
Identification of genes regulated by Dexamethasone in multiple myeloma cells using oligonucleotide...
13
Identification of genes regulated by Dexamethasone in multiple myeloma cells using oligonucleotide arrays Dharminder Chauhan 1,4 , Daniel Auclair 2,4 , Elisabeth K Robinson 2 , Teru Hideshima 1 , Guilan Li 1 , Klaus Podar 1 , Deepak Gupta 1 , Paul Richardson 1 , Robert L Schlossman 1 , Nancy Krett 3 , Lan Bo Chen 2 , Nikhil C Munshi 1 and Kenneth C Anderson* ,1 1 The Jerome Lipper Multiple Myeloma Center, Department of Adult Oncology, Dana Farber Cancer Institute, Harvard Medical School, Boston, Massachusetts, MA 02115, USA; 2 Department of Cancer Biology, Dana Farber Cancer Institute, Harvard Medical School, Boston, Massachusetts, MA 02115, USA; 3 Northwestern University Medical School, Robert H Lurie Cancer Center, Chicago, Illinois, USA Our previous studies have characterized Dexamethasone (Dex)-induced apoptotic signaling pathways in multiple myeloma (MM) cells; however, related transcriptional events are not fully defined. In the present study, gene expression profiles of Dex-treated MM cells were determined using oligonucleotide arrays. Dex triggers early transient induction of many genes involved in cell defense/repair-machinery. This is followed by induction of genes known to mediate cell death and repression of growth/survival-related genes. The molecular and genetic alterations associated with Dex resistance in MM cells are also unknown. We compared the gene expression profiles of Dex-sensitive and Dex-resistant MM cells and identified a number of genes which may confer Dex- resistance. Finally, gene profiling of freshly isolated MM patient cells validates our in vitro MM cell line data, confirming an in vivo relevance of these studies. Collectively, these findings provide insights into the basic mechanisms of Dex activity against MM, as well as mechanisms of Dex-resistance in MM cells. These studies may therefore allow improved therapeutic uses of Dex, based upon targeting genes that regulate MM cell growth and survival. Oncogene (2002) 21, 1346 – 1358. DOI: 10.1038/sj/ onc/1205205 Keywords: multiple myeloma; dexamethasone; interleu- kin-6 Introduction Multiple Myeloma (MM) will have aected 14 000 new individuals in the United States in 2001 and remains incurable, despite all available therapies. Dexametha- sone (Dex) is the most commonly used drug in the treatment of MM (Alexanian et al., 1992). Our prior studies have extensively characterized signaling path- ways activated during Dex-induced apoptosis (Chau- han et al., 1997a,b, 1999, 2000), as well as those cascades mediating Dex-resistance conferred by inter- leukin-6 (IL-6) (Chauhan et al., 2000). To date, however, the genetic events that accompany Dex- triggered apoptosis in MM cells remain largely unknown. Moreover, alterations in the signaling cascades from cell surface 44tyrosine kinases/phos- phatases 44gene transcription 44gene products mediating Dex-resistance in MM cells are also undefined. Identification and characterization of these upstream signaling proteins, as well as downstream eector genes and their products, may identify novel targets and provide the framework for improved therapeutic uses of Dex. In MM, tumor cells are predominantly localized in the bone marrow (BM) microenvironment due to adherence both to extracellular matrix proteins and to bone marrow stromal cells (BMSCs). This interac- tion between tumor cells and BMSCs both triggers production of cytokines mediating autocrine and paracrine growth and survival of MM cells, and protects MM cells against Dex-induced apoptosis (Uchiyama et al., 1993; Chauhan et al., 1996; Anderson, 2001). For example, adherence of tumor cells to BMSCs upregulates Nuclear Factor-kappa B (NF-kB)-dependent interleukin-6 (IL-6) transcription and secretion within BMSCs (Uchiyama et al., 1993; Chauhan et al., 1996); moreover, MM cells in the marrow milieu secrete cytokines, i.e., transforming growth factor-b (TGF-b) and tumor necrosis factor a (TNF-a), which further enhance IL-6 transcription and secretion from BMSCs (Urashima et al., 1996; Hide- shima et al., 2001a). This is of central importance since IL-6 induces growth, survival, and drug resistance in MM cells. Specifically, Dex-induced apoptosis in MM cells is completely abrogated by IL-6 (Lichtenstein et al., 1995; Chauhan et al., 1997a,b, 1999, 2000; Anderson, 2001). However, the transcriptional and signaling cascades whereby IL-6 and other cytokines in Oncogene (2002) 21, 1346 – 1358 ª 2002 Nature Publishing Group All rights reserved 0950 – 9232/02 $25.00 www.nature.com/onc *Correspondence: KC Anderson, Dana-Farber Cancer Institute, 44 Binney Street, Boston, MA 02115, USA; E-mail: [email protected] 4 The first two authors contributed equally to this work. Received 23 May 2001; revised 9 November 2001; accepted 27 November 2001
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of Identification of genes regulated by Dexamethasone in multiple myeloma cells using oligonucleotide...

Identi®cation of genes regulated by Dexamethasone in multiple myelomacells using oligonucleotide arrays
Dharminder Chauhan1,4, Daniel Auclair2,4, Elisabeth K Robinson2, Teru Hideshima1, Guilan Li1,Klaus Podar1, Deepak Gupta1, Paul Richardson1, Robert L Schlossman1, Nancy Krett3,Lan Bo Chen2, Nikhil C Munshi1 and Kenneth C Anderson*,1
1The Jerome Lipper Multiple Myeloma Center, Department of Adult Oncology, Dana Farber Cancer Institute, Harvard MedicalSchool, Boston, Massachusetts, MA 02115, USA; 2Department of Cancer Biology, Dana Farber Cancer Institute, Harvard MedicalSchool, Boston, Massachusetts, MA 02115, USA; 3Northwestern University Medical School, Robert H Lurie Cancer Center,Chicago, Illinois, USA
Our previous studies have characterized Dexamethasone(Dex)-induced apoptotic signaling pathways in multiplemyeloma (MM) cells; however, related transcriptionalevents are not fully de®ned. In the present study, geneexpression pro®les of Dex-treated MM cells weredetermined using oligonucleotide arrays. Dex triggersearly transient induction of many genes involved in celldefense/repair-machinery. This is followed by inductionof genes known to mediate cell death and repression ofgrowth/survival-related genes. The molecular and geneticalterations associated with Dex resistance in MM cellsare also unknown. We compared the gene expressionpro®les of Dex-sensitive and Dex-resistant MM cells andidenti®ed a number of genes which may confer Dex-resistance. Finally, gene pro®ling of freshly isolated MMpatient cells validates our in vitro MM cell line data,con®rming an in vivo relevance of these studies.Collectively, these ®ndings provide insights into the basicmechanisms of Dex activity against MM, as well asmechanisms of Dex-resistance in MM cells. Thesestudies may therefore allow improved therapeutic usesof Dex, based upon targeting genes that regulate MMcell growth and survival.Oncogene (2002) 21, 1346 ± 1358. DOI: 10.1038/sj/onc/1205205
Keywords: multiple myeloma; dexamethasone; interleu-kin-6
Introduction
Multiple Myeloma (MM) will have a�ected 14 000 newindividuals in the United States in 2001 and remainsincurable, despite all available therapies. Dexametha-
sone (Dex) is the most commonly used drug in thetreatment of MM (Alexanian et al., 1992). Our priorstudies have extensively characterized signaling path-ways activated during Dex-induced apoptosis (Chau-han et al., 1997a,b, 1999, 2000), as well as thosecascades mediating Dex-resistance conferred by inter-leukin-6 (IL-6) (Chauhan et al., 2000). To date,however, the genetic events that accompany Dex-triggered apoptosis in MM cells remain largelyunknown. Moreover, alterations in the signalingcascades from cell surface 44tyrosine kinases/phos-phatases 44gene transcription 44gene productsmediating Dex-resistance in MM cells are alsounde®ned. Identi®cation and characterization of theseupstream signaling proteins, as well as downstreame�ector genes and their products, may identify noveltargets and provide the framework for improvedtherapeutic uses of Dex.
In MM, tumor cells are predominantly localized inthe bone marrow (BM) microenvironment due toadherence both to extracellular matrix proteins andto bone marrow stromal cells (BMSCs). This interac-tion between tumor cells and BMSCs both triggersproduction of cytokines mediating autocrine andparacrine growth and survival of MM cells, andprotects MM cells against Dex-induced apoptosis(Uchiyama et al., 1993; Chauhan et al., 1996;Anderson, 2001). For example, adherence of tumorcells to BMSCs upregulates Nuclear Factor-kappa B(NF-kB)-dependent interleukin-6 (IL-6) transcriptionand secretion within BMSCs (Uchiyama et al., 1993;Chauhan et al., 1996); moreover, MM cells in themarrow milieu secrete cytokines, i.e., transforminggrowth factor-b (TGF-b) and tumor necrosis factor a(TNF-a), which further enhance IL-6 transcription andsecretion from BMSCs (Urashima et al., 1996; Hide-shima et al., 2001a). This is of central importance sinceIL-6 induces growth, survival, and drug resistance inMM cells. Speci®cally, Dex-induced apoptosis in MMcells is completely abrogated by IL-6 (Lichtenstein etal., 1995; Chauhan et al., 1997a,b, 1999, 2000;Anderson, 2001). However, the transcriptional andsignaling cascades whereby IL-6 and other cytokines in
Oncogene (2002) 21, 1346 ± 1358ã 2002 Nature Publishing Group All rights reserved 0950 ± 9232/02 $25.00
www.nature.com/onc
*Correspondence: KC Anderson, Dana-Farber Cancer Institute, 44Binney Street, Boston, MA 02115, USA;E-mail: [email protected] ®rst two authors contributed equally to this work.Received 23 May 2001; revised 9 November 2001; accepted 27November 2001

the BM microenvironment protect against drug-induced apoptosis in MM cells are not yet delineated.
