
Facile Electrochemical Hydrogenation and Chlorination of Glassy Carbon to Produce Highly Reactive...
10
& Electrochemical Functionalization Facile Electrochemical Hydrogenation and Chlorination of Glassy Carbon to Produce Highly Reactive and Uniform Surfaces for Stable Anchoring of Thiolated Molecules Ahmed M. Debela, [a] Mayreli Ortiz,* [a] Valerio Beni, [a, c] and Ciara K. O’Sullivan* [a, b] Abstract: Carbon is a highly adaptable family of materials and is one of the most chemically stable materials known, providing a remarkable platform for the development of tunable molecular interfaces. Herein, we report a two-step process for the electrochemical hydrogenation of glassy carbon followed by either chemical or electrochemical chlorination to provide a highly reactive surface for further functionalization. The carbon surface at each stage of the process is characterized by AFM, SEM, Raman, attenuated total reflectance (ATR) FTIR, X-ray photoelectron spectrosco- py (XPS), and electroanalytical techniques. Electrochemical chlorination of hydrogen-terminated surfaces is achieved in just 5 min at room temperature with hydrochloric acid, and chemical chlorination is performed with phosphorus penta- chloride at 50 8C over a three-hour period. A more controlled and uniform surface is obtained using the electrochemical approach, as chemical chlorination is observed to damage the glassy carbon surface. A ferrocene-labeled alkylthiol is used as a model system to demonstrate the genericity and potential application of the highly reactive chlorinated sur- face formed, and the methodology is optimized. This pro- cess is then applied to thiolated DNA, and the functionality of the immobilized DNA probe is demonstrated. XPS reveals the covalent bond formed to be a C ÀS bond. The thermal stability of the thiolated molecules anchored on the glassy carbon is evaluated, and is found to be far superior to that on gold surfaces. This is the first report on the electrochemi- cal hydrogenation and electrochemical chlorination of a glassy carbon surface, and this facile process can be ap- plied to the highly stable functionalization of carbon surfa- ces with a plethora of diverse molecules, finding widespread applications. Introduction Carbon materials are highly adaptable and are among the most chemically stable materials known, so they can provide remarkable platforms for the development of tunable molecu- lar interfaces. [1] Carbon materials can be used for a wide range of applications, and the surface chemistry of these materials is critical for their performances. Carbon-based surfaces are at- tractive substrates for biological interfaces owing to their bio- compatibility, mechanical hardness, and chemical robust- ness. [1, 2] Chemisorption of aryldiazonium salts [3] is a very versatile ap- proach that can be applied to materials of different composi- tions ranging from pure sp 2 materials including graphite [4] and carbon nanotubes [5] to 100% sp 3 materials such as diamond. [6] Chemisorption is mediated by the electrochemical [7] or sponta- neous reduction of an aryldiazonium cation [7a,b] resulting in the dediazonation of the aryl group and the subsequent formation of a robust aryl–substrate linkage. However, the high reactivity of aryldiazonium cations can lead to the formation of multilay- ers and poorly defined organic films. [7a] Various studies [7b,c] have suggested that multilayer formation is regulated by the poten- tial and the charge consumed during the electrografting pro- cess, as well as the composition of the grafting solution and the duration of the grafting step. However, it is not easy to control all these parameters, so good management of the sur- face properties is difficult. [8] These modified carbon surfaces have been demonstrated to have excellent stability, for exam- ple, compared with thiol-based self-assembled monolayers (SAMs) on gold, even at elevated temperatures; [9] the en- hanced stability can be attributed to the greater strength of the C ÀC (348 kJ mol À1 ) and C ÀS (272 kJ mol À1 ) bonds compared with the Au ÀS bond (167 kJ mol À1 ). Alternatively, one of the most powerful and extensively ex- plored ways of activating carbon materials is H-termination, a process commonly performed using hydrogen plasma. [10] This results in the formation of C ÀH dipoles in which the elec- tropositive H is slightly susceptible to nucleophilic attack. [11] Hydrogen-terminated substrates have found applications in [a] A. M. Debela, Dr. M. Ortiz, Dr. V. Beni, Prof. Dr. C. K. O’Sullivan Departament d’Enginyerȷa Quȷmica, Universitat Rovira i Virgili, Avinguda Paı ¨sos Catalans, 26, 43007 Tarragona (Spain) Fax: (+ 34) 977559667/0034977559721 E-mail: [email protected] [email protected] [b] Prof. Dr. C. K. O’Sullivan ICREA, Passeig Lluis Companys 23, 08010 Barcelona (Spain) [c] Dr. V. Beni Current affiliation: Department of Physics, Chemistry and Biology (IFM), Linkoping University, 58183, Linkoping (Sweden) Supporting information for this article is available on the WWW under http ://dx.doi.org/10.1002/chem.201402051. Chem. Eur. J. 2014, 20, 1 – 10 # 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim 1 && These are not the final page numbers! ÞÞ Full Paper DOI: 10.1002/chem.201402051
Transcript of Facile Electrochemical Hydrogenation and Chlorination of Glassy Carbon to Produce Highly Reactive...

& Electrochemical Functionalization
Facile Electrochemical Hydrogenation and Chlorination of GlassyCarbon to Produce Highly Reactive and Uniform Surfaces forStable Anchoring of Thiolated Molecules
Ahmed M. Debela,[a] Mayreli Ortiz,*[a] Valerio Beni,[a, c] and Ciara K. O’Sullivan*[a, b]
Abstract: Carbon is a highly adaptable family of materialsand is one of the most chemically stable materials known,providing a remarkable platform for the development oftunable molecular interfaces. Herein, we report a two-stepprocess for the electrochemical hydrogenation of glassycarbon followed by either chemical or electrochemicalchlorination to provide a highly reactive surface for furtherfunctionalization. The carbon surface at each stage of theprocess is characterized by AFM, SEM, Raman, attenuatedtotal reflectance (ATR) FTIR, X-ray photoelectron spectrosco-py (XPS), and electroanalytical techniques. Electrochemicalchlorination of hydrogen-terminated surfaces is achieved injust 5 min at room temperature with hydrochloric acid, andchemical chlorination is performed with phosphorus penta-chloride at 50 8C over a three-hour period. A more controlledand uniform surface is obtained using the electrochemical
approach, as chemical chlorination is observed to damagethe glassy carbon surface. A ferrocene-labeled alkylthiol isused as a model system to demonstrate the genericity andpotential application of the highly reactive chlorinated sur-face formed, and the methodology is optimized. This pro-cess is then applied to thiolated DNA, and the functionalityof the immobilized DNA probe is demonstrated. XPS revealsthe covalent bond formed to be a C�S bond. The thermalstability of the thiolated molecules anchored on the glassycarbon is evaluated, and is found to be far superior to thaton gold surfaces. This is the first report on the electrochemi-cal hydrogenation and electrochemical chlorination ofa glassy carbon surface, and this facile process can be ap-plied to the highly stable functionalization of carbon surfa-ces with a plethora of diverse molecules, finding widespreadapplications.
