
Proteoglycans: Systems-Level Insight into Their Expression in ...

THE J O U R N A L OF BIOLOGICAL CHEMISTRV 8 1987 by The American Society of Biological Chemists, Inc
Vol. 262, No. 2, Issue of January 15, pp. 854-859, 1987 Printed in U.S.A.
Heparan Sulfate Proteoglycans of Human Lung Fibroblasts STRUCTURAL HETEROGENEITY OF THE CORE PROTEINS OF THE HYDROPHOBIC CELL-ASSOCIATED FORMS*
(Received for publication, June 23, 1986)
Veerle Lories$, Hilde De Boeck, Guido David$, Jean-Jacques Cassiman, and Herman Van Den Berghe From the Center for Human Genetics, University of Leuven, Campus Gasthuisberg Onderwijs en Navorsing, Herestraut, B-3000 Leuven, Belgium
Heparan sulfate proteoglycans (HSPG) were solubi- lized from human lung fibroblast monolayers with de- tergent. Presumptive membrane-associated forms dis- playing hydrophobic properties were purified by gel filtration on Sepharose CL-4B, by ion-exchange chro- matography on Mono Q and by incorporation in lipid vesicles. The HSPG preparations were ’“1-iodinated and treated with heparitinase before sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Five radio- labeled proteins with apparent molecular weights of 125,000, 90,000, 64,000, 48,000, and 35,000 were visualized by autoradiography. A sixth protein, iden- tified in nonreduced 1261-HSPG preparations, appeared as a non-HS chain-bearing M, 35,000 peptide which was disulfide-linked to an HS chain-bearing peptide of similar size. This multiplicity of core proteins did not seem to result from proteolysis during the heparitinase treatment itself, since some of the core proteins mi- grated independently during gel filtration before heparitinase digestion. Moreover, heparitinase diges- tion of ”‘1-HSPG purified by affinity chromatography on an immobilized monoclonal antibody yielded only the M, 64,000 protein. Alternative depolymerizations of the HS chains by heparinase or HNOz also yielded multiple protein bands. These results imply that het- erogeneity of the core protein moiety may be a genuine property of the hydrophobic HSPG of human lung fi- broblasts. The occurrence of multiple integral mem- brane HSPG forms may be relevant for the multiple functions that have been ascribed to cell-surface HSPG.
Heparan sulfate proteoglycans (HSPG)’ have been impli- cated in a variety of cellular processes such as cell attachment and spreading (1-4), maintenance of cell shape (5, 6), growth control (7, 8), anticoagulation (9), ultrafiltration (lo), and matrix assembly (11-13). The HSPG occupy strategic posi-
* This investigation was supported by Grant 3.0030.81 of the Fonds voor Geneeskundig Wetenschappelijk Onderzoek, Belgium and by United States Public Health Service Research Grant HL-31750 (to G. D.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
$ Specialisatiebursaal of the Instituut tot aanmoediging van het Wetenschappelijk Onderzoek in Nijverheid en Landbouw, Belgium.
5 Onderzoeksleider of the Nationaal Fonds voor Wetenschappelijk Onderzoek, Belgium. To whom correspondence should be addressed.
The abbreviations used are: HSPG, heparan sulfate proteogly- can(s); HLF, human lung fibroblasts; GdnHC1, guanidine hydrochlo- ride; SDS, sodium dodecyl sulfate; mAb S1, monoclonal antibody S1.
tions for exercising these multiple functions, occurring both as cell-surface constituents (14, 15) and as integral compo- nents of the extracellular matrix (10, 11). The glycosamino- glycan chains play a crucial role in the biological properties of these proteoglycans, but the core proteins also have impor- tant contributions. First, the structure of the core protein may be an important determinant of the type of glycosami- noglycan chain that is incorporated in the proteoglycan (16). Furthermore, the core proteins of some of the HSPG which occur at the cell surface could supply a hydrophobic anchor that links the proteoglycan to the membrane (14, 15, 17, 18). Other forms, present in the extracellular matrix of the cells, also appear to be retained in their positions by interactions that involve the polypeptide rather than the polysaccharide chains (19), which implies that these proteoglycans may be targeted to their ultimate destinations through molecular addresses that are contained within the core protein. Finally, the HSPG of the cell surface appear to be involved in trans- membrane interactions with the cytoskeleton (5, ZO), indicat- ing that some functions of the core proteins may be regulatory. Some of the membrane-associated forms resemble or may even be cell-surface receptors, and thus may be involved in the transduction of specific messages into the cell interior (21). Substantial core protein structural heterogeneity seems to underlie this functional diversity. Yet, so far, none of the HSPG core proteins has been sufficiently characterized to allow a precise evaluation of this heterogeneity or a determi- nation of structure-function relationships in this moiety of the molecules.
In prior characterizations of the HSPG of human lung fibroblasts (18), we could discriminate three forms of HSPG on the basis of hydrodynamic size, hydrophobic character, and culture distribution: large HSPG which were recovered from the extracellular matrix of the cells, smaller HSPG which were secreted in the culture medium, and small HSPG which were associated with the cellular membranes. The latter had unique protease-sensitive hydrophobic properties and could be incorporated in liposomes. These data confirmed and complemented results obtained previously by others in human fibroblasts (21, 22) and other tissues (14, 15, 17). The coex- istence of hydrophobic and nonhydrophobic forms implied that HSPG isolated from a single tissue were heterogeneous in their peptide moiety. In the present report we describe the purification of the hydrophobic membrane HSPG from cul- tured human lung fibroblasts and the partial characterization of their core proteins. The results suggest that these mem- brane proteoglycans themselves also occur in multiple forms.
