
Evaluation of the binding of the radiolabeled antidepressant drug, 18F-fluoxetine in the rodent...
6
Evaluation of the Binding of the Radiolabeled Antidepressant Drug, 18 F-Fluoxetine in the Rodent Brain: An In Vitro and In Vivo Study Jogeshwar Mukherjee, 1 Malay K. Das, 1 Zhi-Ying Yang 1 and Robert Lew 2 DEPARTMENTS OF 1 RADIOLOGY AND 2 PHARMACOLOGICAL AND PHYSIOLOGICAL SCIENCES, FRANKLIN MCLEAN INSTITUTE, UNIVERSITY OF CHICAGO, CHICAGO, IL 60637, USA ABSTRACT. We have developed 18 F-fluoxetine as a radiotracer analog of the antidepressant drug fluoxetine (Prozac). In vitro saturation experiments of 18 F-fluoxetine were carried out on rat midbrain tissue and citalopram was used for measuring nonspecific binding. A saturation curve for the binding of 18 F-fluoxetine was not obtained. Even when fluoxetine (10 mM) was used for measurements of nonspecific binding, a saturation curve was difficult to obtain. Other compounds, such as deprenyl, clorgyline, amphetamine, and reserpine were also not able to reduce the binding of 18 F-fluoxetine. Ex vivo autoradiographic experiments with 18 F-fluoxetine did not reveal any specific uptake in various brain regions. In vivo administration of 18 F-fluoxetine in rats showed similar uptake in all the brain regions with little regional selectivity. A subcellular analysis of rat brain tissue after intravenous (IV) administration of 18 F-fluoxetine indicated significant amounts of binding in mitochondria and synaptosomes. In summary, in vitro experiments with 18 F-fluoxetine indicate little specific binding. Binding to the serotonin transporter was not identifiable. High nonspecific binding of the tracer resulting from its subcellular nature in the brain masks the ability to detect binding to the serotonin uptake sites in vivo. These findings indicate that a large portion of the binding of 18 F-fluoxetine in rat brains is subcellular and clears slowly out of the cells. Other sites, such as monoamine oxidase, may also play a significant role in the action of fluoxetine. NUCL MED BIOL 25;7:605– 610, 1998. © 1998 Elsevier Science Inc. KEY WORDS. 18 F-Fluoxetine ( 18 F-Prozac), Autoradiography, Serotonin transporter, Monoamine oxidase INTRODUCTION Fluoxetine is a clinically used potent antidepressant (4, 19). Like other antidepressant drugs, the therapeutic effect of fluoxetine and its major metabolite, norfluoxetine, has been reported to depend on their ability to selectively inhibit presynaptic uptake of serotonin (5HT) (3, 14, 20). Radiolabeled fluoxetine may therefore be used as a tracer for the drug and has the potential to be a marker for these serotonin uptake sites. The imaging of these uptake sites would help to monitor them for diagnosis and therapy management of depres- sive illness as well as to probe for neurotoxic damage to serotonin terminals induced by various drugs of abuse. It has been shown that fluoxetine has a moderate affinity for the serotonin transporter sites (5-HTT) in rat brain tissue (4, 12). It is postulated that this pharmacological effect accounts for its thera- peutic effect in cases of depression, whereupon treatment with fluoxetine, synaptic serotonin levels are raised (19). Due to the widespread clinical use of fluoxetine, we have been interested in the study of its sites of action in the brain. To study the nature of binding of fluoxetine in the brain, radiotracer analogs of fluoxetine labeled with carbon-11 have been used in animal studies (15). Biodistribution results on 11 C-fluoxetine in rats and monkeys yielded no identifiable specific binding to the serotonin transporter (15). The moderate affinity of fluoxetine, its high lipophilicity, and binding to other nonspecific sites were considered to be the primary reasons for no specific localization of the radiotracer. Fluoxetine undergoes rapid demethylation [approximately 30 – 40% of demethylated fluoxetine was found in the rat brain in 60 min, following intraperitoneal (IP) injection of fluoxetine] to norfluoxetine (1, 20). Metabolism does not appreciably alter its selectivity or potency (20). Norfluoxetine [particularly the (S)- isomer] also binds strongly at the transporter site (4, 12, 20). Using rat brain synaptosomes, K i for (R)- and (S)-fluoxetine were found to be 33 nM and 21 nM, respectively, whereas the K i for (R)- and (S)-norfluoxetine were found to be 484 nM and 29.8 nM, respec- tively. More recently, norfluoxetine isomers have been reported to be selective 5-HTT inhibitors as well, and it has been suggested that this metabolite of fluoxetine may be responsible for the persistent 5-HTT blockade in vivo (20). In addition to the binding at the 5-HT transporter sites, fluoxetine has also recently been shown to have an inhibitory effect on monoamine oxidase A and B in vitro and it has been suggested that this action of fluoxetine may have the potential to contribute to its antidepressant properties (9). We and others have prepared 18 F-fluoxetine as a potential radiotracer of fluoxetine to help understand its mechanism of action for three reasons: (a) the longer half-life of fluorine-18 (110 min) compared with carbon-11 (20 min) would allow the study of the binding of the radiotracer for longer periods of time (2, 5). This was considered important because it has been suggested that lipophilic- ity of fluoxetine maybe a factor that contributes to the inability to identify specific binding. Therefore, prolonged studies might help in reducing the extent of nonspecific binding and thus assist in the Address correspondence to: Jogeshwar Mukherjee, Ph.D., Department of Nuclear Medicine/PET, Kettering Medical Center, 3535 Southern Boule- vard, Kettering, OH 45429-1298, USA; e-mail: jogeshwar_mukherjee@ ketthealth.com. Received 14 February 1998. Accepted 25 April 1998. Nuclear Medicine & Biology, Vol. 25, pp. 605– 610, 1998 ISSN 0969-8051/98/$19.00 1 0.00 Copyright © 1998 Elsevier Science Inc. PII S0969-8051(98)00043-2
Transcript of Evaluation of the binding of the radiolabeled antidepressant drug, 18F-fluoxetine in the rodent...

