
An allosteric model of the inositol trisphosphate receptor with nonequilibrium binding
Upload
khangminh22Category
view
1download
0
BASIC RESEARCH www.jasn.org
Beneficial Effects of Myo-Inositol OxygenaseDeficiency in Cisplatin-Induced AKI
Rajesh K. Dutta,* Vinay K. Kondeti,* Isha Sharma,* Navdeep S. Chandel,†
Susan E. Quaggin,† and Yashpal S. Kanwar*†
Departments of *Pathology and †Medicine, Northwestern University, Chicago, Illinois
ABSTRACTOverexpression of the proximal tubular enzymemyo-inositol oxygenase (MIOX) induces oxidant stress invitro. However, the relevance of MIOX to tubular pathobiology remains enigmatic. To investigate the roleof MIOX in cisplatin-induced tubular AKI, we generated conditional MIOX-overexpressing transgenic(MIOX-TG)mice andMIOX-knockout (MIOX2/2) mice with tubule-specificMIOX overexpression or knock-out, respectively. Compared with cisplatin-treated wild-type (WT) mice, cisplatin-treated MIOX-TG micehad even greater increases in urea, creatinine, and KIM-1 levels andmore tubular injury and apoptosis, butthese effectswere attenuated in cisplatin-treatedMIOX2/2mice. Similarly,MIOX-TGmice had the highestand MIOX2/2 mice had the lowest renal levels of Bax, cleaved caspase-3, and NADPH oxidase-4 expres-sion and reactive oxygen species (ROS) generation after cisplatin treatment. In vitro, cisplatin dose-dependently increased ROS generation in LLC-PK1 cells. Furthermore, MIOX overexpression in thesecells accentuated cisplatin-induced ROS generation and perturbations in the ratio of GSH to oxidizedGSH, whereas MIOX-siRNA or N-acetyl cysteine treatment attenuated these effects. Additionally, thecisplatin-induced enhancement of p53 activation, NF-kB binding to DNA, andNF-kB nuclear translocationin WTmice was exacerbated in MIOX-TGmice but absent in MIOX2/2mice. In vitro, MIOX-siRNA or NACtreatment reduced the dose-dependent increase in p53 expression induced by cisplatin. We alsoobserved a remarkable influx of inflammatory cells and upregulation of cytokines in kidneys of cisplatin-treated MIOX-TG mice. Finally, analysis of genomic DNA in WT mice revealed cisplatin-induced hypome-thylation of the MIOX promoter. These data suggest that MIOX overexpression exacerbates, whereasMIOX gene disruption protects against, cisplatin-induced AKI.
J Am Soc Nephrol 28: 1421–1436, 2017. doi: https://doi.org/10.1681/ASN.2016070744
AKI is encountered in up to 30% of critically illhospitalized patients, and it is quite often associatedwith acute tubular necrosis (ATN) leading to rapiddeterioration of renal functions.1,2 There are a widerange of clinico-pathologic states that may be asso-ciated with ATN, and the latter can be classifiedunder two broad categories, i.e., ischemic versustubulo-toxic injury.3 The pathophysiologic mecha-nisms leading to AKI between these two categoriesmay differ to a certain extent, and conceivably thismay be one of the factorswhydifferent segments of thenephron are differentially affected, possibly involvingdifferent cellular and molecular mechanisms.4 For in-stance, although all of the three segments of proxi-mal tubules are vulnerable to ischemic injury, the S3segment is highly susceptible to tubulo-toxins by
involving pathogeneticmechanisms that are restrictiveto molecules expressed in this segment of the neph-ron.5 With respect to cellular specificity, past experi-ments have demonstrated that mice with selectiveablation of p53 in proximal tubules are resistant to
Received July 12, 2016. Accepted October 17, 2016.
R.K.D. and V.K.K. contributed equally to this work.
Published online ahead of print. Publication date available atwww.jasn.org.
Correspondence: Dr. Yashpal S. Kanwar, Department of Pathol-ogy, Northwestern University Medical School, 303 E. Chicago Ave-nue, Chicago, Illinois 60611. Email: [email protected]
Copyright © 2017 by the American Society of Nephrology
J Am Soc Nephrol 28: 1421–1436, 2017 ISSN : 1046-6673/2805-1421 1421

tubulo-toxins such as cisplatin, whereas p53 deletion in othersegments of the nephron provided no protection against toxicinjury.6 Intriguingly, repetitive tubulo-toxic assault can lead toboth adaptive as well as maladaptive changes.7 Early adaptivechanges could be beneficial in the form of cytoresistance, whereaslate maladaptive changes may be detrimental; these include upre-gulation of inflammatory and profibrogenic cytokines along withoxidant stress, which ultimately could lead to tubulo-interstitialfibrosis, CKD, and renal failure.7–9
Myo-inositol oxygenase (MIOX), a proximal tubular enzyme,has been implicated in the pathogenesis of diabetic tubulopathy.10
It catabolizesmyo-inositol, that is synthesizedby isomerization anddephosphorylation of glucose 6-phospate, to D-glucuronic acidvia the Glucuronate-Xylulose pathway (G-X pathway).11,12 It hasosmotic-, carbohydrate-, and sterol- response elements in its pro-moter, and its expression is modulated by organic osmolytes, fattyacids, and obesity besides high glucose ambience or diabetes.13,14
In addition, the MIOX promoter region includes antioxidant andoxidant response elements and its transcription is regulated by aredox-sensitive response transcription factor, Nrf2.13 Moreover,MIOX overexpression in vitro accentuates the generation of reac-tive oxygen species (ROS), apparently derived both from mito-chondrial and NADPH oxidase systems.15 The kidney is one ofthemajor organs vulnerable to oxidant damage by ROS because ofits high concentration of long-chain polyunsaturated fatty acids.16
Conceivably, the free radicals or ROS attack long-chain highly-unsaturated fatty acids and promote lipid peroxidation causingoxidative damage. In general, the ROS adversely affect homeostasisof the kidney in a variety of disease states, including diabetes mel-litus, hypertension, obesity, ischemia, and ARF secondary to theadministration of analgesics, antibiotics, and chemotherapeuticagents.16 Some of these states, e.g., secondary to the administrationof therapeutic agents, are associated with AKI, which necessitatesthe development of various animal model systems to delineate themechanisms that are relevant to thepathobiologyof renal tubules.17
Cisplatin-induced nephropathy is a prototype toxic injurymodel which has been extensively utilized to investigate themechanisms relevant to the pathogenesis of AKI.5 Cisplatin istaken up by proximal tubules, and inhibition of organic cationtransporters with cimetidine decreases its cellular uptake.In 1view of the fact that cellular damage is confined to prox-imal tubules in toxin-induced nephropathy, and MIOX, ametabolic enzyme, is expressed in proximal tubules and itsoverexpression in vitro leads to the generation of ROS,studies were initiated to assess the effect of up- or downregu-lation of MIOX on the outcomes of cisplatin-inducednephropathy and associated signaling events in transgenicand knockout mice.
RESULTS
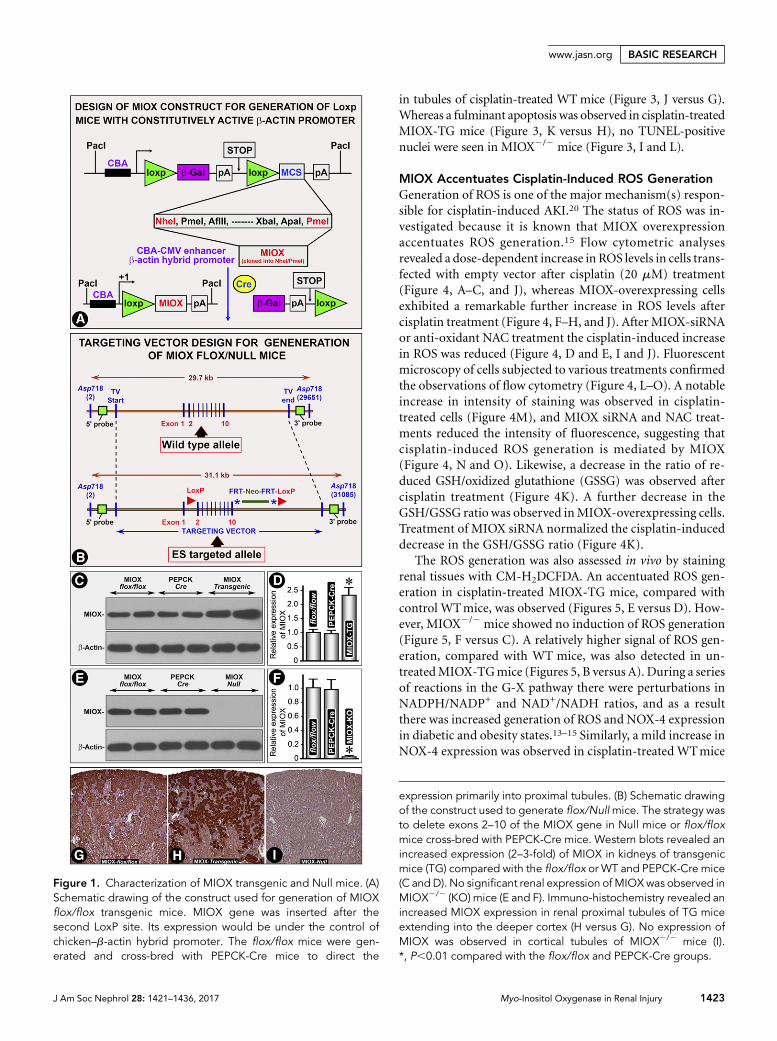
Characterization of MIOX Transgenic and Null MiceWestern blot analyses revealed an increased expression (2–3-fold) of MIOX in kidneys of transgenic mice compared with
the flox/flox or wild-type (WT) and PEPCK-Cre mice (Figure1, C and D). No significant renal expression of MIOX wasobserved in MIOX-knockout (MIOX2/2) mice (Figure 1, Eand F). By immuno-histochemistry a marked increase inMIOX expression was observed in renal proximal tubules inMIOX transgenic (MIOX-TG)mice (Figure 1, H versusG). Noimmuno-histochemical signal of MIOX was observed inMIOX2/2 mice (Figure 1I). Both the mutant mice generatedwere proximal tubule–specific. Moreover, MIOX is a proximaltubule–specific enzyme. We did not observe any MIOX stain-ing in any other renal compartment.