MATERIALS AND METHODS
Cell Culture-Fetal human lung fibroblasts (HLF) were grown on plastic substrata in Dulbecco’s modified Eagle’s minimal essential

Heparan Sulfate Proteoglycan Core Protein Heterogeneity 855
medium (Gibco) containing 10% (v/v) fetal calf serum (13). Confluent monolayers between passages 10 and 17 were incubated for 48 h with 5 pCi of H235S0, (carrier-free)/ml (New England Nuclear) in low sulfate culture medium (13).
Buffers-Triton X-100 buffer contained 150 mM NaCl, 1% (v/v) Triton X-100, 10 mM Na,HPO,, 2 mM KHzP04, pH 7.4, 50 mM 6- aminohexanoic acid, 10 mM EDTA, 5 mM N-ethylmaleimide, 5 mM benzamidine, 1 mM phenylmethanesulfonyl fluoride, and 1 pg/ml pepstatin A. Urea buffer consisted of 6 M urea, 0.5% (v/v) Triton X- 100, and 50 mM Tris-C1, pH 8.0. GdnHCl buffer was composed of 4 M GdnHCl, 100 mM 6-aminohexanoic acid, 10 mM EDTA, 10 mM N- ethylmaleimide, 5 mM benzamidine, 50 pg/ml bovine serum albumin, 10 pg/ml heparin, 10 pg/ml chondroitin sulfate, and 50 mM sodium acetate, pH 5.8. Tris-Cl buffer contained 10 mM octyl glucoside and 100 mM Tris-CI, pH 7.5. Borate buffer was 50 mM octyl glucoside and 100 mM borate, pH 8.5. Pyridine HCl buffer was 1 M pyridine HCl, pH 5.8, 6 M urea, 1 M GdnHC1, and 10% (v/v) Triton X-100. Heparinase buffer contained 50 mM Tris-C1, pH 7.0, 100 mM NaC1, 100 pg/ml bovine serum albumin, 50 mM 6-aminohexanoic acid, 2.5 pg/ml pepstatin A, 1 mM phenylmethanesulfonyl fluoride, and 20 pg/ ml leupeptin. SDS buffer was composed of 1 mg/ml SDS, 350 mM NaC1, 1 mM EDTA, and 50 mM Tris-C1, pH 8.0.
In all instances, the protease inhibitor phenylmethanesulfonyl fluoride was added to these buffers, from a concentrated fresh stock solution in isopropyl alcohol, a few seconds before use (23).
Extraction of Cell-associated HSPG-After the incubation of HLF monolayers with "SOO:-, the medium was recovered from the culture flasks, the flasks were rinsed three times with 10 ml of cold phosphate- buffered saline, and the rinsed HLF monolayers were scraped from the substrate in Triton X-100 buffer (12 m1/150 cmz flask). The suspension was rapidly cooled to 4 "C and was cleared by centrifuga- tion (10,000 X g; 60 min). The proteoglycans in this crude extract (usually -500 ml from 40 flasks) were concentrated by adsorption on a 5-ml DEAE-Sepharose Fast Flow (Pharmacia) column. Bound material was eluted with 5 volumes of urea buffer containing 750 mM NaCl. This eluate was filtered through a Minisart (Sartorius GmbH, Gottingen, West Germany) cellulose acetate membrane of 0.20-pm pore size, diluted %fold in urea buffer and applied on a Mono Q column. After eluting the bulk of the bound proteins from the column with a 0-0.6 M linear NaCl gradient in urea buffer, the retained proteoglycans were eluted in a small volume (1.5-2 ml) of GdnHCl buffer containing 0.5% (v/v) Triton X-100. The "SO:- activity re- covered at this stage was taken to represent 100% of the detergent- extractable label present in the glycosaminoglycan.
The hydrophobic cell-associated HSPG were further purified from this concentrated extract by gel filtration over Sepharose CL-4B, ion- exchange chromatography on Mono Q, and incorporation of the HSPG into liposomes. Prior investigations have indicated that hy- drophobic HSPG represent -15% of the total 35S-glycosaminoglycan and -30-40% of the total =S-heparan sulfate that can be extracted from the cells with detergent (18).
Gel Filtration-Gel filtration over Sepharose CL-4B (Pharmacia) (1 X 100 cm) in GdnHCL buffer, in GdnHCl buffer containing 0.5% (v/v) Triton X-100, or in GdnHCl buffer without heparin, chondroitin sulfate, and bovine serum albumin was performed at 4 "C. The flow rate was 4 ml/h, and 1.6-ml fractions were collected.
Gel filtration over Sepharose CL-4B (1 X 45 cm) in SDS buffer was performed at room temperature. Before chromatography in SDS buffer, the samples were made 2% in SDS and were boiled for 5 min. The flow rate was 6 ml/h, and 0.8-ml fractions were collected. The V, and V, values of the columns were determined with blue dextran and 3H-glucosamine, respectively.
Ion-exchange Chromatography-Samples in urea buffer were ap- plied on a Mono Q column equilibrated in the same buffer. Bound materials were eluted by a linear NaCl gradient (from 0 to 1.2 M) in urea buffer, delivered by a fast protein liquid chromatography system (Pharmacia) at a flow rate of 30 ml/h, at room temperature.
Incorporation of HSPG in Liposomes-After purification of the membrane HSPG by gel filtration and ion-exchange chromatography, the samples were dialyzed against urea buffer and adsorbed on DEAE- Trisacryl M beads (LKB Instruments, Bromma, Sweden). After rins- ing the beads with Tris-CI buffer, bound HSPG were eluted in GdnHCl buffer containing 75 mM octyl glucoside.