Evaluation of the Binding of the RadiolabeledAntidepressant Drug, 18F-Fluoxetine in the Rodent Brain:
An In Vitro and In Vivo StudyJogeshwar Mukherjee,1 Malay K. Das,1 Zhi-Ying Yang1 and Robert Lew2
DEPARTMENTS OF 1RADIOLOGY AND 2PHARMACOLOGICAL AND PHYSIOLOGICAL SCIENCES, FRANKLIN MCLEAN INSTITUTE,
UNIVERSITY OF CHICAGO, CHICAGO, IL 60637, USA
ABSTRACT. We have developed 18F-fluoxetine as a radiotracer analog of the antidepressant drug fluoxetine(Prozac). In vitro saturation experiments of 18F-fluoxetine were carried out on rat midbrain tissue andcitalopram was used for measuring nonspecific binding. A saturation curve for the binding of 18F-fluoxetinewas not obtained. Even when fluoxetine (10 mM) was used for measurements of nonspecific binding, asaturation curve was difficult to obtain. Other compounds, such as deprenyl, clorgyline, amphetamine, andreserpine were also not able to reduce the binding of 18F-fluoxetine. Ex vivo autoradiographic experimentswith 18F-fluoxetine did not reveal any specific uptake in various brain regions. In vivo administration of18F-fluoxetine in rats showed similar uptake in all the brain regions with little regional selectivity. Asubcellular analysis of rat brain tissue after intravenous (IV) administration of 18F-fluoxetine indicatedsignificant amounts of binding in mitochondria and synaptosomes. In summary, in vitro experiments with18F-fluoxetine indicate little specific binding. Binding to the serotonin transporter was not identifiable. Highnonspecific binding of the tracer resulting from its subcellular nature in the brain masks the ability to detectbinding to the serotonin uptake sites in vivo. These findings indicate that a large portion of the binding of18F-fluoxetine in rat brains is subcellular and clears slowly out of the cells. Other sites, such as monoamineoxidase, may also play a significant role in the action of fluoxetine. NUCL MED BIOL 25;7:605–610, 1998.© 1998 Elsevier Science Inc.
KEY WORDS. 18F-Fluoxetine (18F-Prozac), Autoradiography, Serotonin transporter, Monoamine oxidase
INTRODUCTION
Fluoxetine is a clinically used potent antidepressant (4, 19). Likeother antidepressant drugs, the therapeutic effect of fluoxetine andits major metabolite, norfluoxetine, has been reported to depend ontheir ability to selectively inhibit presynaptic uptake of serotonin(5HT) (3, 14, 20). Radiolabeled fluoxetine may therefore be used asa tracer for the drug and has the potential to be a marker for theseserotonin uptake sites. The imaging of these uptake sites would helpto monitor them for diagnosis and therapy management of depres-sive illness as well as to probe for neurotoxic damage to serotoninterminals induced by various drugs of abuse.
It has been shown that fluoxetine has a moderate affinity for theserotonin transporter sites (5-HTT) in rat brain tissue (4, 12). It ispostulated that this pharmacological effect accounts for its thera-peutic effect in cases of depression, whereupon treatment withfluoxetine, synaptic serotonin levels are raised (19). Due to thewidespread clinical use of fluoxetine, we have been interested in thestudy of its sites of action in the brain. To study the nature ofbinding of fluoxetine in the brain, radiotracer analogs of fluoxetinelabeled with carbon-11 have been used in animal studies (15).Biodistribution results on 11C-fluoxetine in rats and monkeysyielded no identifiable specific binding to the serotonin transporter
(15). The moderate affinity of fluoxetine, its high lipophilicity, andbinding to other nonspecific sites were considered to be the primaryreasons for no specific localization of the radiotracer.
Fluoxetine undergoes rapid demethylation [approximately 30–40% of demethylated fluoxetine was found in the rat brain in 60min, following intraperitoneal (IP) injection of fluoxetine] tonorfluoxetine (1, 20). Metabolism does not appreciably alter itsselectivity or potency (20). Norfluoxetine [particularly the (S)-isomer] also binds strongly at the transporter site (4, 12, 20). Usingrat brain synaptosomes, Ki for (R)- and (S)-fluoxetine were found tobe 33 nM and 21 nM, respectively, whereas the Ki for (R)- and(S)-norfluoxetine were found to be 484 nM and 29.8 nM, respec-tively. More recently, norfluoxetine isomers have been reported tobe selective 5-HTT inhibitors as well, and it has been suggested thatthis metabolite of fluoxetine may be responsible for the persistent5-HTT blockade in vivo (20). In addition to the binding at the5-HT transporter sites, fluoxetine has also recently been shown tohave an inhibitory effect on monoamine oxidase A and B in vitroand it has been suggested that this action of fluoxetine may havethe potential to contribute to its antidepressant properties (9).
