
Haploinsufficiency of the Hmga1 Gene Causes Cardiac Hypertrophy and Myelo-Lymphoproliferative...
9
2006;66:2536-2543. Cancer Res Monica Fedele, Vincenzo Fidanza, Sabrina Battista, et al. Mice Hypertrophy and Myelo-Lymphoproliferative Disorders in Gene Causes Cardiac Hmga1 Haploinsufficiency of the Updated version http://cancerres.aacrjournals.org/content/66/5/2536 Access the most recent version of this article at: Material Supplementary http://cancerres.aacrjournals.org/content/suppl/2006/03/16/66.5.2536.DC1.html Access the most recent supplemental material at: Cited Articles http://cancerres.aacrjournals.org/content/66/5/2536.full.html#ref-list-1 This article cites by 35 articles, 22 of which you can access for free at: Citing articles http://cancerres.aacrjournals.org/content/66/5/2536.full.html#related-urls This article has been cited by 12 HighWire-hosted articles. Access the articles at: E-mail alerts related to this article or journal. Sign up to receive free email-alerts Subscriptions Reprints and . [email protected] Department at To order reprints of this article or to subscribe to the journal, contact the AACR Publications Permissions . [email protected] Department at To request permission to re-use all or part of this article, contact the AACR Publications Research. on October 1, 2014. © 2006 American Association for Cancer cancerres.aacrjournals.org Downloaded from Research. on October 1, 2014. © 2006 American Association for Cancer cancerres.aacrjournals.org Downloaded from
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Haploinsufficiency of the Hmga1 Gene Causes Cardiac Hypertrophy and Myelo-Lymphoproliferative...

2006;66:2536-2543. Cancer Res Monica Fedele, Vincenzo Fidanza, Sabrina Battista, et al. MiceHypertrophy and Myelo-Lymphoproliferative Disorders in
Gene Causes CardiacHmga1Haploinsufficiency of the
Updated version
http://cancerres.aacrjournals.org/content/66/5/2536
Access the most recent version of this article at:
Material
Supplementary
http://cancerres.aacrjournals.org/content/suppl/2006/03/16/66.5.2536.DC1.html
Access the most recent supplemental material at:
Cited Articles
http://cancerres.aacrjournals.org/content/66/5/2536.full.html#ref-list-1
This article cites by 35 articles, 22 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/66/5/2536.full.html#related-urls
This article has been cited by 12 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
To request permission to re-use all or part of this article, contact the AACR Publications
Research. on October 1, 2014. © 2006 American Association for Cancercancerres.aacrjournals.org Downloaded from
Research. on October 1, 2014. © 2006 American Association for Cancercancerres.aacrjournals.org Downloaded from

Haploinsufficiency of the Hmga1 Gene Causes Cardiac Hypertrophy
and Myelo-Lymphoproliferative Disorders in Mice
Monica Fedele,1,8Vincenzo Fidanza,
8Sabrina Battista,
1,8Francesca Pentimalli,
1,8
Andres J.P. Klein-Szanto,9Rosa Visone,
1Ivana De Martino,
1Antonio Curcio,
6
Carmine Morisco,2Luigi Del Vecchio,
3Gustavo Baldassarre,
8Claudio Arra,
4
Giuseppe Viglietto,1Ciro Indolfi,
6Carlo M. Croce,
7and Alfredo Fusco
1,5
1Istituto di Endocrinologia ed Oncologia Sperimentale del Consiglio Nazionale delle Ricerche c/o Dipartimento di Biologia e PatologiaCellulare e Molecolare and 2Division of Cardiology, Universita di Napoli ‘‘Federico II’’; 3Servizio di Immunoematologia e MedicinaTrasfusionale, Ospedale A. Cardarelli; 4Istituto dei Tumori di Napoli; 5Naples Oncogenomic Center, CEINGE Biotecnologie Avanzateand European School of Molecular Medicine (Naples Site), Naples, Italy; 6Division of Cardiology, Universita di Catanzaro, Catanzaro,Italy; 7Division of Human Cancer Genetics, Comprehensive Cancer Center, The Ohio State University, Columbus, Ohio; 8KimmelCancer Center, Jefferson Medical College; 9Experimental Histopathology, Fox Chase Cancer Center,Philadelphia, Pennsylvania
Abstract
The HMGA1 protein is a major factor in chromatin architec-ture and gene control. It plays a critical role in neoplastictransformation. In fact, blockage of HMGA1 synthesis preventsrat thyroid cell transformation by murine transformingretroviruses, and an adenovirus carrying the HMGA1 gene inthe antisense orientation induces apoptotic cell death inanaplastic human thyroid carcinoma cell lines, but not innormal thyroid cells. Moreover, both in vitro and in vivostudies have established the oncogenic role of the HMGA1gene. In this study, to define HMGA1 function in vivo, weexamined the consequences of disrupting the Hmga1 gene inmice. Both heterozygous and homozygous mice for theHmga1-null allele show cardiac hypertrophy due to the directrole of HMGA1 on cardiomyocytic cell growth regulation.These mice also developed hematologic malignancies, includ-ing B cell lymphoma and myeloid granuloerythroblasticleukemia. The B cell expansion and the increased expressionof the RAG1/2 endonuclease, observed in HMGA1-knockoutspleen tissues, might be responsible for the high rate ofabnormal IgH rearrangements observed in these neoplasias.Therefore, the data reported here indicate the critical role ofHMGA1 in heart development and growth, and reveal anunsuspected antioncogenic potential for this gene in hemato-logic malignancies. (Cancer Res 2006; 66(5): 2536-43)
Introduction
The high-mobility group A (HMGA) protein family includesthree members: HMGA1a, HMGA1b (both coded for by the samegene through an alternative splicing), and HMGA2 (1). Theseproteins bind the minor groove of AT-rich DNA sequences throughthree short basic repeats, called ‘‘AT-hooks’’, located at the NH2-terminal region of the proteins. By interacting with the transcrip-tion machinery, HMGA1 proteins alter the chromatin structure andthereby regulate the transcriptional activity of several genes either
enhancing or suppressing the ability of the more usual transcrip-tional activators and repressors (2). The HMGA proteins seem to beinvolved in embryonic development (3), adipocytic cell growth, anddifferentiation. In fact, the disruption of the murine Hmga2 geneinduces a pygmy phenotype associated with a drastic reduction offat tissue (4), whereas transgenic mice overexpressing the Hmga2gene are giant and obese (5). Conversely, HMGA1 has an inhibitoryeffect on adipocytic cell growth and is required for differentiation(6). Both genes have a critical role in the process of carcinogenesisbecause they are overexpressed in most human malignantneoplasias (7), and the blockage of their expression has beenshown to prevent thyroid cell transformation and lead malignantcells to death (8, 9). Moreover, in vitro and in vivo studies showedthe oncogenic activity of overexpressed HMGA proteins. In fact,either HMGA1 or HMGA2 overexpression is able to transformmouse and rat fibroblasts (10, 11), and transgenic mice over-expressing either HMGA1 or HMGA2 develop natural killer T-celllymphomas and pituitary adenomas (12–15). To define thephysiologic role of the HMGA1 protein in vivo , we examined theconsequences of disrupting the Hmga1 gene in mice. Here, wereport that both heterozygous and homozygous Hmga1-null micedevelop cardiac hypertrophy and myelo-lymphoid malignancies,including B-cell lymphomas and myeloid leukemia. In vitroexperiments on rat cardiomyocytes showed a direct role for theHMGA1 protein on cardiac cell growth control. As far as the B-celllymphomas are concerned, our data suggest a mechanism bywhich the reduced levels of HMGA1 lead to a dysregulation ofT- and B-cell–specific cytokines, which in turn, causes expansion ofthe B-cell population and increased Rag1/2 levels which mightaccount for the high rate of aberrant IgH rearrangements withconsequent development of B-cell neoplasias.