The recent advent of cDNA microarray technologypermits observation of simultaneous changes in theexpression pattern of a wide variety of genes. Forexample, recent studies have utilized the cDNAmicroarray methodology to improve diagnosis andclassi®cation of cancer (Golub et al., 1999) and tode®ne novel mechanisms of drug actions (Joyce et al.,2001; Kudoh et al., 2001; Zimmermann et al., 2000;Der et al., 1998). In the present study, we utilizedcDNA microarray technology: (1) to identify genesinduced in response to Dex treatment; (2) to comparegene expression pro®les in Dex-sensitive (MM.1S)versus Dex-resistant (MM.1R) MM cells; and (3) toidentify and compare gene expression pro®les in MMpatient cells versus normal BM mononuclear cells(BMMCs). Our study demonstrates that Dex triggersearly transient induction of genes encoding for stressresponse and repair, followed by induction ofapoptotic genes associated with repression of growthand survival genes. Comparative analysis of geneexpression pro®les of Dex-sensitive (MM.1S) versusDex-resistant (MM.1R) cells demonstrates di�erentialexpression of genes that may confer Dex-resistance.Finally, the gene expression pro®les of freshly isolatedpatient MM cells validates our in vitro MM cell linedata, con®rming an in vivo relevance of these studies.These studies elucidate mechanisms of both Dex anti-MM activity and Dex resistance, and provide theframework for novel treatment strategies to enhance itse�cacy and overcome drug resistance.
Results and Discussion
The cellular response to Dex includes growth arrestand induction of apoptosis in MM cells (Chauhan etal., 1997a,b; Feinman et al., 1999); however, the geneticevents that control these events are not fully character-ized. To delineate speci®c alterations in gene expressioninduced by Dex, MM.1S MM cells were treated withDex (10 mM) and harvested at di�erent time intervals.Total cellular RNA was prepared and subjected tocDNA microarray, followed by data analysis usingDNA Chip Analyser (DChip). The results identi®edgenes which are either transiently induced or repressedin response to Dex treatment. To interpret andorganize the data, di�erentially expressed cDNAs weregrouped according to their temporal expression patternusing an hierarchical clustering algorithm (Eisen et al.,1998) (Li and Wong, 2001).
Genes regulated in response to Dex
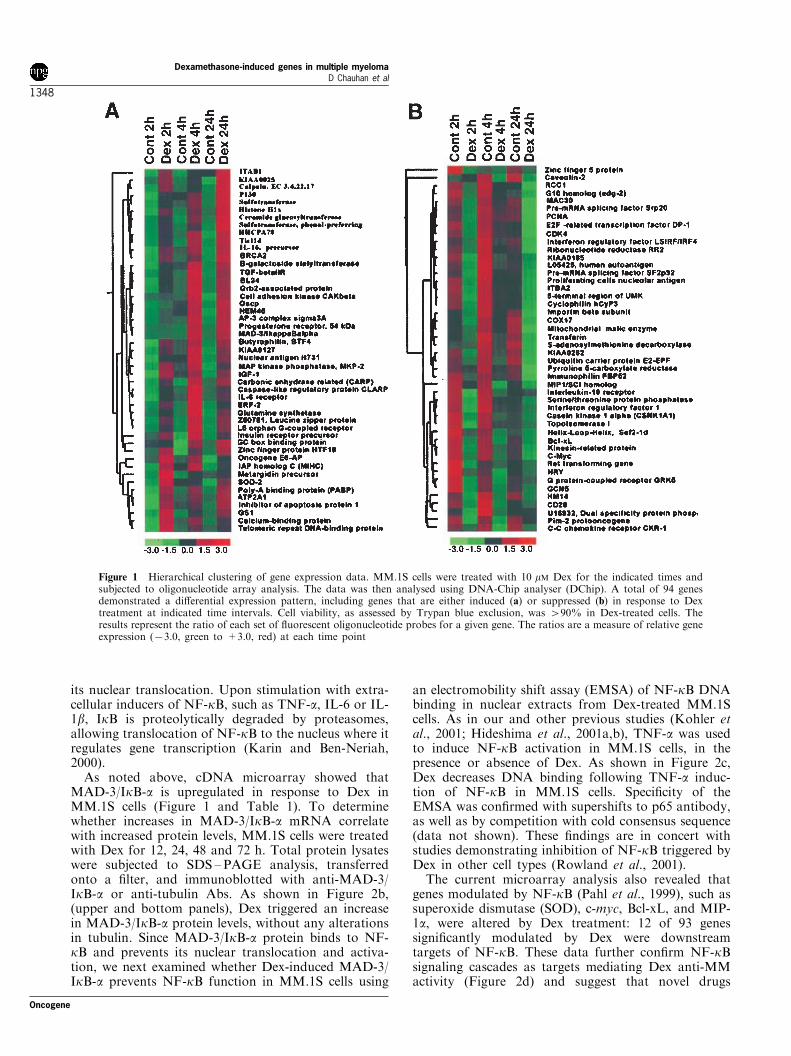
A total of 94 genes demonstrated signi®cantly(P50.05) altered expression pattern, including genesthat are either induced (A) or suppressed (B) inresponse to Dex treatment (Figure 1). Gene expressionpro®les obtained from repeat array experiments servedas internal controls for reproducibility of the observed
temporal changes in gene expression. Most regulatedgenes demonstrated smooth curves of expression ratios,providing further con®rmation of individual timepointmeasurements. The cell viability, as assessed by Trypanblue exclusion, was 490% in Dex-treated cells. As canbe seen in Table 1, treatment of MM.1S cells with Dexfor 24 h signi®cantly alters the expression of genesencoding apoptotic and anti-apoptotic signaling pro-teins, transcription factors, cell surface proteins, as wellas membrane-associated proteins involved in cellularadhesion and signaling.
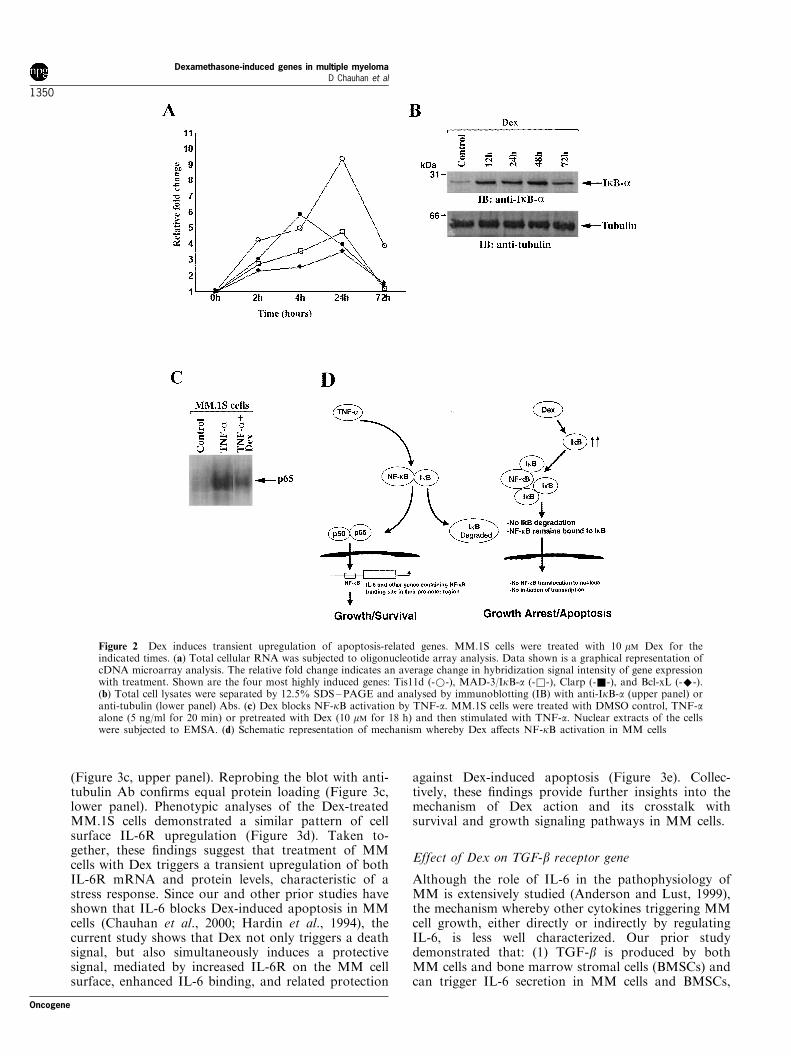
As shown in Figure 1, the expression pattern ofmultiple genes changed progressively with time of Dextreatment. First, relatively few genes were upregulatedearly (4 h) after Dex treatment, suggesting that theinitial stress response is not mediated through geneinduction. Second, various apoptosis-related genes,including caspase-like apoptosis regulator (CLARP),Tis11d, and MAD3/IkB-a are detectable early (2 h)and peak at 24 h (Figure 1, Table 1, and Figure 2a).Third, Dex treatment of MM.1S MM cells alsoinduced genes which a�ect growth and survival (Figure1, and Table 1). For example, Dex induces genesencoding for MAP Kinase phosphatase (MKP), whichinactivates growth-related MAP Kinase. We havepreviously shown that both IL-6 (Ogata et al., 1997)and vascular endothelial growth factor (VEGF) (Podaret al., 2001) trigger MM cell growth via the MAPKsignaling cascade (Chauhan et al., 1997b), and thatDex downregulates these growth kinases in MM cells(Chauhan et al., 1997b). In contrast, anti-apoptoticgenes, such as Bcl-xL, were signi®cantly downregulatedin MM cells in response to Dex treatment (Figure 2a).Fourth, Dex altered expression of many protoonco-genes (Table 1). For example, Dex represses theexpression of both c-myc and FUSE binding protein2 (FBP2), a known transcriptional activator of c-myc.
