
Experimental and bioinformatic investigation of the proteolytic degradation of the C-terminal domain...
9
Experimental and bioinformatic investigation of the proteolytic degradation of the C-terminal domain of a fungal tyrosinase Greta Faccio a, b, ⁎, Mikko Arvas b , Linda Thöny-Meyer a , Markku Saloheimo b a Empa, Swiss Federal Laboratories for Materials Science and Technology, Laboratory for Biomaterials, Lerchenfeldstrasse 5, CH-9014 St. Gallen, Switzerland b VTT Technical Research Centre of Finland, P.O. Box 1000, FI-02044 VTT, 02044 Espoo, Finland abstract article info Article history: Received 6 September 2012 Received in revised form 11 December 2012 Accepted 12 December 2012 Available online 21 December 2012 Keywords: Proteolytic activation Protein processing C-terminal domain Fungal tyrosinase Sensitivity to proteolysis Proteolytic processing is a key step in the production of polyphenol oxidases such as tyrosinases, converting the inactive proenzyme to an active form. In general, the fungal tyrosinase gene codes for a ~60 kDa protein that is, however, isolated as an active enzyme of ~40 kDa, lacking the C-terminal domain. Using the secreted tyrosinase 2 from Trichoderma reesei as a model protein, we performed a mutagenesis study of the residues in proximity of the experimentally determined cleavage site which are possibly involved in the proteolytic process. However, the mutant forms of tyrosinase 2 were not secreted in a full-length form retaining the C-terminal domain, but they were processed to give a ~45 kDa active form. Aiming at explaining this phenomenon, we analysed in silico the properties of the C-terminal domain of tyrosinase 2, of 23 previously retrieved homologous tyrosinase sequences from fungi (C. Gasparetti, G. Faccio, M. Arvas, J. Buchert, M. Saloheimo, K. Kruus, Appl. Microbiol. Biotechnol. 86 (2010) 213–226) and of nine well-characterised polyphenol oxidases. Based on the results of our study, we ex- clude the key role of specific amino acids at the cleavage site in the proteolytic process and report an overall higher sensitivity to proteolysis of the linker region and of the whole C-terminal domain of fungal tyrosinases. © 2012 Elsevier Inc. All rights reserved. 1. Introduction Proteolytic processing is a post-translational modification in- volved in the maturation of different proteins. Among these, polyphe- nol oxidases such as tyrosinases from fungi are usually produced in a truncated form lacking the C-terminal ~ 20 kDa polypeptide sequence also referred to as C-terminal domain. Since tyrosinases, laccase and catechol oxidases share common phenolic substrates, they are gener- ally grouped under the term “polyphenol oxidase”. However, only tyrosinases oxidise mono-phenolic compounds, e.g. tyrosine. Most stud- ied tyrosinases such as the commercial enzyme from the common button mushroom Agaricus bisporus [1–4], from the bread mould Neurospora crassa [5] and the secreted fungal tyrosinase from the cellulolytic fungus Trichoderma reesei [6] are C-terminally processed. As a typical case, the gene of tyrosinase 2 from T. reesei coded for a 61.5 kDa protein. The ma- ture secreted tyrosinase had a significantly lower molecular mass of 43.2 kDa when overexpressed in the native host [6]. The commercially available tyrosinase from A. bisporus is produced as an active 44 kDa pro- tein although the gene codes for a full-length 63.8 kDa inactive protein [2,3,7]. Structurally similar enzymes such as catechol oxidase from sweet potato Ipomoea batatas [8], polyphenol oxidases from broad bean Vicia faba and grapes Vitis vinifera [9,10] are subject to analogous process- ing for maturation. In some cases, the full-length form with a molecular mass of ~ 60 kDa has been isolated and it was reported to undergo activa- tion either upon removal of the C-terminal domain by proteolysis [11–13] or, in a reversible manner, by loosening of the structure, e.g. after denatur- ing agents and detergents were used [14–18]. The function of the C-terminal domain is debated and it has been ascribed to form a shield over the active site and maintain the enzyme inactive [11,12,19]. Polyphe- nol oxidases are structurally related to the oxygen-binding proteins haemocyanins that, interestingly, do not naturally undergo proteolytic processing but acquire polyphenol oxidase activity upon proteolytic treat- ment [20]. Tyrosinases are copper-dependent enzymes able to oxidise mono- and di-phenolic molecules to the corresponding ortho-quinones. Ty- rosinases catalyse the first step of the melanin biosynthesis pathway by oxidising L-DOPA (L-3,4-dihydroxyphenylalanine) or tyrosine [21] that undergo subsequent non-enzymatic polymerisation. Like catechol oxidases and haemocyanins, tyrosinases belong to the class of type-3 copper proteins. These are characterised by the presence of two triads of histidine residues coordinating the two copper ions of the active site (CuA and CuB). Many studies, both experimental and in silico, have identified the residues that are involved in the fold- ing and function of the enzymes [22]. Two sequence motifs have been identified in the proximity of the cleavage region between the core domain and the C-terminal domain of fungal tyrosinases (Fig. 1). While the first motif, the tyrosine motif (Y-motif) [22,23], is highly conserved and it interacts with the N-terminal extremity of the Journal of Inorganic Biochemistry 121 (2013) 37–45 ⁎ Corresponding author at: Empa-Swiss Federal Laboratories for Materials Science and Technology, Lerchenfeldstrasse 5 CH-9014 St. Gallen, Switzerland. Tel.: +41 58 765 7262; fax: +41 58 765 77 88. E-mail addresses: [email protected] (G. Faccio), mikko.arvas@vtt.fi (M. Arvas), [email protected] (L. Thöny-Meyer), markku.saloheimo@vtt.fi (M. Saloheimo). 0162-0134/$ – see front matter © 2012 Elsevier Inc. All rights reserved. http://dx.doi.org/10.1016/j.jinorgbio.2012.12.006 Contents lists available at SciVerse ScienceDirect Journal of Inorganic Biochemistry journal homepage: www.elsevier.com/locate/jinorgbio
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Experimental and bioinformatic investigation of the proteolytic degradation of the C-terminal domain...

Journal of Inorganic Biochemistry 121 (2013) 37–45
Contents lists available at SciVerse ScienceDirect
Journal of Inorganic Biochemistry
j ourna l homepage: www.e lsev ie r .com/ locate / j inorgb io
Experimental and bioinformatic investigation of the proteolytic degradation of theC-terminal domain of a fungal tyrosinase
Greta Faccio a,b,⁎, Mikko Arvas b, Linda Thöny-Meyer a, Markku Saloheimo b
a Empa, Swiss Federal Laboratories for Materials Science and Technology, Laboratory for Biomaterials, Lerchenfeldstrasse 5, CH-9014 St. Gallen, Switzerlandb VTT Technical Research Centre of Finland, P.O. Box 1000, FI-02044 VTT, 02044 Espoo, Finland
⁎ Corresponding author at: Empa-Swiss Federal LaboraTechnology, Lerchenfeldstrasse 5 CH-9014 St. Gallen, S7262; fax: +41 58 765 77 88.
E-mail addresses: [email protected] (G. Faccio),[email protected] (L. Thöny-Meyer), markku.saloh
0162-0134/$ – see front matter © 2012 Elsevier Inc. Allhttp://dx.doi.org/10.1016/j.jinorgbio.2012.12.006
a b s t r a c t
a r t i c l e i n f oArticle history:Received 6 September 2012Received in revised form 11 December 2012Accepted 12 December 2012Available online 21 December 2012
Keywords:Proteolytic activationProtein processingC-terminal domainFungal tyrosinaseSensitivity to proteolysis
Proteolytic processing is a key step in the production of polyphenol oxidases such as tyrosinases, converting theinactive proenzyme to an active form. In general, the fungal tyrosinase gene codes for a ~60 kDa protein that is,however, isolated as an active enzyme of ~40 kDa, lacking the C-terminal domain. Using the secreted tyrosinase 2from Trichoderma reesei as amodel protein, we performed amutagenesis study of the residues in proximity of theexperimentally determined cleavage site which are possibly involved in the proteolytic process. However, themutant forms of tyrosinase 2 were not secreted in a full-length form retaining the C-terminal domain, but theywere processed to give a ~45 kDa active form. Aiming at explaining this phenomenon, we analysed in silico theproperties of the C-terminal domain of tyrosinase 2, of 23 previously retrieved homologous tyrosinase sequencesfrom fungi (C. Gasparetti, G. Faccio, M. Arvas, J. Buchert, M. Saloheimo, K. Kruus, Appl. Microbiol. Biotechnol. 86(2010) 213–226) and of nine well-characterised polyphenol oxidases. Based on the results of our study, we ex-clude the key role of specific amino acids at the cleavage site in the proteolytic process and report an overallhigher sensitivity to proteolysis of the linker region and of the whole C-terminal domain of fungal tyrosinases.