Materials and Methods
Generation of HMGA1-KO mice. Several overlapping clones wereisolated from a EAIXII phage library of a 129SVJ mouse strain (Stratagene,
La Jolla, CA) using standard procedures. Exons III, IV, and V of the murine
Hmga1 gene were replaced by a neomycin-resistant (neo) cassette. The
targeting construct was transfected by electroporation into embryonic stem(ES) AB2.1 cells. Genomic DNA from each G418-resistant clone was digested
with EcoRI and hybridized by standard Southern blot to an external 5Vgenomic fragment that detects 18 and 9 kb fragments corresponding to thewild-type (WT) and mutant alleles, respectively. Two correctly targeted ES
cell lines were injected into C57Bl/6J blastocysts. Both gave rise to germ line
chimeras that were backcrossed to C57Bl/6J females to obtain heterozygous
Note: Supplementary data for this article are available at Cancer Research Online(http://cancerres.aacrjournals.org/).
Requests for reprints: Alfredo Fusco, Dipartimento di Biologia e PatologiaCellulare e Molecolare, Universita di Napoli Federico II, via S. Pansini 5, 80131 Naples,Italy. Phone: 39-81-746-3056; Fax: 39-81-746-3037; E-mail: [email protected].
I2006 American Association for Cancer Research.doi:10.1158/0008-5472.CAN-05-1889
Cancer Res 2006; 66: (5). March 1, 2006 2536 www.aacrjournals.org
Research Article
Research. on October 1, 2014. © 2006 American Association for Cancercancerres.aacrjournals.org Downloaded from

HMGA1-knockout (KO) offspring. HMGA1-KO mice were crossed with eachother to yield homozygous HMGA1-KO animals. All mice were maintained
under specific pathogen-free conditions in our Laboratory Animal Facility
(Istituto dei Tumori di Napoli, Naples, Italy) and all studies were conducted
in accordance with Italian regulations for experiments on animals.Isolation of mRNA and RT-PCR. Total RNA was extracted using TRI-
reagent solution (Molecular Research Center, Cincinnati, OH) according to
the manufacturer’s protocol, and treated with DNase I (Invitrogen, San
Giuliano Milanese, Italy). cDNA was synthesized using random hexamers(100 mmol/L) and MuLV reverse transcriptase (Perkin-Elmer, Boston, MA).
The PCR was run with a 25 AL volume for 20, 25, and 30 cycles (30 seconds
at 94jC, 30 seconds at 55jC, and 30 seconds at 72jC, respectively) using theGene Amp PCR System 9700 (Applied Biosystems, Foster City, CA). As anegative control, RNA samples that had not been reverse-transcribed before
PCR were used. Primer sequences are available on request.
Histologic analysis and immunohistochemistry. For histologic
examination, dissected tissues were fixed by immersion in 10% formalin
and embedded in paraffin. Mounted sections (5 Am thick) were stained
with H&E using routine procedures. To estimate myocyte size, slides
containing abundant myocytes in a cross-sectional orientation were
photomicrographed at �40 magnification. The avidin-biotin-peroxidase
LSAB+ kit (Dako, Glostrup, Denmark) and microwave antigen retrieval was
used for immunoperoxidase staining. The antisera were directed to CD79a
(Abcam, Cambridge, United Kingdom), CD3 (Dako), B220 (PharMingen, San
Diego, CA), Mac-2 (Cedarlane, Tornby, Canada), and MPO (Dako).
Transthoracic echocardiography. Mice were anesthetized by i.p.
injection of 2.5% Avertin (15 AL/g body weight). Echocardiography wasdone with a Hewlett Packard Sonos 5500 ultrasound system fitted with a 12
mHz transducer (described in detail in the Supplemental data).
Primary culture of neonatal rat ventricular myocytes and adeno-virus infection. Primary cultures of cardiac ventricular myocytes from
1-day-old Crl:(WI)BR-Wistar rats (Charles River Laboratories, Calco, Italy)
were prepared as described previously (16). Cells were cultured in the
cardiac myocyte culture medium containing DMEM/F12 supplementedwith 5% HS, 4 Ag/mL transferrin, 0.7 ng/mL sodium selenite, 2 g/L bovine
serum albumin (BSA; fraction V), 3 mmol/L pyruvic acid, 15 mmol/L
HEPES, 100 Amol/L ascorbic acid, 100 Ag/mL ampicillin, 5 Ag/mL linoleic
acid, and 100 Amol/L 5-bromo-2’-deoxyuridine (Sigma-Aldrich, Milan, Italy).We obtained myocyte cultures in which >95% were myocytes, as assessed by
immunofluorescence staining with a monoclonal antibody against sarco-
meric myosin (MF20). Myocytes were plated at a density of 1 � 106 per wellin six-well plates, the culture medium was changed to serum-free medium
at 24 hours and cultured in serum-free conditions for 48 hours prior to
experiments. Cardiomyocytes were exposed to 10 and 100 pfu/cell of the
specific adenovirus and further cultured for 48 hours.Measurement of the total protein content. After insulin-like growth
factor-I (IGF-I) stimulation (17) or adenovirus infection, cardiomyocytes
were rinsed thrice with PBS. The cell layer was scraped with 1 mL of 1� SSC
containing 0.25% (w/v) SDS and vortexed extensively. The total cell proteinand the DNA content were determined by the Lowry method and the
Hoechst dye method, as described previously (18). The protein content was
normalized by the DNA content to correct for differences in the cellnumber.
Analysis of splenocyte cell surface antigens. Spleens removed from
mice were dissociated into single cells and RBCs were lysed by hypotonic
shock using Tris-NH4Cl buffer [NH4Cl, 0.14 mol/L; Tris, 0.017 mol/L (pH7.2)]. Cells were washed in PBS supplemented with 2% FCS, 0.2% sodium
azide, and labeled with FITC-, PE-, and Cy5-conjugated appropriate
antibodies for 30 minutes at 4jC. Cells were then washed and analyzed
on FACSCalibur flow cytometer (Becton Dickinson, Buccinasco, Italy). Allthe antibodies used were obtained from PharMingen/BD Biosciences.
