
Global gene expression of fission yeast in response to cisplatin
Upload
independentCategory
view
0download
0
HMGA1 protein expression sensitizes cells to cisplatin-induced cell death
Gustavo Baldassarre1,2, Barbara Belletti1,2, Sabrina Battista1,3, Milena S Nicoloso2,Francesca Pentimalli1,3, Monica Fedele3, Carlo M Croce1,4 and Alfredo Fusco*,3,5
1Kimmel Cancer Center, Jefferson Medical College, Philadelphia, PA 19107, USA; 2Divisione di Oncologia Sperimentale 2, Centro diRiferimento Oncologico, Aviano 33081, Italy; 3Dipartimento di Biologia e Patologia Cellulare e Molecolare c/o Centro diEndocrinologia ed Oncologia Sperimentale del CNR, Facolta di Medicina e Chirurgia, Universita degli Studi di Napoli ‘Federico II’,Via Pansini 5, 80131 Naples, Italy; 4Division of Human Cancer Genetics,Comprehensive Cancer Center, The Ohio State University,10 West 12th Avenue Columbus, OH 43210, USA; 5NOGEC (Naples Oncogenomic Center)-CEINGE, Centro di BiotecnologieAvanzate, via Comunale Margherita 482, 80145- Naples, Italy
HMGA1 proteins belong to a family of nonhistonechromatin proteins able to bind DNA in AT-rich regionsand to interact with various transcription factors thusenhancing or inhibiting gene transcription by acting asarchitectural proteins. Although their expression is verylow or absent in many adult tissues, HMGA1 proteinshave been frequently found to be upregulated in humancancers and are expressed at high levels during embryo-genesis, suggesting they could have a role in highlyproliferating cells. We have previously demonstrated thatHMGA1 expression in primary breast cancer andmammary carcinoma derived cell lines inversely corre-lated with BRCA1 expression and that HMGA1 is able todownregulate the expression of BRCA1 gene by bindingdirectly to its promoter region. Being BRCA1 proteinexpression strictly linked to the DNA repair activity of thecell, we investigated whether HMGA1 expression wasable to influence cellular responses to DNA damage.Here, we report that high expression levels of HMGA1proteins in MCF-7 or mouse embryonic stem cells resultsin diminished BRCA1 expression and enhanced sensitivityto Cisplatin and Bleomycin. The increased DNA damage-induced cell death in HMGA1-expressing cells is likelydue to a diminished cellular DNA repair activity. There-fore, we propose that high expression of HMGA1 proteinin human malignant neoplasias, acting on BRCA1expression, could contribute to the progression ofmalignant transformation influencing the response of thecells to the damaged DNA.Oncogene (2005) 24, 6809–6819. doi:10.1038/sj.onc.1208831;published online 20 June 2005
Keywords: HMGA1; BRCA1; knockout; ES cells;apoptosis; DNA damage
Introduction
HMGA1a and HMGA1b are structural nonhistonechromatin proteins encoded by a unique gene throughan alternative splicing mechanism. Both isoforms areable to bind DNA in AT-rich regions and interact withvarious transcription factors to enhance or inhibit genetranscription by acting as architectural proteins (Reevesand Beckerbauer, 2001). HMGA1 proteins are abun-dant during embryogenesis, but they are present only atlow levels in normal adult tissues (Chiappetta et al.,1996). Induction of HMGA1 expression occurs inseveral human malignant neoplasias including thyroid(Chiappetta et al., 1998), breast (Baldassarre et al.,2003), prostate (Tamimi et al., 1993), and ovary(Masciullo et al., 2003) carcinomas. Increased expres-sion of the HMGA1 gene has been demonstrated inmouse (Ram et al., 1993) and human (Liu et al., 1999)breast carcinoma derived cell lines, with a directcorrelation between HMGA1 protein levels and themetastatic phenotype of human breast cancer cell lines(Liu et al., 1999).
Inhibition of HMGA protein synthesis prevented theneoplastic transformation induced by the myeloproli-ferative sarcoma virus or by the Kirsten murine sarcomavirus (Berlingieri et al., 1995) and caused death inseveral carcinoma cell lines (Scala et al., 2000), suggest-ing that HMGA overexpression plays a key role in theinduction of the malignant phenotype.
We have previously reported that HMGA1b proteinbinds both in vitro and in vivo to the human and mouseBRCA1 promoters, thus inhibiting their activity, andthat HMGA1 overexpression results in the downregula-tion of BRCA1 mRNA and protein in human mammarycarcinoma cell lines as well as in mouse embryonic stem(ES) cells (Baldassarre et al., 2003). Moreover, acorrelation was found between HMGA1 protein levelsand BRCA1 mRNA expression both in primary breasttumors and breast cancer-derived cell lines, suggestingthat HMGA1 protein overexpression may have a role inthe BRCA1 downregulation observed in aggressivemammary carcinomas (Baldassarre et al., 2003). SinceBRCA1 protein is involved in DNA repair following
Received 14 September 2004; revised 6 May 2005; accepted 11 May 2005;published online 20 June 2005
*Correspondence: A Fusco, Dipartimento di Biologia e PatologiaCellulare e Molecolare c/o Centro di Endocrinologia ed OncologiaSperimentale del CNR, Facolta di Medicina e Chirurgia, Universitadegli Studi di Napoli ‘Federico II’, Via Pansini 5, 80131 Naples, Italy;E-mail: [email protected]
Oncogene (2005) 24, 6809–6819& 2005 Nature Publishing Group All rights reserved 0950-9232/05 $30.00
www.nature.com/onc

several types of DNA damage (Scully and Livingston,2000), we investigated whether there was a correlationbetween HMGA1 expression and cell sensitivity toDNA-damaging agents. It has been proposed that a linkexists between BRCA expression and cis-platinum(cisplatin) sensitivity since exposure to cisplatin deter-mines the upregulation of BRCA1 mRNA expression inmammary carcinoma cells (Andres et al., 1998) andBRCA1 protein levels positively correlate with cisplatin-resistance in breast and ovary carcinoma cells (Husainet al., 1998). More recently, the BRCA1-FANCONIanemia pathway function has been strictly correlatedwith the onset of cisplatin resistance in ovary carcino-mas (Taniguchi et al., 2003). Finally, Brca1 null mouseES cells are more sensitive to cisplatin as compared totheir normal counterpart (Bhattacharyya et al., 2000).Therefore, we investigated the role of HMGA1 proteinfollowing cisplatin-induced DNA damage, both in thebreast carcinoma cell line MCF-7 overexpressing theHMGA1b protein and in ES cells null for the Hmga1gene.
Using these experimental model systems, we demon-strate that HMGA1 expression induces an enhancedapoptosis rate and a decreased cell survival followingexposure to DNA-damaging agents, likely diminishingthe expression levels of BRCA1 protein.
