
A Legislative Proposal to Regulate Timesharing Agreements ...
Upload
independentCategory
view
0download
0
Fludarabine ability to down-regulate Bcl-2 gene productin CD5þ leukaemic B cells: in vitro/in vivo correlations
D. GOTTARDI, A. M. DE LEO, A. ALFARANO, A. STACC HINI, P. CIRCOSTA, M. G. GREGORET TI, L. BE RGUI, M. ARAGNO
AND F. CAL IGARIS-CAPPIO Cattedra di Immunologia Clinica, Dipartimento di Scienze Biomediche e Oncologia Umana,Universita di Torino, Torino, Italy
Received 14 May 1997; accepted for publication 2 July 1997
Summary. CD5þ B-chronic lymphocytic leukaemia (B-CLL)and mantle cell lymphoma (MCL) in leukaemic phase arecharacterized by defects in cell death induction thatprimarily involves the Bcl-2 family of genes. Fludarabine(9-b-D-arabinofuranosyl-2-fluoradenine, F-ara-A) is a potentinducer of apoptosis in CLL cells. This study aimed todetermine whether F-ara-A-induced apoptosis might berelated to Bcl-2 modifications and to evaluate in vitro/invivo correlations.
Peripheral blood lymphocytes from eight B-CLL andfour leukaemic MCL were cultured in the presence ofdifferent concentrations of F-ara-A 6methylprednisolone(MP). F-ara-A down-regulated the expression of Bcl-2 in5/12 cases. mRNA down-regulation was maximal at 48 h;protein down-regulation was prominent after 48 h. Bothevents were dose-dependent. The amount of apoptosis wassignificantly higher in the samples treated with F-ara-A than
in those exposed to MP alone. In the seven remaining cases,no Bcl-2 down-regulation was observed after exposure toF-ara-A and the degree of F-ara-A-induced apoptosis over-lapped that induced by MP.
The in vivo outcome after treatment with three to sixcourses of F-ara-A was evaluable in 10 patients: 4/5 cases,whose cells had shown in vitro Bcl-2 down-regulation andprominent apoptosis after exposure to F-ara-A, had acomplete response (CR) and a partial response (PR) wasobserved in the remaining patient. Of the five patients whosecells had shown no in vitro Bcl-2 modulation after exposureto F-ara-A, two had a PR, but the other three did not showany in vivo clinical response.
Keywords: apoptosis, B-chronic lymphocytic leukaemia,CD5þ B cells, Bcl-2, fludarabine.
Defects in cell death induction are the pathogenetic tenet ofCD5þ B-chronic lymphoid leukaemias, mainly representedby B-chronic lymphocytic leukaemia (B-CLL) and leukaemicmantle cell lymphoma (MCL) (Harris et al, 1994). Thesedefects primarily involve the Bcl-2 gene family, whosemembers encode for important interrelated positive andnegative regulators of apoptosis (Yang & Korsmeyer, 1996).Bcl-2 gene product, the prototype antidote to apoptosis, isconsistently over-expressed in B-CLL CD5þ B cells (Pezzella etal, 1990; Schena et al, 1992). Its levels do not reflect arearrangement of Bcl-2 gene, which is a rare event in CLLcells (Dyer et al, 1994), but may be dependent upon the Bcl-2gene hypomethylation that causes increased transcription(Hanada et al, 1993) or be under the influence of apoptosis-protecting cytokines (Dancescu et al, 1992; van Kooten et al,1992; Buschle et al, 1993). Important determinants of Bcl-2
function are the Bax-encoded protein that may formheterodimers with Bcl-2 (Oltvai et al, 1993) and the splicedforms of Bcl-x, Bcl-xL and Bcl-xS (Boise et al, 1993).Malignant CD5þ B cells are characterized by high levels ofBax and Bcl-xL, whereas Bcl-xS is present in low to traceamounts and in a smaller proportion of cases (Gottardi et al,1996). As Bcl-xL synergizes with Bcl-2, and Bcl-xS inhibitsBcl-2 function (Boise et al, 1993), the pattern of expression ofBcl-2 gene family in malignant CD5þ B cells appears to beshifted toward prevention of apoptosis. Further, B-CLL cellsare Fas (CD95) negative (Mapara et al, 1993) and do notexpress c-myc (Larsson et al, 1987). As Bcl-2 and Fas areinversely related (Moller et al, 1993) and Bcl-2 is able toabrogate c-myc-induced apoptosis (Bissonnette et al, 1992;Fanidi et al, 1992), these data suggest that Bcl-2 ability toblock apoptosis is unrestrained in CD5þ B-CLL cells and isresponsible for extended cell survival and in vivo accumula-tion of malignant cells. Accordingly, the quantitation ofintracellular Bcl-2 protein has been shown to provide
British Journal of Haematology, 1997, 99, 147–157
147q 1997 Blackwell Science Ltd
Correspondence: Dr F. Caligaris-Cappio, Cattedra di ImmunologiaClinica, Via Genova 3, 10126 Torino, Italy.

prognostic information in CLL, the highest expression beingan adverse feature (Robertson et al, 1996).
Paradoxically, short-term cultures indicate that in vitrosome cases of B-CLL are unusually prone to apoptosis. Thisoccurs both spontaneously (Collins et al, 1989), suggestingthat the culture systems lack a cytokine that in vivo ensuressurvival (Mainou-Fowler et al, 1996), or upon exposure toglucocorticoid hormones (McConkey et al, 1991) or drugsused clinically such as fludarabine (9-b-D-arabinofuranosyl-2-fluoradenine, F-ara-A), a purine nucleoside analogueresistant to adenosine deaminase (Robertson et al, 1993;Zinzani et al, 1994; McConkey et al, 1996). F-ara-A is ofgreat interest because of its strong therapeutic activity inlow-grade lymphoid malignancies (Keating et al, 1989).Though F-ara-A is a potent inducer of apoptosis in CLL cell(Robertson et al, 1993; Zinzani et al, 1994; McConkey et al,1996), the precise molecular mechanism through which itexerts this effect has not yet been fully established (Robertsonet al, 1992; Miyashita & Reed, 1992, 1993; Menzel et al,1996). Recently, it has been reported that apoptosissensitivity in CLL cells is determined by endogenousendonuclease content and by the relative expression ofBcl-2 and Bax (McConkey et al, 1996; Petersen et al, 1996;Thomas et al, 1996).
The aim of this study was to define whether F-ara-Ainduction of apoptosis of fresh malignant CD5þ samplesmight be related to Bcl-2 modifications and to exploit in vitro/in vivo correlations.