Transient transfections and luciferase activity.WTand Hmga1�/� ES
cells have been previously described (19). Before transfections, feeder
fibroblasts were removed. WT or double knockout ES cells (4 � 105) wereplated in six-well plates and transfected, 48 hours later, with 1 Ag of reporterplasmid using FuGene6 (Roche, Monza, Italy). Where indicated, 5 Ag of a
plasmid expressing HMGA1b (6) was cotransfected. Cells were harvested
48 hours posttransfection and lysates were analyzed for luciferase activityusing the Dual Light kit (Applied Biosystems, Monza, Italy). Transfection
efficiency was normalized according to h-galactosidase activity. All the
assays were done in triplicate and repeated in three independent
experiments.IgH gene rearrangements. Southern blot of DNA digested with StuI
was prepared following conventional methods and hybridized with the32P-labeled DNA probe PJ3 representing the JH4 region of the IgH locus. The
probe was synthesized by PCR amplification from mouse DNA as previouslydescribed (20).
Protein extraction and Western blot. Tissues were lysed in 1% NP40
buffer, 1 mmol/L EDTA, 50 mmol/L Tris-HCl (pH 7.5), and 150 mmol/L
NaCl, supplemented with complete protease inhibitors mixture (Roche).Total proteins were separated by SDS-PAGE and transferred to nitrocellu-
lose membrane. Membrane was blocked with 5% BSA in TBS and incubated
with the specific primary antibodies recognizing the following proteins:c-myc (C-19, Santa Cruz Biotechnology, Santa Cruz, CA), HMGA1
(polyclonal antibody raised against a synthetic peptide located in the
NH2-terminal region), CaMKII (M-176, Santa Cruz Biotechnology), and
a-tubulin (clone DM1A, Sigma-Aldrich). Bound antibody was detected bythe appropriate secondary antibody, diluted in 5% nonfat milk in TTBS,
and revealed with an enhanced chemiluminescence system (Amersham
Biosciences, Cologno Monzese, Italy).
Statistics. The results are expressed as the mean F SD. For thecomparison of statistical significance between the two groups, Student’s
t test was used. P < 0.05 was considered statistically significant.
Results
Generation of HMGA1-KO mice. We used gene targetingtechniques in ES cells to generate a null mutation at the murineHmga1 genomic locus (Fig. 1A). Heterozygous progeny of chimericanimals were identified by Southern blot analysis of EcoRI-digestedtail DNA (data not shown), and matings were established toproduce mice heterozygous or homozygous for the Hmga1-nullallele (Fig. 1B). Both heterozygous and homozygous mutant mice
Figure 1. Generation of a loss-of-function allele at the Hmga1 locus.A, schematic representation of the WT and mutant alleles and the targetingvector. Abbreviations: (RI) EcoRI, (N) Not I, (H) HindIII, (K) Kpn I, (X) Xba I,(tk) thymidine kinase, (neo) neomycin. B, Southern blot of pups obtained fromheterozygous intercrosses. C, RT-PCR analysis of total RNA from hearts andspleens of WT (+/+), heterozygous (+/�), and homozygous (�/�) mice.
Heart and Myelo-Lymphoid Diseases in HMGA1-KO Mice
www.aacrjournals.org 2537 Cancer Res 2006; 66: (5). March 1, 2006
Research. on October 1, 2014. © 2006 American Association for Cancercancerres.aacrjournals.org Downloaded from

were viable and fertile. We verified Hmga1 gene inactivation byRT-PCR using total RNA from adult hearts and spleens. Asexpected, homozygous mutants did not express Hmga1 mRNA,whereas heterozygous mutants expressed an intermediate amountof Hmga1 (Fig. 1C). Western blot of cell lysates from embryonicfibroblasts confirmed the suppression of the protein expression inthe Hmga1-null cells (data not shown).HMGA1-KO mice display cardiac hypertrophy. Mice homo-
zygous for the Hmga1-null mutation seem to develop normally, andhistologic examination of serial sections confirms the normalstructure and development of all major organ and tissues (data notshown). Cardiac hypertrophy was clearly evident in nearly all theadult homozygous KO animals (starting from 2 months of age andbecoming more consistent at 12 months of age). The heart weight/body ratio increased by an average of 77% in homozygotes and 35%in heterozygotes compared with age-matched WT controls (P <0.001; Fig. 2A and B). Body weight was the same in KO and WTmice or sometimes slightly less in homozygous KO mice (data notshown). Thus, the increased heart/body weight ratio in the KOmice was due to increased ventricular mass. Histologic examina-tion of H&E stained heart sections showed marked myocardialhypertrophy and an abnormal myocyte architecture in heterozy-gous and homozygous KO mice (Fig. 2C ; data not shown). Toevaluate whether increased cardiac size in KO mice was due toenlarged myocytes, we examined cross-sections of myocytes at ahigher magnification. Myocytes were larger in the hypertrophichearts (outsets, Fig. 2C) indicating that cardiac hypertrophy wasdue to myocyte hypertrophy as opposed to increased interstitialcontent. To evaluate further abnormalities in cardiac structure(as observed in histologic sections) and to correlate them withcardiac function, we measured wall thickness, ventricular diameter,
and cardiac function by transthoracic echocardiography in both2- and 11- to 12-month-old KO mice and WT littermates (Fig. 2D ;data not shown). End-diastolic and end-systolic left ventricularchamber diameters were increased 42% and 51%, respectively, inKO animals versus age-matched WT controls. Furthermore, leftventricular posterior wall thickness was increased 19%, and thecalculated left ventricular mass was increased 70% in homozygousKO mice versus controls. Although cardiac function, measured byfractional shortening, was not significantly different in WT and KOmice, a trend toward decreased fractional shortening was observedin KO mice (Supplementary Table S1). The echocardiographicvariables of heterozygous KO mice were intermediate betweenthose from homozygous and WT mice (Fig. 2D ; SupplementaryTable S1). No hypertension was observed in KO mice (data notshown). This result rules out that the hypertrophy occurring inHMGA1-KO mice might be dependent on hemodynamic changes,but clearly suggests a direct role of the HMGA1 proteins in theregulation of the growth properties of cardiomyocytes.Subsequently, using semiquantitative RT-PCR, we evaluated the
expression of a panel of hypertrophic genes in the hearts of bothheterozygous and homozygous HMGA1-null mice. As shown inSupplementary Fig. S1, there was a modest but significant increasein myosin heavy chain-h, atrial natriuretic factor, brain natriureticpeptide, skeletal actin, and myosin light chain levels, whereasmyosin heavy chain-a and cardiac a-actin levels were not changed.Interestingly, the class II calcium/calmodulin-dependent proteinkinase (CaMKII), that has recently been shown to play a crucial rolein inducing cardiac hypertrophy (21), showed a drastic increase.Also, mRNA levels of cardiotrophin-1 (Ctf1 ; ref. 22), GATA4 , and thetwo AP1 genes, c-fos and junB , whose expression is associated withcardiac stress and hypertrophy (23), were significantly increased.