Results
ES cells null for the Hmga1 gene are less sensitive tocisplatin with respect to the wild-type ES cells
Mouse ES cells express very high levels of HMGA1mRNA and protein (Battista et al., 2003), making theES cells knocked-out in both the Hmga1 gene alleles auseful tool to evaluate the role of HMGA1 proteins inseveral experimental conditions (Baldassarre et al., 2003;Battista et al., 2003). In mouse ES cells, deletion of boththe Brca1 alleles results in a fivefold higher sensitivity tothe DNA crosslinking agent cisplatin compared to thewild-type cells (Bhattacharyya et al., 2000). Therefore,we first used ES cells to investigate the role of HMGA1protein following cisplatin-induced DNA damage. Wild-type (WT) mouse ES cells exposed to 5, 10, 25 mMcisplatin or vehicle for 1 h in serum-free medium wereallowed to recover in complete medium for 2 h and,then, the expression of Brca1 and Hmga1 gene productswas evaluated by Western blot (Figure 1a and c), RT–PCR (Figure 1b) and Northern blot (Figure 1d).BRCA1 mRNA and protein levels increased abouttwofolds in cells treated with 25 mM cisplatin and theseincreases were accompanied by a significant reduction ofHMGA1 protein levels (Figure 1a). However, theHmga1 mRNA expression did not significantly change(Figure 1d), suggesting that post-transcriptional or post-translational modifications contributed to cisplatin-induced downregulation of HMGA1 protein levels. Asexpected, we observed a 2.5-fold increase of Brca1expression inHmga1�/�, with respect to parental ES cellsboth at protein (Figure 1c) and at RNA level
(Figure 1b). Cisplatin treatment did not affect Brca1gene expression in Hmga1 null ES cells (Figures 1b andc), suggesting a role for Hmga1 protein in the regulationof Brca1 expression following cisplatin treatment.
Subsequently, we evaluated the apoptotic rate ofcisplatin-treated ES cells by analysing the cleavage ofthe poly ADP-ribose polymerase protein (PARP), whichrepresents a critical molecular event when cells undergoapoptosis (Soldani and Scovassi, 2002). Exposure ofWT ES cells to 25 mM cisplatin resulted in the cleavageof PARP protein, as revealed by the presence of the85-kDa band corresponding to a cleaved form ofPARP, whereas only the major band of 112 kDa, corres-ponding to the uncleaved PARP protein, was detectedin Hmga1�/� ES cells after the same treatment(Figure 1e), suggesting that ES cells unable to expressthe HMGA protein are more resistant to cisplatin-induced apoptosis.
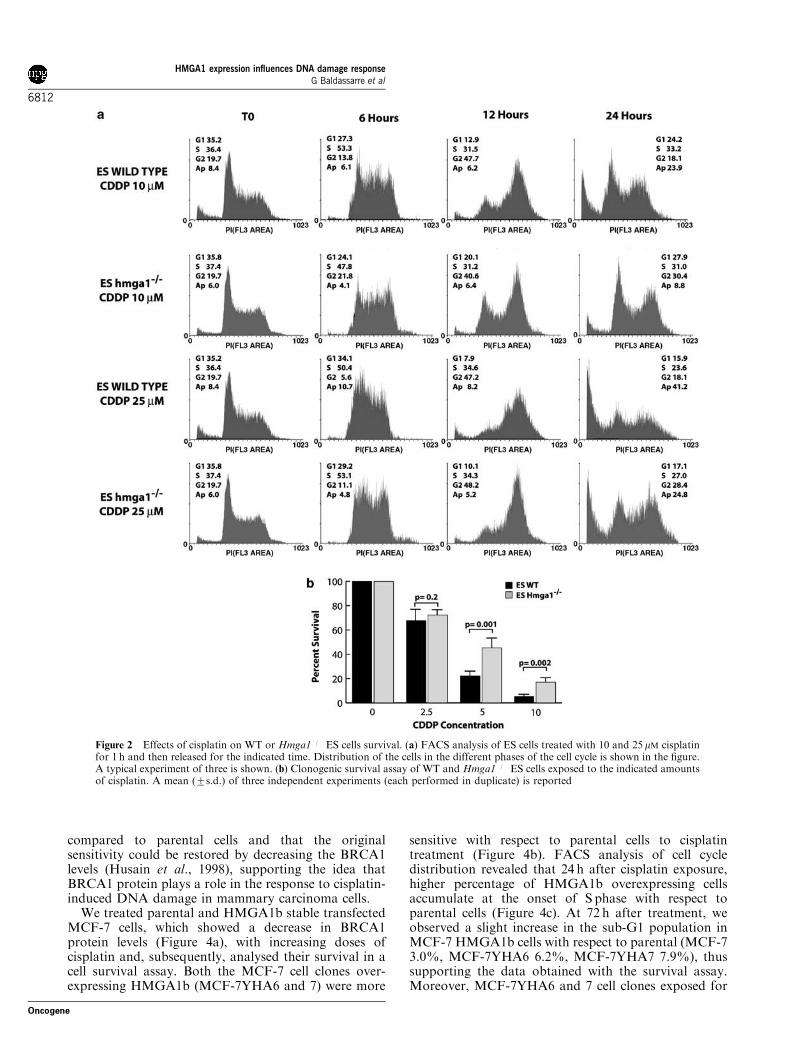
FACS analysis of DNA content of ES cells treatedwith cisplatin confirmed the different cisplatin sensitivitybetween WT and Hmga1�/� cells (Figure 2a). In thisexperiment, the ES cells were treated for 1 h with 10 or25 mM cisplatin and analysed after 6, 12 and 24 h. WT EScells accumulate in S phase 6 h after treatment and inG2/Mphase after 12 h. After 24 h, cells restart to cycleor undergo apoptosis as indicated by their accumulationin sub-G1 phase of the cell cycle. These effects weredose-dependent since the percentage of apoptotic cellswas 2475 and 4174% for 10 and 25 mM cisplatin,respectively. In contrast, although Hmga1�/� ES cellsaccumulate with similar extent in S and in G2/Mphaseat 6 and 24 h post-treatment, they failed to undergoapoptosis. In fact, only a small percentage of cells wasfound in the sub-G1 population after 48 h (873 and2476% for 10 and 25 mM, respectively), demonstratinga statistically significant lower presence of sub-G1 cellspopulation (Po0.01 for both 10 and 25mM treatmentswith respect to WT cells using the Student’s t-test).Next, WT and Hmga1�/� ES cells were treated withincreasing amount of cisplatin (2.5, 5, 10 and 25 mM),and their ability to survive this treatment was evaluated.Accordingly with FACS analysis, WT cells resultedtwofold more sensitive to cisplatin with respect to theHmga1 null cells (Figure 2b).
