
\"Apocalypse and Redemption in Early Christianity: From John of Patmos to Augustine of Hippo.\"
Upload
independentCategory
view
0download
0
2014;13:1457-1467. Published OnlineFirst April 2, 2014.Mol Cancer Ther Dipanjan Basu, Robert Lettan, Krishnan Damodaran, et al. Pathways
, and Wnt SignalingβSmall Molecule Inhibitor of Hippo, TGF-Identification, Mechanism of Action, and Antitumor Activity of a
Updated version
10.1158/1535-7163.MCT-13-0918doi:
Access the most recent version of this article at:
Material
Supplementary
http://mct.aacrjournals.org/content/suppl/2014/04/03/1535-7163.MCT-13-0918.DC1.html
Access the most recent supplemental material at:
Cited Articles
http://mct.aacrjournals.org/content/13/6/1457.full.html#ref-list-1
This article cites by 50 articles, 12 of which you can access for free at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
To request permission to re-use all or part of this article, contact the AACR Publications Department at
on November 3, 2014. © 2014 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst April 2, 2014; DOI: 10.1158/1535-7163.MCT-13-0918
on November 3, 2014. © 2014 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst April 2, 2014; DOI: 10.1158/1535-7163.MCT-13-0918

Small Molecule Therapeutics
Identification, Mechanism of Action, and Antitumor Activityof a Small Molecule Inhibitor of Hippo, TGF-b, and WntSignaling Pathways
Dipanjan Basu1, Robert Lettan3, Krishnan Damodaran2, Susan Strellec3, Miguel Reyes-Mugica1, andAbdelhadi Rebbaa1
AbstractEmbryonic signaling pathways, in particular thosemediated byWnt andTGF-b, are known to play key roles
in tumor progression through the induction of epithelial–mesenchymal transition (EMT). Their simultaneous
targeting could therefore represent a desirable anticancer strategy.On the basis of recent findings that bothWnt
and TGF-b–associated pathways are regulated by Hippo signaling in mammalian cells, we reasoned that
targeting the latter would be more effective in inhibiting EMT. In a search for such inhibitors, we identified a
small molecule (C19) with remarkable inhibitory activity not only against Hippo, but also against Wnt and
TGF-b pathways. C19 inhibited cancer cell migration, proliferation, and resistance to doxorubicin in vitro, and
exerted strong antitumor activity in a mouse tumor model. Mechanistically, C19 induced GSK3-b–mediated
degradation of the Hippo transducer TAZ, through activation of the Hippo kinases Mst/Lats and the tumor
suppressor kinase AMPK upstream of the degradation complex. Overall, this study identified C19 as a multi-
EMT pathway inhibitor with a unique mechanism of action. The findings that both AMPK and Mst/Lats
mediate the antitumor activity of C19 shed light on a potential cross-talk between metabolic and organ size
control pathways in regulating cancer progression. By simultaneously targeting these two pathways, C19may
represent a new type of agents to suppress cancer progression and/or its recurrence. Mol Cancer Ther; 13(6);
1457–67. �2014 AACR.
IntroductionThe development of solid tumors implicates a series of
intrinsic and extrinsic events that prompt cells to prolif-erate out of control and to acquire survival and invasivecapabilities (1). These characteristics are induced by acellular trans-differentiation process termed epithelial–mesenchymal transition (EMT; refs. 2, 3) by which epi-thelial cells undergo changes in morphology and cell–celljunction to detach fromeach other and becomepotentiallyinvasive (2, 4). EMT was first characterized in develop-ment during the formation of earlymesoderm anddelam-ination of neural crest cells from the primitive neuroe-pithelium (5). Later, it was found to play key roles inwound healing, tissue fibrosis, and cancer (6–8).The mes-
enchymal-like state of cancer cells is believed to confer notonly invasive ability, but also protection from cell death,immune escape, and resistance to therapy (9).On the basisof this, approaches to inhibit EMTmayhold great promisefor cancer treatment. However, due to the multifactorialaspect of this process and that existing therapies onlytarget individual pathways (10–12), the identification ofnew agents capable of simultaneously silencing majorEMT pathways would be desirable.
TGF-b and members of its family (2) are well knownfor their ability to induce and maintain EMT in a varietyof biologic systems and pathophysiological contextsthrough activation of the Smad transcriptional regula-tors (13). Another pathway that received much attentionlately with respect to its role in EMT is Wnt signaling(14). In fact, evidence from expression studies in patienttumor specimens, transgenic animals, and in vitroexperiments, all confirmed inappropriate activation ofthe canonical Wnt signaling as a major feature in humanneoplasia (15). Interestingly, both TGF-b and Wnt sig-naling were recently found to be regulated by a devel-opmental pathway called Hippo (16–18). The latter wasinitially described in drosophila for its ability to controlorgan size (19). Its role in EMT was demonstrated bystudies in which nonphosphorylated (active) Hippotransducers YAP, TAZ, and TEAD were found to acteither together, in conjunction with b-catenin/TCF, or
Authors' Affiliations: 1Department of Pathology, University of Pittsburghand the Children's Hospital of Pittsburgh of UPMC; 2Department of Chem-istry, University of Pittsburgh; and 3Department of Chemistry, ChathamUniversity, Pittsburgh, Pennsylvania
Note: Supplementary data for this article are available at Molecular CancerTherapeutics Online (http://mct.aacrjournals.org/).
Corresponding Author: Abdelhadi Rebbaa, Department of Pathology,University of Pittsburgh, Children's Hospital of Pittsburgh of UPMC, 45thStreet & Penn Avenue, Room B269, Pittsburgh, PA 15201. Phone: 412-692-7304; Fax: 412-692-6550; E-mail: [email protected]
doi: 10.1158/1535-7163.MCT-13-0918
�2014 American Association for Cancer Research.
MolecularCancer
Therapeutics
www.aacrjournals.org 1457
on November 3, 2014. © 2014 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst April 2, 2014; DOI: 10.1158/1535-7163.MCT-13-0918

with Smads (20, 21) to induce genes that regulate cellmorphology, migration, and cancer metastasis. Phos-phorylation (inactivation) of the Hippo transducers iscarried out by upstream kinases (Mst/Lats) resulting intheir cytoplasmic retention and either proteasomal deg-radation (22) by the GSK3-b–associated degradationcomplex or sequestration of b-catenin and Smads there-by inhibiting the trans-activation of these transcriptionfactors (16–18).
Deregulation of Hippo signaling has been shown to beassociatedwith carcinogenesis (23). This is best illustratedby studies inwhich lats1knockout inmice led to soft tissuesarcomas and ovarian stromal cell tumors (24). Moreover,expression of TAZ showed an exceptionally strong asso-ciation with poor patient survival from non–small lungcancer and thyroid carcinoma (25, 26). Alterations in thisgene and/or its molecular partners YAP and TEAD havealso been reported in cancers derived from colon, lung,liver, or esophagus (27–29). A very recent study byCamargo and his group has shown that the tumor sup-pressor LKB1, which is known for its ability to regulatecellular metabolism through AMPK activation, also reg-ulates Hippo signaling (30).
Because of the central role of Hippo pathway in regu-lation of EMT, we initiated a search for small molecules toinhibit its transduction. This led to the identification of acompound (C19)with a potent inhibitory activity not onlyon Hippo, but also on Wnt and TGF-b signalling path-ways. As a result, C19 inhibited cancer cell migration,proliferation, resistance to doxorubicin in vitro, and exhib-ited strong antitumor activity in amouse tumor xenograftmodel with no apparent toxicity. Mechanistically, C19was found to act as an activator of the tumor suppressorkinases, Mst/Lats and AMPK, both upstream of theGSK3-b–associated degradation complex, which in turndegrades the Hippo transducer TAZ. Potential applica-tions for the use of C19 and its analogs as putative agentsto treat EMT-associated diseases such as cancer and fibro-sis or metabolic disorders such as diabetes are discussed.
Materials and MethodsCell lines and reagents
Humanmelanoma (WM115 andWM266), breast cancer(MCF7), colon cancer (SW480), and neuroblastoma (SKN-SH) cell lines were purchased from American Type Cul-ture Collection, and 293 human kidney cells from Clon-tech.The above-mentioned cell lines were procured morethan 6 months ago and have not been tested recently forauthentication in our laboratory. Dulbecco’s ModifiedEagle Medium (DMEM), MEM, RPMI, Horse serum, andFBS were from BioWhittaker. The following drugs andreagents were purchased from the companies cited: The8xGTIIC luciferase reporter construct (31) fromAddgene;Doxorubicin, and antibody to b-actin from Sigma-Aldrich; Antibodies to Zeb1 (sc-25388, Santa Cruz Bio-technology), Snail (ab53519, Abcam), cyclin D, Myc andHes, Phospho-MST1, MST1, phospho-Lats1, Lats1, phos-
pho-AMPK, AMPK, YAP, TAZ, and TEAD from CellSignaling Technology; secondary antibodies conjugatedto horseradish peroxidase from Jackson ImmunoresearchLab Inc.; enhanced chemiluminescence reagents andImmobilon-P transfer membrane for Western blot analy-ses from Millipore. Reagents for DNA transfection wereobtained from Life Technologies.