© 2012 Elsevier Inc. All rights reserved.
1. Introduction
Proteolytic processing is a post-translational modification in-volved in the maturation of different proteins. Among these, polyphe-nol oxidases such as tyrosinases from fungi are usually produced in atruncated form lacking the C-terminal ~20 kDa polypeptide sequencealso referred to as C-terminal domain. Since tyrosinases, laccase andcatechol oxidases share common phenolic substrates, they are gener-ally grouped under the term “polyphenol oxidase”. However, onlytyrosinases oxidise mono-phenolic compounds, e.g. tyrosine. Most stud-ied tyrosinases such as the commercial enzyme from the common buttonmushroom Agaricus bisporus [1–4], from the bread mould Neurosporacrassa [5] and the secreted fungal tyrosinase from the cellulolytic fungusTrichoderma reesei [6] are C-terminally processed. As a typical case, thegene of tyrosinase 2 from T. reesei coded for a 61.5 kDa protein. The ma-ture secreted tyrosinase had a significantly lower molecular mass of43.2 kDa when overexpressed in the native host [6]. The commerciallyavailable tyrosinase from A. bisporus is produced as an active 44 kDa pro-tein although the gene codes for a full-length 63.8 kDa inactive protein[2,3,7]. Structurally similar enzymes such as catechol oxidase fromsweet potato Ipomoea batatas [8], polyphenol oxidases from broad bean
tories for Materials Science andwitzerland. Tel.: +41 58 765
[email protected] (M. Arvas),[email protected] (M. Saloheimo).
rights reserved.
Vicia faba and grapes Vitis vinifera [9,10] are subject to analogous process-ing for maturation. In some cases, the full-length form with a molecularmass of ~60 kDa has been isolated and it was reported to undergo activa-tion either upon removal of the C-terminal domain by proteolysis [11–13]or, in a reversiblemanner, by loosening of the structure, e.g. after denatur-ing agents and detergents were used [14–18]. The function of theC-terminal domain is debated and it has been ascribed to form a shieldover the active site andmaintain the enzyme inactive [11,12,19]. Polyphe-nol oxidases are structurally related to the oxygen-binding proteinshaemocyanins that, interestingly, do not naturally undergo proteolyticprocessing but acquire polyphenol oxidase activity uponproteolytic treat-ment [20].
Tyrosinases are copper-dependent enzymes able to oxidise mono-and di-phenolic molecules to the corresponding ortho-quinones. Ty-rosinases catalyse the first step of the melanin biosynthesis pathwayby oxidising L-DOPA (L-3,4-dihydroxyphenylalanine) or tyrosine[21] that undergo subsequent non-enzymatic polymerisation. Likecatechol oxidases and haemocyanins, tyrosinases belong to the classof type-3 copper proteins. These are characterised by the presenceof two triads of histidine residues coordinating the two copper ionsof the active site (CuA and CuB). Many studies, both experimentaland in silico, have identified the residues that are involved in the fold-ing and function of the enzymes [22]. Two sequence motifs have beenidentified in the proximity of the cleavage region between the coredomain and the C-terminal domain of fungal tyrosinases (Fig. 1).While the first motif, the tyrosine motif (Y-motif) [22,23], is highlyconserved and it interacts with the N-terminal extremity of the

Fig. 1. Sequence motifs and residues identified in tyrosinases. Distances and residue po-sitions are related to the tyrosinase 2 from T. reesei. The 18-residue-long signal sequenceis reported as a thick line. The core domain is delimited by the N-terminal arginine andthe C-terminal tyrosine motif [25]. The YG motif is retained in the mature enzyme andits role has not been established yet [6,13,22]. The copper binding motifs A (CuA, H81–
X21–C103–P–H105–X8–H114) and B (CuB, H271–X3–H275–X24–H300) are indicated.
38 G. Faccio et al. / Journal of Inorganic Biochemistry 121 (2013) 37–45
globular core in the three-dimensional structure [8,24], the secondmotif is the tyrosine-glycine motif (YG-motif) that is not always pres-ent and whose role is not clear [22]. The YG-motif is especially foundin fungal polyphenol oxidases [22].
The active core domain of polyphenol oxidases is characterised bythe presence of highly conserved residues such as a histidine patternfor cofactor binding and additional aromatic residues [23,25]. On theother hand, the C-terminal protein region following the Y-motif showsa significantly lower level of homology and its length significantly variesamong proteins of different origin. To our knowledge, no study hasreported the isolation of the C-terminal domain in a stable form andits characterisation. Although the three-dimensional structures of fivetyrosinase and catechol oxidase have been solved, no structurecontaining the C-terminal domain could be obtained [26–30]. However,tyrosinase from A. bisporus was crystallised in a complex with a second,smaller protein that had a lectin-like structure [26], and the bacterialtyrosinase from Streptomyces castaneoglobisporus in complex with aso-called caddie protein ORF378 [31]. Previously, Inlow et al. haveanalysed the sequence features of 11 polyphenol oxidases from plantand 5 from fungi, e.g. the two intracellular enzymes from A. bisporus,the shiitake mushroom Lentinula edodes and the filamentous fungiN. crassa and Podospora anserina [22,25]. This study led to the identifica-tion of the site of proteolytic processing within a linker region that islocated between the core and the C-terminal domain of fungal polyphe-nol oxidases; it also predicted the presence of elements of secondarystructure in the C-terminal domain [25].
Table 1Oligonucleotides and plasmids used in this study.
Mutant Sequence in the proteina Mutation P
TrTyr2 Y395GPNSG↓KKRNAPR407 – pTrTyr2-NoCleav Y395GPNSGGSGSNAPS407 K401G,
K402S,R403G,R407S
p
TrTyr2-FactorXa Y395GPNSGGSGIEGR↓DF409(native R407 as cleavage site)
K401G,K402S,R403G,N404I,A405E,P406G
p
TrTyr2-noLys Y395GPNSGGRRNAPR407 K401G,K402R
p
TrTyr2-STOP Y395GPNSG400 K401Stop p
TrTyr2-YG A395AGPNSGKKRNAPR407 Y395A,G396A
p
a The cleavage site in T. reesei is indicated by an arrow.
In this study, we first aimed at producing a full-length form of tyros-inase 2 from T. reesei to investigate the proteolytic processing and forstructure determination. Mutant variants of tyrosinase 2 were lackingthe residues that are located in proximity of the cleavage site and thatare possibly recognised by specific proteases. These mutant proteinswere produced in T. reesei in a secreted form and, opposed to prediction,were still lacking the C-terminal domain. Suspecting an overall instabil-ity and sensitivity to proteolysis of the whole C-terminal domain, weanalysed whether this domain possessed sequence features that couldexplain the phenomenon. Since tyrosinase 2 is the only secreted fungaltyrosinase characterised to date, we compared the features of itsC-terminal domain to the ones of 22 similar predicted tyrosinases andnine well-characterised polyphenol oxidases described in literature.The presence of sequence features typical of unstable proteins, of intrin-sically disordered regions and of proteolytic sites was considered.