Introduction
Carbon materials are highly adaptable and are among themost chemically stable materials known, so they can provideremarkable platforms for the development of tunable molecu-lar interfaces.[1] Carbon materials can be used for a wide rangeof applications, and the surface chemistry of these materials iscritical for their performances. Carbon-based surfaces are at-tractive substrates for biological interfaces owing to their bio-compatibility, mechanical hardness, and chemical robust-ness.[1, 2]
Chemisorption of aryldiazonium salts[3] is a very versatile ap-proach that can be applied to materials of different composi-
tions ranging from pure sp2 materials including graphite[4] andcarbon nanotubes[5] to 100 % sp3 materials such as diamond.[6]
Chemisorption is mediated by the electrochemical[7] or sponta-neous reduction of an aryldiazonium cation[7a,b] resulting in thedediazonation of the aryl group and the subsequent formationof a robust aryl–substrate linkage. However, the high reactivityof aryldiazonium cations can lead to the formation of multilay-ers and poorly defined organic films.[7a] Various studies[7b,c] havesuggested that multilayer formation is regulated by the poten-tial and the charge consumed during the electrografting pro-cess, as well as the composition of the grafting solution andthe duration of the grafting step. However, it is not easy tocontrol all these parameters, so good management of the sur-face properties is difficult.[8] These modified carbon surfaceshave been demonstrated to have excellent stability, for exam-ple, compared with thiol-based self-assembled monolayers(SAMs) on gold, even at elevated temperatures ;[9] the en-hanced stability can be attributed to the greater strength ofthe C�C (348 kJ mol�1) and C�S (272 kJ mol�1) bonds comparedwith the Au�S bond (167 kJ mol�1).
Alternatively, one of the most powerful and extensively ex-plored ways of activating carbon materials is H-termination,a process commonly performed using hydrogen plasma.[10]
This results in the formation of C�H dipoles in which the elec-tropositive H is slightly susceptible to nucleophilic attack.[11]
Hydrogen-terminated substrates have found applications in
[a] A. M. Debela, Dr. M. Ortiz, Dr. V. Beni, Prof. Dr. C. K. O’SullivanDepartament d’Enginyer�a Qu�mica, Universitat Rovira i Virgili, AvingudaPaı̈sos Catalans, 26, 43007 Tarragona (Spain)Fax: (+ 34) 977559667/0034977559721E-mail : [email protected]
[b] Prof. Dr. C. K. O’SullivanICREA, Passeig Lluis Companys 23, 08010 Barcelona (Spain)
[c] Dr. V. BeniCurrent affiliation : Department of Physics, Chemistry and Biology (IFM),Linkoping University, 58183, Linkoping (Sweden)
Supporting information for this article is available on the WWW underhttp ://dx.doi.org/10.1002/chem.201402051.
Chem. Eur. J. 2014, 20, 1 – 10 � 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim1 &&
These are not the final page numbers! ��
Full PaperDOI: 10.1002/chem.201402051

the highly stable photofunctionalization of surfaces with al-kenes,[1, 12] azides,[13] and alkylthiols.[14]
Halogenation with thionyl chloride (SOCl2) or phosphorustri/pentachloride (PCl3/5) in organic solvents has recently beenreported as ways of increasing the reactivity of H-terminatedsurfaces. These brominated or chlorinated surfaces were thenmodified through the formation of C�C bonds between thesurface and different types of alkyl Grignard reagents[15] oralkylthiols,[9b] and the formed monolayers were demonstratedto be extremely stable even at temperatures above 200 8C.[9b]
This chemical halogenation represents a straightforward ap-proach compared with previously used methods such asatomic beams,[16] and a new approach based on the use ofcarbon tetrachloride cold plasma has been reported for the ex-tensive chlorination of carbon nanotubes.[17] However, thischlorination process suffers from an inherent requirement fortoxic materials as well as the difficulty of controlling the reac-tion, which can result in etching and damaging of the sub-strates.[18] An alternative approach exploits the photochemicalgrafting of organic alkenes to link functional organic moleculesto carbon surfaces including diamond,[1] amorphous carbon,[15a]
glassy carbon (GC),[19] highly ordered pyrolitic graphite(HOPG),[13a, 14] and carbon nanofibers.[20] Recently, the feasibilityof attaching iodine atoms to carbon films formed by photore-sist pyrolysis with iodine plasma and the subsequent linking ofboth alkenes and alkynes to the iodinated surface has beendemonstrated.[21]
As another alternative, electrochemical methods for eitherthe chlorination or hydrogenation of surfaces are attractive be-cause of their simplicity, the ability to control the modificationprocess, and reduced surface damage. Electrochemical hydro-genation of boron-doped diamond[22a] and germanium[22b] hasbeen reported, and there have been several reports on theelectrochemical conversion of C�H to C�X (X = F or Cl) for thefunctionalization of organic molecules.[23]
In this report, we describe a two-step process for electro-chemical hydrogenation and subsequent chemical/electro-chemical chlorination, leading to the formation of a highly re-active surface with wide-ranging potential applications. As anexample of the potential use of this activated carbon surfacefor linking thiolated molecules, a ferrocene-labeled alkylthiolwas used as a model system for optimization of the immobili-zation process. The developed methodology was then appliedto a thiolated DNA probe, and the functionality of the DNAprobe following immobilization was demonstrated. The modi-fied surfaces were characterized using AFM, Raman, attenuatedtotal reflectance (ATR) FTIR, XPS, and SEM techniques. The ther-mal stability and functionality of the immobilized DNA wereevaluated and compared with the stability of self-assembledmonolayers on gold. The developed methodology is extremelyrobust and cost-effective, and is applicable to scale-up witha simple two-step process to produce a highly reactive carbonsurface that can undergo subsequent functionalization witha plethora of different molecules.
Results and Discussion
A facile methodology for the electrochemical hydrogenationand chlorination of glassy carbon surfaces, yielding a highly re-active surface for the anchoring of a plethora of diverse mole-cules, has been developed (Figure 1). To demonstrate the ge-neric applicability of the produced reactive surface, we appliedit to the immobilization of thiolated molecules.
Characterization of modified surfaces by FTIR, Raman, andX-ray photoelectron spectroscopies
The different steps of the preparation of the glassy carbon sur-face were followed by using XPS, ATR-FTIR and Raman spec-troscopies. The combination of these techniques is commonlyused to gain a better understanding of surface pro-cesses.[15a, 24, 25] X-Ray photoelectron spectroscopy (XPS) is prob-ably the most appropriate tool for determining the chemicalcomposition of organic thin layers[9b, 26] because it analyzes justthe first 10 nm of the surface, and for this reason it is widelyused for evaluating the percentage of atoms covalently boundto surfaces.[17, 27]
Neither hydrogen nor helium can be detected by XPS,[17] soa low value of the oxygen/carbon (O/C) atomic ratio[28] wasused as an indication of the successful hydrogenation ofa carbon surface. After electrochemical hydrogenation, the O/Catomic ratio was found to be around 4.4 %, which is compara-ble to the 1–4 % reported for plasma-hydrogenated glassycarbon surfaces, and considerably lower than that obtained forpolished glassy carbon (8–15 %).[24]
In a more direct way, the presence of hydrogen atomslinked to the carbon in the hydrogen-terminated glassy carbonsurface is demonstrated by the FTIR bands corresponding tothe asymmetric and symmetric vibration modes of an sp3 C�Hbond at 2913 and 2843 cm�1, respectively. In the chlorinatedGC generated through both the chemical and electrochemical
Figure 1. Schematic representation of surface preparation: a) electrochemi-cal hydrogenation, b) chemical chlorination of hydrogen-terminated surface,c) electrochemical chlorination of hydrogen-terminated surface, d) immobili-zation of thiol molecules on chlorinated GC surface (produced by b), ande) immobilization of thiol molecules on chlorinated GC surface (produced byc) at 50 8C in the presence of a chemical chlorination agent (PCl5). The highlyreactive chlorinated surface was subsequently used for the anchoring ofthiolated molecules, and the modified surfaces were examined through a va-riety of characterization techniques.
