
ICOS deficiency in patients with common variable immunodeficiency
7
ICOS deficiency in patients with common variable immunodeficiency Ulrich Salzer a , Andrea Maul-Pavicic a , Charlotte Cunningham-Rundles b , Simon Urschel c , Bernd H. Belohradsky c , Jiri Litzman d , Are Holm e , Jose ´ Luis Franco f , Alessandro Plebani g , Lennart Hammarstrom h , Andrea Skrabl i , Wolfgang Schwinger i , Bodo Grimbacher a, * a Division of Rheumatology and Clinical Immunology, University of Freiburg, 79106 Freiburg, Germany b Pediatrics and Immunobiology, Mount Sinai School of Medicine, New York, 10029, United States c Division of Infectious Diseases and Immunology, University Childrens Hospital, University of Munich, Germany d Department of Clinical Immunology and Allergology, St. Anne University Hospital, Masaryk University, Brno, Czech Republic e Research Institute for Internal Medicine, National Hospital, Oslo, Norway f Grupo de Immunodeficiencias Primarias, Departamento de Microbiologı ´a y Parasitologı ´a, Facultad de Medicina, Universidad de Antioquia, Medellin, Colombia g Clinica Pediatrica, Universita ` di Brescia and Istituto Medicina Molecolare bAngelo NocivelliQ, Spedali Civili, Brescia, Italy h Division of Clinical Immunology, IMPI, Karolinska Institute at Huddinge Hospital, Stockholm, Sweden i Division of Pediatric Hemato-Oncology, Department of Pediatrics, University of Graz, Austria Received 18 April 2004; accepted 6 July 2004 Available online 17 September 2004 Abstract Common variable immunodeficiency (CVID) is the most frequent clinically significant primary antibody deficiency in man, predisposing to recurrent bacterial infections. Recently, we showed that the homozygous loss of the inducible costimulator (ICOS) on activated T cells may result in an adult onset form of CVID with autosomal recessive inheritance (AR-CVID). We screened 181 sporadic CVID patients and 13 CVID patients from nine families with AR-CVID for mutations in ICOS by genomic DNA sequencing. In the AR-CVID families, the genomic integrity of the ligand for ICOS (ICOS-L) was also evaluated. In two of the nine AR-CVID families, we identified five individuals with ICOS deficiency, carrying the identical large genomic deletion of ICOS as previously described. In the remaining seven AR-CVID families, we subsequently sequenced the coding region of the ICOS ligand but found no mutations. The incidence of ICOS deficiency among patients with CVID is less than 5%. Worldwide, there are now a total of nine patients diagnosed with ICOS deficiency most likely due to a common founder. ICOS-L deficiency could not be identified in families with AR-CVID. D 2004 Elsevier Inc. All rights reserved. Keywords: CVID; ICOS; Immunodeficiency Introduction The diagnosis of common variable immunodeficiency (CVID) is based on markedly reduced serum levels for IgG and IgA or IgM, an impaired ability to specific antibody production after vaccination or exposure, and exclusion of secondary causes for antibody deficiency [1]. Although it is a rare disease with an estimated prevalence of 1 in 25,000 in the Western population, it is the second most frequent primary immunodeficiency (PID) after selective IgA deficiency (IgAD) and the most frequent PID requiring medical attention. Most cases of CVID are sporadic, however, about 10% are familial with a predominance of autosomal dominant over autosomal recessive inheritance [2,3]. CVID and IgAD 1521-6616/$ - see front matter D 2004 Elsevier Inc. All rights reserved. doi:10.1016/j.clim.2004.07.002 * Corresponding author. Division of Rheumatology and Clinical Immunology, University Hospital Freiburg, University of Freiburg, Hugstetterstr. 55, 79106 Freiburg, Germany. Fax: +49 761 270 3531. E-mail address: [email protected] (B. Grimbacher). Clinical Immunology 113 (2004) 234 – 240 www.elsevier.com/locate/yclim
-
Upload
meduni-graz -
Category
Documents
-
view
3 -
download
0
Transcript of ICOS deficiency in patients with common variable immunodeficiency

www.elsevier.com/locate/yclim
Clinical Immunology 1
ICOS deficiency in patients with common variable immunodeficiency
Ulrich Salzera, Andrea Maul-Pavicica, Charlotte Cunningham-Rundlesb, Simon Urschelc,
Bernd H. Belohradskyc, Jiri Litzmand, Are Holme, Jose Luis Francof, Alessandro Plebanig,
Lennart Hammarstromh, Andrea Skrabli, Wolfgang Schwingeri, Bodo Grimbachera,*
aDivision of Rheumatology and Clinical Immunology, University of Freiburg, 79106 Freiburg, GermanybPediatrics and Immunobiology, Mount Sinai School of Medicine, New York, 10029, United States
cDivision of Infectious Diseases and Immunology, University Childrens Hospital, University of Munich, GermanydDepartment of Clinical Immunology and Allergology, St. Anne University Hospital, Masaryk University, Brno, Czech Republic
eResearch Institute for Internal Medicine, National Hospital, Oslo, NorwayfGrupo de Immunodeficiencias Primarias, Departamento de Microbiologıa y Parasitologıa, Facultad de Medicina,
Universidad de Antioquia, Medellin, ColombiagClinica Pediatrica, Universita di Brescia and Istituto Medicina Molecolare bAngelo NocivelliQ, Spedali Civili, Brescia, Italy
hDivision of Clinical Immunology, IMPI, Karolinska Institute at Huddinge Hospital, Stockholm, SwedeniDivision of Pediatric Hemato-Oncology, Department of Pediatrics, University of Graz, Austria
Received 18 April 2004; accepted 6 July 2004
Available online 17 September 2004
Abstract
Common variable immunodeficiency (CVID) is the most frequent clinically significant primary antibody deficiency in man, predisposing
to recurrent bacterial infections. Recently, we showed that the homozygous loss of the inducible costimulator (ICOS) on activated T cells may
result in an adult onset form of CVID with autosomal recessive inheritance (AR-CVID).
