
Crosstalk among Bcl-2 family members in B-CLL: seliciclib acts via the Mcl-1/Noxa axis and gradual...
177
Downloaded from UvA-DARE, the Institutional Repository of the University of Amsterdam (UvA) http://dare.uva.nl/document/116938 Description Thesis File ID 116938 Filename thesis.pdf SOURCE, OR PART OF THE FOLLOWING SOURCE: Type Dissertation Title 'From the cradle to the grave' : novel therapeutic approaches to attack the microenvironment in chronic lymphocytic leukemia Author D. Hallaert Faculty Faculty of Medicine Year Pages 176 FULL BIBLIOGRAPHIC DETAILS: http://dare.uva.nl/record/284714 Copyrights It is not permitted to download or to forward/distribute the text or part of it without the consent of the copyright holder (usually the author), other then for strictly personal, individual use. UvA-DARE is a service provided by the Library of the University of Amsterdam (http://dare.uva.nl)
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Crosstalk among Bcl-2 family members in B-CLL: seliciclib acts via the Mcl-1/Noxa axis and gradual...

Downloaded from UvA-DARE, the Institutional Repository of the University of Amsterdam (UvA)http://dare.uva.nl/document/116938
Description ThesisFile ID 116938Filename thesis.pdf
SOURCE, OR PART OF THE FOLLOWING SOURCE:Type DissertationTitle 'From the cradle to the grave' : novel therapeutic approaches to attack the microenvironment in
chronic lymphocytic leukemiaAuthor D. HallaertFaculty Faculty of MedicineYearPages 176
FULL BIBLIOGRAPHIC DETAILS: http://dare.uva.nl/record/284714
Copyrights It is not permitted to download or to forward/distribute the text or part of it without the consent of the copyright holder(usually the author), other then for strictly personal, individual use. UvA-DARE is a service provided by the Library of the University of Amsterdam (http://dare.uva.nl)

Delfi ne Hallaert
‘From the cradle to the grave’
Novel therapeutic approaches to attack the microenvironment
in Chronic Lymphocytic Leukemia
Thesis_Final_2_01092008.indd 1Thesis_Final_2_01092008.indd 1 01-09-2008 11:08:0101-09-2008 11:08:01

Hallaert, Delfi ne
Novel therapeutic approaches to attack the microenvironment in Chronic
Lymphocytic Leukemia
Thesis, University of Amsterdam with references
© 2008 D.Y.H. Hallaert, Amsterdam, The Netherlands
Layout by Caroline Op de beeck, Wingene, Belgium
Cover design by Norbert Hallaert and Lindsay Vandenabeele, Brugge, Belgium
Printed by Buijten & Schipperheijn
Printing of this thesis was financially supported by:
University of Amsterdam
Ortho Biotech
Thesis_Final_2_01092008.indd 2Thesis_Final_2_01092008.indd 2 01-09-2008 11:08:0301-09-2008 11:08:03

‘From the cradle to the grave’
Novel therapeutic approaches to attack the
microenvironment in Chronic Lymphocytic Leukemia
ACADEMISCH PROEFSCHRIFT
ter verkrijging van de graad van doctor
aan de Universiteit van Amsterdam
op gezag van de Rector Magnifi cus
prof.dr. D.C. van den Boom
ten overstaan van een door het college voor promotie ingestelde
commissie, in het openbaar te verdedigen in de Aula der Universiteit
op woensdag 15 oktober 2008, te 14:00 uur
door
Delfi ne Yolande Hugo Hallaert
geboren te Brugge, België
Thesis_Final_2_01092008.indd 3Thesis_Final_2_01092008.indd 3 01-09-2008 11:08:0301-09-2008 11:08:03

PROMOTIECOMMISSIE
Promotor: Prof.dr. M.H.J. van Oers
Co-promotor: dr. E. Eldering
Overige leden: Prof.dr. J. Borst
Prof.dr. A. Hagenbeek
Prof.dr. M.H.H. Kramer
Prof.dr. J.P. Medema
Prof.dr. S.T. Pals
Faculteit der Geneeskunde
Thesis_Final_2_01092008.indd 4Thesis_Final_2_01092008.indd 4 01-09-2008 11:08:0301-09-2008 11:08:03

Aan wie ik lief heb
Thesis_Final_2_01092008.indd 5Thesis_Final_2_01092008.indd 5 01-09-2008 11:08:0301-09-2008 11:08:03

CONTENTS
Chapter 1 General introduction
Chapter 2 Crosstalk among Bcl-2 family members in B-CLL:
seliciclib acts via the Mcl-1/Noxa axis and gradual
exhaustion of Bcl-2 protection
Chapter 3 γ-secretase inhibitor (GSI)-1 induces apoptosis in CLL
cells via proteasome inhibition and Noxa upregulation
Chapter 4 Differential Noxa/Mcl-1 balance in peripheral versus
lymph node chronic lymphocytic leukemia cells
correlates with survival capacity
Chapter 5 Persistent Mcl-1/Bim protein signature after CD40
ligation in Chronic Lymphocytic Leukemia is associated
with specifi c drug sensitivity
Chapter 6 c-Abl Kinase Inhibitors Overcome CD40-Mediated Drug
Resistance in CLL; Implications for Therapeutic
Targeting of Chemoresistant Niches
Chapter 7 Concluding remarks
Chapter 8 Summary
Chapter 9 Nederlandse samenvatting
Dankwoord
Curriculum vitae
List of publications
Color fi gures
9
31
55
75
99
117
143
151
155
163
169
173
175
Thesis_Final_2_01092008.indd 6Thesis_Final_2_01092008.indd 6 01-09-2008 11:08:0301-09-2008 11:08:03

Thesis_Final_2_01092008.indd 7Thesis_Final_2_01092008.indd 7 01-09-2008 11:08:0301-09-2008 11:08:03

Thesis_Final_2_01092008.indd 8Thesis_Final_2_01092008.indd 8 01-09-2008 11:08:0301-09-2008 11:08:03

General introduction
1chapter
Thesis_Final_2_01092008.indd 9Thesis_Final_2_01092008.indd 9 01-09-2008 11:08:0301-09-2008 11:08:03

Thesis_Final_2_01092008.indd 10Thesis_Final_2_01092008.indd 10 01-09-2008 11:08:0301-09-2008 11:08:03

Ch
ap
ter 1
▪ Genera
l intro
ductio
n
11INTRODUCTION
CLL: CLINICAL AND PATHOLOGICAL FEATURES
Chronic lymphocytic leukemia (CLL) is a lymphoproliferative disorder characterized
by monoclonal small, mature CD5+/CD19+ B cells which continue to accumulate in the
peripheral blood (PB), bone marrow (BM) and lymphoid organs1. There is a remarkable
clinical variability in patients with CLL2. Following diagnosis, some patients have
asymptomatic disease that may not progress for many years. Others are diagnosed
with advanced-stage disease, or have early-stage disease that progresses rapidly
and requires treatment. Clinical symptoms include peripheral blood lymhocytosis,
lymphadenopathy and hepatosplenomegaly. When the disease progresses many
patients develop hypogammaglobulinemia and neutropenia, resulting in an increased
susceptibility to bacterial infections. Furthermore, up to 25% of the patients develop
autoimmune cytopenias, mostly autoimmune hemolytic anemia and/or autoimmune
thrombocytopenia. The clinical variability is associated with the following molecular
markers: cytogenetic abnormalities3, immunoglobulin VH gene mutational status4;5,
ZAP-70 expression6;7 and CD38 expression8. How these factors infl uence the biology
of CLL is still subject of intensive research. Immunoglobulin gene mutational status
and ZAP-70 expression are associated with the capacity of CLL cells to respond
to signals delivered through the B-cell receptor (BCR)9. Loss of 17p10 or 11q11 may
cause dysfunction of p53 DNA damage pathways12.
Classically, CLL cells were thought to derive from naive B lymphocytes and to behave
as inert cells that passively accumulate13. However, mRNA studies showed that at the
level of gene expression both IgVH mutated and unmutated CLL cells are most similar
to memory B cells14;15. Moreover, stable isotope labeling studies with deuterated water
(D2O) have shown that CLL cells divide at a considerable rate, suggesting a more
dynamic disease than previously appreciated16. Survival and proliferation of the CLL
cells are infl uenced by interactions with non-leukemic cells in the microenvironment
of lymph nodes (LN), bone marrow and spleen1;17;18. Still, deregulated apoptosis
is considered to be a key factor and a major barrier to effective treatment of CLL.
Understanding apoptosis pathways and the impact of the microenvironment on
these pathways is therefore clinically relevant, because it may open new avenues to
effective treatment of CLL.
Thesis_Final_2_01092008.indd Sec1:11Thesis_Final_2_01092008.indd Sec1:11 01-09-2008 11:08:0301-09-2008 11:08:03

12THERAPY IN CLL
Extensive discussion of the treatment of CLL is beyond the scope of this thesis.
Detailed reviews have recently been published19;20. Past and current therapeutic
approaches to CLL will be summarized here.
About twenty years ago, treatment consisted of alkylating agents, usually chlorambucil.
Remission rates were variable (50 – 70%). However, complete remission was rarely
obtained21. Combination treatment, such as CVP (cyclophosphamide, vincristine
and prednisone) and CHOP (cyclophosphamide, doxorubicine, vincristine and
prednisone), did not improve overall survival22. The introduction of the purine analog
fl udarabine has brought new impulse to the research of CLL treatments. Clinical trials
showed fl udarabine to induce higher complete remission rates and longer progression
free survival. Again, no survival advantage was achieved21. Until recently, the goal of
therapy has been palliation with minimal toxicity. Introduction of new therapies and
novel combinatorial approaches have initiated a paradigm shift and made potential
cure the goal of therapy. Three randomized trials, comparing FC (fl udarabine +
cyclophosphamide) with fl udarabine alone, have been published, all showing higher
response rates with prolongation of progression-free survival in the FC arm23-25.
Nevertheless, thus far no overall survival benefi t was obtained. Studies with the
most potent combination FC + Rituximab (anti-CD20 chimeric monoclonal antibody)
showed high overall and complete remission rate and prolonged progression-free
survival, in both previously untreated26 and relapsed27 CLL patients. Alemtuzumab
(anti-CD52 humanized antibody) has been shown to be effective also in the treatment
of p53 dysfunctional CLL patients28. Finally, Reduced Intensity allogeneic Stem cell
Transplantation (RIST) or ‘mini transplants’ have broadened the use of allogeneic stem
cell transplants in CLL patients. By this approach a long-term relapse-free survival
has been achieved 29, but the associated morbidity remains a serious obstacle for
wide application30;31. Thus, despite recent progress there remains a strong need for
novel effective and less toxic treatment options.
APOPTOSIS
Programmed cell death or apoptosis, is essential for tissue homeostasis, and disturbed
regulation of this process underlies many diseases, including cancer. Activation of
apoptosis is important for the removal of infected, transformed or damaged cells.
Apoptotic cells are defi ned according to morphological characteristics such as cellular
Thesis_Final_2_01092008.indd Sec1:12Thesis_Final_2_01092008.indd Sec1:12 01-09-2008 11:08:0301-09-2008 11:08:03

Ch
ap
ter 1
▪ Genera
l intro
ductio
n
13shrinkage, nuclear condensation, membrane blebbing and eventually fragmentation
into membrane bound apoptotic bodies32. During apoptosis, asymmetry of the cell
membrane results in exposure of phosphatidylserine (PS) on the cell surface. PS
functions as an ‘eat me’ signal on apoptotic cells which results in direct recognition,
engulfment and degradation by phagocytes33.
Caspases
Apoptosis involves a family of aspartate-specifi c cysteine proteases, called caspases34.
Caspases are synthesized as precursors (pro-caspases), and are converted into
mature enzymes upon apoptosis signals. Caspases can be divided into three groups
based on their structure and role in apoptosis. First, initiator caspases, consisting of
caspase-2, -8, -9 and -10 that cleave inactive pro-forms of effector caspases, leading
to activation. Second, effector caspases, consisting of caspase-3, -6 and -7, which in
turn cleave other protein substrates within the cell resulting in the apoptotic process35.
Finally, a third group of caspases is described, caspase-1, -4 and -5, which are not
directly involved in apoptosis execution, but play important roles in infl ammatory
cytokine activation and release36.
Pathways of apoptosis induction
Two major apoptotic signaling pathways are recognized in mammals: the extrinsic
or death receptor mediated pathway and the intrinsic or mitochondrial pathway. Key
players in the apoptosis signaling pathway are outlined in Figure 1.
The extrinsic pathway is activated upon ligand binding to the tumor necrosis receptor
(TNF-R) family members such as Fas (CD95/APO-1), TNF-R and TRAIL-R. This
results in receptor trimerization and recruitment of intracellular adaptor proteins,
TRADD or FADD, leading to the assembly of the death-inducing signaling complex
(DISC)37;38, and subsequent recruitment and assembly of initiator caspase-8. Caspase-
10 can also be recruited to the DISC in a similar manner, but it cannot functionally
substitute for caspase-839;40. Subsequently, activated caspase-8 is released into the
cytosol where it can activate effector caspases. In addition, caspase-8 can process
the BH3-only Bcl-2 family member Bid to the truncated form tBid41. Subsequently,
tBid can translocate to the mitochondria to exert its pro-apoptotic activity41;42.
The intrinsic pathway is initiated in response to cellular signals resulting from DNA
damage, cell cycle defects, growth factor withdrawal, hypoxia, or other types of severe
cell stress and is tightly regulated by Bcl-2 family of proteins. Progression through the
Thesis_Final_2_01092008.indd Sec1:13Thesis_Final_2_01092008.indd Sec1:13 01-09-2008 11:08:0301-09-2008 11:08:03

14pathway leads to mitochondrial outer membrane permeabilization (MOMP), resulting
in cytochrome c release and assembly of the apoptosome complex. The apoptosome
is a multimeric holoenzyme consisting of apoptotic protease-activating factor 1 (Apaf1),
pro-caspase-9 and dATP43. Other apoptosis promoting factors are also released
from the mitochondria, including Smac/DIABLO, Omi/HtrA2, endonuclease G and
apoptosis inducing factor (AIF). Smac/DIABLO and Omi/HtrA2 promote caspase
activation by interacting with inhibitors of apoptosis (IAP) family44;45. Endonuclease G
and AIF translocate to the nucleus and induce DNA degradation46;47.
Figure 1. See color fi gures. The two main pathways leading to apoptosis. The extrinsic pathway is
triggered by ligation of cell surface receptors, such as Fas, resulting in activation of caspase-8. The
intrinsic pathway is activated by cytotoxic stimuli, such as DNA damage, which leads to the release of
apoptosis promoting factors from the mitochondria. In the cytosol, cytochrome c results in the activation
of caspase-9. This pathway is regulated by the Bcl-2 family of proteins. Activated caspase-8 and -9 in
turn activate effector caspase-3, -6 and -7. Cross-talk between the pathways occurs through Bid, which is
cleaved by caspase-8 and then can activate the intrinsic pathway.
Thesis_Final_2_01092008.indd Sec1:14Thesis_Final_2_01092008.indd Sec1:14 01-09-2008 11:08:0401-09-2008 11:08:04

Ch
ap
ter 1
▪ Genera
l intro
ductio
n
15Apoptosis regulators
Bcl-2 family
The Bcl-2 (B cell lymphoma 2) family in mammals consists of pro- and anti-apoptotic
proteins. An overview of the Bcl-2 family protein members is presented in Table
1. They all share at least one conserved Bcl-2 homology (BH) domain and are
considered to act mainly on the mitochondria48. Based on structural and functional
features, they can be divided into three subfamilies: the anti-apoptotic subfamily
comprising of Bfl -1, Bcl-2, Bcl-W, Bcl-xL and Mcl-1 (Myeloid-cell leukemia sequence
1) and two other subfamilies which promote cell death: the Bax-like death family
and BH3-only family (in Table 1 also several ‘BH3-only contenders’ are mentioned).
Members of the Bax-like death family include Bax (Bcl-2-associated X protein), Bak
(Bcl-2-antagonist/killer) and Bok (Bcl-2-related ovarian killer), which contain three
BH domains. The BH3-only protein family includes at least 8 members including Bad
(Bcl-2-antagonist of cell death), Bik (Bcl-2-interacting killer), Bid (Bcl-2 interacting
domain), Hrk (Harakiri, also known as DP5), Bim (Bcl-2-interacting mediator of
cell death), Noxa, Puma (p53-upregulated modulator of apoptosis) and Bmf (Bcl-
2-modifying factor). They all have only a short BH3 motif. Biochemical studies
demonstrated that BH3-only proteins interact selectively and with varying affi nities
with anti-apoptotic counterparts. Whereas Bim and tBid bind avidly to all the pro-
survival proteins, the other BH3-only proteins associate only with selected subsets.
For example Noxa binds to Mcl-1 and Bfl -1, while Bad binds to Bcl-2, Bcl-XL and
Bcl-W49. For Noxa, however, others reported that Mcl-1 is the only binding partner50.
Promiscuous binders, like Bim, are much more potent killers than those that cannot
engage all the pro-survival proteins49. This indicates that the multiple anti-apoptotic
proteins all have a different function.
It is the complex interplay between the three Bcl-2 subfamilies that determines the
commitment of MOMP and subsequent apoptosis51. However, the detailed molecular
mechanism remains controversial. The BH3-only proteins act upstream of Bax and
Bak, because they cannot induce apoptosis in cells lacking both Bax and Bak52. How
they induce activation of Bax and Bak is addressed by two distinct models. The direct
activation model proposes that the BH3-only proteins termed ‘activators’ (Bid, Bim
and perhaps Puma) are capable of binding to and enabling the conformational change
and pore formation of Bax/Bak50;53-57. Other BH3-only proteins, termed ‘sensitizers’
(e.g. Noxa and Bad), can displace the activators from anti-apoptotic proteins. In
this model, which suggests the existence of a functional hierarchy within the BH3-
only subfamily, survival is the default. The indirect activation model (also referred as
displacement model), on the other hand, suggests that all BH3-only proteins bind to
Thesis_Final_2_01092008.indd Sec1:15Thesis_Final_2_01092008.indd Sec1:15 01-09-2008 11:08:0401-09-2008 11:08:04

16their specifi c anti-apoptotic relatives, which are inactivated, thus indirectly enabling
the Bax/Bak lethal function. Based on this model, in which death is the default, Bim,
Bid and Puma are the most potent apoptosis inducers simply because they are the
only ones able to engage all pro-survival proteins49;54;58;59. A strong argument for this
scenario was made using murine cells without Bim and Bid, and containing reduced
Puma. These cells were still capable of undergoing apoptosis55, something which is
hard to reconcile with the direct activation model.
So far most of this work has been done in artifi cial systems in vitro using isolated
mitochondria56, gene ablated murine cells, and transfected or virally transduced cell
lines49. These approaches leave open the important matter: how these proteins and
pathways function in human healthy tissues and primary cancer cells.
The Bcl-2 family is subdivided in three main categories: Anti-apoptotic Bcl-2-like proteins (A), pro-apoptotic
Bax-like proteins (B) and BH3-only proteins (C). Members for which solid evidence has been obtained are
reviewed in detail elsewhere58;60. In addition, other ‘candidates’, containing at least a conserved BH3-only
or BH3-like domain have been described and are presented in (D). (Adapted from Alves61)
Prosurvival Pro-apoptotic BH3-like contenders
(A) Bcl-2-like (B) Bax-like (C) BH3-only (D)
Bfl -1 Bak Bad Bcl-G(S)
Bcl-2 Bax Bik BRCC2
Bcl-W Bok Bid MAP-1
Bcl-XL
Hrk Mule
Mcl1 Bim NIP3
Noxa Nix
Puma Spike
Bmf
IAPs
The human IAP family has been implicated in cell division, cell cycle progression,
signal transduction62 and consists of at least 8 members, and includes XIAP, cIAP1,
cIAP2, NIAP, and survivin. Overexpression of several IAPs has been detected in
various cancers63. Of all IAPs, XIAP is most probably the only actual inhibitor of
caspases64. IAP1 and -2 associate with TNF-receptor family members and recent
data have demonstrated that their presence in fact blocks NF-κB signaling. When
unleashed, this pathway leads to TNF production which can kill cancer cells in an
autocrine fashion65. Survivin has been implicated in both apoptosis and cell division, but
compelling evidence now points to a specifi c role in chromosome segregation66;67.
Table 1. Overview of Bcl-2 family members and relatives
Thesis_Final_2_01092008.indd Sec1:16Thesis_Final_2_01092008.indd Sec1:16 01-09-2008 11:08:0401-09-2008 11:08:04

Ch
ap
ter 1
▪ Genera
l intro
ductio
n
17FLIP
c-FLIP (c-FLICE-inhibitory protein) is a family of alternatively spliced protein variants,
and primarily exists as long (c-FLIPL) and short (c-FLIP
S) isoforms in human cells.
c-FLIPL is homologous to caspase-8 but cannot become actived. Both FLIP variants
can be recruited to the DISC, where they can block pro-caspase-8 activation and
protect cells from death receptor mediated apoptosis68.
APOPTOSIS DEREGULATION IN CLL
Apoptosis regulatory proteins
In CLL several mechanisms contribute to resistance towards apoptosis. First,
Bcl-2 is expressed 1.7 – 25-fold higher in CLL than in normal lymphocytes, and
CLL cells isolated from patients refractory to standard chemotherapy show an
increased Bcl-2/Bax ratio69;70. The high expression of Bcl-2 has been postulated to
be related to deletions of two Bcl-2 suppressing miRNAs, miR-15a and miR-16-1,
located in a cluster at 13q14.3, which is deleted in ∼65% of the CLL patients71;72.
Furthermore, a polymorphism in the promoter region of the BCL-2 gene (-938C>A)
was reportedly associated with inferior clinical course and with increased expression
of Bcl-273. However, these initial fi ndings could not be corroborated in two follow-up
studies74;75. Secondly, high Mcl-1 levels were found to be associated with a failure to
achieve complete remission following chemotherapy in CLL76. Furthermore, Mcl-1
downregulation correlated with in vitro apoptosis induced by various therapeutics
in CLL77-79. Signifi cant Mcl-1 upregulation and subsequent protection against
spontaneous apoptosis was induced upon in vitro co-culture of CLL cells with CD40
ligand (CD154) expressed on fi broblasts80 and with follicular dendritic cells (FDC)81.
Thirdly, a nucleotide polymorphism, -248G>A, in the 5’ promoter region of Bax
was found in CLL, causing a reduced protein expression82. However, the outcome
of the disease did not seem to be infl uenced by this polymorphism, and, thus the
clinical impact was uncertain83. Finally, IAPs such as XIAP, cIAP1 and cIAP2 may
also be overexpressed in CLL84. XIAP inhibitors, which can potentially de-repress
caspase activity in malignant cells, are currently viewed as promising novel treatment
options85.
Death receptors
CLL cells express barely detectable levels of Fas on their surface, although transcripts
for both Fas and FasL (CD178) are commonly detected. Several stimuli have been
shown to up-regulate Fas expression, e.g. IFN and CD40-ligation, although CLL cells
Thesis_Final_2_01092008.indd Sec1:17Thesis_Final_2_01092008.indd Sec1:17 01-09-2008 11:08:0401-09-2008 11:08:04

18remain resistant to Fas-mediated apoptosis86. However, CLL cells become sensitive to
Fas-mediated apoptosis upon prolonged stimulation with CD40 ligand, concomitantly
with FLIP down-regulation and up-regulation of FADD87.
Death-inducing receptors for TNF-related apoptosis-inducing ligand (TRAIL), TRAIL-
R1, TRAIL-R2, TRAIL-R3 and TRAIL-R4 are expressed on primary CLL cells at
a low level. Nevertheless, CLL cells are resistant to TRAIL-induced apoptosis. In
correlation with low TRAIL receptor surface expression, DISC formation is hampered
and further caspase-8 activation is prevented88. Sensitivity to TRAIL was induced
when CLL cells were pretreated with nontoxic concentrations of histone deacetylase
(HDAC) inhibitors89. Furthermore, CD40 ligation induces expression of the pro-
apoptotic BH3-only protein Bid and TRAIL-R5. Death receptors CD95 and TRAIL-
R5 can act synergistically to induce caspase-dependent apoptosis of CLL cells and
Bid can facilitate cross-talk between mitochondrial-dependent and death receptor
inducing pathways90.
p53
p53 plays a protective role in normal somatic tissues by preventing division of damaged
cells91;92. The locus of the p53 gene is 17p13.1. The p53 protein acts in many cellular
processes, including cell-cycle checkpoints, DNA repair, senescence, apoptosis and
the surveillance of genomic integrity93;94. Stress stimuli such as DNA-damaging drugs
rapidly induce a transient increase in p53 protein95. Wild type (wt) p53 inhibits cancer
development by inducing transcription of a number of target genes involved in cell
cycle arrest and apoptosis. A p53-inducible gene involved in cell cycle arrest is p21
(also Cyclin-dependent kinase inhibitor 1A, CDKN1A). On the other hand, p53 can
trigger apoptosis via Bax96, Puma-97;98 and Noxa-99 gene transcription.
Deletions of p53 occur in about 10-15% of the CLL cases10;100. However, p53
dysfunction can also occur via alternative mechanisms, such as the inactivation of
ataxia telangiectasia mutated (ATM) gene12. ATM, encoded at chromosome 11q22.3,
is a kinase controlling p53 activation in response to DNA double-strand breaks.
Dysfunction of p53 by inactivation of ATM accounts for an additional abnormality in
15-20% of the patients101;102. In general, defects in p53 function occur in approximately
20% of CLL patients101. However, the frequency of p53 dysfunction increases to
nearly 50% as the disease progresses following initial therapy103;104. CLL patients with
p53 dysfunction do not respond to conventional therapy and tend to have a rapidly
progressive disease3.
Many therapeutic strategies require an active p53. Consequently, loss of p53 function
results in a selective resistance to DNA-damaging therapies, including alkylating
agents and fl udarabine103. Therefore, an important area of research is devoted to
Thesis_Final_2_01092008.indd Sec1:18Thesis_Final_2_01092008.indd Sec1:18 01-09-2008 11:08:0401-09-2008 11:08:04

Ch
ap
ter 1
▪ Genera
l intro
ductio
n
19the identifi cation of new treatment strategies that function independently of the p53
pathway.
ROLE OF THE MICROENVIRONMENT IN CLL
Although CLL cells show characteristics consistent with a defect in programmed
cell death and exhibit prolonged survival in vivo, during in vitro culture CLL cells
isolated from peripheral blood can rapidly undergo spontaneous apoptosis105;106.
This dichotomy highlights the relevance of an in vivo microenvironment capable of
delivering survival signals. Survival-supporting factors might rescue leukemia cells
from cytotoxic therapy107;18 and this may be the basis for subsequent relapse.
The CLL proliferating compartment is represented by focal aggregates of proliferating
lymphocytes that give rise to the so-called pseudofollicles or proliferation centers. In
the LN of CLL patients, the pseudo-follicles represent the histological hallmark, as
well as in the white pulp of the spleen and in the BM. Immunohistochemistry studies
have shown that pseudofollicles are clusters of CD5+ Ki67+ B cells surrounded by
new vessels108;109. In the LN and BM there are CD3+ T cells, most of them belonging to
the CD4+ T helper subset. The CD4+ T cells tend to concentrate around and within the
proliferating pseudofollicles and many of them express CD40L implying that they are
in an activated state. CLL cells retain the capacity to respond to CD40L, expressed
by the CD4+ T cells, thus resulting in their activation110. Furthermore, CLL cells
purifi ed from LN and PB constitutively express mRNA for T cell attracting chemokines
(namely CCL17 and CCL22) and the same holds true for CLL cells stimulated by in
vitro CD40-crosslinking110. These fi ndings indicate that physiological signals in the
tumor microenvironment, such as CD40L, give CLL cells chemo-attracting capacity to
activated CD4+ T cells, which in turn are able to deliver survival signals to the CLL cells.
Other accessory cells, such as bone marrow stromal cells111, FDCs81 and so-called
nurse-like cells112 that can be obtained from peripheral blood, may also be involved in
cross-talk between malignant cells and the microenvironment. Furthermore, various
pro-survival cytokines (IL-2, IL-4, IL-8, TNF-α, IFN-α, IFN-γ and VEGF) and their
respective receptors are found on CLL cells (reviewed in Kay 2002113). However, they
remain unresponsive to most mitogens that induce proliferation of normal B cells.
The pro-survival signaling pathways elicited in CLL appear to depend on both paracrine
and autocrine mechanisms. In some cases it is unclear whether the signal comes
entirely from the cell in the microenvironment or as a consequence of deregulation
in the CLL cells themselves. Alterations in several signaling pathways have been
suggested in CLL.
For example, NF-κB is a transcription factor that can promote survival through the
Thesis_Final_2_01092008.indd Sec1:19Thesis_Final_2_01092008.indd Sec1:19 01-09-2008 11:08:0501-09-2008 11:08:05

20induction of several anti-apoptotic proteins, such as Bfl -1, Bcl-X
L, IAPs and Flip114.
Higher levels of constitutive NF-κB are seen in unstimulated CLL cells compared to
healthy peripheral blood B cells115. Engagement with the TNF-superfamily members
CD40L, BAFF (B cell-activating factor of tumor necrosis factor (TNF) family, also
known as BlyS) and APRIL (A proliferation-inducing ligand), enhances this activity115;116.
However, whilst it has been shown that APRIL is overexpressed in some cases of
CLL117, the APRIL driving pro-survival signaling is also derived from nurse-like cells
in the tumor niche118. The activation of the canonical NF-κB pathway also contrasts
with the alternative pathway in response to BAFF116, which also suggests intrinsic
deregulation of the responses to certain cytokines. Additionally, recent studies
showed an association between the expression of NF-κB subunit Rel A and in vitro
survival of CLL cells119.
Also, the PI3K/AKT, Raf/MEK/ERK and JNK/STAT signaling pathways are essential
for cell survival, and are frequently deregulated in malignancy120. They control
expression and function of many proteins, including apoptosis regulatory proteins.
Several studies done in CLL suggested that also these pathways may contribute to
survival of the malignant cells121;122;123. Relatively new in the CLL fi eld is the nonreceptor
tyrosine kinase c-Abl. Oncogenic fusion versions of c-Abl drive malignancy in chronic
myeloid leukemia, and are the target for successful therapy with kinase inhibitors120. A
recent study highlighted the importance c-Abl itself in CLL survival124 and furthermore
it was reported that c-Abl becomes active upon CD40 triggering125. The relevance of
these signaling events for the in vivo biology of CLL, and especially in the context of
survival niches, is an important aspect that remains to be established.
THERAPEUTIC APROACH TO ATTACK THE
MICROENVIRONMENT
As mentioned above, despite improvements in treatment effects for CLL by a
combination of chemo- and immuno-therapy, the disease will invariably relapse.
Clearly, the clearance of the CLL peripheral blood pool which usually occurs is
not suffi cient. The likely hypothesis is that the proliferation centers in LN, BM and
spleen constitute “germinative” foci, afford resistance to drugs, and will replenish the
peripheral blood after a successful cycle of therapy.
Therefore, drugs that target bystander cells and/or their protective effects on CLL
cells in the microenvironmental “niche”, would provide more effi cacious options to
the treatment of CLL patients. Accordingly, the microenvironment may be targeted
by interfering with various cytokines or by modulating immune effector cells. Clinical
Thesis_Final_2_01092008.indd Sec1:20Thesis_Final_2_01092008.indd Sec1:20 01-09-2008 11:08:0501-09-2008 11:08:05

Ch
ap
ter 1
▪ Genera
l intro
ductio
n
21trials in refractory or relapsed CLL cases have reported encouraging results with
immunomodulating agents, i.e. thalidomide and its analog lenalidomide. Although the
exact antitumour activity of such compounds remains uncertain, they do not exert a
direct cytotoxic effect on CLL cells126;127. In addition, therapies aiming to target CLL
cells in the protective “niches” by counteracting the pro-survival changes provided at
these sites, could be a novel and potentially effective strategy. In this regard, small-
molecule BH3 mimetics such as ABT-737 which is a potent and specifi c Bcl-2/Bcl-XL/
Bcl-W inhibitor are now in preclinical or clinical development128.
To conclude, CLL cells possess and rely on various different ways to escape
apoptosis. Direct cell-to-cell contact between CLL cells and bystander cells creates a
microenvironment in which both membrane-bound and soluble factors collaborate in
protecting CLL cells from apoptosis. Learning how to manipulate the microenvironment,
or to specifi cally target the leukemic cells residing in these niches, may reveal new
strategies for restoring apoptosis sensitivity and improving therapeutic outcome.
SCOPE
The aims of this thesis are:
1. To gain insight into the mechanism of action of potential novel drugs with activity
independent of the p53 pathway, with emphasis on effects on apoptosis regulatory
molecules.
2. To obtain insight into the molecular basis of apoptosis (dys-) regulation and survival
of CLL cells both in peripheral blood and in the lymph node microenvironment in
connection with drug sensitivity.
In Chapters 2 and 3 the apoptosis pathway of two p53-independent drugs are
investigated; the cyclin dependent kinase (CDK) inhibitor roscovitine (Seliciclib,
cyc202), and a novel proteasome inhibitor. In the following section the infl uence of
the microenvironment on CLL survival is addressed. In chapter 4 a comparative study
of CLL cells in the peripheral blood and the lymph node compartments with respect
to mRNA and protein expression levels of various apoptosis regulatory molecules is
presented. In Chapter 5, in order to model the in vivo LN setting of CLL, the infl uence
of the CD40-signaling on the expression of apoptosis regulatory proteins is studied, in
relation to sensitivity to various current and novel chemotherapeutic drugs. In chapter
6 we describe an approach to overcome CD40L induced drug resistance in CLL using
cAbl kinase inhibitors. Finally, in chapter 7 an integrated discussion is presented and
future directions are suggested.
Thesis_Final_2_01092008.indd Sec1:21Thesis_Final_2_01092008.indd Sec1:21 01-09-2008 11:08:0501-09-2008 11:08:05

22
Thesis_Final_2_01092008.indd Sec1:22Thesis_Final_2_01092008.indd Sec1:22 01-09-2008 11:08:0501-09-2008 11:08:05

Ch
ap
ter 1
▪ Genera
l intro
ductio
n
23REFERENCE LIST
1. Caligaris-Cappio F, Hamblin TJ. B-cell
chronic lymphocytic leukemia: a bird
of a different feather. J.Clin.Oncol.
1999;17:399-408.
2. Rozman C, Montserrat E. Chronic
lymphocytic leukemia. N.Engl.J.Med.
1995;333:1052-1057.
3. Dohner H, Stilgenbauer S, Benner A et
al. Genomic aberrations and survival
in chronic lymphocytic leukemia.
N.Engl.J.Med. 2000;343:1910-1916.
4. Damle RN, Wasil T, Fais F et al. Ig
V gene mutation status and CD38
expression as novel prognostic
indicators in chronic lymphocytic
leukemia. Blood 1999;94:1840-1847.
5. Hamblin TJ, Davis Z, Gardiner A,
Oscier DG, Stevenson FK. Unmutated
Ig V(H) genes are associated with
a more aggressive form of chronic
lymphocytic leukemia. Blood
1999;94:1848-1854.
6. Rassenti LZ, Huynh L, Toy TL
et al. ZAP-70 compared with
immunoglobulin heavy-chain gene
mutation status as a predictor of
disease progression in chronic
lymphocytic leukemia. N.Engl.J.Med.
2004;351:893-901.
7. Wiestner A, Rosenwald A, Barry TS
et al. ZAP-70 expression identifi es a
chronic lymphocytic leukemia subtype
with unmutated immunoglobulin
genes, inferior clinical outcome, and
distinct gene expression profi le. Blood
2003;101:4944-4951.
8. Hamblin TJ, Orchard JA, Ibbotson
RE et al. CD38 expression and
immunoglobulin variable region
mutations are independent prognostic
variables in chronic lymphocytic
leukemia, but CD38 expression may
vary during the course of the disease.
Blood 2002;99:1023-1029.
9. Chen L, Widhopf G, Huynh L et al.
Expression of ZAP-70 is associated
with increased B-cell receptor signaling
in chronic lymphocytic leukemia. Blood
2002;100:4609-4614.
10. Dohner H, Fischer K, Bentz M et al.
p53 gene deletion predicts for poor
survival and non-response to therapy
with purine analogs in chronic B-cell
leukemias. Blood 1995;85:1580-1589.
11. Dohner H, Stilgenbauer S, James
MR et al. 11q deletions identify a new
subset of B-cell chronic lymphocytic
leukemia characterized by extensive
nodal involvement and inferior
prognosis. Blood 1997;89:2516-2522.
12. Pettitt AR, Sherrington PD, Stewart G
et al. p53 dysfunction in B-cell chronic
lymphocytic leukemia: inactivation
of ATM as an alternative to TP53
mutation. Blood 2001;98:814-822.
13. Dameshek W. Chronic lymphocytic
leukemia: an accumulative disease
of immunolgically incompetent
lymphocytes. Blood 1967;29:Suppl-84.
14. Klein U, Tu Y, Stolovitzky GA et al.
Gene expression profi ling of B cell
chronic lymphocytic leukemia reveals
a homogeneous phenotype related
to memory B cells. J.Exp.Med.
2001;194:1625-1638.
15. Rosenwald A, Alizadeh AA, Widhopf
G et al. Relation of gene expression
phenotype to immunoglobulin mutation
genotype in B cell chronic lymphocytic
leukemia. J.Exp.Med. 2001;194:1639-
1647.
16. Messmer BT, Messmer D, Allen SL et
al. In vivo measurements document
the dynamic cellular kinetics of chronic
lymphocytic leukemia B cells. J.Clin.
Invest 2005;115:755-764.
Thesis_Final_2_01092008.indd Sec1:23Thesis_Final_2_01092008.indd Sec1:23 01-09-2008 11:08:0501-09-2008 11:08:05

24 17. Caligaris-Cappio F. Cellular
interactions, immunodefi ciency and
autoimmunity in CLL. Hematol.Cell
Ther. 2000;42:21-25.
18. Ghia P, Caligaris-Cappio F. The
indispensable role of microenvironment
in the natural history of low-grade
B-cell neoplasms. Adv.Cancer Res.
2000;79:157-173.
19. Chiorazzi N, Rai KR, Ferrarini M.
Chronic lymphocytic leukemia.
N.Engl.J.Med. 2005;352:804-815.
20. Auer RL, Gribben J, Cotter FE.
Emerging therapy for chronic
lymphocytic leukaemia. Br.J.Haematol.
2007;139:635-644.
21. Rai KR, Peterson BL, Appelbaum
FR et al. Fludarabine compared with
chlorambucil as primary therapy
for chronic lymphocytic leukemia.
N.Engl.J.Med. 2000;343:1750-1757.
22. Leporrier M, Chevret S, Cazin B
et al. Randomized comparison of
fl udarabine, CAP, and ChOP in 938
previously untreated stage B and C
chronic lymphocytic leukemia patients.
Blood 2001;98:2319-2325.
23. Catovsky D, Richards S, Matutes
E et al. Assessment of fl udarabine
plus cyclophosphamide for patients
with chronic lymphocytic leukaemia
(the LRF CLL4 Trial): a randomised
controlled trial. Lancet 2007;370:230-
239.
24. Eichhorst BF, Busch R, Hopfi nger G et
al. Fludarabine plus cyclophosphamide
versus fl udarabine alone in fi rst-line
therapy of younger patients with
chronic lymphocytic leukemia. Blood
2006;107:885-891.
25. Flinn IW, Neuberg DS, Grever MR et
al. Phase III trial of fl udarabine plus
cyclophosphamide compared with
fl udarabine for patients with previously
untreated chronic lymphocytic
leukemia: US Intergroup Trial E2997.
J.Clin.Oncol. 2007;25:793-798.
26. Keating MJ, O’Brien S, Albitar
M et al. Early results of a
chemoimmunotherapy regimen of
fl udarabine, cyclophosphamide, and
rituximab as initial therapy for chronic
lymphocytic leukemia. J.Clin.Oncol.
2005;23:4079-4088.
27. Wierda W, O’Brien S, Wen S et
al. Chemoimmunotherapy with
fl udarabine, cyclophosphamide, and
rituximab for relapsed and refractory
chronic lymphocytic leukemia. J.Clin.
Oncol. 2005;23:4070-4078.
28. Stilgenbauer S. Advances in the use
of alemtuzumab in CLL. Clin.Adv.
Hematol.Oncol. 2008;6:23-24.
29. Dreger P, Brand R, Hansz J et al.
Treatment-related mortality and
graft-versus-leukemia activity after
allogeneic stem cell transplantation
for chronic lymphocytic leukemia
using intensity-reduced conditioning.
Leukemia 2003;17:841-848.
30. Gribben JG. Stem-cell transplantation
in chronic lymphocytic leukaemia.
Best.Pract.Res.Clin.Haematol.
2007;20:513-527.
31. Gine E, Moreno C, Esteve J,
Montserrat E. The role of stem-cell
transplantation in chronic lymphocytic
leukemia risk-adapted therapy. Best.
Pract.Res.Clin.Haematol. 2007;20:529-
543.
32. Kerr JF, Wyllie AH, Currie AR.
Apoptosis: a basic biological
phenomenon with wide-ranging
implications in tissue kinetics.
Br.J.Cancer 1972;26:239-257.
33. Savill J, Fadok V. Corpse clearance
defi nes the meaning of cell death.
Nature 2000;407:784-788.
34. Alnemri ES, Livingston DJ, Nicholson
DW et al. Human ICE/CED-3 protease
nomenclature. Cell 1996;87:171.
Thesis_Final_2_01092008.indd Sec1:24Thesis_Final_2_01092008.indd Sec1:24 01-09-2008 11:08:0501-09-2008 11:08:05

Ch
ap
ter 1
▪ Genera
l intro
ductio
n
25 35. Riedl SJ, Shi Y. Molecular mechanisms
of caspase regulation during apoptosis.
Nat.Rev.Mol.Cell Biol. 2004;5:897-907.
36. Kuida K, Lippke JA, Ku G et al.
Altered cytokine export and apoptosis
in mice defi cient in interleukin-1
beta converting enzyme. Science
1995;267:2000-2003.
37. Kischkel FC, Hellbardt S, Behrmann
I et al. Cytotoxicity-dependent APO-1
(Fas/CD95)-associated proteins form
a death-inducing signaling complex
(DISC) with the receptor. EMBO J.
1995;14:5579-5588.
38. Muzio M, Chinnaiyan AM, Kischkel
FC et al. FLICE, a novel FADD-
homologous ICE/CED-3-like protease,
is recruited to the CD95 (Fas/APO-1)
death--inducing signaling complex. Cell
1996;85:817-827.
39. Kischkel FC, Lawrence DA, Tinel
A et al. Death receptor recruitment
of endogenous caspase-10 and
apoptosis initiation in the absence
of caspase-8. J.Biol.Chem.
2001;276:46639-46646.
40. Sprick MR, Rieser E, Stahl H et
al. Caspase-10 is recruited to and
activated at the native TRAIL and
CD95 death-inducing signalling
complexes in a FADD-dependent
manner but can not functionally
substitute caspase-8. EMBO J.
2002;21:4520-4530.
41. Li H, Zhu H, Xu CJ, Yuan J. Cleavage
of BID by caspase 8 mediates
the mitochondrial damage in the
Fas pathway of apoptosis. Cell
1998;94:491-501.
42. Wei MC, Lindsten T, Mootha VK
et al. tBID, a membrane-targeted
death ligand, oligomerizes BAK to
release cytochrome c. Genes Dev.
2000;14:2060-2071.
43. Li P, Nijhawan D, Budihardjo I et
al. Cytochrome c and dATP-dependent
formation of Apaf-1/caspase-9
complex initiates an apoptotic protease
cascade. Cell 1997;91:479-489.
44. Du C, Fang M, Li Y, Li L, Wang X.
Smac, a mitochondrial protein that
promotes cytochrome c-dependent
caspase activation by eliminating IAP
inhibition. Cell 2000;102:33-42.
45. Suzuki Y, Imai Y, Nakayama H et al.
A serine protease, HtrA2, is released
from the mitochondria and interacts
with XIAP, inducing cell death. Mol.Cell
2001;8:613-621.
46. Li LY, Luo X, Wang X. Endonuclease
G is an apoptotic DNase when
released from mitochondria. Nature
2001;412:95-99.
47. Susin SA, Lorenzo HK, Zamzami
N et al. Molecular characterization
of mitochondrial apoptosis-inducing
factor. Nature 1999;397:441-446.
48. Tsujimoto Y. Cell death regulation
by the Bcl-2 protein family in
the mitochondria. J.Cell Physiol
2003;195:158-167.
49. Chen L, Willis SN, Wei A et al.
Differential targeting of prosurvival
Bcl-2 proteins by their BH3-only
ligands allows complementary
apoptotic function. Mol.Cell
2005;17:393-403.
50. Certo M, Del GM, V, Nishino M et
al. Mitochondria primed by death
signals determine cellular addiction to
antiapoptotic BCL-2 family members.
Cancer Cell 2006;9:351-365.
51. Green DR, Kroemer G. The
pathophysiology of mitochondrial cell
death. Science 2004;305:626-629.
Thesis_Final_2_01092008.indd Sec1:25Thesis_Final_2_01092008.indd Sec1:25 01-09-2008 11:08:0501-09-2008 11:08:05