MIOX Gene Disruption Ameliorates Cisplatin-InducedAKICisplatin (20mg/kg) induced significantweight loss inMIOX-TGand WT mice, whereas MIOX2/2 mice had minimal bodyweight loss (Figure 2A). Maximal renal functional changeswere seen in cisplatin-treated MIOX-TG mice. Both serumcreatinine and urea were markedly elevated in MIOX-TGmice (Figure 2, B and C). A minimal increase in creatinineor urea levels was seen in MIOX2/2 mice. A significant in-crease in expression of Kim-1 mRNA, a marker of AKI,18 wasobserved in kidneys of cisplatin-treated MIOX-TGmice (Fig-ure 2D).WTmice showed cytolysis of tubular cells with loss ofbrush border (Figure 2, H versus E). These changes were ac-centuated in MIOX-TG mice (Figure 2, I versus F). Interest-ingly, the kidneys of cisplatin-treated MIOX2/2 mice had mildcytolytic changes compared with WTor TG strains (Figure 2, Jversus G).
MIOX Gene Disruption Inhibits, Whereas ItsOverexpression Accentuates, Renal Tubular CellApoptosis in Cisplatin-Induced AKIBy immuno-histochemistry and immunoblot analyses an in-creased Bax expression was seen in tubules of cisplatin-treatedWT mice (Figures 3, D versus A, and M). It was markedlyincreased in MIOX-TG mice (Figure 3, E versus B, and M).No significant increase was observed in MIOX2/2 mice(Figure 3, F versus C, and M). On the other hand, Bcl-2 expres-sion remarkably decreased in both WT and MIOX-TG mice,whereas it remained unchanged in MIOX2/2 mice (Figure3M). The status of another executioner of the apoptosis path-way, known as caspase-3, was investigated. It is activated aftercleavage and catalyzes specific cleavage of multiple cellularproteins and thus activates apoptosis.19 An increased expres-sion of cleaved caspase-3 was observed in kidneys of cisplatin-treated WTmice (Figure 3M). This increase was accentuatedin MIOX-TG mice. No expression of cleaved caspase-3 was ob-served in MIOX2/2mice (Figure 3M). An increase in caspase-3activity in kidneys of cisplatin-treated WT mice was observed(Figure 3N), but it was accentuated in MIOX-TG mice. A mildincrease in its activitywas observed in cisplatin-treatedMIOX2/2
mice (Figure 3N).Using the terminal deoxynucleotidyl transferase–mediated digoxigenin-deoxyuridine nick-end labeling(TUNEL) procedure, a notable degree of apoptosis was observed
1422 Journal of the American Society of Nephrology J Am Soc Nephrol 28: 1421–1436, 2017
BASIC RESEARCH www.jasn.org
in tubules of cisplatin-treated WTmice (Figure 3, J versus G).Whereas a fulminant apoptosis was observed in cisplatin-treatedMIOX-TG mice (Figure 3, K versus H), no TUNEL-positivenuclei were seen in MIOX2/2 mice (Figure 3, I and L).
MIOX Accentuates Cisplatin-Induced ROS GenerationGeneration of ROS is one of the major mechanism(s) respon-sible for cisplatin-induced AKI.20 The status of ROS was in-vestigated because it is known that MIOX overexpressionaccentuates ROS generation.15 Flow cytometric analysesrevealed a dose-dependent increase in ROS levels in cells trans-fected with empty vector after cisplatin (20 mM) treatment(Figure 4, A–C, and J), whereas MIOX-overexpressing cellsexhibited a remarkable further increase in ROS levels aftercisplatin treatment (Figure 4, F–H, and J). AfterMIOX-siRNAor anti-oxidant NAC treatment the cisplatin-induced increasein ROS was reduced (Figure 4, D and E, I and J). Fluorescentmicroscopy of cells subjected to various treatments confirmedthe observations of flow cytometry (Figure 4, L–O). A notableincrease in intensity of staining was observed in cisplatin-treated cells (Figure 4M), and MIOX siRNA and NAC treat-ments reduced the intensity of fluorescence, suggesting thatcisplatin-induced ROS generation is mediated by MIOX(Figure 4, N and O). Likewise, a decrease in the ratio of re-duced GSH/oxidized glutathione (GSSG) was observed aftercisplatin treatment (Figure 4K). A further decrease in theGSH/GSSG ratio was observed inMIOX-overexpressing cells.Treatment of MIOX siRNA normalized the cisplatin-induceddecrease in the GSH/GSSG ratio (Figure 4K).
The ROS generation was also assessed in vivo by stainingrenal tissues with CM-H2DCFDA. An accentuated ROS gen-eration in cisplatin-treated MIOX-TG mice, compared withcontrol WTmice, was observed (Figures 5, E versus D). How-ever, MIOX2/2 mice showed no induction of ROS generation(Figure 5, F versus C). A relatively higher signal of ROS gen-eration, compared with WT mice, was also detected in un-treatedMIOX-TGmice (Figures 5, B versus A). During a seriesof reactions in the G-X pathway there were perturbations inNADPH/NADP+ and NAD+/NADH ratios, and as a resultthere was increased generation of ROS and NOX-4 expressionin diabetic and obesity states.13–15 Similarly, a mild increase inNOX-4 expression was observed in cisplatin-treated WTmice
Figure 1. Characterization of MIOX transgenic and Null mice. (A)Schematic drawing of the construct used for generation of MIOXflox/flox transgenic mice. MIOX gene was inserted after thesecond LoxP site. Its expression would be under the control ofchicken–b-actin hybrid promoter. The flox/flox mice were gen-erated and cross-bred with PEPCK-Cre mice to direct the
expression primarily into proximal tubules. (B) Schematic drawingof the construct used to generate flox/Nullmice. The strategy wasto delete exons 2–10 of the MIOX gene in Null mice or flox/floxmice cross-bred with PEPCK-Cre mice. Western blots revealed anincreased expression (2–3-fold) of MIOX in kidneys of transgenicmice (TG) compared with the flox/flox or WT and PEPCK-Cre mice(C andD). No significant renal expression of MIOXwas observed inMIOX2/2 (KO) mice (E and F). Immuno-histochemistry revealed anincreased MIOX expression in renal proximal tubules of TG miceextending into the deeper cortex (H versus G). No expression ofMIOX was observed in cortical tubules of MIOX2/2 mice (I).*, P,0.01 compared with the flox/flox and PEPCK-Cre groups.
J Am Soc Nephrol 28: 1421–1436, 2017 Myo-Inositol Oxygenase in Renal Injury 1423
www.jasn.org BASIC RESEARCH

(Figures 5, J versus G, and M), and it was accentuated inMIOX-TG mice (Figure 5, K versus H, and M). No significantincrease in NOX-4 expressionwas observed inMIOX2/2mice(Figure 5, L versus I, and M).
Modulation of ROS-Dependent p53 Induction inCisplatin-Induced AKICisplatin induces p53 expression in tubular cells via ROS gen-eration resulting in apoptosis.5,20,21 Because MIOX facilitatesROS generation, which in turn could modulate the inductionof p53, we evaluated the expression of p53 in WT, MIOX-TG,and MIOX2/2 mice and cultured cells. In cisplatin-treatedWTmice a notable increase in the nuclear expression of p53in renal tubules was observed (Figure 6, D versus A, arrow-heads andG), and it was highly accentuated inMIOX-TGmice(Figure 6, E versus B, arrowheads and G). A very mild increasein nuclear p53 expression was observed in cisplatin-treatedMIOX2/2 mice (Figure 6, F versus C, and G). Likewise, cisplatin-treated HK-2 cells revealed a dose-dependent increase in theexpression of p53 and it was notably reduced by NAC orMIOX-siRNA treatment (Figure 6G, right panel), suggestingthat p53 activation is related to ROS generation partlymediatedvia upregulation of MIOX.
MIOX Overexpression Promotes NF-kB ActivationWhile Dampened by Its Gene Disruption in Cisplatin-Induced AKIActivation of p53 by NF-kB is one of the major mechanism(s)in the pathogenesis of folate-induced AKI.22 Conceivably,NF-kBbinds to the p53 promoter andmodulates various patho-biologic processes; on the other hand p53 is regarded as theregulator of NF-kB repression.22–25 This suggests an intricatecrosstalk between p53 and NF-kB.22 In view of the intercon-nectivity of these various signaling events, we investigated thepathobiology of NF-kB among three strains of MIOX mice.Electrophoretic mobility shift assays (EMSA) demonstrated anincreased binding of NF-kB–specific probe in cisplatin-treated WTmice (Figure 7A, lane 3, arrowhead). The bindingwas accentuated in cisplatin-treated MIOX-TG mice (Figure7A, lane 5). No significant increase in NF-kB binding wasobserved inMIOX2/2mice (Figure 7A, lane 7). The specificity
Figure 2. MIOX overexpression exacerbates cisplatin-inducedAKI, whereas its gene disruption ameliorates AKI. Cisplatintreatment (20 mg/kg) led to a significant weight loss in MIOXtransgenic (TG) and WT mice, whereas MIOX2/2 (KO) mice had alesser degree of body weight loss (A). Cisplatin administrationalso led to a marked elevation of both serum creatinine and urealevels in TG mice compared with WT and KO mice (B and C). The
KO mice had minimal increase in creatinine and urea levels. A re-markable increase in Kim-1 mRNA expression was observed inkidneys of MIOX-TG mice compared with WT or KO mice (D).Kidney sectionsofWTmice that hadundergone cisplatin treatmentshowed cytolysis of tubular cells with detachment from the un-derlying basal lamina and loss of brush border (H versus E). Thesechanges were accentuated in MIOX-TG mice receiving cisplatin,andamarkedvacuolizationof tubular cellswasobserved (I versusF).The glomeruli had collapse of the capillary loops. The kidneys ofMIOX2/2 KO mice had a relatively lesser degree of cellular dam-age comparedwith theWTorMIOX-TG strains ofmice (J versusG).