Chem. Eur. J. 2014, 20, 1 – 10 www.chemeurj.org � 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim2&&
�� These are not the final page numbers!
Full Paper
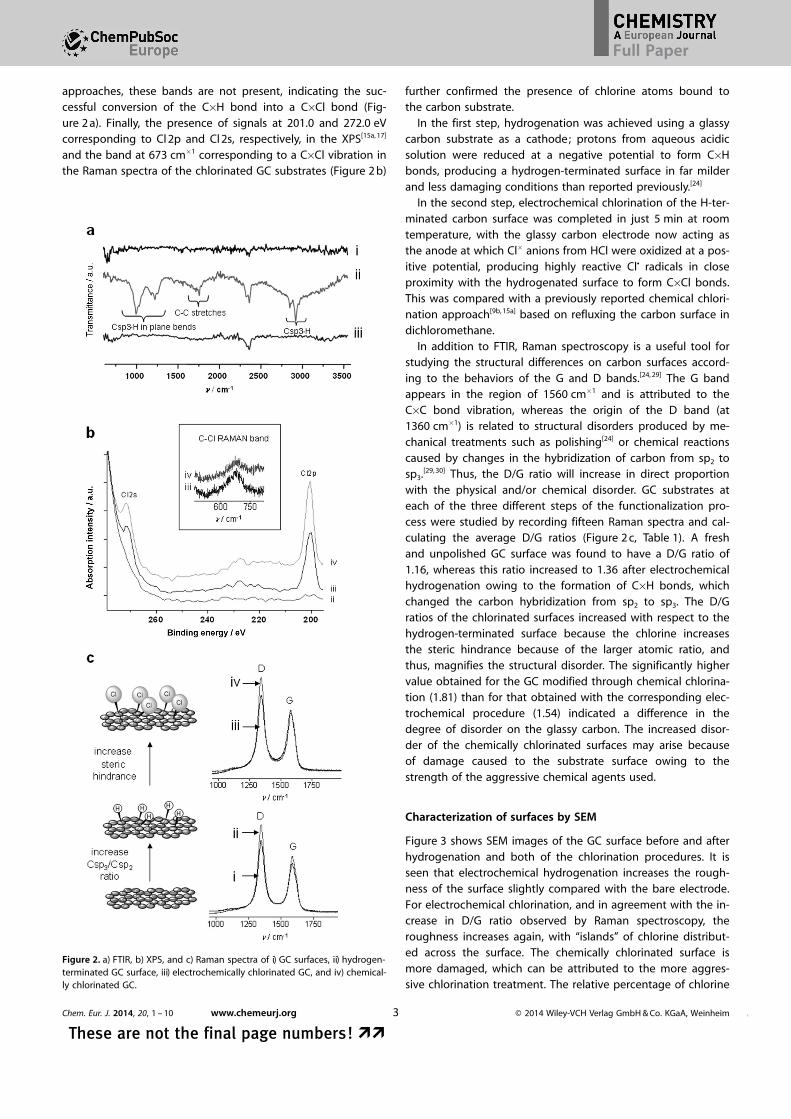
approaches, these bands are not present, indicating the suc-cessful conversion of the C�H bond into a C�Cl bond (Fig-ure 2 a). Finally, the presence of signals at 201.0 and 272.0 eVcorresponding to Cl 2p and Cl 2s, respectively, in the XPS[15a, 17]
and the band at 673 cm�1 corresponding to a C�Cl vibration inthe Raman spectra of the chlorinated GC substrates (Figure 2 b)
further confirmed the presence of chlorine atoms bound tothe carbon substrate.
In the first step, hydrogenation was achieved using a glassycarbon substrate as a cathode; protons from aqueous acidicsolution were reduced at a negative potential to form C�Hbonds, producing a hydrogen-terminated surface in far milderand less damaging conditions than reported previously.[24]
In the second step, electrochemical chlorination of the H-ter-minated carbon surface was completed in just 5 min at roomtemperature, with the glassy carbon electrode now acting asthe anode at which Cl� anions from HCl were oxidized at a pos-itive potential, producing highly reactive ClC radicals in closeproximity with the hydrogenated surface to form C�Cl bonds.This was compared with a previously reported chemical chlori-nation approach[9b, 15a] based on refluxing the carbon surface indichloromethane.
In addition to FTIR, Raman spectroscopy is a useful tool forstudying the structural differences on carbon surfaces accord-ing to the behaviors of the G and D bands.[24, 29] The G bandappears in the region of 1560 cm�1 and is attributed to theC�C bond vibration, whereas the origin of the D band (at1360 cm�1) is related to structural disorders produced by me-chanical treatments such as polishing[24] or chemical reactionscaused by changes in the hybridization of carbon from sp2 tosp3.[29, 30] Thus, the D/G ratio will increase in direct proportionwith the physical and/or chemical disorder. GC substrates ateach of the three different steps of the functionalization pro-cess were studied by recording fifteen Raman spectra and cal-culating the average D/G ratios (Figure 2 c, Table 1). A freshand unpolished GC surface was found to have a D/G ratio of1.16, whereas this ratio increased to 1.36 after electrochemicalhydrogenation owing to the formation of C�H bonds, whichchanged the carbon hybridization from sp2 to sp3. The D/Gratios of the chlorinated surfaces increased with respect to thehydrogen-terminated surface because the chlorine increasesthe steric hindrance because of the larger atomic ratio, andthus, magnifies the structural disorder. The significantly highervalue obtained for the GC modified through chemical chlorina-tion (1.81) than for that obtained with the corresponding elec-trochemical procedure (1.54) indicated a difference in thedegree of disorder on the glassy carbon. The increased disor-der of the chemically chlorinated surfaces may arise becauseof damage caused to the substrate surface owing to thestrength of the aggressive chemical agents used.
Characterization of surfaces by SEM
Figure 3 shows SEM images of the GC surface before and afterhydrogenation and both of the chlorination procedures. It isseen that electrochemical hydrogenation increases the rough-ness of the surface slightly compared with the bare electrode.For electrochemical chlorination, and in agreement with the in-crease in D/G ratio observed by Raman spectroscopy, theroughness increases again, with “islands” of chlorine distribut-ed across the surface. The chemically chlorinated surface ismore damaged, which can be attributed to the more aggres-sive chlorination treatment. The relative percentage of chlorine
Figure 2. a) FTIR, b) XPS, and c) Raman spectra of i) GC surfaces, ii) hydrogen-terminated GC surface, iii) electrochemically chlorinated GC, and iv) chemical-ly chlorinated GC.
Chem. Eur. J. 2014, 20, 1 – 10 www.chemeurj.org � 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim3 &&
These are not the final page numbers! ��
Full Paper

bound is higher in the case of the chemically prepared surfa-ces (0.62 %) than for the electrochemically prepared surfaces(0.55 %), with both values being comparable to the value of0.75 % reported previously for diamond.[31] It is generally ac-cepted that electron-transfer reactions such as the reductionof H+ from acid media are more likely to occur in the “defects”on the planar structure where the charge concentration ishigher. It is thus expected, and indeed observed, that theanodic oxidation of chlorine anions has a greater probability ofoccurring at these points, with removal of hydrogen by theproduced chlorine radicals and the concomitant formation ofC�Cl bonds.