We screened 181 sporadic CVID patients and 13 CVID patients from nine families with AR-CVID for mutations in ICOS by genomic
DNA sequencing. In the AR-CVID families, the genomic integrity of the ligand for ICOS (ICOS-L) was also evaluated.
In two of the nine AR-CVID families, we identified five individuals with ICOS deficiency, carrying the identical large genomic deletion
of ICOS as previously described. In the remaining seven AR-CVID families, we subsequently sequenced the coding region of the ICOS
ligand but found no mutations.
The incidence of ICOS deficiency among patients with CVID is less than 5%. Worldwide, there are now a total of nine patients diagnosed
with ICOS deficiency most likely due to a common founder. ICOS-L deficiency could not be identified in families with AR-CVID.
D 2004 Elsevier Inc. All rights reserved.
Keywords: CVID; ICOS; Immunodeficiency
Introduction
The diagnosis of common variable immunodeficiency
(CVID) is based on markedly reduced serum levels for IgG
1521-6616/$ - see front matter D 2004 Elsevier Inc. All rights reserved.
doi:10.1016/j.clim.2004.07.002
* Corresponding author. Division of Rheumatology and Clinical
Immunology, University Hospital Freiburg, University of Freiburg,
Hugstetterstr. 55, 79106 Freiburg, Germany. Fax: +49 761 270 3531.
E-mail address: [email protected]
(B. Grimbacher).
and IgA or IgM, an impaired ability to specific antibody
production after vaccination or exposure, and exclusion of
secondary causes for antibody deficiency [1]. Although it is a
rare disease with an estimated prevalence of 1 in 25,000 in the
Western population, it is the second most frequent primary
immunodeficiency (PID) after selective IgA deficiency
(IgAD) and the most frequent PID requiring medical
attention. Most cases of CVID are sporadic, however, about
10% are familial with a predominance of autosomal dominant
over autosomal recessive inheritance [2,3]. CVID and IgAD
13 (2004) 234–240

Table 2
Clinical features of CVID patients (n = 194)
Sex Female 106 (55%)
Male 88 (45%)
Age b10 years 11 (6%)
10–30 years 41 (21%)
31–50 years 96 (49%)
N50 years 39 (20%)
Unknown 7 (4%)
Age of diagnosis
of CVID
b10 years 34 (17%)
10–30 years 85 (44%)
31–50 years 52 (27%)
N50 years 11 (6%)
Unknown 12 (6%)
Clinical presentation Recurrent sinusitis 123 (63%)
Recurrent bronchitis 130 (67%)
Recurrent pneumonia 98 (50%)
Bronchiectasis 52 (27%)
Recurrent gastrointestinal
infections
36 (18%)
Autoimmune phenomenaa 42 (21%)
splenomegaly 74 (38%)
Granuloma formation 14 (7%)
Malignancyb 10 (5%)
a Including idiopathic thrombocytopenic purpura, autoimmune hemolytic
anemia, pernicious anemia, rheumatoid arthritis, psoriasis, vitiligo, diabetes
mellitus, and systemic lupus erythematosus.b Including thymoma, Hodgkin lymphoma, non-Hodgkin lymphoma,
glioma, acute lymphatic leucemia, and cutaneous T cell lymphoma.
U. Salzer et al. / Clinical Immunology 113 (2004) 234–240 235
may occur in the same family, which suggests a possible
common genetic basis for these two disease entities [4].
The age of onset of CVID has two major peaks: the first
between 5 and 10 years of age, the second in early
adulthood, ages 20–30. The phenotype of CVID is
characterized by the sequelae of hypogammaglobulinemia
with recurrent upper respiratory infections by encapsulated
bacteria. In addition to these infections, the phenotype of
CVID varies and may be accompanied by autoimmune
phenomena, splenomegaly, granulomas, and the develop-
ment of certain types of cancer [1,5].
The inducible costimulator (ICOS) belongs to the family
of costimulatory T cell molecules such as CD28 and CTLA-
4 [6]. All three genes were mapped to chromosome 2q33.
ICOS spans over 25 kb and is divided into five exons and
four introns [7,8]. Fifty SNPs have been reported for ICOS:
16 lie within the promoter region, 13 are intronic, 1 is within
exon 5, 1 SNP lies within an exon–intron boundary, and the
remaining 19 SNPs are in the 3V UTR [8–10].