Cell culture and transfectionsMelanoma and breast cancer cell lines were cultured in
MEM supplemented with 10% FBS as described by thesupplier. Colon cancer cells were maintained in RPMIsupplemented with 10% FBS, 293 and SKN-SH cells werecultured in DMEM supplemented with 10%FBS penicil-lin/streptomycin and nonessential amino acids (LifeTechnologies). Transfections were carried out in 6-wellplates using a Lipofectamine Kit (Life Technologies) asdescribed by the manufacturer. Briefly, 3 mg of DNAwere mixed in 100 mL of transfection solution containing90 mL of serum-free culture medium and 10 mL lipofec-tamine. After 20-minute incubation at room temperature,the mixture was added to cells cultured in well platecontaining 2 mL of culture medium per well and incu-bated for 5 hours. The medium was then replaced beforeinhibitors were added to the corresponding wells andincubated for an additional 24 hours. Protein extractswere harvested and processed for Western blot analysisor luciferase assay as described below. siRNA transfec-tion were carried out as described previously (32). Brief-ly, transfection was carried out using a GeneSilencersiRNA Transfection Reagent Kit according to the man-ufacturer’s protocol (Gentherapy systems). For this, 25mL of 20 mmol/L stock solution of siRNA duplexes wereadded to cells incubated in 2 mL of culture medium perwell in a 6-well plate. After 48 hours of incubation,proteins were extracted for analysis by Western blotanalysis as described above.
Western blot analysisProteins were extracted from cells cultivated in mono-
layers in 25 cm2 flasks using 100 mL of lysis buffer (50mmol/L HEPES, pH 7.4, 150 mmol/L NaCl, 100 mmol/LNaF, 1 mmol/LMgCl2, 1.5 mmol/L EGTA, 10% glycerol,1% Triton X100, 1 mg/mL leupeptin, 1 mmol/L phenyl-methylsulfonylfluoride; PMSF). For nuclear and cyto-plasmic fractionation (33), cells cultivated in 75 cm2 flakswere resuspended in 400 mL of buffer A (10 mmol/LHEPES, pH 7.9; 10 mmol/L KCl; 0.1 mmol/L EDTA;0.1 mmol/L EGTA; 1 mmol/L DTT; 0.5 mmol/L PMSF).The mixture was incubated for 30 minutes on ice, then25 mL of a 10% solution of Nonidet NP-40 was added,and the homogenate centrifuged for 30 seconds. Equalquantities of protein were separated by electrophoresison a 12% SDS-PAGE gel and transferred to Immobilon-P membranes. Proteins of interest were identified byreaction with specific primary and secondary antibo-dies linked to horseradish peroxidase and detected bychemiluminescence.
Basu et al.
Mol Cancer Ther; 13(6) June 2014 Molecular Cancer Therapeutics1458
on November 3, 2014. © 2014 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst April 2, 2014; DOI: 10.1158/1535-7163.MCT-13-0918

qPCRTotal RNA was extracted using the RNeasy Mini Kit
(Qiagen), and first-strand cDNAwas synthesized accord-ing to manufacturer’s instructions using ThermoScriptRT-PCR system (Life Technologies). Gene expressionwasmeasured by real-time PCR (RT-PCR) using the MaximaSyber green Master Mix (Fermentas) on ABI 7500 instru-ment (Applied Biosystems). Quantitative PCR (qPCR)primers are as follows: smooth muscle actin: forward50-AGT TACGAGTTGCCTGATGG -30, reverse 50- GAGGTC CTT CCT GAT GTC AA -30; fibronectin: forward 50-CGTGGTCCACACTATCTTCC-30, reverse 50- TTCCTTGCTCTC TTGGAT TG -30; collagen A2: forward 50- CGAAGT GCT CGT CCA GTA TT -30, reverse 50- TTC CAGCTC CTT CTG TTC AC -30; CTGF: forward 50-TTT GGCCCA GAC CCA ACT AT-30, reverse: 50-GTG CAG CCAGAAAGC TCAAA -30. Gene expression was normalizedto that of glyceraldehyde-3-phosphate dehydrogenase(forward 50-GAGTCAACGGATTTGGTCGT-30, reverse50- TTGATTTTGGAGGGATCTCG-30) used as internalcontrol.
Reporter activity assayThe 8xGTIIC-luciferase reporter, which contains 8
TEAD-binding sites 8xGTIIC (31), was used to measureactivation of the Hippo pathway, 8xTCF-luciferase STF(34) to measure Wnt signaling and A3Lux (35) for TGF-breporter activity. A construct containing luciferase drivenby the cytomegalovirus (CMV) promoter was used ascontrol. The plasmids were transfected transiently intocells using the lipofectamine kit as described above. After5-hour incubation with DNA, the medium was replacedbefore inhibitors were added to the corresponding wells.After incubation for an additional 24 hours, cells werelysed and protein extracts used as a source of luciferase.For each test, the luminescence reading obtained with8xGTIIC-luciferase was divided by the one obtained withCMV-driven luciferase. In control samples, this ratio isconsidered as 100% of activity.
Kinase activity assaysGSK3-b, AMPK, Mst1, and Lats1 were immunopreci-
pitated from WM266 cells and the immunoprecipitatesresuspended in phosphorylation buffer (20 mmol/LTris-HCl (pH 7.5), 5 mmol/L MgCl2, 5 mmol/L MnCl2,1 mmol/L phosphatase inhibitor) and divided into equalsamples for in vitro kinase reactions as follows: ForGSK3-b, the samples were incubated with 5 mg of theenzyme substrate (Catalog Number G8545, Sigma-Aldrich) in the absence or the presence of C19 or theGSK inhibitor TWS119 (Sigma-Aldrich) in a total vol-ume of 50 mL of kinase buffer [20 mmol/L Tris-HCl (pH7.5), 5 mmol/L MgCl2, 5 mmol/L MnCl2, 1 mmol/Lphosphatase inhibitor, 2 mmol/L DTT, 10 mmol/L ATP,5 mCi of [g-32P] ATP] as described previously (36). Afterincubation for 30 minutes at 30�C, the reaction wasstopped by adding 7 mL of a 5 mmol/L SDS solutionand spotted on a P81 circle filter paper (Whatman),
washed four times with 1% phosphoric acid, dried, andsubjected to cpm radioactivity counting. The relativekinase activity of a given sample was calculated as thepercentage of cpm relative to that of nontreated control.Each reaction was carried out in triplicate and the datawere plotted as the mean of enzymatic activity� SE. ForAMPK, Mst1, and Lats1, the immunoprecipitated sam-ples were incubated in the absence or the presence ofC19 at 10 mmol/L or 5-amino-1-b-D-ribofuranosyl-imid-azole-4-carboxamide (AICAR) at 40 mmol/L in the samekinase reaction conditions described above withoutradioactive ATP. At the end of reactions, equal volumeof Laemmli buffer was added to the samples boiled andprocessed by Western blot analysis using specific anti-phospho antibodies.
Cell viability assayCells were incubated in a 96-well plate (100 mL of medi-
um/well) with the tested compounds for 96 hours and cellviability was quantitatively determined by a colorimetricMTT assay as described previously (37). MTT (10 mL of 5mg/mL solution) was added to each well of the titrationplate containing 100 mL of medium per well and incubatedfor 4 hours at 37�C. The precipitate was then solubilizedby addition of 100 mL of 10% SDS/0.01 mol/L HCl to thewells already containing 100 mL of medium per well andincubation for 15 hours at 37�C. Optical density of eachwell was determined in an ELISA plate reader using anactivationwavelength of 570 nm and referencewavelengthof 650 nm. The percentage of viable cells was determinedby comparison with untreated control used as 100%.
In vivo studiesThe in vivo experiments were carried out according to
an approved protocol by the Institutional Animal Careand Use Committee of the University of Pittsburgh (Pitts-burgh, PA). Nude (nu/nu) mice (Charles River Labora-tories) ages 6 weeks were used. The melanoma cell lineWM266 cultivated in MEM containing 10% FBS wereharvested from subconfluent cultures by trypsinization,washed twicewith serumcontainingmedium, suspendedin PBS (106 cells/200 mL), and inoculated subcutaneouslyinto the right flank of mice to produce tumors at theinoculation site. When tumors become palpable (about5 mm in diameter), the animals were divided into controland treatment groups and injected intraperitoneally asfollows: control group (n ¼ 7) were treated with vehiclesolution (100 mL of DMEM containing 5% dimethyl sulf-oxide). Treatment groups (7 animals per group) consistedof: C19dissolved in the vehicle solution at 5mg/kg,C19 at10 mg/kg, and C19 at 20 mg/kg. Injections were carriedout three times separated by 3 days. Mice health waschecked daily and tumor volumes measured using theformula L�W2/2 (where L is length of the tumor, W is itswidth). The experiment was stopped 2 months afterinoculation. Tumors samples were harvested for bio-chemical and gene expression analysis by Western blotanalysis as described above.
A Small Molecule Inhibitor of EMT Pathways
www.aacrjournals.org Mol Cancer Ther; 13(6) June 2014 1459
on November 3, 2014. © 2014 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst April 2, 2014; DOI: 10.1158/1535-7163.MCT-13-0918
Statistical analysisGraph data are presented as mean � SE. All analyses
were performed by using a two-way ANOVA and valuesin the treated samples were compared with the corre-sponding controls. P < 0.05 was considered statisticallysignificant. Statistical calculations were performed withSPSS 16.0 for Windows (SPSS).