Phosphatidylcholine was added to a concentration of 5 mg/ml from a stock solution (25 mg/ml) in GdnHCl buffer containing 75 mM octyl glucoside. Liposomes were formed by dialyzing the HSPG-lipid mixture against GdnHCl buffer without detergent.
lZ5I Iodination of the Core Proteins-Purified HSPG were dialyzed
against urea buffer and adsorbed on a small volume ( 4 5 0 pl) DEAE- Trisacryl M beads. After careful rinsing with Tris-C1 buffer to remove all Triton X-100, the pelleted beads were mixed with 10 pl (1 mCi) of Na1251 (Amersham Corp.) and 10 pl of chloramine T (1 mg/ml). After 5 min at room temperature the labeling was stopped by adding 100 pl of K2SZ05 (5 mM) and KI (10 mM). The DEAE suspension was extensively washed with Tris-C1 and with urea buffer to remove free label. The IZ5I-HSPG were eluted from the DEAE beads in urea buffer containing 1 M NaCl. The presence of the DEAE matrix during the labeling procedure had no influence on the pattern of labeled bands obtained (not shown). Other HSPG samples were iodinated using the Bolton-Hunter reagent (Amersham). HSPG solubilized in borate buffer (40 pl) were added to dried labeling reagent, and the reaction mixture was agitated for 15 min at 4 "C (24). Excess reagent was reacted with 0.2 M glycine in borate buffer (5 min at 4 "C) and was separated from the Iz5I-HSPG by adsorption of the latter on DEAE beads.
To evaluate the labeling reactions, T - H S P G were again submitted to ion-exchange chromatography on Mono Q and gel filtration over Sepharose CL-4B, as described above. Finally, the fractions contain- ing the Iz5I-HSPG were dialyzed against urea buffer and were ad- sorbed on a small volume of DEAE-Trisacryl M beads. Bound "'I- HSPG were eluted in a small volume of 10-fold-concentrated hepa- rinase buffer (without protease inhibitors) and were stored at -20 "C until further use.
Heparime and Heparitinase Digestion-After appropriate dilution and addition of protease inhibitors, the Iz5I-HSPG samples in hepa- rinase buffer were digested with 1 mIU (22 mIU/ml) heparitinase (heparitin-sulfate lyase, EC 4.2.2.8) (2 h, 37 "C) or heparinase (EC. 4.2.2.7) (2 h, 30 "C). Both enzymes were obtained from Miles Labo- ratories. Control samples were incubated in buffer in the absence of enzyme. Alternatively, and with similar results, the Iz5I-HSPG sam- ples were precipitated with 75% (v/v) ethanol in the presence of 50 pg/ml chondroitin sulfate, dissolved in heparinase buffer, and di- gested as described above.
To test whether the heparitinase digestion was complete within 2 h (the time period for which it was normally performed), "'I-HSPG samples were treated with heparitinase for time periods ranging from 15 to 180 min. SDS-polyacrylamide gel electrophoresis and autora- diography of these treated samples showed that the reaction was complete already after 30 min. Moreover, the addition of fresh enzyme during the incubation did not affect the pattern of protein bands which was obtained (results not shown).
Protease K Digestion-Heparitinase-digested '"I-HSPG were made 0.5% (w/v) in SDS and were boiled for 5 min. Protease K digestion of the denatured samples was performed at 60 "C for 40 min at a concentration of 70 pg/ml protease K. A control sample was treated similarly, but in the absence of protease K.
SDS-Polyacrylamide Gel Electrophoresis-SDS-polyacrylamide gel electrophoresis was performed on gradient (4-16% T, 10% C) poly- acrylamide gels using the buffer system of Laemmli (25). [methyl- L4C]phosphorylase b (97 kDa), [methyl-14C]albumin (69 kDa), [methyl-14C]ovalbumin (46 kDa), and [methyl-"C]carbonic anhydrase (30 kDa) were used as molecular mass standards (New England Nuclear). Before electrophoresis, samples were made 2% (w/v) in SDS and boiled for 5 min. Samples run under reducing conditions were supplemented with 2% P-mercaptoethanol.
After electrophoresis, the gels were stained with 0.2% Coomassie Brilliant Blue R-250 in 12.5% (w/v) trichloroacetic acid, 45% (v/v) ethanol, destained in ethanol/acetic acid/water (265:69), and dried under vacuum. Kodak XAR-5 film was used for autoradiography.
N-Des~lfation-'*~I-HSPG were applied to a small DEAE-Trisa- cry1 M column and were eluted in pyridine HCl buffer. After the addition of 19 volumes of dimethyl sulfoxide, the mixture was incu- bated at 37 "C for 4 h. Under these conditions, dimethyl sulfoxide selectively removes N-SOg- from the pyridinium salts of HS (26). The reaction was terminated by removing the dimethyl sulfoxide by dialysis.
Reduction and Alkylation of Disulfide Bonds-HSPG samples were taken up in GdnHCl buffer without N-ethylmaleimide and buffered with 50 mM Tris-C1 at pH 8.0. Reduction with dithiothreitol (10 mM, 4 h at room temperature) was followed by alkylation with iodoaceta- mide (40 mM, overnight at room temperature).