Characterization of modified surfaces using contact-anglemeasurements
The relationship between the hydrophilicity of a surface andthe value of the static contact angle of a water drop depositedon it is generally accepted, and is used to assess the transfor-mation or modification of a substrate on which the static con-tact angles increase with the hydrophobicity of the surface.[24]
The contact angle obtained for the bare GC surfacewas 478 owing to the presence of polar carbonoxides on the surface.[24] The cathodic hydrogenationdecreased the hydrophilic character of the surface byforming less polar C�H bonds, and the contact angleincreased to 728. Subsequent electrochemical chlori-nation of the surface decreased the contact angleslightly to 708 owing to the formation of the morepolar C�Cl bonds, which confer more hydrophiliccharacter. In agreement with the results obtainedfrom Raman and SEM studies, the physical damage
of the surface upon chemical chlorination resulted in an in-creased roughness, which gave rise to a more hydrophobicsurface reflected in an increase in the contact angle to 778.(Table 1; see also Figure S2, Supporting Information).
Functionalization of modified glassy carbon with (6-(ferro-cenyl)-hexanethiol)
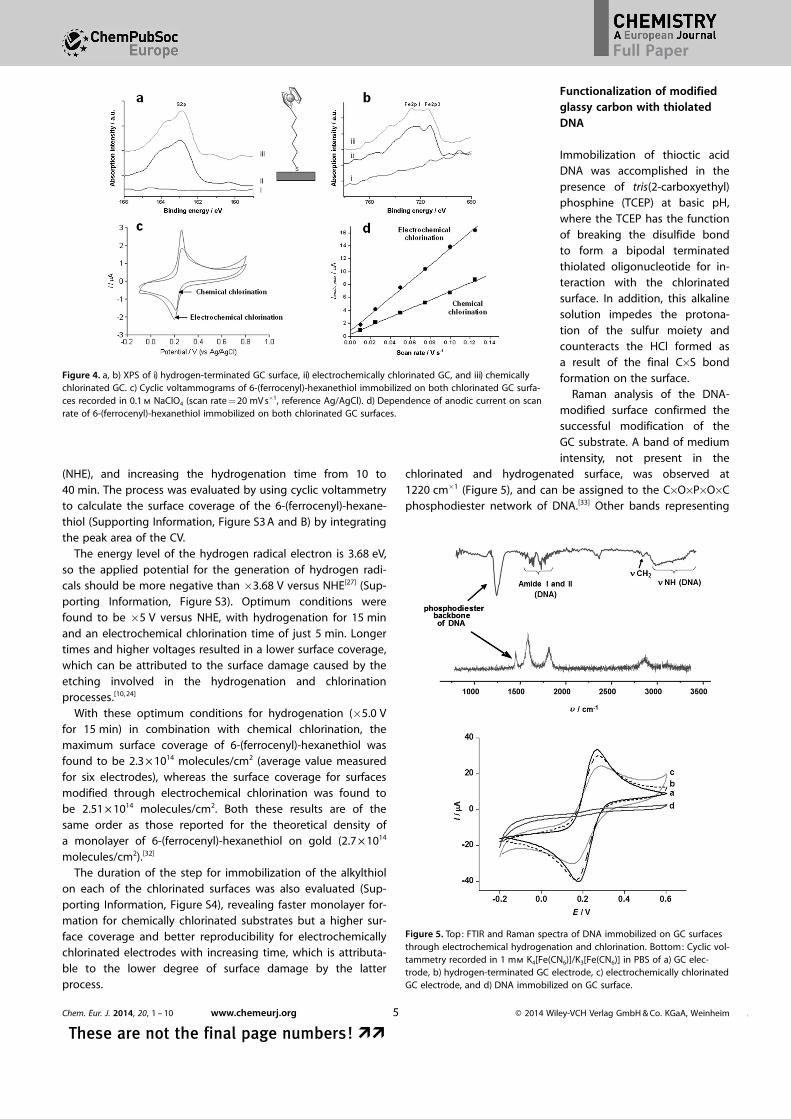
The GC substrates modified through both the chemical andelectrochemical chlorination methods were then compared assupports for the immobilization of thiolated molecules. Forthis purpose, a model thiol molecule bearing an electroactiveferrocene moiety (6-(ferrocenyl)-hexanethiol) was selected. Thepresence of S 2p1 and S 2p3 XPS signals at 162 and 163 eV isindicative of the chemical binding of the alkylthiol to thecarbon surface through the formation of a C�S covalent bond(Figure 4 a). In addition, the signals at 712 and 727 eV corre-spond to the ferrocene moiety, further supporting the linkingof the ferrocenylated alkylthiol molecule[26] (Figure 4 b). Theseobservations are in agreement with reports of an arylthiol im-mobilized on a previously chlorinated surface[9b] as well asthrough self-assembly on gold.[26] A different situation was ob-served when an alkylthiol was tethered photochemically tohighly ordered pyrolytic graphite (HOPG), with no sulfur peakfound on the surface following the immobilization process ;therefore, a C�S covalent bond was not formed, which was ex-plained by a mechanism in which the grafting of alkyl chainson the HOPG involved the formation of alkyl radicals, with thesulfhydryls acting as a sacrificial species.[14]
Cyclic voltammetry (CV) of the modified surfaces revealedwell-defined oxidation and reduction waves with DEp around20 mV (Figure 4 c), with a higher current obtained for the GCmodified through electrochemical chlorination. The confine-ment of the 6-(ferrocenyl)-hexanethiol on the surface was con-firmed by the linearity of the plot of anodic peak currentversus scan rate (Figure 4 d). As a control experiment, an un-modified GC electrode was exposed to the ferrocenyl alkylthioland negligible redox waves were observed.
Optimization of electrochemical surface preparation
According to the mechanism proposed by Hoffman,[27] hydro-gen radicals are generated at negative potentials and reactwith the carbon surface. The potential and duration of the hy-drogenation step were optimized by varying the potentialfrom 0 to �12.5 V versus the normal hydrogen electrode
Table 1. D/G ratio values from Raman spectra and static contact angles for the GCsubstrate at different reaction steps.
D/G Raman Static contact anglesubstrate band ratio [8] (n = 4)
glassy carbon electrode 1.16 47.0�2.0hydrogenated GCE 1.36 72.1�4.8electrochemically chlorinated GCE 1.54 70.0�3.5chemically chlorinated GCE 1.81 77.4�5.5
Figure 3. SEM images of i) GC surface, ii) hydrogen-terminated GC surface,iii) electrochemically chlorinated GC, and iv) chemically chlorinated GC. Scalebar = 50 mm. (See Figure S1 for a colored figure showing chlorine distribu-tion on the surfaces).
Chem. Eur. J. 2014, 20, 1 – 10 www.chemeurj.org � 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim4&&
�� These are not the final page numbers!
Full Paper
(NHE), and increasing the hydrogenation time from 10 to40 min. The process was evaluated by using cyclic voltammetryto calculate the surface coverage of the 6-(ferrocenyl)-hexane-thiol (Supporting Information, Figure S3 A and B) by integratingthe peak area of the CV.
