
Expression and structure-function characterisation of ... - DIVA
76
1 Expression and structure-function characterisation of herpesviral proteins Sue-Li Dahlroth
-
Upload
khangminh22 -
Category
Documents
-
view
1 -
download
0
Transcript of Expression and structure-function characterisation of ... - DIVA

1
Expression and structure-function characterisation of
herpesviral proteins
Sue-Li Dahlroth

2
Doctoral thesis at Stockholm University
Department of Biochemistry and Biophysics
©Sue-Li Dahlroth, Stockholm 2008
ISBN 978-91-7155-755-1 pp 1-76
Printed in Sweden by Universitetsservice AB, Stockholm 2008
Distributor: Department of Biochemistry and Biophysics, Stockholm University
Papers I-III are reprinted with permission from the publisher.

3
Abstract
Human viruses coexist with their hosts, sometimes silently and sometimes
by causing a vast range of symptoms. To fully understand these seemingly
simple particles, how they have evolved, their pathogenesis, to be able to
develop new drugs and potentially new vaccines and diagnostic tools we
need to study the individual viral proteins both functionally and structurally.
In order to determine and study a protein structure, large amounts of it is
needed. The easiest way to obtain a protein is to recombinantly overexpress
it in the well-studied bacterium Escherichia coli. However, this expression
host has one major disadvantage, overexpressed proteins might not be folded
or be insoluble. Within the field of structural genomics, protein production
has become one of the most challenging problems and the recombinant
overexpression of viral proteins has in particular proven to be very difficult.
The first part of the thesis concerns the recombinant overexpression of
troublesome proteins in E. coli. A new method has been developed to screen
for soluble overexpression in E. coli at the colony level, making it suitable
for screening large gene collections. This method was used to successfully
screen deletion libraries of troublesome mammalian proteins as well as
complete ORFeomes from five herpesviruses. As a result soluble expression
of previously insoluble mammalian proteins was obtained as well as crystals
of three proteins from two oncogenic human herpesviruses, all linked to
DNA synthesis of the viral genome. The second part of the work presented
here concerns the structural studies of three herpesviral proteins. SOX from
Kaposi’s sarcoma associated herpesvirus is involved in processing and
maturation of the viral genome. Recently SOX has also been implicated in
host shutoff at the mRNA level. With this structure, we propose a substrate
binding site and a likely exonucleolytic mechanism. The holoenzyme
ribonucleotide reductase is solely responsible for the production of
deoxyribonucleotides and regulates the nucleotide pool of the cell. The small
subunit, R2, has been solved from both Epstein Barr virus and Kaposi’s
sarcoma associated herpesvirus. Both structures show disordered secondary
structure elements in their apo-and mono metal forms, located close to the
iron binding sites in similarity to the p53 induced R2 indicating that these
two R2 proteins might play a similar and important role.

4
Table of Contents
INTRODUCTION 8
HUMAN HERPESVIRUSES 11
HERPESVIRUS STRUCTURE 12 HERPESVIRUS SUBFAMILIES 13 ALPHA HERPESVIRUSES 14 BETA HERPESVIRUSES 15 GAMMA HERPESVIRUSES 15
STRUCTURAL GENOMICS 17
PROTEIN PRODUCTION 19
PHYSICAL PARAMETERS 20 FUSION PROTEINS AND PROTEIN TAGS 21 CONSTRUCT DESIGN 22
SCREENING FOR RECOMBINANT SOLUBLE OVEREXPRESSION
23
COLONY SCREENING METHODS 24 THE COFI BLOT (PAPER I AND III) 28
LIBRARY METHODS 31
DELETION LIBRARIES SCREENED WITH THE COFI BLOT (PAPER II) 32
EXPRESSION SCREENING COMPLETE GENOMES 36
STRUCTURAL GENOMICS AND HUMAN PATHOGENS 38
THE DAILY SCOOP (PAPER IV) 39 SCOOP AND OTHER HERPESVIRUS ORFEOMES 41
MOVING FURTHER DOWN THE PIPELINE 42
HOST SHUTOFF IN HHV 45
SOX 46 STRUCTURAL STUDIES OF THE SOX PROTEIN FROM KSHV (PAPER V) 48 SUBSTRATE BINDING 50 SOX AS AN RNASE? 51
NUCLEOTIDE SYNTHESIS 53
RIBONUCLEOTIDE REDUCTASE 53

5
STRUCTURAL STUDIES OF THE R2 SUBUNIT OF THE RIBONUCLEOTIDE
REDUCTASE FROM EBV AND KSHV (MANUSCRIPT IN PREPARATION) 54
FUTURE PROSPECTS 57
ACKNOWLEDGEMENTS 58
REFERENCES 60

6
List of papers
This thesis is based on the following papers, referred to in the text by their
roman numerals.
I. Cornvik, T., Dahlroth, S-L, Magnusdottir, A., Herman, M.D, Knaust,
R., Ekberg, M. and Nordlund P.
Colony-Filtration blot, A new screening method for soluble protein
expression in E. coli.
Nature Methods. 2005, 2(7):507-9.
II. Cornvik, T, Dahlroth, S-L., Magnusdottir, A., Flodin, S., Engvall, B.,
Lieu, V., Ekberg, M. and Nordlund, P.
An efficient and generic strategy for producing soluble human
proteins and domains in E.coli by screening construct libraries.
Proteins: Structure, Function and Bioinformatics. 2006, 1;65(2):266-
73.
III. Dahlroth, S-L., Nordlund, P. and Cornvik, T.
Colony filtration blot for screening soluble expression in Escherichia
coli.
Nature Protocols, 2006, 1(1):253-8.
IV. Dahlroth, S-L., Lieu., V, Haas, J. and Nordlund, P.
Screening Colonies of Pooled ORFeomes, SCOOP: A rapid and
efficient strategy for expression screening ORFeomes in E. coli.
Submitted
V. Dahlroth, S-L., Gurmu, D., Schmitzberger, F., Erlandsen, H. and
Nordlund, P.
Structure of the shutoff and exonuclease protein from the oncogenic
Kaposi’s sarcoma associated herpesvirus
Manuscript

7
Abbreviations
2D-gel Two-dimensional gel
AE Alkaline exonuclease
AIDS Acquired immune deficiency syndrome
CAT Chloramphenicol acetyltransferase
CMV Cytomegalovirus
CoFi blot Colony filtration blot
EBV Epstein Barr virus
GFP Green fluorescent protein
HHV Human herpesvirus
HIV Human immunodeficiency virus
HSV-1, 2 Herpes simplex 1 and 2
IMAC Immobilised metal affinity chromatography
IPTG Isopropyl -D-1-thiogalactopyranoside
KS Kaposi’s sarcoma
KSHV Kaposi’s sarcoma associated herpes virus
MCD Multicentric Castleman’s disease
mCMV Murine Cytomegalovirus
MS Mass spectrometry
NMR Nuclear magnetic resonance
ORF Open reading frame
ORFeome The complete collection of ORFs from one organism
PEL Pleural effusion lymphoma
RNR Ribonucleotide reductase
SAD Single wavelength anomalous dispersion
SCOOP Screening colonies of ORFeome pools
SG Structural genomics
SOX Shut off and exonuclease
VZV Varicella zooster virus
WHO World health organisation
UNICEF United nations children’s fund
8
a) b)
c) d)
e)
Introduction
Viruses are small biological entities, existing on the border of life as we
define it. It cannot survive on its own and behaves as a molecular parasite,
making use of its host’s cellular machinery to create infectious progeny.
They come in many different and diverse forms and have been around since
ancient times, evolving with its surroundings (1). Viruses are divided into
families, subfamilies, genus and strains and can vary in both shape and size.
The smallest known virus belongs to the Parvovirdae family with a size of
20-25 nm and the largest known virus is the Mimivirus of 400 nm (2). They
will carry their genetic material of only a few genes to hundreds as either
DNA or RNA, which is encapsulated in a protective layer of proteins
(capsid) and sometimes a membrane consisting of lipids and proteins.
Proteins in this outer shell will determine their mode of infection and which
hosts they infect, be it bacteria, plants, fungi, animals or humans (1, 2).
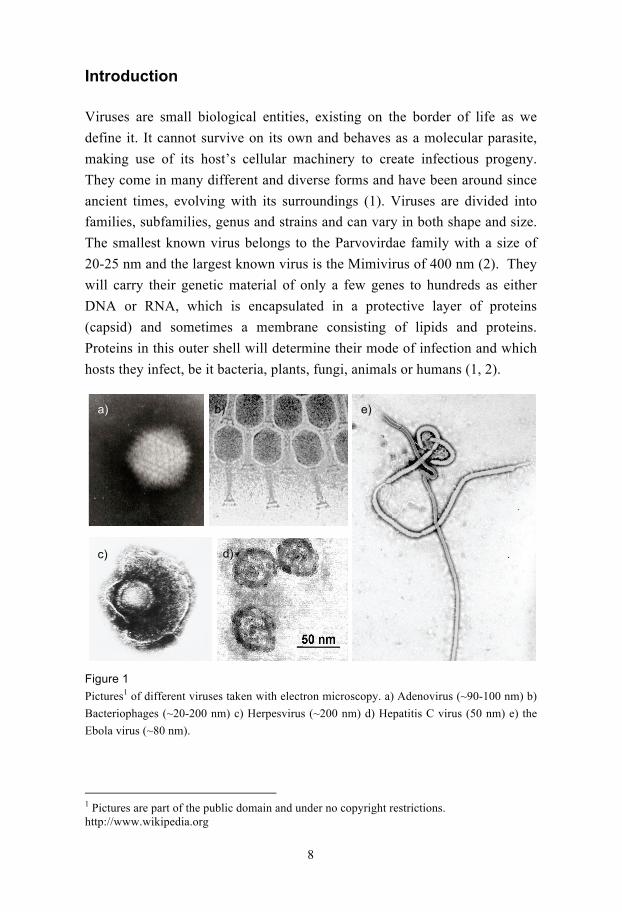
Figure 1
Pictures1 of different viruses taken with electron microscopy. a) Adenovirus (~90-100 nm) b)
Bacteriophages (~20-200 nm) c) Herpesvirus (~200 nm) d) Hepatitis C virus (50 nm) e) the
Ebola virus (~80 nm).
1 Pictures are part of the public domain and under no copyright restrictions. http://www.wikipedia.org

9
Certain types of viruses that infect plants can destroy entire harvests of
certain crops causing enormous economic damage each year (3). In humans
viral infections can be life long or temporary and can cause symptoms that
can range from anything like a common cold, diarrhoea, the flu, chicken
pox, and measles to hepatitis, polio, cancer, AIDS (acquired immune
deficiency syndrome), encephalitis, Ebola hemorrhagic fever and so on.
Viruses can roughly be divided into DNA or RNA viruses depending on how
they carry their genetic material. For both classes, the genome can be double
stranded (ds) single stranded (ss), circular or linear. The life cycle of a virus
(Figure 2) can be divided into several stages, attachment to the target cell,
entry (by endocytosis, fusion or genetic injection), replication and shedding
(the process when new viral particles leave the cell). Shedding occurs either
through lysis, budding, apoptosis or exocytosis (2).
After host cell entry, DNA viruses must move its genome into the host cells
nucleus, the site for DNA replication and transcription. The RNA, from
RNA viruses, can either remain in the cell cytoplasm, which will then be the
scene for its life cycle, or it can convert its RNA into DNA that will move
into the nucleus and fuse with the hosts’ genome. These latter types of RNA
viruses, of which the best known is HIV (human immunodeficiency virus)
are called retroviruses and cause lifelong infections. An interesting fact is
that up to 8% of the human genome is believed to be remnants of retroviral
infections (4) and although we carry what is referred to as proviruses in our
genome, they do not as far as we know cause disease.
It is however not only retroviruses that can cause lifelong infections. Many
additional elements determine weather or not a viral infection will persist,
such as the target cell, the individual’s immune system and how the viral
genome is maintained and replicated once inside its host. For instance
herpesviruses are relatively large dsDNA viruses infecting a wide range of
host cells. Herpesviral infections are life long due to target cell type, genome
maintenance and a cunning strategy to evade the immune system (5).

10
c)
d)
a) e)
Figure 2
The general life cycles of viruses from attachment to shedding. a) A DNA virus, that enters
through fusion. The DNA is exported to the nucleus where it is replicated and transcribed.
The mRNA is transported to the cytoplasm and translated into viral proteins. New virions are
shed through exocytosis. b) An RNA virus enters through endocytosis and is stripped. The
RNA is either c) translated directly into viral proteins and new virions are made or d) the
RNA is reversibly transcribed into DNA that enters the nucleus and fuses with the host
genome. This provirus is transcribed and translated into virions that are shed through budding.
e) The RNA genome is injected into the host cell and is translated into viral proteins, which
assemble into new infectious virions that are shed through host cell lysis.
Since viruses are the causative agents for numerous mild symptoms like
colds but also very brutal diseases like cervical cancer, Burkitt’s lymphoma,
liver cancer etc they are intensely studied. Their mode of infection,
pathogenesis and epidemiology as well as their molecular structure and
cellular interactions are of huge interest with the goal of developing
diagnostic tools, vaccines and antiviral drugs. As huge as the discovery and
development of penicillin and other antibiotics, as a treatment for bacterial
infections in the late 1920’s, is the discovery of vaccination (from the Latin
word vacca meaning cow) in the late 18th century. In 1796 Edward Jenner
used the cowpox virus to vaccinate humans, which resulted in protection
b)

11
against the two smallpox viruses that can cause blister-like scarring in the
face, blindness and even death. Smallpox was officially declared eradicated
in 1979. Polio, caused by the poliovirus was for a very long time a dreaded
childhood disease that can cause paralysis, meningitis and even death.
Vaccines against polio were developed by Jonas Salk in 1952 and Albert
Sabin in 1962. Since the start of a global vaccination effort in 1988 by
WHO, UNICEF and the Rotary Foundation, the number of reported cases
has dropped from hundreds of thousands to only thousands each year2.
Several continents are today declared as polio-free and a global eradication
has been proposed, although is still persists in some developing countries
(6). Even though many viral infections can be stopped by vaccinations there
are still many viruses, such as HIV, Hepatitis C, Dengue fever, for which
vaccines do not exist.
Human herpesviruses
An immense number of books and scientific articles have been written about
a wide range of topics concerning herpesviruses, from the overall virion
structure, the mode of transmission, prevalence in ethnic and social groups,
to the regulation of specific proteins in an infected cell. The main purpose of
this thesis is not to give an absolute introduction to herpesviruses, but just a
sneak peak and enough information about them for the reader to understand
the importance of this work as well as give a general grasp of the complex
relationship between these viruses and their hosts.
To date, hundreds of herpesviruses have been identified but only 8 are
known human pathogens and for the remainder of this thesis the focus will
be on the human herpesviruses and they will be referred to by their common
names or abbreviations (Table 1). All herpesviruses are large dsDNA viruses
that share a common overall structure and similar life cycle. Depending on
their mode of infection, the length of the life cycle and target cells, they are
further divided into subfamilies (2, 5). These subfamilies are further divided
into genera, although these will not be referred to or mentioned in this text.
2 http://www.who.int/en/

12
Table 1
A list of the 8 human herpesviruses, their subfamily, formal names, common names and
abbreviations. In the text, they will henceforth be referred to either by their common name or
abbreviations in column 5, except for HHV-6 and HHV-7.
Subfamily Formal name Abbrev Common name Abbrev
Alpha herpesvirus Human
herpesvirus 1
HHV-1 Herpes simplex virus-1 HSV-1
Alpha herpesvirus Human
herpesvirus 2
HHV-2 Herpes simplex virus-2 HSV-2
Alpha herpesvirus Human
herpesvirus 3
HHV-3 Varicella-zoster virus VZV
Gamma herpesvirus Human
herpesvirus 4
HHV-4 Epstein-Barr virus EBV
Beta herpesvirus Human
herpesvirus 5
HHV-5 Human
cytomegalovirus
HCMV
Beta herpesvirus Human
herpesvirus 6
HHV-6 - -
Beta herpesvirus Human
herpesvirus 7
HHV-7 - -
Gamma herpesvirus Human
herpesvirus 8
HHV-8 Kaposi’s sarcoma-
associated herpesvirus
KSHV
Most of what we know today about herpesviruses has been based on studies
of the herpes simplex virus-1 (HSV-1) due to its early identification and easy
cultivation in cell cultures. Recently, however, significant progress has been
made in understanding the structure, biology and pathogenesis of the other
human herpesviruses.
Herpesvirus structure
As already mentioned, the herpesvirus family is a group of dsDNA viruses,
which are ~200 nm in diameter with a genome size of ~130-250 kbp (70-170
genes). What also unites herpesviruses is the common architecture of the
infectious particles (Figure 3). In the infectious virion, the genome is linear
and is wrapped around a core of proteins. This genome is contained within
an icosahedral shell, the capsid, which is made up of two types of oligomeric
proteins, hexon and penton capsomers. Surrounding the capsid is the poorly
characterized tegument, an amorphous mass consisting of various essential

13
and non-essential proteins that are delivered to the cells at the very initial
stage if infection (5). Amino acid sequencing and MS analyses have been
carried out to determine the protein content of the tegument. Apart from
cellular proteins (that might be specifically or non-specifically packaged into
the virions) (7-9) the tegument contains more than 20 for HSV-1 and more
than 30 for HCMV virus-encoded proteins that aid in viral replication and
immune evasion (8, 10, 11). The tegument is surrounded by a membrane of
lipids and glycoproteins, (each of the herpesviruses encodes a set of 20-80
glycoproteins) used for target cell recognition, attachment and entry (5, 12,
13).
Figure 3
Schematic picture of the overall structure of a herpesvirus. The DNA core is surrounded by
the capsid, the tegument and a lipid envelope containing glycoproteins.
Herpesvirus subfamilies
All herpesviruses belong to one of three subfamilies, alpha, beta and gamma,
depending on their host range, length of reproductive cycle and target cells.
In addition to their overall structure, the three herpesvirus subfamilies share
the mode of infection and life cycle (5, 14). A herpesvirus life cycle has two
distinct phases, a latent and a lytic phase. After attachment to the target cell,
the lipid membrane is fused with the cellular membrane and the capsid and
tegument proteins are released into the cell. The capsid is dissolved and the
DNA is transported into the nucleus where is circularises into an episome.
The viral episome is replicated and maintained with the host genome. Only a
very small set of genes is expressed during the latent phase, and their
products block apoptotic pathways and aid in immune evasion and genome
maintenance. During the latent phase, the infected individual shows no
symptoms of infection. At a given signal, for instance a weakened immune
system due to a cold, the virus is reactivated and goes into lytic phase. The

14
molecular signals that cause reactivation of herpesviruses are still not
entirely known, but HSV reactivation has been ascribed to physical damage,
ultraviolet light, hormones, or even fever. In the lytic phase, the virus will
take over host gene expression and shuts it down. It will then start
replication of its own genome and subsequently produce viral proteins. New
infectious virions will assemble and leave the host cell (5). A common
characteristic of herpesvirus infections is that they rarely pose any real
threats to a healthy person and although infection is life long a person can go
through life without even knowing that they are infected. The real problem
occurs in people with weakened immune systems where infection can lead to
organ failure and consequently death (15-18).
Alpha herpesviruses
HSV-1 is the prototype of the alpha herpesvirus family. The alpha
herpesviruses HSV-1/2 causes cold sores and genital herpes while VZV
causes chicken pox and shingles (14). Symptoms of infection will show in
epithelial cells such as skin and mucosa and these cells will consequently be
targeted by the immune system. However, the target destination is sensory
neurons in the brain. Infection starts at the mucosal surface, where the virus
will undergo lytic replication in the surrounding epithelial cells. After this it
enters a nearby sensory neuron, where it will establish a lifelong albeit latent
infection. The capsid travels up the axon on microtubules to the nucleus
where the genome enters the nucleus and circularises into an episome (19,
20). Upon reactivation, the virus will travel back down the axon to epithelial
cells, where further lytic spread will result in symptoms and a hopeful
transmission to new hosts. Although about 90 % of the general population is
infected with HSV it is only very rarely that this will cause any symptoms
other than cold sores (5). However, alpha herpesvirus infections can result in
encephalitis most commonly in children, the elderly, and people with
weakened immune systems (i.e. those with HIV/AIDS or cancer) although
this is very rare and only occasionally fatal (21).

