
Etude fonctionnelle de l’inactivation du chromosome X au moyen de délétions ciblées dans le...
290
Thèse de Doctorat En vue de l’obtention du diplôme de DOCTEUR de l’UNIVERSITÉ PARIS VI – PIERRE ET MARIE CURIE École Doctorale : Logique du Vivant Spécialité : Biologie du Développement Présentée par Sébastien Vigneau « Etude fonctionnelle de l’inactivation du chromosome X au moyen de délétions ciblées dans le centre d’inactivation du chromosome X murin. » Thèse dirigée par le Docteur Philippe Clerc Soutenue le 19 septembre 2007 à l’Institut Pasteur de Paris Membres du jury : Président: Pierre Netter Examinateurs: Déborah Bourc'his et Michel Cohen-Tannoudji Rapporteurs: Jérôme Cavaillé et Saadi Khochbin Travaux de recherche réalisés au sein de l'unité « Génétique Moléculaire Murine » dirigée par le Pr. Philip Avner Département de Biologie du Développement de l’Institut Pasteur de Paris
-
Upload
dana-farber -
Category
Documents
-
view
0 -
download
0
Transcript of Etude fonctionnelle de l’inactivation du chromosome X au moyen de délétions ciblées dans le...

Thèse de Doctorat
En vue de l’obtention du diplôme de
DOCTEUR de l’UNIVERSITÉ PARIS VI – PIERRE ET MARIE CURIE
École Doctorale : Logique du Vivant
Spécialité : Biologie du Développement
Présentée par
Sébastien Vigneau
« Etude fonctionnelle de l’inactivation du chromosome X au moyen de délétions ciblées dans le centre d’inactivation du chromosome X murin. »
Thèse dirigée par le Docteur Philippe Clerc
Soutenue le 19 septembre 2007 à l’Institut Pasteur de Paris
Membres du jury :
Président: Pierre Netter
Examinateurs: Déborah Bourc'his et Michel Cohen-Tannoudji
Rapporteurs: Jérôme Cavaillé et Saadi Khochbin
Travaux de recherche réalisés au sein de l'unité « Génétique Moléculaire Murine »
dirigée par le Pr. Philip Avner
Département de Biologie du Développement de l’Institut Pasteur de Paris


« La plupart du temps, ce n'est pas la structure qui fait la fonction, mais la fonction qui capture des structures différentes. Comme quand on utilise un livre comme pressepapiers un jour de grand vent. »
(Antoine Danchin, cité dans Le Monde, printemps 2007)
« La complexité n'est jamais simple et il est permis de le regretter. »
(chronique de JeanLouis Ezine sur France Culture, printemps 2007)


Remerciements
Il m'est difficile de remercier tout ceux qui m'ont accompagné, par leur amitié d'abord, durant
ces années de thèse et ces années parisiennes. Difficile, car ils sont nombreux et vouloir résumer en
seulement quelques phrases ne leur rendrait pas justice. Années complexes, de découvertes, de
joies, et quelques fois de doutes. Mais années enrichissantes surtout. Un bout de chemin à travers
lequel j'ai beaucoup appris, et dont les enseignements m'accompagneront longtemps.
Évidemment, mes remerciements s'adressent à ma famille et à mes amis, qui m'ont toujours
soutenu dans mes choix. Mais aussi à la possibilité qui m'a été donnée d'accomplir ma thèse dans
les meilleures conditions, matérielles certes, mais surtout d'encadrement. Philippe, je t'en suis
infiniment reconnaissant, ainsi qu'à tous les membres du laboratoire, passé et présent, avec qui j'ai
eu plaisir à travailler. Philip aussi, pour faire de ce laboratoire ce qu'il est, dynamique et reconnu, un
tremplin en somme lorsqu'on a la chance d'y passer ses années de thèse.
Une page se tourne donc, mais l'une des choses que j'ai apprises durant ces années, c'est de
rester lié avec les personnes qui comptent, malgré les distances, géographiques, qui ne manqueront
pas de nous séparer. Keep in touch!


RésuméChez les mammifères femelles, chaque cellule somatique de l'individu adulte exprime les gènes
portés par un seul des deux chromosomes X. Le second chromosome X est maintenu
transcriptionnellement inactif par un mécanisme épigénétique complexe appelé inactivation du
chromosome X. Cette inactivation permet la compensation de dose des gènes liés au chromosome X
entre les sexes, en assurant un niveau équivalent d'expression de ces gènes chez les femelles (XX)
et les mâles (XY). Une fois établie, elle est transmise de manière clonale au cours des divisions
cellulaires. Chez la souris, dans l'embryon préimplantatoire et dans les tissus extraembryonnaires,
l'inactivation est soumise à l'empreinte parentale, et seul le chromosome d'origine paternelle est
inactivé. Au contraire, après réactivation du chromosome X paternel dans la masse cellulaire interne
des blastocystes, chacun des deux chromosomes X peut être choisi pour être inactivé dans les
cellules somatiques de l'embryon au sens strict. Ce mode d'inactivation, qualifié d'aléatoire,
commence à être mis en place vers 5 jours de gestation, ce qui correspond au début de
l'implantation. L'inactivation aléatoire est récapitulée lors de la différenciation in vitro de cellules
souche embryonnaires murines femelles, les cellules souches embryonnaires mâles étant quant à
elles, normalement, incapables de réaliser l'inactivation de leur unique chromosome X.
Le chromosome X inactif est recouvert par un long ARN non codant, Xist, dont dépend
initialement le recrutement de marques épigénétiques stabilisant la répression transcriptionnelle.
Xist est exprimé à partir d'une région du chromosome X appelée centre d'inactivation du
chromosome X (Xic), qui est nécessaire et suffisante pour initier le processus d'inactivation. Le Xic
contrôle en particulier deux mécanismes, le comptage et le choix, essentiels à l'inactivation
aléatoire. Le comptage détermine le nombre de chromosomes X destinés à demeurer actif, en
évaluant le rapport entre le nombre de chromosomes X et le nombre d'autosomes. Il permet
notamment de maintenir actif l'unique chromosome X chez les mâles, et l'un des deux chromosomes
X chez les femelles. Le choix consiste à décider sur quel chromosome X sera mise en place
l'inactivation. La région en 3' de Xist joue un rôle essentiel pour ces deux processus. Elle contient
notamment la région 5' du gène Tsix, transcrit antisens à Xist et capable de réprimer l'expression de
Xist en cis. Des travaux antérieurs ont démontré que Tsix participait au processus de choix. Le
travail réalisé durant cette thèse a permis de montrer qu'il est également un élément de la voie de
comptage, en utilisant comme modèle des cellules souches embryonnaires mâles, dans lesquelles
une série de délétions ciblées en 3' de Xist ont été générées par recombinaison homologue au locus
endogène. En effet, dans les cellules où Tsix a été invalidé, l'inactivation de l'unique chromosome X
est observée en cours de différenciation, ce qui traduit un défaut de comptage.

La fonction de Tsix dans le comptage semble dépendre de la répression transcriptionnelle du
gène Xist en cours de différenciation, consécutive à l'hétérochromatinisation du promoteur de Xist
lorsque Tsix est exprimé, dans les cellules indifférenciées. La caractérisation du profil épigénétique
dépendant de Tsix au niveau du Xic nous a permis de mettre en évidence une activité opposée de
Tsix dans le corps du gène Xist, consistant à empêcher la mise en place d'une marque de type
hétérochromatique (H3K27me3). Elle a également révélé que Tsix régulait sur une région étendue
en 5' de Xist le niveau d'enrichissement pour cette marque et le niveau d'expression de plusieurs
gènes.
En utilisant la même approche de délétion ciblée, nous avons également caractérisé le
minisatellite DXPas34, situé à proximité du promoteur majoritaire de Tsix, comme étant un
régulateur transcriptionnel majeur de ce dernier dans les cellules souches embryonnaires mâles.
Lorsque DXPas34 est délété, ces cellules sont hypomorphes pour Tsix, avec une expression
résiduelle de l'ordre de 10 % de celle observée dans les cellules sauvages. Cela se traduit, en cours
de différenciation, par l'initiation ectopique de l'inactivation de manière moins efficace que pour une
invalidation complète de Tsix. DXPas34 est donc un élément de la voie de comptage, probablement
du fait de la régulation qu'il exerce sur Tsix. Les cellules porteuses de la délétion de DXPas34 ont
été utilisées pour générer des souris recombinantes, de manière à pouvoir étudier la fonction de
DXPas34 in vivo, en particulier dans le mécanisme de choix et dans l'inactivation soumise à
l'empreinte parentale. Contrairement aux lignées mutantes pour Tsix publiées par d'autres
laboratoires, l'absence de DXPas34 n'a pas d'effet significatif sur le choix et l'inactivation soumise à
l'empreinte parentale. Cela suggère que la régulation exercée par DXPas34 sur Tsix pourrait être
variable en fonction des lignages ou des stades embryonnaires, et/ou que le niveau d'expression de
Tsix pourrait être interprété avec un effet de seuil pour ces deux mécanismes. Des expériences sont
en cours pour adresser ces interrogations.
Mots clés: épigénétique, développement, transcription, ARN, inactivation, minisatellite

Titre et résumé en anglais
Functional study of Xchromosome inactivation using targeted deletions in the mouse Xchromosome inactivation center
In mammals, one of the two X chromosomes is inactivated in females to enable dosage
compensation for X-linked gene products. X-chromosome inactivation (XCI) can occur on either X
in embryo proper after the blastocyst stage in mice, and is thereafter clonally transmitted. Random
XCI is also recapitulated during in vitro differentiation of embryonic stem cells. Due to a counting
process, one X is always maintained active for each diploid autosomal complement. XCI is
initiated by coating of the future inactive X by Xist RNA expressed in cis. Xist is repressed by its
antisense RNA counterpart Tsix, which was shown to be required for determination of the inactive
X in random XCI. To identify genomic elements involved in the regulation of Tsix expression and in
counting, we generated a series of deletions targeted in the 5' part of the Tsix gene within a
previously defined candidate region for counting in male ES cells. These experiments led to the
identification of the DXPas34 tandem repeat as an essential regulator of Tsix expression, and to the
demonstration that Tsix is essential to prevent XCI in males and, therefore, may participate in
counting. Histone modification profiling at the Xist/Tsix locus and 5' to Xist also revealed that Tsix
regulates chromatin configuration and gene expression over an extended region including but not
limited to Xist, and suggests that, in addition to repressing Xist expression in cis, Tsix creates a
chromatin configuration permissive to Xist upregulation in differentiating cells. Finally, we
generated recombinant mice bearing the DXPas34 deletion and we are currently investigating how
this affects Tsix expression and XCI in the embryo.
Key words: epigenetics, development, transcription, RNA, inactivation, minisatellite


Table des matières
Avantpropos.........................................................................................................................................5Introduction..........................................................................................................................................9
Partie 1 Introduction à l'inactivation du chromosome X................................................................9Chapitre 1 Déterminisme sexuel et compensation de dose........................................................9
1.1 Déterminisme sexuel et évolution du chromosome X.....................................................91.2 Le chromosome X, un chromosome atypique?..............................................................121.3 Compensation de dose chez les mammifères.................................................................14
1.3.1 L'inactivation du chromosome X: principes généraux et aperçu historique...........141.3.2 Compensation de dose du chromosome X actif et des autosomes.........................16
1.4 Compensation de dose chez d'autres espèces.................................................................18Chapitre 2 Epigénétique du chromosome X inactif.................................................................23
2.1 L'ARN Xist....................................................................................................................242.2 Réorganisation du territoire chromosomique................................................................352.3 Modifications de la chromatine.....................................................................................38
2.3.1 Le code histone......................................................................................................382.3.2 Marques épigénétiques du chromosome X inactif: stabilité et dynamique............412.3.3 Lecteurs et effecteurs des marques épigénétiques sur le chromosome X inactif...47
2.4 Réplication asynchrone.................................................................................................492.5 Gènes échappant à l'inactivation...................................................................................50
Chapitre 3 Plasticité épigénétique et régulation de l'inactivation au cours du développement533.1 Epigénétique et développement embryonnaire..............................................................533.2 Inactivation méiotique des chromosomes sexuels.........................................................553.3 Inactivation soumise à l'empreinte parentale.................................................................59
3.3.1 Découverte.............................................................................................................593.3.2 Mise en place de l'inactivation empreintée après la fécondation...........................603.3.3 Réactivation du chromosome X paternel dans la masse cellulaire interne du blastocyste........................................................................................................................623.3.4 Inactivation empreintée dans le trophectoderme...................................................633.3.5 Inactivation empreintée dans l'endoderme primitif...............................................643.3.6 Nature de l'empreinte parentale.............................................................................653.3.7 Inactivation soumise à l'empreinte parentale chez l'homme..................................68
3.4 Inactivation aléatoire dans l'embryon............................................................................693.4.1 Mise en place.........................................................................................................693.4.2 Compétence du chromosome X à être reprogrammé.............................................71
3.5 Réactivation du chromosome X inactif dans la lignée germinale.................................723.6 Modèles cellulaires........................................................................................................73
1

Partie 2 Le centre d'inactivation du chromosome X.....................................................................77Chapitre 1 Fonctions et caractérisation du centre d'inactivation..............................................77
1.1 Comptage, choix et initiation en cis de l'inactivation....................................................771.1.1 Comptage................................................................................................................771.1.2 Choix......................................................................................................................83
1.2 Caractérisation du centre d'inactivation du chromosome X..........................................881.2.1 Définition cytogénétique........................................................................................881.2.2 Définition par transgenèse.....................................................................................881.2.3 Composition du Xic...............................................................................................911.2.4 Epigénétique du Xic: propriétés de la région en 5' de Xist....................................93
Chapitre 2 Dissection génétique du centre d'inactivation........................................................952.1 Rôle de Xist dans le choix et le comptage.....................................................................952.2 Le gène Tsix..................................................................................................................98
2.2.1 Variété des transcrits Tsix......................................................................................982.2.2 Expression de Tsix...............................................................................................102
2.2.2.1 Dans les cellules ES....................................................................................102 2.2.2.2 Dans l'embryon............................................................................................103
2.3 Fonctions de Tsix et dissection génétique en 3' de Xist...............................................1042.3.1 Implication de Tsix dans l'inactivation soumise à l'empreinte parentale.............1042.3.2 Tsix et la région en 3' de Xist dans le choix.........................................................107
2.3.2.1 Rôle de Tsix dans le choix...........................................................................107 2.3.2.2 Rôle de la région en 3' de Xist dans le choix..............................................108
2.3.3 Tsix et la région en 3' de Xist dans le comptage..................................................110 2.3.3.1 Rôle de Tsix dans le comptage.....................................................................110 2.3.3.2 Rôle de la région en 3' de Xist dans le comptage........................................111
Chapitre 3 Régulation de l'expression de Tsix et minisatellite DXPas34...............................1153.1 Facteurs généraux de transcription et marques chromatiniennes au promoteur de Tsix............................................................................................................................................1153.2 Régulateurs transcriptionnels de Tsix..........................................................................1163.3 Le minisatellite DXPas34.............................................................................................116
Chapitre 4 Régulation de l'expression de Xist par Tsix..........................................................1214.1 Régulation posttranscriptionnelle...............................................................................1214.2 Régulation transcriptionnelle.......................................................................................1234.3 Remodelage de la chromatine associé à l'expression de Tsix......................................127
Résultats............................................................................................................................................131Chapitre 1 Tsix et le minisatellite DXPas34 jouent un rôle essentiel dans le comptage........131
1.1 Article 1: An essential role for the DXPas34 tandem repeat and Tsix transcription in the counting process of X chromosome inactivation..........................................................1321.2 Tsix est un élément essentiel de la voie de comptage dans les cellules ES mâles.......147
1.2.1 Stabilité de l'inactivation dans les cellules mâles.................................................1471.2.2 Niveau d'expression de Xist et formation de domaines d'ARN Xist....................150
1.3 DXPas34 régule l'expression de Tsix...........................................................................151
2

1.3.1 DXPas34 et autres éléments régulant l'expression de Tsix...................................1511.3.2 Comparaison avec un second mutant de DXPas34..............................................152
Chapitre 2 Resultats préliminaires sur la fonction de DXPas34 dans l'embryon...................1552.1 Transmission maternelle de la délétion DXPas34 et rôle de DXPas34 dansΔ l'inactivation empreintée.....................................................................................................1552.2 DXPas34 et le comptage dans l'embryon.....................................................................1582.3 DXPas34 et le choix dans l'embryon...........................................................................1592.4 Discussion et comparaison avec la délétion DXPas34 générée par Cohen et al.Δ .....160
Chapitre 3 Rôle de Tsix dans la régulation épigénétique du Xic...........................................1653.1 Article 2: Tsix is a boundary element controlling H3K27 trimethylation at the Xinactivation center prior to Xinactivation..........................................................................1663.2 Principales conclusions................................................................................................2103.3 Bivalence des fonctions de Tsix dans le Xic................................................................2113.4 Régulation globale du Xic par Tsix.............................................................................213
Discussion générale..........................................................................................................................215Chapitre 1 Conservation de Tsix et de DXPas34....................................................................215
1.1 Conservation du gène Tsix...........................................................................................2151.2 Conservation des éléments régulant l'expression de Tsix............................................220
Chapitre 2 Le Xic, cas particulier ou paradigme?..................................................................2252.1 ARN non codants et expression monoallélique...........................................................2252.2 Centres d'empreinte et répétitions en tandem..............................................................229
2.2.1 Comparaison entre DXPas34 et les centres d'empreinte......................................2292.2.2 Fonction de la répétition en tandem au niveau de DXPas34...............................233
Chapitre 3 Choix et comptage: une distinction illusoire?......................................................235Chapitre 4 Perspectives du travail de thèse............................................................................239
3

4

Avant-proposChez les mammifères, le sexe est généralement déterminé au moyen de chromosomes sexuels
hétéromorphes, X et Y. La présence d'un chromosome Y détermine le sexe mâle (un chromosome Y
et un seul chromosome X), son absence le sexe femelle (deux chromosomes X). Des évènements
complexes, et parfois distincts suivant les espèces, ont contribué à façonner le contenu génétique,
mais aussi le comportement épigénétique des chromosomes sexuels et, particulièrement, du
chromosome X. La présence des gènes liés à ce dernier en deux copies chez la femelle est
compensée par l'extinction transcriptionnelle, plus ou moins complète, de l'un des deux
chromosomes X. Bien que l'existence d'une telle « inactivation du chromosome X » ait été établie
par Mary Lyon dès 1963 (Lyon, 1963), la compréhension de ce mécanisme essentiel est encore très
partielle. A l'inactivation s'ajoute désormais un second mécanisme de compensation de dose, mis en
évidence récemment chez les mammifères (Nguyen et Disteche, 2006), qui permet l'expression à un
niveau comparable des gènes du chromosome X et des gènes autosomiques, alors qu'un seul allèle
des premiers n'est actif. L'existence de ces deux modes de compensation de dose ouvre des
perspectives évolutives intéressantes dont nous discuterons brièvement au chapitre 1 de la première
partie de l'introduction, après avoir donné un aperçu des relations entre système de déterminisme
sexuel et principes généraux de compensation de dose.
Nous verrons au chapitre 2 de la première partie de l'introduction que l'inactivation du
chromosome X peut, à bien des égards, être considérée comme un paradigme de régulation
épigénétique. En effet, elle a pour conséquence, chez les femelles, un comportement radicalement
opposé des deux chromosomes X qui pourtant, tout au moins dans un organisme modèle comme la
souris, peuvent être génétiquement identiques. L'essor récent de l'épigénétique est en grande partie
lié à la compréhension émergente du rôle des ARN non codants et des combinaisons particulières de
modifications des queues d'histones (l'hypothèse du « code histone »). A ce titre aussi, l'inactivation
du chromosome X est exemplaire, car elle fait appel à l'ARN non codant Xist, qui en recouvrant le
chromosome X inactif initie sa recomposition en marques épigénétiques, parmi lesquelles plusieurs
modifications des queues d'histones. D'autres marques, dont la méthylation de l'ADN, participent
également à maintenir l'un des chromosomes X inactif, et la manière dont la combinaison de
l'ensemble de ces marques est établie et interprétée est un domaine très actif de recherche.
5