PATIENTS
Twelve patients, eight with B-CLL and four with MCL inleukaemic phase, were studied. Seven were male and fivefemale, and the median age was 63 years (range 59–70years). Clinical staging was performed according to the Raistaging system (Rai et al, 1975): three patients were stage II,two stage III and seven stage IV. All patients were refractoryto standard chemotherapy and 11/12 were treated withF-ara-A (25 mg/m2 intravenously (i.v.) daily for 5 d) plusmethylprednisolone (MP: 50 mg i.v. for 5 d). Patientsreceived therapy at 4-week intervals for a total of three tosix courses.
Response criteria. To evaluate the clinical response thenumber of peripheral blood lymphocytes (PBL) as well asliver, spleen and lymphnode size were considered (Robertsonet al, 1992). The criteria for complete response (CR) were:PBL < 4 × 109/ml, liver and spleen not palpable, no pathologicnodes. The criteria for partial response (PR) were: PBLdecrease >50%, liver and spleen span decrease>50%,decrease in node size >50%. CR and PR patients also hadneutrophil counts >1.5 × 109/l, platelet counts >100 × 109/land an untransfused haemoglobin level >11 g/dl. Patientswere considered to have resistant disease if they had notachieved PR criteria after six courses of therapy or hadprogressive disease at any stage of treatment.
MATERIAL AND METHODS
Plan of the study. The plan of the study was as follows:
(a) prior to F-ara-A treatment, PBL were cultured in thepresence of different concentrations of F-ara-A 6MP, har-vested and analysed at different times; (b) in vitro results werecompared to the clinical outcome of the in vivo administra-tion of the drugs.
Cell separation and phenotyping. PBL were obtained byseparation on Ficoll-Hypaque (FH) gradient. Cells werewashed twice in phosphate-buffered saline (PBS). An aliquotwas resuspended in PBS containing 1% bovine serumalbumin (BSA) and 0.02% sodium azide (staining medium)at the concentration of 10 × 106/ml to analyse cellphenotype. An appropriate amount of fluorochrome-labelledantibody (Ab), at optimal concentration, was added to100 ml cell suspension. Negative controls were incubatedwith isotype irrelevant Abs. After 30 min incubation at 48C,cells were washed twice with 2 ml of staining medium andresuspended for flow cytometric analysis.
Different combinations of Abs were used in directimmunofluorescence (IF). Goat (G) antisera to human (h)IgM, IgD, k and l chains, directly conjugated withfluorescein-isothiocyanate (FITC, Tago, Burlingame, Calif.,cat. nos. 4202, 4205, 4206, 4208). The monoclonal (M)Abs used were CD5-FITC (Leu1, Becton Dickinson, MountainView, Calif., cat. no. 7303), CD19-phycoerythrin (PE)(Leu12, Becton Dickinson, cat. no. 9209), CD23-PE(Leu20, Becton Dickinson, cat. no. 7797), CD25-PE (Dako,Glostrup Denmark, cat. no. R0811), CD3-FITC (Leu 4,Beckton Dickinson, cat. no. 92-0001), HLA-DR-PE (BectonDickinson, cat. no. 7367), FMC7-FITC (Serotec, Oxford, U.K.,cat. no. MCA792F).
Permeabilization and cytoplasmic staining. To detect cyto-plasmic Bcl-2 and Bax proteins, cells were fixed andpermeabilized. The cell pellet containing 106 cells, resus-pended in 400 ml of 2% paraformaldehyde (Sigma ChemicalCo., St Louis, Mo.), was placed in ice for 10 min. A 1.0%solution of saponin in PBS (50 ml) (Sigma) was added to thecells which were vortexed and placed in ice for another5 min. Fixed cells were washed once in PBS containing 0.1%saponin (PBS-S), resuspended in PBS-S and preincubated for10 min with 1% heat-inactivated human AB serum toprevent non-specific binding to Fc receptors. The directstaining for Bcl-2 was performed by incubating permeabi-lized cells with 10 ml of anti-Bcl-2 MAb FITC (Dakopatts, cat.no. F053) or anti-IgG1 (isotypic control, Dako, cat. no.X0927) for 30 min at 48C. For indirect Bax staining thepermeabilized cells were first incubated with 10 ml of pre-diluted (1:10) anti-Bax polyclonal rabbit (R) Ab (Santa CruzBiotechnology, Santa Cruz, Calif., cat. no. sc526) for 30 minat 48C, followed by washing and further incubation withswine anti-R-Ig-FITC (SaR-Ig-FITC, Dako, cat. no. F205,1:100 dilution) for 30 min at 48C. Negative controls wereperformed by incubating cells with normal R serum. All Abswere used at pretitrated, near saturating amounts.
Flow cytometric analysis. All samples were analysed using aFACScan Research cytometer (Becton Dickinson) equippedwith a 488 nm argon ion laser (BDIS). Data acquisition wasperformed using the FACScan Research Software (BDIS).Forward light scattering, orthogonal light scattering, andtwo fluorescence signals were determined for each cell and
q 1997 Blackwell Science Ltd, British Journal of Haematology 99: 147–157
148 D. Gottardi et al

149F-ara-A-induced Bcl-2 Down-regulation in CD5þ Leukaemic B Cells
q 1997 Blackwell Science Ltd, British Journal of Haematology 99: 147–157
stored in listmode data files. Each measurement contained atleast 5000 cells. In all samples a gate was used on both lightscattering parameters to obtain more events of lymphocytepopulations.
Cell cultures. Cells washed twice in PBS were cultured at afinal concentration of 106/ml in RPMI-1640 mediumsupplemented with 10% heat-inactivated fetal calf serum(FCS), 100 IU/ml penicillin, 100 mg/ml streptomycin,2 mmol/l glutamine in presence of: (a) F-ara-A 6 mg/ml; (b)F-ara-A 30 mg/ml, (c) F-ara-A 30 mg/ml plus MP 1 mM; (d)MP 1 mM. For each case, a standard control culture withoutthe addition of any drug was performed.
Cells were analysed at time 0 and after 24, 48, 72 h ofculture and the following parameters were evaluated: (a) theviability by Trypan blue dye exclusion; (b) the apoptotic cellrate by morphology on May-Grunwald-Giemsa (MGG)stained slides, FACS and DNA fragmentation analysis; (c)the expression of Bcl-2, Bax and Bcl-x RNA by reversetranscriptase-polymerase chain reaction (RT-PCR) and, in afew cases by Northern blot (NB) analysis; (d) the expressionof Bcl-2 and Bax proteins by cytofluorograph analysis. Insome cases, mean fluorescence intensity (MFI) of anti-Bcl-2staining was determined using the flow cytometer. MFI wasexpressed in arbitrary units which were converted in linearunits (LU) according to the formula:
Number of channelsLogarithm decades
× MFI
Northern blot analysis. Total cellular RNA was isolated bythe RNAzolB procedure (Biotecx Laboratories, U.S.A.).Denaturated total RNA samples (15 mg/well) were fraction-ated on a 1% formaldehyde containing agarose gel,transferred to a nitrocellulose filter, and hybridized with32P-labelled probes in 3 × SSC (1 × SSC ¼ 0.15 M NaCl,0.015 M NaCitrate, pH 7.0), 0.2% SDS, 1 × Denhart’ssolution, 100 mg/ml denaturated salmon sperm DNA, and50% formamide, at 428C for 18 h. The filters, washed twiceunder stringent conditions (0.1% SSC, 0.1% SDS) at 548C,were exposed to Kodak X-OMAT films for 1–14 d at ¹808C.