15
Beta herpesviruses
Beta herpesviruses, such as HCMV, HHV-6 and HHV-7 replicate more
slowly than alpha herpesviruses and establish latency in progenitor cells of
the bone marrow, monocytes and T-cells, which are all part of the immune
system (5).
The best characterised member in this subfamily is HCMV, which replicates
in a vast number of cells i.e. macrophages, dendritic cells, colonic and retinal
cells, endothelial cells (22). It has been estimated that >60% of the general
population carries CMV (5) and infection usually goes unnoticed in healthy
adults. It is only in immunocompromised individuals (like HIV patients,
organ transplant recipients as well as unborn babies) that serious conditions
such as pneumonia, encephalitis and retinitis arise (22, 23). HCMV infection
is considered the major cause of these conditions and the subsequent
mortality among immunosuppressed individuals (24, 25).
Gamma herpesviruses
There are two human herpesviruses that belong to the gamma herpesvirus
subfamily, EBV and KSHV. A key feature of these viruses is their capacity
to induce lymphoproliferation and cancers (5, 26). EBV’s major targets are
epithelial cells and B-cells where it also establishes latency. KSHV infects a
wide range of cells in vivo and in vitro for example endothelial cells, B-cells
epithelial cells and fibroblasts (27). After infection the linear genome
circularises into an episome, which tethers itself to the host chromosome by
a specific protein and replicates in concert with the hosts genome (5, 28, 29).
EBV causes infectious mononucleosis, better known as “kissing disease”
and it has been estimated that >90% of the worlds adult population carries
EBV3. EBV was the first human tumour virus discovered and is associated
with Burkitt’s lymphoma and nasopharyngeal carcinoma and to several other
malignancies for instance several types of Hodgkin’s lymphoma and gastric
carcinoma. In AIDS patients EBV causes a number of other lymphomas and
tumours (5, 30). In addition, there is a growing body of evidence, although
very controversial, that suggests a connection between EBV infection and
3 http://www.who.int/en/

16
liver and breast cancer as well as certain types of auto-immune diseases like
multiple sclerosis, rheumatoid arthritis and diabetes (31, 32)
Kaposi’s sarcoma (KS) was first described in 1872 as a rare purplish-
pigmented sarcoma of the skin typically found in elderly men of Jewish and
Mediterranean descent. However, during the onset of AIDS in the 1980’s
there was a noticeable increase of KS and in 1994 the cause was identified as
a new human herpesvirus subsequently called Kaposi’s sarcoma associated
herpesvirus (KSHV) (33).
Besides causing KS, KSHV has been associated with some rare but lethal
lymphomas, pleural effusion lymphoma (PEL) and multicentric Castleman’s
disease (MCD) (5, 34). In Europe and the US <3% of the general population
are infected with KSHV. However, in some sub-Saharan countries where KS
was almost unknown before HIV, >50% are carriers (35) and in these
countries KS has become one of the most clinically described neoplasms. As
with EBV, KSHV has also been linked to other more controversial
conditions like sarcoidosis (5, 36, 37) and multiple myeloma (38, 39).
What really sets KSHV aside from the rest of the human herpesviruses is the
amount of cellular genes that is has copied throughout evolution, in
something called molecular mimicry or molecular piracy, where more than a
dozen cellular genes have been copied. Furthermore, several of these genes
have potential tumour related functions, so called oncogenes, meaning that
they can affect the cell cycle, apoptosis and other types of cell signalling
(40-42). EBV, on the other hand, encodes several highly evolved
transcriptions factors and signalling proteins that induce many of the same
cellular genes that KSHV has pirated into its own genome (5).
Herpesviruses have coevolved with their hosts to establish lifelong
infections in various cell types (43, 44). They are the major cause of several
minor syndromes and major malignancies in humans. Whether or not the
symptoms are manageable with today’s treatments or more severe is
determined by the individual’s immune system and genetic predispositions.
In immunocompromised patients, such as organ transplant recipients, cancer
patients, and AIDS patients a herpesviral infection can cause major
complications which could result in death.
To fully understand these viruses the individual proteins, such as virion
proteins and proteins from various stages of the viral life cycle, can be

17
studied. However, some of these proteins can be hard to come by, since they
are not present or expressed in sufficient amounts in the virion particle or
target cells. The easiest way to obtain these proteins would therefore be to
recombinantly overexpress them. Recombinant proteins from herpesviruses
could help in creating vaccines, yield in high-resolution protein structures
that can help in understanding the viral life cycle, evolution, and
pathogenicity and might serve as potential drug targets.
Structural genomics
It is broadly accepted that the function of a proteins is dependant on the
amino acid sequence and how this chain of amino acids is folded in the
three-dimensional space. It is also widely accepted that the information for
the folding pattern of a protein resides in its linear DNA sequence. However,
this folding information is at present much too hidden from us and in order
to understand how a protein works at the molecular level we must study its
three-dimensional structure, preferably at high resolution.
In the wake of the genomics efforts, the effort to determine the complete
genomic sequence of all organisms (45-48), new emerging fields have risen
with aims to, on a-full-organism-scale determine patterns, trends, functions
and pathways among and within these genes and their corresponding gene
products, be it at the transcriptional, translational or degradation level. Huge
databases with massive amounts of information have become available for
searches, which in turn have yielded in vast amounts of new results and data.
Structural genomics (SG), the effort to structurally determine a large number
of proteins from one organism/genome, is such a field (49). In Table 2, the
approximate number of genes for some organisms and viruses are shown and
the number of unique protein structures for each of them4.
To date less than 5% of the human herpesviral proteins have been
structurally characterised and even less have been solved within the context
of SG (50, 51).
4 http://www.rcsb.org/pdb/home/home.do
18
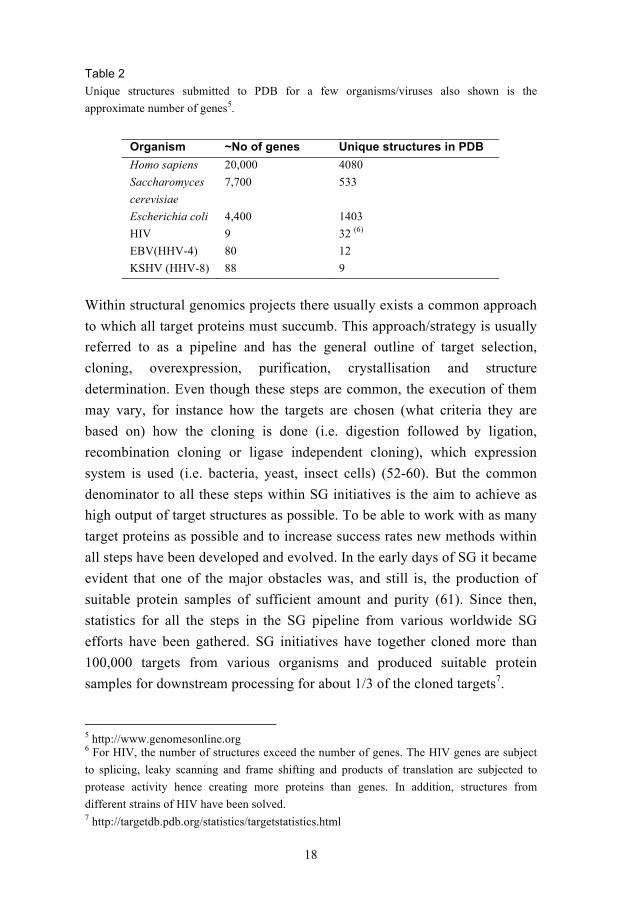
Table 2
Unique structures submitted to PDB for a few organisms/viruses also shown is the
approximate number of genes5.
Organism ~No of genes Unique structures in PDB
Homo sapiens 20,000 4080
Saccharomyces
cerevisiae
7,700 533
Escherichia coli 4,400 1403
HIV 9 32 (6)
EBV(HHV-4) 80 12
KSHV (HHV-8) 88 9
Within structural genomics projects there usually exists a common approach
to which all target proteins must succumb. This approach/strategy is usually
referred to as a pipeline and has the general outline of target selection,
cloning, overexpression, purification, crystallisation and structure
determination. Even though these steps are common, the execution of them
may vary, for instance how the targets are chosen (what criteria they are
based on) how the cloning is done (i.e. digestion followed by ligation,
recombination cloning or ligase independent cloning), which expression
system is used (i.e. bacteria, yeast, insect cells) (52-60). But the common
denominator to all these steps within SG initiatives is the aim to achieve as
high output of target structures as possible. To be able to work with as many
target proteins as possible and to increase success rates new methods within
all steps have been developed and evolved. In the early days of SG it became
evident that one of the major obstacles was, and still is, the production of
suitable protein samples of sufficient amount and purity (61). Since then,
statistics for all the steps in the SG pipeline from various worldwide SG
efforts have been gathered. SG initiatives have together cloned more than
100,000 targets from various organisms and produced suitable protein
samples for downstream processing for about 1/3 of the cloned targets7.
5 http://www.genomesonline.org 6 For HIV, the number of structures exceed the number of genes. The HIV genes are subject
to splicing, leaky scanning and frame shifting and products of translation are subjected to
protease activity hence creating more proteins than genes. In addition, structures from
different strains of HIV have been solved. 7 http://targetdb.pdb.org/statistics/targetstatistics.html

19
Protein production
Since certain proteins are present only at very low levels and at certain time
points in a living organism, the most ethical and effective way of obtaining a
protein of interest is to recombinantly overexpress it. High-resolution
structures of biological macromolecules, such as proteins, can be determined
with several methods. The most convenient and efficient methods are X-ray
crystallography and NMR. For X-ray crystallography, the dominating
method, an absolute requirement is well diffracting protein crystals.
Numerous crystallisation trials and optimisations might be necessary to
produce a well diffracting crystal and therefore large amounts of pure and
soluble protein is needed.
The most widely used expression host to date for recombinant
overexpression is the well-studied bacterium E. coli. The major reason for
using E. coli is that with great ease and low cost, large amounts of biomass
can be generated (62-64). E. coli also possesses other advantages beneficial
for structural biologists. For instance, it will produce a very homogenous
protein sample since it lacks the machinery to create certain covalent
modifications like glycosylations.
In principal, E. coli has the same protein production machinery as any other
cell, although is differs in ways that might create problems. For instance,
when trying to overexpress a protein, the bacterium might form large
insoluble aggregates called inclusion bodies (62, 65, 66). Inclusion bodies
consist of unfolded or partly folded proteins and might take up a very large
volume of the cell (67, 68), although there have been situations where
correctly folded and active proteins have been found in inclusion bodies
(69). The already mentioned lack of covalent modifications, which might be
needed for proper protein folding and function, could be one reason for
inclusion body formation. E. coli may also lack certain important tRNAs
that could lead to halted translation as well as lacking certain folding
partners such as chaperones (67, 68, 70-73). Another problem when
overexpressing proteins is in vivo degradation by indigenous proteases or
that the target protein in itself is toxic to the bacterium (74). The
overexpression of eukaryotic proteins in E. coli is especially troublesome
(75), although even when trying to overexpress indigenous E. coli proteins,
in vivo proteolysis occurs as well as inclusion body formation (65, 66, 76).

20
In contrast to prokaryotic proteins, eukaryotic proteins tend to be, on an
average, larger and consist of more domains that are connected by flexible
linker regions (77) and it has been shown that the size of a protein correlates
with the success rate of its soluble expression in E. coli (53). As already
mentioned, the official success rates from SG efforts for soluble production
of prokaryotic proteins, is 50% and 30% for eukaryotic proteins when a
basic SG pipeline is used (53, 60). However, in these statistics there is no
information of the success rates in regard to full-length protein expression or
protein size.
To increase the likelihood of obtaining soluble protein, when using E. coli,
there are generally two approaches: i) either change physical parameters of
the experiment (like the bacterial strains, culture conditions, promoters or
fusion partners or ii) change the properties of the target protein.
To ensure high success rates and low costs, the soluble recombinant
expression should be screened before proceeding with large-scale expression
and purification.
Physical parameters
To compensate for rare codons, tRNAs can be co-expressed in E. coli (78,
79) and numerous strains are commercially available that can co-express
these tRNAs. Typically these strains also lack certain proteases that could
lead to protein degradation (80). Even strains that should be more resistant to
toxic proteins and strains that provide the right oxidizing conditions,
permitting disulfide bonds to be formed, have been created (81-83). Another
parameter that can influence the recombinant expression of a target protein
is the culture conditions (62). The growth medium can be changed as well as
the growth temperature. Although very few systematic studies have been
reported (84, 85), it is still believed that the expression medium could
influence soluble expression. In regard to expression temperatures, more
support exists for its influence on the expression (62, 72) than for the
expression medium. For several proteins it has been shown that by
decreasing the temperature, target proteins could be rescued from the fate of
inclusion bodies (62, 85-87). How the solubility of a protein in a bacterium
correlates with a decrease in expression temperature is not fully understood
and might be due to a combination of factors involved in the transcription

21
and translation as well as folding of the protein. When the transcription and
translation machineries slow down, due to decreased temperature, the
protein might have time to fold in a proper way. The attractive forces,
between hydrophobic parts, that could lead to protein aggregation are
potentially weaker at low temperatures (88). It has also been shown that the
expression of several indigenous chaperones are induced at lower
temperatures (89).
Although new bacterial strains have been created and culture conditions are
varied, the problem of inclusion bodies and proteolysis still persists,
especially for mammalian proteins.
Fusion proteins and protein tags
Fusion proteins are often large soluble proteins that are subcloned upstream
or downstream of the target protein. It has been shown that by adding a large
soluble fusion protein, the folding propensity and therefore the solubility of
the target protein itself can be increased (75, 90-94). The most widely used
fusion proteins are glutathione S-transferase (GST), maltose binding protein
(MBP) and thioredoxin (TrxA). Fusion proteins do not only serve as
solubilising factors but can also aid in purification and detection of the target
protein. Although there are obvious benefits of adding a large soluble
protein, there are some clear limitations to it. For instance, the fusion protein
should preferably be removed before crystallisation trials either by laborious
recloning or digestions. Large fusion partners might also alter the solubility
of the target protein in a negative way and removal could therefore result in
an unpleasant surprise, such as protein aggregation and precipitation (94-98).
Instead of adding a large fusion protein for detection and purification, that
could potentially alter the solubility of the target protein, small peptide tags
can be used instead. The most commonly used peptide tag is the His-tag. A
stretch of six histidines is added, in the cloning step either upstream or
downstream of the target protein, which has the ability to bind divalent
cations, typically Ni2+ or Co2+. If these cations are immobilised on a gel
resin, the target protein can be caught and separated from non-histidine-
tagged proteins. This method, which we today refer to as IMAC

22
(immobilised metal affinity chromatography), was first described in the late
1980’s (99) and has since then revolutionised recombinant protein
purification. Commercially available antibodies and probes, conjugated with
horseradish peroxidase (HRP) or alkaline phosphatase (AP), directed
towards His-tags have since then been generated and therefore a His-tag can
be used for target protein detection based on immunochemicals.
Construct design
The second approach to increase the chances of obtaining recombinant
soluble expression is to change the characteristics of the target protein. As
already mentioned the expression of prokaryotic proteins can be problematic
and success rates for soluble expression of such a protein is approximately
~50 %. However, for eukaryotic proteins the success rates drop significantly
to ~30%8. This could be due to the larger size of these proteins, the number
of domains and flexible linker regions (which might be protease sensitive),
the requirement of specific chaperones for folding and the requirement for
post-translational modifications. A natural reaction to these problems has
been the attempts to clone and express the individual domains of the target
protein, which has been proven to be very useful (57, 75, 100-103). This
strategy is based on the theory that if the full-length protein fails to express
or crystallise, perhaps its individual domains might. Domains can be
predicted either experimentally, with limited proteolysis coupled with MS
analysis (101, 102), deuterium exchange MS (104) or with special computer
programs (105).
The latter strategy, designing new constructs partly with help of domain
predictions, has successfully been employed within SG-initiatives where
several expression constructs for one target protein are generated (Figure 4).
It has been shown that by using this approach the probability of generating
soluble protein increased two-fold (100). Since these types of domain
prediction programs have a fair degree of uncertainty, several expression
constructs have to be designed that start close to the predicted domains.
8 http://targetdb.pdb.org/statistics/targetstatistics.html

23
b)
Figure 4
Construct design. a) A schematic picture of a result from a domain prediction program of a
multi-domain protein. b) New expression constructs are designed to define domain borders in
hope of finding a better expressing construct.
The solubility of a protein can also be increased by making random or
focused mutations or deletions. This approach is called in vitro evolution and
will be described later in the text. Whether new expression constructs are
generated in a focused or random approach generate, all of them have to be
screened for soluble expression.
Screening for recombinant soluble overexpression
The aim of a screen, no matter how it is executed, is to rapidly reduce a large
number of clones/targets to a more easily handled number.
The traditional approach when screening for soluble expression is usually
done with liquid cultures in individual vials and more recently in a 96-well
format. 1 ml cultures are grown and induced in parallel, cells are harvested,
lysed and soluble material is separated from insoluble by centrifugation
and/or purification. The soluble fraction is usually analysed by SDS-PAGE
gels (53, 60, 85, 106-108). Robots can perform certain steps in this process
while others still have to be done manually.
A couple of years ago an effort at genome wide expression screening was
attempted. Some 10,167 ORFs from the nematode Caenorhabditis elegans
was screened for soluble expression in E. coli in a 96-well format. Soluble
expression could be detected for 1,356 ORFs corresponding to a success rate
of 13% (109). This number is very much lower than more recently reported
success rates for eukaryotic proteins and it was later shown that many ORFs
a)