Hmga1�/� ES cells show a higher DNA integritycompared to the WT ES cells after cisplatin treatment
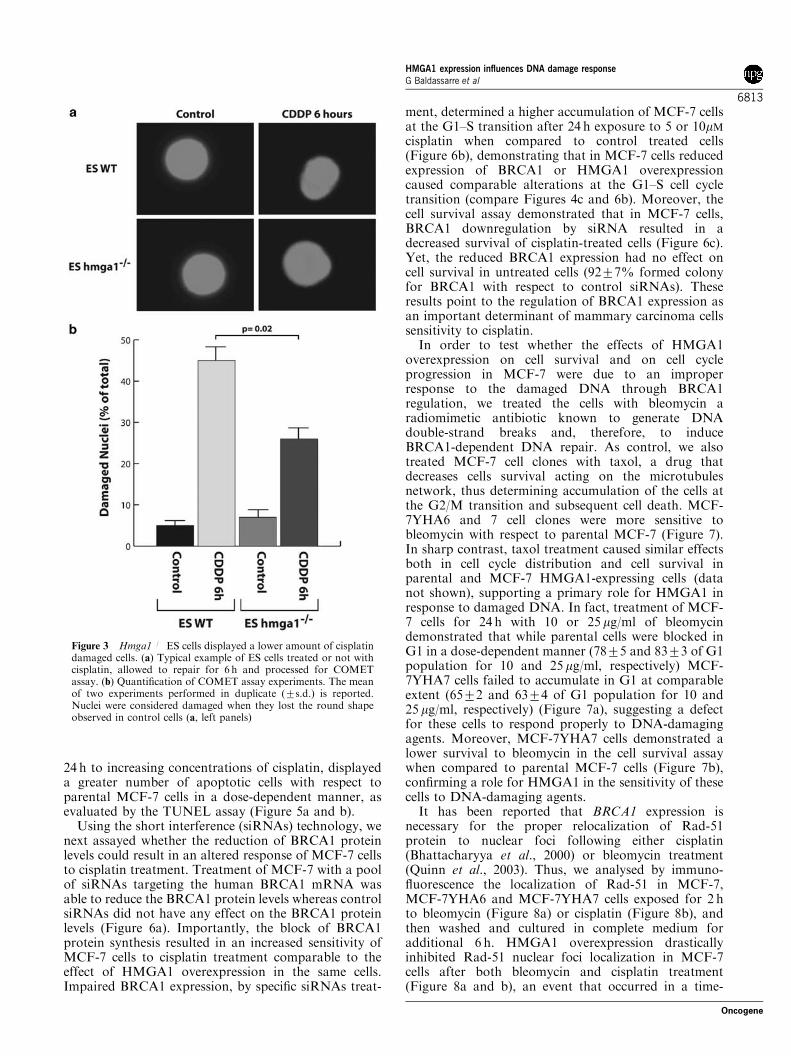
On the basis of these results, we investigated the extentof damaged DNA in Hmga1 WT and null cells using thesingle-cell gel electrophoresis COMET assay. The assayis based upon the ability of denatured cleaved DNAfragments to migrate out of the cell nucleus under theinfluence of an electric field, while the undamaged DNAremains within the nucleus. Thus, only undamagednuclei retain their round shape when visualized byfluorescence microscopy (Figure 3a). Although cisplatinforms DNA adducts and the formation of the tail is lessevident with respect to other treatments like cellularirradiation, this assay has been successfully used toidentify cisplatin damaged cells (Buschfort-Papewalis
HMGA1 expression influences DNA damage responseG Baldassarre et al
6810
Oncogene

et al., 2002). To quantify the DNA damage induced bycisplatin in this system, we evaluated the percentage ofdamaged cells identified by the loss of the round-shapenuclear morphology (Figure 3a). Cells treated or notwith 10 mM cisplatin were allowed to repair the DNA for6 h in complete medium and then processed for theCOMET assay. The statistical analysis clearly demon-strated a higher number of damaged cells in WT respectto Hmga1�/� cells (Figure 3b). Moreover, we observedthat the distance between the nucleoid and the migrating
DNA was consistently higher in WT with respect toHmga1 null ES cells suggesting the possibility thatqualitative differences in DNA repair activity existbetween the WT and Hmga1�/� ES cells.
MCF-7 cells overexpressing the HMGA1 protein aremore sensitive to cisplatin than the parental cells
It has been previously published that the MCF-7 cellsresistant to cisplatin displayed higher levels of BRCA1
Figure 1 Effects of cisplatin on BRCA1 and HMGA1 expression in ES cells. (a) Western blot analysis of BRCA1 (upper panel) andHMGA1 expression (middle panel) in mouse ES cells treated with the indicated amounts of cisplatin. Tubulin expression (lower panel)was used to normalize the amount of loaded proteins in each well. Data, expressed as fold change with respect to control, represent themean of two independent experiments and were obtained by densitometric scanning of the blots, normalized to tubulin levels. (b) RT–PCR analysis of Brca1 mRNA levels (upper panel) in ES cells WT or Hmga1�/� treated with 10 or 25 mM cisplatin. As internal controlof RNA status and quantity Gapdh expression was evaluated (lower panel). Data, expressed as fold change respect to control,represent the mean of two independent experiments and were obtained by densitometric scanning of the blots, normalized to Gapdhlevels. (c) Western blot analysis of BRCA1 (upper panel) and HMGA1 expression (middle panel) in WT or Hmga1�/� ES cells treatedwith 25mM cisplatin. Tubulin expression (lower panel) was used to normalize the amount of loaded proteins in each well. (d) Northernblot analysis of Hmga1 mRNA levels (upper panel) in ES cells WT or Hmga1�/� treated with 5, 10 or 25 mM cisplatin. As internalcontrol of RNA status and quantity 18 and 28S ribosomal RNAs were shown (lower panel). (e) Western blot analysis of total PARP(upper panel) and cleaved PARP expression (middle panel) in WT or Hmga1�/� ES cells treated with indicated amounts of cisplatin.cdc2 expression (lower panel) was used to normalize the amount of loaded proteins in each well
HMGA1 expression influences DNA damage responseG Baldassarre et al
6811
Oncogene
compared to parental cells and that the originalsensitivity could be restored by decreasing the BRCA1levels (Husain et al., 1998), supporting the idea thatBRCA1 protein plays a role in the response to cisplatin-induced DNA damage in mammary carcinoma cells.