Nitrous Acid Degrudation-1261-HSPG were treated with 0.4 M HNOZ, pH 1.5, for 10 min at room temperature. The reaction was stopped by the addition of excess ammonium sulfamate and neutral-
Heparan Sulfate Proteoglycan Core Protein Heterogeneity
Kav
3 1
z2
6 tb
Z 9,
X
FRACTION NUMBER Kav
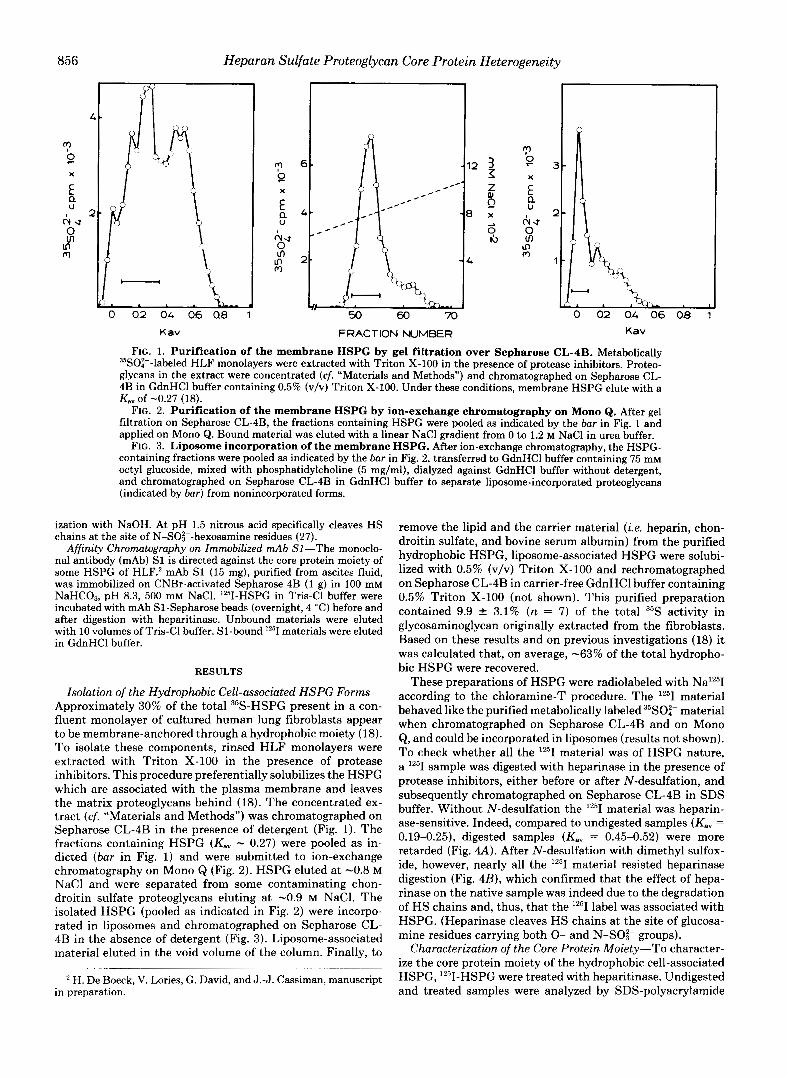
FIG. 1. Purification of the membrane HSPG by gel filtration over Sepharose CL-4B. Metabolically 35SOi--labeled HLF monolayers were extracted with Triton X-100 in the presence of protease inhibitors. Proteo- glycans in the extract were concentrated (cf. "Materials and Methods") and chromatographed on Sepharose CL- 4B in GdnHCl buffer containing 0.5% (v/v) Triton X-100. Under these conditions, membrane HSPG elute with a Kav of -0.27 (18).
FIG. 2. Purification of the membrane HSPG by ion-exchange chromatography on Mono Q. After gel filtration on Sepharose CL-4B, the fractions containing HSPG were pooled as indicated by the bar in Fig. 1 and applied on Mono Q. Bound material was eluted with a linear NaCl gradient from 0 to 1.2 M NaCl in urea buffer.
FIG. 3. Liposome incorporation of the membrane HSPG. After ion-exchange chromatography, the HSPG- containing fractions were pooled as indicated by the bar in Fig. 2, transferred to GdnHCl buffer containing 75 mM octyl glucoside, mixed with phosphatidylcholine (5 mg/ml), dialyzed against GdnHCl buffer without detergent, and chromatographed on Sepharose CL-4B in GdnHCl buffer to separate liposome-incorporated proteoglycans (indicated by bar) from nonincorporated forms.
ization with NaOH. At pH 1.5 nitrous acid specifically cleaves HS chains at the site of N-SO$--hexosamine residues (27).
Affinity Chromatography on Immobilized mAb SI-The monoclo- nal antibody (mAb) S1 is directed against the core protein moiety of some HSPG of HLF.' mAb S1 (15 mg), purified from ascites fluid, was immobilized on CNBr-activated Sepharose 4B (1 g) in 100 mM NaHC03, pH 8.3, 500 mM NaCl. lZ5I-HSPG in Tris-C1 buffer were incubated with mAb S1-Sepharose beads (overnight, 4 "C) before and after digestion with heparitinase. Unbound materials were eluted with 10 volumes of Tris-C1 buffer. S1-bound Iz5I materials were eluted in GdnHCl buffer.
RESULTS
Isolation of the Hydrophobic Cell-associated HSPG Forms- Approximately 30% of the total 35S-HSPG present in a con- fluent monolayer of cultured human lung fibroblasts appear to be membrane-anchored through a hydrophobic moiety (18). To isolate these components, rinsed HLF monolayers were extracted with Triton X-100 in the presence of protease inhibitors. This procedure preferentially solubilizes the HSPG which are associated with the plasma membrane and leaves the matrix proteoglycans behind (18). The concentrated ex- tract (cf. "Materials and Methods") was chromatographed on Sepharose CL-4B in the presence of detergent (Fig. 1). The fractions containing HSPG (Kav - 0.27) were pooled as in- dicted (bar in Fig. 1) and were submitted to ion-exchange chromatography on Mono Q (Fig. 2). HSPG eluted at -0.8 M NaCl and were separated from some contaminating chon- droitin sulfate proteoglycans eluting a t -0.9 M NaCl. The isolated HSPG (pooled as indicated in Fig. 2) were incorpo- rated in liposomes and chromatographed on Sepharose CL- 4B in the absence of detergent (Fig. 3). Liposome-associated material eluted in the void volume of the column. Finally, to
H. De Boeck, V. Lories, G. David, and J.-J. Cassiman, manuscript in preparation.
remove the lipid and the carrier material (i.e. heparin, chon- droitin sulfate, and bovine serum albumin) from the purified hydrophobic HSPG, liposome-associated HSPG were solubi- lized with 0.5% (v/v) Triton X-100 and rechromatographed on Sepharose CL-4B in carrier-free GdnHCl buffer containing 0.5% Triton X-100 (not shown). This purified preparation contained 9.9 f 3.1% (n = 7 ) of the total 35S activity in glycosaminoglycan originally extracted from the fibroblasts. Based on these results and on previous investigations (18) it was calculated that, on average, -63% of the total hydropho- bic HSPG were recovered.