The energy level of the hydrogen radical electron is 3.68 eV,so the applied potential for the generation of hydrogen radi-cals should be more negative than �3.68 V versus NHE[27] (Sup-porting Information, Figure S3). Optimum conditions werefound to be �5 V versus NHE, with hydrogenation for 15 minand an electrochemical chlorination time of just 5 min. Longertimes and higher voltages resulted in a lower surface coverage,which can be attributed to the surface damage caused by theetching involved in the hydrogenation and chlorinationprocesses.[10, 24]
With these optimum conditions for hydrogenation (�5.0 Vfor 15 min) in combination with chemical chlorination, themaximum surface coverage of 6-(ferrocenyl)-hexanethiol wasfound to be 2.3 � 1014 molecules/cm2 (average value measuredfor six electrodes), whereas the surface coverage for surfacesmodified through electrochemical chlorination was found tobe 2.51 � 1014 molecules/cm2. Both these results are of thesame order as those reported for the theoretical density ofa monolayer of 6-(ferrocenyl)-hexanethiol on gold (2.7 � 1014
molecules/cm2).[32]
The duration of the step for immobilization of the alkylthiolon each of the chlorinated surfaces was also evaluated (Sup-porting Information, Figure S4), revealing faster monolayer for-mation for chemically chlorinated substrates but a higher sur-face coverage and better reproducibility for electrochemicallychlorinated electrodes with increasing time, which is attributa-ble to the lower degree of surface damage by the latterprocess.
Functionalization of modifiedglassy carbon with thiolatedDNA
Immobilization of thioctic acidDNA was accomplished in thepresence of tris(2-carboxyethyl)phosphine (TCEP) at basic pH,where the TCEP has the functionof breaking the disulfide bondto form a bipodal terminatedthiolated oligonucleotide for in-teraction with the chlorinatedsurface. In addition, this alkalinesolution impedes the protona-tion of the sulfur moiety andcounteracts the HCl formed asa result of the final C�S bondformation on the surface.
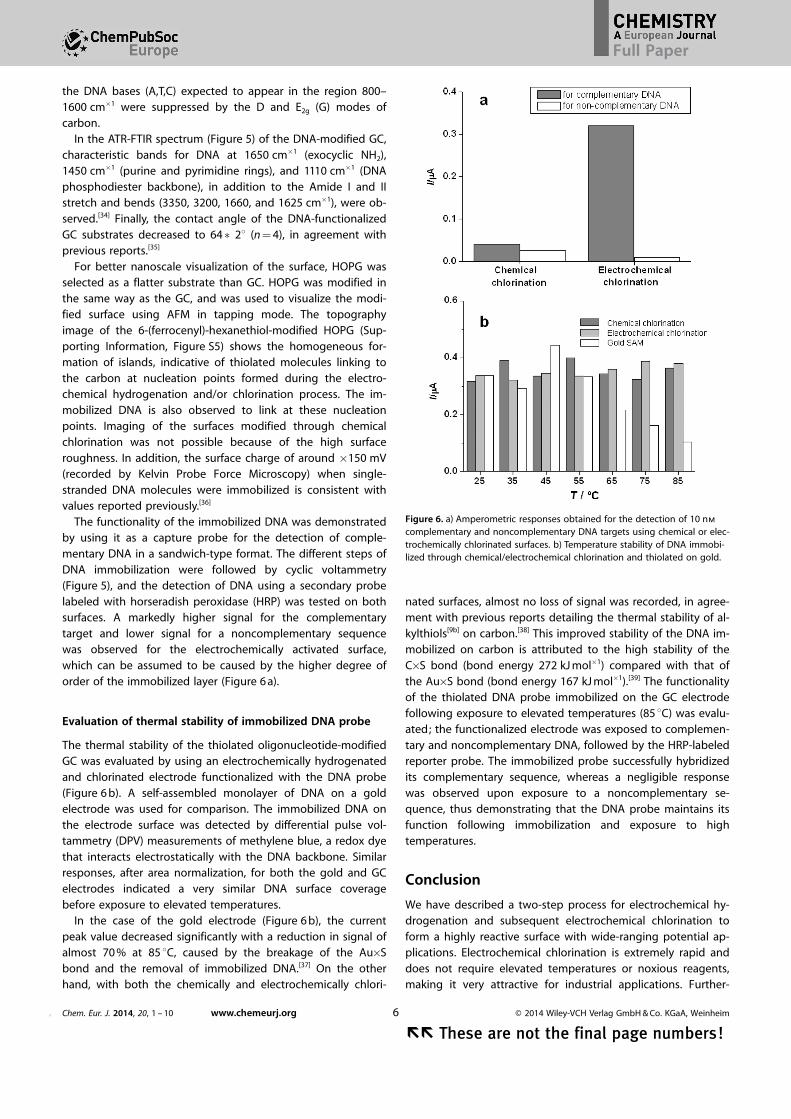
Raman analysis of the DNA-modified surface confirmed thesuccessful modification of theGC substrate. A band of mediumintensity, not present in the
chlorinated and hydrogenated surface, was observed at1220 cm�1 (Figure 5), and can be assigned to the C�O�P�O�Cphosphodiester network of DNA.[33] Other bands representing
Figure 4. a, b) XPS of i) hydrogen-terminated GC surface, ii) electrochemically chlorinated GC, and iii) chemicallychlorinated GC. c) Cyclic voltammograms of 6-(ferrocenyl)-hexanethiol immobilized on both chlorinated GC surfa-ces recorded in 0.1 m NaClO4 (scan rate = 20 mV s�1, reference Ag/AgCl). d) Dependence of anodic current on scanrate of 6-(ferrocenyl)-hexanethiol immobilized on both chlorinated GC surfaces.
Figure 5. Top: FTIR and Raman spectra of DNA immobilized on GC surfacesthrough electrochemical hydrogenation and chlorination. Bottom: Cyclic vol-tammetry recorded in 1 mm K4[Fe(CN6)]/K3[Fe(CN6)] in PBS of a) GC elec-trode, b) hydrogen-terminated GC electrode, c) electrochemically chlorinatedGC electrode, and d) DNA immobilized on GC surface.
Chem. Eur. J. 2014, 20, 1 – 10 www.chemeurj.org � 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim5 &&
These are not the final page numbers! ��
Full Paper
the DNA bases (A,T,C) expected to appear in the region 800–1600 cm�1 were suppressed by the D and E2g (G) modes ofcarbon.
In the ATR-FTIR spectrum (Figure 5) of the DNA-modified GC,characteristic bands for DNA at 1650 cm�1 (exocyclic NH2),1450 cm�1 (purine and pyrimidine rings), and 1110 cm�1 (DNAphosphodiester backbone), in addition to the Amide I and IIstretch and bends (3350, 3200, 1660, and 1625 cm�1), were ob-served.[34] Finally, the contact angle of the DNA-functionalizedGC substrates decreased to 64�28 (n = 4), in agreement withprevious reports.[35]
For better nanoscale visualization of the surface, HOPG wasselected as a flatter substrate than GC. HOPG was modified inthe same way as the GC, and was used to visualize the modi-fied surface using AFM in tapping mode. The topographyimage of the 6-(ferrocenyl)-hexanethiol-modified HOPG (Sup-porting Information, Figure S5) shows the homogeneous for-mation of islands, indicative of thiolated molecules linking tothe carbon at nucleation points formed during the electro-chemical hydrogenation and/or chlorination process. The im-mobilized DNA is also observed to link at these nucleationpoints. Imaging of the surfaces modified through chemicalchlorination was not possible because of the high surfaceroughness. In addition, the surface charge of around �150 mV(recorded by Kelvin Probe Force Microscopy) when single-stranded DNA molecules were immobilized is consistent withvalues reported previously.[36]
The functionality of the immobilized DNA was demonstratedby using it as a capture probe for the detection of comple-mentary DNA in a sandwich-type format. The different steps ofDNA immobilization were followed by cyclic voltammetry(Figure 5), and the detection of DNA using a secondary probelabeled with horseradish peroxidase (HRP) was tested on bothsurfaces. A markedly higher signal for the complementarytarget and lower signal for a noncomplementary sequencewas observed for the electrochemically activated surface,which can be assumed to be caused by the higher degree oforder of the immobilized layer (Figure 6 a).