ICOS is only expressed on activated T cells and
coinduces the secretion of IL-4, IL-5, IL-6, GM-CSF,
TNF-a, and IFN-g but is pivotal for the superinduction of
IL-10 [11]. Highest expression of ICOS is found within the
T cell zones of secondary lymphoid organs and in the
apical light zones of germinal centers [6,11]. This
expression pattern and the cytokines induced by ICOS
point to an important role of ICOS:ICOS-L interaction in
mediating T–B cell cooperation and promoting the
terminal differentiation of B cells into memory cells and
plasma cells. This is further supported by findings in the
ICOS and ICOS-L knockout mice, both showing a defect
in germinal center formation and impaired humoral
immune responses, especially after secondary immuniza-
tions [12–14].
ICOS-L is the unique ligand for ICOS and is
constitutively expressed on both lymphoid and nonlym-
phoid tissues. ICOS-L can be up-regulated by inflamma-
tory stimuli such as TNF-a, IFN-g, and lipopolysaccharide
(LPS) [15]. ICOS-L shares 20% homology with CD80 and
Table 1
Amplification and sequencing primers for ICOS and ICOS-L
Forward primer (5VY3V)
ICOS Exon 1 TGC CTT TCA TAT AGC CAT TG
Exon 2 GCA ACA GAG ATG ACT TTA TGC ATT GA
Exon 3 AAG CCC TTT CTA TAA ACC ACA TT
Exon 4 TGG GGGATA TTC TTT TTG GTC
Exon 5 GTG TAT GAA AGG CAA TGG AGA GG
ICOS-L Exon 1 GCG GGA GCG CAG TTA GAG C
Exon 2 CAC GGG CAC GCC TGA TGT TCT
Exon 3 GGA CCT CAC CAT GAA ATG TC
Exon 4 CCA TGT CGG GGC ACA ATG
Exon 5 CCT GGC TGT GGT TGG GGT TAT C
Exon 6 GGT GTT TAG GGG GCT GCT GAG AGC
Exon 7 GGC CGA CCG CAG AAA CGC ACT T
CD86 (the ligands of CD28 and CTLA4). The gene
encoding ICOS-L is located on chromosome 21q22.3 and
consists of seven exons [16]. ICOS-L is expressed as two
splice variants designated as hGL50 and B7H2, with
hGL50 showing an expression pattern restricted to
lymphoid organs [17,18]. ICOS-L expression on naRve B
cells is down regulated via B cell receptor and ICOS
engagement or IL-4 signaling, but it can be sustained by
stimulation via CD40 [19].
We recently identified homozygous deletions in ICOS in
4 of 32 patients with CVID [20]. Therefore, the aim of this
study was to determine the incidence of ICOS deficiency
Reverse primer (5VY3V) Annealing
temperature (8C)
AGT CAT TTT GCC TTT CAT CTT T 55
CTG CAT CTA AGT GAA CTC CAA 58
TGT CTC CCT GTT GGT CAA A 55
TCT AGA ATT AGG CCT TGG AGA TGT T 58
AGA ATG CTG GCC CAT TAA AGA TGA T 62
CTC ACA GGT TCA CCC AGG GG 58
GTG CTG CGG TGC TTC GTC CTT ATT 63
GCT AAC TAA CTC AAG AGG CAA 63
GCG GTG GTT CTC ACT GTG 63
CTT GGC AGT GTC AGG AAT G 58
ACA GGC CGG CCG TGT TTT TAG C 58
GGA ACA GCC GAG GGA CAT TG 63

Table 3
Genotype frequencies of SNPs at the ICOS-locus
SNPa Genotype frequency, n/(%)
p1203 CVID T/T 5 (3) C/T 28 (17) C/C 128 (80) n = 161
HDb T/T 0 (0) C/T 15 (14) C/C 95 (86) n = 110
T-repeat CVID T[13] 130 (91) T[14] 13 (9) T[15] 0 (0) n = 145
HDb T[13] 99 (93) T[14] 5 (5) T[15] 2 (2) n = 106
p3990 CVID T/T 32 (20) G/T 63 (41) G/G 62 (39) n = 157
HDb T/T 18 (16) G/T 55 (50) G/G 37 (34) n = 110
P4031 CVID C/C 15 (10) A/C 34 (22) A/A 106 (68) n = 155
HDb C/C 3 (3) A/C 35 (32) A/A 72 (65) n = 110
a Single nucleotide polymorphism.b HD = healthy donor, refers to Ref. [10].
U. Salzer et al. / Clinical Immunology 113 (2004) 234–240236
(OMIM #607594) among patients with CVID. In addition,
we screened autosomal recessive CVID (AR-CVID) families
for disease causing mutations in the ICOS-L gene.
Patients and methods
Patients
Patients were diagnosed with CVID according to the
ESID criteria (www.esid.org). One hundred and twenty-
three patients were recruited from central Europe, 16
patients were from Scandinavia, 48 patients were from the
United States, and 7 patients were from Colombia.
Informed written consent was obtained from each indivi-
dual before participation under the internal ethics review
board-approved clinical study protocol (#239/99). Ten
milliliters of heparinized blood was obtained from each
participant of the study for isolation of genomic DNA
using DNA Blood isolation kit reagents (Puregene DNA
isolation kit; Gentra Systems, Minneapolis, USA).