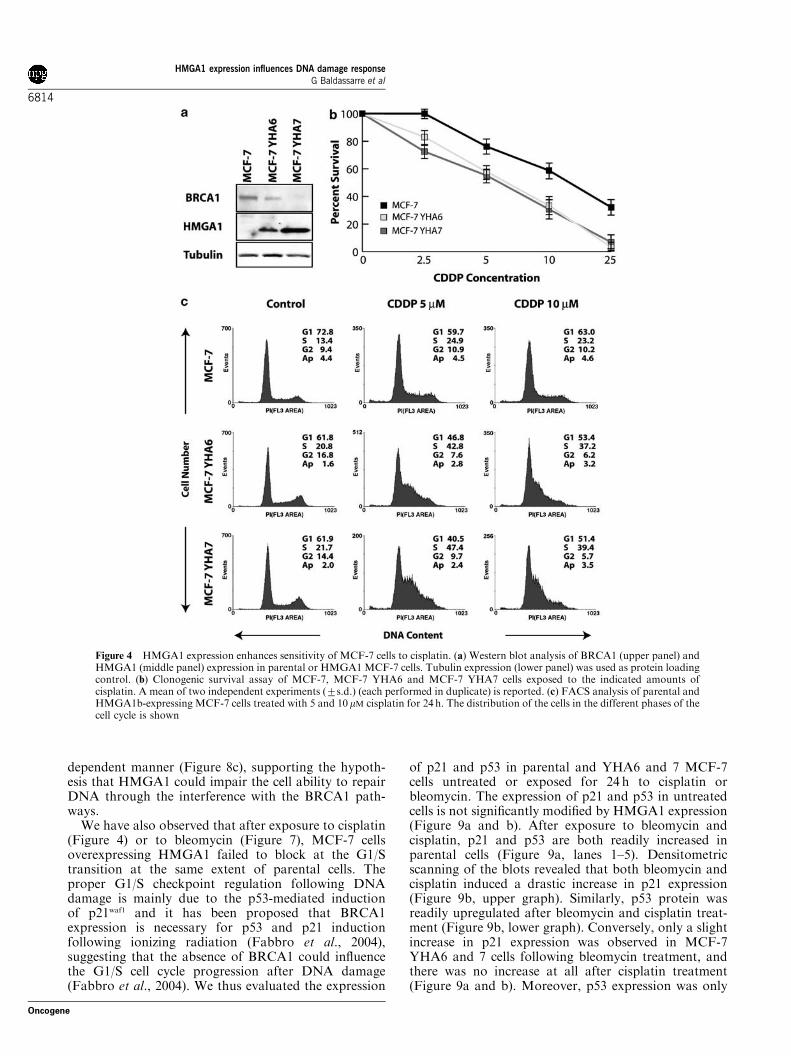
We treated parental and HMGA1b stable transfectedMCF-7 cells, which showed a decrease in BRCA1protein levels (Figure 4a), with increasing doses ofcisplatin and, subsequently, analysed their survival in acell survival assay. Both the MCF-7 cell clones over-expressing HMGA1b (MCF-7YHA6 and 7) were more
sensitive with respect to parental cells to cisplatintreatment (Figure 4b). FACS analysis of cell cycledistribution revealed that 24 h after cisplatin exposure,higher percentage of HMGA1b overexpressing cellsaccumulate at the onset of S phase with respect toparental cells (Figure 4c). At 72 h after treatment, weobserved a slight increase in the sub-G1 population inMCF-7 HMGA1b cells with respect to parental (MCF-73.0%, MCF-7YHA6 6.2%, MCF-7YHA7 7.9%), thussupporting the data obtained with the survival assay.Moreover, MCF-7YHA6 and 7 cell clones exposed for
Figure 2 Effects of cisplatin on WT or Hmga1�/� ES cells survival. (a) FACS analysis of ES cells treated with 10 and 25 mM cisplatinfor 1 h and then released for the indicated time. Distribution of the cells in the different phases of the cell cycle is shown in the figure.A typical experiment of three is shown. (b) Clonogenic survival assay of WT and Hmga1�/� ES cells exposed to the indicated amountsof cisplatin. A mean (7s.d.) of three independent experiments (each performed in duplicate) is reported
HMGA1 expression influences DNA damage responseG Baldassarre et al
6812
Oncogene
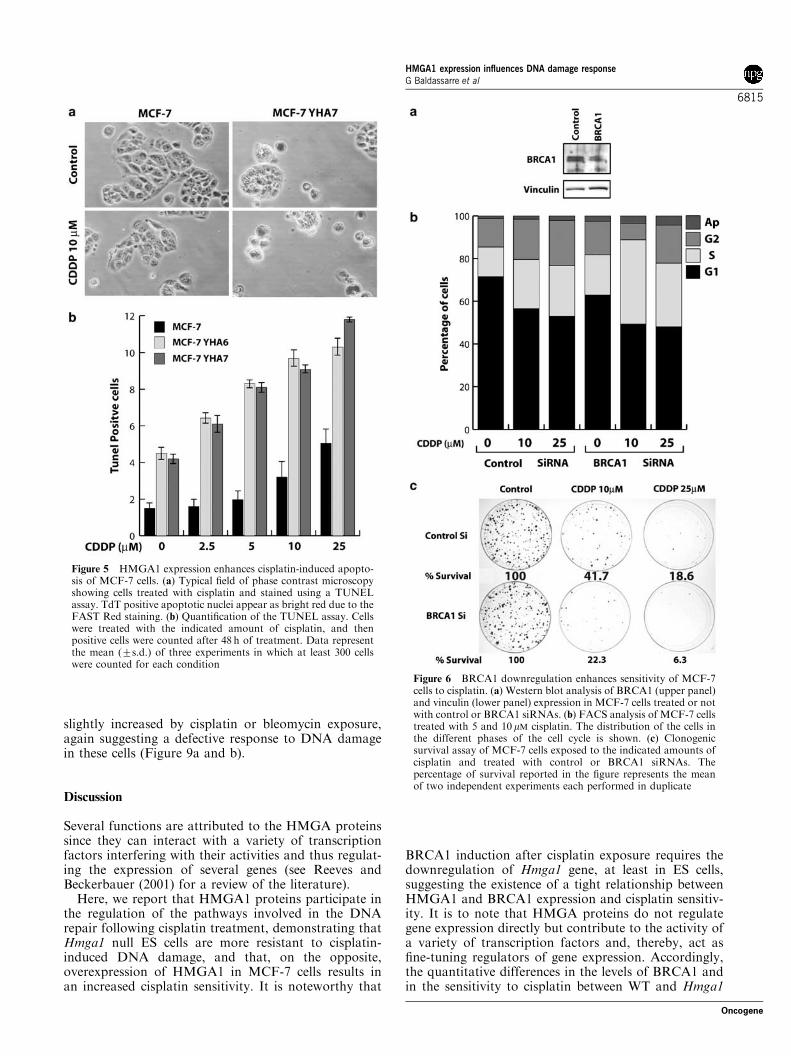
24 h to increasing concentrations of cisplatin, displayeda greater number of apoptotic cells with respect toparental MCF-7 cells in a dose-dependent manner, asevaluated by the TUNEL assay (Figure 5a and b).
Using the short interference (siRNAs) technology, wenext assayed whether the reduction of BRCA1 proteinlevels could result in an altered response of MCF-7 cellsto cisplatin treatment. Treatment of MCF-7 with a poolof siRNAs targeting the human BRCA1 mRNA wasable to reduce the BRCA1 protein levels whereas controlsiRNAs did not have any effect on the BRCA1 proteinlevels (Figure 6a). Importantly, the block of BRCA1protein synthesis resulted in an increased sensitivity ofMCF-7 cells to cisplatin treatment comparable to theeffect of HMGA1 overexpression in the same cells.Impaired BRCA1 expression, by specific siRNAs treat-
ment, determined a higher accumulation of MCF-7 cellsat the G1–S transition after 24 h exposure to 5 or 10mMcisplatin when compared to control treated cells(Figure 6b), demonstrating that in MCF-7 cells reducedexpression of BRCA1 or HMGA1 overexpressioncaused comparable alterations at the G1–S cell cycletransition (compare Figures 4c and 6b). Moreover, thecell survival assay demonstrated that in MCF-7 cells,BRCA1 downregulation by siRNA resulted in adecreased survival of cisplatin-treated cells (Figure 6c).Yet, the reduced BRCA1 expression had no effect oncell survival in untreated cells (9277% formed colonyfor BRCA1 with respect to control siRNAs). Theseresults point to the regulation of BRCA1 expression asan important determinant of mammary carcinoma cellssensitivity to cisplatin.