We and others have prepared 18F-fluoxetine as a potentialradiotracer of fluoxetine to help understand its mechanism of actionfor three reasons: (a) the longer half-life of fluorine-18 (110 min)compared with carbon-11 (20 min) would allow the study of thebinding of the radiotracer for longer periods of time (2, 5). This wasconsidered important because it has been suggested that lipophilic-ity of fluoxetine maybe a factor that contributes to the inability toidentify specific binding. Therefore, prolonged studies might help inreducing the extent of nonspecific binding and thus assist in the
Address correspondence to: Jogeshwar Mukherjee, Ph.D., Department ofNuclear Medicine/PET, Kettering Medical Center, 3535 Southern Boule-vard, Kettering, OH 45429-1298, USA; e-mail: [email protected].
Received 14 February 1998.Accepted 25 April 1998.
Nuclear Medicine & Biology, Vol. 25, pp. 605–610, 1998 ISSN 0969-8051/98/$19.00 1 0.00Copyright © 1998 Elsevier Science Inc. PII S0969-8051(98)00043-2
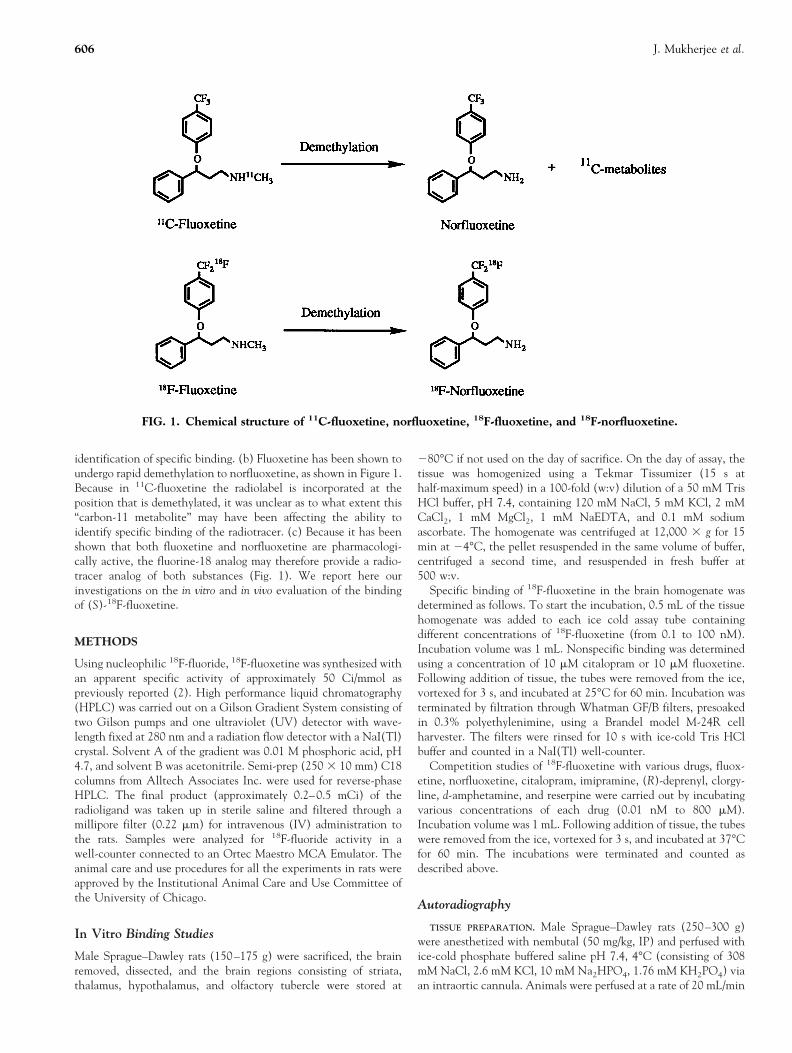
identification of specific binding. (b) Fluoxetine has been shown toundergo rapid demethylation to norfluoxetine, as shown in Figure 1.Because in 11C-fluoxetine the radiolabel is incorporated at theposition that is demethylated, it was unclear as to what extent this“carbon-11 metabolite” may have been affecting the ability toidentify specific binding of the radiotracer. (c) Because it has beenshown that both fluoxetine and norfluoxetine are pharmacologi-cally active, the fluorine-18 analog may therefore provide a radio-tracer analog of both substances (Fig. 1). We report here ourinvestigations on the in vitro and in vivo evaluation of the bindingof (S)-18F-fluoxetine.
METHODS
Using nucleophilic 18F-fluoride, 18F-fluoxetine was synthesized withan apparent specific activity of approximately 50 Ci/mmol aspreviously reported (2). High performance liquid chromatography(HPLC) was carried out on a Gilson Gradient System consisting oftwo Gilson pumps and one ultraviolet (UV) detector with wave-length fixed at 280 nm and a radiation flow detector with a NaI(Tl)crystal. Solvent A of the gradient was 0.01 M phosphoric acid, pH4.7, and solvent B was acetonitrile. Semi-prep (250 3 10 mm) C18columns from Alltech Associates Inc. were used for reverse-phaseHPLC. The final product (approximately 0.2–0.5 mCi) of theradioligand was taken up in sterile saline and filtered through amillipore filter (0.22 mm) for intravenous (IV) administration tothe rats. Samples were analyzed for 18F-fluoride activity in awell-counter connected to an Ortec Maestro MCA Emulator. Theanimal care and use procedures for all the experiments in rats wereapproved by the Institutional Animal Care and Use Committee ofthe University of Chicago.