24
were wrongly annotated, had mispredicted gene boundaries and were out of
frame (110). Although only one vector and one expression strain was used
and some steps were automated, the workload of this effort is likely to have
been very large.
In our lab, we have previously developed a method that utilises filtration in
order to separate soluble material from insoluble called FiDo (filtration dot
blot) (111). Liquid cultures are grown and induced in a 96-well plate. A
small fraction of the culture is then transferred to another 96-well plate with
a low protein binding submicron filter in the bottom. The liquid media is
removed by vacuum and a bacterial pellet formed on top of the filter. The
pellet is either resuspended in a denaturing lysis buffer (solubilising all
proteins in the bacteria) or a native lysis buffer (only releasing the soluble
proteins). Vacuum is reapplied and the filtrate is collected in a collector
plate. The filtrate is then used to make dots on a membrane with a high
protein affinity, like nitrocellulose. The nitrocellulose is then blocked and
probed with an antibody or probe and developed like a Western blot.
The FiDo screen has also been modified to accommodate for an affinity
purification step to be able to determine the purifying ability of the target
protein (112).
In order to secure a high output of structures in an SG pipeline the best
strategy would be to generate multiple constructs subcloned with different
fusion proteins/tags, which would then be expressed in different strains and
at different temperatures. A quick calculation shows that working with 96
targets, creating 10 variants of each (based on domain predictions) cloned
with 2 different fusion proteins/tags and expressed in two different strains at
two different temperatures, would generate 7,680 different experiments and
although most of the work would be done in a 96-well format, it would be
quite labour intense. Therefore these types of combinatorial experiments are
currently not pursued within SG initiatives due to high costs and heavy
workload.
Colony screening methods
As already mentioned, solubility can be increased by adding a large fusion
protein. The solubility would then subsequently be screened based on the
physical characteristics of a soluble protein, such as its ability to be

25
separated from insoluble protein by centrifugation, filtration or purification
of liquid cultures.
However, solubility of a protein is intimately connected to its folding and
activity and could therefore be monitored by fusing the target protein to a
reporter protein, that when folded correctly would give rise to an easily
monitored phenotype. The theory relies on, that if the reporter protein is well
folded, hence soluble, the target protein should also be soluble and well
folded (Figure 5).
Figure 5
Schematic picture of a target protein (grey) fused to a reporter protein (black). a) If the target
protein is well folded and soluble, the reporter protein will fold and the phenotype can be
observed. b) If the target protein misfolds, the reporter protein will misfold and no phenotype
will be seen.
Solubility would not have to be screened in liquid cultures but instead at
colony level and would therefore lift the heavy burden of handling liquid
cultures since thousands of colonies, if they all carried different constructs,
could potentially be screened on one colony plate.
Waldo et al described in 1999 a method to monitor folding and therefore
solubility in colonies by fusing the target protein to GFP (green fluorescent
protein). Only bacterial colonies that fluoresce would have complete read-
through and express a well-folded and soluble target protein and vice versa
(113).
Another method relying on the same theory, presented by Maxwell et al, is
to fuse CAT (chloramphenicol acetyltransferase) to the protein of interest.
Only a bacterium that can grow in the presence of the antibiotic
chloramphenicol would express the target protein (114). Both these methods
are easy to use and allow thousands of colonies to be screened in one
experiment.
a) b)

26
a)
b)
Nevertheless, these types of methods have potential drawbacks. Firstly, the
reporter protein might affect the solubility of the target as already
mentioned. Secondly, false positives are seen for example when GFP is used
(115, 116), and thirdly, the reporter protein has to be removed before the
target protein can be used for structural studies.
In order to avoid the drawbacks of adding a large soluble protein, several
methods only relying on a small reporter peptide have been developed. In
practice, these methods rely on splitting a large reporter protein in two,
where none of the parts are active on their own. The target protein is fused to
a small peptide, corresponding to a vital part of the reporter protein. If the
target protein is soluble and well folded the phenotype should be detectable
when the “rest” of the reporter protein is added or co-expressed and vice
versa (Figure 6).
Figure 6
The theory behind a split reporter protein. The target (grey) is fused with a part of the reporter
protein (dark grey). If the target protein folds the tag will subsequently fold and when
combined with the rest of the reporter protein (black), the phenotype will show. If the target
protein is insoluble, the tag will be unfolded and no phenotype will be seen when adding the
rest of the reporter protein.
Wigley et al (117) used -galactosidase in this manner. -galactosidase was
split into a 52 amino acid -fragment, which was fused to different control
targets, and an -fragment corresponding to the rest of the protein. When the
control targets were expressed, colonies would turn blue or white depending
on the solubility. This method was effectively used to screen a hybrid gene
library of the human P450 for proper folding and solubility. Even though
this method seems to work well, especially when wanting to monitor slow
folding processes, a certain degree of false positives and negatives could be

27
observed. Additionally, in a review by G.S Waldo from 2003 it was claimed
that the -fragment could render proteins insoluble (118).
The developers of the previously described GFP-method have recently
developed it to better suit the demands of a smaller tag (119). Only part of
GFP, representing -strand 11, is fused to the target protein via a flexible
linker. This approach has successfully been used to screen mutation libraries
of proteins from Mycobacterium tuberculosis (115).
Both these methods have approached the problem, that reporter proteins can
remain active although it is fused to an insoluble target protein as well as
that they might alter protein solubility, by splitting the reporter protein in
two pieces. Both these methods have the advantage that they work in vivo
and in vitro making it easy to monitor solubility of the protein in a cell
lysate. A small complication is however, that in order for the rest of the
reporter protein to be present in the cell, two plasmids have to be used and
the protein has to be co-expressed.
A method that can directly monitor folding has been described by DeLisa et
al (120). It relies on that the twin-arginine translocation (Tat) pathway only
moves well-folded proteins across the inner membrane to the periplasmic
space of the E. coli cell (121). By fusing the target protein to a Tat
exporting-signal at the N-terminal and to -lactamase (which only confers
antibiotic resistance in the periplasm) at the C-terminal, the folding of the
target protein can be directly monitored. This method was tested on a
number of well-characterised proteins known to be soluble in the cytoplasm
(like GST, MBP, GFP, TrxA etc) as well as some cytoplasmic unstable
proteins and a good correlation could be seen. Potentially, -lactamase could
influence the folding in some unforeseen way and there were no reports on
any upper size-limit of the proteins that could be exported with the Tat-
pathway. In addition, before the target protein can be used the tag and/or the
-lactamase have to be removed either by recloning or digestions.
By fusing a gene to a C-terminal biotin acceptor peptide and therefore
enabling biotinylation in vivo by E. coli biotin ligase, BirA (122), detection
and affinity purification can be used based on the very strong binding of
biotin to the protein avidin (123).
Tarendeau et al (124) used this type of approach at the colony level.
Colonies carrying an expression construct with a C-terminal biotin acceptor
peptide (Avi-tag) are arrayed very closely, by a robot, on a nitrocellulose

28
membrane and induced for expression. The Avi-tag will be biotinylated in
vivo and after lysis, colonies expressing biotinylated proteins can be detected
on the nitrocellulose by probing with a fluorescent streptavidin conjugate. A
deletion library of almost 27,000 constructs (and with a seven-fold
oversampling!) of the influenza virus polymerase PB2 was successfully
screened in this manner and the approach was called ESPRIT (expression of
soluble proteins by incremental truncation). A major advantage is (at least in
theory) that since neither recloning nor digestions should be needed,
identified constructs can, if a plate replica were to be made, go directly in to
scale up experiments and subsequent affinity purifications based on biotins
affinity for avidin could be done. In addition no reports have so far surfaced
on any potential negative side effects by adding an Avi-tag, whether it
affects the solubility or if aggregated protein could potentially be
biotinylated and misinterpreted as soluble. Although this is a very powerful
method in its present implementation it still relies on expensive robotics,
which would not be part of standard laboratory equipment, and the use of
costly streptavidin-magnetic beads.
Several colony-based fusion protein or fusion tag screens have been
developed and evolved into well performing strategies. They are all elegant
methods that allow for a swift and easy way to identify targets that could be
suitable for large-scale protein production. Most importantly, however, is
that when it comes to colony-based screens they are best put to use when the
desire is to screen large collections of gene variants.
The CoFi blot (paper I and III)
We wanted to develop a method that works as a solubility screen at the
colony level but neither relies on a reporter protein that could potentially
affect the solubility of the target protein nor makes use of a tag that needs a
reporter protein to be co-expressed. Since we had very good experiences
with the previously described FiDo screen we adapted it in such a way so it
would allow us to screen soluble expression at the colony level.
In this method colonies are grown on a plate, called the master plate, and are
transferred to a submicron low protein-binding filter that can separate
inclusion bodies from soluble protein. Colonies on the master plate are

29
a) b)
c) d)
e) f)
g) h)
regrown and colonies on the filter are induced for expression by placing the
filter on a plate containing IPTG. After induction the filter is used to make a
filter sandwich (Figure 7). Upon lysis, soluble protein will diffuse through
the filter and attach to a high protein-binding membrane, such as
nitrocellulose. Detection is done by incubating the nitrocellulose with probes
or antibodies directed at the target protein and using standard
immunochemicals.
Figure 7
A schematic picture of the CoFi-blot method. a) Colonies are grown on a master plate, which
are b) transferred to a Durapore filter and c) expression is induced on a plate containing IPTG.
d) The filter with the colonies is then used to make a filter sandwich consisting of the
Durapore on top of a nitrocellulose and a Whatman paper with lysis buffer. e) Close up of
sandwich. f) Upon lysis, by repeated freeze thawing, soluble protein will diffuse through the
filter and bind to the nitrocellulose. g) The nitrocellulose is then blocked and incubated with
probes or antibodies directed at the target protein. h) The signals are detected by
chemiluminescence.
In our case we tend to use His-tag fusions, which efficiently can be probed
with Ni2+ conjugates as well as be used for purification. Colonies that give
rise to signals are picked from the master plate and can either go directly into
scale-up experiments or be further analysed. We chose to name this method
the CoFi blot (colony filtration blot).

30
In order to verify how well the CoFi blot works we decided to compare it to
the traditional method of growing liquid cultures and separating soluble
protein from insoluble by centrifugation.
32 eukaryotic and 24 E. coli proteins were subcloned in two different
expression vectors yielding either an N-terminal His- or FLAG-tag. Targets
were both grown and induced as colonies, which were subjected to the CoFi
blot, and as liquid cultures. The bacteria in the liquid cultures were
harvested, lysed and centrifuged. Dots were made on a nitrocellulose of both
total and soluble protein content and developed in the same way as for the
CoFi blot.
Figure 8
The CoFi blots performance was compared to traditional way of screening for soluble
expression, centrifugation. In the picture the results and correlation between the two methods
are shown for 32 eukaryotic proteins. Targets have been noted with (+), (-) or (0). (+) for
where the two methods are in agreement, (-) for disagreement and (0) for when there is no
total expression. Constructs with no total expression have been excluded from the statistics.
We have shown that the two methods are in 84% agreement with a fairly
good correlation between expression levels. We have also shown that the
CoFi blot is reproducible by re-screening clones in quadruplicates.
Differences between the two methods can be due to the different metabolic
states for a bacteria growing in a liquid culture as opposed to growing on a
solid support. Another factor that could explain the deviances is the affinity
of the filter for the proteins. Even though the filter is a low-protein binding

31
filter, some proteins might still stick and therefore the CoFi blot would not
work as a detection method for these particular proteins.
In summary, we have developed a colony expression screen, called the CoFi
blot, with good reproducibility and correlation to a more traditional
expression screen. The CoFi blot can be applied to any type of protein to
which antibodies have been generated or that contain a detectable tag, such
as a His-tag. Since it utilises standard molecular biology reagents and
equipment, it is a method suitable for any laboratory. Another advantage is
that the CoFi blot only utilises a small affinity tag for detection and not a
large fusion protein and therefore there is little risk that the tag will influence
the solubility/folding of the target protein. In addition, colonies containing
constructs that yield soluble protein, can be directly picked from the master
plate and be subjected to scale up experiments. One major advantage is that
the CoFi blot detects solubility after lysis and separation of cell debris,
meaning that the target protein has survived two additional steps required for
scale-up purification and is therefore, potentially, a better indicator of a
useful protein as compared to other methods where solubility is detected in
the cell before lysis. The CoFi blot also carries the same advantage of other
colony-based screens; the ability to screen thousands of colonies in a single
experiment.
Library methods
A successful strategy to make a protein soluble is by making mutations,
amino acid substitutions and deletions that could favour protein expression
and/or folding (115, 125). These types of alterations could be made in a
focused manner like changing the design of the expression construct based
on domain predictions (generating only a limited number of variants), or by
randomly generating thousands of variants (by error prone PCR, DNA
shuffling, truncations etc) creating a library. However, beneficial mutations
can be quite rare and one could potentially end up with a needle in a
haystack scenario. A colony screen is therefore an efficient mean to lift the
heavy burden of screening overexpression of such a library with traditional
methods.

32
As already mentioned, compared to prokaryotic proteins eukaryotic are often
larger and consist of more domains connected by flexible linker regions. As
discussed, a very common approach to increase a protein’s solubility is also
to change the expression construct by predicting domains. Domains are
predicted by computer programs, and expression constructs close to
predicted domain borders are cloned and tried for soluble expression. Even
though this approach has shown to be very successful, domain predictions
based on computer programs are not always accurate.
Domain borders can also be determined experimentally by limited
proteolysis coupled to MS, but this approach poses a kind of catch-22
situation since it relies on soluble expression from start. We wanted to
develop a strategy with which we could obtain or enhance soluble protein
expression and potentially map domain borders at the same time.
Deletion libraries screened with the CoFi blot (paper II)
We decided to produce deletion libraries of 21 human proteins that
previously, as determined in paper I, had been insoluble or did not express
protein at all to in an effort render them soluble and at the same time
investigate if we could experimentally map domain borders. Deletion
libraries were generated at DNA-level from the 5’ end of the target gene by a
method described by Henikoff et al (126) and is now available as a kit called
Erase-a-Base® (Promega). The target gene has to be cloned into a plasmid,
which is linearised and incubated with exonuclease III that removes
nucleotides from one strand of the DNA at a constant rate. By removing
timed aliquots a deletion library spanning the entire length of the gene can
be created.
In order for this method to work, two restriction sites have to be introduced
in front of the target gene to ensure unidirectional deletion. One to create a
5’-overhang that is susceptible to exonuclease III digestion and one that
generates a protecting 3’-overhang at the opposite end of the gene. The
remaining single strand is removed by S1 nuclease and the ends are flushed
by Klenow polymerase. The linear blunt-ended fragments are then re-ligated
and transformed into a cloning strain (Figure 9). Colonies are grown and
harvested and the library is extracted and transformed into an expression
33
strain. The library inside the expression strain is then screened for soluble
expression with the CoFi blot method.
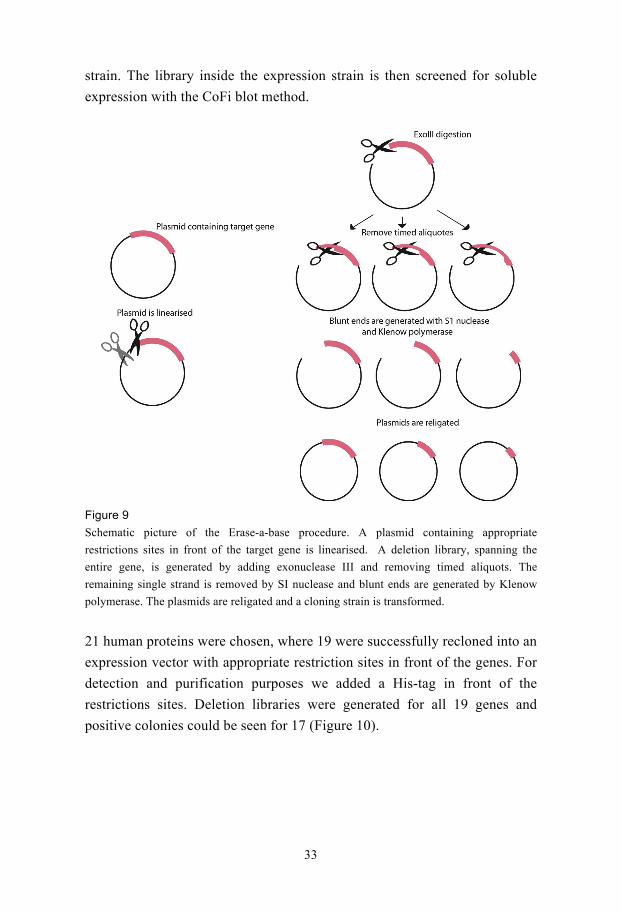
Figure 9
Schematic picture of the Erase-a-base procedure. A plasmid containing appropriate
restrictions sites in front of the target gene is linearised. A deletion library, spanning the
entire gene, is generated by adding exonuclease III and removing timed aliquots. The
remaining single strand is removed by SI nuclease and blunt ends are generated by Klenow
polymerase. The plasmids are religated and a cloning strain is transformed.
21 human proteins were chosen, where 19 were successfully recloned into an
expression vector with appropriate restriction sites in front of the genes. For
detection and purification purposes we added a His-tag in front of the
restrictions sites. Deletion libraries were generated for all 19 genes and
positive colonies could be seen for 17 (Figure 10).