26 52. Zong WX, Lindsten T, Ross AJ,
MacGregor GR, Thompson CB. BH3-
only proteins that bind pro-survival
Bcl-2 family members fail to induce
apoptosis in the absence of Bax and
Bak. Genes Dev. 2001;15:1481-1486.
53. Cartron PF, Gallenne T, Bougras G et
al. The fi rst alpha helix of Bax plays
a necessary role in its ligand-induced
activation by the BH3-only proteins Bid
and PUMA. Mol.Cell 2004;16:807-818.
54. Kim H, Rafi uddin-Shah M, Tu HC
et al. Hierarchical regulation of
mitochondrion-dependent apoptosis
by BCL-2 subfamilies. Nat.Cell Biol.
2006;8:1348-1358.
55. Kuwana T, Bouchier-Hayes L, Chipuk
JE et al. BH3 domains of BH3-only
proteins differentially regulate Bax-
mediated mitochondrial membrane
permeabilization both directly and
indirectly. Mol.Cell 2005;17:525-535.
56. Letai A, Bassik MC, Walensky LD
et al. Distinct BH3 domains either
sensitize or activate mitochondrial
apoptosis, serving as prototype cancer
therapeutics. Cancer Cell 2002;2:183-
192.
57. Walensky LD, Pitter K, Morash J et
al. A stapled BID BH3 helix directly
binds and activates BAX. Mol.Cell
2006;24:199-210.
58. Willis SN, Chen L, Dewson G et al.
Proapoptotic Bak is sequestered by
Mcl-1 and Bcl-xL, but not Bcl-2, until
displaced by BH3-only proteins. Genes
Dev. 2005;19:1294-1305.
59. Willis SN, Fletcher JI, Kaufmann
T et al. Apoptosis initiated when
BH3 ligands engage multiple Bcl-2
homologs, not Bax or Bak. Science
2007;315:856-859.
60. Strasser A. The role of BH3-only
proteins in the immune system. Nat.
Rev.Immunol. 2005;5:189-200.
61. Alves NL, van Lier RA, Eldering E.
Withdrawal symptoms on display: Bcl-2
members under investigation. Trends
Immunol. 2007;28:26-32.
62. Schimmer AD. Inhibitor of apoptosis
proteins: translating basic knowledge
into clinical practice. Cancer Res.
2004;64:7183-7190.
63. Nachmias B, Ashhab Y, Ben-Yehuda
D. The inhibitor of apoptosis protein
family (IAPs): an emerging therapeutic
target in cancer. Semin.Cancer Biol.
2004;14:231-243.
64. Eckelman BP, Salvesen GS, Scott FL.
Human inhibitor of apoptosis proteins:
why XIAP is the black sheep of the
family. EMBO Rep. 2006;7:988-994.
65. Vince JE, Wong WW, Khan N et al.
IAP antagonists target cIAP1 to induce
TNFalpha-dependent apoptosis. Cell
2007;131:682-693.
66. Stauber RH, Mann W, Knauer SK.
Nuclear and cytoplasmic survivin:
molecular mechanism, prognostic,
and therapeutic potential. Cancer Res.
2007;67:5999-6002.
67. Lens SM, Vader G, Medema RH. The
case for Survivin as mitotic regulator.
Curr.Opin.Cell Biol. 2006;18:616-622.
68. Scaffi di C, Schmitz I, Krammer
PH, Peter ME. The role of
c-FLIP in modulation of CD95-
induced apoptosis. J.Biol.Chem.
1999;274:1541-1548.
69. Schena M, Larsson LG, Gottardi D
et al. Growth- and differentiation-
associated expression of bcl-2 in
B-chronic lymphocytic leukemia cells.
Blood 1992;79:2981-2989.
70. Pepper C, Hoy T, Bentley DP. Bcl-2/
Bax ratios in chronic lymphocytic
leukaemia and their correlation with in
vitro apoptosis and clinical resistance.
Br.J.Cancer 1997;76:935-938.
Thesis_Final_2_01092008.indd Sec1:26Thesis_Final_2_01092008.indd Sec1:26 01-09-2008 11:08:0501-09-2008 11:08:05

Ch
ap
ter 1
▪ Genera
l intro
ductio
n
27 71. Calin GA, Dumitru CD, Shimizu M
et al. Frequent deletions and down-
regulation of micro- RNA genes
miR15 and miR16 at 13q14 in chronic
lymphocytic leukemia. Proc.Natl.Acad.
Sci.U.S.A 2002;99:15524-15529.
72. Cimmino A, Calin GA, Fabbri M et al.
miR-15 and miR-16 induce apoptosis
by targeting BCL2. Proc.Natl.Acad.
Sci.U.S.A 2005;102:13944-13949.
73. Nuckel H, Frey UH, Bau M et al.
Association of a novel regulatory
polymorphism (-938C>A) in the
BCL2 gene promoter with disease
progression and survival in chronic
lymphocytic leukemia. Blood
2007;109:290-297.
74. Kaderi MA, Norberg M, Murray F et
al. The BCL-2 promoter (-938C>A)
polymorphism does not predict clinical
outcome in chronic lymphocytic
leukemia. Leukemia 2008;22:339-343.
75. Majid A, Tsoulakis O, Walewska
R et al. BCL2 expression in
chronic lymphocytic leukemia:
lack of association with the BCL2
938A>C promoter single nucleotide
polymorphism. Blood 2008;111:874-
877.
76. Kitada S, Andersen J, Akar S et al.
Expression of apoptosis-regulating
proteins in chronic lymphocytic
leukemia: correlations with In vitro
and In vivo chemoresponses. Blood
1998;91:3379-3389.
77. Byrd JC, Lin TS, Dalton JT et al.
Flavopiridol administered using a
pharmacologically derived schedule is
associated with marked clinical effi cacy
in refractory, genetically high-risk
chronic lymphocytic leukemia. Blood
2007;109:399-404.
78. Kitada S, Zapata JM, Andreeff M, Reed
JC. Protein kinase inhibitors fl avopiridol
and 7-hydroxy-staurosporine down-
regulate antiapoptosis proteins in
B-cell chronic lymphocytic leukemia.
Blood 2000;96:393-397.
79. Hahntow IN, Schneller F, Oelsner M et
al. Cyclin-dependent kinase inhibitor
Roscovitine induces apoptosis in
chronic lymphocytic leukemia cells.
Leukemia 2004;18:747-755.
80. Kitada S, Zapata JM, Andreeff M,
Reed JC. Bryostatin and CD40-
ligand enhance apoptosis resistance
and induce expression of cell
survival genes in B-cell chronic
lymphocytic leukaemia. Br.J.Haematol.
1999;106:995-1004.
81. Pedersen IM, Kitada S, Leoni LM
et al. Protection of CLL B cells
by a follicular dendritic cell line is
dependent on induction of Mcl-1. Blood
2002;100:1795-1801.
82. Saxena A, Moshynska O, Sankaran
K, Viswanathan S, Sheridan DP.
Association of a novel single
nucleotide polymorphism, G(-248)A,
in the 5’-UTR of BAX gene in chronic
lymphocytic leukemia with disease
progression and treatment resistance.
Cancer Lett. 2002;187:199-205.
83. Skogsberg S, Tobin G, Krober A et
al. The G(-248)A polymorphism in
the promoter region of the Bax gene
does not correlate with prognostic
markers or overall survival in chronic
lymphocytic leukemia. Leukemia
2006;20:77-81.
84. Munzert G, Kirchner D, Stobbe
H et al. Tumor necrosis factor
receptor-associated factor 1 gene
overexpression in B-cell chronic
lymphocytic leukemia: analysis of
NF-kappa B/Rel-regulated inhibitors of
apoptosis. Blood 2002;100:3749-3756.
85. Schimmer AD, Dalili S, Batey RA, Riedl
SJ. Targeting XIAP for the treatment
of malignancy. Cell Death.Differ.
2006;13:179-188.
86. Osorio LM, Jondal M, guilar-Santelises
M. Regulation of B-CLL apoptosis
through membrane receptors and
Bcl-2 family proteins. Leuk.Lymphoma
1998;30:247-256.
Thesis_Final_2_01092008.indd Sec1:27Thesis_Final_2_01092008.indd Sec1:27 01-09-2008 11:08:0501-09-2008 11:08:05

28 87. Chu P, Deforce D, Pedersen IM et
al. Latent sensitivity to Fas-mediated
apoptosis after CD40 ligation may
explain activity of CD154 gene therapy
in chronic lymphocytic leukemia. Proc.
Natl.Acad.Sci.U.S.A 2002;99:3854-
3859.
88. MacFarlane M, Harper N, Snowden
RT et al. Mechanisms of resistance to
TRAIL-induced apoptosis in primary
B cell chronic lymphocytic leukaemia.
Oncogene 2002;21:6809-6818.
89. Inoue S, MacFarlane M, Harper N
et al. Histone deacetylase inhibitors
potentiate TNF-related apoptosis-
inducing ligand (TRAIL)-induced
apoptosis in lymphoid malignancies.
Cell Death.Differ. 2004;11 Suppl
2:S193-S206.
90. Dicker F, Kater AP, Fukuda T,
Kipps TJ. Fas-ligand (CD178)
and TRAIL synergistically induce
apoptosis of CD40-activated chronic
lymphocytic leukemia B cells. Blood
2005;105:3193-3198.
91. Lane DP. Cancer. p53, guardian of the
genome. Nature 1992;358:15-16.
92. Levine AJ. p53, the cellular gatekeeper
for growth and division. Cell
1997;88:323-331.
93. Evan GI, Vousden KH. Proliferation,
cell cycle and apoptosis in cancer.
Nature 2001;411:342-348.
94. Ko LJ, Prives C. p53: puzzle and
paradigm. Genes Dev. 1996;10:1054-
1072.
95. Giaccia AJ, Kastan MB. The complexity
of p53 modulation: emerging patterns
from divergent signals. Genes Dev.
1998;12:2973-2983.
96. Miyashita T, Reed JC. Tumor
suppressor p53 is a direct
transcriptional activator of the human
bax gene. Cell 1995;80:293-299.
97. Yu J, Zhang L, Hwang PM, Kinzler KW,
Vogelstein B. PUMA induces the rapid
apoptosis of colorectal cancer cells.
Mol.Cell 2001;7:673-682.
98. Nakano K, Vousden KH. PUMA, a
novel proapoptotic gene, is induced by
p53. Mol.Cell 2001;7:683-694.
99. Oda E, Ohki R, Murasawa H et al.
Noxa, a BH3-only member of the
Bcl-2 family and candidate mediator
of p53-induced apoptosis. Science
2000;288:1053-1058.
100. Wattel E, Preudhomme C, Hecquet
B et al. p53 mutations are associated
with resistance to chemotherapy
and short survival in hematologic
malignancies. Blood 1994;84:3148-
3157.
101. Austen B, Powell JE, Alvi A et al.
Mutations in the ATM gene lead to
impaired overall and treatment-free
survival that is independent of IGVH
mutation status in patients with B-CLL.
Blood 2005;106:3175-3182.
102. Stankovic T, Stewart GS, Fegan C
et al. Ataxia telangiectasia mutated-
defi cient B-cell chronic lymphocytic
leukemia occurs in pregerminal center
cells and results in defective damage
response and unrepaired chromosome
damage. Blood 2002;99:300-309.
103. Sturm I, Bosanquet AG, Hermann S
et al. Mutation of p53 and consecutive
selective drug resistance in B-CLL
occurs as a consequence of prior
DNA-damaging chemotherapy. Cell
Death.Differ. 2003;10:477-484.
104. Lozanski G, Heerema NA, Flinn IW et
al. Alemtuzumab is an effective therapy
for chronic lymphocytic leukemia with
p53 mutations and deletions. Blood
2004;103:3278-3281.
105. Collins RJ, Verschuer LA, Harmon BV
et al. Spontaneous programmed death
(apoptosis) of B-chronic lymphocytic
leukaemia cells following their culture
in vitro. Br.J.Haematol. 1989;71:343-
350.
Thesis_Final_2_01092008.indd Sec1:28Thesis_Final_2_01092008.indd Sec1:28 01-09-2008 11:08:0501-09-2008 11:08:05

Ch
ap
ter 1
▪ Genera
l intro
ductio
n
29 106. McConkey DJ, guilar-Santelises M,
Hartzell P et al. Induction of DNA
fragmentation in chronic B-lymphocytic
leukemia cells. J.Immunol.
1991;146:1072-1076.
107. Caligaris-Cappio F. Role of the
microenvironment in chronic
lymphocytic leukaemia. Br.J.Haematol.
2003;123:380-388.
108. Pileri SA, Ascani S, Sabattini E et al.
The pathologist’s view point. Part I--
indolent lymphomas. Haematologica
2000;85:1291-1307.
109. Pileri SA, Ascani S, Sabattini E et
al. The pathologist’s view point.
Part II --aggressive lymphomas.
Haematologica 2000;85:1308-1321.
110. Ghia P, Granziero L, Chilosi M,
Caligaris-Cappio F. Chronic B cell
malignancies and bone marrow
microenvironment. Semin.Cancer Biol.
2002;12:149-155.
111. Lagneaux D, Huhtinen M, Koskinen E,
Palmer E. Effect of anti-freeze protein
(AFP) on the cooling and freezing
of equine embryos as measured by
DAPI-staining. Equine Vet.J.Suppl
199785-87.
112. Burger JA, Tsukada N, Burger M et al.
Blood-derived nurse-like cells protect
chronic lymphocytic leukemia B cells
from spontaneous apoptosis through
stromal cell-derived factor-1. Blood
2000;96:2655-2663.
113. Kay NE, Hamblin TJ, Jelinek DF et
al. Chronic lymphocytic leukemia.
Hematology.Am.Soc.Hematol.Educ.
Program. 2002193-213.
114. Takada Y, Kobayashi Y, Aggarwal BB.
Evodiamine abolishes constitutive
and inducible NF-kappaB activation
by inhibiting IkappaBalpha kinase
activation, thereby suppressing
NF-kappaB-regulated antiapoptotic
and metastatic gene expression,
up-regulating apoptosis, and
inhibiting invasion. J.Biol.Chem.
2005;280:17203-17212.
115. Furman RR, Asgary Z, Mascarenhas
JO, Liou HC, Schattner EJ. Modulation
of NF-kappa B activity and apoptosis in
chronic lymphocytic leukemia B cells.
J.Immunol. 2000;164:2200-2206.
116. Endo T, Nishio M, Enzler T et al. BAFF
and APRIL support chronic lymphocytic
leukemia B-cell survival through
activation of the canonical NF-kappaB
pathway. Blood 2007;109:703-710.
117. Planelles L, Carvalho-Pinto CE,
Hardenberg G et al. APRIL promotes
B-1 cell-associated neoplasm. Cancer
Cell 2004;6:399-408.
118. Nishio M, Endo T, Tsukada N et al.
Nurselike cells express BAFF and
APRIL, which can promote survival of
chronic lymphocytic leukemia cells via
a paracrine pathway distinct from that
of SDF-1alpha. Blood 2005;106:1012-
1020.
119. Hewamana S, Alghazal S, Lin TT et
al. The NF-kappaB subunit Rel A is
associated with in vitro survival and
clinical disease progression in chronic
lymphocytic leukemia and represents
a promising therapeutic target. Blood
2008;111:4681-4689.
120. Steelman LS, Pohnert SC, Shelton
JG et al. JAK/STAT, Raf/MEK/ERK,
PI3K/Akt and BCR-ABL in cell cycle
progression and leukemogenesis.
Leukemia 2004;18:189-218.
121. Ringshausen I, Schneller F, Bogner
C et al. Constitutively activated
phosphatidylinositol-3 kinase (PI-3K)
is involved in the defect of apoptosis in
B-CLL: association with protein kinase
Cdelta. Blood 2002;100:3741-3748.
122. Barragan M, Bellosillo B, Campas C
et al. Involvement of protein kinase
C and phosphatidylinositol 3-kinase
pathways in the survival of B-cell
chronic lymphocytic leukemia cells.
Blood 2002;99:2969-2976.
Thesis_Final_2_01092008.indd Sec1:29Thesis_Final_2_01092008.indd Sec1:29 01-09-2008 11:08:0601-09-2008 11:08:06

30 123. Wickremasinghe RG, Ganeshaguru
K, Jones DT et al. Autologous
plasma activates Akt/protein kinase
B and enhances basal survival and
resistance to DNA damage-induced
apoptosis in B-chronic lymphocytic
leukaemia cells. Br.J.Haematol.
2001;114:608-615.
124. Lin K, Glenn MA, Harris RJ et
al. c-Abl expression in chronic
lymphocytic leukemia cells: clinical and
therapeutic implications. Cancer Res.
2006;66:7801-7809.
125. Dicker F, Kater AP, Prada CE et al.
CD154 induces p73 to overcome the
resistance to apoptosis of chronic
lymphocytic leukemia cells lacking
functional p53. Blood 2006;108:3450-
3457.
126. Chanan-Khan A, Porter CW.
Immunomodulating drugs for chronic
lymphocytic leukaemia. Lancet Oncol.
2006;7:480-488.
127. Ferrajoli A, Lee BN, Schlette EJ et
al. Lenalidomide induces complete
and partial remissions in patients
with relapsed and refractory chronic
lymphocytic leukemia. Blood
2008;111:5291-5297.
128. Oltersdorf T, Elmore SW, Shoemaker
AR et al. An inhibitor of Bcl-2 family
proteins induces regression of solid
tumours. Nature 2005;435:677-681.
Thesis_Final_2_01092008.indd Sec1:30Thesis_Final_2_01092008.indd Sec1:30 01-09-2008 11:08:0601-09-2008 11:08:06

chapter 33
Cross-talk among Bcl-2 family members in B-CLL: seliciclib acts via the Mcl-1/Noxa axis and gradual exhaustion of Bcl-2 protection
Delfi ne Y.H. Hallaert1,2, René Spijker1,2, Margot Jak1, Ingrid A.M. Derks2, Nuno L.
Alves2, Felix M. Wensveen2, Jan Paul de Boer3, Daphne de Jong3, Simon R Green4,
Marinus H.J. van Oers1 and Eric Eldering2
1 Dept. of Hematology, AMC, Amsterdam, The Netherlands2 Dept. of Experimental Immunology, AMC, Amsterdam, The Netherlands3 Dept. of Pathology, The Netherlands Cancer Institute/Antoni van Leeuwenhoek
Hospital, Amsterdam, The Netherlands4 Cyclacel Ltd., Dundee, UK
Cell Death and Differentiation, 2007; 14: 1958-1967
2
Thesis_Final_2_01092008.indd Sec1:31Thesis_Final_2_01092008.indd Sec1:31 01-09-2008 11:08:0601-09-2008 11:08:06

ABSTRACT
Seliciclib (R-roscovitine) is a cyclin-dependent kinase inhibitor in clinical development.
It triggers apoptosis by inhibiting de novo transcription of the short-lived Mcl-1 protein,
but it is unknown how this leads to Bax/Bak activation that is required for most forms
of cell death. Here, we studied the effects of seliciclib in B cell chronic lymphocytic
leukemia (B-CLL), a malignancy with aberrant expression of apoptosis regulators.
Although Seliciclib induced Mcl-1 degradation within 4 hrs, Bax/Bak activation
occurred between 16 and 20 hrs. During this period, no transcriptional changes in
apoptosis-related genes occurred. In untreated cells, pro-survival Mcl-1 was engaged
by the pro-apoptotic proteins Noxa and Bim. Upon drug treatment, Bim was quickly
released. The contribution of Noxa and Bim as a specifi c mediator of seliciclib-
induced apoptosis was demonstrated via RNAi. Signifi cantly, 16 hrs after seliciclib
treatment, there was accumulation of Bcl-2, Bim, and Bax in the ‘mitochondria-
rich’ insoluble fraction of the cell. This suggests that after Mcl-1 degradation, the
remaining apoptosis neutralising capacity of Bcl-2 is gradually overwhelmed, until
Bax forms large multimeric pores in the mitochondria. These data demonstrate in
primary leukemic cells hierarchical binding and cross-talk among Bcl-2 members,
and suggest that their functional interdependence can be exploited therapeutically.
32
Thesis_Final_2_01092008.indd Sec1:32Thesis_Final_2_01092008.indd Sec1:32 01-09-2008 11:08:0601-09-2008 11:08:06

33
Ch
ap
ter 2
▪ Selic
iclib
acts
via
the M
cl-1
/No
xa a
xis
INTRODUCTION
B cell chronic lymphocytic leukemia (B-CLL) is a heterogeneous disease, typifi ed by
the accumulation of long-lived monoclonal B cells arrested at the G0/G1 phase of the
cell cycle1. B-CLL is considered a malignancy involving deregulated apoptosis. High
levels of pro-survival Bcl-2 and Mcl-1 proteins can be detected in B-CLL, which are
associated with aggressive disease and refractoriness to chemotherapy2;3. Contrasting
with these clinical observations, CLL cells succumb easily to ‘spontaneous’ apoptosis
during in vitro culture. So it remains unclear whether overexpression of these
anti-apoptosis proteins is strictly required to block imminent apoptosis, or affords
protection during stress or drug treatment. This aspect is particularly relevant in light
of the concept of ‘oncogene addiction’, which holds that each cancer type may strictly
depend on just a few particular oncogenes, like Bcl-24.
A pro-apoptotic subgroup within the Bcl-2 family consists of so called BH3-only proteins
and includes Bid, Bim, Noxa, Puma, Bad, Bmf, Bik and Hrk. By forming heterodimeric
complexes at the mitochondria, the interplay between BH3-only proteins and pro-
survival Bcl-2-like counterparts determines the activation of pro-apoptotic Bax-like
members and subsequent cell death5. Paradoxically, Noxa and Bmf are overexpressed
in circulating B-CLL cells6 although the functional signifi cance of these observations
remains unclear. Puma, a bona-fi de p53 target gene7;8, is rapidly induced in B-CLL
cells obtained from the majority of patients upon treatment with therapeutic drugs
such as fl udarabine and chlorambucil6. Still, it has been diffi cult to demonstrate an
improvement in overall survival upon treatment with these chemotherapeutic drugs
and disease relapse is inevitable in virtually all patients1. Up to 30% of B-CLL tumors
have a defect in the p53 pathway, which is often associated with resistance to therapy.
Therefore, there is an urgent need for novel drugs that work independently of p53.
Recently, the cyclin-dependent kinase (CDK) inhibitor seliciclib (CYC202,
R-roscovitine), has emerged as a potential drug for treatment of B cell malignancies
including B-CLL9. Another CDK inhibitor, fl avopiridol has recently been tested in CLL
with encouraging results10. Seliciclib induces apoptosis, most likely by inhibiting RNA
polymerase II dependent transcription and thereby resulting in loss of the short-lived
pro-survival Mcl-1 protein11. Yet, it remains unresolved how the decrease in Mcl-1
levels is coupled to apoptosis induction. Despite the notion that Bax and Bak are
indispensable for apoptosis12, the molecular cascade that mediates their activation is
presently a matter of debate. New developments have shed light on the interaction
between members of the Bcl-2 family. Specifi cally, the pivotal pro-survival Mcl-1
interacts, amongst others, with Noxa, Bim, Puma and Bak in model cell lines13;14.
These fi ndings may be relevant in connection with our observation of elevated
Thesis_Final_2_01092008.indd Sec1:33Thesis_Final_2_01092008.indd Sec1:33 01-09-2008 11:08:0601-09-2008 11:08:06

34expression of Noxa in B-CLL6, as well as in light of the above mentioned concept of
‘oncogene addiction’.
In the present study, we investigated the molecular mechanism underlying apoptosis
elicited by seliciclib in B-CLL, with emphasis on protein interactions between Bcl-2
family members. We show for the fi rst time that Mcl-1 is associated with Noxa in
primary leukemic cells and that Noxa is required to trigger the mitochondrial apoptosis
pathway upon seliciclib treatment. Furthermore, despite rapid proteasome-dependent
degradation of Mcl-1 (within 4 hrs), Bax/Bak activation and apoptosis are substantially
delayed (16-20 hrs). During this time Bim and Bax become associated with Bcl-2 and
these proteins eventually translocate to a mitochondria-rich insoluble fraction. Our
data suggest that in absence of Mcl-1, the remaining apoptosis neutralizing capacity
of Bcl-2 is gradually overwhelmed, resulting in Bax/Bak multimerisation and pore
formation in the mitochondria.
MATERIALS AND METHODS
B-CLL patients and isolation of leukemic B cells
Peripheral blood samples were obtained from B-CLL patients from the outpatient
clinic of the department of Hematology of the Academic Medical Center, Amsterdam;
the department of Internal Medicine, Meander Medical Center, Amersfoort and the
department of Internal Medicine, The Netherlands Cancer Institute, Amsterdam.
Informed consent was obtained according to the guidelines of the local Ethical
Review Board. Clinical characteristics of patients are presented in Table 1. This
study was conducted in accordance with the ethical standards in our institutional
medical committee on human experimentation, as well as in agreement with the
Helsinki Declaration of 1975, revised in 1983. Peripheral blood mononuclear cells
(PBMC), obtained via density-gradient centrifugation were frozen in 15% fetal calf
serum (FCS) containing 10% dimethyl sulphoxide (DMSO; Sigma Chemical Co., St
Louis, MO, USA) and stored in liquid nitrogen. Expression of CD5, CD19 and CD23
(all Becton Dickinson (BD) Biosciences, San Jose, CA, USA) on leukemic cells was
assessed by fl ow cytometry (FACScalibur, BD Biosciences) and CellQuest software
(BD Biosciences)6.
Thesis_Final_2_01092008.indd Sec1:34Thesis_Final_2_01092008.indd Sec1:34 01-09-2008 11:08:0601-09-2008 11:08:06

Ch
ap
ter 2
▪ Selic
iclib
acts
via
the M
cl-1
/No
xa a
xis
35Cell lines, retroviral constructs and transduction
Cell lines were cultured in Iscove’s modifi ed Dulbecco’s medium (IMDM; Gibco Life
Technologies, Paisley, Scotland), supplemented with 10% (v/v) heat-inactivated
fetal calf serum (FCS) (ICN Biomedicals GmbH, Meckenhein, Germany), 5 mg/l
gentamycin and 5 mM L-glutamine (Gibco). The Burkitt lymphoma Ramos FSA clone
with enhanced response to CD95, the caspase-9-DN (C9DN) overexpressing Ramos
FSA cell line, Ramos 2G6 clone and the Bcl-2 overexpressing Ramos 2G6 cell line
have been described previously15;16. The B-CLL cell line Mec-117 was a kind gift of
Dr. Caligaris-Cappio (Milano, Italy). Mock and two Noxa siRNA sequences (N7 or
N8) were retrovirally transduced into the Ramos FSA, Mec-1 and J16 cell lines as
described previously18. Furthermore, siRNA targeting Bim (pSuper-Bim) was kindly
provided by Andreas Villunger (Biocenter, Innsbrück, Austria) and subcloned in the
EcoRI/Xho sites of pRetroSuper. After retroviral transduction of J16 cell line, the
B18 and B22 subclones were obtained after limiting dilution. The pcDNA3.1 vector
encoding Myc-hMcl-1 was obtained from Professor J. Borst (Netherlands Cancer
Institute). After digestion with BamH1 and Xba the product was inserted into the
BamH1 and SnaB1 blunt ended sites of the LZRS-IRES-GFP vector, resulting in
LZRS-Myc-hMcl-1-IRES-GFP.
Reagents
Seliciclib was obtained from Cyclacel Ltd (Dundee, UK). Fludarabine, staurosporine,
etoposide, carbonyl cyanide m-chlorophenylhydrazone (CCCP), Taxol and propidium
iodide (PI) were purchased from Sigma Chemical Co. Anti-human Fas10 (agonistic
antibody to the CD95 receptor) and anti-human IgM mAbs (CLB/MH15) were a kind
gift from Prof. Dr. L. Aarden (Sanquin, Amsterdam, The Netherlands). The pan-
caspase inhibitor zVAD-fmk was obtained from Alexis Biochemicals (Läuferlingen,
Switzerland). APC-labeled Annexin V was from IQ Products (Groningen, The
Netherlands) and MitoTracker Orange was from Molecular Probes (Leiden, The
Netherlands). Proteasome inhibitors MG132 and bortezomib, were obtained from
Calbiochem, (Amsterdam, The Netherlands) and Janssen-Cilag, (Tilburg, The
Netherlands), respectively.
Analysis of apoptosis
For cell stimulations, B-CLL cells (>90% CD19+CD5+ PBMC suspensions) were used
at a concentration of 1.7×106 cells/ml in 24-well plate, Ramos and Mec-1 cell lines
at a concentration of 5×105 cells/ml. Cells were harvested and washed in ice-cold
HEPES buffer (10 mM HEPES, 150 mM KCl, 1 mM MgCl2 and 1,3 mM CaCl
2, pH 7.4),
Thesis_Final_2_01092008.indd Sec1:35Thesis_Final_2_01092008.indd Sec1:35 01-09-2008 11:08:0601-09-2008 11:08:06

36and incubated with APC-labeled Annexin-V (BD Biosciences) for 20 minutes. Prior to
analyses, PI was added (fi nal concentration 5 µg/ml). Viable cells were defi ned by
Annexin V-/PI- staining. Alternatively, cells were incubated with 200 mM MitoTracker
for 30 mins at 37˚C. To assess activation of Bax and Bak, B-CLL cells (1×106) were
washed in PBS prior to fi xation in 4% formaldehyde for 5 min at room temperature.
Cells were washed in PBS and incubated in the presence of 1 µg/ml of anti-Bax (Ab-
6, clone 6A7, Calbiochem) or anti-Bak (Ab-1, Calbiochem), diluted in permeabilisation
buffer (0.5% BSA + 500 µg/ml digitonin in PBS) for 30 min at room temperature. Cells
were washed in PBA (PBS + 0.2% BSA + 0.02% NaN3) and incubated with goat
anti-mouse PE conjugated secondary antibody (Jackson ImmunoResearch Europe
Ltd., Suffolk, England), diluted 1/200 in permeabilization buffer for 30 min at room
temperature. Cells were washed with PBA, resuspended in PBS and analysed using
the FACS.
Western blotting and immuno-precipitation
Cell lysates for Western blotting were separated by 13% sodium dodecyl sulphate
polyacrylamide gel electrophoresis (SDS-PAGE) followed by western blotting as
described previously16. Blots were probed with the following antisera: monoclonal
anti-Mcl-1 (BD Pharmingen), monoclonal anti-Bax (BD Pharmingen), Bak monoclonal
antibody (mAb) (Upstate Biotechnology, Inc. Lake Placid, NY, USA), monoclonal anti-
Bim (Fig. 2 and Fig. 5A; Chemicon, Temecula, CA, USA), polyclonal anti-Bim (Fig.
3Aiii and Fig. 5B; Nventa, San Diego, CA, USA) monoclonal anti-Noxa from Imgenex
(San Diego, CA, USA), polyclonal anti-Bcl-2 (Alexis Biochemicals), polyclonal cleaved
caspase 3 (Asp175) antibody, polyclonal anti-p44/42-MAP Kinase (Cell signaling
Technology Inc., Danvers, MA, USA) antibody and antiserum to β-actin was from
Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA).
For immuno-precipitation (IP) of Mcl-1, 50×106 cells were washed twice in ice-
cold PBS (supplemented with 10 mM Na3VO
4) and lysed in 500 µL ice-cold Triton
X-100 lysis buffer (1% Triton-X100, 20 mM Tris-Cl (pH=7.4), 135 mM NaCl, 1.5 mM
MgCl2,10% Glycerol, 2 µg/ml Leupeptin, 1 mM PMSF and 2 µg/ml Trypsin inhibitor).
Whole cell lysates were precleared with normal rabbit serum precoupled to protein
A-sepharose and incubated for 3 hr with 5 µg polyclonal anti-Mcl-1 (BD Pharmingen)
precoupled to protein A-sepharose. Beads were pelleted, washed six times with lysis
buffer and boiled in SDS sample buffer, and further processed for Western blotting.
CHAPS lysates were used in certain experiments to avoid detergent-induced
conformational changes of Bax19, by lysing 50×106 cells (for IP) or 15×106 cells (for
Western Blot) for 30 min on ice in lysis buffer (1% Chaps, 20 mM Tris-Cl (pH=7.4),
Thesis_Final_2_01092008.indd Sec1:36Thesis_Final_2_01092008.indd Sec1:36 01-09-2008 11:08:0601-09-2008 11:08:06

Ch
ap
ter 2
▪ Selic
iclib
acts
via
the M
cl-1
/No
xa a
xis
37135 mM NaCl, 1.5 mM MgCl
2,10% Glycerol, 1 mM EGTA, 2 mM Na
3VO
4, 10 mM
NaF, 2 µg/ml Leupeptin, 1 mM PMSF, 0.1 mM TLCK and 2 µg/ml Trypsin inhibitor).
After centrifugation (15 min; 13.000 × g; 4°C), the supernatant was used for IP or as
‘CHAPS soluble fraction’ on Western Blot. Pellets were solubilized in SDS sample
buffer and used in Western Blotting as the ‘CHAPS insoluble fraction’. Immuno-
precipitation of Bim (monoclonal anti-Bim (Chemicon)) was performed as described
above in 500 µL ice-cold CHAPS and lysates were precleared with normal rat serum
precoupled to protein G-sepharose.
Immunocytochemistry and confocal analysis
B-CLL cells were incubated with 200 nM MitoTracker for 24 hrs at 37˚C and treated
as described above (intracellular fl ow cytometry procedure). Cells (1 × 106) were
resuspended in 25 µl vectashield (Hardset with DAPI; Vector Laboratories, CA, USA)
and were spotted on cover slips. Images were independently captured by confocal
laser microscopy (model TCS 4D, Leica, Heidelberg, Germany) at the wavelengths
488 and 594 nm lines of the krypton/argon laser used for the excitation of FITC and
MitoTracker (Alexa594), respectively. Excitation of DAPI was by 594 nm.
Statistical analyses
The student’s t-test was used for analysis of differences between 2 groups. P-values
< 0.01 were considered statistically signifi cant.
RESULTS
Seliciclib induces apoptosis and rapid proteasome-dependent
degradation of Mcl-1
The effect of seliciclib on cell viability was investigated in 20 B-CLL samples. B-CLL
cells were >90% pure as determined by fl ow cytometry. Consistent with previous
reports20, in vitro culture of B-CLL cells was accompanied by moderate spontaneous
increase of apoptosis. Increasing doses (10, 25 and 50 µM) of seliciclib resulted in
dose-dependent increase of apoptosis (Fig. 1A). In comparison, cells treated with
high dose of fl udarabine (100 µM) showed a lower level of apoptosis at 24 hrs of
treatment (Fig. 1A), but this increased over time (data not shown). IgVH mutated
and unmutated B-CLL patients (n=8 vs. n=12) showed no difference in apoptosis
induction by seliciclib (data not shown).
Seliciclib was reported to result in rapid proteasomal breakdown of Mcl-1 protein11.
Thesis_Final_2_01092008.indd Sec1:37Thesis_Final_2_01092008.indd Sec1:37 01-09-2008 11:08:0601-09-2008 11:08:06

38
Mcl-1 is also described as a substrate for caspase-mediated degradation21. We
tested which of these pathways predominates in B-CLL. Seliciclib was applied in
combination with zVAD-fmk and the proteasome inhibitors MG132 and bortezomib.
As shown in Figure 1B, the degradation of Mcl-1 in B-CLL cells was at best partially
affected by zVAD-fmk (lane 2 vs. 3). In contrast, the proteasome inhibitors MG132
and bortezomib almost fully prevented Mcl-1 degradation (lane 2 vs. 4 and 5). Thus,
in B-CLL cells Mcl-1 is largely degraded already 4 hrs after addition of seliciclib,
predominantly via the proteasome.
In B-CLL, Mcl-1 associates with Noxa and Bim but not with Bak
Prior studies in model cell lines indicate that Mcl-1 selectively interacts with Bim,
Bak, Puma and Noxa, whereas Noxa has a strong preference for Mcl-1 and possibly
Bfl -113;14;22. In primary B-CLL cells the majority of Noxa protein was associated with
Mcl-1 (Fig. 2, compare IP with cleared lysate lanes). As also reported for normal tonsil
B cells23, Bim was clearly but not exclusively associated with Mcl-1. Cells were then
analysed after 2 hrs of Seliciclib treatment, i.e. before extensive degradation of Mcl-1
and zVAD-fmk or MG132 were added to block caspases or the proteasome. Rapid
dissociation of Bim from Mcl-1 could be observed, while Noxa was still largely bound
(Fig. 2, left panels).
Previous studies in HeLa cells indicated that Mcl-1 directly sequesters Noxa, Bim and
Bak, and upon UV treatment, Bak is displaced by Noxa24 . In B-CLL cells, Mcl-1 was
1 2 3 4 5 6 7 WB:
Mcl-1
β-actin
- + + + + - - Seliciclib (25 μM)
- - zVAD MG132 bort MG132 bort Inhibitors
48 kD —
37 kD —
B-CLL24
Figure 1. Apoptosis and Mcl-1 degradation in B-CLL upon seliciclib treatment.
(A) B-CLL cells (n=20) were treated with seliciclib (0, 10, 25 or 50µM) or fl udarabine (100 µM). Apoptosis
was determined via annexin-VAPC/PI staining at 0 and 24 hrs.
(B) B-CLL cells were untreated (lanes 1, 6 and 7) or treated with 25 µM seliciclib (lanes 2-5) for 4 hrs.
Where indicated, cells were pre-treated for 1 hr with 100 µM zVAD-fmk, 10 µM MG132 or 2 µM bortezomib.
Samples containing 30 µg of protein were analyzed by Western blotting for the presence of Mcl-1 (top panel)
and β-actin (bottom panel) as loading control. Experiments were repeated twice with similar results.
0 0 10 25 50 100
0
25
50
75
100
sel [mM] fluda [mM]
24 hrs0 hrs
% A
po
pto
sis
A B
Thesis_Final_2_01092008.indd Sec1:38Thesis_Final_2_01092008.indd Sec1:38 01-09-2008 11:08:0601-09-2008 11:08:06

Ch
ap
ter 2
▪ Selic
iclib
acts
via
the M
cl-1
/No
xa a
xis
39associated with Noxa and Bim, but we consistently could not detect association of
Mcl-1 with Bak, irrespective of seliciclib treatment, although Bak was present in CLL
lysates (Fig. 2, right panel). As expected, proteasome inhibition by MG132 resulted
in increased Mcl-1 protein, but also increased Noxa levels (Fig. 2, right panels). Bax
protein levels were unaffected by either seliciclib or MG132 treatment. In summary,
in primary B-CLL cells undergoing seliclib treatment Noxa remained bound to Mcl-1
while Bim dissociated.
Figure 2. In seliciclib treated B-CLL cells, Mcl-1 engages Noxa and Bim but not Bak.
B-CLL cells were treated with 25 μM seliciclib for 2 hrs. The pan-caspase inhibitors zVAD-fmk (100
μM) and proteasome inhibitor MG132 (10 μM) were added as indicated. Cell lysates were subjected to
immunoprecipitation (IP) assays using control Ab (CAb) or anti-Mcl-1 antibody. Bound (IP) and unbound
fractions (cleared lysate = 5% supernatant of lysate after IP) as well as total cell fractions (5% of total lysate)
were analysed by Western blotting to evaluate protein associations with Mcl-1. The blotting membrane was
rehybridized with anti-Bim, anti-Bak, anti-Bax and anti- -actin antibodies as indicated. In the case of Bim,
the data derive from a separate experiment. Experiments were repeated three times with similar results.
Noxa defi cient cells exhibit resistance to seliciclib-induced apoptosis
To test whether the association between Noxa and Mcl-1 was refl ected at a functional
level, the endogenous levels of Noxa and Mcl-1 were manipulated in model systems
via RNAi and overexpression. A signifi cant reduction of Noxa in Ramos FSA cells was
achieved with two different siRNA sequences (N7 and N8; Fig. 3Ai). Functionally, Noxa
RNAi conferred a signifi cant resistance to apoptosis induced by seliciclib compared
to the mock transduced cells (Fig. 3B), which correlated with the knock-down in Noxa
protein. These data were confi rmed in the Mec-1 cell line (Fig. 3C), originally derived
from a B-CLL patient, and the Jurkat T cell line18 (Fig. 3D). In contrast, upon several
other death stimuli like fl udarabine, staurosporine, CD95 and BCR stimulation,
no signifi cant effect of reduced Noxa levels was observed (Fig. 3B). Conversely,
Thesis_Final_2_01092008.indd Sec1:39Thesis_Final_2_01092008.indd Sec1:39 01-09-2008 11:08:0701-09-2008 11:08:07

40increased Mcl-1 levels (Fig. 3Aii) conferred resistance to seliciclib, but also to several
other apoptotic stimuli (Fig 3E). In agreement with the Mcl-1 association studies in
B-CLL presented in fi gure 2, knock-down of Bim also resulted in increased resistance
against seliciclib. As might be expected from its broader interaction range compared
to Noxa, Bim knock-down conferred resistance against diverse apoptotic stimuli,
among which taxol, in agreement with earlier studies25. In conclusion, these data
indicate that Noxa functions specifi cally in case of Mcl-1 degradation, such as after
seliciclib treatment.
Ai
Aii
Aiii
Figure 3. Noxa RNAi selectively affords resistance
to seliciclib, compared to Bim RNAi and Mcl-1
overexpression.
Noxa knock down was obtained using two different
Noxa RNAi constructs (N7 and N8) in Ramos and
Mec-1 cells. Ramos FSA was transduced with LZRS-
Mcl-1-IRES-GFP vector to obtain overexpression of
Mcl-1. Bim knock down in Jurkat T cells was achieved
with a Bim RNAi contstruct, yielding two independent
clones B18 and B22. Transduction with empty vectors
(M) served as control. Total cell lysates derived from
GFP+ cells were separated by SDS-PAGE.
(A) Noxa, exogenous Mcl-1 and Bim protein levels
were monitored by Western blotting in Ramos FSA,
Mec-1 and Jurkat T cells (J16). Mcl-1 was c-myc
tagged, which could be characterized with an anti-
c-myc antibody. -actin and a nonspecifi c band (*)
served as a loading control.
(B) Ramos FSA (M) (black), N7 (gray) and N8 (white)
were cultured 24 hrs in the absence or presence of
25 μM seliciclib, 100 μM fl udarabine (fl uda), 0.25 μM
staurosporine (stauro), 5 μg/ml α-CD95 or α-BCR
antibodies.
(C) Mec-1 (M) (black) and N8 (gray) were cultured 24
hrs in the absence or presence of 50 μM seliciclib,
0.25 μM staurosporine (stauro) and 5 μg/ml α-CD95.
Aiii
B C
Thesis_Final_2_01092008.indd Sec1:40Thesis_Final_2_01092008.indd Sec1:40 01-09-2008 11:08:0801-09-2008 11:08:08

D E41
Ch
ap
ter 2
▪ Selic
iclib
acts
via
the M
cl-1
/No
xa a
xis
F
(D) J16 (M) (black), N7 (gray) and N8 (white) were cultured as described above with 25 μM seliciclib, 0.25
μM staurosporine (stauro) and 5 μg/ml α-CD95.
(E) Ramos FSA (M) (black) and Mcl-1 (gray) were cultured in the absence or presence of 25 μM seliciclib,
100 μM fl udarabine (fl uda), 0.25 μM staurosporine (stauro), 5 μg/ml α-CD95 or α-BCR antibodies.
(F) J16 (M) (black), J16-B18 (gray) and J16-B22 (white) were cultured 24 hrs in de absence or presence of
25 μM seliciclib, 5 μg/ml α-CD95, 3.1 μM etoposide, 25 μM CCCP, or 5 nM Taxol. Apoptosis was assessed
by fl ow cytometry analysis. Data represent mean ± SD from three or more independent experiments,
except in E; the CD95 and BCR triggering was done twice and no statistics were performed. P values
< 0.01 with Student’s T–test are indicated by an asterisk (*). In case of Bim knock-down (F) , only those
stimuli where both clones yielded P values <0.01 are marked.
Delay in Bax/Bak activation upon seliciclib treatment
Most forms of apoptosis ultimately require activation of Bax and/or Bak, which can
be monitored with conformation-specifi c antibodies26. Surprisingly, we observed that
at 4 hrs after seliciclib treatment, when Mcl-1 degradation was almost complete as
shown above, activation of Bax/Bak was still minimal (see Fig. 4,upper panel). At 12
hrs, Bax/Bak activation was still modest (middle panel). Only after 20 hrs substantial
conformational changes of Bax/Bak occurred. At all timepoints the mitochondrial
membrane potential as measured via MitoTracker staining mirrored the pattern of
Thesis_Final_2_01092008.indd Sec1:41Thesis_Final_2_01092008.indd Sec1:41 01-09-2008 11:08:0801-09-2008 11:08:08

Bax/Bak activation, and substantial damage occurred only at 20 hrs. These data
were obtained in various B-CLL samples tested (n=3) and demonstrate that there
was a substantial delay between Mcl-1 degradation and the actual onset of apoptosis.
During this period, there were no signifi cant changes in apoptosis-regulatory genes,
such as p53-responsive Puma, as detected by RT-MLPA6 (data not shown).
42
Figure 4. Seliciclib induced cell
death in B-CLL involves loss of the
mitochondrial membrane potential
and Bax/Bak conformational change.
B-CLL cells were incubated in the absence
(control, gray peak) or presence of 25 μM
seliciclib and monitored every 4 hrs up to
24 hrs. Only 4, 12 and 20 hr time points
are shown. Bax/Bak conformational
changes were determined by intracellular
fl ow cytometry using anti-Bax and anti-
Bak monoclonal antibodies (solid black
lines). For analysis of the mitochondrial
membrane potential (∆Ψm), cells were
incubated with MitoTracker Orange for
30 minutes.
Translocation of Bim, Bax and Bcl-2 to the mitochondria-rich
insoluble fraction subsequent to Mcl-1 degradation
In order to elucidate events occurring between Mcl-1 degradation and the onset of
apoptosis, we monitored candidate proteins to be potentially involved in this process.
This was done by co-immunoprecipitation of CHAPS-soluble lysates taken 0, 4
and 16 hrs after seliciclib treatment. The data shown in fi gure 2 suggested early
dissociation of Bim from Mcl-1. Since Bim can also bind Bcl-2 which is in general
highly expressed in B-CLL, we studied Bim associations over time. Bim was bound
to Bcl-2 at 0 hrs, and this association declined following prolonged seliciclib treatment
(Fig. 5A, left panel). These fi ndings were confi rmed in the reverse experiment by
immunoprecipitation of Bcl-2 (data not shown). Signifi cantly, at 16 hrs of treatment,
Bim, Bcl-2 and Bax were detected in the insoluble fraction of the cell (right panel).
Moreover, we could not detect association between Bim and Bax or Bak (the latter
is generally expressed at low levels in B-CLL cells) in viable nor in apoptotic cells.
In accordance with published fi ndings13;14, we could not detect association between
Bcl-2 and Noxa. The transition of Bim, Bax and Bcl-2 to the CHAPS-insoluble cell
Thesis_Final_2_01092008.indd Sec1:42Thesis_Final_2_01092008.indd Sec1:42 01-09-2008 11:08:0901-09-2008 11:08:09