ResultsIdentification of C19 and determination of its effectson Hippo, Wnt, and TGF-b signaling
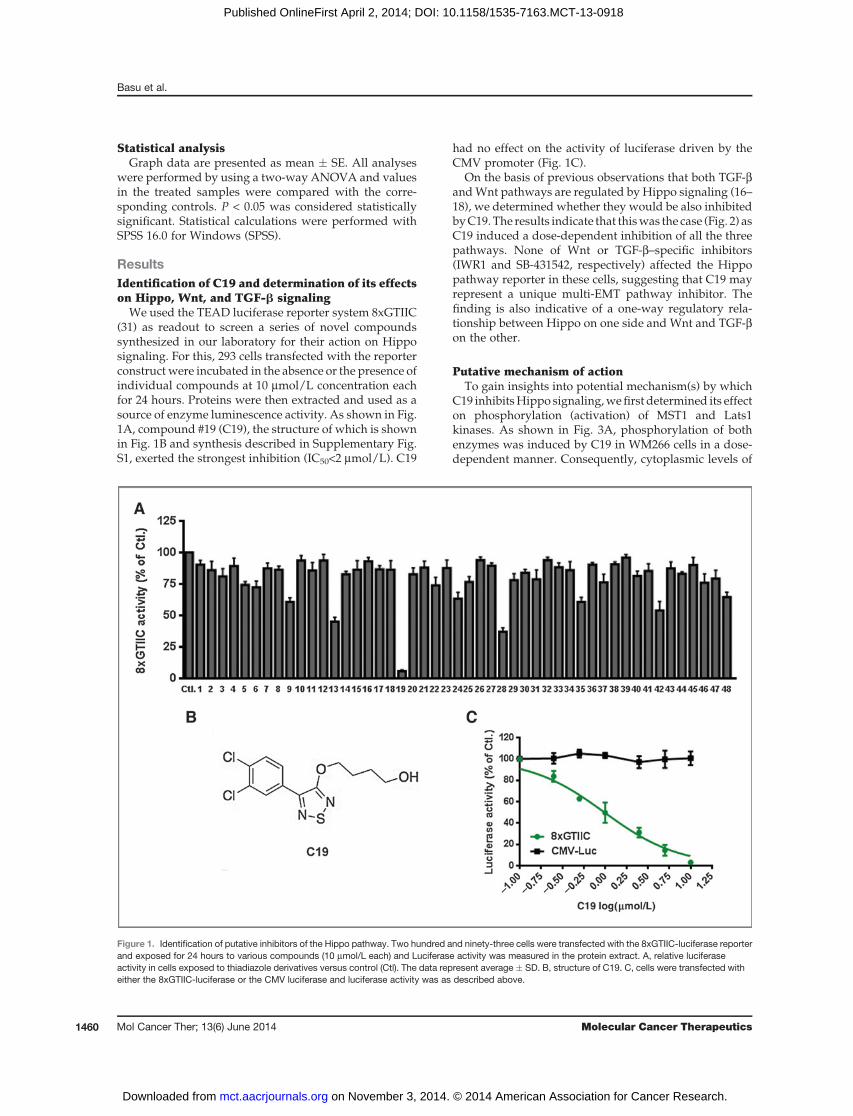
We used the TEAD luciferase reporter system 8xGTIIC(31) as readout to screen a series of novel compoundssynthesized in our laboratory for their action on Hipposignaling. For this, 293 cells transfected with the reporterconstruct were incubated in the absence or the presence ofindividual compounds at 10 mmol/L concentration eachfor 24 hours. Proteins were then extracted and used as asource of enzyme luminescence activity. As shown in Fig.1A, compound #19 (C19), the structure of which is shownin Fig. 1B and synthesis described in Supplementary Fig.S1, exerted the strongest inhibition (IC50<2 mmol/L). C19
had no effect on the activity of luciferase driven by theCMV promoter (Fig. 1C).
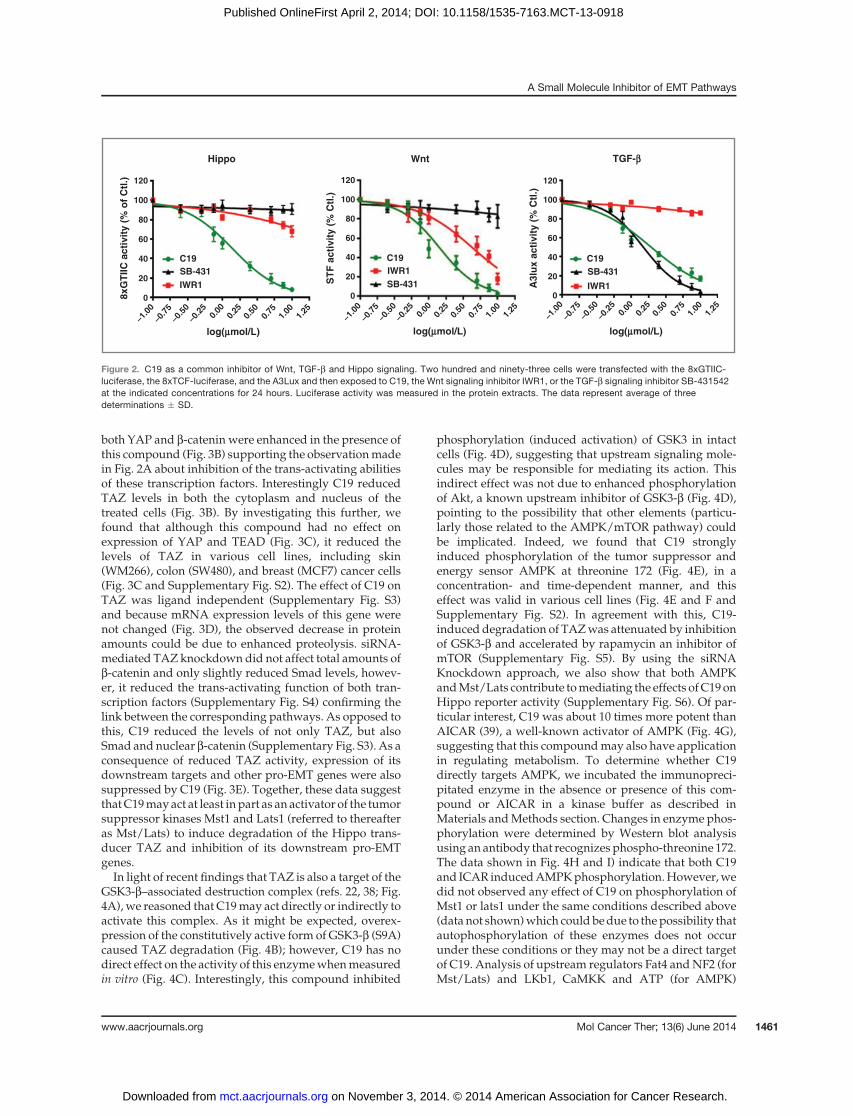
On the basis of previous observations that both TGF-bandWnt pathways are regulated by Hippo signaling (16–18), we determined whether they would be also inhibitedbyC19. The results indicate that thiswas the case (Fig. 2) asC19 induced a dose-dependent inhibition of all the threepathways. None of Wnt or TGF-b–specific inhibitors(IWR1 and SB-431542, respectively) affected the Hippopathway reporter in these cells, suggesting that C19 mayrepresent a unique multi-EMT pathway inhibitor. Thefinding is also indicative of a one-way regulatory rela-tionship between Hippo on one side and Wnt and TGF-bon the other.
Putative mechanism of actionTo gain insights into potential mechanism(s) by which
C19 inhibitsHippo signaling,wefirst determined its effecton phosphorylation (activation) of MST1 and Lats1kinases. As shown in Fig. 3A, phosphorylation of bothenzymes was induced by C19 in WM266 cells in a dose-dependent manner. Consequently, cytoplasmic levels of
Figure 1. Identification of putative inhibitors of the Hippo pathway. Two hundred and ninety-three cells were transfected with the 8xGTIIC-luciferase reporterand exposed for 24 hours to various compounds (10 mmol/L each) and Luciferase activity was measured in the protein extract. A, relative luciferaseactivity in cells exposed to thiadiazole derivatives versus control (Ctl). The data represent average � SD. B, structure of C19. C, cells were transfected witheither the 8xGTIIC-luciferase or the CMV luciferase and luciferase activity was as described above.
Basu et al.