34
Figure 11
SDS-PAGE of constructs
identified with the CoFi blot
method a) The best expressing
constructs were purified by small
scale IMAC and the eluate was
run on SDS-PAGE. b) 6 different
constructs from two proteins
showing the difference in
construct length and their level
Figure 10
CoFi blots of 6 targets. Positive colonies can be seen as black spots on a, b, c, d, and f and
negative colonies are seen as very faint grey spots. Since this is a deletion library from the 5’-
end, only one third of the colonies can in theory be positive. e) Example of a completely
negative CoFi blot. Positive colonies were picked for analysis and identification purposes.
From each target, 24 positive colonies were picked and used to inoculate
small liquid cultures that were induced for expression. The soluble fraction
was purified by IMAC and the eluate was analysed as Dot blots. At this
stage 14 targets gave soluble expression and out of these, 11 could be seen
as a band on an SDS-PAGE gel (Figure 11). The remaining three proteins
with no soluble protein detected were either extracellular or membrane
associated.
35
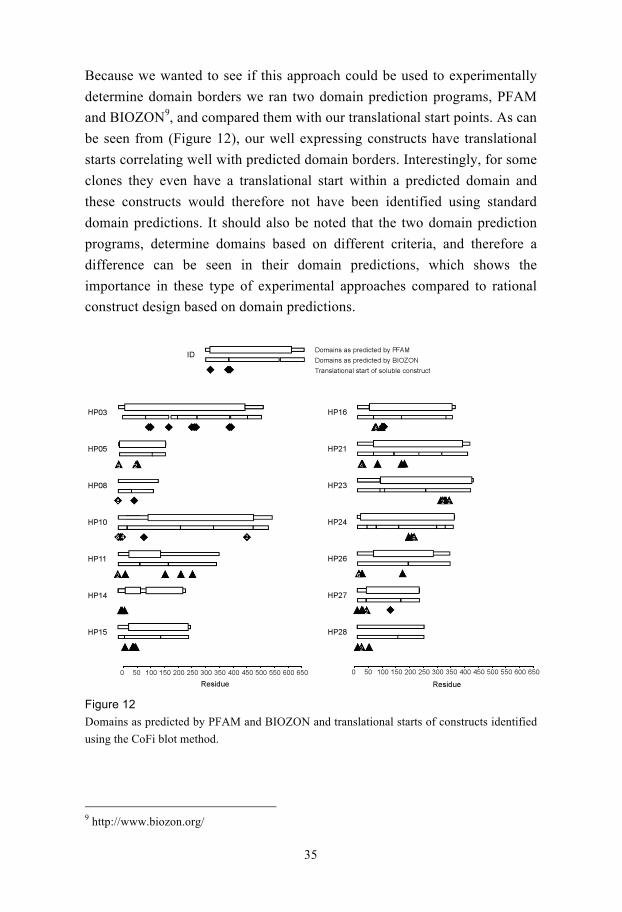
Because we wanted to see if this approach could be used to experimentally
determine domain borders we ran two domain prediction programs, PFAM
and BIOZON9, and compared them with our translational start points. As can
be seen from (Figure 12), our well expressing constructs have translational
starts correlating well with predicted domain borders. Interestingly, for some
clones they even have a translational start within a predicted domain and
these constructs would therefore not have been identified using standard
domain predictions. It should also be noted that the two domain prediction
programs, determine domains based on different criteria, and therefore a
difference can be seen in their domain predictions, which shows the
importance in these type of experimental approaches compared to rational
construct design based on domain predictions.
Figure 12
Domains as predicted by PFAM and BIOZON and translational starts of constructs identified
using the CoFi blot method.
9 http://www.biozon.org/

36
Another very intriguing observation is that for most targets there are clones
that express soluble protein that is full length or close to full length. This
could be the result of favourable changes in these regions that affect the
transcription, translation and/or folding. This would be in concurrence with a
report that regions downstream of the initiation codon can act as
translational enhancers (127).
From the initial set used in paper I, where only 30% of the mammalian
proteins could be expressed we increased the success to 60%.
In retrospect we have developed an experimental method, by generating
random deletions to increase soluble expression and at the same time define
domain borders.
The CoFi blot is routinely used in our lab and has now also been adapted to
allow the screening of recombinant overexpression of integral membrane
proteins. It was verified by screening random mutation libraries of nine
integral membrane proteins (128) and has now been used to screen a very
large part of all human membrane proteins (Unpublished results).
Expression screening complete genomes
The main objective of an expression screen is to slim down the number of
samples from a larger set. It could be applied to a library of different variants
of the same gene, as earlier described, or a collection of genes representing
for example a protein family, a pathway or a whole genome. Particularly the
handling of an entire genome and the cloning into an expression vector could
become quite troublesome, and new strategies are needed to handle very
large gene collections for expression studies. A good start point, for such
studies are Gateway® adapted entry clones allowing for the easy subcloning
of a gene into an expression vector through recombination (Figure 13).
Today several ORFeomes (a collection of all ORFs from one
organism/genome) for a number of genomes are available as Gateway clones
(110, 129-132).

37
Figure 13
Schematic picture of Gateway® cloning from Invitrogen. A PCR product is generated flanked
by specific recombination sites that are complementary with a vector, pENTR. The PCR
product is moved through recombination into the pENTR creating an Entry clone with new
recombination sites. The Entry clone is incubated with an expression vector containing
recombination sites complementary to the new sites. An expression clone is created.
As earlier mentioned a genome wide expression screen was performed on
the C. elegans ORFeome v 1.1 with very low success rates. This screen was
done in the more traditional 96-well format. Even though all ORFs were
available as Gateway clones and some steps were automated, this work was
probably very labour intense. Another attempt at handling and screening a
part of the C .elegans ORFeome was reported by Gillette et al (133). This
strategy named POET (pooled ORFeome expression technology) was based
on pooling 688 C. elegans ORFs and in a single recombination reaction
transfer them into an expression vector with a His-tag. An expression strain
was subsequently transformed with this pooled ORFeome library. The
transformation reaction was then used to inoculate a larger volume to make a
large expression culture. This culture was induced for expression, harvested
and lysed. To separate soluble protein from insoluble, the lysate was purified
by IMAC. To analyse expression and separate the different overexpressed
proteins from each other, the eluate was run on a 2D-gel. Intense and deviant
spots were picked for MS analysis. 165 spots were picked and 50 were
identified as C. elegans proteins by MS. Out of these 50 proteins, 12 were

38
chosen for small-scale expression and 6 out of these for large-scale
expression.
This method acts as a rough sieve to identify strong expressers in a large
gene collection. It also makes the handling, from cloning to expression
screening simpler. However, individual DNA measurements and the pooling
and subpooling of equal amounts must have been tiresome. In addition, this
method might not be readily used in labs with more modest equipment,
referring to the access of 2D-gel electrophoresis or MS equipment. POET is
also haunted by some limiting factors, such as the step of converting a
transformation reaction into a large culture and the iso-electric focusing
range on the gel. In a culture containing several variants of a bacterial strain
there is always a risk that one or a few variants take over the entire culture
and several targets might be lost in this step. The iso-electric range of pH 4-7
will result in that only proteins with a pI within this range will be separated,
excluding a vast amount of potentially overexpressed proteins. In the end,
anything identified must be fished out from the original collection and
recloned for any downstream processes.
Structural genomics and human pathogens
In the beginning of this thesis, a general introduction was given to human
pathogens in the form of viruses. Although recombinant protein and protein
structures from viruses could aid in the development of vaccines and drugs,
relatively few protein structures of viral proteins have been solved10.
Especially from SG initiatives submission of viral protein structures in PDB
have been modest (53). This could partly be explained by that the focus,
within these initiatives, is on other types of proteins. In two articles (50, 51),
where the focus or part of the focus was on viral proteins, poor success rates
were reported. The main problem was low expression and low solubility in
E. coli. Success was predominantly achieved when each target was treated
individually or expressed in insect cells.
10 http://www.rcsb.org/pdb/home/home.do

39
The daily SCOOP (Paper IV)
We wanted to use the CoFi blot as a homing method to, within a genome,
identify easily expressed targets and targets in need of special care. We
designed a general strategy and applied it to the ORFeome of KSHV. KSHV
consist of approximately 90 ORFs, which were supplied to us as 113 entry
clones (132). ORFs coding for integral membrane proteins were cloned as
full-length constructs, as well as into separate non-transmembrane domains.
In similarity to POET, our approach also relied on pooling of Gateway®
adapted entry clones and a single recombination reaction. However, instead
of tedious DNA measurements we grew separate cultures of bacteria
containing the individual entry clones over night. OD600 measurements
showed similar density for all of them and equal amounts were pooled.
Plasmids from this pool were extracted and ORFs were moved, in one
recombination reaction, into an expression vector with an N-terminal His-
tag. A cloning strain was transformed with the reaction and plated. The
library was harvested and an expression strain was transformed and plated.
4,000 colonies were subjected to the CoFi blot procedure. Positive colonies
were analysed based on their purifying propensity with small-scale
purification. Identification was done through regular Sanger sequencing. 23
unique constructs could be identified as expressing soluble protein with this
approach. 11 out of these 23 constructs were randomly chosen for large-
scale expression and purification.
In order to benchmark this method we compared it to the more traditional
handling and screening; individual subcloning, expression screening and
subsequent affinity purification all in a 96-well format. With the more
traditional setup 25 constructs could be identified as soluble expressers.
When comparing our new approach, now called SCOOP (screening colonies
of ORFeome pools), with the more traditional approach 23 and 25
constructs, respectively, were identified as yielding soluble protein in E. coli.
20 out of these 23 and 25 were the same for the two experiments. To
elucidate why 5 constructs were missed with SCOOP, their expression at the
colony level was investigated using the CoFi blot. Soluble expression at the
colony level could not be detected for 3/5. The additional 3 constructs that
were only identified with the CoFi blot were grown and induced for
expression in liquid cultures. The soluble fraction was affinity purified and

40
the eluate was run on SDS-PAGE gels and very faint bands could be seen on
the gel, indicating that these constructs were expressed at low levels.
It has long been known that a bacterium growing on solid support in a
colony behaves differently from bacteria growing in a liquid culture (134,
135). In this context we therefore propose that either the metabolic state of
the colony, compared to bacteria growing in liquid, does not allow for
soluble expression of some ORFs and vice versa, or that the CoFi blot
simply does not work for these proteins. In addition, when making pools of
this kind, some targets are inevitable lost along the way, perhaps accounting
for the additional two not identified by the CoFi blot. Success rates will
correlate to the overall coverage/normalisation of the library and although
we find several constructs more often than others, we must conclude that our
library is well dispersed since we cover almost all soluble expressers.
Although 113 constructs were screened by SCOOP, sequencing at a very late
stage revealed empty vectors, mutations leading to frame shifts and early
stop codons for 31 constructs. A TMHMM11 search of the remaining
constructs predicted 8 ORFs to code for transmembrane proteins with at
least 2 transmembrane regions. Of the 113 constructs used in the original
experiment only 74 could be expected to produce proteins of the expected
size.
In a matter of days we have been able to screen the ORFeome from a human
pathogen for soluble recombinant expression in E. coli using SCOOP
(Figure 14). Once again we have shown the general applicability and
advantages of the CoFi blot method, by which constructs identified can
directly be moved further down the SG pipeline. A normalised library is
created through pooling of bacterial cultures instead of at DNA-level, thus
avoiding tedious DNA measurements. Subcloning into an expression vector
is done in one recombination reaction and the subsequent screening is done
with the CoFi blot. Identification of positive constructs is done by
sequencing. From this very small set of targets, soluble expression in one
strain and at one temperature was roughly about 30%, which was also
verified by SCOOP. This number is a bit lower than the SG statistics of
37%, although this corresponds to proteins from all types of viruses and
11 http://www.cbs.dtu.dk/services/TMHMM-2.0/

41
there is no information about full-length expression nor expression
conditions (53).
Figure 14
Flow scheme of SCOOP.
SCOOP has also been applied to deletion libraries of the KSHV ORFeome.
Pools were generated based on restriction enzyme compatibility for the
Erase-a-base® protocol. Deletion libraries were generated and expression
was screened using the CoFi blot. Initial results show that several other
targets with widely different functions can be made to express in E. coli with
this approach. We have also screened the overexpression of all integral
membrane proteins from KSHV using the membrane protein CoFi-blot.
SCOOP and other herpesvirus ORFeomes
As mentioned, structure determination of viral proteins has been suffering, in
part, due to poor soluble expression in E. coli and therefore these types of
proteins have been considered as “difficult”. As a result only 525 unique
virus protein structures have been submitted to the PDB, a number that
includes proteins from all types of viruses, from SARS and HIV to

42
herpesviruses. Out of these 525 structures only 31 have been submitted by
SG initiatives, worldwide. SCOOP was applied to four additional
ORFeomes from herpesviruses, HSV-1, VZV, EBV and mCMV available as
Gateway® adapted entry clones. By using one expression strain and one
temperature we were able to identify 75 unique constructs as expressing
soluble protein. These constructs corresponded to proteins of various
functions like DNA metabolism, immune evasion and tegument proteins as
well as proteins of unknown function. For 65 there was no protein structure
in PDB. Large-scale expression, purification and crystallisation trials have
been attempted for all 65. So far 3 protein structures have been solved.
Moving further down the pipeline
One area that has been intensely studied for human herpesviruses is DNA
metabolism due to the high degree of conservation among the proteins
involved and the importance of DNA synthesis (5, 136). In fact the most
effective drug, available on the market today, targets the viral infection at the
level of DNA synthesis and therefore proteins involved in DNA metabolism
would be effective as antiviral drug targets. Acyclovir is an acyclic
nucleoside analogue used to treat alpha herpesvirus infections, which targets
the viral thymidine kinase that phosphorylates acyclovir to acyclovir-
monophosphate. Downstream phosphorylations, by cellular enzymes,
convert this monophosphate to a triphosphate that when incorporated in to
the growing DNA chain of the virus, by the viral DNA polymerase, causes
chain termination. Acyclovir and its related compounds represent a success
story in the general treatment of HSV, VZV and CMV infections (25, 137).
It can be administered as a lotion on symptomatic tissue, orally or
intravenously depending on the severity of infection. However, resistant
strains of these viruses have been identified, especially in HIV-infected
persons and almost all significant resistance has been seen in
immunocompromised patients (25, 137, 138). For these patients more
aggressive antiviral remedies have to be used.
In short, all herpesviruses encode a core set of six DNA synthesis enzymes
(Table 4); a DNA polymerase, a DNA polymerase processivity subunit,
three helicase primase subunits and a single strand DNA binding protein

43
(aka ICP8). In addition to their importance in DNA synthesis they also
mediate homologous recombination during viral replication (5, 139).
A second set of proteins with links to DNA synthesis, more or less
conserved in the herpesvirus family includes the thymidine kinase, alkaline
endo-exonuclease, ribonucleotide reductase, uracil N-glycosylase and a
dUTPase (Table 4). These proteins are considered non-essential for
replication in cultured cells but several appear essential for “normal”
behaviour of the virus in animal models (5).
Table 4
The proteins involved in DNA replication and synthesis, their abbreviations and their gene
names in all the human herpesviruses. White rows are proteins directly part of DNA synthesis
and grey rows are proteins with links to DNA synthesis.
Enzyme HSV-1/-2 VZV HCMV HHV6/7 EBV KSHV
DNA polymerase,
POL
UL30 ORF28 UL54 U38 BALF5 ORF9
DNA polymerase
processivity subunit,
PPS
UL42 ORF16
UL44 U27 BMRF1 ORF59
Helicase-primase
ATPase subunit, HP1
UL5 ORF55 UL105 U77 BBLF4 ORF44
Helicase-primase
RNA pol subunit B,
HP2
UL52 ORF6 UL70 U43 BSLF1 ORF56
Helicase-primase
subunit C, HP3
UL8 ORF52 UL102 U74 BBLF2/3 ORF40/41
Single strand DNA
binding protein, ICP8
UL29 ORF29 UL57 U41 BALF2 ORF6
Thymidine kinase, TK UL23 ORF36 - - BXLF1 ORF21
Alkaline exonuclease,
AE
UL12 ORF48 UL98 U70 BGLF5 ORf37
Deoxyuridine
triphosphate, dUTPase
UL50 ORF8 UL72 U45 BLLF3 ORF54
Uracil-DNA
glycosidase, UNG
UL2 ORF59 UL114 U81 BKRF3 ORF46
Ribonucleotide
reductase, large
subunit, R1
UL39 ORF19 UL45 U28 BORF2 ORF61
Ribonucleotide
reductase, small
subunit, R2
UL40 ORF18 - - BaRF1 ORF60

44
The alkaline endo-exonuclase (AE) is conserved in all herpes viruses and is
involved in the maturation and packaging of viral DNA (140, 141). In HSV
this protein interacts with ICP8 (142). These two proteins can also work
together to promote strand exchange similar to recombination events in
bacteriophage (143). Recently it was shown that the AE from both EBV
and KSHV is involved in host shutoff at the mRNA level, an activity that
has not been shown for the other HHV AEs (140, 144, 145).
The well-known allosterically regulated enzyme ribonucleotide reductase
(RNR) is solely responsible for the de novo synthesis of all four
deoxyribonucleotides. RNR is a heterotetramer of two subunits, R1 and R2
(146). All HHV carries homologues to R1 and R2 except beta herpesviruses,
which lack the R2 (5). Interestingly, the RNR of HSV is not negatively
regulated by high TTP pools as their cellular counterparts and seems
necessary for viral growth in “resting” cells (147, 148). There are reports
that this viral enzyme has an accessory function to protect HSV-1 infected
cells against cytokine induced apoptosis (149).
Uracil N-glycosylase functions as a repair enzyme that cleaves mutated U
residues from the DNA backbone (150, 151).
The virally encoded dUTPase breaks down dUTP, thus preventing its
incorporation into viral DNA(152, 153).
More understanding of proteins involved in DNA synthesis and replication,
their mode of action and interactions, amongst themselves but also with
cellular proteins, could aid in the development of new drugs targeting this
important step of the viral life cycle.
The fundamental mission of all viruses is to replicate, spread and persist in
the host environment to which they have become adapted. Viruses differ
with respect to the mechanisms by which they accomplish their objectives.
The difference is reflected not only in the basic mechanisms of viral entry,
synthesis of proteins, nucleic acid synthesis, virion assembly and egress but
also with respect to the basic strategies by which they counteract the
enormous recourses of the host cell and of the multicellular organism aiming
at total viral extinction. In principal, as a virus, you want no reports to the
immune system or to the apoptotic machinery of your presence and you will
take necessary precautions to achieve this. However, the immune system and
the apoptotic machinery are not so easily tricked with many back up systems

45
to fend off and terminate any invader. The infected cell, must in order to
pass immune system control, show several signs of being “normal”,
therefore certain mechanisms need to be upregulated while others need to be
blocked. Anything remotely suspicious will result in an attack by the
immune system. Immune evasion in the host is a major feature of
herpesviruses. To persist and replicate in a host cell the virus has to launch
several anti-host functions. For herpesviruses these functions tend to fall,
more or less, within four categories, i) selectively block synthesis of new
proteins, ii) block the function of preexisting host proteins activated after
infection, iii) selectively degrade cellular proteins and iv) block signalling to
the host immune system indicating that the cell is infected.
Host shutoff in HHV
Viruses i.e. polio, influenza, SARS-CoV etc frequently induce host shutoff
to avoid competition from host cell transcripts during the mass-production of
their own proteins and to prevent expression of factors involved in
stimulating an immune response (154).
One way to take over a cell is to down regulate cellular protein synthesis by
degrading RNA (155, 156). Not only are host proteins not synthesised due to
low mRNA levels but pre-existing polyribosomes are also degraded and
splicing is impaired. Host shutoff is a consequence of infection of alpha
herpesviruses and gamma herpesviruses and occurs a few hours after
infection (5, 157). One of the consequences of host shutoff is a block in the
synthesis of HLA class I and II molecules, revealed by reduced levels of
these antigen presenting complexes at the surface of cells in the EBV lytic
phase. This effect could lead to escape of T-cell recognition and elimination
(157).
In alpha herpesviruses, mRNA degradation is a result of the tegument
protein vhs (virion host shutoff) protein encoded by UL41 (158). However,
although vhs functions as an RNase it cannot succeed in complete host
shutoff by itself and collaborates with ICP27, inhibiting splicing of mRNA
(5, 159). Intriguingly, it appears that some mRNAs are more resistant to
degradation than others (160). Vhs homologues are found in all alpha
herpesviruses but not in beta-or gamma herpesviruses (157).