The positive controls were total RNA obtained from Nalm-6 cell line for Bcl-2 expression (Schena et al, 1992).
Probe. The Bcl-2 probe (kind gift of C. Croce, Philadelphia,U.S.A.) was a 2.8 kb EcoRI–Hind III fragment correspondingto the ‘major breakpoint region’ (MBR) in the 30 untrans-lated region of Bcl-2 gene that was radiolabelled using arandom primer labelling kit (Multiprime DNA LabellingSystem, Amersham, U.K.) with [a-32P]dCTP.
The actin probe was mouse a-actin cDNA clone p91.Reverse transcriptase-polymerase chain reaction (RT-PCR)
analysis. 2 mg of total cellular RNA were reverse-transcribedusing 15 U of AMV-RT (Promega, U.S.A.) and 1 mg of (oligo)dTas primer for 30 min at 428C. The PCR was performed in a100 ml reaction mixture containing 10 ml of the obtainedcDNA fragments, 50 pmol of each mRNA sequence-specificsynthetic primers, 200 mmol of each deoxynucleotide triphos-phate and 1 U of Thermus aquaticus (Taq) polymerase(AmpliTaq-DNA polymerase, Perkin Elmer, U.S.A.).
Bcl-2 amplification was performed for 35 cycles (1 mindenaturing at 948C, 30 s annealing at 658C, 30 s extension
at 728C) in a DNA thermal cycler (Perkin Elmer Cetus). Ascontrol the b2 microglobulin (b2M) amplification wasperformed in the same conditions for 35 cycles. Baxamplification was performed for 35 cycles (2 min denaturingat 948C, 90 s annealing at 658C, 90 s extension at 728C).Bcl-x amplification was performed for 40 cycles (1 mindenaturing at 948C, 1 min annealing at 588C, 1 minextension at 728C).
The sequences of Bcl-2 specific primers were: 50-TTCGCC-GAGATGTCCAGCC-; 30-TCACTTGTGGCCCAGATAGG-. Thesequences of Bax specific primers were: 50-TTTATGGACGGG-TCCGGGGA-; 30-TGTCCAGCCCATGATGGTTCT-. The sequencesof Bcl-x specific primers were: 50-TTGGACAATGGACTGG-TTGA-; 30-GTAGAGTGGATGGTCAGTG-. These two primersidentify the short (S) and the long (L) forms of Bcl-x. Aspositive controls we used the amplification product obtainedfrom two different plasmids carrying the two cDNA,respectively Bcl-xL and Bcl-xS (kind gift of Dr C. B.Thompson, Chicago, Illinois). The sequences of b2M specificprimers were: 50-CTCGCGCTACTCTCTCTTTCTGG-; 30-GCTTA-CATGTCTCGATCCCACTTAA.
Amplified products were analysed by 1.5% agaroseelectrophoresis gel and visualized under UV rays afterethidium bromide staining. The Bcl-2 amplified productwas 387 nucleotide (nt) long, Bax product 420 nt long,Bcl-xL 800 nt long, Bcl-xS 600 nt long, and b2M product334 nt long.
Detection of DNA fragmentation. 0.5 × 106 cells werewashed twice in PBS and lysed at 508C for 1 h in lysissolution (EDTA 10 mM, Tris pH 8, 50 mM, laurylsarcosine0.5% w/v, proteinase A 0.5 mg/ml). RNAse 10 ml (0.5 mg/ml) was added and cells were incubated for 1 h at 508C. 10 mlof sample buffer (EDTA 10 mM pH 8, low melting agarose 1%,Bromophenoleblue 0.25 w/v, saccharose 40% w/v) warmedto 728C were added at each sample. Samples wereelectrophoresed on 2% agarose gel, stained with ethidiumbromide, in TBE 1 × buffer.
Flow cytometric evaluation of apoptotic cells. Cells werecentrifuged (200 g, 5 min) and the cell pellet carefullyresuspended in 1.5 ml hypotonic solution (propidiumiodide (PI) 50 mg/ml in 0.1% sodium citrate plus 0.2%Triton X-100 and RNAse A 100 mg/ml, Sigma Chemical Co.,St Louis, Mo.). The tubes were placed at 48C in the darkovernight before flow cytometric analysis.
The PI fluorescence of individual nuclei was measuredusing a FACScan flow cytometer (Becton Dickinson, Moun-tain View, Calif., U.S.A.) equipped with a 488 nm Argon laserand the data were registered on a linear scale on the FL2channel. Cell debris was excluded by appropriately raisingthe forward-scatter threshold. At least 104 cells of eachsample were analysed.
All measurements were performed under the sameinstrument setting.
Statistical analysis. Results are expressed as themean 6 standard deviation (SD). Analysis of the fluorescencehistograms was performed by the Kolmogorov-Smirnov (KS)test (Young, 1977). The test was used for the calculation ofthe D value with the FACScan software, version 2.1-3/89(Becton Dickinson). The D value corresponded to the

maximum difference between the two summation curvescomputed from the histograms, whereas D/S(n) indicated thesimilarity of the two noncompared curves. The closer D/S(n)was to zero, the more alike the two curves were. The formulawas: S(n) ¼ (n1 þ n2)/n1 × n2, where n1 equalled the number ofevents in the first graph and n2 equalled the number of eventsin the second graph. Samples were considered positive when aD/S (n) value >10 was obtained.
RESULTS
General features of the cells studiedThe major immunologic and molecular features of thepatients studied are shown in Table I. 10 cases were kþ andtwo were lþ; CD5 was present in all the cases but one(patient 1, Table I). The proportion of monoclonal B cells wasin the range 90–95% and that of CD3þ T cells did not exceed8% in all the cases studied.
High levels of Bcl-2 mRNA were observed by Northern blotand high levels of Bcl-2 protein were detected by cytofluoro-graph analysis with a specific MAb in all 12 cases (Table I).10 cases were tested for Bax, Bcl-xL and Bcl-xS expression byRT-PCR. 9/10 proved to be Bax positive; in all samples thepresence of Bax protein was confirmed by cytofluorographanalysis (Table I). Bcl-xL was detectable in 5/10 cases at highlevels, wheras Bcl-xS was found in low to trace amounts in3/10 cases (Table I).