Ch
ap
ter 2
▪ Selic
iclib
acts
via
the M
cl-1
/No
xa a
xis
43fraction was confi rmed by Western blotting of 4 additional B-CLL samples taken after
0, 4 and 16 hrs of seliciclib treatment (Fig. 5B). Together, these data demonstrate that
Bim, Bcl-2 and Bax associations change during drug treatment, and this correlates
with transition to an insoluble state. The mitochondrial membrane protein porin/
VDAC2 was predominantly expressed in the ‘mitochondia rich’ insoluble fraction.
Presumably, after release from Mcl-1 and subsequently Bcl-2, Bim is responsible for
activation of Bax, although this is not detectable via direct interaction in the soluble
cell fraction.
As Bax is translocated to the membrane fraction during seliciclib-induced apoptosis,
we assessed its subcellular distribution using confocal microscopy. Figure 5C shows
that Bax is localized throughout the cytoplasm in untreated B-CLL cells. Following 16
hrs seliciclib treatment (+ pretreatment with zVAD), Bax showed a punctate distribution
and co-localization with the mitochondria, suggesting that Bax multimerizes at the
mitochondria.
A
B
Figure 5. Bim associates with Bcl-2 and Mcl-1
and shifts to the mitochondria-rich insoluble
fraction over time in B-CLL cells.
See color fi gures.
B-CLL cells were treated with 25 μM seliciclib for
0, 4 and 16 hrs.
(A) Cell lysates were subjected to immunopre-
cipitation (IP) assays using a Bim antibody. The
bound (IP) fraction (left panel) was analysed by
Western blotting to evaluate the association of
Bcl-2, Mcl-1, Bak and Bax with Bim over time. To-
tal cell fractions (5% of total lysate; middle panel)
and insoluble fractions (right panel) were moni-
tored by Western blotting. The blotting membrane
was hybridized with Bcl-2, Mcl-1, Bak, Bax, Bim,
-actin and Porin antibodies as indicated. (* Im-
munoglobulin heavy chain; ** Immunoglobulin
light chain)
(B) CHAPS- soluble and -insoluble fractions from
4 other B-CLL patients were analysed by Western
blotting to evaluate the localization of Bim, Bax
and Bcl-2 over time.
(C) Immunocytochemical staining for Bax of B-CLL
cells incubated in the absence or presence of 25
μM seliciclib for 24 hrs. MitoTracker Orange was
used for localization of the mitochondria.
Thesis_Final_2_01092008.indd Sec1:43Thesis_Final_2_01092008.indd Sec1:43 01-09-2008 11:08:1001-09-2008 11:08:10

C
Bcl-2 confers a temporary block to selicilib-induced apoptosis
To directly determine the role of Bcl-2 in the delayed onset of seliciclib-induced
apoptosis, we used a Ramos model cell line overexpressing Bcl-216. In parental
Ramos cells treated with seliciclib, Mcl-1 degradation (Fig. 6A; upper panel) occurred
within 4 hrs, similar to B-CLL cells (Fig. 1B). The decline in Mcl-1 levels occurred
independently of caspase activation, as demonstrated by overexpression of C9DN.
In contrast to B-CLL, Mcl-1 degradation was almost simultaneous with apoptosis
induction, shown by cleavage of effector caspase 3 (Fig. 6A; panel 3), and annexin-V
exposure (Fig. 6B). Overexpression of Bcl-2 in Ramos cell lines recapitulated
the delayed onset of apoptosis after seliciclib treatment observed in B-CLL cells.
Clearly, apoptosis was blocked after 4 hrs of treatment, whereas at 24 hrs virtually
all cells were apoptotic, despite the presence of Bcl-2 (Fig. 6B). In comparison, time
course analysis of Ramos cells with Noxa knock-down or Mcl-1 overexpression also
showed that the block in seliciclib induced apoptosis was not absolute. Here, the
fraction of apoptotic cells increased more gradually over time up to the last point
measured (48 hrs; Fig. 6C and 6D). The effect of seliciclib on Mcl-1 produced from
a recombinant vector may be less than on the endogenous transcript. However, the
recently described phosphorylation of Mcl-1 at Ser64 which enhances its apoptotic
activity is apparently also prevented by seliciclib27, therefore a complete block of
seliciclib-induced apoptosis of overexpressed Mcl-1 is not expected. Together, our
data indicate that manipulation of individual Bcl-2 family members is insuffi cient to
warrant full protection to seliciclib, which suggest they are subject to an intricate
functional interplay.
44
Thesis_Final_2_01092008.indd Sec1:44Thesis_Final_2_01092008.indd Sec1:44 01-09-2008 11:08:1001-09-2008 11:08:10

Ch
ap
ter 2
▪ Selic
iclib
acts
via
the M
cl-1
/No
xa a
xis
45A B
DC
Figure 6. Bcl-2 overexpression in Ramos cell line recapitulates delayed onset of apoptosis.
(A) Ramos 2G6 and Ramos FSA C9DN (DN9c) cells were treated with 25 μM seliciclib. At the indicated
time points samples were analyzed by Western blotting for the presence of Mcl-1 (top panel), Noxa (sec-
ond panel), activated caspase-3 (third panel) and -actin (bottom panel) as loading control.
(B) Ramos 2G6 and Bcl-2 cells (5×105/ml) were left untreated or treated with 50 μM seliciclib for 4 and 24
hrs.
(C) Ramos FSA and N8 cells (5×105/ml) were left untreated or treated with 25 μM seliciclib for 4, 24 and
48 hr.
(D) Ramos FSA and Mcl-1 cells (5×105/ml) were left untreated or treated with 25 μM seliciclib for 4, 24 and
48 hr. Viability in B, C and D was assessed by means of MitoTracker staining. Data represent mean ± SD
from three independent experiments.
Thesis_Final_2_01092008.indd Sec1:45Thesis_Final_2_01092008.indd Sec1:45 01-09-2008 11:08:1001-09-2008 11:08:10

DISCUSSION
In hematological cells, the primary apoptotic trigger ascribed to the novel cyclin-
dependent kinase inhibitor seliciclib is a rapid decrease in Mcl-1 levels independent
of p53 function9;11. This is of clinical importance in view of the poor prognosis of
B-CLL with functional defi ciency in the p53 pathway28 and the outgrowth of p53-
defi cient clones during treatment1. Furthermore, there is broad interest in the exact
mechanism of action of the various Bcl-2 family members, as it has become clear
that their protective function can be targeted also directly by pharmacological
compounds29. However, in the absence of a functional link to Bax and/or Bak
activation, drug-induced Mcl-1 decrease is in itself not suffi cient to explain cell death.
Our fi ndings emphasize two new aspects in the chain of events triggered in B-CLL
cells upon seliciclib treatment and Mcl-1 degradation. We found previously that the
BH3-only protein Noxa is expressed at high levels in B-CLL6, and here we establish
that the molecular effects of seliciclib rely on Noxa. Secondly, subsequent to Mcl-1
degradation, we observed a gradual deterioration of the protection afforded by Bcl-2,
which was most probably caused by liberated Bim.
Biochemical studies have shown that distinct BH3-only proteins have differential
binding affi nities to pro-survival Bcl-2 proteins13;14;30. Reportedly, Noxa has a strong
preference for binding to Mcl-1 which we confi rm here in primary B-CLL cells (Fig.
2). At present, the precise role of Noxa is not entirely understood, and our data point
to a new and specifi c role in drug-induced cell death. This was demonstrated by a
resistance to seliciclib in Noxa knock-down leukemic cell lines (Fig. 3). Difference in
Noxa expression did not affect apoptosis by other stimuli (fl udarabine, staurosporine,
CD95 and BCR). Together, these observations imply that the lethal effect of seliciclib
in B-CLL cells specifi cally involves Noxa function. In agreement with their broader
interaction range, Mcl-1 overexpression or Bim knock-down had an inhibitory effect
on multiple apoptotic stimuli.
Although Noxa levels are 5-10 times higher in B-CLL than in normal B cells6, this is
apparently in itself not a direct trigger for apoptosis, which fi ts well with the current
view of its function as an indirect apoptosis mediator13;14. The available evidence
indicates that Noxa acts by displacing pro-apoptotic Bcl-2 members from Mcl-124;30.
We have studied potential involvement of two pro-apoptotic candidates Bak and Bim
in seliciclib-mediated apoptosis in B-CLL. In agreement with studies in HeLa cells24,
we found that Noxa can liberate Bak from Mcl-1 in leukemic cell lines (data not shown).
However, we consistently could not detect Mcl-1 associated with Bak in primary B-CLL
cells. Instead, we observed that the level of Bim associated with Mcl-1 rapidly declined
upon seliciclib treatment, while Noxa remained bound (Fig. 2). This is consistent with
46
Thesis_Final_2_01092008.indd Sec1:46Thesis_Final_2_01092008.indd Sec1:46 01-09-2008 11:08:1101-09-2008 11:08:11

Ch
ap
ter 2
▪ Selic
iclib
acts
via
the M
cl-1
/No
xa a
xis
47the differential binding affi nities among the BH3-only members reported by others13;22.
Once liberated from Mcl-1, Bim is reportedly capable of directly activating Bax and/
or Bak14;30. Direct association between Bax or Bak with purported direct BH3-only
activators such as Bid and Bim in model systems has been described31;32. Yet, direct
association of endogenous proteins under physiological conditions is diffi cult to
observe. We were also unable in various co-immunoprecipitation approaches to fi nd
Bim complexed with Bax or Bak in early or late apoptotic B-CLL cells. The aspect of
direct vs. indirect activation of Bax (or Bak) by BH3-only members such as Bim is
currently the subject of intense research and debate19;30;33. Although it is in fact still
unknown what actually causes transition of cytosolic Bax34, its activation is known to
result in multimerization and pore formation in mitochondria35. Therefore, we looked
for shifts from soluble to insoluble fraction upon apoptosis initiation, and could observe
transition of both Bim and Bax to the mitochondria-rich insoluble cell fraction (Fig.
5). Our fi ndings in primary B-CLL cells suggest that any direct interaction between
Bim and Bax is presumably short-lived, and/or induces rapid multimerization and
insertion of Bax in the mitochondrial membrane. At this point the interacting proteins
become insoluble and can no longer be detected by standard co-immunoprecipitation
approaches. This possible sequence of events upon seliciclib treatment of B-CLL
cells is schematically depicted in fi gure 7.
The proposed sequence of events from Mcl-1 degradation, Noxa-dependent Bim
displacement and liberation, and fi nally Bax activation is compatible with recent data
and interpretations concerning the hierarchical involvement of BH3-only members in
initiation of the mitochondrial apoptosis pathway13;30. Moreover, a functional linkage
between Noxa and Bim was very recently reported in cell lines36. During the period
between Mcl-1 degradation and eventual Bax/Bak activation in B-CLL cells, there
were no signifi cant changes in gene transcription of Bcl-2 family members (data not
shown). The available data suggest that after Mcl-1 degradation, Bcl-2 is temporarily
capable of withstanding the pro-apoptotic action of Bim and Bax, but is progressively
overwhelmed. In support of this, overexpression of Bcl-2 in a model system could
indeed delay but not prevent apoptosis induced by seliciclib treatment (Fig. 6). The
gradual undermining of Bcl-2 function could either be a direct effect of seliciclib by
an as yet unknown pathway, or indirectly result from shifts in the distribution of and
interactions between Bcl-2 family members.
The fi ndings reported here agree with and complement recent fi ndings concerning
the mechanism of action of the BH3-mimetic ABT-73737;38. This compound can
counteract the protective function of Bcl-2, Bcl-XL and Bcl-w and has potential as a
cancer therapeutic29. Cells overexpressing Mcl-1 are however resistant to ABT-737,
and strategies to target Mcl-1 levels enhance its effi cacy37;38. Based on our fi ndings
Thesis_Final_2_01092008.indd Sec1:47Thesis_Final_2_01092008.indd Sec1:47 01-09-2008 11:08:1101-09-2008 11:08:11

48with B-CLL cells reported here and elsewhere39, both direct targeting of Mcl-1 by drugs
such as seliciclib as well as indirect targeting by means of bortezomib to increase Noxa
levels, may be a promising combination with ABT-737 or similar compounds. Indeed,
it has been shown very recently that seliciclib treatment dramatically increased ABT-
737 lethality in human leukemia cell lines40. Overall, the functional cross-talk between
Bcl-2 family members suggest that the ‘addiction’ of leukemic cells to overexpression
of anti-apoptosis proteins such as Bcl-2 and Mcl-1 could be specifi cally targeted and
exploited in a therapeutic setting.
Figure 7. Model for the steps in apoptosis triggered by seliciclib in B-CLL cells.
In the initial phase (< 4hrs) Mcl-1 is degraded rapidly in a proteasome dependent manner. As Mcl-1 levels
fall, Bim is released while Noxa remains bound. Liberated Bim can be absorbed by Bcl-2 for a while, but
gradually there is a transition to a situation where Bcl-2 is no longer capable of binding excess Bim. What
is actually causing this transition is unknown. Both disrupting the balance between Bcl-2 family members
or unknown signaling events might eventually lead to Bcl-2 dysfunction. From that point on, Bim is able to
activate Bax, leading to multimerization and apoptosis. As indicated in the main text, whether Bim activates
Bax directly, indirectly or via transient interactions is currently the subject of controversy. Therefore, this
scheme only emphasizes that all three direct participants (Bcl-2, Bim and Bax) shift to an insoluble fraction
of the cell, which in the case of multimeric Bax is presumably mitochondrial.
Thesis_Final_2_01092008.indd Sec1:48Thesis_Final_2_01092008.indd Sec1:48 01-09-2008 11:08:1101-09-2008 11:08:11

Ch
ap
ter 2
▪ Selic
iclib
acts
via
the M
cl-1
/No
xa a
xis
49
Of 24 patients the following characteristics were determined at time of inclusion in the study; sex (M=male,
F=female); age (in years); Rai stage: 0 (good prognosis), I-II (intermediate prognosis), and III-IV (poor
prognosis); IgVH mutation status (UM = unmutated IgV
H genes, M = mutated IgV
H genes) and treatment.
ND = not determined
Patient no. Sex Age (years) Rai (stage) lgVH
status Treatment
1 M 63 1 M ND
2 M 51 2 UM Yes
3 M 79 2 UM Yes
4 M 58 2 M No
5 M 81 0 M No
6 F 56 2 UM Yes
7 M 65 4 M No
8 M 64 ND UM Yes
9 F 81 0 M ND
10 M 75 0 M No
11 M 72 1 UM ND
12 M 56 2 UM Yes
13 F 49 1 M ND
14 M 71 1 M ND
15 M 73 2 M ND
16 M 70 2 UM ND
17 M 65 4 UM Yes
18 M 60 2 UM Yes
19 M 50 0 UM Yes
20 M 56 2 UM Yes
21 M 65 1 M Yes
22 M 65 ND M ND
23 F 71 0 M ND
24 F 75 3 M Yes
Table 1. Patient’s characteristics.
Thesis_Final_2_01092008.indd Sec1:49Thesis_Final_2_01092008.indd Sec1:49 01-09-2008 11:08:1201-09-2008 11:08:12

50 ACKNOWLEDGEMENT
We thank Bart de Goeij for technical assistance. In addition we thank the patients for
their blood donations. We also acknowledge Dr. Kramer, Dr. Wittebol and Dr. Baars
for including B-CLL patients in this study.
Thesis_Final_2_01092008.indd Sec1:50Thesis_Final_2_01092008.indd Sec1:50 01-09-2008 11:08:1201-09-2008 11:08:12

Ch
ap
ter 2
▪ Selic
iclib
acts
via
the M
cl-1
/No
xa a
xis
51REFERENCE LIST
1. Byrd JC, Stilgenbauer S, Flinn IW.
Chronic lymphocytic leukemia.
Hematology.(Am.Soc.Hematol.Educ.
Program.) 2004163-183.
2. Robertson LE, Plunkett W, McConnell
K, Keating MJ, McDonnell TJ. Bcl-2
expression in chronic lymphocytic
leukemia and its correlation with the
induction of apoptosis and clinical
outcome. Leukemia 1996;10:456-459.
3. Kitada S, Andersen J, Akar S et al.
Expression of apoptosis-regulating
proteins in chronic lymphocytic
leukemia: correlations with In vitro
and In vivo chemoresponses. Blood
1998;91:3379-3389.
4. Weinstein IB, Joe AK. Mechanisms
of disease: Oncogene addiction--a
rationale for molecular targeting in
cancer therapy. Nat.Clin.Pract.Oncol.
2006;3:448-457.
5. Strasser A. The role of BH3-only
proteins in the immune system. Nat.
Rev.Immunol. 2005;5:189-200.
6. Mackus WJ, Kater AP, Grummels A
et al. Chronic lymphocytic leukemia
cells display p53-dependent drug-
induced Puma upregulation. Leukemia
2005;19:427-434.
7. Nakano K, Vousden KH. PUMA, a
novel proapoptotic gene, is induced by
p53. Mol.Cell 2001;7:683-694.
8. Yu J, Zhang L, Hwang PM, Kinzler KW,
Vogelstein B. PUMA induces the rapid
apoptosis of colorectal cancer cells.
Mol.Cell 2001;7:673-682.
9. Hahntow IN, Schneller F, Oelsner M et
al. Cyclin-dependent kinase inhibitor
Roscovitine induces apoptosis in
chronic lymphocytic leukemia cells.
Leukemia 2004;18:747-755.
10. Byrd JC, Lin TS, Dalton JT et al.
Flavopiridol administered using a
pharmacologically derived schedule is
associated with marked clinical effi cacy
in refractory, genetically high-risk
chronic lymphocytic leukemia. Blood
2007;109:399-404.
11. MacCallum DE, Melville J, Frame S et
al. Seliciclib (CYC202, R-Roscovitine)
induces cell death in multiple myeloma
cells by inhibition of RNA polymerase
II-dependent transcription and down-
regulation of Mcl-1. Cancer Res.
2005;65:5399-5407.
12. Wei MC, Zong WX, Cheng EH et al.
Proapoptotic BAX and BAK: a requisite
gateway to mitochondrial dysfunction
and death. Science 2001;292:727-730.
13. Chen L, Willis SN, Wei A et al.
Differential targeting of prosurvival
Bcl-2 proteins by their BH3-only ligands
allows complementary apoptotic
function. Mol.Cell 2005;17:393-403.
14. Kuwana T, Bouchier-Hayes L, Chipuk
JE et al. BH3 domains of BH3-only
proteins differentially regulate Bax-
mediated mitochondrial membrane
permeabilization both directly and
indirectly. Mol.Cell 2005;17:525-535.
15. Eldering E, Mackus WJ, Derks IA et
al. Apoptosis via the B cell antigen
receptor requires Bax translocation and
involves mitochondrial depolarization,
cytochrome C release, and
caspase-9 activation. Eur.J.Immunol.
2004;34:1950-1960.
16. van der Kolk LE, Evers LM, Omene C
et al. CD20-induced B cell death can
bypass mitochondria and caspase
activation. Leukemia 2002;16:1735-
1744.
Thesis_Final_2_01092008.indd Sec1:51Thesis_Final_2_01092008.indd Sec1:51 01-09-2008 11:08:1201-09-2008 11:08:12

52 17. Stacchini A, Aragno M, Vallario A et al.
MEC1 and MEC2: two new cell lines
derived from B-chronic lymphocytic
leukaemia in prolymphocytoid
transformation. Leuk.Res.
1999;23:127-136.
18. Alves NL, Derks IA, Berk E et al. The
Noxa/Mcl-1 axis regulates susceptibility
to apoptosis under glucose limitation in
dividing T cells. Immunity. 2006;24:703-
716.
19. Willis SN, Fletcher JI, Kaufmann
T et al. Apoptosis initiated when
BH3 ligands engage multiple Bcl-2
homologs, not Bax or Bak. Science
2007;315:856-859.
20. Collins RJ, Verschuer LA, Harmon BV
et al. Spontaneous programmed death
(apoptosis) of B-chronic lymphocytic
leukaemia cells following their culture
in vitro. Br.J.Haematol. 1989;71:343-
350.
21. Herrant M, Jacquel A, Marchetti S et
al. Cleavage of Mcl-1 by caspases
impaired its ability to counteract
Bim-induced apoptosis. Oncogene
2004;23:7863-7873.
22. Certo M, Del GM, V, Nishino M et
al. Mitochondria primed by death
signals determine cellular addiction to
antiapoptotic BCL-2 family members.
Cancer Cell 2006;9:351-365.
23. Gomez-Bougie P, Bataille R, Amiot
M. Endogenous association of Bim
BH3-only protein with Mcl-1, Bcl-xL
and Bcl-2 on mitochondria in human B
cells. Eur.J.Immunol. 2005;35:971-976.
24. Willis SN, Chen L, Dewson G et al.
Proapoptotic Bak is sequestered by
Mcl-1 and Bcl-xL, but not Bcl-2, until
displaced by BH3-only proteins. Genes
Dev. 2005;19:1294-1305.
25. Sunters A, Fernandez de MS, Stahl M
et al. FoxO3a transcriptional regulation
of Bim controls apoptosis in paclitaxel-
treated breast cancer cell lines. J.Biol.
Chem. 2003;278:49795-49805.
26. Dewson G, Snowden RT, Almond JB,
Dyer MJ, Cohen GM. Conformational
change and mitochondrial translocation
of Bax accompany proteasome
inhibitor-induced apoptosis of chronic
lymphocytic leukemic cells. Oncogene
2003;22:2643-2654.
27. Kobayashi S, Lee SH, Meng XW et al.
Serine 64 phosphorylation enhances
the antiapoptotic function of Mcl-1.
J.Biol.Chem. 2007; 282: 18407-18417.
28. Lin K, Sherrington PD, Dennis M et al.
Relationship between p53 dysfunction,
CD38 expression, and IgV(H) mutation
in chronic lymphocytic leukemia. Blood
2002;100:1404-1409.
29. Oltersdorf T, Elmore SW, Shoemaker
AR et al. An inhibitor of Bcl-2 family
proteins induces regression of solid
tumours. Nature 2005;435:677-681.
30. Kim H, Rafi uddin-Shah M, Tu HC
et al. Hierarchical regulation of
mitochondrion-dependent apoptosis
by BCL-2 subfamilies. Nat.Cell Biol.
2006;8:1348-1358.
31. Harada H, Quearry B, Ruiz-Vela A,
Korsmeyer SJ. Survival factor-induced
extracellular signal-regulated kinase
phosphorylates BIM, inhibiting its
association with BAX and proapoptotic
activity. Proc.Natl.Acad.Sci.U.S.A
2004;101:15313-15317.
32. Cartron PF, Gallenne T, Bougras G et
al. The fi rst alpha helix of Bax plays
a necessary role in its ligand-induced
activation by the BH3-only proteins Bid
and PUMA. Mol.Cell 2004;16:807-818.
33. Uren RT, Dewson G, Chen L et al.
Mitochondrial permeabilization relies
on BH3 ligands engaging multiple
prosurvival Bcl-2 relatives, not Bak.
J.Cell Biol. 2007;177:277-287.
34. Wolter KG, Hsu YT, Smith CL et al.
Movement of Bax from the cytosol to
mitochondria during apoptosis. J.Cell
Biol. 1997;139:1281-1292.
Thesis_Final_2_01092008.indd Sec1:52Thesis_Final_2_01092008.indd Sec1:52 01-09-2008 11:08:1201-09-2008 11:08:12

Ch
ap
ter 2
▪ Selic
iclib
acts
via
the M
cl-1
/No
xa a
xis
53 35. Antonsson B, Conti F, Ciavatta
A et al. Inhibition of Bax channel-
forming activity by Bcl-2. Science
1997;277:370-372.
36. Han J, Goldstein LA, Hou W,
Rabinowich H. Functional linkage
between noxa and bim in mitochondrial
apoptotic events. J.Biol.Chem. 2007
282: 16223-16231.
37. Konopleva M, Contractor R, Tsao T et
al. Mechanisms of apoptosis sensitivity
and resistance to the BH3 mimetic
ABT-737 in acute myeloid leukemia.
Cancer Cell 2006;10:375-388.
38. van Delft MF, Wei AH, Mason KD
et al. The BH3 mimetic ABT-737
targets selective Bcl-2 proteins and
effi ciently induces apoptosis via Bak/
Bax if Mcl-1 is neutralized. Cancer Cell
2006;10:389-399.
39. Smit LA, Hallaert DY, Spijker R et al.
Differential Noxa/Mcl-1 balance in
peripheral versus lymph node chronic
lymphocitic leukemia cells correlates
with survival capacity. Blood 2007; 109:
1660-1668.
40. Chen S, Dai Y, Harada H, Dent P,
Grant S. Mcl-1 down-regulation
potentiates ABT-737 lethality by
cooperatively inducing Bak activation
and Bax translocation. Cancer Res.
2007;67:782-791.
Thesis_Final_2_01092008.indd Sec1:53Thesis_Final_2_01092008.indd Sec1:53 01-09-2008 11:08:1201-09-2008 11:08:12

54
Thesis_Final_2_01092008.indd Sec1:54Thesis_Final_2_01092008.indd Sec1:54 01-09-2008 11:08:1201-09-2008 11:08:12

chapter
3 -secretase inhibitor (GSI)-1 induces apoptosis in CLL cells via proteasome inhibition and Noxa upregulation
Delfi ne Y.H. Hallaert1,2, Heike Schmidlin3, Marinus H.J. van Oers1 and Eric
Eldering2
1 Dept of Hematology, AMC, Amsterdam, The Netherlands
2 Dept of Experimental Immunology, AMC, Amsterdam, The Netherlands
3 Dept of Cell Biology, AMC, Amsterdam, The Netherlands
Submitted for publication
γ
Thesis_Final_2_01092008.indd Sec1:55Thesis_Final_2_01092008.indd Sec1:55 01-09-2008 11:08:1201-09-2008 11:08:12

56 ABSTRACT
Deregulation of the Notch pathway has been suggested to contribute to the
pathogenesis of B cell chronic lymphocytic leukemia (CLL). γ-Secretase inhibitors
(GSIs) can block intracellular processing of all four different Notch receptors. We
found that GSI-1 (z-Leu-Leu-Nle-CHO) was a potent inducer of apoptosis in CLL,
but this appeared not to be due to inhibition of Notch signaling. Instead, effi cient
blocking of proteasomal activity by GSI-1 was observed, equivalent to established
proteasome inhibitors bortezomib and MG-132. In contrast with GSI-1, another
γ-secretase inhibitor GSI-9/DAPT neither affected proteasome activity nor induced
apoptosis in CLL. GSI-1-induced apoptosis was associated with a transcription-
independent accumulation of the BH3-only protein Noxa. The pivotal role of Noxa
in GSI-1 mediated apoptosis was demonstrated via RNAi in model systems. Our
data offer an explanation for certain confl icting notions on Notch signaling in B cell
malignancies, and suggest that GSI-1 or related compounds may hold promise for
therapeutic application in CLL.
Thesis_Final_2_01092008.indd Sec1:56Thesis_Final_2_01092008.indd Sec1:56 01-09-2008 11:08:1201-09-2008 11:08:12

Ch
ap
ter 3
▪ GS
I-1 a
cts
as a
pro
teaso
me in
hib
itor in
CL
L
57INTRODUCTION
B cell chronic lymphocytic leukemia (CLL) is characterized by the relentless
accumulation of monoclonal B cells that coexpress CD5, CD19 and CD23. The
malignant B cells have a low proliferative activity, but prolonged cell survival. Although
over the last few years new (immuno)chemotherapy regimens have resulted in
strongly improved quality of remissions and progression free survival, at present
there still is no curative treatment1.
A prominent feature of CLL cells is the overexpression of the transmembrane
glycoprotein CD232. It was reported that Notch signaling is involved in the up-
regulation of the CD23a isoform in CLL cells, and that this may be linked with the
aberrant apoptosis regulation in CLL3;4. The Notch gene family consists of four
evolutionarily conserved transmembrane receptors that play a fundamental role in
cell fate decisions including cell proliferation, differentiation and apoptosis. Notch
signaling is initiated by receptor-ligand interaction resulting in two successive
proteolytic cleavages of the Notch receptor by TACE (TNF-α-converting enzyme) and
the γ-secretase/presenilin complex, which liberates the cytoplasmic domain of the
Notch receptor (Notch intracellular domain; NIC). The NIC enters the nucleus leading
to transcriptional activation of downstream target genes5. Recent reports suggest
that deregulated Notch signaling is associated with various malignancies, in which
Notch may function either as an oncogene or as a tumor suppressor6. Variable mRNA
expression levels of Notch1 and Notch2 (receptors); Delta-like 1 (ligand); Deltex
(a regulator molecule) and Hairy/Enhancer of Split-1 (Hes-1, the most well-known
target gene of Notch5) were found in CLL7. In general, studies into the role of Notch
signaling in B cell maligancies have led to disparate reports, with suggestions that
Notch activity induces apoptosis8, while others have indicated that in fact inhibition of
Notch signaling induces apoptosis9.
Various studies implicate the ubiquitin-proteasome pathway in the control of Notch
activity4;10;11, and proteasome inhibitors such as bortezomib induce apoptosis.
Following the successful application of proteasome inhibitors in multiple myeloma
(MM)12;13, this topic has attracted broad interest as novel treatment strategy for
cancer. Exposure to proteasome inhibitors results among others in induction of
the pro-apoptotic BH3-only protein Noxa, which was preceded in melanoma and
myeloma cells by enhanced transcription of Noxa mRNA14. We recently showed that
Bortezomib-mediated apoptosis in CLL also involves Noxa protein accumulation15.
In the present study we investigated the possible role of Notch signaling in CLL by
examining the effect of γ-secretase inhibitors. These chemical inhibitors can block
processing of all Notch receptors16;17. Unexpectedly, we observed that in CLL GSI-1
Thesis_Final_2_01092008.indd Sec1:57Thesis_Final_2_01092008.indd Sec1:57 01-09-2008 11:08:1201-09-2008 11:08:12

58is a potent inducer of apoptosis without discernable involvement of Notch signaling.
Instead, the compound effi ciently inhibits proteasomal activity, leading to accumulation
of the pro-apoptotic BH3-only protein Noxa, which has a pivotal role in GSI-1-induced
cell death.
MATERIALS & METHODS
Isolation of leukemic, normal B and normal T lymphocytes
Peripheral blood samples were obtained from CLL patients from the outpatient clinic
of the department of Hematology of the Academic Medical Center, Amsterdam;
the department of Internal Medicine, Meander Medical Center, Amersfoort and the
department of Internal Medicine, The Netherlands Cancer Institute, Amsterdam.
Informed consent was obtained according to the guidelines of the local Ethical
Review Board. Clinical characteristics of patients are presented in Table 1. This study
was conducted in accordance with the ethical standards in our institutional medical
committee on human experimentation, as well as in agreement with the Helsinki
Declaration of 1975, revised in 1983. Peripheral blood mononuclear cells (PBMC) from
CLL patients and healthy donors, obtained via density-gradient centrifugation were
frozen in 15% fetal calf serum (FCS) containing 10% dimethyl sulphoxide (DMSO;
Sigma Chemical Co., St Louis, MO, USA) and stored in liquid nitrogen before use or
directly used for experiments. Expression of CD3, CD5, CD19 (all antibodies from
Becton Dickinson (BD) Biosciences, San Jose, CA, USA) and CD23 (clone MHM6
from DAKO, Glostrup, Denmark) on leukemic cells were assessed by fl ow cytometry
(FACScalibur, BD Biosciences) and CellQuest software (BD Biosciences)15.
Cell lines
Cell lines were cultured in Iscove’s modifi ed Dulbecco’s medium (IMDM; Gibco Life
Technologies, Paisley, Scotland), supplemented with 10% (v/v) heat-inactivated fetal
FCS (ICN Biomedicals GmbH, Meckenhein, Germany), 100 U/ml penicillin, 100 µg/
ml streptomycin and 5 mM L-glutamine (Gibco). The RPMI myeloma cell line was
kindly provided by Dr. Spaargaren (Department of Pathology, AMC, Amsterdam, The
Netherlands). The CLL cell line Mec-1 was a kind gift of Dr. Caligaris-Cappio (Milano,
Italy).
Mock and Noxa shRNA sequence (N7, N818) were retrovirally transduced into Ramos
FSA (with enhanced sensitivity to CD95)19, Mec-1 and Jurkat (J16) cell lines. To
improve knockdown of Noxa, a limiting dilution of the Ramos FSA N8 cell population
was performed according to standard procedures. Resulting clones were selected
Thesis_Final_2_01092008.indd Sec1:58Thesis_Final_2_01092008.indd Sec1:58 01-09-2008 11:08:1201-09-2008 11:08:12

59for increased resistance to bortezomib and further characterized by Western blot to
assess reduced Noxa expression. Clone N8G10 was used for experiments in fi gure
7.
Reagents and antibodies
Roscovitine, fl udarabine and propidium iodide (PI) were purchased from Sigma
Chemical Co. (St. Louis, MO, USA). The pan-caspase inhibitor zVAD-fmk was
obtained from Alexis Biochemicals (Läuferlingen, Switzerland). APC-labeled Annexin
V was from IQ Products (Groningen, The Netherlands) and MitoTracker Orange was
from Molecular Probes (Leiden, The Netherlands). Proteasome inhibitors MG-132
and bortezomib, were obtained from Calbiochem, (Amsterdam, The Netherlands) and
Janssen-Cilag, (Tilburg, The Netherlands), respectively. -secretase inhibitors, GSI-
120 (Z-LLNle-CHO – Cat.nr. 565750) and GSI-921 (DAPT (Difl uorophenacetyl-L-alanyl)-
S-phenylglycine t-Butyl Ester) - Cat.nr. 565770) were purchased from Calbiochem.
Anti-human Fas10 (agonistic antibody to the CD95 receptor) was a kind gift from
Prof. Dr. L. Aarden (Sanquin, Amsterdam, The Netherlands). Monoclonal antibody
to CD123 (IL-3Rα) conjugated to PE was purchased from Becton Dickinson. Anti-
BDCA2-APC was obtained from Miltenyi Biotec (Bergisch Gladbach, Germany).
Analysis of apoptosis & Western Blot
For the analysis of apoptosis of CLL cells (>90% CD19+CD5+ PBMC suspensions)
and PBMCs from healthy donors, 5×106 cells/ml were incubated for indicated time
points with GSIs or other drugs. Apoptosis of CD19+ CLL and normal B cells and
CD3+ T cells was measured by Annexin-V22 or MitoTracker staining as described
before23.
Western blotting was performed as described previously24. Blots were probed with
monoclonal anti-Noxa from Imgenex (San Diego, CA, USA), monoclonal anti-Bim
Chemicon, Temecula, CA, USA), monoclonal anti-Mcl-1 (BD Biosciences) and
antiserum to β-actin from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA).
Anti-ubiquitin was a kind gift from Prof. Rapoport (Department of Cell Biology,
Harvard Medical School, Boston, MA, USA). p53 functionality of CLL samples was
screened by the effect of radiation on the expression of p53 and p21 by Western Blot
analysis25.
Ch
ap
ter 3
▪ GS
I-1 a
cts
as a
pro
teaso
me in
hib
itor in
CL
L
Thesis_Final_2_01092008.indd Sec1:59Thesis_Final_2_01092008.indd Sec1:59 01-09-2008 11:08:1201-09-2008 11:08:12

60Notch-dependent differentiation assay
Isolation of CD34+ cells from postnatal thymus tissue was described previously
by Dontje et al 26. The development of pDCs was assessed by coculturing 5 ×
104 CD34+CD1a- progenitor cells with 5 × 104 OP9 cells in MEMα (Invitrogen Life
Technologies, Carlsbad, CA, USA) with 20% FCS (Hyclone, Logan, UT,USA), 5 ng/
mL IL-7 and 5 ng/mL Flt3L. Differentiation assays for pDCs were analyzed after 1
week of coculture. Flow cytometric analyses were performed on an LSRII FACS
analyzer (Becton Dickinson).
Reverse transcription (RT) PCR
Total RNA was isolated using the Nucleospin RNA isolation kit (Macherey-nagel,
Düren, Germany). For RT-PCR (Reverse Transcriptase Polymerase Chain Reaction),
total RNA was extracted as described above, and cDNA was synthesized by means
of oligodeoxythymidine (oligo(dT)) and Superscript II RNase H-reverse trancriptase
(Invitrogen Life Technologies). Noxa, Notch1 and Notch2 transcripts were amplifi ed
by PCR as described before3;18. Products were electrophoresed on 1% agarose
gel. PCR for Hes-1 was performed on an iCycler PCR (Bio-Rad, Hercules, CA) as
described before26.
Proteasome activity assay
Cytoplasmic extracts (Assay buffer: 250 mM HEPES (pH7.5), 5 mM EDTA, 0.5%
NP-40 and 0.01% SDS) from freshly isolated PBMCs from CLL patients were used to
measure proteasome activity using a 20S proteasome activity assay kit (Chemicon,
part of Millipore; Billerica, USA) following the manufacturer’s instructions. The
assay is based on detection of the fl uorophore 7-amino-4-methylcoumarin (AMC)
after cleavage from the labeled substrate LVVY-AMC. The free AMC fl uorescence
was quantifi ed using a 380/460-nm fi lter set in a VICTOR2 D fl uorometer (Wallac-
PerkinElmer) PerkinElmer, Massachusetts, USA). Proteasomal activity was calculated
from the changes in fl uorescence over time and expressed per μg of protein in the
extract.
Thesis_Final_2_01092008.indd Sec1:60Thesis_Final_2_01092008.indd Sec1:60 01-09-2008 11:08:1301-09-2008 11:08:13

61RESULTS
GSI-1 is a potent, p53-independent inducer of apoptosis of primary
CLL cells
Notch expression has been characterized previously in CLL3;7, and we could
confi rm expression of Notch1 and Notch2 in CLL, while Notch3 and Notch4 were
not present in the samples tested (data not shown). γ-Secretase inhibitors (GSIs)
are pharmacological agents able to block Notch signaling16;17. GSI-1 (0.25 – 50 µM)
induced a signifi cant dose-dependent increase in apoptosis of CLL cells after 24
hours (Fig. 1A). Similar experiments were performed with GSI-9/DAPT, another class
of small molecule inhibitors of γ-secretase activity. DAPT, however, had no effect on
apoptosis in CLL (1.0 – 50 µM) (Fig. 1A). To determine the effect on normal B and T
lymphocytes, PBMCs from 3 healthy individuals were treated for 24 hrs with GSI-1 at
concentrations ranging from 0.5 to 50 µM. The cytotoxicity of GSI-1 towards normal
CD19+ B cells was comparable to that observed for CLL cells, while normal CD3+ T
cells were less sensitive to GSI-1 (Fig. 1B), and showed a maximum of approximately
50% apoptotic cells. Next, CLL samples of patients lacking functional p53 were
analyzed following incubation with GSI-1, in comparison with induction of apoptosis
by γ-irradiation (γ-IR). Both p53 defi cient (n=2) CLL samples showed prominent cell
death following treatment with 5 µM GSI-1, comparable to WT samples (see also
Fig. 1A), whereas they resisted radiation-induced apoptosis (Fig. 2). Taken together,
these results indicate that Notch1 and Notch2 are expressed in primary CLL cells and
treatment with GSI-1 but not DAPT results in rapid, p53-independent cell death.
Figure 1: Apoptosis of CLL cells, normal B and T lymphocytes upon GSI-1 treatment.
(A) Apoptosis response as analyzed by MitoTracker staining of CLL cells upon 24 hours incubation with
GSI-1 ( ; n=13; 0.25 - 50 µM) or DAPT ( ; n=3; 1 - 50 µM). Data represent mean ± SD.
(B) Apoptosis response as analyzed by MitoTracker staining of PBMCs from healthy donors (HD) (n=3).
CD19+ normal B cells ( ) and CD3+ normal T cells ( ) were treated with 1 – 50 µM GSI-1. Data represent
mean ± SD from 3 independent experiments.
Ch
ap
ter 3
▪ GS
I-1 a
cts
as a
pro
teaso
me in
hib
itor in
CL
L
A BA
Treatment [μM]
Thesis_Final_2_01092008.indd Sec1:61Thesis_Final_2_01092008.indd Sec1:61 01-09-2008 11:08:1301-09-2008 11:08:13

62
Notch signaling does not contribute to CLL cell survival in vitro,
and GSI-1 cannot block Notch signaling
To test whether triggering via Notch infl uenced in vitro survival of CLL cells, freshly
isolated CLL cells were evaluated in two co-culture systems; murine fi broblast L cells27
or stromal OP-9 cells transfected with the Notch ligands Jag1 or DL126. CLL survival
was evaluated every day up to day 7. No signifi cant or reproducible difference in
survival compared to co-culture with untransfected control cells or in medium could
be observed (11 CLL samples tested at various cell densities, data not shown).
To address whether Notch signaling can be specifi cally blocked by GSI-1, we applied
an established cell culture system where lineage decision between T cells and
CD123+BDCA2+ plasmacytoid dendritic cells (pDCs) depends on Notch signals26.
The OP-9 cell line expressing human DL1 inhibits the development of pDCs from
CD34+CD1a- thymic progenitor cells, but as mentioned above it has no effect on
CLL survival. Addition of DAPT could overcome the inhibition of pDC development,
whereas GSI-1 could not (Fig. 3A). In fact, as can be seen from the FACS plots, GSI-
1 caused massive apoptosis at high concentration (5 µM). The effect of GSI-1 and
DAPT on the expression of the Notch specifi c target gene Hes-1 was evaluated by
quantitative RT-PCR (Fig. 3B). The mRNA levels were normalized to expression in
untreated CLL cells (relative value of 1) using β-actin as internal control. Expression of
Hes-1 shows large variation among untreated CLL cases and the median expression
of Hes-1 was lower than in normal B cells (0.1-17.5 % of the average of normal B
cells)7. However, incubation of CLL cells (n=2) with DAPT (1 or 10 µM) blocked the
basal Hes-1 expression, whereas GSI-1 (1 or 10 µM) showed no clear inhibitory
effect. Thus, triggering of Notch receptors on CLL did not affect their survival and
in contrast to DAPT, GSI-1 was unable to prevent Notch signaling in an established
system of lineage decision.
Figure 2: Apoptotic response to GSI-1 does
not require functional p53.
Apoptosis response of 3 CLL patients: CLL17
(black bar; p53 wt); CLL27 and CLL28 (gray bar
and white bar; p53 dysfunctional), to GSI-1 (5 µM)
and γ-IR (5Gy) assessed by MitoTracker staining
at 24 hours. (med = medium)
Thesis_Final_2_01092008.indd Sec1:62Thesis_Final_2_01092008.indd Sec1:62 01-09-2008 11:08:1301-09-2008 11:08:13
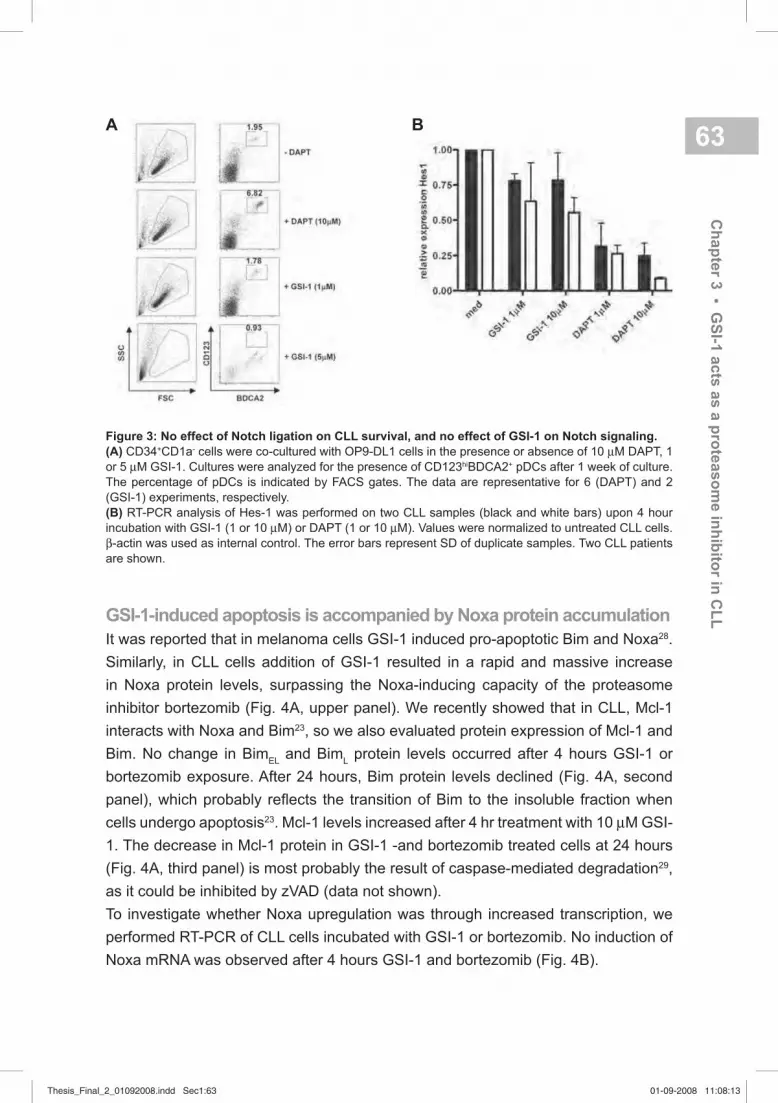
63
GSI-1-induced apoptosis is accompanied by Noxa protein accumulation
It was reported that in melanoma cells GSI-1 induced pro-apoptotic Bim and Noxa28.
Similarly, in CLL cells addition of GSI-1 resulted in a rapid and massive increase
in Noxa protein levels, surpassing the Noxa-inducing capacity of the proteasome
inhibitor bortezomib (Fig. 4A, upper panel). We recently showed that in CLL, Mcl-1
interacts with Noxa and Bim23, so we also evaluated protein expression of Mcl-1 and
Bim. No change in BimEL
and BimL protein levels occurred after 4 hours GSI-1 or
bortezomib exposure. After 24 hours, Bim protein levels declined (Fig. 4A, second
panel), which probably refl ects the transition of Bim to the insoluble fraction when
cells undergo apoptosis23. Mcl-1 levels increased after 4 hr treatment with 10 µM GSI-
1. The decrease in Mcl-1 protein in GSI-1 -and bortezomib treated cells at 24 hours
(Fig. 4A, third panel) is most probably the result of caspase-mediated degradation29,
as it could be inhibited by zVAD (data not shown).
To investigate whether Noxa upregulation was through increased transcription, we
performed RT-PCR of CLL cells incubated with GSI-1 or bortezomib. No induction of
Noxa mRNA was observed after 4 hours GSI-1 and bortezomib (Fig. 4B).
Figure 3: No effect of Notch ligation on CLL survival, and no effect of GSI-1 on Notch signaling.
(A) CD34+CD1a- cells were co-cultured with OP9-DL1 cells in the presence or absence of 10 µM DAPT, 1
or 5 µM GSI-1. Cultures were analyzed for the presence of CD123hiBDCA2+ pDCs after 1 week of culture.
The percentage of pDCs is indicated by FACS gates. The data are representative for 6 (DAPT) and 2
(GSI-1) experiments, respectively.
(B) RT-PCR analysis of Hes-1 was performed on two CLL samples (black and white bars) upon 4 hour
incubation with GSI-1 (1 or 10 µM) or DAPT (1 or 10 µM). Values were normalized to untreated CLL cells.
β-actin was used as internal control. The error bars represent SD of duplicate samples. Two CLL patients
are shown.
A B
Ch
ap
ter 3
▪ GS
I-1 a
cts
as a
pro
teaso
me in
hib
itor in
CL
L
Thesis_Final_2_01092008.indd Sec1:63Thesis_Final_2_01092008.indd Sec1:63 01-09-2008 11:08:1301-09-2008 11:08:13