Le champ disciplinaire que représente l'épigénétique est encore mal défini. Nous tenterons d'en
cerner les contours au fil du texte et, particulièrement au chapitre 3 de la première partie de
l'introduction, où nous situerons l'inactivation du chromosome X dans le contexte du
développement embryonnaire. Nous verrons que les modalités de l'inactivation sont variables en
fonction des stades et de l'appartenance à l'un ou l'autre des premiers lignages embryonnaires et
extraembryonnaires. Nous aborderons également dans ce chapitre la question de la stabilité de
l'inactivation. Bien que généralement transmis inchangé au cours des divisions cellulaires, l'état
inactif peut en effet être déstabilisé dans certaines lignages cellulaires ou pour certaines conditions
expérimentales.
L'initiation de l'inactivation est contrôlée par une région du chromosome X, baptisée centre
d'inactivation du chromosome X. En particulier, y sont contrôlés deux processus essentiels, celui
qualifié de comptage, qui empêche l'inactivation d'avoir lieu sur un chromosome X par complément
diploïde d'autosomes, et le processus de choix, qui décide lequel des chromosomes X sera inactivé
ou maintenu actif. Nous présenterons ces notions en détail dans la seconde partie de l'introduction,
en mettant l'accent sur l'identification au sein du centre d'inactivation d'éléments contrôlant le
comptage et le choix. Deux acteurs essentiels en sont le gène Xist et un gène antisens à celui-ci,
nommé Tsix, dont nous détaillerons les fonctions.
Si la participation de Tsix au choix a été établie au moment de sa découverte, son rôle dans le
comptage est par contre sujet à controverse. Le travail de thèse a permis de préciser cette
contribution de Tsix au comptage (Résultats, chapitre 1), mais aussi de décrire plus précisément la
manière dont Tsix interagit avec le reste du centre d'inactivation (Résultats, chapitre 3).
Parallèlement, nous avons caractérisé un minisatellite, appelé DXPas34, situé à proximité du
promoteur de Tsix, comme élément essentiel de la régulation transcriptionnelle de ce dernier
(Résultats, chapitre 1). La plus grande partie de ce travail a été réalisé en mettant à profit un modèle
cellulaire, mais la caractérisation de la fonction de DXPas34 est désormais poursuivie dans la souris
(Résultats, chapitre 2).
De nombreuses zones d'ombres subsistent néanmoins concernant la fonction de Tsix ou de
DXPas34. Une difficulté majeure est d'arriver à concilier des résultats, parfois divergents, obtenus
par différentes approches. Néanmoins, un certain nombre de découvertes ont permis de faire
évoluer, depuis quelques années, le cadre conceptuel dans lequel nous envisageons l'inactivation.
D'une part, parce que la compréhension d'autres systèmes de régulation épigénétique a progressé de
pair avec celle de l'inactivation, et nous offre désormais de nombreux points de comparaison qui
6

alimentent notre réflexion. D'autre part, parce que de nouveaux modèles ont émergé pour concilier
entre elles les données expérimentales. Nous donnerons un aperçu des ces perspectives dans la
discussion générale.
7

8

Introduction
Partie 1 Introduction à l'inactivation du chromosome X
Chapitre 1 Déterminisme sexuel et compensation de dose
1.1 Déterminisme sexuel et évolution du chromosome X
Chez les vertébrés, le sexe des individus peut être déterminé en fonction de facteurs
environnementaux ou au moyen de chromosomes sexuels hétéromorphes chez la femelle (système
ZZ/ZW) ou chez le mâle (système XX/XY). Ainsi, le système XX/XY est étendu à la plupart des
mammifères, tandis que le système ZZ/ZW l'est à la plupart des oiseaux (pour revue, Schartl, 2004).
Chez les mammifères, le contenu du chromosome X est étroitement lié à l'évolution du
chromosome Y, dont on pense qu'il résulte de l'altération d'un autosome ancestral (Ohno, 1967).
Chez l'homme, par exemple, le contenu génique du Y est quantitativement huit fois inférieur à celui
du X (140 gènes contre 1142, respectivement; source: http://www.ensembl.org/)1. L'évènement
originel ayant conduit à une telle perte de gènes sur le Y pourrait être l'acquisition d'un gène
participant à la détermination du sexe mâle, probablement l'ancêtre du gène SRY (fig. 2B, p. 17)
(Lahn et Page, 1999a). La suppression, par des inversions successives, des recombinaisons entre les
chromosomes d'une même paire portant ou non ce gène aurait alors constitué un avantage sélectif
en prévenant l'apparition de phénotypes sexuels anormaux (Charlesworth, 1996; Charlesworth et
al., 2005; Lahn et Page, 1999a; Rice, 1996). Non seulement l'absence de recombinaisons
interchromosomiques, mais aussi le nombre plus important de divisions cellulaires requises pour
produire un spermatozoïde comparativement à un ovule, ainsi que la présence d'un environnement
1 Ce ratio pourrait être réévalué à la baisse dans la mesure où les séquences correspondant à environ la moitié du chromosome Y humain n'ont pas encore été assemblées.
9

10
Fig. 1: Evolution du chromosome X humain (adapté d'après Graves, 2006)
Légende page 11

oxidatif, combiné à l'absence d'enzymes de réparations, dans les spermatozoïdes, sont autant
d'explications au taux important de mutations observé sur le Y et à sa rapide divergence vis à vis du
X (Aitken et Marshall Graves, 2002).
D'après la comparaison du contenu génique du chromosome X dans différentes espèces de
mammmifères, il semblerait que la structure d'ensemble du chromosome X des marsupiaux ait peu
divergé de celle du chromosome X ancestral des mammifères placentaires, contrairement au
11
Légende de la figure 1, p. 10:(1) Arbre évolutif des mammifères. Les périodes estimées d'apparition des différents phyla sont précisées en millions d'années (Ma)(2) Strates évolutives des chromosomes X et Y humains(2A) Le positionnement de gènes orthologues d'autres mammmifères sur le chromosomes X humain a permis de caractériser une région conservée sur le chromosome X chez tous les mammifères (XCR pour Xchromosome conserved region, en bleu) et une région additionnelle (XAR pour Xchromosome added region, en jaune), qui est autosomique chez les marsupiaux et les monotremes, et a par conséquent été ajoutée au chromosome des euthériens il y a 100 à 180 millions d'années.(2B) Les homologues chez le poulet des gènes humains du chromosome X se répatissent sur trois régions autosomiques. Ils conduisent à distinguer trois strates évolutives qui correspondent à la région XAR (strate 3), et subdivisent la région XCR en deux (strates 1 et 2).(2C) La comparaison des séquences nucléotidiques entre les gènes du chromosome Y humain et les gènes homologues sur le chromosome X a conduit à grouper les gènes du chromosome X en cinq ensembles. Les ensembles 1 et 2 correspondent à la région XCR. La région XAR contient les ensembles 3 et 4, ainsi que la région pseudoautosomique (PAR).(2D) L'analyse comparative directe du chromosome Y humain et de celui des marsupiaux, et l'analogie avec les strates évolutives du chromosome Y, a permis de subdiviser le chromosome Y en régions d'apport ancien ou récent. La région la plus ancienne (YCR) est très petite, et la plupart du chromosome Y dérive d'ajouts plus récents (YAR). Quelques gènes (en orange) ont également été transposés à partir d'autres chromosomes. La région grisée est mal connue.(3) Origine de la région XCR du chromosome X humain(3A) Le platypus (monotrème) a 5 chromosomes X et 5 chromosomes Y qui s'assemblent en une chaîne dite « de translocation » lors de la méïose. Les extrémités de chaque chromosome avec une sonde (rose) qui hybride avec les télomères. Le chromosome X à une extrémité de la chaîne est homologue à la région XCR du chromosome X des mammifères, et le chromosome X à l'aute extrémité de la chaîne partage des gènes avec les système ZW de déterminisme sexuel chez le poulet.(3B) Une sonde (jaune) constituée à partir du chromosome X du kangourou (marsupial) hybride seulement avec le bras long et la région péricentrique du chromosome X humain, identifiant ainsi la région XCR.

chromosome X des euthériens, dont une part importante résulte de translocations successives de
matériel autosomique (Watson et al., 1991). Le chromosome X humain a ainsi cinq strates
évolutives d'après la comparaison des couples résiduels de gènes sur le X et sur le Y, la plus récente
d'entre elles étant la région pseudo-autosomique (pseudo-autosomal region, PAR) (fig. 1, p. 10 et
2B, p. 17) (Lahn et Page, 1999a; Ross et al., 2005). Ce schéma d'ensemble doit être enrichi de
données récentes sur l'organisation des chromosomes sexuels chez le monotrème Platypus. Ce
dernier possède en effet cinq chromosomes X et cinq chromosomes Y, qui forment ensemble une
chaîne unique lors de la méïose (fig. 1-3A, p. 10)(Grutzner et al., 2004). Le chromosome X présent
au début de cette chaîne (X1) contient des gènes typiques du couple XY des mammifères (Watson
et al., 1990), tandis que celui à l'extrémité opposée (X5) abrite le gène DMRT1, présent sur le
chromosome Z des oiseaux et candidat pour déterminer le sexe de ces derniers. En revanche, aucun
orthologue du gène SRY n'a pu être trouvé et aucune homologie entre le chromosome Y1 de
Platypus et le chromosome Y des mammifères placentaires n'a pu être mise en évidence. Plusieurs
modèles ont été proposés pour expliquer la formation et le maintien de cette configuration atypique
des chromosomes sexuels observée chez Platypus, vraisemblablement à la suite de translocations
répétées entre chromosomes sexuels et autosomes (Grutzner et al., 2004). Quoiqu'il en soit, elle
illustre la coexistence, au cours de l'évolution, de deux systèmes de chromosomes sexuels,
caractéristiques des oiseaux et des mammifères et ne présentant apparemment aucune homologie
entre eux (Nanda et al., 1999; Shetty et al., 1999). Ceci ouvre des possibilités inédites pour
comprendre comment a eu lieu la transition entre ces deux modes de déterminisme sexuel.
1.2 Le chromosome X, un chromosome atypique?
Le séquençage complet du génome humain a permis de conforter l'idée selon laquelle le
chromosome X serait à certains égards atypique, comparé aux autosomes. Le chromosome X
humain a ainsi une densité en gènes légèrement inférieure à la moyenne du génome, contrepartie
d'une densité en éléments répétés légèrement plus élevée. Il est enrichi en LINEs (Long
Interspersed Repeats), et particulièrement leur sous-classe L1, mais a une plus faible densité de
SINEs (Short Interspersed Repeats) (Bailey et al., 2000). La localisation préférentielle des
répétitions Alu dans les régions riches en GC et en gènes y est plus prononcée (Jurka et al., 2004).
Les LTR, de même que les éléments répétés inversés, y sont plus nombreux (Warburton et al.,
2004), ce qui n'est cependant pas le cas des minisatellites . Ces derniers ont toutefois un taux de
mutation inférieur à celui observé sur les autosomes, probablement lié au fait que le chromosome X
12

séjourne seulement un tiers du temps dans la lignée germinale mâle, où ont lieu la majorité des
mutations transmises (Kumar et Subramanian, 2002). Sur 12 SSR (simple sequence repeats) de trois
nucleotides documentés, 3 sont présents sur le chromosome X (pour revue, Chow et al., 2005). Le
chromosome X murin est moins bien décrit dans la littérature mais, tout au moins pour le cas
particulier de l'enrichissement en LINEs, dont nous verrons plus loin l'importance (cf p. 33), un
enrichissement similaire à celui du chromosome X humain est observé (pour revue, Lyon, 2003). La
proportion de rétrogènes est par ailleurs anormalement élevée sur les chromosome X humain et
murin, leur expression étant retrouvée le plus souvent dans les testicules et absente chez les femelles
(Emerson et al., 2004), ce qui a pu suggérer un rôle de ces derniers dans l'inactivation du
chromosome X lors de la méïose (Chow et al., 2005). De manière générale, le chromosome X des
mammifères est enrichi en gènes participant aux fonctions reproductives (pour revue Hurst, 2001)
mais aussi, de manière plus surprenante, exprimés dans le cerveau (Khil et al., 2004; Ross et al.,
2005; Simpson et al., 2005; Vallender et Lahn, 2004; Zechner et al., 2001) ou dans les muscles
(Bortoluzzi et al., 1998). A l'inverse, les gènes exprimés dans le placenta y sont sous-représentés
(Ko et al., 1998).
L'enrichissement du chromosome X en gènes dont l'expression est différente en fonction du
sexe avait été prédit il y a plus de vingt ans (Rice, 1987), en prenant l'hypothèse d'un gène récessif
avantageux pour les mâles mais délétère pour les femelles. La fréquence d'un tel gène au sein d'une
population doit augmenter rapidement car l'avantage qu'il procure aux mâles est immédiat du fait de
son hémizygotie, tandis que le désavantage créé chez les femelles est masqué par son caractère
récessif. Lorsque la fréquence de ce gène est suffisamment importante, ses effets négatifs chez les
femelles doivent cependant être annulés par l'acquisition d'une expression limitée à la lignée
germinale mâle, ou par l'accumulation sur le chromosome X de gènes avantageux pour les femelles.
C'est bien ce que l'on observe, une proportion anormalement élevée de gènes liés au chromosome X
ayant leur expression limitée aux spermatogonies (Wang et al., 2001b) ou participant à l'ovogenèse
(Graves, 2006; Rice, 1987).
L'enrichissement du chromosome X en gènes exprimés dans le cerveau ou dans les muscles est
plus difficile à expliquer. Il est vraisemblable que ces gènes n'aient acquis leur fonction actuelle
qu'après avoir été recrutés sur le chromosome X, compte-tenu de la synténie entre l'homme et
l'oiseau (Graves, 2006). Plusieurs hypothèses ont été proposées afin de rendre compte de
l'acquisition préférentielle d'une expression dans le cerveau (Graves, 2006). A l'instar des gènes
participant aux mécanismes reproductifs, les gènes contribuant aux fonctions cérébrales auraient pu
conférer, au moins transitoirement, un avantage préférentiel pour l'un des deux sexes. Il faudrait
13

alors s'attendre à leur expression différentielle en fonction du sexe, ce qui, pour certains d'entre eux,
semble être le cas (Nguyen et Disteche, 2006). En influant sur les comportements, ils auraient pu
contribuer, au même titre que les gènes exprimés dans les gonades, à l'établissement de barrières
reproductives lors des processus de spéciation (Graves et al., 2002). La sélection de fonctions
avantageuses principalement chez l'adulte aurait conduit, en retour, à l'éviction de gènes dont le rôle
est essentiellement embryonnaire, comme ceux exprimés dans le placenta. Il a également été
proposé que les gènes participant à des mécanismes généraux (par exemple le remodelage de la
chromatine pour SOX3 et ATRX, ou le métabolisme des ARN pour RBMX) pourraient avoir une
propension accrue à être recrutés aussi bien pour le développement du cerveau (dans le cas de
SOX3, ATRX et RBMX) que pour celui des gonades (dans le cas des gènes homologues SRY, ATRY
et RBMY, situés sur le chromosome Y) (Graves, 2006). On peut enfin imaginer qu'il existe des
mécanismes communs de régulation de l'expression des gènes entre ces deux lignages. A cet égard,
le système de compensation de dose mis en oeuvre au niveau du chromosome X n'est pas neutre.
L'existence d'un dispositif régulant la transcription au niveau d'un chromosome entier offrirait un
moyen ad hoc pour contrôler l'expression de gènes localisés sur ce chromosome et partageant une
même fonction, ce qui semble être le cas dans le cerveau (Nguyen et Disteche, 2006).
Parallèlement, l'incorporation progressive des gènes dans le système de compensation de dose
(Charlesworth, 1996) expliquerait la grande conservation du contenu génique du chromosome X
parmi les euthériens (Ohno, 1967; Rugarli et al., 1995).
1.3 Compensation de dose chez les mammifères
1.3.1 L'inactivation du chromosome X: principes généraux et aperçu
historique
C'est en 1961 que les bases théoriques de l'inactivation du chromosome X ont été pour la
première fois posées par Mary Lyon (Lyon, 1961), suivant de peu la découverte du système de
déterminisme sexuel chez les mammifères (Welshons et Russell, 1959).
L'élément déterminant a été la découverte, chez la souris, de mutants pour des gènes de pelage
liés au chromosome X, tabby (Falconer, 1953) et mottled (Fraser et al., 1953). Chez les femelles
hétérozygotes, ces mutations conduisent à la formation d'un pelage bigarré (mottled en anglais)
consistant en la juxtaposition de zones de pelage de phénotype sauvage et de zones de phénotype
14

mutant, tandis que chez les mâles porteurs de l'une ou l'autre de ces mutations, le pelage est toujours
uni. Un troisième mutant de même phénotype, lié au chromosome X, est caractérisé à l'issu d'un
crible par Mary Lyon en 1960 (Lyon, 1961). Le même type de mosaïcisme est observé dans le cas
de translocations impliquant le chromosome X (Russell et Bangham, 1961; Russell Bangham,
1959). La même année a lieu la découverte de souris femelles aneuploïdes (XO) viables et fertiles,
révélant que la détermination du sexe mâle dépend de la présence du chromosome Y (et non pas du
nombre de chromosomes X comme chez la drosophile), et qu'un unique chromosome X suffit à un
développement normal (Welshons et Russell, 1959). Le modèle que proposa alors Mary Lyon pour
expliquer ces observations consistait en 1) l'inactivation d'un chromosome X chez les femelles à un
stade précoce du développement embryonnaire; 2) le choix aléatoire du chromosome X maternel
(Xm) ou paternel (Xp) pour cette inactivation; 3) la stabilité de l'inactivation, une fois établie, lors
des divisions somatiques (Lyon, 1961; Lyon, 1962). Ce modèle rendait compte de l'effet clonal
observé pour les mutations ou translocations liées au chromosome X. Il faisait également le lien
avec des observations cytogénétiques antérieures. Ainsi, Barr avait décrit en 1949 la présence d'une
structure condensée, associée à la membrane nucléaire et révélée par la coloration de Nissl, dans des
noyaux interphasiques de motoneurones de chattes (Barr et Bertram, 1949). Cette « chromatine
sexuelle », dès lors appelée corpuscule de Barr, fut rapidement utilisée comme un moyen simple de
détermination du sexe sur des préparations histologiques, avant d'être finalement associée à un état
hétérochromatique de l'un des deux chromosomes X (OHNO et al., 1959; OHNO et HAUSCHKA,
1960). Elle pouvait être observée dès le stade blastocyste tardif dans de nombreuses espèces comme
le chat, le singe et l'homme (Austin et Amoroso, 1957; PARK, 1957), en agrément avec le modèle
proposé par Mary Lyon. Plus tard, la caratérisation de nombreuses marques épigénétiques associées
au domaine formé par le chromosome X inactif donnera une consistance nouvelle à ce corpuscule et
permettra de mieux situer, au cours de l'embryogenèse, la mise en place de l'inactivation (cf
intoruction, partie 1, chapitre 2, p. 23).
Le modèle proposé par Mary Lyon fut rapidement affiné pour tenir compte d'observations
nouvelles. Il fut en effet montré que des individus multisomiques pour le chromosome X, mais chez
qui le nombre d'autosomes restait inchangé (AA:XXY; AA:XXXX; AA:XXXXY), avaient
l'ensemble de leurs chromosomes X condensés, à l'exception d'un seul (Barr, 1963; Giannelli, 1963;
Grumbach et al., 1963). Ainsi, il s'agissait plutôt de maintenir actif un seul chromosome X par lot
d'autosome, ce qui introduisait une notion de comptage (Lyon, 1962; OHNO et al., 1964). D'autre
part, contrairement à ce qui avait été observé chez la souris, les femmes aneuploïdes XO présentent
des altérations phénotypiques sévères, correspondant au syndrome de Turner. Il fut alors proposé
15