In Vitro Binding Studies
Male Sprague–Dawley rats (150–175 g) were sacrificed, the brainremoved, dissected, and the brain regions consisting of striata,thalamus, hypothalamus, and olfactory tubercle were stored at
280°C if not used on the day of sacrifice. On the day of assay, thetissue was homogenized using a Tekmar Tissumizer (15 s athalf-maximum speed) in a 100-fold (w:v) dilution of a 50 mM TrisHCl buffer, pH 7.4, containing 120 mM NaCl, 5 mM KCl, 2 mMCaCl2, 1 mM MgCl2, 1 mM NaEDTA, and 0.1 mM sodiumascorbate. The homogenate was centrifuged at 12,000 3 g for 15min at 24°C, the pellet resuspended in the same volume of buffer,centrifuged a second time, and resuspended in fresh buffer at500 w:v.
Specific binding of 18F-fluoxetine in the brain homogenate wasdetermined as follows. To start the incubation, 0.5 mL of the tissuehomogenate was added to each ice cold assay tube containingdifferent concentrations of 18F-fluoxetine (from 0.1 to 100 nM).Incubation volume was 1 mL. Nonspecific binding was determinedusing a concentration of 10 mM citalopram or 10 mM fluoxetine.Following addition of tissue, the tubes were removed from the ice,vortexed for 3 s, and incubated at 25°C for 60 min. Incubation wasterminated by filtration through Whatman GF/B filters, presoakedin 0.3% polyethylenimine, using a Brandel model M-24R cellharvester. The filters were rinsed for 10 s with ice-cold Tris HClbuffer and counted in a NaI(Tl) well-counter.
Competition studies of 18F-fluoxetine with various drugs, fluox-etine, norfluoxetine, citalopram, imipramine, (R)-deprenyl, clorgy-line, d-amphetamine, and reserpine were carried out by incubatingvarious concentrations of each drug (0.01 nM to 800 mM).Incubation volume was 1 mL. Following addition of tissue, the tubeswere removed from the ice, vortexed for 3 s, and incubated at 37°Cfor 60 min. The incubations were terminated and counted asdescribed above.
Autoradiography
TISSUE PREPARATION. Male Sprague–Dawley rats (250–300 g)were anesthetized with nembutal (50 mg/kg, IP) and perfused withice-cold phosphate buffered saline pH 7.4, 4°C (consisting of 308mM NaCl, 2.6 mM KCl, 10 mM Na2HPO4, 1.76 mM KH2PO4) viaan intraortic cannula. Animals were perfused at a rate of 20 mL/min
FIG. 1. Chemical structure of 11C-fluoxetine, norfluoxetine, 18F-fluoxetine, and 18F-norfluoxetine.
606 J. Mukherjee et al.

for a period of 12 min. Blood clearance from peripheral tissues suchas kidney and liver were used as indicators of adequate bloodclearance from brain. After perfusion, brain and kidney wereremoved and frozen in isopentane previously immersed in dry ice.Coronal tissue sections (10 mm) of rat kidney and brain were cut ona Riechert–Jung Cryocut 1800 cryostat and thaw-mounted on tosubbed (i.e., gelatin/chromalum) microscope slides previouslycleaned in chromic acid. Tissue sections were then stored at 220°Cuntil required.
LABELING WITH 18F-FLUOXETINE. Tissue sections were removedfrom storage and were equilibrated to room temperature (22–25°C)over a period of 30–60 min. Following equilibration, tissue sectionswere placed in 50 mM Tris HCl buffer (pH 7.4, 37°C) containing120 mM NaCl and 5 mM KCl and incubated with 25 nM18F-fluoxetine (specific activity 5 50 Ci/mmol) for 60 min at 37°C.Nonspecific binding was defined as the binding remaining in thepresence of 1 mM citalopram. Following incubation, tissue sectionswere briefly rinsed and washed for 2 3 2 min periods at 37°C in theabove buffer. After washing, tissue sections were rinsed in distilledwater and dried under a cool stream of air for X-ray film autora-diography. Tissue sections were apposed to Kodak X-omat AR X-rayfilm and exposed for 0.5–2 h. Autoradiograms were photographi-cally developed in Dektol developer (5 min) and fixed in Kodakrapid fixer (5 min).
In Vivo Biodistribution Studies
Groups of rats (Sprague–Dawley, 150–175 g) were injected IV viatail vein with 20 mCi of 18F-fluoxetine in 200 mL of saline. Ratswere sacrificed at 30, 60, and 120 min postinjection by decapitation.The brain was then rapidly removed, rinsed in ice-cold saline, andplaced on an ice block. Brain regions such as olfactory tubercles,thalamus, hypothalamus, striatum, and frontal cortex that havebeen reported to contain significant concentrations of the serotonintransporter sites were dissected (13). Reference regions such ascerebellum, bone, and blood, which are known to contain fewerserotonin transporter sites, were also isolated. Tissue samples weretaken in preweighed glass vials and counted in a NaI(Tl) well-counter. All counting data were corrected for decay and expressedas percentage injected dose per gram of tissue. For each time pointthree rats were taken.
