
High expression of ErbB family members and their ligands in lung adenocarcinomas that are sensitive...
9
High Expression of ErbB Family Members and Their Ligands in Lung Adenocarcinomas That Are Sensitive to Inhibition of Epidermal Growth Factor Receptor Nobukazu Fujimoto, 1 Marie Wislez, 1,4 Jie Zhang, 1 Kentaro Iwanaga, 1 Jennifer Dackor, 5 Amy E. Hanna, 1 Shailaja Kalyankrishna, 1 Dianna D. Cody, 2 Roger E. Price, 2 Mitsuo Sato, 6 Jerry W. Shay, 6 John D. Minna, 6 Michael Peyton, 6 Ximing Tang, 1 Erminia Massarelli, 1 Roy Herbst, 1 David W. Threadgill, 5 Ignacio I. Wistuba, 1,3 and Jonathan M. Kurie 1 Departments of 1 Thoracic/Head and Neck Medical Oncology, 2 Imaging Physics, and 3 Pathology, The University of Texas M.D. Anderson Cancer Center, Houston, Texas; 4 UPRESEA 3493, CHU Saint-Antoine, Universite Paris VI, Paris, France; 5 Department of Genetics, Curriculum in Genetics and Molecular Biology, Lineberger Cancer Center, University of North Carolina, Chapel Hill, North Carolina; and 6 Harold Simmons Cancer Center, University of Texas Southwestern Medical Center, Dallas, Texas Abstract Recent findings in tumor biopsies from lung adenocarcinoma patients suggest that somatic mutations in the genes encoding epidermal growth factor receptor (EGFR ) and Kirsten ras (KRAS ) confer sensitivity and resistance, respectively, to EGFR inhibition. Here, we provide evidence that these genetic mutations are not sufficient to modulate the biological response of lung adenocarcinoma cells to EGFR inhibition. We found high expression of ErbB family members, ErbB ligands, or both in three models that were sensitive to EGFR inhibition, including alveolar epithelial neoplastic lesions in mice that develop lung adenocarcinoma by oncogenic KRAS , human lung adenocarcinoma cell lines, and tumor biopsies from lung adenocarcinoma patients. Thus, lung adenocarci- noma cells that depend on EGFR for survival constitutively activate the receptor through a combination of genetic mutations and overexpression of EGFR dimeric partners and their ligands. (Cancer Res 2005; 65(24): 11478-85) Introduction Somatic mutations in epidermal growth factor receptor (EGFR ), the gene encoding the EGFR, have been found in the tumors of 10% to 40% of patients with non–small cell lung cancer (NSCLC; refs. 1–4). These mutations activate the EGFR tyrosine kinase and are associated with adenocarcinoma histology, female gender, and a nonsmoking history. Lung cancer patients with EGFR mutations frequently experience rapid and sustained shrinkage of primary and metastatic disease after treatment with the EGFR tyrosine kinase inhibitors (TKI) gefitinib or erlotinib (1–4). In a phase III trial, treatment with erlotinib conferred a survival benefit (5), but the proportion of patients who experienced this benefit exceeded the expected frequency of EGFR mutations. In addition, a small proportion of patients whose tumors shrank in response to EGFR TKIs had no evidence of EGFR mutations (1–5). Together, these findings suggest that factors other than EGFR mutations confer sensitivity to EGFR inhibition. EGFR forms homodimers and heterodimers with the other ErbB family members ErbB2, ErbB3, and ErbB4 (6). These dimeric complexes have distinct ligand binding and signaling activities (7). For example, ErbB3 is unique in that it lacks a functional kinase domain. Despite this deficiency, ErbB3 undergoes transphosphor- ylation in complex with other ErbBs and activates downstream kinases, such as phosphatidylinositol 3-kinase (PI3K), in response to ligand binding. Recent studies have implicated ErbB3 in the sensitivity of NSCLC cell lines to EGFR inhibition (8, 9). Together, these findings suggest that aberrant expression of ErbB family members contributes to the responsiveness of NSCLC cells to EGFR inhibition. A recent report (10) linked de novo resistance to treatment with EGFR TKIs in lung adenocarcinoma patients with somatic mutations in Kirsten ras (KRAS ). A growing body of evidence indicates that KRAS mutations are important in the development of lung adenocarcinoma. They occur in 30% to 50% of lung adenocarcinomas (11, 12) and are mutually exclusive from mutations in EGFR (13). Mice that express mutant KRAS develop lung adenocarcinoma rapidly and with high penetrance (14–17). In this study, we investigated the role of KRAS mutations in the resistance of lung adenocarcinoma to EGFR TKIs. We examined Kras LA1 mice, which develop lung adenocarcinoma through somatic activation of a KRAS allele carrying an activating mutation in codon 12 (G12D; ref. 17). Alveolar epithelial cells in this mouse model recapitulate the series of morphologic stages through which human atypical alveolar hyperplasia (AAH) evolves into adenocar- cinoma. Our findings suggest that the presence of KRAS mutations is not sufficient to confer resistance to EGFR inhibition and reveal a novel mechanism of response to EGFR inhibition that is potentially relevant to lung cancer patients. Materials and Methods Animal experiments. Animal experiments were compliant with the guidelines of The University of Texas M.D. Anderson Cancer Center. The Kras LA1 mice were provided by Dr. Tyler Jacks (Massachusetts Institute of Technology, Cambridge, MA), and gefitinib was provided by AstraZeneca (Wilmington, DE). Four-month-old Kras LA1 mice were randomly allocated to treatment with vehicle (PBS with 0.05% Tween 80 in PBS by oral gavage daily), low-dose gefitinib (100 mg/kg/d), or high-dose gefitinib (250 mg/kg/d). Treatment was given for 28 days with a 4-day intermission at 14 days to minimize dermatologic toxicity. Mice treated with high-dose Note: N. Fujimoto and M. Wislez contributed equally to this work. M. Wislez is a postdoctoral fellow of La Fondation de France and La Societe de Pneumonologie de Langue Francaise. Supplementary data for this article are available at Cancer Research Online (http:// cancerres.aacrjournals.org/). Requests for reprints: Jonathan M. Kurie, The University of Texas M.D. Anderson Cancer Center, Box 432, 1515 Holcombe Boulevard, Houston, TX 77030. Phone: 713- 792-6363; Fax: 713-796-8655; E-mail: [email protected]. I2005 American Association for Cancer Research. doi:10.1158/0008-5472.CAN-05-1977 Cancer Res 2005; 65: (24). December 15, 2005 11478 www.aacrjournals.org Research Article Research. on August 24, 2015. © 2005 American Association for Cancer cancerres.aacrjournals.org Downloaded from
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of High expression of ErbB family members and their ligands in lung adenocarcinomas that are sensitive...