64After 24 hours of treatment with these agents, mRNA could hardly be extracted
because of the high proportion of dead cells (data not shown). In addition, Figure
4 B illustrates that in the myeloma cell line RPMI 8226, bortezomib does however
induce upregulation of Noxa mRNA at intermediate concentration, in agreement
with a previous report30. In conclusion, addition of GSI-1 to CLL cells causes a rapid
accumulation of Noxa protein levels, independent of transcriptional regulation.
A
B
GSI-1 inhibits proteasome activity and induces accumulation of
polyubiquitinated proteins
Given the aforementioned results, demonstrating Noxa protein accumulation upon
treatment with GSI-1 and bortezomib, we determined the effect of GSI-1 on the total
level of ubiquitinated proteins in HeLa cells and the proteasomal activity in primary CLL
samples. First, HeLa cells were incubated for 16 hours with GSI-1 or acknowledged
proteasome inhibitors bortezomib and MG-132. Figure 5A (upper panel) illustrates
that addition of GSI-1, bortezomib or MG-132 led to accumulation of polyubiquitinated
proteins. Concomitant with increased levels of ubiquitinated proteins, Noxa protein
levels accumulated over time (Fig. 5A; middle panel). Second, incubation of lysates
from freshly isolated CLL cells with GSI-1, bortezomib or MG-132 resulted in clear
inhibition of proteasome activity (Fig. 5B). In contrast, roscovitine and fl udarabine, both
chemotherapeutic agents capable of inducing apoptosis in CLL cells, or the gamma
secretase inhibitor DAPT had no measurable effect on proteasomal activity (Fig. 5B).
Figure 4: Effect of GSI-1 and
bortezomib on Noxa mRNA and
protein levels.
(A) CLL cells were treated for 4 and
24 hrs with GSI-1 (1 or 10 µM) or
bortezomib (B) (10 or 30 nM). Cell
lysates were probed for expression of
Noxa, Bim, Mcl1 and -actin protein
by Western Blot. Apoptosis of cells
was determinated by Annexin-VAPC/
PI staining and is indicated above the
lanes. Results are representative for 2
separate experiments.
(B) CLL cells or the myeloma cell
line RPMI 8226 were incubated for 4
hours with GSI-1 or bortezomib. RNA
was isolated and RT-PCR for Noxa
and GAPDH was performed. Results
are representative for 2 separate
experiments.
Thesis_Final_2_01092008.indd Sec1:64Thesis_Final_2_01092008.indd Sec1:64 01-09-2008 11:08:1401-09-2008 11:08:14

65In addition, proteasomal activity was measured in lysates obtained from CLL cells
after 4 and 24 hours incubation with various drugs. Again, GSI-1, bortezomib and
MG-132 showed blocking of the proteasome, whereas roscovitine did not (Fig. 5C).
Note that upon 24 hours after triggering with roscovitine proteasomal activity was
also undetectable, probably as a result of massive apoptosis, and subsequent block
in proteasomal activity31. Apoptosis responses under these various conditions are
represented in fi gure 5D. In conclusion, GSI-1 blocked proteasomal activity, leading
to accumulation of polyubiquitinated proteins, similar to the effects of well known
proteasome inhibitors such as MG-132 and bortezomib.
Figure 5: Effect of GSI-1 on ubiquitin / proteasome system.
(A) Accumulation of polyubiquitinated proteins after MG-132, bortezomib or GSI-1 treatment. HeLa cells
were treated with 10 µM MG-132, 10 nM bortezomib or 5 µM GSI-1 for16 hours. Lysates were probed for
expression of ubiquitinated proteins, Noxa and β-actin by Western blot.
(B) The enzymatic activity of the 20S proteasome was measured after adding bortezomib (20 nM), MG-132
(0.5 µM), GSI-1 (5 µM), DAPT (5 µM), roscovitine (25 µM) or fl udarabine (100 µM) to cytoplasmic extracts
from freshly isolated cells from 3 CLL patients. After 15 minutes incubation, the fl uorogenic proteasome
substrate LVVY-AMC was added. Results are expressed as change in AMC fl uorescence per minute per
μg protein.
(C) CLL cells were treated for 4 (black bar) and 24 (white bar) hrs with GSI-1 (1 or 5 µM), bortezomib (20
nM), MG-132 (0.5 µM) and roscovitine (25 µM). 20 S proteasomal activity was measured as described
above (4B). Data represent mean ± SD from four different CLL patients.
(D) Apoptosis response of CLL cells from the experiments described in C were measured by MitoTracker
staining after 4 (black bar) and 24 (white bar) hours.
A
C
B
D
Ch
ap
ter 3
▪ GS
I-1 a
cts
as a
pro
teaso
me in
hib
itor in
CL
L
Thesis_Final_2_01092008.indd Sec1:65Thesis_Final_2_01092008.indd Sec1:65 01-09-2008 11:08:1401-09-2008 11:08:14

66Surface expression of CD23 is reduced upon proteasomal inhibition
Exposure of CLL cells to the proteasome inhibitors bortezomib and MG-132
induced apoptosis and decreased expression of CD23, which was proposed to
be a consequence of Notch inhibition4. To test the alternative possibility that CD23
down-regulation is a direct consequence of proteasomal inhibition, we incubated CLL
cells (n=3) with GSI-1, proteasome inhibitors bortezomib and MG-132, or the Notch
inhibitor DAPT. As can be seen in fi gure 6, GSI-1 led to a reduction of CD23 surface
expression similar to that seen with bortezomib or MG-132, while CD23 expression
remained essentially unchanged during 24 hours incubation with DAPT.
Figure 6: CD23 expression on CLL cells upon incubation with GSI-
1. CLL cells were cultured for 24 hours with 30 nM bortezomib (n=2),
0.5 μM MG-132 (n=2), 5 μM GSI-1 (n=4) and 5 μM DAPT (n=3). The
percentage CD23-positive cells was measured by FACS analysis: within
the viable population, CD5 and CD19 double positive cells were gated.
Data represent mean from ± SD.
Noxa knock-down results in decreased sensitivity to GSI-1
The previous experiments showed that Noxa protein accumulates upon GSI-1
treatment, but do not establish whether this is required to induce apoptosis. To
investigate a direct role for Noxa in GSI-1 mediated apoptosis, we studied various
cell lines transduced with retroviral vectors encoding two different Noxa shRNA
sequences (N7 and N8). Figure 7A illustrates signifi cant but not complete knock-down
of Noxa protein in the different cell lines. Incubation with GSI-1 (+/- pre-treatment
with zVAD-fmk) clearly raised Noxa protein levels, and this in fact still occurred in
the knock-down lines. Ramos FSA cells expressing two types of shRNA, exhibited a
decreased sensitivity to GSI-1-induced apoptosis compared to the mock transduced
cells, similar to results obtained with bortezomib (Fig. 7B).The effects of Noxa shRNA
on GSI-1-mediated cell death were reproducible in other cell lines, Mec-1 and Jurkat
(Fig. 7C&D). Consistent with previous results23, no effect of Noxa protein reduction was
observed on apoptosis triggered via other pathways, such as fl udarabine treatment
or triggering of the CD95 receptor (Fig. 7E). In all cases resistance to GSI-1-mediated
Thesis_Final_2_01092008.indd Sec1:66Thesis_Final_2_01092008.indd Sec1:66 01-09-2008 11:08:1501-09-2008 11:08:15

67apoptosis was not complete, most probably due to the fact that remaining levels of
Noxa protein in the knock-down lines were still augmented by GSI-1 (Fig. 7A). In
summary, these data demonstrate that decreased expression of Noxa has a direct
and specifi c effect on the susceptibility to apoptosis induced by GSI-1.
A
C
B
D E
Figure 7: Knockdown of Noxa with RNAi
specially prevents apoptosis induction
by GSI-1.
(A) Ramos FSA-M (=Mock, lane 1-4),
Ramos FSA-N7 (lane 5-8) and Ramos
FSA-N8G10 cells (lane 9-12) were treated
with the pan-caspase inhibitor zVAD-fmk
(100 µM) and GSI-1 (1µM) as indicated.
Noxa protein levels were monitored by
Western blotting. Noxa knockdown in Mec-
1 cells and Jurkat T (J16) cells has been
described previously 18;23. Equal protein
loading is shown by reprobing for β-actin.
(B) Mock ( ), N7 (▼) and N8.G10 ( )
Ramos FSA cells were incubated for
24 hours in the presence of indicated
concentrations of GSI-1 and bortezomib.
Viability was assessed by MitoTracker
staining and FACS analysis. Data
represent mean ± SD from 3 independent
experiments, and one experiment with N7
and bortezomib.
(C) Mock ( ) and N8 ( ) transduced Mec-
1 cells were incubated and analyzed as in
panel B. Data represent mean ± SD from
6 independent experiments for GSI-1 and
1 experiment for bortezomib treatment.
(D) Mock ( ) and N8 ( ) transduced J16
cells were incubated for 24 hours in the
presence of indicated concentrations of
GSI-1 and analyzed as in panel B. Data
represent mean ± SD from 3 independent
experiments.
(E) Mock ( ) and N8.G10 ( ) transduced
Ramos FSA cells were incubated for
24 hours in the presence of 100 µM
fl udarabine (F) and 0.25 µM α-CD95, and
analyzed as in panel B. Medium = M. Data
represent mean ± SD from 3 independent
experiments.
Ch
ap
ter 3
▪ GS
I-1 a
cts
as a
pro
teaso
me in
hib
itor in
CL
L
Thesis_Final_2_01092008.indd Sec1:67Thesis_Final_2_01092008.indd Sec1:67 01-09-2008 11:08:1501-09-2008 11:08:15

68 DISCUSSION
The major fi ndings of our study are that GSI-1 is a potent inducer of apoptosis in CLL
and that this response does not involve Notch signaling. Rather, effi cient inhibition of
proteasomal activity and subsequent Noxa accumulation seems to play a key role.
Dysregulation of the Notch pathway has been suggested in CLL, mainly because of
the observation that overexpression of CD23 in CLL cells is regulated by Notch23.
In agreement with other reports3;7 we found that Notch1 and Notch2 receptors are
present in CLL cells. To explore a possible functional role of the Notch pathway in
CLL we performed two kinds of experiments. First we co-cultured primary CLL cells
with either L cells or OP9 cells expressing the Notch ligands DL1 and Jagged1.
This did not affect cell survival in CLL cells. To exclude that this observation is due
to already optimal autocrine or paracrine Notch stimulation in CLL we performed
a set of experiments aimed at blocking Notch signaling. To this end we used the
inhibitors DAPT21 and GSI-120, both assumed to specifi cally block γ-secretases.
Here we encountered a surprising dichotomy: whereas DAPT was unable to induce
apoptosis in CLL, it did inhibit pDC development in the presence of the Notch ligand
DL1, as described previously26. In contrast, this was not the case for GSI-1, which
was capable of potent induction of apoptosis in CLL. A similar dichotomy has been
decribed with respect to Notch2 dependent CD23 upregulation. Treatment of CLL
cells with proteasome inhibitors led to inhibition of nuclear Notch2 intracellular domain
(Notch2IC) activity and downregulation of CD234. However, CD23 down-regulation
could not be achieved with DAPT, as we have also observed. With respect to CD23
down-regulation we could confi rm that this is the result of proteasome- rather than
Notch inhibition (Fig. 6). CD23 is a known transcriptional target of NF-κB32, and
NFκB-p52 constitutively binds to the CD23 promotor region in murine cells33. Thus, a
possible explanation for the down-regulation of CD23 by proteasome inhibitors could
be their well-known negative effect on NF-κB signaling34. However, in experiments
using specifi c inhibitors of NF-κB signaling35 we observed no infl uence on the
expression of CD23 (data not shown).
One of the best characterized Notch targets is the HES family of transcription factors5.
The majority of the CLL patients display substantially lower HES-1 transcripts
compared to normal B cells7. In agreement, we could detect low HES-1 in two CLL
samples, which could further be decreased by DAPT but not signifi cantly by GSI-1
(Fig. 3B). Our data agree with previous studies showing Noxa upregulation and
apoptosis upon GSI in melanoma cell lines, melanoma xenografts and myeloma
cells9;14. To our knowledge, no data are available that would directly demonstrate
repression of Noxa by HES-1.
Thesis_Final_2_01092008.indd Sec1:68Thesis_Final_2_01092008.indd Sec1:68 01-09-2008 11:08:1501-09-2008 11:08:15

69In CLL, GSI-1-induced apoptosis was accompanied by rapid and extensive
accumulation of Noxa protein, due to blocking of proteasomal function, similar to
earlier fi ndings in CLL and other cell types that proteasome inhibitors like bortezomib
and MG-132 effectively block the proteasome and upregulate Noxa protein15. Effects
of GSI-1 on BH3-only proteins Noxa and Bim in melanoma cells14, were apparently
also regulated at transcriptional level14;36. The latter is clearly not the case in CLL,
since Noxa mRNA levels do not increase upon treatment with bortezomib or GSI-1. In
support of a direct role for Noxa accumulation, Noxa shRNA experiments demonstrated
a clear reduction in apoptosis mediated by GSI-1, whereas other apoptosis pathways
were unaffected.
The demonstration that GSI-1 acts as a proteasome inhibitor is perhaps not surprising
since the structure of GSI-1 (Z-LLNle-CHO) resembles that of other chymotrypsin
inhibitors and also that of MG-132 (z-LLL-CHO). Others have also remarked that the
aldehyde group on the GSI-1 peptide is probably able to covalently inhibit certain
serine protease20. Our data agree with that view and we propose that many, if not all,
of the apoptosis-inducing activities ascribed to GSI-1, and related compounds such
as GSI-12 (Z-IL-CHO)9 are probably due to proteasome rather than Notch inhibition.
An intriguing remaining question concerns the mechanism behind the rapid and
massive upregulation of Noxa protein by proteasome inhibition. Accumulation of
higher molecular weight species of Noxa, indicative of (poly)ubiquitination, were
never observed by us. Also, efforts to detect ubiquitination of Noxa in cells transiently
transfected with differentially tagged Noxa and ubiquitin, and treated with proteasome
inhibitors, were unsuccesfull (data not shown). A possible explanation for this might
be that Noxa accumulation occurs because its binding partner Mcl-1 is ubiquitinated
and degraded37;38, whereby Noxa dissociates and passively accumulates. Support
for such a chaperone-like role for Noxa in controlling the degradation of Mcl-1 has
indeed been obtained39.
In CLL, new therapies that-act independently of p53 are urgently required. We observed
that GSI-1 is effective in CLL cells from p53 dysfunctional patients, and a variety of
compounds related to GSI are already in the clinic for the treatment of Alzheimer’s
disease40. In that setting, side effects are limited41;42. Bortezomib has proven effi cacy
as single agent in multiple myeloma and some forms of non-Hodgkin’s lymphoma43-
45, although a number of toxicities are described46;47. For unknown reasons, clinical
trials with bortezomib in patients with fl udarabine-refractory CLL showed only limited
responses although biological activity was observed48. We propose that our studies
provide a basis to continued studies into alternative proteasome inihibitors such as
GSI-1 or related compounds as potential treatment for chemoresistant CLL.
Ch
ap
ter 3
▪ GS
I-1 a
cts
as a
pro
teaso
me in
hib
itor in
CL
L
Thesis_Final_2_01092008.indd Sec1:69Thesis_Final_2_01092008.indd Sec1:69 01-09-2008 11:08:1501-09-2008 11:08:15

70 ACKNOWLEDGEMENTS
We thank René Spijker for technical assistance, and the patients for their blood
donations. We thank Bianca Blom for critical reading of the manuscript, and
acknowledge Dr. Kramer, Dr. Wittebol and Dr. Baars for including CLL patients in this
study.
Thesis_Final_2_01092008.indd Sec1:70Thesis_Final_2_01092008.indd Sec1:70 01-09-2008 11:08:1601-09-2008 11:08:16

71REFERENCE LIST
1. Byrd JC, Stilgenbauer S, Flinn IW.
Chronic lymphocytic leukemia.
Hematology.(Am.Soc.Hematol.Educ.
Program.) 2004163-183.
2. Fournier S, Yang LP, Delespesse G
et al. The two CD23 isoforms display
differential regulation in chronic
lymphocytic leukaemia. Br.J.Haematol.
1995;89:373-379.
3. Hubmann R, Schwarzmeier JD,
Shehata M et al. Notch2 is involved in
the overexpression of CD23 in B-cell
chronic lymphocytic leukemia. Blood
2002;99:3742-3747.
4. Duechler M, Shehata M, Schwarzmeier
JD et al. Induction of apoptosis by
proteasome inhibitors in B-CLL cells
is associated with downregulation
of CD23 and inactivation of Notch2.
Leukemia 2005;19:260-267.
5. Jarriault S, Brou C, Logeat F
et al. Signalling downstream of
activated mammalian Notch. Nature
1995;377:355-358.
6. Radtke F, Raj K. The role of Notch
in tumorigenesis: oncogene or
tumour suppressor? Nat.Rev.Cancer
2003;3:756-767.
7. Hajdu M, Sebestyen A, Barna G et
al. Activity of the notch-signalling
pathway in circulating human
chronic lymphocytic leukaemia cells.
Scand.J.Immunol. 2007;65:271-275.
8. Zweidler-McKay PA, He Y, Xu L et al.
Notch signaling is a potent inducer of
growth arrest and apoptosis in a wide
range of B-cell malignancies. Blood
2005;106:3898-3906.
9 . Nefedova Y, Sullivan DM, Bolick SC,
Dalton WS, Gabrilovich DI. Inhibition
of Notch signaling induces apoptosis
of myeloma cells and enhances
sensitivity to chemotherapy. Blood
2008;111:2220-2229.
10. Qiu L, Joazeiro C, Fang N et al.
Recognition and ubiquitination of Notch
by Itch, a hect-type E3 ubiquitin ligase.
J.Biol.Chem. 2000;275:35734-35737.
11. Wu G, Lyapina S, Das I et al. SEL-10
is an inhibitor of notch signaling that
targets notch for ubiquitin-mediated
protein degradation. Mol.Cell Biol.
2001;21:7403-7415.
12. Elliott PJ, Ross JS. The proteasome:
a new target for novel drug therapies.
Am.J.Clin.Pathol. 2001;116:637-646.
13. Kanagasabaphy P, Morgan GJ, Davies
FE. Proteasome inhibition and multiple
myeloma. Curr.Opin.Investig.Drugs
2007;8:447-451.
14. Qin JZ, Ziffra J, Stennett L et al.
Proteasome inhibitors trigger NOXA-
mediated apoptosis in melanoma
and myeloma cells. Cancer Res.
2005;65:6282-6293.
15. Smit LA, Hallaert DY, Spijker R et al.
Differential Noxa/Mcl-1 balance in
peripheral versus lymph node chronic
lymphocitic leukemia cells correlates
with survival capacity. Blood 2006
16. De Strooper B., Annaert W, Cupers P
et al. A presenilin-1-dependent gamma-
secretase-like protease mediates
release of Notch intracellular domain.
Nature 1999;398:518-522.
17. Josien H. Recent advances in the
development of gamma-secretase
inhibitors. Curr.Opin.Drug Discov.
Devel. 2002;5:513-525.
Ch
ap
ter 3
▪ GS
I-1 a
cts
as a
pro
teaso
me in
hib
itor in
CL
L
Thesis_Final_2_01092008.indd Sec1:71Thesis_Final_2_01092008.indd Sec1:71 01-09-2008 11:08:1601-09-2008 11:08:16

72 18. Alves NL, Derks IA, Berk E et al. The
Noxa/Mcl-1 axis regulates susceptibility
to apoptosis under glucose limitation in
dividing T cells. Immunity. 2006;24:703-
716.
19. Lens SM, den Drijver BF, Potgens AJ
et al. Dissection of pathways leading
to antigen receptor-induced and Fas/
CD95-induced apoptosis in human B
cells. J.Immunol. 1998;160:6083-6092.
20. Curry CL, Reed LL, Golde TE et al.
Gamma secretase inhibitor blocks
Notch activation and induces apoptosis
in Kaposi’s sarcoma tumor cells.
Oncogene 2005;24:6333-6344.
21. Dovey HF, John V, Anderson JP et al.
Functional gamma-secretase inhibitors
reduce beta-amyloid peptide levels in
brain. J.Neurochem. 2001;76:173-181.
22. Koopman G, Reutelingsperger
CP, Kuijten GA et al. Annexin V
for fl ow cytometric detection of
phosphatidylserine expression on B
cells undergoing apoptosis. Blood
1994;84:1415-1420.
23. Hallaert DY, Spijker R, Jak M et al.
Crosstalk among Bcl-2 family members
in B-CLL: seliciclib acts via the Mcl-1/
Noxa axis and gradual exhaustion
of Bcl-2 protection. Cell Death.Differ.
2007
24. van der Kolk LE, Evers LM, Omene C
et al. CD20-induced B cell death can
bypass mitochondria and caspase
activation. Leukemia 2002;16:1735-
1744.
25. Pettitt AR, Sherrington PD, Stewart G
et al. p53 dysfunction in B-cell chronic
lymphocytic leukemia: inactivation
of ATM as an alternative to TP53
mutation. Blood 2001;98:814-822.
26. Dontje W, Schotte R, Cupedo T et al.
Delta-like1-induced Notch1 signaling
regulates the human plasmacytoid
dendritic cell versus T-cell lineage
decision through control of GATA-3 and
Spi-B. Blood 2006;107:2446-2452.
27. Hicks C, Johnston SH, diSibio G et al.
Fringe differentially modulates Jagged1
and Delta1 signalling through Notch1
and Notch2. Nat.Cell Biol. 2000;2:515-
520.
28. Qin JZ, Stennett L, Bacon P et al.
p53-independent NOXA induction
overcomes apoptotic resistance of
malignant melanomas. Mol.Cancer
Ther. 2004;3:895-902.
29. Herrant M, Jacquel A, Marchetti S et
al. Cleavage of Mcl-1 by caspases
impaired its ability to counteract
Bim-induced apoptosis. Oncogene
2004;23:7863-7873.
30. Gomez-Bougie P, Wuilleme-Toumi S,
Menoret E et al. Noxa up-regulation
and Mcl-1 cleavage are associated
to apoptosis induction by bortezomib
in multiple myeloma. Cancer Res.
2007;67:5418-5424.
31. Sun XM, Butterworth M, MacFarlane
M et al. Caspase activation inhibits
proteasome function during apoptosis.
Mol.Cell 2004;14:81-93.
32. Tinnell SB, Jacobs-Helber SM,
Sterneck E, Sawyer ST, Conrad DH.
STAT6, NF-kappaB and C/EBP in
CD23 expression and IgE production.
Int.Immunol. 1998;10:1529-1538.
33. Debnath I, Roundy KM, Weis JJ,
Weis JH. Defi ning in vivo transcription
factor complexes of the murine
CD21 and CD23 genes. J.Immunol.
2007;178:7139-7150.
34. Nencioni A, Grunebach F, Patrone
F, Ballestrero A, Brossart P.
Proteasome inhibitors: antitumor
effects and beyond. Leukemia
2007;21:30-36.
35. Pickering BM, de MS, Lee M et al.
Pharmacological inhibitors of NF-
kappaB accelerate apoptosis in
chronic lymphocytic leukaemia cells.
Oncogene 2007;26:1166-1177.
Thesis_Final_2_01092008.indd Sec1:72Thesis_Final_2_01092008.indd Sec1:72 01-09-2008 11:08:1601-09-2008 11:08:16

73 45. O’Connor OA, Wright J, Moskowitz
C et al. Phase II clinical experience
with the novel proteasome inhibitor
bortezomib in patients with indolent
non-Hodgkin’s lymphoma and
mantle cell lymphoma. J.Clin.Oncol.
2005;23:676-684.
46. Lonial S, Waller EK, Richardson
PG et al. Risk factors and kinetics
of thrombocytopenia associated
with bortezomib for relapsed,
refractory multiple myeloma. Blood
2005;106:3777-3784.
47. Richardson PG, Briemberg H,
Jagannath S et al. Frequency,
characteristics, and reversibility of
peripheral neuropathy during treatment
of advanced multiple myeloma
with bortezomib. J.Clin.Oncol.
2006;24:3113-3120.
48. Faderl S, Rai K, Gribben J et al. Phase
II study of single-agent bortezomib
for the treatment of patients with
fl udarabine-refractory B-cell chronic
lymphocytic leukemia. Cancer
2006;107:916-924.
36. Fernandez Y, Verhaegen M, Miller TP
et al. Differential regulation of noxa in
normal melanocytes and melanoma
cells by proteasome inhibition:
therapeutic implications. Cancer Res.
2005;65:6294-6304.
37. Warr MR, Acoca S, Liu Z et al. BH3-
ligand regulates access of MCL-1 to its
E3 ligase. FEBS Lett. 2005;579:5603-
5608.
38. Zhong Q, Gao W, Du F, Wang X. Mule/
ARF-BP1, a BH3-only E3 ubiquitin
ligase, catalyzes the polyubiquitination
of Mcl-1 and regulates apoptosis. Cell
2005;121:1085-1095.
39. Czabotar PE, Lee EF, van Delft MF
et al. Structural insights into the
degradation of Mcl-1 induced by BH3
domains. Proc.Natl.Acad.Sci.U.S.A
2007;104:6217-6222.
40. Wolfe MS. Secretase as a target for
Alzheimer’s disease. Curr.Top.Med.
Chem. 2002;2:371-383.
41. Siemers E, Skinner M, Dean RA et
al. Safety, tolerability, and changes
in amyloid beta concentrations
after administration of a gamma-
secretase inhibitor in volunteers. Clin.
Neuropharmacol. 2005;28:126-132.
42. Siemers ER, Quinn JF, Kaye J et al.
Effects of a gamma-secretase inhibitor
in a randomized study of patients
with Alzheimer disease. Neurology
2006;66:602-604.
43. Richardson PG, Mitsiades C,
Hideshima T, Anderson KC.
Proteasome inhibition in the treatment
of cancer. Cell Cycle 2005;4:290-296.
44. Goy A, Younes A, McLaughlin P et al.
Phase II study of proteasome inhibitor
bortezomib in relapsed or refractory
B-cell non-Hodgkin’s lymphoma. J.Clin.
Oncol. 2005;23:667-675.
Ch
ap
ter 3
▪ GS
I-1 a
cts
as a
pro
teaso
me in
hib
itor in
CL
L
Thesis_Final_2_01092008.indd Sec1:73Thesis_Final_2_01092008.indd Sec1:73 01-09-2008 11:08:1601-09-2008 11:08:16

74
Thesis_Final_2_01092008.indd Sec1:74Thesis_Final_2_01092008.indd Sec1:74 01-09-2008 11:08:1601-09-2008 11:08:16

chapter
4Differential Noxa/Mcl-1 balance in peripheral versus lymph node chronic lymphocytic leukemia cells correlates with survival capacity
Laura A. Smit1,4, Delfi ne Y.H. Hallaert2,3,4, René Spijker2,3, Bart de Goeij3, Annelieke
Jaspers2,3, Arnon P. Kater2, Marinus H.J. van Oers2, Carel J.M. van Noesel1, and
Eric Eldering2,3
1Dept. of Pathology, AMC, Amsterdam, the Netherlands
2Dept. of Hematology, AMC, Amsterdam, the Netherlands
3Dept. of Experimental Immunology, AMC, Amsterdam, The Netherlands
4These authors contributed equally to the manuscript
Blood, 2007; 109 (4): 1660-1668.
Thesis_Final_2_01092008.indd Sec1:75Thesis_Final_2_01092008.indd Sec1:75 01-09-2008 11:08:1601-09-2008 11:08:16

76 ABSTRACT
The gradual accumulation of chronic lymphocytic leukemia (B-CLL) cells is presumed
to derive from proliferation centers in lymph nodes and bone marrow. To what extent
these cells possess the purported anti-apoptotic phenotype of peripheral B-CLL cells
is unknown. Recently, we have described that in B-CLL samples from peripheral
blood, aberrant apoptosis gene expression was not limited to protective changes but
also included increased levels of pro-apoptotic BH3-only member Noxa. Here, we
compare apoptosis gene profi les from peripheral blood B-CLL (n=15) with lymph node
B-CLL (>90% CD5+/CD19+/CD23+ lymphocytes with Ki67+ centers; n=9). Apart from
expected differences in Survivin and Bcl-XL, a prominent distinction with peripheral
B-CLL cells was the decreased averaged level of Noxa in lymph nodes. Mcl-1 protein
expression showed a reverse trend. Noxa expression could also be reduced in vitro
by CD40 stimulation of peripheral blood B-CLL. Direct manipulation of Noxa protein
levels was achieved by proteasome inhibition in B-CLL and via RNAi in model cell
lines. In each instance, cell viability was directly linked with Noxa levels. These data
indicate that suppression of Noxa in the lymph node environment contributes to the
persistence of B-CLL at these sites and suggest that therapeutic targeting of Noxa
might be benefi cial.
Thesis_Final_2_01092008.indd Sec1:76Thesis_Final_2_01092008.indd Sec1:76 01-09-2008 11:08:1601-09-2008 11:08:16

Ch
ap
ter 4
▪ Ap
op
tosis
gen
es in
CL
L ly
mp
h n
od
e v
ers
us p
erip
hery
77INTRODUCTION
B-cell chronic lymphocytic leukemia (B-CLL) is characterized by a progressive
accumulation of monoclonal CD5+ CD23+ mature B cells in the secondary lymphoid
tissues, bone marrow, and blood1. Previously, it was assumed that B-CLL is
associated with a defective regulation of programmed cell death (apoptosis), rather
than uncontrolled cell proliferation2. Indeed, high expression of the anti-apoptotic
proteins Bcl-2 and Mcl-1 has been associated with rapid disease progression and
a poor response to chemotherapy3;4. Paradoxically, investigation of virtually all
direct apoptosis regulators known at present revealed that, in addition to these anti-
apoptotic alterations, the pro-apoptotic proteins Noxa and Bmf are also abundantly
expressed in B-CLL5;6. How the elevated expression of these pro-apoptotic proteins
is associated with the reputed increased life span of the B-CLL cells is currently
unknown.
The vast majority of the circulating B-CLL cells is arrested in G0/G1 phase of the cell
cycle7, which has contributed to the view that B-CLL is an indolent disease. However,
isotopic labeling of leukemic cells in vivo revealed that a substantial fraction of the
B-CLL cells does proliferate8. It seems logical to assume that the generation of new
cells takes place in so called proliferation centers frequently found in lymph nodes
and bone marrow of B-CLL patients. This is supported by the numerous Ki67+ and
Survivin+ cells present in these structures1;9. The microenvironment not only plays an
essential role in the induction of proliferation but presumably also in the suppression
of apoptosis. In vitro experiments revealed that various cell types can support the
survival of B-CLL cells. Apart from follicular dendritic cells (FDC), bone marrow
stromal cells, IL-6 producing endothelial cells, VCAM-1 and SDF-producing nurse-
like cells, CD4+ T cells can also aid in providing a microenvironment where B-CLL
cells can survive and proliferate10-14. The importance of the microenvironment for
the survival of B-CLL cells is also shown by the fi nding that despite the relentless
accumulation of the B-CLL cells in vivo, culturing the leukemic cells in vitro results in
spontaneous apoptosis15;16. In vitro culture of B-CLL cells in the presence of CD40L
rescues the cells from spontaneous and drug-induced apoptosis, suggesting that
such co-stimulatory signals play a role in the survival of B-CLL cells in vivo and even
in the response to treatment9;17-19.
To date, B-CLL is an incurable disease. Although multi-agent treatment can result
in a profound peripheral lymphocyte depletion, the B-CLL cells in the bone marrow
and/or lymph nodes are less effectively targeted20. Persistence of B-CLL cells in
the bone marrow is associated with an increased risk of relapse21. Therefore, more
molecular data about the B-CLL cells in the lymphoid tissues and bone marrow are
Thesis_Final_2_01092008.indd Sec1:77Thesis_Final_2_01092008.indd Sec1:77 01-09-2008 11:08:1601-09-2008 11:08:16

78necessary, preferably coupled with assessment of effi cacy of therapeutics towards
B-CLL residing in those niches. We here initiated such an effort by comparing a large
panel of apoptosis regulators in circulating B-CLL cells and B-CLL cells residing in
lymph nodes. Although the expression of most apoptosis regulators was remarkably
comparable, a prominent difference was the expression of the BH3-only protein Noxa.
Furthermore, we demonstrate that CD40 engagement of peripheral B-CLL cells can
largely reproduce the altered apoptosis profi le found in lymph node B-CLL cells.
Finally, we show that in vitro manipulation of Noxa expression has a signifi cant and
direct effect on B-CLL cell survival. Together, these data provide a new link between
the anti-apoptotic microenvironment in the lymph nodes and suppression of Noxa,
which suggests that drugs that increase Noxa levels, such as proteasome inhibitors22-25,
may be of therapeutic benefi t in B-CLL.
MATERIAL EN METHODS
Patient material and cell lines
Patient material was obtained after routine diagnostic or follow-up procedures at
the departments of Hematology and Pathology of the Academic Medical Center
Amsterdam. All patients were diagnosed according to the WHO classifi cation system1.
Lymph node (LN) material diffusely infi ltrated by B-CLL cells was freshly frozen in
liquid nitrogen directly after surgical removal. Immuno-histochemical analysis (see
below) of these lymph nodes revealed that more than 90% of the tissue consisted
of tumor cells. Peripheral blood (PB) mononuclear cells (PBMC) of B-CLL patients
were obtained after Ficoll density centrifugation (Pharmacia Biotech, Roosendaal,
the Netherlands). PBMCs from B-CLL patients contained >75% CD5+, CD19+ cells
as assessed by fl ow cytometry and were stored in liquid nitrogen as cell suspensions
in 10% DMSO (Merck, Darmstadt, Germany) in heat-inactivated FCS (Invitrogen,
Breda, The Netherlands). Clone FSA of the Burkitt’s lymphoma cell line Ramos with
enhanced response to CD95 has been described previously24. Cell lines were cultured
in Iscove’s modifi ed Dulbecco’s medium (IMDM; Invitrogen), supplemented with 10%
(v/v) heat-inactivated FCS (ICN Biomedicals GmbH, Meckenheim, Germany), 100 U/
ml penicillin, 100 μg/ml streptomycin and 5 mM L-glutamine (Invitrogen).This study
was conducted in accordance with the ethical standards in our institutional medical
committee on human experimentation, as well as in agreement with the Helsinki
Declaration of 1975, revised in 1983.
Thesis_Final_2_01092008.indd Sec1:78Thesis_Final_2_01092008.indd Sec1:78 01-09-2008 11:08:1601-09-2008 11:08:16

79RNA isolation and reverse transcription-multiplex ligation-
dependent probe amplifi cation assay (RT-MLPA)
Total RNA was isolated using the Nucleospin RNA isolation kit (Macherey-nagel,
Düren, Germany). RT-MLPA procedure was performed as described previously5;25.
Briefl y, 100 ng total RNA was reverse transcribed using a gene-specifi c probe mix.
The resulting cDNA was annealed overnight at 60°C to the MLPA probes. Annealed
oligonucleotides were covalently linked by Ligase-65 at 54°C (MRC, Amsterdam, The
Netherlands). Ligation products were amplifi ed by polymerase chain reaction (PCR;
33 cycles, 30 seconds at 95°C, 30 seconds at 60°C and 1 minute at 72°C) using
one unlabelled and one 6-carboxy-fl uorescein (FAM)-labeled primer (10 pM). PCR
products were run on an ABI 3100 capillary sequencer in the presence of 1pM ROX
500 size standard (Applied biosystems, Warrington, UK). Results were analyzed
using the programs Genescan analysis and Genotyper (Applied Biosystems).
Category tables containing the area for each assigned peak (scored in arbitrary
units) were compiled in Genotyper and exported for further analysis with Microsoft
Excel spreadsheet software. Data were normalised by setting the sum of all signals
at 100%, and expressing individual peaks relative to the 100% value.
Immunohistochemistry
Monoclonal antibodies specifi c for CD5 (clone 4C7 Lab vision, Neomarkers, Fremont,
CA), CD3 (clone SP7), CD23 (clone 1B12), Bcl-6 (clone PG-B67), were used on
formalin-fi xed paraffi n-embedded lymph node specimens. When necessary, antigen
retrieval was achieved using a TRIS-EDTA buffer pH 9.2. Antibody detection was
performed with the Powervision+ system (ImmunoVision Technologies, Daly City, CA)
which was succeeded, for the single antibody staining, by peroxidase visualization
with 3,3’-diaminobenzidine (DAB) (Sigma), 0.03% H2O
2 in Tris-HCl pH 7.6. Finally, the
sections were counterstained with haematoxylin, dehydrated and mounted in pertex.
For the CD3/Ki67 double stainings the Ki67 MIB-1 clone (Dako, Glostrup, Denmark)
was used, for the CD20/Ki67 double staining the Ki67 SP6 clone (Neomarkers),
and the L26 clone from Dako. After antibody detection with the Powervision+
system (ImmunoVision) the Liquid permanent red kit (Dako) was used, followed by
peroxidase visualization with DAB (Sigma). Finally the slides were counterstained
with haematoxylin and mounted in Vectamount.
Flow cytometry
Purifi ed B-CLL cells were incubated FITC- or PE-conjugated mAbs directed against
CD5 (Sanquin), CD19 (Sanquin) and CD3 (Becton and Dickinson, San Jose, CA) and
Ch
ap
ter 4
▪ Ap
op
tosis
gen
es in
CL
L ly
mp
h n
od
e v
ers
us p
erip
hery
Thesis_Final_2_01092008.indd Sec1:79Thesis_Final_2_01092008.indd Sec1:79 01-09-2008 11:08:1601-09-2008 11:08:16

80analyzed by fl ow cytometry with the CellQuest program on a FACS Calibur (Becton
and Dickinson).
In vitro CD40 stimulation
B-CLL samples were enriched to >95% purity from PBMCs via negative depletion as
described previously26. In brief, T cells, monocytes and granulocytes were depleted
using anti-CD3, anti-CD14 and anti-CD16 immunomagnetic beads on a magnetic
particle concentrator (Dynal A.S. Oslo, Norway). The B-CLL cells were stimulated for
three days in culture-treated 24-wells plates (Costar, Corning NY, USA). Each well
contained 5 x 106 B-CLL cells and 1.5 x 105 irradiated (30 Gy) CD40L-transfected or
untransfected fi broblast (NIH3T3).
Retroviral constructs and transduction
To knock-down Noxa, pRetro-super was used, which contains the polymerase III H1-
RNA promoter (pol3) for transcription of the siRNA probe and the phosphoglycerin
kinase (pgk)1 promoter driving GFP expression27. The siRNA sequences were: N7
5’GAAGGTGCATTCATGGTG3’ and N8 5’GTAATTATTGACACATTTC3’. The retroviral
plasmids were transfected into the helper virus amhotropic producer cell line Phoenix
with Fugen-6 (Roche Diagnostics, Almere, The Netherlands). For transduction,
Ramos-FSA cells were exposed overnight to viral supernatant (containing vector
GFP-only or one of the two Noxa RNAi-targeting sequences) on retronectin-coated
(Takara Shuzo, Otso, Japan) 24-well plates. GFP-positive cells were sorted using a
FACS-Aria (BD Biosciences) cell sorter to >90% purity for further experiments.
Analysis of apoptosis
PB B-CLL cells were stimulated at a concentration of 5x106 cells/ml with 20 nM
bortezomib (Janssen-Cilag, Tilburg, The Netherlands) for four hours. The cells were
washed twice with IMDM (when indicated) and incubated at given time points with
FITC-labeled Annexin-V (IQ products, Groningen, The Netherlands) for 20 minutes.
Prior to analyses, PI was added (fi nal concentration 5 µg/ml). Viable cells were
defi ned by Annexin V-/PI- staining. Ramos.FSA clones expressing either control-GFP
or Noxa-RNAi (N7 or N8) were stimulated at a concentration of 5x105 cells/ml with
30 nM bortezomib for 24 hours, harvested and incubated with 200 nM MitoTracker
Orange (Molecular Probes, Leiden, The Netherlands) for 30 minutes at 37°C,
washed and double-stained with APC-labeled Annexin-V (IQ products). Fludarabine,
staurosporine and propidium iodide (PI) were purchased from Sigma Chemical Co.
(St. Louis, MO, USA). Anti-human Fas10 (agonistic antibody to the CD95 receptor)
were a kind gift from Prof. Dr. L. Aarden (Sanquin, Amsterdam, The Netherlands).
Thesis_Final_2_01092008.indd Sec1:80Thesis_Final_2_01092008.indd Sec1:80 01-09-2008 11:08:1601-09-2008 11:08:16

81Western blotting
Western blotting was done as described previously5. Protein samples were separated
by 13% sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE)
followed by western blotting. Blots were probed with the following antisera: polyclonal
Mcl-1 (cat. no 554103, Pharmingen, BD Biosciences), monoclonal anti-Noxa (clone
114C307.1, Imgenex, San Diego, CA, USA), monoclonal anti-Bim (clone 14A8,
Chemicon, Temecula, CA, USA) and antiserum to -actin (clone I-19, Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA). Protein bands were quantifi ed using high
resolution (1200 dpi) scanned images of exposed fi lms and AIDA image analyzer
software v3.5 (Raytest Gmbh; Straubenhardt, Germany). Exposed fi lms were only
considered when software indicated that bands were not overexposed. In each
sample, background corrected intensity of Mcl-1 or Noxa bands were normalized for
actin.
Statistical analyses
The Mann Whitney U test was used to analyze if differences in gene expression
between the PB and LN B-CLL were statistically signifi cant. P-values < 0.01 were
considered statistically signifi cant. Densitometric scans of western blots and MLPA
analyses of CD40-triggered CLL cells were analyzed with Student’s T-test. P-values
<0.05 were considered statistically signifi cant.
RESULTS
Patients characteristics and immunohistochemistry
Lymph nodes from 9 B-CLL patients and peripheral blood samples from 15 B-CLL
patients were included in the study. From 2 patients (B-CLL25 and B-CLL31) both
lymph node (LN) tissue and a peripheral blood (PB) sample was available (Table 1).
All B-CLL expressed CD5, CD23 and CD19/CD20. The B-CLL cells of the patients
with LN involvement expressed unmutated immunoglobulin heavy chain (IgVH)
genes. Of the peripheral blood B-CLL, 10 expressed mutated IgVH genes and 8
unmutated IgVH genes (Table 1). In the peripheral blood samples, at least 75% of
the leucocytes were lymphocytes. Due to low levels of CD5 expression, the standard
FACS gating yielded low percentage of CD5/CD19+ cells in some patients; Patient
30 had in addition low numbers of circulating cells and was fi rst diagnosed as small
lymphocytic leukemia (SLL). Immunohistochemistry demonstrated that >90% of the
LNs consisted of leukemic lymphocytes. Ki67+ cells were present in all LNs, either
diffusely or in proliferation centers. These cells were of B cell origin, as demonstrated
by double staining which showed that all Ki67+ cells were also CD20+. In contrast, the
Ch
ap
ter 4
▪ Ap
op
tosis
gen
es in
CL
L ly
mp
h n
od
e v
ers
us p
erip
hery
Thesis_Final_2_01092008.indd Sec1:81Thesis_Final_2_01092008.indd Sec1:81 01-09-2008 11:08:1601-09-2008 11:08:16

82scattered CD3+ T cells were generally negative for Ki67. Absence of clusters of Bcl-6+
or CD21+ (data not shown) cells excluded the presence of germinal center remnants
in these LNs (Fig.1).
Figure 1: Histology of lymph node infi ltrated by B-CLL cells. See color fi gures.
Ubiquitously present B-CLL cells were positive for CD23 and CD5. Scattered CD3+ T cells were present
throughout the LN. The absence of clusters of BCl-6+ cells excluded the presence of germinal center
remnants in the LNs. Ki67/CD20 and Ki67/CD3 double staining indicate that all cycling Ki67+ cells (pink)
were of CD20+ (brown) origin - see also inset -, while the CD3+ T cells were predominantly Ki67 negative.
Magnifi cation 40x.
Profi ling of apoptosis genes in peripheral blood and LN samples of B-CLL
The relative expression 34 known apoptosis regulators was investigated by RT-
MLPA5;25;28 in PB samples of 13 B-CLL patients, LN samples of 7 B-CLL patients (Fig.
2A) and paired PB and LN samples of 2 B-CLL patients (Fig. 2B and data not shown).
The relative expression of the majority of the investigated genes was remarkably
comparable between the PB and LN samples. We have described previously that,
compared to normal tonsillar B cell fractions, in PB B-CLL several anti- and pro-
apoptosis genes (e.g. Flip, Bcl-2, Noxa and Bmf) are aberrantly expressed5, and this
was also found in LN samples of B-CLL. Interestingly, 3 genes were differentially
expressed in PB B-CLL cells as compared to LN samples (Fig. 2C). In agreement
with previous reports, the IAP family member Survivin was not expressed in any of
the PB B-CLL samples whereas it was clearly expressed in LN B-CLL5;9 (P=0.0005).
Also, the anti-apoptotic Bcl-2-family member Bcl-XL was more abundantly expressed
in the LN samples (P=0.0003). The most striking difference in expression was
observed for the BH3-only member Noxa. As found previously, this apoptogenic gene
is abundantly expressed in PB B-CLL cells5, but its expression was clearly lower in LN
Thesis_Final_2_01092008.indd Sec1:82Thesis_Final_2_01092008.indd Sec1:82 01-09-2008 11:08:1701-09-2008 11:08:17