Metabolite analysis of 18F-fluoxetine were carried out in bloodplasma, urine, and brain after IV administration of 18F-fluoxetine.The blood samples (1 mL) were centrifuged at 1,000 3 g for 4 minto separate plasma from blood cells and the plasma was removed forfurther analyses. Sodium bicarbonate (5% solution, 100 mL) wasadded to the plasma (500 mL) along with 30 mg of unlabeledfluoxetine. This mixture was extracted three times with 500 mLethyl acetate each time. The combined ethyl acetate extracts werefiltered and evaporated to dryness, and the residue was redissolvedin methanol for thin layer chromatographic (TLC) analysis. TheTLC plates were developed in chloroform-methanol (9:1) and cutinto 1-cm wide strips, which were then counted in a well counter.The location of 18F-fluoxetine and 18F-norfluoxetine was deter-mined by co-spotting unlabeled fluoxetine and norfluoxetine. Sim-ilarly, the excised brain was homogenized with 5% sodium bicar-bonate (500 mL) and centrifuged at 1,000 3 g for 10 min. Theaqueous layer was removed and extracted three times with ethylacetate (500 mL each time) and the solutions were counted andanalysed by TLC and the Rfs compared with fluoxetine andnorfluoxetine as described above. Urine (200 mL) was treated with
5% sodium bicarbonate (50 mL) and was extracted with ethylacetate and analyzed as described above.
For in vivo inhibition studies, unlabeled fluoxetine, citalopram,(R)-deprenyl, and saline (for control rats) were administered IP 5min prior to the administration of 18F-fluoxetine. Each rat receivedfluoxetine (1 mg/kg), citalopram (1 mg/kg), and (R)-deprenyl (10mg/kg) and 20 mCi of 18F-fluoxetine. For competition studies onlytwo time points (60 and 120 min) were investigated. The rats weresacrificed at 60 and 120 min after injection and brain tissue sampleswere prepared as described above.
Subcellular Analysis of 18F-Fluoxetine Binding
Groups of rats were administered IP saline, deprenyl (10 mg/kg),and citalopram (10 mg/kg) 5 min prior to the administration of 80mCi of 18F-fluoxetine. After 60 min, the rats were sacrificed and thebrains isolated. The cerebrum of each rat was homogenized in 10mL of 0.32 M sucrose. Using the reported procedure (18), variousfractions (S1, P1, and P2) were obtained by centrifugation. Theseparate fractions were counted and weighed to determine theamount of radioactivity present.
RESULTSIn Vitro Binding Studies
The results of in vitro binding studies with 18F-fluoxetine showsaturable binding sites for 18F-fluoxetine in the rat brain homoge-nate. Nonspecific binding was measured in the presence of 10 mMfluoxetine. The extent of specific binding was small, amounting toapproximately 12% of the total binding (Fig. 2). A saturation plotof the binding of 18F-fluoxetine could not be obtained whencitalopram (10 mM) was used for the measurement of nonspecificbinding in the same homogenate. Total binding of 18F-fluoxetineand binding in the presence of citalopram were not different,indicating that citalopram was unable to displace 18F-fluoxetine atthe various concentrations.
Competition studies were carried out with MAO inhibitors,(R)-deprenyl and clorgyline; serotonin uptake blockers, fluoxetine,norfluoxetine, and citalopram; and d-amphetamine and reserpine,which are known to release presynaptic neurotransmitters. Bindingof 18F-fluoxetine in the rat brain homogenates was not displaced byany of the drugs. On the contrary, most of the drugs increased thebinding of 18F-fluoxetine in the brain tissue.
In Vitro Autoradiography
Results of the following regions were analyzed: (a) striata; (b)hippocampus, hypothalamus, thalamus; (c) mid-brain, hippocam-pus; (d) raphe-nucleus; (e) cerebellum, brain stem; and (f) kidney.Both total binding and nonspecific binding results indicated thelarge amount of nonspecific binding of 18F-fluoxetine (results notshown). Regional differences in the binding of 18F-fluoxetine werenot evident. Also, autoradiographs in the presence of 1 mMcitalopram did not provide any significant changes in the totalbinding. The subcellular nature of binding of fluoxetine may be oneimportant issue that masks any specific binding to the serotonintransporter.
In Vivo Rat Studies
Time–activity graphs were plotted for the various regions and areshown in Figure 3. 18F-fluoxetine showed uptake of 0.4–0.8%
18F-Fluoxetine Binding in the Brain 607

injected dose per gram of brain tissue. Radioactivity in the variousbrain regions, olfactory tubercle, hypothalamus, thalamus, frontalcortex, striata, and cerebellum were generally similar. Marginal
differences in activity in the olfactory tubercles and striata at latertime points were observed. At 120 min postinjection, the highestamount of radioactivity was obtained in olfactory tubercles, fol-lowed by hypothalamus and thalamus. Relatively little activitywashed out from the brain regions during the 30–120-min period.Significant amounts of radioactivity also remained in the blood.Bone showed uptake of radioactivity comparable to blood levels.The lipophilic nature of 18F-fluoxetine is evident from the relativelylarge radioactivity levels in regions such as cerebellum where thereare fewer serotonin uptake sites compared with receptor-rich re-gions such as hypothalamus (13). These findings of little in vivoselectivity, particularly for olfactory tubercles and hypothalamus,were reproducible in two separate experiments (n 5 3 for each timepoint in each experiment).
In the blocking studies [preinjection with fluoxetine 1 mg/kg,citalopram 1 mg/kg, and (R)-deprenyl 10 mg/kg], results wereinconsistent in all brain regions. No significant lowering of uptakeof 18F-fluoxetine was found for the brain regions compared with thecontrol experiments.
Metabolite analysis of 18F-fluoxetine in blood plasma and thebrain indicated the presence of a large amount of 18F-fluoxetine and18F-norfluoxetine in the brain tissue. The urine was comprisedprimarily of polar metabolites with trace amounts of 18F-fluoxetine.Similarly, the blood plasma contained very small amounts of18F-fluoxetine compared with the brain.