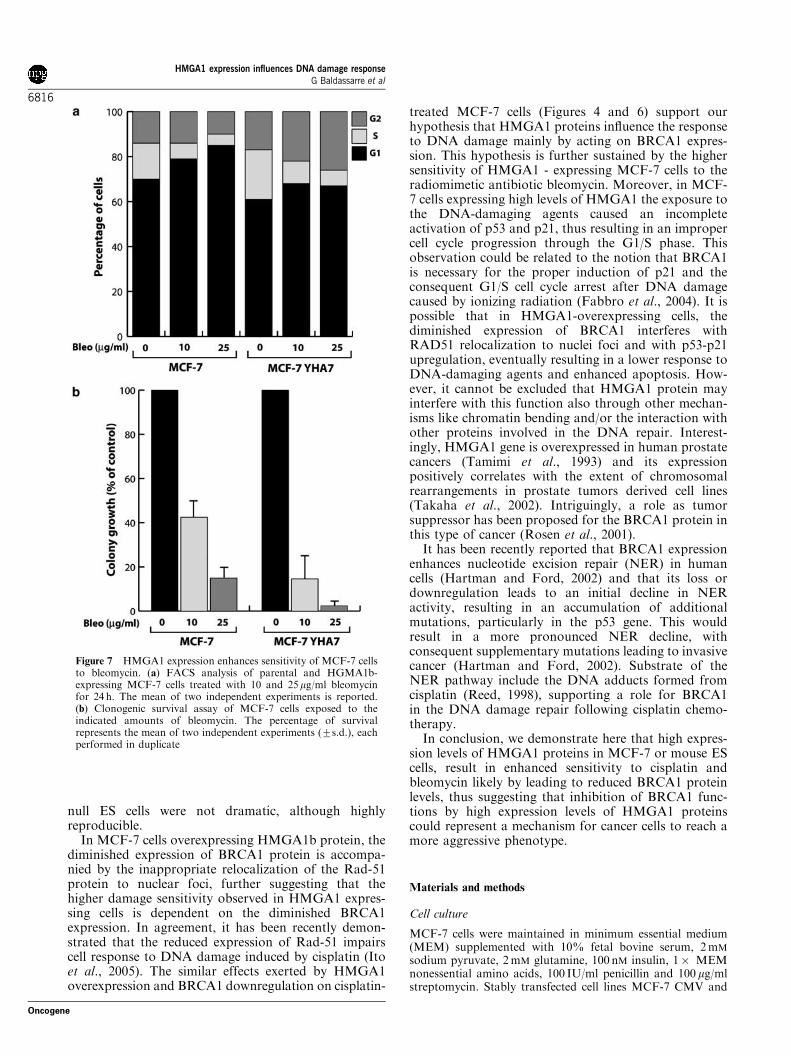
In order to test whether the effects of HMGA1overexpression on cell survival and on cell cycleprogression in MCF-7 were due to an improperresponse to the damaged DNA through BRCA1regulation, we treated the cells with bleomycin aradiomimetic antibiotic known to generate DNAdouble-strand breaks and, therefore, to induceBRCA1-dependent DNA repair. As control, we alsotreated MCF-7 cell clones with taxol, a drug thatdecreases cells survival acting on the microtubulesnetwork, thus determining accumulation of the cells atthe G2/M transition and subsequent cell death. MCF-7YHA6 and 7 cell clones were more sensitive tobleomycin with respect to parental MCF-7 (Figure 7).In sharp contrast, taxol treatment caused similar effectsboth in cell cycle distribution and cell survival inparental and MCF-7 HMGA1-expressing cells (datanot shown), supporting a primary role for HMGA1 inresponse to damaged DNA. In fact, treatment of MCF-7 cells for 24 h with 10 or 25 mg/ml of bleomycindemonstrated that while parental cells were blocked inG1 in a dose-dependent manner (7875 and 8373 of G1population for 10 and 25 mg/ml, respectively) MCF-7YHA7 cells failed to accumulate in G1 at comparableextent (6572 and 6374 of G1 population for 10 and25 mg/ml, respectively) (Figure 7a), suggesting a defectfor these cells to respond properly to DNA-damagingagents. Moreover, MCF-7YHA7 cells demonstrated alower survival to bleomycin in the cell survival assaywhen compared to parental MCF-7 cells (Figure 7b),confirming a role for HMGA1 in the sensitivity of thesecells to DNA-damaging agents.
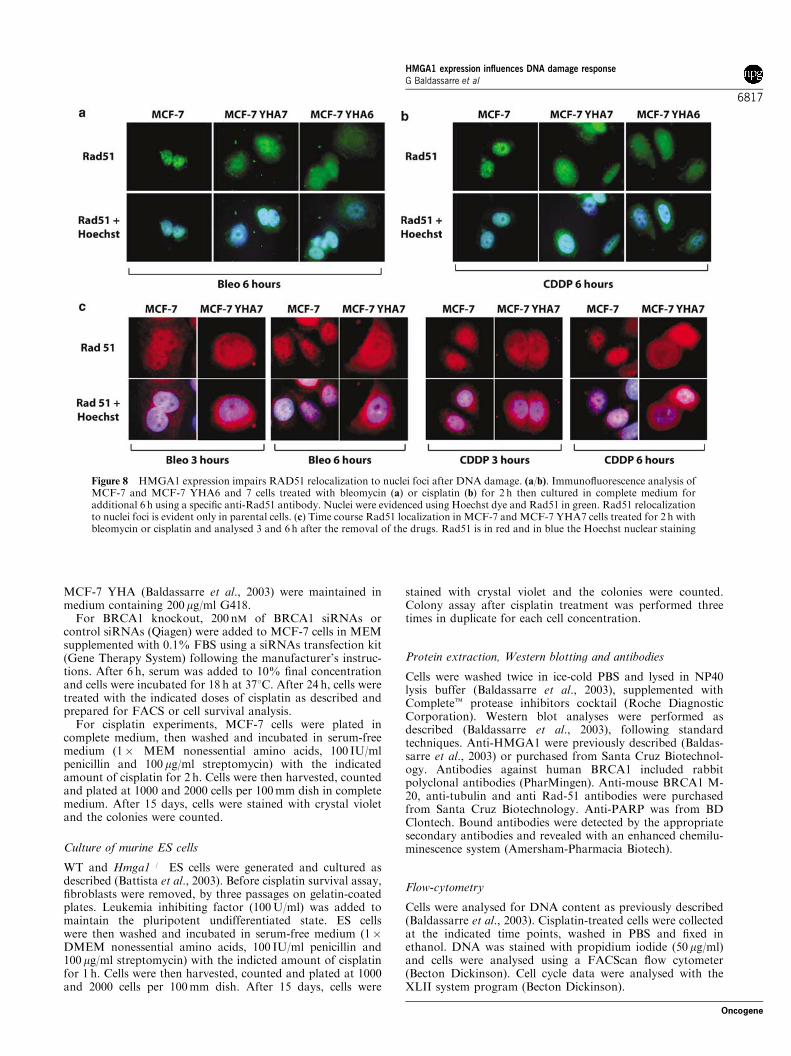
It has been reported that BRCA1 expression isnecessary for the proper relocalization of Rad-51protein to nuclear foci following either cisplatin(Bhattacharyya et al., 2000) or bleomycin treatment(Quinn et al., 2003). Thus, we analysed by immuno-fluorescence the localization of Rad-51 in MCF-7,MCF-7YHA6 and MCF-7YHA7 cells exposed for 2 hto bleomycin (Figure 8a) or cisplatin (Figure 8b), andthen washed and cultured in complete medium foradditional 6 h. HMGA1 overexpression drasticallyinhibited Rad-51 nuclear foci localization in MCF-7cells after both bleomycin and cisplatin treatment(Figure 8a and b), an event that occurred in a time-
Figure 3 Hmga1�/� ES cells displayed a lower amount of cisplatindamaged cells. (a) Typical example of ES cells treated or not withcisplatin, allowed to repair for 6 h and processed for COMETassay. (b) Quantification of COMET assay experiments. The meanof two experiments performed in duplicate (7s.d.) is reported.Nuclei were considered damaged when they lost the round shapeobserved in control cells (a, left panels)
HMGA1 expression influences DNA damage responseG Baldassarre et al
6813
Oncogene
dependent manner (Figure 8c), supporting the hypoth-esis that HMGA1 could impair the cell ability to repairDNA through the interference with the BRCA1 path-ways.