High Expression of ErbB Family Members and Their Ligands in
Lung Adenocarcinomas That Are Sensitive to Inhibition of
Epidermal Growth Factor Receptor
Nobukazu Fujimoto,1Marie Wislez,
1,4Jie Zhang,
1Kentaro Iwanaga,
1Jennifer Dackor,
5
Amy E. Hanna,1Shailaja Kalyankrishna,
1Dianna D. Cody,
2Roger E. Price,
2Mitsuo Sato,
6
Jerry W. Shay,6John D. Minna,
6Michael Peyton,
6Ximing Tang,
1Erminia Massarelli,
1
Roy Herbst,1David W. Threadgill,
5Ignacio I. Wistuba,
1,3and Jonathan M. Kurie
1
Departments of 1Thoracic/Head and Neck Medical Oncology, 2Imaging Physics, and 3Pathology, The University of Texas M.D. AndersonCancer Center, Houston, Texas; 4UPRESEA 3493, CHU Saint-Antoine, Universite Paris VI, Paris, France; 5Department of Genetics,Curriculum in Genetics and Molecular Biology, Lineberger Cancer Center, University of North Carolina, Chapel Hill,North Carolina; and 6Harold Simmons Cancer Center, University of Texas Southwestern Medical Center, Dallas, Texas
Abstract
Recent findings in tumor biopsies from lung adenocarcinomapatients suggest that somatic mutations in the genes encodingepidermal growth factor receptor (EGFR) and Kirsten ras(KRAS) confer sensitivity and resistance, respectively, to EGFRinhibition. Here, we provide evidence that these geneticmutations are not sufficient to modulate the biologicalresponse of lung adenocarcinoma cells to EGFR inhibition.We found high expression of ErbB family members, ErbBligands, or both in three models that were sensitive to EGFRinhibition, including alveolar epithelial neoplastic lesions inmice that develop lung adenocarcinoma by oncogenic KRAS ,human lung adenocarcinoma cell lines, and tumor biopsiesfrom lung adenocarcinoma patients. Thus, lung adenocarci-noma cells that depend on EGFR for survival constitutivelyactivate the receptor through a combination of geneticmutations and overexpression of EGFR dimeric partners andtheir ligands. (Cancer Res 2005; 65(24): 11478-85)
Introduction
Somatic mutations in epidermal growth factor receptor (EGFR),the gene encoding the EGFR, have been found in the tumors of 10%to 40% of patients with non–small cell lung cancer (NSCLC; refs.1–4). These mutations activate the EGFR tyrosine kinase and areassociated with adenocarcinoma histology, female gender, and anonsmoking history. Lung cancer patients with EGFR mutationsfrequently experience rapid and sustained shrinkage of primaryand metastatic disease after treatment with the EGFR tyrosinekinase inhibitors (TKI) gefitinib or erlotinib (1–4). In a phase IIItrial, treatment with erlotinib conferred a survival benefit (5), butthe proportion of patients who experienced this benefit exceededthe expected frequency of EGFR mutations. In addition, a smallproportion of patients whose tumors shrank in response to EGFRTKIs had no evidence of EGFR mutations (1–5). Together, these
findings suggest that factors other than EGFR mutations confersensitivity to EGFR inhibition.EGFR forms homodimers and heterodimers with the other ErbB
family members ErbB2, ErbB3, and ErbB4 (6). These dimericcomplexes have distinct ligand binding and signaling activities (7).For example, ErbB3 is unique in that it lacks a functional kinasedomain. Despite this deficiency, ErbB3 undergoes transphosphor-ylation in complex with other ErbBs and activates downstreamkinases, such as phosphatidylinositol 3-kinase (PI3K), in responseto ligand binding. Recent studies have implicated ErbB3 in thesensitivity of NSCLC cell lines to EGFR inhibition (8, 9). Together,these findings suggest that aberrant expression of ErbB familymembers contributes to the responsiveness of NSCLC cells toEGFR inhibition.A recent report (10) linked de novo resistance to treatment with
EGFR TKIs in lung adenocarcinoma patients with somaticmutations in Kirsten ras (KRAS). A growing body of evidenceindicates that KRAS mutations are important in the developmentof lung adenocarcinoma. They occur in 30% to 50% of lungadenocarcinomas (11, 12) and are mutually exclusive frommutations in EGFR (13). Mice that express mutant KRAS developlung adenocarcinoma rapidly and with high penetrance (14–17). Inthis study, we investigated the role of KRAS mutations in theresistance of lung adenocarcinoma to EGFR TKIs. We examinedKrasLA1 mice, which develop lung adenocarcinoma throughsomatic activation of a KRAS allele carrying an activating mutationin codon 12 (G12D; ref. 17). Alveolar epithelial cells in this mousemodel recapitulate the series of morphologic stages through whichhuman atypical alveolar hyperplasia (AAH) evolves into adenocar-cinoma. Our findings suggest that the presence of KRAS mutationsis not sufficient to confer resistance to EGFR inhibition and reveala novel mechanism of response to EGFR inhibition that ispotentially relevant to lung cancer patients.
Materials and Methods
Animal experiments. Animal experiments were compliant with the
guidelines of The University of Texas M.D. Anderson Cancer Center. The
KrasLA1 mice were provided by Dr. Tyler Jacks (Massachusetts Institute ofTechnology, Cambridge, MA), and gefitinib was provided by AstraZeneca
(Wilmington, DE). Four-month-old KrasLA1 mice were randomly allocated
to treatment with vehicle (PBS with 0.05% Tween 80 in PBS by oralgavage daily), low-dose gefitinib (100 mg/kg/d), or high-dose gefitinib
(250 mg/kg/d). Treatment was given for 28 days with a 4-day intermission at
14 days to minimize dermatologic toxicity. Mice treated with high-dose
Note: N. Fujimoto and M. Wislez contributed equally to this work. M. Wislez is apostdoctoral fellow of La Fondation de France and La Societe de Pneumonologie deLangue Francaise.
Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Requests for reprints: Jonathan M. Kurie, The University of Texas M.D. AndersonCancer Center, Box 432, 1515 Holcombe Boulevard, Houston, TX 77030. Phone: 713-792-6363; Fax: 713-796-8655; E-mail: [email protected].
I2005 American Association for Cancer Research.doi:10.1158/0008-5472.CAN-05-1977
Cancer Res 2005; 65: (24). December 15, 2005 11478 www.aacrjournals.org
Research Article
Research. on August 24, 2015. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from

gefitinib or vehicle were imaged by micro–computed tomography (micro-CT) at the beginning and completion of treatment to examine changes in
lesion size as described previously (18). Mice were killed at the completion
of therapy. At that time, an investigator who was blinded to the identity of
the treatment groups counted lesions visible on lung pleural surfaces. Onelung from each mouse was then frozen for protein extraction, and the other
was formalin fixed for immunohistochemical analysis.
Cell lines. The LKR-13 and LKR-10 cell lines (provided by Dr. Tyler Jacks)
were derived by serial passage of minced lung adenocarcinoma tissues fromtwo tumors isolated from separate lobes of the same KrasLA1 mouse. These
cells and the human NSCLC cell lines (H1299, HCC827, H3255, H1819,
H4006, and HCC2279) were passaged in RPMI 1640 supplemented with 10%
fetal bovine serum on standard Falcon (Bedford, MA) plasticware.Immortalized human bronchial epithelial cells (HBEC) and KRAS/HBECs
were cultured with keratinocyte serum-free medium containing bovine
pituitary extract and recombinant EGF (Gibco). KRAS/HBECs were derivedby stably infecting parental HBECs with the retroviral vector pBabe-
hyg-KRAS2-V12. H1299 cells were transfected with a vector (pcDNA3.1)
expressing wild-type or mutant (D746-750) EGFR , and single-cell subclones
were selected in G418. Expression of the exogenous (wild-type or mutant)EGFR mRNA was confirmed in single-cell subclones by real-time PCR (RT-
PCR) analysis of total cellular RNA (1 Ag) using primers spanning exons 18
and 19 and by direct sequencing of the PCR products. PCR products of the
D746-750 EGFR transfectants lacked 15 bp corresponding to the deletionmutation. EGFR-transfected H1299 cells were subsequently transfected with
a vector expressing ErbB3, and single-cell subclones of the double
transfectants (EGFR and ErbB3) were selected in G418 and hygromycin.All cells were maintained at 37jC in a humidified atmosphere containing
5% CO2.
Antibodies. For Western blotting and immunohistochemical analysis,
we used antibodies specific for EGFR (NeoMarkers, Fremont, CA), ErbB2 andErbB3 (Santa Cruz Biotechnology, Santa Cruz, CA), h-actin (Sigma, St. Louis,
MO), and Tyr1289-phosphorylated ErbB3 (pErbB3), Ser473-phosphorylated
AKT (pAKT), actin, and AKT (Cell Signaling Technologies, Beverly, MA).