Subcellular Analysis of 18F-Fluoxetine Binding
Binding of 18F-fluoxetine was found to occur substantially tosubcellular components. Figure 4 shows the binding of 18F-fluox-etine to the mitochondrial fractions (fraction P2) amounted toapproximately 60–70%, which is similar to the reported findings
FIG. 2. Saturation curve for 18F-fluoxetine measured using rat brain tissue. Inset shows a magnified view of the specificbinding.
FIG. 3. Rat brain distribution of 18F-fluoxetine. Male Spra-gue–Dawley rats were injected with 20 mCi 18F-fluoxetineand sacrificed at times shown. Data are the mean 6 SD ofthree animals.
608 J. Mukherjee et al.
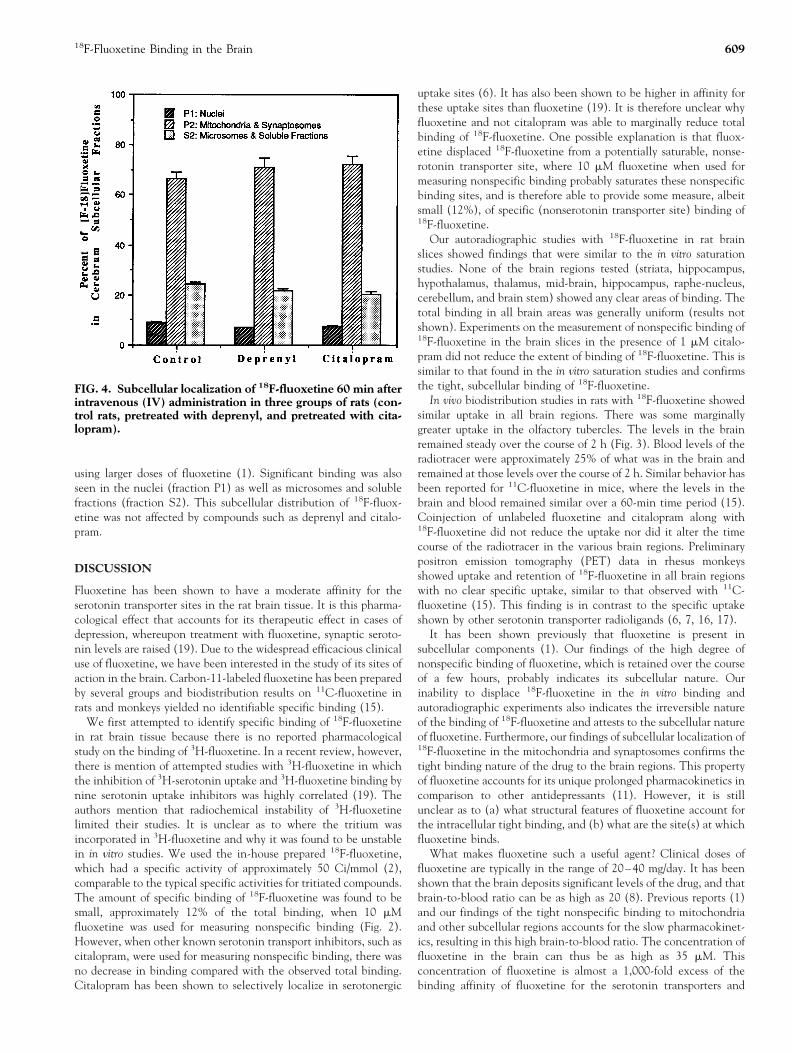
using larger doses of fluoxetine (1). Significant binding was alsoseen in the nuclei (fraction P1) as well as microsomes and solublefractions (fraction S2). This subcellular distribution of 18F-fluox-etine was not affected by compounds such as deprenyl and citalo-pram.
DISCUSSION
Fluoxetine has been shown to have a moderate affinity for theserotonin transporter sites in the rat brain tissue. It is this pharma-cological effect that accounts for its therapeutic effect in cases ofdepression, whereupon treatment with fluoxetine, synaptic seroto-nin levels are raised (19). Due to the widespread efficacious clinicaluse of fluoxetine, we have been interested in the study of its sites ofaction in the brain. Carbon-11-labeled fluoxetine has been preparedby several groups and biodistribution results on 11C-fluoxetine inrats and monkeys yielded no identifiable specific binding (15).
We first attempted to identify specific binding of 18F-fluoxetinein rat brain tissue because there is no reported pharmacologicalstudy on the binding of 3H-fluoxetine. In a recent review, however,there is mention of attempted studies with 3H-fluoxetine in whichthe inhibition of 3H-serotonin uptake and 3H-fluoxetine binding bynine serotonin uptake inhibitors was highly correlated (19). Theauthors mention that radiochemical instability of 3H-fluoxetinelimited their studies. It is unclear as to where the tritium wasincorporated in 3H-fluoxetine and why it was found to be unstablein in vitro studies. We used the in-house prepared 18F-fluoxetine,which had a specific activity of approximately 50 Ci/mmol (2),comparable to the typical specific activities for tritiated compounds.The amount of specific binding of 18F-fluoxetine was found to besmall, approximately 12% of the total binding, when 10 mMfluoxetine was used for measuring nonspecific binding (Fig. 2).However, when other known serotonin transport inhibitors, such ascitalopram, were used for measuring nonspecific binding, there wasno decrease in binding compared with the observed total binding.Citalopram has been shown to selectively localize in serotonergic
uptake sites (6). It has also been shown to be higher in affinity forthese uptake sites than fluoxetine (19). It is therefore unclear whyfluoxetine and not citalopram was able to marginally reduce totalbinding of 18F-fluoxetine. One possible explanation is that fluox-etine displaced 18F-fluoxetine from a potentially saturable, nonse-rotonin transporter site, where 10 mM fluoxetine when used formeasuring nonspecific binding probably saturates these nonspecificbinding sites, and is therefore able to provide some measure, albeitsmall (12%), of specific (nonserotonin transporter site) binding of18F-fluoxetine.