qu'une partie du chromosome X humain ait une région homologue sur le chromosome Y et ne
nécessite donc pas de compensation de dose (Lyon, 1962). L'inactivation du chromosome X, telle
que nous venons de la décrire, s'applique essentiellement aux euthériens. En effet, chez les
marsupiaux, l'inactivation est partielle, variable selon les tissus, et ne concerne que le chromosome
X transmis par le père (Cooper et al., 1993; Wakefield et al., 1997).
Les bases de l'inactivation étant ainsi posées, il restait donc à en comprendre les mécanismes
moléculaires, aussi bien ceux permettant l'hétérochromatinisation et l'extinction transcriptionnelle
d'un chromosome entier, que ceux contrôlant la mise en place de l'inactivation de manière
opportune, au cours du développement embryonnaire. La caractérisation du centre d'inactivation du
chromosome X, puis de l'ARN Xist, dans les années quatre-vingt dix, puis l'essor de l'épigénétique
au tournant du siècle, ont refaçonné notre vision, sans pour autant remettre en cause les principes
généraux énoncés par Mary Lyon en 1961. Nous reviendrons sur ces différents aspects dans les
chapitres qui suivent.
1.3.2 Compensation de dose du chromosome X actif et des autosomes
Mise au regard de l'évolution, l'inactivation du chromosome X envisagée comme seul mode de
compensation de dose pose problème. En effet, sa conséquence logique aurait été de diminuer de
moitié l'expression des gènes liés au chromosome X vis-à-vis des autosomes. Or, il a été démontré,
tout au moins chez Drosophila melanogaster, qu'être haploïde pour seulement 1 % du génome
suffisait à réduire la viabilité, et devenait léthal lorsque 3 % du génome était concernés (Lindsley et
al., 1972). Dès 1967, Ohno suggérait que « ...dans le cours de l'évolution, un ancêtre des
mammifères placentaires a dû échapper au péril résultant de la présence à l'état hémizygote, chez les
mâles, des gènes liés au chromosome X, en doublant, pour chaque gène lié au chromosome X, la
quantité de produit de leur expression » (Ohno, 1967) (fig. 2A, p. 17). Pourtant, il fallut attendre
1997 pour avoir un premier indice que cela avait pu effectivement être le cas, lorsqu'il fut montré
qu'un gène (Clcn4-2), localisé sur le chromosome X chez la souris Mus spretus, y était exprimé à un
niveau double comparativement à un allèle du même gène, autosomal chez la souris de laboratoire
Mus musculus. Très récemment, deux équipes ont étendu la portée de cette observation à l'ensemble
du chromosome X, en mesurant chez la souris un niveau moyen d'expression des gènes liés au
chromosome X équivalent au niveau moyen d'expression des gènes autosomaux, alors que les
premiers ne sont exprimés qu'à partir d'un seul chromosome X (l'unique chromosome X chez le
mâle, le chromosome X actif chez la femelle) (Gupta et al., 2006; Nguyen et Disteche, 2006). Une
16

telle surexpression des gènes liés au chromosome X modifie profondément la manière dont nous
envisageons désormais l'inactivation du chromosome X. Bien que, aujourd'hui, ces deux
mécanismes de compensation de dose soient également indispensables, il n'est pas dit qu'ils aient
évolué initialement de manière concomitante. A l'instar d'autres espèces telles que Drosophila
melanogaster, on peut tout à fait imaginer que le premier mécanisme apparu pour compenser
l'hémizygotie des gènes du chromosomes X chez les mâles ait reposé seulement sur leur
17
Fig. 2: Evolution des chromosomes sexuels et compensation de dose (adapté d'après Heard et Disteche, 2006)
Légende page 18

surexpression. Il sera intéressant de voir dans quelle mesure une telle surexpression pourra être
retrouvée chez des mammifères phylogénétiquement distants, mais aussi de déterminer si
surexpression et inactivation sont moléculairement coordonnés ou non.
1.4 Compensation de dose chez d'autres espèces
Des systèmes de compensation de dose, conséquences du système de déterminisme sexuel, ont
été caractérisés, et étudiés en détail, dans de nombreuses espèces. Non seulement, comme nous
venons de le présenter, chez les mammifères, mais encore chez la drosophile (Drosophila
melanogaster) et le nématode (Caenorhabditis elegans). Notre propos n'est pas de faire ici un
exposé exhaustif de ce qu'il en est dans ces deux espèces. Il s'agit plutôt, à titre d'introduction,
d'esquisser les similitudes dans les stratégies qui ont été mises en oeuvre pour tirer le meilleur profit
d'un certain nombre de fonctions nucléaires préexistantes. C'est bien en ce sens que les mécanismes
de compensation de dose peuvent être paradigmatiques des régulations épigénétiques.
Chez D. melanogaster, comme chez les mammifères, les chromosomes sexuels sont
dimorphiques, et les femelles sont caractérisées par la présence de deux chromosomes X, tandis que
les mâles n'ont qu'un seul chromosome X, accompagné d'un chromosome Y. Ce dernier est le
produit de nombreux réarrangements, ayant abouti à la perte de nombreux gènes. Chez C. elegans,
ce processus a été poussé à son terme avec la disparition complète du chromosome Y, de telle sorte
que les hermaphrodites ont un génotype XO (un seul chromosome X) tandis que les mâles ont un
génotype XX. Dans les deux espèces, la valeur du ratio du nombre de chromosomes X et
d'autosomes est déterminante pour le choix du sexe.
18
Légende de la figure 2, p. 17: (A) Les chromosomes sexuels dérivent d'une paire de chromosomes homomorphes, ou protochromosomes sexuels. Lorsque le sexe commence à être déterminé par le gène SRY, présent sur le chromosome Y, la recombinaison entre les chromosomes sexuels disparaît. Chez les mâles, le chromosome Y diverge du chromosome Y à la fois par la perte de gènes et par l'accumulation de gènes avantageux pour les mâles (en bleu clair) autour du gène SRY (en bleu foncé), tandis que le chromosome X commence à être surexprimé (orange). Chez les femelles, le chromosome X actif (Xa) commence à être surexprimé (orange), alors que le second chromosome X (Xi) est soumis à l'inactivation du chromosome X (noir). (B) Translocation de matériel autosomique sur les chromosomes sexuels. La translocation de matériel autosomique sur la région pseudoautosomiques (PAR) des chromosomes sexuels est suivies de sa disparition ou différenciation (points bleus) sur le chromosome Y, et de sa surexpression (points oranges) ou son inactivation (points noirs) sur le chromosome X.

Les gènes portés par l'unique chromosome X des mâles voient leur expression doublée chez D.
melanogaster, grâce à l'expression spécifiquement chez les mâles, dans les tissus somatiques, de la
protéine MSL2 (Hamada et al., 2005; Straub et al., 2005), composante essentielle du complexe de
compensation de dose (Dosage Compensation Complex, DCC). Une autre composante du DCC est
l'ARN non-codant roX (RNA on the X), dont l'expression est elle-même activée par le DCC chez les
mâles (Bai et al., 2004; Rattner et Meller, 2004; Straub et al., 2005). L'existence d'un dispositif de
compensation de dose impliquant une surexpression du chromosome X mais ne dépendant pas de
MSL2 a également été mis en évidence dans la lignée germinale (Gupta et al., 2006).
Chez C. elegans, de même que chez les mammifères, l'expression des gènes liés aux
chromosomes X est doublée comparativement aux autosomes, aussi bien chez les individus
possédant deux chromosomes X (XX:AA, femelles chez les mammifères ou hermaphrodites chez
C. elegans) que chez ceux n'en possédant qu'un seul (XY:AA ou XO:AA, mâles chez les
mammifères et C. elegans, respectivement). Mais alors que chez les mammifères, l'un des deux
chromosomes X est pour l'essentiel transcriptionnellement inactif chez les femelles, chez C. elegans
les deux chromosomes X ont leur niveau d'expression diminué de moitié chez les hermaphrodites,
grâce à l'action d'un complexe de compensation de dose désormais bien caractérisé (pour revue,
Meyer et al., 2004). Il est intéressant de voir qu'une variante de ce dernier régule aussi le gène
autosomique her-1 (hermaphrodisation of XO animals), qui est un suppresseur de la différentiation
en hermaphrodite (Chu et al., 2002), illustrant le liant étroit entre compensation de dose et
déterminisme sexuel.
Cependant, il existe dans toutes les espèces des variations notables des niveaux d'expression des
gènes liés chromosome X pris individuellement, traduisant certainement leur parcours évolutif
différent et, notamment, leur intégration plus ou moins récente ou prononcée aux systèmes de
compensation de dose. Ceux-ci devraient donc être pensés plutôt comme une modulation
additionnelle de l'activité des gènes, suffisante pour garantir la viabilité des individus.
Il ne semble pas, pour l'instant, y avoir de conservation des mécanismes mis en oeuvre pour la
compensation de dose entre D. melanogaster, C. elegans et les mammifères. En revanche, des
fonctions analogues du métabolisme nucléaire semblent bien avoir été recrutées indépendamment.
Nous reviendrons plus en détail sur le cas des mammifères dans les chapitres ultérieurs. Aussi bien
chez la drosophile que le nématode, la régulation du niveau de compaction de la chromatine paraît
jouer un rôle essentiel, mais dans le premier cas au moyen de combinaisons de modifications
d'histones (auxquelles contribue l'acétylase MOF, pour male on first) et dans le second,
probablement, grâce à l'utilisation de condensines (pour revue, Lucchesi et al., 2005). Une seconde
19

constante est la capacité des DCC à recouvrir le chromosome cible de manière à créer,
potentiellement, un compartiment nucléaire aux propriétés particulières. Chez D. melanogaster, les
ARN non-codants RoX1 et RoX2, dont les gènes sont sur le chromosome X, sont nécessaires au
ciblage du DCC sur ce dernier (nous verrons, chez les mammifères, le rôle central de l'ARN non-
codant Xist). Des régions de plus forte affinité pour le complexe, pouvant servir de centres de
nucléation, sont réparties sur toute la longueur du chromosome. Une répartition biaisée de certains
codons en faveur du chromosome X pourrait servir au ciblage, mais tout autant être une
conséquence des contraintes de sélection particulières appliquées au chromosome X (Singh et al.,
2005). Le DCC est en général plus fortement enrichi au niveau de séquences transcrites
(Alekseyenko et al., 2006; Gilfillan et al., 2006), suggérant un rôle de la transcription en tant que
telle, ou des marques épigénétiques associées à cette dernière, dans le ciblage du complexe. D'autre
part, le DCC interagit fonctionnellement avec des composantes du complexe de pore nucléaire, dont
la présence est requise pour la compensation de dose, et qui pourraient participer à la formation d'un
domaine nucléaire particulier centré sur le chromosome X. Une distinction similaire entre sites de
ciblage primaires, de forte affinité pour le DCC, et secondaires, de plus faible affinité, a été décrite
20
Fig. 3: Compensation de dose chez D. melanogaster, C. elegans et les mammifères (adapté d'après Cheng et Disteche, 2006)
Un niveau équivalent d'expression des gènes du chromosome X et des gènes autosomiques est obtenu, dans les cellules somatiques mâles de D. melanogaster, C. elegans et des mammifères, en surexprimant les gènes de l'unique chromosome X (bleu, flèche montante). Dans les cellules somatiques femelles de D. melanogaster, les deux chromosomes X sont exprimés et n'ont donc pas besoin d'être surexprimés (rose). Dans les cellules somatiques hermaphrodites de C.elegans, la surexpression des deux chromosomes X se surimpose à un mécanisme permettant de diminuer de moitié l'expression de chaque chromosome X par rapport aux cellules mâles (rose, double flèche). Dans les cellules somatiques femelles des mammifères, l'un des deux chromomes X est actif et surexprimé (rose, flèche montante), tandis que le second est inactif (blanc, flèche descendante).

chez C. elegans (pour revue, Csankovszki et al., 2004; Nusinow et Panning, 2005). La séquence de
motifs enrichis dans les premiers, appelés rex (recruitment elements on the X), a été élucidée
récemment (McDonel et al., 2006).
On remarquera que, dans tous les organismes chez lesquels la compensation de dose a été
étudiée, les travaux se sont naturellement portés vers des systèmes modèles, tels les chromosomes
géants des glandes salivaires chez la drosophile, à la recherche de principes généraux. La mise en
contexte de ces mécanismes au cours du développement embryonnaire et de la vie des individus
reste, pour l'essentiel, à réaliser. Mais elle contribuera très certainement à mieux comprendre
comment l'épigénétique réalise ses deux missions contradictoires que sont la permanence et la
plasticité de l'expression génique.
21

22

Chapitre 2 Epigénétique du chromosome X inactif
Nous l'avons vu, les systèmes de compensation de dose ont évolué de manière à garantir
l'homéostasie génique, tandis qu'étaient mis en place des mécanismes de déterminisme sexuel
s'appuyant sur un nombre différent de chromosome X entre les sexes. L'exigence d'une telle
homéostasie est maintenue tout au long de la vie des individus. Il a donc fallu inventer, ou adapter,
des mécanismes garantissant la stabilité, au cours des divisions cellulaires, de la compensation de
dose. Désormais, le fil de notre discussion portera sur un exemple particulier de compensation de
dose, l'inactivation du chromosome X des mammifères, qui a été au centre de ce travail de thèse.
Dans ce chapitre, nous discuterons comment est créé puis maintenu l'état « silencieux » du
chromosome X inactif. Nous y présenterons des mécanismes concernant le chromosome X dans son
ensemble, caractérisant en quelque sorte l'état épigénétique du chromosome X inactif. L'inactivation
du chromosome X a plusieurs facettes. Nous ne ferons dans un premier temps référence qu'à l'une
d'entre elle, celle qui a été découverte par Mary Lyon et s'applique aux cellules somatiques, de
l'embryon au sens strict à l'individu adulte. La description des propriétés de ce X inactif a
grandement bénéficié de l'utilisation d'un modèle cellulaire, les cellules souches embryonnaires
murine (nommées communément cellules ES d'après l'acronyme anglais pour embryonic stem
cells), capables de récapituler in vitro la mise en place de l'inactivation (Rastan et Robertson, 1985).
Lorsqu'elles sont cultivées dans des conditions permettant de conserver leur pluripotence, ces
cellules ont leurs chromosomes X actifs. Mais dans des conditions de culture les conduisant à se
différencier (sans qu'aucun type cellulaire ne soit a priori favorisé), les cellules ES femelles voient
l'un de leurs deux chromosome X être inactivé, l'unique chromosome X des mâles restant lui actif,
conformément au modèle de Mary Lyon. C'est grâce aux cellules ES que le recrutement séquentiel
de certaines « marques » épigénétiques sur le chromosome X inactif, ou en train d'être inactivé, a
d'abord et surtout était caractérisé. C'est également en nous appuyant sur ce modèle que nous
souhaitons l'exposer dans ce chapitre. Par soucis de simplicité, nous attendrons le chapitre suivant
pour présenter la mise en place de l'inactivation dans son véritable contexte, le développement
embryonnaire, une fois que les bases nécessaires à la compréhension auront été posées.
23

2.1 L'ARN Xist
Le gène Xist (X inactive specific transcript) est le premier gène à avoir été caractérisé comme
n'étant exprimé qu'à partir du chromosome X inactif (Xi) dans les cellules somatiques. Il fut
découvert en 1991, successivement chez l'homme (on l'écrit dans ce cas en lettres capitales: XIST)
(Borsani et al., 1991), puis chez la souris (Brown et al., 1991). Son profil d'expression unique
suggérait une implication dans l'inactivation du chromosome X, qui fut démontrée en 1996 par
Penny et al. (Penny et al., 1996).
Xist dérive, au moins en partie, du gène codant Lnx3, et est devenu non codant après la
divergence entre marsupiaux et euthériens (Duret et al., 2006). Huit exons ont été caractérisés chez
l'homme (Brockdorff et al., 1992; Brown et al., 1992; Chureau et al., 2002), la vache (Chureau et
al., 2002), la souris (Chureau et al., 2002; Sheardown et al., 1997a; Simmler et al., 1996) et le
campagnol (Nesterova et al., 2001b). L'alignement des séquences entre l'homme, la souris et la
vache, indique que 7 des 8 exons présents chez la souris sont conservés dans les trois espèces.
Cependant deux des exons (h2 et h7) inclus dans les transcrits XIST chez l'homme ne sont pas
retrouvés chez la souris et la vache. De plus, l'exon m2 présent chez la souris et la vache n'est
apparemment pas inclus chez l'homme (Chureau et al., 2002) (fig. 3, p. 25). L'identité de séquence
des exons est de 66 % entre la souris et l'homme, et de 62 % entre la souris et la vache (Chureau et
al., 2002), ce qui est proche du degré moyen de conservation observé pour les extrémités 3' et 5' non
traduites (3' et 5' UTR, untranslated region) des gènes codants orthologues entre la souris et
l'homme (Makalowski et Boguski, 1998). Contrairement aux exons, et à l'exception de quelques
blocs de séquences, les introns de Xist sont quant à eux peu conservés (Chureau et al., 2002).
Il existe également des variants de terminaison de XIST/Xist n'incluant pas l'exon 8 chez la
souris, et les exons 7 et 8 chez l'homme (Brockdorff et al., 1992; Hong et al., 1999; Hong et al.,
2000). Chez la souris et l'homme, les formes les plus longues mesurent 19,3 kb et 17,9 kb,
respectivement, et se terminent avec l'exon 7 chez la souris et l'exon 6, homologue, chez l'homme.
Les variants d'épissage incluant les derniers exons (m8, h7 et h8, voir fig. 3, p. 25) sont plus courts
d'environ 5 kb (Chureau et al., 2002). Chez la souris, plusieurs variants de terminaison dans l'exon 7
ont été décrits: une forme courte (forme S) polyadénylée mesurant 17,0 kb, qui est présente dans
les cellules ES femelles différenciées femelles, mais pas dans les cellules ES indifférenciées, et la
forme longue de 17,9 kb (forme L), qui inclut en réalité au moins cinq variants de terminaison non
polyadénylés, peu différents en taille, dans les cellules ES femelles différenciées, mais un seul dans
les cellules ES indifférenciées (Hong et al., 1999; Ma et Strauss, 2005; Memili et al., 2001).
24

Chez la souris, une source supplémentaire de diversité des transcrits Xist tient à l'existence de
deux promoteurs alternatifs (P1 et P2), situés à seulement 1,5 kb l'un de l'autre, les transcrits
provenant de P2 représentant, dans les cellules somatiques femelles, environ 75 % des transcrits
totaux (Johnston et al., 1998).
La présence de l'ARN Xist n'a été détectée que dans le noyau, que cela soit chez la souris
(Brockdorff et al., 1992) ou chez l’homme (Brown et al., 1992). Son marquage par RNA-FISH a
révélé qu'il était, dans les cellules somatiques, localisé sous la forme d'un domaine compact
correspondant au territoire du Xi (Clemson et al., 1996; Clemson et al., 1998; Panning et al., 1997).
Dans les cellules ES murines pluripotentes, Xist est très faiblement exprimé sur chacun des
chromosomes X: il est présent en moyenne à raison de 10 copies par cellule femelle et 4 copies par
cellule mâle (Sun et al., 2006). L'ARN Xist peut alors être visualisé à son site de transcription par
RNA-FISH sous la forme d'un signal ponctuel de faible intensité. Néanmoins, ce signal est très
variable selon les cellules, et Xist pourrait ne pas être exprimé dans un proportion importante d'entre
elles (Clerc, communication personnelle). Lors de la différenciation in vitro de ces cellules,
l'expression de Xist est perdue chez les mâles ainsi que sur l'un des deux chromosomes X chez les
femelles. L'expression dans les femelles devient alors monoallélique, et plus de 300 copies de
l'ARN Xist contribuent au recouvrement du chromosome X destiné à être inactivé (Buzin et al.,
1994; Sun et al., 2006).
Des expériences de perte et de gain de fonction ont permis de mieux comprendre le rôle
essentiel joué par Xist dans l'inactivation du chromosome X. Les premières d'entre elles ont eu pour
but d'étudier les conséquences, en cis, de l'absence complète de transcrits Xist, soit après avoir retiré
la région promotrice et le premier exon (Penny et al., 1996) (XistΔProm-1, fig. 5, p. 27), soit après
25
Fig. 3: Comparaison de la structure intron-exon de Xist chez la souris, la vache et l'homme (d'après Chureau et al., 2002)
Les introns et le premier exon ne sont pas représentés à l'échelle. Les signaux consensus d'épissage qui correspondent à des exons connus sont en majuscule. Le préfixe "h" identifie les exons chez l'homme, et le préfixe "m" les exons chez la souris. Les commentaires sont donnés dans le texte.