Mol Cancer Ther; 13(6) June 2014 Molecular Cancer Therapeutics1460
on November 3, 2014. © 2014 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst April 2, 2014; DOI: 10.1158/1535-7163.MCT-13-0918
both YAP and b-catenin were enhanced in the presence ofthis compound (Fig. 3B) supporting the observationmadein Fig. 2A about inhibition of the trans-activating abilitiesof these transcription factors. Interestingly C19 reducedTAZ levels in both the cytoplasm and nucleus of thetreated cells (Fig. 3B). By investigating this further, wefound that although this compound had no effect onexpression of YAP and TEAD (Fig. 3C), it reduced thelevels of TAZ in various cell lines, including skin(WM266), colon (SW480), and breast (MCF7) cancer cells(Fig. 3C and Supplementary Fig. S2). The effect of C19 onTAZ was ligand independent (Supplementary Fig. S3)and because mRNA expression levels of this gene werenot changed (Fig. 3D), the observed decrease in proteinamounts could be due to enhanced proteolysis. siRNA-mediated TAZ knockdown did not affect total amounts ofb-catenin and only slightly reduced Smad levels, howev-er, it reduced the trans-activating function of both tran-scription factors (Supplementary Fig. S4) confirming thelink between the corresponding pathways. As opposed tothis, C19 reduced the levels of not only TAZ, but alsoSmad andnuclear b-catenin (Supplementary Fig. S3). As aconsequence of reduced TAZ activity, expression of itsdownstream targets and other pro-EMT genes were alsosuppressed by C19 (Fig. 3E). Together, these data suggestthatC19mayact at least inpart as an activator of the tumorsuppressor kinases Mst1 and Lats1 (referred to thereafteras Mst/Lats) to induce degradation of the Hippo trans-ducer TAZ and inhibition of its downstream pro-EMTgenes.In light of recent findings that TAZ is also a target of the
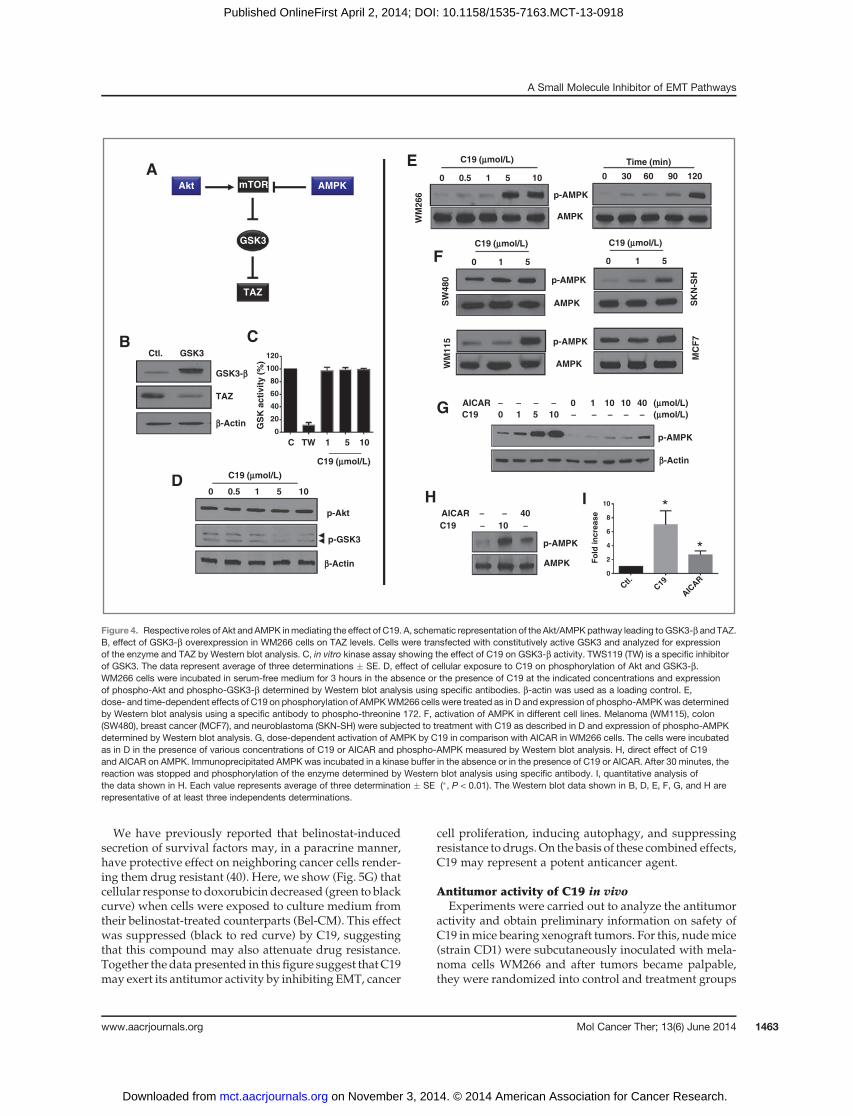
GSK3-b–associated destruction complex (refs. 22, 38; Fig.4A),we reasoned that C19may act directly or indirectly toactivate this complex. As it might be expected, overex-pression of the constitutively active form of GSK3-b (S9A)caused TAZ degradation (Fig. 4B); however, C19 has nodirect effect on the activity of this enzymewhenmeasuredin vitro (Fig. 4C). Interestingly, this compound inhibited
phosphorylation (induced activation) of GSK3 in intactcells (Fig. 4D), suggesting that upstream signaling mole-cules may be responsible for mediating its action. Thisindirect effect was not due to enhanced phosphorylationof Akt, a known upstream inhibitor of GSK3-b (Fig. 4D),pointing to the possibility that other elements (particu-larly those related to the AMPK/mTOR pathway) couldbe implicated. Indeed, we found that C19 stronglyinduced phosphorylation of the tumor suppressor andenergy sensor AMPK at threonine 172 (Fig. 4E), in aconcentration- and time-dependent manner, and thiseffect was valid in various cell lines (Fig. 4E and F andSupplementary Fig. S2). In agreement with this, C19-induced degradation of TAZwas attenuated by inhibitionof GSK3-b and accelerated by rapamycin an inhibitor ofmTOR (Supplementary Fig. S5). By using the siRNAKnockdown approach, we also show that both AMPKandMst/Lats contribute tomediating the effects ofC19onHippo reporter activity (Supplementary Fig. S6). Of par-ticular interest, C19 was about 10 times more potent thanAICAR (39), a well-known activator of AMPK (Fig. 4G),suggesting that this compoundmay also have applicationin regulating metabolism. To determine whether C19directly targets AMPK, we incubated the immunopreci-pitated enzyme in the absence or presence of this com-pound or AICAR in a kinase buffer as described inMaterials andMethods section. Changes in enzyme phos-phorylation were determined by Western blot analysisusing an antibody that recognizes phospho-threonine 172.The data shown in Fig. 4H and I) indicate that both C19and ICAR inducedAMPKphosphorylation.However,wedid not observed any effect of C19 on phosphorylation ofMst1 or lats1 under the same conditions described above(data not shown)which could bedue to thepossibility thatautophosphorylation of these enzymes does not occurunder these conditions or they may not be a direct targetof C19. Analysis of upstream regulators Fat4 andNF2 (forMst/Lats) and LKb1, CaMKK and ATP (for AMPK)
8x
GT
IIC
ac
tiv
ity
(%
of
Ctl
.)
ST
F a
cti
vit
y (
% C
tl.)
A3
lux
ac
tiv
ity
(%
Ctl
.)
120
100
80
60
40
20
0
120
100
80
60
40
20
0
120
100
80
60
40
20
0
log(mmol/L) log(mmol/L) log(mmol/L)
C19 C19 C19
SB-431
SB-431
SB-431
IWR1
IWR1
IWR1
Hippo Wnt TGF-b
-1.0
0-0
.75
-0.5
0-0
.25
0.00
0.25
0.50
1.00
1.25
0.75
-1.0
0-0
.75
-0.5
0-0
.25
0.00
0.25
0.50
1.00
1.25
0.75
-1.0
0-0
.75
-0.5
0-0
.25
0.00
0.25
0.50
1.00
1.25
0.75
Figure 2. C19 as a common inhibitor of Wnt, TGF-b and Hippo signaling. Two hundred and ninety-three cells were transfected with the 8xGTIIC-luciferase, the 8xTCF-luciferase, and the A3Lux and then exposed to C19, the Wnt signaling inhibitor IWR1, or the TGF-b signaling inhibitor SB-431542at the indicated concentrations for 24 hours. Luciferase activity was measured in the protein extracts. The data represent average of threedeterminations � SD.
A Small Molecule Inhibitor of EMT Pathways
www.aacrjournals.org Mol Cancer Ther; 13(6) June 2014 1461
on November 3, 2014. © 2014 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst April 2, 2014; DOI: 10.1158/1535-7163.MCT-13-0918
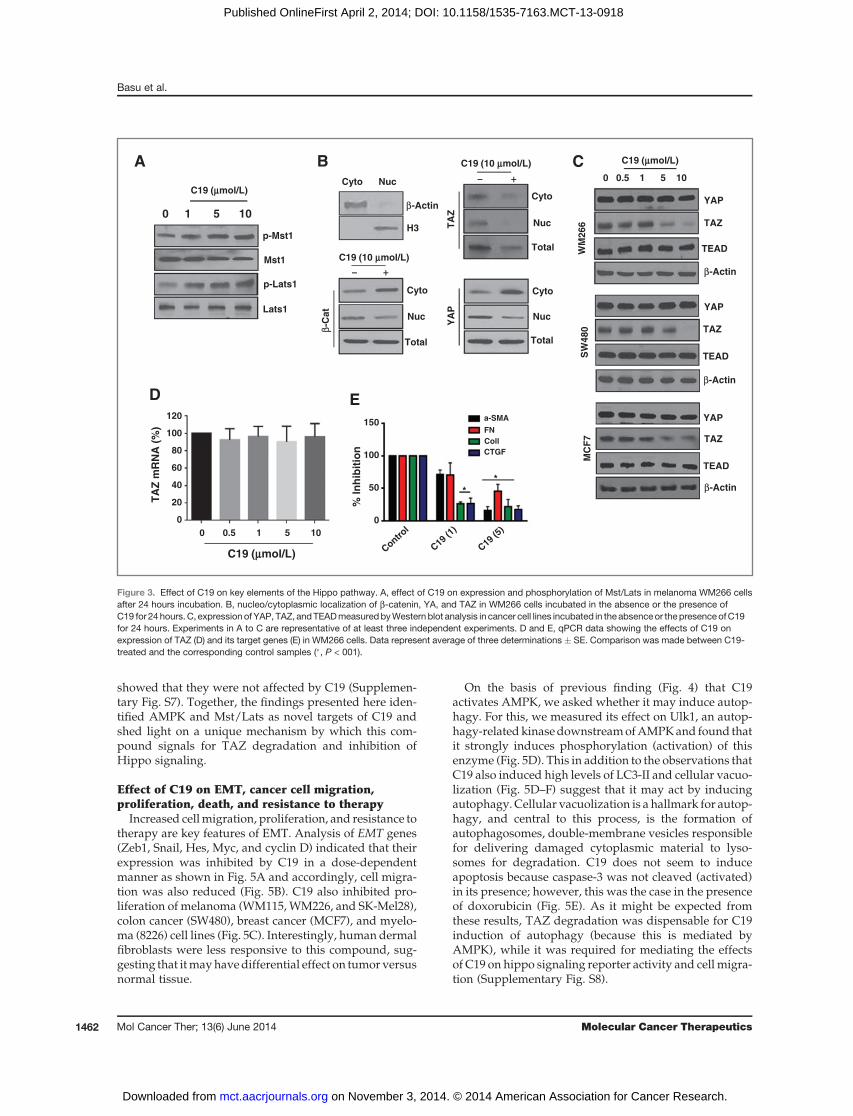
showed that they were not affected by C19 (Supplemen-tary Fig. S7). Together, the findings presented here iden-tified AMPK and Mst/Lats as novel targets of C19 andshed light on a unique mechanism by which this com-pound signals for TAZ degradation and inhibition ofHippo signaling.
