
Reversible inactivation of dihydrolipoamide dehydrogenase by Angeli's salt

ISSN: 1524-4571 Copyright © 2008 American Heart Association. All rights reserved. Print ISSN: 0009-7330. Online
TX 72514Circulation Research is published by the American Heart Association. 7272 Greenville Avenue, Dallas,
DOI: 10.1161/CIRCRESAHA.107.169953 published online Jun 26, 2008; Circ. Res.
Avkiran and Michael S. Marber Clark, Sebastien Jacquet, Ajay M. Shah, Tetsuji Miura, Derek M. Yellon, Metin Yasuhiro Nishino, Ian G. Webb, Sean M. Davidson, Aminul I. Ahmed, James E.
Preconditioning or Postconditioning in the MouseGlycogen Synthase Kinase-3 Inactivation Is Not Required for Ischemic
http://circres.ahajournals.org/cgi/content/full/CIRCRESAHA.107.169953/DC1Data Supplement (unedited) at:
http://circres.ahajournals.org
located on the World Wide Web at: The online version of this article, along with updated information and services, is
http://www.lww.com/reprintsReprints: Information about reprints can be found online at
[email protected]. E-mail:
Fax:Kluwer Health, 351 West Camden Street, Baltimore, MD 21202-2436. Phone: 410-528-4050. Permissions: Permissions & Rights Desk, Lippincott Williams & Wilkins, a division of Wolters
http://circres.ahajournals.org/subscriptions/Subscriptions: Information about subscribing to Circulation Research is online at
by on May 10, 2011 circres.ahajournals.orgDownloaded from

Glycogen Synthase Kinase-3 Inactivation Is Not Requiredfor Ischemic Preconditioning or Postconditioning
in the MouseYasuhiro Nishino, Ian G. Webb, Sean M. Davidson, Aminul I. Ahmed, James E. Clark,
Sebastien Jacquet, Ajay M. Shah, Tetsuji Miura, Derek M. Yellon, Metin Avkiran, Michael S. Marber
Abstract—The inactivation of glycogen synthase kinase-3� (GSK-3�) is proposed as the event integrating protectivepathways initiated by preconditioning and other interventions. The inactivation of GSK-3 is thought to decrease theprobability of opening of the mitochondrial permeability transition pore. The aim of this study was to verify the role ofGSK-3 using a targeted mouse line lacking the critical N-terminal serine within GSK-3� (Ser9) and the highlyhomologous GSK-3� (Ser21), which when phosphorylated results in kinase inactivation. Postconditioning with 10cycles of 5 seconds of reperfusion/5 seconds of ischemia and preconditioning with 6 cycles of 4 minutes of ischemia/6minutes of reperfusion, similarly reduced infarction of the isolated perfused mouse heart in response to 30 minutes ofglobal ischemia and 120 minutes of reperfusion. Preconditioning caused noticeable inactivating phosphorylation ofGSK-3. However, both preconditioning and postconditioning still protected hearts of homozygous GSK-3 doubleknockin mice. Moreover, direct pharmacological inhibition of GSK-3 catalytic activity with structurally diverseinhibitors before or after ischemia failed to recapitulate conditioning protection. Nonetheless, cyclosporin A, a directmitochondrial permeability transition pore inhibitor, reduced infarction in hearts from both wild-type and homozygousGSK-3 double knockin mice. Furthermore, in adult cardiac myocytes from GSK-3 double knockin mice, insulinexposure was still as effective as cyclosporin A in delaying mitochondrial permeability transition pore opening. Ourresults, which include a novel genetic approach, suggest that the inhibition of GSK-3 is unlikely to be the keydeterminant of cardioprotective signaling in either preconditioning or postconditioning in the mouse. (Circ Res.2008;103:0-0.)
Key Words: postconditioniong � preconditioning � GSK-3 � mPTP
Ischemic preconditioning is a powerful and establishedmodulator of intrinsic myocardial resistance to lethal
ischemia.1 Its clinical application in patients with acutemyocardial infarction is limited as a result of the inability topredict the moment of coronary artery occlusion. Morerecently, Zhao et al described the concept of postconditioningas a novel strategy for protecting the heart againstreperfusion-injury.2 They demonstrated that a similar regimenof brief periods of ischemia after a lethal index ischemicinsult was as protective as preconditioning. This phenomenonhas subsequently been confirmed in a variety of animalmodels, both in vivo and in isolated heart preparations,3 aswell as in preliminary clinical interventional studies.4,5
The endogenous cellular and molecular mechanisms in-volved in cardioprotection against ischemia have been exten-sively investigated in the field of preconditioning.6 Becausepre- and postconditioning are temporally remote, the signal-
ing pathways might be expected to differ considerably.2,7
However, recent evidence suggests that the early reperfusionphase after lethal ischemia is crucial in mediating the cardio-protective effects of preconditioning.8,9 Furthermore, many ofthe mimetics and kinases of preconditioning have also beenimplicated in postconditioning.10,11 Finally, there is increas-ing evidence that the final step of both signaling pathways isinhibition of the mitochondrial permeability transition pore(mPTP),12–14 whereby pore opening results in cell death.Glycogen synthase kinase-3� (GSK-3�) has been reported asa common target of converging protective signals in precon-ditioning, immediately proximal to the mPTP, that inhibitspore opening in cardiomyocytes.15 Using both adult ratcardiomyocytes and neonatal rat cardiomyocytes, Juhaszovaet al15 demonstrated that numerous preconditioning pathwaysconverged to inactivate GSK-3� by Ser9 phosphorylation.Although the importance of GSK-3 in preconditioning is
Original received December 13, 2007; revision received June 9, 2008; accepted June 12, 2008.From the King’s College London BHF Centre (Y.N., I.G.W., A.I.A., J.E.C., S.J., A.M.S., M.A., M.S.M.), Cardiovascular Division, The Rayne Institute,
St. Thomas’ Hospital, UK; The Hatter Cardiovascular Institute (S.M.D., D.M.Y.), University College London Hospital and Medical School, UK; andSecond Department of Internal Medicine (T.M.), Sapporo Medical University School of Medicine, Japan.
Correspondence to Prof Michael S. Marber, Department of Cardiology, King’s College London, The Rayne Institute, St. Thomas’ Hospital, LambethPalace Rd, London SE1 7EH, United Kingdom. E-mail [email protected]
© 2008 American Heart Association, Inc.
Circulation Research is available at http://circres.ahajournals.org DOI: 10.1161/CIRCRESAHA.107.169953
1 by on May 10, 2011 circres.ahajournals.orgDownloaded from