Figure 2. Cardiac hypertrophy in HMGA1-KO mice.A, representative gross morphology of hearts from Hmga1+/+ (left )and Hmga1�/� (right ) mice at 50 weeks of age. B, heartweight/body weight ratio at 48 to 54 weeks of age. Columns ,means of a cohort of four WT, four heterozygous, and fourhomozygous HMGA1-KO mice; bars , F SD (*, P < 0.01versus WT). C, representative histologic analysis of WT (left ),Hmga1+/�, (middle ), and Hmga1�/� (right ) mice at 44 weeks ofage, cut at the midsagittal level and parallel to the base. Outsets ,hearts at a higher magnification, showing cardiomyocyteenlargement in heterozygous and homozygous KO hearts. lv, leftventricle; rv, right ventricle. D, representative M-mode tracingsfrom transthoracic echocardiography in the mice from (C ).
Cancer Research
Cancer Res 2006; 66: (5). March 1, 2006 2538 www.aacrjournals.org
Research. on October 1, 2014. © 2006 American Association for Cancercancerres.aacrjournals.org Downloaded from
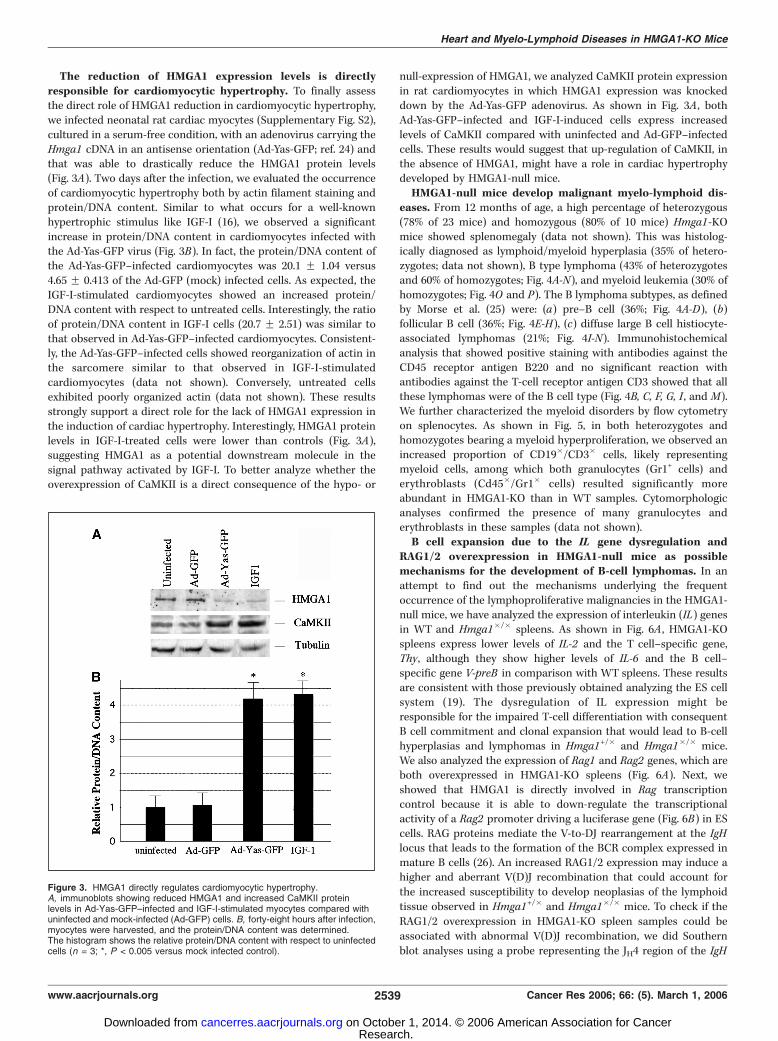
The reduction of HMGA1 expression levels is directlyresponsible for cardiomyocytic hypertrophy. To finally assessthe direct role of HMGA1 reduction in cardiomyocytic hypertrophy,we infected neonatal rat cardiac myocytes (Supplementary Fig. S2),cultured in a serum-free condition, with an adenovirus carrying theHmga1 cDNA in an antisense orientation (Ad-Yas-GFP; ref. 24) andthat was able to drastically reduce the HMGA1 protein levels(Fig. 3A). Two days after the infection, we evaluated the occurrenceof cardiomyocytic hypertrophy both by actin filament staining andprotein/DNA content. Similar to what occurs for a well-knownhypertrophic stimulus like IGF-I (16), we observed a significantincrease in protein/DNA content in cardiomyocytes infected withthe Ad-Yas-GFP virus (Fig. 3B). In fact, the protein/DNA content ofthe Ad-Yas-GFP–infected cardiomyocytes was 20.1 F 1.04 versus4.65 F 0.413 of the Ad-GFP (mock) infected cells. As expected, theIGF-I-stimulated cardiomyocytes showed an increased protein/DNA content with respect to untreated cells. Interestingly, the ratioof protein/DNA content in IGF-I cells (20.7 F 2.51) was similar tothat observed in Ad-Yas-GFP–infected cardiomyocytes. Consistent-ly, the Ad-Yas-GFP–infected cells showed reorganization of actin inthe sarcomere similar to that observed in IGF-I-stimulatedcardiomyocytes (data not shown). Conversely, untreated cellsexhibited poorly organized actin (data not shown). These resultsstrongly support a direct role for the lack of HMGA1 expression inthe induction of cardiac hypertrophy. Interestingly, HMGA1 proteinlevels in IGF-I-treated cells were lower than controls (Fig. 3A),suggesting HMGA1 as a potential downstream molecule in thesignal pathway activated by IGF-I. To better analyze whether theoverexpression of CaMKII is a direct consequence of the hypo- or
null-expression of HMGA1, we analyzed CaMKII protein expressionin rat cardiomyocytes in which HMGA1 expression was knockeddown by the Ad-Yas-GFP adenovirus. As shown in Fig. 3A , bothAd-Yas-GFP–infected and IGF-I-induced cells express increasedlevels of CaMKII compared with uninfected and Ad-GFP–infectedcells. These results would suggest that up-regulation of CaMKII, inthe absence of HMGA1, might have a role in cardiac hypertrophydeveloped by HMGA1-null mice.HMGA1-null mice develop malignant myelo-lymphoid dis-
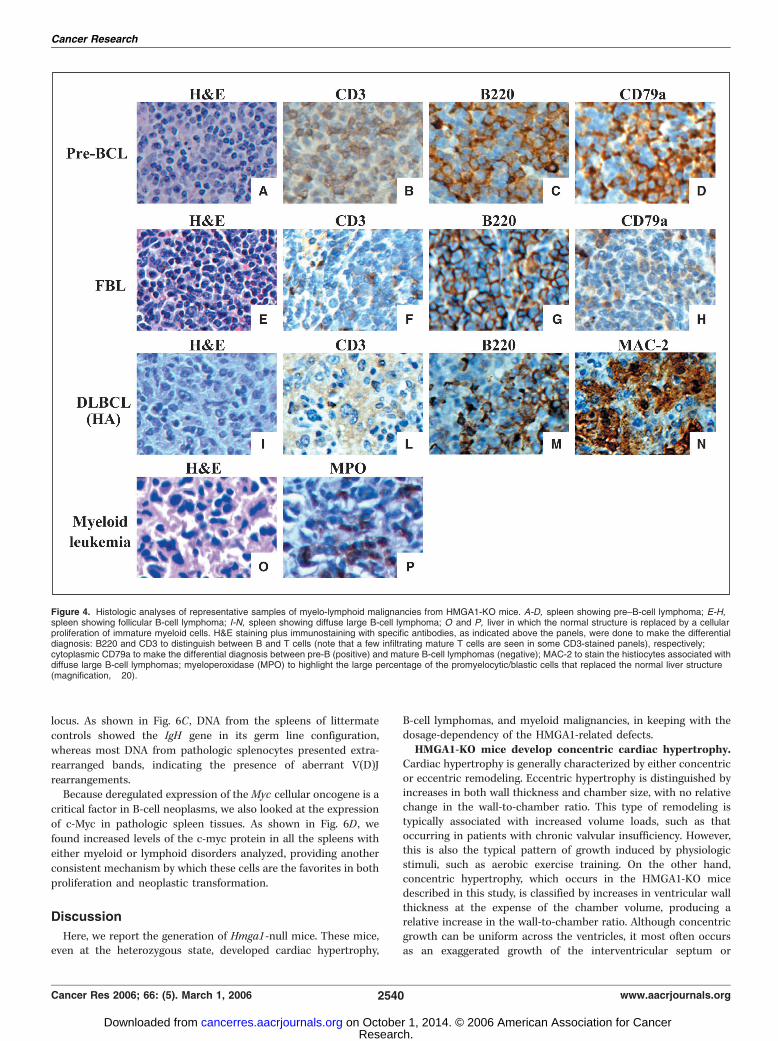
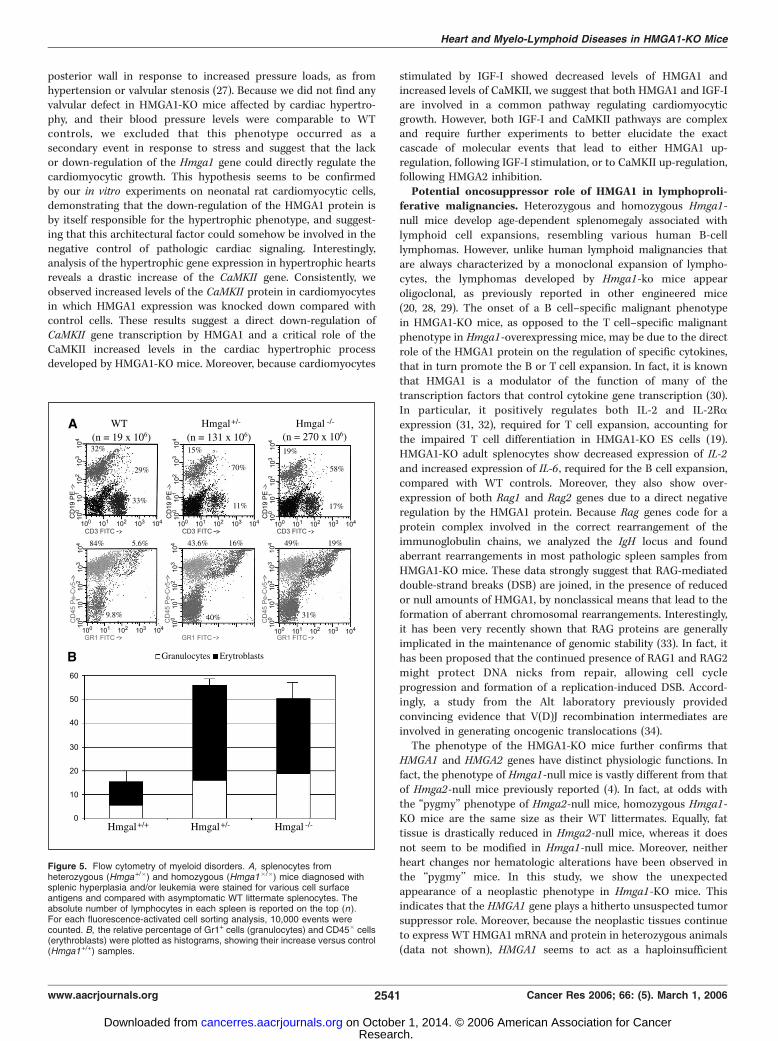