Effect of C19 on EMT, cancer cell migration,proliferation, death, and resistance to therapy
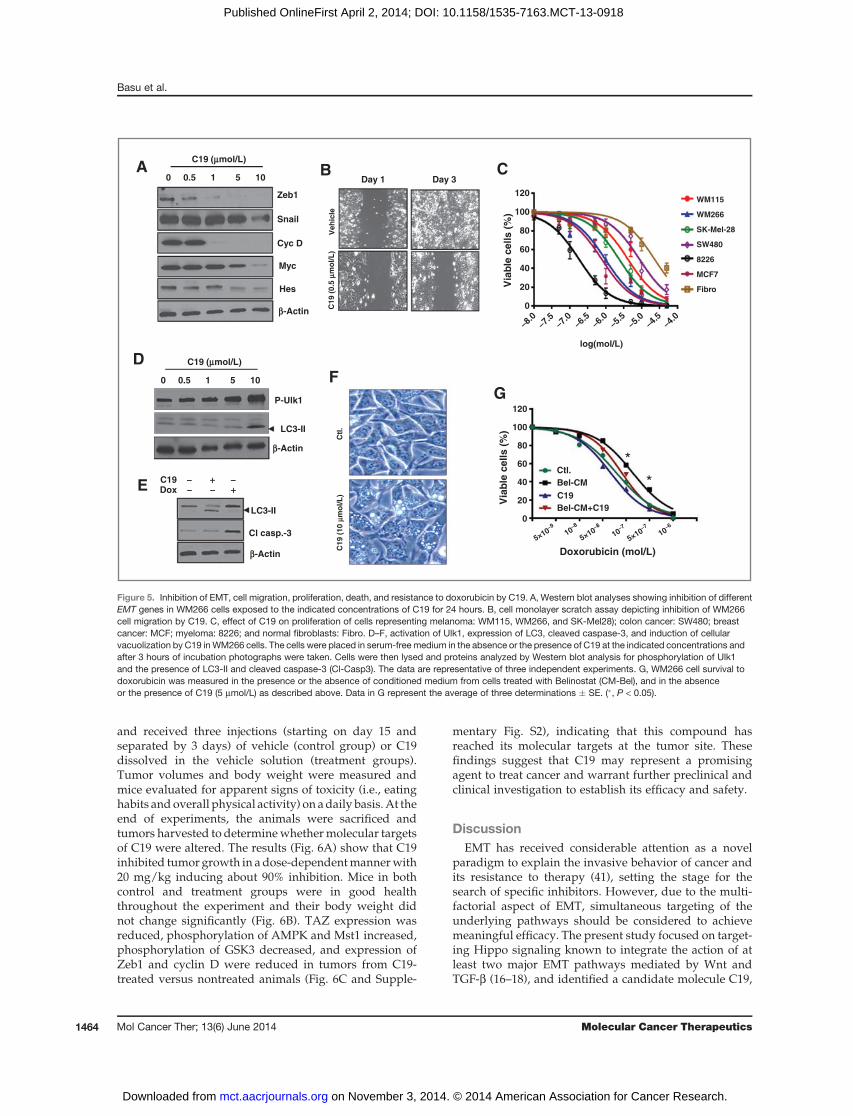
Increased cellmigration, proliferation, and resistance totherapy are key features of EMT. Analysis of EMT genes(Zeb1, Snail, Hes, Myc, and cyclin D) indicated that theirexpression was inhibited by C19 in a dose-dependentmanner as shown in Fig. 5A and accordingly, cell migra-tion was also reduced (Fig. 5B). C19 also inhibited pro-liferation of melanoma (WM115,WM226, and SK-Mel28),colon cancer (SW480), breast cancer (MCF7), and myelo-ma (8226) cell lines (Fig. 5C). Interestingly, human dermalfibroblasts were less responsive to this compound, sug-gesting that itmayhavedifferential effect on tumor versusnormal tissue.
On the basis of previous finding (Fig. 4) that C19activates AMPK, we asked whether it may induce autop-hagy. For this, we measured its effect on Ulk1, an autop-hagy-relatedkinasedownstreamofAMPKand found thatit strongly induces phosphorylation (activation) of thisenzyme (Fig. 5D). This in addition to the observations thatC19 also induced high levels of LC3-II and cellular vacuo-lization (Fig. 5D–F) suggest that it may act by inducingautophagy. Cellular vacuolization is a hallmark for autop-hagy, and central to this process, is the formation ofautophagosomes, double-membrane vesicles responsiblefor delivering damaged cytoplasmic material to lyso-somes for degradation. C19 does not seem to induceapoptosis because caspase-3 was not cleaved (activated)in its presence; however, this was the case in the presenceof doxorubicin (Fig. 5E). As it might be expected fromthese results, TAZ degradation was dispensable for C19induction of autophagy (because this is mediated byAMPK), while it was required for mediating the effectsof C19 on hippo signaling reporter activity and cell migra-tion (Supplementary Fig. S8).
p-Mst1
p-Lats1
0 1 5 10
ββ-C
at
YA
P
Cyto
Nuc
Cyto
Nuc
- +
TotalTotal
Mst1
Lats1
A B
D E
% I
nh
ibit
ion
TA
Z m
RN
A (
%)
**
C19 (10 mmol/L)
C19 (mmol/L)
C19 (mmol/L) C19 (10 mmol/L)
C19 (mmol/L)
TAZ
β-Actin
0 0.5 1 5 10
WM
26
6
C
YAP
TEAD
TA
Z
H3
Cyto Nuc
β-ActinCyto
Nuc
Total
- +
SW
48
0M
CF
7
β-Actin
YAP
TEAD
TAZ
β-Actin
YAP
TEAD
TAZ
120
100
80
60
40
20
0
150
100
50
0
Contr
ol
C19
(1)
C19
(5)0 0.5 1 5 10
a-SMA
FN
Coll
CTGF
Figure 3. Effect of C19 on key elements of the Hippo pathway. A, effect of C19 on expression and phosphorylation of Mst/Lats in melanoma WM266 cellsafter 24 hours incubation. B, nucleo/cytoplasmic localization of b-catenin, YA, and TAZ in WM266 cells incubated in the absence or the presence ofC19 for 24 hours. C, expression of YAP, TAZ, andTEADmeasuredbyWestern blot analysis in cancer cell lines incubated in the absenceor the presenceofC19for 24 hours. Experiments in A to C are representative of at least three independent experiments. D and E, qPCR data showing the effects of C19 onexpression of TAZ (D) and its target genes (E) in WM266 cells. Data represent average of three determinations � SE. Comparison was made between C19-treated and the corresponding control samples (�, P < 001).
Basu et al.
Mol Cancer Ther; 13(6) June 2014 Molecular Cancer Therapeutics1462
on November 3, 2014. © 2014 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst April 2, 2014; DOI: 10.1158/1535-7163.MCT-13-0918
We have previously reported that belinostat-inducedsecretion of survival factors may, in a paracrine manner,have protective effect on neighboring cancer cells render-ing them drug resistant (40). Here, we show (Fig. 5G) thatcellular response to doxorubicin decreased (green to blackcurve) when cells were exposed to culture medium fromtheir belinostat-treated counterparts (Bel-CM). This effectwas suppressed (black to red curve) by C19, suggestingthat this compound may also attenuate drug resistance.Together the data presented in this figure suggest that C19may exert its antitumor activity by inhibiting EMT, cancer
cell proliferation, inducing autophagy, and suppressingresistance to drugs.On the basis of these combined effects,C19 may represent a potent anticancer agent.