These preparations of HSPG were radiolabeled with Na 'T according to the chloramine-T procedure. The material behaved like the purified metabolically labeled "SOO," material when chromatographed on Sepharose CL-4B and on Mono Q , and could be incorporated in liposomes (results not shown). To check whether all the '''1 material was of HSPG nature, a '"I sample was digested with heparinase in the presence of protease inhibitors, either before or after N-desulfation, and subsequently chromatographed on Sepharose CL-4B in SDS buffer. Without N-desulfation the material was heparin- ase-sensitive. Indeed, compared to undigested samples (Kav = 0.19-0.25), digested samples (Kav = 0.45-0.52) were more retarded (Fig. 4A). After N-desulfation with dimethyl sulfox- ide, however, nearly all the lZ5I material resisted heparinase digestion (Fig. 4B), which confirmed that the effect of hepa- rinase on the native sample was indeed due to the degradation of HS chains and, thus, that the lz5I label was associated with HSPG. (Heparinase cleaves HS chains at the site of glucosa- mine residues carrying both 0- and N-SOi- groups).
Characterization of the Core Protein Moiety-To character- ize the core protein moiety of the hydrophobic cell-associated HSPG, lz5I-HSPG were treated with heparitinase. Undigested and treated samples were analyzed by SDS-polyacrylamide
Heparan Sulfate Proteoglycan Core Protein Heterogeneity 85 7
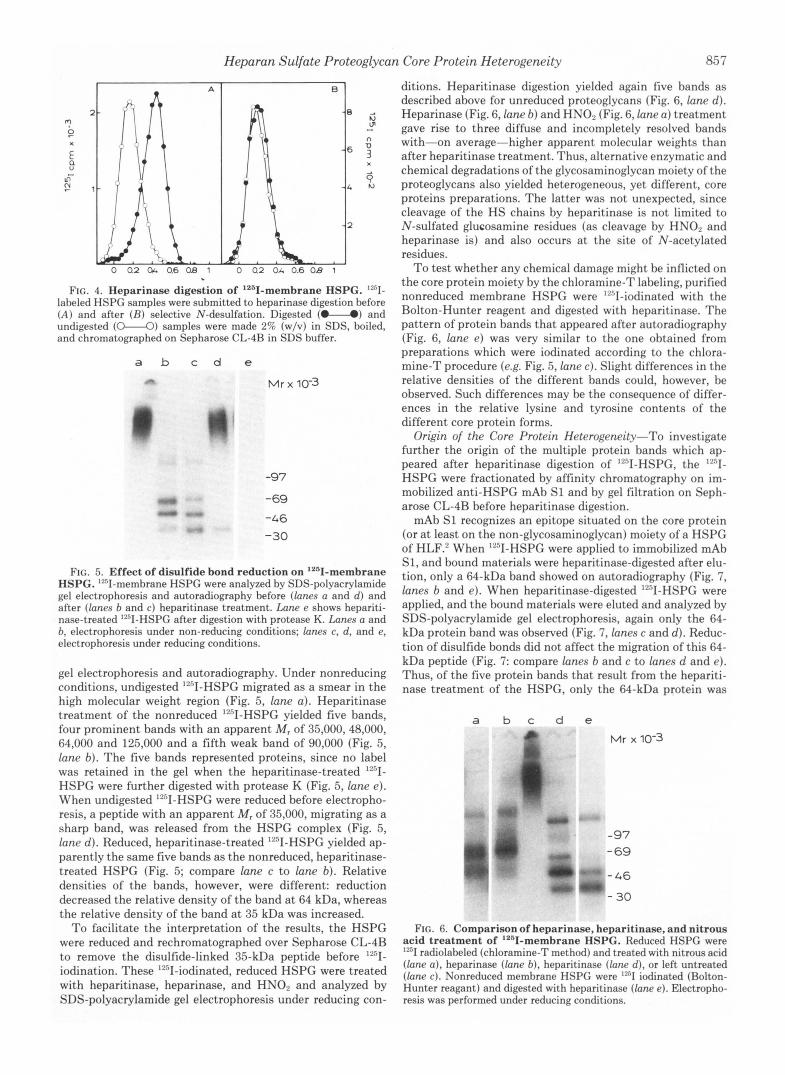
FIG. 4. Heparinase digestion of '2611-membrane HSPG. "'1- labeled HSPG samples were submitted to heparinase digestion before ( A ) and after (H) selective N-desulfation. Digested (U) and undigested (o"-o) samples were made 2% (w/v) in SDS, ))oiled, and chromatographed on Sepharose CL-4R in SDS buffer.
a b c d e
Mr x 10-3
-97
-69
-46 -30
FIG. 5. Effect of disulfide bond reduction on '261-membrane HSPG. "sI-membrane HSPC were analyzed by SDS-polyacrylamide gel electrophoresis and autoradiography before (lanes a and d ) and after (lanes b and c ) heparitinase treatment. Lane e shows hepariti- nase-treated "'1-HSPG after digestion with protease K. Lunes a and b, electrophoresis under non-reducing conditions; lanes c, d , and e, electrophoresis under reducing conditions.
gel electrophoresis and autoradiography. Under nonreducing conditions, undigested '"I-HSPG migrated as a smear in the high molecular weight region (Fig. 5, lane a ) . Heparitinase treatment of the nonreduced ""I-HSPG yielded five bands, four prominent bands with an apparent M , of 35,000,48,000, 64,000 and 125,000 and a fifth weak band of 90,000 (Fig. 5, lane b ) . The five bands represented proteins, since no label was retained in the gel when the heparitinase-treated ""'1- HSPG were further digested with protease K (Fig. 5, lane e ) . When undigested "'I-HSPG were reduced before electropho- resis, a peptide with an apparent M , of 35,000, migrating as a sharp band, was released from the HSPG complex (Fig. 5, lane d) . Reduced, heparitinase-treated "'I-HSPG yielded ap- parently the same five bands as the nonreduced, heparitinase- treated HSPG (Fig. 5; compare lane c to lane b) . Relative densities of the bands, however, were different: reduction decreased the relative density of the band at 64 kDa, whereas the relative density of the band at 35 kDa was increased.