F-ara-A may induce Bcl-2 down-regulation in vitroWe first evaluated the possibility that culturing CD5þ
malignant cells in vitro in presence of F-ara-A might down-regulate the expression of Bcl-2. A significant down-regulation of Bcl-2 was evident in 5/12 cases. Even ifmRNA and protein down-regulation were not alwaysproperly synchronized, mRNA down-regulation was evidentat 24 h and maximal at 48 h (Fig 1) and Bcl-2 protein
down-regulation was prominent after 48 h (Fig 2). Bothevents were dose-dependent and took place in the samplestreated with F-ara-A 30 mg/ml or with F-ara-A 30 mg/ml þ MP 1 mM. In 4/5 cases Bcl-2 protein levels wereregistered and expressed in LU. In the presence of F-ara-Athe Bcl-2 LU levels dropped to values which were 60–70% ofthose expressed by control culture cells. The addition of MPto the culture did not modify the entity of Bcl-2 down-regulation F-ara-A induced. The mRNA and protein expres-sion of Bax (Fig 1), Bcl-xL and Bcl-xS (data not shown),evaluated by RT-PCR and flow cytometric analysis in threecases, was unaffected by the addition of F-ara-A and/or MP.
In the remaining seven cases no Bcl-2 mRNA nor Bcl-2protein down-regulation were observed irrespective of theconcentration of F-ara-A and/or the addition of MP.
F-ara-A-induced Bcl-2 down-regulation significantly increases invitro apoptosisAs expected, the investigation of apoptosis in culturedmalignant CD5þ B cells was complicated for two reasons.First, cultured cells underwent a certain degree of spon-taneous apoptosis which, as described (Collins et al, 1989),was increased in the presence of MP. Second, prominentinterpatient variations occurred. Therefore we studied theoccurrence of apoptosis by different techniques. DNAfragmentation analysis of the five cases where F-ara-Atreatment in vitro had been associated with Bcl-2 down-regulation revealed that, after 24–48 h of culture, apoptosiswas definitely more pronounced in the samples treated withF-ara-A than in those treated with MP (Fig 3). At 72 h thedifference between F-ara-A and MP-treated samples virtuallydisappeared (Fig 3). The combination of F-ara-A 6 mg/ml þMP 1 mM appeared to have an additive effect and gave originto a pattern similar to that seen in samples treated withF-ara-A 30 mg/ml alone, which was not modified by thecombination of F-ara-A 30 mg/ml þ MP 1 mM (Fig 3). The
q 1997 Blackwell Science Ltd, British Journal of Haematology 99: 147–157
150 D. Gottardi et alTable I. General features of the patients studied.
Bcl-x‡Age
Patients (yr) Rai stage sIg L chain CD5 CD23 Bcl-2† Bax† S L
1 (MCL) 61 II M/D þþ * k ¹ þ¹ þ þ ¹ ¹
2 (CLL) 60 II M/D þ¹ k þ n.d. þ þ ¹ ¹
3 (CLL) 59 II M/D þ¹ k þ þ þ þ þ– þ
4 (CLL) 66 III M/D þ¹ l þ þ þ þ ¹ ¹
5 (CLL) 61 III M þ¹ l þ þ þ n.d. n.d. n.d.6 (MCL) 58 IV M/D þþ k þ þ þ þ þ þ
7 (MCL) 62 IV M þþ k þ ¹ þ n.d. n.d. n.d.8 (CLL) 67 IV M/D þ¹ k þ þ þ þ ¹ þ
9 (CLL) 62 IV M þ¹ k þ þ þ þ þ– þ
10 (MCL) 62 IV M þþ k þ ¹ þ ¹ ¹ ¹
11 (CLL) 70 IV M/D ¹ k þ þ þ þ ¹ þ
12 (CLL) 65 IV M/D þ¹ k þ þ þ þ ¹ ¹
* Degree of sIg intensity: þþ: strong expression; þ¹: weak expression; ¹: undetectable.† Positivity evaluated at both mRNA and protein level.‡ Positivity evaluated by RT-PCR; þ¹: low to trace amounts.

151F-ara-A-induced Bcl-2 Down-regulation in CD5þ Leukaemic B Cells
q 1997 Blackwell Science Ltd, British Journal of Haematology 99: 147–157
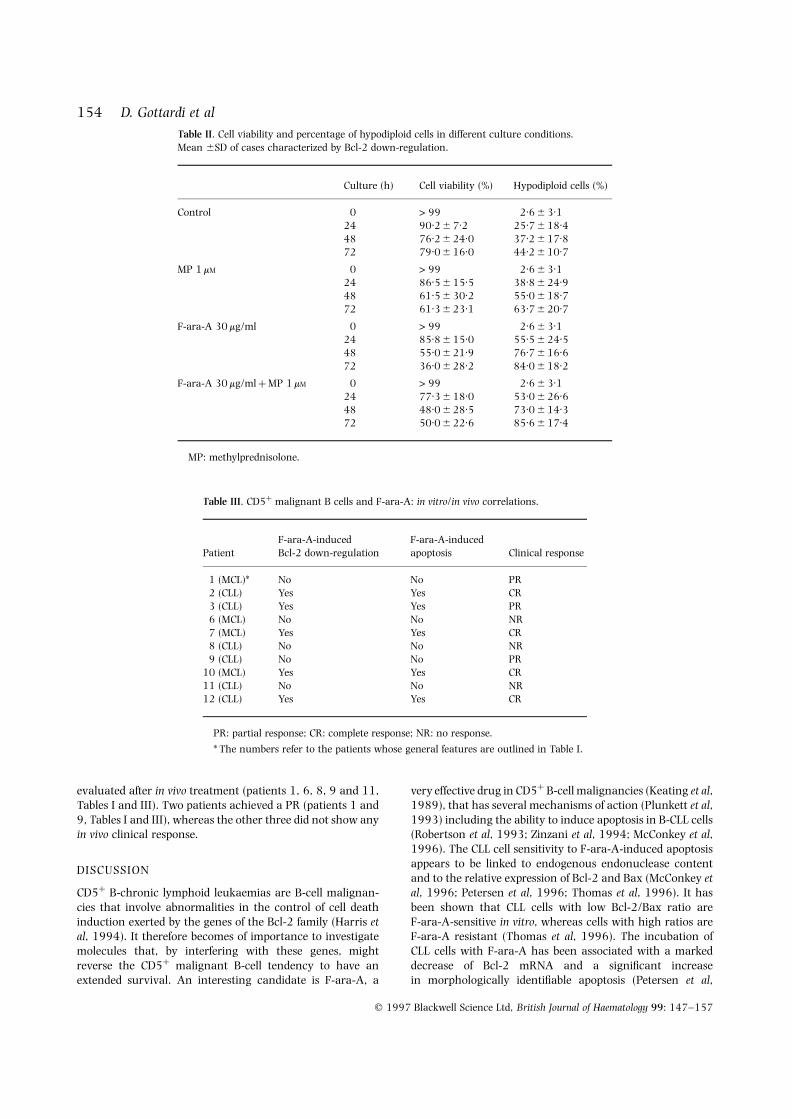
different apoptotic tendency was confirmed by cytofluoro-graph analysis which demonstrated that, after 24–48 h,apoptosis was significantly higher in the samples exposed toF-ara-A 30 mg/ml or F-ara-A 30 mg/ml þ MP 1 mM than inthose treated with MP alone (Fig 4). The morphology ofMGG-stained cytospins showed the same trend, even if thedifferences of apoptosis at different times were less promi-nent. When the PI fluorescence of individual nuclei wasmeasured in four cases to calculate the percentage ofapoptotic cells, the values showed a trend in favour of F-ara-A (or F-ara-A þ MP) treated versus MP-treated samples;however, the differences did not reach statistical significance.As shown in Table II, apoptosis in F-ara-A-treated samplesranged from 55.5 6 24.5% at 24 h of culture to76.7 6 16.6% at 48 h, to 84.0 6 18.2% at 72 h as opposedto 38.8 6 21.2% at 24 h, 55.0 6 18.7% at 48 h,63.7 6 20.7% at 72 h in the MP-treated samples(P ¼ 0.316 at 24 h, 0.133 at 48 h, 0.191 at 72 h). TheF-ara-A/MP combination did not modify the entity of
F-ara-A-induced apoptosis (53.0 6 32.5% at 24 h;69.0 6 14.5% at 48 h; 82.3 6 19.7% at 72 h). The lownumber of cases studied together with the importantindividual variations and the possibility that PI stainingmay include not only apoptotic but also dead cells mayaccount for this discrepancy.