Finally, cDNA microarray also demonstrated altera-tions in novel genes encoding for nuclear proteins,which have not previously been associated with Dexresponses in MM cells (Table 1). For example, Dexinduces the expression of Nuclear antigen H731/Pdcd4gene, which inhibits tumor promoter-induced neoplas-tic transformation and promotes tumor progression(Yang et al., 2001). Dex also induces mRNA for AP3Sigma complex, a clarithin-associated adaptor complexknown to mediate the tra�cking of membrane proteinto lysosome-related organelles (Dell'Angelica et al.,1997). The alterations in the expression of these genessuggests their role during Dex-induced apoptosis inMM cells.
Effects of Dex on NF-kB pathway
NF-kB is ubiquitously expressed and involved inactivation of various genes in response to in¯ammation,infections, and other stress agents. NF-kB is normallysequestered in the cytoplasm of unstimulated cells andrequires translocation into the nucleus to function. Thesubcellular location of NF-kB is controlled by a family ofIkB inhibitory proteins, which bind to NF-kB and block
Oncogene
Dexamethasone-induced genes in multiple myelomaD Chauhan et al
1347
its nuclear translocation. Upon stimulation with extra-cellular inducers of NF-kB, such as TNF-a, IL-6 or IL-1b, IkB is proteolytically degraded by proteasomes,allowing translocation of NF-kB to the nucleus where itregulates gene transcription (Karin and Ben-Neriah,2000).
As noted above, cDNA microarray showed thatMAD-3/IkB-a is upregulated in response to Dex inMM.1S cells (Figure 1 and Table 1). To determinewhether increases in MAD-3/IkB-a mRNA correlatewith increased protein levels, MM.1S cells were treatedwith Dex for 12, 24, 48 and 72 h. Total protein lysateswere subjected to SDS ±PAGE analysis, transferredonto a ®lter, and immunoblotted with anti-MAD-3/IkB-a or anti-tubulin Abs. As shown in Figure 2b,(upper and bottom panels), Dex triggered an increasein MAD-3/IkB-a protein levels, without any alterationsin tubulin. Since MAD-3/IkB-a protein binds to NF-kB and prevents its nuclear translocation and activa-tion, we next examined whether Dex-induced MAD-3/IkB-a prevents NF-kB function in MM.1S cells using
an electromobility shift assay (EMSA) of NF-kB DNAbinding in nuclear extracts from Dex-treated MM.1Scells. As in our and other previous studies (Kohler etal., 2001; Hideshima et al., 2001a,b), TNF-a was usedto induce NF-kB activation in MM.1S cells, in thepresence or absence of Dex. As shown in Figure 2c,Dex decreases DNA binding following TNF-a induc-tion of NF-kB in MM.1S cells. Speci®city of theEMSA was con®rmed with supershifts to p65 antibody,as well as by competition with cold consensus sequence(data not shown). These ®ndings are in concert withstudies demonstrating inhibition of NF-kB triggered byDex in other cell types (Rowland et al., 2001).
The current microarray analysis also revealed thatgenes modulated by NF-kB (Pahl et al., 1999), such assuperoxide dismutase (SOD), c-myc, Bcl-xL, and MIP-1a, were altered by Dex treatment: 12 of 93 genessigni®cantly modulated by Dex were downstreamtargets of NF-kB. These data further con®rm NF-kBsignaling cascades as targets mediating Dex anti-MMactivity (Figure 2d) and suggest that novel drugs
Figure 1 Hierarchical clustering of gene expression data. MM.1S cells were treated with 10 mM Dex for the indicated times andsubjected to oligonucleotide array analysis. The data was then analysed using DNA-Chip analyser (DChip). A total of 94 genesdemonstrated a di�erential expression pattern, including genes that are either induced (a) or suppressed (b) in response to Dextreatment at indicated time intervals. Cell viability, as assessed by Trypan blue exclusion, was 490% in Dex-treated cells. Theresults represent the ratio of each set of ¯uorescent oligonucleotide probes for a given gene. The ratios are a measure of relative geneexpression (73.0, green to +3.0, red) at each time point
Dexamethasone-induced genes in multiple myelomaD Chauhan et al
1348
Oncogene

targeting MAD-3/IkB-a may have additive e�ects withDex. Our recent study has utilized PS-341, a protea-some inhibitor which prevents MAD-3/IkB-a degrada-tion and inhibits NF-kB activity, to enhance theinhibitory e�ects of Dex on NF-kB inhibition andapoptosis in MM cells (Hideshima et al., 2001b).
Effect of Dex on interleukin-6 receptor (IL-6R) gene
In the present study, we focused on genes that are knownto play an important role in MM pathogenesis, includinggenes for interleukin-6 receptor (IL-6R) and transform-ing growth factor receptor type II (TGFbRII). Speci®-cally, we and others have shown that IL-6 mediatesautocrine and paracrine growth of MM cells (Andersonet al., 1989; Klein et al., 1989; Urashima et al., 1996;Chauhan et al., 1996; Uchiyama et al., 1993; Levy et al.,1996). IL-6 not only triggers MM cell growth via theMAPK signaling cascade (Ogata et al., 1997), but alsoblocks Dex-induced apoptosis via activation of PI-3kinase/Akt signaling (Hideshima et al., 2001b) and atyrosine phosphatase, SHP2 (Chauhan et al., 2000).Based upon these observations, multiple therapies forMM have attempted to block IL-6 signaling, includingthe use of mAbs to IL-6 (Bataille et al., 1995), as well asIL-6R blockade (Tassone et al., 2000; Smith et al., 1998;Juge-Morineau et al., 1995).
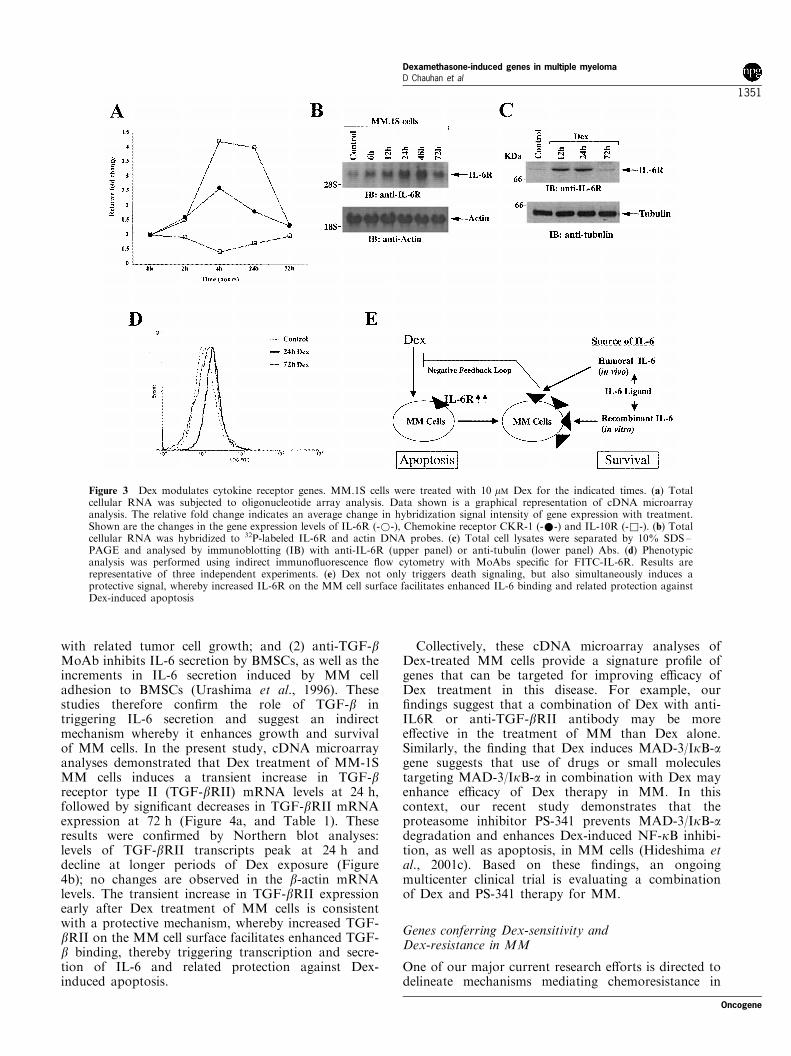
In the present study, cDNA microarray analysisdemonstrates that Dex triggers a transient upregulationIL-6R mRNA in MM.1S cells: peak levels of IL-6R ofmRNA were observed at 4 and 24 h after Dextreatment, followed by a decrease at 72 h (Figure 3a).These results were con®rmed by Northern blotanalyses: low levels of IL-6R mRNA were observedin untreated MM.1S cells (Figure 3b, upper panel);Dex induced a signi®cant increase (10 ± 12-fold, asmeasured by densitometric analysis) in IL-6R mRNAat 12 and 24 h (Figure 3b, upper panel); and IL-6RmRNA levels decreased at longer exposures i.e., 72 h,to Dex. No changes in b-actin mRNA levels wereobserved (Figure 3b, lower panel). Our ®nding thatDex enhances the expression of IL-6R mRNA levels isin concert with previous studies (Mori et al., 1998;Fischer et al., 1996).
