when the barrier is transiently opened byany means. It is also possible that glia preferentiallyexpress heme transporters and thus play an important rolein sequestering extracellular hemeproteins.
We demonstrate both astrocytic and microglial induc-w xtion of HO-1 after hyperosmotic insult. Matz et al. 26,27
have studied the induction of HO-1 after intracisternaladministration of either lysed or whole blood. Exposure of

( )J.D. Richmon et al.rBrain Research 780 1998 108–118 117
the brain to these heme proteins results in an almostexclusive induction in microglia. In the present study,HO-1 induction in microglia may likewise be a response toextravasated heme proteins. The nature of the astrocyticinduction after hyperosmotic insult is unclear. Nimura et
w xal. 32 have reported HO-1 induction primarily in astro-cytes after ischemic injury. Thus, it appears that astrocyticinduction of HO-1 is associated with selective neuronalinjury. Taken together, these findings suggest that induc-tion of HO-1 in microglia and astrocytes may reflectdifferent cellular responses to injury.
4.4. Induction of HO-1: functional significance
Whereas HO-1 has been shown to be induced in avariety of pathologic states, it is not clear if this responseconfers cytoprotection or is detrimental to the brain. HO-1catalyzes heme to biliverdin, carbon monoxide and iron,and is the rate limiting step in the formation of bilirubinw x24 . HO-1 is therefore thought to protect cells from oxida-tive stress by increasing the metabolism of heme to form
w xthe antioxidant bilirubin 28,42 as well as by allowing thew xcell to sequester catalytically active iron with ferritin 4,5 .
In support of a role in cytoprotection, HO-1 has beenshown to protect the kidney against damage after ischemia
w xand reperfusion 25 .There are contrary data which suggest a harmful role of
HO-1 in the setting of ischemic and traumatic brain injury.w xRecent studies by Pannizon et al. 34 have demonstrated
that metalloporphyrins, inhibitors of heme oxygenase, at-tenuate cellular injury after hypoxic injury in the hip-pocampal slice preparation as well as after traumatic brain
w xinjury. Similarly, Kadoya et al. 16 have reported thatadministration of zinc protoporphyrin prior to focal cere-bral ischemia reduces infarct size and edema formation.These studies are somewhat difficult to interpret, however,because these inhibitors are not specific to heme oxyge-nase.
We have previously demonstrated prominent barrierbreakdown and widespread induction of HO-1 after trau-
w xmatic brain injury 12,13,45,46 . The present study pro-vides the first evidence that barrier breakdown itself maybe a contributing factor in the induction of HO-1 aftertraumatic brain injury. Whether induction of HO-1 reflectsa protective response or is detrimental to the brain remainsto be defined.
Acknowledgements
This research was supported by NIH grants NS23324Ž . Ž . Ž .LJN , NS28167 FRS , NS14543 LJN, FRS , HL53040Ž .SSP, FRS the Merit Review Program of the Department
Ž .of Veterans Affairs FRS and the Department of DefenseŽ .SSP, FRS .
References
w x1 J. Alam, D. Zhining, Distal AP-1 binding sites mediate basal levelenhancement and TPA induction of the mouse heme oxygenase-1
Ž .gene, J. Biol. Chem. 267 1992 21894–21900.w x2 J. Anathan, A. Goldberg, R. Voellmy, Abnormal proteins serve as
eukaryotic stress signals and trigger the activation of heat shockŽ .genes, Science 232 1986 522–524.
w x3 L. Applegate, P. Luscher, R. Tyrrell, Induction of heme oxygenase:a general response to oxidant stress in cultured mammalian cells,
Ž .Cancer Res. 51 1991 974–978.w x4 J. Balla, H. Jacob, G. Balla, K. Nath, J. Eaton, G. Vercellotti,
Endothelial-cell heme uptake from heme proteins: induction ofsensitization and desensitization to oxidant damage, Proc. Natl.
Ž .Acad. Sci. USA 90 1993 9285–9289.w x5 G. Balla, H. Jacob, J. Balla, M. Rosenberg, K. Nath, F. Apple, J.
Eaton, G. Vercellotti, Ferritin: a cytoprotective antioxidant stratagemŽ .of endothelium, J. Biol. Chem. 267 1992 18148–18153.
w x6 M. Bergeron, D. Ferriero, H. Vreman, D. Stevenson, F. Sharp,Hypoxia–ischemia, but not hypoxia alone, induce the expression of
Ž .heme oxygenase-1 HSP32 in newborn rat brain, J. Cereb. BloodŽ .Flow Metab. 17 1997 647–658.
w x7 B. Dywer, R. Nishimura, A. Alcaraz, Transient induction of hemeŽ .oxygenase after cortical stab wound injury, Brain Res. 38 1996
251–259.w x8 R. Eisentein, M. Garcia, W. Pettingell, H. Munro, Regulation of
ferritin and heme oxygenase synthesis in rat fibroblasts by differentŽ .forms of iron, Proc. Natl. Acad. Sci. USA 88 1991 688–692.