To facilitate the interpretation of the results, the HSPG were reduced and rechromatographed over Sepharose CL-4B to remove the disulfide-linked 35-kDa peptide before "'I- iodination. These "'I-iodinated, reduced HSPG were treated with heparitinase, heparinase, and HNO, and analyzed by SDS-polyacrylamide gel electrophoresis under reducing con-
ditions. Heparitinase digestion yielded again five bands as described above for unreduced proteoglycans (Fig. 6, lane d). Heparinase (Fig. 6, lane b ) and HNO, (Fig. 6, lane a ) treatment gave rise to three diffuse and incompletely resolved bands with-on average-higher apparent molecular weights than after heparitinase treatment. Thus, alternative enzymatic and chemical degradations of the glycosaminoglycan moiety of the proteoglycans also yielded heterogeneous, yet different, core proteins preparations. The latter was not unexpected, since cleavage of the HS chains by heparitinase is not limited to N-sulfated glucosamine residues (as cleavage by HN02 and heparinase is) and also occurs at the site of N-acetylated residues.
To test whether any chemical damage might be inflicted on the core protein moiety by the chloramine-T labeling, purified nonreduced membrane HSPG were "'I-iodinated with the Bolton-Hunter reagent and digested with heparitinase. The pattern of protein bands that appeared after autoradiography (Fig. 6, lane e ) was very similar to the one obtained from preparations which were iodinated according to the chlora- mine-?' procedure (e.g. Fig. 5, lane c) . Slight differences in the relative densities of the different bands could, however, be observed. Such differences may be the consequence of differ- ences in the relative lysine and tyrosine contents of the different core protein forms.
Origin of the Core Protein Heterogeneity-To investigate further the origin of the multiple protein bands which ap- peared after heparitinase digestion of "'I-HSPG, the I2"I-
HSPG were fractionated by affinity chromatography on im- mobilized anti-HSPG mAb S1 and by gel filtration on Seph- arose CL-4B before heparitinase digestion.
mAb S1 recognizes an epitope situated on the core protein (or at least on the non-glycosaminoglycan) moiety of a HSPG of HLF.' When "'I-HSPG were applied to immobilized mAb SI, and bound materials were heparitinase-digested after elu- tion, only a 64-kDa band showed on autoradiography (Fig. 7, lanes b and e ) . When heparitinase-digested "'I-HSPG were applied, and the bound materials were eluted and analyzed by SDS-polyacrylamide gel electrophoresis, again only the 64- kDa protein band was observed (Fig. 7, lanes c and d) . Reduc- tion of disulfide bonds did not affect the migration of this 64- kDa peptide (Fig. 7: compare lanes b and c to lanes d and e ) . Thus, of the five protein bands that result from the hepariti- nase treatment of the HSPG, only the 64-kDa protein was
a b c d e v l r x 10-3
97 69
46
30
FIG. 6. Comparison of heparinase, heparitinase, and nitrous acid treatment of ""I-membrane HSPG. Reduced HSPG were '"1 radiolaheled (chloramine-T method) and treated with nitrous acid (lane a) , heparinase (lane b) , heparitinase (lane d), or left untreated (lane c). Nonreduced membrane HSPC were iodinated (Bolton- Hunter reagant) and digested with heparitinase (lane e ) . Electropho- resis was performed under reducing conditions.

858 Heparan Sulfate Proteoglycan Core Protein Heterogeneity
a b c d e f
Mr x 10-3
.97
,69 .46
30
FIG. 7. Fractionation of 1251-membrane HSPG by mAb SI. Nonreduced 1251-membrane HSPG were applied on CNBr-Sepharose immobilized mAb S1 before (lanes b and e ) or after (lanes c and d ) heparitinase digestion. The 12511-HSPG that were applied and bound as nondigested HSPG were treated with heparitinase before electro- phoresis. Lanes a and f, starting sample, applied on mAb S1 without prior heparitinase digestion; lanes b-e, bound samples, eluted from mAb S1; lanes a-c, electrophoresis under nonreducing conditions; lanes d-f, electrophoresis under reducing conditions.
recognized by mAb SI. Since heparitinase digestion of the HSPG which was bound to mAb S1 as native HSPG also yielded only a 64-kDa band, it was concluded that the 64-kDa core protein forms are derived from a distinct subset of membrane proteoglycans. This conclusion was supported by results observed from the digestion of 9 - H S P G t h a t had been fractionated by gel filtration on Sepharose CL-4B in GdnHCl buffer containing 0.5% Triton X-100. The broad HSPG peak (maximum a t K,, - 0.27; not shown) was subdi- vided in six pools, ranging from materials eluting a t K., - 0.1 to materials eluting a t Kay - 0.5. When equal amounts of material of these different pools were digested with hepariti- nase and analyzed by SDS-polyacrylamide gel electrophoresis under nonreducing (Fig. 8A) and reducing (Fig. 8B) condi- tions, autoradiography showed that the composition of the HSPG fractions changed with increasing Kav. The 125-, 90-, and 48-kDa protein bands decreased in relative density with increasing Kav, whereas the 64- and 35-kDa bands increased. Thus, at least some of the different core protein forms mi- grated independently during gel filtration before heparitinase digestion. It was concluded, also in view of the results obtained with mAb S1, that the multiplicity of the core protein bands is not simply explained by proteolytic activity during the heparitinase treatment.