46
Host shutoff at the mRNA level has also been observed in gamma
herpesviruses. However, this occurrence has been ascribed to another
protein, the already mentioned alkaline exonucleases (AEs) of these viruses
(140, 144, 145). A screen of KSHV genes for ability of their corresponding
gene products to, in vivo, diminish expression of a GFP reporter protein lead
to the identification of the gene product of ORF37 as a host shutoff factor.
Since this protein functions both as an exo- and endonuclease as well as a
shutoff protein it was named SOX (shutoff and exonuclease) (140).
SOX
SOX is conserved in all herpesviruses as an alkaline exonuclease involved in
DNA maturation and packaging, but it is only the gamma herpesvirus
homologues that display a shutoff activity in vivo. SOX expression initiates
8 hours after lytic reactivation, which precisely coincides with the beginning
of host shutoff (161). Unlike vhs, SOX is not a tegument protein (162) and
the co-expression with GFP resulted in loss of GFP fluorescence, which was
the result of the GFP mRNA degradation. This discovery was further
verified by siRNA knockdowns (140, 144).
In vitro studies of the herpesviral AEs exhibit a 5’-3’ and a 3’-5’ (10-fold
weaker) dsDNA exonuclease activity and an endonuclease activity (163)
They work best at an alkaline pH and have an absolute requirement for Mg2+
(although Mn2+ can work, equally well for HSV-1/2 AE at some
concentrations) (164-166). For the HSV null mutant in gene UL12, growth is
severely restricted but is still able to replicate and encapsidate considerable
amounts of viral DNA. Capsids, as a result from this mutant, are impaired in
the egress from the nucleus to the cytoplasm (167). In addition the HSV AE,
UL12, exhibits strand exchange activity, in vitro, and acts together with
ICP8 as a recombinase (143, 168). In contrast to UL12, which only localises
to the nucleus, SOX localises both to the nucleus and cytoplasm (144).
Complex sequence alignments showed that all herpesviral AEs belong to the
PD-(D/E)XK superfamily, which comprises endonucleases (i.e. EcoRI,
EcoRV BamHI etc), nicking enzymes and one exonuclease (169-172).
Typically, sequence similarity between these proteins is so low that it is only
upon structure determination that they are assigned to this superfamily.
Although low sequence similarity, it has been proposed that all these

47
proteins share a common core of a 4-5-stranded -sheet that brings together
the charged residues in the conserved motif PDX10-30(D/E)XK that is
involved in Mg2+ binding and catalysis (169).
When aligning all herpesviral AEs, seven conserved linear motifs can be
identified (173). Both random and focused mutations identified single and
double function mutants. Single function mutants lacked either DNase or
shutoff activity in vitro or in vivo respectively, whereas double function
mutants had neither. When linearly mapping these mutations it was revealed
that mutations that abolished shutoff activity located outside of these
conserved motifs (except one, which is not conserved) whereas double
function mutations and single function mutations affecting only DNase
activity located within the conserved linear motifs (Table 5, Figure 15)
(144).
Table 5
Summary of locations of mutations that affect SOX activity (140, 144)
Mutation DNase activity Shutoff activity Conserved motif Conserved
residue in HHV
AEs
T24I Yes No No No
A61T Yes No No No
Q129H No Yes I Yes
P176S Yes No No No
G217A No No II Yes
D221E No No II Yes
V369I Yes No VI No
D474N Yes No No No
Y477Stop Yes No No No
Figure 15
Schematic figure of the seven (I-VII) conserved motifs of SOX and the mutations affecting
the activity. Black box indicates a double function mutation. Grey box indicates single
function mutations abolishing DNase activity and white boxes indicate single function
mutations abolishing shutoff activity.

48
As a result it was proposed that SOX is a bifunctional protein that exhibits
DNase activity in the nucleus and shutoff activity in the cytoplasm, inducing
host shutoff. Because mutations that abolished shutoff activity located
outside of conserved linear motifs, it was proposed that this activity had
been acquired later in evolution (140). However, since the shutoff activity
only could be shown in vivo, it was suggested that SOX might need an
additional cellular or viral-binding partner to induce this action.
The EBV homologue, BGLF5, has been shown to possess the same features
as SOX by similar characterisations (145, 166, 174, 175). With an amino
acid sequence identity of 42% it has now been shown that both these
proteins down regulate expression of HLA molecules, which impairs the
recognition of infected cells, by T-cells and that this is not a feature of the
other herpesviral AEs (175).
Structural studies of the SOX protein from KSHV (Paper V)
One of the proteins identified as positive with SCOOP was the KSHV SOX
protein. Catalytically active SOX, both native and selenomethionine
labelled, was expressed with an N-terminal His-tag in E. coli BL21[DE3]
and purified to homogeneity with our generic two-step purification scheme
by IMAC and gel filtration. Well diffracting crystals were obtained through
vapour diffusion. Native crystals diffracted to 1.85 Å and phases were
obtained by SAD.
SOX consists of 487 residues with a molecular mass of 55 kDa. It consists of
two domains giving it a rough bowl shape. The two domains, the N-terminal
domain and the core domain are connected by a partly unmodelled region
called the bridge. The N-terminal domain consists of -helices and the core
domain consist of a 5 stranded -sheet flanked by several helices in
similarity to the rest of the members in the PD(D/E)-XK superfamily. The
two domains form a crevice where the bottom is made up of a U-shaped
loop, called the U-loop, stretching from core domain into the N-terminal
domain (Figure 16). This crevice is positively charged, consists of highly
conserved amino acids in all herpesviral AEs as well as the residues in the
already mentioned conserved sequence motif of the PD(D/E)-XK
superfamily.

49
U-loop
Figure 16
Overall model of SOX. The N-terminal domain in light and the core domain in dark. Three sulphates can be seen in the density and have therefore been modelled. Density for what we believe is a magnesium ion could be seen in the cleft and has therefore been modelled. The bridge connecting the N-terminal domain with the core domain is indicated by a dotted line
Densities can be seen for what we believe are three sulphates, where two are
coordinated by totally or semi-conserved residues proposed to be binding the
site for the substrate backbone, and one is involved in crystal contacts. A
magnesium ion has also been modelled in the crevice. This magnesium ion
is partly coordinated by residues in the PD(D/E)XK motif. A DALI search
revealed a surprisingly similar fold to the only known exonuclease within
the superfamily, the -exonuclease (Figure 17) (176).
Figure 17 Superposition of a monomer from the -exonuclease (dark) on SOX (light). -exonuclease is also a two-domain protein. The two domains in -exonuclease are connected by a completely modelled bridge. The partly modelled bridge in SOX is an ordered -helix and the U-loop has small -sheet in the -exonuclease. The bridge in -exonuclease is completely modelled and a sulphate in SOX is bound in the same location as a phosphate in -exonuclease. In addition, the magnesium ion in SOX is located in the same position as a manganese ion in -exonuclease (not shown).

50
Substrate binding
In a superposition of SOX and the -exonuclease, we can see that one of the
sulphates is located and coordinated in the same way as a phosphate in the -
exonuclease structure. The magnesium ion in SOX is also located and
coordinated as a manganese ion in the -exonuclease structure as well as
other divalent ions in other proteins belonging to the same superfamily.
Since this area in SOX is highly conserved and both the tertiary structure as
well as the aligning of ligands with the -exonuclease, we propose that this
is the site for substrate binding. However, although great similarities are
observed in both the overall fold of these two enzymes, dissimilarities can be
seen. For instance, SOX is larger than -exonuclease and where SOX has
partly unstructured and unmodelled regions (such as the bridge and the U-
loop) -exonuclease has an ordered secondary structure. We believe that the
-exonuclease structure represents what for SOX would be a substrate bound
state, where conformational changes lead to ordered secondary structures in
the bridge and the U-loop. Another major difference between these two
proteins is their quaternary structure, where the -exonuclease is active as a
trimer (176), which could explain its processivity. No prior reports or our
results indicate that SOX functions as anything but a monomer (166).
The second modelled sulphate in SOX, located at the beginning of the
crevice, is coordinated by totally or semi-conserved residues, which would
indicate that this is a true substrate backbone binding site. To elucidate
whether this sulphate could represent part of the substrate backbone, we
modelled a 4bp oligo by aligning the phosphates in the backbone to the two
sulphates in the model. As can be seen (Figure 18) this oligo fits nicely in
this highly conserved crevice and aligns very well with our sulphates.
The second phosphate in the oligo backbone places itself very close to the
modelled magnesium ion. It is possible that the phosphodiester bond is
cleaved by a nucleophilic attack by an activated water molecule close to the
magnesium, which stabilises negative charges in this area, cleaving off one
nucleotide at a time. Since the substrate of SOX can be both double stranded
and single stranded SOX must have a type of unwinding activity. We
therefore believe that SOX separates the strands of the dsDNA and the single
stranded substrate destined for digestion is guided into the active site by the
bridge and is stabilised by binding to conserved and partly conserved

51
aromatic residues in and around the crevice. The backbone is cleaved and
the free nucleotide is released on the opposite side of the “tunnel” created
under the bridge. The remaining free single strand of the substrate is directed
away from the protein surface.
Figure 18
Conserved residues in SOX as determined by sequence alignments with all other HHV AEs.
Totally conserved residues are depicted in red and least conserved residues are light blue. A
4bp oligo was modelled in the crevice based on the location of the two sulphates.
SOX as an RNase?
An intriguing question is whether SOX can mediate host shut off through an
intrinsic RNase activity. As already mentioned, functional characterisations
of SOX in vivo, show that it affects the total mRNA content of the host cell
and although host shutoff is observed in alpha herpesviruses, the other
herpesviral AE’s cannot induce this type of degradation.
Figure 19
Mapping mutations that affect the activity of SOX. Red deletes overall activity. Orange
deletes DNase activity and pink the shutoff activity.

52
In addition, single function mutations abolishing the shutoff activity of SOX
locate outside of conserved linear sequence motifs found in all herpesviral
AE’s. It has therefore been proposed that the host shutoff trait of SOX, and
BGLF5 from EBV, has been acquired later in evolution. It has also been
proposed that SOX might need an additional cellular binding partner or that
it does not have any intrinsic RNase activity and only activates some sort of
RNA decay pathway (140, 144).
With the structure in hand we can now map known mutations, which affect
the shutoff function. As can be seen (Figure 19), mutations that abolish
shutoff function are spread all over the protein, with predominance in the N-
terminal domain. The mutations locate both on the exposed surface and in
buried regions although they are mostly found close to the surface.
Since SOX has a nuclease type active site, with an extended pocket that
appear well suited for binding nucleic acids, it could be argued that both the
DNase and a potential shutoff RNase activity are carried out in the same
active site, since no other obvious binding or active site can be seen in the
structure. As support for this theory, both double-function mutations
(G217A, D221E), located in the U-loop, as well as one shutoff mutation
(P176S), located in the end of the unmodelled bridge are either directly
involved in metal binding or reside very close to the substrate binding and
active site. As in most mutational studies carried out in vivo, it cannot be
ruled out that these mutations only affect the folding and stability of SOX,
cancelling out activity all together. Even so, these mutations emphasize the
importance of this region. However, since RNase activity cannot be detected
for SOX in vitro, additional proteins are likely to be required enabling the
nuclease activity for mRNA degradation. Possibly this can be viral or host
factors mediating the interaction of SOX with conserved elements of
mRNA.
What would support the hypothesis of an additional cellular binding partner
for shutoff activity is the predominance of mutations affecting shutoff in the
N-terminal domain, especially in parts that are not present in the -
exonuclease. Perhaps this domain, which also carries a more overall positive
charge than the rest of the protein, will play a role in cellular protein binding.
In this scenario, whether or not it is SOX that carries out the RNA
degradation or the potential binding partner cannot be determined presently.

53
Nucleotide synthesis
For every living cell, cell division is a highly regulated process and even the
slightest stray could lead to serious consequences, where cancer is a classic
sign. In a multicellular organism, there has to be a strict balance between
cells that rest, divide and die. The cell cycle is a series of events that lead to
cell division and is under strict control of key enzymes called cyclins and
cyclin dependent kinases. The cell cycle is roughly divided into four phases;
G1-, S-, G2- and M-phase. During G1, cells synthesise RNA and proteins
that are needed for S-phase, which is characterised by DNA synthesis and
chromosome replication. In the G2-phase, proteins needed for cell division
are synthesised, which is followed by actual cell division during the M-
phase. An additional phase exists for cells, called G0, this phase is entered by
cells in G1 and is the state of cells that are non-dividing or are fully
differentiated. The actual time period a cell is present in G0 varies with cell
type some, i.e. neurons, never leave. The passage from one phase to another
is under rigorous regulation and has certain check points that need to be
cleared in order for safe cell division. Deoxyribonucleotides are the building
blocks of DNA and the synthesis of deoxyribonucleotides peaks during S-
phase. The enzyme responsible for the production of deoxyribonucleotides is
the extremely well studied and strictly regulated enzyme ribonucleotide
reductase (146). However, this enzyme is also important for the synthesis of
deoxyribonucleotides in resting cells, as a response to DNA damage. Both
alpha and gamma herpesviruses encode their own RNR while beta
herpesviruses only encode one of the RNR subunits (R1) (5).
Ribonucleotide reductase
RNR is an enzyme that can be found in all kingdoms of life as well as in
viruses. As already mentioned, RNR performs the chemically difficult
reduction of all four ribonucleotides to their corresponding
deoxyribonucleotides in a reaction involving a free radical (177). The
allosterically regulated RNR is the only enzyme that can perform this
reaction and it therefore controls the entire dNTP pool in the cell.

54
Fig 20
The reaction catalysed by RNR.
RNR’s have been divided based on their radical cofactors into three classes,
I, II and III, where class I further is divided into subclasses Ia and Ib. A
strictly balanced supply of deoxyribonucleotides is essential for accurate
DNA replication and repair (146). RNR could therefore be a potential drug
target for the treatment of different types of cancer and viral infections (178-
182).
RNR is a heterotetramer consisting of two homodimers, R1 and R2 and has
an absolute requirement for iron in the form of a di-iron site located in the
R2 subunit. A free radical is generated, by reductive cleavage of molecular
oxygen at the di-iron site of the R2 subunit, which is transported to the R1
subunit to activate the nucleotide substrate for catalysis (177).
RNR activity is under strict transcriptional and postranscriptional control
and in replicating mammalian cells enzyme activity is regulated by control
of R2 protein stability (146, 177). However, to produce
deoxyribonucleotides as a response to DNA damage in resting cells, the
expression of another cellular R2 is induced, which lies under the control of
the transcription factor and tumour suppressor p53, hence it has been named
p53R2 (183, 184). p53R2 also appears to play a role in providing
deoxyribonucleotides for mitochondrial DNA synthesis (188)
Structural studies of the R2 subunit of the ribonucleotide
reductase from EBV and KSHV (manuscript in preparation)
The EBV genome encodes for a class I RNR R2 subunit with low amino
acid sequence identity to bacterial and eukaryotic R2s. This R2, encoded by
the BaRF1 gene was identified as positive by SCOOP of the EBV ORFeome
and is different to R2s of other organisms in that it has a glutamate (Glu61)
in the position of a metal-coordinating aspartate common to most R2s. The
structure of EBV R2 was solved in its apo, mono metal and di-metal form

55
(Figure 21) at 1.9, 1.6 and 2.0 Å respectively and their radical generating
abilities were investigated using electron paramagnetic resonance (EPR).
The monomer of the EBV R2 is an -helix bundle in similarity to other R2s,
but with several interesting differences in the region around the metal site
(Figure 22). In one of the monomers in the apo and monometal structures,
residues 62-73 of -helix B are disordered and Glu61 is either disordered
or oriented away from the metal binding site. Additionally, there is no
traceable density for residues 120-143 in 2 and D indicating that these
elements too are disordered. These stretches seem to order themselves only
when the di-ferrous metal site is fully formed, where Glu61 participates in
metal coordination. This large conformational flexibility upon iron site
formation has not previously been described for any R2 subunits. Recently,
however, disorder in this particular region is found in three R2 structures
also recently available in PDB; of H. sapiens p53R2 (2VUX) in Bacillus
halodurans (2RCC), Plasmodium vivax (2O1Z) and S. cerevisiae (2RCC).
It is likely that the extra flexibility of helix B affects the rate of iron
binding and therefore the timing of the radical generation reaction. EPR
confirms that EBV R2 is slower in generating the radical and that the radical
site is more accessible to radical scavengers. These observed deviances in
the different metal bound forms of EBV R2, become all the more interesting
when put into a wider virus-host cell context. p53R2 also displays this
disordered region. In nonproliferating cells, DNA damage induces
expression of the p53R2 and also the R1 protein. Together they form an
active RNR complex supplying the resting cell with dNTPs for DNA repair
(183, 186). Since the host cell already encodes for its own RNR it is
therefore believed that the viral RNR supplies nucleotides in a resting host
cell (187).
The mobility of these regions in both EBV R2 and p53R2 may be a relevant
adaptation to particular chemical environments in a resting cell, and may
have a functional role in modulating iron and oxygen access, respectively to
the active site.
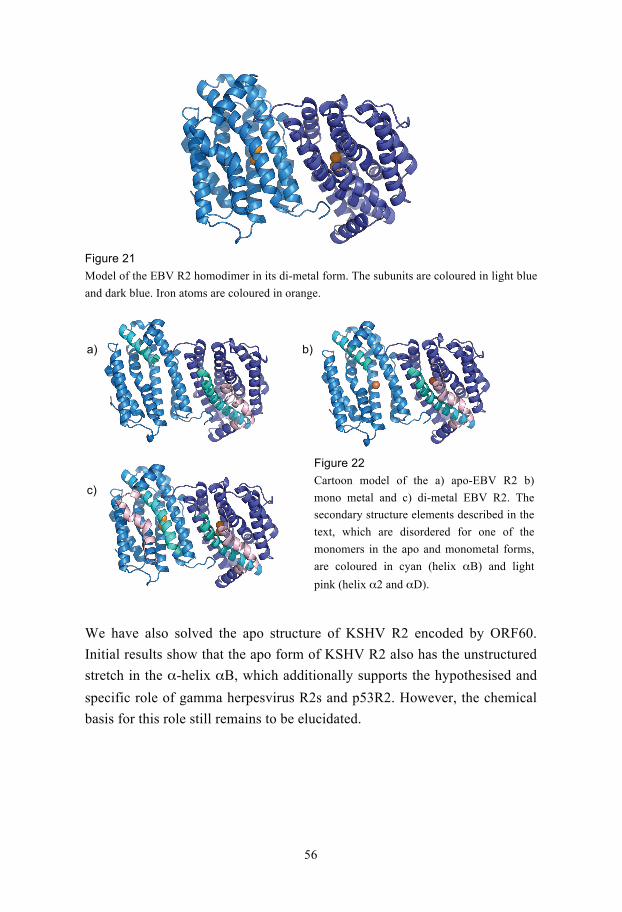
56
a) b)
c)
Figure 21
Model of the EBV R2 homodimer in its di-metal form. The subunits are coloured in light blue
and dark blue. Iron atoms are coloured in orange.
We have also solved the apo structure of KSHV R2 encoded by ORF60.
Initial results show that the apo form of KSHV R2 also has the unstructured
stretch in the -helix B, which additionally supports the hypothesised and
specific role of gamma herpesvirus R2s and p53R2. However, the chemical
basis for this role still remains to be elucidated.
Figure 22
Cartoon model of the a) apo-EBV R2 b)
mono metal and c) di-metal EBV R2. The
secondary structure elements described in the
text, which are disordered for one of the
monomers in the apo and monometal forms,
are coloured in cyan (helix B) and light
pink (helix 2 and D).

57
Future prospects
Needless to say, viral biology is an interesting field. To thoroughly
understand its evolution, pathogenesis, to develop new drugs and potentially
new vaccines and diagnostic assays etc we need to study the individual viral
proteins both functionally and structurally. Unfortunately, the recombinant
overexpression of viral proteins has so far proven to be very difficult and
therefore very few viral proteins have been structurally determined. We have
developed methods and strategies to speed up the structural genomics
pipeline and increase the throughput. Recombinant viral proteins are being
routinely pushed through our pipeline and protein structures are coming out
as a result. Three structures have so far been solved from proteins involved
in DNA metabolism, which is an area of both scientific as well as medical
interest.
However, to really comprehend a virus’ interaction with its host, structural
studies of protein complexes are necessary and this will in the future be
pursued. The CoFi blot and SCOOP are routinely used in our lab, screening
gene collections of various kinds. The CoFi blot is evolving and we are
finding new applications for it.