1424 Journal of the American Society of Nephrology J Am Soc Nephrol 28: 1421–1436, 2017
BASIC RESEARCH www.jasn.org

Figure 3. MIOX gene disruption inhibits, whereas its overexpression accentuates, renal tubular cell apoptosis. Expression of proa-poptogenic Bax was increased after cisplatin treatment in kidney tubules of WT mice (D versus A, and M). It was highly accentuated inMIOX-TG mice (E versus B, and M). No increase was observed in MIOX-KO mice (F versus C, and M). Intriguingly, at times a milddecrease in BAX expression was noted in KO mice (M), whereas antiapoptogenic Bcl-2 expression drastically decreased in both the WTandMIOX-TGmice, and it was unchanged in KOmice (M). An increased expression of cleaved caspase-3 was observed in kidneys of WTmice after cisplatin treatment (M), which was accentuated inMIOX-TGmice. No expression of cleaved caspase-3 was observed in treatedor untreated MIOX-KO mice (M). After cisplatin treatment, a marked increase in caspase-3 activity was observed in MIOX-TG mice,whereas a moderate increase was also observed inWTmice (N). No significant increase in caspase-3 was observed in kidneys of KOmice.Along these lines, a fulminant degree of apoptosis was observed in kidneys of MIOX-TG mice (K versus H), although a mild-to-moderatedegree of apoptosis was also observed in WT mice (J versus G), and no apoptosis was detected in MIOX-KO mice (I and L).
J Am Soc Nephrol 28: 1421–1436, 2017 Myo-Inositol Oxygenase in Renal Injury 1425
www.jasn.org BASIC RESEARCH

Figure 4. MIOX accentuates cisplatin-induced ROS generation in vitro. LLC-PK1 cells transfected with empty vector and MIOX pcDNA(overexpressing proximal tubular cell line) were treated with cisplatin (20 mM), and ROS generation was assessed by CM-H2DCF-DAstaining and flow cytometric analyses. The analyses revealed a dose-dependent increase in ROS levels in cells transfected with emptyvector (A–C, and J). MIOX-overexpressing cells had higher ROS levels after cisplatin treatment (F–H, and J). After MIOX-siRNA orantioxidant NAC treatment the cisplatin-induced increase in ROS was notably reduced (D, E, I, and J). Fluorescent microscopy of cells
1426 Journal of the American Society of Nephrology J Am Soc Nephrol 28: 1421–1436, 2017
BASIC RESEARCH www.jasn.org

of the NF-kB probe binding was confirmed by incubatingDNA-protein complexes of renal extracts from WT cisplatin-treated mice with antibody against the p65 subunit of NF-kB.Another high molecular band indicating supershift was ob-served after incubation with the antibody (Figure 7B, lane 3,
arrow). Western blot analyses revealed a translocation of p65into the nucleus from cytoplasm in cisplatin-treated WTmice,and it was accentuated inMIOX-TGmice (Figure 7C, lanes 2 and4). No significant p65 translocation was observed in MIOX2/2
mice (Figure 7C, lanes 5 and 6). Cisplatin-treated HK-2 cells
confirmed the observations made by flow cytometric analyses (L–O). Cellular redox was quantified by assessing the levels of GSSG andreduced GSH (K). A decrease in the ratio of GSH/GSSG was seen after cisplatin treatment, which was further reduced in MIOX-over-expressing cells. MIOX siRNA treatment largely normalized the GSH/GSSG ratio (K). *, P,0.01 compared with the control group;#, P,0.05 compared with the control group.
Figure 5. MIOX accentuates cisplatin-induced ROS generation andNOX-4 expression in vivo. The ROS generation was assessed by stainingrenal tissues of mice with CM-H2DCFDA (DCFA) after cisplatin treatment. A mild increase in DCFA staining was observed in WT type mice (Dversus A), which was accentuated in MIOX-TG mice (E versus B). The MIOX-KO mice showed no induction of ROS generation after cisplatintreatment (F versus C). Compared with WT mice, a relatively higher degree of signal of DCFA staining was observed in TG mice that did notreceive the cisplatin treatment (B versus A). Likewise, NOX-4 expression was markedly increased in MIOX-TG mice compared with WT mice(J versus G, K versus H, and M). No significant increase in the expression was observed in MIOX-KO mice (L versus I, and M).
J Am Soc Nephrol 28: 1421–1436, 2017 Myo-Inositol Oxygenase in Renal Injury 1427
www.jasn.org BASIC RESEARCH

also showed cytoplasm-to-nuclear translocation of the p65, thusestablishing the role of NF-kB p65 in AKI (Figure 7D).
MIOX Modulates the Expression of InflammatoryCytokines in Cisplatin-Induced AKINF-kB plays a role in inflammation via induction of proin-flammatory cytokines.26,27 Because they are implicated in thepathogenesis of AKI, we investigated the expression of variouscytokines, e.g., IL-1b, IL-6, TNF-a, and MCP-1, in differentstrains of mice (Figures 8, A –D). Overall, the changes in theirexpression were similar. A remarkable increase in expression ofall of the cytokines was observed in cisplatin-treated MIOX-TGmice, although a moderate increase in TNF-a expression wasobserved in cisplatin-treated WT mice (Figure 8C). The sourceof these cytokines in AKI could be from the tubular or resident
dendritic cells or cells influxed into the tubulo-interstitium.5,28,29
Keeping in view these possibilities we assessed if there was adifferential increase in infiltrating cells among various strainsof mice. Normally, very few resident inflammatory cells wereseen in the interstitium (Figure 8E, arrowhead). After cisplatintreatment, a mild-to-moderate increase in the influxed inflam-matory cells was observed (Figure 8, F–H, arrowheads). A re-markable increase was seen in cisplatin-treated MIOX-TGmice (Figure 8G, arrowheads). The inset shows the morphol-ogy of infiltrating inflammatory cells (green arrowheads). A fewtubular cells undergoing apoptosis were also seen in MIOX-TGmice (Figure 8G, arrow).
Cisplatin Increases MIOX Expression via Demethylationof Its PromoterHere, we addressed the question of whether cisplatin can in-crease the expression of MIOX and if as a result there is anexcessive ROS generation, which would further accentuate re-nal injury. The answer seems to be affirmative because cisplatinadministration caused an increased MIOX expression (Figure9, A–C). The increase was remarkable and was confined totubules (Figure 9B). Next, mechanism(s) that induced the in-creased MIOX expression were explored. Genomic DNA sam-ples were isolated from kidneys of WT and cisplatin-treatedmice. They were subjected to bisulfite treatment, followed byPCR and nucleotide sequencing to assess the efficiency of con-version (cytosine to thymine) of CpG dinucleotides. Theirnucleotide analyses revealed that the MIOX promoter isdevoid of CpG islands but it has eight CpG dinucleotidesat 219, 222, 242, 248, 273, 2424, 2436, and 2456 bp sites.InWTcontrolmiceCpGat222 and2436bpwereunmethylated,whereas six others were methylated (Figure 9, D and E). Thesamples from cisplatin-treated mice had all of the CpG dinu-cleotides unmethylated, except for 2424 and 2456. Overall,the efficiency of conversion of C to T nucleotide was .95%.The analyses of percentage of methylation of individual dinu-cleotide residues between control and cisplatin-treated micewere plotted as bar graphs, which suggested that cisplatin-induced hypomethylation is most likely responsible for theMIOX upregulation in AKI (Figures 9F).
DISCUSSION
Several years ago, MIOX was identified in proximal tubules ofnewborn mice.30 Since then its relevance in the pathogenesisof diabetic nephropathy has been described in several publi-cations.10,13–15 These studies highlight its biochemical char-acteristics pertaining to transcriptional, translational, andpost-translational events that modulate MIOX expression.However, its relevance to the pathobiology of tubules remainsenigmatic, and that warranted generation of mice with itsgenetic overexpression or deletion. We employed Cre/loxPtechnology to direct its overexpression in renal tubules bycross-breeding floxed mice with PEPCK-Cre mice. These
Figure 6. MIOX accentuates cisplatin-induced ROS-dependentp53 induction in vivo and in vitro. A moderate increase in the nu-clear expression of p53 was observed in WT mice after cisplatintreatment (D versus A, arrowheads, and G, left). The p53 nuclearexpression was markedly accentuated in MIOX-TG mice (E versusB, arrowheads, and G, left). A very mild increase in the p53 nuclearexpression was observed in MIOX-KO mice (F versus C, and G,left). After cisplatin treatment a dose-dependent increase in p53expression was observed in HK-2 cells, and it was reduced afterMIOX-siRNA or antioxidant NAC treatment (G, right).
1428 Journal of the American Society of Nephrology J Am Soc Nephrol 28: 1421–1436, 2017
BASIC RESEARCH www.jasn.org

mice express Cre recombinase gene, and the latter is under thecontrol of mutated PEPCK promoter and thus would expressMIOXmainly in proximal tubules.31 Incidentally, the PEPCKCretransgene is expressed on the X chromosome, and thereforemalemice exhibit greater penetrance than females. In view of this, weselected male mice for our studies. Nevertheless, both male andfemale mice exhibited identical phenotypes, i.e., MIOX overex-pression in proximal tubules of MIOX-TG mice and no expres-sion in MIOX2/2 mice (Figure 1).