2. Experimental
2.1. Cloning and overexpression of tyrosinase 2 mutants
The expression plasmid pMS190 carrying the gene for tyrosinase 2between the cellobiohydrolase I (cbh1) promoter and terminator wasused as template for the introduction of the desired mutations byoverlap-extension PCR [32]. Two mutagenic oligonulceotides with op-posite direction were designed for each desired mutation (Table 1).First, two overlapping mutated fragments were produced by PCRusing a mutagenic oligonucleotide (Table 1) and an oligonucleotideannealing to either the cbh1 promoter (cbh1-prom, and including a re-striction site SnaBI site), or to the cbh1 terminator (cbh1-term, and in-cluding a SpeI site). PCR reactions were carried out with the followingconditions: 1 min of denaturation at 98 °C and 18 cycles 18 cycles of98 °C for 1 min, annealing at 65 °C–1 °C per cycle for 50 s and exten-sion at 72 °C for 1 min. In a second step, the fragments producedwere isolated and combined. The PCR reaction included (i) 7 cycles ofdenaturation at 98 °C for 30 s, (ii) annealing at 65 °C–0.5 °C/cycle for45 s and (iii) extension at 72 °C for 1 min 20 s. Oligonucleotidescbh1-prom and cbh1-term were then added and 18 additional cycleswere carried out to produce the full-length mutated sequence. Ampli-fication was performed with Phusion polymerase (New EnglandBiolabs, Espoo, Finland). After double-digestion with SnaBI and SpeI,the fragments were cloned into pMS186 to give the corresponding ex-pression plasmids (Table 1). Mutagenised plasmids were firsttransformed into E. coli DH5α and the presence of the desiredmutationwas checked by sequencing. Correct plasmids were transformed intothe T. reesei strain VTT-D-00775 with a protoplast-based method
lasmid Sequence of oligos
MS190 –
GF013 FW: ggg ccc aac tcg ggc ggc tcc ggc aac gcc ccg tcc gac ttc ttg agc;REV: gct caa gaa gtc gga cgg ggc gtt gcc gga gcc gcc cga gtt ggg ccc
GF014 FW: ggg ccc aac tcg ggc ggc tcc ggc atc gag ggc cgc gac ttc ttg agc;REV: gct caa gaa gtc gcg gcc ctc gat gcc gga gcc gcc cga gtt ggg ccc
GF015 FW: ggg ccc aac tcg ggc ggc cgc cgc aac gcc ccg cgc gac ttc ttg agc;REV: gct caa gaa gtc gcg cgg ggc gtt gcg gcg gcc gcc cga gtt ggg ccc
GF016 FW: ggg ccc aac tcg ggc taa aag cgc aac gcc ccg cgc gac ttc ttg agc;REV:gct caa gaa gtc gcg cgg ggc gtt gcg ctt tta gcc cga gtt ggg ccc
GF017 FW: cca gct ggc cgc ccc caa tcg gg;REV: ccc gag ttg ggg gcg gcc agc tgg

Table 2Sequences of secreted long fungal tyrosinases analysed in this work.
Organism Nr. and Sequencea Length (aa)
Botrytis cinerea 1. GenBank ID: EDN17470.1 (BC1G.00048) 5952. GenBank ID: XP_001556707.1 (BC1G.04092) 5323. GenBank ID: XP_001546493.1 (BC1G.14990) 670
Fusarium graminearum 4. GenBank ID: XP_385804.1 (FG05628) 5565. GenBank ID: XP_389698.1 (FG09522.1) 575
Fusarium oxysporum 6. GenBank ID: EGU82344.1 (FOXG.06218) 5727. FOXG.09259b 578
Fusarium verticillioides 8. FVEG.04071b 5729. FVEG.06859b 579
Histoplasma capsulatum 10. GenBank ID: XP_001540422.1 (HCAG.04262.1) 636Magnaporthe grisea 11. GenBank ID: A4RK91 640Nectria haematococca 12. GenBank ID: XP_003048894.1 (NECHA0047204) 600
13. GenBank ID: XP_003046195.1 (NECHA0066755) 566Phaeosphaeria nodorum 14. GenBank ID: Q0UNZ8 691Coccidioides immitis 15. GenBank ID: Q1DQ30 658Chaetomium globosum 16. GenBank ID: Q2HEC0 644Neurospora crassa 17. GenBank ID: Q7S218 (NCU05978.1) 767
18. GenBank ID: Q7SFK3 (NCU08615.1) 692Sclerotinia sclerotiorum 19. GenBank ID: XP_001597382.1 (SS1G.01576.1) 602
20. GenBank ID: XP_001594917.1 (SS1G.04725.1) 59321. GenBank ID: XP_001585847.1 (SS1G.13364.1) 636
Trichoderma reesei 22. GenBank ID: CAL90884.1 (tre45445, TrTyr2) 56123. GenBank ID: EGR46236.1 (tre50793) 490
a In parenthesis as reported in [34].b Sequences available at http://www.broadinstitute.org/.
39G. Faccio et al. / Journal of Inorganic Biochemistry 121 (2013) 37–45
essentially as described in [33]. 200 transformants were selected firstby streaking them twice on plates containing 125 μg/mL hygromycinB. After purification of the transformants to single-spore clones, posi-tives were grown on plates containing L-tyrosine (trichoderma mini-mal medium TrMM, 2% lactose, 1% potassium phthalate at pH 5.5,0.1 mM CuSO4 and 1% L-tyrosine). Selected transformants weregrown in small shake-flasks containing 50 mL of TrMM [33], 2% spentgrain, 4% lactose and 1 mM CuSO4 for 9 days monitoring growth asdrop in the pH of the medium. Activity in the cell-free culture mediumwas assayed spectrophotometrically as absorbance at 475 nm after9 min of incubation with 15 mM L-DOPA as substrate in 0.1 M sodiumphosphate buffer at pH 7.0. Proteins present in the culture mediumwere analysed by SDS PAGE (12% acrylamide) and by Western blotwith anti-tyrosinase 2 antibodies. Specific antibodies His-Probe forthe six-histidine tag were purchased from Santa Cruz biotechnologyInc. (Santa Cruz, USA).
2.2. Sequences analysed
Twenty-three sequences including tyrosinase 2 were previouslyidentified as secreted ‘long tyrosinases’ in a genome mining study[34]; all were from Ascomycota, carried a C-terminal domain and in-cluded the well-studied tyrosinase 2 from T. reesei (sequence 22 inTable 2) [6,35]. All proteins carried a central Interpro tyrosinasemotif (IPR002227) and signal peptide for the secretory pathway[36]. Accession numbers of the 23 protein sequences analysed
Table 3Sequences of characterised polyphenol oxidases and haemocyanins analysed.
Origin Organism Proteina
Fungus Agaricus bisporus PPO1Fungus Agaricus bisporus PPO2Fungus Pholiota nameko TYRFungus Trametes sanguineus TYRPlant Ipomoea batatas COFungus Neurospora crassa TYRPlant Vicia faba TYRPlant Vitis vinifera PPOBacterium Verrucomicrobium spinosum TYRAnimal Octopus dofleini Hc
a Abbreviations: PPO polyphenol oxidase, TYR tyrosinase, CO catechol oxidase and Hc ha
(Table 2) and the well-studied polyphenol oxidases reported toundergo proteolytic processing are summarised in Table 3.
2.3. Bioinformatic tools
All bioinformatic softwares used in this work are freely availableand have been used with the default parameters, unless otherwisestated. The presence of regions of intrinsic disorder was predictedwith the recently developed software PrDOS (http://prdos.hgc.jp[37], 5% prediction false positive rate and a two-state prediction)and the meta-predictor PONDR-FIT [38]. Low complexity regionswere identified with SMART (http://smart.embl-heidelberg.de) [39].The instability index of whole protein and protein fraction was calcu-lated with Protein Instability Index Calculator (http://www.ux1.eiu.edu/~grperiyannan/protein_instability.html) [40]. Sequence align-ments were performed with CLUSTALW [41] and the elements of sec-ondary structure were predicted from the alignment with the JPredsoftware embedded in the Jalview suite [42]. Trypsin and chymotryp-sin cleavage sites were detected with the improved version ofPeptidecutter specifically designed for trypsin, and predicted siteswith a cleavage probability higher than 70% were selected [43,44].Pairwise sequence alignments for the determination of identity levelsbetween sequences were performed with EMBOSS needle–pairwisesequence alignment from EMBL-EBI (Gap open penalty 10.0, gap ex-tension penalty: 0.5, gap closing penalty 10.0, http://www.ebi.ac.uk/Tools/psa/emboss_needle). According to [40], dipeptides with a
Sequence Length Reference
Genbank ID: Q00024.1 568 [2,26,67]Genbank ID: CAA11562.1 556 [2,26,67]Genbank ID: BAF74396.1 625 [11,13]Genbank ID: AAX46018.1 618 [68]Genbank ID: CAC83609.1 496 [14,69]Genbank ID: AAA33618.1 621 [5,69,70,71]Genbank ID: Q06215 606 [9,16]Genbank ID: P43311 607 [29]Genbank ID: ZP_02925214 518 [12]Genbank ID: 1JS8_A 394 [72]
emocyanins.

Fig. 2. Western blot analysis of culture medium expressing TrTyr2-FactorXa (lane 1),TrTyr2-noLys (lane 2), TrTyr2-NoCleav (lane 3), TrTyr2-YG (lane 8) and originalTrTyr2 (lane 5) and in purified form (2.5 μg, lane 7). Culture medium from the parentalstrain (lane 6) and molecular weight markers (lane 4, in kDa) are also indicated. TheWestern blot was hybridised with anti-TrTyr2 antibodies and the position of tyrosinase2 is indicated by an arrow.