Fig. 1. Pedigrees of newly identified patients with ICOS deficiency; circles, female; squares, male; slashed rhombuses, deceased; filled symbols, affected; open
symbols, unaffected. Individuals II.1 and II.2 died due to complications at birth as premature neonates. No autopsies were performed. Long-range PCR analysis
of ICOS. Genomic DNA from members of family C and D was amplified with primers covering ICOS exons 2 and 3. From DNA of a healthy donor, a 3-kb
PCR fragment could be amplified, in the heterozygote parents (family D) both a 3-kb and a shortened 1.2-kb fragment was present, and in all affected
individuals only a 1.2-kb fragment could be detected, indicating a genomic deletion.
Sequencing of ICOS and ICOS-L
The coding sequence of the five exons and the adjacent
intron–exon boundaries of ICOS was amplified from
genomic DNA using primer pairs and conditions as
indicated in Table 1. Long-range PCR from genomic
DNA was performed using Takara LA Taq (Cambrex
Bioscience Rockland Inc., Rockland, USA) with the
following primers: LRfw (5VY3V) TGG GGC TTT ATC
TTT ATT ATC AGG, LRrv (5VY3V) TGG GCG CTATCT
CAT TCT CT. The coding sequence of seven exons and
adjacent intron–exon boundaries of ICOS-L was amplified
from genomic DNA using the primer pairs and conditions as
indicated in Table 1.
PCR products were sequenced with the PCR amplifica-
tion primers. After gel electrophoresis on an ABI Prismk377 DNA Sequencer (PE Applied Biosystems, Foster City,
USA), the data were analyzed with aid of the DNA
Sequencing Analysis software, version 3.4 (PE Applied
Biosystems) and Sequenchertk version 3.4.1 (Gene Codes
Corporation, Ann Arbor, USA).
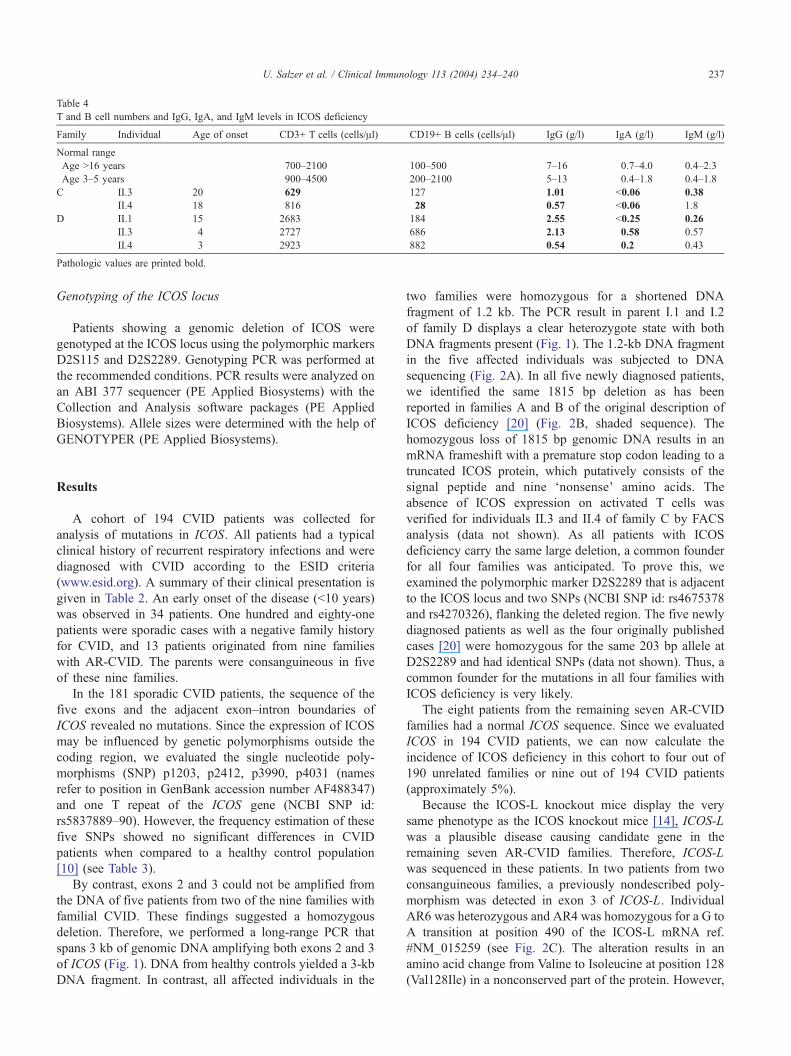
Table 4
T and B cell numbers and IgG, IgA, and IgM levels in ICOS deficiency
Family Individual Age of onset CD3+ T cells (cells/Al) CD19+ B cells (cells/Al) IgG (g/l) IgA (g/l) IgM (g/l)
Normal range
Age N16 years 700–2100 100–500 7–16 0.7–4.0 0.4–2.3
Age 3–5 years 900–4500 200–2100 5–13 0.4–1.8 0.4–1.8
C II.3 20 629 127 1.01 b0.06 0.38
II.4 18 816 28 0.57 b0.06 1.8
D II.1 15 2683 184 2.55 b0.25 0.26
II.3 4 2727 686 2.13 0.58 0.57
II.4 3 2923 882 0.54 0.2 0.43
Pathologic values are printed bold.