Our autoradiographic studies with 18F-fluoxetine in rat brainslices showed findings that were similar to the in vitro saturationstudies. None of the brain regions tested (striata, hippocampus,hypothalamus, thalamus, mid-brain, hippocampus, raphe-nucleus,cerebellum, and brain stem) showed any clear areas of binding. Thetotal binding in all brain areas was generally uniform (results notshown). Experiments on the measurement of nonspecific binding of18F-fluoxetine in the brain slices in the presence of 1 mM citalo-pram did not reduce the extent of binding of 18F-fluoxetine. This issimilar to that found in the in vitro saturation studies and confirmsthe tight, subcellular binding of 18F-fluoxetine.
In vivo biodistribution studies in rats with 18F-fluoxetine showedsimilar uptake in all brain regions. There was some marginallygreater uptake in the olfactory tubercles. The levels in the brainremained steady over the course of 2 h (Fig. 3). Blood levels of theradiotracer were approximately 25% of what was in the brain andremained at those levels over the course of 2 h. Similar behavior hasbeen reported for 11C-fluoxetine in mice, where the levels in thebrain and blood remained similar over a 60-min time period (15).Coinjection of unlabeled fluoxetine and citalopram along with18F-fluoxetine did not reduce the uptake nor did it alter the timecourse of the radiotracer in the various brain regions. Preliminarypositron emission tomography (PET) data in rhesus monkeysshowed uptake and retention of 18F-fluoxetine in all brain regionswith no clear specific uptake, similar to that observed with 11C-fluoxetine (15). This finding is in contrast to the specific uptakeshown by other serotonin transporter radioligands (6, 7, 16, 17).
It has been shown previously that fluoxetine is present insubcellular components (1). Our findings of the high degree ofnonspecific binding of fluoxetine, which is retained over the courseof a few hours, probably indicates its subcellular nature. Ourinability to displace 18F-fluoxetine in the in vitro binding andautoradiographic experiments also indicates the irreversible natureof the binding of 18F-fluoxetine and attests to the subcellular natureof fluoxetine. Furthermore, our findings of subcellular localization of18F-fluoxetine in the mitochondria and synaptosomes confirms thetight binding nature of the drug to the brain regions. This propertyof fluoxetine accounts for its unique prolonged pharmacokinetics incomparison to other antidepressants (11). However, it is stillunclear as to (a) what structural features of fluoxetine account forthe intracellular tight binding, and (b) what are the site(s) at whichfluoxetine binds.
What makes fluoxetine such a useful agent? Clinical doses offluoxetine are typically in the range of 20–40 mg/day. It has beenshown that the brain deposits significant levels of the drug, and thatbrain-to-blood ratio can be as high as 20 (8). Previous reports (1)and our findings of the tight nonspecific binding to mitochondriaand other subcellular regions accounts for the slow pharmacokinet-ics, resulting in this high brain-to-blood ratio. The concentration offluoxetine in the brain can thus be as high as 35 mM. Thisconcentration of fluoxetine is almost a 1,000-fold excess of thebinding affinity of fluoxetine for the serotonin transporters and
FIG. 4. Subcellular localization of 18F-fluoxetine 60 min afterintravenous (IV) administration in three groups of rats (con-trol rats, pretreated with deprenyl, and pretreated with cita-lopram).
18F-Fluoxetine Binding in the Brain 609

therefore is more than enough to inhibit serotonin transporters.However, because fluoxetine is located primarily in intracellularregions, the synaptic concentration maybe be smaller. In addition tothe serotonin transporter, using radiolabeled monoamine oxidaseinhibitors, we have recently shown that fluoxetine has an inhibitoryeffect on monoamine oxidase B (10).
CONCLUSION
Our results show that 18F-fluoxetine becomes intracellular andbinds nonspecifically to the mitochondria, synaptosomes, and othercellular organelles. This intracellular nature of 18F-fluoxetine cir-cumvents the potential of the radiotracer to localize preferentiallyin regions of the rat brain that are known to contain serotoninuptake sites (hypothalamus, olfactory tubercles, etc.). The nonse-lective uptake is also evident from in vitro autoradiographs using18F-fluoxetine. 18F-Fluoxetine does show saturable binding charac-teristics in the rat brain homogenate. However, it is unlikely thatthis represents binding to the serotonin transporter sites. Thebinding of 18F-fluoxetine in the rat brain tissue was found to belargely irreversible, which accounts for the slow pharmacokineticsof fluoxetine.
We thank the U.S. Department of Energy, Office of Health andEnvironmental Research (DE-FG02-86ER60438 and DE-FG02-94ER61840) for financial support. We thank Dr. E. Cook for providinga sample of fluoxetine. We also thank Drs. M. Cooper, E. Cook and L.Seiden for helpful discussions and acknowledge Robert Mintzer andXiaohu Ouyang for technical assistance.