When the same kind of analysis with different technicalapproaches was applied to the seven remaining cases, whereno Bcl-2 down-regulation had been observed after in vitroexposure to F-ara-A, the degree of F-ara-A-induced apoptosis
Fig 1. RT-PCR of B-CLL cells cultured at different times in thefollowing conditions: lane 1: control; lane 2: control; lane 3:methylprednisolone (MP) 1 mM; lane 4: F-ara-A 30 mg/ml; lane 5: F-ara-A 30 mg/ml þ MP 1 mM; lane 6: control; lane 7: MP 1 mM; lane 8:F-ara-A 30 mg/ml; lane 9: F-ara-A 30 mg/ml þ MP 1 mM; M:molecular weight marker VI.
Fig 2. Cytofluorograph assessment of Bcl-2 protein down-regulationafter in vitro treatment with F-ara-A. Solid line: control; dotted line:F-ara-A 30 mg/ml.

overlapped the degree of apoptosis induced by MP asobserved both by DNA fragmentation and by cytofluoro-graph analysis. The morphology revealed a lower number ofscattered apoptotic cells and the PI index was super-imposable in F-ara-A-treated (30.9 6 19.7% at 24 h;47.4 6 18.5% at 48 h) and in MP-treated (28.4 6 22.4%at 24 h; 49.8 6 15.5% at 48 h) samples.
B-CLL and F-ara-A: in vitro/in vivo correlationsThe in vitro data were then correlated with the in vivooutcome after treatment with F-ara-A; 10 patients were
evaluable (Table III). Four out of the five cases whose cellshad shown in vitro Bcl-2 down-regulation and prominentapoptosis after the cells had been exposed to F-ara-A(patients 2, 7, 10 and 12, Tables I and III), exhibited a CRafter in vivo treatment; a PR being observed in the remainingpatient (no. 3, Tables I and III). The CR was independent ofthe patient’s diagnosis, as both B-CLL patients (nos. 2 and12, Tables I and III) and MCL patients (nos. 7 and 10, TablesI and III) responded equally well.
Five of the seven patients whose cells had shown no in vitromodulation of Bcl-2 after exposure to F-ara-A, could be
q 1997 Blackwell Science Ltd, British Journal of Haematology 99: 147–157
152 D. Gottardi et al
Fig 3. Gel electrophoresis of internucleosomal cleavage ofDNA from B-CLL cells cultured at different times in thefollowing conditions: lane 1: control; lane 2:methylprednisolone (MP) 1 mM; lane 3: F-ara-A 6 mg/ml;lane 4: F-ara-A 6 mg/ml þ MP 1 mM; lane 5: F-ara-A 30 mg/ml; lane 6: F-ara-A 30 mg/ml þ MP 1 mM. M: molecularweight marker III and VI; t0 ¼ time zero.

153F-ara-A-induced Bcl-2 Down-regulation in CD5þ Leukaemic B Cells
q 1997 Blackwell Science Ltd, British Journal of Haematology 99: 147–157
Fig
4.P
ropi
diu
mio
dide
(PI)
stai
nin
gan
dD
NA
fluor
esce
nce
flow
cyto
met
ric
anal
ysis
ofB
-CLL
nu
clei
afte
rin
vitr
ocu
ltu
rew
ith
orw
ith
out
met
hylp
redn
isol
one
(MP
)1
mM
and
F-ar
a-A
30
mg/
ml.
evaluated after in vivo treatment (patients 1, 6, 8, 9 and 11,Tables I and III). Two patients achieved a PR (patients 1 and9, Tables I and III), whereas the other three did not show anyin vivo clinical response.
DISCUSSION
CD5þ B-chronic lymphoid leukaemias are B-cell malignan-cies that involve abnormalities in the control of cell deathinduction exerted by the genes of the Bcl-2 family (Harris etal, 1994). It therefore becomes of importance to investigatemolecules that, by interfering with these genes, mightreverse the CD5þ malignant B-cell tendency to have anextended survival. An interesting candidate is F-ara-A, a
very effective drug in CD5þ B-cell malignancies (Keating et al,1989), that has several mechanisms of action (Plunkett et al,1993) including the ability to induce apoptosis in B-CLL cells(Robertson et al, 1993; Zinzani et al, 1994; McConkey et al,1996). The CLL cell sensitivity to F-ara-A-induced apoptosisappears to be linked to endogenous endonuclease contentand to the relative expression of Bcl-2 and Bax (McConkey etal, 1996; Petersen et al, 1996; Thomas et al, 1996). It hasbeen shown that CLL cells with low Bcl-2/Bax ratio areF-ara-A-sensitive in vitro, whereas cells with high ratios areF-ara-A resistant (Thomas et al, 1996). The incubation ofCLL cells with F-ara-A has been associated with a markeddecrease of Bcl-2 mRNA and a significant increasein morphologically identifiable apoptosis (Petersen et al,
q 1997 Blackwell Science Ltd, British Journal of Haematology 99: 147–157
154 D. Gottardi et alTable II. Cell viability and percentage of hypodiploid cells in different culture conditions.Mean 6SD of cases characterized by Bcl-2 down-regulation.