We have also observed that after exposure to cisplatin(Figure 4) or to bleomycin (Figure 7), MCF-7 cellsoverexpressing HMGA1 failed to block at the G1/Stransition at the same extent of parental cells. Theproper G1/S checkpoint regulation following DNAdamage is mainly due to the p53-mediated inductionof p21waf1 and it has been proposed that BRCA1expression is necessary for p53 and p21 inductionfollowing ionizing radiation (Fabbro et al., 2004),suggesting that the absence of BRCA1 could influencethe G1/S cell cycle progression after DNA damage(Fabbro et al., 2004). We thus evaluated the expression
of p21 and p53 in parental and YHA6 and 7 MCF-7cells untreated or exposed for 24 h to cisplatin orbleomycin. The expression of p21 and p53 in untreatedcells is not significantly modified by HMGA1 expression(Figure 9a and b). After exposure to bleomycin andcisplatin, p21 and p53 are both readily increased inparental cells (Figure 9a, lanes 1–5). Densitometricscanning of the blots revealed that both bleomycin andcisplatin induced a drastic increase in p21 expression(Figure 9b, upper graph). Similarly, p53 protein wasreadily upregulated after bleomycin and cisplatin treat-ment (Figure 9b, lower graph). Conversely, only a slightincrease in p21 expression was observed in MCF-7YHA6 and 7 cells following bleomycin treatment, andthere was no increase at all after cisplatin treatment(Figure 9a and b). Moreover, p53 expression was only
Figure 4 HMGA1 expression enhances sensitivity of MCF-7 cells to cisplatin. (a) Western blot analysis of BRCA1 (upper panel) andHMGA1 (middle panel) expression in parental or HMGA1MCF-7 cells. Tubulin expression (lower panel) was used as protein loadingcontrol. (b) Clonogenic survival assay of MCF-7, MCF-7 YHA6 and MCF-7 YHA7 cells exposed to the indicated amounts ofcisplatin. A mean of two independent experiments (7s.d.) (each performed in duplicate) is reported. (c) FACS analysis of parental andHMGA1b-expressing MCF-7 cells treated with 5 and 10 mM cisplatin for 24 h. The distribution of the cells in the different phases of thecell cycle is shown
HMGA1 expression influences DNA damage responseG Baldassarre et al
6814
Oncogene
slightly increased by cisplatin or bleomycin exposure,again suggesting a defective response to DNA damagein these cells (Figure 9a and b).
Discussion
Several functions are attributed to the HMGA proteinssince they can interact with a variety of transcriptionfactors interfering with their activities and thus regulat-ing the expression of several genes (see Reeves andBeckerbauer (2001) for a review of the literature).
Here, we report that HMGA1 proteins participate inthe regulation of the pathways involved in the DNArepair following cisplatin treatment, demonstrating thatHmga1 null ES cells are more resistant to cisplatin-induced DNA damage, and that, on the opposite,overexpression of HMGA1 in MCF-7 cells results inan increased cisplatin sensitivity. It is noteworthy that
BRCA1 induction after cisplatin exposure requires thedownregulation of Hmga1 gene, at least in ES cells,suggesting the existence of a tight relationship betweenHMGA1 and BRCA1 expression and cisplatin sensitiv-ity. It is to note that HMGA proteins do not regulategene expression directly but contribute to the activity ofa variety of transcription factors and, thereby, act asfine-tuning regulators of gene expression. Accordingly,the quantitative differences in the levels of BRCA1 andin the sensitivity to cisplatin between WT and Hmga1
Figure 5 HMGA1 expression enhances cisplatin-induced apopto-sis of MCF-7 cells. (a) Typical field of phase contrast microscopyshowing cells treated with cisplatin and stained using a TUNELassay. TdT positive apoptotic nuclei appear as bright red due to theFAST Red staining. (b) Quantification of the TUNEL assay. Cellswere treated with the indicated amount of cisplatin, and thenpositive cells were counted after 48 h of treatment. Data representthe mean (7s.d.) of three experiments in which at least 300 cellswere counted for each condition
Figure 6 BRCA1 downregulation enhances sensitivity of MCF-7cells to cisplatin. (a) Western blot analysis of BRCA1 (upper panel)and vinculin (lower panel) expression in MCF-7 cells treated or notwith control or BRCA1 siRNAs. (b) FACS analysis of MCF-7 cellstreated with 5 and 10 mM cisplatin. The distribution of the cells inthe different phases of the cell cycle is shown. (c) Clonogenicsurvival assay of MCF-7 cells exposed to the indicated amounts ofcisplatin and treated with control or BRCA1 siRNAs. Thepercentage of survival reported in the figure represents the meanof two independent experiments each performed in duplicate
HMGA1 expression influences DNA damage responseG Baldassarre et al
6815
Oncogene
null ES cells were not dramatic, although highlyreproducible.