Using these mutant mice we exploredadditional functionalities of MIOX in thecontext of pathobiology of proximal tu-bules. The latter are adversely affected invarious disease states that result in AKI. AprototypeofAKI is cisplatin-inducedneph-rotoxicity, where proximal tubules bear thebrunt of injury, possibly due to their uptakeof cisplatin.32 In this scenario, in whichproximal tubules are targeted and whereMIOX is expressed, it would indicate thatthe cisplatin-induced AKI is an appropriatemodel to delineate the pathogenetic role ofMIOX in renal injury. Numerous reportshave documented the cellular toxicity inproximal tubules accompanied with com-promise in renal functions.4,5,32 Likewise,we observed vacuolization and necrosis oftubular cells accompanied with a rise in se-rum urea and creatinine, and loss of bodyweight in C57BL/6J mice (Figure 2). Thesechanges were highly accentuated inMIOX-TGmice. Interestingly, the MIOX2/2 micehad amuch lesser degree of cisplatin-inducedtubular toxicity and minimal elevation ofserum creatinine. An affirmation of theseresults was derived by assessing the statusof KIM-1, a marker of tubular injury.18 Aremarkable elevation of KIM-1 levels wasobserved in MIOX-TG mice, whereas therewas a modest increase in control mice anda minimal rise in MIOX2/2 mice. Besidesnecrosis, cellular apoptosis has been welldescribed in cisplatin nephrotoxicity, whichapparently relates to the activation of proa-poptotic protein Bax and caspases whilediminishing the expression/activity of an-tiapoptotic Bcl-2.20,21,33,34 Interestingly,apoptosis, Bax activation, and expressionof cleaved caspase-3 and its activity weremarkedly accentuated in MIOX-TG micecompared with control animals (Figure 3).Notably, the MIOX2 /2 mice hadno apoptosis or Bax activation or expres-sion of cleaved caspase-3, and only a mar-ginal increase in the caspase-3 activity,
whereas a marked downregulation of antiapoptotic proteinBcl-2 was observed in MIOX-TG after cisplatin treatment.Taken together, these data indicate that MIOX overexpressionaccentuates AKI, whereas its gene disruption is protective forkidney homeostasis.
The next question that was addressed was how MIOX ac-centuates AKI. Besides necrosis/apoptosis, oxidant stress hasbeen regarded as one of the factors for induction of nephro-toxicity.16 Previous studies indicate that MIOX promoter
Figure 7. MIOX overexpression promotes NF-kB activation while dampened by itsgene disruption in AKI. EMSA revealed an increased binding of the NF-kB–specificprobe with the nuclear extracts isolated from kidneys of WT mice treated with cisplatin(A, lane 3, arrowhead). The binding was further accentuated in MIOX-TG mice, asgauged by the intensity of the band (A, lane 5). Minimal binding of NF-kB probe wasobserved in MIOX-KO mice receiving cisplatin (A, lane 7). The specificity of the NF-kBprobe binding was confirmed by incubating renal nuclear extracts of WT mice treatedwith anti-p65 antibody before EMSA. An additional high molecular band indicatingsupershift was observed after incubation with antibody against the p65 subunit ofNF-kB (B, lane 3, arrow). Subcellular compartmentalization of the transcriptionalsubunit of NF-kB p65 after cisplatin treatment demonstrated the activation of NF-kB.Western blot analyses revealed a translocation of p65 from the cytoplasm into thenucleus after cisplatin administration in WT mice (C, lane 2), and the translocation wasfurther accentuated in MIOX-TG mice, whereas no significant translocation was ob-served in MIOX-KO mice after cisplatin treatment (C, lanes 4 and 6). The b-actin andlamin–b-1 served as controls. The cisplatin-induced p65 translocation was confirmedby in vitro experiments in HK-2 cells treated with cisplatin (D).
J Am Soc Nephrol 28: 1421–1436, 2017 Myo-Inositol Oxygenase in Renal Injury 1429
www.jasn.org BASIC RESEARCH

includes oxidant response elements, and its overexpression invitro accentuates generation of ROS.10,14 The current data re-garding accentuated expression, as well as activity, of caspase-3points towardmitochondria as the source of excessive generationof ROS in MIOX-TGmice. In this regard, MIOX overexpressionin LLC-PK1 cells has been reported to cause exaggerated mito-chondrial dysfunctions, such as cytochrome C release, DNA
fragmentation, and mitochondrial fission, under high glucoseambience.14 However, in cisplatin-induced nephropathy the in-creased ROS generation in MIOX-overexpressing cells could bealso related to the pathobiology of the NADPH oxidase system.In this regard, perturbations in NAD+/NADH ratios have beendescribed in the MIOX-initiated G-X pathway.14 In support ofthe latter contention our in vitro data indicate a perturbed GSH(oxidized)/GSSG (reduced) ratio after cisplatin treatment,which was largely normalized by the concomitant administra-tion of MIOX-siRNA (Figure 4). Likewise, MIOX-siRNA treat-ment of LLC-PK1 cells markedly reduced the generation ofROS after cisplatin exposure, as assessed by FACS analysesand immunofluorescence microscopy after DCFDA staining(Figure 4). The production of ROS was markedly accentuatedin the renal tubular compartment of MIOX-TG mice, whereasMIOX2/2 mice had minimal generation of ROS, even aftercisplatin treatment (Figure 5). Further support for the involve-ment of NADPH oxidase in the current model comes fromexpression studies on the induction of tubular–NOX-4. Aprominent expression of NOX-4 was observed after cisplatintreatment in MIOX-TG mice, whereas it was barely detectablein WT and MIOX2/2 mice (Figure 5). Taking these data to-gether, one can conclude that MIOX accentuates cisplatin-induced AKI, most likely via boosting ROS generation.
Besides ROS and apoptosis, various interlinked p53-associatedpathways and molecules are also involved in the pathogen-esis of cisplatin toxicity. They may include p53 upregulatedmodulator of apoptosis (PUMA), histone deacetylase inhibitors,taurine transporter (TauT) gene, and Sirt1.20,21,35–37 The induc-tion of PUMA is dependent on the activation of p53 becauseinhibition of p53 with pifithrin or its genetic ablation reducesapoptosis.21 Interestingly,N-acetyl-cyteine (NAC) or dimethylth-iourea administration reduces hydroxyl radical generation, i.e.,oxidant stress, p53 activation, and suppression of PUMA-a andapoptosis.20 Along these lines the TauT transgenic mice exhibitattenuation of cisplatin-induced apoptosis by dampening the ac-tivation of p53.36 Similarly, Sirt1, anNAD-dependent deacetylase,has been reported to reduce ROS-induced apoptosis most likelyvia deacetylation of p53, thereby reducing its transcriptional ac-tivation.38 On the other hand, histone deacetylase inhibitor tri-chostatin Awas administered alongwith cisplatin, and although itenhances tumor cell death it limits cisplatin-induced toxicity orapoptosis in the kidney, probably by increasing phosphorylationof cAMP-response element binding protein and reversing cAMP-response element binding protein–mediated gene repression.39 Inline with these literature data we observed increased p53 expres-sion after cisplatin administration, especially in MIOX-TG micehaving accentuated ROS generation (Figure 6). The question ofwhether MIOX-generated ROS induces p53 expression wasassessed by in vitro experiments. The cisplatin-treated cells hadincreased expression of p53, which was abrogated with MIOX-siRNAorNAC treatment, suggesting thatMIOXaccentuates p53-mediated injury via excessive ROS generation (Figure 6).
Another pathobiologic process that plays a noteworthy rolegermane to cisplatin-induced AKI relates to inflammation.28
Figure 8. MIOX modulates the expression of inflammatory cy-tokines in cisplatin-induced AKI. A remarkable increase in theexpression of IL-1b, IL-6, TNF-a, and MCP-1 was observed inMIOX-TG mice receiving cisplatin compared with WT or MIOX-KOmice or untreated control mice (A–D), although a moderateincrease of TNF-a was observed in WT mice after cisplatintreatment (C). Examination of kidneys revealed a remarkableincrease of interstitial inflammatory cells in the MIOX-TG micereceiving cisplatin (G, arrowheads). The inset shows the mor-phologic nature of the infiltrating inflammatory cells (greenarrowheads). A mild increase was also observed in cisplatin-treated WT mice (F, arrowheads). The MIOX-KO mice had nosignificant increase of inflammatory cells compared with the un-treated WT control (H versus E, arrowheads). A few foci of apo-ptosis were also seen in MIOX-TG mice (G, arrow). *, P,0.01compared with the control group; #, P,0.05 compared with thecontrol group.