U. Salzer et al. / Clinical Immunology 113 (2004) 234–240 237
Genotyping of the ICOS locus
Patients showing a genomic deletion of ICOS were
genotyped at the ICOS locus using the polymorphic markers
D2S115 and D2S2289. Genotyping PCR was performed at
the recommended conditions. PCR results were analyzed on
an ABI 377 sequencer (PE Applied Biosystems) with the
Collection and Analysis software packages (PE Applied
Biosystems). Allele sizes were determined with the help of
GENOTYPER (PE Applied Biosystems).
Results
A cohort of 194 CVID patients was collected for
analysis of mutations in ICOS. All patients had a typical
clinical history of recurrent respiratory infections and were
diagnosed with CVID according to the ESID criteria
(www.esid.org). A summary of their clinical presentation is
given in Table 2. An early onset of the disease (b10 years)
was observed in 34 patients. One hundred and eighty-one
patients were sporadic cases with a negative family history
for CVID, and 13 patients originated from nine families
with AR-CVID. The parents were consanguineous in five
of these nine families.
In the 181 sporadic CVID patients, the sequence of the
five exons and the adjacent exon–intron boundaries of
ICOS revealed no mutations. Since the expression of ICOS
may be influenced by genetic polymorphisms outside the
coding region, we evaluated the single nucleotide poly-
morphisms (SNP) p1203, p2412, p3990, p4031 (names
refer to position in GenBank accession number AF488347)
and one T repeat of the ICOS gene (NCBI SNP id:
rs5837889–90). However, the frequency estimation of these
five SNPs showed no significant differences in CVID
patients when compared to a healthy control population
[10] (see Table 3).
By contrast, exons 2 and 3 could not be amplified from
the DNA of five patients from two of the nine families with
familial CVID. These findings suggested a homozygous
deletion. Therefore, we performed a long-range PCR that
spans 3 kb of genomic DNA amplifying both exons 2 and 3
of ICOS (Fig. 1). DNA from healthy controls yielded a 3-kb
DNA fragment. In contrast, all affected individuals in the
two families were homozygous for a shortened DNA
fragment of 1.2 kb. The PCR result in parent I.1 and I.2
of family D displays a clear heterozygote state with both
DNA fragments present (Fig. 1). The 1.2-kb DNA fragment
in the five affected individuals was subjected to DNA
sequencing (Fig. 2A). In all five newly diagnosed patients,
we identified the same 1815 bp deletion as has been
reported in families A and B of the original description of
ICOS deficiency [20] (Fig. 2B, shaded sequence). The
homozygous loss of 1815 bp genomic DNA results in an
mRNA frameshift with a premature stop codon leading to a
truncated ICOS protein, which putatively consists of the
signal peptide and nine dnonsenseT amino acids. The
absence of ICOS expression on activated T cells was
verified for individuals II.3 and II.4 of family C by FACS
analysis (data not shown). As all patients with ICOS
deficiency carry the same large deletion, a common founder
for all four families was anticipated. To prove this, we
examined the polymorphic marker D2S2289 that is adjacent
to the ICOS locus and two SNPs (NCBI SNP id: rs4675378
and rs4270326), flanking the deleted region. The five newly
diagnosed patients as well as the four originally published
cases [20] were homozygous for the same 203 bp allele at
D2S2289 and had identical SNPs (data not shown). Thus, a
common founder for the mutations in all four families with
ICOS deficiency is very likely.
The eight patients from the remaining seven AR-CVID
families had a normal ICOS sequence. Since we evaluated
ICOS in 194 CVID patients, we can now calculate the
incidence of ICOS deficiency in this cohort to four out of
190 unrelated families or nine out of 194 CVID patients
(approximately 5%).
Because the ICOS-L knockout mice display the very
same phenotype as the ICOS knockout mice [14], ICOS-L
was a plausible disease causing candidate gene in the
remaining seven AR-CVID families. Therefore, ICOS-L
was sequenced in these patients. In two patients from two
consanguineous families, a previously nondescribed poly-
morphism was detected in exon 3 of ICOS-L. Individual
AR6 was heterozygous and AR4 was homozygous for a G to
A transition at position 490 of the ICOS-L mRNA ref.