83B-CLL cells (averaged relative expression of 15.6 ± 9.8 in PB B-CLL versus 3.0±1.1
in LN B-CLL; P<0.0001 Fig. 2C). Of note, a difference in Noxa expression was also
observed between the paired PB- and LN samples of an individual patient (relative
expression 9.6 in the PB sample versus 3.4 in the LN sample) (Fig. 2B). Western
blot analyses confi rmed that the differences in Bcl-XL and Noxa mRNA expression
were also present at the protein level. The B-CLL LN samples generally expressed
lower levels of Noxa than the PB B-CLL samples, and the reverse was observed for
A
Ch
ap
ter 4
▪ Ap
op
tosis
gen
es in
CL
L ly
mp
h n
od
e v
ers
us p
erip
hery
B
C
Figure 2: Apoptosis gene expression profi le of B-CLL cells in peripheral blood and lymph nodes.
(A) Relative expression of 34 apoptosis regulators was investigated in 15 PB B-CLL (black bars) and 9
LN B-CLL (grey bars). Results of individual apoptosis regulatory genes are shown as expression relative
to the total signal in the sample, with standard deviation. Non-apoptosis genes included as housekeeping
genes are 2-microglobulin (B2M), Ferritin Light chain (FLT), -glucoronidase (GUS), and poly(A)-specifi c
ribonuclease (PARN).
(B) RT-MLPA data from PB and LN sample of B-CLL-25.
(C) The expression of Noxa, Survivin, Bcl-XL and Mcl-1 in individual patients are depicted as dots. Asterix (*)
indicates statistical signifi cance (P<0.001) of differences in gene expression between PB and LN B-CLL.
Thesis_Final_2_01092008.indd Sec1:83Thesis_Final_2_01092008.indd Sec1:83 01-09-2008 11:08:1701-09-2008 11:08:17

84
Figure 3: Comparison of Noxa, Mcl-1 and Bcl-XL protein in PB vs. LN B-CLL.
Protein lysates of 7 PB samples and 6 LN samples were subjected to Western blot analyses.
(A) Blots were stained with antibodies directed against Noxa, Mcl-1 or Bcl-XL, and reprobed with an
antibody against -actin as a loading control. In case of Bcl-XL, aspecifi c staining at the upper cutting edge
of the blot is visible and precluded analysis of the rightmost two samples.
(B) Densitometric scanning was performed, and averaged Noxa/actin and Mcl-1/actin ratios are plotted in
PB and LN samples. Unpaired T-test showed that Noxa ratios were statistically signifi cant (P=0.0026), and
Mcl-1 ratios showed a non-signifi cant trend.
A
B
Bcl-XL (Fig. 3). Comparison of the RT-MLPA data with the Western blot data revealed
a clear correlation between the levels of Noxa mRNA and Noxa protein. As reported
previously, no differences were observed in expression of these apoptosis genes
among IgVH-mutated versus unmutated cases5.
Since Noxa can selectively interact with the anti-apoptotic protein Mcl-1 and this may
infl uence the degradation of Mcl-129, we investigated the expression of this Bcl-2
family member in PB B-CLL and LN B-CLL. Although RT-MLPA showed no difference
in mRNA expression (Fig. 2C), Western blot analyses revealed that in most LN
B-CLL, where Noxa levels were low, Mcl-1 was slightly elevated. Furthermore, the
PB samples that expressed higher levels of Noxa showed a decreased expression
of Mcl-1 (Fig. 3). This is further illustrated by a paired PB/LN protein sample, where
in fact Noxa expression was almost equal, but in this case Mcl-1 protein levels were
clearly higher in the LN compartment. Densitometric scanning of Noxa and Mcl-1
protein levels also showed divergence between LN and PB, of which the differences
in Noxa levels were statistically signifi cant (Fig. 3B). In summary, the majority of the
apoptotic regulators are expressed equally in PB - and LN B-CLL, but a novel and
prominent distinction in Noxa level was found.
Thesis_Final_2_01092008.indd Sec1:84Thesis_Final_2_01092008.indd Sec1:84 01-09-2008 11:08:1801-09-2008 11:08:18

85Noxa expression is modulated by CD40 engagement in B-CLL cells
In the LNs the CD40+ B-CLL cells are in close contact with T cells that may express
CD40L2 (Fig. 1). To investigate the effect of this interaction on the expression of
the apoptotic regulators, PB B-CLL samples (n = 11) were co-cultured for 1-5
days with CD40L-transfected or untransfected 3T3 fi broblasts (Fig. 4). As reported
previously, CD40 stimulation resulted in increased expression of Bcl-XL, A1/Bfl -1,
Bid and Survivin9;17;18. Interestingly, in accordance with our fi ndings in the LN B-CLL
cells, CD40L-stimulated PB B-CLL cells also showed a diminished expression of
Noxa (Fig. 4A). The effects were observed after one day of CD40 stimulation and
RT-MLPA performed at day three and day fi ve showed that the expression of the
apoptosis regulators did not alter signifi cantly after that (Fig. 4B). It should be noted
that the levels of Noxa mRNA as compared to t=0 also decreased after culture on
the control 3T3 cells (p=0.019). The reason for this is not known, however a stronger
decline in Noxa levels was consistently observed after CD40 ligation (p=0.004), and
the difference between control and CD40-treated cells was statistically signifi cant
(p=0.016). These differences were further investigated at the protein level for three
patients (see Fig. 4C). Concordant with RT-MLPA analyses, Noxa levels decreased
after 3 days culture in presence of CD40L-expressing cells, and Bcl-XL levels
increased. Mcl-1 protein levels also clearly increased upon CD40-triggering, although
this was not observed via RT-MLPA. So, similar to fi ndings in LN samples (Fig. 3),
Mcl-1 levels were apparently under post-transcriptional control.
A prominent distinction between CD40-stimulated B-CLL and LN B-CLL was found
for expression of the apoptogenic BH3-only protein Bid. In contrast to LN B-CLL,
CD40L- stimulated B-CLL showed a strong and continuous induction of Bid (Fig. 4B).
Thus, the altered gene expression in LNs can be mimicked largely but not entirely by
in vitro CD40 engagement of B-CLL cells.
A
Ch
ap
ter 4
▪ Ap
op
tosis
gen
es in
CL
L ly
mp
h n
od
e v
ers
us p
erip
hery
Thesis_Final_2_01092008.indd Sec1:85Thesis_Final_2_01092008.indd Sec1:85 01-09-2008 11:08:1901-09-2008 11:08:19

86B
C
Figure 4: CD40 stimulation of peripheral blood B-CLL results in an apoptosis gene expression
profi le similar to lymph node B-CLL.
(A) Apoptosis gene expression profi le was investigated by RT-MLPA in PB samples of 11 freshly isolated
B-CLL patients without culturing (black bars) and after three days of culturing on either 3T3 cells (grey bars)
or CD40L-transfected 3T3 cells (white bars). Data plus standard deviation are presented as in fi gure 2.
(B) The expression of Bcl-XL, Bfl -1/A1, Bid and Noxa are shown at day 1, 3 and 5 of culturing on 3T3
cells (white triangles) or CD40L-transfected 3T3 cells (black dots). Statistical analysis of day 0 vs. day 1
samples showed that in all cases the CD40L treated values were signifi cantly different (P<0.01). In case
of Noxa, there was also a small but signifi cant decrease for the 3T3 control cells (P=0.019), and a more
pronounced effect for CD40L treated cells (P=0.004; difference between 3T3 and CD40L-treated cells
P=0.0159, indicated by **).
(C) Western blot of t=0 samples in comparison of CD40L treated cells at day 3 for Noxa, Mcl-1 and Bcl-
XL showed that Noxa protein levels decrease while Mcl-1 and Bcl-X increase. For B-CLL sample 226 the
Mcl-1 levels at t=0 were in fact undetectable (see also Fig. 3).
Thesis_Final_2_01092008.indd Sec1:86Thesis_Final_2_01092008.indd Sec1:86 01-09-2008 11:08:1901-09-2008 11:08:19

87Bortezomib-induced Noxa upregulation causes apoptosis of PB
B-CLL cells
To establish a functional relationship between Noxa expression levels and apoptosis
sensitivity of B-CLL cells, we made use of recent fi ndings that proteasome inhibitors
rapidly and specifi cally upregulate Noxa22;23;30. To reduce a widespread impact of
proteasome inhibition on protein levels and transcription dependent processes31,
PB B-CLL cells were transiently exposed to bortezomib for 4 hours.The reversible
proteasome inhibitor was then either washed away or incubation was continued. As
expected, a pulse of bortezomib treatment already caused a rise in Noxa protein, and
this was suffi cient to impair survival of B-CLL cells (Fig. 5). Continuous exposure to
bortezomib resulted in a massive increase in Noxa levels that was accompanied by
almost 100% cell death at 48 hrs. Over the course of this experiment, Mcl-1 protein
levels fi rst increased (4 hr timepoint), most likely due to proteasome inhibition, and
then declined when cells went into apoptosis. This decline could be prevented by
blocking caspase activity with z-VAD (data not shown), and is thus in accord with
reports that Mcl-1 is a caspase substrate32;33. Next, we investigated whether the level
of Bim, another pro-apoptotic binding partner of Mcl-134, was also subject to change
upon bortezomib treatment, and might thereby trigger apoptosis. However, Bim levels
were unaffected, both as detected by RT-MLPA (data not shown), and by Western
blotting (Fig. 5A). Thus, pharmacological manipulation of the levels of Noxa protein in
B-CLL cells appeared to be directly related to viability in an in vitro setting.
Figure 5: Noxa upregulation via transient treatment with bortezomib impacts CLL survival.
Freshly isolated peripheral blood B-CLL cells were treated for 4 hrs with 20 nM of the proteasome inhibitor
bortezomib. Cells were then washed and cultured in fresh medium, or incubation was continued.
(A) At the indicated timepoints, cell lysates were prepared and probed for expression of Noxa, Mcl-1, Bim
and Actin protein by Western blot. Indicated below the lanes: untreated (M), bortezomib washed away
after 4 hrs (B+), and bortezomib without washing (B-). The decrease in Mcl-1 levels in bortezomib treated
cells at 24 and 48 hrs could be inhibited by the pan-caspase inhibitor z-VAD (data not shown). Due to
massive cell death after 48 hrs in the presence of bortezomib, these lysates did not yield suffi cient protein
for analysis.
(B) Apoptosis of cells was determined via AnnexinV staining. Spontaneous apoptosis in medium was
approximately 50%, which was increased by bortezomib treatment. Results are representative for 3
separate experiments.
A B
Ch
ap
ter 4
▪ Ap
op
tosis
gen
es in
CL
L ly
mp
h n
od
e v
ers
us p
erip
hery
Thesis_Final_2_01092008.indd Sec1:87Thesis_Final_2_01092008.indd Sec1:87 01-09-2008 11:08:2001-09-2008 11:08:20

88Noxa-defi cient cells exhibit resistance to bortezomib-induced cell death
Apoptosis regulatory genes as detected via RT-MLPA were not affected during the
short-term bortezomib treatment in the previous experiments (data not shown). Yet,
it can not be excluded that other genes and proteins besides Noxa that might impact
survival were affected by bortezomib. Therefore, to investigate a direct role for Noxa in
bortezomib-induced apoptosis, we employed a model system. Ramos B cells (clone
FSA)24 were transduced with distinct retroviral constructs encoding Noxa siRNAs (N7
or N8) or control-GFP. GFP-positive cells were sorted and Western blot analysis
revealed a suppression of Noxa-levels to approximately 50-75% compared to the
control-GFP (Fig. 6A). Both Ramos FSA cell lines expressing Noxa RNAi exhibited
a signifi cant resistance to bortezomib-induced apoptosis compared to the mock-
transduced cells (Fig. 6B). The partial resistance to proteasome inhibitor-mediated
apoptosis matched the partial knock-down of Noxa protein. Of note, also in Noxa
RNAi cells, bortezomib treatment caused a rapid increase in Noxa protein (data not
shown), thus explaining that apoptosis still occurred at higher concentration of the
drug. These data are in good agreement with effects of Noxa knock-down in other
celltypes (melanoma, mantle cell lymphoma and T cell leukemia)22;23;30. In addition,
we obtained similar fi ndings with another protease inhibitor (MG132; data not
shown). In contrast, no effect of Noxa protein reduction was observed on apoptosis
triggered via other pathways such as fl udarabine or staurosporin treatment, or
triggering of the CD95 receptor (Fig. 6C). In conclusion, these data demonstrate that
decreased expression of Noxa has a direct and specifi c impact on the susceptibility
to apoptosis induced by proteasome inhibitors. Conversely, the death-inducing effect
of proteasome inhibition observed in B-CLL cells may therefore rely predominantly
on shifts in Noxa expression.
Thesis_Final_2_01092008.indd Sec1:88Thesis_Final_2_01092008.indd Sec1:88 01-09-2008 11:08:2101-09-2008 11:08:21

89
DISCUSSION
There is increasing awareness that the B-CLL population in lymphoid proliferation
centers differs fundamentally from the well studied fraction in PB and that this
distinction may have clinical relevance8;35. Here, we present a fi rst direct comparison
of these two populations, focusing on the expression of 34 apoptosis regulatory
genes. Apart from expected differences in proliferation-related genes (Survivin
and Ki67) and anti-apoptotic Bcl-XL, we observed a prominent divergence in the
expression of pro-apoptotic Noxa. Previously we described that, compared to non-
malignant tonsil or peripheral B-cell fractions, B-CLL cells in the periphery display
Ch
ap
ter 4
▪ Ap
op
tosis
gen
es in
CL
L ly
mp
h n
od
e v
ers
us p
erip
hery
Figure 6: Noxa reduction via RNAi specifi cally
prevents apoptosis induction by proteasome
inhibitors.
Ramos Burkitt lymphoma cells were retrovirally
transduced with two RNAi constructs targeting
Noxa (N7 or N8), or GFP control.
(A) Western blot demonstrating reduced Noxa
expression in Ramos-N7 and -N8. Equal protein
loading is shown by reprobing for β-Actin.
(B) Mock, N7 and N8 transduced Ramos FSA
cells were cultured 24 hours in the presence of in-
dicated concentration of bortezomib. Viability was
assessed by AnnexinV/mitotracker staining and
FACS analysis. Data represent mean ± SD from
three independent experiments.
(C) Cells were incubated for 24 hrs in medium
containing 100 µM fl udarabine (fl uda), 0.25 µM
staurosporine (stauro), or 5 µg/ml α-CD95, and
analysed as in B.
A
B
C
Thesis_Final_2_01092008.indd Sec1:89Thesis_Final_2_01092008.indd Sec1:89 01-09-2008 11:08:2101-09-2008 11:08:21

90signifi cantly increased levels of this BH3-only member of the Bcl-2 family, in a p53-
independent manner5. The high levels of Noxa and another BH3-only member Bmf6,
contrasted with the purported anti-apoptotic phenotype of B-CLL cells2 but remained
functionally unexplained. Our new fi ndings show that the Noxa level is considerably
lower in LN CLL and that this is linked with survival capacity. Therefore, targeting
Noxa expression or function could be of clinical benefi t, also in p53 defi cient cases.
In vitro CD40 stimulation of PB B-CLL cells resulted in a clear reduction of Noxa
expression. Within the LN microenvironment, CD40 stimulation is most likely
delivered by CD40L+ T cells. Several groups have investigated the effects of in
vitro CD40 engagement in B-CLL cells9;14;17;18;36-39 but an effect on Noxa expression
was not yet reported. It is well known that CD40-stimulated B-CLL cells are more
resistant to spontaneous or drug-induced apoptosis. This is most probably due to the
induction the transcription factor nuclear factor κB (NFκB) and as a consequence, the
expression of various anti-apoptotic genes, such as Bcl-XL, cIAP2, A20 and Flip36.
Previously, Noxa was proposed to be a p53-response gene40, but in B-CLL cells,
Noxa is apparently not under control of p53, as illustrated by the clearly divergent
expression of Puma and Noxa upon p53 stimuli5;41. Later, various transcription factors
were proposed to regulate Noxa such as E2F1, p73 and hypoxia inducible factor
HIF-1α42-45. Therefore at present it is diffi cult to defi nitely assign a specifi c signaling
route that mediates Noxa expression. Very recently though, it was reported that HIF-
1α is overexpressed in peripheral B-CLL cells45, which may constitute a potential link
to the increased Noxa levels in B-CLL.
Although CD40 stimulation of PB B-CLL cells resulted in a similar apoptosis gene
expression profi le to LN B-CLL, several genes deviated from this profi le, most
prominently Bid, as reported previously17, but also A1/Bfl -1. This indicates that in the
LN, B-CLL cells also receive other stimuli than CD40. Indeed, apart from CD4+ T
cells expressing CD40L, other cell types can support survival of B-CLL cells. In vitro
culture with an FDC cell line or dendritic cells protects B-CLL cells from spontaneous
apoptosis14;46. FDC-mediated survival was reported to depend on the expression of
the Bcl-2 family member Mcl-114 and in vitro experiments revealed that Mcl-1 levels
decline in B-CLL cells undergoing apoptosis3;47. Interestingly, recent data indicate
that Mcl-1 is a preferred binding partner of Noxa34, and we have indeed observed
association of Mcl-1 with Noxa in primary B-CLL samples (D. Hallaert; manuscript
in preparation). Furthermore, in 293T cells, Noxa has been described to mediate
the degradation of Mcl-129. If this mechanism also holds true for B-CLL cells, it may
explain the increase in Mcl-1 protein we observed in LN B-CLL, which was not
accompanied by an increase in Mcl-1 mRNA. Accordingly, augmented Mcl-1 protein
levels are a consequence of the downregulation of Noxa in the LNs, rather than a
Thesis_Final_2_01092008.indd Sec1:90Thesis_Final_2_01092008.indd Sec1:90 01-09-2008 11:08:2101-09-2008 11:08:21

91environmental effect on Mcl-1 RNA expression. In addition, in vitro triggering of CD40
on B-CLL cells also infl uenced Mcl-1 levels in a post-transcriptional fashion (Fig. 4).
Thus, the differences in protein levels observed by us for Noxa, Mcl-1 and Bcl-XL
levels in the B-CLL LN environment, correspond with current models based on the
differential interaction potential of these Bcl-2 family members29;34, and support the
anti-apoptosis phenotype of B-CLL cells at this location compared to PB. In addition,
spontaneous apoptosis in vitro of B-CLL cells may be connected with the high levels
of Noxa which eventually saturate the short-lived Mcl-1 protein29;48.
Two separate experimental approaches supported a direct role for Noxa in survival
capacity of B-CLL cells. First, we used the recently discovered rapid and direct effect of
bortezomib on Noxa levels22;23;30 to demonstrate that short term bortezomib exposure
also quickly induced Noxa protein in B-CLL cells, with a corresponding decrease
in viability (Fig. 5). Although the levels of Bim did not change upon bortezomib
treatment, a role for Bim during the actual triggering phase of apoptosis cannot be
excluded. In model systems, Bim is capable of actively triggering Bax activation, while
Noxa functions in as ‘sensitiser’49;50. Secondly, a complementary experiment was
performed in a model system where only Noxa levels were modifi ed via RNAi. Here,
a clear inhibitory effect of Noxa reduction towards apoptosis mediated by proteasome
inhibition was observed, while other apoptosis pathways were unaffected (Fig. 6).
Taken together, our data support a model where the viability of the malignant B-CLL
clone within the LNs and possibly also the bone marrow corresponds with low levels
of Noxa and an upregulation of Bcl-XL and Mcl-1. In addition to these anti-apoptotic
gene expression alterations, the B-CLL cells also receive proliferative stimuli as
indicated by the Ki-67+ and Survivin+ cells. When the B-CLL cells enter the circulation
these stimuli are lost, Noxa is upregulated, and Bcl-XL, Mcl-1 and Survivin are
downregulated. As a result, the B-CLL cells may become prone to apoptosis, which
can however still be prevented by the continuous high expression of Bcl-2. To what
extent circulating B-CLL are actually undergoing apoptosis is diffi cult to detect directly.
Freshly isolated CLL cells are mostly non-apoptotic but undergo rapid ‘spontaneous’
apoptosis in vitro, and recent calculations point to appreciable in vivo death rates8.
Together, this suggests that apoptotic B-CLL cells are rapidly cleared from circulation
in vivo. It is generally assumed that in the LNs and bone marrow the B-CLL cells are
relatively protected against therapeutic drugs20. The circulating B-CLL cells that are
already prone to apoptosis are more easily targeted, but the residual B-CLL cells in
the LN/bone marrow will eventually lead to a relapse. Currently, there is much interest
in application of novel, p53-independent drugs to treat B-CLL51-53. Our data provide
new insight into the regulation of the apoptotic behavior of B-CLL cells, and also
afford new clues for therapeutic intervention by targeting Noxa expression.
Ch
ap
ter 4
▪ Ap
op
tosis
gen
es in
CL
L ly
mp
h n
od
e v
ers
us p
erip
hery
Thesis_Final_2_01092008.indd Sec1:91Thesis_Final_2_01092008.indd Sec1:91 01-09-2008 11:08:2101-09-2008 11:08:21

ACKNOWLEDGEMENTWe are grateful to the patients for donating samples and the clinicians involved for
their collaboration. This study was initiated after suggestions from Professor Steven
Pals (Dept of Pathology of the AMC) that investigation into survival and apoptosis of
B-CLL cells should include lymph nodes. The authors would like to thank JBG Mulder
and AR Musler for immunohistochemical stainings.
92
Thesis_Final_2_01092008.indd Sec1:92Thesis_Final_2_01092008.indd Sec1:92 01-09-2008 11:08:2101-09-2008 11:08:21

93
Ch
ap
ter 4
▪ Ap
op
tosis
gen
es in
CL
L ly
mp
h n
od
e v
ers
us p
erip
hery
LN indicates lymph node; PB, peripheral blood; ND, not done.
*Lymphocytes were investigated in the lymph node samples by immunohistochemistry and in the peripheral
blood samples by FACS analysis.
†IgVH mutations were positive if 2% of the IgV
H gene was mutated.
‡These samples were used only for Western blot analyses.
§These samples displayed low CD5 staining; therefore, the combined CD5/CD19 gate yielded low
values.
Patient Location Age,
year
Rai
stage
Lymph,*
%
CD5+CD19+,
%
CD19+,
%
CD3+,
%
IgVH
mutations †
B-CLL24 LN 52 >90 ND ND ND -
B-CLL27 LN 64 1 >90 76 76 10 ND
B-CLL28 LN 80 4 >90 64 93 8 -
B-CLL30 LN 46 3 >90 37# 72 26 -
B-CLL33 LN 76 2 >90 ND ND ND -
B-CLL35 LN 67 2 >90 ND ND ND -
B-CLL36 LN 59 4 >90 93 93 5 -
B-CLL37 PB 62 1 ND ND ND ND +
B-CLL38 PB 62 0 98 97 97 2 +
B-CLL39 PB 72 3 ND 94 98 3 +
B-CLL40 PB 70 3 93 48# 98 1 -
B-CLL41 PB ND 4 94 99 99 1 -
B-CLL42 PB 48 0 69 76 76 17 -
B-CLL43 PB 54 2 91 96 96 4 -
B-CLL44 PB 55 2 86 91 91 0 +
B-CLL45 PB 63 4 82 98 98 2 +
B-CLL46 PB ND ND 82 ND ND ND ND
B-CLL47 PB 54 2 83 ND ND 7 -
B-CLL48 PB 59 2 75 89 89 4 +
B-CLL50 PB 69 0 ND 99 99 3 +
B-CLL167‡ PB 70 0 62 84 86 11 +
B-CLL183‡ PB 78 0 47 94 95 4 +
B-CLL226‡ PB 64 1 70 98 98 1 -
B-CLL261‡ PB 77 4 32 92 92 4 +
B-CLL25 PB 68 2 ND ND 78 13 -
LN 2 >90 82 83 20 -
B-CLL31 PB 72 2 ND 81 81 8 -
LN 2 >90 72 81 12 -
Table 1. Patient and B-CLL sample characteristics.
Thesis_Final_2_01092008.indd Sec1:93Thesis_Final_2_01092008.indd Sec1:93 01-09-2008 11:08:2101-09-2008 11:08:21

94 REFERENCES
1. Jaffe ES SH and Vardiman JW. World
health organization classifi cation of
tumours. Vol. 39283224116. 2001.
Lyon, IARC press
2. Caligaris-Cappio F, Hamblin TJ. B-cell
chronic lymphocytic leukemia: a bird
of a different feather. J.Clin.Oncol.
1999;17:399-408.
3. Kitada S, Andersen J, Akar S et al.
Expression of apoptosis-regulating
proteins in chronic lymphocytic
leukemia: correlations with In vitro
and In vivo chemoresponses. Blood
1998;91:3379-3389.
4. Saxena A, Viswanathan S, Moshynska
O et al. Mcl-1 and Bcl-2/Bax ratio are
associated with treatment response
but not with Rai stage in B-cell chronic
lymphocytic leukemia. Am.J.Hematol.
2004;75:22-33.
5. Mackus WJ, Kater AP, Grummels A
et al. Chronic lymphocytic leukemia
cells display p53-dependent drug-
induced Puma upregulation. Leukemia
2005;19:427-434.
6. Morales AA, Olsson A, Celsing F et
al. Expression and transcriptional
regulation of functionally distinct Bmf
isoforms in B-chronic lymphocytic
leukemia cells. Leukemia 2004;18:41-
47.
7. Andreeff M, Darzynkiewicz Z,
Sharpless TK, Clarkson BD, Melamed
MR. Discrimination of human leukemia
subtypes by fl ow cytometric analysis
of cellular DNA and RNA. Blood
1980;55:282-293.
8. Messmer BT, Messmer D, Allen SL et
al. In vivo measurements document
the dynamic cellular kinetics of chronic
lymphocytic leukemia B cells. J.Clin.
Invest 2005;115:755-764.
9. Granziero L, Ghia P, Circosta P
et al. Survivin is expressed on
CD40 stimulation and interfaces
proliferation and apoptosis in B-cell
chronic lymphocytic leukemia. Blood
2001;97:2777-2783.
10. Burger JA, Tsukada N, Burger M et al.
Blood-derived nurse-like cells protect
chronic lymphocytic leukemia B cells
from spontaneous apoptosis through
stromal cell-derived factor-1. Blood
2000;96:2655-2663.
11. Firestein GS, Kipps TJ. Fibroblast-
like synoviocytes support B-cell
pseudoemperipolesis via a stromal
cell-derived factor-1- and CD106
(VCAM-1)-dependent mechanism.
J.Clin.Invest 2001;107:305-315.
12. Lagneaux L, Delforge A, Bron D, De
BC, Stryckmans P. Chronic lymphocytic
leukemic B cells but not normal B cells
are rescued from apoptosis by contact
with normal bone marrow stromal cells.
Blood 1998;91:2387-2396.
13. Panayiotidis P, Jones D, Ganeshaguru
K, Foroni L, Hoffbrand AV. Human
bone marrow stromal cells prevent
apoptosis and support the survival of
chronic lymphocytic leukaemia cells in
vitro. Br.J.Haematol. 1996;92:97-103.
14. Pedersen IM, Kitada S, Leoni LM
et al. Protection of CLL B cells
by a follicular dendritic cell line is
dependent on induction of Mcl-1. Blood
2002;100:1795-1801.
15. Collins RJ, Verschuer LA, Harmon BV
et al. Spontaneous programmed death
(apoptosis) of B-chronic lymphocytic
leukaemia cells following their culture
in vitro. Br.J.Haematol. 1989;71:343-
350.
Thesis_Final_2_01092008.indd Sec1:94Thesis_Final_2_01092008.indd Sec1:94 01-09-2008 11:08:2201-09-2008 11:08:22

95 16. McConkey DJ, guilar-Santelises M,
Hartzell P et al. Induction of DNA
fragmentation in chronic B-lymphocytic
leukemia cells. J.Immunol.
1991;146:1072-1076.
17. Kater AP, Evers LM, Remmerswaal
EB et al. CD40 stimulation of B-cell
chronic lymphocytic leukaemia cells
enhances the anti-apoptotic profi le, but
also Bid expression and cells remain
susceptible to autologous cytotoxic
T-lymphocyte attack. Br.J.Haematol.
2004;127:404-415.
18. Kitada S, Zapata JM, Andreeff M,
Reed JC. Bryostatin and CD40-
ligand enhance apoptosis resistance
and induce expression of cell
survival genes in B-cell chronic
lymphocytic leukaemia. Br.J.Haematol.
1999;106:995-1004.
19. Romano MF, Lamberti A, Tassone P et
al. Triggering of CD40 antigen inhibits
fl udarabine-induced apoptosis in B
chronic lymphocytic leukemia cells.
Blood 1998;92:990-995.
20. Robertson LE, Huh YO, Butler JJ et
al. Response assessment in chronic
lymphocytic leukemia after fl udarabine
plus prednisone: clinical, pathologic,
immunophenotypic, and molecular
analysis. Blood 1992;80:29-36.
21. Provan D, Bartlett-Pandite L, Zwicky C
et al. Eradication of polymerase chain
reaction-detectable chronic lymphocytic
leukemia cells is associated with
improved outcome after bone marrow
transplantation. Blood 1996;88:2228-
2235.
22. Fernandez Y, Verhaegen M, Miller TP
et al. Differential regulation of noxa in
normal melanocytes and melanoma
cells by proteasome inhibition:
therapeutic implications. Cancer Res.
2005;65:6294-6304.
23. Perez-Galan P, Roue G, Villamor
N et al. The proteasome inhibitor
bortezomib induces apoptosis in
mantle-cell lymphoma through
generation of ROS and Noxa activation
independent of p53 status. Blood
2006;107:257-264.
24. Lens SM, den Drijver BF, Potgens AJ
et al. Dissection of pathways leading
to antigen receptor-induced and Fas/
CD95-induced apoptosis in human B
cells. J.Immunol. 1998;160:6083-6092.
25. Eldering E, Spek CA, Aberson HL et al.
Expression profi ling via novel multiplex
assay allows rapid assessment of
gene regulation in defi ned signalling
pathways. Nucleic Acids Res.
2003;31:e153.
26. Lens SM, Drillenburg P, den Drijver BF
et al. Aberrant expression and reverse
signalling of CD70 on malignant B
cells. Br.J.Haematol. 1999;106:491-
503.
27. Brummelkamp TR, Bernards R, Agami
R. A system for stable expression of
short interfering RNAs in mammalian
cells. Science 2002;296:550-553.
28. Schouten JP, McElgunn CJ, Waaijer
R et al. Relative quantifi cation of 40
nucleic acid sequences by multiplex
ligation-dependent probe amplifi cation.
Nucleic Acids Res. 2002;30:e57.
29. Willis SN, Chen L, Dewson G et al.
Proapoptotic Bak is sequestered by
Mcl-1 and Bcl-xL, but not Bcl-2, until
displaced by BH3-only proteins. Genes
Dev. 2005;19:1294-1305.
30. Qin JZ, Ziffra J, Stennett L et al.
Proteasome inhibitors trigger NOXA-
mediated apoptosis in melanoma
and myeloma cells. Cancer Res.
2005;65:6282-6293.
31. Lipford JR, Deshaies RJ. Diverse roles
for ubiquitin-dependent proteolysis in
transcriptional activation. Nat.Cell Biol.
2003;5:845-850.
Ch
ap
ter 4
▪ Ap
op
tosis
gen
es in
CL
L ly
mp
h n
od
e v
ers
us p
erip
hery
Thesis_Final_2_01092008.indd Sec1:95Thesis_Final_2_01092008.indd Sec1:95 01-09-2008 11:08:2201-09-2008 11:08:22

96 32. Gomez-Bougie P, Oliver L, Le GS,
Bataille R, Amiot M. Melphalan-
induced apoptosis in multiple myeloma
cells is associated with a cleavage
of Mcl-1 and Bim and a decrease in
the Mcl-1/Bim complex. Oncogene
2005;24:8076-8079.
33. Han J, Goldstein LA, Gastman BR,
Rabinovitz A, Rabinowich H. Disruption
of Mcl-1.Bim complex in granzyme
B-mediated mitochondrial apoptosis.
J.Biol.Chem. 2005;280:16383-16392.
34. Chen L, Willis SN, Wei A et al.
Differential targeting of prosurvival
Bcl-2 proteins by their BH3-only ligands
allows complementary apoptotic
function. Mol.Cell 2005;17:393-403.
35. Chiorazzi N, Rai KR, Ferrarini M.
Chronic lymphocytic leukemia.
N.Engl.J.Med. 2005;352:804-815.
36. Gricks CS, Zahrieh D, Zauls AJ et
al. Differential regulation of gene
expression following CD40 activation of
leukemic compared to healthy B cells.
Blood 2004;104:4002-4009.
37. Craxton A, Shu G, Graves JD et al. p38
MAPK is required for CD40-induced
gene expression and proliferation
in B lymphocytes. J.Immunol.
1998;161:3225-3236.
38. Wang Z, Karras JG, Howard RG,
Rothstein TL. Induction of bcl-x by
CD40 engagement rescues sIg-
induced apoptosis in murine B cells.
J.Immunol. 1995;155:3722-3725.
39. Sarma V, Lin Z, Clark L et al. Activation
of the B-cell surface receptor CD40
induces A20, a novel zinc fi nger protein
that inhibits apoptosis. J.Biol.Chem.
1995;270:12343-12346.
40. Oda E, Ohki R, Murasawa H et al.
Noxa, a BH3-only member of the
Bcl-2 family and candidate mediator
of p53-induced apoptosis. Science
2000;288:1053-1058.
41. Stankovic T, Hubank M, Cronin D et al.
Microarray analysis reveals that. Blood
2004;103:291-300.
42. Hershko T, Ginsberg D. Up-regulation
of Bcl-2 homology 3 (BH3)-only
proteins by E2F1 mediates apoptosis.
J.Biol.Chem. 2004;279:8627-8634.
43. Flinterman M, Guelen L, Ezzati-Nik S
et al. E1A activates transcription of p73
and Noxa to induce apoptosis. J.Biol.
Chem. 2005;280:5945-5959.
44. Kim JY, Ahn HJ, Ryu JH, Suk K,
Park JH. BH3-only protein Noxa is a
mediator of hypoxic cell death induced
by hypoxia-inducible factor 1alpha.
J.Exp.Med. 2004;199:113-124.
45. Yean K., Lee AKS, Nancy D.Bone,
Linda E.Wellik, D.A.Chan, A.J.Giaccia,
Debabrata Mukhopadhyay, and Neil
E.Kay. Hypoxia Inducible Factor-
1{alpha} Is over Expressed in CLL
B Cells Because of an Impaired
Proteasome Pathway Associated with
Defective Interaction with von Hippel-
Landau Protein. ASH. 106(11). 2005.
46. Chilosi M, Pizzolo G, Caligaris-
Cappio F et al. Immunohistochemical
demonstration of follicular dendritic
cells in bone marrow involvement of
B-cell chronic lymphocytic leukemia.
Cancer 1985;56:328-332.
47. Hanada M, Delia D, Aiello A,
Stadtmauer E, Reed JC. bcl-2
gene hypomethylation and high-
level expression in B-cell chronic
lymphocytic leukemia. Blood
1993;82:1820-1828.
48. Packham G, Stevenson FK.
Bodyguards and assassins: Bcl-2
family proteins and apoptosis control
in chronic lymphocytic leukaemia.
Immunology 2005;114:441-449.
Thesis_Final_2_01092008.indd Sec1:96Thesis_Final_2_01092008.indd Sec1:96 01-09-2008 11:08:2201-09-2008 11:08:22

49. Kuwana T, Bouchier-Hayes L, Chipuk
JE et al. BH3 domains of BH3-only
proteins differentially regulate Bax-
mediated mitochondrial membrane
permeabilization both directly and
indirectly. Mol.Cell 2005;17:525-535.
50. Letai A, Bassik MC, Walensky LD
et al. Distinct BH3 domains either
sensitize or activate mitochondrial
apoptosis, serving as prototype cancer
therapeutics. Cancer Cell 2002;2:183-
192.
51. Alvi AJ, Austen B, Weston VJ et
al. A novel CDK inhibitor, CYC202
(R-roscovitine), overcomes the defect
in p53-dependent apoptosis in B-CLL
by down-regulation of genes involved
in transcription regulation and survival.
Blood 2005;105:4484-4491.
52. Carew JS, Nawrocki ST, Krupnik YV
et al. Targeting endoplasmic reticulum
protein transport: a novel strategy to
kill malignant B cells and overcome
fl udarabine resistance in CLL. Blood
2006;107:222-231.
53. Castro JE, Prada CE, Loria O et al.
ZAP-70 is a novel conditional heat
shock protein 90 (Hsp90) client:
inhibition of Hsp90 leads to ZAP-70
degradation, apoptosis, and impaired
signaling in chronic lymphocytic
leukemia. Blood 2005;106:2506-2512.
97
Ch
ap
ter 4
▪ Ap
op
tosis
gen
es in
CL
L ly
mp
h n
od
e v
ers
us p
erip
hery
Thesis_Final_2_01092008.indd Sec1:97Thesis_Final_2_01092008.indd Sec1:97 01-09-2008 11:08:2201-09-2008 11:08:22

98
Thesis_Final_2_01092008.indd Sec1:98Thesis_Final_2_01092008.indd Sec1:98 01-09-2008 11:08:2201-09-2008 11:08:22

chapter
5
11
Persistent Mcl-1/Bim protein signature after CD40 ligation in Chronic Lymphocytic Leukemia is associated with specific drug sensitivity
Delfi ne Y.H. Hallaert1,2, Annelieke Jaspers1, Arnon P. Kater1, Marinus H.J. van Oers1,
Eric Eldering2
1Dept. of Hematology, AMC, Amsterdam, the Netherlands
2Dept. of Experimental Immunology, AMC, Amsterdam, the Netherlands
Manuscript in preparation
Thesis_Final_2_01092008.indd Sec1:99Thesis_Final_2_01092008.indd Sec1:99 01-09-2008 11:08:2201-09-2008 11:08:22

100ABSTRACT
Relapse in CLL might originate from a niche where CLL cells proliferate and are
protected from chemotherapeutic drugs. It is therefore important to understand how
CLL cells from lymph nodes (LN) differ from the cells which have moved into the
circulation. To mimic this in vivo LN setting we used CLL cells stimulated via CD40.
The aim of the present study was to monitor expression of apoptosis regulatory genes,
in relation to sensitivity for various types of drugs (fl udarabine, bortezomib, GSI-1 and
roscovitine) during and following a CD40-ligand stimulus. CD40 ligation resulted in
enhanced NF-κB activity and increased expression of target genes Bcl-XL and Bfl -
1. Furthermore, Mcl-1 and BimEL
protein, but not RNA levels, were increased and
decreased respectively. Four days after cessation of CD40L stimulation there was a
dichotomy: NF-κB activity, Bcl-XL and Bfl -1 gene expression gradually declined, but
Mcl-1 and BimEL
protein changes persisted. This was accompanied by reversal of
resistance to drugs, except for roscovitine, a cyclin dependent kinase (CDK) inhibitor,
where the apoptosis-inducing capacity strongly depends on Mcl-1 and Bim. These
data illustrate the long-lasting, non-transcriptional effects of CD40 signals on Mcl-1
and Bim in CLL.
Thesis_Final_2_01092008.indd Sec1:100Thesis_Final_2_01092008.indd Sec1:100 01-09-2008 11:08:2201-09-2008 11:08:22

Ch
ap
ter 5
▪ Pro
lon
ged
an
ti-ap
op
totic
pro
tein
sig
natu
re a
fter C
D40 lig
atio
n in
CL
L
INTRODUCTION
Chronic lymphocytic leukemia (CLL) remains an incurable disease and although
multi-agent treatment can result in peripheral lymphocyte depletion, the CLL cells in
the protected microenvironmental niches (lymph node (LN), bone marrow (BM) and
spleen) are less effectively targeted. Therefore, it is important to assess the effi cacy
of therapeutics directed towards CLL cells residing in those niches1.
CLL cells in the peripheral blood (PB) are arrested in the G0/G
1 phase of the cell
cycle, and replication occurs in proliferation centers (pseudofollicles). Recently it was
shown that the disease process is more dynamic than previously considered and is
characterized by proliferating as well as dying cells2. Selected microenvironmental
signals delivered by accessory cells, such as follicular dendritic cells (FDCs), BM
stromal cells, IL-6-producing endothelial cells, SDF-producing nurse-like cells, or
CD40 ligand (CD40L/CD154) expressing CD4+ T cells, have been shown to increase
the apoptotic threshold in vitro3-7. CD40L is expressed on activated T cells and plays
an important role in B cell activation, proliferation, isotype switching, normal germinal
centre formation8;9 and normal specifi c antibody responses10;11. In B cells, the response
to CD40 is complex and it is well established that CD40 can support cell survival
through upregulation of the expression of genes encoding antiapoptotic proteins such
as Bcl-XL and Bfl -112-15. The roles of the phosphoinositide 3-kinase (PI3K), mitogen-
activated protein kinase (MAPK), and nuclear factor-kappa B (NF-κB) pathways in
mediating CD40 stimulation are well described16. NF-κB signaling is divided into
two pathways: the canonical pathway and the alternative pathway. Activation of
the canonical pathway proceeds through the degradation of phosporylated IκBα
and subsequent nuclear translocation of active heterodimers (composed of p50,
p65, and/or c-Rel). Activation of the alternative NF-κB pathway results in nuclear
translocation of p52 along with RelB. Translocation of active heterodimers to the
nucleus infl uences the expression of various genes17;18. Studies suggest that CLL
cells may have constitutively activated canonical NF-κB activity, which enhances cell
survival19;20.
The Bcl-2 family of proteins plays an important role in the response to chemotherapeutic
drugs and includes anti-apoptotic and pro-apoptotic members. The Bcl-2 family
members Bax and Bak directly trigger cytochrome c release from the mitochondria.
In turn, this is under the control of the anti-apoptotic Bcl-2 family members (Bcl-2,
Bfl -1, Mcl-1 and Bcl-XL). This balance can be tipped by the pro-apoptotic BH3-only
subgroup of the family21;22.
In a recent comparative ex vivo study of apoptosis regulatory genes and proteins in
neoplastic B cells derived from CLL LN proliferation centers and from PB, we observed
101
Thesis_Final_2_01092008.indd Sec1:101Thesis_Final_2_01092008.indd Sec1:101 01-09-2008 11:08:2201-09-2008 11:08:22

specifi c changes in mRNA expression: Bcl-XL and Bfl -1 were upregulated and Noxa
was downregulated. Furthermore, although Mcl-1 mRNA levels were unaltered,
protein expression was upregulated in LN samples14. Studies on Bcl-2 family members
in CLL cells stimulated in vitro with CD40L showed upregulation in mRNA expression
of Bcl-XL, Bfl -1, Bid, and survivin and downregulation of Noxa12-15. Again, Mcl-1 was
clearly increased upon CD40 triggering13;14, on the protein level, but not at the RNA
level. Thus, in vitro CD40L stimulated CLL and ex vivo LN CLL cells displayed a highly
analogous anti-apoptotic phenotype compared to PB CLL. Therefore, in vitro CD40L
stimulation of CLL cells seems to be a useful model to mimic the in vivo LN setting.
Mapping this transit from LN to PB at the molecular level in relation to drug sensitivity
could provide new insights into the effi cacy of chemotherapeutic regimens. We
observed a distinctive trend where reversible transcriptional changes are contrasted
with long-lasting post-transcriptional modulation of Mcl-1 and Bim proteins. Together,
these changes dictate short- and long-term effects with respect to drug sensitivity.
MATERIALS AND METHODS
CLL cells
PB from CLL patients was obtained in the setting of routine diagnostic or follow-
up procedures at the department of Hematology of the Academic Medical Center
Amsterdam. Patients had to give informed consent and the study was approved by the
AMC Ethical Review Board. This study was conducted in accordance with the ethical
standards in our institute and in agreement with the Helsinki Declaration of 1975,
revised in 1983. Peripheral blood (PB) mononuclear cells (PBMCs) obtained after Ficoll
density gradient centrifugation (Pharmacia Biotech, Roosendaal, The Netherlands)
were frozen in Iscove’s Modifi ed Dulbecco’s Medium (IMDM) supplemented with
L-Glutamine, 25 mM HEPES (Biowhittaker, Europe) containing, 2mM L-Glutamine
(Invitrogen), 50 mg Gentamycine (Invitrogen) 3.57 x 10-4% (v/v) -mercapto ethanol
(Merck, Darmstadt, Germanyand), 10 % dimethyl sulphoxide (DMSO; Sigma Chemical
Co., St. Louis, MO, USA) and 15% fetal calf serum, (FCS; ICN Biomedicals GnbH,
Mecken heim, Germany), and stored in liquid nitrogen. Expression of CD5 and CD19
(both Bekton Dickinson (BD) Biosciences, San Jose, CA, USA) on leukemic cells
(>90% purity) was assessed by fl ow cytometry (FACScalibur, BD Biosciences) and
analysed with CellQuest software (BD Biosciences).
102
Thesis_Final_2_01092008.indd Sec1:102Thesis_Final_2_01092008.indd Sec1:102 01-09-2008 11:08:2201-09-2008 11:08:22