In MCF-7 cells overexpressing HMGA1b protein, thediminished expression of BRCA1 protein is accompa-nied by the inappropriate relocalization of the Rad-51protein to nuclear foci, further suggesting that thehigher damage sensitivity observed in HMGA1 expres-sing cells is dependent on the diminished BRCA1expression. In agreement, it has been recently demon-strated that the reduced expression of Rad-51 impairscell response to DNA damage induced by cisplatin (Itoet al., 2005). The similar effects exerted by HMGA1overexpression and BRCA1 downregulation on cisplatin-
treated MCF-7 cells (Figures 4 and 6) support ourhypothesis that HMGA1 proteins influence the responseto DNA damage mainly by acting on BRCA1 expres-sion. This hypothesis is further sustained by the highersensitivity of HMGA1 - expressing MCF-7 cells to theradiomimetic antibiotic bleomycin. Moreover, in MCF-7 cells expressing high levels of HMGA1 the exposure tothe DNA-damaging agents caused an incompleteactivation of p53 and p21, thus resulting in an impropercell cycle progression through the G1/S phase. Thisobservation could be related to the notion that BRCA1is necessary for the proper induction of p21 and theconsequent G1/S cell cycle arrest after DNA damagecaused by ionizing radiation (Fabbro et al., 2004). It ispossible that in HMGA1-overexpressing cells, thediminished expression of BRCA1 interferes withRAD51 relocalization to nuclei foci and with p53-p21upregulation, eventually resulting in a lower response toDNA-damaging agents and enhanced apoptosis. How-ever, it cannot be excluded that HMGA1 protein mayinterfere with this function also through other mechan-isms like chromatin bending and/or the interaction withother proteins involved in the DNA repair. Interest-ingly, HMGA1 gene is overexpressed in human prostatecancers (Tamimi et al., 1993) and its expressionpositively correlates with the extent of chromosomalrearrangements in prostate tumors derived cell lines(Takaha et al., 2002). Intriguingly, a role as tumorsuppressor has been proposed for the BRCA1 protein inthis type of cancer (Rosen et al., 2001).
It has been recently reported that BRCA1 expressionenhances nucleotide excision repair (NER) in humancells (Hartman and Ford, 2002) and that its loss ordownregulation leads to an initial decline in NERactivity, resulting in an accumulation of additionalmutations, particularly in the p53 gene. This wouldresult in a more pronounced NER decline, withconsequent supplementary mutations leading to invasivecancer (Hartman and Ford, 2002). Substrate of theNER pathway include the DNA adducts formed fromcisplatin (Reed, 1998), supporting a role for BRCA1in the DNA damage repair following cisplatin chemo-therapy.
In conclusion, we demonstrate here that high expres-sion levels of HMGA1 proteins in MCF-7 or mouse EScells, result in enhanced sensitivity to cisplatin andbleomycin likely by leading to reduced BRCA1 proteinlevels, thus suggesting that inhibition of BRCA1 func-tions by high expression levels of HMGA1 proteinscould represent a mechanism for cancer cells to reach amore aggressive phenotype.
Materials and methods
Cell culture
MCF-7 cells were maintained in minimum essential medium(MEM) supplemented with 10% fetal bovine serum, 2mM
sodium pyruvate, 2mM glutamine, 100 nM insulin, 1� MEMnonessential amino acids, 100 IU/ml penicillin and 100 mg/mlstreptomycin. Stably transfected cell lines MCF-7 CMV and
Figure 7 HMGA1 expression enhances sensitivity of MCF-7 cellsto bleomycin. (a) FACS analysis of parental and HGMA1b-expressing MCF-7 cells treated with 10 and 25mg/ml bleomycinfor 24 h. The mean of two independent experiments is reported.(b) Clonogenic survival assay of MCF-7 cells exposed to theindicated amounts of bleomycin. The percentage of survivalrepresents the mean of two independent experiments (7s.d.), eachperformed in duplicate
HMGA1 expression influences DNA damage responseG Baldassarre et al
6816
Oncogene
MCF-7 YHA (Baldassarre et al., 2003) were maintained inmedium containing 200mg/ml G418.For BRCA1 knockout, 200 nM of BRCA1 siRNAs or
control siRNAs (Qiagen) were added to MCF-7 cells in MEMsupplemented with 0.1% FBS using a siRNAs transfection kit(Gene Therapy System) following the manufacturer’s instruc-tions. After 6 h, serum was added to 10% final concentrationand cells were incubated for 18 h at 371C. After 24 h, cells weretreated with the indicated doses of cisplatin as described andprepared for FACS or cell survival analysis.For cisplatin experiments, MCF-7 cells were plated in
complete medium, then washed and incubated in serum-freemedium (1� MEM nonessential amino acids, 100 IU/mlpenicillin and 100 mg/ml streptomycin) with the indicatedamount of cisplatin for 2 h. Cells were then harvested, countedand plated at 1000 and 2000 cells per 100mm dish in completemedium. After 15 days, cells were stained with crystal violetand the colonies were counted.
Culture of murine ES cells
WT and Hmga1�/� ES cells were generated and cultured asdescribed (Battista et al., 2003). Before cisplatin survival assay,fibroblasts were removed, by three passages on gelatin-coatedplates. Leukemia inhibiting factor (100U/ml) was added tomaintain the pluripotent undifferentiated state. ES cellswere then washed and incubated in serum-free medium (1�DMEM nonessential amino acids, 100 IU/ml penicillin and100mg/ml streptomycin) with the indicted amount of cisplatinfor 1 h. Cells were then harvested, counted and plated at 1000and 2000 cells per 100mm dish. After 15 days, cells were
stained with crystal violet and the colonies were counted.Colony assay after cisplatin treatment was performed threetimes in duplicate for each cell concentration.
Protein extraction, Western blotting and antibodies
Cells were washed twice in ice-cold PBS and lysed in NP40lysis buffer (Baldassarre et al., 2003), supplemented withCompletet protease inhibitors cocktail (Roche DiagnosticCorporation). Western blot analyses were performed asdescribed (Baldassarre et al., 2003), following standardtechniques. Anti-HMGA1 were previously described (Baldas-sarre et al., 2003) or purchased from Santa Cruz Biotechnol-ogy. Antibodies against human BRCA1 included rabbitpolyclonal antibodies (PharMingen). Anti-mouse BRCA1 M-20, anti-tubulin and anti Rad-51 antibodies were purchasedfrom Santa Cruz Biotechnology. Anti-PARP was from BDClontech. Bound antibodies were detected by the appropriatesecondary antibodies and revealed with an enhanced chemilu-minescence system (Amersham-Pharmacia Biotech).
Flow-cytometry
Cells were analysed for DNA content as previously described(Baldassarre et al., 2003). Cisplatin-treated cells were collectedat the indicated time points, washed in PBS and fixed inethanol. DNA was stained with propidium iodide (50 mg/ml)and cells were analysed using a FACScan flow cytometer(Becton Dickinson). Cell cycle data were analysed with theXLII system program (Becton Dickinson).
Figure 8 HMGA1 expression impairs RAD51 relocalization to nuclei foci after DNA damage. (a/b). Immunofluorescence analysis ofMCF-7 and MCF-7 YHA6 and 7 cells treated with bleomycin (a) or cisplatin (b) for 2 h then cultured in complete medium foradditional 6 h using a specific anti-Rad51 antibody. Nuclei were evidenced using Hoechst dye and Rad51 in green. Rad51 relocalizationto nuclei foci is evident only in parental cells. (c) Time course Rad51 localization in MCF-7 and MCF-7 YHA7 cells treated for 2 h withbleomycin or cisplatin and analysed 3 and 6 h after the removal of the drugs. Rad51 is in red and in blue the Hoechst nuclear staining
HMGA1 expression influences DNA damage responseG Baldassarre et al
6817
Oncogene

TUNEL assay
Tunel assay was performed essentially as previously described(Baldassarre et al., 1999). Briefly, MCF-7 cells were treated
with increasing doses of cisplatin and after 72 h fixed in 3%paraformaldehyde (PFA) and permeabilized with 0.2% TritonX-100 in PBS. After blocking of nonspecific sites with 0.1%BSA in PBS for 18 h at 41C, cells were stained using the ‘CellDeath Detection Kit AP’ (Roche) following manufacturer’sinstructions.