58
Acknowledgements When wanting to write the acknowledgements I realised that what I appreciate the most, with
all the people that have surrounded me during this journey, is their fantastic patience and their
never fading trust and support. Many people have been part and I would especially like to
thank a few of them.
Pär, för ditt förtoende. Inte bara när att du antog mig som student utan också för din
övertygelse om att mitt projekt skulle fungera. För att du lät mig åka ända till Kina. Du har
skapat en inspirerande miljö och jag har lärt mig mycket, både om forskning och mig själv.
Stefan Nordlund, för att du alltid lyssnar tålmodigt på en students predikament.
Alla på DBB, speciellt Anki, Kicki, Lotta och Ann, som hjälpt mig många gånger med
administrativa saker.
Past and present members of PN group especially: Martin A, Vickan, Susanne F, Helena,
Susanne VdB, Deb, Tove, Karl-Magnus, Agnes, Martin M, Martin Hä, Amin, Benita,
Anders T, Jessica A, Andreas, Herwig, Albert, Ken, Lina-Beth, Alberto, Marie, Pelle,
Hanna, Henrik, Anna, Florian, Elisabeth and Marina.
Monica, för alla visdomsord om livet.
Said, för ditt fantastiska tålamod och för att du tar hand om labbet!!
Ulrika, U som i ullig, L för labrador!!!! För att du är en fantastisk vän.
Audur, för sjukt roliga stunder både i och utanför labbet! Och all hjälp!!!!
Lola, for always sharing ranting experiences and listening to my ranting.
Heidi, för prat och stöd.
Martin, Stina, Pål och Emma, för upplyftande diskussioner som berör alla möjliga ämnen!
Daniel M-M, för att du alltid kan få mig att skratta.
Karin, för vänskap på labbet och utanför.
Daniel G, för din humor.
Damian, för att du tålmodigt lyssnar och hjälper mig med mina dataproblem.
Maria D, för alla leenden.
People at Beida, especially Xiao Dong, Erik, Nan Jie, Xiaoyan, Koh and Dr Gao! Xie xie.
Sunny “shi pa” and family, your generosity and curiosity overwhelm me. I am certain we
will meet again!
Mina Kina-kompisar, Anders K, Kim, Tove, Anna, Sara, Anders L, Ola och Maria!!!!
Stockholms biovetenskapliga forskarskola, med Eva och Camilla i spetsen.
Mina fosko-kompisar, speciellt Anton, Anna och Linda. Med era historier inser jag att jag
inte är själv!

59
Mia och Linda för att ni är helt utanför labblivet, men alltid är på min sida!
Jenny, för att du alltid är redo att luncha.
Crista and Jill, for your never seizing friendship although you are so far away.
Jasmine and Stephanie, for great times and really being there for me!
Familjerna Nolvi, Jansson/Umana, Tronström och Sundesson.
My entire family in Singapore, who means the world to me, see you soon!
Familjen Cornvik, för att ni alltid lyssnar på mig och mitt eviga tjatande om allt och inget.
Anna-Karin, det finns inga ord som beskriver vår vänskap. Du är världens bästa och ingen
kan ersätta dig!
Kim, världens bästa bror, som alltid ser till att ta ner mig på jorden och som alltid ställer upp
för mig.
Mamma och Pappa, för att ni helt utan tvekan står vid mig. Ni ger mig perspektiv och
försöker alltid förstå vad jag håller på med, både på labbet och med mitt liv.
Tobias, för den fantastiska person du är. För det du betytt, betyder och alltid kommer att
betyda för mig. Du gör mig komplett i alla avseenden. DOJ, DOJ!

60
References
1. Koonin, E. V., Senkevich, T. G., and Dolja, V. V. (2006) The ancient Virus World and evolution of cells, Biol Direct 1, 29.
2. Cann, A. J. (2005) Principles of Molecular Virology (Standard Edition), Fourth Edition.
3. Abo, M. E., and Sy, A. A. (1998) Rice Virus Diseases: Epidemiology and Management Strategies, J. Sust. Agri. 11, 113-134.
4. Khodosevich, K., Lebedev, Y., and Sverdlov, E. (2002) Endogenous retroviruses and human evolution, Comp. Funct. Genomics 3, 494-498.
5. Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P. S., Roizman, B., Whitley, R., and Yamanishi, K. (2007) Human
Herpesviruses, Biology, Therapy and Immunoprophylaxis, First Edition.
6. Dutta, A. (2008) Epidemiology of poliomyelitis-Options and update, Vaccine.
7. Zhu, F. X., Chong, J. M., Wu, L., and Yuan, Y. (2005) Virion proteins of Kaposi's sarcoma-associated herpesvirus, J. Virol. 79, 800-811.
8. Baldick, C. J., Jr., and Shenk, T. (1996) Proteins associated with purified human cytomegalovirus particles, J. Virol. 70, 6097-6105.
9. Johannsen, E., Luftig, M., Chase, M. R., Weicksel, S., Cahir-McFarland, E., Illanes, D., Sarracino, D., and Kieff, E. (2004) Proteins of purified Epstein-Barr virus, Proc. Natl. Acad. Sci. U. S.
A. 101, 16286-16291. 10. Loret, S., Guay, G., and Lippe, R. (2008) Comprehensive
characterization of extracellular herpes simplex virus type 1 virions, J. Virol. 82, 8605-8618.
11. Kalejta, R. F. (2008) Tegument proteins of human cytomegalovirus, Microbiol. Mol. Biol. Rev. 72, 249-265.
12. Heldwein, E. E., and Krummenacher, C. (2008) Entry of herpesviruses into mammalian cells, Cell. Mol. Life Sci. 65, 1653-1668.
13. Spear, P. G. (2004) Herpes simplex virus: receptors and ligands for cell entry, Cell. Microbiol. 6, 401-410.
14. Mori, I., and Nishiyama, Y. (2005) Herpes simplex virus and varicella-zoster virus: why do these human alphaherpesviruses behave so differently from one another?, Rev. Med. Virol. 15, 393-406.
15. Lin, A., Xu, H., and Yan, W. (2007) Modulation of HLA expression in human cytomegalovirus immune evasion, Cell. Mol. Immunol. 4, 91-98.

61
16. Laurent, C., Meggetto, F., and Brousset, P. (2008) Human herpesvirus 8 infections in patients with immunodeficiencies, Hum.
Pathol. 39, 983-993. 17. Klein, E., Kis, L. L., and Klein, G. (2007) Epstein-Barr virus
infection in humans: from harmless to life endangering virus-lymphocyte interactions, Oncogene 26, 1297-1305.
18. Griffiths, P. D. (2006) CMV as a cofactor enhancing progression of AIDS, J. Clin. Virol. 35, 489-492.
19. Sandri-Goldin, R. M. (2003) Replication of the herpes simplex virus genome: does it really go around in circles?, Proc. Natl. Acad. Sci.
U. S. A. 100, 7428-7429. 20. Deshmane, S. L., and Fraser, N. W. (1989) During latency, herpes
simplex virus type 1 DNA is associated with nucleosomes in a chromatin structure, J. Virol. 63, 943-947.
21. Kennedy, P. G., and Chaudhuri, A. (2002) Herpes simplex encephalitis, J. Neurol. Neurosurg. Psychiatry 73, 237-238.
22. Landolfo, S., Gariglio, M., Gribaudo, G., and Lembo, D. (2003) The human cytomegalovirus, Pharmacol. Ther. 98, 269-297.
23. De Bolle, L., Naesens, L., and De Clercq, E. (2005) Update on human herpesvirus 6 biology, clinical features, and therapy, Clin.
Microbiol. Rev. 18, 217-245. 24. Vancikova, Z., and Dvorak, P. (2001) Cytomegalovirus infection in
immunocompetent and immunocompromised individuals--a review, Curr. Drug Targets Immune Endocr. Metabol. Disord. 1, 179-187.
25. Mercorelli, B., Sinigalia, E., Loregian, A., and Palu, G. (2008) Human cytomegalovirus DNA replication: antiviral targets and drugs, Rev. Med. Virol. 18, 177-210.
26. Du, M. Q., Bacon, C. M., and Isaacson, P. G. (2007) Kaposi sarcoma-associated herpesvirus/human herpesvirus 8 and lymphoproliferative disorders, J. Clin. Pathol. 60, 1350-1357.
27. Barozzi, P., Potenza, L., Riva, G., Vallerini, D., Quadrelli, C., Bosco, R., Forghieri, F., Torelli, G., and Luppi, M. (2007) B cells and herpesviruses: a model of lymphoproliferation, Autoimmun. Rev.
7, 132-136. 28. Cotter, M. A., 2nd, and Robertson, E. S. (1999) The latency-
associated nuclear antigen tethers the Kaposi's sarcoma-associated herpesvirus genome to host chromosomes in body cavity-based lymphoma cells, Virology 264, 254-264.
29. Leight, E. R., and Sugden, B. (2000) EBNA-1: a protein pivotal to latent infection by Epstein-Barr virus, Rev Med Virol 10, 83-100.
30. Thorley-Lawson, D. A., Duca, K. A., and Shapiro, M. (2008) Epstein-Barr virus: a paradigm for persistent infection - for real and in virtual reality, Trends Immunol. 29, 195-201.
31. Lunemann, J. D., and Munz, C. (2007) Epstein-Barr virus and multiple sclerosis, Curr. Neurol. Neurosci. Rep. 7, 253-258.

62
32. Posnett, D. N. (2008) Herpesviruses and autoimmunity, Curr. Opin.
Investig. Drugs 9, 505-514. 33. Chang, Y., Cesarman, E., Pessin, M. S., Lee, F., Culpepper, J.,
Knowles, D. M., and Moore, P. S. (1994) Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma, Science 266, 1865-1869.
34. Verma, S. C., and Robertson, E. S. (2003) Molecular biology and pathogenesis of Kaposi sarcoma-associated herpesvirus, FEMS
Microbiol. Lett. 222, 155-163. 35. Dedicoat, M., and Newton, R. (2003) Review of the distribution of
Kaposi's sarcoma-associated herpesvirus (KSHV) in Africa in relation to the incidence of Kaposi's sarcoma, Br. J. Cancer 88, 1-3.
36. Haburchak, D. R., Thomason, J. W., Edelman, D. C., and Constantine, N. T. (2001) Negative human herpesvirus 8 serology in sarcoidosis, J. Hum. Virol. 4, 111.
37. Di Alberti, L., Piattelli, A., Artese, L., Favia, G., Patel, S., Saunders, N., Porter, S. R., Scully, C. M., Ngui, S. L., and Teo, C. G. (1997) Human herpesvirus 8 variants in sarcoid tissues, Lancet 350, 1655-1661.
38. Sjak-Shie, N. N., Vescio, R. A., and Berenson, J. R. (1999) The role of human herpesvirus-8 in the pathogenesis of multiple myeloma, Hematol. Oncol. Clin. North Am. 13, 1159-1167.
39. Zong, J. C., Arav-Boger, R., Alcendor, D. J., and Hayward, G. S. (2007) Reflections on the interpretation of heterogeneity and strain differences based on very limited PCR sequence data from Kaposi's sarcoma-associated herpesvirus genomes, J. Clin. Virol. 40, 1-8.
40. Choi, J., Means, R. E., Damania, B., and Jung, J. U. (2001) Molecular piracy of Kaposi's sarcoma associated herpesvirus, Cytokine Growth Factor Rev. 12, 245-257.
41. Fujimuro, M., Hayward, S. D., and Yokosawa, H. (2007) Molecular piracy: manipulation of the ubiquitin system by Kaposi's sarcoma-associated herpesvirus, Rev. Med. Virol. 17, 405-422.
42. Neipel, F., Albrecht, J. C., and Fleckenstein, B. (1997) Cell-homologous genes in the Kaposi's sarcoma-associated rhadinovirus human herpesvirus 8: determinants of its pathogenicity, J. Virol. 71, 4187-4192.
43. McGeoch, D. J., and Cook, S. (1994) Molecular phylogeny of the alphaherpesvirinae subfamily and a proposed evolutionary timescale, J. Mol. Biol. 238, 9-22.
44. Davison, A. (2002) Comments on the phylogenetics and evolution of herpesviruses and other large DNA viruses, Virus Res. 82, 127-132.
45. Fleischmann, R. D., Adams, M. D., White, O., Clayton, R. A., Kirkness, E. F., Kerlavage, A. R., Bult, C. J., Tomb, J. F., Dougherty, B. A., Merrick, J. M., McKenney, K., Sutton, G.,

63
FitzHugh, W., Fields, C., Gocayhe, J.D., Scott, J., Shirley, R., Liu, L-I., Glodek, A., Kelley, J.M, Weidman, J.F., Phillips, C.A., Spriggs, T., Hedblom, E., Cotton, M.D., Herback, T., Hanna, M.C., Nguyen, D.T., Saudek, D.M., Brandon, R.C., Fine, L.D., Frichtman, J.L., Fuhrmann, J.L., Geoghagen, N.S.M., Gnehm, C.C., McDonald, L.A., Small, K.V., Fraser, C.M., Smith, H.O., Venter, J.C. (1995) Whole-genome random sequencing and assembly of Haemophilus
influenzae Rd, Science 269, 496-512. 46. (2001) Initial sequencing and analysis of the human genome, Nature
409, 860-921. 47. Venter, J. C., Adams, M. D., Myers, E. W., Li, P. W., Mural, R. J.,
Sutton, G. G., Smith, H. O., Yandell, M., Evans, C. A., Holt, R. A., Gocayne, J. D., Amanatides, P., Ballew, R. M., Huson, D. H., Wortman, J. R., Zhang, Q., Kodira, C. D., Zheng, X. H., Chen, L., Skupski, M., Subramanian, G., Thomas, P. D., Zhang, J., Gabor Miklos, G. L., Nelson, C., Broder, S., Clark, A. G., Nadeau, J., McKusick, V. A., Zinder, N., Levine, A. J., Roberts, R. J., Simon, M., Slayman, C., Hunkapiller, M., Bolanos, R., Delcher, A., Dew, I., Fasulo, D., Flanigan, M., Florea, L., Halpern, A., Hannenhalli, S., Kravitz, S., Levy, S., Mobarry, C., Reinert, K., Remington, K., Abu-Threideh, J., Beasley, E., Biddick, K., Bonazzi, V., Brandon, R., Cargill, M., Chandramouliswaran, I., Charlab, R., Chaturvedi, K., Deng, Z., Francesco, V. D., Dunn, P., Eilbeck, K., Evangelista, C., Gabrielian, A. E., Gan, W., Ge, W., Gong, F., Gu, Z., Guan, P., Heiman, T. J., Higgins, M. E., Ji, R.-R., Ke, Z., Ketchum, K. A., Lai, Z., Lei, Y., Li, Z., Li, J., Liang, Y., Lin, X., Lu, F., Merkulov, G. V., Milshina, N., Moore, H. M., Naik, A. K., Narayan, V. A., Neelam, B., Nusskern, D., Rusch, D. B., Salzberg, S., Shao, W., Shue, B., Sun, J., Wang, Z. Y., Wang, A., Wang, X., Wang, J., Wei, M.-H., Wides, R., Xiao, C., Yan, C., Yao, A., Ye, J., Zhan, M., Zhang, W., Zhang, H., Zhao, Q., Zheng, L., Zhong, F., Zhong, W., Zhu, S. C., Zhao, S., Gilbert, D., Baumhueter, S., Spier, G., Carter, C., Cravchik, A., Woodage, T., Ali, F., An, H., Awe, A., Baldwin, D., Baden, H., Barnstead, M., Barrow, I., Beeson, K., Busam, D., Carver, A., Center, A., Cheng, M. L., Curry, L., Danaher, S., Davenport, L., Desilets, R., Dietz, S., Dodson, K., Doup, L., Ferriera, S., Garg, N., Gluecksmann, A., Hart, B., Haynes, J., Haynes, C., Heiner, C., Hladun, S., Hostin, D., Houck, J., Howland, T., Ibegwam, C., Johnson, J., Kalush, F., Kline, L., Koduru, S., Love, A., Mann, F., May, D., McCawley, S., McIntosh, T., McMullen, I., Moy, M., Moy, L., Murphy, B., Nelson, K., Pfannkoch, C., Pratts, E., Puri, V., Qureshi, H., Reardon, M., Rodriguez, R., Rogers, Y.-H., Romblad, D., Ruhfel, B., Scott, R., Sitter, C., Smallwood, M., Stewart, E., Strong, R., Suh, E., Thomas, R., Tint, N. N., Tse, S., Vech, C., Wang, G., Wetter, J., Williams, S.,

64
Williams, M., Windsor, S., Winn-Deen, E., Wolfe, K., Zaveri, J., Zaveri, K., Abril, J. F., Guigo, R., Campbell, M. J., Sjolander, K. V., Karlak, B., Kejariwal, A., Mi, H., Lazareva, B., Hatton, T., Narechania, A., Diemer, K., Muruganujan, A., Guo, N., Sato, S., Bafna, V., Istrail, S., Lippert, R., Schwartz, R., Walenz, B., Yooseph, S., Allen, D., Basu, A., Baxendale, J., Blick, L., Caminha, M., Carnes-Stine, J., Caulk, P., Chiang, Y.-H., Coyne, M., Dahlke, C., Mays, A. D., Dombroski, M., Donnelly, M., Ely, D., Esparham, S., Fosler, C., Gire, H., Glanowski, S., Glasser, K., Glodek, A., Gorokhov, M., Graham, K., Gropman, B., Harris, M., Heil, J., Henderson, S., Hoover, J., Jennings, D., Jordan, C., Jordan, J., Kasha, J., Kagan, L., Kraft, C., Levitsky, A., Lewis, M., Liu, X., Lopez, J., Ma, D., Majoros, W., McDaniel, J., Murphy, S., Newman, M., Nguyen, T., Nguyen, N., Nodell, M., Pan, S., Peck, J., Peterson, M., Rowe, W., Sanders, R., Scott, J., Simpson, M., Smith, T., Sprague, A., Stockwell, T., Turner, R., Venter, E., Wang, M., Wen, M., Wu, D., Wu, M., Xia, A., Zandieh, A., and Zhu, X. (2001) The Sequence of the Human Genome, Science 291, 1304-1351.
48. Venter, J. C., Remington, K., Heidelberg, J. F., Halpern, A. L., Rusch, D., Eisen, J. A., Wu, D. Y., Paulsen, I., Nelson, K. E., Nelson, W., Fouts, D. E., Levy, S., Knap, A. H., Lomas, M. W., Nealson, K., White, O., Peterson, J., Hoffman, J., Parsons, R., Baden-Tillson, H., Pfannkoch, C., Rogers, Y. H., and Smith, H. O. (2004) Environmental genome shotgun sequencing of the Sargasso Sea, Science 304, 66-74.
49. Kim, S. H. (1998) Shining a light on structural genomics, Nat.
Struct. Biol. 5 Suppl, 643-645. 50. Tarbouriech, N., Buisson, M., Geoui, T., Daenke, S., Cusack, S., and
Burmeister, W. P. (2006) Structural genomics of the Epstein-Barr virus, Acta Crystallogr. D. Biol. Crystallogr. 62, 1276-1285.
51. Fogg, M. J., Alzari, P., Bahar, M., Bertini, I., Betton, J. M., Burmeister, W. P., Cambillau, C., Canard, B., Corrondo, M. A., Coll, M., Daenke, S., Dym, O., Egloff, M. P., Enguita, F. J., Geerlof, A., Haouz, A., Jones, T. A., Ma, Q., Manicka, S. N., Migliardi, M., Nordlund, P., Owens, R. J., Peleg, Y., Schneider, G., Schnell, R., Stuart, D. I., Tarbouriech, N., Unge, T., Wilkinson, A. J., Wilmanns, M., Wilson, K. S., Zimhony, O., and Grimes, J. M. (2006) Application of the use of high-throughput technologies to the determination of protein structures of bacterial and viral pathogens, Acta Crystallogr. D. Biol. Crystallogr. 62, 1196-1207.
52. Marsden, R. L., and Orengo, C. A. (2008) Target selection for structural genomics: an overview, Meth. Mol. Biol. 426, 3-25.
53. Graslund, S., Nordlund, P., Weigelt, J., Hallberg, B. M., Bray, J., Gileadi, O., Knapp, S., Oppermann, U., Arrowsmith, C., Hui, R., Ming, J., dhe-Paganon, S., Park, H. W., Savchenko, A., Yee, A.,