délétion de la majorité de l'exon 1, ainsi que des exons 2 à 5 (Marahrens et al., 1997) (XistΔ1-5,
fig. 5, p. 27). Dans les deux cas, le chromosome X muté n'est jamais inactivé, contrairement au
chromosome X sauvage. Le gène Xist est donc nécessaire en cis pour que l'inactivation ait lieu. Il
est désormais acquis que cette fonction est médiée par l'ARN Xist en tant que tel, et non par les
séquences génomiques correspondantes, en particulier compte-tenu du fait que, lorsque l'expression
d'un transgène formé de l'ADNc de Xist est, induite de manière ectopique, elle est capable de
récapituler la mise en place de l'inactivation. Dans un tel contexte d'expression d'un fort niveau de
Xist, ce dernier est suffisant pour conduire à l'extinction transcriptionnelle de gènes présents sur le
même chromosome, qu'il s'agisse du chromosome X ou d'un autosome, y compris dans des cellules
ES non différenciées, femelles ou mâles (Wutz et Jaenisch, 2000). Néanmoins, si l'extinction
26
Fig. 4: Expression de Xist/Tsix dans les cellules ES
Panneaux du haut: apparence en microscopie à transmission de cultures de cellules ESÀ gauche: cellules ES indifférenciées. Les cellules ES forment en se multipliant des colonies d'aspect arrondi, dans lesquelles elles se répartissent dans les trois dimensions.À droite: cellules ES différenciées après induction par l'acide rétinoïque. Les cellules forment une couche unique.Panneaux du milieu et du bas: Marquage par RNAFISH (en vert) des ARN issus du locus Xist. La sonde marque également les ARN issus du gène Tsix antisens à Xist. Les noyaux sont colorés en bleu au DAPI.À gauche: cellules indifférenciées. Les transcrits Tsix sont quantitativement prépondérants par rapport aux transcrits Xist (Sun et al., 2006), et
l'essentiel du signal en FISH provient par conséquent des transcrits Tsix. Le marquage Xist/Tsix est observé sur chaque chromosome X sous la forme d'un signal ponctuel correspondant aux transcrits primaires (au milieu, cellules mâles; en bas, cellules femelles).À droite: cellules différenciées. L'expression de Xist et Tsix est perdue dans les cellules mâles (au milieu) en cours de différenciation. Dans les cellules femelles (en bas), l'expression de Tsix est perdue sur les deux chromosomes X., l'expression de Xist est également perdue sur le chromosome X actif, mais les transcrits Xist s'accumulent au niveau du chromosome X inactif pour former un domaine d'ARN Xist coincident avec le territoire chromosomique (Chaumeil et al., 2006).
transcriptionnelle a bien lieu, il semblerait que des étapes plus tardives d'hétérochromatinisation du
chromosome X requièrent des facteurs additionnels régulés au cours de la différentiation cellulaire
(Rasmussen et al., 2001).
27
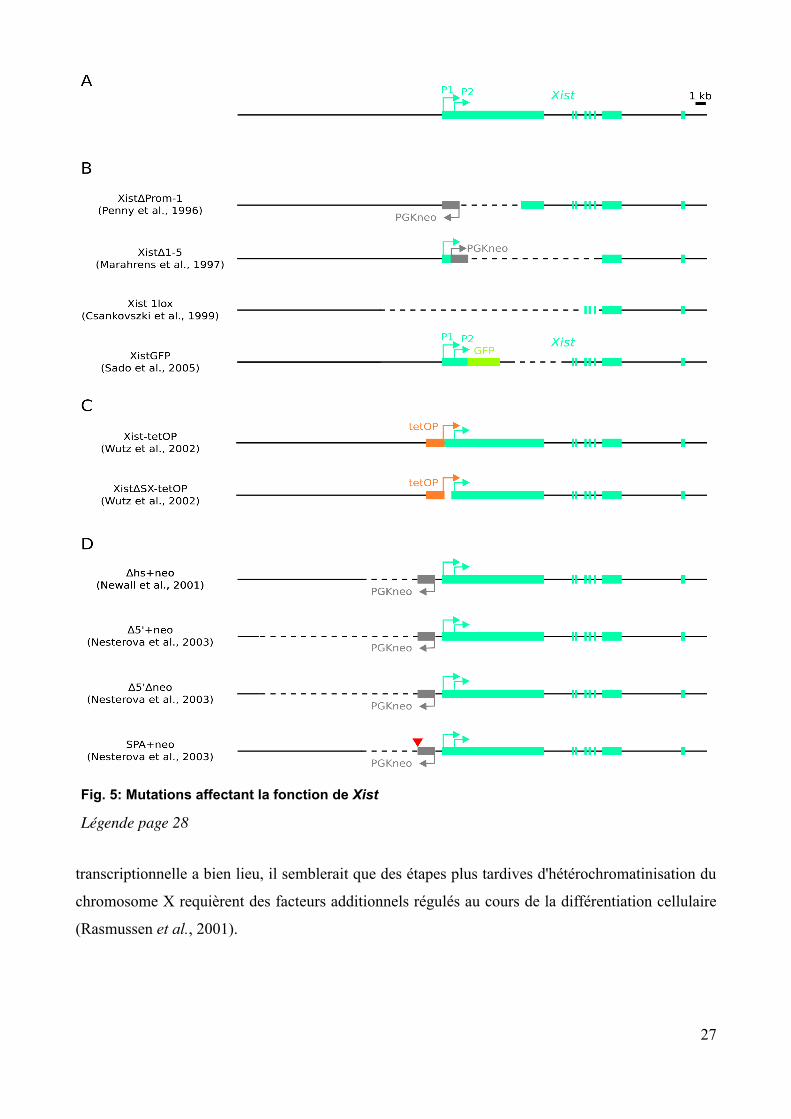
Fig. 5: Mutations affectant la fonction de Xist
Légende page 28

Xist contient six éléments répétés, numérotés de A à F, et bien conservés entre l'homme et la
souris (Brown et al., 1992) (fig. 6A, p. 29). La fonction de ces répétitions a été testée par A. Wutz
en 2002, en insérant en simple copie au locus Hprt, sur le chromosome X de cellules ES mâles, des
transgènes inductibles du cDNA de Xist dans lesquels différentes portions de ce dernier étaient
délétées (Wutz et al., 2002). Ces expériences, ainsi que la délétion de la répétition A au locus
endogène, ont permis de caractériser cette dernière comme essentielle à l'extinction
transcriptionnelle des gènes liés au chromosome X, mais pas à la localisation de Xist sous forme
d'un domaine, ni à l'extinction transcriptionnelle de séquences répétées intergéniques et introniques
du chromosome X (Chaumeil et al., 2006; Clemson et al., 2006) (fig. 6B et 6C, p. 29; Xist-tetOP et
XistΔSXtetOP fig. 5C, p. 27). La localisation correcte de l'ARN Xist est quant à elle dépendante de
séquences réparties sur le reste de sa longueur (et incluant la répétition A), mais de manière
redondante (Wutz et al., 2002) (fig. 6B et 6C, p. 29). Cette redondance est également illustrée par
l'absence d'effet d'une délétion de l'exon 4, pourtant très conservé entre l'homme et la souris, sur la
capacité de Xist à recouvrir le chromosome X et à provoquer l'inactivation en cis (Caparros et al.,
2002). Si la localisation correcte de Xist n'est pas conditionnée par sa capacité à « éteindre »
l'expression génique, l'inverse n'est pas nécessairement vrai. En effet, parmi les transgènes
inductibles de Xist générés par Wutz et al., il semble exister une corrélation entre la capacité des
formes délétées de Xist à être localisées sur le chromosome X et l'inactivation de ce dernier (fig. 6B
28
Légende de la figure 5, p.27: Différentes mutations générées au locus endogène de Xist et altérant sa fonction ou modifiant son expression. Les mutations et leurs phénotypes sont décrits dans le texte. Les exons de Xist sont représentés en vert. La forme à 8 exons est ici utilisée comme référence (cf p. 24). Les deux promoteurs P1 et P2 sont également représentés. La ligne continue noire représente la séquence génomique. Elle est en pointillés lorsque la séquence correspondante a été délétée. Les cassettes de sélection plaçant le gène de résistance à la néomycine sous contrôle du promoteur fort PGK sont représentées en gris et leur orientation est donnée par celle de leur promoteur. (A) Allèle sauvage de Xist. (B) Déletions au locus Xist, se traduisant pas une perte de fonction. La délétion Xist1lox est inductible en présence de recombinase Cre et seul l'allèle après excision est représenté. Dans l'allèle XistGFP, la GFP est exprimée sous contrôle du promoteur de Xist. (C) Allèles inductibles de Xist. tetOP désigne le promoteur inductible de la tétracyline (en jaune), qui remplace le promoteur P1. Dans les mêmes cellules, le transactivateur nlsrtTA est exprimé à partir du locus ROSA26, permettant l'induction de Xist après ajout de doxycycline dans le milieu de culture. Dans l'allèle XistΔSXtetOP, la répétition A (cf p. 28), située entre les promoteurs P1 et P2, a été délétée. (D) Mutations générées en 5' de Xist et conduisant à une expression ectopique de ce dernier. Ces mutations seront décrites à la section 2.1 de la seconde partie de l'introduction (p. 95). Le triangle rouge représente un signal d'arrêt de transcription.

et 6C, p. 29). D'autre part, des expériences de « cartographie d'interférence » (interference mapping)
au moyen de PNA (pour Peptid Nucleic Acids, molécules synthétiques s'hybridant spécifiquement
aux ARN et interférant avec leur fonction) dirigés contre les répétitions B, C et D ont montré que
lorsque la première d'entre elles était ciblée, l'ARN Xist était aussi bien incapable de former un
domaine que de conduire à l'inactivation (Beletskii et al., 2001). Ce résultat est néanmoins en
contradiction avec la redondance des différentes répétitions de Xist vis-à-vis de sa localisation,
montrée par Wutz et al. Cela pourrait suggérer un effet dominant-négatif des PNA, à l'appui d'un
rôle essentiel joué par la structure de l'ARN Xist pour lui permettre d'accomplir sa fonction. La
structuration des répétitions sous forme de tiges-boucles (Wutz et al., 2002) pourrait ainsi être
déterminante pour la formation de complexes autour de l'ARN Xist.
29
Fig. 6: Analyse stucturale et fonctionnelle de l'ARN Xist (adapté de Brockdorff et al., 1992, et Wutz et al., 2002)
Légende page 30

Ainsi donc, le recouvrement du domaine chromosomique par l'ARN Xist est un prérequis. Mais
comment un tel domaine peut-il être formé? Les travaux dont le but était de répondre à cette
question se sont naturellement organisés autour de deux hypothèses de travail qui, d'ailleurs, ne sont
pas exclusives l'une de l'autre.
La première de ces hypothèses consiste à envisager le domaine formé par l'ARN Xist comme un
compartiment nucléaire répressif pour la transcription en général. Dans ce contexte, il importe
d'abord de prouver l'association de l'ARN Xist à des molécules permettant de structurer un tel
domaine, d'une part, et de réprimer la transcription, d'autre part. Nous parlerons des « marques
épigénétiques » réprimant la transcription à la section 2.3 de la première partie de l'introduction
(p. 38). Mais nous pouvons dès à présent donner une idée de de la manière dont serait structuré un
tel domaine, en nous référant à des expériences de préparation de matrice nucléaire (Clemson et al.,
1996), montrant que XIST est retrouvé dans la même fraction insoluble que cette dernière. La même
30
Légende de la figure 6, p. 29: (A) Éléments répétés dans l'ARN Xist/XIST, d'après (Brockdorff et al., 1992). Les répétitions sont figurées par des rectangles bleus, en mettant en visàvis les régions orthologues chez l'homme et la souris. Les répétitions A et B sont très conservées entre l'homme et la souris. Dans les répétitions C, D et E le nombre d'éléments répétés varie au contraire beaucoup entre les deux espèces. Les rectangles grisés sous l'ARN Xist représentent les éléments importants pour la formation d'un domaine d'ARN Xist, tels qu'ils ont été caractérisés par Wutz et al. (Wutz et al., 2002). L'intensité de la coloration grisée traduit l'importance plus ou moins grande que ces éléments ont pour la localisation correcte de l'ARN Xist. (B) Représentation de certains des transgènes générés par Wutz et al. pour caractériser les éléments fonctionnels de l'ARN Xist. Les traits continus réprésentent les séquences insérées, les interruptions correspondent à des délétions dans la séquence de l'ADNc de Xist. (C) Phénotypes associés au différents transgènes. RNAFISH avec une sonde marquant les transcrits Xist (en rouge), les noyaux étant colorés au DAPI (en bleu). Les transgènes Xist et ΔSX, correspondant au cDNA complet de Xist et à la délétion de la répétition A, respectivement, forment un domaine d'ARN Xist indiscernable du domaine observé naturellement dans les cellules somatiques femelles. Les transgènes PΔ , SaCΔ et EΔ expriment une forme de Xist partiellement délocalisée, la délocalisation étant plus importante pour EΔ et moins importante pour PΔ . Le degré de localisation des transcrits Xist, tel qu'estimé par Wutz et al., est représenté par le nombre de signes +. Le pourcentage exprimé sur chaque photographie correspond à la proportion de cellules survivant à l'expression ectopique de Xist, traduisant la fréquence avec laquelle l'unique chromosome X est inactivé. Le transgène ΔSX dans lequel a répétition A est absente ne conduit donc pas à l'inactivation, car le pourcentage de survie est proche de celui observé dans les lignées contrôle (insertion d'un vecteur vide). Dans les autres lignées, le taux de survie est d'autant plus élevé (et l'inactivation d'autant moins efficace) que l'ARN Xist est délocalisé. Se référer au texte (p. 28) pour plus de détails.

étude montre également que la majorité de XIST n'interagit pas directement avec l'ADN, puisque sa
localisation est insensible à un traitement par la RNAse H, digérant les duplexes ARN/ADN. Il a par
la suite était observé que la protéine de matrice nucléaire SAF-A était enrichie sur le Xi et que cet
enrichissement dépendait de son domaine d'interaction avec les ARN, laissant ouverte la possibilité
d'une interaction fonctionnelle avec Xist (Fackelmayer, 2005; Helbig et Fackelmayer, 2003). Il sera
intéressant de tester si la structure particulière de l'ARN Xist pourrait jouer un rôle dans cette
interaction éventuelle.
L'idée d'un compartiment nucléaire répressif associé au domaine de Xist a été renforcée par des
travaux récents examinant la position des gènes du chromosome X respectivement au domaine
chromosomique et à celui formé par l'ARN Xist (Chaumeil et al., 2006). Nous y reviendrons à la
section 2.2 de la première partie de l'introduction (p. 35).
Enfin, certaines observations suggèrent la participation de la protéine suppresseur de tumeur
Brca1/BRCA1 à la localisation correcte de Xist/XIST sur le Xi, chez l'homme et la souris (Ganesan
et al., 2002; Ganesan et al., 2004; Silver et al., 2007). Une telle conclusion, résulte de trois
catégories d'observations, toutes sujettes à controverse. Premièrement, la protéine BRCA1/Brca1
colocaliserait avec le Xi en phase S tardive (Chadwick et Lane, 2005; Diaz-Perez et al., 2006;
Ganesan et al., 2002; Ganesan et al., 2004; Ouyang et al., 2005; Pageau et al., 2007; Pageau et
Lawrence, 2006; Silver et al., 2007). Cependant, cette colocalisation est extrêmement partielle et ne
semble pas spécifique du Xi, mais correspondrait plutôt à une association générale de BRCA1 à
l'héterochromatine constitutive au niveau des centromères (Pageau et al., 2007; Pageau et
Lawrence, 2006; Xiao et al., 2007). Deuxièmement, un domaine d'ARN XIST n'est plus observé
dans plusieurs lignées ou cultures primaires de cellules tumorales provenant de cancers du sein ou
des ovaires et dans lesquelles BRCA1/Brca1 est muté (Ganesan et al., 2002; Ganesan et al., 2004;
Silver et al., 2007; Sirchia et al., 2005; Vincent-Salomon et al., 2007; Xiao et al., 2007). Il semble
s'agir alors d'un défaut de localisation des ARN XIST/Xist, dans la mesure où leur niveau
d'expression n'est pas modifié (Ganesan et al., 2002; Ganesan et al., 2004). Ce défaut conduit, avec
une fréquence faible, à la réactivation d'un transgène localisé sur le Xi (Ganesan et al., 2002;
Ganesan et al., 2004). Il cependant été montré que l'absence de domaine d'ARN XIST pourrait
résulter, dans certains cas, de la perte du Xi, éventuellement associée à la duplication du Xa (Sirchia
et al., 2005; Vincent-Salomon et al., 2007). Cela serait à mettre en relation avec la fréquence élevée
des réarrangements génomiques dans les cellules cancéreuses, ainsi qu'avec la fonction de
BRCA1/Brca1 lui-même dans la réparation de l'ADN et le contrôle de la réplication (pour revue,
Gudmundsdottir et Ashworth, 2006). D'autre part, la présence d'une forme mutée de BRCA1/Brca1
31

ne conduit pas toujours à l'absence de domaine d'ARN XIST/Xist, et ces domaines sont
fréquemment absents dans des cellules tumorales provenant de cancer du sein non causés par la
mutation de BRCA1 (Ganesan et al., 2005; Richardson et al., 2006; Silver et al., 2007; Sirchia et
al., 2005; Vincent-Salomon et al., 2007; Xiao et al., 2007). Il a toutefois été remarqué que l'effet sur
la localisation de Xist de différents knock-down ou knock-out de Brca1 chez la souris est très
dépendant du niveau d'activité résiduelle de Brca1. L'absence de domaine d'ARN Xist n'est ainsi
observée que dans les knock-down par RNAi les plus efficaces ou dans des knock-out supprimant
complètement la fonction de Brca1 (Silver et al., 2007; Xiao et al., 2007). Un troisième argument
en faveur d'une fonction de BRCA1 dans la localisation de XIST est la restauration de cette
localisation dans une lignée de cellules cancéreuses mutées pour BRCA1, lorsque ce dernier est
exprimé de manière ectopique (Ganesan et al., 2002; Ganesan et al., 2004; Silver et al., 2007).
Néanmoins, l'expression ectopique de BRCA1 a apparemment pour effet la surexpression de XIST,
qui ne serait plus présent à des niveaux physiologique (Pageau et al., 2007). Au final, il est donc peu
probable que BRCA1/Brca1 ait un rôle direct dans la localisation correcte de XIST/Xist. Par contre,
il pourrait être un élément important dans une voie de régulation affectée dans les cancers du sein et
des ovaires, et qui jouerait un rôle essentiel dans la stabilité du Xi et, particulièrement, dans la
structuration d'un compartiment nucléaire répressif.
Une seconde orientation donnée aux travaux de recherche a consisté à contourner la question de
la formation d'un compartiment nucléaire et à se demander comment l'ARN Xist pouvait être ciblé
spécifiquement sur le chromosome X, et non pas sur les autosomes adjacents. Que l'interaction avec
le chromosome X soit directe ou indirecte importe ici finalement peu. Pour répondre à cette
question, l'expression ectopique de Xist à partir d'autosomes donne des résultats a priori
contradictoires. Lorsqu'il s'agit de transgènes insérés en multicopie, résultant en une expression de
Xist à des niveaux vraisemblablement non physiologiques, Xist est capable de recouvrir
efficacement l'autosome à partir duquel il est exprimé et de conduire à l'extinction génique (Lee et
al., 1996; Wutz et Jaenisch, 2000). Il n'y aurait donc pas de spécificité du chromosome X, ce qui
importerait serait plutôt l'expression en cis sur ce dernier. Cependant, dans le cas de translocations
sur un autosome d'une portion du chromosome X contenant Xist, le recouvrement de l'autosome et
l'extinction des gènes qui lui sont liés sont atténués, si on les compare à un chromosome X inactif,
aussi bien chez la souris (Duthie et al., 1999; Rastan, 1983; Shao et Takagi, 1991) que chez
l'homme (Hall et al., 2002; Keohane et al., 1999; Sharp et al., 2002; White et al., 1998). Cette
incapacité relative des autosomes à être recouverts par Xist pourrait être liée à un défaut de
propagation de l'ARN Xist sur ces derniers, ou à un défaut de maintien de l'état inactif, une fois
32