Antitumor activity of C19 in vivoExperiments were carried out to analyze the antitumor
activity and obtain preliminary information on safety ofC19 inmice bearing xenograft tumors. For this, nudemice(strain CD1) were subcutaneously inoculated with mela-noma cells WM266 and after tumors became palpable,they were randomized into control and treatment groups
SW
480 p-AMPK
SK
N-S
HM
CF
7
0 0.5 1 5 10 0 30 60 90 120
Time (min)
0 1 5
WM
115
WM
266
AMPK
p-AMPK
AMPK
p-AMPK
AMPK
A
C
0 1 5
E
F
D
mTOR
GSK3
Akt AMPK
TAZ
B
TAZ
GSK3-b
b-Actin
b-Actin
b-Actin
Ctl. GSK3
C19 (mmol/L)
C19 (mmol/L)
C19 (mmol/L)
C19 (mmol/L) C19 (mmol/L)
p-GSK3
p-Akt
0 0.5 1 5 10
G
C TW 1 5 10
GS
K a
cti
vit
y (
%)
p-AMPK
AICAR - - - - 0 1 10 10 40 (mmol/L)
C19 0 1 5 10 - - - - - (mmol/L)
AMPK
p-AMPK
AICAR - - 40
C19 - 10 -
H I *
*
120
100
80
60
40
20
0
Fo
ld in
cre
ase
10
8
6
4
2
0
Ctl.
C19
AIC
AR
Figure 4. Respective roles of Akt and AMPK inmediating the effect of C19. A, schematic representation of the Akt/AMPKpathway leading toGSK3-b and TAZ.B, effect of GSK3-b overexpression in WM266 cells on TAZ levels. Cells were transfected with constitutively active GSK3 and analyzed for expressionof the enzyme and TAZ by Western blot analysis. C, in vitro kinase assay showing the effect of C19 on GSK3-b activity. TWS119 (TW) is a specific inhibitorof GSK3. The data represent average of three determinations � SE. D, effect of cellular exposure to C19 on phosphorylation of Akt and GSK3-b.WM266 cells were incubated in serum-free medium for 3 hours in the absence or the presence of C19 at the indicated concentrations and expressionof phospho-Akt and phospho-GSK3-b determined by Western blot analysis using specific antibodies. b-actin was used as a loading control. E,dose- and time-dependent effects of C19 on phosphorylation of AMPKWM266 cells were treated as in D and expression of phospho-AMPKwas determinedby Western blot analysis using a specific antibody to phospho-threonine 172. F, activation of AMPK in different cell lines. Melanoma (WM115), colon(SW480), breast cancer (MCF7), and neuroblastoma (SKN-SH) were subjected to treatment with C19 as described in D and expression of phospho-AMPKdetermined by Western blot analysis. G, dose-dependent activation of AMPK by C19 in comparison with AICAR in WM266 cells. The cells were incubatedas in D in the presence of various concentrations of C19 or AICAR and phospho-AMPK measured by Western blot analysis. H, direct effect of C19and AICAR on AMPK. Immunoprecipitated AMPK was incubated in a kinase buffer in the absence or in the presence of C19 or AICAR. After 30 minutes, thereaction was stopped and phosphorylation of the enzyme determined by Western blot analysis using specific antibody. I, quantitative analysis ofthe data shown in H. Each value represents average of three determination � SE (�, P < 0.01). The Western blot data shown in B, D, E, F, G, and H arerepresentative of at least three independents determinations.
A Small Molecule Inhibitor of EMT Pathways
www.aacrjournals.org Mol Cancer Ther; 13(6) June 2014 1463
on November 3, 2014. © 2014 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst April 2, 2014; DOI: 10.1158/1535-7163.MCT-13-0918
and received three injections (starting on day 15 andseparated by 3 days) of vehicle (control group) or C19dissolved in the vehicle solution (treatment groups).Tumor volumes and body weight were measured andmice evaluated for apparent signs of toxicity (i.e., eatinghabits andoverall physical activity) on adaily basis.At theend of experiments, the animals were sacrificed andtumors harvested to determinewhethermolecular targetsof C19 were altered. The results (Fig. 6A) show that C19inhibited tumor growth in adose-dependentmannerwith20 mg/kg inducing about 90% inhibition. Mice in bothcontrol and treatment groups were in good healththroughout the experiment and their body weight didnot change significantly (Fig. 6B). TAZ expression wasreduced, phosphorylation of AMPK and Mst1 increased,phosphorylation of GSK3 decreased, and expression ofZeb1 and cyclin D were reduced in tumors from C19-treated versus nontreated animals (Fig. 6C and Supple-
mentary Fig. S2), indicating that this compound hasreached its molecular targets at the tumor site. Thesefindings suggest that C19 may represent a promisingagent to treat cancer and warrant further preclinical andclinical investigation to establish its efficacy and safety.
DiscussionEMT has received considerable attention as a novel
paradigm to explain the invasive behavior of cancer andits resistance to therapy (41), setting the stage for thesearch of specific inhibitors. However, due to the multi-factorial aspect of EMT, simultaneous targeting of theunderlying pathways should be considered to achievemeaningful efficacy. The present study focused on target-ing Hippo signaling known to integrate the action of atleast two major EMT pathways mediated by Wnt andTGF-b (16–18), and identified a candidate molecule C19,
Zeb1
Snail
Cyc D
Myc
b-Actin
b-Actin
b-Actin
A
Veh
icle
Day 1 Day 3
Hes
Doxorubicin (mol/L)
**
0 0.5 1 5 10
C19 (mmol/L)
C1
9 (
0.5
mm
ol/
L)
C1
9 (
10
mm
ol/
L)
Ctl
.
C19 (mmol/L)
B C
0 0.5 1 5 10
P-Ulk1
LC3-II
DF
LC3-II
Cl casp.-3
C19 - + -Dox - - +E
G
log(mol/L)
120
100
80
60
40
20
0
120
100
80
60
40
20
0
Via
ble
cells (
%)
Via
ble
cells (
%)
Ctl.
Bel-CM
Bel-CM+C19
C19
5×10-9
5×10-8
5×10-7
10-8
10-7
10-6
-8.0
-7.5
-7.0
-6.5
-6.0
-5.5
-5.0
-4.5
-4.0
WM115
WM266
SW480
SK-Mel-28
8226
MCF7
Fibro
Figure 5. Inhibition of EMT, cell migration, proliferation, death, and resistance to doxorubicin by C19. A, Western blot analyses showing inhibition of differentEMT genes in WM266 cells exposed to the indicated concentrations of C19 for 24 hours. B, cell monolayer scratch assay depicting inhibition of WM266cell migration by C19. C, effect of C19 on proliferation of cells representing melanoma: WM115, WM266, and SK-Mel28); colon cancer: SW480; breastcancer: MCF; myeloma: 8226; and normal fibroblasts: Fibro. D–F, activation of Ulk1, expression of LC3, cleaved caspase-3, and induction of cellularvacuolization by C19 inWM266 cells. The cells were placed in serum-free medium in the absence or the presence of C19 at the indicated concentrations andafter 3 hours of incubation photographs were taken. Cells were then lysed and proteins analyzed by Western blot analysis for phosphorylation of Ulk1and the presence of LC3-II and cleaved caspase-3 (Cl-Casp3). The data are representative of three independent experiments. G, WM266 cell survival todoxorubicin was measured in the presence or the absence of conditioned medium from cells treated with Belinostat (CM-Bel), and in the absenceor the presence of C19 (5 mmol/L) as described above. Data in G represent the average of three determinations � SE. (�, P < 0.05).
Basu et al.