In order to further de®ne the functional sequelae ofchanges in IL-6R mRNA expression, we used bothWestern blot and ¯ow cytometric analysis to assay forassociated changes in IL-6R protein expression.MM.1S cells were treated with Dex (10 mM), harvestedat di�erent time intervals, and total cell lysates thensubjected to immunoblot analyses with anti-IL-6R Ab.Dex induced a transient increase in IL-6R protein,which was maximal at 12 and 24 h, followed by adecline at longer intervals of Dex exposure (72 h)
Table 1 Genes modulated by Dex in MM.1S MM cells
Gene Fold change GenBank# Gene Fold change GenBank#
Apoptosis Kinases and phosphatasesTis11d 5.6 U07082 MAP kinase phosphatase (MKP-2) 1.6 U48807HHCPA78 4.8 S73591 G-protein-coupled receptor kinase (GRK5) 71.6 L15388MAD-3/IkB-a 4.2 M69043 Scrine/threonine-protein kinase PRP4h 71.7 U48736IAP1 3.2 U45878 Casein kinase I alpha isoform (CSNK1A1) 71.8 L37042Clarp 2.1 AF005775 Cyclin-dependent kinase 4 (CDK4) 71.8 U37022MAC30 72.7 L19183 Dual-specificity protein phosphatase 71.9 U15932Bcl-XL 73.9 Z23115 OthersCytokines and growth factors Butyrophilin (BTF4) 20.2 U90546TGF betaIIR alpha 5.4 D50683 Orphan G protein-coupled receptor L5 6.9 L06797Insulin receptor precursor 4.0 X02160 Immunophilin (FKBP54) 6.6 U42031IL-6 receptor 2.1 M20566 Propyl 4-hydroxylase alpha subunit 4.6 M24486Insulin like growth factor 1 2.0 X57025 Glutamine synthetase 3.9 X59834Chemokine receptor (CKR-1) 1.9 L09230 Cap-binding protein 3.8 M15353IL-16 protein precursor 1.7 M90391 Sulfotransferase 3.5 U20499LSIR 71.6 L05072 B cell activation gene BL34 3.2 S59049IL-10 receptor 71.7 U00672 Beta galactoside alpha-2,6 sialyltransferase 2.7 X62822MIP1/SCI 71.9 M23178 P130 2.1 X76061Oncogenes and protooncogene HEM45 2.0 U88964Oncogene E6-AP 2.4 HG884-HT-884 Grb2-associated binder-1 1.9 U43885Ret transforming gene 71.5 HG2825-HT2949 Carbonic anhydrase related protein (CARP) 1.7 L04656Pim-2 protooncogene 71.5 U77735 Ceramide glucosyltransferase 1.6 D50840Proto-oncogene c-myc 73.9 HG3523-HT4899 Calcium activated neutral protease 1.6 X04366Nuclear proteins ITBA1 1.6 X92475Nuclear antigen H731 5.3 U83908 metergidin 1.5 U41767BRCA2 4.5 U50527 Poly (A)-binding protein (PABP) 1.5 U68105AP3 complex sigma3A 3.2 U91932 Kinesin-related protein 71.5 D14678Leucine zipper protein 2.8 Z50781 Autoantigen mRNA 71.5 L05425Kruppel related HTF10 2.5 L11672 Immunophilin (FKBP52) 71.5 M88279GC box binding protein 2.1 D31716 ITBA2 71.5 X92896RNA binding protein Etr-3 1.9 U69546 G10 homolog (edg-2) 71.5 U11861DNA-binding protein (PIN2) 1.6 U74382 GCN5 71.5 U57316FUSE bindinh protein 2 (FBP2) 72.0 U69126 Importin beta 71.6 L38951
Fold change shown are the average ratio of gene expression in Dex-treated vs control MM.1S cells
Oncogene
Dexamethasone-induced genes in multiple myelomaD Chauhan et al
1349
(Figure 3c, upper panel). Reprobing the blot with anti-tubulin Ab con®rms equal protein loading (Figure 3c,lower panel). Phenotypic analyses of the Dex-treatedMM.1S cells demonstrated a similar pattern of cellsurface IL-6R upregulation (Figure 3d). Taken to-gether, these ®ndings suggest that treatment of MMcells with Dex triggers a transient upregulation of bothIL-6R mRNA and protein levels, characteristic of astress response. Since our and other prior studies haveshown that IL-6 blocks Dex-induced apoptosis in MMcells (Chauhan et al., 2000; Hardin et al., 1994), thecurrent study shows that Dex not only triggers a deathsignal, but also simultaneously induces a protectivesignal, mediated by increased IL-6R on the MM cellsurface, enhanced IL-6 binding, and related protection
against Dex-induced apoptosis (Figure 3e). Collec-tively, these ®ndings provide further insights into themechanism of Dex action and its crosstalk withsurvival and growth signaling pathways in MM cells.
Effect of Dex on TGF-b receptor gene
Although the role of IL-6 in the pathophysiology ofMM is extensively studied (Anderson and Lust, 1999),the mechanism whereby other cytokines triggering MMcell growth, either directly or indirectly by regulatingIL-6, is less well characterized. Our prior studydemonstrated that: (1) TGF-b is produced by bothMM cells and bone marrow stromal cells (BMSCs) andcan trigger IL-6 secretion in MM cells and BMSCs,
Figure 2 Dex induces transient upregulation of apoptosis-related genes. MM.1S cells were treated with 10 mM Dex for theindicated times. (a) Total cellular RNA was subjected to oligonucleotide array analysis. Data shown is a graphical representation ofcDNA microarray analysis. The relative fold change indicates an average change in hybridization signal intensity of gene expressionwith treatment. Shown are the four most highly induced genes: Tis11d (-*-), MAD-3/IkB-a (-&-), Clarp (-&-), and Bcl-xL (-^-).(b) Total cell lysates were separated by 12.5% SDS±PAGE and analysed by immunoblotting (IB) with anti-IkB-a (upper panel) oranti-tubulin (lower panel) Abs. (c) Dex blocks NF-kB activation by TNF-a. MM.1S cells were treated with DMSO control, TNF-aalone (5 ng/ml for 20 min) or pretreated with Dex (10 mM for 18 h) and then stimulated with TNF-a. Nuclear extracts of the cellswere subjected to EMSA. (d) Schematic representation of mechanism whereby Dex a�ects NF-kB activation in MM cells
Dexamethasone-induced genes in multiple myelomaD Chauhan et al
1350
Oncogene
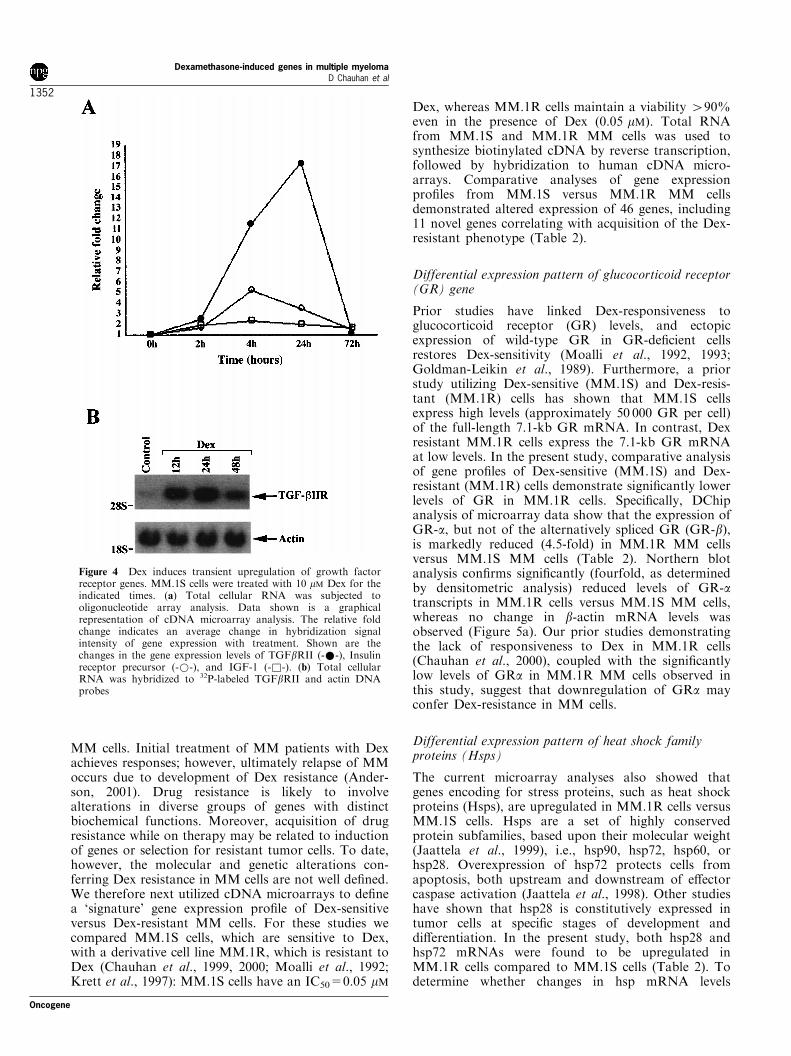
with related tumor cell growth; and (2) anti-TGF-bMoAb inhibits IL-6 secretion by BMSCs, as well as theincrements in IL-6 secretion induced by MM celladhesion to BMSCs (Urashima et al., 1996). Thesestudies therefore con®rm the role of TGF-b intriggering IL-6 secretion and suggest an indirectmechanism whereby it enhances growth and survivalof MM cells. In the present study, cDNA microarrayanalyses demonstrated that Dex treatment of MM-1SMM cells induces a transient increase in TGF-breceptor type II (TGF-bRII) mRNA levels at 24 h,followed by signi®cant decreases in TGF-bRII mRNAexpression at 72 h (Figure 4a, and Table 1). Theseresults were con®rmed by Northern blot analyses:levels of TGF-bRII transcripts peak at 24 h anddecline at longer periods of Dex exposure (Figure4b); no changes are observed in the b-actin mRNAlevels. The transient increase in TGF-bRII expressionearly after Dex treatment of MM cells is consistentwith a protective mechanism, whereby increased TGF-bRII on the MM cell surface facilitates enhanced TGF-b binding, thereby triggering transcription and secre-tion of IL-6 and related protection against Dex-induced apoptosis.
Collectively, these cDNA microarray analyses ofDex-treated MM cells provide a signature pro®le ofgenes that can be targeted for improving e�cacy ofDex treatment in this disease. For example, our®ndings suggest that a combination of Dex with anti-IL6R or anti-TGF-bRII antibody may be moree�ective in the treatment of MM than Dex alone.Similarly, the ®nding that Dex induces MAD-3/IkB-agene suggests that use of drugs or small moleculestargeting MAD-3/IkB-a in combination with Dex mayenhance e�cacy of Dex therapy in MM. In thiscontext, our recent study demonstrates that theproteasome inhibitor PS-341 prevents MAD-3/IkB-adegradation and enhances Dex-induced NF-kB inhibi-tion, as well as apoptosis, in MM cells (Hideshima etal., 2001c). Based on these ®ndings, an ongoingmulticenter clinical trial is evaluating a combinationof Dex and PS-341 therapy for MM.