40 G. Faccio et al. / Journal of Inorganic Biochemistry 121 (2013) 37–45
instability weight value (DIWV) above 10 such as MH, YM, LW, HY,HI, NI, SP, RR, MS, QS, IS, EE, MP, KR, KM, CM, CH, CT, RW, EC, AC,SC, IE, LQ, CW and LW were considered as recurrent in unstable pro-teins. Dipeptides with a negative DIWV were considered characteris-tic for stable proteins [40]. The presence of PEST (Pro–Glu–Ser–Thr)sequences was identified with the software Epestfind by EMBOSS6.3.1 (http://bioweb2.pasteur.fr/docs/EMBOSS/epestfind.html) [45].The isoelectric point of proteins was calculated with the ProtParamtool from Expasy [44] (http://web.expasy.org/protparam/). Box-and-whiskers plots were prepared with Excel (Microsoft Office, version2011) and outliers were defined as values below the first quartileminus 1.5-fold the interquartile value, or above the third quartileplus 1.5-fold the interquartile value. Outliers were not consideredfor the statistical analysis of the result that was performed with thenon-parametric Wilcoxon–Mann–Whitney rank sum test.
3. Results and discussion
3.1. Mutagenesis study and protein expression
Aiming at producing the T. reesei tyrosinase 2 in the unprocessed,full-length form of 543 amino acids (lacking the signal peptide of 18amino acids), and assuming the truncation took place at the proteinlevel and not at themRNA level, we identified the residues surroundingthe site of C-terminal proteolytic processing in T. reesei and the onespossibly involved in the determination of the final C-terminal end, i.e.the YG-motif [22]. The C-terminal residue of the mature tyrosinase 2in T. reesei was G400 [6], located 30 and 4 residues C-terminally tothe Y-motif and YG-motif, respectively. The cleavage site was foundto be followed by two K residues and few R residues. K and R residuesare potential cleavage sites for lysyl-endopeptidases (LysC or LysN),trypsin-like proteases and Kex2-like proteases (serine protease) thatare present in fungi [46–49]. Assuming that the presence of these res-idues would be the only determining factor for the removal of theC-terminal domain, we conducted a site-directed mutagenesis studywherein these residues were substituted by either G or S residues(Table 2). Thus, we constructed forms of tyrosinase 2 mutated in thisregion while lacking K (TrTyr2–noLys) to evaluate the importance ofthis residue and possibly discriminate between the role of a LysN anda Kex2-like protease. Moreover, a mutant lacking both K and R residues(TrTyr2–NoCleav) and having the whole cleavage site replaced by aFactor Xa recognition site (TrTYr2–FactorXa) was constructed. Thislatter form was predicted to be produced in full-length and it shouldallow assessing how the removal of the C-terminal domain affects theenzymatic activity in vitro. In order to assess the role of the wholeC-terminal region on protein maturation, we constructed a mutant ty-rosinase 2 lacking the C-terminal domain completely by placing a STOPcodon after G400 (TrTyr2–STOP). Aiming at clarifying the role of theYG-motif, these two amino acids were substituted by an alanine–alanine dipeptide in mutant TrTyr2-YG.
Mutations were introduced in the expression plasmid pMS190containing the tyrosinase 2 gene by overlap-extension PCR. Mutationsin the resulting plasmids pGF013-17 were confirmed by sequencingand these were then transformed in the same T. reesei host strain usedfor the production of native tyrosinase 2 (strain VTT-D-00775).Transformants were selected for resistance to antibiotic by streakingthem twice on plates containing hygromycin B (125 μg/mL). Positiveswere tested on tyrosine-containing plates and cultivated in small shakeflasks (culture volume 50 mL) in inducing medium supplementedwith 1 mM copper. Unexpectedly, activity could be detected in fewtransformants on tyrosine-containing plates and in the cell-freemediumof all cultures. The number of positive transformants obtained per con-struct was: 14 for the TrTYR2-NoCleav, 10 for TrTYR2-FactorXa, 6 forTrTYR2-noLys and 5 for TrTyr2-YG. Activity was due to the productionof the mutant tyrosinases 2 in good amounts but in a C-terminallycleaved form, as SDS PAGE and Western Blot analyses of the secreted
proteins showed (Fig. 2). No additional protein band was visible bySDS PAGE analyses of the culture medium of clones positive to antibioticselection and negative on tyrosine-containing plates, especially at60 kDa (data not shown). All mutant forms of tyrosinase 2 were pro-duced with a similar molecular weight that corresponded to the one ofthe native enzyme, as was shown by SDS PAGE (not shown) and West-ern Blot (Fig. 2). For purification purposes, tyrosinase 2 carried asix-histidine tag at the C-terminus that, however, could not be exploiteddue to the removal of the whole C-terminal domain during protein pro-duction [6]. No signal was detected in Western blot analyses of culturemedia containing the mutant forms of tyrosinase 2 produced in thisstudy with specific anti-histidine tag antibodies.
Additional processing sites were clearly present in the C-terminaldomain and possibly in the surroundings of G400 of tyrosinase 2. Thisshowed that the residues involved in the proteolytic processing ofthe C-terminal domain were not uniquely the ones surrounding theexperimentally determined C-terminus, nor nearby R or K residues,and not the YG-motif. No transformant was obtained for the TrTyr2-STOP construct and the procedure was performed twice.
3.2. Properties of the sequences analysed
With the aim of analysing the conserved features of the C-terminaldomain of tyrosinase 2, we first performed a sequence alignment to po-tentially secreted tyrosinases from fungi previously identified with a ge-nomemining study [34]. All 22 sequences of secreted fungal tyrosinasespreviously identified carried a tyrosinase Interpro domain (IPR002227)and a C-terminal stretch following the Y-motif (C-terminal domain).The proteins had the conserved six-histidine pattern HA1–X20–23–
HA2–X8–HA3 and HB1–X3–HB2–X22–35–HB3, where X is any aminoacid, typical of fungal tyrosinases carrying a C-terminal domain. Thecharacteristics of the N-terminal region and active domain of theseproteins were discussed in [34].
The YG-motif is not found in tyrosinases from plant and bacteria. Amore general version of the YG-motif such as ΦX-motif, where Φ is anaromatic residue and X is any amino acid, most often G, could thus beidentified. The traditional YG-motif was conserved in the majority ofthe sequences analysed but corresponding YN, YK, WG and YS dipep-tides were present in some sequences instead. Interestingly, wheneverthe first residue was not a Y but aW residue, themotif was preceded byan aromatic W residue (Fig. S1 C). Nevertheless, we will keep referringto this motif as YG-motif for clarity purposes throughout the manu-script. All sequences carried the YG-motif, or variants of it.
Well-characterised tyrosinases reported in the literature to undergoproteolytic processing have also been analysed and the results com-pared. The 23 sequences shared an average of 26.9% and 26.5% sequence

41G. Faccio et al. / Journal of Inorganic Biochemistry 121 (2013) 37–45
identity with the well-studied fungal enzymes PPO1 from A. bisporusand tyrosinase from Pholiota nameko and a lower 18.8% and 15.9% se-quence identity with the bacterial tyrosinase from Verrucomicrobiumspinosum and the plant enzyme from V. vinifera, respectively.
Sequences including tyrosinase 2 from T. reesei (Table 2) were firstanalysed in terms of length and domain composition and compared toother known tyrosinases (Table 3). Sequences had an average length of608±60 amino acids with an average 370-residue long core domain(lacking the N-terminal signal sequence) and a 218±52-residueslong C-terminal domain after the Y-motif (36% of the whole sequence,Fig. S1 panels A and B). These values also represented the features ofwell-studied intracellular tyrosinases such as PPO1 and PPO2 fromA. bisporus that had a core region of 355 and 351 amino acids and aC-terminal extension of 213 and 215 residues, respectively. Similarly,tyrosinase from N. crassa had a core domain made up by 377 aminoacids and a C-terminal domain of 244 residues that is partially removedduring the production of the active enzyme. The C-terminal domainconstituted up to 30–40% of the amino acidic sequence of the tyrosi-nase. For example, a 213-residue long sequence following the tyrosinemotif in tyrosinase PPO1 from A. bisporus accounted for 43% of the pre-cursor protein sequence. In tyrosinase 2 from T. reesei the C-terminaldomain represented 34% of the full-length protein.