w x10 J. Ewing, M. Maines, Rapid induction of heme oxygenase 1 mRNAand protein by hyperthermia in rat brain: heme oxygenase 2 is not a
Ž .heat shock protein, Proc. Natl. Acad. Sci. USA 88 1991 5364–5368.w x11 J. Ewing, M. Maines, Glutathione depletion induces heme oxyge-
Ž .nase-1 HSP32 mRNA and protein in rat brain, J. Neurochem. 60Ž .1993 1512–1519.
w x12 K. Fukuda, S.S. Panter, F.R. Sharp, L.J. Noble, Induction of hemeŽ .oxygenase-1 HO-1 after traumatic brain injury in the rat, Neurosci.
Ž .Lett. 199 1995 127–130.w x13 K. Fukuda, J.D. Richmon, M. Sato, F.R. Sharp, S.S. Panter, L.J.
Ž .Noble, Induction of heme oxygenase-1 HO-1 in glia after traumaticŽ .brain injury, Brain Res. 736 1996 8–75.
w x14 J. Geddes, L. Pettigrew, M. Holtz, S. Craddock, M. Maines, Perma-nent focal and transient global cerebral ischemia increase glial andneuronal expression of heme oxygenase-1, but not heme oxygenase-
Ž .2, protein in rat brain, Neurosci. Lett. 210 1996 205–208.w x15 H.J. Houthoff, K.G. Go, P.O. Gerrits, The mechanisms of blood–
brain barrier impairment by hyperosmolar perfusion, Acta Neu-Ž .ropath. 56 1982 99–112.
w x16 C. Kadoya, E. Domino, G. Yang, J. Stern, A. Betz, Preischemic butnot postischemic zinc protoporphyrin treatment reduces infarct sizeand edema accumulation after temporary focal cerebral ischemia in
Ž .rats, Stroke 26 1995 1035–1038.w x17 M.J. Karnovsky, The ultrastructural basis of capillary permeability
Ž .studied with peroxidase as a tracer, J. Cell Biol. 35 1967 213–236.w x18 S. Keyse, R. Tyrell, Heme oxygenase is the major 32-kDa stress
protein induced in human skin fibroblasts by UVA radiation, hydro-gen peroxide, and sodium arsenate, Proc. Natl. Acad. Sci. USA 86Ž .1989 99–103.
w x19 S. Keyse, R. Tyrell, Induction of the heme oxygenase gene in humanŽ .skin fibroblasts by hydrogen peroxide and UVA 365 nm radiation:
evidence for the involvement of the hydroxyl radical, CarcinogenesisŽ .11 1990 787–791.
w x20 A. Koong, E. Chen, N. Mivechi, N. Denki, P. Stambrook, A.Giaccia, Hypoxic activation of nuclear factor-kappa B is mediatedby a Ras and Raf signalling pathway and does not involve MAP
Ž . Ž .kinase ERK1 or ERK2 , Cancer Res. 54 1994 5273–5279.w x21 Y. Lavrovsky, M. Schwartzman, N. Abraham, Novel regulatory sites

( )J.D. Richmon et al.rBrain Research 780 1998 108–118118
of the human heme oxygenase-1 promoter region, Biochem. Bio-Ž .phys. Res. Commun. 196 1993 336–341.
w x22 Y. Lavrovsky, M. Schwartzman, R. Levere, Kappa, N. Abraham,Identification of binding sites for transcription factors NF-kappa Band AP-2 in the promoter region of the human heme oxygenase 1
Ž .gene, Proc. Natl. Acad. Sci. USA 91 1994 5987–5991.w x23 M.D. Maines, New developments in the regulation of heme
metabolism and their implications, CRC Crit. Rev. Toxicol. 12Ž .1984 241–314.
w x24 M.D. Maines, Heme oxygenase: function, multiplicity, regulatoryŽ .mechanisms, and clinical application, FASEB J. 2 1988 2557–2568.
w x25 M. Maines, R. Mayer, J. Ewing, W. McCoubrey, Induction ofŽ .kidney heme oxygenase-1 HSP32 mRNA and protein by is-
chemiarreperfusion: possible role of heme as both promotor oftissue damage and regulator of HSP32, J. Pharmacol. Exp. Ther. 264Ž .1993 457–462.
w x26 P. Matz, C. Turner, P. Weinstein, S. Mass, S. Panter, F. Sharp,HO-1 hemeoxygenase induction in glia throughout rat brain follow-
Ž .ing experimental subarachnoid haemorrhage, Brain Res. 713 1996211–222.