65
Edwards, A., Vincentelli, R., Cambillau, C., Kim, R., Kim, S. H., Rao, Z., Shi, Y., Terwilliger, T. C., Kim, C. Y., Hung, L. W., Waldo, G. S., Peleg, Y., Albeck, S., Unger, T., Dym, O., Prilusky, J., Sussman, J. L., Stevens, R. C., Lesley, S. A., Wilson, I. A., Joachimiak, A., Collart, F., Dementieva, I., Donnelly, M. I., Eschenfeldt, W. H., Kim, Y., Stols, L., Wu, R., Zhou, M., Burley, S. K., Emtage, J. S., Sauder, J. M., Thompson, D., Bain, K., Luz, J., Gheyi, T., Zhang, F., Atwell, S., Almo, S. C., Bonanno, J. B., Fiser, A., Swaminathan, S., Studier, F. W., Chance, M. R., Sali, A., Acton, T. B., Xiao, R., Zhao, L., Ma, L. C., Hunt, J. F., Tong, L., Cunningham, K., Inouye, M., Anderson, S., Janjua, H., Shastry, R., Ho, C. K., Wang, D., Wang, H., Jiang, M., Montelione, G. T., Stuart, D. I., Owens, R. J., Daenke, S., Schutz, A., Heinemann, U., Yokoyama, S., Bussow, K., and Gunsalus, K. C. (2008) Protein production and purification, Nat. Methods 5, 135-146.
54. Lesley, S. A., Kuhn, P., Godzik, A., Deacon, A. M., Mathews, I., Kreusch, A., Spraggon, G., Klock, H. E., McMullan, D., Shin, T., Vincent, J., Robb, A., Brinen, L. S., Miller, M. D., McPhillips, T. M., Miller, M. A., Scheibe, D., Canaves, J. M., Guda, C., Jaroszewski, L., Selby, T. L., Elsliger, M. A., Wooley, J., Taylor, S. S., Hodgson, K. O., Wilson, I. A., Schultz, P. G., and Stevens, R. C. (2002) Structural genomics of the Thermotoga maritima proteome implemented in a high-throughput structure determination pipeline, Proc. Natl. Acad. Sci. U. S. A. 99, 11664-11669.
55. Bussow, K., Scheich, C., Sievert, V., Harttig, U., Schultz, J., Simon, B., Bork, P., Lehrach, H., and Heinemann, U. (2005) Structural genomics of human proteins--target selection and generation of a public catalogue of expression clones, Microb. Cell. Fact. 4, 21.
56. Heinemann, U., Bussow, K., Mueller, U., and Umbach, P. (2003) Facilities and methods for the high-throughput crystal structural analysis of human proteins, Acc. Chem. Res. 36, 157-163.
57. Banci, L., Bertini, I., Cusack, S., de Jong, R. N., Heinemann, U., Jones, E. Y., Kozielski, F., Maskos, K., Messerschmidt, A., Owens, R., Perrakis, A., Poterszman, A., Schneider, G., Siebold, C., Silman, I., Sixma, T., Stewart-Jones, G., Sussman, J. L., Thierry, J. C., and Moras, D. (2006) First steps towards effective methods in exploiting high-throughput technologies for the determination of human protein structures of high biomedical value, Acta Crystallogr. D.
Biol. Crystallogr. 62, 1208-1217. 58. Aricescu, A. R., Assenberg, R., Bill, R. M., Busso, D., Chang, V. T.,
Davis, S. J., Dubrovsky, A., Gustafsson, L., Hedfalk, K., Heinemann, U., Jones, I. M., Ksiazek, D., Lang, C., Maskos, K., Messerschmidt, A., Macieira, S., Peleg, Y., Perrakis, A., Poterszman, A., Schneider, G., Sixma, T. K., Sussman, J. L., Sutton, G., Tarboureich, N., Zeev-Ben-Mordehai, T., and Jones, E. Y.

66
(2006) Eukaryotic expression: developments for structural proteomics, Acta Crystallogr. D. Biol. Crystallogr. 62, 1114-1124.
59. Gong, W. M., Liu, H. Y., Niu, L. W., Shi, Y. Y., Tang, Y. J., Teng, M. K., Wu, J. H., Liang, D. C., Wang, D. C., Wang, J. F., Ding, J. P., Hu, H. Y., Huang, Q. H., Zhang, Q. H., Lu, S. Y., An, J. L., Liang, Y. H., Zheng, X. F., Gu, X. C., and Su, X. D. (2003) Structural genomics efforts at the Chinese Academy of Sciences and Peking University, J. Struct. Funct. Genomics 4, 137 - 139.
60. Alzari, P. M., Berglund, H., Berrow, N. S., Blagova, E., Busso, D., Cambillau, C., Campanacci, V., Christodoulou, E., Eiler, S., Fogg, M. J., Folkers, G., Geerlof, A., Hart, D., Haouz, A., Herman, M. D., Macieira, S., Nordlund, P., Perrakis, A., Quevillon-Cheruel, S., Tarandeau, F., van Tilbeurgh, H., Unger, T., Luna-Vargas, M. P., Velarde, M., Willmanns, M., and Owens, R. J. (2006) Implementation of semi-automated cloning and prokaryotic expression screening: the impact of SPINE, Acta Crystallogr. D.
Biol. Crystallogr. 62, 1103-1113. 61. Edwards, A. M., Arrowsmith, C. H., Christendat, D., Dharamsi, A.,
Friesen, J. D., Greenblatt, J. F., and Vedadi, M. (2000) Protein production: feeding the crystallographers and NMR spectroscopists, Nat. Struct. Biol. 7, 970-972.
62. Sorensen, H., and Mortensen, K. (2005) Soluble expression of recombinant proteins in the cytoplasm of Escherichia coli, Microb.l
Cell Fact. 4, 1-8. 63. Makrides, S. C. (1996) Strategies for achieving high-level
expression of genes in Escherichia coli, Microbiol. Rev. 60, 512-538.
64. Hannig, G., and Makrides, S. C. (1998) Strategies for optimizing heterologous protein expression in Escherichia coli, Trends
Biotechnol. 16, 54-60. 65. Prouty, W. F., and Goldberg, A. L. (1972) Fate of abnormal proteins
in E. coli accumulation in intracellular granules before catabolism, Nature New Biol. 240, 147-150.
66. Prouty, W. F., Karnovsky, M. J., and Goldberg, A. L. (1975) Degradation of abnormal proteins in Escherichia coli. Formation of protein inclusions in cells exposed to amino acid analogs, J. Biol.
Chem. 250, 1112-1122. 67. Bowden, G. A., Paredes, A. M., and Georgiou, G. (1991) Structure
and morphology of inclusion bodies in Escherichia coli,
Biotechnology. (N. Y). 9, 725-730. 68. Taylor, G., Hoare, M., Gray, D. R., and Martson, F. A. O. (1986)
Size and density of inclusion bodies, Biotechnology. (N. Y). 4, 553-557.
69. Garcia-Fruitos, E., Gonzalez-Montalban, N., Morell, M., Vera, A., Ferraz, R., Aris, A., Ventura, S., and Villaverde, A. (2005)

67
Aggregation as bacterial inclusion bodies does not imply inactivation of enzymes and fluorescent proteins, Microb. Cell Fact.
4, 27. 70. Carrio, M. M., Corchero, J. L., and Villaverde, A. (1998) Dynamics
of in vivo protein aggregation: building inclusion bodies in recombinant bacteria, FEMS Microb. Lett. 169, 9-15.
71. Carrio, M. M., and Villaverde, A. (2003) Role of molecular chaperones in inclusion body formation, FEBS Lett. 537, 215-221.
72. Baneyx, F., and Mujacic, M. (2004) Recombinant protein folding and misfolding in Escherichia coli, Nat. Biotechnol. 22, 1399-1408.
73. Baneyx, F., and Palumbo, J. L. (2003) Improving heterologous protein folding via molecular chaperone and foldase co-expression, Methods Mol. Biol. 205, 171-197.
74. Sorensen, H. P., and Mortensen, K. K. (2005) Advanced genetic strategies for recombinant protein expression in Escherichia coli, J.
Biotechnol. 115, 113-128. 75. Dyson, M. R., Shadbolt, S. P., Vincent, K. J., Perera, R. L., and
McCafferty, J. (2004) Production of soluble mammalian proteins in Escherichia coli: identification of protein features that correlate with successful expression, BMC Biotechnol 4, 32.
76. Vincentelli, R., Bignon, C., Gruez, A., Canaan, S., Sulzenbacher, G., Tegoni, M., Campanacci, V., and Cambillau, C. (2003) Medium-scale structural genomics: strategies for protein expression and crystallization, Acc. Chem. Res. 36, 165-172.
77. Brocchieri, L., and Karlin, S. (2005) Protein length in eukaryotic and prokaryotic proteomes, Nucleic Acids Res 33, 3390-3400.
78. Sorensen, H. P., Sperling-Petersen, H. U., and Mortensen, K. K. (2003) Production of recombinant thermostable proteins expressed in Escherichia coli: completion of protein synthesis is the bottleneck, J. Chromatogr. B Analyt. Technol. Biomed. Life. Sci.
786, 207-214. 79. Hua, Z., Wang, H., Chen, D., Chen, Y., and Zhu, D. (1994)
Enhancement of expression of human granulocyte-macrophage colony stimulating factor by argU gene product in Escherichia coli,
Biochem. Mol. Biol. Int. 32, 537-543. 80. Baneyx, F., and Georgiou, G. (1991) Construction and
characterization of Escherichia coli strains deficient in multiple secreted proteases: protease III degrades high molecular weight substrates in vivo, J. Bacteriol. 173, 2696-2703.
81. Studier, F. W., and Moffatt, B. A. (1986) Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes, J. Mol. Biol. 189, 113-130.
82. Miroux, B., and Walker, J. E. (1996) Over-production of proteins in Escherichia coli: Mutant hosts that allow synthesis of some

68
membrane proteins and globular proteins at high levels, J. Mol. Biol.
260, 289-298. 83. Bessette, P. H., Aslund, F., Beckwith, J., and Georgiou, G. (1999)
Efficient folding of proteins with multiple disulfide bonds in the Escherichia coli cytoplasm, Proc. Natl. Acad. Sci. USA 96, 13703-13708.
84. Abergel, C., Coutard, B., Byrne, D., Chenivesse, S., Claude, J.-B., Deregnaucourt, C. l., Fricaux, T., Gianesini-Boutreux, C., Jeudy, S., Lebrun, R. g., Maza, C., Notredame, C. d., Poirot, O., Suhre, K., Varagnol, M., and Claverie, J.-M. (2003) Structural genomics of highly conserved microbial genes of unknown function in search of new antibacterial targets, J. Struct. Funct. Genom. 4, 141-157.
85. Berrow, N. S., Bussow, K., Coutard, B., Diprose, J., Ekberg, M., Folkers, G. E., Levy, N., Lieu, V., Owens, R. J., Peleg, Y., Pinaglia, C., Quevillon-Cheruel, S., Salim, L., Scheich, C., Vincentelli, R., and Busso, D. (2006) Recombinant protein expression and solubility screening in Escherichia coli: a comparative study, Acta
Crystallogr. D. Biol. Crystallogr. 62, 1218-1226. 86. Vasina, J. A., and Baneyx, F. (1997) Expression of aggregation-
prone proteins at low temperatures: a comparative study of the E.
coli cspA and tac promoter systems, Protein Expr. Purif. 9, 211-218. 87. Vasina, J. A., and Baneyx, F. (1997) Expression of Aggregation-
Prone Recombinant Proteins at Low Temperatures: A Comparative Study of the Escherichia coli cspA and tac Promoter Systems, Protein Expr. Purif. 9, 211-218.
88. Kiefhaber, T., Rudolph, R., Kohler, H. H., and Buchner, J. (1991) Protein aggregation in vitro and in vivo: a quantitative model of the kinetic competition between folding and aggregation, Biotechnology. (N. Y). 9, 825-829.
89. Ferrer, M., Chernikova, T. N., Yakimov, M. M., Golyshin, P. N., and Timmis, K. N. (2003) Chaperonins govern growth of Escherichia coli at low temperatures, Nat. Biotechnol. 21, 1266-1267.
90. Kapust, R. B., and Waugh, D. S. (1999) Escherichia coli maltose-binding protein is uncommonly effective at promoting the solubility of polypeptides to which it is fused, Protein Sci. 8, 1668-1674.
91. Hammarstrom, M., Hellgren, N., Van den Berg, S., Berglund, H., and Hard, T. (2002) Rapid screening for improved solubility of small human proteins produced as fusion proteins in Escherichia
coli, Protein Sci. 11, 313-321. 92. Braun, P., Hu, Y., Shen, B., Halleck, A., Koundinya, M., Harlow,
E., and LaBaer, J. (2002) Proteome-scale purification of human proteins from bacteria, Proc. Natl. Acad. Sci. USA 99, 2654-2659.
93. Braun, P., and LaBaer, J. (2003) High throughput protein production for functional proteomics, Trends Biotechnol. 21, 383-388.

69
94. Waugh, D. S. (2005) Making the most of affinity tags, Trends
Biotechnol. 23, 316-320. 95. Nallamsetty, S., and Waugh, D. S. (2007) A generic protocol for the
expression and purification of recombinant proteins in Escherichia
coli using a combinatorial His6-maltose binding protein fusion tag, Nat. Protoc. 2, 383-391.
96. Jeon, W., Aceti, D., Bingman, C., Vojtik, F., Olson, A., Ellefson, J., McCombs, J., Sreenath, H., Blommel, P., Seder, K., Burns, B., Geetha, H., Harms, A., Sabat, G., Sussman, M., Fox, B., and Phillips, G. (2005) High-throughput Purification and Quality Assurance of Arabidopsis thaliana Proteins for Eukaryotic Structural Genomics, J. Struct. Funct. Genom. 6, 143-147.
97. Ashraf, S. S., Benson, R. E., Payne, E. S., Halbleib, C. M., and Gron, H. (2004) A novel multi-affinity tag system to produce high levels of soluble and biotinylated proteins in Escherichia coli, Protein Expr. Purif. 33, 238-245.
98. Nallamsetty, S., and Waugh, D. S. (2006) Solubility-enhancing proteins MBP and NusA play a passive role in the folding of their fusion partners, Protein Expr. Purif. 45, 175-182.
99. Hochuli, E., Bannwarth, W., Dobeli, H., Gentz, R., and Stuber, D. (1988) Genetic Approach to Facilitate Purification of Recombinant Proteins with a Novel Metal Chelate Adsorbent, Nat. Biotechnol. 6,
1321-1325. 100. Graslund, S., Sagemark, J., Berglund, H., Dahlgren, L. G., Flores,
A., Hammarstrom, M., Johansson, I., Kotenyova, T., Nilsson, M., Nordlund, P., and Weigelt, J. (2008) The use of systematic N- and C-terminal deletions to promote production and structural studies of recombinant proteins, Protein Expr. Purif. 58, 210-221.
101. Cohen, S. L. (1996) Domain elucidation by mass spectrometry, Structure 4, 1013-1016.
102. Gao, X., Bain, K., Bonanno, J., Buchanan, M., Henderson, D., Lorimer, D., Marsh, C., Reynes, J., Sauder, J., Schwinn, K., Thai, C., and Burley, S. (2005) High-throughput Limited Proteolysis/Mass Spectrometry for Protein Domain Elucidation, J. Struct. Funct.
Genom. 6, 129-134. 103. Bjornsson, A., Mottagui-Tabar, S., and Isaksson, L. A. (1996)
Structure of the C-terminal end of the nascent peptide influences translation termination, EMBO J. 15, 1696-1704.
104. Pantazatos, D., Kim, J. S., Klock, H. E., Stevens, R. C., Wilson, I. A., Lesley, S. A., and Woods, V. L., Jr. (2004) Rapid refinement of crystallographic protein construct definition employing enhanced hydrogen/deuterium exchange MS, Proc. Natl. Acad. Sci. U. S. A.
101, 751-756. 105. Bateman, A., Coin, L., Durbin, R., Finn, R. D., Hollich, V.,
Griffiths-Jones, S., Khanna, A., Marshall, M., Moxon, S.,

70
Sonnhammer, E. L. L., Studholme, D. J., Yeats, C., and Eddy, S. R. (2004) The Pfam protein families database, Nucleic Acids Res. 32, D138-D141.
106. Sauder, M. J., Rutter, M. E., Bain, K., Rooney, I., Gheyi, T., Atwell, S., Thompson, D. A., Emtage, S., and Burley, S. K. (2008) High throughput protein production and crystallization at NYSGXRC, Meth. Mol. Biol. 426, 561-575.
107. Puri, M., Robin, G., Cowieson, N., Forwood, J. K., Listwan, P., Hu, S. H., Guncar, G., Huber, T., Kellie, S., Hume, D. A., Kobe, B., and Martin, J. L. (2006) Focusing in on structural genomics: the University of Queensland structural biology pipeline, Biomol. Eng.
23, 281-289. 108. Peti, W., and Page, R. (2007) Strategies to maximize heterologous
protein expression in Escherichia coli with minimal cost, Protein
Expr. Purif. 51, 1-10. 109. Luan, C. H., Qiu, S., Finley, J. B., Carson, M., Gray, R. J., Huang,
W., Johnson, D., Tsao, J., Reboul, J., Vaglio, P., Hill, D. E., Vidal, M., Delucas, L. J., and Luo, M. (2004) High-throughput expression of C. elegans proteins, Genome Res. 14, 2102-2110.
110. Lamesch, P., Milstein, S., Hao, T., Rosenberg, J., Li, N., Sequerra, R., Bosak, S., Doucette-Stamm, L., Vandenhaute, J., Hill, D. E., and Vidal, M. (2004) C. elegans ORFeome version 3.1: increasing the coverage of ORFeome resources with improved gene predictions, Genome Res. 14, 2064-2069.
111. Knaust, R. K. C., and Nordlund, P. (2001) Screening for soluble expression of recombinant proteins in a 96-well format, Anal.
Biochem. 297, 79-85. 112. Eshaghi, S., Hedren, M., Nasser, M. I. A., Hammarberg, T.,
Thornell, A., and Nordlund, P. (2005) An efficient strategy for high-throughput expression screening of recombinant integral membrane proteins, Protein Sci. 14, 676-683.
113. Waldo, G. S., Standish, B. M., Berendzen, J., and Terwilliger, T. C. (1999) Rapid protein-folding assay using green fluorescent protein, Nat. Biotechnol. 17, 691-695.
114. Maxwell, K. L., Mittermaier, A. K., Forman-Kay, J. D., and Davidson, A. R. (1999) A simple in vivo assay for increased protein solubility, Protein Sci. 8, 1908-1911.
115. Cabantous, S., Pédelacq, J.-D., Mark, B. L., Naranjo, C., Terwilliger, T., and Waldo, G. (2005) Recent Advances in GFP Folding Reporter and Split-GFP Solubility Reporter Technologies. Application to Improving the Folding and Solubility of Recalcitrant Proteins from Mycobacterium tuberculosis, J. Struct. Funct. Genom.
6, 113-119. 116. Denis-Quanquin, S., Lamouroux, L., Lougarre, A., Maheo, S.,
Saves, I., Paquereau, L., Demange, P., and Fournier, D. (2007)