3.3. Amino acidic and dipeptide composition
As early as 1986, specific amino acids have been found to be morerecurrent in the sequence of unstable proteins and protein regions
Fig. 3. Box-and-whiskers plot showing the presence of specific residues (A–C) and dipeptiderally occurring in low complexity sequences such as QSPEK (B) and residues commonly founanalysed. Content of dipeptides recurrent in stable proteins (D) and highly recurrent inC-terminal domain are shown. Either the YG-motif or Y-motif was considered as landmarkdifferences (p-valueb0.01 in Wilcoxon–Mann–Whitney rank sum test) are indicated by ast
that preferably undergo degradation [50]. Among them, N, K and Gare more present in stable proteins andM, Q, P, E and S in unstable pro-teins [40,50]. Residues S, G and E are reported to recur in linker regions,which are regions with pronounced flexibility [51]. We thus analysedthe distribution of these residues in the full-length protein, in the coreregion (delimited by the cleavage site of the signal sequence and eitherthe Y-motif or the YG-motif) and in the respective C-terminal region.
Amino acids such as E were found significantly enriched in theC-terminal domain, with an average of 5.3–5.4% vs. 3.3–3.5% in thecore region, and a p-valueb0.001 in Wilcoxon–Mann–Whitney ranksum test (Fig. S2, panel A). Residues such as P, T and Q were underrep-resented in this domain (Fig. S2, panels B, C and F). Consideringproteolysis-sensitive residues such as K and R, the former was morerepresented in the C-terminal region (with an average of 2.5–2.7% inthe core vs. 5.3–5.4% in the C-terminal domain, p-valueb0.01 inWilcoxon–Mann–Whitney rank sum test) whereas no difference wasevident for the latter (Fig. S2, panels D and E). When specific aminoacids were analysed in groups, a certain degree of variability among se-quences and no statistical difference in the presence of PEST (P, E, S andT) residues was found between core and C-terminal region (Fig. 3,panel A). However, two sequences were predicted to carry a PESTsequence in the C-terminal domain by the Epestfind Software, e.g. a42-amino acid long PEST sequence starting at position 632 inQ0UNZ8 from Phaeosphaeria nodorum and a 15-amino acid long PESTsequence starting at position 589 in Q1DQ30 from Coccidioides immitis.Moreover, a statistically lower content of hydrophobic amino acidssuch as V, L, I, M, F, W, Y and a higher presence of polar and charged
es (D, E). The content of PEST residues common in unstable protein (A), residues gen-d in structured protein regions such as VLIMFWY (C) are reported for the 23 sequencesunstable proteins (E) in the sequence of the core (lacking the signal sequence) ands between the core (lacking the signal sequence) and C-terminal domain. Significanterisks and outliers by dots.
42 G. Faccio et al. / Journal of Inorganic Biochemistry 121 (2013) 37–45
amino acids such as Q, S, P, E and K indicated the presence of regionscharacterised by intrinsic disorder in the C-terminal domains(p-valueb0.01 in Wilcoxon–Mann–Whitney rank sum test, Fig. 3,panels B and C) [51]. According to this, the C-terminal domain wascharacterised by intrinsic disorder, when compared to the core region,and this finding was further investigated.
The distribution of dipeptides varies between stable and unstableprotein independently from their physiological role [40]. The coreand C-terminal region were analysed for the presence of dipeptidesdescribed as common in stable or unstable proteins. The C-terminal re-gion and especially the region between the Y-motif and the YG-motif(part of the linker region) carried a higher amount of dipeptidescharacteristic of highly unstable proteins a lower amount of dipeptidestypical of stable proteins (p-valueb0.01 in Wilcoxon–Mann–Whitneyrank sum test, Fig. 3 panels D and E). This did not affect the calculationof the overall instability index that showed no significant differencebetween the core domain and the C-terminal domain, e.g. average38.8±5.3 in the core domain until the Y-motif and 39.5±6.8 in theC-terminal region (not shown).
3.4. Low complexity, intrinsically disordered regions and secondarystructure prediction
Sequences were analysed by SMART software for the presence ofregions characterised by low complexity. 12 of the proteins analysedcarried a low-complexity region localised in the C-terminal domain.In five of them, including tyrosinase 2, this was localised in proximityof the YG-motif, and seven of them had two or more (Fig. S3).Low-complexity regions ranged from 8 to 32 amino acids in lengthwith an average of 15. Remarkably, they constituted 23% and 22% ofthe C-terminal domain of proteins Q7S218 and Q7SFK3 from N. crassa.Low-complexity regions were also predicted in the core domain of nineof the sequences analysed (Fig. S3).
The portion of protein characterised by intrinsic disorder, as predict-ed by PrDOS, was significantly higher in the C-terminal domain. An av-erage of 27% of the C-terminal domain was included in intrinsicallydisordered regions, compared to an average of 7% in the core region.
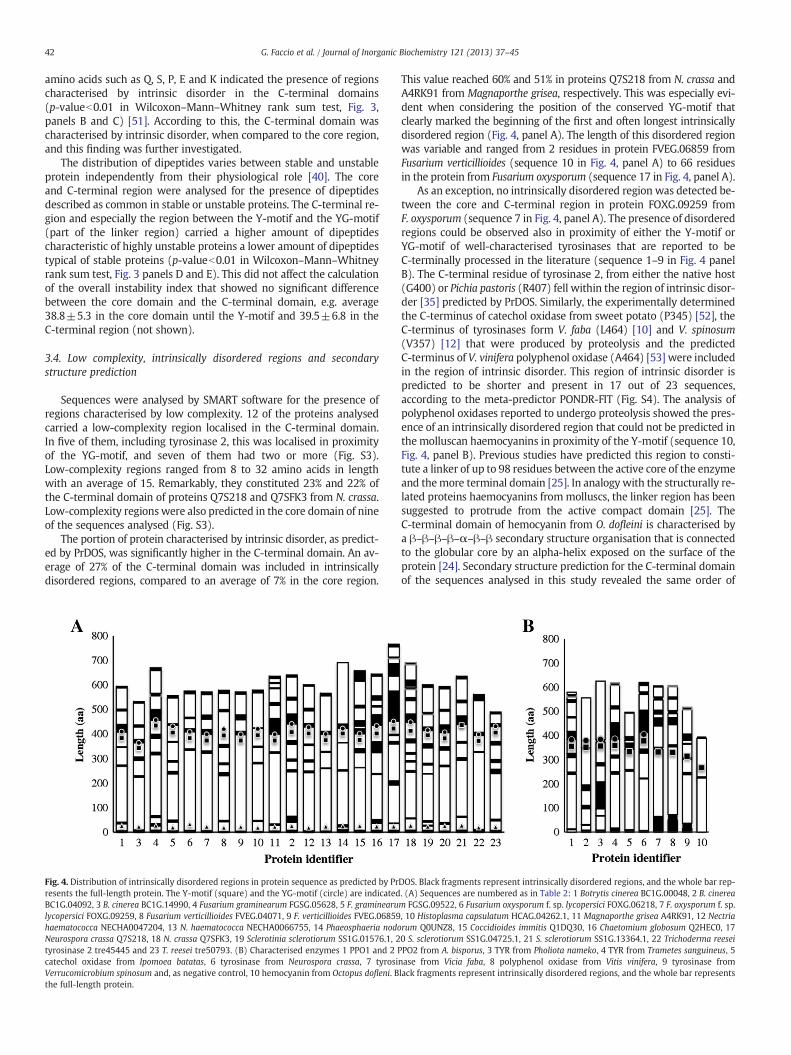
Fig. 4. Distribution of intrinsically disordered regions in protein sequence as predicted by Prresents the full-length protein. The Y-motif (square) and the YG-motif (circle) are indicatedBC1G.04092, 3 B. cinerea BC1G.14990, 4 Fusarium graminearum FGSG.05628, 5 F. graminearulycopersici FOXG.09259, 8 Fusarium verticillioides FVEG.04071, 9 F. verticillioides FVEG.06859haematococca NECHA0047204, 13 N. haematococca NECHA0066755, 14 Phaeosphaeria nodoNeurospora crassa Q7S218, 18 N. crassa Q7SFK3, 19 Sclerotinia sclerotiorum SS1G.01576.1, 2tyrosinase 2 tre45445 and 23 T. reesei tre50793. (B) Characterised enzymes 1 PPO1 and 2 Pcatechol oxidase from Ipomoea batatas, 6 tyrosinase from Neurospora crassa, 7 tyrosiVerrucomicrobium spinosum and, as negative control, 10 hemocyanin from Octopus dofleni. Bthe full-length protein.