RNA isolation and RT-MLPA
Total RNA was isolated using the GenElute Mammalian Total RNA Miniprep Kit (Sigma
Aldrich). Reverse transcription–multiplex ligation-dependent probe amplifi cation
assay (RT-MLPA) procedure was performed as described previously23;24.
Reagents
The proteasome inhibitor bortezomib was obtained from Janssen-Cilag (Tilburg the
Netherlands). GSI-125 (gamma-secretase inhibitor-1; Z-LLNle-CHO – Cat.nr. 565750)
and cycloheximide were obtained from Calbiochem (Amsterdam, the Netherlands).
Roscovitine and fl udarabine were purchased from Sigma Chemical Co. (St. Louis,
MO, USA).
Analysis of apoptosis, Western blot and antibodies
For apoptosis induction, 5×106 CLL/ml cells were incubated with 100μM fl udarabine
(48 hrs), 30nM bortezomib, 25 μM roscovitine or 5 μM GSI-1 (24 hrs), and stained
with 200 nM MitoTracker Orange (Molecular Probes, Leiden, The Netherlands) for
30 minutes at 37°C and analysed by FACS. Western blotting was performed as
described previously23. Cells were lysed in Laemmli Sample Buffer, and samples
(10-30 μg protein) were separated by 13% sodium dodecyl sulfate polyacrylamide
gel electrophoresis (10% gels for Erk). Blots were probed with the following
antibodies: polyclonal anti-Mcl-1 (catalog no. 554103; Pharmingen, BD Biosciences),
monoclonal anti-Noxa (clone 114C307.1; Imgenex, San Diego, CA), monoclonal anti-
Bim (clone 14A8; Chemicon, Temecula, CA), antiserum to -actin (clone I-19; Santa
Cruz Biotechnology, Santa Cruz, CA), polyclonal anti-Bcl-XL (catalog no. 620211,
BD Biosciences), polyclonal anti-Bcl-2 (catalog no. 210-701-C100, Kordia, Leiden
the Netherlands) polyclonal anti-phospho-Erk (catalog no. #9102 Cell Signaling),
polyclonal anti-Erk (catalog no. #9101 Cell Signaling), monoclonal anti-phospho-IκBα
(clone 5A5, Cell Signaling), polyclonal anti-IκBα (catalog no. #9242, Cell Signaling),
polyclonal anti-p100/p52 (catalog no. #4882, Cell Signaling), polyclonal anti-phospho-
GSK-3β (sc-11757, Santa Cruz Biotechnology) and polyclonal anti-GSK-3β (clone
1H8, Santa Cruz Biotechnology).
In vitro CD40 stimulation
CLL cells were stimulated by co-culture with fi broblasts (NIH3T3) that had been
stably transfected with a plasmid encoding human CD40L (3T40L). Fibroblasts
were irradiated (30Gy) and plated in culture-treated 6-wells plates (6x105 cells/well).
CLL cells were thawed and 5x106 cells per well were added (day-3) to the adhered
103
Ch
ap
ter 5
▪ Pro
lon
ged
an
ti-ap
op
totic
pro
tein
sig
natu
re a
fter C
D40 lig
atio
n in
CL
L
Thesis_Final_2_01092008.indd Sec1:103Thesis_Final_2_01092008.indd Sec1:103 01-09-2008 11:08:2201-09-2008 11:08:22

fi broblasts in 3 ml IMDM containing 10% FCS and incubated at 37oC. After 3 days
(day 0) CLL cells were gently removed and transferred to new 6-well plates and
incubated for 4 days (day 1-4) in 3 ml IMDM containing 10% FCS. There were no
residual fi broblasts in the transferred CLL cell culture.
Proteasome activity assay
Cytoplasmic extracts (Assay buffer: 250 mM HEPES (pH7.5), 5 mM EDTA, 0.5%
NP-40 and 0.01% SDS) from freshly isolated PBMCs from CLL patients (CD40L
stimulation and time point of lyses is indicated in legend) were used to measure
proteasome activity using a 20S proteasome activity assay kit (Chemicon, part of
Millipore; Billerica, USA) following the manufacturer’s instructions. The assay is based
on detection of the fl uorophore 7-amino-4-methylcoumarin (AMC) after cleavage
from the labeled substrate LVVY-AMC. The free AMC fl uorescence was quantifi ed
using a 380/460-nm fi lter set in a VICTOR2 D fl uorometer (Wallac-PerkinElmer,
Massachusetts, USA). Proteasomal activity was calculated from the changes in
fl uorescence over time and expressed per μg of protein in the extract.
Statistical analysis
The Mann Whitney U test was used to analyze weather differences in proteasomal
activity between control 3T3 and 3T40L stimulated CLL cells at day 0 and day 4
(4 days after cessation of CD40 stimulation) were statistically signifi cant. P values
below 0.05 were considered statistically signifi cant.
RESULTS
The anti-apoptotic expression profi le resets after termination of
CD40 stimulation
CLL cells were co-cultured with non transfected 3T3 cells (control), or with 3T3 cells
stably transfected with a huCD40L-encoding plasmid (3T40L). We evaluated mRNA
expression profi les of different apoptosis genes by means of RT-MLPA14;24 from cells
of CLL patients during, and at different time points after, CD40 triggering. Cells were
stimulated for three days (day -3 to day 0), followed by four days culture in the absence
of CD40 signals (day 1-4). As reported previously, CD40L stimulation resulted in
increased mRNA expression of Bcl-XL, Bfl -1, Bid and Survivin and decreased mRNA
expression of Noxa (Fig. 1; day 0)12-14;26. Expression of Bcl-XL, Bfl -1 and Bid mRNA
declined gradually to pre-stimulation levels after cessation of CD40 stimulation. Noxa
downregulation, however, was not only observed in CLL cells cultured on 3T40L cells
104
Thesis_Final_2_01092008.indd Sec1:104Thesis_Final_2_01092008.indd Sec1:104 01-09-2008 11:08:2201-09-2008 11:08:22

Figure 1. Apoptosis gene expression profi le of CLL cells after termination of CD40 stimulation.
Changes in the mRNA expression of Bcl-XL, Bfl -1, Mcl-1, Noxa, Bim, Bid, survivin and GUS were
investigated by RT-MLPA. CLL cells were co-cultured for 3 days on control 3T3 cells (dashed lines) or
CD40L-transfected 3T3 cells (solid lines). After detachment and washing, cells were incubated in medium
for 4 days without stimulation. mRNA expression is shown at day -3 (prior to stimulation), 0 (3 days co-
culture), 1, 2, 3 and 4 (days after termination of stimulation). Data represent mean ± SD from 5 independent
experiments.
Ch
ap
ter 5
▪ Pro
lon
ged
an
ti-ap
op
totic
pro
tein
sig
natu
re a
fter C
D40 lig
atio
n in
CL
L
105
Thesis_Final_2_01092008.indd Sec1:105Thesis_Final_2_01092008.indd Sec1:105 01-09-2008 11:08:2201-09-2008 11:08:22

A
but also on 3T3 cells and the mRNA levels remained low under both conditions. Mcl-1
and Bim mRNA expression was not altered upon CD40, similar to the housekeeping
gene GUS which showed stable expression throughout the experimental period. In
conclusion, the altered gene expression by in vitro CD40 engagement of CLL cells
was in agreement with previous results12-14;26. Cessation of CD40 stimulation resulted
in a gradual decline to baseline expression.
CLL cells selectively regain chemosensitivity after termination of
CD40 stimulation
Next, sensitivity to different chemotherapeutic drugs was tested, in relation to
106
Figure 2. Apoptosis response after termination of CD40 stimulation in CLL.
(A) CLL cells were co-cultured for 3 days on 3T3 cells (dashed lines) or CD40L-transfected 3T3 cells
(solid lines). After detachment and washing, cells were directly or after 1-4 days of additional CD40L free
culture, incubated with bortezomib (30 nM) or GSI-1 (5 µM) for 24 hours and with fl udarabine (100 µM) for
48 hours. Apoptosis response was analyzed by MitoTracker staining. Data represent mean ± SD from 6
independent experiments.
(B) Apoptosis response upon 24 hours incubation with roscovitine (25 µM) of CLL cells cultured as
described in A. Data represent mean ± SD from 4 independent experiments.
(C) CLL cells were co-cultured for 3 days on 3T3 cells (black bars) or CD40L-transfected 3T3 cells (white
bars). After detachment and washing, cells were incubated with fl udarabine (F) (100 µM) for 48 hours or
roscovitine (R) (25 µM), bortezomib (B) (30 nM) and GSI-1 (G) (5 µM) for 24 hours. Also the following
combinations of drugs were used: bortezomib (30 nM) + roscovitine (25 µM) (B30/R25) or GSI-1 (5 µM) +
roscovitine (25 µM) (G5/R25). M=medium. Apoptosis response was analyzed by MitoTracker staining. %
Apoptosis is shown at day 0 (+/- 3 days CD40 stimulation). Data represent mean ± SD from 6 independent
experiments.
Thesis_Final_2_01092008.indd Sec1:106Thesis_Final_2_01092008.indd Sec1:106 01-09-2008 11:08:2301-09-2008 11:08:23

the culture regimen described above. Figure 2 shows that at day 0 CLL cells had
become resistant to all drugs tested: the purine analog fl udarabine, the proteasome
inhibitors bortezomib and GSI-1 (gamma-secretase inhibitor-1), and the CDK inhibitor
roscovitine. Also the combination of roscovitine with bortezomib, GSI-1 (Fig. 2C)
or fl udarabine (data not shown), did not induce apoptosis at day 0. After cessation
of CD40 stimulation CLL cells became gradually sensitive again to fl udarabine,
bortezomib and GSI-1, but not to roscovitine (Fig. 2A&B; day0-4). This surprising
dichotomy was further investigated.
Prolonged changes in Mcl-1 and BimEL protein expression after
termination of CD40 triggering in CLL
Previous research showed that roscovitine induced apoptosis via the Mcl-1/Noxa
axis. Rapid (4 hours) Mcl-1 degradation is the initial event in roscovitine-induced
apoptosis27;28 and the BH3-only proteins Noxa and Bim are involved as specifi c
mediators in this process27.
Changes in Mcl-1, Bcl-XL and Noxa were determined in CLL cells treated for 24
hours with roscovitine or bortezomib at two different time points; after three days of
CD40L stimulation (day 0) and four days after termination of CD40L stimulation (day
4). In accord with previous data12-14;27;28, Mcl-1 and Bcl-XL protein expression was
upregulated after CD40L stimulation at day 0 compared to unstimulated (3T3) CLL
cells (Fig. 3A). At day 4, the levels of Bcl-XL were declined compared to day 0, whereas
Mcl-1 protein expression was still upregulated. Also, Noxa protein expression was
higher after CD40L stimulation at day 0 and 4, in contrast with mRNA levels observed
in fi gure 1. Finally, CD40 stimulation increases proteasomal degradation of BimEL
.
Figure 3B illustrated the decrease in BimEL
protein, which persisted up to day 4.
Concerning drug responses in this setup, roscovitine was functional, as measured
by Mcl-1 degradation, at day 4 but not at day 0. Nevertheless, at both time points
107
Ch
ap
ter 5
▪ Pro
lon
ged
an
ti-ap
op
totic
pro
tein
sig
natu
re a
fter C
D40 lig
atio
n in
CL
L
B C
Thesis_Final_2_01092008.indd Sec1:107Thesis_Final_2_01092008.indd Sec1:107 01-09-2008 11:08:2401-09-2008 11:08:24

A
B
108
Figure 3. Protein expression of Mcl-1, Bcl-XL, Noxa and Bim after termination of CD40 stimulation
in CLL.
(A) CLL cells were co-cultured with control 3T3 or CD40L-transfected 3T3 cells for 3 days (day 0). After
detachment and washing, cells were directly (day 0) or after 4 days (day 4) of CD40L free culture, incubated
with the indicated drugs (24 hours). Lysates were probed for Mcl-1, Noxa and Bcl-XL as indicated and actin
as loading control.
(B) CLL cells were co-cultured as in A. Lysates were probed for Bim as indicated and actin as loading
control.
Thesis_Final_2_01092008.indd Sec1:108Thesis_Final_2_01092008.indd Sec1:108 01-09-2008 11:08:2401-09-2008 11:08:24

roscovitine did not induce apoptosis. Furthermore, as expected bortezomib induced
accumulation of Noxa and Mcl-114;29, at both time points (Fig 3A).
In conclusion, Mcl-1 and Bcl-XL protein expression were increased after CD40
triggering. After termination of CD40 triggering, Bcl-XL expression gradually decreased,
however, Mcl-1 and Noxa remained high. Also the Erk-mediated BimEL
degradation
continued after cessation of the CD40 signal.
CD40 signaling results in NF-κB, in GSK-3β and long-lasting ERK
activation
Various signaling pathways are known to affect levels of Bcl-2 family members. First,
we examined the canonical and alternative NF-κB activity in three CLL samples
stimulated with CD40L. Figure 4A illustrates the phosphorylation of IκBα and the
processing of p100 to p52 at day 0, demonstrating activation of the canonical
and alternative NF-κB pathway upon CD40 triggering. At day 4, activation of the
canonical pathway was decreased to baseline expression in CLL 4&5 and reduced
approximately 50% in CLL 6. The alternative pathway was decreased in CLL 4&6, but
still increased in CLL 5.
Next, it is known that Erk signaling mediates the proteasomal degradation of BimEL
in model systems30;31. Figure 4B shows that Erk is phosphorylated upon CD40
stimulation and this was maintained and even increased at day 4.
Third, Mcl-1 is a known target for phosphorylation and subsequent proteasomal
degradation. Cytokine withdrawal in murine cell lines causes decreased PI3K-Akt/
PKB signaling to activate GSK-3β which in turn phosporylates Mcl-1, thus marking
it for proteasomal degradation32. In CLL, CD40 ligation induced both total and
phosphorylated (inactive) forms of GSK-3β (Fig. 4C).
Taken together, these data demonstrated activation of various CD40L-induced survival
pathways in CLL. After cessation of the CD40 signal, NF-κB in general returns to
baseline, as also indicated by mRNA data (indicated by Bcl-XL and Bfl -1 expression).
Erk remained signifi cantly activated, while GSK-3β appeared persistently blocked.
Proteasomal activity does not account for dysregulated protein
expression after CD40 stimulation in CLL
Next, we tested whether the altered protein profi le (of Mcl-1 and BimEL
) observed
after CD40 stimulation was a consequence of altered proteasomal activity. In fi gure
5 proteasomal activity was measured in CLL cell lysates obtained after 3 days
incubation of the CLL cells with CD40L (day 0; n=13) and four days after cessation
of CD40L stimulation (day 4; n=4). Proteasomal activity was signifi cantly increased
after CD40 stimulation at day 0. This persisted four days after termination of CD40
stimulation.
Ch
ap
ter 5
▪ Pro
lon
ged
an
ti-ap
op
totic
pro
tein
sig
natu
re a
fter C
D40 lig
atio
n in
CL
L
109
Thesis_Final_2_01092008.indd Sec1:109Thesis_Final_2_01092008.indd Sec1:109 01-09-2008 11:08:2501-09-2008 11:08:25

B
A
C
Figure 4. Activity of NF-κB, Erk and GSK-3β upon CD40 engagement in CLL.
(A) CLL cells were co-cultured with control 3T3 or CD40L-transfected 3T3 cells for 3 days (day0). After
detachment and washing (day 0), cells were incubated for 4 days in medium (day 4). Lysates were probed
for IκBα, p-IκBα and p100/52 as indicated. Shown are representative examples of 3 CLL patients.
(B) CLL cells were co-cultured with control 3T3 or CD40L-transfected 3T3 cells for 3 days (day0). Lysates
were probed for Erk and p-Erk as indicated and actin as loading control. Shown are representative
examples of 3 CLL patients.
(C) CLL cells were co-cultured with control 3T3 or CD40L-transfected 3T3 cells for 3 days (day 0). Lysates
were probed for GSK-3β and p-GSK-3β as indicated. Shown are representative examples of 2 CLL
patients.
110
Figure 5. Proteasomal activity in CLL cells upon
CD40 stimulation.
CLL cells were co-cultured with control 3T3 or 3T40L
cells for 3 days. After detachment and washing (day 0).
CLL cells were incubated for 4 days in medium (day
4). At day 0 (n=13) and 4 (n=4), CLL cells were lysed,
the fl uorogenic proteasome substrate LVVY-AMC was
added and the enzymatic activity of the 20S proteasome
was measured as the amount of AMC fl uorescence per
minute (counts/min). Results are expressed as change
in AMC fl uorescence per minute per μg protein.
P<0.01 with the Mann Whitney U test are indicated by
an asterisk (*).
NS = not signifi cant.
Thesis_Final_2_01092008.indd Sec1:110Thesis_Final_2_01092008.indd Sec1:110 01-09-2008 11:08:2501-09-2008 11:08:25

A
BFigure 6. Mcl-1 turnover upon CD40
stimulation.
(A) CLL cells were co-cultured with 3T40L cells for
3 days. After detachment and washing, cells were
incubated with 5 µg/ml cycloheximide. Cells were
harvested at indicated time points (0, 30, 60, 120,
240 and 360 minutes). Lysates were probed for
Mcl-1 and actin as loading control.
(B) The experiment described in A, was quantifi ed
with AIDA evaluation software. Results are
presented as the percentage of Mcl-1 expression
at indicated time points after addition of
cycloheximide (5 µg/ml).
111
Ch
ap
ter 5
▪ Pro
lon
ged
an
ti-ap
op
totic
pro
tein
sig
natu
re a
fter C
D40 lig
atio
n in
CL
L
CD40 ligation does not infl uence Mcl-1 and Bim protein turnover
in CLL
Finally, we examined whether the half-life of Mcl-1 changed upon CD40 ligation.
Therefore, the stability of Mcl-1 was assessed following inhibition of protein synthesis
by treatment with cycloheximide. Previous reports in cell lines showed that Mcl-1
protein was rapidly lost with a half-life of around 30 minutes33;34. In CLL, after CD40
stimulation, the half-life of Mcl-1 was similar (Fig. 6). These results suggest that CD40
ligation does not considerably infl uence the half-life of Mcl-1 in CLL cells.
DISCUSSION
The aim of this study was to mimic the transition of CLL cells as they egress from LN
to PB, and relate this to drug sensitivity. The major fi nding of the present work was
that CD40 stimulation has long-lasting effects on protein expression of Mcl-1 and Bim,
and this was paralleled by prolonged resistance to the CDK inhibitor roscovitine.
In CLL, CD40 ligation affects various signaling pathways with subsequent transcriptional
consequences16;20;35. We observed that both the canonical and alternative NF-κB
pathways were stimulated, and as a result, the NF-κB target genes Bcl-XL and Bfl -
Thesis_Final_2_01092008.indd Sec1:111Thesis_Final_2_01092008.indd Sec1:111 01-09-2008 11:08:2601-09-2008 11:08:26

1, were upregulated. This expression signature was accompanied by broad drug
resistance. Termination of CD40 signals resulted in diminished NF-κB activity,
subsequent Bcl-XL and Bfl -1 downregulation and pre-sensitization to proteasome
inhibitors and fl udarabine. Importantly, the resistance to the CDK inhibitor roscovitine
persisted. Recent studies by us, and others, have shown that Noxa, Bim and Mcl-1
are crucial mediators in roscovitine-induced apoptosis27. Therefore, the persistent
increased Mcl-1/Bim protein ratio observed after cessation of CD40 triggering might
mediate the resistance to roscovitine. Remarkably, although roscovitine did initiate
Mcl-1 degradation at day 4, this obviously did not trigger apoptosis. We speculate that
the Mcl-1 reservoir still dominates and thus continuously neutralizes pro-apoptotic
proteins, such as Bim and Noxa. In previous studies we demonstrated the role of Mcl-
1, Bim and Noxa in roscovitine-induced cell death27. It would be interesting; however,
to test the specifi c role of these proteins in the resistance to roscovitine through gene
knockdown or overexpression experiments in CD40 stimulated CLL cells. Additionally,
there might be unknown mechanisms of action of roscovitine, other than the described
fast effect on Mcl-127. In this context, it would be interesting to test the effects of the BH3-
mimetic ABT-73736;37 or similar compounds, in combination with roscovitine, as it has
been shown that roscovitine increased ABT-737 toxicity in human leukemia cell lines38.
Mcl-114 and Bim protein levels in CD40 triggered CLL are under posttranscriptional
control. Both proteins are known targets for phosphorylation and proteasomal
degradation, but via different upstream routes. Activated Erk can mediate BimEL
degradation30;39;40, whereas termination of PKB signaling has been linked with GSK-
3β-mediated Mcl-1 phosphorylation and subsequent degradation32. Since both
phosphorylated Erk and reduced BimEL
protein levels persist after termination of
CD40 signaling (Fig. 3B), it appears likely that this pathway is indeed continuously
active in the CLL cells. The control of Mcl-1 protein levels seems less clear. In the
two CLL samples tested, CD40 stimulation induced phosphorylation of GSK-3 ,
thereby inactivating it. In growth factor withdrawal-mediated apoptosis, this depends
on decreased PKB activity and lead to decreased Mcl-1 turnover32. PKB activity
was however unaffected upon CD40 stimulation in CLL and the PI3 kinase inhibitor
LY294002 did not induce apoptosis (Hallaert, unpublished observation). More
surprisingly, although protein levels were persistently increased, the half-life of Mcl-1
was still quite short (<45 minutes). This value is close to the reported basal half-life
of approximately 30 minutes in other cells33;34. It would seem therefore, that CD40
triggering does not result in a drastically increased half-life of Mcl-1, although direct
comparison with un-triggered CLL cells was not feasible due to very low Mcl-1 levels
in those cells. In view of these observations, another possible control mechanism
may be involved. Other studies suggested translational regulation of Mcl-1 through
112
Thesis_Final_2_01092008.indd Sec1:112Thesis_Final_2_01092008.indd Sec1:112 01-09-2008 11:08:2601-09-2008 11:08:26

Ch
ap
ter 5
▪ Pro
lon
ged
an
ti-ap
op
totic
pro
tein
sig
natu
re a
fter C
D40 lig
atio
n in
CL
L
113the initiation factor eIF41;42. It is known that phosphorylation eIF2α can repress
translation of Mcl-1 resulting in downregulation of Mcl-1 protein41. Whether Mcl-1
protein expression after CD40 triggering in CLL is also regulated via eIF2α or other
posttranscriptional mechanisms remains to be determined.
Our earlier work has shown that Noxa is highly expressed in circulating CLL levels23,
but is low in LN14;23This can be mimicked to a certain extent by in vitro CD40 signals,
but some inconsistencies concerning Noxa protein levels have come to light in the
present studies. First, prolonged in vitro culture of CLL on 3T40L cells but surprisingly
also 3T3 cells caused persistent lowering of Noxa mRNA as detected by MLPA. In
various CLL samples however, a rise of Noxa protein was observed upon prolonged
CD40 stimulation. Since the regulation of Noxa at transcriptional level and especially
protein level is uncertain43-46, we can only speculate about the underlying mechanism.
First, the rapid suppressive effect of CD40 triggering as well as the slower effect of
prolonged co-culture with 3T3 cells on Noxa mRNA indicates that in vivo circulating
CLL cells lack signals that might suppress this high basal transcriptional rate. In support
of this, the basal Noxa mRNA levels are low in LN niches14. This difference is unlikely
to be due to CD40 signals, since we observed that prolonged in vitro CD40 triggering
can in fact cause a gradual increase in Noxa protein. The underlying mechanism of
this puzzling discrepancy between Noxa mRNA and protein remains unexplained. It
could on one hand be caused by unknown modes of post-transcriptional regulation of
Noxa, or also result from the various forms of regulation of its binding partner Mcl-1.
If the latter were true, the observed changes in Noxa are not an intrinsic property, but
would result merely from passive accumulation as Mcl-1 protein levels change. More
research is required to discriminate among these different options.
Taken together, prolonged CD40 triggering induced not only reversible changes
in transcription patterns, but also seems to have long-lasting effects on post-
transcriptional regulation of Mcl-1 and Bim. Using this model system to mimic the in
vivo LN setting could help to predict responses to new drugs.
Thesis_Final_2_01092008.indd Sec1:113Thesis_Final_2_01092008.indd Sec1:113 01-09-2008 11:08:2601-09-2008 11:08:26

REFERENCE LIST
1. Chanan-Khan A, Porter CW.
Immunomodulating drugs for chronic
lymphocytic leukaemia. Lancet Oncol.
2006;7:480-488.
2. Messmer BT, Messmer D, Allen SL et
al. In vivo measurements document
the dynamic cellular kinetics of chronic
lymphocytic leukemia B cells. J.Clin.
Invest 2005;115:755-764.
3. Burger JA, Tsukada N, Burger M et
al. Blood-derived nurse-like cells
protect chronic lymphocytic leukemia
B cells from spontaneous apoptosis
through stromal cell-derived factor-1.
Blood 2002;100:1795-1801.
4. Burger JA, Zvaifl er NJ, Tsukada N,
Firestein GS, Kipps TJ. Fibroblast-
like synoviocytes support B-cell
pseudoemperipolesis via a stromal
cell-derived factor-1- and CD106
(VCAM-1)-dependent mechanism.
J.Clin.Invest 2001;107:305-315.
5. Pedersen IM, Kitada S, Leoni LM
et al. Protection of CLL B cells
by a follicular dendritic cell line is
dependent on induction of Mcl-1. Blood
2002;100:1795-1801.
6. Lagneaux L, Delforge A, Bron D, De
BC, Stryckmans P. Chronic lymphocytic
leukemic B cells but not normal B cells
are rescued from apoptosis by contact
with normal bone marrow stromal cells.
Blood 1998;91:2387-2396.
7. Panayiotidis P, Jones D, Ganeshaguru
K, Foroni L, Hoffbrand AV. Human
bone marrow stromal cells prevent
apoptosis and support the survival of
chronic lymphocytic leukaemia cells in
vitro. Br.J.Haematol. 1996;92:97-103.
8. Allen RC, Armitage RJ, Conley ME
et al. CD40 ligand gene defects
responsible for X-linked hyper-IgM
syndrome. Science 1993;259:990-993.
114
9. DiSanto JP, Bonnefoy JY, Gauchat JF,
Fischer A, de Saint BG. CD40 ligand
mutations in x-linked immunodefi ciency
with hyper-IgM. Nature 1993;361:541-
543.
10. Banchereau J, Bazan F, Blanchard D
et al. The CD40 antigen and its ligand.
Annu.Rev.Immunol. 1994;12:881-922.
11. Nonoyama S, Hollenbaugh D, Aruffo
A, Ledbetter JA, Ochs HD. B cell
activation via CD40 is required for
specifi c antibody production by
antigen-stimulated human B cells.
J.Exp.Med. 1993;178:1097-1102.
12. Kater AP, Evers LM, Remmerswaal EB
et al. CD40 stimulation of B-cell
chronic lymphocytic leukaemia cells
enhances the anti-apoptotic profi le, but
also Bid expression and cells remain
susceptible to autologous cytotoxic
T-lymphocyte attack. Br.J.Haematol.
2004;127:404-415.
13. Kitada S, Zapata JM, Andreeff M,
Reed JC. Bryostatin and CD40-
ligand enhance apoptosis resistance
and induce expression of cell
survival genes in B-cell chronic
lymphocytic leukaemia. Br.J.Haematol.
1999;106:995-1004.
14. Smit LA, Hallaert DY, Spijker R et al.
Differential Noxa/Mcl-1 balance in
peripheral versus lymph node chronic
lymphocitic leukemia cells correlates
with survival capacity.
Blood 2007; 109: 1660-1668
15. Willimott S, Baou M, Naresh K,
Wagner SD. CD154 induces a switch
in pro-survival Bcl-2 family members
in chronic lymphocytic leukaemia.
Br.J.Haematol. 2007;138:721-732.
16. Gricks CS, Zahrieh D, Zauls AJ et
al. Differential regulation of gene
expression following CD40 activation of
leukemic compared to healthy B cells.
Blood 2004;104:4002-4009.
Thesis_Final_2_01092008.indd Sec1:114Thesis_Final_2_01092008.indd Sec1:114 01-09-2008 11:08:2601-09-2008 11:08:26

17. Bonizzi G, Karin M. The two NF-
kappaB activation pathways and their
role in innate and adaptive immunity.
Trends Immunol. 2004;25:280-288.
18. Karin M. Nuclear factor-kappaB in
cancer development and progression.
Nature 2006;441:431-436.
19. Hewamana S, Alghazal S, Lin TT et
al. The NF-kappaB subunit Rel A is
associated with in vitro survival and
clinical disease progression in chronic
lymphocytic leukemia and represents
a promising therapeutic target. Blood
2008;111:4681-4689.
20. Cuni S, Perez-Aciego P, Perez-Chacon
G et al. A sustained activation of PI3K/
NF-kappaB pathway is critical for
the survival of chronic lymphocytic
leukemia B cells. Leukemia
2004;18:1391-1400.
21. Chipuk JE, Green DR. How do BCL-2
proteins induce mitochondrial outer
membrane permeabilization? Trends
Cell Biol. 2008;18:157-164.
22. Strasser A. The role of BH3-only
proteins in the immune system. Nat.
Rev.Immunol. 2005;5:189-200.
23. Mackus WJ, Kater AP, Grummels A
et al. Chronic lymphocytic leukemia
cells display p53-dependent drug-
induced Puma upregulation. Leukemia
2005;19:427-434.
24. Eldering E, Spek CA, Aberson HL et al.
Expression profi ling via novel multiplex
assay allows rapid assessment of
gene regulation in defi ned signalling
pathways. Nucleic Acids Res.
2003;31:e153.
25. Curry CL, Reed LL, Golde TE et al.
Gamma secretase inhibitor blocks
Notch activation and induces apoptosis
in Kaposi’s sarcoma tumor cells.
Oncogene 2005;24:6333-6344.
26. Granziero L, Ghia P, Circosta P
et al. Survivin is expressed on
CD40 stimulation and interfaces
proliferation and apoptosis in B-cell
chronic lymphocytic leukemia. Blood
2001;97:2777-2783.
27. Hallaert DY, Spijker R, Jak M et al.
Crosstalk among Bcl-2 family members
in B-CLL: seliciclib acts via the Mcl-1/
Noxa axis and gradual exhaustion
of Bcl-2 protection. Cell Death.Differ.
2007
28. MacCallum DE, Melville J, Frame S et
al. Seliciclib (CYC202, R-Roscovitine)
induces cell death in multiple myeloma
cells by inhibition of RNA polymerase
II-dependent transcription and down-
regulation of Mcl-1. Cancer Res.
2005;65:5399-5407.
29. Perez-Galan P, Roue G, Villamor
N et al. The proteasome inhibitor
bortezomib induces apoptosis in
mantle-cell lymphoma through
generation of ROS and Noxa activation
independent of p53 status. Blood
2006;107:257-264.
30. Ley R, Balmanno K, Hadfi eld K,
Weston C, Cook SJ. Activation of the
ERK1/2 signaling pathway promotes
phosphorylation and proteasome-
dependent degradation of the BH3-
only protein, Bim. J.Biol.Chem.
2003;278:18811-18816.
31. Luciano F, Jacquel A, Colosetti P et al.
Phosphorylation of Bim-EL by Erk1/2
on serine 69 promotes its degradation
via the proteasome pathway and
regulates its proapoptotic function.
Oncogene 2003;22:6785-6793.
32. Maurer U, Charvet C, Wagman AS,
Dejardin E, Green DR. Glycogen
synthase kinase-3 regulates
mitochondrial outer membrane
permeabilization and apoptosis by
destabilization of MCL-1. Mol.Cell
2006;21:749-760.
Ch
ap
ter 5
▪ Pro
lon
ged
an
ti-ap
op
totic
pro
tein
sig
natu
re a
fter C
D40 lig
atio
n in
CL
L
115
Thesis_Final_2_01092008.indd Sec1:115Thesis_Final_2_01092008.indd Sec1:115 01-09-2008 11:08:2601-09-2008 11:08:26

33. Zhong Q, Gao W, Du F, Wang X. Mule/
ARF-BP1, a BH3-only E3 ubiquitin
ligase, catalyzes the polyubiquitination
of Mcl-1 and regulates apoptosis. Cell
2005;121:1085-1095.
34. Nijhawan D, Fang M, Traer E et al.
Elimination of Mcl-1 is required for
the initiation of apoptosis following
ultraviolet irradiation. Genes Dev.
2003;17:1475-1486.
35. Furman RR, Asgary Z, Mascarenhas
JO, Liou HC, Schattner EJ. Modulation
of NF-kappa B activity and apoptosis
in chronic lymphocytic leukemia B
cells. J.Immunol. 2000;164:2200-2206.
36. Konopleva M, Contractor R, Tsao T et
al. Mechanisms of apoptosis sensitivity
and resistance to the BH3 mimetic
ABT-737 in acute myeloid leukemia.
Cancer Cell 2006;10:375-388.
37. van Delft MF, Wei AH, Mason KD
et al. The BH3 mimetic ABT-737
targets selective Bcl-2 proteins and
effi ciently induces apoptosis via Bak/
Bax if Mcl-1 is neutralized. Cancer Cell
2006;10:389-399.
38. Chen S, Dai Y, Harada H, Dent P,
Grant S. Mcl-1 down-regulation
potentiates ABT-737 lethality by
cooperatively inducing Bak activation
and Bax translocation. Cancer Res.
2007;67:782-791.
39. Iglesias-Serret D, de FM,
Santidrian AF et al. Regulation of
the proapoptotic BH3-only protein
BIM by glucocorticoids, survival
signals and proteasome in chronic
lymphocytic leukemia cells. Leukemia
2007;21:281-287.
40. Ley R, Ewings KE, Hadfi eld K, Cook
SJ. Regulatory phosphorylation of Bim:
sorting out the ERK from the JNK. Cell
Death.Differ. 2005;12:1008-1014.
41. Fritsch RM, Schneider G, Saur D,
Scheibel M, Schmid RM. Translational
repression of MCL-1 couples stress-
induced eIF2 alpha phosphorylation
to mitochondrial apoptosis initiation.
J.Biol.Chem. 2007;282:22551-22562.
42. Rahmani M, Davis EM, Bauer C,
Dent P, Grant S. Apoptosis induced
by the kinase inhibitor BAY 43-9006
in human leukemia cells involves
down-regulation of Mcl-1 through
inhibition of translation. J.Biol.Chem.
2005;280:35217-35227.
43. Flinterman M, Guelen L, Ezzati-Nik S
et al. E1A activates transcription of p73
and Noxa to induce apoptosis. J.Biol.
Chem. 2005;280:5945-5959.
44. Hershko T, Korotayev K, Polager
S, Ginsberg D. E2F1 modulates
p38 MAPK phosphorylation via
transcriptional regulation of ASK1 and
Wip1. J.Biol.Chem. 2006;281:31309-
31316.
45. Kim H, Rafi uddin-Shah M, Tu HC
et al. Hierarchical regulation of
mitochondrion-dependent apoptosis
by BCL-2 subfamilies. Nat.Cell Biol.
2006;8:1348-1358.
46. Oda E, Ohki R, Murasawa H et al.
Noxa, a BH3-only member of the
Bcl-2 family and candidate mediator
of p53-induced apoptosis. Science
2000;288:1053-1058.
116
Thesis_Final_2_01092008.indd Sec1:116Thesis_Final_2_01092008.indd Sec1:116 01-09-2008 11:08:2601-09-2008 11:08:26

chapter
6c-Abl Kinase Inhibitors Overcome CD40Mediated Drug Resistance in CLL; Implications for Therapeutic Targeting of Chemoresistant Niches
Delfi ne Y.H. Hallaert1,3, Annelieke Jaspers1, Carel J.M. van Noesel2, Marinus H.J.
van Oers1, Arnon P. Kater1, Eric Eldering3
1Dept. of Hematology, AMC, Amsterdam, the Netherlands
2Dept. of Pathology, AMC, Amsterdam, the Netherlands
3Dept. of Experimental Immunology, AMC, Amsterdam, the Netherlands
Blood: accepted for publication
Thesis_Final_2_01092008.indd Sec1:117Thesis_Final_2_01092008.indd Sec1:117 01-09-2008 11:08:2601-09-2008 11:08:26

ABSTRACT
In lymph node (LN) proliferation centers in chronic lymphocytic leukemia (CLL), the
environment protects from apoptotic and cytotoxic triggers. Here, we aimed to defi ne
the molecular basis for the increased drug resistance and searched for novel strategies
to circumvent it. The situation in CLL LN could be mimicked by prolonged in vitro
CD40 stimulation, which resulted in upregulation of anti-apoptotic Bcl-xL, A1/Bfl -1
and Mcl-1 proteins, and afforded resistance to various classes of drugs (fl udarabine,
bortezomib, roscovitine). CD40 stimulation also caused ERK-dependent reduction of
BimEL
protein, but ERK inhibition did not prevent drug resistance. Drugs combined with
sublethal doses of the BH3-mimetic ABT-737 displayed partial and variable effects
per individual CD40-stimulated CLL. The anti-apoptotic profi le of CD40-triggered CLL
resembled BCR-Abl-dependent changes seen in CML, which prompted application
of c-Abl inhibitors imatinib or dasatinib. Both compounds, but especially dasatinib,
prevented the entire anti-apoptotic CD40 program in CLL cells, and restored drug
sensitivity. These effects also occurred in CLL samples with dysfunctional p53.
Importantly, ex vivo CLL LN samples also displayed strong ERK activation together
with high Bcl-xL and Mcl-1 but low BimEL
levels. These data indicate that CLL cells in
chemoresistant niches may be sensitive to therapeutic strategies that include c-Abl
inhibitors.
118
Thesis_Final_2_01092008.indd Sec1:118Thesis_Final_2_01092008.indd Sec1:118 01-09-2008 11:08:2701-09-2008 11:08:27

INTRODUCTION
Chronic lymphocytic leukemia (CLL) is a CD5+ B-cell malignancy that is still considered
incurable, although novel treatment combinations of monoclonal antibodies and
chemotherapy1 seem promising. Many patients eventually develop drug resistance
through several pathways including mutation of the p53 tumor suppressor gene, or
involving the gene encoding the ataxia telangectasia mutated (ATM), which is a kinase
required for p53 function. Such genetic lesions are uncommon in CLL at diagnosis,
but increase in frequency as the disease progresses2. Since the cytoreductive
activity of most current chemotherapeutic agents requires functional p53, loss of p53
is associated with drug resistance and poor prognosis3. Because of these aspects,
different agents with p53-independent modes of action are clearly needed.
CLL has been considered a smoldering disease characterized by long-lived tumor
cells arrested in the G0/G
1-phase of the cell cycle and possessing intrinsic apoptosis
defects4. This concept was largely based on analyses of peripheral blood derived
CLL cells. A study of in vivo cellular kinetics however suggested that CLL is a dynamic
disease with substantial proliferation rates as well as increased death rates compared
to normal B-cells5. Prior to this, it was already known that proliferation and especially
increased survival of the malignant B-cells may not result primarily from intrinsic
defects, but appear to depend largely on interactions with microenvironmental
bystander cells. Interactions between CLL cells and follicular dendritic cells, bone
marrow stromal cells, IL-6-producing endothelial cells, SDF-producing nurselike cells,
or CD40L expressing CD4+ T cells cells6-9 have been shown to increase the apoptotic
threshold in vitro. In a recent comparative survey of apoptosis regulatory genes and
proteins in neoplastic B-cells derived from CLL lymph node (LN) proliferation centers
and from peripheral blood10, we observed specifi c changes including increased
expression of anti-apoptotic proteins such as Mcl-1, Bcl-xL and A1/Bfl -1 in LN cells.
Extended cell survival of tumor cells within the LN microenvironment may create an
intracellular milieu permissive for genetic instability and for the accumulation of gene
mutations that favors disease progression. Furthermore, these microenvironmental
interactions may provide a safe haven from cytotoxic anticancer drugs, thus serving
as a tumor reservoir from which relapse occurs (reviewed by Pedersen and Reed11).
This concept is supported by the observation that prolonged CD40 activation, which
to a large extent recapitulates the anti-apoptotic expression profi le of LN derived
CLL cells, renders CLL cells resistant to current chemotherapeutics12;13. The currently
widely applied drug fl udarabine relies on an intact p53 response which induces
expression of the Bcl-2 member Puma, thereby triggering apoptosis14-16. Alternative,
p53-independent drugs such as the proteasome inhibitor bortezomib or the cyclin
Ch
ap
ter 6
▪ c-A
bl in
hib
itors
pre
ven
t dru
gre
sis
tan
ce in
CL
L
119
Thesis_Final_2_01092008.indd Sec1:119Thesis_Final_2_01092008.indd Sec1:119 01-09-2008 11:08:2701-09-2008 11:08:27

dependent kinase inhibitor roscovitine engage other pro-apoptotic Bcl-2 members
such as Noxa and Bim. Especially Bim is a potent pro-apoptosis member of the BH3-
only subgroup of the Bcl-2 family, engaged by a variety of apoptotic triggers17-20. A
potential means of suppressing the lethal capacity of Bim involves the pro-survival
kinase ERK. In model systems activation of ERK leads to phosphorylation and
subsequent proteasomal degradation of the BimEL
splice variant21;22.
In the present study we used in vitro CD40 stimulation as a model for chemoresistant
LN CLL, and searched for means to circumvent it. CD40 stimulation of CLL cells
strongly induced Bcl-xL, Mcl-1 and A1/Bfl -1 proteins, resulting in a broad drug
resistance. Various aspects of this anti-apoptotic program also occur in chronic
myeloid leukemia (CML), a disease for which current treatment includes kinase
inhibitors that were developed to target BCR-Abl signaling23. Therefore, we next
applied the c-Abl inhibitors imatinib (Gleevec, STI-571) or dasatinib (Sprycel, BMS-
354825) in conjunction with CD40. Both drugs caused a profound reversal of the
protective CD40 effects, and restored drug sensitivity. Probing of LN CLL samples
demonstrated that in these protective niches similar pro-survival signaling pathways
are active as upon CD40 triggering in vitro. Collectively, these data suggest that
CLL cells residing in LN might be therapeutically targeted by drug combinations that
include c-Abl inhibitors.
MATERIALS AND METHODS
Patient material
Patient material was obtained after routine diagnostic or follow-up procedures at
the departments of Hematology and Pathology of the Academic Medical Center
Amsterdam. This study was conducted after informed consent and approved by the
AMC medical committee on human experimentation, in agreement with the Helsinki
Declaration of 1975, revised in 1983. Lymph node (LN) material, diffusely infi ltrated
by CLL cells, was freshly frozen in liquid nitrogen directly after surgical removal.
Immunohistochemical analysis of these lymph nodes revealed that greater than 90%
of the tissue consisted of tumor cells10. Peripheral blood (PB) mononuclear cells
(PBMCs) of patients with CLL, obtained after Ficoll density gradient centrifugation
(Pharmacia Biotech, Roosendaal, The Netherlands) were frozen in Iscove’s Modifi ed
Dulbecco’s Medium (IMDM) supplemented with L-Glutamine, 25 mM HEPES
(Biowhittaker, Europe) containing, 2mM L-Glutamin (Invitrogen), 50 mg Gentamycin
(Invitrogen) 3.57 x 10-4% (v/v) -mercapto ethanol (Merck, Darmstadt, Germany), 10
120
Thesis_Final_2_01092008.indd Sec1:120Thesis_Final_2_01092008.indd Sec1:120 01-09-2008 11:08:2701-09-2008 11:08:27