Mol Cancer Ther; 13(6) June 2014 Molecular Cancer Therapeutics1464
on November 3, 2014. © 2014 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst April 2, 2014; DOI: 10.1158/1535-7163.MCT-13-0918
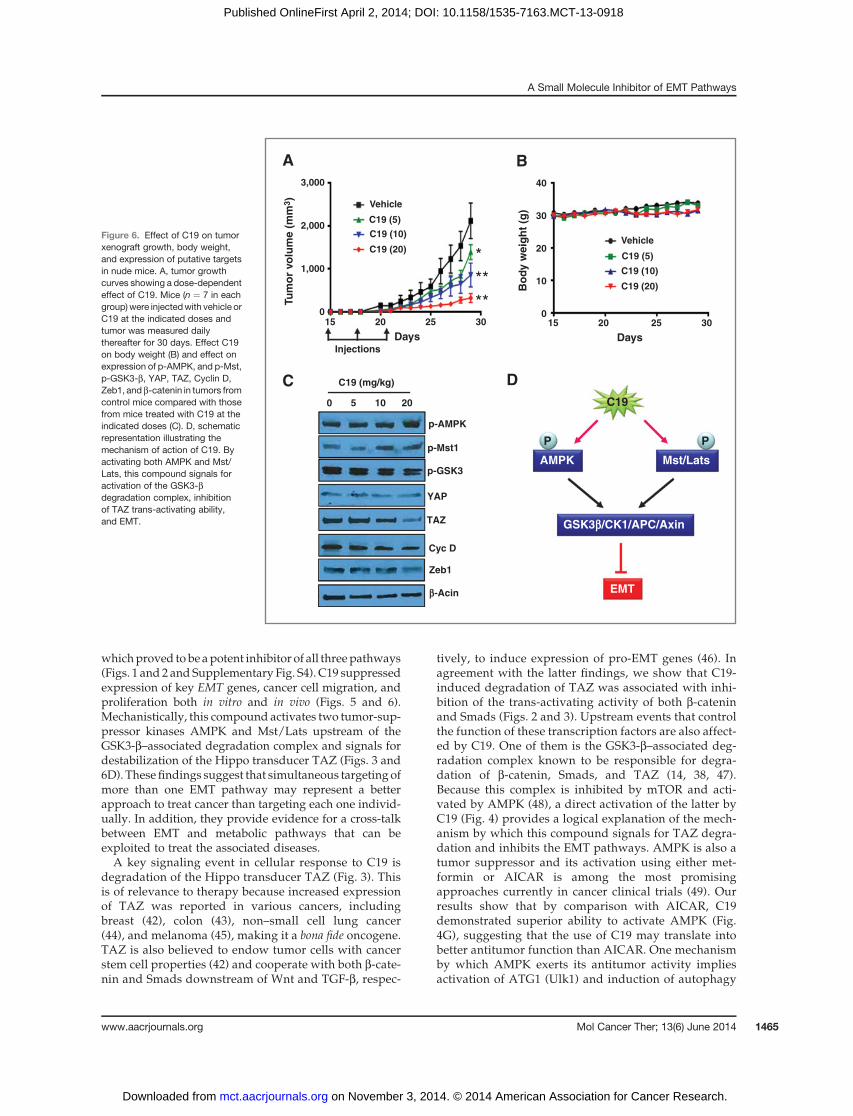
whichproved to be apotent inhibitor of all threepathways(Figs. 1 and 2 and Supplementary Fig. S4). C19 suppressedexpression of key EMT genes, cancer cell migration, andproliferation both in vitro and in vivo (Figs. 5 and 6).Mechanistically, this compound activates two tumor-sup-pressor kinases AMPK and Mst/Lats upstream of theGSK3-b–associated degradation complex and signals fordestabilization of the Hippo transducer TAZ (Figs. 3 and6D). These findings suggest that simultaneous targeting ofmore than one EMT pathway may represent a betterapproach to treat cancer than targeting each one individ-ually. In addition, they provide evidence for a cross-talkbetween EMT and metabolic pathways that can beexploited to treat the associated diseases.A key signaling event in cellular response to C19 is
degradation of the Hippo transducer TAZ (Fig. 3). Thisis of relevance to therapy because increased expressionof TAZ was reported in various cancers, includingbreast (42), colon (43), non–small cell lung cancer(44), and melanoma (45), making it a bona fide oncogene.TAZ is also believed to endow tumor cells with cancerstem cell properties (42) and cooperate with both b-cate-nin and Smads downstream of Wnt and TGF-b, respec-
tively, to induce expression of pro-EMT genes (46). Inagreement with the latter findings, we show that C19-induced degradation of TAZ was associated with inhi-bition of the trans-activating activity of both b-cateninand Smads (Figs. 2 and 3). Upstream events that controlthe function of these transcription factors are also affect-ed by C19. One of them is the GSK3-b–associated deg-radation complex known to be responsible for degra-dation of b-catenin, Smads, and TAZ (14, 38, 47).Because this complex is inhibited by mTOR and acti-vated by AMPK (48), a direct activation of the latter byC19 (Fig. 4) provides a logical explanation of the mech-anism by which this compound signals for TAZ degra-dation and inhibits the EMT pathways. AMPK is also atumor suppressor and its activation using either met-formin or AICAR is among the most promisingapproaches currently in cancer clinical trials (49). Ourresults show that by comparison with AICAR, C19demonstrated superior ability to activate AMPK (Fig.4G), suggesting that the use of C19 may translate intobetter antitumor function than AICAR. One mechanismby which AMPK exerts its antitumor activity impliesactivation of ATG1 (Ulk1) and induction of autophagy
Injections
*****
A B
C
AMPK Mst/Lats
GSK3b/CK1/APC/Axin
EMT
C19
PP
D
p-AMPK
p-Mst1
YAP
TAZ
0 5 10 20
b-Acin
p-GSK3
Zeb1
C19 (mg/kg)
Cyc D
Tu
mo
r vo
lum
e (
mm
3)
3,000
2,000
1,000
0
Days Days
15 20 25 30 15 20 25 30
Bo
dy
we
igh
t (g
)
40
30
20
10
0
Vehicle
Vehicle
C19 (5)
C19 (5)
C19 (10)
C19 (10)
C19 (20)
C19 (20)
Figure 6. Effect of C19 on tumorxenograft growth, body weight,and expression of putative targetsin nude mice. A, tumor growthcurves showing a dose-dependenteffect of C19. Mice (n ¼ 7 in eachgroup)were injectedwith vehicle orC19 at the indicated doses andtumor was measured dailythereafter for 30 days. Effect C19on body weight (B) and effect onexpression of p-AMPK, and p-Mst,p-GSK3-b, YAP, TAZ, Cyclin D,Zeb1, and b-catenin in tumors fromcontrol mice compared with thosefrom mice treated with C19 at theindicated doses (C). D, schematicrepresentation illustrating themechanism of action of C19. Byactivating both AMPK and Mst/Lats, this compound signals foractivation of the GSK3-bdegradation complex, inhibitionof TAZ trans-activating ability,and EMT.
A Small Molecule Inhibitor of EMT Pathways
www.aacrjournals.org Mol Cancer Ther; 13(6) June 2014 1465
on November 3, 2014. © 2014 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst April 2, 2014; DOI: 10.1158/1535-7163.MCT-13-0918

(50). C19 was found to induce phosphorylation (activa-tion) of Ulk1 and autophagy as indicated by increasedlevels of LC3 and enhanced cellular vacuolization incells exposed to this compound (Fig. 5D–F). This, inaddition to the observations that C19 inhibits expressionof various EMT genes, cell migration, proliferation, andresistance to doxorubicin (Fig. 5A–C), shed light on asecond mechanism, in addition to autophagy, by whichAMPK could inhibit tumor progression, and that is bysignaling for TAZ degradation and inhibition of EMT-associated features.
In vivo studies indicated that C19 potently suppresstumor growth in nudemice bearingmelanoma xenografts(Fig. 6A) and this was accompanied by increased phos-phorylation of bothAMPK andMst1 and decreased levelsof TAZandothermarkers of EMT in tumors from theC19-treated mice (Fig. 6C). As judged by body weight andanimal fitness, C19 did not induce any apparent signs oftoxicity, suggesting that this compoundmay represent aneffective and potentially safe agent to treat cancer. Furtherefficacy and toxicity studies in other animalmodels and inrelation to various tumor types are however warranted.
Overall, findings from this study shed light on theimportance of multi-EMT pathway targeting for efficientsuppression of cancer progression and identified a chem-ical tool (C19) to test this concept. On the basis of themechanismdescribed above (Fig. 6D) bywhichC19 exertsits activity, it is anticipated that this compound and itsanalogs could have applications for cancer therapy or at
least as tools to investigate molecular links betweenmetabolism, autophagy, and EMT signaling pathways.Also, the interesting finding that C19 activates AMPKopens new possibilities for this compound to treat notonly EMT-related pathologies such as cancer and tissuefibrosis, but also other diseases associated with AMPK,including obesity and diabetes.
Disclosure of Potential Conflicts of InterestNo potential conflicts of interest were disclosed.
Authors' ContributionsConception and design: R. Lettan, S. Strellec, A. RebbaaDevelopment of methodology: R. Lettan, S. Strellec, A. RebbaaAcquisition of data (provided animals, acquired and managed patients,provided facilities, etc.): D. Basu, R. Lettan, K. DamoderanAnalysis and interpretation of data (e.g., statistical analysis, biostatis-tics, computational analysis): D. Basu, A. RebbaaWriting, review, and/or revision of the manuscript: M. Reyes-Mugica,A. RebbaaAdministrative, technical, or material support (i.e., reporting or orga-nizing data, constructing databases): K. Damoderan, S. StrellecStudy supervision: M. Reyes-Mugica, A. Rebbaa
Grant SupportThis work was supported by a startup fund (toM. Reyes-Mugica) from
the University of Pittsburgh (Pittsburgh, PA).The costs of publication of this article were defrayed in part by the
payment of page charges. This article must therefore be hereby markedadvertisement in accordance with 18 U.S.C. Section 1734 solely to indicatethis fact.
Received October 24, 2013; revised March 26, 2014; accepted March 27,2014; published OnlineFirst April 2, 2014.
References1. Martin-Belmonte F, Perez-MorenoM. Epithelial cell polarity, stem cells
and cancer. Nat Rev Cancer 2012;12:23–38.2. Thiery JP. Epithelial-mesenchymal transitions in development and
pathologies. Curr Opin Cell Biol 2003;15:740–6.3. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The
epithelial-mesenchymal transition generates cells with properties ofstem cells. Cell 2008;133:704–15.
4. Acloque H, Adams MS, Fishwick K, Bronner-Fraser M, Nieto MA.Epithelial-mesenchymal transitions: the importance of changingcell state in development and disease. J Clin Invest 2009;119:1438–49.
5. Morales AV, Barbas JA, Nieto MA. How to become neural crest: fromsegregation to delamination. Semin Cell Dev Biol 2005;16:655–62.
6. Oft M, Heider KH, Beug H. TGFbeta signaling is necessary for carci-noma cell invasiveness and metastasis. Curr Biol 1998;8:1243–52.
7. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transi-tion. J Clin Invest 2009;119:1420–8.
8. Thiery JP, Chopin D. Epithelial cell plasticity in development and tumorprogression. Cancer Metastasis Rev 1999;18:31–42.
9. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymaltransitions in development and disease. Cell 2009;139:871–90.
10. Waaler J, Machon O, Tumova L, Dinh H, Korinek V, Wilson SR, et al. Anovel tankyrase inhibitor decreases canonical Wnt signaling in coloncarcinoma cells and reduces tumor growth in conditional APC mutantmice. Cancer Res 2012;72:2822–32.