supported by the literature,16,17 these data predominantly relyon pharmacological manipulation, with all the inherent prob-lems of target selectivity. There is, therefore, a need to verifythe role of GSK-3 in both ischemic preconditioning andpostconditioning in the whole heart by using genetic manip-ulation of the signaling axis.
GSK-3 exists as 2 isoforms, � and �, which are highlyhomologous, similarly inhibited by protein kinase (PK)B/Akt-mediated phosphorylation of a critical N-terminal serineresidue, and share common substrates and sensitivity topharmacological inhibitors.18,19 Thus, the primary aim of thisstudy was to clarify the role of GSK-3 in ischemic pre- andpostconditioning in the whole heart, through the use of micein which all GSK-3 alleles encode inactivation resistantkinases lacking the PKB/Akt-targeted Ser21(�) and Ser9 (�).
Materials and MethodsDetailed methodology is provided in the online data supplement,available at http://circres.ahajournals.org. Key techniques involvedadaptations of previously published protocols, including those for theperfusion and assessment of infarction in isolated murine hearts,20,21
immunoblot analysis,20,21 isolation of murine ventricular myocytes,22
and assay of the mitochondrial permeability transition.23
GSK-3 Knockin MiceThe targeting strategy used to generate GSK-3�/� knockin (KI)mice, in which the PKB/Akt phosphorylation sites on GSK-3� (Ser21) and GSK-3� (Ser9) are changed to Ala, has been describedpreviously.18
Pre- and Postconditioning Experiments
Protocol 1Our objective in this protocol was to assess the potential forpostconditioning (PostC) to limit infarct size and compare it topreconditioning (PreC) in our model. C57BL/6 mice were dividedinto 4 study groups, as shown in Figure 1A. In the control group,there was no additional intervention. Hearts in the PreC groupunderwent 4 cycles of 4 minutes of ischemia/6 minutes of reperfu-sion before the 30 minutes of global ischemia. Hearts in the PostCgroup were subjected to 1 of 2 protocols to determine the optimumstrategy: in the 30 seconds PostC and 5 seconds PostC groups, 4cycles of 30 seconds of reperfusion/30 seconds of ischemia and 10cycles of 5 seconds of reperfusion/5 seconds of ischemia wereperformed at the end of 30 minutes of index ischemia, respectively.
Protocol 2GSK-3�/� KI mice were used to determine the dependence of pre-and postconditioning cardioprotection on GSK-3 inhibition. GSK-3wild-type (WT) and KI mice were assigned to control, PreC, andPostC with repetitive 5 seconds of reperfusion/ischemia groups asdescribed in protocol 1.
Protocol 3In these experiments, the effects on infarct size of direct pharmaco-logical inhibition of GSK-3 activity before and after ischemia wereexamined (Figure 1B). Two inhibitors, SB216763 (3 �mol/L)(Sigma) and 6-bromoindirubin-3�-oxime (BIO) (100 nmol/L) (Cal-biochem), were used based on concentrations required to achievedephosphorylation of glycogen synthase. C57BL/6 mice were di-vided into five groups (Figure 1B): (1) standard ischemia/reperfusiongroup (control); pretreatment with (2) 3 �mol/L SB216763 (Pre SB)or (3) 100 nmol/L BIO (Pre BIO) for the last 15 minutes ofstabilization; and treatment postischemia with (4) 3 �mol/LSB216763 for the first 15 minutes of reperfusion (Post SB15) or (5)3 �mol/L SB216763 for 2 hours (Post SB120). SB216763 and BIOwere dissolved in dimethyl sulfoxide (DMSO) and then diluted withperfusion buffer so that the final concentration of DMSO was 0.05%.
Protocol 4These experiments set out to determine whether the cardioprotectiveeffect of direct pharmacological inhibition of mPTP opening atreperfusion is preserved in the GSK-3�/� KI mouse. After 30minutes of global ischemia, GSK-3 WT and KI hearts were subjectedto either infusion of 0.2 �mol/L cyclosporin A (CsA) (Sigma) for 10minutes at the moment of reperfusion or normal buffer (control)(Figure 1B). CsA was dissolved in DMSO at a final concentration ofless than 0.05%. Similar experiments were also performed in isolatedadult murine myocytes to visualize mPTP opening directly.
ResultsPre- and Postconditioning the Isolated MurineHeart: Protocol 1The PreC protocol was as used previously in this model.20
Postconditioning was examined using 2 reperfusion patternsto determine the optimum strategy (30 seconds PostC and 5seconds PostC). The protocols, described in Figure 1A,resulted in significant reductions of infarction only in the caseof PreC and 5 seconds PostC (see protocol 1 in Table II in theonline data supplement), which was reflected by improvedcontractile recovery in these groups (see protocol 1 ofsupplemental Table I).
PreC
Control Pre SBPre BIO
30sec PostC
Post SB120Post SB15
Ischaemia Reperfusion
5sec PostC
CsA
0 02151520304-Time in minutesTime in minutes
0 02151010351-
Ischaemia ReperfusionA B
SB or BIO
SBSB
CsA
Figure 1. Experimental protocols for assessment of infarction and GSK-3 signaling. A, In all experiments, hearts were subjected to 30minutes of global ischemia and 120 minutes of reperfusion. Preconditioning (PreC) was performed with 4 cycles of 4 minutes of ische-mia and 6 minutes of reperfusion. Postconditioning was performed with 10 cycles of 5 seconds of reperfusion/ischemia (5 secondsPostC) and with 4 cycles of 30 seconds of reperfusion/ischemia (30 seconds PostC). Arrows indicate time points for protein analysis.Hearts were harvested before ischemia, at the end of ischemia, and at the indicated time after reperfusion with or without pre- andpostconditioning. B, Hearts in the Pre SB and Pre BIO groups were perfused with 3 �mol/L SB216763 and 100 nmol/L BIO for 15 min-utes before ischemia, respectively. Post SB15 and Post SB120 hearts were perfused with 3 �mol/L SB216763 for 15 minutes and 120minutes after ischemia, respectively. Hearts in the CsA group were perfused with 0.2 �mol/L CsA for 10 minutes after ischemia.
2 Circulation Research August 1, 2008
by on May 10, 2011 circres.ahajournals.orgDownloaded from

Despite improved recovery in the PreC and 5 secondsPostC groups, there was only a slight effect on GSK-3�/�phosphorylation (see Figure 2A and 2B, respectively). Onclose examination GSK-3�, but not GSK-3�, was phosphor-ylated after preconditioning (phospho-/total GSK-3�335.3�38.1% of control baseline; P�0.05, n�5) and thendephosphorylated during ischemia to a level similar to that ofcontrol (150.5�82.4% of control baseline and 95.1�52.1%of time-matched control ischemia; P�NS, n�5 for both).During reperfusion, GSK-3� demonstrated some rephosphor-ylation that was comparable in preconditioned and control-reperfused hearts (Figure 2A) and far less than that seen withinsulin. Postconditioning did not significantly alter GSK-3�/� Ser21/9 phosphorylation compared to control (Figure2B). Furthermore, we examined the phosphorylation status ofTyr 279/216 of GSK-3�/� and found these sites similarlyphosphorylated under all conditions (data not shown).
Thus, robust methods exist to pre- and postcondition theisolated mouse heart on the C57BL/6 background. Theseforms of conditioning were associated with variable, but lowlevels, of inhibitory phosphorylation of GSK-3�/�. We nextexamined whether this phosphorylation, albeit at a lowlevel, had any influence on conditioning of WT and KImice from the GSK-3 colony, ostensibly on the sameC57BL/6 background.
Characterization of GSK-3�/�-DependentSignaling in Inactivation-Resistant HeartsGSK-3 KI mice were phenotypically similar to WT animalsfrom the same colony. In particular, there were no differencesin baseline developed pressure or coronary flow (see protocol2 of supplemental Table I) or body weight and heart size (seeprotocol 2 of supplemental Table II). Myocardial content ofPKB/Akt, GSK-3, and glycogen synthase protein was alsocomparable between genotypes (see Figure 3), consistentwith existing data.24 The � and � isoforms of GSK-3 seemapproximately equally abundant and unchanged by targeting.Insulin induced robust phosphorylation of PKB/Akt at Ser473in both genotypes (Figure 3). As expected, GSK-3�/� wasphosphorylated at serines 21 and 9, respectively, but only inWT hearts. However, because these epitopes are disrupted inthe KI hearts, we examined glycogen synthase phosphoryla-tion as an index of GSK-3 activity. Under basal conditions,glycogen synthase remains phosphorylated at serine 641 and645 in both WT and KI hearts because of constitutive GSK-3activity. Importantly, GSK-3 inhibition by insulin-inducedactivation of PKB/Akt results in glycogen synthase dephos-phorylation, presumably through unopposed phosphatase ac-tivity in WT, but not in KI, hearts. Moreover, the serial natureof the GSK-3 consensus sites on glycogen synthase results innoticeable changes in migration between phospho- and de-phosphoforms recognized by the total glycogen synthaseantibody (see lowermost bands). In conclusion, GSK-3� and-� are both resistant to inactivation by PKB/Akt in the KI, butnot WT, heart.
Pre- and Postconditioning of GSK-3�/�Inactivation-Resistant Heart: Protocol 2Having determined that GSK-3 within KI hearts is resistant toinactivation even in response to insulin (see Figure 3), they
were next subjected to pre- and postconditioning. We usedthe protocols (PreC and 5 seconds PostC within protocol 1 ofsupplemental Tables I and II) optimized to reduce infarction,which were associated with residual GSK-3 phosphorylation(albeit at a much lower level than occurs with insulin; seeFigure 2).
Surprisingly, both pre- and postconditioning protectedhearts with inactivation-resistant (KI) forms of GSK-3 just aswell as within colony controls containing inactivation-sensitive forms of GSK-3 (WT) (see protocol 2 of supple-mental Tables I and II). The protection by both forms ofconditioning was evident in infarction size on both WT andKI backgrounds. However, the WT mice were more sensitiveto infarction than the KI mice and also probably outsourcedC57BL/6 controls.
Based on these data, it seems that inhibition of GSK-3�/�through phosphorylation of the regulatory N-terminal serinesis not essential to either pre- or postconditioning. However,given the indirect supporting evidence within the literature, itis possible that GSK-3 inhibition by another means contrib-uted to protection. To examine this possibility we determinedthe effect of pharmacological inhibition of GSK-3 on infarc-tion and postischemic recovery.
Pharmacological Inhibition ofGSK-3�/�: Protocol 3To determine whether direct inhibition of GSK-3 mimicsconditioning, we used 2 structurally diverse ATP-competitivesmall molecule inhibitors. As seen in Figure 2, comparinginsulin to other lanes, the majority of GSK-3 is in a nonphos-phorylated active form and thereby amenable to inhibition.
In Figure 4, we used the same readout of GSK-3 activityas in Figure 3, ie, the maintenance of glycogen synthasephosphorylation. Exposure to 3 �mol/L SB216763 or 100nmol/L BIO for 15 minutes resulted in complete dephos-phorylation of the GSK-3 sites within glycogen synthase,suggesting that these concentrations completely inhibitGSK-3 activity. We therefore used these concentrations andexposure times immediately before global ischemia to deter-mine the relevance of the GSK-3 phosphorylation seen withpreconditioning (see Figure 2A). Neither regime had aconsistent effect on postischemic recovery or infarction size(see protocol 3 Pre BIO and Pre SB of supplemental TablesI and II). Next, we examined the relevance of the low levelsof GSK-3 phosphorylation seen during reperfusion withpostconditioning in Figure 2B by determining whether pro-tection could be mimicked by GSK-3 inhibition duringreperfusion. Because it was uncertain for how long the lowlevels of GSK-3 phosphorylation persisted during reperfu-sion, we examined infarction in response to 3 �mol/LSB216763 for the first 15 minutes (Post SB15) and through-out (Post SB120) reperfusion. As can be seen from the resultsin protocol 3 of supplemental Tables I and II, neither durationof GSK-3 inhibition recapitulated the protection seen withpostconditioning.
The Effect of CsA: Protocol 4Although we were able to pre- and postcondition the mouseheart, none of our pharmacological or genetic manipulations
Nishino et al GSK3 in Preconditioning and Postconditioning 3
by on May 10, 2011 circres.ahajournals.orgDownloaded from