% dimethyl sulphoxide (DMSO; Sigma Chemical Co., St. Louis, MO, USA) and 15%
fetal calf serum, (FCS; ICN Biomedicals GnbH, Meckenheim, Germany), and stored
in liquid nitrogen. Expression of CD5 and CD19 (antibodies obtained from Bekton
Dickinson (BD) Biosciences, San Jose, CA, USA) on leukemic cells was assessed by
fl ow cytometry (FACScalibur, BD Biosciences) and analysed with CellQuest software
(BD Biosciences).
RNA isolation and RT-MLPA
Total RNA was isolated using the GenElute Mammalian Total RNA Miniprep Kit (Sigma
Aldrich). Reverse transcription–multiplex ligation-dependent probe amplifi cation
assay (RT-MLPA) procedure was performed as described previously16;24.
Reagents
The proteasome inhibitor bortezomib was obtained from Janssen-Cilag (Tilburg
the Netherlands). The -secretase inhibitor GSI-1, the Erk inhibitor PD-98059, the
NF-κB inhibitor BAY-11-7082 and the proteasome inhibitor MG132 were obtained
from Calbiochem (Amsterdam, the Netherlands). Roscovitine and fl udarabine
(F-Ara-A) were purchased from Sigma Chemical Co. (St. Louis, MO, USA). ABT-
737 was obtained under MTA from Abbott (courtesy Dr S Rosenberg, Abbott Park,
Illinois, USA). The kinase inhibitors, imatinib and dasatinib were from Novartis
(Basel, Switzerland) and Bristol-Myers Squibb (New York, NY, USA) respectively.
Analysis of apoptosis, Western Blot and antibodies
For apoptosis induction, cells at a density of 1.5.106/ml in culture medium were
treated with 100μM fl udarabine (48 hrs), 30nM bortezomib, 25 μM roscovitine or 5
μM GSI1 (24 hrs), and stained with 200 nM MitoTracker Orange (Molecular Probes,
Leiden, The Netherlands) for 30 minutes at 37°C and analysed by FACS. Western
blotting was performed as described previously16. Cells were lysed in Laemmli
Sample Buffer, and samples (10-30 μg protein) were separated by 13% sodium
dodecyl sulfate polyacrylamide gel electrophoresis (10% gels for ERK). To screen for
p53 functionality, cells were irradiated (5Gy) and after O/N incubation tested for the
expression of p53 and p21 by western blot analysis as described before25. Blots were
probed with polyclonal anti-Mcl-1 (catalog no. 554103; Pharmingen, BD Biosciences),
monoclonal anti-Noxa (clone 114C307.1; Imgenex, San Diego, CA), monoclonal anti-
Bim (clone 14A8; Chemicon, Temecula, CA), antiserum to -actin (clone I-19; Santa
Cruz Biotechnology, Santa Cruz, CA), polyclonal anti-Bcl-xL (catalog no. 620211,
BD Biosciences), polyclonal anti-Bcl-2 (catalog no 210-701-C100, Kordia, Leiden
Ch
ap
ter 6
▪ c-A
bl in
hib
itors
pre
ven
t dru
gre
sis
tan
ce in
CL
L
121
Thesis_Final_2_01092008.indd Sec1:121Thesis_Final_2_01092008.indd Sec1:121 01-09-2008 11:08:2701-09-2008 11:08:27

the Netherlands) polyclonal anti-phospho-Erk (catalog no. #9102 Cell Signaling),
polyclonal anti-Erk (catalog no. #9101 Cell Signaling), polyclonal antibodies against
A1/Bfl -1 and Bid were a kind gift of Prof. Dr. J. Borst (The Netherlands Cancer
Institute, Amsterdam, The Netherlands).
In vitro CD40 stimulation and cell lines
BCR-Abl positive K562 cells and NIH3T3 fi broblasts were cultured in IMDM as
described above for CLL cells. CD40-ligand (CD40L, CD154) was expressed on
NIH3T3 fi broblasts, stably transfected with a plasmid encoding human CD40L.
Fibroblasts were irradiated (30Gy) and plated in culture-treated 6-wells plates (6x 105
cells/well). CLL cells were thawed and 5 x 106 cells per well were added to the adhered
fi broblasts in 3 ml IMDM containing 10% FCS and incubated for 48 hrs at 37oC. To
test the effect of c-Abl kinase inhibitors, imatinib and dasatinib, and the effect of Erk-
inhibitor PD-58059, CLL cells were pre-treated with 80 μM imatinib or dasatinib, or
50 μM PD-58059 for 30 minutes. After pre-incubation CLL cells were stimulated for
48 hrs at 37oC with CD40L with or without 30 μM imatinib or dasatinib or 50 μM PD-
58059. In the case of dasatinib, also other regimens and concentrations were used,
where CLL cells were fi rst co-cultured for 48 hrs with CD40L-expressing or control
3T3 fi broblasts, detached and washed and subsequently incubated in medium for an
additional 48 hrs in the presence of varying dasatinib concentrations (30nM-30μM),
followed by testing sensitivity to cytotoxic drugs, as described above.
RESULTS
Prolonged CD40 stimulation of CLL cells results in broad drug
resistance, which is independent of ERK- mediated decrease in
BimEL levels
In vitro stimulation via CD40 renders CLL cells resistant to fl udarabine and induces
expression of various anti-apoptotic proteins such as Bcl-xL and A1/Bfl -1 via de
novo transcription12;13;26. In addition to previously described transcriptional effects
of prolonged CD40 triggering, several novel effects on protein levels of various
apoptosis regulators were observed. In particular, the BimEL
splice variant decreased
while Mcl-1 levels increased (see Fig. 1A).
Since it is known that ERK signaling can affect BimEL
protein levels in model
systems21;27 this aspect was investigated further. Over the course of several days of
CD40 stimulation, a signifi cant reduction in BimEL
protein levels occurred, although
Bim mRNA levels remained constant10 (see also Fig. 4 below). In Figure 1B, the larger
122
Thesis_Final_2_01092008.indd Sec1:122Thesis_Final_2_01092008.indd Sec1:122 01-09-2008 11:08:2701-09-2008 11:08:27

two Bim species represent BimEL
and most presumably a splice variant Bimα128,
which became visible in certain samples upon prolonged migration in SDS-PAGE.
Under the experimental conditions applied a short-lived phosphorylated form of Bim
(p-Bim) is probably also present21, but in our hands this form of Bim could not be
observed in primary samples either with the antibody used here, nor with commercial
antibodies specifi cally generated against p-Bim. The activation status of ERK upon
CD40 triggering was increased, and addition of the specifi c ERK inhibitor PD-98059
during CD40 stimulation prevented the reduction of BimEL
(Fig. 1C). Addition of the
proteaseome inhibitor MG132 after CD40 stimulation demonstrated that BimEL
levels
Figure 1. Anti-apoptotic changes in CLL cells upon CD40 stimulation include ERK- mediated
decrease in BimEL
levels.
(A) Changes in expression of apoptosis regulators upon 48 hrs CD40 triggering were monitored by Western
blot for the indicated proteins. Results are from a representative CLL sample from >10 patients studied.
Equal protein loading was confi rmed by staining for actin as loading control.
(B) Time course of BimEL
decrease monitored in 2 CLL samples. Shown are samples taken on consecutive
days of co-culture in absence or presence of CD40 stimulation. In sample 11-166 a decrease in BimEL
can
be observed on day 2, and in sample 12 on day 1. Position of BimEL
is indicated by a triangle, the faster
migrating species is probably the Bimα1 splice variant.
(C) Effects of ERK inhibition on BimEL
levels and phosphorylated ERK. CLL cells were stimulated with CD40
in the presence of ERK inhibitor as indicated, and lysates were probed for Bim protein, phosphorylated
and total ERK levels.
were controlled via increased protein turnover, confi rming previous reports21;22;29 (data
not shown).
Next, CLL cells triggered via CD40 in the absence or presence of ERK inhibition were
A B
C
Ch
ap
ter 6
▪ c-A
bl in
hib
itors
pre
ven
t dru
gre
sis
tan
ce in
CL
L
123
Thesis_Final_2_01092008.indd Sec1:123Thesis_Final_2_01092008.indd Sec1:123 01-09-2008 11:08:2701-09-2008 11:08:27

investigated for sensitivity to drugs that are in current clinical use or in preclinical
development. As can be seen in fi gure 2. prolonged CD40 stimulation rendered
the cells resistant to fl udarabine, as observed before12;13,the proteasome inhibitor
bortezomib and the cyclin-dependent kinase inhibitor roscovitine. In addition, the
-secretase inhibitor GSI-1 was included, which is considered to be an inhibitor of
Notch signaling30. We have recently observed that GSI-1 is in fact an inhibitor of the
proteasome and a potent inducer of apoptosis in CLL (Hallaert et al., submitted).
CD40 triggering also rendered CLL cells resistant to GSI-1. For multiple CLL isolates
tested, addition of ERK inhibitors did not alleviate the broad drug resistance afforded
Figure 2. Broad drug resistance of CLL cells upon CD40 stimulation is not prevented by ERK
inhibition.
CLL cells were co-cultured with control 3T3 (control) or CD40L-expressing cells for 48 hrs, in the presence
of ERK inhibitor PD-98059 as indicated. After detachment and washing, cells were incubated with the
indicated drugs as described in detail in Methods, and analyzed for apoptosis by MitoTracker staining
after 24 hrs (roscovitine, bortezomib and GSI-1) or 48 hrs (fl udarabine). Cells cultured on 3T3 cells (black
bars) are sensitive to all drugs, but CD40 stimulation (white bars) confers broad drug resistance and this
is maintained when ERK is inhibited (grey bars). The data shown for untreated samples (medium) were
measured at 24hr. Apoptosis levels of medium samples at 48hrs were comparable.
via prolonged CD40 stimulation (Fig. 2). Together these data indicate that although
CD40 signaling activates ERK and thereby causes a decline in BimEL
levels, this is
not the cause for the observed broad drug resistance.
c-Abl inhibors prevent the anti-apoptotic protein profi le of CD40-
treated CLL cells
Another aspect of prolonged CD40 triggering of CLL cells was an increase in Mcl-1
protein (see Fig. 1A) which was, similar to the changes in Bim, independent from
increased transcription (see also below). Mcl-1 has recently been recognized as
promising target for drugs31, and has been implicated in anti-apoptotic signaling via
BCR-Abl in chronic myeloid leukemia32-34. Furthermore, other anti-apoptotic changes
124
Thesis_Final_2_01092008.indd Sec1:124Thesis_Final_2_01092008.indd Sec1:124 01-09-2008 11:08:2701-09-2008 11:08:27

in our in vitro CD40-CLL system, such as increased Bcl-xL and decreased Bim, have
also been implicated in BCR-Abl signaling32;35-37. Lastly, it was recently reported that
c-Abl protein expression correlates positively with tumor burden and disease stage
in CLL38. Therefore, we next tested the c-Abl inhibitor STI-571/gleevec/imatinib as a
potential suppressor of CD40-mediated pro-survival effects in CLL cells. In fi gure 3 it
can be seen that imatinib caused a clear reversal of almost all effects of CD40 stimu-
lation regarding Bcl-xL, Mcl-1, A1/Bfl -1 and BimEL
levels (left panel). This was also
observed for the second generation Abl inhibitor sprycel/dasatinib (middle panel).
A B
Figure 3. Anti-apoptotic gene and protein profi le of CLL
induced by CD40 stimulation is reversed by kinase
inhibitors imatinib and dasatinib.
(A) CLL cells were co-cultured with control 3T3 or CD40L-
expressing cells for 48 hrs, in the presence of PD-98059,
imatinib or dasatinib as indicated. Lysates were probed for
Bim, Mcl-1, Bcl-xL, A1/Bfl -1 and Bcl-2 as indicated and actin
as loading control. Shown are representative examples of 2
CLL samples with WT p53 function (left and middle panel),
and 1 CLL with p53 dysfunction (right panel; note different
order of samples). The upregulation of Mcl-1, Bcl-xL and
A1/Bfl -1 is not affected by ERK inhibition, but prevented by
imatinib or dasatinib, irrespective of p53 functionality.
(B) RNA was collected from CLL cells stimulated for 48
hrs with CD40 and inhibitors as indicated, and assayed for
expression of 34 apoptosis genes by MLPA. Shown are
averaged relative expression levels ± SD (in percentage
of total normalized signal) of selected genes in samples
from p53 WT (n=4) and p53 dysfunctional (n=3) CLL cells.
The CD40-mediated positive effects on transcription of
A1/Bfl -1 and Bcl-xL are reversed by Abl kinase inhibitors.
Examples of genes that are not signifi cantly affected
at the transciptional level are Mcl-1, Bim and GUS
( -Glucuronidase, a housekeeping gene).
Ch
ap
ter 6
▪ c-A
bl in
hib
itors
pre
ven
t dru
gre
sis
tan
ce in
CL
L
125
Rela
tive e
xpre
ssio
n
Thesis_Final_2_01092008.indd Sec1:125Thesis_Final_2_01092008.indd Sec1:125 01-09-2008 11:08:2701-09-2008 11:08:27

This compound has a higher specifi c activity towards c-Abl, but is also less specifi c
for Abl kinase and targets other kinases such as Btk, Lyn and Tec23;39. The effects
of imatinib and dasatinib with respect to reversing the CD40 effects on pro-survival
parameters were also observed in CLL cells with a dysfunction in the p53 pathway
(Fig. 3, right panel).
The various kinase inhibitors were also monitored for their effects on transcription
using a multiplex assay able to quantify expression of 34 apoptosis regulatory genes.
As described previously, prolonged in vitro CD40 stimulation of CLL cells induces
transcription of Bcl-xL and A1/Bfl -1, as well as a reduction in Noxa10;13. For the ERK
inhibitor PD-98059 no effects on transcription of these genes were found. In contrast,
the c-Abl inhibitors prevented upregulation of Bcl-xL and A1/Bfl -1 transcripts, while for
example Mcl-1 and Bim transcripts were hardly affected by these drugs (Fig. 3B black
bars), although they did display changes at the protein level (Fig. 3A). The effects of
the Abl kinase inhibitors on Bcl-xL and A1/Bfl -1 were similar to those observed when
CLL cells were exposed to NF-κB inhibitor BAY-11-7082 during stimulation via CD40
(Supplementary Fig. 1). The inhibitory effects of especially dasatinib on Bcl-xL and
A1/Bfl -1 transcription were also detected in cells with a dysfunctional p53 response
(Fig. 3B white bars). In these cases, the effects of imatinib on CD40-induced gene
transcription were limited, suggesting that perhaps the suppressive effects of imatinib
may require p53 function. The complete dataset for all genes interrogated by the MLPA
probe set is represented in supplementary fi gure 2. Together these data demonstrate
that imatinib/dasatinib have a clear impact on signaling pathways leading to gene
transcription such as NF-κB, and also on mechanisms controlling protein turnover of
Mcl-1 and Bim.
Contribution to drug resistance of pro-survival proteins probed
by ABT-737
Anti-apoptotic Bcl-2 family members can be counteracted by BH3 mimetics such
as ABT-737, a widely studied compound in preclinical development40. ABT-737 is
very effective against Bcl-2 and Bcl-xL, but does not bind to Mcl-1 or A1/Bfl -131;41.
As reported before42, CLL cells are quite sensitive to ABT-737, but upon stimulation
with CD40 this is reduced approximately 100-fold (Fig.4 A&B). We tested whether
sublethal doses of ABT-737 could synergize with other drugs in this setting. There
was a slight increase in apoptosis of CD40-stimulated cells when 0.1μM ABT-737
was combined with various other drugs. This was further restored to levels observed
in medium or control cultures with 3T3 cells by using 1 μM ABT-737. In fi gure 4C
the averaged data from 4 CLL patients are shown. Individual sample responses to
126
Thesis_Final_2_01092008.indd Sec1:126Thesis_Final_2_01092008.indd Sec1:126 01-09-2008 11:08:2801-09-2008 11:08:28

ABT-737 showed divergent patterns, with some patient’s cells displaying full reversal
of drug sensitivity at 1.0 μm ABT-737 for all drugs tested, while others displayed
different patterns depending on the drug tested (Supplemental Fig. 3). This seemed
consistent with the clear patient to patient variation in the degree of upregulation of
Mcl-1 and A1/Bfl -1 (Fig. 3A). These results indicate that the contribution of Bcl-2 and
Bcl-xL to the observed drug resistance in this in vitro model is substantial, but could
generally be counteracted by ABT-737. The effi ciency and effective dose with which
ABT-737 acts differs per patient sample and this probably correlates with the degree
of increase in Mcl-1, and possibly A1/Bfl -1, obtained with CD40 stimulation.
c-Abl kinase inhibitors prevent drug resistance of CD40-treated
CLL cells
In a similar fasion as above for ABT-737, the effect of c-Abl kinase inhibitors on the
drug resistance afforded by CD40 triggering was measured. The apoptosis-inducing
effects of the Abl inhibitors themselves on control samples co-cultured with 3T3 cells
and CD40L-expressing cells were minimal (Fig. 5A, medium samples). Only at high
Figure 4. Contribution of Mcl-1 to drug resistance probed by ABT-737.
(A) CLL cells were treated immediately after thawing with the indicated concentrations of ABT-737 or the
inactive enantiomer. After 24 hrs, apoptosis was measured by MitoTracker staining.
(B) CLL were cultured for 2 days in medium, with 3T3 control cells or with 3T40L cells before treatment with
ABT-737 as above. Data in A and B represent averages ± SD from 3 different CLL samples.
(C) Sublethal doses of ABT-737 after CD40 stimulation as determined in B) (0.1 and 1.0μM) were combined
with various other drugs as indicated to test synergy in reversal of drug resistance. Data are averages ± SD
from 5 (0.1 μM) or 4 (1μM) patient samples, tested in 3 independent experiments.
A
C
B
Ch
ap
ter 6
▪ c-A
bl in
hib
itors
pre
ven
t dru
gre
sis
tan
ce in
CL
L
127
Thesis_Final_2_01092008.indd Sec1:127Thesis_Final_2_01092008.indd Sec1:127 01-09-2008 11:08:2801-09-2008 11:08:28

levels and upon prolonged exposure did imatinib and dasatinib induce signifi cant
apoptosis in CLL cells, in contrast to e.g. K562 cells which are very sensitive due
to their dependence on the BCR-Abl fusion protein for survival (Supplemental Fig. 4).
Remarkably however, imatinib and especially dasatinib prevented the resistance
towards various drugs normally observed upon CD40 treatment of CLL cells. This
appeared true for CLL samples with mutated as well as unmutated IgVH gene
sequences (both n=2). The sensitising effect of these inhibitors was also seen in CLL
cells with a dysfunctional p53 pathway (Fig. 5B). Especially the cytotoxic effect of
128
A C
B D
Figure 5. Drug resistance of CD40-stimulated CLL cells is reversed by c-Abl kinase inhibitors.
(A) CLL samples (n=4) were cultured on 3T3 (control) or CD40L-expressing cells in the presence of the
indicated inhibitors for 48 hrs, and after detachment and washing cultured for 24 hrs in medium or with the
cytotoxic drugs. Average results for apoptosis measured via MitoTracker staining are shown.
(B) The same as in A) for an experiment with p53 dysfunctional cells. Data are representative for 3 similar
experiments performed; the variation among samples in particular for background apoptosis in the absence
of external stimuli precluded averaging.
(C) A similar experiment as in A) was performed with decreasing concentrations of dasatinib as indicated.
Drug susceptibility was assessed by incubation with 5 μM GSI-1 for 24 hrs. Results represent averages of
4 experiments or 2 where indicated. At 3 nM there was no effect of dasatinib detectable (not shown).
(D) Sequential CD40 stimulation followed by incubation with c-Abl kinase inhibitors. CLL cells were co-
cultured with 3T3 cells expressing CD40L for 48 hrs, detached and washed before addition of dasatinib
(300 nM) for an additional 48 hrs, and were then tested for drug susceptibility. Results represent average
data of 3 experiments.
Thesis_Final_2_01092008.indd Sec1:128Thesis_Final_2_01092008.indd Sec1:128 01-09-2008 11:08:2901-09-2008 11:08:29

proteasome inhibitors (bortezomib and GSI-1) was potentiated by treatment of CLL
cells with c-Abl inhibitors during CD40 exposure. In general, the effects of dasatinib
were stronger than those of imatinib at the concentrations used (30 μM), as was
also observed for the effects on protein levels (see Fig. 3). Since dasatinib has a
higher specifi c activity towards its target kinases than imatinib23;43 we also tested its
effects over a lower range of concentrations. The capacity of dasatinib to modulate
the drug sensitivity of CD40-treated CLL cells could also be observed at substantially
lower concentrations (30nM-3μM). This is demonstrated in fi gure 5C for the results
obtained with GSI-1, which was in general the strongest inducer of apoptosis in CLL
cells among the drugs tested.
The results thusfar were obtained with simultaneous administration of CD40 signals
and kinase inhibitors. To better refl ect the actual situation of LN CLL cells already
exposed to a protective environment, isolated PB CLL cells were fi rst stimulated
via CD40 for 48 hrs, followed by separate addition of dasatinib (0.3μM) and drug-
sensitivity tests. Also in this set-up, a reversal of resistance towards various drugs
(fl udarabine, bortezomib, roscovitine) could be observed (Fig. 5D). Thus, dasatinib
has a clear capacity to interfere with the protective effects afforded by prolonged
CD40 stimulation.
Similar apoptosis protein signature in ex vivo LN samples as
upon in vitro CD40 triggering
To relate the effects of in vitro CD40 stimulation with the in vivo situation, samples
from CLL lymph nodes were lysed directly in SDS-containing sample buffer and
probed for the presence of proteins involved in apoptosis regulation. As observed
before, a clear increase of Bcl-xL protein was present in LN samples compared to
peripheral blood (PB) samples10. This was also found for Mcl-110 and A1/Bfl -1 (data
not shown). Regarding the expression levels of other signature proteins involved
in CD40-mediated anti-apoptosis pathways, a strong increase in both total and
phosphorylated ERK was found, concomitant with decreased levels of BimEL
(Fig. 6).
These fi ndings indicate that in CLL lymph nodes similar survival pathways are
operational as those that can be induced in peripheral blood CLL cells by prolonged
in vitro CD40 stimulation.
Ch
ap
ter 6
▪ c-A
bl in
hib
itors
pre
ven
t dru
gre
sis
tan
ce in
CL
L
129
Thesis_Final_2_01092008.indd Sec1:129Thesis_Final_2_01092008.indd Sec1:129 01-09-2008 11:08:2901-09-2008 11:08:29

DISCUSSION
Previous reports have described effects of inhibitors of BCR-Abl kinase on single
anti-apoptosis proteins (predominantly Mcl-1, Bcl-xL or Bim) in CML or model cell
lines35-37. This study provides an overview on the effects of c-Abl inhibitors on all Bcl-2
members in the context of CD40 signaling in CLL cells. The rationale for the present
study was two-fold. First, there is the growing concept that CLL is a dynamic disease,
with proliferation centers in LN and possibly also BM. These protective niches, where
cells are prone to be more drug resistant, are presumably the source of relapsing
clones. Second, the potential of novel drugs such as kinase inhibitors, to target pro-
survival signaling pathways to which malignant cells have become addicted.
We have observed that our in vitro CLL culture model setting provides strong and
probably supra-physiological CD40 signals, with longlasting protective effects
which continue after detachment of CLL cells from CD40L cells (data not shown).
Nevertheless, comparison between LN samples and PB CLL cells stimulated in vitro
via CD40 indicated the presence of a comparable pro-survival signature as implied
by ERK activation and BimEL
levels. Previously, we have shown that in LN samples
also increased levels of Bcl-xL and Mcl-1 are detectable10. Together, the available
data indicate that the pro-survival signature triggered via CD40 stimulation in vitro is
also found in CLL lymph nodes, and imply that our experimental data hold promise
for extrapolation towards a therapeutic setting.
With respect to post-transcriptional effects of CD40 stimulation on CLL cells, both Bim
and Mcl-1 proteins are known targets for phosphorylation and subsequent increased
proteasomal degradation. Cytokine withdrawal in murine cell lines causes decreased
PI3K-Akt/PKB signaling to activate GSK3 which in turn phosporylates Mcl-1, thus
marking it for proteasomal degradation44. In the case of CLL cells, our data indicate
that upon CD40 stimulation PKB phosphorylation was undetectable, the PI3 kinase
130 Figure 6. Anti-apoptotic protein
signature in CLL lymph nodes.
Protein lysates obtained from
peripheral blood (PB, n= 5) and
lymph node (LN, n=6) were probed
for phosphorylated-ERK, total
ERK, Bim and actin as indicated.
The expression of these proteins in
ex vivo LN was similar to changes
observed upon in vitro stimulation
of PB CLL cells with CD40.
Thesis_Final_2_01092008.indd Sec1:130Thesis_Final_2_01092008.indd Sec1:130 01-09-2008 11:08:2901-09-2008 11:08:29

inhibitor LY294002 did not trigger apoptosis, and the rate of Mcl-1 protein turnover was
not changed (unpublished observations). Since Mcl-1 transcription in CLL cells was
also not affected by CD40, this suggests that the increase in Mcl-1 protein is possibly
controlled at the level of translation by a non-PKB dependent mechanism. Recent
evidence from other experimental systems indeed points at translational repression
of Mcl-1 via eIF initiation factors as yet another point of regulation45;46. If this system
is operational under our experimental conditions and whether it may be connected
with the other recently described pathway implicating antigen receptor/PI3-K/PKB
signaling in affecting Mcl-1 levels47 remains to be determined. In contrast to the
situation in AML cells41, in primary CLL cells the ERK pathway seems not responsible
for increased Mcl-1 protein, as the ERK inhibitor PD-98059 did not block its increase,
and did not affect drug susceptibility (Fig. 2&3). Whether or not increased Mcl-1 plays
an important role in vivo in survival of CLL in lymph nodes seems an important issue
with respect to therapeutic application of ABT-737. Our data and those of others31;41
indicate that variations in Mcl-1 and possibly also A1/Bfl -1 levels will determine the
effective dose of ABT-737 both as a single agent and in drug combinations. Of note,
the combination of ABT-737 with roscovitine, which should counteract Bcl-2, Bcl-xL as
well as Mcl-131, was not effective in all patients (Supplemental Fig. 4). This suggests
that either roscovitine is unable to reduce Mcl-1 in this setting or that perhaps in these
samples A1/Bfl -1 is a dominant factor. Our observations on increased BimEL
turnover
are in accord with an established pathway of ERK-mediated phosphorylation and
proteasomal degradation21;22;29. To our knowledge, this is the fi rst example of this
pathway operating in primary tumor cells upon CD40 stimulation, and in CLL LN
samples.
In our experience, neither imatinib nor dasatinib are effi cient inducers of apoptosis as
single agents, in contrast to their effects on K562 cells, which depend for survival on
the BCR-Abl fusion oncogene (Supplemental Fig. 3). In a recent study, considerable
variation in apoptosis susceptibility in untreated and dasatinib-treated peripheral
blood samples was found using 5 μM dasatinib, and the response was correlated
with IgVH mutation and ZAP70 status48. This and other studies performed to date
agree that, in CLL cells from peripheral blood, dasatinib has a strong synergistic
effect in combination with p53-pathway dependent and -independent agents48-50.
Transcriptional effects of imatinib and dasatinib on Bcl-xL and A1/Bfl -1 were similar
to those of inhibitors of NF-κB. We noted that the effects of the c-Abl inhibitors on
reversing ERK phosphorylation status, and the corresponding changes in BimEL
levels varied among patients, without apparent correlation with prognostic factors
such as mutation or p53 status (data not shown and Fig. 3). These signaling
pathways are affected/reversed by imatinib and dasatinib although the actual
Ch
ap
ter 6
▪ c-A
bl in
hib
itors
pre
ven
t dru
gre
sis
tan
ce in
CL
L
131
Thesis_Final_2_01092008.indd Sec1:131Thesis_Final_2_01092008.indd Sec1:131 01-09-2008 11:08:2901-09-2008 11:08:29

target(s) remains unknown. Recent analyses of the spectrum of kinase targets of
these compounds points to various candidates involved in T- and/or B-cell activation
such as Src kinases including Lck and Fyn, Btk and Tec kinase23;39. The spectrum of
non-Abl kinases targeted by dasatinib is in fact quite broad (>20 kinases), and an
immunosuppressive effect was predicted23, and recently confi rmed for T cells51. Our
preliminary analyses do not show a similar inhibitory effect of dasatinib on in vitro
B-cell proliferation however (A. Jaspers, unpublished observation). From the kinases
targeted by dasatinib no obvious candidate(s) for exclusive participation in the CD40
pathway is apparent, although the Ser/Thr kinase p38α and upstream MAP kinases
appear likely as participants. A clue for the participation of Btk or Tec kinases comes
from a recent report that their expression level is regulated via NF-κB in a positive
feedback-loop. This loop can be interrupted by proteasome inhibitors52, which fi ts
with our observation that the combination of bortezomib or GSI-1 with dasatinib has
the strongest effect on apoptosis of CD40-stimulated CLL cells (Fig. 5A&B).
Obviously, c-Abl kinase itself may very well be involved, and there is evidence
that levels of c-Abl protein expression correlate positively with tumor burden and
disease stage in CLL53. Another study reported that c-Abl becomes active upon CD40
triggering and then induces p7354. This signaling route is predicted to bypass p53
and may therefore be therapeutically relevant. Both these studies used imatinib and/
or introduction of recombinant c-Abl, so they cannot provide defi nitive evidence of
endogenous c-Abl kinase activity in CLL. The majority of studies on activity have
been done with the BCR-Abl-positive cell line K562 or primary CML samples where
expression levels of the oncogenic fusion protein are augmented. Our preliminary
efforts to detect active endogenous c-Abl either in un-stimulated, CD40-triggered or
LN CLL cells by western blotting with commercial antibodies were inconsistent.
At present, two independent mechanisms are attributed to the development of
chemoresistance in CLL. The fi rst is a shift in the balance between pro- and anti-
apoptotic regulators, and both Mcl-155 and Bfl -1/A156 have been associated with
resistance to chemotherapy. Signifi cantly, these hallmarks are very similar to the
CD40-activated CLL phenotype we use as a model. The second mechanism is based
on acquired mutations resulting in a dysfunctional p53 response3. A recent phase
II evaluation of dasatinib as single agent in relapsed and refractory CLL showed
limited effects, but in good correlation with our data a reduction of lymph node size
was observed in a major fraction of patients57. Our data indicate that c-Abl inhibitors,
notably dasatinib, overcome the protective profi le within the micro-environment
resulting in susceptibility to p53 pathway dependent drugs (fl udarabine) as well as
to p53-independent agents (roscovitine, bortezomib, ABT-737). Thus, from a clinical
perspective it may be more effective to apply combination strategies of dasatinib
132
Thesis_Final_2_01092008.indd Sec1:132Thesis_Final_2_01092008.indd Sec1:132 01-09-2008 11:08:3001-09-2008 11:08:30

with other drugs. Our data provide a rationale to combine dasatinib both with purine-
analogues but also with drug regimens that do not exclusively rely on p53 function
for effi cacy.
ACKNOWLEDGEMENTS
We thank Dr. M. Kramer, Dr. S. Wittebol, (department of Internal Medicine, Meander
Medical Centre, Amersfoort) and Dr. J. Baars (department of Internal Medicine, The
Netherlands Cancer Institute, Amsterdam) for including CLL patients in this study, and
René van Lier for his insightful comments and critical reading of the manuscript.
This work was supported by the Dutch Cancer Foundation (DCF) grant nr. UVA2004-
3039. A.P.K. is supported by a ‘Veni’ grant from ZonMw (The Netherlands Organization
for Health Research and Development).
Ch
ap
ter 6
▪ c-A
bl in
hib
itors
pre
ven
t dru
gre
sis
tan
ce in
CL
L
133
Thesis_Final_2_01092008.indd Sec1:133Thesis_Final_2_01092008.indd Sec1:133 01-09-2008 11:08:3001-09-2008 11:08:30

134
Patient Rai
stage
WBC
10E9/L
Lymphocytes
(%)CD5+/
CD19+2
CD3+
(%)
IgVH
3
mut.
status
p53
status4
CD38
(CD20+)
Chromosomal
abnomalities5
CLL01 l 113.0 90.0 88.3 4.4 + functional 81 none
CLL07 II 93.3 86.9 96.8 2.4 + functional 87 11q-
CLL08 0 58.5 91.0 92.7 4.3 + ND 24 none
CLL11 II 49.3 88.8 91.4 5.5 - functional 92 13q-
CLL12 I 72.2 86.3 93.0 5.0 + functional 33 none
CLL16 II 80.1 73.7 92.0 6.0 + ND 39 13q-
CLL17 II 102.0 92.1 93.3 4.1 - ND 47 none
CLL18 0 48.0 89.2 90.5 7.7 + ND 29 ND
CLL20 II 61.4 89.0 95.0 4.0 + functional 20 ND
CLL21 IV 60.6 89.8 86.7 3.1 - ND 24 ND
CLL22 I 60.1 94.4 81.7 6.5 - dysfunc-
tional
44 17p-
CLL23 IV 137.0 94.8 83.5 3.8 + dysfunc-
tional
57 17p-;13q-
CLL25 I 79.0 93.3 84.7 3.4 - dysfunc-
tional
39 17p-
CLL29 I 117.0 95.8 95.0 1.5 + ND 9.5 ND
CLL31 II 68.8 94.7 90.8 4.7 + ND 1.9 13q-
CLL32 0 73.8 86.9 85.2 7.2 + ND 2.4 ND
CLL40 IV 232.0 96.1 98.5 1.2 - ND 97 ND
1 Patient data for the lymph node samples used in this study can be found in ref 10.2 Percentage of cells positive for CD5 and CD19 surface expression.3 Mutated IgV
H gene (+) denotes >2% mutations compared to germline sequence.
4 P53 functional status was measured via radiation-induced RNA expression of p53 target genes Puma
and Bax, or by p53 and p21 protein upregulation via western blot, as described in refs 16 and 25. Patient
25 had a so-called type A dysfunction. (ND = not determined)5 As determined by FISH. Probes for 11q22.3 (ATM), 13q14 (D13S319) and 17p13 (TP53) were obtained
from Abbott-Vysis. Samples with >10% aberrant signals were considered abnormal.
Table 1. Patient characteristics (peripheral blood samples)1.
Thesis_Final_2_01092008.indd Sec1:134Thesis_Final_2_01092008.indd Sec1:134 01-09-2008 11:08:3001-09-2008 11:08:30

Ch
ap
ter 6
▪ c-A
bl in
hib
itors
pre
ven
t dru
gre
sis
tan
ce in
CL
L
135REFERENCES
1. Keating MJ, O’Brien S, Albitar
M et al. Early results of a
chemoimmunotherapy regimen of
fl udarabine, cyclophosphamide, and
rituximab as initial therapy for chronic
lymphocytic leukemia. J.Clin.Oncol.
2005;23:4079-4088.
2. Grever MR, Lucas DM, Dewald GW
et al. Comprehensive assessment
of genetic and molecular features
predicting outcome in patients with
chronic lymphocytic leukemia: results
from the US Intergroup Phase III Trial
E2997. J.Clin.Oncol. 2007;25:799-804.
3. Oscier DG, Gardiner AC, Mould SJ et
al. Multivariate analysis of prognostic
factors in CLL: clinical stage, IGVH
gene mutational status, and loss
or mutation of the p53 gene are
independent prognostic factors. Blood
2002;100:1177-1184.
4. Caligaris-Cappio F, Hamblin TJ. B-cell
chronic lymphocytic leukemia: a bird
of a different feather. J.Clin.Oncol.
1999;17:399-408.
5. Messmer BT, Messmer D, Allen SL et
al. In vivo measurements document
the dynamic cellular kinetics of chronic
lymphocytic leukemia B cells. J.Clin.
Invest 2005;115:755-764.
6. Pedersen IM, Kitada S, Leoni LM
et al. Protection of CLL B cells
by a follicular dendritic cell line is
dependent on induction of Mcl-1. Blood
2002;100:1795-1801.
7. Panayiotidis P, Jones D, Ganeshaguru
K, Foroni L, Hoffbrand AV. Human
bone marrow stromal cells prevent
apoptosis and support the survival of
chronic lymphocytic leukaemia cells in
vitro. Br.J.Haematol. 1996;92:97-103.
8. Lagneaux L, Delforge A, Bron D, De
BC, Stryckmans P. Chronic lymphocytic
leukemic B cells but not normal B cells
are rescued from apoptosis by contact
with normal bone marrow stromal cells.
Blood 1998;91:2387-2396.
9. Burger JA, Tsukada N, Burger M et al.
Blood-derived nurse-like cells protect
chronic lymphocytic leukemia B cells
from spontaneous apoptosis through
stromal cell-derived factor-1. Blood
2000;96:2655-2663.
10. Smit LA, Hallaert DY, Spijker R et al.
Differential Noxa/Mcl-1 balance in
peripheral versus lymph node chronic
lymphocitic leukemia cells correlates
with survival capacity. Blood 2006
11. Pedersen IM, Reed JC.
Microenvironmental interactions
and survival of CLL B-cells. Leuk.
Lymphoma 2004;45:2365-2372.
12. Kitada S, Zapata JM, Andreeff M,
Reed JC. Bryostatin and CD40-
ligand enhance apoptosis resistance
and induce expression of cell
survival genes in B-cell chronic
lymphocytic leukaemia. Br.J.Haematol.
1999;106:995-1004.
13. Kater AP, Evers LM, Remmerswaal
EB et al. CD40 stimulation of B-cell
chronic lymphocytic leukaemia cells
enhances the anti-apoptotic profi le, but
also Bid expression and cells remain
susceptible to autologous cytotoxic
T-lymphocyte attack. Br.J.Haematol.
2004;127:404-415.
14. Wattel E, Preudhomme C, Hecquet
B et al. p53 mutations are associated
with resistance to chemotherapy
and short survival in hematologic
malignancies. Blood 1994;84:3148-
3157.
Thesis_Final_2_01092008.indd Sec1:135Thesis_Final_2_01092008.indd Sec1:135 01-09-2008 11:08:3001-09-2008 11:08:30

13615. Rosenwald A, Chuang EY, Davis
RE et al. Fludarabine treatment of
patients with chronic lymphocytic
leukemia induces a p53-dependent
gene expression response. Blood
2004;104:1428-1434.
16. Mackus WJ, Kater AP, Grummels A
et al. Chronic lymphocytic leukemia
cells display p53-dependent drug-
induced Puma upregulation. Leukemia
2005;19:427-434.
17. Gomez-Bougie P, Wuilleme-Toumi S,
Menoret E et al. Noxa up-regulation
and Mcl-1 cleavage are associated
to apoptosis induction by bortezomib
in multiple myeloma. Cancer Res.
2007;67:5418-5424.
18. Hallaert DY, Spijker R, Jak M et al.
Crosstalk among Bcl-2 family members
in B-CLL: seliciclib acts via the Mcl-1/
Noxa axis and gradual exhaustion
of Bcl-2 protection. Cell Death.Differ.
2007
19. Strauss SJ, Higginbottom K, Juliger
S et al. The proteasome inhibitor
bortezomib acts independently of p53
and induces cell death via apoptosis
and mitotic catastrophe in B-cell
lymphoma cell lines. Cancer Res.
2007;67:2783-2790.
20. Qin JZ, Ziffra J, Stennett L et al.
Proteasome inhibitors trigger NOXA-
mediated apoptosis in melanoma
and myeloma cells. Cancer Res.
2005;65:6282-6293.
21. Ley R, Balmanno K, Hadfi eld K,
Weston C, Cook SJ. Activation of the
ERK1/2 signaling pathway promotes
phosphorylation and proteasome-
dependent degradation of the BH3-
only protein, Bim. J.Biol.Chem.
2003;278:18811-18816.
22. Ley R, Ewings KE, Hadfi eld K, Cook
SJ. Regulatory phosphorylation of Bim:
sorting out the ERK from the JNK. Cell
Death.Differ. 2005;12:1008-1014.
23. Rix U, Hantschel O, Durnberger G
et al. Chemical proteomic profi les
of the BCR-ABL inhibitors imatinib,
nilotinib, and dasatinib reveal novel
kinase and nonkinase targets. Blood
2007;110:4055-4063.
24. Eldering E, Spek CA, Aberson HL et al.
Expression profi ling via novel multiplex
assay allows rapid assessment of
gene regulation in defi ned signalling
pathways. Nucleic Acids Res.
2003;31:e153.
25. Pettitt AR, Sherrington PD, Stewart G
et al. p53 dysfunction in B-cell chronic
lymphocytic leukemia: inactivation
of ATM as an alternative to TP53
mutation. Blood 2001;98:814-822.
26. Willimott S, Baou M, Naresh K,
Wagner SD. CD154 induces a switch
in pro-survival Bcl-2 family members
in chronic lymphocytic leukaemia.
Br.J.Haematol. 2007;138:721-732.
27. Luciano F, Jacquel A, Colosetti P et al.
Phosphorylation of Bim-EL by Erk1/2
on serine 69 promotes its degradation
via the proteasome pathway and
regulates its proapoptotic function.
Oncogene 2003;22:6785-6793.
28. Mami U, Miyashita T, Shikama Y,
Tadokoro K, Yamada M. Molecular
cloning and characterization of six
novel isoforms of human Bim, a
member of the proapoptotic Bcl-2
family. FEBS Lett. 2001;509:135-141.
29. Iglesias-Serret D, de FM, Santidrian
AF et al. Regulation of the
proapoptotic BH3-only protein BIM by
glucocorticoids, survival signals and
proteasome in chronic lymphocytic
leukemia cells. Leukemia 2007;21:281-
287.
30. Nickoloff BJ, Hendrix MJ, Pollock
PM et al. Notch and NOXA-related
pathways in melanoma cells.
J.Investig.Dermatol.Symp.Proc.
2005;10:95-104.
Thesis_Final_2_01092008.indd Sec1:136Thesis_Final_2_01092008.indd Sec1:136 01-09-2008 11:08:3001-09-2008 11:08:30

38. Lin K, Glenn MA, Harris RJ et
al. c-Abl expression in chronic
lymphocytic leukemia cells: clinical and
therapeutic implications. Cancer Res.
2006;66:7801-7809.
39. Bantscheff M, Eberhard D, Abraham Y
et al. Quantitative chemical proteomics
reveals mechanisms of action of
clinical ABL kinase inhibitors. Nat.
Biotechnol. 2007;25:1035-1044.
40. Oltersdorf T, Elmore SW, Shoemaker
AR et al. An inhibitor of Bcl-2 family
proteins induces regression of solid
tumours. Nature 2005;435:677-681.
41. Konopleva M, Contractor R, Tsao T et
al. Mechanisms of apoptosis sensitivity
and resistance to the BH3 mimetic
ABT-737 in acute myeloid leukemia.
Cancer Cell 2006;10:375-388.
42. Del GM, V, Brown JR, Certo M et
al. Chronic lymphocytic leukemia
requires BCL2 to sequester prodeath
BIM, explaining sensitivity to BCL2
antagonist ABT-737. J.Clin.Invest
2007;117:112-121.
43. Hantschel O, Rix U, Schmidt U
et al. The Btk tyrosine kinase is a
major target of the Bcr-Abl inhibitor
dasatinib. Proc.Natl.Acad.Sci.U.S.A
2007;104:13283-13288.
44. Maurer U, Charvet C, Wagman AS,
Dejardin E, Green DR. Glycogen
synthase kinase-3 regulates
mitochondrial outer membrane
permeabilization and apoptosis by
destabilization of MCL-1. Mol.Cell
2006;21:749-760.
45. Rahmani M, Davis EM, Bauer C,
Dent P, Grant S. Apoptosis induced
by the kinase inhibitor BAY 43-9006
in human leukemia cells involves
down-regulation of Mcl-1 through
inhibition of translation. J.Biol.Chem.
2005;280:35217-35227.
Ch
ap
ter 6
▪ c-A
bl in
hib
itors
pre
ven
t dru
gre
sis
tan
ce in
CL
L
137 31. van Delft MF, Wei AH, Mason KD
et al. The BH3 mimetic ABT-737
targets selective Bcl-2 proteins and
effi ciently induces apoptosis via Bak/
Bax if Mcl-1 is neutralized. Cancer Cell
2006;10:389-399.
32. Aichberger KJ, Mayerhofer M, Krauth
MT et al. Identifi cation of mcl-1 as
a BCR/ABL-dependent target in
chronic myeloid leukemia (CML):
evidence for cooperative antileukemic
effects of imatinib and mcl-1
antisense oligonucleotides. Blood
2005;105:3303-3311.
33. Mow BM, Chandra J, Svingen PA et al.
Effects of the Bcr/abl kinase inhibitors
STI571 and adaphostin (NSC 680410)
on chronic myelogenous leukemia cells
in vitro. Blood 2002;99:664-671.
34. Nguyen TK, Rahmani M, Harada H,
Dent P, Grant S. MEK1/2 inhibitors
sensitize Bcr/Abl+ human leukemia
cells to the dual Abl/Src inhibitor BMS-
354/825. Blood 2007;109:4006-4015.
35. Horita M, Andreu EJ, Benito A et
al. Blockade of the Bcr-Abl kinase
activity induces apoptosis of chronic
myelogenous leukemia cells by
suppressing signal transducer and
activator of transcription 5-dependent
expression of Bcl-xL. J.Exp.Med.
2000;191:977-984.
36. Kuribara R, Honda H, Matsui H
et al. Roles of Bim in apoptosis
of normal and Bcr-Abl-expressing
hematopoietic progenitors. Mol.Cell
Biol. 2004;24:6172-6183.
37. Aichberger KJ, Mayerhofer M, Krauth
MT et al. Low-level expression of
proapoptotic Bcl-2-interacting mediator
in leukemic cells in patients with
chronic myeloid leukemia: role of BCR/
ABL, characterization of underlying
signaling pathways, and reexpression
by novel pharmacologic compounds.
Cancer Res. 2005;65:9436-9444.
Thesis_Final_2_01092008.indd Sec1:137Thesis_Final_2_01092008.indd Sec1:137 01-09-2008 11:08:3001-09-2008 11:08:30

55. Kitada S, Andersen J, Akar S et al.
Expression of apoptosis-regulating
proteins in chronic lymphocytic
leukemia: correlations with In vitro
and In vivo chemoresponses. Blood
1998;91:3379-3389.
56. Morales AA, Olsson A, Celsing F et al.
High expression of bfl -1 contributes to
the apoptosis resistant phenotype in
B-cell chronic lymphocytic leukemia.
Int.J.Cancer 2005;113:730-737.
57. Amrein PC, Takvorian T, Hochberg EP
et al. A Phase II Study of Dasatinib
in Relapsed and Refractory Chronic
Lymphocytic Leukemia (CLL/SLL).
Blood 2007;110 (11).
58. Eldering E, Spek CA, Aberson HL et al.
Expression profi ling via novel multiplex
assay allows rapid assessment of
gene regulation in defi ned signalling
pathways. NAR 2003: 31 (23)
138 46. Fritsch RM, Schneider G, Saur D,
Scheibel M, Schmid RM. Translational
repression of MCL-1 couples stress-
induced eIF2 alpha phosphorylation
to mitochondrial apoptosis initiation.
J.Biol.Chem. 2007;282:22551-22562.
47. Longo PG, Laurenti L, Gobessi
S et al. The Akt/Mcl-1 pathway
plays a prominent role in mediating
antiapoptotic signals downstream
of the B-cell receptor in chronic
lymphocytic leukemia B cells. Blood
2008;111:846-855.
48. Veldurthy, A., Patz, M., Hagist, S.,
Pallasch, C. P., Wendtner, C. M.,
Hallek, M., and Krause, G. The kinase
inhibitor dasatinib induces apoptosis in
chronic lymphocytic leukemia cells in
vitro with preference for a subgroup of
patients with unmutated IgVH genes.
Blood Prepublished online Jun 12,
2008. 12-6-2008.
49. Aguillon, R. A., Llanos, C. A., Suarez,
C. J., Loria, O. J., Kipps, T. J., and
Castro, J. E. Dasatinib Induces
Apoptosis in Chronic Lymphocytic
Leukemia and Enhances the Activity
of Rituximab and Fludarabine. Blood
110(11). 2007.
50. Sigal D.S., Nieva J, Diver J, Romeo E,
Lee F. Signifi cant Activity of Dasatinib
in CLL Demonstrated by Ex-Vivo
Assay. Blood 110(11) 2007.
51. Schade AE, Schieven GL, Townsend
R et al. Dasatinib, a small-molecule
protein tyrosine kinase inhibitor, inhibits
T-cell activation and proliferation. Blood
2008;111:1366-1377.
52. Yu L, Mohamed AJ, Simonson OE
et al. Proteasome dependent auto-
regulation of Bruton’s tyrosine kinase
(Btk) promoter via NF-{kappa}B. Blood
2008;111:4617-26
53. Lin K, Glenn MA, Harris RJ et
al. c-Abl expression in chronic
lymphocytic leukemia cells: clinical and
therapeutic implications. Cancer Res.
2006;66:7801-7809.
54. Dicker F, Kater AP, Prada CE et al.
CD154 induces p73 to overcome the
resistance to apoptosis of chronic
lymphocytic leukemia cells lacking
functional p53. Blood 2006;108:3450-
3457.
Thesis_Final_2_01092008.indd Sec1:138Thesis_Final_2_01092008.indd Sec1:138 01-09-2008 11:08:3001-09-2008 11:08:30

SUPPLEMENTAL FIGURE LEGENDS
Figure S1. NF-кB inhibitor BAY-11-7082 blocks CD40-mediated Bcl-xL and Bfl -1 upregulation.
CLL cells were stimulated with or without CD40L for 48 hrs in the presence of ERK inhibitor PD-98059 or
of BAY-11-7082, an irreversible inhibitor of IκB-α phosphorylation (1 and 5 μM). Cells were lysed in SDS-
containing buffer and analyzed by western blot. Ch
ap
ter 6
▪ c-A
bl in
hib
itors
pre
ven
t dru
gre
sis
tan
ce in
CL
L
139
Figure S2. RT-MLPA analysis of p53 WT and p53 dysfunctional CLL cells, stimulated via CD40 in
absence or presence of kinase inhibitors.
Cells were stimulated for 48 hrs as in fi gure 3b, and apoptotic gene expression profi le was analyzed by
RT-MLPA. Results were normalized and expressed relative to total signal. Housekeeping genes included
in this assay were, GUS, B2M, FLT and PARN. (Note that probes for IAP2 are non functional58). Graphs
represent averaged data ± SD of n=4 p53 functional (A) and n=3 dysfunctional (B) samples.
A
B
Thesis_Final_2_01092008.indd Sec1:139Thesis_Final_2_01092008.indd Sec1:139 01-09-2008 11:08:3001-09-2008 11:08:30

Figure S3. Apoptosis sensitivity of individual CLL samples after co-culture with 3T3 or 3T40L cells,
followed by treatment with various drugs in conjunction with ABT-737.
CLL samples were cultured as described in legend to fi gure 4, followed by incubation with the indicated
drugs in the presence of 1μM ABT-737. Apoptosis was determined after 24 hrs by Mito Tracker staining.
140
Figure S4. Peripheral blood-derived and CD40-stimulated CLL cells are insensitive to imatinib or
dasatinib.
(A,B) Peripheral blood-derived CLL cells (n=3) were stimulated with increasing concentration of imatinib
or dasatinib for 24, 48 or 72 hrs.
(C,D) CLL cells (n=4) were cultured for 48 hrs in medium or together with 3T3, or 3T40L cells, followed
by stimulation with different concentrations imatinib or dasatinib for 48 hrs. The BCR-Abl positive cell
line K562 was used as a positive control for imatinib and dasatinib-induced apoptosis. Apoptosis was
determined by MitoTracker staining.
A
C
B
D
Thesis_Final_2_01092008.indd Sec1:140Thesis_Final_2_01092008.indd Sec1:140 01-09-2008 11:08:3201-09-2008 11:08:32