RNA analysis. Total RNA was prepared from whole mouse lung tissues
and cell lines using a RNeasy mini kit (Qiagen, Valencia, CA) according to
the manufacturer’s protocol. Quantitative RT-PCR (Q-PCR) was done using
1 Ag of each RNA sample, which was reverse transcribed and used in a
PCR reaction to measure the transcript levels of each gene. Levels were
normalized relative to those of h-glucuronidase or glyceraldehyde-3-
phosphate dehydrogenase (GAPDH) in each sample. Gene expression was
assayed using Assays-On-Demand (Applied Biosystems, Foster City, CA)
according to the manufacturer’s protocol using a Stratagene (La Jolla, CA)
Mx3000P RT-PCR machine.
Cell proliferation. Cell proliferation was measured using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma) color-
imetric dye reduction method. Cells were seeded at a density of 1 � 103 to
5 � 103 per well (10 replicate wells per assay condition) in 0.1 mL medium,treated for 5 days with different dosages of gefitinib, and subjected to MTT
assays.
Immunoblot analysis. Whole lung tissues were homogenized using a
Tearor homogenizer in Reingeauld lysis buffer [200 mmol/L Tris (pH 7.4),
137 mmol/L NaCl, 2 mmol/L EDTA, 10% glycerol, 1% Triton X-100, 100
Amol/L phenylmethylsulfonyl fluoride, 100 nmol/L okadaic acid, 1 mmol/
L sodium orthovanadate, and protease inhibitor cocktail]. Cell lines were
washed twice with ice-cold PBS and scraped in radioimmunoprecipitation
assay buffer [50 mmol/L Tris-HCl (pH 7.4), 150 mmol/L NaCl, 1 mmol/L
EDTA, 0.25% sodium deoxycholate, 1% NP40, 1 mmol/L phenylmethyl-
sulfonyl fluoride, 1 mmol/L sodium orthovanadate, 1 mmol/L NaF, and
protease inhibitor cocktail]. Lysates were cleared by centrifugation, and
protein concentrations were measured with a protein assay kit (Bio-Rad,
Hercules, CA). Equal amounts of protein were resolved by SDS-PAGE and
transferred to polyvinylidene difluoride membranes. The membranes were
then immunoblotted overnight at 4jC with primary antibodies in TBS
containing 5% nonfat dry milk. Antibody binding was detected with an
enhanced chemiluminescence kit (Amersham, Piscataway, NJ) according
to the manufacturer’s directions.
Mutational analysis of tumor samples and cell lines. Approximately103 tumor cells were microdissected from sequential 8 Am-thick tissue
sections that were stained with H&E, fixed in formalin, and embedded in
paraffin. DNA was extracted from tumor samples and cell lines using 25 ALPico Pure DNA extraction solution (Arcturus, Mountain View, CA)containing proteinase K and incubated at 65 jC for 24 hours. Exons 18
to 21 of EGFR and codon 12 of KRAS were amplified by PCR using intron-
based primers as described previously (13). All PCR products were directly
sequenced using the Applied Biosystems PRISM dye terminator cyclesequencing method (Perkin-Elmer, Wellesley, CA). All sequence variants
were confirmed by independent PCR amplifications from at least two
independent microdissections and were sequenced in both directions.
Tissue microarrays. Tissue microarrays were constructed with coresfrom formalin-fixed, paraffin-embedded blocks, and triplicate core sampleswere included for each tumor. To examine changes in the expression ofErbB3 and pAKT with malignant progression, an array was constructed withspecimens of mouse normal lung tissue (n = 30), AAH (n = 40), adenoma(n = 206), and adenocarcinoma (n = 11) according to the histologic criteriaestablished (17). To examine changes in the expression of ErbB3 and pAKTresulting from gefitinib treatment, an array was constructed from all lesionsidentified by histologic analysis from the mice treated with gefitinib orvehicle. Each lesion was sampled with a single core 1 mm in diameter, asthe lesions were too small to permit multiple cores to be obtained.
For human studies, we constructed an array using tumor samples frompatients before oral administration of gefitinib (250 mg/d) at The Universityof Texas M.D. Anderson Cancer Center. Only patients for whom pertinentclinical data were available were included. Response to treatment wasmeasured with the Response Evaluation Criteria in Solid Tumors, andpatients with more than one lesion were categorized according to the lesionthat achieved the best response. Stabilization of disease was defined as anabsence of change in lesion size for at least 6 months following treatmentinitiation.
Immunohistochemistry. For immunohistochemical analyses, 4-Amsections were deparaffinized, rehydrated, and washed with PBS as described
previously (18). Antigens were retrieved with 0.01 mol/L citrate buffer (pH 6;DakoCytomation) for 30 minutes in a steamer. Samples were blocked for
endogenous activity with 3% hydrogen peroxide/PBS, avidin/biotin solution
(Zymed, Carlsbad, CA), and Dako serum-free protein block (DakoCytoma-
tion, Carpinteria, CA) before incubation with the primary antibodiesovernight at 4jC. Standard avidin/biotin immunoperoxidase methods, with
diaminobenzidine as the chromogen, were used for detection. As negative
controls for the specificity of the immunostaining results, we pretreated
the samples with blocking peptides. ErbB3-blocking peptides were the17 COOH-terminal amino acids.
Staining was quantified by two investigators (M.W. and I.I.W.) who wereblinded to the identity of the treatment groups. Staining in each sample wasquantified from a single tissue section using a combined score based onstaining intensity � extension. Staining intensity was graded as undetect-able (0), weak (1), medium (2), or strong (3). Staining extension was gradedas the percentage of positive cells per square at �20 magnification. Thetumor score was defined as the highest of the three core samples, andresults were presented as mean (SE).
Statistical analysis. The immunostaining scores, patient age, tumornumbers, and tumor volumes were considered continuous variables andwere compared using the Kruskal-Wallis test followed by the Mann-Whitneynonparametric test with Bonferroni correction. P = 0.05 was consideredsignificant for two pair-wise comparisons, 0.017 for three pair-wisecomparisons, and 0.012 for four pair-wise comparisons. Fisher’s exact testwas used to compare categorical variables, and P = 0.05 was consideredsignificant. Data were processed with StatView and Survival Tools softwareversion 5.0 (Abacus Concepts, Berkeley, CA).