1430 Journal of the American Society of Nephrology J Am Soc Nephrol 28: 1421–1436, 2017
BASIC RESEARCH www.jasn.org

In this realm there may be activation of transcription factors,generation of cytokines, and influx of inflammatory cells. Oneof the important transcription factors that is induced afteroxidant stress includes NF-kB.40 It modulates diverse biologicprocesses, including inflammatory responses, while at thesame time modulating p53 expression.22 Interestingly, recentdata also suggest that p53 itself can regulate NF-kB repres-sion.25 This would suggest an intricate reciprocal crosstalkbetween these two molecules during an inflammatory re-sponse in AKI.22 The role of NF-kB has been ascribed to di-verse diseases associated with renal inflammation.27 Also, itsrole in the folic acid–induced toxic injury model is wellknown, where folic acid administration was shown to con-comitantly upregulate expression of p53, NF-kB, and its ac-tive transcriptional subunit, relA/p65.22 Administration ofpyrrolidine dithio-carbamate ammonium, an NF-kB inhibi-tor, reduced the upregulated expression of all of these mole-cules along with improvement in renal functions. Along theselines, we investigated the role of NF-kB in cisplatin-inducedinjury in MIOX-TG and MIOX2/2 mice. By EMSA, an in-creasedNF-kB binding was seen in cisplatin-treatedWTmice,and it was accentuated in MIOX-TG, although no significantbinding was seen in MIOX2/2 mice (Figure 7). The role ofNF-kB in cisplatin-induced nephropathy was further suppor-ted by accentuated translocation of p65 into the nuclear com-partment in renal cells ofMIOX-TGmice. The downstream ofNF-kB activation would include upregulation of variousinflammatory cytokines, including TNF-a.41 In fact, TNF-ageneration after the activation of NF-kB in cisplatin-inducednephropathy iswell described.42 In addition, there is an increasedexpression of other cytokines, including IL-1b, MCP-1, andRANTES. We also observed an upregulation of inflammatorycytokines after cisplatin administration, especially in MIOX-TGmice (Figure 8). The concomitant rise of different cytokines maysuggest that they synergistically stimulate one another’s produc-tion in MIOX-TG mice that are endowed with increased gener-ation of ROS in their kidneys. Some of these cytokines, e.g.,MCP-1, are chemotactic for a variety of inflammatory cells, in-cluding neutrophils, monocytes, and lymphocytes.42 In concur-rence with the production of cytokines an accentuated influx ofinflammatory cells into renal parenchyma was observed in
Figure 9. Cisplatin treatment per se upregulates MIOX via de-methylation of its promoter. WT mice treated with cisplatin hadan increased renal expression of MIOX, and it was confined tothe tubules (A–C). No significant MIOX expression was seen inglomeruli. Genomic DNA was isolated from kidneys of WT andcisplatin-treated mice and subjected to bisulfite treatment. Pro-moter fragments were isolated by PCR, cloned into pGEM-Tplasmid, and transformed in DH5a cells. Ten clones from each
variable were selected and subjected to nucleotide sequencing toassess the efficiency of conversion (cytosine to thymine) of CpGdinucleotides. Nucleotide analyses revealed eight CpG dinucle-otides at 219, 222, 242, 248, 273, 2424, 2436 and 2456 bpsites. In WT, untreated control mice samples, CpG at 222 and 2436 bp were found to be unmethylated, whereas six others weremethylated (D and E). The samples from cisplatin-treated micewere found to have all of the CpG dinucleotides unmethylated,except for2424 and2456. Overall, the efficiency of conversion ofC to T nucleotide was .95%. The percentages of methylation ofindividual dinucleotide residues are presented as bar graphs,which indicate that cisplatin induces hypomethylation that is likelyresponsible for the MIOX upregulation (F).
J Am Soc Nephrol 28: 1421–1436, 2017 Myo-Inositol Oxygenase in Renal Injury 1431
www.jasn.org BASIC RESEARCH

MIOX-TGmice (Figure 8). No significant influx was observed inMIOX2/2mice, suggesting that increasedMIOXexpression leadsto an activation of various inflammatory responses.
Several publications indicate that in a variety of pathobiologicstates the increasedexpressionofMIOXseems tobe the resultof itstranscriptional modulation, where conceivably various transcrip-tion factors bind to specific consensus sequences localized withinits promoter.10,13–15,43 Therefore, the question that warrants ananswer is, whether there is an increased expression of MIOX aftercisplatin treatment, and if so, what are the mechanism(s) respon-sible for such an upregulation? Indeed, there was increased ex-pression of MIOX after cisplatin treatment (Figure 9). Becausethere are no known transcription factors which could bind to theMIOX promoter and induce its upregulation in cisplatin toxicity,we therefore proceeded to search for an alternative mechanism.One such mechanism that we explored pertains to the epigeneticregulation of MIOX, i.e., methylation/demethylation of itspromoter. Epigenetic modification of various genes has been im-plicated in various renal diseases with consequential altered ex-pression.44 In this regard, hypomethylation/demethylation hasbeen reported in AKI induced by ischemia reperfusion or contactfreezing of the kidney surface.45–47Althoughamultitude ofmech-anisms have been described in cisplatin-induced AKI, the studiesrelating to epigenetic modifications are sparsely reported.5,32,48 Inlinewith the observationsmade in the ischemia reperfusion injurymodel, we observed a marked demethylation of the CpG dinu-cleotides in the MIOX promoter (Figure 9), which could explainits increased expression and ultimately exacerbation of cisplatin-induced injury in MIOX-TG mice.
In summary, this investigation highlights the worsening ofAKI in cisplatin-induced injury via a multitude of mechanismsin states of overexpression of MIOX, although its gene disrup-tion seems to ameliorate toxic renal proximal tubular injury.
CONCISE METHODS
ReagentsReagents were purchased from the following vendors. Sigma-Aldrich:
cisplatin (# P4394), anti–b-actin antibody (# A5441), Caspase-3 assay
kit, Colorimetric (# CASP-3C), poly-(deoxyinosinic-deoxycytidylic)
acid sodium salt (# P4929), NAC (# A7250); BioAssay Systems: Quanti-
Chrom creatinine assay kit (# DICT-500) and QuantiChrom urea
assay kit (# DIUR-500); Life Technologies: Power SYBR Green PCR
Master Mix (# 4367659), Chloromethyl derivative of 29, 79-dichloro-
fluorescein diacetate (CM-H2DCF-DA, # C6827), 49-6-diamidino-2-
phenylindole (DAPI, # D1306), and TRIzol Reagent (# 15596026);
Cell Signaling Technology: anti–cleaved caspase-3 antibody
(# D175), anti-p53 (1C12) antibody (# 2524), and anti-p65 antibody
(# D14E12); Abcam: anti–lamin B1(ab16048); Enzo Life Sciences:
DNA methylation gold kit (# D5005); Santa Cruz Biotechnology:
anti-Bax antibody (# SC-526), anti–NOX-4 antibody (# SC-21860),
anti-p53 antibody (# SC-6243); Roche Diagnostic: In Situ Cell Death
Detection Kit, Fluorescein (# 11684795910); Cayman Chemicals:
GSH assay kit (# 703002); Promega Corporation: NF-kB EMSA oligo
(# E329A) and pGEM-T plasmid vector; Perkin Elmer: ATP g-32p
(# BLU002A); and Thermo Scientific: polynucleotide kinase
(EK0032). OriGene Technologies: MIOX siRNA (# SR310776).
Anti-MIOX polyclonal antibody was prepared in our laboratory using
recombinant mouse MIOX as the immunogen.10
Generation of MIOX Transgenic and Null MiceThe transgenic CBA-flox mice were generated using the pCMV flox
vector (a generous gift fromDr.Holzman).49 TheCMVpromoter was
replaced with the cytomegalovirus–chicken b-actin hybrid promoter
which contains the cytomegalovirus immediate early enhancer linked
to chicken b-actin promoter and a chimeric intron of b-globin and
immunoglobin genes.50,51 This hybrid promoter demonstrates en-
hanced activity in vivo and was used to drive the expression of a
flanked b-gal-stop-codon cassette.52 The MIOX cDNA was inserted
downstream of the b-gal-stop-codon cassette at the NheI and PmeI
sites (Figure 1A). This construct enables the expression of the b-gal
gene (as verified by X-gal staining in LLC-PK1 cells), but it does not
express the MIOX gene. After recombination of the Loxp sites in the
presence of Cre recombinase, the b-gal gene is deleted and MIOX is
expressed under the regulation of the b-actin hybrid promoter. The
CBA-flox construct was released from the plasmid vector by diges-
tion with PacI and microinjected into the pronucleus of fertilized
ova from C57BL/6 F1 mice. The litter was screened for the presence
of transgene using b-gal–specific primers as previously described.52
The founders were cross-bred with PEPCK-Cre C57BL/6J mice
(a generous gift from Dr. Volker Hasse)31 to direct expression pri-
marily into the proximal tubules. The mice were then screened for
the MIOX transgene using primers encompassing the NheI and
PmeI restriction sites. Their kidneys were tested for the extent of
MIOX expression by Western blot analyses and immuno-histochemical
techniques.
MIOXnullmicewere generated atCellMolecularTechnology Inc.,
Phillipsburg, New Jersey. The design of the targeting vector for gen-
eration of MIOX knockout mice is included in Figure 1B. Briefly, the
targeting vector was designed to generate both Null and Flox mice
lines. A 26 kb BAC subclone was cloned by applying Gene Bridges
technology.53 A 59 LoxP site was placed in intron 1 at the AfIII site
and a FRT-Neo-FRT-39 LoxP cassette at the AatII site after exon 10. In
doing so, Cre-recombinase would excise exons 2–10 along with the
Neo cassette leaving behind a single LoxP site to generate Null mice
lines; whereas FLP recombinase would excise only the Neo cassette
leaving exons 2–10 and Loxp sites intact to generate the conditional
(Flox) line in embryonic stem cell (ES) recombinants. The targeting
vector was electroporated into 129/SVEV ES cells. The ES recombi-
nant clones were picked and tested by PCR tomake sure the excisions
had taken place. After injection of ES recombinants into blastocysts
and subsequent implantation, chimeras were generated and mouse
lines expanded. The Flox mouse lines were also cross-bred with
PEPCK-Cre mice to selectively delete the MIOX gene from the prox-
imal tubules. Both the mouse lines were backcrossed with C57BL/6J
for seven generations to generate mouse lines with a homogenous
background. Their kidneys were harvested to confirm deletion of
MIOX genes by Western blot analyses and immuno-histochemical
techniques.14,15
1432 Journal of the American Society of Nephrology J Am Soc Nephrol 28: 1421–1436, 2017
BASIC RESEARCH www.jasn.org

Design of In Vivo ExperimentsFor AKI experiments, 8-week-old male WTmice and sex- and age-
matched MIOX2/2 (KO) and MIOX-overexpressing transgenic
(MIOX-TG) mice were utilized. Initially, various dosages (5–25
mg/kg body wt) of cisplatin (single intraperitoneal injection) were
tested for tolerance among these three strains of mice. A dose of 20
mg/kg (1 mg/ml solution in sterile normal saline) was tolerated by all
three of the strains without having significant mortality, and this dose
was chosen for all of the subsequent experiments (n=6). The control
mice were injected with normal saline only. Mice were euthanized 3
days after the cisplatin treatment. Blood samples were collected for
measuring levels of serum creatinine and urea using QuantiChrom
Creatinine and QuantiChrom Urea assay kits (BioAssay Systems).