References1. Caccia S., Cappi M., Fracasso C. and Garattini S. (1990) Influence of
dose and route of administration of fluoxetine and its metabolitenorfluoxetine in the rat. Psychopharmacology 100, 509–514.
2. Das M. K. and Mukherjee J. (1993) Radiosynthesis of [F-18]fluoxetineas a potential radiotracer for serotonin reuptake sites. Appl. Radiat. Isot.44, 835–842.
3. Fuller R. W. (1987) Pharmacologic properties of serotonergic agentsand antidepressant drugs. J. Clin. Psychiatr. 48, (suppl. 3), 5–11.
4. Fuller R. W., Wong D. T. and Robertson D. W. (1991) Fluoxetine, aselective inhibitor of serotonin uptake. Med. Res. Rev. 11, 17–34.
5. Hammadi A. and Crouzel C. (1993) Synthesis of fluorine-18 (S)-fluoxetine: A selective serotonin uptake inhibitor. J. Label. Compds.Radiopharm. 33, 703–710.
6. Hume S. P., Pascali C., Pike V. W., Turton D. R., Ahier R. G., MyersR., Bateman D. M., Cremer J. E., Manjil L. G. and Dolan R. (1991)Citalopram: Labelling with carbon-11 and evaluation in rat as apotential radioligand for in vivo PET studies of 5-HT re-uptake sites.Nucl. Med. Biol. 18, 339–351.
7. Jagust W. J., Eberling J. L., Roberts J. A., Brennan K. M., HanrahanS. M., VanBrocklin H., Enas J. D., Biegon A. and Mathis C. A. (1993)In vivo imaging of the 5-hydroxytryptamine reuptake site in primatebrain using single photon emission computed tomography and [123I]5-iodo-6-nitroquipazine. Eur. J. Pharmacol. 242, 189–193.
8. Karson C. N., Newton J. E. O., Livingston R., Jolly J. B., Cooper T. B.,Sprigg J. and Komoroski R. A. (1993) Human brain fluoxetine concen-trations. J. Neuropsychiatry Clin. Neurosci. 5, 322–329.
9. Leonardi E. T. K. and Azmitia E. C. (1994) MDMA (ecstasy) inhibitionof MAO type A and type B: Comparisons with fenfluramine andfluoxetine (Prozac). Neuropsychopharmacology 10, 231–238.
10. Mukherjee J. and Yang Z. Y. (1997) Evaluation of monoamine oxidaseB inhibition by fluoxetine (Prozac): An in vitro and in vivo study. Eur.J. Pharmacol. 337, 111–114.
11. Richelson E. (1994) Pharmacology of antidepressants–-Characteristicsof the ideal drug. Mayo Clin. Proc. 69, 1069–1081.
12. Robertson D. W., Krushinski J. H., Fuller R. W. and Leander J. D.(1988) Absolute configuration and pharmacological activities of theoptical isomers of fluoxetine, a selective serotonin-uptake inhibitor.J. Med. Chem. 31, 1412–1417.
13. Scheffel U. and Hartig P. R. (1989) In vivo labeling of serotonin uptakesites with [3H]paroxetine. J. Neurochem. 52, 1605–1612.
14. Schmidt M. J., Fuller R. W. and Wong D. T. (1988) Fluoxetine, ahighly selective serotonin reuptake inhibitor: A review of preclinicalstudies. Br. J. Psychiatr. 153, (suppl. 3), 40–46.
15. Shiue C. Y., Shiue G. G., Cornish K. G. and O’Rourke M. F. (1995)PET study of the distribution of [11C]fluoxetine in a monkey brain.Nucl. Med. Biol. 22, 613–616.
16. Suehiro M., Scheffel U., Dannals R. F., Ravert H. T., Ricaurte G. A.and Wagner Jr. H. N. (1993) A PET radiotracer for studying serotoninuptake sites: Carbon-11-McN-5652Z. J. Nucl. Med. 34, 120–127.
17. Szabo Z., Kao P. F., Scheffel U., Suehiro M., Mathews W. B., RavertH. T., Musachio J. L., Marenco S., Kim S. E. and Ricaurte G. A. (1995)Positron emission tomography imaging of serotonin transporters in thehuman brain using [11C](1)McN5652. Synapse 20, 37–43.
18. Whittaker V. P. and Barker L. A. (1972) The subcellular fractionationof brain tissue with special reference to the preparation of synaptosomesand their component organelles. In: Methods of Neurochemistry, Vol. 2(Edited by Fried R.), pp. 1–50. Marcel Dekker, New York.
19. Wong D. T., Bymaster F. P. and Engleman E. A. (1995) Prozac(fluoxetine, Lilly 110140), the first selective serotonin uptake inhibitorand an antidepressant drug: Twenty years since its first publication. LifeSci. 57, 411–441.
20. Wong D. T., Bymaster F. P., Reid L. R., Mayle D. A., Krushinski J. H.and Robertson D. W. (1993) Norfluoxetine enantiomers as inhibitors ofserotonin uptake in rat brain. Neuropsychopharmacology 8, 337–344.
610 J. Mukherjee et al.

















![A rapid solid-phase extraction method for measurement of non-metabolised peripheral benzodiazepine receptor ligands, [18F]PBR102 and [18F]PBR111, in rat and primate plasma](https://static.fdokumen.com/doc/165x107/63349cad6c27eedec605ce97/a-rapid-solid-phase-extraction-method-for-measurement-of-non-metabolised-peripheral.jpg)