Evaluation of thermal stability of immobilized DNA probe
The thermal stability of the thiolated oligonucleotide-modifiedGC was evaluated by using an electrochemically hydrogenatedand chlorinated electrode functionalized with the DNA probe(Figure 6 b). A self-assembled monolayer of DNA on a goldelectrode was used for comparison. The immobilized DNA onthe electrode surface was detected by differential pulse vol-tammetry (DPV) measurements of methylene blue, a redox dyethat interacts electrostatically with the DNA backbone. Similarresponses, after area normalization, for both the gold and GCelectrodes indicated a very similar DNA surface coveragebefore exposure to elevated temperatures.
In the case of the gold electrode (Figure 6 b), the currentpeak value decreased significantly with a reduction in signal ofalmost 70 % at 85 8C, caused by the breakage of the Au�Sbond and the removal of immobilized DNA.[37] On the otherhand, with both the chemically and electrochemically chlori-
nated surfaces, almost no loss of signal was recorded, in agree-ment with previous reports detailing the thermal stability of al-kylthiols[9b] on carbon.[38] This improved stability of the DNA im-mobilized on carbon is attributed to the high stability of theC�S bond (bond energy 272 kJ mol�1) compared with that ofthe Au�S bond (bond energy 167 kJ mol�1).[39] The functionalityof the thiolated DNA probe immobilized on the GC electrodefollowing exposure to elevated temperatures (85 8C) was evalu-ated; the functionalized electrode was exposed to complemen-tary and noncomplementary DNA, followed by the HRP-labeledreporter probe. The immobilized probe successfully hybridizedits complementary sequence, whereas a negligible responsewas observed upon exposure to a noncomplementary se-quence, thus demonstrating that the DNA probe maintains itsfunction following immobilization and exposure to hightemperatures.
Conclusion
We have described a two-step process for electrochemical hy-drogenation and subsequent electrochemical chlorination toform a highly reactive surface with wide-ranging potential ap-plications. Electrochemical chlorination is extremely rapid anddoes not require elevated temperatures or noxious reagents,making it very attractive for industrial applications. Further-
Figure 6. a) Amperometric responses obtained for the detection of 10 nm
complementary and noncomplementary DNA targets using chemical or elec-trochemically chlorinated surfaces. b) Temperature stability of DNA immobi-lized through chemical/electrochemical chlorination and thiolated on gold.
Chem. Eur. J. 2014, 20, 1 – 10 www.chemeurj.org � 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim6&&
�� These are not the final page numbers!
Full Paper

more, the surface obtained following electrochemical chlorina-tion was compared to that obtained after chemical chlorina-tion, and was observed to be markedly less damaged, and con-sequently, more organized. The highly reactive surface ob-tained could be used for the anchoring of a plethora of mole-cules ranging from olefins to thiols, but because of the hugerange of easily available diverse thiolated molecules, we decid-ed to use this as a model system. In the first instance, a ferro-cene-labeled alkylthiol was used to optimize the immobiliza-tion process in terms of the hydrogenation and chlorinationtimes, applied potential, and duration of immobilization. Thedeveloped methodology was then used in an example of an“application” of the formed reactive surface for the anchoringof a thiolated DNA probe, and the functionality of the DNAprobe following immobilization was demonstrated. The ther-mal stability and functionality of the immobilized DNA wasevaluated and compared to the stability of self-assembledmonolayers on gold. The modified surfaces were characterizedusing AFM, Raman, ATR-FTIR, XPS, and SEM techniques, the re-sults of which concurred on the superior surface obtainedthrough electrochemical chlorination, as well as confirming thepresence of a C�S bond. The developed methodology is ex-tremely robust, cost-effective, and applicable to scale-up, witha simple two-step electrochemical process for the productionof a highly reactive carbon surface that can undergo subse-quent functionalization with a plethora of different molecules.
Experimental Section
Materials and methods
Hydrochloric acid, sulfuric acid, potassium dihydrogen phosphate,sodium chloride, and potassium chloride were purchased fromScharlau (Barcelona Spain); methylene blue, potassium hexacyano-ferrate (II) and (III) (K4[Fe(CN6)]/K3[Fe(CN6)]) were obtained fromFluka (Barcelona Spain). Trizma base, tris (2-carboxyethyl) phos-phine (TCEP), 6-(ferrocenyl) hexanethiol, hexa-amine ruthenium (III)chloride ([Ru(NH3)6]Cl3), phosphorous pentachloride (PCl5), benzoylperoxide, sodium perchlorate, sodium carbonate and 3,3’,5,5’-tetra-methyl benzidine (TMB) for enzyme-linked immunosorbent assay(ELISA) were supplied by Sigma Aldrich. Sodium hydroxide pelletswere from Panreac and benzene from Acros Organics. Highly or-dered pyrolytic graphite (HOPG) was supplied by SPI, Japan. Thio-ctic acid DNA was purchased from Biomers as lyophilized powderand reconstituted in phosphate buffered saline solution (PBS,pH 7.4).
DQA1*0301 probe for immobilization on glassy carbon surface:
Thioctic acid-5’CAAATCTAAGTCTGTGGA-3’
DQA1*0301 target:
5’ GAG AGG AAG GAG ACT GTC TGG AAG TTG CCT CTG T TC CACAGA CTT AGA TTT G AC CCG CAA TTT GCA CTG ACA AAC ATC GCTGTG CTA AAA CATA 3’
Reporting sequence:
HRP-5’CCCCAGTGTTTCAGAAGA3’
Chemicals and reagents were of analytical grade and used withoutfurther purification. All solutions were prepared in ultrapure water(18 MW cm) obtained using a Simplicity Water Purification System(Millipore, France).
All electrochemical measurements were made with an Autolabmodel PGSTAT 12 potentiostat/galvanostat controlled with theGeneral Purpose Electrochemical System (GPES) software (EcoChemie B.V., The Netherlands). A classical three-electrode configu-ration was used: Ag/AgCl reference electrode, Pt wire counter elec-trode, and glassy carbon electrodes (diameter 3 mm, ALS Japan) orgold electrodes (diameter 2 mm, CHI Instruments, Inc.) as theworking electrode. All the potentials were recorded with respect tothe reference electrode.
Surface characterization
Glassy carbon sheets of 1.0 � 10 � 10 mm (ALS, Japan) were used tocharacterize the surface of the GC electrode during the differentsteps of the functionalization process. Kelvin probe force microsco-py images were acquired in noncontact mode with the chromium-platinum-coated conductive silicon microcantilever tip set 5 nmabove the surface. A resonant frequency of 75 kHz and a constantforce of 3 N m�1 were applied to the tip using a 5420 atomic forcemicroscope (AFM) from Agilent Technologies (USA). Scans wereexecuted by applying a potential of 1 V to the tip at a scan speedof 1.5 lines/s. The images were processed using WSxM 5.0 Develop3.2 and Pico View 1.8. AFM images were recorded with the samemicroscope in tapping mode with a tip of a resonant frequency of75 kHz.