DISCUSSION
Exploiting the typical solubility characteristics, high charge density, hydrodynamic size, and hydrophobic properties of the membrane HSPG of human lung fibroblasts, we were able to obtain sufficiently purified preparations to analyze the protein moiety of these molecules after '"1 radiolabeling. The results indicate extensive structural heterogeneity and imply that the membrane HSPG of human lung fibroblasts occur in multiple forms.
Part of the membrane HSPG of human lung fibroblasts consists of single polypeptide core proteins of varying size carrying one or more HS chains. Others, in contrast, appear to have a disulfide-bonded dimer structure. Indeed, hepariti- nase treatment of the "'I-HSPG yielded five iodinated pro- teins with apparent M, of 125,000, 90,000,64,000, 48,000, and 35,000 (Fig. 5 , lanes b and c). Comparing nonreduced to reduced core protein patterns (Fig. 5, lane b to lane c; Fig. 7, lane a to lane f ; Fig. 8, A and H), it was obvious that the relative density of the 64-kDa band was higher in the nonre- duced samples than in the reduced samples. Structural het-
A a b c d e f
Mr x 10-3
Y -! 37 59
46 30
/Ir x 10-3
97 69 46
30
t FIG. 8. Fractionation of 1251-membrane HSPG by gel filtra-
tion over Sepharose CL-4B. Purified "'1-HSPG, chromato- graphed over Sepharose CL-4B in GdnHCl buffer containing 0.5% (v/v) Triton X-100 elute as a broad peak with maximum a t K., - 0.27 (not shown). Such an HSPG peak was subdivided in six pools: Kav = 0.083-0.150 (lane a), K., = 0.150-0.217 (lane b) , Kav = 0.217- 0.283 (lane c), K., = 0.283-0.350 (lane d), K., = 0.350-0.417 (lane e), and K.. = 0.417-0.483 (lane f ) . Equal amounts of radiolabel were precipitated with ethanol (75% v/v) and digested with heparitinase before SDS-polyacrylamide gel electrophoresis and autoradiography. A, electrophoresis under nonreducing conditions; R, electrophoresis under reducing conditions.
erogeneity, rather than incompleteness of the reaction, seems to account for the 64-kDa band remaining after reduction. Indeed, the migration of the 64-kDa core protein which is recognized by the monoclonal antibody S1 is not affected by reduction of disulfide bonds (Fig. 7). In the reduced samples the 35-kDa protein band became more prominent. This sug- gested that in addition to the five core protein forms described above, a dimeric complex consisting of two disulfide-linked peptides, each with an apparent M, of about 35,000, was present in nonreduced samples. Some of these 35-kDa pep- tides did not carry H S chains, since they were released from the HSPG complex by reduction of disulfide bonds (Fig. 5, lane d ) and since heparitinase treatment apparently did not reduce their size.
The structure of the membrane HSPG of HLF appears distinct from that reported for HSPG isolated from other cultured cells or tissues. Disulfide-bonded multimers of HSPG have also been described for human skin fibroblasts (28) and for mouse 3T3 cells (29), but the subunit structure and the participation of a non-HS chain-bearing polypeptide in the formation of a disulfide-bonded HSPG dimer, seem to be unique features of the HSPG of human lung fibroblasts. In human skin fibroblasts the core protein moiety of the HSPG is formed by a disulfide-bonded homodimer of M, 180,000 (21). Although one of the core peptides of the HSPG of HLF had an apparent M, of 90,000 and the 64-kDa band may harbor disulfide-bonded homodimers, no evidence for the

Heparan Sulfate Proteoglycan Core Protein Heterogeneity a59
occurrence of a form mimicking the organization described for skin fibroblasts was obtained here. In Swiss mouse 3T3 cells, in contrast, HSPG monomers of M , 20,000 form disul- fide-linked aggregates of M, > 700,000 (29). HSPG isolated from other in vitro cultured tissues also seem to be structurally distinct in their core protein moiety. Characterization studies have led to variable core protein M, estimates: 240,000 for human colon carcinoma cells (30), 80,000 for PC-12 cells (31), and 53,000 for mouse mammary epithelial cells (32). A core protein of 53-kDa has also been identified in HSPG directly isolated from bovine lungs (33). Finally, structural heteroge- neity occurring within a single tissue has been reported for the core protein moiety of HSPG isolated from baby hamster kidney cells (34). Nitrous acid degradation of the cell-associ- ated HSPG of these fibroblasts revealed three main polypep- tides having apparent molecular weights of 65,000, 85,000, and 120,000. Interestingly, rather similar M, estimates were obtained here for nitrous acid-degraded IZ5I-HSPG of HLF (Fig. 6, lane a) . The high variability of reported size estimates might be an indication of species and tissue specificities of the HSPG structure. On the other hand, methodological dif- ferences and disulfide bridge formation between HSPG mon- omers may account for some of these variations (29). Alkyla- tion of the thiol groups during extraction, a precaution which was taken here, should minimize this possibility of artificial cross-linking.