eases. From 12 months of age, a high percentage of heterozygous(78% of 23 mice) and homozygous (80% of 10 mice) Hmga1-KOmice showed splenomegaly (data not shown). This was histolog-ically diagnosed as lymphoid/myeloid hyperplasia (35% of hetero-zygotes; data not shown), B type lymphoma (43% of heterozygotesand 60% of homozygotes; Fig. 4A-N), and myeloid leukemia (30% ofhomozygotes; Fig. 4O and P). The B lymphoma subtypes, as definedby Morse et al. (25) were: (a) pre–B cell (36%; Fig. 4A-D), (b)follicular B cell (36%; Fig. 4E-H), (c) diffuse large B cell histiocyte-associated lymphomas (21%; Fig. 4I-N). Immunohistochemicalanalysis that showed positive staining with antibodies against theCD45 receptor antigen B220 and no significant reaction withantibodies against the T-cell receptor antigen CD3 showed that allthese lymphomas were of the B cell type (Fig. 4B, C, F, G, I , and M).We further characterized the myeloid disorders by flow cytometryon splenocytes. As shown in Fig. 5, in both heterozygotes andhomozygotes bearing a myeloid hyperproliferation, we observed anincreased proportion of CD19�/CD3� cells, likely representingmyeloid cells, among which both granulocytes (Gr1+ cells) anderythroblasts (Cd45�/Gr1� cells) resulted significantly moreabundant in HMGA1-KO than in WT samples. Cytomorphologicanalyses confirmed the presence of many granulocytes anderythroblasts in these samples (data not shown).B cell expansion due to the IL gene dysregulation and
RAG1/2 overexpression in HMGA1-null mice as possiblemechanisms for the development of B-cell lymphomas. In anattempt to find out the mechanisms underlying the frequentoccurrence of the lymphoproliferative malignancies in the HMGA1-null mice, we have analyzed the expression of interleukin (IL) genesin WT and Hmga1�/� spleens. As shown in Fig. 6A , HMGA1-KOspleens express lower levels of IL-2 and the T cell–specific gene,Thy, although they show higher levels of IL-6 and the B cell–specific gene V-preB in comparison with WT spleens. These resultsare consistent with those previously obtained analyzing the ES cellsystem (19). The dysregulation of IL expression might beresponsible for the impaired T-cell differentiation with consequentB cell commitment and clonal expansion that would lead to B-cellhyperplasias and lymphomas in Hmga1+/� and Hmga1�/� mice.We also analyzed the expression of Rag1 and Rag2 genes, which areboth overexpressed in HMGA1-KO spleens (Fig. 6A). Next, weshowed that HMGA1 is directly involved in Rag transcriptioncontrol because it is able to down-regulate the transcriptionalactivity of a Rag2 promoter driving a luciferase gene (Fig. 6B) in EScells. RAG proteins mediate the V-to-DJ rearrangement at the IgHlocus that leads to the formation of the BCR complex expressed inmature B cells (26). An increased RAG1/2 expression may induce ahigher and aberrant V(D)J recombination that could account forthe increased susceptibility to develop neoplasias of the lymphoidtissue observed in Hmga1+/� and Hmga1�/� mice. To check if theRAG1/2 overexpression in HMGA1-KO spleen samples could beassociated with abnormal V(D)J recombination, we did Southernblot analyses using a probe representing the JH4 region of the IgH
Figure 3. HMGA1 directly regulates cardiomyocytic hypertrophy.A, immunoblots showing reduced HMGA1 and increased CaMKII proteinlevels in Ad-Yas-GFP–infected and IGF-I-stimulated myocytes compared withuninfected and mock-infected (Ad-GFP) cells. B, forty-eight hours after infection,myocytes were harvested, and the protein/DNA content was determined.The histogram shows the relative protein/DNA content with respect to uninfectedcells (n = 3; *, P < 0.005 versus mock infected control).