Ischemia 30 mins Reperfusion 15 mins
PreC
-Bas
e
PreC
-Bas
e
Con
-Bas
e
Insu
lin
Con
-Isch
PreC
-Isch
Con
-Isch
PreC
-isch
PreC
-Rep
Con
-Rep
Con
-Rep
PreC
-Rep
pSer9-GSK-3
total-GSK3-
total-GSK-3
pSer21/9-GSK-3
phos
ho/to
tal G
SK-3
β
Ischemia30 min
Reperfusion15 min
Con
Bas
e
Insu
lin
PreC
Bas
elin
e
Con
-Isch
PreC
-Isch
Con
-Rep
PreC
-Rep
0.0
0.2
0.4
0.6
0.8 *
*
Reperfusion 2 mins Reperfusion 5 mins
Con
-Rep
Isch
30
Con
-Bas
e
Insu
lin
Post
C-R
ep
Con
-Rep
Post
C-R
ep
Con
-Rep
Con
-Rep
Post
C-R
ep
Post
C-R
ep
Con
-Rep
Con
-Rep
Post
C-R
ep
Post
C-R
ep
pSer9-GSK-3
total-GSK3-
total-GSK-3
pSer21/9-GSK-3
Reperfusion 15 mins
phos
ho/to
tal G
SK-3
β
Reperfusion15 mins
Reperfusion5 mins
Reperfusion2 mins
Con
-Bas
e
Insu
lin
Isch
30
Con
-Rep
Post
C-R
ep
Con
-Rep
Post
C-R
ep
Con
-Rep
Post
C-R
ep
0.0
0.2
0.4
0.6
0.8
1.0
*
A
B
Figure 2. A, GSK-3 phos-phorylation time course inbuffer-perfused C57BL/6hearts subjected to precon-ditioning (PreC) or controlperfusion (Con). Heart sam-ples were collected justbefore ischemia (PreC-Baseor Con-Base), at the end of30 minutes of ischemia(PreC-Isch30 or Con-Isch30) and at 15 minutesof reperfusion (PreC-Rep orCon-Rep) with or withoutischemic preconditioning.Perfusion of a nonischemicheart with insulin (5 U/L;Actrapid, Novo Nordisk) for15 minutes was used aspositive control for GSK-3phosphorylation. Heartsamples were snap frozenin liquid nitrogen for immu-noblotting analysis. Repre-sentative immunoblots areshown with quantitativeanalyses of repeat experi-ments expressed as theratio of phosphorylated tototal protein. Data areexpressed as themeans�SEM (n�5).*P�0.05 vs control base-line. B, GSK-3 phosphoryla-tion time course in buffer-perfused C57BL/6 heartssubjected to postcondition-ing (PostC-Rep) or controlreperfusion (Con-Rep).Heart samples were col-lected just before ischemia(Con-Base), at the end of30 minutes of ischemia(Isch-30), and at 2, 5, and15 minutes of reperfusion,with or without ischemicpostconditioning (PostC-Rep or Con-Rep). Perfusionof a nonischemic heart withinsulin (5 U/L; Actrapid,Novo Nordisk) for 15 min-utes was used as positivecontrol for GSK-3 phos-phorylation. Heart sampleswere snap frozen in liquidnitrogen for immunoblottinganalysis. Representativeimmunoblots are shownwith quantitative analysesof repeat experimentsexpressed as the ratio ofphosphorylated to total pro-tein. Data are expressed asthe means�SEM (n�5).*P�0.05 vs controlbaseline.
4 Circulation Research August 1, 2008
by on May 10, 2011 circres.ahajournals.orgDownloaded from

of GSK-3 activity had an effect on postischemic recovery orinfarction. The mouse heart has been used by a variety ofinvestigators to elucidate cardioprotective signaling, but weremained concerned that the GSK-33mPTP signaling axiswas not operative in our model and in particular on thebackground of the GSK-3 colony. We therefore examined theeffect of CsA. Once again, the WT hearts had greater infarctsthan KI hearts (see protocol 4 of supplemental Table II).However, CsA reduced infarction and improved contractilerecovery similarly in both genotypes (see protocol 4 ofsupplemental Tables I and II), suggesting mPTP opening atreperfusion does contribute to infarction in this model but isnot influenced by varied manipulations of GSK-3 activity.
To confirm more directly that GSK-3 inactivation did notinfluence mPTP, the timing of opening was assayed incardiomyocytes isolated from adult KI mice using confocallaser analysis, as previously described.23 The time to mPTPopening was significantly increased in cells exposed to CsAin both GSK-3 WT cardiomyocytes (from 91.6�6.6 to144.2�6.4 seconds; P�0.05) and KI cardiomyocytes (from127.4�10.3 to 196.3�18.7 seconds; P�0.05). Insulin expo-sure was as effective as CsA in delaying mPTP opening inboth genotypes (WT, 136.0�6.8 seconds; KI, 194.5�17.2seconds; P�0.05 compared to control baseline). This dem-onstrates that, even in the absence of inactivatable GSK-3,
signals downstream of insulin are still able to act on themPTP. Furthermore, the time to mPTP opening in isolatedcardiomyocytes subjected to laser-induced oxidative stresswas longer in KI cells compared to WT cells. This was trueat baseline and after treatment with insulin or CsA (P�0.05in each study group). This is consistent with the results thatthe KI heart is more resistant to global ischemia than WT.
DiscussionThe purpose of the present study was to use a novel mouseline to confirm that the inhibition of GSK-3 is a necessarystep in the signal transduction cascade that leads to protectionin pre- and postconditioning. Contrary to our expectations,mouse hearts with only noninhibitable forms of GSK-3 couldstill be protected by either form of conditioning. Furthermore,in myocytes isolated from these mice, insulin was still able todelay mPTP opening. Moreover, documented pharmacologi-cal inhibition of GSK-3 with structurally diverse agentspresent before, or after, ischemia failed to recapitulate theprotection observed with conditioning. Thus, in the murineheart, the inhibition of GSK-3 is neither necessary norsufficient to cause pre- or postconditioning.
Notably, our results differ from those of Juhaszova et al15
who previously demonstrated that isolated cardiomyocytesfrom transgenic mice expressing a noninhibitable isoform of
total-glycogensynthase
total-GSK-3
pSer641/645-glycogensynthase
pSer473-Akt
total-Akt
pSer21/9-GSK-3
pSer9-GSK-3
Con Insulin Con Insulin
KIWT
phos
pho/
tota
lA
kt
0
0.2
0.4
0.6
0.8
Con Insulin Con InsulinKIWT
0
0.2
0.4
0.6
0.8
1.0
phos
pho/
tota
l G
SK-3
β
N/DN/D
0
0.1
0.2
0.3
0.4
0.5
phos
pho/
tota
lgl
ycog
en s
ynth
ase
* *
*
*
†
Figure 3. Effects of insulin on PKB/Akt–GSK-3–glycogen synthase signaling cascade in GSK-3 WT and KI hearts. GSK-3 WT and KIhearts were harvested at 40 minutes after intraperitoneal saline (Con) or insulin injection. Representative immunoblots are shown withquantitative analyses of repeat experiments expressed as the ratio of phosphorylated to total protein. Data are expressed as themeans�SEM (n�3). *P�0.05 vs control in each protocol; †P�0.05 KI vs WT in each protocol. N/D indicates not detectable.
Nishino et al GSK3 in Preconditioning and Postconditioning 5
by on May 10, 2011 circres.ahajournals.orgDownloaded from