11. Zubeldia IG, Bleau AM, Redrado M, Serrano D, Agliano A, Gil-Puig C,et al. Epithelial to mesenchymal transition and cancer stem cellphenotypes leading to liver metastasis are abrogated by the novelTGFbeta1-targeting peptides P17 and P144. Exp Cell Res 2013;319:12–22.
12. Fang Y, Chen Y, Yu L, Zheng C, Qi Y, Li Z, et al. Inhibition of breastcancer metastases by a novel inhibitor of TGFbeta receptor 1. J NatlCancer Inst 2013;105:47–58.
13. Zavadil J, Bottinger EP. TGF-beta and epithelial-to-mesenchymaltransitions. Oncogene 2005;24:5764–74.
14. Doble BW, Woodgett JR. Role of glycogen synthase kinase-3 in cellfate and epithelial-mesenchymal transitions. Cells Tissues Organs2007;185:73–84.
15. Smalley MJ, Dale TC. Wnt signalling in mammalian development andcancer. Cancer Metastasis Rev 1999;18:215–30.
16. Heallen T, ZhangM,Wang J, Bonilla-Claudio M, Klysik E, Johnson RL,et al. Hippo pathway inhibits Wnt signaling to restrain cardiomyocyteproliferation and heart size. Science 2011;332:458–61.
17. Imajo M, Miyatake K, Iimura A, Miyamoto A, Nishida E. A molecularmechanism that links Hippo signalling to the inhibition of Wnt/beta-catenin signalling. EMBO J 2012;31:1109–22.
18. Varelas X, Samavarchi-Tehrani P, Narimatsu M, Weiss A, Cockburn K,Larsen BG, et al. The Crumbs complex couples cell density sensing toHippo-dependent control of the TGF-beta-SMAD pathway. Dev Cell2010;19:831–44.
19. Pan D. Hippo signaling in organ size control. Genes Dev 2007;21:886–97.
20. Varelas X, Sakuma R, Samavarchi-Tehrani P, Peerani R, Rao BM,Dembowy J, et al. TAZcontrolsSmadnucleocytoplasmic shuttling andregulates human embryonic stem-cell self-renewal. Nat Cell Biol2008;10:837–48.
21. Avruch J, Zhou D, Bardeesy N. YAP oncogene overexpression super-charges colon cancer proliferation. Cell Cycle 2012;11:1090–6.
22. Huang W, Lv X, Liu C, Zha Z, Zhang H, Jiang Y, et al. The N-terminalphosphodegron targets TAZ/WWTR1 protein for SCFbeta-TrCP-
Basu et al.
Mol Cancer Ther; 13(6) June 2014 Molecular Cancer Therapeutics1466
on November 3, 2014. © 2014 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst April 2, 2014; DOI: 10.1158/1535-7163.MCT-13-0918

dependent degradation in response to phosphatidylinositol 3-kinaseinhibition. J Biol Chem 2012;287:26245–53.
23. HarveyK, TaponN. TheSalvador-Warts-Hippo pathway - an emergingtumour-suppressor network. Nat Rev Cancer 2007;7:182–91.
24. St JohnMA, TaoW, Fei X, FukumotoR,CarcangiuML,BrownsteinDG,et al. Mice deficient of Lats1 develop soft-tissue sarcomas, ovariantumours and pituitary dysfunction. Nat Genet 1999;21:182–6.
25. Zhou Z, Hao Y, Liu N, Raptis L, Tsao MS, Yang X. TAZ is a noveloncogene in non-small cell lung cancer. Oncogene 2011;30:2181–6.
26. de Cristofaro T, Di Palma T, Ferraro A, Corrado A, Lucci V, Franco R,et al. TAZ/WWTR1 is overexpressed in papillary thyroid carcinoma. EurJ Cancer 2011;47:926–33.
27. KonsavageWMJr, Kyler SL, Rennoll SA, JinG, YochumGS.Wnt/beta-catenin signaling regulatesYes-associated protein (YAP) gene expres-sion in colorectal carcinoma cells. J Biol Chem 2012;287:11730–9.
28. WangY, DongQ, ZhangQ, Li Z,Wang E,Qiu X. Overexpression of yes-associated protein contributes to progression and poor prognosis ofnon-small-cell lung cancer. Cancer Sci 2010;101:1279–85.
29. Zender L, Spector MS, Xue W, Flemming P, Cordon-Cardo C, Silke J,et al. Identification and validation of oncogenes in liver cancer using anintegrative oncogenomic approach. Cell 2006;125:1253–67.
30. MohseniM, Sun J, Lau A, Curtis S, Goldsmith J, Fox VL, et al. A geneticscreen identifies an LKB1-MARK signalling axis controlling the Hippo-YAP pathway. Nat Cell Biol 2014;16:108–17.
31. Dupont S,Morsut L, AragonaM, Enzo E, Giulitti S, CordenonsiM, et al.Role of YAP/TAZ in mechanotransduction. Nature 2011;474:179–83.
32. Zheng X, Chou PM, Mirkin BL, Rebbaa A. Senescence-initiated rever-sal of drug resistance: specific role of cathepsin L. Cancer Res 2004;64:1773–80.
33. Rebbaa A, Chou PM, Mirkin BL. Factors secreted by human neuro-blastoma mediated doxorubicin resistance by activating STAT3 andinhibiting apoptosis. Mol Med 2001;7:393–400.
34. Veeman MT, Slusarski DC, Kaykas A, Louie SH, Moon RT. Zebrafishprickle, a modulator of noncanonical Wnt/Fz signaling, regulatesgastrulation movements. Curr Biol 2003;13:680–5.
35. Kretzschmar M, Doody J, Timokhina I, Massague J. A mechanism ofrepression of TGFbeta/ Smad signaling by oncogenic Ras. Genes Dev1999;13:804–16.
36. Hastie CJ, McLauchlan HJ, Cohen P. Assay of protein kinases usingradiolabeled ATP: a protocol. Nat Protoc 2006;1:968–71.
37. Mirkin BL, Clark S, Zheng X, Chu F, White BD, Greene M, et al.Identification of midkine as a mediator for intercellular transfer of drugresistance. Oncogene 2005;24:4965–74.
38. Azzolin L, Zanconato F, Bresolin S, Forcato M, Basso G, Bicciato S,et al. Role of TAZ as mediator of Wnt signaling. Cell 2012;151:1443–56.
39. Corton JM, Gillespie JG, Hawley SA, Hardie DG. 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur J Biochem 1995;229:558–65.
40. Basu D, Reyes-Mugica M, Rebbaa A. Histone acetylation-mediatedregulation of the hippo pathway. PLoS ONE 2013;8:e62478.
41. Nauseef JT, Henry MD. Epithelial-to-mesenchymal transition in pros-tate cancer: paradigm or puzzle? Nat Rev Urol 2011;8:428–39.
42. LeiQY,ZhangH,ZhaoB,ZhaZY,Bai F, Pei XH, et al. TAZpromotes cellproliferation and epithelial-mesenchymal transition and is inhibited bythe hippo pathway. Mol Cell Biol 2008;28:2426–36.
43. Wang L, Shi S, GuoZ, ZhangX, HanS, YangA, et al. Overexpression ofYAP and TAZ is an independent predictor of prognosis in colorectalcancer and related to the proliferation and metastasis of colon cancercells. PLoS ONE 2013;8:e65539.
44. Xie M, Zhang L, He CS, Hou JH, Lin SX, Hu ZH, et al. Prognosticsignificance of TAZ expression in resected non-small cell lung cancer.J Thorac Oncol 2012;7:799–807.
45. Nallet-Staub F, Marsaud V, Li L, Gilbert C, Dodier S, Bataille V, et al.Pro-invasive activity of the hippo pathway effectors YAP and TAZ incutaneous melanoma. J Invest Dermatol 2014;134:123–32.
46. Attisano L, Wrana JL. Signal integration in TGF-beta, WNT, and Hippopathways. F1000Prime Rep 2013;5:17.
47. Ha NC, Tonozuka T, Stamos JL, Choi HJ, Weis WI. Mechanism ofphosphorylation-dependent binding of APC to beta-catenin and itsrole in beta-catenin degradation. Mol Cell 2004;15:511–21.
48. Cross DA, Alessi DR, Vandenheede JR, McDowell HE, Hundal HS,Cohen P. The inhibition of glycogen synthase kinase-3 by insulin orinsulin-like growth factor 1 in the rat skeletal muscle cell line L6 isblocked by wortmannin, but not by rapamycin: evidence that wort-mannin blocks activation of the mitogen-activated protein kinasepathway in L6 cells between Ras and Raf. Biochem J 1994;303 (Pt1):21–6.
49. Ben Sahra I, Le Marchand-Brustel Y, Tanti JF, Bost F. Metformin incancer therapy: a new perspective for an old antidiabetic drug? MolCancer Ther 2010;9:1092–9.
50. EganDF, ShackelfordDB,MihaylovaMM,GelinoS, KohnzRA,MairW,et al. Phosphorylation of ULK1 (hATG1) by AMP-activated proteinkinase connects energy sensing to mitophagy. Science 2011;331:456–61.
www.aacrjournals.org Mol Cancer Ther; 13(6) June 2014 1467
A Small Molecule Inhibitor of EMT Pathways
on November 3, 2014. © 2014 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst April 2, 2014; DOI: 10.1158/1535-7163.MCT-13-0918
Copyright © 2022 FDOKUMEN




