w x27 P. Matz, S. Massa, P. Weinstein, C. Turner, S. Panter, F. Sharp,Focal hyperexpression of hemeoxygenase-1 protein and messengerRNA in rat brain caused by cellular stress following subarachnoid
Ž .injections of lysed blood, J. Neurosurg. 85 1996 892–900.w x Ž .28 A. McDonagh, Is bilirubin good for you?, Clin. Perinatol. 17 1990
359–369.w x29 R. Muller, H. Taguchi, S. Shibahara, Nucleotide sequence and
organization of the rat heme-oxygenase gene, J. Biol. Chem. 262Ž .1987 6795–6802.
w x31 E.A. Neuwelt, J. Minna, E. Frenkel, P.A. Barnett, C.I. McCormick,Osmotic blood–brain barrier opening to IgM monoclonal antibody
Ž .in the rat, Am. J. Physiol. 250 1986 R875–R883.w x32 T. Nimura, P. Weinsten, S. Massa, S. Panter, F. Sharp, Heme
Ž .oxygenase-1 HO-1 protein induction in rat brain following focalŽ .ischemia, Mol. Brain Res. 37 1996 201–208.
w x33 S. Okinaga, S. Shibahara, Identification of a nuclear protein thatconstitutively recognizes the sequence containing a heat-shock ele-ment: Its binding properties and possible function modulating heat-shock induction of the rat heme oxygenase gene, Eur. J. Biochem.
Ž .212 1993 167–175.w x34 K.L. Panizzon, B.E. Dwyer, R.N. Nishimura, R.A. Wallis, Neuro-
protection against CA1 injury with metalloporphyrins, Neuroreport 7Ž .1996 662–666.
w x35 W. Paschen, A. Uto, B. Djuricic, J. Schmitt, Hemeoxygenase ex-
pression after reversible ischemia of rat brain, Neurosci. Lett. 180Ž .1994 5–8.
w x36 H. Pelham, Speculations on the functions of the major heat shockŽ .and glucose-related proteins, Cell 46 1986 959–961.
w x37 S.I. Rapoport, W.R. Fredericks, K. Ohno, K.D. Pettigrew, Quantita-tive aspects of reversible osmotic opening of the blood–brain bar-
Ž .rier, Am. J. Physiol. 238 1980 R421–R431.w x39 J.D. Richmon, K. Fukuda, F.R. Sharp, L.J. Noble, Induction of
HSP-70 after hyperosmotic opening of the blood–brain barrier in theŽ .rat, Neurosci. Lett. 202 1995 1–4.
w x40 B. Schlosshauer, The blood–brain barrier: morphology, molecules,Ž .and neurothelin, BioEssays 15 1993 341–346.
¨w x41 S. Shibahara, R. MYller, H. Taguchi, Transcriptional control of ratŽ .heme oxygenase by heat shock, J. Biol. Chem. 262 1987 12889–
12892.w x42 R. Stocker, Y. Yamamoto, A. McDonagh, A. Glazer, B. Ames,
Bilirubin is an antioxidant of possible physiological importance,Ž .Science 235 1987 1043–1046.
w x43 A. Takeda, T. Kimpara, H. Onodera, Y. Itoyama, S. Shibahara, K.Kogure, Regional difference in induction of heme oxygenase-1protein following rat transient forebrain ischemia, Neurosci. Lett.
Ž .205 1996 169–172.w x44 A. Takeda, H. Onodera, A. Sugimoto, Y. Itoyama, K. Kogure, S.
Shibahara, Increased expression of heme oxygenase mRNA in ratŽ .brain following transient forebrain ischemia, Brain Res. 666 1994
120–124.w x45 H. Tanno, R.P. Nockels, L.H. Pitts, L.J. Noble, Breakdown of the
blood–brain barrier after fluid percussive head injury in the rat: PartI. Distribution and time course of protein extravasation, J. Neuro-
Ž .trauma 9 1992 21–32.w x46 H. Tanno, R.P. Nockels, L. Pitts, L.J. Noble, Immunolocalization of
heat shock protein after fluid percussive brain injury and relationshipto breakdown of the blood–brain barrier, J. Cereb. Blood Flow
Ž .Metab. 13 1993 116–124.w x47 G. Trakshel, R. Kutty, M. Maines, Purification and characterization
of the major constitutive form of the testicular heme oxygenase, J.Ž .Biol. Chem. 261 1986 11131–11137.
w x49 R.M. Tyrrell, S. Basu-Modak, Transient enhancement of hemeoxygenase 1 mRNA accumulation: a marker of oxidative stress in
Ž .eukaryotic cells, Meth. Enzymol. 234 1994 224–235.w x50 S.R. Vincent, S. Das, M. Maines, Brain heme oxygenase isoenzymes
and nitric oxide synthase are co-localized in select neurons, Neu-Ž .rosci. 63 1994 223–231.




