GSK-3� were resistant to the protective effects of ischemicand pharmacological preconditioning.15 Transgenic GSK-3�expression in these hearts was approximately 8-fold greaterthan the physiological endogenous level introducing thepossibility of loss of physiological substrate selectivity andinduction of a misfolded protein response, whereas allelicallyencoded GSK-3� still remained potentially inhibitable byN-terminal phosphorylation. Furthermore, in these mice at 4weeks, transgene expression is accompanied by a 4- to 5-foldupregulation of both atrial natriuretic factor and brain natri-uretic peptide,25 which are cardioprotective peptides.26–28
Moreover, transgenic overexpression of GSK-3� has beenassociated with metabolic derangement29 and myocardialcontractile dysfunction.30 These factors may in part explainthe divergence in findings between the 2 genetic approachestaken to examine the same hypothesis.
The reason we adopted a genetic approach that differedfrom that taken by Juhaszova et al15 included the concernsoutlined above, as well as the fact that little is known aboutthe action of GSK-3� in the heart. The most notable differ-ence between the 2 isoforms is that GSK-3� possesses aglycine-rich N-terminal extension of unknown function thatis absent in GSK-3�, resulting in 5-kDa difference in molec-ular mass between isoforms.31 However, the isoforms sharemore than 98% homology within their kinase domain,19 seemapproximately equally abundant in cardiac myocytes,32 inkeeping with our findings, are identically inhibited by PKB/Akt,31 cannot be selectively inhibited pharmacologically,31
and, most importantly, exhibit functional redundancy onsystematic allelic disruption.31 Thus, although the emphasisto date has been on GSK-3�, it seems probable that inhibitionof GSK-3� could similarly lead to protection or even thatcoincident inhibition of both kinases is required. To cover allof these possibilities, we chose to use GSK-3�/� double KI
mice in our initial experiments to confirm their role inconditioning, before progressing to mice homologous fortargeted alleles encoding noninhibitable kinases of eachindividual isoform. Because conditioning still occurred on thedouble KI background, however, the additional experimentsbecame redundant.
Despite the evidence of functional redundancy betweenGSK-3� and GSK-3� in early mammalian development,31
the situation in the heart maybe more complex. For example,in zebrafish, a morphilino-based knockdown of GSK-3�results in a cardiac phenotype that differs from GSK-3�knockdown and cannot be rescued by GSK-3� overexpres-sion.33 Similarly, although cardiac-restricted expression ofGSK-3� and GSK-3� in mice results in a similar phenotype,there are biochemical distinctions in the coupled downstreamsignals and in the response to hemodynamic stress.34 Thus, itis possible GSK-3� and GSK-3� have distinct, and perhapsopposing, roles during myocardial ischemia that complicateour observations in a mouse line in which both alleles havebeen targeted in all cell types and tissues. Thus, our findingsmay have differed if we had examined mice in which onlyGSK-3� had been targeted. However, in other species atleast, it seems such opposition does not prevent the reductionin infarction seen with pharmacological inhibitors that do notdiscriminate between the GSK-3 isoforms.16,17,35,36
Downstream of GSK-3�/� there are proposed links withcertain components of the mPTP, including the voltage-dependent anion channel and adenine nucleotide transloca-tor.15,37 GSK-3�/� is suggested as the immediate point ofconvergence for multiple preconditioning signaling path-ways.15 However, more recent studies have suggested thatvoltage-dependent anion channel and adenine nucleotidetranslocator are not essential to functional mPTP forma-tion38,39 and that there is no mitochondrial GSK-3� detectable
BIO
100
nM
BIO
50
nM
Insu
lin
Con
trol
SB1.
0 µM
SB 3
.0 µ
M
total-GSK-3
pSer9-GSK-3 phos
ho/to
tal
GSK
-3β
0
0.2
0.4
0.6
0.8
1.0
Insu
lin
Con
trol
BIO
50
nM
BIO
100
nM
SB 1
.0
SB 3
.0
0.1
0.2
0.3
0.4
0
0.5
phos
ho/to
tal
glyc
ogen
syn
thas
e
total-glycogensynthase
pSer641/645-glycogensynthase
*
**
* **
Figure 4. Effects of GSK-3 inhibition on GSK-3 and glycogen synthase levels.C57BL/6 hearts were harvested after 15 minutes of perfusion with normalbuffer (Control), insulin, SB216763 (SB), and BIO. Representative immuno-blots are shown with quantitative analyses of repeat experiments expressedas the ratio of phosphorylated to total protein. Data are expressed as themeans�SEM (n�3). *P�0.05 vs control.
6 Circulation Research August 1, 2008
by on May 10, 2011 circres.ahajournals.orgDownloaded from

after preconditioning stimuli,14 raising doubt more than thedependency of mPTP function on upstream GSK-3 activity.CsA administered at the point of reperfusion is believed toinhibit mPTP opening by preventing binding of cyclophilin Dto the adenine nucleotide translocator40 and has been shownsignificantly to reduce infarct size by numerous independentinvestigators.3,41–44 This correlates with the cardioprotectionafforded by CsA in our study, demonstrated in both WT andKI animals, supporting the independence of mPTP functionand its own candidacy as end effector in protective signaling.
Surprisingly, GSK-3 KI hearts appeared less sensitive toinfarction than WT mice, both in response to global ischemiain the whole heart and at the cellular level in response tooxidative stress. One possible explanation for this may beattributable to compensatory signaling pathways other thanGSK-3 in KI mice. However, we did not observe anydifference between genotypes in Akt, p38, extracellularsignal-regulated kinase, p70S6 kinase, and PKC-� signaling,all of which have been reported as important kinases incardioprotective signaling (data not shown).10,11
Our results using pharmacological inhibitors of GSK-3 inthe mouse heart differ from those previously reported in otherspecies (rats16,17,35 or rabbit36). Most importantly, this mayreflect species difference, because we observed a protectiveeffect in parallel studies of the isolated ischemic rat heartpretreated with BIO and SB216763 (percentage infarct size;29.7�2.9% [P�0.05] and 38.4�4.3% versus 43.6�3.9% incontrol, respectively) (unpublished data). Other potentialexplanations for the discrepancy, including experimentalpreparations (global versus regional ischemia17,35,36 or exvivo versus in vivo17) and inhibitor concentration (eg,3 �mol/L versus 1 �mol/L SB21676336), should be consid-ered and inhibitors titrated based on their effects on signalingin that specific model and species. However, the use of 2structurally diverse pharmacological inhibitors, together withthe persistence of the conditioning effects, in the double KImice would support the validity of our results in the mouse.
In the present study, the isolated Langendorff-perfusedmurine heart model was used to obtain contractile parametersof cardiac function, without confounding influences of neu-rohormonal and other compensatory factors. We acknowl-edge the use of isolated hearts with relatively short durationsof reperfusion as a limitation of the study which might affect5-triphenyl tetrazolium chloride staining for infarct measure-ment. Two postconditioning regimens were tested against anestablished murine preconditioning protocol. Postcondition-ing with 5-second cycles of reperfusion/ischemia was equallyeffective as preconditioning in limiting infarct size andpreserving hemodynamic parameters in response to globalischemia, whereas 30-second cycles of postconditioning,known to be protective in some species,2,45–47 proved inef-fective. These findings provide confirmatory evidence thatthe effectiveness of ischemic postconditioning is highlydependent on the number and duration of the interruptions toreperfusion, with a trend toward longer reperfusion-ischemiacycle lengths required in animals of increasing size (ie, 5 to10 seconds in mice7,42 versus 1 minute in humans4).
In conclusion, our findings based on a combination ofgenetic and pharmacological interventions do not support the
premise that GSK-3 inhibition is essential to pre- or postcon-ditioning of isolated mouse hearts.
AcknowledgmentsWe thank Dario Alessi (Medical Research Council Protein Phos-phorylation Unit, School of Life Sciences, University of Dundee,Scotland) for providing the GSK-3–targeted mice.
Sources of FundingThis work was funded by a Wellcome Trust Project grant (074653)and British Heart Foundation fellowships 07/032 (to I.G.W.) and06/026 (to J.E.C.).
DisclosuresNone.
References1. Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a
delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136.
2. Zhao ZQ, Corvera JS, Halkos ME, Kerendi F, Wang NP, Guyton RA,Vinten-Johansen J. Inhibition of myocardial injury by ischemic postcon-ditioning during reperfusion: comparison with ischemic preconditioning.Am J Physiol Heart Circ Physiol. 2003;285:H579–H588.
3. Cohen MV, Yang XM, Downey JM. The pH hypothesis of postcondi-tioning: staccato reperfusion reintroduces oxygen and perpetuates myo-cardial acidosis. Circulation. 2007;115:1895–1903.
4. Staat P, Rioufol G, Piot C, Cottin Y, Cung TT, L’Huillier I, Aupetit JF,Bonnefoy E, Finet G, Andre-Fouet X, Ovize M. Postconditioning thehuman heart. Circulation. 2005;112:2143–2148.
5. Laskey WK. Brief repetitive balloon occlusions enhance reperfusionduring percutaneous coronary intervention for acute myocardialinfarction: a pilot study. Catheter Cardiovasc Interv. 2005;65:361–367.
6. Downey JM, Davis AM, Cohen MV. Signaling pathways in ischemicpreconditioning. Heart Fail Rev. 2007;12:181–188.
7. Heusch G, Buchert A, Feldhaus S, Schulz R. No loss of cardioprotectionby postconditioning in connexin 43-deficient mice. Basic Res Cardiol.2006;101:354–356.
8. Kuno A, Critz SD, Cui L, Solodushko V, Yang XM, Krahn T, AlbrechtB, Philipp S, Cohen MV, Downey JM. Protein kinase C protects precon-ditioned rabbit hearts by increasing sensitivity of adenosine A(2b)-dependent signaling during early reperfusion. J Mol Cell Cardiol. 2007;43:262–271.
9. Hausenloy DJ, Wynne AM, Yellon DM. Ischemic preconditioning targetsthe reperfusion phase. Basic Res Cardiol. 2007;102:445–452.
10. Zhao ZQ, Vinten-Johansen J. Postconditioning: reduction of reperfusion-induced injury. Cardiovasc Res. 2006;70:200–211.
11. Hausenloy DJ, Yellon DM. Survival kinases in ischemic preconditioningand postconditioning. Cardiovasc Res. 2006;70:240–253.
12. Halestrap AP, Clarke SJ, Javadov SA. Mitochondrial permeability tran-sition pore opening during myocardial reperfusion–a target for cardiopro-tection. Cardiovasc Res. 2004;61:372–385.
13. Gateau-Roesch O, Argaud L, Ovize M. Mitochondrial permeability tran-sition pore and postconditioning. Cardiovasc Res. 2006;70:264–273.
14. Halestrap AP, Clarke SJ, Khaliulin I. The role of mitochondria in pro-tection of the heart by preconditioning. Biochim Biophys Acta. 2007;1767:1007–1031.
15. Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu Q, Fishbein KW, ZimanBD, Wang S, Ytrehus K, Antos CL, Olson EN, Sollott SJ. Glycogensynthase kinase-3beta mediates convergence of protection signaling toinhibit the mitochondrial permeability transition pore. J Clin Invest.2004;113:1535–1549.
16. Tong H, Imahashi K, Steenbergen C, Murphy E. Phosphorylation ofglycogen synthase kinase-3beta during preconditioning through aphosphatidylinositol-3-kinase-dependent pathway is cardioprotective.Circ Res. 2002;90:377–379.
17. Gross ER, Hsu AK, Gross GJ. Opioid-induced cardioprotection occursvia glycogen synthase kinase beta inhibition during reperfusion in intactrat hearts. Circ Res. 2004;94:960–966.
18. McManus EJ, Sakamoto K, Armit LJ, Ronaldson L, Shpiro N, MarquezR, Alessi DR. Role that phosphorylation of GSK3 plays in insulin and
Nishino et al GSK3 in Preconditioning and Postconditioning 7
by on May 10, 2011 circres.ahajournals.orgDownloaded from