Genes conferring Dex-sensitivity andDex-resistance in MM
One of our major current research e�orts is directed todelineate mechanisms mediating chemoresistance in
Figure 3 Dex modulates cytokine receptor genes. MM.1S cells were treated with 10 mM Dex for the indicated times. (a) Totalcellular RNA was subjected to oligonucleotide array analysis. Data shown is a graphical representation of cDNA microarrayanalysis. The relative fold change indicates an average change in hybridization signal intensity of gene expression with treatment.Shown are the changes in the gene expression levels of IL-6R (-*-), Chemokine receptor CKR-1 (-*-) and IL-10R (-&-). (b) Totalcellular RNA was hybridized to 32P-labeled IL-6R and actin DNA probes. (c) Total cell lysates were separated by 10% SDS±PAGE and analysed by immunoblotting (IB) with anti-IL-6R (upper panel) or anti-tubulin (lower panel) Abs. (d) Phenotypicanalysis was performed using indirect immuno¯uorescence ¯ow cytometry with MoAbs speci®c for FITC-IL-6R. Results arerepresentative of three independent experiments. (e) Dex not only triggers death signaling, but also simultaneously induces aprotective signal, whereby increased IL-6R on the MM cell surface facilitates enhanced IL-6 binding and related protection againstDex-induced apoptosis
Oncogene
Dexamethasone-induced genes in multiple myelomaD Chauhan et al
1351
MM cells. Initial treatment of MM patients with Dexachieves responses; however, ultimately relapse of MMoccurs due to development of Dex resistance (Ander-son, 2001). Drug resistance is likely to involvealterations in diverse groups of genes with distinctbiochemical functions. Moreover, acquisition of drugresistance while on therapy may be related to inductionof genes or selection for resistant tumor cells. To date,however, the molecular and genetic alterations con-ferring Dex resistance in MM cells are not well de®ned.We therefore next utilized cDNA microarrays to de®nea `signature' gene expression pro®le of Dex-sensitiveversus Dex-resistant MM cells. For these studies wecompared MM.1S cells, which are sensitive to Dex,with a derivative cell line MM.1R, which is resistant toDex (Chauhan et al., 1999, 2000; Moalli et al., 1992;Krett et al., 1997): MM.1S cells have an IC50=0.05 mM
Dex, whereas MM.1R cells maintain a viability 490%even in the presence of Dex (0.05 mM). Total RNAfrom MM.1S and MM.1R MM cells was used tosynthesize biotinylated cDNA by reverse transcription,followed by hybridization to human cDNA micro-arrays. Comparative analyses of gene expressionpro®les from MM.1S versus MM.1R MM cellsdemonstrated altered expression of 46 genes, including11 novel genes correlating with acquisition of the Dex-resistant phenotype (Table 2).
Differential expression pattern of glucocorticoid receptor(GR) gene
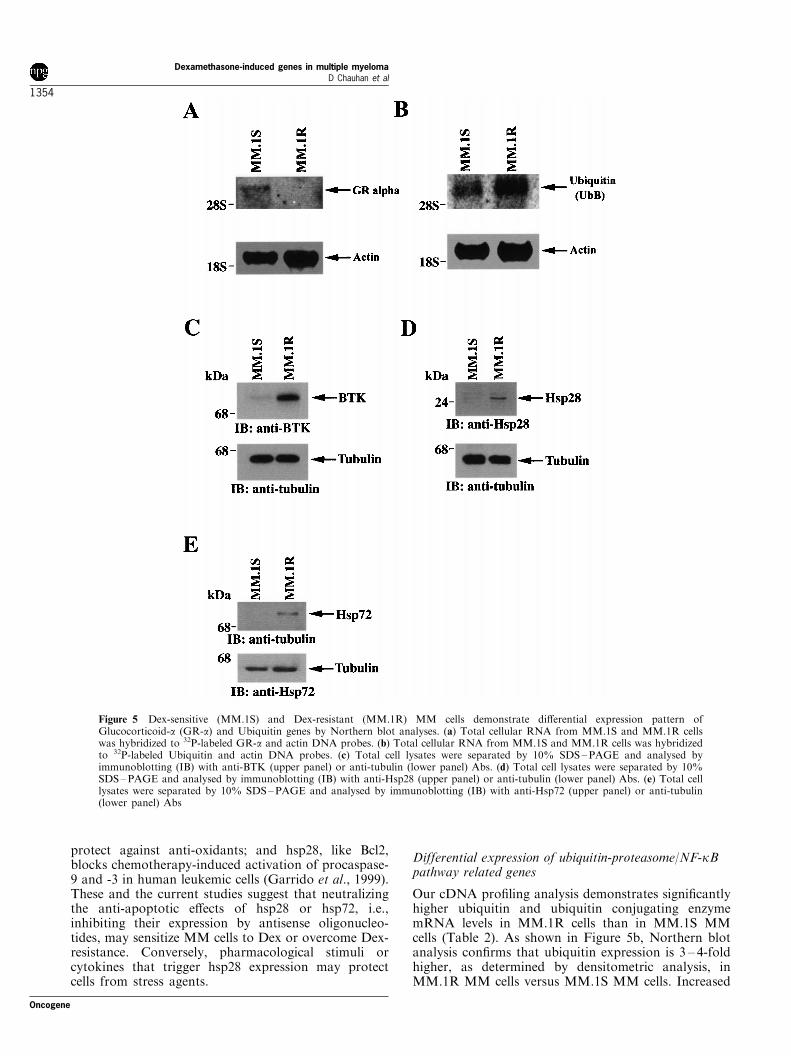
Prior studies have linked Dex-responsiveness toglucocorticoid receptor (GR) levels, and ectopicexpression of wild-type GR in GR-de®cient cellsrestores Dex-sensitivity (Moalli et al., 1992, 1993;Goldman-Leikin et al., 1989). Furthermore, a priorstudy utilizing Dex-sensitive (MM.1S) and Dex-resis-tant (MM.1R) cells has shown that MM.1S cellsexpress high levels (approximately 50 000 GR per cell)of the full-length 7.1-kb GR mRNA. In contrast, Dexresistant MM.1R cells express the 7.1-kb GR mRNAat low levels. In the present study, comparative analysisof gene pro®les of Dex-sensitive (MM.1S) and Dex-resistant (MM.1R) cells demonstrate signi®cantly lowerlevels of GR in MM.1R cells. Speci®cally, DChipanalysis of microarray data show that the expression ofGR-a, but not of the alternatively spliced GR (GR-b),is markedly reduced (4.5-fold) in MM.1R MM cellsversus MM.1S MM cells (Table 2). Northern blotanalysis con®rms signi®cantly (fourfold, as determinedby densitometric analysis) reduced levels of GR-atranscripts in MM.1R cells versus MM.1S MM cells,whereas no change in b-actin mRNA levels wasobserved (Figure 5a). Our prior studies demonstratingthe lack of responsiveness to Dex in MM.1R cells(Chauhan et al., 2000), coupled with the signi®cantlylow levels of GRa in MM.1R MM cells observed inthis study, suggest that downregulation of GRa mayconfer Dex-resistance in MM cells.
Differential expression pattern of heat shock familyproteins (Hsps)
The current microarray analyses also showed thatgenes encoding for stress proteins, such as heat shockproteins (Hsps), are upregulated in MM.1R cells versusMM.1S cells. Hsps are a set of highly conservedprotein subfamilies, based upon their molecular weight(Jaattela et al., 1999), i.e., hsp90, hsp72, hsp60, orhsp28. Overexpression of hsp72 protects cells fromapoptosis, both upstream and downstream of e�ectorcaspase activation (Jaattela et al., 1998). Other studieshave shown that hsp28 is constitutively expressed intumor cells at speci®c stages of development anddi�erentiation. In the present study, both hsp28 andhsp72 mRNAs were found to be upregulated inMM.1R cells compared to MM.1S cells (Table 2). Todetermine whether changes in hsp mRNA levels
Figure 4 Dex induces transient upregulation of growth factorreceptor genes. MM.1S cells were treated with 10 mM Dex for theindicated times. (a) Total cellular RNA was subjected tooligonucleotide array analysis. Data shown is a graphicalrepresentation of cDNA microarray analysis. The relative foldchange indicates an average change in hybridization signalintensity of gene expression with treatment. Shown are thechanges in the gene expression levels of TGFbRII (-*-), Insulinreceptor precursor (-*-), and IGF-1 (-&-). (b) Total cellularRNA was hybridized to 32P-labeled TGFbRII and actin DNAprobes
Dexamethasone-induced genes in multiple myelomaD Chauhan et al
1352
Oncogene

correlate with altered protein levels, total proteinlysates from MM.1S cells and MM.1R cells weresubjected to SDS ±PAGE analysis, transferred onto a®lter, and immunoblotted with anti-hsp28, anti-hsp72,or anti-tubulin Abs. A 4 ± 6-fold higher level of bothhsp28 and hsp72 protein expression was present inDex-resistant MM.1R cells compared to Dex-sensitiveMM.1S cells (Figure 5d,e, upper panels respectively).Reprobing the immunoblot with anti-tubulin Abcon®rmed equal protein loading (Figure 5d,e, lowerpanels).