Heart and Myelo-Lymphoid Diseases in HMGA1-KO Mice
www.aacrjournals.org 2539 Cancer Res 2006; 66: (5). March 1, 2006
Research. on October 1, 2014. © 2006 American Association for Cancercancerres.aacrjournals.org Downloaded from
locus. As shown in Fig. 6C , DNA from the spleens of littermatecontrols showed the IgH gene in its germ line configuration,whereas most DNA from pathologic splenocytes presented extra-rearranged bands, indicating the presence of aberrant V(D)Jrearrangements.Because deregulated expression of theMyc cellular oncogene is a
critical factor in B-cell neoplasms, we also looked at the expressionof c-Myc in pathologic spleen tissues. As shown in Fig. 6D , wefound increased levels of the c-myc protein in all the spleens witheither myeloid or lymphoid disorders analyzed, providing anotherconsistent mechanism by which these cells are the favorites in bothproliferation and neoplastic transformation.
Discussion
Here, we report the generation of Hmga1-null mice. These mice,even at the heterozygous state, developed cardiac hypertrophy,
B-cell lymphomas, and myeloid malignancies, in keeping with thedosage-dependency of the HMGA1-related defects.HMGA1-KO mice develop concentric cardiac hypertrophy.
Cardiac hypertrophy is generally characterized by either concentricor eccentric remodeling. Eccentric hypertrophy is distinguished byincreases in both wall thickness and chamber size, with no relativechange in the wall-to-chamber ratio. This type of remodeling istypically associated with increased volume loads, such as thatoccurring in patients with chronic valvular insufficiency. However,this is also the typical pattern of growth induced by physiologicstimuli, such as aerobic exercise training. On the other hand,concentric hypertrophy, which occurs in the HMGA1-KO micedescribed in this study, is classified by increases in ventricular wallthickness at the expense of the chamber volume, producing arelative increase in the wall-to-chamber ratio. Although concentricgrowth can be uniform across the ventricles, it most often occursas an exaggerated growth of the interventricular septum or
Figure 4. Histologic analyses of representative samples of myelo-lymphoid malignancies from HMGA1-KO mice. A-D, spleen showing pre–B-cell lymphoma; E-H,spleen showing follicular B-cell lymphoma; I-N, spleen showing diffuse large B-cell lymphoma; O and P, liver in which the normal structure is replaced by a cellularproliferation of immature myeloid cells. H&E staining plus immunostaining with specific antibodies, as indicated above the panels, were done to make the differentialdiagnosis: B220 and CD3 to distinguish between B and T cells (note that a few infiltrating mature T cells are seen in some CD3-stained panels), respectively;cytoplasmic CD79a to make the differential diagnosis between pre-B (positive) and mature B-cell lymphomas (negative); MAC-2 to stain the histiocytes associated withdiffuse large B-cell lymphomas; myeloperoxidase (MPO) to highlight the large percentage of the promyelocytic/blastic cells that replaced the normal liver structure(magnification, �20).
Cancer Research
Cancer Res 2006; 66: (5). March 1, 2006 2540 www.aacrjournals.org
Research. on October 1, 2014. © 2006 American Association for Cancercancerres.aacrjournals.org Downloaded from
posterior wall in response to increased pressure loads, as fromhypertension or valvular stenosis (27). Because we did not find anyvalvular defect in HMGA1-KO mice affected by cardiac hypertro-phy, and their blood pressure levels were comparable to WTcontrols, we excluded that this phenotype occurred as asecondary event in response to stress and suggest that the lackor down-regulation of the Hmga1 gene could directly regulate thecardiomyocytic growth. This hypothesis seems to be confirmedby our in vitro experiments on neonatal rat cardiomyocytic cells,demonstrating that the down-regulation of the HMGA1 protein isby itself responsible for the hypertrophic phenotype, and suggest-ing that this architectural factor could somehow be involved in thenegative control of pathologic cardiac signaling. Interestingly,analysis of the hypertrophic gene expression in hypertrophic heartsreveals a drastic increase of the CaMKII gene. Consistently, weobserved increased levels of the CaMKII protein in cardiomyocytesin which HMGA1 expression was knocked down compared withcontrol cells. These results suggest a direct down-regulation ofCaMKII gene transcription by HMGA1 and a critical role of theCaMKII increased levels in the cardiac hypertrophic processdeveloped by HMGA1-KO mice. Moreover, because cardiomyocytes
stimulated by IGF-I showed decreased levels of HMGA1 andincreased levels of CaMKII, we suggest that both HMGA1 and IGF-Iare involved in a common pathway regulating cardiomyocyticgrowth. However, both IGF-I and CaMKII pathways are complexand require further experiments to better elucidate the exactcascade of molecular events that lead to either HMGA1 up-regulation, following IGF-I stimulation, or to CaMKII up-regulation,following HMGA2 inhibition.Potential oncosuppressor role of HMGA1 in lymphoproli-
ferative malignancies. Heterozygous and homozygous Hmga1-null mice develop age-dependent splenomegaly associated withlymphoid cell expansions, resembling various human B-celllymphomas. However, unlike human lymphoid malignancies thatare always characterized by a monoclonal expansion of lympho-cytes, the lymphomas developed by Hmga1-ko mice appearoligoclonal, as previously reported in other engineered mice(20, 28, 29). The onset of a B cell–specific malignant phenotypein HMGA1-KO mice, as opposed to the T cell–specific malignantphenotype in Hmga1-overexpressing mice, may be due to the directrole of the HMGA1 protein on the regulation of specific cytokines,that in turn promote the B or T cell expansion. In fact, it is knownthat HMGA1 is a modulator of the function of many of thetranscription factors that control cytokine gene transcription (30).In particular, it positively regulates both IL-2 and IL-2Raexpression (31, 32), required for T cell expansion, accounting forthe impaired T cell differentiation in HMGA1-KO ES cells (19).HMGA1-KO adult splenocytes show decreased expression of IL-2and increased expression of IL-6 , required for the B cell expansion,compared with WT controls. Moreover, they also show over-expression of both Rag1 and Rag2 genes due to a direct negativeregulation by the HMGA1 protein. Because Rag genes code for aprotein complex involved in the correct rearrangement of theimmunoglobulin chains, we analyzed the IgH locus and foundaberrant rearrangements in most pathologic spleen samples fromHMGA1-KO mice. These data strongly suggest that RAG-mediateddouble-strand breaks (DSB) are joined, in the presence of reducedor null amounts of HMGA1, by nonclassical means that lead to theformation of aberrant chromosomal rearrangements. Interestingly,it has been very recently shown that RAG proteins are generallyimplicated in the maintenance of genomic stability (33). In fact, ithas been proposed that the continued presence of RAG1 and RAG2might protect DNA nicks from repair, allowing cell cycleprogression and formation of a replication-induced DSB. Accord-ingly, a study from the Alt laboratory previously providedconvincing evidence that V(D)J recombination intermediates areinvolved in generating oncogenic translocations (34).The phenotype of the HMGA1-KO mice further confirms that