Wnt signalling defined by knockin analysis. EMBO J. 2005;24:1571–1583.
19. Meijer L, Flajolet M, Greengard P. Pharmacological inhibitors ofglycogen synthase kinase 3. Trends Pharmacol Sci. 2004;25:471–480.
20. Saurin AT, Pennington DJ, Raat NJ, Latchman DS, Owen MJ, MarberMS. Targeted disruption of the protein kinase C epsilon gene abolishesthe infarct size reduction that follows ischaemic preconditioning ofisolated buffer-perfused mouse hearts. Cardiovasc Res. 2002;55:672–680.
21. Tanno M, Bassi R, Gorog DA, Saurin AT, Jiang J, Heads RJ, Martin JL,Davis RJ, Flavell RA, Marber MS. Diverse mechanisms of myocardialp38 mitogen-activated protein kinase activation: evidence for MKK-independent activation by a TAB1-associated mechanism contributing toinjury during myocardial ischemia. Circ Res. 2003;93:254–261.
22. Bellahcene M, Jacquet S, Cao XB, Tanno M, Haworth RS, Layland J,Kabir AM, Gaestel M, Davis RJ, Flavell RA, Shah AM, Avkiran M,Marber MS. Activation of p38 mitogen-activated protein kinase con-tributes to the early cardiodepressant action of tumor necrosis factor. J AmColl Cardiol. 2006;48:545–555.
23. Hausenloy DJ, Yellon DM, Mani-Babu S, Duchen MR. Preconditioningprotects by inhibiting the mitochondrial permeability transition. Am JPhysiol Heart Circ Physiol. 2004;287:H841–H849.
24. Mora A, Sakamoto K, McManus EJ, Alessi DR. Role of the PDK1-PKB-GSK3 pathway in regulating glycogen synthase and glucose uptakein the heart. FEBS Lett. 2005;579:3632–3638.
25. Antos CL, McKinsey TA, Frey N, Kutschke W, McAnally J, Shelton JM,Richardson JA, Hill JA, Olson EN. Activated glycogen synthase-3 betasuppresses cardiac hypertrophy in vivo. Proc Natl Acad Sci U S A. 2002;99:907–912.
26. D’Souza SP, Yellon DM, Martin C, Schulz R, Heusch G, Onody A,Ferdinandy P, Baxter GF. B-type natriuretic peptide limits infarct size inrat isolated hearts via KATP channel opening. Am J Physiol Heart CircPhysiol. 2003;284:H1592–H1600.
27. Okawa H, Horimoto H, Mieno S, Nomura Y, Yoshida M, Shinjiro S.Preischemic infusion of alpha-human atrial natriuretic peptide elicitsmyoprotective effects against ischemia reperfusion in isolated rat hearts.Mol Cell Biochem. 2003;248:171–177.
28. Yang XM, Philipp S, Downey JM, Cohen MV. Atrial natriuretic peptideadministered just prior to reperfusion limits infarction in rabbit hearts.Basic Res Cardiol. 2006;101:311–318.
29. Pearce NJ, Arch JR, Clapham JC, Coghlan MP, Corcoran SL, Lister CA,Llano A, Moore GB, Murphy GJ, Smith SA, Taylor CM, Yates JW,Morrison AD, Harper AJ, Roxbee-Cox L, Abuin A, Wargent E, HolderJC. Development of glucose intolerance in male transgenic mice overex-pressing human glycogen synthase kinase-3beta on a muscle-specificpromoter. Metabolism. 2004;53:1322–1330.
30. Michael A, Haq S, Chen X, Hsich E, Cui L, Walters B, Shao Z, Bhat-tacharya K, Kilter H, Huggins G, Andreucci M, Periasamy M, SolomonRN, Liao R, Patten R, Molkentin JD, Force T. Glycogen synthasekinase-3beta regulates growth, calcium homeostasis, and diastolicfunction in the heart. J Biol Chem. 2004;279:21383–21393.
31. Doble BW, Patel S, Wood GA, Kockeritz LK, Woodgett JR. Functionalredundancy of GSK-3alpha and GSK-3beta in Wnt/beta-catenin signalingshown by using an allelic series of embryonic stem cell lines. Dev Cell.2007;12:957–971.
32. Markou T, Cullingford TE, Giraldo A, Weiss SC, Alsafi A, Fuller SJ,Clerk A, Sugden PH. Glycogen synthase kinases 3alpha and 3beta in
cardiac myocytes: regulation and consequences of their inhibition. CellSignal. 2007;20:206–218.
33. Lee HC, Tsai JN, Liao PY, Tsai WY, Lin KY, Chuang CC, Sun CK,Chang WC, Tsai HJ. Glycogen synthase kinase 3 alpha and 3 beta havedistinct functions during cardiogenesis of zebrafish embryo. BMC DevBiol. 2007;7:93.
34. Zhai P, Gao S, Holle E, Yu X, Yatani A, Wagner T, Sadoshima J.Glycogen synthase kinase-3alpha reduces cardiac growth and pressureoverload-induced cardiac hypertrophy by inhibition of extracellularsignal-regulated kinases. J Biol Chem. 2007;282:33181–33191.
35. Park SS, Zhao H, Mueller RA, Xu Z. Bradykinin prevents reperfusioninjury by targeting mitochondrial permeability transition pore throughglycogen synthase kinase 3beta. J Mol Cell Cardiol. 2006;40:708–716.
36. Forster K, Paul I, Solenkova N, Staudt A, Cohen MV, Downey JM, FelixSB, Krieg T. NECA at reperfusion limits infarction and inhibits formationof the mitochondrial permeability transition pore by activating p70S6kinase. Basic Res Cardiol. 2006;101:319–326.
37. Pastorino JG, Hoek JB, Shulga N. Activation of glycogen synthase kinase3beta disrupts the binding of hexokinase II to mitochondria by phosphor-ylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Res. 2005;65:10545–10554.
38. Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP,MacGregor GR, Wallace DC. The ADP/ATP translocator is not essentialfor the mitochondrial permeability transition pore. Nature. 2004;427:461–465.
39. Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD. Voltage-dependent anion channels are dispensable for mitochondrial-dependentcell death. Nat Cell Biol. 2007;9:550–555.
40. Woodfield K, Ruck A, Brdiczka D, Halestrap AP. Direct demonstrationof a specific interaction between cyclophilin-D and the adenine nucleo-tide translocase confirms their role in the mitochondrial permeabilitytransition. Biochem J. 1998;336:287–290.
41. Hausenloy DJ, Maddock HL, Baxter GF, Yellon DM. Inhibiting mito-chondrial permeability transition pore opening: a new paradigm for myo-cardial preconditioning? Cardiovasc Res. 2002;55:534–543.
42. Lim SY, Davidson SM, Hausenloy DJ, Yellon DM. Preconditioning andpostconditioning: the essential role of the mitochondrial permeabilitytransition pore. Cardiovasc Res. 2007;75:530–535.
43. Krolikowski JG, Bienengraeber M, Weihrauch D, Warltier DC, KerstenJR, Pagel PS. Inhibition of mitochondrial permeability transitionenhances isoflurane-induced cardioprotection during early reperfusion:the role of mitochondrial KATP channels. Anesth Analg. 2005;101:1590–1596.
44. Argaud L, Gateau-Roesch O, Muntean D, Chalabreysse L, Loufouat J,Robert D, Ovize M. Specific inhibition of the mitochondrial permeabilitytransition prevents lethal reperfusion injury. J Mol Cell Cardiol. 2005;38:367–374.
45. Yang XM, Proctor JB, Cui L, Krieg T, Downey JM, Cohen MV. Multiple,brief coronary occlusions during early reperfusion protect rabbit hearts bytargeting cell signaling pathways. J Am Coll Cardiol. 2004;44:1103–1110.
46. Halkos ME, Kerendi F, Corvera JS, Wang NP, Kin H, Payne CS, Sun HY,Guyton RA, Vinten-Johansen J, Zhao ZQ. Myocardial protection withpostconditioning is not enhanced by ischemic preconditioning. AnnThorac Surg. 2004;78:961–969.
47. Iliodromitis EK, Georgiadis M, Cohen MV, Downey JM, Bofilis E,Kremastinos DT. Protection from post-conditioning depends on thenumber of short ischemic insults in anesthetized pigs. Basic Res Cardiol.2006;101:502–507.
8 Circulation Research August 1, 2008
by on May 10, 2011 circres.ahajournals.orgDownloaded from