Results
Epidermal growth factor receptor inhibition suppresses theexpansion of alveolar neoplasia in KrasLA1 mice. We treatedKrasLA1 mice with the TKI gefitinib. Mice were treated daily by
ErbB and Their Ligands in Gefitinib Sensitivity
www.aacrjournals.org 11479 Cancer Res 2005; 65: (24). December 15, 2005
Research. on August 24, 2015. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from
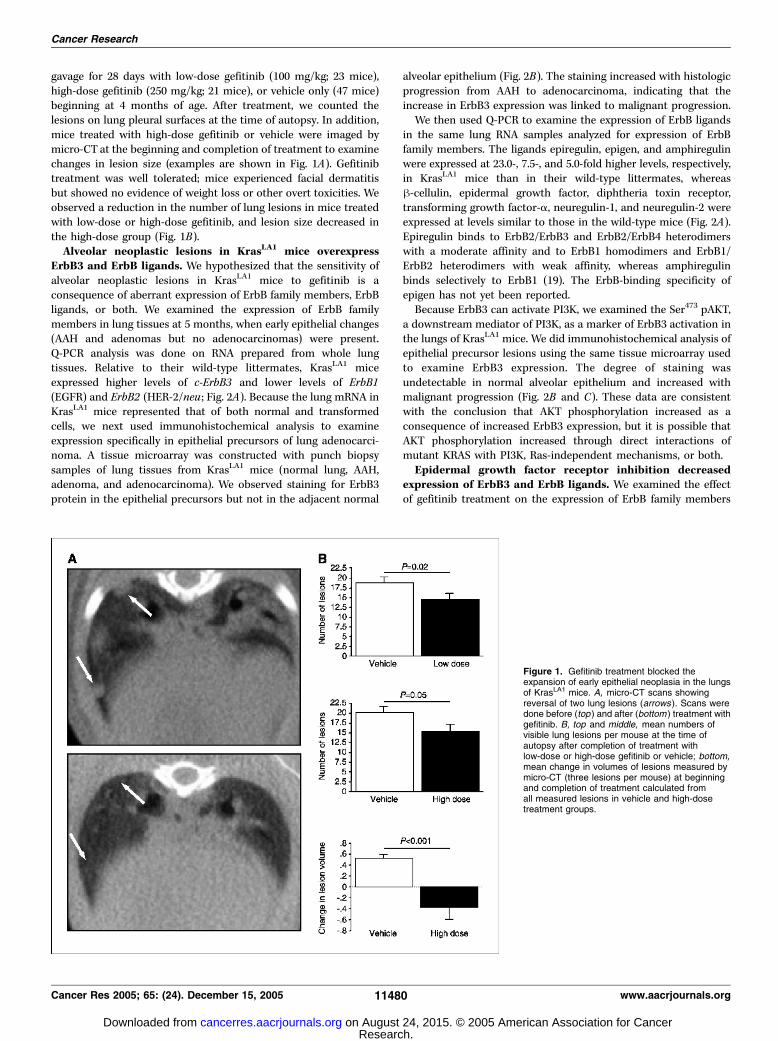
gavage for 28 days with low-dose gefitinib (100 mg/kg; 23 mice),high-dose gefitinib (250 mg/kg; 21 mice), or vehicle only (47 mice)beginning at 4 months of age. After treatment, we counted thelesions on lung pleural surfaces at the time of autopsy. In addition,mice treated with high-dose gefitinib or vehicle were imaged bymicro-CT at the beginning and completion of treatment to examinechanges in lesion size (examples are shown in Fig. 1A). Gefitinibtreatment was well tolerated; mice experienced facial dermatitisbut showed no evidence of weight loss or other overt toxicities. Weobserved a reduction in the number of lung lesions in mice treatedwith low-dose or high-dose gefitinib, and lesion size decreased inthe high-dose group (Fig. 1B).Alveolar neoplastic lesions in KrasLA1 mice overexpress
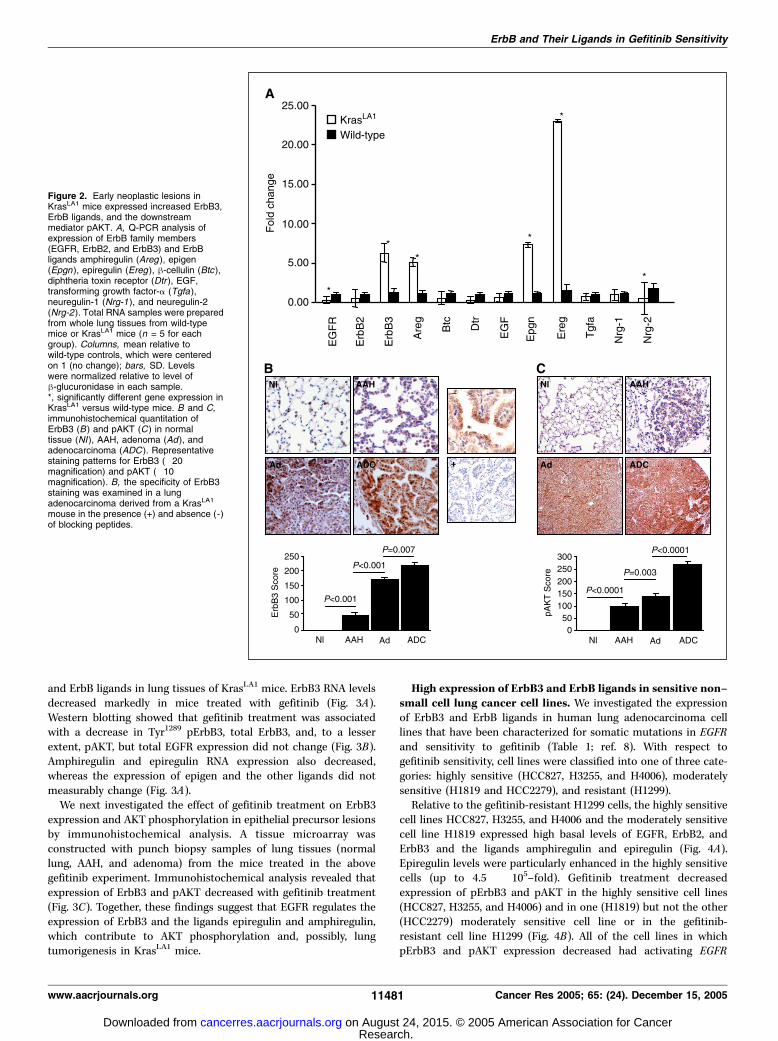
ErbB3 and ErbB ligands. We hypothesized that the sensitivity ofalveolar neoplastic lesions in KrasLA1 mice to gefitinib is aconsequence of aberrant expression of ErbB family members, ErbBligands, or both. We examined the expression of ErbB familymembers in lung tissues at 5 months, when early epithelial changes(AAH and adenomas but no adenocarcinomas) were present.Q-PCR analysis was done on RNA prepared from whole lungtissues. Relative to their wild-type littermates, KrasLA1 miceexpressed higher levels of c-ErbB3 and lower levels of ErbB1(EGFR) and ErbB2 (HER-2/neu ; Fig. 2A). Because the lung mRNA inKrasLA1 mice represented that of both normal and transformedcells, we next used immunohistochemical analysis to examineexpression specifically in epithelial precursors of lung adenocarci-noma. A tissue microarray was constructed with punch biopsysamples of lung tissues from KrasLA1 mice (normal lung, AAH,adenoma, and adenocarcinoma). We observed staining for ErbB3protein in the epithelial precursors but not in the adjacent normal
alveolar epithelium (Fig. 2B). The staining increased with histologicprogression from AAH to adenocarcinoma, indicating that theincrease in ErbB3 expression was linked to malignant progression.We then used Q-PCR to examine the expression of ErbB ligands
in the same lung RNA samples analyzed for expression of ErbBfamily members. The ligands epiregulin, epigen, and amphiregulinwere expressed at 23.0-, 7.5-, and 5.0-fold higher levels, respectively,in KrasLA1 mice than in their wild-type littermates, whereash-cellulin, epidermal growth factor, diphtheria toxin receptor,transforming growth factor-a, neuregulin-1, and neuregulin-2 wereexpressed at levels similar to those in the wild-type mice (Fig. 2A).Epiregulin binds to ErbB2/ErbB3 and ErbB2/ErbB4 heterodimerswith a moderate affinity and to ErbB1 homodimers and ErbB1/ErbB2 heterodimers with weak affinity, whereas amphiregulinbinds selectively to ErbB1 (19). The ErbB-binding specificity ofepigen has not yet been reported.Because ErbB3 can activate PI3K, we examined the Ser473 pAKT,
a downstream mediator of PI3K, as a marker of ErbB3 activation inthe lungs of KrasLA1 mice. We did immunohistochemical analysis ofepithelial precursor lesions using the same tissue microarray usedto examine ErbB3 expression. The degree of staining wasundetectable in normal alveolar epithelium and increased withmalignant progression (Fig. 2B and C). These data are consistentwith the conclusion that AKT phosphorylation increased as aconsequence of increased ErbB3 expression, but it is possible thatAKT phosphorylation increased through direct interactions ofmutant KRAS with PI3K, Ras-independent mechanisms, or both.Epidermal growth factor receptor inhibition decreased
expression of ErbB3 and ErbB ligands. We examined the effectof gefitinib treatment on the expression of ErbB family members
Figure 1. Gefitinib treatment blocked theexpansion of early epithelial neoplasia in the lungsof KrasLA1 mice. A, micro-CT scans showingreversal of two lung lesions (arrows ). Scans weredone before (top ) and after (bottom ) treatment withgefitinib. B, top and middle, mean numbers ofvisible lung lesions per mouse at the time ofautopsy after completion of treatment withlow-dose or high-dose gefitinib or vehicle; bottom,mean change in volumes of lesions measured bymicro-CT (three lesions per mouse) at beginningand completion of treatment calculated fromall measured lesions in vehicle and high-dosetreatment groups.