établi. Des travaux récents sont venus à l'appui de la première de ces hypothèses en montrant que,
dans le cas d'une translocation (T(X;4)37H) impliquant le chromosome 4, le recouvrement par
l'ARN Xist était incapable de se propager au-delà du point de cassure (Popova et al., 2006).
Ce type de comportement est à mettre en relation avec la proposition faite par Gartler et Riggs
de l'existence de centres « relais » ou « tremplins » (way stations et booster elements, dans les
termes des auteurs) spécifiques du chromosome X et permettant au recouvrement par Xist de n'être
propagé que sur le chromosome X (Gartler et Riggs, 1983). Une telle vision est d'ailleurs très
proche du modèle en vigueur pour la compensation de dose chez D. melanogaster (cf introduction,
partie 1, section 1.4, p. 18). En l'absence de tels centres relais, sur les autosomes, seule la
surexpression de Xist à un niveau non physiologique, dans le cas de transgènes, permettrait malgré
tout d'initier l'inactivation. Encore faudrait-il déterminer quelles sont les cibles de plus forte affinité,
qui doivent répondre au critère d'être retrouvées préférentiellement sur le chromosome X, ou d'être
distribuées de manière particulière sur ce dernier. Il a été proposé par Mary Lyon que les éléments
répétés LINE-1 (L1) pourraient jouer un tel rôle (Lyon, 1998). Il a par la suite été confirmé que, sur
le chromosome X humain, leur densité était double de celle retrouvée sur les autosomes (Bailey et
al., 2000; Ross et al., 2005). Elle est, de plus, particulièrement élevée autour du gène XIST (Bailey
et al., 2000) et, à l'inverse, conforme à la moyenne du génome au niveau des gènes échappant à
l'inactivation (Ross et al., 2005). Des observations similaires ont été faites chez la souris (Allen et
al., 2003; Chureau et al., 2002; Nesterova et al., 2001b). De manière intéressante, la région
autosomique à proximité du point de cassure dans la translocation T(X;4)37H est particulièrement
pauvre en L1 (Popova et al., 2006). D'autre part, l'ARN Xist est enrichi au niveau des bandes R
(négatives à la coloration de Giemsa) des chromosomes X métaphasiques, alors même que ces
bandes sont connues pour être enrichies en L1 (Duthie et al., 1999). Les éléments L1 sont, d'une
manière générale, plus nombreux au niveau des gènes dont l'expression est monoallélique (Allen et
al., 2003), ce qui laisse penser qu'ils pourraient servir de centre de nucléation pour
l'hétérochromatinisation, et seulement de manière indirecte pour la propagation de l'ARN Xist qui
interagirait avec l'hétérochromatine nouvellement formée. Dans la même ligne de pensée qui a
alimenté l'hypothèse de Mary Lyon concernant les éléments L1, une approche computationnelle est
désormais utilisée pour caractériser des « mots » de nucléotides associés à l'inactivation. De tels
mots de 12 nt associés soit aux gènes soumis à l'inactivation, soit aux gènes échappant à
l'inactivation (cf introduction, partie 1, section 2.5, p. 50), ont d'ores et déjà pu être identifiés et
utilisés avec une valeur prédictive sur le chromosome X humain (Carrel et al., 2006). De manière
intéressant, la plupart de ces 12-mer associés au gènes soumis à l'inactivation sont retrouvés dans
33

des séquences L1, et plus particulièrement dans des portions de ces dernières sous-représentées dans
le génome. Une approche similaire a montré l'enrichissement des gènes du chromosome X humain
dans son ensemble, ainsi que des gènes échappant à l'inactivation, en motif (GATA)n (McNeil et al.,
2006). Les implications de ces observations concernant les gènes échappant à l'inactivation seront
discutées plus loin (cf introduction, partie 1, section 2.5, p. 50). Quant à la portée d'une telle
approche computationnelle, il est probable qu'on ne la mesurera réellement que lorsqu'on aura
commencé à comprendre comment de tels mots peuvent être « lus ».
Que Xist soit capable de recouvrir le chromosome destiné à devenir inactif n'est pourtant pas
suffisant. Il faut aussi qu'il soit exprimé à un niveau adéquat au moment voulu, et tout laisse à
penser que cela est régulé de manière liée à la différenciation cellulaire. Selon les moments, il est
probable que des régulations d'ordre transcriptionnel ou post-transcriptionnel prennent le pas et cela
a intimement à voir avec l'organisation et l'activité du locus auquel est présent le gène Xist, appelé
centre d'inactivation du chromosome X (dont l'acronyme est Xic, pour X-chromosome inactivation
center). Nous y reviendrons plus tard et en détail, à partir de la seconde partie de l'introduction, car
cette interrogation a alimenté de manière centrale le travail de thèse. Contentons-nous pour l'heure
de mentionner que, au cours de la différenciation, la transcription de Xist est fortement augmentée
(Navarro et al., 2006; Sun et al., 2006) et que, bien qu'une stabilisation de l'ARN Xist dans les
cellules différenciées ait été montrée par le passé (Panning et al., 1997; Sheardown et al., 1997a),
elle a été remise en cause récemment (Sun et al, 2006). Par contre, l’efficacité du processus
d’épissage, régulée dans ce cas par des protéines de la voie NMD (Nonsense-Mediated Decay),
semble jouer un rôle déterminant (Ciaudo et al., 2006). L'importance fonctionnelle de l'expression
d'isoformes de Xist différentes dans les cellules différenciées et non différenciées n'a pour l'instant
pas encore fait l'objet d'une exploration détaillée, mais nous noterons au passage que le transcrit issu
du promoteur P2, donné pour prépondérant dans les cellules différenciées (Johnston et al., 1998), ne
comporte pas la répétition A, pourtant essentielle à l'activité « inactivatrice » de Xist. Il reste
cependant environ 25 % de transcrits issus de P1 et, d'autre part, , bien que Xist continue à être
exprimé durant la vie entière des individus, lorsqu'il est délété de manière conditionnelle dans des
cellules ayant déjà mis en place l'inactivation, la réactivation du Xi n'est observée qu'à une
fréquence extrêmement faible (Csankovszki et al., 1999). Ainsi se pose la nécessité, pour le Xi, de
mécanismes supplémentaires stabilisant l'extinction transcriptionnelle de ses gènes. Nous allons
voir lesquels ils sont ou pourraient être.
34

2.2 Réorganisation du territoire chromosomique
Savoir s'il existe un lien entre l'activité transcriptionnelle des gènes et leur positionnement vis-à-
vis des territoires chromosomiques est une question débattue (pour revue, Cremer et Cremer, 2001;
Williams, 2003), parfois avec virulence. Cela tient d'ailleurs beaucoup à l'imprécision de la
définition des territoires chromosomiques, sur laquelle nous ne reviendrons pas. D'une manière
générale, les gènes sont souvent localisés à la périphérie de ces derniers (Kurz et al., 1996; Moen et
al., 2004; Nogami et al., 2000; Scheuermann et al., 2004; Zirbel et al., 1993). Il ne semble pas y
avoir, pour l'instant, de consensus quant au comportement de gènes considérés individuellement, qui
peuvent se retrouver aussi bien à l'intérieur, à la périphérie ou à l'extérieur des territoires
chromosomiques lorsqu'ils sont exprimés (Mahy et al., 2002a; Mahy et al., 2002b; Osborne et al.,
2004; Parada et Misteli, 2002). Toutefois, la situation prête moins à controverse lorsqu'on s'intéresse
à des régions riches en gènes, prises dans leur ensemble, dont la relocalisation vers l'extérieur des
territoires chromosomiques corrèle bien avec leur expression (Dietzel et al., 1999; Mahy et al.,
2002a; Mahy et al., 2002b; Volpi et al., 2000; pour revue, Chambeyron et Bickmore, 2004).
Mais qu'en est-il des gènes portés par le chromosome X inactif? La question se pose avec
d'autant plus d'acuité que, dans ce cas, le positionnement se fait non seulement vis-à-vis du territoire
chromosomique, mais également par rapport au domaine coïncidant d'ARN Xist (Clemson et al.,
1996; Clemson et al., 1998; Panning et al., 1997). Les premières observations dans cette direction
ont porté sur deux gènes liés au chromosome X, et montraient que celui d'entre eux qui était sujet à
l'inactivation avait une localisation plus interne au territoire chromosomique correspondant, dans ce
cas formé par le Xi (Dietzel et al., 1999). Toutefois, une telle conclusion n'a pas été confirmée dans
des travaux plus récents, portant sur un plus grand nombre de gènes liés au chromosome X, et
montrant que leur positionnement était toujours au bord des territoires formés par le territoire
chromosomique du Xi ou le domaine d'ARN Xist, sans distinction possible entre les gènes assujettis
et échappant à l'inactivation (Clemson et al., 2006). Néanmoins, ces observations ont été réalisées
dans des cellules somatiques humaines adultes et ne renseignent pas sur un éventuel réarrangement
du chromosome X au moment où l'inactivation est mise en place.
Dans ce cas, en effet, une étude récente a montré qu'il en allait autrement (Chaumeil et al.,
2006), en établissant une cinétique précise des premiers évènements suivant la mise en place du
domaine d'ARN Xist, lors de la différenciation in vitro de cellules ES murines femelles. On assiste
ainsi rapidement à l'exclusion de la polymérase II et de facteurs généraux de transcription hors du
domaine de Xist alors même que les gènes liés au chromosome X sont toujours exprimés. Ceci
35

corrèle avec le positionnement de ces derniers à la périphérie ou à l'extérieur du domaine. Leur
extinction transcriptionnelle est par la suite associée à leur relocalisation à l'intérieur de celui-ci.
Dans le fenêtre de temps correspondante, le territoire chromosomique coïncide toujours avec le
domaine de Xist et ne connaît pas de compaction notable. Le domaine d'exclusion de la polymérase
II apparaît comme essentiellement formé de séquences répétées non géniques (incluant notamment
les répétitions L1), dont l'expression est réprimée à l'intérieur du domaine de Xist (Chaumeil et al.,
2006; Clemson et al., 2006). De manière significative, un telle répression transcriptionnelle, de
même que la mise en place du domaine de Xist, n'est pas affectée par l'absence de la répétition A (cf
introduction, partie 1, section 2.1, p. 28) de l'ARN Xist, qui est par contre requise à la fois pour
l'inactivation des gènes du chromosome X et leur relocalisation (Chaumeil et al., 2006). Il est ainsi
possible de distinguer deux étapes, à la fois temporellement et fonctionnellement: la formation d'un
compartiment répressif et le recrutement des gènes dans ce compartiment, prérequis au remodelage
de la chromatine, que nous présentons à la section suivante.
La localisation ultérieure du chromosome X inactif dans le noyau pourrait également jouer un
rôle dans le maintien de l'inactivation. De longue date, la localisation du Xi à proximité de
l'enveloppe nucléaire (Barton et al., 1964; Belmont et al., 1986; Dyer et al., 1989; Eils et al., 1996),
ainsi qu'à proximité du nucléole (Barr et Bertram, 1949; Borden et Manuelidis, 1988; Bourgeois et
al., 1985), a été observée. Les exemples décrits dans la littérature correspondaient à l'observation de
cellules somatiques humaines (en général des fibroblastes), à l'exception de la description originelle
de Barr, qui a été réalisée dans des motoneurones de chatte (Barr et Bertram, 1949). Ce n'est que
très récemment qu'une étude plus systématique du positionnement du Xi chez la souris a été
publiée, à partir d'observations réalisées lors de la différenciation in vitro de cellules ES (Zhang et
al., 2007). En cours de différenciation, après la formation du domaine d'ARN Xist, le chromosome
X nouvellement inactivé est ainsi retrouvé de plus en plus fréquemment à proximité du nucléole,
avant que la proportion de cellules témoignant de cette localisation ne se stabilise, probablement
lorsque la totalité des cellules sont différenciées, vers 8 jours de différenciation en corps
embryonnaires (cf introduction, partie 1, section 3.6, p. 73). Dans des fibroblastes embryonnaires
murins en culture asynchrone, 30 % des cellules ont leur Xi accolé au nucléole mais à distance de
l'enveloppe nucléaire, 35 % des cellules ont leur Xi à proximité de l'enveloppe nucléaire sans qu'il
soit associé au nucléole, et 25 % des cellules ont leur Xi non seulement à proximité de l'enveloppe
nucléaire, mais aussi du nucléole (Zhang et al., 2007). Le Xa est quant à lui également observé à la
périphérie du noyau, tout au moins chez l'homme (Eils et al., 1996), mais n'est par contre pas
localisé de manière préférentielle à proximité du nucléole (Zhang et al., 2007). La proximité du Xi
36

37
Fig. 7: Inactivation par relocalisation dans le domaine d'ARN Xist (d'après Chaumeil et al., 2006)
(A) Reconstitution tridimensionnelle montrant la relocalisation d'un gène lié au chromosome X, marqué par DNAFISH (en rouge), à l'intérieur du domaine d'ARN Xist, marqué par RNAFISH (en vert), au cours de la différenciation de cellules ES femelles. (B) Modèle proposé par Chaumeil et al. (Chaumeil et al., 2006). Dans les cellules ES femelles indifférenciées, les deux chromsomes X sont actifs (en jaune) (1) En début de différenciation, l'ARN Xist commence à recouvrir le chromosome X choisi pour être inactivé. (2) La formation du domaine d'ARN Xist est associée à l'exclusion de la machinerie de transcription et à la répression transcriptionnelle des séquences répétées, toutes deux indépendantes de la répétition A de Xist. (3) L'inactivation des gènes liés au chromosome X est réalisée par un processus dépendant de la répétition A de Xist, concomitant à la mise en place de marques de type hétérochromatique sur le Xi. (4) Les gènes du chromosome X sont relocalisés dans le domaine d'ARN Xist. Les gènes échappant à l'inactivation sont maintenus en dehors de ce domaine, peutêtre du fait d'éléments insulateurs fixant CTCF. On ne sait pas si la relocalisation des gènes dans le domaine d'ARN Xist est simultanée ou postérieure à leur inactivation. Ici, les deux évènements ont été distingués.

vis-à-vis de l'enveloppe nucléaire ne varie pas au cours du cycle cellulaire, contrairement à
l'accolement au nucléole, qui est observé presque exclusivement en milieu ou fin de phase S (dans
ce cas, dans 90 % des cellules), c'est-à-dire lorsque le Xi est répliqué (cf introduction, partie 1,
section 2.4, p. 49 ).
Dans la mesure où la même localisation périnucléaire semble observée pour le Xa et pour le Xi,
il paraît difficile de lui donner une signification particulière. Par contre, de manière intéressante, le
Xi colocalise dans la région périnucléolaire avec un enrichissement en protéine Snf2h (Zhang et al.,
2007), qui est l'unité catalytique du complexe de remodelage de la chromatine ACF1-ISWI, requis
pour la progression de la fourche de réplication dans l'hétérochromatine (Collins et al., 2002). La
localisation dans un « compartiment » périnucléolaire pourrait donc participer au maintien en cours
de réplication des marques hétérochromatiques associées au Xi, que nous aborderons bientôt. Elle
dépend en tout cas de l'expression de Xist, et n'est plus visible après la délétion de Xist, dans un
mutant conditionnel (Zhang et al., 2007) (Xist1lox, fig. 5, p. 27). La réactivation de certains gènes
et des régions intergénique est alors observée, mais de manière très partielle et variable selon les
clones, ce qui est cohérent avec l'effet limité de l'absence de Xist sur les marques associées au Xi,
une fois l'inactivation établie ((Csankovszki et al., 2001; Zhang et al., 2007)). Le maintien de ces
marques pourrait donc être réalisé, au moins en partie, indépendamment de la localisation
périnucléolaire.
2.3 Modifications de la chromatine
2.3.1 Le code histone
Dans le noyau, la molécule d'ADN n'est pas seule à déterminer l'information, y compris de
nature essentiellement génétique, transmise par une cellule à ses descendantes. Au cours du
développement embryonnaire, ou lors de la vie normale des individus, l'expression de certains
gènes doit pouvoir être modifiée rapidement, pour permettre un comportement approprié face à un
environnement changeant. Lorsque cela a lieu au niveau transcriptionnel, les facteurs impliqués sont
généralement rangés dans la catégorie des facteurs de transcription. Pourtant, la combinaison des
gènes exprimés à un moment donné définit une identité cellulaire particulière, et changer trop
souvent d'identité peut avoir, à l'échelle d'un organisme, des conséquences dévastatrices. Sont là
pour en témoigner de nombreuses maladies dont, particulièrement, les cancers. Il importe donc,
38

entre une cellule et ses descendantes, de conserver l'essentiel de la combinaison des gènes exprimés.
Cela peut être fait à l'échelle de gènes individuels ou, ce qui est plus facile à caractériser, au niveau
de territoires chromosomiques étendus. L'hétérochromatinisation du Xi, sous une forme également
qualifiée de corpuscule de Barr, en est un exemple, et s'inscrit dans la catégorie plus vaste de
l'hétérochromatine facultative, c'est-à-dire ayant « décidé » de le devenir (cette « décision » est
souvent restreinte à certaines lignages cellulaires), mais ne l'ayant pas toujours été et pouvant, à
certains moments particuliers, « changer d'avis ». Il existe une seconde catégorie
d'hétérochromatine, dite constitutive. Comme son nom l'indique, elle est un attribut quasi permanent
(indépendant du type cellulaire) d'une région donnée, qui est souvent péricentromérique ou
télomérique. Un autre exemple en est donné pour d'importantes portions du chromosome Y. Ces
notions ont déjà été utilisées en filigrane mais il est utile, pour la suite de notre exposé, de revenir
dessus un peu plus en détail.
L'hétérochromatine est souvent définie par opposition à l'euchromatine associée à la portion du
génome riche en gènes, en général transcriptionnellement active et répliquée précocement au cours
de la phase S du cycle cellulaire. L'hétérochromatine correspond à une organisation de la
chromatine plus compacte, moins accessible aux facteurs et machineries de transcription. L'unité de
base de la chromatine est le nucléosome, formé par l'enroulement de l'ADN autour d'un octamère
protéique, lui-même constitué de deux exemplaires de chacune des histones H2A, H2B, H3 et H4.
L'histone H1 joue quant à elle un rôle dans l'espacement des nucléosomes et leur compaction
(Zlatanova et van Holde, 1998). La résolution cristallographique de la structure des nucléosomes
(Luger et al., 1997) a révélé que les histones étaient structurées en un domaine C-terminal
globulaire (appelé histone fold), qui intéragit directement avec l'ADN et les autres histones, et une
queue N-terminale, qui se déploie hors du nucléosome et est par conséquent accessible à d'autres
facteurs. Suite à des expériences de transcription in vitro à partir d'ADN nu ou de chromatine
reconstituée, il est commun de voir l'ADN nu comme un environnement permissif et la chromatine
comme un environnement répressif vis-à-vis de l'expression des gènes (Izban et Luse, 1992;
Knezetic et Luse, 1986). Pourtant, ce n'est qu'en présence d'histones que l'expression des gènes peut
être régulée d'une manière qui donne aux facteurs de transcription toute leur efficacité (Cote et al.,
1994; Kraus et al., 1999). La possibilité de modifications covalentes des queues des histones joue à
ce titre un rôle particulier, en ce sens qu'elles ont la capacité de conférer une identité et un
comportement propre à un locus donné. Ainsi, leurs résidus sérine et thréonine peuvent être
phosphorylés, leurs arginines méthylées et leurs lysines acétylées, méthylées, ADP-ribosylées,
ubiquitinylées ou sumoylés avec, selon les cas, un effet activateur ou répresseur sur l'activité des
39