#NM_015259 (see Fig. 2C). The alteration results in an
amino acid change from Valine to Isoleucine at position 128
(Val128Ile) in a nonconserved part of the protein. However,
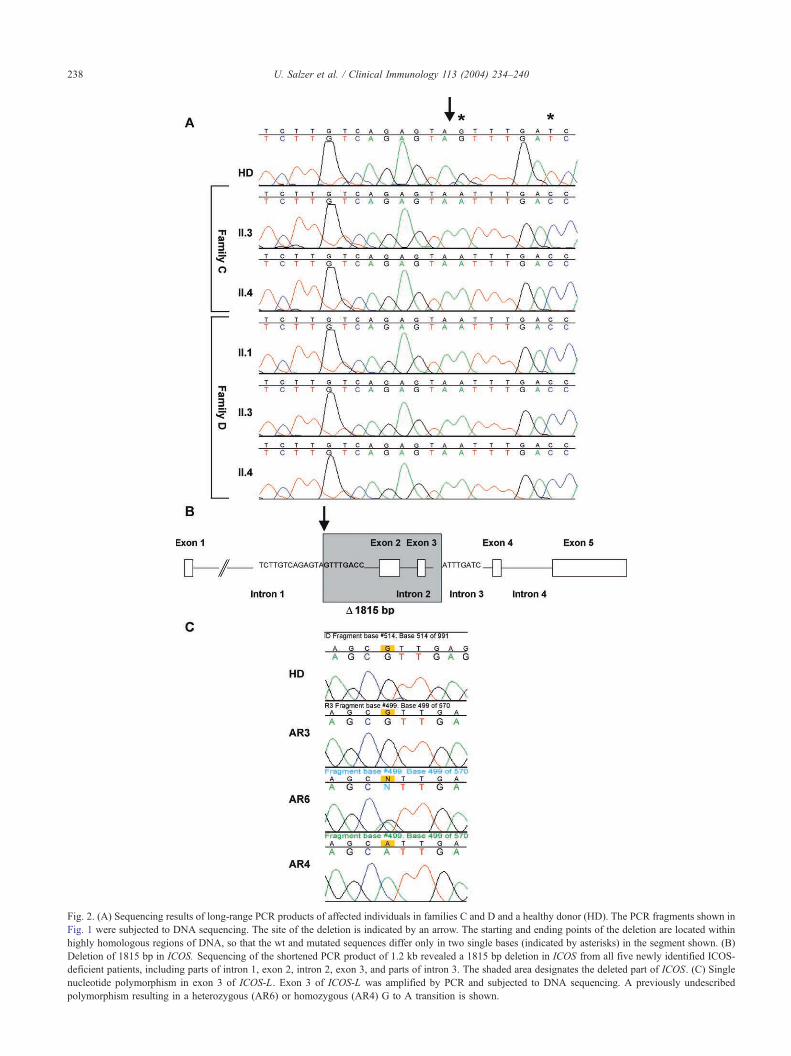
Fig. 2. (A) Sequencing results of long-range PCR products of affected individuals in families C and D and a healthy donor (HD). The PCR fragments shown in
Fig. 1 were subjected to DNA sequencing. The site of the deletion is indicated by an arrow. The starting and ending points of the deletion are located within
highly homologous regions of DNA, so that the wt and mutated sequences differ only in two single bases (indicated by asterisks) in the segment shown. (B
Deletion of 1815 bp in ICOS. Sequencing of the shortened PCR product of 1.2 kb revealed a 1815 bp deletion in ICOS from all five newly identified ICOS
deficient patients, including parts of intron 1, exon 2, intron 2, exon 3, and parts of intron 3. The shaded area designates the deleted part of ICOS. (C) Single
nucleotide polymorphism in exon 3 of ICOS-L. Exon 3 of ICOS-L was amplified by PCR and subjected to DNA sequencing. A previously undescribed
polymorphism resulting in a heterozygous (AR6) or homozygous (AR4) G to A transition is shown.
U. Salzer et al. / Clinical Immunology 113 (2004) 234–240238
)
-

U. Salzer et al. / Clinical Immunology 113 (2004) 234–240 239
we could detect this polymorphism also in control individ-
uals at a similar frequency for both heterozygotes and
homozygotes.
Therefore, we concluded that neither ICOS nor ICOS-L
is the disease causing gene in those remaining seven CVID
families.
Discussion
The CVID phenotype appears to origin from genetic
conditions resulting in the cardinal symptoms of primary
antibody deficiency. In the past, patients with other mono-
genic primary immunodeficiencies such as X-linked Agam-
maglobulinemia (XLA), X-linked lymphoproliferative
Syndrome (XLP), or hyper-IgM-syndrome (HIgM) have
been misdiagnosed as CVID since these disorders can
mimic the phenotype of CVID [21–23].
ICOS deficiency represents the first single gene defect
identified to solely cause the CVID phenotype [20]. Thus,
we screened a cohort of 194 CVID patients for mutations of
ICOS to determine how frequent a defect in ICOS causes
CVID.
The originally described four ICOS deficiency patients
were homozygous for a large genomic deletion in ICOS
[20]. The patients came from two CVID families with an
autosomal recessive trait. Heterozygote parents and siblings
had no clinical signs of immunodeficiency. The immuno-
logic laboratory findings in the heterozygote carriers were
also normal, despite a slightly weaker surface staining for
ICOS on their activated T cells [20]. We therefore concluded
that both alleles of ICOS had to be affected by mutations to
cause the phenotype of CVID. In a previous study [24],
ICOS was evaluated in 47 unrelated CVID patients by
SSCP analysis and no mutations were found. However,
SSCP has a sensitivity of about 80% and thus possible
mutations in this cohort may have been missed.
In a group of 181 sporadic CVID patients, we could not
detect any mutations in the coding regions of ICOS. The
frequency of a known polymorphism (p4031 AYC) located
in the located in the 3VUTR two bp after the stop codon was
determined to be comparable to that in control populations
[10]. Lee et al. [25] found that there was no difference in the
binding affinity to B7RP-1 between the different allelic
variants of this polymorphism. However, we cannot exclude
alterations or deficiency in ICOS expression due to mutations
in the promoter region of ICOS or putative regulatory intronic
regions in the CVID patients we analyzed since protein
expression of ICOS on activated T cells could only be
assessed by flow cytometry in 28 patients.