Cancer Research
Cancer Res 2005; 65: (24). December 15, 2005 11480 www.aacrjournals.org
Research. on August 24, 2015. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from
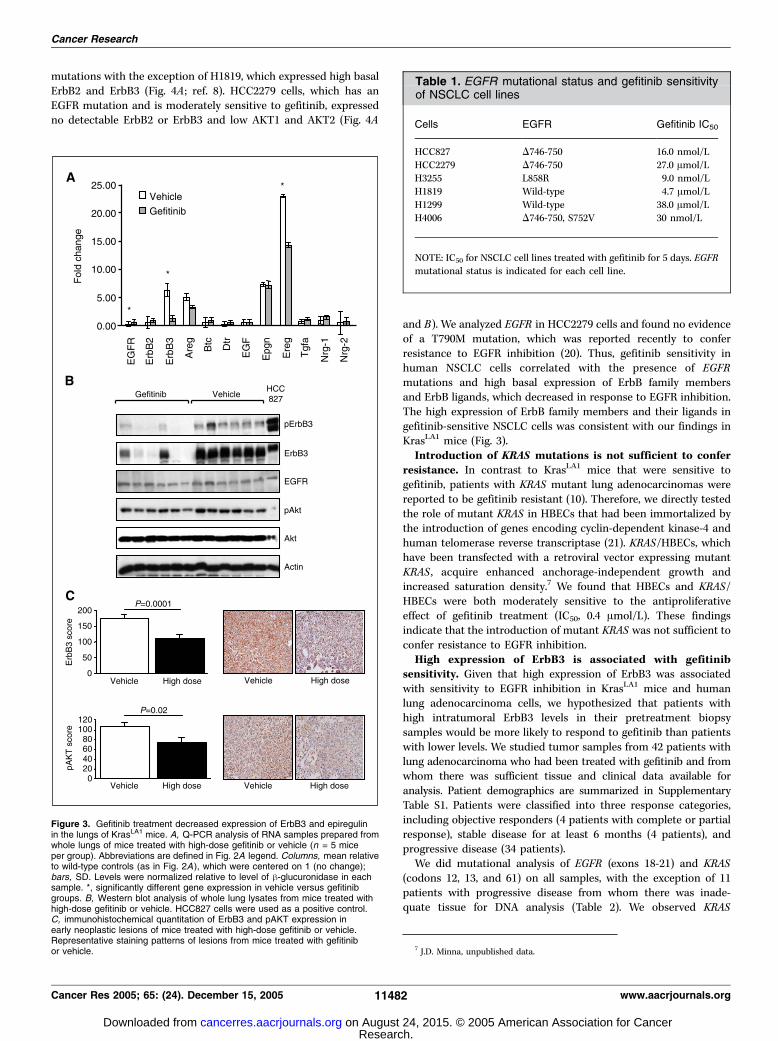
and ErbB ligands in lung tissues of KrasLA1 mice. ErbB3 RNA levelsdecreased markedly in mice treated with gefitinib (Fig. 3A).Western blotting showed that gefitinib treatment was associatedwith a decrease in Tyr1289 pErbB3, total ErbB3, and, to a lesserextent, pAKT, but total EGFR expression did not change (Fig. 3B).Amphiregulin and epiregulin RNA expression also decreased,whereas the expression of epigen and the other ligands did notmeasurably change (Fig. 3A).We next investigated the effect of gefitinib treatment on ErbB3
expression and AKT phosphorylation in epithelial precursor lesionsby immunohistochemical analysis. A tissue microarray wasconstructed with punch biopsy samples of lung tissues (normallung, AAH, and adenoma) from the mice treated in the abovegefitinib experiment. Immunohistochemical analysis revealed thatexpression of ErbB3 and pAKT decreased with gefitinib treatment(Fig. 3C). Together, these findings suggest that EGFR regulates theexpression of ErbB3 and the ligands epiregulin and amphiregulin,which contribute to AKT phosphorylation and, possibly, lungtumorigenesis in KrasLA1 mice.
High expression of ErbB3 and ErbB ligands in sensitive non–small cell lung cancer cell lines. We investigated the expressionof ErbB3 and ErbB ligands in human lung adenocarcinoma celllines that have been characterized for somatic mutations in EGFRand sensitivity to gefitinib (Table 1; ref. 8). With respect togefitinib sensitivity, cell lines were classified into one of three cate-gories: highly sensitive (HCC827, H3255, and H4006), moderatelysensitive (H1819 and HCC2279), and resistant (H1299).Relative to the gefitinib-resistant H1299 cells, the highly sensitive
cell lines HCC827, H3255, and H4006 and the moderately sensitivecell line H1819 expressed high basal levels of EGFR, ErbB2, andErbB3 and the ligands amphiregulin and epiregulin (Fig. 4A).Epiregulin levels were particularly enhanced in the highly sensitivecells (up to 4.5 � 105–fold). Gefitinib treatment decreasedexpression of pErbB3 and pAKT in the highly sensitive cell lines(HCC827, H3255, and H4006) and in one (H1819) but not the other(HCC2279) moderately sensitive cell line or in the gefitinib-resistant cell line H1299 (Fig. 4B). All of the cell lines in whichpErbB3 and pAKT expression decreased had activating EGFR
Figure 2. Early neoplastic lesions inKrasLA1 mice expressed increased ErbB3,ErbB ligands, and the downstreammediator pAKT. A, Q-PCR analysis ofexpression of ErbB family members(EGFR, ErbB2, and ErbB3) and ErbBligands amphiregulin (Areg ), epigen(Epgn ), epiregulin (Ereg ), h-cellulin (Btc),diphtheria toxin receptor (Dtr ), EGF,transforming growth factor-a (Tgfa),neuregulin-1 (Nrg-1 ), and neuregulin-2(Nrg-2 ). Total RNA samples were preparedfrom whole lung tissues from wild-typemice or KrasLA1 mice (n = 5 for eachgroup). Columns, mean relative towild-type controls, which were centeredon 1 (no change); bars, SD. Levelswere normalized relative to level ofh-glucuronidase in each sample.*, significantly different gene expression inKrasLA1 versus wild-type mice. B and C,immunohistochemical quantitation ofErbB3 (B ) and pAKT (C ) in normaltissue (NI), AAH, adenoma (Ad ), andadenocarcinoma (ADC ). Representativestaining patterns for ErbB3 (�20magnification) and pAKT (�10magnification). B, the specificity of ErbB3staining was examined in a lungadenocarcinoma derived from a KrasLA1
mouse in the presence (+) and absence (- )of blocking peptides.