Various stress stimuli not only trigger apoptotic ordeath signals, but also simultaneously induce protective
signaling pathways. For example, oxidative stress,staurosprine, and cytotoxic drugs (Mehlen et al.,1996; Garrido et al., 1997) induce overexpression ofhsp28 in tumor cells, thereby protecting againstapoptosis and increasing their tumorigenesis (Garridoet al., 1998). In the present study, we therefore nextexamined whether treatment of MM.1S cells with Dexinduces hsp28. Our cDNA microarray data showed aninduction of hsp28 in MM.1S cells in response to Dex(Table 1), which was associated with an increase inhsp28 protein levels (data not shown). Severalmechanisms for anti-apoptotic activity of hsp28 havebeen reported: hsp28 increases glutathione level to
Table 2 Gene expression pro®le of Dex-resistant (MM.1R) vs Dex sensitive (MM.1S) MM cells
Genes Fold change GenBank#
ReceptorsG-coupled receptor V28 41.6 U20350Peripheral benzodiazepine receptor (hpbs) 1.6 M36035Glucocorticoid receptor alpha 77.3 M10901gp130 associated protein GAM 6.3 AF072902Ubiquitin and stress proteinsNedd-4-like ubiquitin-protein ligase 3.3 U96114Heat shock protein 28 kDa 3.0 Z23090Ubiquitin conjugating enzyme Ubch5 2.7 U39317Hsp72 2.4 S67070Ubiquitin 2.1 X04803Surface and secreted proteinsHuman hematopoietic neural membrane protein 3.1 U87947Pregnancy-specific glycoprotein 13 (PSG13) 1.9 U25988Integrin alpha 6 72.2 X53586Transmembrane protein rnp24 72.4 X92098viral semaphorin protein (VESPR) 72.8 AF030339OthersMIC2 6.7 M16279Human lymphocyte-specific protein 1 (LSP1) 4.9 M33552Human autoimmunogenic antigen NY-ESO-1 4.6 U87459Brutons tyrosine kinase (BTK) 4.4 U78027Ca2-activated neutral protease large subunit (CANP) 2.9 M23254Aryl sulfotransferase (ST1A3) 2.0 X78783Myc-associated zinc-finger protein 1.8 D85131ld-2H 1.8 D13891Human ADP/ATP translocase 1.8 J03592PDGF receptor beta-like tumor suppressor (PRLTS) 1.7 D37965Follistatin-related protein (FRP) 71.6 D89937AP-3 complex sigma3A 71.7 U91932GM3 synthase 71.9 AB018356Insulin induced protein 1 (INSIG1) 72.2 U96876Retinoic acid hydroxylase 72.4 AF005418Replication factor C 72.5 L07540Protein disulfide isomerase 72.5 Z49835Synaptosome associated 23kD protein 72.6 AJ011915Ras-Like Protein Tc4 72.7 M31469Viral semaphorin protein (VESPR) 72.8 AF030339Unknown genesIMAGE-2295060 clone 8.1 AI635895IMAGE-2546059 clone 7.3 AI951946Clone 24742 72.0 AF07064434b1 72.2 W26633BAC 15E1 clone on chromosome 12 72.3 AL021546KIAA0152 72.4 D6348632gl1 72.4 W26521Clone-PDL-108 72.5 AB019409IMAGE-2062868 clone 72.6 AI347155IMAGE-2258697 clone 72.7 AI620381KIAA0147 73.6 D63481
Fold changes shown represent the ratio of gene expression in MM.1R vs MM.1S cells
Oncogene
Dexamethasone-induced genes in multiple myelomaD Chauhan et al
1353
protect against anti-oxidants; and hsp28, like Bcl2,blocks chemotherapy-induced activation of procaspase-9 and -3 in human leukemic cells (Garrido et al., 1999).These and the current studies suggest that neutralizingthe anti-apoptotic e�ects of hsp28 or hsp72, i.e.,inhibiting their expression by antisense oligonucleo-tides, may sensitize MM cells to Dex or overcome Dex-resistance. Conversely, pharmacological stimuli orcytokines that trigger hsp28 expression may protectcells from stress agents.
Differential expression of ubiquitin-proteasome/NF-kBpathway related genes
Our cDNA pro®ling analysis demonstrates signi®cantlyhigher ubiquitin and ubiquitin conjugating enzymemRNA levels in MM.1R cells than in MM.1S MMcells (Table 2). As shown in Figure 5b, Northern blotanalysis con®rms that ubiquitin expression is 3 ± 4-foldhigher, as determined by densitometric analysis, inMM.1R MM cells versus MM.1S MM cells. Increased
Figure 5 Dex-sensitive (MM.1S) and Dex-resistant (MM.1R) MM cells demonstrate di�erential expression pattern ofGlucocorticoid-a (GR-a) and Ubiquitin genes by Northern blot analyses. (a) Total cellular RNA from MM.1S and MM.1R cellswas hybridized to 32P-labeled GR-a and actin DNA probes. (b) Total cellular RNA from MM.1S and MM.1R cells was hybridizedto 32P-labeled Ubiquitin and actin DNA probes. (c) Total cell lysates were separated by 10% SDS±PAGE and analysed byimmunoblotting (IB) with anti-BTK (upper panel) or anti-tubulin (lower panel) Abs. (d) Total cell lysates were separated by 10%SDS±PAGE and analysed by immunoblotting (IB) with anti-Hsp28 (upper panel) or anti-tubulin (lower panel) Abs. (e) Total celllysates were separated by 10% SDS±PAGE and analysed by immunoblotting (IB) with anti-Hsp72 (upper panel) or anti-tubulin(lower panel) Abs
Dexamethasone-induced genes in multiple myelomaD Chauhan et al
1354
Oncogene

expression of ubiquitin-related genes is an early event intumorigenesis and has been associated with a drug-resistant phenotype in other cell systems (Desai et al.,2000; Spataro et al., 1998). For example, it has beenreported that some ubiquitin-conjugating enzymes,most notably cdc34, speci®cally degrade IkB andmediate resistance to Dex-induced apoptosis (Wu and
Ghosh, 1999). It is possible that some of the ubiquitinconjugating enzymes identi®ed in our study maysimilarly protect against Dex by activating the degrada-tion of Dex-induced MAD-3/IkB-a in MM cells.
Since multiple studies have linked NF-kB activationwith survival and proliferation in tumor cells (Bours etal., 2000; Barkett and Gilmore, 1999), we next
Table 3 Gene expression pro®le of patient MM vs normal bone marrow cells
Gene GenBank# Average fold change
Cell surface proteinsCD68 S57235 4.6MEM-102/CD48 M37766 3.7CD24 L33930 724.1CD18 X64072 712.7LFA-1 M15395 711.5CD36 M98399 77.8CD19 M28170 73.9CytokinesMacrophage inhibitory factor (MIF) L19686 5.9MIP-1a M23178 2.1Interleukin-6 receptor X58298 3.6Adhesion moleculesSyndecan-1 Z48199 6.4Integrin b5 X53002 2.1N-CAM S71824 2.0Apoptosis-relatedFas/Apo 1 X83492 2.7Perforin M31951 2.5Ubiquitin/proteasome pathwayPGP 9.5 X04741 25.1UbA52 X56997 3.2Proteasome subunit HSN3 D26600 2.8Ubiquitin fusion degradation protein U64444 2.3Cell proliferationProhibitin S85655 12.6Proliferation associated antigen (pag) X67951 4.2Proliferation associated ATP binding D64158 3.2BTG1 X61123 75.0Transcription factorsMADS/MEF2 related L8895 3.9ESE-1 U73843 3.6NF45 U10323 2.8Protein kinasesMixed lineage kinase 2 X90846 7.7Nucleotide diphosphate kinase Nm23 X17620 4.8IL-1 receptor associated kinase IRAK L76191 4.4ERK3 X80692 3.9Bruton's tyrosine kinase (BTK) U78027 2.3Stat5A U43185 77.6Cytoskeletal proteinsCollagen M55998 729.9Vinculin M33308 711.8Spectrin a M61826 711.4OthersPre-B cell stimulatory factor L36033 748.6Myeloid cells differentiation factor M81750 719.0Inteferon regulatory factor U52682 13.1Tumor suppressor Mac25 HG987-HT987 713.0Tumor specific MAGE-3 antigen U03735 8.7Receptor 4-1BB ligand U03398 6.2Adenosine deaminase M13792 5.8Endogeneous retroviral protease M27826 4.9Human LTR Mrna M92449 3.6Immunophilin FKBP52 M88279 3.3IGF-II S73149 3.2Tubby related TULP1 U82468 3.2Human autoantigen Ge-1 L26339 2.4
Fold changes shown represent the ratio of gene expression in MM Patients' vs Normal BMMN cells
Oncogene
Dexamethasone-induced genes in multiple myelomaD Chauhan et al
1355

examined the gene pro®les of MM.1S and MM.1Rcells for genes that a�ect NF-kB. For example, arecent study has shown that Bruton's Tyrosine kinase(BTK), a member of the Tec family of tyrosinekinases, regulates activation of NF-kB (Petro andKhan, 2001; Bajpai et al., 2000). In the present study,both mRNA (Table 1) as well as protein levels (Figure5c) of BTK were upregulated in MM.1R cellscompared to MM.1S cells, suggesting that it may alsocontribute to Dex-resistance and enhanced survival ofMM.1R cells.
Our microarray results also showed that G-proteincoupled receptor V28 or CX3CR1 mRNA is morehighly expressed in MM.1R cells compared to MM.1Scells (Table 2). CX3CR1 is recognized by the unique,membrane bound chemokine Fractalkine (FKN,CX3CL1) (Combadiere et al., 1998), and a recentstudy demonstrated that activation of CX3CR1 byFKN mediates both leukocyte migration and adhesion(Fraticelli et al., 2001; Volin et al., 2001). Theobservation that adhesion of MM cells to BMSCstriggers NF-kB dependent IL-6 secretion in BMSCsand confers drug-resistance (Anderson, 2001; Chauhanet al., 1997b), coupled with the present demonstrationthat MM.1R cells express higher CX3CR1 mRNAlevels than MM.1S cells, implicate CX3CR1-FKNinteractions in mediating Dex resistance.
Taken together, these results demonstrate that the`signature' gene expression pro®les of Dex-sensitive andDex-resistant MM cells are distinct. They provide thebasis for further elucidation of mechanisms of Dexresistance, as well as the framework for noveltherapeutic approaches based upon targeting alteredgenes to overcome the Dex-resistant phenotype.