Culture (h) Cell viability (%) Hypodiploid cells (%)
Control 0 > 99 2.6 6 3.124 90.2 6 7.2 25.7 6 18.448 76.2 6 24.0 37.2 6 17.872 79.0 6 16.0 44.2 6 10.7
MP 1 mM 0 > 99 2.6 6 3.124 86.5 6 15.5 38.8 6 24.948 61.5 6 30.2 55.0 6 18.772 61.3 6 23.1 63.7 6 20.7
F-ara-A 30 mg/ml 0 > 99 2.6 6 3.124 85.8 6 15.0 55.5 6 24.548 55.0 6 21.9 76.7 6 16.672 36.0 6 28.2 84.0 6 18.2
F-ara-A 30 mg/ml þ MP 1 mM 0 > 99 2.6 6 3.124 77.3 6 18.0 53.0 6 26.648 48.0 6 28.5 73.0 6 14.372 50.0 6 22.6 85.6 6 17.4
MP: methylprednisolone.
Table III. CD5þ malignant B cells and F-ara-A: in vitro/in vivo correlations.
F-ara-A-induced F-ara-A-inducedPatient Bcl-2 down-regulation apoptosis Clinical response
1 (MCL)* No No PR2 (CLL) Yes Yes CR3 (CLL) Yes Yes PR6 (MCL) No No NR7 (MCL) Yes Yes CR8 (CLL) No No NR9 (CLL) No No PR
10 (MCL) Yes Yes CR11 (CLL) No No NR12 (CLL) Yes Yes CR
PR: partial response; CR: complete response; NR: no response.
* The numbers refer to the patients whose general features are outlined in Table I.

155F-ara-A-induced Bcl-2 Down-regulation in CD5þ Leukaemic B Cells
q 1997 Blackwell Science Ltd, British Journal of Haematology 99: 147–157
1996). The in vitro drug chemosensitivity has also beenassociated with the CLL cell ability to up-regulate theexpression of an 18 kD bax protein and to form bax dimers(Thomas et al, 1996).
The aim of the present work was to study whether theability of F-ara-A to induce apoptosis in fresh CD5þ
malignant B cells was related to Bcl-2 modifications andwhether in vitro data might be related to the in vivo outcomeof F-ara-A-treated patients. To this end a two-fold approachwas devised. PBL from patients who were going to be treatedwith F-ara-A were cultured in vitro in presence of differentconcentrations of F-ara-A 6 MP and the effect of differentculture conditions on Bcl-2 gene product and apoptosis wasanalysed. In vitro results were then compared to the clinicaloutcome of the in vivo administration of the drugs. Theevaluation of the apoptotic effects of different cultureconditions in CD5þ malignant B cells was complicated bythe observation that these cells had a variable degree ofspontaneous apoptosis and that pronounced interpatientvariations occurred. To minimize these problems we used anapproach based upon the concomitant use of differenttechniques currently used to evaluate apoptosis. The resultsindicate that, in a proportion of patients, F-ara-A was able toinduce Bcl-2 down-regulation which was followed in vitro bymassive apoptosis and in vivo was associated with asignificantly better clinical response. As the vast majorityof cells in the populations exposed to F-ara-A were malignantB cells, it appears that these were the major source of Bcl-2down-regulation and apoptosis. The question may be raisedwhether Bcl-2 down-regulation contributes to, or is aconsequence of, apoptosis. Increased relative levels of Baxcompared to Bcl-2 might suggest that more Bax–Baxhomodimers are produced and favour apoptosis (Yang &Korsmeyer, 1996), whereas down-regulation of Bax andBcl-2 at the same time would suggest that the phenomenonis a consequence of apoptosis. The RT-PCR we have used tomonitor the levels of Bcl-2, Bax and Bcl-x transcripts is not areally quantitative assay, so that the absence of modificationsin the levels of Bax PCR products might simply depend on thesaturation kinetics of the reaction. We tried to overcome thequantitative limits of RT-PCR by measuring Bax proteinswith a flow cytometer. This analysis failed to reveal a down-regulation of Bax protein. These findings are in keeping withliterature data on Bax expression in CLL cells exposed toF-ara A (Thomas et al, 1996) and suggest that Bcl-2 down-regulation contributes to apoptosis. Still, they have beenobtained in a limited number of cases and need to beextended to a larger number of patients to produce more firmconclusions.
Taken together, our findings indicate that, when theexposure of CD5þ malignant B cells to F-ara-A causes thedown-regulation of Bcl-2, a tendency to undergo apoptosisis triggered. As in vivo patient’s cells are exposed to thedrug for a number of days, it is not unreasonable toconclude that, in these cases, a progressively increasingnumber of cells will down-regulate Bcl-2 and acquire thepropensity to die. The interpatient variation and theheterogeneity of in vitro response suggest caution ininterpreting the in vitro data as predictive of the in vivo
outcome of F-ara-A treatment in CD5þ B-cell malignan-cies. Also, the progressive and rather late down-regulationof Bcl-2 might be a secondary consequence of F-ara-A-induced apoptosis, rather than a primary indication ofsensitivity to the drug. Still, it is tempting to suggest that,whenever F-ara-A is able to interfere in vitro with theapoptosis-related genes by down-regulating Bcl-2, thepatient’s chance to obtain a desirable clinical response isincreased. The down-regulation of Bcl-2 protein (and thestable levels of Bax protein) in the presence of F-ara-Acan be easily measured in vitro by cytofluorographtechniques and may be added to the range of preclinicalinvestigations.
A number of points have to be discussed when analysingthis study. First, this is a pilot study where all patients had along-standing disease which had become refractory toconventional treatment. Therefore it is conceivable thatadditional genetic damage might have occurred in the cellsstudied and explain why only in some cases cells exposed toF-ara-A down-regulate Bcl-2. In agreement with Thomaset al (1996), preliminary data rule out p53 abnormalities inall but one of the patients studied (unpublished). Also, even ifthe half-life of Bcl-2 mRNA in B-CLL is the same as in Nalm-6cells and normal B cells (Caligaris-Cappio et al, 1992), we stilldo not know whether the Bcl-2 protein half-life in B-CLL isprolonged and might thus contribute to an altered response(Reed, 1996). Further, a different pattern of phosphorylation(Haldar et al, 1995) might help to bring about a differentialregulation of Bcl-2 protein in responsive versus non-responsive cases. Taken together, these possibilities indicatethat our experimental approach needs to be extended topreviously untreated cases.