Raman spectra were acquired with a Renishaw 2003 spectrometeroperating at a wavelength of 514 nm. The spectra were analyzedusing Wire 3.2 software (Renishaw plc, United Kingdom).
The ATR-FTIR spectra were recorded with a Jasco FT/IR-600 Plusspectrometer. Typically, 128 scans were recorded at a resolution of2 cm�1. During measurement, the cell compartment was flushedwith dry nitrogen gas.
The X-ray photoelectron spectroscopy (XPS) measurements wererecorded with a PHI ESCA-5500 spectrometer with an aluminum X-ray source. The samples were analyzed at an ultra-high-vacuum(UHV) chamber pressure between 5 � 10�9 and 2 � 10�8 torr.
Static contact angles of DI water on the various glassy carbon sub-strates were measured with an OCA15EC instrument equippedwith DropImage Standard software. Measurements were made bydelivering a 3 mL drop of Milli-Q water from a microsyringe ontothe surface of the sample mounted on an illuminated horizontalstage. The reported contact angle results are the average of fourmeasurements taken at different positions on each GC surface.
Electrode preparation
The glassy carbon electrodes (GCEs) were polished sequentiallywith 1.0, 0.3, and 0.05 mm alumina for approximately 5 min, andwere sonicated in water between each polishing step. GCE activa-tion was performed in saturated sodium carbonate solution for5 min at a potential of 1.2 V versus Ag/AgCl to eliminate any hy-drocarbon adsorbed from the environment; this was followed bysweeping of the potential for 40 cycles between 0 and 1.0 V versusAg/AgCl at 250 mV s�1 in 0.1 m sodium perchlorate.
Hydrogenation of GCE: H-termination of the GCEs was performedelectrochemically as described previously for boron-doped dia-mond substrates[21] with some minor modifications. Briefly, the GCEwas immersed in 2 m HCl and used as the cathode, and a platinumwire, immersed in 2 m H2SO4 solution, was used as the anode. Thesolutions were separated by a glass tube with a porous membrane,and a potential of �5 V and 20 mA current were applied for15 min. During the electrolytic process the solutions were stirredto prevent the accumulation of bubbles at the electrode surface.
Chem. Eur. J. 2014, 20, 1 – 10 www.chemeurj.org � 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim7 &&
These are not the final page numbers! ��
Full Paper

Chemical chlorination of hydrogenated GCE: H-termination wasfollowed by chemical chlorination of the GCE.[9b, 15a] The H-terminat-ed electrodes were immersed in a vial containing anhydrous ben-zene with a catalytic amount of benzoyl peroxide and 100 mm
phosphorus pentachloride for 3 h at 50 8C. Following the chlorina-tion reaction, the electrodes were rinsed with ethanol and water,dried under nitrogen, and used immediately for analysis or modifi-cation with the thiolated molecules.
Electrochemical chlorination of hydrogenated GCE: In this pro-cess, the H-terminated GCE was used as an anode and immersedin a 3:1 ratio of 2 m HCl/2 m HNO3. A platinum wire was used asthe cathode and immersed in 2 m H2SO4. The solutions were sepa-rated by a glass tube with a porous membrane. A potential of+ 2 V and 20 mA current were applied for 5 min.
6-(ferrocenyl)hexanethiol immobilization on modified GCE: Forthe functionalization of the surface with thiolated molecules, theelectrochemically hydrogenated and chlorinated GCEs were incu-bated in an ethanolic solution of 6-(ferrocenyl)hexanethiol over-night at room temperature; this was followed by extensive wash-ing and sonication in ethanol and isopropanol to remove any non-specifically adsorbed molecules.
DNA probe immobilization on GCE: A thioctic acid DNA probe(50 mL, 5 mm) in the presence of 0.5 mm TCEP in Trizma base (pH 8)was pipetted onto the surface of a chlorinated GCE and incubatedfor 24 h at room temperature. The electrodes were then rinsedwith Tris-HCl buffer (pH 7.4) and dried under a stream of nitrogen.For the test of the functionality of the immobilized probe, specificand nonspecific synthetic oligonucleotide targets (in Tris-HCl,pH 7.4 containing 1 m NaCl) were hybridized with the on-surface-immobilized DNA probes for 1 h. Following the target capture,a secondary reporting probe modified with an enzymatic label(HRP) was introduced. The presence of captured target oligonucle-otides was detected electrochemically by monitoring the reductionof the product produced enzymatically from the TMB substrateusing fast amperometric detection at �0.2 V versus Ag/AgCl.
DNA probe immobilization on gold: A clean gold electrode wasimmersed in a thioctic acid DNA probe (50 mL, 5 mm) in 1.0 m
KH2PO4 for 24 h. After chemisorption, the electrode was rinsedwith water, dried under nitrogen, and used immediately.
Thermal stability studies of the oligonucleotide monolayers
The DNA-modified GC and gold electrodes were washed for10 min, under gentle shaking, with an aliquot of 10 mm Tris-HClbuffer (pH 7.4), and thermostated to the desired temperature usinga thermomixer (Eppendorf Iberica, Spain), in the range 25–85 8C insteps of 10 8C. Following thermal incubation and subsequent thor-ough washing, the electrodes were immersed in 10 mm Tris-HCl(pH 7.4) containing 20 mm methylene blue at 25 8C for 5 min. Final-ly, the electrodes were transferred to 10 mm Tris-HCl (pH 7.4) with1 m NaCl, and DPV was performed. The experimental conditions forDPV were: potential between 0.1 and �0.6 V; amplitude of 0.1 V;and scan rate of 100 mV s�1.
Keywords: carbon · chlorination · electrochemistry ·hydrogenation · thiolated
[1] C. Stavis, T. L. Clare, J. E. Butler, A. D. Radadia, R. Carre, H. Zeng, W. P.King, J. A. Carlisle, A. Aksimentiev, R. Bashir, R. J. Hamers, Proc. Natl.Acad. Sci. USA 2011, 108, 983 – 988.
[2] B. Sun, E. P. Colavita, H. Kim, M. Lockett, S. M. Marcus, L. Smith, J. R.Hamers, Langmuir 2006, 22, 9598 – 9605.
[3] S. Mahouche-Chergui, S. Gam-Derouich, C. Mangeney, M. M. Chehimi,Chem. Soc. Rev. 2011, 40, 4143 – 4166.
[4] D. M. Murphy, R. J. Cullen, D. R. Jayasundara, E. M. Scanlan, P. E. Colavita,RSC Adv. 2012, 2, 6527 – 6534.
[5] C. D. Doyle, J. D. Rocha, R. B. Weisman, J. M. Tour, J. Am. Chem. Soc.2008, 130, 6795 – 6800.
[6] a) R. H. M. Delamar, J. Pinson, J. Am. Chem. Soc. 1992, 114, 5883 – 5884;b) H. Uetsuka, D. Shin, N. Tokuda, K. Saeki, C. E. Nebel, Langmuir 2007,23, 3466 – 3472.
[7] a) J. K. Kariuki, M. T. McDermott, Langmuir 2001, 17, 5947 – 5951; b) P. Al-longue, C. Henry de Villeneuve, G. Cherouvrier, R. Cortes, J. Electroanal.Chem. 2003, 550, 161 – 174; c) A. Ricci, C. Bonazzola, E. J. Calvo, Phys.Chem. Chem. Phys. 2006, 8, 4297 – 4299.