gènes (pour revue, Jenuwein et Allis, 2001; Margueron et al., 2005; Strahl et Allis, 2000). Une telle
corrélation avec la transcription est bien établie pour certaines d'entre elles, qui ont été analysées à
l'échelle de chromosomes ou génomes entiers (Bernstein et al., 2005; Pokholok et al., 2005;
Schubeler et al., 2004). En simplifiant, l'hyperacétylation de résidus sur les histones H3 et H4, mais
également l'hyper-méthylation de certaines lysines (en particulier K4) de H3, sont généralement
associés à des gènes transcrits, tandis que l'hyperméthylation de résidus sur les histones H3 et H4
(en particulier K9 et K27 de H3, et à quelques exceptions près, dont K4 de H3) correspond en
général à des gènes transcriptionnellement inactifs (pour revue, Margueron et al., 2005). Bien
qu'apparemment plus marginales quant à leurs effets, des modifications des queues des histones
H2A et H2B ont également été caractérisées.
Deux hypothèses de travail ont été avancées pour expliquer la fonction des modifications des
queues des histones. Elles pourraient, du simple fait de la modification des propriétés physico-
chimiques du nucléosome, rendre les séquences régulatrices plus accessibles à la machinerie de
transcription (Turner, 1993). La difficulté de modéliser, de cette manière, les conséquences de
combinaisons complexes d'histones a conduit Brian Strahl et Davis Allis à faire l'hypothèse d'un
« code histone » (Strahl et Allis, 2000). Voici comment ils l'inroduisaient en 2000: « We propose
that distinct histone modifications, on one or more tails, act sequentially or in combination to form
a 'histone code' that is, read by other proteins to bring about distinct downstream events. » (« Nous
proposons que des modications distinctes des histones, sur une ou plusieurs queues, agissent
séquentiellement ou de manière combinatoire pour former un « code histone », c'est-à-dire lu par
d'autres protéines et entraînant des évènements consécutifs distincts. »). Le terme « séquentiel »
employé ici est important, car il ouvre la porte à un code plus instructif (ou générateur) que
descriptif, c'est-à-dire ne spécifiant qu'un petit nombre d'actions possibles, qui en auront d'autres en
conséquence, aboutissant in fine à un paysage favorable ou non à l'expression des gènes. On
retrouve esquissé l'ancien débat entre épigenèse et préformisme, qui avait animé la biologie au
tournant du XIXème et du XXème siècle. L'image que nous nous faisons aujourd'hui du code histone
est clairement du ressort de la première (incidemment, on retrouve la continuité de concepts entre
épigenèse et épigénétique), mais la possibilité d'une alternative plus figée, en quelque sorte
préformiste, est souvent source d'incompréhensions. Ainsi donc, toute l'information n'est pas
nécessairement contenue dans la photographie des marques portées par les histones à un moment
donné. Au contraire, elle peut émerger de la lecture de ces marques par d'autres protéines qui
agiront en conséquence, par exemple en déposant de nouvelles modifications sur les queues des
histones ou en en supprimant d'anciennes. Il faut ajouter que les histones elles-mêmes peuvent être
40

remplacées (c'est bien entendu le cas lors de la réplication de l'ADN, mais pas seulement), ce qui
pose toute la question de la transmission de l'information associée à ces modifications. Plusieurs
domaines capables de lire les modifications des histones ont été caractérisés (bromodomaines
reconnaissant les lysines acétylées ou chromodomaines reconnaissant les lysines méthylées, pour ne
citer que ceux-là) et, de plus en plus, est faite la distinction entre protéines, ou domaines protéiques,
lecteurs et effecteurs du code histone. Pour finir, ajoutons que, sur un même résidu peuvent être
déposées des modifications successives (par exemple di- ou tri-méthylation de K4 sur l'histone H3),
qui seront reconnues par des protéines différentes et auront donc des conséquences distinctes.
2.3.2 Marques épigénétiques du chromosome X inactif: stabilité et
dynamique
Au vu de ce qui précède, l'acquisition de « marques » (modifications covalentes de leurs
queues) par les histones est intimement liée à deux notions essentielles: la stabilité de l'information
portée par la chromatine et transmise au cours des divisions cellulaires d'une part, et le recours à
une séquence d'évènements distincts mais coordonnés pour établir une mémoire épigénétique,
d'autre part. Parler d'épigénétique du Xi revient nécessairement à traiter de ces deux aspects, à en
comprendre l'articulation.
Qu'en est-il de la stabilité? Nous l'avons mentionné plus haut, passée l'étape de mise en place de
l'inactivation, Xist est dispensable à son maintien (Csankovszki et al., 2001; Marahrens et al., 1998;
Penny et al., 1996). Par ailleurs, le Xi est associé à plusieurs marques épigénétiques: modifications
des queues des histones (enrichissement en H3K27m3, H3K9me2, H3K9me3, H4K20m1,
H2AK119ub1 , diminution de H3K9ac, H3K4m2, H3K4m3, déacétylation de H4), association à des
variants d'histone (macroH2A, ce qui inclut en réalité les variants macroH2A1a, macroH2A1b et
macroH2A2), méthylation des îlots CpG (Boggs et al., 1996; Boggs et al., 2002; Chadwick et
Willard, 2003; Chaumeil et al., 2002; Costanzi et al., 2000; Costanzi et Pehrson, 1998; Heard et al.,
2001; Jeppesen et Turner, 1993; Keohane et al., 1996; Kohlmaier et al., 2004; Mermoud et al.,
1999; Mermoud et al., 2002; Norris et al., 1991; Peters et al., 2002; Plath et al., 2003; Rougeulle et
al., 2004; Silva et al., 2003; de Napoles et al., 2004) (fig. 8, p. 42 et fig. 9, p. 44). De manière
significative, il a été montré pour la plupart de ces marques qu'elles restaient associées au
chromosome en métaphase, ce qui est cohérent avec leur participation supposée à la transmission de
l'état inactif (Boggs et al., 2002; Chadwick et Willard, 2003; Chaumeil et al., 2002; Mak et al.,
2002; Mermoud et al., 2002; Peters et al., 2002).
41

Pourtant, les gènes portés par le Xi ne sont pas ou peu réactivés lorsque sont altérés,
individuellement, l'hypoacétylation des histones, en utilisant l'inhibiteur de déacétylase TSA (pour
trichostatin A) (Csankovszki et al., 2001; Yoshida et al., 1995), ou l'association avec Xist, dans un
mutant conditionnel de Xist (Xist1lox, fig. 5, p. 27) qui a également pour effet de voir disparaître
sur le Xi l'enrichissement en macroH2A1 (Csankovszki et al., 1999; Csankovszki et al., 2001) et en
H3K27me3, ainsi que sa localisation périnucléolaire en milieu et fin de phase S (Zhang et al.,
2007)(cf introduction, partie 1, section 2.2, p. 35). Ainsi, après délétion de Xist dans des
fibroblastes, le taux de réactivation d'un transgène situé sur le Xi et exprimant la GFP n'est que
doublé par rapport au taux spontané (0,04 % contre 0,01 à 0,02 %), mais il n'est pas modifié après
traitement par la TSA. La déméthylation de l'ADN a un effet plus important (Csankovszki et al.,
2001; Graves, 1982; Mohandas et al., 1981; Sado et al., 2000). Lorsqu'elle est réalisée de manière
partielle après traitement à la 5-azacytidine, la réactivation du transgène GFP évoqué précédemment
est augmentée d'un facteur 20 (0,2 %), mais cette augmentation est d'un facteur 1500 (15 %) dans
un mutant conditionnel de la méthylase Dnmt1, conduisant à une déméthylation complète de l'ADN
(Csankovszki et al., 2001). Il existe toutefois une synergie entre Xist, l'hypoacétylation des histones
et la méthylation de l'ADN pour maintenir l'état inactif, ce dont rend compte une réactivation plus
forte lorsque deux ou trois de ces facteurs sont altérés de pair (Csankovszki et al., 2001). Le taux de
réactivation est ainsi augmenté, par rapport au taux spontané, d'un facteur 40 (0,4 %) lorsque la
délétion de Xist est combinée à un traitement à la 5-azacytidine, d'un facteur 80 (0,8 %) lorsque un
42
Fig. 8: Modifications des queues d'histones associées au Xa et au Xi (d'après Heard et Disteche, 2006)
Les cœurs d'histones sont représentés sous forme d'ovales. Les résidus des queues d'histones sont indiqués avec les modifications qui peuvent leur être associées sur le Xa (en haut) et sur le Xi (en bas).

traitement additionnel à la TSA est réalisé, et d'un facteur 3000 (30 %) après l'induction simultanée
des délétions de Xist et de Dnmt1 (Csankovszki et al., 2001). Des résultats comparables sont
obtenus par les mêmes auteurs en utilisant comme rapporteur le gène Hprt, également localisé sur le
chromosome X, mais la réactivation de Hprt et du transgène GFP n'est pas toujours coïncidente, ce
qui dénote son caractère partiel (Csankovszki et al., 2001). Quoiqu'il en soit, la méthylation de
l'ADN en tant que telle semble jouer, pour la stabilité de l'état inactif, un rôle plus important que
l'expression de Xist ou l'hypoacétylation des histones (bien que l'on ne connaise pas l'efficacité du
traitement à la TSA). Une seconde conclusion est que, une fois établie, l'inactivation est
extrêmement stable et semble faire appel, pour garantir cette stabilité, à un combinaison de marques
épigénétiques agissant de concert, mais parmi lesquelles aucune n'est vraiment indispensable.
La méthylation des îlots CpG, mais aussi l'association aux variants d'histone macroH2A, sont en
particulier deux marques qui apparaissent tardivement sur le Xi, ce qui privilégie pour elles un rôle
plus strictement dans la maintenance de l'état inactif. Il faut néanmoins remarquer que, à l'exception
de Dnmt1, la mauvaise connaissance des enzymes responsables des différentes marques présentes
sur le Xi, ainsi que leur possible redondance ou interdépendance, n'a pas permis jusqu'à maintenant
une étude détaillée du rôle individuel de chacune d'entre elle (cf section suivante).
Le recouvrement par Xist du futur Xi est suivi, dans un délai de un à deux cycles cellulaires, par
l'extinction transcriptionnelle des gènes de ce dernier (Kay et al., 1993; Okamoto et al., 2004;
Panning et al., 1997; Sheardown et al., 1997a; Wutz et Jaenisch, 2000). Nous l'avons vu,
l'extinction génique dépend de la répétition A de Xist (Wutz et al., 2002) mais ce n'est le cas, ni pour
l'extinction des séquences répétées intergéniques (Chaumeil et al., 2006), ni pour la formation du
domaine de Xist (Wutz et al., 2002) (cf introduction, partie 1, section 2.2, p. 35). Ce dernier, qui
coïncide avec le territoire chromosomique (Chaumeil et al., 2006), est rapidement associé à la perte
de marques de type euchromatique, telles que H3K9ac, H3K4me2 et H3K4me3 (Chaumeil et al.,
2002; Goto et al., 2002; Heard et al., 2001; Okamoto et al., 2004), bientôt suivies par
l'hypoacétylation des histones H4 (Chaumeil et al., 2002; Heard et al., 2001; Keohane et al., 1996)
et l'enrichissement en H3K27me3 (Okamoto et al., 2004; Plath et al., 2003; Silva et al., 2003),
H3K9me2 (Heard et al., 2001; Okamoto et al., 2004), H4K20 me1 (Kohlmaier et al., 2004) et
H2AK119ub1 (Fang et al., 2004; Hernandez-Munoz et al., 2005; Smith et al., 2004; de Napoles et
al., 2004) (fig. 9). De manière intéressante, l'absence de la répétition A, et donc d'extinction
génique, n'a pas d'effet sur l'apparition des marques H3K27m3 et H4K20m1 (Kohlmaier et al.,
43

44
Fig. 9: Mise en place des marques épigénétiques sur le Xi durant la différenciation de cellules ES (adapté de Heard et Disteche, 2006)
Légende page 45

2004) en cours de différenciation (mais la répétition A de Xist est bel et bien requise pour permettre
l'acquisition de ces marques lorsque l'expression de Xist est induite dans des cellules non
différenciées, ce qui relève cependant d'un contexte moins physiologique).
On peut supposer que l'ambivalence des fonctions de Xist – dépendant ou non de la
répétition A – refléterait la nécessité de disposer de différentes marques avec des cinétiques
particulières, une autre possibilité étant la compartimentalisation du chromosome X en différentes
regions pour l'inactivation desquelles les mécanismes mis en jeu diffèrent.
A l'appui de la première idée, l'élégant travail initié par A. Wutz et repris par ses collaborateurs,
tirant parti de transgènes inductibles de Xist, et démontrant l'existence de périodes de temps
successives et non équivalentes quant aux possibilités de parvenir à l'inactivation du chromosome
X, au cours de la différenciation in vitro de cellules ES (Kohlmaier et al., 2004; Wutz et al., 2002).
Ainsi, Xist n'est-il capable de conduire à l'inactivation que dans les cellules non différenciées ou
durant les premières 24 h de différenciation (induite par l'acide rétinoïque), dites fenêtre d'initiation.
L'inactivité du chromosome X devient irréversible, en l'absence d'expression de Xist, après 48 h de
différenciation (Wutz et al., 2002). Il reste donc une fenêtre critique de 24 h durant laquelle la
décision d'inactiver un chromosome X doit être consolidée. On notera au passage que chacune de
ces périodes de 24 h correspond peu ou prou à un cycle de division cellulaire. Dans le modèle
expérimental utilisé, l'expression de Xist est rapidement associée à la mise en place de H3K27m3
(ainsi que de H4K20m1 mais, curieusement, pas de H3K9me2 et H3K9me3), qui est perdu peu
après l'expression de Xist, lorsque l'induction de ce dernier n'est pas maintenue. Pourtant, quand
Xist est ré-exprimé tardivement, la méthylation H3K27me3 est efficacement restaurée, mais
uniquement si Xist avait été préalablement exprimé au moins durant les premières 48 à 72 h de
différenciation, c'est-à-dire durant la fenêtre d'initiation et la fenêtre critique (Kohlmaier et al.,
2004). Pour expliquer ces observations, les auteurs estiment qu'une mémoire épigénétique, de
nature inconnue, a dû être mise en place au moment de la transition vers une inactivation
45
Légende de la figure 9, page 44: Les jours sont indiquées sur la flèche de temps à gauche. A droite, RNAimmunoFISH permettant de visualiser les différentes marques associées au Xi. Première rangée à droite: le domaine d'ARN Xist coïncide avec l'exclusion de la polymérase II et l'absence de transcription de séquences répétées non géniques, marquées en RNAFISH par une sonde Cot1, vers 2 jours de différenciation (Chaumeil et al., 2006). Deuxième rangée à droite:le Xi est associée à la protéine Eed et à l'exclusion de la marque euchromatique H3K4me2, vers 23 jours de différenciation (Silva et al., 2003). Troisième rangée à droite: enrichissement du Xi en H3K27me3, vers 23 jours de différenciation (Rougeulle et al., 2004). Quatrième rangée à droite:enrichissement du Xi en macroH2A1.2 dans des fibroblastes (Csankovszki et al., 1999).

irréversible, et il est tentant d'imaginer, d'après les cinétiques, que l'acquisition de H3K27me3
pourrait jouer un rôle essentiel dans l'établissement d'une telle mémoire (fig. 10). Pouvoir associer
fonctionnellement fenêtres et mémoire, telles que définies par l'équipe de A. Wutz, aux nombreuses
marques épigénétiques mises en place précocément lors de l'inactivation offrirait, à n'en pas douter,
un cadre conceptuel particulièrement séduisant.
La cinétique de mise en place de ces marques pourrait donc être au moins aussi importante que
leur combinatoire, envisagée de manière plus statique. Cependant, toute conclusion doit être
nuancée par la distribution précise des marques, à l'échelle du chromosome X, mais aussi de chaque
gène. Par exemple, il a été montré par immunofluorescence que le chromosome X inactif humain
était subdivisé en deux types d'hétérochromatine facultative, alternés sur la longueur du
46
Fig. 10: Transition entre initiation et maintenance de l''inactivation (d'après Kohlmaier et al., 2004)
Modèle proposé par Kohlmaier et al. (Kohlmaier et al., 2004). Les jours de différenciation sont indiqués sur la flèche de temps. (A) Dans les cellules ES indifférenciées, le recrutement efficace de H3K27me3 dépend à la fois de la formation d'un domaine de Xist et de l'extinction transcriptionnelle des gènes du chromosome X, via la répétition A (triangles noirs). (B) En début de différenciation, l'extinction génique n'est pas indispensable à l'enrichissement de H3K27me3 (flèches pointillées). (C) Début de la fenêtre critique durant laquelle Xist perd sa capacité à recruter H3K27me3 (flèche pointillées) et à initier l'inactivation des gènes du chromosome X. Une mémoire chromosomique est supposée être alors mise en place (lettre M dans l'ovale noir) grâce à H3K27me3 et à Xist. Cette mémoire est complètement mise en place sur le Xi lorsque l'inactivation devient irréversible et indépendante de Xist. (D) Durant la phase de maintenance, la mémoire chromosomique permet à Xist de recruter H3K27me3 de manière efficace sur le Xi.

chromosome (ce qui exclut a priori une corrélation stricte avec les régions pseudo-autosomiques
échappant à l'inactivation) et retrouvés aussi bien en interphase qu'en métaphase. Le premier type
est défini par la présence conjointe de l'ARN XIST, macroH2A et H3K27me3, le second par la
présence de H3K9me3, H4K20me3 et la protéine HP1 (Chadwick, 2007), connue pour se fixer à
H3K9me3 (Bannister et al., 2001; Jacobs et al., 2001; Lachner et al., 2001). Dans une autre étude
réalisée sur des fibroblastes de souris, l'examen par immunoprécipitation de chromatine (ChIP) des
marques H3K27me3 et H3K9me2 révèle des profils partiellement chevauchant, l'enrichissement de
H3K27me3 étant observé au promoteur et dans le corps des gènes étudiés, sur le chromosome X
inactif, mais pas sur le chromosome X actif, tandis que H3K9me2 est présent dans les deux cas dans
le corps des gènes, et enrichi sur le chromosome X inactif uniquement au niveau des promoteurs
(Rougeulle et al., 2004). La généralisation d'approches de type ChIP on chip utilisant des puces à
hautes résolutions (tiling arrays) devrait permettre à l'avenir de mieux nous représenter la
distribution de telles marques, en combinant résolution et vision d'ensemble. Le parallèle pourrait
également être fait entre l'observation de types distincts d'hétérochromatine et l'existence d'un
compartiment formé, au niveau du domaine d'ARN Xist, dans un premier temps par les séquence
intergéniques, rejointes seulement plus tard par les gènes du futur Xi (cf introduction, partie 1,
section 2.2, p. 35). Espérons que, là aussi, les nouvelles technologies pourront être informatives.
2.3.3 Lecteurs et effecteurs des marques épigénétiques sur le chromosome X
inactif
Pour compléter le panorama dressé jusqu'à présent des marques épigénétiques du Xi, il nous
reste à présenter les facteurs dont on pense qu'ils pourraient être responsables de leur déposition,
mais également capables de les « lire » (on retrouve ici la définition originelle du code histone,
présentée plus haut) (fig. 9, p. 44). La protéine Ezh2 appartenant à la famille Polycomb semble être
l'histone méthyltransférase responsable de la triméthylation de H3K27 (Plath et al., 2003; Silva et
al., 2003). Son activité requiert les protéines Eed et Suz12, membres avec elle du complexe PRC2
(pour revue, Cao et Zhang, 2004), et toutes trois sont recrutées au niveau du chromosome X inactif
en même temps que H3K27me3 ((Kohlmaier et al., 2004; Plath et al., 2003; de Napoles et al.,
2004; de la Cruz et al., 2005). Toutefois, il n'est pas clair que le même complexe soit responsable du
maintien de H3K27me3, une fois établi, car l'association de Eed et Ezh2 avec le Xi est transitoire
47