Their kidneys were utilized for various morphologic, biochemical,
and molecular biology studies. All animal procedures used in this
study were approved by the Animal Care and Use committee of
Northwestern University.
Cell Culture StudiesHK-2 cells and LLC-PK1 cells were purchased fromATCC (Manassas,
VA) and used in this study. The HK-2 cells were grown in low glucose
DMEM medium supplemented with 5% heat-inactivated FBS,
100 U/ml penicillin, and 100 mg/ml streptomycin. LLC-PK1 cells were
grown in low glucose M199 medium supplemented with 5% heat-
inactivated FBS, 100 U/ml penicillin, and 100 mg/ml streptomycin.
The cells in quadruplicate plates were maintained in a humidified at-
mosphere of 5% CO2, 95% air, at 37°C. In addition, MIOX-overexpressing
LLC-PK1 cells were also used in some of the experiments. The gener-
ation of overexpressing cell lines has been detailed in previous publi-
cations.15 Approximately 13 106 cells were seeded per well in six-well
collagen-coated plates and allowed to adhere to the culture plates
overnight. The cells were then treated with various concentrations
of cisplatin (5–20 mM) for 12–24 hours, and then processed for var-
ious morphologic and biochemical studies. In addition, MIOX siRNA
(50 nM) and ROS scavenger (NAC, 10 mM) studies were also carried
out. The LLCPK1 cells were transfected with siRNA against MIOX14
or scramble (serve as a control) using Lipofectamine 2000 reagent
(Invitrogen) in the presence of 20 mM of cisplatin. Forty-eight hours
later the ROS generation, GSH/GSGG ratios, and p53 expression were
assessed in the cells.
Renal Tissues’ Histologic and Immuno-HistochemicalStudiesKidneys from control (WT) and experimental (TG/KO) mice were
harvested, and 3–4 mm thick slices were prepared. They were fixed in
10% buffered formalin, dehydrated by graded concentrations (50%–
100%) of ethanols, and then transferred into xylene and embedded in
paraffin. For immuno-histochemistry, 4 mM thick sections were pre-
pared and mounted on glass slides. The tissue sections were air-dried
overnight. They were then deparaffinized, hydrated, and then pro-
cessed for antigen retrieval. After antigen retrieval, endogenous per-
oxidase activity in the tissues was quenched by immersing glass slides
with tissue sections in 3% H2O2 for 3 minutes. Tissue sections were
immersed in 3% BSA for 1 hour and incubated in a primary antibody
overnight at 4°C. After washing with PBS the sections were incubated
with horse radish peroxidase (HRP)–conjugated anti-rabbit IgG,
immPRESS (Vector Laboratories), for 1 hour. The sections were sub-
jected to another PBS wash and then incubated with HRP substrate
diaminobenzadine for color development. Sections were rewashed
and counterstained with hematoxylin to delineate the cellular details.
The sections were then dehydrated in a graded series of ethanols,
treated with xylene, and coverslip-mounted after immersion in a
drop of mounting media (Vector Labs). Slides were then viewed
and photographed.
Immunoblotting Analyses of Relevant ProteinsExpressed in Kidney TissuesKidney tissues from control and cisplatin-treatedWT,MIOX-TG, and
MIOX2/2 mice were harvested and lysed in RIPA buffer. Superna-
tants were collected and protein concentration was measured using
Bradford reagent. Equal amounts of protein for each sample were
mixed with 53 SDS sample buffer and boiled for 5 minutes. The
proteins in the lysates were fractionated by SDS-PAGE and electro-
blotted onto PVDF membranes. The blots were then probed with
various primary antibodies, followed by incubation with secondary
antibodies conjugated withHRP. Autoradiograms were developed for
detection of the bands using an Enhanced ChemiLuminence kit
(Thermoscientific). Equal loading of the samples was confirmed by
probing the immunoblots with mouse b-actin or lamin-b1 anti-
bodies.15
TUNEL Staining Studies in Kidney TissuesThe extent of apoptosis in kidney tissues was determined by estab-
lished TUNEL procedures.14 Deparaffinized kidney sections were hy-
drated and then digested with Proteinase K (240 unit/ml; Promega,
Madison) for 30 minutes at 22°C. After washing tissue sections with
PBS, they were incubated with TUNEL reagent for enzymatic labeling
of the nicked DNA with a fluorescent nucleotide probe (Roche Ap-
plied Science).
Assessment of Caspase-3 Activity in Kidney TissuesThe assay was performed using a Colorimetric caspase-3 assay kit
(Sigma), as previously described.15 Kidney tissues from WT,
MIOX-TG, and MIOX2/2 mice were harvested. Their cortices were
dissected and homogenized in a lysis solution provided in the kit. The
tissue lysates were then centrifuged at 16,0003 g for 10 minutes at 4°
C. Supernatants were transferred into a new tube and protein con-
centration measured by using the Biorad Bradford Reagent. After
adjusting the protein concentration to 5 mg/ml the caspase-3 activity
was determined. Each sample was assayed in duplicate in a 96-well
plate in 200 ml reaction volume. The reaction mixture included 10 ml
of lysate containing 50 mg of protein per sample, 170 ml of 13 assay
buffer, and 20ml of caspase-3 substrate. Wells were then covered with
adhesive film and incubated at 37°C for 3 hours. The sample absor-
bance was read at 405 nm.
Assessment of ROS in Tissues and CellsFor determination of ROS in kidneys, tissues from both control and cis-
platin-treatedWT,MIOX-TG,andMIOX2/2micewereembeddedinOCT
compound and processed for frozen sectioning. Eight-micrometer-thick
J Am Soc Nephrol 28: 1421–1436, 2017 Myo-Inositol Oxygenase in Renal Injury 1433
www.jasn.org BASIC RESEARCH

frozen sections of kidney were prepared, transferred onto glass slides, and
air dried for 5minutes. The slideswere then rinsedwith PBS to remove the
OCT compound from tissue sections. Tissue sections were then stained
with CM-H2DCFDA for 15 minutes at 37°C in the dark and visualized
using a fluorescent microscope equipped with UV illumination.
ROS levels were determined in cisplatin-treated cells by CM-
H2DCFDA staining using flow cytometry and fluorescent micros-
copy.14,15 For flow cytometry, cells transfected with empty vector
and MIOX-overexpressing LLCPK1 cells were seeded onto six-well
plates and treated overnight with different concentrations of cis-
platin, as indicated above. Cells were then washed in PBS and de-
tached from the plate using BD Accutase cell detachment solution.
Cells were then stained with of CM-H2DCFDA (5 mM) for 15 min-
utes at 37°C in dark. After washing twice with PBS the cells were
resuspended in 300 ml of PBS and processed for acquisition of fluo-
rescence with a flow cytometer. After which, mean fluorescent inten-
sity of the CM-H2DCFDA was measured using standard operational
procedures of flow cytometry and employing FACSDiva software
(Becton Dickinson). For fluorescence microscopy, HK-2 cells were
seeded onto coverslips and treated with different concentrations of
cisplatin (10 and 20 mM) for 12 hours. Cells were then washed and
stained with 5 mM of CM-H2DCFDA for 15 minutes at 37°C in the
dark. After a brief rewash with PBS the coverslips with attached cells
were inverted and mounted on the glass slides after placing a drop of
DAKO mounting medium. The cells were then examined with a UV
microscope.
Determination of GSH/GSSG Ratio in Cultured CellsThiswasmeasuredusingGSHassay kit (CaymenChemicals) using the
manufacturer’s instructions, as previously described.15 Both empty
vector control LLCPK1 cells andMIOX-overexpressing LLCPK1 cells
were used in this study. Cells that had undergone various treatments
were collected and sonicated in 500ml of MES buffer. Lysed cells were
then centrifuged to collect supernatant. Protein concentration in su-
pernatant (#1) wasmeasured using the Biorad Bradford Reagent. The
supernatants were then deproteinated by using equal volumes of
freshly prepared 10% metaphosphoric acid. The precipitates were
allowed to settle and supernatants (#2) were utilized for GSH mea-
surement. Fifty microliters of triethanolamine reagent was added to
each 1 ml sample of the supernatants (#2) and mixed by immediate
vortexing. Fifty microliters of mixture was used for determination of
GSH. For GSSG determination, 10 ml of 1 M 2-vinylpyridine was
added to 1 ml of triethanolamine reagent mixture, as prepared above.
The mixture was vortexed and incubated at 22°C for 1 hour. Fifty
microliters of this mixture was added to the 150 ml of assay solution
provided in the kit and gently shaken in the dark on an orbital shaker
for 45minutes to achieve full color development. Spectrophotometer
readings were made from four experiments at 415 nm, and concen-
trations of GSH and GSSG were measured using a standard curve.
Preparation of Cytosolic and Nuclear Extracts fromKidney Tissues for EMSAKidney tissues harvested from control and cisplatin-treated WT,
MIOX-TG, and MIOX2/2 mice were first homogenized in cytoplas-
mic extraction buffer (10 mM HEPES, 10 mM KCl, 0.1 mM EDTA,
and 0.1 mM EGTA) and incubated for 15 minutes on ice. Then, 12.5
ml of 10% NP-40 per 400 ml of lysis buffer was added and vortexed
vigorously for 10 seconds to lyse the cells. The lysate was then centri-
fuged for 1 minute, and the supernatant was collected and designated
as the cytosolic extract. Pellets were then processed for nuclear extract
preparation. They were incubated in nuclear extraction buffer
(20 mM HEPES, 400 mM NaCl, 1 mM EDTA, and 1 mM EGTA)
for 30 minutes on ice with intermittent vigorous vortexing to lyse
nuclei. Nuclear extract was collected as supernatant after 5minutes of
centrifugation, and protein concentration in various samples was
measured. The nuclear extracts prepared above from various kidney
tissues were utilized for EMSA to assess the NF-kB activation and its
nuclear translocation. Ten micrograms of nuclear extract from each
sample was incubated with 32P-labeled double stranded NF-kB
probe (Promega) for 15 minutes at 37°C. The DNA-protein complex
formed was subjected to 6.6% native PAGE. Gels were dried and
exposed to x-ray film and autoradiograms prepared. Specificity of
binding of NF-kB oligo probe with NF-kB protein was assessed by
performing a supershift assay. Nuclear extract was prepared from
cisplatin-treated WT mice kidneys. It was incubated with antibody
against the p65 subunit of NF-kB for 15 minutes at 22°C before incuba-
tion with labeled NF-kB probe. The shift in the DNA-protein-antibody
complex migration in the gel was visualized after autoradiography.