Online Supplement Nishino et al., GSK3 in preconditioning and postconditioning
Materials and Methods
All experiments were performed in accordance with United Kingdom Home Office
Guidance on the Operation of Animals (Scientific Procedures) Act 1986, published by Her
Majesty’s Stationary Office, London.
GSK-3 knock-in mice
The targeting strategy used to generate GSK-3α/β knock-in mice, in which the PKB/Akt
phosphorylation sites on GSK-3α (Ser 21) and GSK-3β (Ser9) are changed to Ala, has been
described previously.1 GSK-3 wild type (WT) and homozygous GSK-3 double knock-in mice
(KI) were in-bred, and from the same colony, but for practical reasons not littermates.
Efficacy of GSK-3 modulation was assessed by insulin stimulation of the PKB/Akt-GSK-3
signalling pathway.
Mice were fasted for 8 hours and injected intraperitoneally with insulin (150 mU/g;
Actrapid, Novo Nordisk) or saline solution. At 40 min after injection hearts were rapidly
extracted and snap frozen in liquid nitrogen. Immunoblotting was performed for PKB/Akt,
GSK-3 and glycogen synthase.
Perfusion of isolated murine hearts
Adult male mice were anesthetized with pentobarbital (300 mg/kg) and heparin (150 units)
intraperitoneally. Hearts were rapidly isolated, mounted onto a Langendorff apparatus and
retrogradely perfused at a constant pressure of 80 mmHg with Krebs-Hensleit buffer (in
mmol/L: 118.5 NaCl, NaHCO3, 4.75 KCl, 0.18 KH2PO4, 1.19 MgSO4, 11.0 D-glucose, and
1.41 CaCl2) equilibrated with 95% O2 and 5% CO2 at 37°C. A fluid-filled balloon inserted
into the left ventricular cavity monitored contractile function. The balloon was gradually
inflated until the left ventricular end-diastolic pressure (LVEDP) was between 2 and 8
mmHg. Atrial pacing was performed at 580 bpm and coronary flow (CF) measured by timed
collection of perfusate.
Infarction assessment in isolated murine hearts
Hearts were stabilised for 30 min after initiation of retrograde perfusion. For inclusion, all
hearts had to fulfill the following criteria:- CF between 1.5 and 4.5 mL/min, initial heart rate
> 300 bpm (unpaced), left ventricular developed pressure (LVDP) > 55 mmHg, time from
by on May 10, 2011 circres.ahajournals.orgDownloaded from

Online Supplement Nishino et al., GSK3 in preconditioning and postconditioning thoracotomy to aortic cannulation < 3 min, and no persistent dysrhythmias during the
stabilization period. All hearts underwent 30 min of global ischemia followed by 2 hours of
reperfusion. At the end of reperfusion hearts were perfused for 1 min with 5 mL of 1 %
triphenyl tetrazolium chloride (TTC) in PBS and then placed in an identical solution at 37 °C
for 10 min. The atria were then removed, and the hearts were blotted dry, weighed, and stored
at -20 °C. Hearts were subsequently thawed, placed in 2.5 % glutaraldehyde for 1 min and set
in 5 % agarose. The agarose heart blocks were then sectioned from apex to base in 0.75 mm
slices using a vibratome (Agar Scientific). Sections were compressed between glass plates
and scan-imaged (Epson model G850A). After magnification, planimetry was carried out
using image analysis software (Adobe Photoshop 7.0). Risk and infarct volumes were
calculated from surface area analysis of whole myocardium and TTC-negative myocardium,
respectively, multiplied by tissue thickness. After summation of individual slices, infarct
volume was expressed as a percentage of ventricular volume. Infarct analysis was performed
in all cases by an investigator blinded to the group assignments.
Pre- and post-conditioning experiments
Protocol 1: Our objective in this protocol was to assess the potential for postconditioning
(PostC) to limit infarct size, and compare it to preconditioning (PreC) in our model. C57BL/6
mice were divided into 4 study groups, as shown in Figure 1A. In the Control group there
was no additional intervention. Hearts in the PreC group underwent 4 cycles of 4 min
ischemia/6 min reperfusion before the 30 min global ischemia. Hearts in the PostC group
were subjected to one of two protocols in order to determine the optimum strategy:- in the
30sec PostC and 5sec PostC groups, 4 cycles of 30 sec reperfusion/30 sec ischemia and 10
cycles of 5 sec reperfusion/5 sec ischemia were performed at the end of 30 min index
ischemia, respectively.
Protocol 2: GSK-3α/β knock-in mice were used to determine the dependence of pre- and
post-conditioning cardioprotection on GSK-3 inhibition. GSK-3 WT and KI mice were
assigned to Control, PreC and PostC with repetitive 5sec reperfusion/ischemia groups as
described in Protocol 1.
Protocol 3: In these experiments the effects on infarct size of direct pharmacological
inhibition of GSK-3 activity before and after ischemia were examined (Figure 1B). Two
inhibitors, SB216763 (3 µmol/L) (Sigma) and 6-bromoindirubin-3’-oxime (BIO) (100
by on May 10, 2011 circres.ahajournals.orgDownloaded from