Among nine CVID families most likely affected by an
autosomal recessive trait, we identified two new unrelated
families with five individuals carrying the identical large
genomic deletion in ICOS as previously published [20].
Further genetic analysis of all nine ICOS deficiency patients
revealed that they share identical homozygous haplotypes in
the ICOS locus, indicating that the mutation was passed on
to them deriving from a common founder. This is further
supported by the fact that three of the four families with
ICOS deficiency originate from the same village, which is
linked to the origin of the fourth family by the long river of
Danube. The Danube was a frequently used route of
emigration in the 18th century. The observation that the
deleted region is flanked by highly homologous stretches of
DNA including a 13-bp identical segment suggests that the
deletion resulted from an aberrant recombination event.
Family C showed a childhood onset of the disease, which
was not observed in the other three families. The flow
cytometry analysis of T cells and their proliferative response
to mitogens gave similar results as in the originally
described patients [20]. The B cell compartment showed a
reduction in memory B cell subsets (Table 4), which
confirms the results obtained in the previously described
ICOS-deficient patients, a phenotype also found in about
three quarters of CVID patients.
After exclusion of mutations in ICOS, we subsequently
evaluated the ICOS-L in the remaining seven AR-CVID
families. In two patients, we could identify a previously
undescribed polymorphism located in exon 3 of ICOS-L
giving rise to an amino acid substitution from Valine to
Isoleucine. However, as both amino acids are aliphatic and
the exchange is located within a nonconserved part of the
protein [16], the structure or function of ICOS-L are
unlikely to be impaired, which is further supported by the
observation of the same polymorphism in healthy controls at
a similar frequency.
We suggest mutations of ICOS be sought in patients with
an recessive pattern of inheritance of CVID. While we did
not identify this mutation in the sporadic cases investigated
here, additional cases arising from the same or other ICOS
mutations may be identified.
Acknowledgments
We thank Dfrte Thiel, Judith Deimel, and Cristina
Wfllner for excellent technical assistance and the patients’
physicians Dr. Mezger and Prof. Dr. Vaith. This work was
supported by the Deutsche Forschungsgemeinschaft (DFG)
grant GR 1617/3 and SFB 620/project C2 to B.G and by
grants from the National Institutes of Health, AI-467320,
AI-48693, and contract N01-AI-30070, NIH-NIAID-DAIT
03-22 to C.C.
References
[1] C. Cunningham-Rundles, C. Bodian, Common variable immunode-
ficiency: clinical and immunological features of 248 patients, Clin.
Immunol. 92 (1999) 34–48.
[2] R.F. Ashman, F.M. Schaffer, J.D. Kemp, W.M. Yokoyama, Z.B. Zhu,
M.D. Cooper, J.E. Volanakis, Genetic and immunologic analysis of a
family containing five patients with common variable immune

U. Salzer et al. / Clinical Immunology 113 (2004) 234–240240
deficiency or selective IgA deficiency, J. Clin. Immunol. 12 (1992)
406–414.
[3] I. Vorechovsky, M. Cullen, M. Carrington, L. Hammarstrom, A.D.
Webster, Fine mapping of IGAD1 in IgA deficiency and common
variable immunodeficiency: identification and characterization of
haplotypes shared by affected members of 101 multiple-case families,
J. Immunol. 164 (2000) 4408–4416.
[4] P.D. Burrows, M.D. Cooper, IgA deficiency, Adv. Immunol. 65
(1997) 245–276.
[5] L. Mellemkjaer, L. Hammarstrom, V. Andersen, J. Yuen, C.
Heilmann, T. Barington, J. Bjorkander, J.H. Olsen, Cancer risk
among patients with IgA deficiency or common variable immunode-
ficiency and their relatives: a combined Danish and Swedish study,
Clin. Exp. Immunol. 130 (2002) 495–500.
[6] A. Hutloff, A.M. Dittrich, K.C. Beier, B. Eljaschewitsch, R. Kraft, I.
Anagnostopoulos, R.A. Kroczek, ICOS is an inducible T-cell co-
stimulator, structurally and functionally related to CD28, Nature 397
(1999) 263–266.
[7] A.J. Coyle, S. Lehar, C. Lloyd, J. Tian, T. Delaney, S. Manning, T.
Nguyen, T. Burwell, H. Schneider, J.A. Gonzalo, M. Gosselin, L.R.
Owen, C.E. Rudd, J.C. Gutierrez-Ramos, The CD28-related molecule
ICOS is required for effective T cell-dependent immune responses,
Immunity 13 (2000) 95–105.
[8] K. Ihara, S. Ahmed, F. Nakao, N. Kinukawa, R. Kuromaru, N.
Matsuura, I. Iwata, S. Nagafuchi, H. Kohno, K. Miyako, T. Hara,
Association studies of CTLA-4, CD28, and ICOS gene polymor-
phisms with type 1 diabetes in the Japanese population, Immunoge-
netics 53 (2001) 447–454.
[9] K.E. Haimila, J.A. Partanen, P.M. Holopainen, Genetic polymorphism
of the human ICOS gene, Immunogenetics 53 (2002) 1028–1032.
[10] A.D. Haaning Andersen, M. Lange, S.T. Lillevang, Allelic variation
of the inducible costimulator (ICOS) gene: detection of polymor-
phisms, analysis of the promoter region, and extended haplotype
estimation, Tissue Antigens 61 (2003) 276–285.