Cellular Translocation of NF-kBTo assess the translocation of NF-kB from cytoplasm to the nucleus in
HK-2 cells, immunofluorescence studies were performed. Briefly, 5
3 105 cells were seeded onto coverslips in a six-well plate and allowed
to attach overnight. Cells were then treated with 20 mM of cisplatin
for 12 hours. After treatment, the cells were fixed with 4% parafor-
maldehyde for 5 minutes in PBS and washed three times with PBS.
They were then permeabilized with ice cold methanol for 10 minutes
at 220°C. Cells were rewashed twice with PBS and incubated in a
blocking solution (13 PBS/1% BSA/0.3% Triton X-100) for 1 hour.
After blocking, fixed cells were incubated with anti-p65 antibody di-
luted in blocking solution for 2 hours at 22°C. Cells were thenwashed
three times with PBS and incubated with FITC-conjugated secondary
antibody for 2 hours in the dark. Cells were then rewashed with PBS,
and nuclei were stained with 0.5 mg/ml of DAPI for 5 minutes. After
another PBS wash, a drop of DAKO fluorescent mounting media was
placed on the coverslips, and they were then inverted and mounted
onto the glass slides. The images were captured using a Nikon A1R
Spectral Confocal microscope.
Assessment of Tissue Expression of InflammatoryCytokines and Kidney Injury Biomarkers by Real-TimePCRTotal RNAwas isolated from kidneys using TRIzol Reagent (Invitro-
gen) and cDNAwas synthesized using Go Script reverse transcription
system (Promega). ThemRNA levels of various genes were quantified
using Step One Plus System Real Time PCR (Applied Biosystems). A
20 ml total reaction mix included 100 ng of cDNA, 50 nmol/L for-
ward/reverse primers, and 13 Fast SYBR Green Master Mix (Life
Techologies). GAPDHwas used as an internal control and the amount of
mRNA was calculated by comparative CTmethod. PCR reactions were
1434 Journal of the American Society of Nephrology J Am Soc Nephrol 28: 1421–1436, 2017
BASIC RESEARCH www.jasn.org

run in quadruplicate. Primers used were, Kim 1: Forward, 59-GGAAG-
TAAAGGGGGTAGTGGG-39, Reverse, 59-AAGCAGAAGATGGGCATTGC-
39; IL-1b: Forward, 59-ACCTGTCCTGTGTAATGAAAGACG-39, Reverse,
59-TGGGTATTGCTTGGG ATCC-39; MCP-1: Forward, 59-CCCAATGAG-
TAGGCTGGAGAG-39, Reverse, 59-TGGTTG AAAAGGTAGTGGATG-39;
TNF-a: Forward, 59-AGAAGAGGCACTCCCCCAAAAG-39, Reverse,
59-TTCAGTAGACAGAAGAGCGTGGTG-39; GAPDH: Forward,
59-GAATACGGCTACAGCAACAGG 39, Reverse, 59-GGTCTGGGATG-
GAAATTGTG-39; and IL-6: Forward, 59-CCGGAGAGGAGACTTCA-
CAG-39, Reverse, 59-CAGAATTGCCATTGCACAAC-39.
Analysis of MIOX Promoter Methylation/Demethylation in Kidney Tissues after CisplatinTreatmentKidneys of mice treated with cisplatin were harvested for isolation of
DNA. Their cortices were dissected, and an aliquot of cortex (20 mg)
was homogenized in 550 ml of lysis buffer (50 mM Tris-HCI, 10 mM
EDTA, 100 Mm NaCl, 1% SDS) containing RNAase1 (1 mg/ml). The
homogenate was kept at 55°C for 2 hours to achieve complete solu-
bilization of cellular proteins. After a phenol-choloroform extraction
the DNA was precipitated with the addition of isopropanol at 4°C.
The precipitated DNAwas pelleted by a centrifugation at 10,0003 g
for 5 minutes. The DNA pellet was washed with 70% ethanol, air-
dried, and dissolved in nuclease-free water. The isolated DNA (2 mg)
was subjected to bisulphite modification, using EZ DNAmethylation
Gold Kit (Zymo Res. Irvine, CA), and by following the methodology
originally described by Frommer et al.54 In doing so, the sodium
bisulphite in an acidic environment will convert all cytosine residues
in single stranded DNA to uracil, excluding the methylated cytosine.
The bisulfite-treated DNAwas loaded into spin columns provided in
the kit and eluted with 20 ml of elution buffer. This purified and
bisulfite-treated DNAwas used to determine the frequency of meth-
ylation of CpG islands within the MIOX promoter, using bisulfite-
specific PCR primers. These primers were devoid of CpG sites. For
amplification of the mouseMIOX promoter, 2ml of treated DNAwas
amplified in a reaction mixture containing: 4 mM primers (Forward-
59-GTTTTTATAGTAAAAGGTGGG-39 Reverse- 59-CCTAAAAAAA-
CAAACACC-39), 13 PCR buffer, 0.4 mM dNTP mix, 1 U Taq
polymerase (1 U/ml), and the final volume of the reaction mixture
was adjusted to 25ml with nuclease-free water. Amplification conditions
were as follows: 94°C for 5minutes, 39 cycles at 94°C for 30 seconds, 55°
C for 45 seconds, 70°C 2 minutes, and final extension at 70°C for 7
minutes. Amplified PCR products were subjected to 1.5% agarose gel
electrophoresis, and the relevant band was eluted using QIA quick gel
extraction kit (Qiagen). The PCRproduct was then cloned into pGEM-T
Vector System I. After transformation in the DH5a strain of E.coli, the
transfectants were plated on ampicillin agar plates. Ten positive colonies
of each sample were amplified for preparation and purification of plas-
mid DNA containing the MIOX promoter. For sequencing of plasmid
DNA, T7 promoter and SP6 promoter primers were used.
Statistical AnalysesResults were expressed asmean6SD after statistical analyses. t test was
used to compare the data between groups. A P value of ,0.05 was
considered statistically significant.
ACKNOWLEDGMENTS
We are thankful to Dr. Anupam Agarwal, Dr. Subhashini Bolisetty,
Dr. Volker Hasse, and Dr. Larry Holzman for providing the plasmid
constructs and PEPCK mice.
Supported by National Institutes of Health grants DK60635,
DK78314, and T32 DK007139.
DISCLOSURESNone.
REFERENCES
1. Bellomo R, Kellum JA, Ronco C: Acute kidney injury. Lancet 380: 756–766, 2012
2. de Mendonça A, Vincent JL, Suter PM, Moreno R, Dearden NM,Antonelli M, Takala J, Sprung C, Cantraine F: Acute renal failure in theICU: Risk factors and outcome evaluated by the SOFA score. IntensiveCare Med 26: 915–921, 2000
3. Racusen L, Kashgarian M: Ischemic and toxic acute tubular injury andother ischemic renal injury. In: Pathology of the Kidney, Vol. II, 6th Ed.,edited by Jennette JC, Olson JL, Schwartz MM, Silva FG, Philadelphia,Lippincott-Williams & Wilkins, 2007, pp 1139–1198
4. Agarwal A, Dong Z, Harris R, Murray P, Parikh SM, Rosner MH, KellumJA, Ronco C; Acute Dialysis Quality Initiative XIII Working Group:Cellular and molecular mechanisms of AKI. J Am Soc Nephrol 27:1288–1299, 2016
5. Ozkok A, Edelstein CL: Pathophysiology of cisplatin-induced acutekidney injury. BioMed Res Int 2014: 967826, 2014
6. Zhang D, Liu Y, Wei Q, Huo Y, Liu K, Liu F, Dong Z: Tubular p53 reg-ulates multiple genes to mediate AKI. J Am Soc Nephrol 25: 2278–2289, 2014
7. Zager RA: ‘Biologic memory’ in response to acute kidney injury: Cy-toresistance, toll-like receptor hyper-responsiveness and the onset ofprogressive renal disease.Nephrol Dial Transplant 28: 1985–1993, 2013
8. Nath KA, Croatt AJ, Haggard JJ, Grande JP: Renal response to re-petitive exposure to heme proteins: Chronic injury induced by an acuteinsult. Kidney Int 57: 2423–2433, 2000
9. Bydash JR, Ishani A: Acute kidney injury and chronic kidney disease: Awork in progress. Clin J Am Soc Nephrol 6: 2555–2557, 2011
10. Nayak B, Xie P, Akagi S, Yang Q, Sun L, Wada J, Thakur A, Danesh FR,Chugh SS, Kanwar YS: Modulation of renal-specific oxidoreductase/myo-inositol oxygenase by high-glucose ambience. Proc Natl Acad SciUSA 102: 17952–17957, 2005
11. Charalampous FC, Lyras C: Biochemical studies on inositol. IV. Con-version of inositol to glucuronic acid by rat kidney extracts. J Biol Chem228: 1–13, 1957
12. Charalampous FC: Biochemical studies on inositol. V. Purification andproperties of the enzyme that cleaves inositol to D-glucuronic acid. JBiol Chem 234: 220–227, 1959
13. Nayak B, Kondeti VK, Xie P, Lin S, Viswakarma N, Raparia K, Kanwar YS:Transcriptional and post-translational modulation of myo-inositol oxy-genase by high glucose and related pathobiological stresses. J BiolChem 286: 27594–27611, 2011