[8] J. J. Gooding, Electroanalysis 2008, 20, 573 – 582.[9] a) L. Civit, A. Fragoso, C. K. O’Sullivan, Electrochem. Commun. 2010, 12,
1045 – 1048; b) R. M. Lockett, M. L. Smith, J. Phys. Chem. C 2010, 114,12635 – 12641; c) L. Y. Zhong, P. K. Loh, Chem. Asian J. 2010, 5, 1532 –1540.
[10] R. DeClements, G. M. Swain, T. Dallas, M. W. Holtz, R. D. Herrick II, J. L.Stickney, Langmuir 1996, 12, 6578 – 6586.
[11] C. E. Nebel, H. Kato, B. Rezek, D. Shin, D. Takeuchi, H. Watanabe, T. Ya-mamoto, Diam. Relat. Mater. 2006, 15, 264 – 268.
[12] a) L. T. Lasseter, W. Cai, J. R. Hamers, Analyst 2004, 129, 3 – 8; b) P. E. Co-lavita, B. Sun, K. Y. Tse, J. R. Hamers, J. Am. Chem. Soc. 2007, 129,13554 – 13565.
[13] a) M. Tanaka, T. Sawaguchi, Y. Sato, K. Yoshioka, O. Niwa, Langmuir 2011,27, 170 – 178; b) N. Dontha, W. B. Nowall, W. G. Kuhr, Anal. Chem. 1997,69, 2619 – 2625.
[14] L. Soldi, R. J. Cullen, D. R. Jayasundara, E. M. Scanlan, S. Giordani, P. E.Colavita, J. Phys. Chem. C 2011, 115, 10196 – 10204.
[15] a) R. M. Lockett, M. L. Smith, Langmuir 2009, 25, 3340 – 3343; b) M.Wang, M. R. Das, V. G. Praig, F. LeNormand, M. Li, R. Boukherroub, S.Szunerits, Chem. Commun. 2008, 6294 – 6296.
[16] A. Freedman, Diam. Relat. Mater. 1995, 4, 216 – 219.[17] V. K. Abdelkader, S. Scelfo, C. Garcia-Gallar�n, M. L. Godino-Salido, M.
Domingo-Garc�a, F. J. L�pez-Garz�n, M. P�rez-Mendoza, J. Phys. Chem. AJ. Phys. Chem. C. 2013, 117, 16677 – 16685.
[18] T. Ando, M. Nishitani-Gamo, R. E. Rawles, K. Yamamoto, M. Kamo, Y.Sato, Diam. Relat. Mater. 1996, 5, 1136 – 1142.
[19] S. C. S. Yu, J. A. Downard, Langmuir 2007, 23, 4662 – 4668.[20] C. E. Landis, L. K. Klein, A. Liao, E. Pop, K. D. Hensley, V. A. Melechko, J. R.
Hamers, Chem. Mater. 2010, 22, 2357 – 2366.[21] C. Fairman, M. Chockalingam, G. Liu, A. H. Soeriyadi, J. J. Gooding, Lang-
muir 2014, 30, 332 – 339.[22] a) R. Hoffmann, A. Kriele, H. Obloh, J. Hees, M. Wolfer, W. Smirnov, N.
Yang, E. C. Nebel, Appl. Phys. Lett. 2010, 97, 052101 – 052103; b) H. C.CHoi, J. M. Buriak, Chem. Commun. 2000, 1669 – 1670.
[23] a) B. V. Lyalin, V. A. Petrosyan, Russian Journal of Electrochemistry 2013,49, 497 – 529; b) T. Fuchigami, S. Inagi, Chem. Commun. 2011, 47,10211 – 10223.
[24] T. C. Kuo, L. R. McCreery, Anal. Chem. 1999, 71, 1553 – 1560.[25] R. M. Lockett, M. L. Smith, Langmuir 2010, 26, 16642 – 16646.[26] T. Kitagawa, H. Matsubara, K. Komatsu, K. Hirai, T. Okazaki, T. Hase, Lang-
muir 2013, 29, 4275 – 4282.[27] E. Papirer, R. Lacroix, J.-B. Donnet, G. Nans�, P. Fioux, Carbon 1995, 33,
63 – 72.[28] A. Deslandes, J. G. Shapter, E. L. Lawrance, J. S. ,Quinton, Nanoscience
and Nanotechnology,. ICONN ‘06. International Conference on. E-ISBN :1 – 4244 – 0453 – 3, Brisbane, Qld. 2006.
[29] A. Jorio, International Scholarly Research Network ISRN Nanotechnology2012, doi :10.5402/2012/234216.
[30] B. Li, L. Zhou, D. Wu, H. Peng, K. Yan, Y. Zhou, Z. Liu, ACS Nano 2011, 5,5957 – 5961.
[31] T. I. Hukka, T. A. Pakkanen, M. P. D’Evelyn, J. Phys. Chem. 1995, 99, 4710 –4719.
[32] a) M. M. Walczak, D. D. Popenoe, R. S. Deinhammer, B. D. Lamp, C.Chung, M. D. Porter, Langmuir 1991, 7, 2687 – 2693; b) C. E. D. Chidsey,C. R. Bertozzi, T. M. Putvinski, A. M. Mujsce, J. Am. Chem. Soc. 1990, 112,4301 – 4306.
[33] Y. Guan, C. J. Wurrey, G. J. J. Thomas, Biophys. J. 1994, 66, 225 – 235.[34] P. He, L. He, Biomacromolecules 2009, 10, 1804 – 1809.
Chem. Eur. J. 2014, 20, 1 – 10 www.chemeurj.org � 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim8&&
�� These are not the final page numbers!
Full Paper

[35] T. Sakata, S. Maruyama, A. Ueda, H. Otsuka, Y. Miyahara, Langmuir 2007,23, 2269 – 2272.
[36] C. Leung, H. Kinns, W. B. Hoogenboom, S. Howorka, P. Mesquida, NanoLett. 2009, 9, 2769 – 2773.
[37] M. D. Shewchuk, T. M. McDermott, Langmuir 2009, 25, 4556 – 4563.[38] H. Sabbah, S. Ababou-Girard, A. Zebda, D. David, B. Fabre, S. D�putier,
A. Perrin, M. Guilloux-Viry, F. Solal, C. Gode, Diam. Relat. Mater. 2009, 18,1074 – 1080.
[39] E. de La Llave, A. Ricci, E. J. Calvo, D. A. Scherlis, J. Phys. Chem. C 2008,112, 17611 – 17617.
Received: February 5, 2014
Published online on && &&, 0000
Chem. Eur. J. 2014, 20, 1 – 10 www.chemeurj.org � 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim9 &&
These are not the final page numbers! ��
Full Paper

FULL PAPER
& Electrochemical Functionalization
A. M. Debela, M. Ortiz,* V. Beni,C. K. O’Sullivan*
&& –&&
Facile Electrochemical Hydrogenationand Chlorination of Glassy Carbon toProduce Highly Reactive and UniformSurfaces for Stable Anchoring ofThiolated Molecules
Carbon functionalization : A two-stepprocess for the electrochemical hydro-genation of glassy carbon followed by
electrochemical chlorination providesa highly reactive surface for furtherfunctionalization (see figure).
Chem. Eur. J. 2014, 20, 1 – 10 www.chemeurj.org � 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim10&&
�� These are not the final page numbers!
Full Paper




