This value reached 60% and 51% in proteins Q7S218 from N. crassa andA4RK91 from Magnaporthe grisea, respectively. This was especially evi-dent when considering the position of the conserved YG-motif thatclearly marked the beginning of the first and often longest intrinsicallydisordered region (Fig. 4, panel A). The length of this disordered regionwas variable and ranged from 2 residues in protein FVEG.06859 fromFusarium verticillioides (sequence 10 in Fig. 4, panel A) to 66 residuesin the protein from Fusarium oxysporum (sequence 17 in Fig. 4, panel A).
As an exception, no intrinsically disordered region was detected be-tween the core and C-terminal region in protein FOXG.09259 fromF. oxysporum (sequence 7 in Fig. 4, panel A). The presence of disorderedregions could be observed also in proximity of either the Y-motif orYG-motif of well-characterised tyrosinases that are reported to beC-terminally processed in the literature (sequence 1–9 in Fig. 4 panelB). The C-terminal residue of tyrosinase 2, from either the native host(G400) or Pichia pastoris (R407) fell within the region of intrinsic disor-der [35] predicted by PrDOS. Similarly, the experimentally determinedthe C-terminus of catechol oxidase from sweet potato (P345) [52], theC-terminus of tyrosinases form V. faba (L464) [10] and V. spinosum(V357) [12] that were produced by proteolysis and the predictedC-terminus of V. vinifera polyphenol oxidase (A464) [53] were includedin the region of intrinsic disorder. This region of intrinsic disorder ispredicted to be shorter and present in 17 out of 23 sequences,according to the meta-predictor PONDR-FIT (Fig. S4). The analysis ofpolyphenol oxidases reported to undergo proteolysis showed the pres-ence of an intrinsically disordered region that could not be predicted inthe molluscan haemocyanins in proximity of the Y-motif (sequence 10,Fig. 4, panel B). Previous studies have predicted this region to consti-tute a linker of up to 98 residues between the active core of the enzymeand themore terminal domain [25]. In analogy with the structurally re-lated proteins haemocyanins frommolluscs, the linker region has beensuggested to protrude from the active compact domain [25]. TheC-terminal domain of hemocyanin from O. dofleini is characterised bya β–β–β–β–α–β–β secondary structure organisation that is connectedto the globular core by an alpha-helix exposed on the surface of theprotein [24]. Secondary structure prediction for the C-terminal domainof the sequences analysed in this study revealed the same order of
DOS. Black fragments represent intrinsically disordered regions, and the whole bar rep-. (A) Sequences are numbered as in Table 2: 1 Botrytis cinerea BC1G.00048, 2 B. cineream FGSG.09522, 6 Fusarium oxysporum f. sp. lycopersici FOXG.06218, 7 F. oxysporum f. sp., 10 Histoplasma capsulatum HCAG.04262.1, 11 Magnaporthe grisea A4RK91, 12 Nectriarum Q0UNZ8, 15 Coccidioides immitis Q1DQ30, 16 Chaetomium globosum Q2HEC0, 170 S. sclerotiorum SS1G.04725.1, 21 S. sclerotiorum SS1G.13364.1, 22 Trichoderma reeseiPO2 from A. bisporus, 3 TYR from Pholiota nameko, 4 TYR from Trametes sanguineus, 5nase from Vicia faba, 8 polyphenol oxidase from Vitis vinifera, 9 tyrosinase fromlack fragments represent intrinsically disordered regions, and the whole bar represents

43G. Faccio et al. / Journal of Inorganic Biochemistry 121 (2013) 37–45
elements of secondary structure (Fig. S1, panel C). The linker region be-tween the Y-motif and the first beta-strand could thus be identified.The YG-motif was predicted to be located exactly at the C-terminalend of the connecting alpha-helix (Fig. S1). The location of theYG-motif at the end of the connecting alpha-helix and at the beginningof a region of intrinsic disorder, possibly susceptible to proteolyticattack, is in agreement with the recently resolved structure of the ty-rosinase PPO3 from A. bisporus where the C-terminal domain is absentbut the connecting alpha-helix is retained and it terminates with theYG-motif (that constitutes the C-terminus) [26]. The function of theconnecting alpha-helix seems to be closely related to the presence ofthe C-terminal domain as this helix is absent in enzymes lacking theC-terminal domain such as the catechol oxidase from A. oryzae whosesequence ends with the Y-motif [34].
3.5. Proteolytic sites
Proteolytic enzymes have been used to remove the active domain ofthe tyrosinase from the full-length protein. For example, trypsin(cutting before a K or R) has been reported to efficiently remove theC-terminal domain from the bacterial tyrosinase from V. spinosum [12],chymotrypsin (cutting before W, Y and F) for tyrosinase fromP. nameko [11] and thermolysin for polyphenol oxidase from V. faba[10]. Limited proteolysis of the proenzyme of V. faba polyphenol oxidasewith trypsin produced fragments of 18, 14, and 12 kDa from the 18 kDaC-terminal domain that clearly underwent proteolytic degradation [10].Similarly, the C-terminal domain of the tyrosinase MelB from A. oryzaecould not be detected and was completely degraded upon treatmentwith trypsin [19]. In vivo studies showed how the proteolytic processwas inhibited by inhibitors of serine proteases in sweet potato cells[14]. Proteases with trypsin-like activity are common in fungi [54,55].
When tyrosinase 2 from T. reesei was overexpressed in the yeastP. pastoris the enzyme had a different C-terminus and retainedseven additional amino acids [35]. This suggested that both cleavagesites were located in a region exposed on the surface of the moleculeand rich in proteolytic sites; it also indicated that the position of thecleavage site was not crucial for the development of the enzymaticactivity. This phenomenon indicated that the whole C-terminal do-main could be rich in proteolytic sites and that it could undergo com-plete degradation for enzyme activation. In order to assess thesensitivity to proteolysis of the whole tyrosinase sequences and theC-terminal region in particular, we focused first on a representativeserine protease such as trypsin. Long tyrosinase sequences wereanalysed for the presence of potential recognition sites for trypsinwith PeptideCutter, and only cutting sites with a probability higher
Fig. 5. Box-and-whiskers plot showing the content of trypsin and chymotrypsin recog-nition sites among the core region, the C-terminal region (after the Y-motif) and thelinker region of the sequences of potentially secreted tyrosinases analysed. Significantdifferences (p-valueb0.01 in Wilcoxon–Mann–Whitney rank sum test) are indicatedby asterisks and outliers by dots.
than 70% were considered. As seen in Fig. 5 (for further details seeFig. S5), tryptic sites are more frequent in the C-terminal regionsand especially in the linker region. An average of 9.16 tryptic sitesper 100 residues is found in the C-terminus (after the Y-motif) com-pared to 6.2 found in the core regions of the 23 sequences analysed(p-valueb0.01 in Wilcoxon–Mann–Whitney rank sum test). Thehighest value was from protein NECHA0047204 from Nectriahaematococca with 12.1 sites per 100 residues in the C-terminal re-gion and 6.1 sites per 100 residues in the core region (sequence 12in Fig. S5). Similar results were obtained analysing the sequences ofknown polyphenol oxidases (Fig. S5) that, however, were intracellu-lar enzymes. Recognition sites for trypsin were detected also in thecore region. However these might be not accessible to protease dueto the globular folding of the protein, e.g. the core region of tyrosinaseMelB from A. oryzae (R19-Y419, GenBank ID: XP_001818671.2)carried 38 potential cleavage sites for trypsin (prediction confi-dence>70%, PeptideCutter), but only K312 was recognised whenthe proenzyme was subjected to in vitro activation with trypsin [19].
The lysine residues that were found enriched in the C-terminal do-main are not only recognition sites for trypsin but also for proproteinconvertases, when recurring in the R/K–X2/4/6–R/K pattern [56]. De-tailed analysis revealed the presence of numerous potential recogni-tion sites for proprotein convertases in the C-terminal domains(Fig. S1). Although not all of these sites might be recognised by theenzyme, we cannot exclude that these Golgi-resident enzymes areinvolved in the removal of the C-terminal domain of tyrosinases.