(Plath et al., 2003; Silva et al., 2003). D'autre part, Eed n'est pas indispensable pour que
l'inactivation ait lieu de manière correcte dans l'embryon proprement dit, entre E5,5 et E8,5, c'est à
dire pendant et peu après sa mise en place (Kalantry et Magnuson, 2006).
Un second complexe polycomb, PRC1, est retrouvé associé au Xi. Il inclut en particulier les
protéines Ring1a et Ring1b, responsables de la mono-ubiquitination de H2A sur la lysine 119
(H2AK119ub) (de Napoles et al., 2004). La composition précise de ce complexe varie au cours du
processus d'inactivation et en fonction du type cellulaire (Plath et al., 2004; pour revue, Heard,
2005). Par exemple, la protéine Phc1 pourrait jouer un rôle lors de la phase d'initiation, avant d'être
progressivement remplacée par Phc2, en association notamment avec Bmi-1 et Cbx2 (Plath et al.,
2004). Il est tentant d'imaginer, sur le modèle des complexes homologues étudiés chez D.
melanogaster (Fischle et al., 2003; Min et al., 2003), que PRC1 serait recruté sur le Xi en s'ancrant
sur la marque H3K27me3, qui serait elle-même mise en place par PRC2. Pourtant, si un tel modèle
n'est pas à exclure, il ne peut rendre compte de la totalité de la présence de PRC1, puisque celui-ci
est retrouvé dans des régions dépourvues de H3K27me3 (Plath et al., 2004).
Les ubiquitines ligases CULLIN3 et SPOP sont responsables de l'ubiquitinylation de BMI1 (qui
fait partie du complexe PRC1) et de macroH2A1. En l'absence de l'une ou l'autre de ces enzymes,
macroH2A1 n'est plus localisé à l'emplacement du domaine d'ARN Xist qui, lui, reste intact
(Hernandez-Munoz et al., 2005). Comme le complexe PRC1 a lui aussi une activité ubiquitine
ligase, il serait intéressant de voir si celle-ci pourrait également contribuer à la localisation de
macroH2A sur le Xi. Notons que aussi bien PRC1 que macroH2A requièrent l'expression continue
de Xist pour pouvoir être observés sur le Xi, ce qui a pu être montré en utilisant un mutant
conditionnel de Xist (Csankovszki et al., 1999; Plath et al., 2004). MacroH2A1, en tant que
composant des nucléosomes, en modifie la mobilité, et réduit en conséquence l'accessibilité de la
chromatine, dans des tests in vitro (Angelov et al., 2003). Il est par ailleurs requis pour l'activité de
la Poly ADP-ribose polymerase 1 (PARP1), qui elle-même contribue à la stabilité de l'inactivation
(Nusinow et al., 2007).
La diméthylation de H3K9 apparaît sur le chromosome X inactif plus tardivement que
H3K27me3 (Okamoto et al., 2004; Rougeulle et al., 2004). D'autre part, la répartition de ces deux
marques n'est pas identitique (cf section précédente). C'est pourquoi il est peu probable que Ezh2
soit la méthylase responsable de H3K9me2 dans le cas qui nous occupe, bien qu'une telle activité ait
été rapportée par ailleurs. L'invalidation d'un deuxième candidat, l'histone methyl transférase G9a,
n'a pas d'effet sur l'inactivation du chromosome X, mais il n'a pas été regardé si la présence de
H3K9me2 était altérée au niveau du Xi (Ohhata et al., 2004). Les methylase Suv39h1 et Suv39h2,
48

généralement responsable de la méthylation de H3K9 dans l'hétéchromatine facultative, ne sont pas
pas non plus requise pour la méthylation de H3K9 sur le Xi (Peters et al., 2002). La méthylation par
Suv39h est en général associée à la reconnaissance de l'histone méthylée par la protéine HP1. Celle-
ci n'est pas observée au niveau du Xi chez la souris, mais y est associée chez l'homme (Chadwick et
Willard, 2001), ce qui pourrait suggérer des mécanismes différents dans les deux espèces.
L'enzyme PR-Set7, qui réalise la monométhylation de H4K20 chez la drosophile (Karachentsev
et al., 2005), pourrait être un bon candidat pour déposer cette marque sur le Xi, mais cela reste à
vérifier.
Enfin, la méthylation des îlots CpG du Xi dépend de Dnmt1, et en l'absence de celui-ci, la
réactivation de gènes liés au chromosome X a pu être observée (Csankovszki et al., 2001; Sado et
al., 2000). Citons également un autre candidat, Momme D1, identifié à l'issu d'un crible recherchant
des modificateurs de variégation, et conduisant, lorsqu'il est muté à l'état homozygote, à une
diminution importante de la méthylation des îlots CpG sur le Xi (Blewitt et al., 2006). A défaut
d'avoir pu identifier, pour l'instant, le gène correspondant, cette étude illustre comment une
approche par mutagenèse chez la souris peut être utile pour caractériser de nouveaux facteurs
impliqués dans des processus épigénétiques. Remarquons que l'inactivation du chromosome X se
prête particulièrement bien à une telle approche qui, en retour, peut être fructueuse lorsqu'il s'agit de
mieux comprendre les spécificités du Xi, par rapport à des processus soumis à des contraintes
développementales différentes.
2.4 Réplication asynchrone
Chez les métazoaires, la réplication tardive en phase S des régions non transcrites semble être
une tendance générale (Gilbert, 2002). Dans les cellules somatiques des mammifères, le Xi ne fait
pas exception. Tandis que le Xa est répliqué en début de phase S, le Xi l'est à la fin de cette
dernière, cette propriété étant très bien conservée chez les mammifères (Evans et al., 1965; Galton
et Holt, 1965; Priest et al., 1967; Sharman, 1971; Takagi et al., 1982; Taylor, 1968; Taylor et Miner,
1968). Plutôt que de réplication tardive, il serait peut-être préférable, dans le cas de l'inactivation du
chromosome X, de parler de réplication asynchrone car, comme nous le verrons bientôt, pour ce qui
est de l'inactivation, embryon et tissus extraembryonnaires présentent des différences notables et, en
particulier, la réplication du Xi se fait plus tôt que celle du Xa dans les derniers (Sugawara et al.,
1983).
49

Dans tous les cas, la réplication asynchrone suit de peu l'apparition de la plupart des
modifications d'histones (Chaumeil et al., 2002; Okamoto et al., 2005), et pourrait donc être la
conséquence des modifications induites par elles sur l'organisation de la chromatine. Ce qui ne
signifie pas nécessairement qu'elle soit dénuée de fonctions, bien que cela reste difficile (voire
impossible) à tester sans toucher en même temps aux autres marques du Xi. La réplication
asynchrone pourrait par exemple permettre une ségrégation temporelle des chromosomes, de façon
à minimiser le contact avec la machinerie de transcription, ou bien à maximiser l'accessibilité aux
enzymes de modification de la chromatine, et contribuer ainsi au maintien de l'état inactif. A cet
égard, il est intéressant de remarquer que les origines de réplication sont les mêmes sur le Xi et le
Xa, et semblent être utilisées avec la même efficacité (Cohen et al., 2003; Gomez et Brockdorff,
2004), ce qui souligne bien que la différence essentielle est dans le moment où la réplication est
initiée, et non dans le mécanisme de réplication en tant que tel. Ségrégation temporelle est spatiale
pourraient être coordonnées, ce que semble illustrer la localisation préférentielle du Xi (mais pas du
Xa) à proximité du nucléole en milieu et fin de phase S (Zhang et al., 2007) (cf introduction,
partie 1, section 2.2, p. 36). Toutefois, une fois l'inactivation mise en place, la délétion
conditionnelle de Xist sur le Xi n'a pas d'effet sur la réplication tardive de ce dernier (Csankovszki
et al., 1999), bien que sa localisation à proximité du nucléole ne soit plus observée (Zhang et al.,
2007). Par contre, la délétion conditionnelle de Xist (Xist1lox, fig. 5, p. 27) sur le Xa conduit à une
réplication retardée de celui-ci, indépendamment de la présence d'un second chromosome X (Diaz-
Perez et al., 2005). Cette observation suggère que le chromosome X n'est pas actif par défaut, mais
que le locus Xist joue un rôle important sur le Xa pour maintenir un comportement distinct de celui
du Xi. Dans cette optique, il serait intéressant de connaître la localisation nucléaire du Xa dans
lequel a été délété Xist, notamment vis-à-vis du nucléole (cf introduction, partie 1, section 2.2,
p. 36).
2.5 Gènes échappant à l'inactivation
Nous avons déjà mentionné que certains gènes du chromosome X échappaient à l'inactivation.
Ils sont plus ou moins nombreux selon les espèces: chez la souris, il y en a sept, tandis que chez
l'homme, il s'agirait de 15 % des gènes du chromosome X (Brown et Greally, 2003; Carrel et al.,
1999). Discuter les causes de cette différence est hors de notre propos, mais cela tient certainement
à l'histoire évolutive du chromosome X dans les deux espèces (à ce sujet, voir (Brown et Greally,
2003; Disteche et al., 2002; Tsuchiya et al., 2004)). Les gènes échappant à l'inactivation ont en
50

règle générale un paralogue sur le chromosome Y et, tout au moins chez l'homme, ils sont
concentrés sur les strates évolutives les plus récentes, de telle sortes qu'ils sont regroupés en régions
échappant à l'hétérochromatinisation (Carrel et Willard, 2005; Jegalian et Page, 1998; Lahn et Page,
1999b). Une telle distribution est cohérente avec l'idée que ces gènes n'auraient pas encore été
incorporés au système de compensation de dose (Charlesworth, 1996). Il est assez remarquable
qu'aux 15 % de gènes échappant à l'inactivation s'ajoutent 10 % de gènes soumis à l'inactivation de
manière variable selon les individus (Anderson et Brown, 1999; Carrel et Willard, 2005), mais il
reste à évaluer si cela est associé à une variabilité phénotypique.
Dans les régions échappant à l'inactivation la densité de LTR (long terminal repeat) est plus
faible (Tsuchiya et al., 2004), mais il n'est pas certain que cela soit le cas pour les éléments L1, qui
font débat (Bailey et al., 2000; Carrel et Willard, 2005; Tsuchiya et al., 2004). En revanche, nous
avons déjà mentionné que certaine motifs, généralement sous-représentés dans les éléments L1,
étaient plus fréquents dans les éléments L1 associés aux gènes soumis à l'inactivation (Carrel et al.,
2006) (cf introduction, partie 1, section 2.1, p. 33). D'autre part, le même type d'approche
computationnelle ayant servi à caractériser de tels motifs a permis d'en identifier d'autres enrichis de
manière spécifique au niveau des gènes échappant à l'inactivation (cf introduction, partie 1,
section 2.1, p. 33). Pour l'un d'entre eux, (GATA)n, l'enrichissement est observé non seulement vis-
à-vis des gènes soumis à l'inactivation, mais également par rapport aux autosomes (McNeil et al.,
2006). Cela suggère la coévolution d'éléments génomiques favorisant l'inactivation et empêchant
celle-ci, selon les gènes, sur le chromosome X humain. On ne sait toutefois pas à quel point ce
type de motifs témoigne de l'ajout évolutivement récent des régions concernées sur le chromosome
X ou est véritablement apte à empêcher l'inactivation. Mais la valeur prédictive de certains de ces
motifs pour des gènes échappant ou non l'inactivation provenant d'une même strate évolutive tend à
privilégier, dans leur cas, la seconde option (Carrel et al., 2006).
Il a par ailleurs été montré, dans des cellules ES murines indifférenciées, que le profil de
H3K4me2 au niveau de gènes échappant à l'inactivation était comparable à celui de gènes
autosomiques, mais distinct de celui des gènes pouvant être inactivés (Rougeulle et al., 2004). Ce
profil particulier pourrait jouer un rôle déterminant pour empêcher l'inactivation, mais cela reste à
déterminer. Dans les cellules somatiques différenciées, la chromatine au niveau des gènes
échappant à l'inactivation ne semble avoir acquis aucune des marques associées aux gènes inactifs
(Boggs et al., 2002; Filippova et al., 2005; Gilbert et Sharp, 1999; Goodfellow et al., 1988). Mais
une caractérisation plus détaillée devrait être réalisée pour déterminer si le profil chromatinien des
51

gènes échappant à l'inactivation et celui de gènes exprimés à partir des autosomes sont en tout point
comparables ou pourraient différer pour certaines marques, comme semble le suggérer le profil de
H3K4me2 au niveau du gène échappant à l'inactivation Smcx (Rougeulle et al., 2004).
Une explication alternative à l'existence d'éléments ou marques répartis sur toute la longueur des
régions échappant à l'inactivation pourrait être la présence d'éléments insulateurs en bordure des
gènes ou régions à protéger. Ce cas de figure est en tout cas vraisemblable pour les gènes murins
Jarid1c et Eif2s3x et pour le gène humain EIF2S3X, à l'extrémité 5' desquels se fixe la protéine
CTCF, connue pour avoir une fonction insulatrice. De manière significative, la fixation de CTCF n'a
pas été observée à proximité du gène JARID1C qui, chez l'homme, est entouré de gènes échappant
également à l'inactivation, contrairement à son orthologue murin Jarid1c (Filippova et al., 2005).
Cependant, la présence de sites de fixation de CTCF autour d'un transgène inséré sur le
chromosome X n'est pas suffisante pour lui permettre d'échapper à l'inactivation (Ciavatta et al.,
2006). Il faut donc envisager d'autres facteurs, ou considérer d'autres contraintes que celles prises en
compte dans cette dernière expérience. Par exemple, il a été montré que le gène Jarid1c demeurait à
l'extérieur ou sur la bordure externe du domaine d'ARN Xist tout au long de la différenciation de
cellules ES, au cours de laquelle il n'était jamais inactivé (Chaumeil et al., 2006). Cela suppose
probablement la formation d'une boucle de chromatine pour sortir du domaine de Xist et, avec elle,
un certain nombre de contraintes stériques. On remarquera que le gène Jarid1c a été décrit pour être
inactivé de manière transitoire au cours du développement (Lingenfelter et al., 1998), ce qui n'est
pas le cas lors de la différenciation de cellule ES (Chaumeil et al., 2006). Ceci pourrait refléter des
limitations intrinsèques du modèle des cellules ES, dont nous aurons l'occasion de rediscuter.
52

Chapitre 3 Plasticité épigénétique et régulation de l'inactivation
au cours du développement
3.1 Epigénétique et développement embryonnaire
Nous avons fait jusqu'à maintenant l'économie d'une définition de ce que pouvait être
l'épigénétique, tout en introduisant des notions ayant trait à celle-ci, lorsque nous en avions besoin.
Le moment est désormais venu de regarder plus en détail ce que recouvre ce mot mystérieux. Une
mise en perspective historique (une esquisse seulement, car là n'est pas notre propos) est d'autant
plus nécessaire que, si l'on en croit certains journaux, l'épigénétique serait née il y a à peine
quelques années. Cela est pourtant bien loin de la réalité, tant les questionnements sous-jacents au
concept d'épigénitique sont constitutifs des interrogations ayant trait à la biologie, et ce depuis les
origines.
L'épigénétique est une notion dérivée, et assez proche (de plus en plus, en fait), de la notion
d'épigenèse. La question essentielle se résume en fait à savoir comment un individu entièrement
nouveau (ou presque) peut-être formé. Au IVème siècle avant J.-C., Aristote proposa deux
hypothèses: 1) l'individu était préformé dès le commencement et ne résultait que d'un accroissement
de taille à partir d'une forme miniature originelle, 2) l'individu était constitué grâce à la « formation
par dessus », c'est-à-dire l'épigenèse, de nouvelles structures, qu'il comparait à la « construction d'un
filet ». C'est cette dernière idée qu'il défendait. L'existence d'une forme miniature (un homunculus,
dans le cas de l'homme), contenue et transmise par les êtres vivants depuis la Création, supposait en
particulier qu'un seul des parents (l'homme, de préférence), puisse en réaliser la transmission. Cette
idée fut battue en brèche par deux découvertes majeures de la biologie au XIXème siècle. Tout
d'abord, celle des cellules, en tant qu'élément de base de tous les êtres vivants et, notoirement, la
découverte que les gamètes sont elles-même des cellules uniques. Dans ce cas, où loger un
homunculus? Peu après furent posées les bases de la génétique moderne, décrivant l'égale
contribution des deux parents à l'identité génétique de leurs enfants, ce qui rendit plus incongru
encore l'image d'un homonculus logé dans la tête de tout spermatozoïde.
53

La découverte du support moléculaire de l'information génétique, en particulier avec la
démonstration de la structure de l'ADN par Crick et Watson en 1953, allait par la suite permettre
l'essor de la génétique moléculaire dans la seconde moitié du XXème siècle, en s'appuyant sur la
notion de programme génétique, elle-même étroitement liée à la découverte du code génétique.
Comme nous l'avons vu à propos du code histone, toute idée de code ou de programme est souvent
ambivalente en biologie. C'est qu'ici, deux visions du monde s'affrontent. Dans les faits, pourtant,
codes ou programmes biologiques sont bien plus souvent instructifs (ou générateurs) que
descriptifs, en ce sens qu'ils ne procurent pas une vision de ce qu'est un individu adulte, mais plutôt
permettent l'exécution d'un petit jeu d'instructions, déterminantes pour la cascade d'évènements dont
elles sont à l'origine. C'est la combinaison de ces instructions, et leur séquence, qui façonnent le
développement d'un individu.
Si le programme génétique est essentiellement instructif, il importe, d'une certaine manière, de
garder la mémoire des instructions déjà réalisées, sans quoi tout serait toujours à recommencer et
aucun organisme multicellulaire correctement formé ne verrait le jour. C'est là le rôle de
l'épigénétique. Waddington, qui la définit pour la première fois en 1942, le fit ainsi: « branche de la
biologie qui étudie les relations de cause à effet entre les gènes et leurs produits, faisant apparaître
le phénotype ». Une définition plus cocasse était proposée par Denise Barlow récemment:
« L'épigénétique a toujours été l'ensemble de ces choses bizarres et merveilleuses que la génétique
ne sait pas expliquer ». En effet, à quelques exceptions près (dont celle de la maturation des
lymphocytes), la spécification des lignages cellulaires ne dépend pas de réarrangements au sein du
génome. L'épigénétique mérite donc bien son nom: elle vient « au dessus » des gènes.
Une définition concurrente de l'épigénétique aurait été possible, fondée sur la distinction entre
lignée germinale et cellules somatiques. La génétique serait alors du domaine de l'héritabilité, c'est-
à-dire de la première, tandis que l'épigénétique s'appliquerait à l'individu en tant que tel, et
concernerait les secondes. Mais cette définition est difficile à tenir dès le moment où l'on considère,
à juste titre, l'ADN comme le support de l'information génétique. Ainsi, des réarrangements
génomiques participent à définir les lignages lymphocytaire sans pour autant contribuer à la lignée
germinale, tandis que des caractères acquis peuvent être hérités, parfois sur plusieurs générations,
sans que la séquence nucléotidique ait été modifiée (on parle dans ce cas d'épimutation ou de
paramutation). C'est pourquoi l'utilisation pratique des termes « génétique » et « épigénétique » est
devenue, de nos jours, le plus souvent orthogonale à la notion d'héritabilité, et s'attache d'avantage
aux aspects moléculaires.
54

C'est que, comme ce fut le cas pour la génétique, l'épigénétique a bénéficié d'un intérêt
renouvelé dès lors qu'ont pu lui être associés différents supports moléculaires, que nous avons
abordés, pour certains d'entre eux, au chapitre précédent. L'embryologie fut associée dès le début à
ce renouveau avec, en particulier, les découvertes liées des gènes homéotiques et des complexes
protéiques polycomb chez D. melanogaster. Chez les mammifères, cependant, des obstacles en
partie d'ordre technique ont conduit, pendant longtemps, à une évolution essentiellement parallèle
de l'embryologie, rebaptisée biologie du développement, et de l'épigénétique, dont l'essor a été
rendu possible par l'utilisation habile de modèles cellulaires. C'est beaucoup moins le cas
aujourd'hui, et les échanges entre les deux disciplines (constituées comme telles, bien que cela soit
un peu artificiel) sont particulièrement féconds. Comme nous allons le voir, l'exemple de
l'inactivation du chromosome X est là pour en témoigner. L'inactivation du chromosome X illustre
d'autant mieux les liens entre épigénétique et développement que sa mise en place puis sa réversion
ont lieu à de multiples reprises au cours de l'embryogenèse ainsi que, plus rarement, après la
naissance et dans l'individu adulte.
3.2 Inactivation méiotique des chromosomes sexuels
L'un des deux chromosomes X est transcriptionnellement inactif dans les cellules somatiques
femelles, nous l'avons vu. Dans les gamètes mâles, les chromosomes sexuels (X et Y) sont
également inactivés, plus précisément lors de la méiose, au stade pachytène (Lifschytz et Lindsley,
1972; McCarrey et al., 1992; Monesi, 1965) (fig. 11, p. 56). Une telle inactivation méiotique des
chromosomes sexuels (MSCI, pour meiotic sex chromosome inactivation) a lieu dans la plupart des
espèces possédant des chromosomes sexuels hétéromorphes, dont D. melanogaster et C. elegans
(pour revue, Graves, 2006).
La MSCI est, en effet, consécutive au non-appariement des chromosomes sexuels (Turner et al.,
2006) et peut être généralisée en parlant d'inactivation méiotique des chromosome non synaptiques
(MSUC, pour meiotic silencing of unsynapsed chromosomes), qui s'applique également aux régions
autosomiques hétéromorphes, au même stade et (en l'état des connaissances) de la même manière
que pour les chromosomes sexuels (Schimenti, 2005; Turner, 2005; Turner et al., 2006).
Au stade leptotène a lieu, à de multiples positions, la cassure double brin des chromosomes,
destinée à permettre leur appariement et leur recombinaison, à travers la formation et la résolution
de jonctions de Holliday (pour revue, Turner, 2007). L'appariement ou synapse des autosomes (hors
régions hétéromorphes, lorsqu'elles existent) est complet au stade pachytène, mais impossible pour
55