ErbB and Their Ligands in Gefitinib Sensitivity
www.aacrjournals.org 11481 Cancer Res 2005; 65: (24). December 15, 2005
Research. on August 24, 2015. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from
mutations with the exception of H1819, which expressed high basalErbB2 and ErbB3 (Fig. 4A ; ref. 8). HCC2279 cells, which has anEGFR mutation and is moderately sensitive to gefitinib, expressedno detectable ErbB2 or ErbB3 and low AKT1 and AKT2 (Fig. 4A
and B). We analyzed EGFR in HCC2279 cells and found no evidenceof a T790M mutation, which was reported recently to conferresistance to EGFR inhibition (20). Thus, gefitinib sensitivity inhuman NSCLC cells correlated with the presence of EGFRmutations and high basal expression of ErbB family membersand ErbB ligands, which decreased in response to EGFR inhibition.The high expression of ErbB family members and their ligands ingefitinib-sensitive NSCLC cells was consistent with our findings inKrasLA1 mice (Fig. 3).Introduction of KRAS mutations is not sufficient to confer
resistance. In contrast to KrasLA1 mice that were sensitive togefitinib, patients with KRAS mutant lung adenocarcinomas werereported to be gefitinib resistant (10). Therefore, we directly testedthe role of mutant KRAS in HBECs that had been immortalized bythe introduction of genes encoding cyclin-dependent kinase-4 andhuman telomerase reverse transcriptase (21). KRAS/HBECs, whichhave been transfected with a retroviral vector expressing mutantKRAS , acquire enhanced anchorage-independent growth andincreased saturation density.7 We found that HBECs and KRAS/HBECs were both moderately sensitive to the antiproliferativeeffect of gefitinib treatment (IC50, 0.4 Amol/L). These findingsindicate that the introduction of mutant KRAS was not sufficient toconfer resistance to EGFR inhibition.High expression of ErbB3 is associated with gefitinib
sensitivity. Given that high expression of ErbB3 was associatedwith sensitivity to EGFR inhibition in KrasLA1 mice and humanlung adenocarcinoma cells, we hypothesized that patients withhigh intratumoral ErbB3 levels in their pretreatment biopsysamples would be more likely to respond to gefitinib than patientswith lower levels. We studied tumor samples from 42 patients withlung adenocarcinoma who had been treated with gefitinib and fromwhom there was sufficient tissue and clinical data available foranalysis. Patient demographics are summarized in SupplementaryTable S1. Patients were classified into three response categories,including objective responders (4 patients with complete or partialresponse), stable disease for at least 6 months (4 patients), andprogressive disease (34 patients).We did mutational analysis of EGFR (exons 18-21) and KRAS
(codons 12, 13, and 61) on all samples, with the exception of 11patients with progressive disease from whom there was inade-quate tissue for DNA analysis (Table 2). We observed KRAS
Figure 3. Gefitinib treatment decreased expression of ErbB3 and epiregulinin the lungs of KrasLA1 mice. A, Q-PCR analysis of RNA samples prepared fromwhole lungs of mice treated with high-dose gefitinib or vehicle (n = 5 miceper group). Abbreviations are defined in Fig. 2A legend. Columns, mean relativeto wild-type controls (as in Fig. 2A), which were centered on 1 (no change);bars, SD. Levels were normalized relative to level of h-glucuronidase in eachsample. *, significantly different gene expression in vehicle versus gefitinibgroups. B, Western blot analysis of whole lung lysates from mice treated withhigh-dose gefitinib or vehicle. HCC827 cells were used as a positive control.C, immunohistochemical quantitation of ErbB3 and pAKT expression inearly neoplastic lesions of mice treated with high-dose gefitinib or vehicle.Representative staining patterns of lesions from mice treated with gefitinibor vehicle.
Table 1. EGFR mutational status and gefitinib sensitivityof NSCLC cell lines
Cells EGFR Gefitinib IC50
HCC827 D746-750 16.0 nmol/L
HCC2279 D746-750 27.0 Amol/LH3255 L858R 9.0 nmol/L
H1819 Wild-type 4.7 Amol/L
H1299 Wild-type 38.0 Amol/LH4006 D746-750, S752V 30 nmol/L
NOTE: IC50 for NSCLC cell lines treated with gefitinib for 5 days. EGFRmutational status is indicated for each cell line.
7 J.D. Minna, unpublished data.
Cancer Research
Cancer Res 2005; 65: (24). December 15, 2005 11482 www.aacrjournals.org
Research. on August 24, 2015. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from

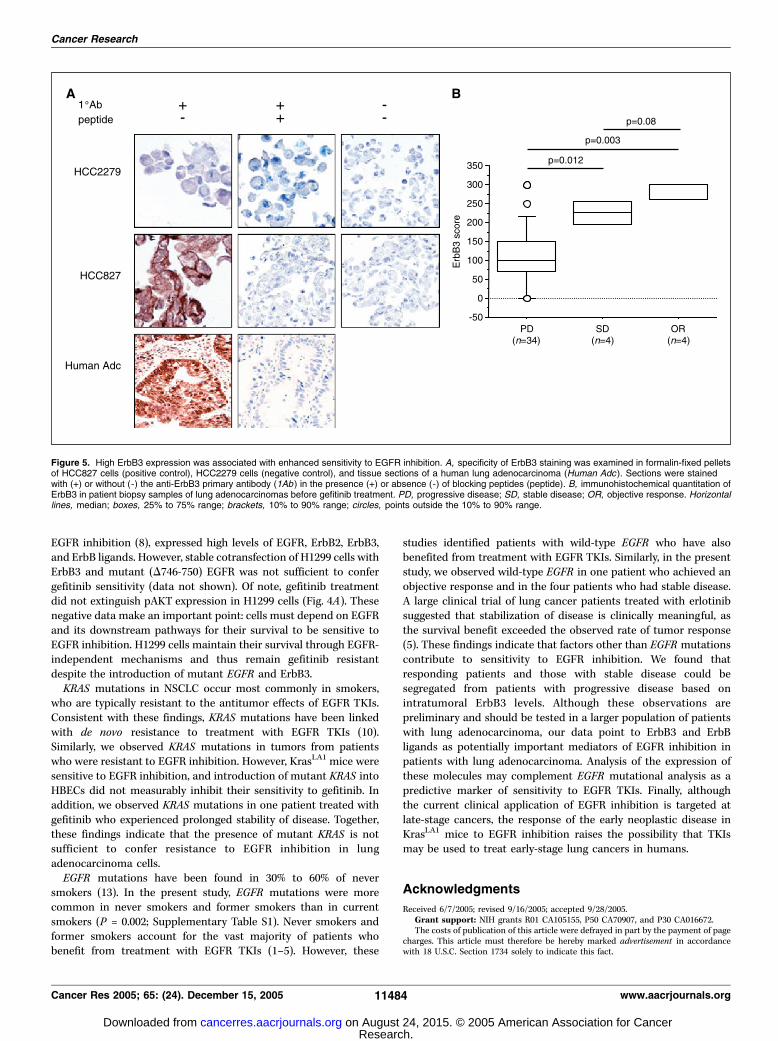
mutations in tumors from patients with progressive disease [6 of23 (26%)] and stable disease [1 of 4 (25%)] but not in respondingtumors. We observed EGFR mutations in responding tumors [3 of4 (75%)] and those from patients with progressive disease [2 of 23(8%)] but not in those from patients with stable disease.Immunohistochemical analysis was done to quantify ErbB3expression in biopsy samples using conditions that specificallydetected ErbB3 (Fig. 5A). ErbB3 expression was higher in tumorsfrom patients who achieved an objective response or stabilizationof disease than in those with progressive disease (Fig. 5B). Thus,in this group of patients, EGFR mutational analysis did notidentify one of the four patients who achieved an objectiveresponse or any of those whose disease stabilized. In contrast,ErbB3 expression clearly segregated patients with progressivedisease from those who achieved tumor response or diseasestabilization after treatment.