Gene profiles from MM patient and normal donor bonemarrow mononuclear cells
Having demonstrated the cDNA pro®le of Dex-treatedMM.1S cells and compared cDNA pro®les of Dex-sensitive versus Dex-resistant MM cells, we next askedwhether puri®ed MM cells obtained from patientsresistant to Dex have similar gene expression pro®les.Microarray analysis of six freshly isolated patient MMcells were compared to normal donor BMMCs (Table3). A number of genes whose expression was alteredwith Dex treatment of MM.1S cells in vitro, includingIL-6R, MIP-1a, syndecan-1, fas/APO-1, and ubiquitinligase, were also more highly expressed in these patientMM samples than in normal BMMCs. Genes relatedto ubiquitin-proteasome pathway were also highlyexpressed, consistent with the drug-resistant phenotypein MM patient cells. The ®ndings that Ubiquitin-Proteasome pathway related genes are upregulated inresponse to Dex treatment of MM.1S cells, coupledwith their high expression in Dex-resistant patient MMcells, further supports a role for the Ubiquitin-Proteasome pathway in mediating Dex-resistance.Comparison of microarray pro®les of Dex sensitiveversus Dex resistant patient MM cells is ongoing tofurther address this issue.
Our microarray comparison also demonstrated thatgenes encoding for growth-related signaling proteins,such as ERK3 and BTK, were more highly expressedin MM patient cells than in normal BMMCs. Incontrast, the expression of anti-proliferative genes, suchas B cell translocation gene-1 (BTG-1) (Matsuda et al.,2001), was signi®cantly reduced in MM patient cells.Genes for known MM cell adhesion molecules, i.e.,syndecan-1 and integrin-b5 (Uchiyama et al., 1993),were more highly expressed (6.4- and 2.1-fold,respectively) in MM patient cells than BMMCs. Sincewe and others have shown that adherence of patientMM cells to BMSCs promotes their growth andsurvival (Chauhan and Anderson, 2001), microarraystudies focusing on MM cell-BMSC interactions mayfurther suggest mechanisms regulating MM cell growthand survival in the BM milieu.
In summary, we used cDNA microarray analysis toidentify Dex-responsive genes in MM cells and todemonstrate a di�erential expression pattern of genesin Dex-sensitive versus Dex-resistant cells. Thesestudies have provided the framework for ongoingpro®ling to further identify genes predicting for Dexresponsiveness or conferring resistance to Dex therapy.Ultimately, they will provide the rationale for optimaluse of Dex, alone or in combination treatmentapproaches, to improve patient outcome in MM.
Materials and methods
Cell culture and reagents
Human MM.1 cell line was established from the peripheralblood of a 42-year-old woman with an IgA-l MM (Gold-man-Leikin et al., 1989). MM.1 cells have been characterizedby cytochemical, ¯ow cytometric, immunohistochemical, aswell as Southern and Northern blotting analyses and shownto be an Epstein-Barr virus (EBV)-negative MM cell linewhich exclusively secretes lambda light chain (Goldman-Leikin et al., 1989). MM.1S, a sensitive clone of MM.1(IC50=0.05 mM), was isolated using the Salmon Hamburgersoft agar technique (Salmon, 1984). A population ofMM.1 cells resistant to glucocorticoid-induced cytolysisderived by continuous exposure to Dex (MM.1R cells)maintains 490% viability when cultured in the presence ofDex (0.05 mM) (Moalli et al., 1992). MM.1S (Dex-sensitive)and MM.1R (Des-resistant) cells (Moalli et al., 1992, 1993;Krett et al., 1997) were grown in RPMI 1640 mediumsupplemented with 10% heat inactivated fetal-bovine serum,100 units/ml penicillin, 100 mg/ml streptomycin, and 2 mM L-glutamine. MM.1S cells were treated with Dex (SigmaChemical Co, St. Louis, MO, USA) for 2, 4, 24, 48, or72 h, and harvested. Total RNA was isolated from 107 lysedcells using TRIzol Reagent (Life Technologies, Rockville,MD, USA), and double-stranded cDNA was prepared from5 mg of total RNA using Life Technologies Superscript choicesystem and an oligo (dT) anchored T7 primer. Cell viabilitywas con®rmed by Trypan blue exclusion.Mononuclear cells were isolated from six patients with
MM by Ficoll-Hypaque density gradient centrifugation andincubated with HB-7 (anti-CD38) MoAb-biotin-streptavidinand 2H4 (anti-CD45RA) MoAb-¯uorescein isothiocyanateon ice (Chauhan et al., 1997b). Tumor cells (96+2%
Dexamethasone-induced genes in multiple myelomaD Chauhan et al
1356
Oncogene

CD38+45RA7) were isolated using an Epics C cell sorter(Coulter Electronics, Hialeah, FL, USA), washed, andresuspended in regular growth medium.
Preparation of biotinylated probes and hybridization onmicroarrays
A�ymetrix huGene FLTM arrays (Santa Clara, CA, USA)containing 6800 genes were used for mRNA expressionpro®ling. Biotinylated RNA was synthesized using theBioArray RNA transcript labeling kit (Enzo, Farmingdale,NY, USA) with biotin-11-CTP and biotin-16-UTP for 5 h at378C. In vivo transcription products were puri®ed usingRNeasy columns (Quiagen, Valenica, CA, USA). Biotiny-lated RNA was then treated for 35 min at 948C in a bu�ercontaining 200 mM Tris acetate, pH 8.1, 500 mM potassiumacetate, and 150 mM magnesium acetate.A�ymetrix huGene FLTM and U95A arrays were hybri-
dized with biotinylated in vivo transcription products (10 mg/Chip) for 16 h at 458C using the manufacturer's hybridiza-tion bu�er. Fluidic station 400 (A�ymetrix, Santa Clara, CA,USA) was used for washing and staining the arrays. Due tothe size-reduced hybridization features for the huGene FLTM
array, a three-step protocol was used to enhance detection ofthe hybridized biotinylated RNA: incubation with streptavi-din-phycoerythrin conjugate, labeling with an anti-streptavi-din goat biotinylated Ab (Vector Laboratories, Burlingam,CA, USA); and staining again with streptavidin-phycoery-thrin conjugate. DNA Chips were then analysed using theGene Array Scanner (A�ymetrix, Santa Clara, CA, USA).The excitation source was an argon ion laser, and theemission was detected by a photomultiplier tube through a570-nm long pass ®lter.Digitized image data were processed using version 4.0
GeneChip software (A�ymetrix), and image data ®les wereprocessed using the DNA Chip Analyser (DChip) (Li andWong, 2001). Genes di�erentially modulated with a 95%con®dence level were considered signi®cant. For MM patientdata, GeneChip Microarray Suite 4.0 software was used, and5twofold changes were considered signi®cant. Internalcontrols of housekeeping genes and a test chip trial wererun prior to evaluation of MM cell line and patient samples.
Northern blot analyses
Northern blot analyses were performed as previously described(Chauhan et al., 1994). Brie¯y, total RNA (20 mg/lane) wassubjected to electrophoresis through a 1% agarose/2.2 mol/L
formaldehyde gel, transferred to nitrocellulose ®lters, andhybridized to 32P-labeled DNA probes for IL-6R (Barut et al.,1993), TGF-brII (Chaudhry et al., 1994), ubiquitin, glucocorti-coid receptor (GR), and chicken b-actin probe (Chauhan et al.,1993). Hybridizations were performed at 428C for 24 h in 50%(vol/vol) formamide, 26SSC (SSC: 0.15 mol/L sodiumchloride, 0.015 mol/L sodium citrate), 16Denhardt's solution,0.1% (wt/vol) sodium dodecyl sulfate (SDS), and 200 mg/mlsalmon sperm DNA. The ®lters were washed twice in 26SSC-0.1% SDS at room temperature and then in 0.16SSC-0.1%SDS at 608C for 30 min, before exposure to Kodak X-OmatXAR ®lm (Eastman Kodak, Rochester, NY, USA) using anintensifying screen. The autoradiograms were scanned using anLKB producter (Bromma, Sweden) Ultrascan XL laserdensitometer and analysed with the Gelscan software package.Signal intensity was determined in a linear range and normal-ized to that for actin.
Western blot analyses
Cell lysates were prepared as previously described (Chauhan etal., 1997a). Brie¯y, equal amounts of proteins (250 ± 300 mg)were resolved by 10% SDS±PAGE and transferred tonitrocellulose membranes. Proteins were then transferred tonitrocellulose ®lters, blocked by incubation in 5% dry milk inPBST (0.05% Tween-20 in PBS), and probed with the indicatedAbs. Blots were then developed by enhanced chemilumines-cence (ECL; Amersham, Arlington Heights, IL, USA).
Flow cytometric analyses
Cell surface antigen expression was analysed by directimmuno¯uorescence (FACScan, Becton Dickinson, Moun-tain View, CA, USA). Cells were washed twice with culturemedia and incubated for 30 min on ice with each test mAbdiluted to the optimal concentration for immunostaining.Labeled cells were then washed, ®xed in 1% paraformalde-hyde, and analysed for ¯uorescence staining using FITC-IL-6R mAbs (Coulter/Immunotech, Miami, FL, USA).
AcknowledgmentsThis investigation was supported by NIH CA 78373awarded by the National Cancer Institute, MultipleMyeloma Research Foundation Senior Research ScientistAwards (D Chauhan, T Hideshima), and a Doris DukeDistinguished Clinical Research Scientist Award (KCAnderson).
References
Alexanian R, Dimopoulos MA, Delasalle K and Barlogie B.(1992). Blood, 80, 887 ± 890.
Anderson KC, Jones RM, Morimoto C, Leavitt P and BarutBA. (1989). Blood, 73, 1915 ± 1924.
Anderson KC and Lust JL. (1999). Semin. Hematol., 36, 14 ±20.
Anderson KC. (2001). Semin. Hematol., 38, 286 ± 294.Barut B, Chauhan D, Uchiyama H and Anderson KC.
(1993). J. Clin. Invest., 92, 2346 ± 2352.Bataille R, Barlogie B, Lu ZY, Rossi JF, Lavabre-Bertrand
T, Beck T, Wijdenes J, Brochier J and Klein B. (1995).Blood, 86, 685 ± 691.