14. Tominaga T, Dutta RK, Joladarashi D, Doi T, Reddy JK, Kanwar YS:Transcriptional and translational modulation ofmyo-inositol oxygenase(MIOX) by fatty acids: Implications in renal tubular injury induced inobesity and diabetes. J Biol Chem 291: 1348–1367, 2016
15. Sun L, Dutta RK, Xie P, Kanwar YS: myo-Inositol oxygenase over-expression accentuates generation of reactive oxygen species andexacerbates cellular injury following high glucose ambience: A NEW
J Am Soc Nephrol 28: 1421–1436, 2017 Myo-Inositol Oxygenase in Renal Injury 1435
www.jasn.org BASIC RESEARCH

MECHANISM RELEVANT TO THE PATHOGENESIS OF DIABETICNEPHROPATHY. J Biol Chem 291: 5688–5707, 2016
16. Ozbek E: Induction of oxidative stress in kidney. Int J Nephrol 2012:465897, 2012
17. SinghAP,JunemannA,MuthuramanA,JaggiAS,SinghN,GroverK,DhawanR: Animal models of acute renal failure. Pharmacol Rep 64: 31–44, 2012
18. Han WK, Bailly V, Abichandani R, Thadhani R, Bonventre JV: KidneyInjury Molecule-1 (KIM-1): A novel biomarker for human renal proximaltubule injury. Kidney Int 62: 237–244, 2002
19. Gross A, McDonnell JM, Korsmeyer SJ: BCL-2 family members and themitochondria in apoptosis. Genes Dev 13: 1899–1911, 1999
20. JiangM,WeiQ, Pabla N, DongG,WangCY, Yang T, Smith SB, Dong Z:Effects of hydroxyl radical scavenging on cisplatin-induced p53 acti-vation, tubular cell apoptosis and nephrotoxicity. Biochem Pharmacol73: 1499–1510, 2007
21. Jiang M, Wei Q, Wang J, Du Q, Yu J, Zhang L, Dong Z: Regulation ofPUMA-alpha by p53 in cisplatin-induced renal cell apoptosis. Onco-gene 25: 4056–4066, 2006
22. Kumar D, Singla SK, Puri V, Puri S: The restrained expression of NF-kB inrenal tissue ameliorates folic acid induced acute kidney injury in mice.PLoS One 10: e115947, 2015
23. Wu H, Lozano G: NF-kB activation of p53. A potential mechanism forsuppressing cell growth in response to stress. J Biol Chem 269: 20067–20074, 1994
24. Ryan KM, Ernst MK, Rice NR, Vousden KH: Role of NF-kB in p53-mediated programmed cell death. Nature 404: 892–897, 2000
25. Murphy SH, Suzuki K, Downes M, Welch GL, De Jesus P, Miraglia LJ,Orth AP, Chanda SK, Evans RM, Verma IM: Tumor suppressor protein(p)53, is a regulator of NF-kB repression by the glucocorticoid receptor.Proc Natl Acad Sci USA 108: 17117–17122, 2011
26. Guijarro C, Egido J: Transcription factor-kB (NF-kB) and renal disease.Kidney Int 59: 415–424, 2001
27. Sanz AB, Sanchez-Niño MD, Ramos AM, Moreno JA, Santamaria B,Ruiz-OrtegaM, Egido J, Ortiz A: NF-kB in renal inflammation. J Am SocNephrol 21: 1254–1262, 2010
28. Akcay A, Nguyen Q, Edelstein CL: Mediators of inflammation in acutekidney injury. Mediators Inflamm 2009: 137072, 2009
29. RabbH, GriffinMD,McKay DB, Swaminathan S, Pickkers P, Rosner MH,Kellum JA, Ronco C; Acute Dialysis Quality Initiative Consensus XIIIWork Group: Inflammation in AKI: Current understanding, key ques-tions and knowledge gaps. J Am Soc Nephrol 27: 371–379, 2016
30. Yang Q, Dixit B, Wada J, Tian Y, Wallner EI, Srivastva SK, Kanwar YS:Identification of a renal-specific oxido-reductase in newborn diabeticmice. Proc Natl Acad Sci USA 97: 9896–9901, 2000
31. Rankin EB, Tomaszewski JE, Haase VH: Renal cyst development in micewith conditional inactivation of the von Hippel-Lindau tumor suppres-sor. Cancer Res 66: 2576–2583, 2006
32. Pabla N, Dong Z: Cisplatin nephrotoxicity: Mechanisms and re-noprotective strategies. Kidney Int 73: 994–1007, 2008
33. Cummings BS, Schnellmann RG: Cisplatin-induced renal cell apopto-sis: Caspase 3-dependent and -independent pathways. J PharmacolExp Ther 302: 8–17, 2002
34. Yang C, Kaushal V, Haun RS, Seth R, Shah SV, Kaushal GP: Transcrip-tional activation of caspase-6 and -7 genes by cisplatin-induced p53and its functional significance in cisplatin nephrotoxicity. Cell DeathDiffer 15: 530–544, 2008
35. Sridevi P,NhiayiMK,Wang JY:Genetic disruption of Abl nuclear importreduces renal apoptosis in a mouse model of cisplatin-induced neph-rotoxicity. Cell Death Differ 20: 953–962, 2013
36. Han X, Yue J, Chesney RW: Functional TauT protects against acutekidney injury. J Am Soc Nephrol 20: 1323–1332, 2009
37. Hasegawa K, Wakino S, Yoshioka K, Tatematsu S, Hara Y, Minakuchi H,Sueyasu K,WashidaN, TokuyamaH, TzukermanM, Skorecki K, HayashiK, ItohH: Kidney-specific overexpression of Sirt1 protects against acutekidney injury by retaining peroxisome function. J Biol Chem 285:13045–13056, 2010
38. Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK,GuarenteL, Weinberg RA: hSIR2(SIRT1) functions as an NAD-dependent p53deacetylase. Cell 107: 149–159, 2001
39. Arany I, Herbert J, Herbert Z, Safirstein RL: Restoration of CREB functionameliorates cisplatin cytotoxicity in renal tubular cells. Am J PhysiolRenal Physiol 294: F577–F581, 2008
40. Chandel NS, Trzyna WC, McClintock DS, Schumacker PT: Role of oxi-dants inNF-kB activation and TNF-alpha gene transcription induced byhypoxia and endotoxin. J Immunol 165: 1013–1021, 2000
41. Monaco C, Andreakos E, Kiriakidis S, Mauri C, Bicknell C, Foxwell B,Cheshire N, Paleolog E, Feldmann M: Canonical pathway of nuclearfactor kB activation selectively regulates proinflammatory and pro-thrombotic responses in human atherosclerosis. Proc Natl Acad SciUSA 101: 5634–5639, 2004
42. Ramesh G, Reeves WB: TNF-a mediates chemokine and cytokine ex-pression and renal injury in cisplatin nephrotoxicity. J Clin Invest 110:835–842, 2002
43. Prabhu KS, Arner RJ, Vunta H, Reddy CC: Up-regulation of humanmyo-inositol oxygenase by hyperosmotic stress in renal proximal tubularepithelial cells. J Biol Chem 280: 19895–19901, 2005
44. Susztak K: Understanding the epigenetic syntax for the genetic al-phabet in the kidney. J Am Soc Nephrol 25: 10–17, 2014
45. Pratt JR, Parker MD, Affleck LJ, Corps C, Hostert L, Michalak E, LodgeJP: Ischemic epigenetics and the transplanted kidney. Transplant Proc38: 3344–3346, 2006
46. Huang N, Tan L, Xue Z, Cang J, Wang H: Reduction of DNA hydrox-ymethylation in the mouse kidney insulted by ischemia reperfusion.Biochem Biophys Res Commun 422: 697–702, 2012
47. Endo K, Kito N, Fukushima Y, Weng H, Iwai N: A novel biomarker foracute kidney injury using TaqMan-based unmethylated DNA-specificpolymerase chain reaction. Biomed Res 35: 207–213, 2014
48. Miller RP, Tadagavadi RK, Ramesh G, Reeves WB: Mechanisms ofCisplatin nephrotoxicity. Toxins (Basel) 2: 2490–2518, 2010
49. Moeller MJ, Soofi A, Sanden S, Floege J, Kriz W, Holzman LB: Anefficient system for tissue-specific overexpression of transgenes inpodocytes in vivo. Am J Physiol Renal Physiol 289: F481–F488,2005
50. Xu ZL, Mizuguchi H, Ishii-Watabe A, Uchida E, Mayumi T, Hayakawa T:Optimization of transcriptional regulatory elements for constructingplasmid vectors. Gene 272: 149–156, 2001
51. Chen S, Agarwal A,GlushakovaOY, JorgensenMS, Salgar SK, Poirier A,Flotte TR, Croker BP, Madsen KM, Atkinson MA, Hauswirth WW, BernsKI, Tisher CC: Gene delivery in renal tubular epithelial cells using re-combinant adeno-associated viral vectors. J Am Soc Nephrol 14: 947–958, 2003
52. Hull TD, Bolisetty S, DeAlmeida AC, Litovsky SH, Prabhu SD, AgarwalA, George JF: Heme oxygenase-1 expression protects the heart fromacute injury caused by inducible Cre recombinase. Lab Invest 93: 868–879, 2013
53. Testa G, Vintersten K, Zhang Y, Benes V, Muyrers JP, Stewart AF: BACengineering for the generation of ES cell-targeting constructs andmouse transgenes. Methods Mol Biol 256: 123–139, 2004
54. Frommer M, McDonald LE, Millar DS, Collis CM, Watt F, Grigg GW,Molloy PL, Paul CL: A genomic sequencing protocol that yields apositive display of 5-methylcytosine residues in individual DNA strands.Proc Natl Acad Sci USA 89: 1827–1831, 1992
1436 Journal of the American Society of Nephrology J Am Soc Nephrol 28: 1421–1436, 2017
BASIC RESEARCH www.jasn.org
Copyright © 2022 FDOKUMEN




