71
Protein expression from synthetic genes: selection of clones using GFP, J. Biotechnol. 131, 223-230.
117. Wigley, W. C., Stidham, R. D., Smith, N. M., Hunt, J. F., and Thomas, P. J. (2001) Protein solubility and folding monitored in
vivo by structural complementation of a genetic marker protein, Nat.
Biotechnol. 19, 131-136. 118. Waldo, G. S. (2003) Genetic screens and directed evolution for
protein solubility, Curr. Opin. Chem. Biol. 7, 33-38. 119. Cabantous, S., Terwilliger, T. C., and Waldo, G. S. (2005) Protein
tagging and detection with engineered self-assembling fragments of green fluorescent protein, Nat. Biotechnol. 23, 102-107.
120. DeLisa, M. P., Samuelson, P., Palmer, T., and Georgiou, G. (2002) Genetic analysis of the twin arginine translocator secretion pathway in bacteria, J. Biol. Chem. 277, 29825-29831.
121. DeLisa, M. P., Tullman, D., and Georgiou, G. (2003) Folding quality control in the export of proteins by the bacterial twin-arginine translocation pathway, Proc. Natl. Acad. Sci. U. S. A. 100, 6115-6120.
122. Beckett, D., Kovaleva, E., and Schatz, P. J. (1999) A minimal peptide substrate in biotin holoenzyme synthetase-catalyzed biotinylation, Protein Sci. 8, 921-929.
123. Scott, C. J., Martin, S. L., Wallace, A., Curran, M. D., and Walker, B. (2000) Characterization of the affinity of streptavidin toward a peptide sequence previously identified as a target substrate for biotinylation by the Escherichia coli biotin holoenzyme synthetase, BirA, Anal. Biochem. 284, 416-417.
124. Tarendeau, F., Boudet, J., Guilligay, D., Mas, P. J., Bougault, C. M., Boulo, S., Baudin, F., Ruigrok, R. W., Daigle, N., Ellenberg, J., Cusack, S., Simorre, J. P., and Hart, D. J. (2007) Structure and nuclear import function of the C-terminal domain of influenza virus polymerase PB2 subunit, Nat. Struct. Mol. Biol. 14, 229-233.
125. van den Berg, S., Lofdahl, P. A., Hard, T., and Berglund, H. (2006) Improved solubility of TEV protease by directed evolution, J.
Biotechnol. 121, 291-298. 126. Henikoff, S. (1984) Unidirectional digestion with exonuclease III
creates targeted breakpoints for DNA sequencing, Gene 28, 351-359.
127. Etchegaray, J. P., and Inouye, M. (1999) Translational enhancement by an element downstream of the initiation codon in Escherichia
coli, J. Biol. Chem. 274, 10079-10085. 128. Martinez Molina, D., Cornvik, T., Eshaghi, S., Haeggstrom, J. Z.,
Nordlund, P., and Sabet, M. I. (2008) Engineering membrane protein overproduction in Escherichia coli, Protein Sci. 17, 673-680.
129. Labaer, J., Qiu, Q., Anumanthan, A., Mar, W., Zuo, D., Murthy, T. V., Taycher, H., Halleck, A., Hainsworth, E., Lory, S., and Brizuela,

72
L. (2004) The Pseudomonas aeruginosa PA01 gene collection, Genome Res. 14, 2190-2200.
130. Lamesch, P., Li, N., Milstein, S., Fan, C., Hao, T., Szabo, G., Hu, Z., Venkatesan, K., Bethel, G., Martin, P., Rogers, J., Lawlor, S., McLaren, S., Dricot, A., Borick, H., Cusick, M. E., Vandenhaute, J., Dunham, I., Hill, D. E., and Vidal, M. (2007) hORFeome v3.1: a resource of human open reading frames representing over 10,000 human genes, Genomics 89, 307-315.
131. Gong, W., Shen, Y. P., Ma, L. G., Pan, Y., Du, Y. L., Wang, D. H., Yang, J. Y., Hu, L. D., Liu, X. F., Dong, C. X., Ma, L., Chen, Y. H., Yang, X. Y., Gao, Y., Zhu, D., Tan, X., Mu, J. Y., Zhang, D. B., Liu, Y. L., Dinesh-Kumar, S. P., Li, Y., Wang, X. P., Gu, H. Y., Qu, L. J., Bai, S. N., Lu, Y. T., Li, J. Y., Zhao, J. D., Zuo, J., Huang, H., Deng, X. W., and Zhu, Y. X. (2004) Genome-wide ORFeome cloning and analysis of Arabidopsis transcription factor genes, Plant
Physiol. 135, 773-782. 132. Uetz, P., Dong, Y. A., Zeretzke, C., Atzler, C., Baiker, A., Berger,
B., Rajagopala, S. V., Roupelieva, M., Rose, D., Fossum, E., and Haas, J. (2006) Herpesviral protein networks and their interaction with the human proteome, Science 311, 239-242.
133. Gillette, W. K., Esposito, D., Frank, P. H., Zhou, M., Yu, L. R., Jozwik, C., Zhang, X., McGowan, B., Jacobowitz, D. M., Pollard, H. B., Hao, T., Hill, D. E., Vidal, M., Conrads, T. P., Veenstra, T. D., and Hartley, J. L. (2005) Pooled ORF expression technology (POET): using proteomics to screen pools of open reading frames for protein expression, Mol. Cell. Proteomics 4, 1647-1652.
134. Shapiro, J. A. (1998) Thinking about bacterial populations as multicellular organisms, Annu. Rev. Microbiol. 52, 81-104.
135. Vlamakis, H., Aguilar, C., Losick, R., and Kolter, R. (2008) Control of cell fate by the formation of an architecturally complex bacterial community, Genes Dev. 22, 945-953.
136. Boehmer, P. E., and Lehman, I. R. (1997) Herpes simplex virus DNA replication, Annu. Rev. Biochem. 66, 347-384.
137. Wu, J. J., Brentjens, M. H., Torres, G., Yeung-Yue, K., Lee, P., and Tyring, S. K. (2003) Valacyclovir in the treatment of herpes simplex, herpes zoster, and other viral infections, J. Cutan. Med.
Surg. 7, 372-381. 138. Morfin, F., and Thouvenot, D. (2003) Herpes simplex virus
resistance to antiviral drugs, J. Clin. Virol. 26, 29-37. 139. Nishiyama, Y. (2004) Herpes simplex virus gene products: the
accessories reflect her lifestyle well, Rev. Med. Virol. 14, 33-46. 140. Glaunsinger, B., and Ganem, D. (2004) Lytic KSHV infection
inhibits host gene expression by accelerating global mRNA turnover, Mol. Cell 13, 713-723.

73
141. Martinez, R., Sarisky, R. T., Weber, P. C., and Weller, S. K. (1996) Herpes simplex virus type 1 alkaline nuclease is required for efficient processing of viral DNA replication intermediates, J. Virol.
70, 2075-2085. 142. Thomas, M. S., Gao, M., Knipe, D. M., and Powell, K. L. (1992)
Association between the herpes simplex virus major DNA-binding protein and alkaline nuclease, J. Virol. 66, 1152-1161.
143. Reuven, N. B., Staire, A. E., Myers, R. S., and Weller, S. K. (2003) The herpes simplex virus type 1 alkaline nuclease and single-stranded DNA binding protein mediate strand exchange in vitro, J.
Virol. 77, 7425-7433. 144. Glaunsinger, B., Chavez, L., and Ganem, D. (2005) The exonuclease
and host shutoff functions of the SOX protein of Kaposi's sarcoma-associated herpesvirus are genetically separable, J. Virol. 79, 7396-7401.
145. Rowe, M., Glaunsinger, B., van Leeuwen, D., Zuo, J., Sweetman, D., Ganem, D., Middeldorp, J., Wiertz, E. J., and Ressing, M. E. (2007) Host shutoff during productive Epstein-Barr virus infection is mediated by BGLF5 and may contribute to immune evasion, Proc.
Natl. Acad. Sci. U. S. A. 104, 3366-3371. 146. Nordlund, P., and Reichard, P. (2006) Ribonucleotide reductases,
Annu. Rev. Biochem. 75, 681-706. 147. Cohen, G. H. (1972) Ribonucleotide reductase activity of
synchronized KB cells infected with herpes simplex virus, J. Virol.
9, 408-418. 148. Daikoku, T., Yamamoto, N., Maeno, K., and Nishiyama, Y. (1991)
Role of viral ribonucleotide reductase in the increase of dTTP pool size in herpes simplex virus-infected Vero cells, J. Gen. Virol. 72 ( Pt 6), 1441-1444.
149. Langelier, Y., Bergeron, S., Chabaud, S., Lippens, J., Guilbault, C., Sasseville, A. M., Denis, S., Mosser, D. D., and Massie, B. (2002) The R1 subunit of herpes simplex virus ribonucleotide reductase protects cells against apoptosis at, or upstream of, caspase-8 activation, J. Gen. Virol. 83, 2779-2789.
150. Sekino, Y., Bruner, S. D., and Verdine, G. L. (2000) Selective inhibition of herpes simplex virus type-1 uracil-DNA glycosylase by designed substrate analogs, J. Biol. Chem. 275, 36506-36508.
151. Mullaney, J., Moss, H. W., and McGeoch, D. J. (1989) Gene UL2 of herpes simplex virus type 1 encodes a uracil-DNA glycosylase, J.
Gen. Virol. 70 ( Pt 2), 449-454. 152. Pyles, R. B., Sawtell, N. M., and Thompson, R. L. (1992) Herpes
simplex virus type 1 dUTPase mutants are attenuated for neurovirulence, neuroinvasiveness, and reactivation from latency, J.
Virol. 66, 6706-6713.

74
153. Preston, V. G., and Fisher, F. B. (1984) Identification of the herpes simplex virus type 1 gene encoding the dUTPase, Virology 138, 58-68.
154. Aranda, M., and Maule, A. (1998) Virus-induced host gene shutoff in animals and plants, Virology 243, 261-267.
155. Oroskar, A. A., and Read, G. S. (1989) Control of mRNA stability by the virion host shutoff function of herpes simplex virus, J. Virol.
63, 1897-1906. 156. Strom, T., and Frenkel, N. (1987) Effects of herpes simplex virus on
mRNA stability, J. Virol. 61, 2198-2207. 157. Smiley, J. R. (2004) Herpes simplex virus virion host shutoff
protein: immune evasion mediated by a viral RNase?, J. Virol. 78,
1063-1068. 158. Everly, D. N., Jr., Feng, P., Mian, I. S., and Read, G. S. (2002)
mRNA degradation by the virion host shutoff (Vhs) protein of herpes simplex virus: genetic and biochemical evidence that Vhs is a nuclease, J. Virol. 76, 8560-8571.
159. Hardwicke, M. A., and Sandri-Goldin, R. M. (1994) The herpes simplex virus regulatory protein ICP27 contributes to the decrease in cellular mRNA levels during infection, J. Virol. 68, 4797-4810.
160. Corcoran, J. A., Hsu, W. L., and Smiley, J. R. (2006) Herpes simplex virus ICP27 is required for virus-induced stabilization of the ARE-containing IEX-1 mRNA encoded by the human IER3 gene, J.
Virol. 80, 9720-9729. 161. Glaunsinger, B. A., and Ganem, D. E. (2006) Messenger RNA
turnover and its regulation in herpesviral infection, Adv. Virus Res.
66, 337-394. 162. Jenner, R. G., Alba, M. M., Boshoff, C., and Kellam, P. (2001)
Kaposi's sarcoma-associated herpesvirus latent and lytic gene expression as revealed by DNA arrays, J. Virol. 75, 891-902.
163. Hoffmann, P. J., and Cheng, Y. C. (1978) The deoxyribonuclease induced after infection of KB cells by herpes simplex virus type 1 or type 2. I. Purification and characterization of the enzyme, J. Biol.
Chem. 253, 3557-3562. 164. Hoffmann, P. J., and Cheng, Y. C. (1979) DNase induced after
infection of KB cells by herpes simplex virus type 1 or type 2. II. Characterization of an associated endonuclease activity, J. Virol. 32, 449-457.
165. Bronstein, J. C., and Weber, P. C. (1996) Purification and characterization of herpes simplex virus type 1 alkaline exonuclease expressed in Escherichia coli, J. Virol. 70, 2008-2013.
166. Stolzenberg, M. C., and Ooka, T. (1990) Purification and properties of Epstein-Barr virus DNase expressed in Escherichia coli, J. Virol.
64, 96-104.

75
167. Shao, L., Rapp, L. M., and Weller, S. K. (1993) Herpes simplex virus 1 alkaline nuclease is required for efficient egress of capsids from the nucleus, Virology 196, 146-162.
168. Reuven, N. B., and Weller, S. K. (2005) Herpes simplex virus type 1 single-strand DNA binding protein ICP8 enhances the nuclease activity of the UL12 alkaline nuclease by increasing its processivity, J. Virol. 79, 9356-9358.
169. Pingoud, A., Fuxreiter, M., Pingoud, V., and Wende, W. (2005) Type II restriction endonucleases: structure and mechanism, Cell.
Mol. Life Sci. 62, 685-707. 170. Bujnicki, J. M., and Rychlewski, L. (2001) The herpesvirus alkaline
exonuclease belongs to the restriction endonuclease PD-(D/E)XK superfamily: insight from molecular modeling and phylogenetic analysis, Virus Genes 22, 219-230.
171. Kosinski, J., Feder, M., and Bujnicki, J. M. (2005) The PD-(D/E)XK superfamily revisited: identification of new members among proteins involved in DNA metabolism and functional predictions for domains of (hitherto) unknown function, BMC Bioinformatics 6, 172.
172. Pingoud, A., and Jeltsch, A. (2001) Structure and function of type II restriction endonucleases, Nucleic Acids Res 29, 3705-3727.
173. Goldstein, J. N., and Weller, S. K. (1998) The exonuclease activity of HSV-1 UL12 is required for in vivo function, Virology 244, 442-457.
174. Liu, M. T., Hsu, T. Y., Lin, S. F., Seow, S. V., Liu, M. Y., Chen, J. Y., and Yang, C. S. (1998) Distinct regions of EBV DNase are required for nuclease and DNA binding activities, Virology 242, 6-13.
175. Zuo, J., Thomas, W., van Leeuwen, D., Middeldorp, J. M., Wiertz, E. J., Ressing, M. E., and Rowe, M. (2008) The DNase of gammaherpesviruses impairs recognition by virus-specific CD8+ T cells through an additional host shutoff function, J. Virol. 82, 2385-2393.
176. Kovall, R., and Matthews, B. W. (1997) Toroidal structure of lambda-exonuclease, Science 277, 1824-1827.
177. Eklund, H., Uhlin, U., Farnegardh, M., Logan, D. T., and Nordlund, P. (2001) Structure and function of the radical enzyme ribonucleotide reductase, Prog. Biophys. Mol. Biol. 77, 177-268.
178. Wnuk, S. F., and Robins, M. J. (2006) Ribonucleotide reductase inhibitors as anti-herpes agents, Antiviral Res. 71, 122-126.
179. Liuzzi, M., Deziel, R., Moss, N., Beaulieu, P., Bonneau, A. M., Bousquet, C., Chafouleas, J. G., Garneau, M., Jaramillo, J., Krogsrud, R. L., Lagacé, L., McCollum, R.S., Nawoot, S. and Guindon, Y. (1994) A potent peptidomimetic inhibitor of HSV

76
ribonucleotide reductase with antiviral activity in vivo, Nature 372, 695-698.
180. Crute, J. J., Grygon, C. A., Hargrave, K. D., Simoneau, B., Faucher, A. M., Bolger, G., Kibler, P., Liuzzi, M., and Cordingley, M. G. (2002) Herpes simplex virus helicase-primase inhibitors are active in animal models of human disease, Nat. Med. 8, 386-391.
181. Cerqueira, N. M., Pereira, S., Fernandes, P. A., and Ramos, M. J. (2005) Overview of ribonucleotide reductase inhibitors: an appealing target in anti-tumour therapy, Curr. Med. Chem. 12, 1283-1294.
182. Wang, X., Zhenchuk, A., Wiman, K. G., and Albertioni, F. (2008) Regulation of p53R2 and its role as potential target for cancer therapy, Cancer Lett.
183. Tanaka, H., Arakawa, H., Yamaguchi, T., Shiraishi, K., Fukuda, S., Matsui, K., Takei, Y., and Nakamura, Y. (2000) A ribonucleotide reductase gene involved in a p53-dependent cell-cycle checkpoint for DNA damage, Nature 404, 42-49.
184. Nakano, K., Balint, E., Ashcroft, M., and Vousden, K. H. (2000) A ribonucleotide reductase gene is a transcriptional target of p53 and p73, Oncogene 19, 4283-4289.
185. Koshland, D. E., Jr. (1993) Molecule of the year, Science 262, 1953. 186. Guittet, O., Hakansson, P., Voevodskaya, N., Fridd, S., Graslund,
A., Arakawa, H., Nakamura, Y., and Thelander, L. (2001) Mammalian p53R2 protein forms an active ribonucleotide reductase in vitro with the R1 protein, which is expressed both in resting cells in response to DNA damage and in proliferating cells, J. Biol. Chem.
276, 40647-40651. 187. Hakansson, P., Hofer, A., and Thelander, L. (2006) Regulation of
mammalian ribonucleotide reduction and dNTP pools after DNA damage and in resting cells, J. Biol. Chem. 281, 7834-7841.
188. Bourdon, A., Minai, L., Serre, V., Jais, J.P., Sarzi, E., Aubert, S., Chrétien, D., de Lonlay, P., Paquis-Flucklinger, V., Arakawa, H., Nakamura, Y., Munnich,. A and Rötig, A. (2007) Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion. Nat. Genet. 39:703-704.




