Online Supplement Nishino et al., GSK3 in preconditioning and postconditioning nmol/L) (Calbiochem), were employed based on concentrations required to achieve
dephosphorylation of glycogen synthase. C57BL/6 mice were divided into five groups
(Figure 1B):- (i) standard ischemia/reperfusion group (Control), (ii) pretreatment with 3
µmol/L SB216763 (Pre SB) or (iii) 100 nmol/L BIO (Pre BIO) for the last 15 min of
stabilization, (iv) treatment postischemia with 3 µmol/L SB216763 for the first 15 min of
reperfusion (Post SB15) or (v) 3 µmol/L SB216763 for 2 hours (Post SB120). SB216763 and
BIO were dissolved in dimethyl sulfoxide (DMSO) and then diluted with perfusion buffer so
that the final concentration of DMSO was 0.05 %.
Protocol 4: These experiments set out to determine if the cardioprotective effect of direct
pharmacological inhibition of mPTP opening at reperfusion is preserved in the GSK-3α/β
knock-in mouse. After 30 min global ischemia, GSK-3 WT and KI hearts were subjected to
either infusion of 0.2 µmol/L Ciclosporin A (Sigma) (CsA) for 10 min at the moment of
reperfusion, or normal buffer (Control) (Figure 1B). Ciclosporin A was dissolved in DMSO
at a final concentration of less than 0.05 %. Similar experiments were also performed in
isolated adult murine myocytes to visualise mPTP opening directly.
Immunoblotting
Samples were thawed and homogenized in protein extraction buffer (50 mmol/L Tris-HCl;
pH 7.5, 1 mmol/L EDTA, 1 mmol/L EGTA, 1% (w/v) Triton X-100, 0.1 % β-
mercaptoethanol, 1 mmol/L sodium orthovanadate, 50 mmol/L sodium fluoride, 5 mmol/L
sodium pyrophosphate and protease inhibitor tablets (Complete®, Roche Applied Science),
(1 Tab/50 ml of buffer). One millilitre of extraction buffer was used per 100 mg of frozen
tissue. Homogenates were centrifuged at 4 °C for 10 min at 13,000 rpm and the insoluble
fraction discarded. Protein content was estimated using a Bio-Rad Protein Assay Kit. Twenty
micrograms of protein were electrophoresed on a 10 % polyacrylamide gel and then blotted
onto a polyvinylidene difluoride membrane. After blocking with 5 % non-fat milk, the
membranes were incubated with the following primary antibodies at dilutions of 1:1000:-
anti-total-GSK-3β (#9315), anti-phospho-GSK-3α/β (Ser21/9) (#9331), anti-phospho-GSK-
3β (Ser9) (#9336), anti-total-PKB/Akt (#9272), anti-phospho-PKB/Akt (Ser473) (#9271) (all
from Cell Signalling), anti-total-GSK-3α/β (#05-903) (Upstate), anti-total-glycogen synthase
(#MAB3106) (Chemicon), anti-phospho-GSK-3α/β (Tyr 279 and 216) (#44-604G) (#9331)
and anti-phospho-glycogen synthase (Ser 641 and 645) (#44-1092G) (Biosource).
by on May 10, 2011 circres.ahajournals.orgDownloaded from

Online Supplement Nishino et al., GSK3 in preconditioning and postconditioning Appropriate secondary antibodies were used and proteins visualized using an ECL Western
blotting detection kit (Amersham).
Isolation of adult mouse ventricular cardiomyocytes
Adult male mice were injected with heparin sodium (250 units) and anesthetized with
ketamine/xylazine/atropine. The hearts were rapidly excised, cannulated and perfused with
(in mmol/L) 113 NaCl, 4.7 KCl, 0.6 KH2PO4, 0.6 Na2HPO4, 1.2 MgSO4.7H2O, 12 NaHCO3,
10 KHCO3, 30 taurine, 10 HEPES, and 5.5 glucose, saturated with 95% O2-5% CO2 at 37°C.
The hearts were perfused at 3 ml/min with perfusion buffer for 4 min, then with perfusion
buffer containing 0.9 mg/ml collagenase (Worthington type II), 0.125 mg/ml hyaluronidase
and 12.5 μmol/L CaCl2 for 10 min. The ventricles were then cut into several pieces and
shaken at 37oC with oxygenation for 10 min. The supernatant was collected and 5% foetal
calf serum was added. After centrifugation at 600 rpm for 3 min, the cell pellet was
suspended in 10 ml of perfusion buffer containing 12.5 μmol/L CaCl2 and the calcium
concentration was gradually restored to 1 mmol/L over 20 min. The myocytes were
recentrifuged then seeded onto sterilized laminin-coated coverslips in MEM medium
containing 10 units/ml penicillin, 10 μg/ml streptomycin, 5% foetal calf serum for incubation
before use on the same day of isolation.
Assay of mPTP opening
The sensitivity of the mPTP to opening was assayed using a well-characterized and
reproducible cellular model.2 Live isolated myocytes were incubated with the fluorescent
dye, tetra-methyl rhodamine methyl ester (TMRM, 3 μmol/L) for 15 minutes in microscopy
buffer (i.e.: perfusion buffer containing 1.2 mmol/L CaCl2), then washed and imaged using
confocal microscopy. TMRM, a lipophilic cation, accumulates selectively into mitochondria
according to the mitochondrial membrane potential. Continual confocal laser-scanning
generates reactive oxygen species (ROS) from the TMRM within the mitochondria, which,
after several minutes, provokes mPTP opening, as indicated by mitochondrial depolarization.
Some wells were treated with Ciclosporin A (0.2 μmol/L), or with insulin (0.3 U/L) for 10
min before and during analysis. A total of 20-39 cells from 4-6 hearts were analyzed in
separate experiments.
Statistical analysis
by on May 10, 2011 circres.ahajournals.orgDownloaded from

Online Supplement Nishino et al., GSK3 in preconditioning and postconditioning All data are presented as means ± SEM. Comparisons between groups were assessed for
significance by analysis of variance (ANOVA) or repeated measures ANOVA, as appropriate.
When significant differences were detected, individual mean values were compared by
Bonferroni’s post hoc test which allowed for multiple comparisons. All statistical analyses
were performed using SPSS for Windows (v12.0.2; SPSS). P values less than 0.05 were
considered significant.
by on May 10, 2011 circres.ahajournals.orgDownloaded from