Opposite results were found when we considered the distributionof chymotrypsin sites. Chymotrypsin sites were less frequent in theC-terminal domain (average 6.8±1.7 in the C-terminal domaincompared to 10.32±0.7 in the core domain as delimited by the signalsequence cleavage site and the Y-motif, p-valueb0.01 in Wilcoxon–Mann–Whitney rank sum test). Long parts of the C-terminal domainwere free from chymotrypsin sites (Fig. S5), with the longest fragmentbeing 150 residues long in sequence Q7S218 from N. crassa (sequence17 in Fig. S5 panel C) [57]. However one tyrosinase undergoing activa-tion upon treatment with chymotrypsin has been reported [11]. Thelower presence of chymotrypsin sites in the linker and C-terminal re-gion might be explained by the hydrophobic nature of the residuesrecognised by the enzyme (W, Y and F). These residues tend to be bur-ied inside protein molecules and not exposed on the surface where theC-terminal domain and the linker region are expected to be localised.
The production as pro-forms of enyzmes with cross-linking activityon proteins is not uncommon. For example, lysyl oxidase is responsiblefor the formation of cross-links between collagen molecules, and it issynthesised in a pro-form that gets activated upon removal of thepro-peptide by the specific pro-collagen c proteinase. The cross-linking activity of human tissue transglutaminase has also beenreported to be activated by the removal of C-terminal peptides invitro, and treatment with trypsin produces a 50 kDa form, from apro-enzyme of 80 kDa [58]. Similarly to tyrosinases, the size of theC-terminal peptide removed by trypsin is one third of the full-lengthtissue transglutaminase [59]. Transglutaminase is also present inblood plasma where it is involved in the cross-linking of fibrin toform clots. In plasma, transglutaminase is in a zymogen form, also-called Factor XIII, which undergoes activation by proteolysis in theN-terminal region by thrombin during coagulation [60]. The secretedtransglutaminase from Streptoverticillium mobaraense is also producedas zymogen by the bacterium and it is activated by a specific proteasein the extracellular compartment. The active form lacks a 45-aminoacid pre-pro-peptide that has been shown to keep the enzyme inactiveand significantly affect protein thermostability [61]. Processing of thisN-terminal domain in the transglutaminase from S. mobaraense resultsin a mature protein with a higher pI than the proenzyme. An oppositetrend was calculated for the sequences analysed here (Fig. S6)suggesting that this is not a common feature of cross-linking enzymesthat undergo proteolytic maturation.

44 G. Faccio et al. / Journal of Inorganic Biochemistry 121 (2013) 37–45
The production of inactive cross-linking enzymes protects the or-ganism from undesired reactions [61]. The possible toxicity of an ac-tive cross-linking enzyme such as TrTyr2-STOP inside the cell mightexplain why no transformant was obtained. However, polyphenol ox-idases with cross-linking activity and lacking the C-terminal domainhave been reported to be efficiently produced in bacteria. As an exam-ple, the bacterial tyrosinase from Streptomyces sp. REN-21 was activeon awide range of proteins, e.g. gelatin, casein, haemoglobin and in lowerextent, bovine serum albumin and ovalbumin when recombinantlyproduced in E. coli [61]. Bacterial tyrosinases from streptomycetes donot usually carry a C-terminal domain however their production isassisted by the co-expression of a helper, or “caddie”, protein thatmight play an analogous role [63–66], e.g.ORF-393 for tyrosinase fromStreptomyces sp. REN-21 [62].
4. Conclusion
Polyphenol oxidases such as tyrosinases are known to be producedin a C-terminally cleaved form in many organisms. We aimed atproducing an uncleaved form of tyrosinase 2 by mutating the residuessurrounding the cleavage site and the YG-motif. However, the enzymewas still secreted in a ~40 kDa processed form. In order to explain thephenomenon, we analysed the features of the C-terminal domain fromtyrosinase 2, from previously identified secreted fungal tyrosinases andfrom well-studied tyrosinases by bioinformatics means. In comparisonwith the active domain, the C-terminal domain carried a statisticallysignificant higher amount of amino acids and dipeptides that are typi-cal of proteins and protein regions with low stability and prone to deg-radation. Secondary structure prediction suggests that the C-terminaldomain folds as a beta-sandwich structure similarly to the C-terminaldomain of mollusc hemocyanin. A high presence of recognition sitesfor trypsin and numerous recognition sites for pro-protein convertasewere found in the C-terminal domain and in the linker region, in partic-ular. This explains why no C-terminal domain has yet been isolated in astable form. Similarly to hemocyanins, an alpha-helical elementconnecting the globular catalytic domain and the C-terminal domainwas identified and it terminated with the YG-motif that marked alsothe beginning of a region characterised by low-complexity and intrinsicdisorder. We suggest that the proteolytic process responsible for theactivation of fungal tyrosinases is not site-specific and that the wholeC-terminal domain and especially the linker region are subjected toproteolysis.
Abbreviations
CO Catechol oxidaseHc HemocyaninLysC Lysyl endopeptidaseLysN Peptidyl-Lys metalloendopeptidaseL-DOPA L-3,4-dihydroxyphenylalaninepI Isoelectric pointPPO Polyphenol oxidaseTYR TyrosinaseTrMM Trichoderma minimal mediumTrTyr2 Tyrosinase 2 from Trichoderma reeseiY-motif Tyrosine motif (Y/FxY)YG-motif Tyrosine-glycine motif
Acknowledgements
GF was funded by TYROMAT, a project within the Empa Postdocsprogramme that is co-funded by the FP7: People Marie-Curie actionCOFUND. GF was also financially supported by a Marie Curie — EU
project PROENZ (MEST-CT-2005-020924) and by the Finnish CulturalFoundation.
Appendix A. Supplementary data
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.jinorgbio.2012.12.006.
References
[1] J. Wu, H. Chen, J. Gao, X. Liu, W. Cheng, X. Ma, Biotechnol. Lett. 32 (2010)1439–1447.
[2] H.J. Wichers, K. Recourt, M. Hendriks, C.E. Ebbelaar, G. Biancone, F.A. Hoeberichts,H. Mooibroek, C. Soler-Rivas, Appl. Microbiol. Biotechnol. 61 (2003) 336–341.
[3] J.C. Espin, H.J. Wichers, J. Agric. Food Chem. 47 (1999) 3518–3525.[4] H. Wichers, Y.A.M. Gerritsen, C.G.J. Chapelon, Phytochemistry 43 (1996) 333–337.[5] U. Kupper, D.M. Niedermann, G. Travaglini, K. Lerch, J. Biol. Chem. 264 (1989)
17250–17258.[6] E. Selinheimo, M. Saloheimo, E. Ahola, A. Westerholm-Parvinen, N. Kalkkinen,
J. Buchert, K. Kruus, FEBS J. 273 (2006) 4322–4335.[7] A. Flurkey, J. Cooksey, A. Reddy, K. Spoonmore, A. Rescigno, J. Inlow, W.H. Flurkey,
J. Agric. Food Chem. 56 (2008) 4760–4768.[8] T. Klabunde, C. Eicken, J.C. Sacchettini, B. Krebs, Nat. Struct. Biol. 5 (1998)
1084–1090.[9] W.H. Flurkey, Plant Physiol. 91 (1989) 481–483.
[10] S.P. Robinson, I.B. Dry, Plant Physiol. 99 (1992) 317–323.[11] Y. Kawamura-Konishi, S. Maekawa, M. Tsuji, H. Goto, Appl. Microbiol. Biotechnol.
(2010) 227–234.[12] M. Fairhead, L. Thony-Meyer, FEBS J. 277 (2010) 2083–2095.[13] Y. Kawamura-Konishi, M. Tsuji, S. Hatana, M. Asanuma, D. Kakuta, T. Kawano, E.B.
Mukouyama, H. Goto, H. Suzuki, Biosci. Biotechnol. Biochem. 71 (2007)1752–1760.
[14] M. Nozue, D. Arakawa, Y. Iwata, H. Shioiri, M. Kojima, J. Plant Physiol. 155 (1999)297–301.
[15] M. Perez-Gilabert, A. Morte, F. Garcia-Carmona, Plant Sci. 166 (2004) 365–370.[16] T. Swain, L.W. Mapson, D.A. Robb, Phytochemistry 5 (1966) 469–482.[17] S.R. Kanade, B. Paul, A.G.A. Rao, L.R. Gowda, Biochem. J. 395 (2006) 551–562.[18] M. Okot-Kotber, A. Liavoga, K. Yong, K. Bagorogoza, J. Agric. Food Chem. 50 (2002)
2410–2417.[19] N. Fujieda, M. Murata, S. Yabuta, T. Ikeda, C. Shimokawa, Y. Nakamura, Y. Hata, S.