Discussion
The findings presented here provide the first evidence that lungadenocarcinoma cells that depend on EGFR for survivalconstitutively activate the receptor through a combination ofgenetic mutations and overexpression of EGFR dimeric partnersand their ligands. In KrasLA1 mice, high expression of ErbB3,amphiregulin, and epiregulin in the lung was dependent on EGFR
activation. These findings suggest the presence of an autocrineloop in which EGFR activates expression of ErbB3 and ErbBligands, which in turn activate EGFR. However, the introduction ofmutant EGFR into H1299 cells was not sufficient to increase theexpression of ErbB3, amphiregulin, or epiregulin, suggesting thatfactors other than EGFR activation contributed to the highexpression of these proteins in NSCLC cells with somaticallyacquired EGFR mutations.Consistent with our findings in KrasLA1 mice, human tumor cells
with KRAS mutations express high levels of ErbB ligands (22–24).The levels of ErbB3 mRNA are significantly higher in human lungadenocarcinomas than in normal tissues (P < 0.001; ref. 25).However, high ErbB3 expression in human lung adenocarcinomacells does not correlate with the presence of KRAS mutations (25).Thus, lung adenocarcinomas arising in humans and KrasLA1 micediffer with respect to the role of KRAS mutations in the regulationof ErbB3 expression. Nevertheless, the observation that both mouseand human lung adenocarcinomas have high expression of ErbB3underscores its importance in tumorigenesis. Moreover, ErbB3 isrequired to drive breast epithelial cell proliferation, and over-expression of ErbB3 leads to the development of lung adenocarci-nomas in mice and is associated with poor prognosis in patientswith NSCLC (26–28).Several of our findings suggest that EGFR mutations are neither
sufficient nor required for sensitivity to EGFR inhibition and thatother ErbBs and ErbB ligands have an important role in thissensitivity. First, gefitinib-sensitive NSCLC cells had high expressionof ErbB2, ErbB3, and ErbB ligands. Second, pharmacologicinhibition of EGFR in KrasLA1 mice decreased the expression ofErbB3, amphiregulin, and epiregulin and blocked the expansion ofalveolar epithelial neoplasia. Third, HCC2279 cells, which expressundetectable levels of ErbB2 and ErbB3, were only moderatelysensitive to gefitinib despite having an activating mutation in EGFR ;in addition, they did not have the T790M EGFR mutation, whichconfers resistance to EGFR inhibition (20). Fourth, H1819 cells,which have no EGFR mutations and are moderately sensitive to
Figure 4. Gefitinib-sensitive human lung adenocarcinoma cells expressed highlevels of ErbBs and ErbB ligands. A, left, Q-PCR analysis of total RNA. Resultsof Q-PCR were normalized relative to levels of GAPDH in each sample andexpressed relative to wild-type EGFR-expressing H1299 cells, which werecentered on 1 (no change). Columns, mean plotted on log scale; bars, SD.Right, Western blotting of extracts prepared from cell lines. B, Western blottingdone on lung adenocarcinoma cells treated with (+) or without (- ) 1 Amol/Lgefitinib for 48 hours.
Table 2. EGFR and KRAS mutational status of tumorsfrom lung cancer patients treated with gefitinib
Category No. (%) patients
Progressive
disease
(n = 23)
Stable
disease
(n = 4)
Objective
response
(n = 4)
KRAS 6 (26) 1 (25) 0 (0)
G12T 5 1 0G12D 1 0 0
EGFR 2 (9) 0 (0) 3 (75)
D746-750 1 0 3
Leu692Phe 1 0 0
NOTE: Mutational analysis of KRAS (codons 12, 13, and 61) and EGFR
(exons 18-21) was done on tumor DNA as described in Materials and
Methods. Patients were grouped into three categories based onresponse to EGFR inhibitor therapy: progressive disease, stable
disease, and objective response. Specific mutations detected in tumors
are listed.
ErbB and Their Ligands in Gefitinib Sensitivity
www.aacrjournals.org 11483 Cancer Res 2005; 65: (24). December 15, 2005
Research. on August 24, 2015. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from
EGFR inhibition (8), expressed high levels of EGFR, ErbB2, ErbB3,and ErbB ligands. However, stable cotransfection of H1299 cells withErbB3 and mutant (D746-750) EGFR was not sufficient to confergefitinib sensitivity (data not shown). Of note, gefitinib treatmentdid not extinguish pAKT expression in H1299 cells (Fig. 4A). Thesenegative data make an important point: cells must depend on EGFRand its downstream pathways for their survival to be sensitive toEGFR inhibition. H1299 cells maintain their survival through EGFR-independent mechanisms and thus remain gefitinib resistantdespite the introduction of mutant EGFR and ErbB3.KRAS mutations in NSCLC occur most commonly in smokers,
who are typically resistant to the antitumor effects of EGFR TKIs.Consistent with these findings, KRAS mutations have been linkedwith de novo resistance to treatment with EGFR TKIs (10).Similarly, we observed KRAS mutations in tumors from patientswho were resistant to EGFR inhibition. However, KrasLA1 mice weresensitive to EGFR inhibition, and introduction of mutant KRAS intoHBECs did not measurably inhibit their sensitivity to gefitinib. Inaddition, we observed KRAS mutations in one patient treated withgefitinib who experienced prolonged stability of disease. Together,these findings indicate that the presence of mutant KRAS is notsufficient to confer resistance to EGFR inhibition in lungadenocarcinoma cells.EGFR mutations have been found in 30% to 60% of never
smokers (13). In the present study, EGFR mutations were morecommon in never smokers and former smokers than in currentsmokers (P = 0.002; Supplementary Table S1). Never smokers andformer smokers account for the vast majority of patients whobenefit from treatment with EGFR TKIs (1–5). However, these
studies identified patients with wild-type EGFR who have alsobenefited from treatment with EGFR TKIs. Similarly, in the presentstudy, we observed wild-type EGFR in one patient who achieved anobjective response and in the four patients who had stable disease.A large clinical trial of lung cancer patients treated with erlotinibsuggested that stabilization of disease is clinically meaningful, asthe survival benefit exceeded the observed rate of tumor response(5). These findings indicate that factors other than EGFR mutationscontribute to sensitivity to EGFR inhibition. We found thatresponding patients and those with stable disease could besegregated from patients with progressive disease based onintratumoral ErbB3 levels. Although these observations arepreliminary and should be tested in a larger population of patientswith lung adenocarcinoma, our data point to ErbB3 and ErbBligands as potentially important mediators of EGFR inhibition inpatients with lung adenocarcinoma. Analysis of the expression ofthese molecules may complement EGFR mutational analysis as apredictive marker of sensitivity to EGFR TKIs. Finally, althoughthe current clinical application of EGFR inhibition is targeted atlate-stage cancers, the response of the early neoplastic disease inKrasLA1 mice to EGFR inhibition raises the possibility that TKIsmay be used to treat early-stage lung cancers in humans.
Acknowledgments
Received 6/7/2005; revised 9/16/2005; accepted 9/28/2005.Grant support: NIH grants R01 CA105155, P50 CA70907, and P30 CA016672.The costs of publication of this article were defrayed in part by the payment of page
charges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.
Figure 5. High ErbB3 expression was associated with enhanced sensitivity to EGFR inhibition. A, specificity of ErbB3 staining was examined in formalin-fixed pelletsof HCC827 cells (positive control), HCC2279 cells (negative control), and tissue sections of a human lung adenocarcinoma (Human Adc ). Sections were stainedwith (+) or without (-) the anti-ErbB3 primary antibody (1Ab ) in the presence (+) or absence (-) of blocking peptides (peptide). B, immunohistochemical quantitation ofErbB3 in patient biopsy samples of lung adenocarcinomas before gefitinib treatment. PD, progressive disease; SD, stable disease; OR, objective response. Horizontallines, median; boxes, 25% to 75% range; brackets, 10% to 90% range; circles, points outside the 10% to 90% range.