141
Thesis_Final_2_01092008.indd Sec1:141Thesis_Final_2_01092008.indd Sec1:141 01-09-2008 11:08:3201-09-2008 11:08:32

142
Thesis_Final_2_01092008.indd Sec1:142Thesis_Final_2_01092008.indd Sec1:142 01-09-2008 11:08:3301-09-2008 11:08:33

chapter
7Concluding remarks
Thesis_Final_2_01092008.indd Sec1:143Thesis_Final_2_01092008.indd Sec1:143 01-09-2008 11:08:3301-09-2008 11:08:33

144
Thesis_Final_2_01092008.indd Sec1:144Thesis_Final_2_01092008.indd Sec1:144 01-09-2008 11:08:3301-09-2008 11:08:33

One focus of the studies described in this thesis is the mechanism of apoptosis
induction by novel drugs that act independently of the p53 response pathway. In
chapter 2 we discussed the insights obtained from the studies on these drugs in
relation to the current debate regarding the interactions and functional mechanism of
the Bcl-2 family members. Our conclusions are summarized in a model (chapter 2)1.
A second major aspect of this thesis concerned the functional consequences of CD40
triggering and the identifi cation of abnormalities in apoptosis pathways in CLL cells,
in relation to drug sensitivity (chapter 4-5-6). In this fi nal chapter the relevance of our
fi ndings and the implications with respect to future treatment strategies for CLL are
discussed.
CD40L expressed on the surface of fi broblast or epithelial cell lines has been widely
used to enhance the in vitro survival of CLL cells2-9. We have used this system to study
the effects of CD40 ligation on drug sensitivity. The basic premise underlying these
studies is that CD40-stimulated peripheral blood (PB) CLL cells are a representative
model for CLL cells residing in lymph node (LN) proliferation centers, the site where
they receive pro-survival signals from the microenvironment. Thus, a key question is
whether the model is indeed appropriate and relevant. To our opinion the answer to
this question should be affi rmative because of the following arguments.
(1) Histochemical studies have shown that in vivo CLL cells may be exposed to
CD40L. T lymphocytes have been found interspersed between CLL cells in LN and
bone marrow (BM) proliferation centers10;11 and CD40L-positive T lymphocytes have
been detected in LN pseudofollicles12. However, the functional activity of these T cells
remains to be proven. It is known that ex vivo CD40 stimulation provides signifi cant
survival signals to normal tonsil-derived B lymphocytes13;14. Similar experiments with
LN-derived CLL cells have to our knowledge not been published, but it appears likely
that these would yield comparable results.
(2) We and others have shown remarkable similarities between in vitro CD40L
stimulated PB CLL cells and in vivo LN CLL cells regarding changes in expression
pattern of apoptosis regulating proteins. Table 1 summarizes the expression of
important members of the Bcl-2 family (Bcl-XL, Mcl-1, Bid, Bim
EL and Noxa), the
activated form of the MAP kinase ERK (p-ERK), XIAP and survivin. Both in CD40-
triggered PB CLL cells and in LN CLL cells expression of Bcl-XL, Mcl-1, p-ERK, XIAP,
survivin and Bid are increased, whereas Noxa and BimEL
are decreased.
(3) In CD40 stimulated PB CLL cells and in LN CLL cells the expression pattern
of these proteins is comparable not only qualitatively but also quantitatively. In our
experience, the observed changes compared to PB CLL are of the same order of
magnitude. As a corollary, this notion provides an argument against the possibility that
Ch
ap
ter 7
▪ Co
nclu
din
g re
mark
s
145
Thesis_Final_2_01092008.indd Sec1:145Thesis_Final_2_01092008.indd Sec1:145 01-09-2008 11:08:3301-09-2008 11:08:33

the in vitro CD40 system provides unphysiological stimuli. Of course, some caution
must be exerted in directly extrapolating the data to the in vivo situation because it
has to be assumed that in vivo CLL cells will receive a variety of signals.
Protein LN CD40L Reference
Bcl-XL
Up Up [3, 4, 7, 15]
Mcl-1 up Up [3, 4, 7, 15]
Bid up* Up* [2, 7]
BimEL
Down Down Chapter 5 and 6
Noxa Down Down [15]
p-ERK Up Up Chapter 5 and 6
XIAP Up* Up [4, 5]
Survivin Up Up [16]
Protein expression of indicated proteins was evaluated in LN CLL cells and in PB CLL cells after
in vitro CD40 triggering. Changes as compared to non-stimulated PB CLL cells are indicated by
up- or down-regulation.
* (Hallaert, unpublished observations)
These observations have 2 important implications.
The proliferation centers in LN, BM and spleen might be considered as 1.
‘sanctuary sites’ where CLL cells are protected from the effects of cytotoxic
drugs. Thus, these sites probably are the source of the relentless relapses
that characterize the clinical course of CLL, explaining why thus far curative
treatment is lacking. Relapses occur even with recently introduced, very potent
drug combinations (like FCR) which result in minimal residual disease (MRD)
negativity in the PB and BM in a high percentage of patients17. Whereas in
the later phases of the disease (acquired) p53 dysfunction is an important
cause of therapy resistance, at diagnosis only 10-15% of patients have a 17p
deletion18;19. In contrast, the microenvironmentally induced chemo-resistance
is of great importance in all phases of CLL.
Novel therapies should be tested on CD40L stimulated CLL cells. 2. To
date, the potential effi cacy of novel cytotoxic agents is usually assessed
on PB CLL cells. This disregards the potential protective signals from the
microenvironment. As a consequence, various compounds induce signifi cant
apoptosis in vitro, but the responses in vivo remain disappointing.
146
Table 1. Protein expression in LN CLL and in vitro CD40L stimulated PB CLL cells is similar.
Thesis_Final_2_01092008.indd Sec1:146Thesis_Final_2_01092008.indd Sec1:146 01-09-2008 11:08:3301-09-2008 11:08:33

It can be concluded that the ideal treatment of CLL should at the same time be able to
overcome resistance in the sanctuary sites and induce apoptosis independent of p53
function. A promising drug combination in this perspective is the c-Abl kinase inhibitor
with proteasome inhibitors (chapter 6). In clinical trials in CLL however, dasatinib and
bortezomib administered as single agent yielded disappointing effects, underscoring
the need for combination regimens. To explore this, in the near future a clinical trial
with dasatinib plus fl udarabine will be started in fl udarabine refractory CLL.
In the more distant future, the CD40 system might not only be useful for new drug
testing in general but may also enable pre-treatment assessment of the potential
responsiveness to (combinations of) drugs in individual patients, opening possibilities
to attain the important goal of individualized, “tailor made” therapy in CLL.
Ch
ap
ter 7
▪ Co
nclu
din
g re
mark
s
147
Thesis_Final_2_01092008.indd Sec1:147Thesis_Final_2_01092008.indd Sec1:147 01-09-2008 11:08:3301-09-2008 11:08:33

8. Biagi E, Yvon E, Dotti G et al.
Bystander transfer of functional human
CD40 ligand from gene-modifi ed
fi broblasts to B-chronic lymphocytic
leukemia cells. Hum.Gene Ther.
2003;14:545-559.
9. Fluckiger AC, Rossi JF, Bussel A et al.
Responsiveness of chronic lymphocytic
leukemia B cells activated via surface
Igs or CD40 to B-cell tropic factors.
Blood 1992;80:3173-3181.
10. Ghia P, Strola G, Granziero L et al.
Chronic lymphocytic leukemia B
cells are endowed with the capacity
to attract CD4+, CD40L+ T cells by
producing CCL22. Eur.J.Immunol.
2002;32:1403-1413.
11. Schmid C, Isaacson PG. Proliferation
centres in B-cell malignant
lymphoma, lymphocytic (B-CLL):
an immunophenotypic study.
Histopathology 1994;24:445-451.
12. Carbone A, Gloghini A, Gruss
HJ, Pinto A. CD40 ligand is
constitutively expressed in a subset
of T cell lymphomas and on the
microenvironmental reactive T cells of
follicular lymphomas and Hodgkin’s
disease. Am.J.Pathol. 1995;147:912-
922.
13. Hennino A, Berard M, Krammer
PH, Defrance T. FLICE-inhibitory
protein is a key regulator of germinal
center B cell apoptosis. J.Exp.Med.
2001;193:447-458.
14. Lagresle C, Mondiere P, Bella C,
Krammer PH, Defrance T. Concurrent
engagement of CD40 and the
antigen receptor protects naive and
memory human B cells from APO-1/
Fas-mediated apoptosis. J.Exp.Med.
1996;183:1377-1388.
REFERENCE LIST
1. Hallaert DY, Spijker R, Jak M et al.
Crosstalk among Bcl-2 family members
in B-CLL: seliciclib acts via the Mcl-1/
Noxa axis and gradual exhaustion
of Bcl-2 protection. Cell Death.Differ.
2007
2. Dicker F, Kater AP, Prada CE et al.
CD154 induces p73 to overcome the
resistance to apoptosis of chronic
lymphocytic leukemia cells lacking
functional p53. Blood 2006;108:3450-
3457.
3. Kater AP, Evers LM, Remmerswaal
EB et al. CD40 stimulation of B-cell
chronic lymphocytic leukaemia cells
enhances the anti-apoptotic profi le, but
also Bid expression and cells remain
susceptible to autologous cytotoxic
T-lymphocyte attack. Br.J.Haematol.
2004;127:404-415.
4. Kitada S, Zapata JM, Andreeff M,
Reed JC. Bryostatin and CD40-
ligand enhance apoptosis resistance
and induce expression of cell
survival genes in B-cell chronic
lymphocytic leukaemia. Br.J.Haematol.
1999;106:995-1004.
5. Cuni S, Perez-Aciego P, Perez-Chacon
G et al. A sustained activation of PI3K/
NF-kappaB pathway is critical for
the survival of chronic lymphocytic
leukemia B cells. Leukemia
2004;18:1391-1400.
6. Pedersen IM, Kitada S, Leoni LM
et al. Protection of CLL B cells
by a follicular dendritic cell line is
dependent on induction of Mcl-1. Blood
2002;100:1795-1801.
7. Willimott S, Baou M, Naresh K,
Wagner SD. CD154 induces a switch
in pro-survival Bcl-2 family members
in chronic lymphocytic leukaemia.
Br.J.Haematol. 2007;138:721-732.
148
Thesis_Final_2_01092008.indd Sec1:148Thesis_Final_2_01092008.indd Sec1:148 01-09-2008 11:08:3301-09-2008 11:08:33

15. Smit LA, Hallaert DY, Spijker R et
al. Differential Noxa/Mcl-1 balance
in peripheral versus lymph node
chronic lymphocitic leukemia cells
correlates with survival capacity.
Blood 2007; 109: 1660-1668
16. Granziero L, Ghia P, Circosta P
et al. Survivin is expressed on
CD40 stimulation and interfaces
proliferation and apoptosis in B-cell
chronic lymphocytic leukemia. Blood
2001;97:2777-2783.
17. Tam CS, O’Brien S, Wierda W et al.
Long term results of the fl udarabine,
cyclophosphamide & rituximab regimen
as initial therapy of chronic lymphocytic
leukemia. Blood 2008;112:975-980
18. Dohner H, Fischer K, Bentz M et al.
p53 gene deletion predicts for poor
survival and non-response to therapy
with purine analogs in chronic B-cell
leukemias. Blood 1995;85:1580-1589.
19. Wattel E, Preudhomme C, Hecquet
B et al. p53 mutations are associated
with resistance to chemotherapy
and short survival in hematologic
malignancies. Blood 1994;84:3148-
3157.
Ch
ap
ter 7
▪ Co
nclu
din
g re
mark
s
149
Thesis_Final_2_01092008.indd Sec1:149Thesis_Final_2_01092008.indd Sec1:149 01-09-2008 11:08:3301-09-2008 11:08:33

150
Thesis_Final_2_01092008.indd Sec1:150Thesis_Final_2_01092008.indd Sec1:150 01-09-2008 11:08:3301-09-2008 11:08:33

chapter
Summary
8
Thesis_Final_2_01092008.indd Sec1:151Thesis_Final_2_01092008.indd Sec1:151 01-09-2008 11:08:3301-09-2008 11:08:33

152
Thesis_Final_2_01092008.indd Sec1:152Thesis_Final_2_01092008.indd Sec1:152 01-09-2008 11:08:3301-09-2008 11:08:33

Ch
ap
ter 8
▪ Su
mm
ary
153The work described in this thesis can be divided into two parts. The fi rst part focuses
on the mechanism of apoptosis induction by novel drugs that act independently of
the p53 response pathway (chapter 2-3). The second part focuses on the functional
consequences of CD40 stimulation and the understanding of aberrant apoptosis
pathways in CLL. These studies were designed to test drug sensitivity and to fi nd
future treatment strategies for CLL (chapter 4-5-6).
Chapter 1 introduces the current understanding in therapy, apoptosis and the
microenvironment in CLL and provides an outline for the studies presented.
In chapter 2 we investigated the mechanism underlying apoptosis elicited by the
cyclin dependent kinase inhibitor, roscovitine, in CLL. Biochemical analysis showed
that the BH3-only protein Noxa was associated with the Bcl-2 homologue Mcl-1 in
CLL cells. Furthermore, this apoptosis route required the BH3-only member Bim.
Defi ciency of these Bcl-2 family proteins, assessed by RNA interference techniques,
protected CLL cells from roscovitine induced apoptosis. We concluded that CLL
survival depends on the balance between these pro- and anti-apoptotic Bcl-2 family
members,and that roscovitine activates a selective apoptosis pathway, described as
the Mcl-1/Noxa axis.
In chapter 3 we focused on the effect of a chemical compound, γ-secretase inhibitor
(GSI-1), that is used in the treatment of Alzheimer’s disease. We found that GSI-1
was a potent inducer of apoptosis in CLL through effi cient blocking of proteasomal
activity. Furthermore, inhibition of proteasomal activity triggered the accumulation of
the pro-apoptotic molecule Noxa. The role of Noxa was also demonstrated via RNA
interference experiments. We suggest that GSI-1 or related compounds may hold
promise for therapeutic applications in CLL.
In chapter 4 we compared apoptosis gene profi les from peripheral blood with
lymph node material of CLL patients. A prominent difference between CLL cells in
the peripheral blood and the lymph node was the decreased expression of the pro-
apoptotic protein Noxa and the increased expression of anti-apoptotic Mcl-1 and Bcl-
XL in the lymph node. This differential expression pattern was also observed by in
vitro CD40 stimulation of peripheral blood CLL cells. Direct manipulation of Noxa
protein levels was achieved by proteasome inhibition (increase in Noxa expression,
see chapter 3) in CLL cells and RNA interference techniques (decrease in Noxa
expression) in model cell lines. In both experiments, cell viability was directly linked
with the levels of Noxa. We concluded that the suppression of Noxa in the lymph
Thesis_Final_2_01092008.indd Sec1:153Thesis_Final_2_01092008.indd Sec1:153 01-09-2008 11:08:3301-09-2008 11:08:33

154node microenvironment contributes to the resistance of the CLL cells at these sites.
The hypothesis is that proliferating CLL cells in the lymph node are protected from
apoptosis through the modulation of pro- and anti-apoptotic molecules by CD40
signaling. Once CLL cells leave their lymph node ‘cradle’, the nurturing CD40 signal
is lost and a strong increase in Noxa levels occurs, which corresponds with the high
apoptosis rate of peripheral blood-derived CLL in vitro. We propose that Noxa is a
promising therapeutic target to treat CLL.
In chapter 5 we gained more insight in how CLL cells in the lymph node differ from
the CLL cells which have moved into the circulation. We mimicked this transit in vitro
from the lymph node to the peripheral blood by temporary CD40 receptor triggering
of CLL cells (CD40 model) and we monitored the expression of apoptosis regulatory
genes in relation to sensitivity to various types of drugs during and following CD40
triggering. We showed that CD40 induced changes in the apoptosis gene expression
profi le and that the observed broad drug resistance was reversible after cessation
of CD40 stimulation. In contrast, however, Bim and Mcl-1 protein levels remained
unchanged and also roscovitine resistance was sustained. We conclude that this
model system to mimic the in vivo lymph node setting could help to predict responses
of CLL to new drugs.
Chapter 6 aimed to defi ne the molecular basis for the increased drug resistance
observed after CD40 stimulation in CLL and searched for novel strategies to
circumvent this resistance. As shown in chapters 4 and 5, CD40 triggering resulted
in an anti-apoptotic RNA and protein profi le and resulted in resistance to various
chemotherapeutic drugs. The anti-apoptotic profi le of CD40-stimulated CLL cells
resembles BCR-Abl-dependent changes seen in Chronic Myeloid Leukemia (CML).
This abnormality is characterized by a translocation between one chromosome 9 and
one chromosome 22, resulting in the BCR/Abl fusion-gene. This prompted us to use
c-Abl inhibitors imatinib and dasatinib, which prevented the entire anti-apoptotic profi le
of CD40 triggered CLL, and restored drug sensitivity. These effects also occurred
in CLL samples with a dysfunctional p53. We concluded that treatment settings,
combining c-Abl inhibitors such as imatinib or dasatinib with other chemotherapeutic
drugs may be promising for the treatment of CLL.
Thesis_Final_2_01092008.indd Sec1:154Thesis_Final_2_01092008.indd Sec1:154 01-09-2008 11:08:3301-09-2008 11:08:33

chapter
9Nederlandse samenvatting
Thesis_Final_2_01092008.indd Sec1:155Thesis_Final_2_01092008.indd Sec1:155 01-09-2008 11:08:3301-09-2008 11:08:33

156
Thesis_Final_2_01092008.indd Sec1:156Thesis_Final_2_01092008.indd Sec1:156 01-09-2008 11:08:3301-09-2008 11:08:33

Ch
ap
ter 9
▪ Ned
erla
nd
se s
am
en
vattin
g
157ACHTERGROND
Apoptose
Apoptose of geprogrammeerde celdood is een strict gereguleerd proces dat het
organisme beschermt door niet gewenste cellen dood te laten gaan. Verstoring
van apoptose speelt een belangrijke rol bij de maligne transformatie van cellen
en draagt bij tot resistentie tegen chemotherapie. Apoptose geschiedt door middel
van activatie van een specifi eke groep enzymen (caspases). Deze enzymen zetten
de cel aan tot de degradatie van, voor de cel belangrijke, eiwitten, wat uiteindelijk
resulteert in celdood. Er zijn twee belangrijke apoptotische signaleringspaden die
caspases kunnen activeren (zie hoofdstuk 1, Fig.1). Als eerste de intrinsieke route
of mitochondriale apoptoseroute. Hier vindt signalering plaats via de mitochondriën,
de energiecentrale van de cel. Ten tweede is er de extrinsieke route, waarbij
signalering vanaf het celoppervlak plaats vindt. Regulatie van caspase activatie wordt
gecontroleerd door twee families van eiwitten: de Bcl-2- en inhibitor-of-apoptosis
(IAP) familie. De Bcl-2 familie bestaat uit eiwitten met een pro- (stimuleren apoptose)
of anti-apoptotische (remmen apoptose af) functie. Het evenwicht tussen deze twee
groepen eiwitten bepalen uiteindelijk de gevoeligheid van de cel voor apoptotische
signalen. IAP eiwitten hebben een anti-apoptotische werking doordat ze kunnen
binden aan caspases en daarmee hun activiteit remmen.
In verschillende soorten kanker worden afwijkingen gevonden in genen die apoptose
reguleren. Een voorbeeld is het tumor suppressor gen p53 dat bij celschade
de celgroei zal stopzetten (waarbij het DNA hersteld kan worden) of apoptose
zal induceren (indien de schade te erg is). Hierdoor wordt het ontstaan van een
tumor verhinderd. Echter, bij kanker is p53 vaak gemuteerd wat kan leiden tot een
verminderde gevoeligheid voor apoptose en chemoresistentie.
Chronisch Lymfatische Leukemie
Chronisch Lymfatische Leukemie (CLL) is een bij ouderen veel voorkomende
bloedkanker (leukemie), of preciezer: kanker van witte bloedcellen (lymfocyten). Er
zijn drie types lymfocyten en in CLL is het de kwaadaardige B lymfocyt die de ziekte
veroorzaakt. In het bloed kunnen enorme aantallen kwaadaardige B lymfocyten
voorkomen. Eerst werd er verondersteld dat de toename van de B lymfocyten eerder
het gevolg is van defecten in apoptose regulatie waardoor deze cellen niet verwijderd
kunnen worden. Tegenwoordig is deze opvatting hervat en staat het vrijwel vast
dat er ook een langzame celgroei (deling) is die voornamelijk in de lymfeklieren
en het beenmerg, de zogenaamde micro-omgeving, plaatsvindt. Met de huidige
Thesis_Final_2_01092008.indd Sec1:157Thesis_Final_2_01092008.indd Sec1:157 01-09-2008 11:08:3301-09-2008 11:08:33

158chemotherapie is het mogelijk de prognose van de ziekte tijdelijk te verbeteren door
de cellen in het bloed te laten verdwijnen, maar volledige genezing is niet mogelijk.
Er is nog onvoldoende inzicht in de onderliggende oorzaak van de kwaadaardige
groei van de B lymfocyten in lymfeklieren en de resistentie tegen chemotherapeutica.
Daarom is het noodzakelijk onderzoek te doen naar de samenhang tussen deze
aspecten en nieuwe behandelingsmogelijkheden.
DOELSTELLING VAN DIT PROEFSCHRIFT
De studies beschreven in dit proefschrift hebben als doelstelling een beter inzicht te
verwerven in:
Het werkingsmechanisme van nieuwe chemotherapeutica die onafhankelijk 1.
van het p53 pad werken. Hierbij wordt dieper ingegaan op het effect van
chemotherapeutica op de apoptose signalering in CLL.
Het mechanisme van de verstoorde apoptose en de overleving van CLL cellen 2.
die zich in zowel het perifeer bloed als in de lymfeklier bevinden. Aan de hand
van deze bevindingen wordt gezocht naar optimale therapeutische combinaties
om resistentie in CLL tegen te gaan.
In hoofdstukken 2 en 3 werd het apoptose pad onderzocht van twee potentiële
chemotherapeutica: een cyclin-dependent kinase (CDK) inhibitor (remt de celgroei)
roscovitine en een inhibitor van het proteasoom (het eiwit complex in de cel dat
zorgt voor de afbraak van overbodige en beschadigde eiwitten). De invloed van
de micro-omgeving (in de micro-omgeving, zoals de lymfeklier en het beenmerg,
vindt er interactie plaats tussen de tumorcel en niet tumorale cellen) werd in de
volgende hoofdstukken bestudeerd. In hoofdstuk 4 werd onderzocht wat het verschil
is in de expressie van apoptose regulerende eiwitten in CLL cellen in het perifeer
bloed versus de lymfeklier en wat de functionele gevolgen van die verschillen zijn. In
hoofdstuk 5 en 6 werd verder ingegaan op de functionele gevolgen met betrekking
tot het overwinnen van resistentie tegen chemotherapeutica. Hiervoor werd gebruik
gemaakt van een in vitro (buiten het lichaam) cel systeem (model) dat de micro-
omgeving gedeeltelijk nabootst.
Thesis_Final_2_01092008.indd Sec1:158Thesis_Final_2_01092008.indd Sec1:158 01-09-2008 11:08:3401-09-2008 11:08:34

Ch
ap
ter 9
▪ Ned
erla
nd
se s
am
en
vattin
g
159DE RESULTATEN VAN HET PROMOTIE ONDERZOEK
Hoofdstuk 2
In dit hoofdstuk werd de apoptose route van een nieuw apoptose inducerend
chemotherapeuticum, roscovitine, bestudeerd op moleculair vlak in CLL. We vonden
dat in CLL cellen het pro-apoptotisch BH3-only eiwit Noxa gebonden is met het Bcl-2
homologe anti-apoptotisch eiwit Mcl-1. Tevens toonden we dat bij deze apoptose
route ook het pro-apoptotisch BH3-only eiwit Bim betrokken is. Het belang van deze
eiwitten in roscovitine geïnduceerde celdood werd bevestigd met behulp van RNA
interferentie (remmende) technieken. Er kon geconcludeerd worden dat er in CLL
een functionele interactie is tussen pro- en anti-apoptotische eiwitten en dat apoptose
geïnduceerd door roscovitine een selectieve route volgt, genaamd de Mcl-1/Noxa
route.
Hoofdstuk 3
In dit hoofdstuk werd aangetoond dat γ-secretase remmers, een geneesmiddel voor
de behandeling van Alzheimer, ook effectief is in het induceren van apoptose in CLL.
Wij ontdekten dat het toedienen van dergelijk geneesmiddel aan CLL cellen leidt tot
een signifi cante remming van het proteasoom. Het blokkeren van het proteasoom
heeft als gevolg dat eiwitten zich ophopen in de cel, waarbij de accumulatie van Noxa
het meest prominent was. De sleutelrol van Noxa kon worden aangetoond met RNA
interferentie technieken. Deze bevindingen werpen licht op nieuwe geneesmiddelen
voor de behandeling van CLL, maar ook voor andere types kanker. Doordat er
een groot aantal γ-secretase remmers reeds beschikbaar zijn in de kliniek voor de
behandeling van de ziekte van Alzheimer, zal er veel sneller een antwoord zijn voor
de toepassing ervan in kanker.
Hoofdstuk 4
Eerder was er al aangetoond dat de regulatie van celdood van CLL cellen verstoord is.
Er werd een uitgebreid onderzoek gedaan naar een scala van apoptose regulerende
eiwitten. Het resultaat was echter paradoxaal, want naast het waargenomen anti-
apoptotisch profi el was ook de expressie van de pro-apoptotische ‘BH3-only’
eiwitten Noxa en Bmf fors gestegen in CLL B lymfocyten t.o.v. B lymfocyten van
een gezonde donor. Echter, deze analyses zijn gedaan op CLL B lymfocyten uit het
perifeer bloed, die bij kweek in het laboratorium (in vitro) spontaan in apoptose gaan.
Deze spontane apoptose kon voorkomen worden door condities te simuleren zoals
deze voorkomen in lymfeklieren. Dit geschiedde middels het kweken van CLL cellen
in de aanwezigheid van ‘hulp’ cellen (fi broblasten) die de CD40 receptor op CLL
Thesis_Final_2_01092008.indd Sec1:159Thesis_Final_2_01092008.indd Sec1:159 01-09-2008 11:08:3401-09-2008 11:08:34

160cellen stimuleren (CD40 model). Deze vaststelling leidde ertoe dat er (in hoofdstuk
4) in klierweefsel van CLL patienten (ex vivo) werd gekeken naar het expressie
profi el van apoptose regulerende moleculen om zo onderliggende stoornissen
in apoptose genen in deze compartimenten (perifeer bloed en lymfeklier) in kaart
te brengen. We konden zien dat, net als in het in vitro CD40 model, er ook in de
lymfeklier een anti-apoptotisch profi el aanwezig is: de anti-apoptotische eiwiten
(Mcl-1, Bcl-XL en A1) waren hoog en het pro-apoptotische eiwit Noxa was laag.
Onze hypothese is dat op de plek waar groei van CLL cellen plaatsvindt door
omgevingsfactoren, waarschijnlijk in de lymfeklier, de dodelijke apoptose
genen onderdrukt worden waardoor er bij die cellen resistentie optreedt voor
chemotherapie. Wanneer de cellen in het perifeer bloed terecht komen valt die
bescherming weg, waardoor ze gevoelig worden voor apoptose en chemotherapie.
Hoofdstuk 5
Om meer inzicht te verwerven in onze hierboven opgestelde hypothese werd er
in hoofdstuk 5 onderzoek gedaan naar veranderingen in het apoptotische profi el
en de consequenties voor chemotherapie gevoeligheid tussen CLL cellen in de
lymfeklier en cellen die de lymfeklier micro-omgeving hebben verlaten om in de
periferie verder te circuleren. Deze situatie werd nagebootst in vitro door CLL cellen
gedurende een bepaalde periode in kweek te brengen bij ‘hulp’ cellen (het hierboven
beschreven CD40 model). Onze bevindingen toonden aan dat het gewijzigd apoptose
genexpressie profi el dat wordt waargenomen na CD40 stimulatie in het CD40 model
systeem normaliseert na het wegnemen van deze stimulus. Onverwachts werd echter
gevonden dat het pro-apoptotisch eiwit Bim en het anti-apoptotisch eiwit Mcl-1 nog
een lange tijd ontregeld waren en dit had consequenties voor de gevoeligheid voor
sommige chemotherapeutica. CLL cellen werden opnieuw gevoelig voor proteasoom
remmers en voor fl udarabine (tegenwoordig eerstelijns therapie in CLL), maar niet
voor roscovitine. Verondersteld werd dat Bim en Mcl-1 cruciaal zijn voor apoptose
geïnduceerd door roscovitine. Er kon geconcludeerd worden dat de bepaling van het
apoptotisch profi el en de gevoeligheid voor chemotherapeutica in het CD40 in vitro
model waardevolle informatie zou kunnen geven voor de keuze van een geschikte
therapie in CLL.
Hoofdstuk 6
In hoodstuk 6 werd er gezocht naar therapeutische strategieën om resistentie in
CLL te omzeilen. Ook hier werd het CD40 model gebruikt om CLL cellen te
stimuleren. Hier kon opnieuw bevestigd worden dat CLL cellen gestimuleerd via de
CD40 receptor een zelfde anti-apoptotisch eiwit profi el vertonen als in de lymfeklier
Thesis_Final_2_01092008.indd Sec1:160Thesis_Final_2_01092008.indd Sec1:160 01-09-2008 11:08:3401-09-2008 11:08:34

Ch
ap
ter 9
▪ Ned
erla
nd
se s
am
en
vattin
g
161(ex vivo) en dat deze cellen ongevoelig zijn voor alle geteste chemotherapeutica. In
Chronische Myeloide Leukemie (CML) wordt hetzelfde anti-apoptotische eiwit profi el
waargenomen. Bij CML is er sprake van een translocatie tussen het 9e en het 22e
chromosoom, waardoor er het BCR/Abl fusie-gen ontstaat. Dit is een versmelting
tussen het Abl-(Abelson mouse leukemia) gen afkomstig van chromosoom 9 en BCR-
(breakpoint cluster region) gen van chromosoom 22. Veelbelovend in de behandeling
van CML zijn de Abl-inhibitors imatinib en dasatinib. Beide middelen veroorzaakten
een normalisatie van het anti-apoptotisch eiwit profi el, met als gevolg dat de CLL
cellen opnieuw gevoelig werden voor alle geteste chemotherapeutica. Dit was ook
het geval voor cellen van CLL patienten waarvan het p53 eiwit niet functioneel was.
Er kon zo geconcludeerd worden dat combinaties van medicijnen cruciaal zijn in
de behandeling van resistentie in CLL. De combinatie van Abl-inhibitors imatinib of
dasatinib met andere chemotherapeutica lijken een veelbelovende benadering voor
de behandeling van CLL.
Thesis_Final_2_01092008.indd Sec1:161Thesis_Final_2_01092008.indd Sec1:161 01-09-2008 11:08:3401-09-2008 11:08:34

162
Thesis_Final_2_01092008.indd Sec1:162Thesis_Final_2_01092008.indd Sec1:162 01-09-2008 11:08:3401-09-2008 11:08:34

Dankwoord
Thesis_Final_2_01092008.indd Sec1:163Thesis_Final_2_01092008.indd Sec1:163 01-09-2008 11:08:3401-09-2008 11:08:34

164
Thesis_Final_2_01092008.indd Sec1:164Thesis_Final_2_01092008.indd Sec1:164 01-09-2008 11:08:3401-09-2008 11:08:34

Het onderzoek beschreven in dit proefschrift begon 4 jaar geleden ... in een enorme
fi le op de A2 richting Amsterdam. Eenmaal aangekomen op mijn sollicitatiegesprek
was alle fi le-ellende snel vergeten en was ik ten volle overtuigd van dit project en
mijn nieuwe omgeving! Ik was klaar voor de overzet naar het noorden! Het einde van
een periode van hard werken is nu nabij en ik moet eerlijk toegeven dat het niet altijd
mee zat. Dit AIO-schap zorgde voor een bijzondere confrontatie met mezelf. Nu kan
ik oprecht zeggen dat ik erg trots en verheugd ben en dit is te danken aan heel veel
mensen om me heen!
Allereerst de begeleiders, die dit promotieonderzoek hebben gestart en opgevolgd.
Eric, hoewel onze communicatie niet altijd vlekkenloos verliep, heb ik veel van je
bijgeleerd. Je altijd kritische blik op het plannen, uitvoeren en uitwerken van proeven,
hebben mij zeer zeker wetenschappelijk gevormd. Je geduld en toewijding in het tot
stand brengen van kwalitatieve publicaties waren onontbeerlijk voor het succesvol
afronden van het promotietraject.
Rien, ik bewonder hoe je steeds met ethousiasme het onderzoek benadert. Je goede
begeleiding, maar ook je betrokkenheid vormden een belangrijke steun. Ik kon altijd
bij je terecht, hartelijk dank daarvoor.
René van Lier, ook voor jouw inzet in dit werk ben ik erg dankbaar. Elke donderdag,
tijdens de werkbespreking, stond je klaar voor onze CLL groep om de data kritisch te
analyseren en bij te sturen. Je verbaasde me telkens weer met je kennis en snelheid
van denken.
Eric, Rien en René, bedankt dat ik bij jullie kan promoveren!
En dan de onmisbare hulp van Annelieke en René Spijker.
Annelieke, liefste paranimf, ik bewonder je gedrevenheid. Ik wil je bedanken voor je
essentiële bijdragen aan dit proefschrift. Niet alleen onze samenwerking in het CD40
werk, maar ook onze congresbezoekjes en andere spontane gelegenheden waren
altijd geweldig. Ik vergeet nooit onze ‘shopping-acts’, hoe kan ik dat vergeten, mijn
hele huis ligt vol met die zooi! En hopelijk komen er nog veel leuke momenten in de
toekomst.
René, volgens mij ben jij degene aan wie ik het meest vragen heb gesteld tijdens mijn
hele AIO periode. Je bent daarom mijn ‘AMC-Carlo’. Bedankt voor je altijd luisterend
oor en waardevolle discussies over wetenschap. Ook al je inzet in de langdurige
zoektocht naar Bim wil ik hier nog eens in het licht zetten. Zonder jouw IP-kunsten
was hoofdstuk 2 niet wat het nu is. Ik wens je veel succes en geluk toe met je nieuwe
baan.
Dan
kw
oo
rd
165
Thesis_Final_2_01092008.indd Sec1:165Thesis_Final_2_01092008.indd Sec1:165 01-09-2008 11:08:3401-09-2008 11:08:34

Overige leden van de CLL-apoptose groep,
Arnon, je altijd enthousiaste samenwerking heb ik enorm gewaardeerd. Je bent niet
alleen een goede arts en onderzoeker, maar ook een gezellige collega. Ik ben je
erg dankbaar voor je inzet in dit proefschrift. Rogier, we zijn ongeveer tergelijkertijd
begonnen en we eindigen ongeveer samen in oktober in de Lutherse kerk. Wat goed
zeg! Ik wil je bedanken voor de leuke tijd samen. Zowel in het lab als ernaast. Felix,
gezellige altijd vrolijke collega, bedankt voor de leuke tijd en veel success met je
Noxa beestjes (Ik stem alleen niet volledig in met je keuze van ‘decoratie’ in onze
oude AIO kamer). Margot en Jacqueline, jullie kwamen er pas het laatste jaar van
mijn AIO-schap bij, bedankt voor de gezellige samenwerking. Dieuwertje, bedankt
voor het opwerken van alle patiëntenmateriaal. Dankzij jou kunnen wij meer proeven
doen! Ook de hulp van Ingrid was onmisbaar. Bedankt voor de vele keren dat ik
je mocht storen met weer eens ‘een vraagje’. Bovendien wil ik mijn student Bart
bedanken voor het nuttige ‘bartezomib’ werk, alsook de nieuwe studenten Chantal en
Iris. Jullie motivatie en inzet voor het CLL onderzoek vind ik geweldig!
Oude en nieuwe kamergenootjes,
Henrike, liefste paranimf, rechter-kant-in-de-kamer van me, een speciaal woord van
dank voor jou. We zijn echte maatjes geworden! Ik wil je van harte bedanken voor de
waardevolle tijd samen. Onze hilarische ‘hupslesjes’ gaven ons telkens weer nieuwe
energie (zelfs de psychological warfare met die twee keno’s daar). To be continued!
Daphne, ‘panter-verzorgster’, ook voor jou een speciaal woord van dank. Ik heb
genoten van onze gezellige tijd samen zowel binnen als buiten het AMC. Bedankt
voor alle ‘catsitting’ en dat ‘cadeautje’ dat je nooit terug mag geven is misschien wel
het proberen waard?
Niet te vergeten zijn de oude kamergenootjes. Ester van Leeuwen, bedankt om mij
bij de hand te nemen in het eerste jaar van mijn AIO-schap. Je vertrek naar San
Diego heeft me niet koud gelaten. Nuno (Nuni), I admire your qualities as a scientist.
Thank you for all the helpful commentaries and discussions on my work. Ook bedankt
aan alle andere kamergenootjes, Godelieve, Elsa, Kirsten, Marjolein, Madeleine en
Alek.
Uiteraard wil ik alle stafl eden, Kris, Jörg en René Lutter en verder ook Martijn en
Robert bedanken voor jullie waardevol technisch en inhoudelijk advies op mijn werk.
Ook alle andere collega’s van het Laboratorium voor Experimentele Immunologie
en het Laboratorium Speciële Hematologie wil ik bedanken voor hun input en
collegialiteit. Bovendien wil ik Carel en Laura van de afdeling pathologie bedanken
voor de bijdrage aan dit proefschrift, met name voor het tot stand komen van hoofdstuk
166
Thesis_Final_2_01092008.indd Sec1:166Thesis_Final_2_01092008.indd Sec1:166 01-09-2008 11:08:3401-09-2008 11:08:34

4 en de immunohistochemische kleuringen van klierweefsel.
I would like to give a special word of gratitude to Fiona and Fabrizio. I feel lucky for
both meeting you at that interesting apoptosis congress in Rolduc. Fiona, my running
mate, in this last two years of my PhD, you were very helpful to me. Not only on the
level of research but also personal. Thank you so much and I am honoured to be
invited to your wedding in Oxford. It will be amazing! Fabrizio, special friend of mine,
thank you for our interesting discussions, both research and non-research, thank you
for reading parts of this work and for helping me dealing with ‘Kinsky’!
En als laatst de onschatbare waarde van mijn familie.
Liefste mammie en pappie, bedankt voor al wat je mij gegeven hebt. Het resultaat
is er! Jullie kleinste ‘achterkomertje’ waar jullie zo trots op zijn volgt haar tao tot ze
haar doel bereikt en ze is er ontzettend gelukkig om! Mama, ik bewonder je altijd
aanwezige kracht. Papa, ik bewonder je stille bron van intelligentie. Papa, bedankt
voor je ch’an-lesjes.
Dimitri, wonder broer, het bewijs dat we geboren zijn voor wetenschap en geneeskunde
is er. Onze urenlange gesprekken over het ontstaan van het heelal, over ‘oneindig’
(al werd ik er soms helemaal gek van), over biologie, fysiologie, anatomie, en nog zo
veel meer, blijven me eeuwig bij. Bedankt voor al je inzet in mijn vorming. Bedankt
grote broer!
Lientje, van paarden-freaks tot schoonzussen tot soulmates! For ever!
Sophie, mijn grote zus, Thierry, Magalie en Florence, bedankt voor jullie altijd
aanwezige interesse en steun in mijn vak.
Joachim, broere (‘Chipje’, ‘John’), Isabel, Manou en Elias, ook jullie zijn er altijd voor
mij met even grote interesse en waardering. Bedankt!
Ook de belangstelling van mijn schoonfamilie voor mijn werk stel ik ten zeerste op prijs.
Liefste schoonpa en schoonmama, bedankt voor alle mooie ontspannende tijden met
jullie die zeer zeker hebben bijgedragen tot wat ik nu ben. Yves Jr., Charlotte, Céleste
en klein wondertje; Catherine en Mikaël; Caroline en Michaël, bedankt voor jullie
interesse en steun. Met name Caroline, hartelijk dank voor je inzet bij het layouten
van dit werk.
Tot slot, Guy, liefde van mijn leven, hoe zeer ik jou wil bedanken voor alles wat je voor
me doet is eigenlijk onbeschrijfl ijk. Zonder jou had ik het nooit gered! Je bent mijn
wederhelft en ik laat je nooit meer los! Ti amo!!!
Dan
kw
oo
rd
167
Thesis_Final_2_01092008.indd Sec1:167Thesis_Final_2_01092008.indd Sec1:167 01-09-2008 11:08:3401-09-2008 11:08:34

168
Thesis_Final_2_01092008.indd Sec1:168Thesis_Final_2_01092008.indd Sec1:168 01-09-2008 11:08:3501-09-2008 11:08:35

Curriculum vitae
Thesis_Final_2_01092008.indd Sec1:169Thesis_Final_2_01092008.indd Sec1:169 01-09-2008 11:08:3501-09-2008 11:08:35

170
Thesis_Final_2_01092008.indd Sec1:170Thesis_Final_2_01092008.indd Sec1:170 01-09-2008 11:08:3501-09-2008 11:08:35

De auteur van dit proefschrift, Delfi ne Hallaert, werd op 9 december 1977 geboren
te Brugge, België. In 1996 behaalde ze haar middelbaar diploma aan het Sint-
Jozefsinstituut te Brugge. In 1998 startte zij de studie Biomedische Laboratorium
Technologie aan de Katholieke Hoge School, Brugge, België. Tijdens deze studie liep
zij stage bij de afdeling Klinische Chemie aan het Sint-Jan ziekenhuis te Brugge (Prof.
Blaton). In 2001 behaalde zij haar diploma met onderscheiding. Aansluitend studeerde
zij Biomedische Wetenschappen aan de Vrije Universiteit Brussel, te Brussel, België.
Tijdens deze studie liep zij stage bij de afdeling Immunologie aan de Vrije Universiteit
Brussel, België (Prof.dr. Thielemans). In 2004 behaalde zij haar diploma met grote
onderscheiding. In dit zelfde jaar begon zij als assistent in opleiding op de afdeling
Hematologie (hoofd Prof.dr. M.H.J. van Oers) en Experimentele Immunologie (hoofd
Prof.dr. R.A.W. van Lier), aan het Academisch Medisch Centrum, Universiteit van
Amsterdam, te Amsterdam, om onder leiding van Prof.dr. van Oers en dr. Eldering
aan het in dit proefschrift beschreven onderzoek te werken. In september 2008 start
zij met een verkort programma van de opleiding Geneeskunde aan de Universiteit
van Nijmegen.
Cu
rricu
lum
vita
e
171
Thesis_Final_2_01092008.indd Sec1:171Thesis_Final_2_01092008.indd Sec1:171 01-09-2008 11:08:3501-09-2008 11:08:35

172
Thesis_Final_2_01092008.indd Sec1:172Thesis_Final_2_01092008.indd Sec1:172 01-09-2008 11:08:3501-09-2008 11:08:35

173LIST OF PUBLICATIONS
1. Smit L, Hallaert DYH, Spijker R, de Goeij B, Jaspers A, Kater AP, van Oers MHJ,
van Noesel CJ, Eldering E. Differential Noxa/Mcl-1 balance in peripheral versus
lymph node chronic lymphocytic leukemia cells correlates with survival capacity.
Blood, 2007; 109 (4): 1660-1668.
2. Hallaert DYH, Spijker, Jak M, Derks IAM, Alves NL, Wensveen FM, de Boer
JP, de Jong D, Green SR, van Oers MHJ and Eldering E. Cross-talk among Bcl-2
family members in B-CLL: seliciclib acts via the Mcl-1/Noxa axis and gradual
exhaustion of Bcl-2 protection.
Cell Death and Differentiation, 2007; 14: 1958-1967.
3. Hallaert DYH, Jaspers A, van Noesel CJ, van Oers MHJ, Kater AP, Eldering E. c-Abl
Kinase Inhibitors Overcome CD40-Mediated Drug Resistance in CLL; Implications for
Therapeutic Targeting of Chemoresistant Niches. Blood (accepted for publication).
Lis
t of p
ub
licatio
ns
Thesis_Final_2_01092008.indd Sec1:173Thesis_Final_2_01092008.indd Sec1:173 01-09-2008 11:08:3501-09-2008 11:08:35

174
Thesis_Final_2_01092008.indd Sec1:174Thesis_Final_2_01092008.indd Sec1:174 01-09-2008 11:08:3501-09-2008 11:08:35

175
Co
lor fi g
ure
s
COLOR FIGURES
Chapter 1. Figure 1. The two main pathways leading to apoptosis. The extrinsic pathway is
triggered by ligation of cell surface receptors, such as Fas, resulting in activation of caspase-8.
The intrinsic pathway is activated by cytotoxic stimuli, such as DNA damage, which leads to
the release of apoptosis promoting factors from the mitochondria. In the cytosol, cytochrome c
results in the activation of caspase-9. This pathway is regulated by the Bcl-2 family of proteins.
Activated caspase-8 and -9 in turn activate effector caspase-3, -6 and -7. Cross-talk between
the pathways occurs through Bid, which is cleaved by caspase-8 and then can activate the
intrinsic pathway.
Thesis_Final_2_01092008.indd Sec1:175Thesis_Final_2_01092008.indd Sec1:175 01-09-2008 11:08:3501-09-2008 11:08:35

Chapter 2. Figure 5C. Bim and Bax association with Bcl-2 shifts to the mitochondria-rich
insoluble fraction over time in B-CLL cells.
Immunocytochemical staining for Bax of B-CLL cells incubated in the absence or pres-
ence of 25 μM seliciclib for 24 hrs. MitoTracker Orange was used for localization of the
mitochondria.
chapter 4. Figure 1: Histology of lymph node infi ltrated by B-CLL cells.
Ubiquitously present B-CLL cells were positive for CD23 and CD5. Scattered CD3+ T cells were
present throughout the LN. The absence of clusters of BCl-6+ cells excluded the presence of
germinal center remnants in the LNs. Ki67/CD20 and Ki67/CD3 double staining indicate that all
cycling Ki67+ cells (pink) were of CD20+ (brown) origin - see also inset -, while the CD3+ T cells
were predominantly Ki67 negative. Magnifi cation 40x.
176
Thesis_Final_2_01092008.indd Sec1:176Thesis_Final_2_01092008.indd Sec1:176 01-09-2008 11:08:3501-09-2008 11:08:35




