Itoh, ChemBioChem 13 (2012) 193–201.[20] H. Decker, T. Schweikardt, D. Nillius, U. Salzbrunn, E. Jaenicke, F. Tuczek, Gene 398
(2007) 183–191.[21] H.C. Eisenman, A. Casadevall, Appl. Microbiol. Biotechnol. (2012) 931–940.[22] W.H. Flurkey, J.K. Inlow, J. Inorg. Biochem. 102 (2008) 2160–2170.[23] J.C. García-Borrón, F. Solano, Pigment Cell Res. 15 (2002) 162–173.[24] M.E. Cuff, K.I. Miller, K.E. van Holde, W.A. Hendrickson, J. Mol. Biol. 278 (1998)
855–870.[25] C.M. Marusek, N.M. Trobaugh, W.H. Flurkey, J.K. Inlow, J. Inorg. Biochem. 100
(2006) 108–123.[26] W.T. Ismaya, H.J. Rozeboom, A. Weijn, J.J. Mes, F. Fusetti, H.J. Wichers, B.W.
Dijkstra, Biochemistry 50 (2011) 5477–5486.[27] M. Sendovski, M. Kanteev, V.S. Ben-Yosef, N. Adir, A. Fishman, J. Mol. Biol. 405
(2011) 227–237.[28] M. Sendovski, M. Kanteev, V. Shuster Ben-Yosef, N. Adir, A. Fishman, Acta
Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 66 (2010) 1101–1103.[29] V.M. Virador, J.P. Reyes Grajeda, A. Blanco-Labra, E. Mendiola-Olaya, G.M. Smith,
A. Moreno, J.R. Whitaker, J. Agric. Food Chem. 58 (2010) 1189–1201.[30] C. Gerdemann, C. Eicken, B. Krebs, Acc. Chem. Res. 35 (2002) 183–191.[31] Y. Matoba, T. Kumagai, A. Yamamoto, H. Yoshitsu, M. Sugiyama, J. Biol. Chem. 281
(2006) 8981–8990.[32] S.N. Ho, H.D. Hunt, R.M. Horton, J.K. Pullen, L.R. Pease, Gene 77 (1989) 51–59.[33] M. Penttila, H. Nevalainen, M. Ratto, E. Salminen, J. Knowles, Gene 61 (1987)
155–164.[34] C. Gasparetti, G. Faccio, M. Arvas, J. Buchert, M. Saloheimo, K. Kruus, Appl.
Microbiol. Biotechnol. 86 (2010) 213–226.[35] A. Westerholm-Parvinen, E. Selinheimo, H. Boer, N. Kalkkinen, M. Mattinen, M.
Saloheimo, Protein Expr. Purif. 55 (2007) 147–158.[36] O. Emanuelsson, S. Brunak, G. von Heijne, H. Nielsen, Nat. Protoc. 2 (2007)
953–971.[37] T. Ishida, K. Kinoshita, Nucleic Acids Res. 35 (2007) W460–W464.[38] B. Xue, R.L. Dunbrack, R.W.Williams, A.K. Dunker, V.N. Uversky, Biochim. Biophys.
Acta 1804 (2010) 996–1010.[39] I. Letunic, T. Doerks, P. Bork, Nucleic Acids Res. 40 (2012) D302–D305.[40] K. Guruprasad, B.V.B. Reddy, M.W. Pandit, Protein Eng. 4 (1990) 155–161.[41] E. Gasteiger, A. Gattiker, C. Hoogland, I. Ivanyi, R.D. Appel, A. Bairoch, Nucleic
Acids Res. 31 (2003) 3784–3788.[42] M.A. Larkin, G. Blackshields, N.P. Brown, R. Chenna, P.A. McGettigan, H.
McWilliam, F. Valentin, I.M. Wallace, A. Wilm, R. Lopez, J.D. Thompson, T.J.Gibson, D.G. Higgins, Bioinformatics 23 (2007) 2947–2948.
[43] A.M.Waterhouse, J.B. Procter, D.M.A. Martin, M. Clamp, G.J. Barton, Bioinformatics25 (2009) 1189–1191.

45G. Faccio et al. / Journal of Inorganic Biochemistry 121 (2013) 37–45
[44] E. Gasteiger, C.G. Hoogland, A.S. Duvaud, M.R. Wilkins, R.D. Appel, A. Bairoch,Methods Mol. Biol. 112 (1999) 531–552.
[45] P. Rice, I. Longden, A. Bleasby, Trends Genet. 16 (2000) 276–277.[46] T. Nonaka, N. Dohmae, Y. Hashimoto, K. Takio, J. Biol. Chem. 272 (1997)
30032–30039.[47] S.P. Goller, D. Schoisswohl, M. Baron, M. Parriche, C.P. Kubicek, Appl. Environ.
Microbiol. 64 (1998) 3202–3208.[48] D. Julius, A. Brake, L. Blair, R. Kunisawa, J. Thorner, Cell 37 (1984) 1075–1089.[49] O. Bader, Y. Krauke, B. Hube, BMC Microbiol. (2008) 116.[50] S. Rogers, R. Wells, M. Rechsteiner, Science 234 (1986) 364–368.[51] H.J. Dyson, P.E. Wright, Nat. Rev. Mol. Cell Biol. 6 (2005) 197–208.[52] C. Gerdemann, C. Eicken, A. Magrini, H.E. Meyer, A. Rompel, F. Spener, B. Krebs,
Biochim. Biophys. Acta 1548 (2001) 94–105.[53] I.B. Dry, S.P. Robinson, Plant Mol. Biol. 26 (1994) 495–502.[54] L.A. Gzogyan, M.T. Proskuryakov, I.V. Ievleva, T.A. Valueva, Appl. Biochem.
Microbiol. 41 (2005) 538–541.[55] I. Yike, Mycopathologia 171 (2011) 293–323.[56] N.G. Seidah, M. Chrétien, Curr. Opin. Biotechnol. 8 (1997) 602–607.[57] T. Maruhashi, I. Kii, M. Saito, A. Kudo, J. Biol. Chem. 285 (2010) 13294–13303.[58] C.W. van Gelder, W.H. Flurkey, H.J. Wichers, Phytochemistry 45 (1997)
1309–1323.[59] B.M. Fraij, J. Cell. Biochem. 112 (2011) 3469–3481.
[60] L. Lorand, in: Annals of the New York Academy of Sciences - Fibrinogen: XVITHInternational Fibrinogen Workshop, 936, 2001, pp. 291–311.
[61] R. Pasternack, S. Dorsch, J.T. Otterbach, I.R. Robenek, S.F.L. Wolf, Eur. J. Biochem.257 (1998) 570–576.
[62] M. Ito, K. Inouye, J. Biochem. 138 (2005) 355–362.[63] K. Ikeda, T. Masujima, K. Suzuki, M. Sugiyama, Appl. Microbiol. Biotechnol. 45
(1996) 80–85.[64] S. Kawamoto, M. Nakamura, S. Yashima, J. Ferment. Bioeng. 76 (1993) 345–355.[65] P.Y. Kohashi, T. Kumagai, Y. Matoba, A. Yamamoto, M. Maruyama, M. Sugiyama,
Protein Expr. Purif. 34 (2004) 202–207.[66] S. Omura, H. Ikeda, J. Ishikawa, A. Hanamoto, C. Takahashi, M. Shinose, Y.
Takahashi, H. Horikawa, H. Nakazawa, T. Osonoe, H. Kikuchi, T. Shiba, Y. Sakaki,M. Hattori, Proc. Natl. Acad. Sci. U. S. A. 98 (2001) 12215–12220.
[67] J. Van Leeuwen, H.J. Wichers, Mycol. Res. 103 (1999) 413–418.[68] S. Halaouli, M. Asther, K. Kruus, L. Guo, M. Hamdi, J.C. Sigoillot, M. Asther, A.
Lomascolo, J. Appl. Microbiol. 98 (2005) 332–343.[69] C. Eicken, F. Zippel, K. Buldt-Karentzopoulos, B. Krebs, FEBS Lett. 436 (1998)
293–299.[70] M. Huber, K. Lerch, FEBS Lett. 219 (1987) 335–338.[71] K. Lerch, J. Biol. Chem. 257 (1982) 6414–6419.[72] K.I. Miller, M.E. Cuff, W.F. Lang, P. Varga-Weisz, K.G. Field, K.E. van Holde, J. Mol.
Biol. 278 (1998) 827–842.




