Online Supplement Nishino et al., GSK3 in preconditioning and postconditioning
Reference List
(1) McManus EJ, Sakamoto K, Armit LJ, Ronaldson L, Shpiro N, Marquez R, Alessi DR. Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J. 2005;24:1571-83.
(2) Hausenloy DJ, Yellon DM, Mani-Babu S, Duchen MR. Preconditioning protects by inhibiting the mitochondrial permeability transition. Am J Physiol Heart Circ Physiol. 2004;287:H841-H849.
by on May 10, 2011 circres.ahajournals.orgDownloaded from
1
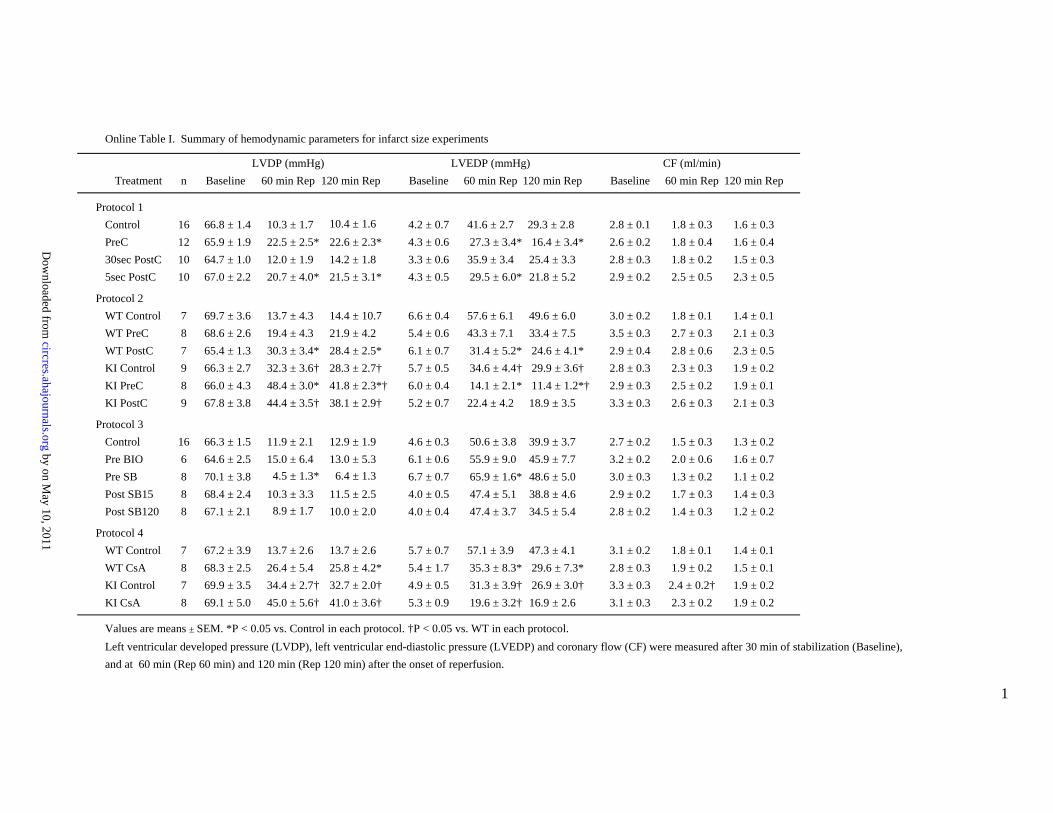
Online Table I. Summary of hemodynamic parameters for infarct size experiments
LVDP (mmHg) LVEDP (mmHg) CF (ml/min)Treatment n Baseline 60 min Rep 120 min Rep Baseline 60 min Rep 120 min Rep Baseline 60 min Rep 120 min Rep
Protocol 1Control 16 66.8 ± 1.4 10.3 ± 1.7 10.4 ± 1.6 4.2 ± 0.7 41.6 ± 2.7 29.3 ± 2.8 2.8 ± 0.1 1.8 ± 0.3 1.6 ± 0.3PreC 12 65.9 ± 1.9 22.5 ± 2.5* 22.6 ± 2.3* 4.3 ± 0.6 27.3 ± 3.4* 16.4 ± 3.4* 2.6 ± 0.2 1.8 ± 0.4 1.6 ± 0.430sec PostC 10 64.7 ± 1.0 12.0 ± 1.9 14.2 ± 1.8 3.3 ± 0.6 35.9 ± 3.4 25.4 ± 3.3 2.8 ± 0.3 1.8 ± 0.2 1.5 ± 0.35sec PostC 10 67.0 ± 2.2 20.7 ± 4.0* 21.5 ± 3.1* 4.3 ± 0.5 29.5 ± 6.0* 21.8 ± 5.2 2.9 ± 0.2 2.5 ± 0.5 2.3 ± 0.5
Protocol 2WT Control 7 69.7 ± 3.6 13.7 ± 4.3 14.4 ± 10.7 6.6 ± 0.4 57.6 ± 6.1 49.6 ± 6.0 3.0 ± 0.2 1.8 ± 0.1 1.4 ± 0.1WT PreC 8 68.6 ± 2.6 19.4 ± 4.3 21.9 ± 4.2 5.4 ± 0.6 43.3 ± 7.1 33.4 ± 7.5 3.5 ± 0.3 2.7 ± 0.3 2.1 ± 0.3WT PostC 7 65.4 ± 1.3 30.3 ± 3.4* 28.4 ± 2.5* 6.1 ± 0.7 31.4 ± 5.2* 24.6 ± 4.1* 2.9 ± 0.4 2.8 ± 0.6 2.3 ± 0.5KI Control 9 66.3 ± 2.7 32.3 ± 3.6† 28.3 ± 2.7† 5.7 ± 0.5 34.6 ± 4.4† 29.9 ± 3.6† 2.8 ± 0.3 2.3 ± 0.3 1.9 ± 0.2KI PreC 8 66.0 ± 4.3 48.4 ± 3.0* 41.8 ± 2.3*† 6.0 ± 0.4 14.1 ± 2.1* 11.4 ± 1.2*† 2.9 ± 0.3 2.5 ± 0.2 1.9 ± 0.1KI PostC 9 67.8 ± 3.8 44.4 ± 3.5† 38.1 ± 2.9† 5.2 ± 0.7 22.4 ± 4.2 18.9 ± 3.5 3.3 ± 0.3 2.6 ± 0.3 2.1 ± 0.3
Protocol 3Control 16 66.3 ± 1.5 11.9 ± 2.1 12.9 ± 1.9 4.6 ± 0.3 50.6 ± 3.8 39.9 ± 3.7 2.7 ± 0.2 1.5 ± 0.3 1.3 ± 0.2Pre BIO 6 64.6 ± 2.5 15.0 ± 6.4 13.0 ± 5.3 6.1 ± 0.6 55.9 ± 9.0 45.9 ± 7.7 3.2 ± 0.2 2.0 ± 0.6 1.6 ± 0.7Pre SB 8 70.1 ± 3.8 14.5 ± 1.3* 16.4 ± 1.3 6.7 ± 0.7 65.9 ± 1.6* 48.6 ± 5.0 3.0 ± 0.3 1.3 ± 0.2 1.1 ± 0.2Post SB15 8 68.4 ± 2.4 10.3 ± 3.3 11.5 ± 2.5 4.0 ± 0.5 47.4 ± 5.1 38.8 ± 4.6 2.9 ± 0.2 1.7 ± 0.3 1.4 ± 0.3Post SB120 8 67.1 ± 2.1 18.9 ± 1.7 10.0 ± 2.0 4.0 ± 0.4 47.4 ± 3.7 34.5 ± 5.4 2.8 ± 0.2 1.4 ± 0.3 1.2 ± 0.2
Protocol 4WT Control 7 67.2 ± 3.9 13.7 ± 2.6 13.7 ± 2.6 5.7 ± 0.7 57.1 ± 3.9 47.3 ± 4.1 3.1 ± 0.2 1.8 ± 0.1 1.4 ± 0.1WT CsA 8 68.3 ± 2.5 26.4 ± 5.4 25.8 ± 4.2* 5.4 ± 1.7 35.3 ± 8.3* 29.6 ± 7.3* 2.8 ± 0.3 1.9 ± 0.2 1.5 ± 0.1KI Control 7 69.9 ± 3.5 34.4 ± 2.7† 32.7 ± 2.0† 4.9 ± 0.5 31.3 ± 3.9† 26.9 ± 3.0† 3.3 ± 0.3 2.4 ± 0.2† 1.9 ± 0.2KI CsA 8 69.1 ± 5.0 45.0 ± 5.6† 41.0 ± 3.6† 5.3 ± 0.9 19.6 ± 3.2† 16.9 ± 2.6 3.1 ± 0.3 2.3 ± 0.2 1.9 ± 0.2
Values are means ± SEM. *P < 0.05 vs. Control in each protocol. †P < 0.05 vs. WT in each protocol.Left ventricular developed pressure (LVDP), left ventricular end-diastolic pressure (LVEDP) and coronary flow (CF) were measured after 30 min of stabilization (Baseline),and at 60 min (Rep 60 min) and 120 min (Rep 120 min) after the onset of reperfusion.
by on May 10, 2011
circres.ahajournals.orgD
ownloaded from

1
Online Table II. Summary of infarct size data
Treatment n Body weight (g) Ventricular weight (mg) Ventricular volume (mm3) Infarct volume (mm3) I/R (%)
Protocol 1Control 16 25.2 ± 0.7 123.0 ± 4.5 196.7 ± 4.2 46.1 ± 3.4 47.2 ± 2.2PreC 12 23.9 ± 1.0 112.5 ± 4.9 185.2 ± 6.0 29.1 ± 3.8* 33.3 ± 2.7* 30sec PostC 10 25.6 ± 0.7 124.0 ± 5.4 193.1 ± 5.0 36.0 ± 3.8 38.3 ± 3.25sec PostC 10 23.5 ± 0.9 116.0 ± 4.8 196.9 ± 3.9 25.9 ± 4.0* 26.4 ± 3.8*
Protocol 2WT Control 7 26.1 ± 1.7 138.6 ± 5.8 101.8 ± 5.6 62.0 ± 4.9 61.1 ± 4.1WT PreC 8 27.4 ± 0.8 134.7 ± 8.8 102.5 ± 7.9 42.6 ± 5.0* 41.8 ± 4.7* WT PostC 7 26.9 ± 0.5 121.5 ± 5.3 191.8 ± 6.1 37.8 ± 6.6* 41.7 ± 7.2* KI Control 9 27.2 ± 1.9 128.4 ± 10.7 196.9 ± 7.9 39.1 ± 5.0† 39.5 ± 2.8† KI PreC 8 27.4 ± 0.9 128.2 ± 6.7 196.6 ± 5.5 21.1 ± 3.3*† 21.9 ± 3.2*†KI PostC 9 28.3 ± 1.7 133.9 ± 9.0 101.4 ± 8.6 22.4 ± 5.3† 22.2 ± 4.3*†
Protocol 3Control 16 25.5 ± 0.7 128.5 ± 6.3 191.1 ± 5.4 40.9 ± 4.1 45.4 ± 3.9Pre BIO 6 26.2 ± 1.9 123.0 ± 8.4 191.0 ± 5.6 37.7 ± 3.4 42.7 ± 4.4Pre SB 8 24.3 ± 0.9 124.1 ± 4.5 193.2 ± 5.0 46.9 ± 6.8 48.8 ± 4.6Post SB15 8 26.1 ± 1.0 126.0 ± 7.2 195.6 ± 7.4 42.9 ± 7.3 44.2 ± 5.6Post SB120 8 26.0 ± 0.6 126.1 ± 4.7 195.2 ± 4.0 39.0 ± 3.1 40.8 ± 2.6
Protocol 4WT Control 7 27.4 ± 1.0 144.4 ± 5.5 105.9 ± 4.3 57.2 ± 4.9 54.4 ± 3.0WT CsA 8 26.9 ± 1.1 133.1 ± 11.2 105.2 ± 8.5 39.3 ± 8.3* 36.2 ± 7.2* KI Control 7 26.4 ± 0.6 126.4 ± 4.3 196.0 ± 4.5 38.3 ± 3.9 39.6 ± 2.7KI CsA 8 27.0 ± 1.0 120.1 ± 6.7 194.1 ± 5.0 22.9 ± 3.4* 24.3 ± 3.5*
Values are means ± SEM. I/R, infarct size as a percentage of area at risk.*P < 0.05 vs. Control in each study group. †P < 0.05 vs. WT in each study group.
by on May 10, 2011
circres.ahajournals.orgD
ownloaded from
Copyright © 2022 FDOKUMEN




