Cancer Research
Cancer Res 2005; 65: (24). December 15, 2005 11484 www.aacrjournals.org
Research. on August 24, 2015. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from

ErbB and Their Ligands in Gefitinib Sensitivity
www.aacrjournals.org 11485 Cancer Res 2005; 65: (24). December 15, 2005
References
1. Kosaka T, Yatanabe Y, Endoh H, Kuwano H, TakahashiT, Mitsudomi T. Mutations of the epidermal growthfactor receptor gene in lung cancer: biological andclinical implications. Cancer Res 2004;64:8919–23.
2. Lynch TJ, Bell DW, Sordella R, et al. Activatingmutations in the epidermal growth factor receptorunderlying responsiveness of non-small-cell lung cancerto gefitinib. N Engl J Med 2004;350:2129–39.
3. Paez JG, Janne PA, Lee JC, et al. EGFR mutations inlung cancer: correlation with clinical response togefitinib therapy. Science 2004;304:1497–500.
4. Pao W, Miller V, Zazowski M, et al. EGF receptor genemutations are common in lung cancers from ‘‘neversmokers’’ and are associated with sensitivity of tumorsto gefitinib and erlotinib. Proc Natl Acad Sci U S A 2004;101:13306–11.
5. Shepherd FA, Periera J, Ciuleanu TE, et al. Erlotinib inpreviously treated non-small cell lung cancer. N Engl JMed 2005;353:123–32.
6. Schlessinger J. Ligand-induced, receptor-mediateddimerization and activation of EGF receptor. Cell 2002;110:669–72.
7. Citri A, Skaria KB, Yarden Y. The deaf and the dumb:the biology of ErbB-2 and ErbB-3. Exp Cell Res 2003;284:54–65.
8. Amann J, Kalyankrishna S, Massion PP, et al. Aberrantepidermal growth factor receptor signaling and en-hanced sensitivity to EGFR inhibitors in lung cancer.Cancer Res 2005;65:226–35.
9. Engelman JA, Yanne PA, Mermel C, et al. ErbB-3mediates phosphoinositide 3-kinase activity in gefitinib-sensitive non-small cell lung cancer cell lines. Proc NatlAcad Sci U S A 2005;102:3788–93.
10. Pao W, Wang TY, Riely GJ, et al. KRAS mutations and
primary resistance of lung adenocarcinomas to gefitinibor erlotinib. PLoS Med 2005;2:57–61.
11. Mitsudomi T, Viallet J, Mulshine JL, Linnoila I,Minna JD, Gazdar AF. Mutations of ras genesdistinguish a subset of non-small-cell lung cancer celllines from small-cell lung cancer cell lines. Oncogene1991;6:1353–62.
12. Sugio K, Kishimoto Y, Virmani AK, Hung JY, GazdarAF. Kras gene mutations as a prognostic marker inadenocarcinoma of the human lung without lymphnode metastasis. Cancer Res 1992;52:2903–6.
13. Shigematsu H, Lin L, Takahashi T, et al. Clinical andbiological features associated with epidermal growthfactor receptor gene mutations in lung cancers. J NatlCancer Inst 2005;97:339–46.
14. Fisher GH, Wellen SL, Klimstra D, et al. Inductionand apoptotic regression of lung adenocarcinomas byregulation of a Kras transgene in the presence andabsence of tumor suppressor genes. Genes Dev 2001;15:3249–62.
15. Guerra C, Mijimolle N, Dhawahir A, et al. Tumorinduction by an endogenous Kras oncogene is highlydependent on cellular context. Cancer Cell 2003;4:111–20.
16. Jackson EL, Willis N, Mercer K, et al. Analysis of lungtumor initiation and progression using conditionalexpression of oncogenic Kras. Genes Dev 2001;15:3243–8.
17. Johnson L, Mercer K, Greenbaum D, et al. Somaticactivation of the Kras oncogene causes early onset lungcancer in mice. Nature 2001;410:1111–6.
18. Wislez M, Spencer ML, Izzo JG, et al. Inhibition ofmammalian target of rapamycin reverses alveolarepithelial neoplasia induced by oncogenic Kras. CancerRes 2005;65:3226–35.
19. Jones JT, Akita RW, Slikowski MX. Binding specific-ities and affinities of EGF domains for ErbB receptors.FEBS Lett 1999;447:227–31.
20. Kobayashi S, Boggon TJ, Dayaram T, et al. EGFRmutation and resistance of non-small cell lung cancer togefitinib. N Engl J Med 2005;352:786–92.
21. Ramirez RD, Sheridan S, Girard L, et al. Immortali-zation of human bronchial epithelial cells in the absenceof viral oncoproteins. Cancer Res 2004;64:9027–34.
22. Baba I, Shirasawa S, Iwamoto R, et al. Involve-ment of deregulated epiregulin expression in tumor-igenesis in vivo through activated Ki-Ras signalingpathway in human colon cancer cells. Cancer Res 2000;60:6886–9.
23. Dlugosz AA, Hansen L, Cheng C, et al. Autocrinetransforming growth factor a is dispensible for v-rasHa-induced epidermal neoplasia: potential involvement ofalternate epidermal growth factor receptor ligands.Cancer Res 1995;55:1883–93.
24. Sweet-Cordero A, Mukherjee S, Subramanian A, et al.An oncogenic KRAS2 gene expression signature identi-fied by cross-species gene expression analysis. NatGenet 2005;37:48–55.
25. Beer DG, Kardia SLR, Huang CC, et al. Geneexpression profiles predict survival of patients withlung adenocarcinoma. Nat Med 2002;8:816–24.
26. Holbro T, Beerli RR, Maurer F, Koziczak M, BarbasCF, Hynes NE. The ErbB2/ErbB3 heterodimer functionsas an oncogenic unit: ErbB2 requires ErbB3 to drivebreast tumor cell proliferation. Proc Natl Acad Sci U S A2003;100:8933–8.
27. Yi ES, Harclerozde D, Gondo M, et al. High c-erbB-3protein expression is associated with shorter survival inadvanced non-small cell lung carcinomas. Mod Pathol1997;10:142–8.
28. Zhou H, Liu L, Lee K, et al. Lung tumorigenesisassociated with erb-B-2 and erb-B-3 overexpression inhuman erb-B-3 transgenic mice is enhanced bymethylnitrosourea. Oncogene 2002;21:8732–40.
Research. on August 24, 2015. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from

2005;65:11478-11485. Cancer Res Nobukazu Fujimoto, Marie Wislez, Jie Zhang, et al. Epidermal Growth Factor Receptorin Lung Adenocarcinomas That Are Sensitive to Inhibition of High Expression of ErbB Family Members and Their Ligands
Updated version
http://cancerres.aacrjournals.org/content/65/24/11478
Access the most recent version of this article at:
Material
Supplementary
http://cancerres.aacrjournals.org/content/suppl/2005/12/21/65.24.11478.DC2.html
http://cancerres.aacrjournals.org/content/suppl/2005/12/12/65.24.11478.DC1.htmlAccess the most recent supplemental material at:
Cited articles
http://cancerres.aacrjournals.org/content/65/24/11478.full.html#ref-list-1
This article cites 28 articles, 14 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/65/24/11478.full.html#related-urls
This article has been cited by 45 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
To request permission to re-use all or part of this article, contact the AACR Publications
Research. on August 24, 2015. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from




