Barkett M and Gilmore TD. (1999). Oncogene, 18, 6910 ±6924.
Bours V, Bentires-Alj M, Hellin AC, Viatour P, Robe P,Delhalle S, Benoit V and Merville MP. (2000). Biochem.Pharmacol., 60, 1085 ± 1089.
Bajpai UD, Zhang K, Teutsch M, Sen R and Wortis HH.(2000). J. Exp. Med., 191, 1735 ± 1744.
Chaudhry A, Oberg K, Gobl A, Heldin CH and Funa K.(1994). Antican. Res., 14, 2085 ± 2091.
Chauhan D and Anderson KC. (2001). Apoptosis, 6, 47 ± 55.Chauhan D, Hideshima T, Pandey P, Treon SP, Teoh G,
Rosen S, Krett N, Husson H, Avraham S, Kharbanda Sand Anderson KC. (1999). Oncogene,18, 6733 ± 6740.
Chauhan D, Kharbanda SM, Rubin E, Barut BA, Mohrba-cher A, Kufe DW and Anderson KC. (1993). Blood, 81,1540 ± 1548.
Oncogene
Dexamethasone-induced genes in multiple myelomaD Chauhan et al
1357

Chauhan D, Kharbanda SM, Uchiyama H, Sukhatme VP,Kufe DW and Anderson KC. (1994). Can. Res., 54, 2234 ±2239.
Chauhan D, Pandey P, Ogata A, Teoh G, Krett N, HalgrenR, Rosen S, Kufe D, Kharbanda S and Anderson KC.(1997a). J. Biol. Chem., 272, 29995 ± 29997.
Chauhan D, Pandey P, Ogata A, Teoh G, Treon S, UrashimaM, Kharbanda S and Anderson KC. (1997b). Oncogene,15, 837 ± 843.
Chauhan D, Pandey P, Hideshima TH, Treon S, Raje N,Davies FE, Shima Y, Tai YT, Rosen S, Avraham S,Kharbanda S and Anderson KC. (2000). J. Biol. Chem.,275, 27845 ± 27850.
Chauhan D, Uchiyama H, Akbarali Y, Urashima M,Yamamoto K, Libermann TA and Anderson KC.(1996). Blood, 87, 1104 ± 1112.
Combadiere C, Salzwedel K, Smith ED, Ti�any HL, BergerEA and Murphy PM. (1998). J. Biol. Chem., 273, 23799 ±23804.
Der SD, Zhou A, Williams BR and Silverman RH. (1998).Proc. Natl. Acad. Sci. USA, 95, 15623 ± 15628.
Desai SD, Mao Y, Sun M, Li TK, Wu J and Liu LF. (2000).Ann. NY Acad. Sci., 922, 306 ± 308.
Dell'Angelica EC, Ohno H, Ooi CE, Rabinovich E, RocheKW and Bonifacino JS. (1997). EMBO J., 16, 917 ± 928.
Eisen MB, Spellman PT, Brown PO and Botstein D. (1998).Proc. Natl. Acad. Sci. USA, 95, 14863 ± 14868.
Feinman R, Koury J, Thames M, Barlogie B, Epstein J andSiegel DS. (1999). Blood, 93, 3044 ± 3052.
Fischer CP, Bode BP, Takahashi K, Tanabe KK and SoubaWW. (1996). Ann. Surg., 223, 610 ± 618; Discussion 618 ±619.
Fraticelli P, Sironi M, Bianchi G, D'Ambrosio D, AlbanesiC, Stoppacciaro A, Chieppa M, Allavena P, Ruco L,Girolomoni G, Sinigaglia F, Vecchi A and Mantovani A.(2001). J. Clin. Invest., 107, 1173 ± 1181.
Goldman-Leikin RE, Salwen HR, Herst CV, Variakojis D,Bian ML, Le Beau MM, Selvanayagan P, Marder R,Anderson R and Weitzman S. (1989). J. Lab. Clin. Med.,113, 335 ± 345.
Garrido C, Ottavi P, Fromentin A, Hammann A, Arrigo AP,Chau�ert B and Mehlen P. (1997). Cancer Res., 57, 2661 ±2667.
Garrido C, Fromentin A, Bonnotte B, Favre N, Moutet M,Arrigo AP, Mehlen P and Solary E. (1998). Cancer Res.,58, 5495 ± 5499.
Golub TR, Slonim D, Tamayo P, Huard C, Gaasenbeek M,Mesirov JP, Coller H, Loh ML, Downing JR, CaligiuriMA, Bloom®eld CD and Lander ES. (1999). Science, 286,531 ± 537.
Garrido C, Bruey JM, Fromentin A, Hammann A, ArrigoAP and Solary E. (1999). FASEB J., 13, 2061 ± 2070.
Hardin J, Macleod S, Grigorieva I, Chang R, Barlogie B,Xiao X and Epstein J. (1994). Blood, 84, 3063 ± 3070.
Hideshima T, Chauhan D, Schlossman R, Richardson P andAnderson KC. (2001a). Oncogene, 20, 4519 ± 4527.
Hideshima T, Nakamura N, Chauhan D and Anderson KC.(2001b). Oncogene, 20, 5991 ± 6000.
Hideshima T, Richardson P, Chauhan D, Palaomabella V,Elliott PJ, Adams J and Anderson KC. (2001c). Con. Res.,61, 3071 ± 3076.
Juge-Morineau N, Francois S, Puthier D, Godard A, BatailleR and Amiot M. (1995). Br. J. Haematol., 90, 707 ± 710.
Jaattela M. (1999). Ann. Med., 31, 261 ± 271.Jaattela M, Wissing D, Kokholm K, Kallunki T and Egeblad
M. (1998). EMBO J., 17, 6124 ± 6134.
Joyce DE, Gelbert L, Ciaccia A, DeHo� B and Grinnell BW.(2001). J. Biol. Chem., 276, 11199 ± 11203.
Karin M and Ben-Neriah Y. (2000). Annu. Rev. Immunol.,18, 621 ± 663.
Klein B, Zhang XG, Jourden M, Content M, Houssiau F,Aarden L, Piechaczyk M and Bataille R. (1989). Blood, 73,517 ± 526.
Kohler HB, Huchzermeyer B, Martin M, De Bruin A, MeierB and Nolte I. (2001). Vet. Dermatol., 12, 129 ± 137.
Krett NL, Zell JL, Halgren RG, Pillay S, Traynor AE andRosen ST. (1997). Clin. Can. Res., 3, 1781 ± 1787.
Kudoh K, Manasi R, Ravatin R, Elkahloun AG, BittnerML, Meltzer PS, Trent JM, Dalton WS and Chin KV.(2001). Can. Res., 60, 4161 ± 4166.
Lichtenstein A, Tu Y, Fady C, Vescio R and Berenson J.(1995). Cell Immunol., 162, 248 ± 255.
Levy Y, Labaume S, Colombel M and Brouet JC. (1996).Clin. Exp. Immunol., 104, 167 ± 172.
Li C and Wong WH. (2001). Proc. Natl. Acad. Sci. USA, 98,31 ± 36.
Matsuda S, Rouault J, Magaud J and Berthet C. (2001).FEBS Lett., 497, 67 ± 72.
Moalli PA, Pillay S, Weiner D, Leikin R and Rosen ST.(1992). Blood, 79, 213 ± 222.
Moalli PA, Pillay S, Krett NL and Rosen ST. (1993). CancerRes., 53, 3877 ± 3879.
Mehlen P, Schulze-Ostho� K and Arrigo AP. (1996). J. Biol.Chem., 271, 16510 ± 16514.
Mori S, Murakami-Mori K and Bonavida B. (1998). Antican.Res., 18, 4403 ± 4408.
Ogata A, Chauhan D, Teoh G, Treon SP, Urashima M,Schlossman RL and Anderson KC. (1997). J. Immunology,159, 2212 ± 2221.
Pahl, HL. (1999). Oncogene, 18, 6853 ± 6866.Petro JB and Khan WN. (2001). J. Biol. Chem., 276, 1715 ±
1719.Podar K, Tai TY, Davies FE, Lentzsch S, Sattler M,
Hideshima T, Lin BK, Gupta D, Chauhan D, MitsiadesC, Raje N, Richardson P and Anderson K. (2001). Blood,98, 428 ± 435.
Rowland TL, McHugh SM, Deighton J, Ewan PW, Dear-man RJ and Kimber I. (2001). Int. Immunopharmacol., 1,49 ± 61.
Salmon, SE. (1984). Cancer Treat. Rep., 68, 117 ± 125.Smith MR, Xie T, Joshi I and Schilder RJ. (1998). Br. J.
Haematol., 102, 1090 ± 1097.Spataro V, Norbury C and Harris AL. (1998). Br. J. Cancer,
77, 448 ± 455.Tassone P, Forciniti S, Galea E, Savino R, Turco MC,
Iacopino P, Tagliaferri P, Morrone G, Ciliberto G andVenuta S. (2000). Cell Death Di�er., 7, 327 ± 328.
Uchiyama H, Barut BA, Mohrbacher AF, Chauhan D andAnderson KC. (1993). Blood, 82, 3712 ± 3720.
Urashima M, Ogata A, Chauhan D, Hatziyanni M, VidrialesMB, Dedera DA, Schlossman RL and Anderson KC.(1996). Blood, 87, 1928 ± 1938.
Volin MV, Woods JM, Amin MA, Connors MA, Harlow LAand Koch AE. (2001). Am. J. Pathol., 159, 1521 ± 1530.
Wu C and Ghosh S. (1999). J. Biol. Chem., 274, 29591 ±29594.
Yang HS, Jansen AP, Nair R, Shibahara K, Verma AK,Cmarik JL and Colburn NH. (2001). Oncogene,20, 669 ±676.
Zimmermann J, Erdmann D, Lalande I, Grossenbacher R,Noorani M and Furst P. (2000). Oncogene,19, 2913 ± 2920.
Dexamethasone-induced genes in multiple myelomaD Chauhan et al
1358
Oncogene




