The origin and significance of the occurrence of multiple core protein forms among membrand proteoglycans isolated from a single tissue remains unclear. Although nitrous acid could cleave some core proteins, the result obtained after chemical cleavage of the glycosaminoglycan chains (Fig. 6, lane a) is consistent with the multiplicity of core proteins obtained after heparinase (Fig. 6, lane b ) and heparitinase (Fig. 6, lune d ) treatments. Moreover, at least part of the core protein moiety appeared to be heterogenous before hepariti- nase digestion. Heparitinase treatment of mAb Sl-immuno- purified HSPG yielded only a 64-kDa protein band (Fig. 7), and some of the different core protein forms migrated inde- pendently during gel filtration before heparitinase digestion (Fig. 8). Thus, proteolysis of a single proteoglycan during the heparitinase treatment (if any proteolysis occurs in the pres- ence of several protease inhibitors) is not likely to be the origin of the multiple protein bands. The possibility that some of the multiple protein bands were generated during the isolation of the HSPG, however, could not definitely be ex- cluded. If so, we would have isolated the hydrophobic terminal of the core protein (by the criterion of incorporation in liposomes) but degraded t o different degrees. This seems unlikely because the heparitinase digestions of five prepara- tions of independently purified HSPG yielded all very similar results. When the core protein moiety was radiolabeled with the Bolton-Hunter reagent, again very similar results were obtained (Fig. 6, lane e ) , indicating that this part of the procedure can be excluded in induction of artifacts. Thus, the heterogeneity of the membrane HSPG might occur in situ. This heterogeneity could be relevant for the multiplicity of the functions that have been ascribed to these HSPG. For example, specialized core protein structures may target dis- tinct proteoglycans into specific membrane domains or spe- cific supramolecular complexes. Alternatively, the different forms may represent different subunits or different metabolic
maturation or processing steps that control the function or turnover of a single proteoglycan species. This will have to be clarified in further experiments.
Acknowledgments-We thank A. Ray6 and M. Willems for their expert technical assistance. We also thank Dr. F. Van Leuven for suggestions and critical reading of the manuscriupt.
REFERENCES 1. Johansson, S., and HWk, M. (1984) J. Cell Biol. 9 8 , 810-817 2. Schwartz, M. A., and Juliano, R. L. (1985) J. Cell. Physiol. 124,
3. Schubert, D., and LaCorbiere, M. (1985) J. Cell Biol. 100,56-63 4. Gill, P. J., Silbert, C. K., and Silbert, J. E. (1986) Biochemistry
5. Rapraeger, A. C., and Bernfield, M. R. (1982) in Extracellulur Matrix (Hawkes, S., and Way, J. L., eds) pp. 265-269, Academic Press, New York
6. Laterra, J., Silbert, J. E., and Culp, L. A. (1983) J. Cell Bwl. 9 6 ,
7. Fritze, L. M. S., Reilly, C. F., and Rosenberg, R. D. (1985) J. Cell
8. Wright, T. C., Johnstone, T. V., Castellot, J. J., and Karnovsky,
9. Marcum, J. A., and Rosenberg, R. D. (1985) Biochem. Biophys.
10. Kanwar, Y. S., and Farquhar, M. G. (1979) Proc. Natl. Acad. Sci.
11. Hedman, K., Johansson, S., Vartio, T., Kjellen, L., Vaheri, A,,
12. David, G., and Bernfield, M. R. (1982) J. Cell. Physiol. 110 , 56-
13. David, G., and Van Den Berghe, H. (1983) J. Biol. Chem. 2 5 8 ,
14. Kjellen, L., Pettersson, I., and Hook, M. (1981) Proc. Natl. Acad.
113-119
25,405-410
112-123
Bioi. 100, 1041-1049
M. J . (1985) J. Cell. Physiol. 125 , 499-506
Res. Commun. 126,365-372
U. S. A. 76,1303-1307
and Hook, M. (1982) Cell 28,663-671
62
7338-7344
Sci. U. S. A. 78,5371-5375 15. Rapraeger, A. C., and Bernfield, M. (1983) J. Biol. Chem. 258 ,
3632-3636 16. Stevens, R. L., and Austen, K. F. (1982) J. Biol. Chem. 257,253-
259 17. Woods, A., Couchman, J . R., and Hook, M. (1985) J. Biol. Chem.
260,10872-10879 18. Lories, V., David, G., Cassiman, J. J., and Van Den Berghe, H.
(1986) Eur. J. Biochem. 158 , 351-360 19. Hook, M., Couchman, J., Woods, A., Robinson, J., and Christe-
ner, J. E. (1984) in Basement Membranes and Cell Movement (Porter, R., and Whelan, J., eds) pp. 44-50, Pitman, London
20. Woods, A., Hook, M., Kjellen, L., Smith, C. G., and Rees, D. A. (1984) J. Cell Biol. 9 9 , 1743-1753
21. Fransson, L. A., Carlstedt, I., Coster, L., and Malmstrom, A. (1984) Proc. Natl. Acad. Sci. U. S. A. 8 1 , 5657-5661
22. Vogel, K. G., and Peterson, D. W. (1981) J. Biol. Chem. 2 5 6 , 13235-13242
23. James, G. T. (1978) Anal. Biochem. 86,574-579 24. Bolton, A. E., and Hunter, W. M. (1973) Biochem. J. 133 , 529-
25. Laemmli, U. K. (1970) Nature 227 , 680-685 26. Inoue, Y., and Nagasawa, K. (1976) Carbohydr. Res. 46,87-95 27. Shively, J. E., and Conrad, H. E. (1976) Biochemistry 15 , 3932-
28. Coster, L., Malmstrom, A., Carlstedt, I., and Fransson, L. A.
29. Lowe-Krentz, L. J., and Keller, J. M. (1984) Biochemistry 2 3 ,
30. Iozzo, R. V. (1984) J. Cell Biol. 9 9 , 403-417 31. Matthew, W.D., Greenspan, R. J., Lander, A. D., and Reichardt,
L. F. (1985) J. Neurosci. 5, 1842-1850 32. Rapraeger, A., Jalkanen, M., Endo, E., Koda, J., and Bernfield,
M. (1985) J. Biol. Chem. 260,11046-11052 33. Radhakrishnamurthy, B., Jeansonne, N. E., and Berenson, G. S.
(1984) Biochim. Biophys. Acta 802,314-320 34. Bretscher, M. S. (1985) EMBO J. 4 , 1941-1944
539
3942
(1983) Biochem. J. 215,417-419
2621-2627
Copyright © 2022 FDOKUMEN




