HMGA1 and HMGA2 genes have distinct physiologic functions. Infact, the phenotype of Hmga1-null mice is vastly different from thatof Hmga2-null mice previously reported (4). In fact, at odds withthe ‘‘pygmy’’ phenotype of Hmga2-null mice, homozygous Hmga1-KO mice are the same size as their WT littermates. Equally, fattissue is drastically reduced in Hmga2-null mice, whereas it doesnot seem to be modified in Hmga1-null mice. Moreover, neitherheart changes nor hematologic alterations have been observed inthe ‘‘pygmy’’ mice. In this study, we show the unexpectedappearance of a neoplastic phenotype in Hmga1-KO mice. Thisindicates that the HMGA1 gene plays a hitherto unsuspected tumorsuppressor role. Moreover, because the neoplastic tissues continueto express WT HMGA1 mRNA and protein in heterozygous animals(data not shown), HMGA1 seems to act as a haploinsufficient
Figure 5. Flow cytometry of myeloid disorders. A, splenocytes fromheterozygous (Hmga+/�) and homozygous (Hmga1�/�) mice diagnosed withsplenic hyperplasia and/or leukemia were stained for various cell surfaceantigens and compared with asymptomatic WT littermate splenocytes. Theabsolute number of lymphocytes in each spleen is reported on the top (n).For each fluorescence-activated cell sorting analysis, 10,000 events werecounted. B, the relative percentage of Gr1+ cells (granulocytes) and CD45� cells(erythroblasts) were plotted as histograms, showing their increase versus control(Hmga1+/+) samples.
Heart and Myelo-Lymphoid Diseases in HMGA1-KO Mice
www.aacrjournals.org 2541 Cancer Res 2006; 66: (5). March 1, 2006
Research. on October 1, 2014. © 2006 American Association for Cancercancerres.aacrjournals.org Downloaded from

tumor suppressor gene. This seems to contrast with the oncogenicpotential of HMGA1 that several studies, both in vitro and in vivo ,have clearly established (10, 13–15). Therefore, the results reportedhere indicate the intriguing effect of a gene that is endowed withboth oncogenic and antioncogenic properties. This might not besurprising given the mechanism of action of HMGA proteins. Infact, they do not exert transcriptional activity per se but theyregulate the expression of several genes by interacting with diversepartners (2). Because the protein partners of the HMGA proteinsmight change depending on the cell type, HMGA1-dependent generegulation may also differ from cell to cell, depending on thecellular context (35), which would also account for the dual effectof the HMGA1 protein on cell transformation.
Acknowledgments
Received 6/7/2005; revised 11/24/2005; accepted 12/13/2005.Grant support: Associazione Italiana per la Ricerca sul Cancro, BioGem, and
Ministero dell’ Universita e della Ricerca Scientifica e Tecnologica, projects ‘‘Terapieantineoplastiche innovative’’ and ‘‘Piani di Potenziamento della Rete Scientifica eTecnologica,’’ the Programma Italia-USA sulla Terapia dei Tumori coordinated by Prof.Cesare Peschle, by the MIUR project ‘‘Terapie antineoplastiche innovative,’’ and the‘‘Ministero della Sanita.’’
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.
We are grateful to A. Affuso for advice on mice management, to A. Lucianofor animal care, to J. Martinez Hoyos for adenovirus amplification, and to B. Rotoli andA. Risitano for cytomorphologic analysis of spleen spreads; to D. Palmieri for his helpand the Associazione Partenopea per le Ricerche Oncologiche for its support; and toJean Ann Gilder for editing the text.
Figure 6. Mechanism of B-cell lymphomagenesis in HMGA1-KO mice. A, RT-PCR analysis showing the expression of IL 2, IL-6, Thy, V-preB, Rag1 , and Rag2genes in Hmga1+/+, Hmga1+/�, and Hmga1�/� spleens. Gapdh expression was evaluated as an internal control of RNA used. B, RT-PCR analysis showing theexpression of the Rag2 gene in Hmga1+/+, Hmga1+/�, and Hmga1�/� ES cells (left). h-Actin expression was evaluated as an internal control of RNA used. Luciferaseactivity assay using the Rag2 promoter region transfected alone or with 5 Ag of HMGA1 expression vectors in the ES cells (right ). Columns , mean results ofthree different experiments; bars , F SE. C, analysis of IgH gene configuration. IgH gene rearrangements were analyzed by Southern blot on Stu I-digestedspleen DNA. Control (+/+) is WT mouse with the genomic 4.7 kb Stu I fragment representing the gene in its germ line configuration. Most KO mice show rearrangedextra-bands (asterisks on the side ). The histologic diagnoses of the tissues are indicated above. D, c-Myc protein expression analysis by Western blot in WT (+/+)and HMGA1-KO (�/�) spleen tissues. As a control for equal protein loading, the blotted proteins were incubated with tubulin-specific antibodies. The histologicdiagnoses of the tissues are indicated above.
Cancer Research
Cancer Res 2006; 66: (5). March 1, 2006 2542 www.aacrjournals.org
Research. on October 1, 2014. © 2006 American Association for Cancercancerres.aacrjournals.org Downloaded from

References1. Fedele M, Battista S, Manfioletti G, Croce CM,Giancotti V, Fusco A. Role of the high mobility groupA proteins in human lipomas. Carcinogenesis 2001;22:1583–91.
2. Thanos D, Maniatis T. Virus induction of human IFNh gene expression requires the assembly of anenhanceosome. Cell 1995;83:1091–100.
3. Chiappetta G, Avantaggiato V, Visconti R, et al. Highlevel expression of the HMGI (Y) gene during embryonicdevelopment. Oncogene 1996;13:2439–46.
4. Zhou X, Benson KF, Ashar HR, Chada K. Mutationresponsible for the mouse pygmy phenotype in thedevelopmentally regulated factor HMGI-C. Nature 1995;376:771–4.