Comet assay
Cells treated or not with 10 mM cisplatin were allowed to repairthe DNA for 6 h in complete medium and then processed forthe COMET assay (Trevigen). Assay was performed followingmanufacturer’s instructions. Briefly, untreated or cisplatin-treated cells were suspended in low melting point agarose at adensity of 1� 105/ml at 421C and immediately 75 ml werepipetted onto a CometSlidet. Slides were incubated at 41C forat least 15min, immersed in prechilled lysis solution for 60minand then in alkali solution for 60min in the dark. After twowashes in TBE 1� , slides were transferred in a horizontalelectrophoresis apparatus, covered with TBE 1� buffer andrun for 10min at 1V/cm. Slides were then incubated in ethanolfor 5min, air-dried, stained with SYBRt Green and analysedwith epifluorescence microscopy. Quantification was per-formed by counting 50 cells per slide.
Immunofluorescence analysis
MCF-7 cells were fixed in 3% PFA and then processed forimmunofluorescence as previously described (Baldassarreet al., 1999). The anti-Rad51 antibody was diluted 1 : 100and the anti-rabbit Texas-Red or FITC-conjugated (JacksonImmunoresearch) was diluted 1 : 500. Hoechst (Sigma) stainingwas used to identify cell nuclei.
Acknowledgements
B Belletti is supported by an EMBO restart fellowship.Supported by Grants P01CA76259 and P30CA56036 fromthe National Cancer Institute to CM Croce and by grants fromthe Associazione Italiana Ricerca sul Cancro (AIRC) to AFusco and G Baldassarre.
References
Andres JL, Fan S, Turkel GJ, Wang JA, Twu NF, Yuan RQ,Lamszus K, Goldberg ID and Rosen EM. (1998). Oncogene,16, 2229–2241.
Baldassarre G, Barone MV, Belletti B, Sandomenico C,Bruni P, Spiezia S, Boccia A, Vento MT, Romano A, PepeS, Fusco A and Viglietto G. (1999). Oncogene, 18,6241–6251.
Baldassarre G, Battista S, Belletti B, Thakur S, Pentimalli F,Trapasso F, Fedele M, Pierantoni G, Croce CM and FuscoA. (2003). Mol. Cell. Biol., 23, 2225–2238.
Battista S, Pentimalli F, Baldassarre G, Fedele M, Fidanza V,Croce CM and Fusco A. (2003). FASEB J., 17, 1496–1498.
Berlingieri MT, Manfioletti G, Santoro M, Bandiera A,Visconti R, Giancotti V and Fusco A. (1995). Mol. Cell.Biol., 15, 1545–1553.
Bhattacharyya A, Ear US, Koller BH, Weichselbaum RR andBishop DK. (2000). J. Biol. Chem., 275, 23899–23903.
Buschfort-Papewalis C, Moritz T, Liedert B and Thomale J.(2002). Blood, 100, 845–853.
Chiappetta G, Avantaggiato V, Visconti R, Fedele M, BattistaS, Trapasso F, Merciai BM, Fidanza V, Giancotti V,
Santoro M, Simeone A and Fusco A. (1996). Oncogene,13, 2439–2446.
Chiappetta G, Tallini G, De Biasio MC, Manfioletti G,Martinez-Tello FJ, Pentimalli F, de Nigris F, Mastro A,Botti G, Fedele M, Berger N, Santoro M, Giancotti V andFusco A. (1998). Cancer Res., 58, 4193–4198.
Fabbro M, Savage K, Hobson K, Deans AJ, Powell SN,McArthur GA and Khanna KK. (2004). J. Biol. Chem., 279,31251–31258.
Hartman AR and Ford JM. (2002). Nat. Genet., 32,180–184.
Husain A, He G, Venkatraman ES and Spriggs DR. (1998).Cancer Res., 58, 1120–1123.
Ito M, Yamamoto S, Nimura K, Hiraoka K, Tamai K andKaneda Y. (2005). J. Gene Med. Mar 8; [E-pub aheadof print].
Liu WM, Guerra-Vladusic FK, Kurakata S, Lupu Rand Kohwi-Shigematsu T. (1999). Cancer Res., 59,
5695–5703.Masciullo V, Baldassarre G, Pentimalli F, Berlingieri MT,Boccia A, Chiappetta G, Palazzo J, Manfioletti G, Giancotti
Figure 9 HMGA1 expression impairs p53 and p21waf1 upregula-tion following DNA damage. (a) Western blot analysis of p21(upper panel), p53 (middle panel) and vinculin (lower panel, usedas loading control) expression in MCF-7 and MCF-7 YHA6 and 7cells untreated (lanes 1, 6 and 11) or treated with the indicatedamounts of bleomycin (lanes 2, 3, 7, 8, 12 and 13) or cisplatin (lanes4, 5, 9, 10, 14 and 15). (b) Densitometric analysis of the blots,representing the mean of two independent experiments. Data areexpressed as protein fold induction with respect to the control(untreated parental MCF-7 was arbitrarily set as ‘1’)
HMGA1 expression influences DNA damage responseG Baldassarre et al
6818
Oncogene

V, Viglietto G, Scambia G and Fusco A. (2003). Carcino-genesis, 24, 1191–1198.
Quinn JE, Kennedy RD, Mullan PB, Gilmore PM, CartyM, Johnston PG and Harkin DP. (2003). Cancer Res., 63,6221–6228.
Ram TG, Reeves R and Hosick HL. (1993). Cancer Res., 53,2655–2660.
Reed E. (1998). Cancer Treat. Rev., 24, 331–344.Reeves R and Beckerbauer L. (2001). Biochem. Biophys. Acta,
1519, 13–29.Rosen EM, Fan S and Goldberg ID. (2001). Cancer Invest., 19,396–412.
Scala S, Portella G, Fedele M, Chiappetta G and Fusco A.(2000). Proc. Natl. Acad. Sci. USA, 97, 4256–4261.
Scully R and Livingston DL. (2000). Nature, 408, 429–432.Soldani C and Scovassi AI. (2002). Apoptosis, 7, 321–328.Takaha N, Hawkins AL, Griffin CA, Isaacs WB and CoffeyDS. (2002). Cancer Res., 62, 647–651.
Tamimi Y, van der Poel HG, Denyn MM, Umbas R,Karthaus HF, Debruyne FM and Schalken JA. (1993).Cancer Res., 3, 5512–5516.
Taniguchi T, Tischkowitz M, Ameziane N, Hodgson SV,Mathew CG, Joenje H, Mok SC and D’Andrea AD. (2003).Nat. Med., 9, 568–574.
HMGA1 expression influences DNA damage responseG Baldassarre et al
6819
Oncogene
Copyright © 2022 FDOKUMEN




