The second point is that we have not eliminated T cellsfrom our cultures reasoning that in vivo the drug wouldcertainly not encounter a purified population of B cells. Inour cases, the low number of residual T cells togetherwith the short culture period indicate that the T-cellremoval is not essential in this kind of experiment. Still,the ability of F-ara-A to influence T cells (Robertson et al,1992; Boldt et al, 1984) as well as the B-CLL T cell abilityto produce apoptosis-protecting cytokines (Dancescu et al,1992; Buschle et al, 1993) call for further T-cell focusedexperiments. Finally, the Bcl-2 gene family is growingquickly and other members of the family including Bad(Yang et al, 1995), Bag (Takayama et al, 1995) and Bak(Chittenden et al, 1995) will have to be added to therange of apoptosis-related genes to be evaluated underF-ara-A pressure. In any case, the data here presenteddemonstrate that in vitro studies may help to define thepatients who will most benefit from F-ara-A treatmentand may be useful in planning the therapy of CD5þ B-cellmalignancies.
ACKNOWLEDGMENTS
This work was supported by A.I.R.C., Milano and PF ACRO,CNR. M.G.G. is recipient of an AIRC fellowship. Thesecretarial assistance of Mrs G. Tessa, Fondazione R.Favretto, is gratefully acknowledged.

REFERENCES
Bissonnette, R.P., Echeverri, F., Mahboubi, A. & Green, D.R. (1992)Apoptotic cell death induced by c-myc is inhibited by bcl-2. Nature,359, 552–554.
Boise, L.H., Gonzales-Garcoıa, M., Postema, C.E., Ding, L., Lindstein,T., Turka, L.A., Mao, X., Nunez, G. & Thompson, C.B. (1993) Bcl-X, a bcl-2 related gene that functions as a dominant regulator ofapoptotic cell death. Cell, 74, 597–608.
Boldt, D.H., Von Hoff, D.D., Kuhn, J.H. & Hersh, M. (1984) Effectson human peripheral lymphocytes of in vivo administration of9-b-D-arabinofuranosyl-2 fluoradenine-50-monophosphate (NSC312887), a new purine antimetabolite. Cancer Research, 44,4661–4668.
Buschle, M., Campana, D., Carding, S.R., Richard, C., Hoffbrand,A.V. & Brenner, M.K. (1993) Interferon g inhibits apoptotic celldeath in B cell chronic lymphocytic leukemia. Journal ofExperimental Medicine, 177, 213–218.
Caligaris-Cappio, F., Ghia, P., Gottardi, D., Parvis, G., Gregoretti,M.G., Nilsson, K. & Schena, M. (1992) The role of BCL-2 in thenatural history of B-chronic lymphocytic leukemia. Current Topicsin Microbiology, 182, 279–286.
Chittenden, T., Flemington C., Houghton, A.B., Ebb, R.G., Gallo, G.J.,Elangovan, B., Chinnadurai, G. & Lutz, R.J. (1995) A conserveddomain of Bak, distinct from BH1 and BH2, mediates cell deathand protein binding fuctions. EMBO Journal, 14, 5589–5596.
Collins, R.J., Verschuer, L.A., Harmon, B.V., Prentice, R.L., Pope, J.H.& Kerr, J.F.R. (1989) Spontaneous programmed death (apoptosis)of B-chronic lymphocytic leukaemia cells following their culture invitro. British Journal of Haematology, 71, 343–350.
Dancescu, M., Rubio-Trujillo, M., Biron, G., Bron, D., Delespesse, G. &Sarfati, M. (1992) Interleukin 4 protects chronic lymphocyticleukemia from death by apoptosis and upregulates Bcl-2expression. Journal of Experimental Medicine, 176, 1319–1326.
Dyer, M.J.S., Zani, V.J., Lu, W.Z., O’Byrne, A., Mould, S., Chapman, R.,Heward, J.M., Kayano, H., Jadayel, D., Matutes, E., Catowsky, D. &Oscier, D.G. (1994) BCL2 translocations in leukemias of mature Bcells. Blood, 83, 3682–3688.
Fanidi, A., Harrington, E.A. & Evan, G.I. (1992) Cooperativeinteraction between c-myc and bcl-2 proto-oncogenes. Nature,359, 554–556.
Gottardi, D., Alfarano, A., De Leo, A.M., Stacchini, A., Aragno, M.,Rigo, A., Veneri, D., Zanotti, R., Pizzolo, G. & Caligaris-Cappio, F.(1996) In leukaemic CD5þ B cells the expression of BCL-2 genefamily is shifted toward protection from apoptosis. British Journalof Haematology, 94, 612–618.
Haldar, S., Jena, N. & Croce, C.M. (1995) Inactivation of Bcl-2 byphosphorylation. Proceedings of the National Academy of Sciences ofthe United States of America, 92, 4507–4511.
Hanada, M., Delia, D., Aiello, A., Stadtmauer, E. & Reed, J.C. (1993)Bcl-2 gene hypomethylation and high-level expression in B-cellchronic lymphocytic leukemia. Blood, 82, 1820–1828.
Harris, N.L., Jaffe, E.S., Stein, H., Banks, P.M., Chan, J.K.C., Cleary,M.L., Delsol, G., De Wolf-Peeters, C., Falini, B., Gatter, K.C.,Grogan, T.M., Isaacson, P.G., Knowles, D.M., Mason, D.Y., Muller-Hermelink, H.K., Pileri, S.A., Piris, M.A., Ralfkiaer, E. & Warnke,R.A. (1994) A revised European–American classification oflymphoid neoplasms: a proposal from the International Lym-phoma Study Group. Blood, 84, 1361–1392.
Keating, M.J., Kantarjian, M., Talpaz, M., Redman, J., Koller, C.,Barlogie, B., Velasquez, W., Plunkett, W., Freireich, E.J. &McCredie, K.B. (1989) Fludarabine: a new agent with majoractivity against chronic lymphocytic leukemia. Blood, 74, 19–25.
Larsson, L.G., Gray, H.E., Totterman, T., Pettersson, U. & Nilsson, K.
(1987) Drastically increased expression of MYC and FOSprotooncogenes during in vitro differentiation of chronic lympho-cytic leukemia cells. Proceedings of the National Academy of Sciencesof the United States of America, 84, 223–225.
Mainou-Fowler, T. & Prentice, A.G. (1996) Modulation of apoptosisby cytokines in B-cell chronic lymphocytic leukemia. Leukemia andLymphoma, 21, 369–377.
Mapara, M.Y., Bargon, R., Zugck, C., Dohner, H., Jonker, R.R.,Krammer, P.H. & Dorken, B. (1993) APO-1 mediated apoptosis orproliferation in human chronic B lymphocytic leukaemia:correlation with bcl-2 oncogene expression. European Journal ofImmunology, 23, 702–708.
McConkey, D.J., Aguilar-Santelises, M., Hartzell, P., Eriksson, I.,Mellstedt, H., Orrenius, S. & Jondal, M. (1991) Induction of DNAfragmentation in chronic B-lymphocytic leukemia cells. Journal ofImmunology, 146, 1072–1076.