5. Battista S, Fidanza V, Fedele M, et al. The expression ofa truncated HMGI-C gene induces gigantism associatedwith lipomatosis. Cancer Res 1999;59:4793–7.
6. Melillo RM, Pierantoni GM, Scala S, et al. Critical roleof the HMGI(Y) proteins in adipocytic cell growth anddifferentiation. Mol Cell Biol 2001;21:2485–95.
7. Tallini G, Dal Cin P. HMGI(Y) and HMGI-C dysregu-lation: a common occurrence in human tumors. AdvAnat Pathol 1999;6:237–46.
8. Berlingieri MT, Pierantoni GM, Giancotti V, Santoro M,Fusco A. Thyroid cell transformation requires theexpression of the HMGA1 proteins. Oncogene 2002;21:2971–80.
9. Scala S, Portella G, Fedele M, Chiappetta G, FuscoA. Adenovirus-mediated suppression of HMGI(Y)protein synthesis as potential therapy of humanmalignant neoplasias. Proc Natl Acad Sci U S A 2000;97:4256–61.
10. Fedele M, Berlingieri MT, Scala S, et al. Truncatedand chimeric HMGI-C genes induce neoplastic trans-formation of NIH3T3 murine fibroblasts. Oncogene1998;17:413–8.
11. Wood LJ, Maher JF, Bunton TE, Resar LM. Theoncogenic properties of the HMG-I gene family. CancerRes 2000;60:4256–61.
12. Baldassarre G, Fedele M, Battista S, et al. Onset ofnatural killer cell lymphomas in transgenic micecarrying a truncated HMGI-C gene by the chronicstimulation of the IL-2 and IL-15 pathway. Proc NatlAcad Sci U S A 2001;98:7970–5.
13. Xu Y, Sumter TF, Bhattacharya R, et al. The HMG-Ioncogene causes highly penetrant, aggressive lymphoidmalignancy in transgenic mice and is overexpressed inhuman leukemia. Cancer Res 2004;64:3371–5.
14. Fedele M, Pentimalli F, Baldassarre G, et al.Transgenic mice overexpressing the wild-type form ofthe HMGA1 gene develop mixed growth hormone/prolactin cell pituitary adenomas and natural killer celllymphomas. Oncogene 2005;24:3427–35.
15. Fedele M, Battista S, Kenyon L, et al. Overexpressionof the HMGA2 gene in transgenic mice leads to theonset of pituitary adenomas. Oncogene 2002;21:3190–8.
16. Aoki H, Izumo S, Sadoshima J. Angiotensin IIactivates RhoA in cardiac myocytes: a critical role ofRhoA in angiotensin II-induced premyofibril formation.Circ Res 1998;81:666–76.
17. Ito H, Hiroe M, Hirata Y, et al. Insulin-like growthfactor-I induces hypertrophy with enhanced expressionof muscle specific genes in cultured rat cardiomyocytes.Circulation 1993;87:1715–21.
18. Sadoshima J, Jahn L, Takahashi T, Kulik TJ, Izumo S.Molecular characterization of the stretch-induced ad-aptation of cultured cardiac cells. An in vitro model ofload-induced cardiac hypertrophy. J Biol Chem 1992;267:10551–60.
19. Battista S, Pentimalli F, Baldassarre G, et al. Loss ofHmga1 gene function affects embryonic stem celllympho-hematopoietic differentiation. FASEB J 2003;17:1496–8.
20. Bichi R, Shinton SA, Martin ES, et al. Human chroniclymphocytic leukemia modeled in mouse by targetedTCL1 expression. Proc Natl Acad Sci U S A 2002;99:6955–60.
21. Zhang T, Johnson EN, Gu Y, et al. The cardiac-specific nuclear y(B) isoform of Ca2+/calmodulin-dependent protein kinase II induces hypertrophy anddilated cardiomyopathy associated with increasedprotein phosphatase 2A activity. J Biol Chem 2002;277:1261–7.
22. Pennica D, King KL, Shaw KJ, et al. Expressioncloning of cardiotrophin 1, a cytokine that inducescardiac myocyte hypertrophy. Proc Natl Acad Sci U S A1995;92:1142–6.
23. Swynghedauw B. Molecular mechanisms of myocar-dial remodeling. Physiol Rev 1999;79:215–62.
24. Masciullo V, Baldassarre G, Pentimalli F, et al.
HMGA1 protein over-expression is a frequent featureof epithelial ovarian carcinomas. Carcinogenesis 2003;24:1191–8.
25. Morse HC III, Anver MR, Fredrickson TN, et al.Bethesda proposal for classification of lymphoid neo-plasms in mice. Blood 2002;100:246–58.
26. Rajewsky K. Clonal selection and learning in theantibody system. Nature 1996;381:751–8.
27. Lorell BH, Carabello BA. Left ventricular hypertro-phy: pathogenesis, detection, and prognosis. Circulation2000;102:470–9.
28. Zapata JM, Krajewska M, Morse HC III, et al. TNFreceptor-associated factor (TRAF) domain and Bcl-2cooperate to induce small B cell lymphoma/chroniclymphocytic leukemia in transgenic mice. Proc NatlAcad Sci U S A 2004;101:16600–5.
29. Katsumata M, Siegel RM, Louie DC, et al. Differentialeffects of Bcl-2 on T and B cells in transgenic mice. ProcNatl Acad Sci U S A 1992;89:11376–80.
30. Shannon MF, Himes SR, Attema J. A role for thearchitectural transcription factors HMGI(Y) in cytokinegene transcription in T cells. Immunol Cell Biol 1998;76:461–6.
31. Himes SR, Reeves R, Attema J, Nissen M, Li Y,Shannon MF. The role of high-mobility group I(Y)proteins in expression of IL-2 and T cell proliferation.J Immunol 2000;164:3157–68.
32. John S, Reeves RB, Lin JX, et al. Regulation ofcell-type-specific interleukin-2 receptor a-chain geneexpression: potential role of physical interactionsbetween Elf-1, HMG-I(Y), and NF-nB family proteins.Mol Cell Biol 1995;15:1786–96.
33. Lee GS, Neiditch MB, Salus SS, Roth DB. RAGproteins shepherd double-strand breaks to a specificpathway. Suppressing error-prone repair, but RAGnicking initiates homologous recombination. Cell 2004;117:171–84.
34. Zhu C, Mills KD, Ferguson DO, et al. Unrepaired DNAbreaks in p53-deficient cells lead to oncogenic geneamplification subsequent to translocations. Cell 2002;109:811–21.
35. Martinez Hoyos J, Fedele M, Battista S, et al.Identification of the genes up- and down-regulated bythe high mobility group A1 (HMGA1) proteins: tissuespecificity of the HMGA1-dependent gene regulation.Cancer Res 2004;64:5728–35.
Heart and Myelo-Lymphoid Diseases in HMGA1-KO Mice
www.aacrjournals.org 2543 Cancer Res 2006; 66: (5). March 1, 2006
Research. on October 1, 2014. © 2006 American Association for Cancercancerres.aacrjournals.org Downloaded from




