[11] K.C. Beier, A. Hutloff, A.M. Dittrich, C. Heuck, A. Rauch, K.
Buchner, B. Ludewig, H.D. Ochs, H.W. Mages, R.A. Kroczek,
Induction, binding specificity and function of human ICOS, Eur. J.
Immunol. 30 (2000) 3707–3717.
[12] A. Tafuri, A. Shahinian, F. Bladt, S.K. Yoshinaga, M. Jordana, A.
Wakeham, L.M. Boucher, D. Bouchard, V.S. Chan, G. Duncan, B.
Odermatt, A. Ho, A. Itie, T. Horan, J.S. Whoriskey, T. Pawson, J.M.
Penninger, P.S. Ohashi, T.W. Mak, ICOS is essential for effective
T-helper-cell responses, Nature 409 (2001) 105–109.
[13] C. Dong, U.A. Temann, R.A. Flavell, Cutting edge: critical role of
inducible costimulator in germinal center reactions, J. Immunol. 166
(2001) 3659–3662.
[14] S.C. Wong, E. Oh, C.H. Ng, K.P. Lam, Impaired germinal center
formation and recall T-cell-dependent immune responses in mice
lacking the costimulatory ligand B7-H2, Blood 102 (2003)
1381–1388.
[15] M.M. Swallow, J.J. Wallin, W.C. Sha, B7h, a novel costimulatory
homolog of B7.1 and B7.2, is induced by TNFalpha, Immunity 11
(1999) 423–432.
[16] V. Ling, P.W. Wu, H.F. Finnerty, K.M. Bean, V. Spaulding, L.A.
Fouser, J.P. Leonard, S.E. Hunter, R. Zollner, J.L. Thomas, J.S.
Miyashiro, K.A. Jacobs, M. Collins, Cutting edge: identification of
GL50, a novel B7-like protein that functionally binds to ICOS
receptor, J. Immunol. 164 (2000) 1653–1657.
[17] A. Aicher, M. Hayden-Ledbetter, W.A. Brady, A. Pezzutto, G.
Richter, D. Magaletti, S. Buckwalter, J.A. Ledbetter, E.A. Clark,
Characterization of human inducible costimulator ligand expression
and function, J. Immunol. 164 (2000) 4689–4696.
[18] V. Ling, P.W. Wu, J.S. Miyashiro, S. Marusic, H.F. Finnerty, M.
Collins, Differential expression of inducible costimulator-ligand splice
variants: lymphoid regulation of mouse GL50-B and human GL50
molecules, J. Immunol. 166 (2001) 7300–7308.
[19] L. Liang, E.M. Porter, W.C. Sha, Constitutive expression of the B7h
ligand for inducible costimulator on naive B cells is extinguished after
activation by distinct B cell receptor and interleukin 4 receptor-
mediated pathways and can be rescued by CD40 signaling, J. Exp.
Med. 196 (2002) 97–108.
[20] B. Grimbacher, A. Hutloff, M. Schlesier, E. Glocker, K. Warnatz, R.
Drager, H. Eibel, B. Fischer, A.A. Schaffer, H.W. Mages, R.A.
Kroczek, H.H. Peter, Homozygous loss of ICOS is associated with
adult-onset common variable immunodeficiency, Nat. Immunol. 4
(2003) 261–268.
[21] A. Soresina, V. Lougaris, S. Giliani, F. Cardinale, L. Armenio, M.
Cattalini, L.D. Notarangelo, A. Plebani, Mutations of the X-linked
lymphoproliferative disease gene SH2D1A mimicking common
variable immunodeficiency, Eur. J. Pediatr. 161 (2002) 656–659.
[22] H. Kanegane, S. Tsukada, T. Iwata, T. Futatani, K. Nomura, J.
Yamamoto, T. Yoshida, K. Agematsu, A. Komiyama, T. Miyawaki,
Detection of Bruton’s tyrosine kinase mutations in hypogammaglo-
bulinaemic males registered as common variable immunodeficiency
(CVID) in the Japanese Immunodeficiency Registry, Clin. Exp.
Immunol. 120 (2000) 512–517.
[23] L. Mouthon, P. Cohen, C. Larroche, M.H. Andre, I. Royer, P.
Casassus, L. Guillevin, Common variable immunodeficiency: one or
multiple illnesses? 3 clinical cases, Ann. Med. Interne 150 (1999)
275–282.
[24] J. Kralovicova, L. Hammarstroem, A. Plebani, D.B. Webster, I.
Vorechovsky, Fine scale mapping at IgAD1 and genome-wide linkage
analysis implicate HLA-DQ/DR as a major susceptibility locus in
selective IgA deficiency and common variable immunodeficiency, J.
Immunol. 170 (2003) 2765–2775.
[25] W.I. Lee, Q. Zhu, E. Gambine, Y. Jin, A.A. Welcher, H.D. Ochs,
Inducible CO-stimulator molecule, a candidate gene for defective
isotype switching, is normal in patients with hyper-IgM syndrome of
unknown molecular diagnosis, J. Allergy Clin. Immunol. 112 (2003)
958–964.




