McConkey, D.J., Chandra, J., Wright, S., Plunkett, W., McDonnell, T.J.,Reed, J.C. & Keating, M. (1996) Apoptosis sensitivity in chroniclymphocytic leukemia is determined by endogenous endonucleasecontent and relative expression of Bcl-2 and Bax. Journal ofImmunology, 156, 2624–2630.
Menzel, T., Rahman, Z., Celleja, E., White, K., Wilson, E.L., Wieder, R.& Gabrilove, J. (1996) Elevated intracellular level of basicfibroblast growth factor correlates with stage of chronic lympho-cytic leukemia and is associated with resistance to fludarabine.Blood, 87, 1056–1063.
Miyashita, T. & Reed, J.C. (1992) Bcl-2 gene transfer increases relativeresistance of S49.1 and WEHI7.2 lymphoid cells to cell death andDNA fragmentation induced by glucocorticoids and multiplechemotherapeutic drugs. Cancer Research, 52, 5407–5411.
Miyashita, T. & Reed, J.C. (1993) Bcl-2 oncoprotein blockschemotherapy-induced apoptosis in a human leukemia cell line.Blood, 81, 151–157.
Moller, P., Henne, C., Leithauser, F., Eichelmann, A., Schmidt, A.,Brudetrlein, S., Dhein, J. & Krammer, P. (1993) Coregulation of theAPO-1 antigen with intercelllar adhesion molecule-1 (CD54) intonsillar B cells and coordinate expression in follicular center Bcells and in follicle center and mediastinal B-cell lymphomas.Blood, 81, 2067–2075.
Oltvai, Z.N., Milliman, C.L. & Korsmeyer, S.J. (1993) Bcl-2heterodimerizes in vivo with a conserved homolog, Bax, thataccelerates programmed cell death. Cell, 74, 609–619.
Petersen, A.J., Brown, R.D., Gibson, J., Pope, B., Luo, X.F., Schutz, L.,Wiley, J.S. & Joshua, D.E. (1996) Nucleoside transporters, bcl-2and apoptosis in CLL cells exposed to nucleoside analogues invitro. European Journal of Haematology, 56, 213–220.
Pezzella, F., Tse, A.G.D., Cordell, J.L., Pulford, K.A.F., Gatter, K.C. &Mason, D.Y. (1990) Expression of the bcl-2 oncogene protein is notspecific for the 14;18 chromosomal translocation. AmericanJournal of Pathology, 137, 225–232.
Plunkett, W., Gandhi, V., Huang, P., Robertson, L.E., Ying-Yang, L.,Gregoire, V., Estey, E. & Keating, M.J. (1993) Fludarabine:pharmacokinetics, mechanisms of action and rationale forcombination therapies. Seminars in Oncology, 20, 2–11.
Rai, K.R., Sawitsky, A., Cronkite, E.P., Chanana, A.D., Levy, R.N. &Pasternack, B.S. (1975) Clinical staging of chronic lymphocyticleukemia. Blood, 46, 219–234.
Reed, J.C. (1996) A day in the life of the Bcl-2 protein: does theturnover rate of Bcl-2 serve as a biological clock for cellularlifespan regulation? Leukemia Research, 20, 109–111.
Robertson, L.E., Chubb, S., Meyn, R.E., Story, M., Ford, R., Hittelman,W.N. & Plunkett, W. (1993) Induction of apoptotic cell death inchronic lymphocytic leukemia by 2-chloro-20-deoxyadenosine and9-b-D-arabinosyl-2-fluoroadenine. Blood, 81, 143–150.
q 1997 Blackwell Science Ltd, British Journal of Haematology 99: 147–157
156 D. Gottardi et al

157F-ara-A-induced Bcl-2 Down-regulation in CD5þ Leukaemic B Cells
q 1997 Blackwell Science Ltd, British Journal of Haematology 99: 147–157
Robertson, L.E., Huh, Y.O., Butler, J.J., Pugh, W.C., Hirsch-Ginsberg,C., Stass, S., Kantarjian, H. & Keating, M.J. (1992) Responseassessment in chronic lymphocytic leukemia after fludarabineplus prednisone clinical, pathologic, immunophenotypic, andmolecular analysis. Blood, 80, 29–36.
Robertson, L.E., Plunkett, W., McConnell, K., Keating, M.J. &McDonnell, T.J. (1996) Bcl-2 expression in chronic lymphocyticleukemia and its correlation with the induction of apoptosis andclinical outcome. Leukemia, 10, 456–459.
Schena, M., Larsson, L.G., Gottardi, D., Gaidano, G.L., Carlsson, M.,Nilsson, K. & Caligaris-Cappio, F. (1992) Growth and differentia-tion-associated expression of bcl-2 in B-chronic lymphocyticleukemia cells. Blood, 79, 2981–2989.
Takayama, S., Sato, T., Krajewski, S., Kochel, K., Irie, S., Millan, J.A.& Reed, J.C. (1995) Cloning and functional analysis of BAG-1:a novel Bcl-2-binding protein with anti-cell death activity. Cell,80, 279–284.
Thomas, A., El Rouby, S., Reed, J.C., Krajewski, S., Silber, R.,Potmesil, M. & Newcomb, E.W. (1996) Drug-induced apoptosis inB-cell chronic lymphocytic leukaemia: relationship between p53
gene mutation and bcl-2/bax proteins in drug resistance.Oncogene, 12, 1055–1062.
van Kooten, C., Rensink, I., Aarden, L. & van Oers, R. (1992)Interleukin-4 inhibits both paracrine and autocrine tumornecrosis factor-a-induced proliferation of B chronic lymphocyticleukemia cells. Blood, 80, 1299–1306.
Yang, E. & Korsmeyer, S.J. (1996) Molecular thanatopsis: a discourseon the Bcl2 family and cell death. Blood, 88, 386–401.
Yang, E., Zha, J., Jockel, J., Boise, L.H., Thompson, C.B. & Korsmeyer,S.J. (1995) Bad, a heterodimeric partner for Bcl-XL and Bcl-2,displaces Bax and promotes cell death. Cell, 80, 285–291.
Young, I.T. (1977) Proof without prejudice: use of the Kalmogorov-Smirnov test for the analysis of the histograms from flow systemsand other sources. Journal of Histochemistry and Cytochemistry, 25,935–941.
Zinzani, P.L., Tosi, P., Visani, G., Martinelli, G., Farabegoli, P., Buzzi,M., Ottoviani, E., Salvucci, M., Bendandi, M., Zaccaria, A. & Tura,S. (1994) Apoptosis induction with three nucleoside analogs onfreshly isolated B-chronic lymphocytic leukemia cells. AmericanJournal of Hematology, 47, 301–306.
Copyright © 2022 FDOKUMEN




