56
Fig. 11: L'inactivation au cours du développement embryonnaire précoce (adapté de Heard et Disteche, 2006)
Pour plus de détails, se référer au texte. Les domaines d'ARN Xist sont représentés en vert, et l'expression des gènes liés au chromosome X sous forme de points rouges.

les chromosomes sexuels, régions pseudo-autosomiques mises à part. C'est alors qu'a lieu la MSCI,
probablement pour empêcher des recombinaisons inappropriées et délétères durant la méiose
(Jablonka et Lamb, 1988) ou, dans un premier temps tout au moins, pour stabiliser l'ADN après
cassure double brin et devant l'impossibilité de former une jonction de Holliday. En effet, la cassure
double brin de l'ADN semble un pré-requis à la mise en place de la MSCI (Barchi et al., 2005;
Bellani et al., 2005), qui est inititiée par des enzymes connues pour participer à la réparation de
l'ADN, BRCA1 (Turner et al., 2004) et ATR (Bellani et al., 2005; Turner et al., 2004). BRCA1
permet la relocalisation d'ATR vers les régions non synaptiques (Turner et al., 2004), ce dernier
phosphorylant alors, de manière locale, le variant d'histone H2AX, constitutif des nucléosomes lors
de la méiose (Baarends et al., 2005; Bellani et al., 2005; Fernandez-Capetillo et al., 2003;
Mahadevaiah et al., 2001; Turner, 2005; Turner et al., 2004). Ces évènements sont concomitants à
la séquestration des chromosomes sexuels dans un compartiment qualifié de vésicule sexuelle (sex-
body ou XY-body, en anglais), elle-même relocalisée au centre du noyau (McKee et Handel, 1993;
Solari, 1974).
L'association des chromosomes sexuels avec BRCA1, ATR et la forme phosphorylée de H2AX
est perdue lors de la transition diplotène-métaphase I (Mahadevaiah et al., 2001; Turner et al.,
2004). Toutefois, la majorité des gènes demeurent transcriptionnellement inactifs jusqu'en fin de
méiose (Greaves et al., 2006; Khil et al., 2004; Namekawa et al., 2006; Turner et al., 2006), c'est-à-
dire dans les spermatides ronds, voire même lors de l'élongation de ses derniers (Namekawa et al.,
2006). Cela s'explique par l'apparition de plusieurs modifications d'histones sur les chromosomes
sexuels dès la fin du stade pachytène de méiose I, conduisant à la formation d'une structure
hétérochromatique répliquée tardivement (Kofman-Alfaro et Chandley, 1970; Odartchenko et
Pavillard, 1970; Priest et al., 1967), nommée, à l'achèvement de la méiose, chromatine sexuelle
postméiotique (PMSC, pour post meiotic sex chromatin) (pour revue, Turner, 2007) . Les
modifications d'histones qui ont pu être caractérisées sont l'ubiquitinylation des histones H2A
(Baarends et al., 1999), la déacétylation des histones H3 et H4 (Khalil et al., 2004), la
diméthylation sur K9 des histones H3 (Khalil et al., 2004) et la sumoylation de cibles pour l'instant
inconnues (Rogers et al., 2004; Vigodner et Morris, 2005). De manière concomitante à leur
apparition, la polymerase II en élongation est exclue de la chromatine sexuelle (Khalil et al., 2004).
Cette exclusion n'est toutefois plus observée dans les spermatides (Khalil et al., 2004), alors que
l'on assiste à l'incorporation des variants d'histone MACROH2A1.2 (également nommé H2AFY)
(Hoyer-Fender et al., 2000) et H2AFZ (Greaves et al., 2006). Les enzymes réalisant les
modifications d'histones dans le cadre de la MSCI sont pour l'essentiel inconnues. Néanmoins, la
57

PMSC est associée aux enzymes UBE2A et UBE2B, qui pourraient être responsables de
l'ubiquitylation de H2A (Baarends et al., 2005), et aux protéines à chromodomaine (lesquels
s'associent en général aux histones méthylés) CBX1 (Metzler-Guillemain et al., 2003; Motzkus et
al., 1999) et CBX3 (Metzler-Guillemain et al., 2003).
La MSCI suscite plusieurs interrogations. Tout d'abord, son existence même semble
contradictoire avec l'enrichissement du chromosome X en gènes requis pour la spermatogenèse, que
nous avions évoqué à la section 1.1 de la première partie de l'introduction (p. 9). Ce paradoxe est
résolu de deux manières. D'une part, la plupart de ces gènes sont exprimés durant la période de
multiplication des spermatogonies, avant que la MSCI n'ait lieu (Khil et al., 2004). D'autre part, une
minorité non négligeable de gènes sont malgré tout exprimés après la méiose, aussi bien sur le X
que sur le Y (Hendriksen, 1999; Hendriksen et al., 1995; McCarrey et al., 1992; Nguyen et
Disteche, 2006; Wang et al., 2005). L'absence d'exclusion de la polymerase II phosphorylée au
niveau de la PMSC (Khalil et al., 2004) dans les spermatides pourrait d'ailleurs suggérer la
transcription, au moins à un niveau faible, des gènes portés par les chromosomes sexuels, à moins
qu'il ne s'agisse de transcription intergénique (d'autres hypothèses non dénuées d'intérêt pourraient
être formulées comme, par exemple, une éventuelle absence de délimitation claire des domaines
chromosomiques à ces stades).
La PMSC possède avec le Xi des cellules somatiques la caractéristique commune d'être
organisée sous forme d'hétérochromatine facultative, avec les modifications d'histones que cela
implique. De manière intéressante, comme le Xi, elle est également associée à des variants d'histone
de la famille macroH2A. Toutefois, cela ne dénote pas nécessairement un lien de parenté entre les
deux mécanismes, car ces derniers semblent contribuer à la régulation de loci plus variés qu'il n'était
initialement envisagé (voir, par exemple, (Agelopoulos et Thanos, 2006)). Quoiqu'il en soit, la
différence essentielle réside dans les mécanismes conduisant à l'hétéchromatinisation qui, dans le
cas de la MSCI, ne requièrent pas la présence du gène Xist (McCarrey et Dilworth, 1992; Turner et
al., 2002) bien qu'il soit, chez les mâles, exprimé spécifiquement dans la lignée germinale (Turner
et al., 2002) et que son ARN recouvre les chromosomes sexuels dans les spermatocytes (Ayoub et
al., 1997). Il faut cependant remarquer que, dans le cas les spermatocytes, l'association des ARN
Xist sur le chromosome X semble plus faible que celle observée dans les cellules somatiques
femelles (Clerc, communication personnelle).
Chez les marsupiaux, l'inactivation du chromosome X concerne exclusivement le chromosome
X d'origine paternelle, et ce dans toute les cellules (pour revue, Grutzner et Graves, 2004). Elle est
réalisée très probablement indépendamment du gène homologue à Xist, qui est codant et partage
58

peu de similarités avec ce dernier (Duret et al., 2006). Dès lors, il est tentant d'imaginer qu'elle
pourrait être initiée dès la MSCI. Dès travaux récents ont, à tout le moins, rendu cette hypothèse
plausible en montrant que l'extinction transcriptionnelle méiotique et post-méiotique était observée
pour plusieurs gènes chez le marsupial Monodelphis domestica (Hornecker et al., communication
orale, 2006; (Namekawa et al., 2007)).
Comme nous allons le voir, une telle possibilité fait également l'objet de débats pour les
mammifères euthériens, chez qui seul le chromosome X d'origine paternelle est inactivé jusqu'au
stade morula, ainsi que, plus tard, dans les lignages extraembryonnaires, contrastant avec
l'inactivation aléatoire exposée jusqu'à présent, qui lors de l'embryogénèse est restreinte aux cellules
somatiques de l'embryon stricto sensus, à partir des stades péri-implantatoires.
3.3 Inactivation soumise à l'empreinte parentale
3.3.1 Découverte
Dans tous les tissus chez les marsupiaux, mais également dans les tissus extraembryonnaires des
mammifères – tout au moins chez la souris, dont nous parlerons ici, ainsi que chez le campagnol
(Shevchenko, communication orale, 2006) et la vache (Dindot et al., 2004; Xue et al., 2002), la
situation étant plus controversée chez l'homme (cf p. 68) – seul le chromosome X d'origine paternel
est inactivé, ce que l'on a qualifié d'inactivation soumise à l'empreinte parentale, ou par
simplification et anglicisme (mais c'est la désignation que nous avons pris l'habitude d'utiliser au
laboratoire et qui sera employée ici) d'inactivation empreintée (Cooper et al., 1993; Sharman, 1971;
Takagi et Sasaki, 1975; West et al., 1977).
Des études cytogénétiques classiques, basées sur la coloration différentielle et la réplication
asynchrone du Xi avaient positionné le début de l'inactivation empreintée un peu avant celui de
l'inactivation aléatoire, distinguant ainsi trois vagues successives d'inactivation. D'inactivation
empreintée, d'abord, dans le trophectoderme dès le stade blastocyste précoce (vers E3,5), puis dans
l'endoderme primitif, lors de sa formation à partir de la masse cellulaire interne (vers E4,5).
D'inactivation aléatoire ensuite, lors de la troisième vague, se déroulant dans l'épiblaste, de manière
plus ou moins concomitante à la gastrulation (Sugawara et al., 1985; Takagi et al., 1982; Takagi et
Sasaki, 1975). Ces résultats convergeaient avec l'analyse de l'expression de deux allozymes (Epstein
et al., 1978a; Epstein et al., 1978b; Kratzer et Gartler, 1978; Monk et Harper, 1979) et de transgènes
59

LacZ (Adler et al., 1977; Lebon et al., 1995; Sugimoto et al., 2000) et GFP (Hadjantonakis et al.,
2001), tous localisés sur le chromosome X. Cependant, il était également connu que Xist était
exprimé de manière monoallélique, uniquement à partir du Xp, dès les stades 2 à 4 cellules (Kay et
al., 1994; Matsui et al., 2001; Nesterova et al., 2001a; Okamoto et al., 2000; Zuccotti et al., 2002),
tandis que l'expression de l'allèle d'origine maternelle est réprimée jusqu'au stade morula inclus
(Nesterova et al., 2001a). Pour ajouter au doute, il avait été montré par RT-PCR que l'expression des
allèles paternels de certains gènes était plus faible que celle de leurs allèles maternels respectifs, et
ce avant que l'inactivation empreintée ne soit, comme on le croyait, mise en place (Latham et
Rambhatla, 1995; Singer-Sam et al., 1992). Le caractère contradictoire de ces observations a
conduit différentes équipes à ré-examiner l'établissement de l'inactivation empreintée, en tirant parti
de progrès techniques mais surtout de la découverte de nouveaux marqueurs associés au
chromosome X inactif (cf introduction, partie 1, section 2.3, p. 38). Il est apparu alors que
l'inactivation avait lieu plus tôt qu'on ne le pensait, était consecutive à l'expression monoallélique de
Xist et par conséquent empreintée dans toutes les cellules, jusqu'à sa réversion dans la masse
cellulaire interne au stade blastocyste intermédiaire, vers E4,5 (Huynh et Lee, 2003; Mak et al.,
2004; Okamoto et al., 2004; Okamoto et al., 2005). Cette réactivation du X inactif dans les cellules
à l'origine de l'embryon stricto sensus permet à l'inactivation aléatoire de se dérouler comme
attendu, alors que l'inactivation reste empreintée dans les tissus extraembryonnaires.
3.3.2 Mise en place de l'inactivation empreintée après la fécondation
Une vision assez précise de la cinétique d'inactivation du Xp et de mise en place des diverses
marques épigénétiques associées a émergé ces dernières années, en particulier grâce au travail de
Okamoto et al. (Okamoto et al., 2004) (fig. 11, p. 56). Selon eux, le Xp est transcriptionnellement
actif immédiatement après l'activation du génome zygotique (stade 2 cellules), comme en témoigne
la présence d'ARN polymérase II, observée par immunofuorescence, et la détection de transcrits
primaires de gènes liés au chromosome X et de transcrits marqués avec des sondes Cot-1
(correspondant à des éléments répétées, principalement dans les régions intergéniques et dans les
introns) par RNA-FISH (Okamoto et al., 2004). Au même moment, Xist est exprimé sur le Xp et
visible sous forme d'un signal ponctuel par RNA-FISH (Okamoto et al., 2004). Mais dès le stade 4
cellules, l'ARN Xist forme un domaine généralement de petite taille sur le Xp, qui est également
associé avec l'hypoacétylation de H3K9, l'hypométhylation de H3K4 et l'exclusion de la polymérase
II. Ces marques sont toutefois observées dans un nombre variable de blastomères (1 ou 2 en
60

général) et conduisent à l'inactivation transcriptionnelle, tout au moins pour le gène Chic1, proche
de Xist sur le chromosome X. Mais dès le stade 16 cellules, le phénomène s'est étendu à l'ensemble
de la morula (Okamoto et al., 2004). Cela correspond en même temps à la consolidation de
l'inactivation et, probablement, à son extension à tout le chromosome avec un domaine d'ARN Xist
de plus grande taille associé, dans un nombre variable de blastomères, aux protéines Eed et Enx1, à
la diméthylation de H3K9, à la triméthylation de H3K27 et au recrutement de macroH2A, qui est
donc ici une marque plus précoce que lors de l'inactivation aléatoire (Costanzi et al., 2000; Mak et
al., 2004; Okamoto et al., 2004). Au stade 32 cellules, ces marques sont observées dans toutes les
cellules, sauf dans le cas de H3K9me2 pour laquelle il faudra attendre le stade blastocyste selon
Okamoto et al., alors que pour Mak et al., cette modification d'histone est au contraire plus précoce
que H3K27me3 (Mak et al., 2004). De telles divergences pourraient sous-entendre l'absence de
causalité entre ces deux marques. Quoiqu'il en soit, dans les blastocystes précoces, toutes les
marques évoquées ici sont présentes et associées à un Xp transcriptionnellement inactif dans toutes
les cellules (Mak et al., 2004; Okamoto et al., 2004).
Ce premier exposé doit être nuancé par une certaine variabilité dans le degré d'inactivation des
gènes et dans la cinétique de cette inactivation. Chic1, qui est localisé à proximité de Xist, est
inactivé dès le stade 4 cellules (Okamoto et al., 2004). Le gène Pgk1 l'est cependant seulement à
partir du stade 16 cellules, tandis que le gène Smc1l1 échappe en partie à cette première vague
d'inactivation (Mak et al., 2004). En fait, l'inactivation semble d'autant plus forte et mise en place
plus rapidement, en moyenne, que la distance au gène Xist est faible (Huynh et Lee, 2003). Cela est
corrélé en métaphase, dès le stade 2 cellules, avec un recouvrement partiel du Xp par l'ARN Xist,
centré sur le locus du gène Xist (Huynh et Lee, 2003). Les mêmes auteurs observent dès ce stade,
dans la plupart des cellules, l'association d'un domaine d'ARN Xist avec l'absence de marquage par
une sonde Cot1 en RNA-FISH. Huynh et al. positionnent en conséquence le début de l'inactivation
plus tôt que Okamoto et al, ce qui les a conduit à proposer une continuité avec la MSCI (cf
introduction, partie 1, section 3.2, p. 55), au cours de laquelle auraient été acquises des marques
(inconnues) constituant une empreinte paternelle. Il est difficile de concilier les deux études quant à
la cinétique d'apparition des domaines de Xist ou la disparition du marquage Cot-1. On pourra
penser à des différences de sensibilité de détection ou de fond génétique... Dans ce dernier cas, cela
relativiserait l'hypothèse d'une continuité fonctionnelle avec la MSCI. Quant à ce qui est de
l'inactivation proprement dite des gènes liés au chromosome X, on peut noter le recours à deux
modes distincts d'analyse. Huynh et al. se sont intéressés au niveau moyen d'expression de ces
gènes mesuré par RT-PCR à l'échelle d'une population de cellules, tandis qu'Okamoto et al. ont
61

examiné l'expression au niveau de cellules individuelles, mais de manière non quantitative, car la
technique de RNA-FISH ne le permet encore que très difficilement. On pourrait donc penser que
l'expression des gènes liés au chromosome X est d'abord diminuée, de manière progressive, avant
d'être perdue complètement, probablement une division cellulaire plus tard. L'impression générale
est que, à ces stades, l'inactivation est plus variable d'un gène à un autre qu'elle ne le sera plus tard,
après le stade blastocyste, ce qui expliquerait que des transgènes LacZ ou GFP, probablement
insérés en multi-copie, aient pu échapper à cette première vague d'inactivation empreintée
(Hadjantonakis et al., 1998; Sugimoto et al., 2000).
3.3.3 Réactivation du chromosome X paternel dans la masse cellulaire
interne du blastocyste
Dans le blastocyste précoce, le Xp est inactif dans toutes les cellules. Or, dans l'embryon (au
sens strict) post-implantatoire comme dans l'individu adulte, l'inactivation est réalisée, selon les
cellules, aussi bien pour le Xp que pour le Xm, de manière aléatoire. Le prérequis à cela est, en
premier lieu, une réactivation du Xp, qui a lieu lors de la maturation des blastocystes, entre E3,5 et
E4,5 (Mak et al., 2004; Okamoto et al., 2004). Elle correspond à la perte concomitante des
domaines d'ARN Xist et de l'association avec Eed et Enx1. Les autres marques suivront, avec
cependant un retard pour ce qui est de K27me3 et K9me2 qui, dans le blastocyste tardif sont encore
observés dans quelques cellules (Mak et al., 2004; Okamoto et al., 2004).
Dans la masse cellulaire interne des blastocystes, de même que dans les cellules ES qui en sont
dérivées, l'expression biallélique du gène Tsix (qui sera au coeur de notre exposé à partir du
chapitre 2 de la seconde partie de l'introduction, p. 95), antisens à Xist, est observée dans toutes les
cellules. Elle est associée à la déposition de H3K4me2, le long de son unité de transcription, qui
inclut celle du gène Xist, à la manière de poupées russes (Morey et al., 2004; Navarro et al., 2005;
Navarro et al., 2006). La transcription de Tsix s'oppose également à la propagation de H3K27me3 à
partir de la région en amont du gène Xist, comme nous le montrerons plus tard (cf résultats,
chapitre 3, p. 165). Il a été proposé que de telles modifications de la chromatine associées à
l'expression de Tsix permettaient de rendre épigénétiquement équivalents les deux allèles du gène
Xist, en effaçant ainsi toute marque d'empreinte parentale, ce qui permettrait la transition d'un mode
empreinté vers un mode aléatoire d'inactivation (Navarro et al., 2005). Il est également possible que
62

la réactivation du Xp participe d'un processus plus général, dans la masse cellulaire interne, de
reprogrammation épigénétique de l'ensemble des gènes dont la fonction sera requise pour la
formation des différents feuillets puis lignages embryonnaires.
3.3.4 Inactivation empreintée dans le trophectoderme
Alors que le Xp est réactivé dans les cellules à l'origine de l'embryon au sens strict,
l'inactivation reste empreintée dans les lignage extraembryonnaires, dérivés du trophectoderme et
de l'endoderme primitif. Le trophectoderme est clairement distinct de la masse cellulaire interne au
stade blastocyste, vers E3,5. Néanmoins, la spécification des cellules à l'origine du trophectoderme
pourrait être réalisée plus tôt, précédant la réorganisation en cellules internes et externes lors de la
compaction, qui a lieu au stade 16 cellules (Rossant, communication personnelle; Hadkjantonakis,
communication personnelle). Connaissant cela, on peut se demander si le caractère variable de
l'inactivation aux stades 4 à 8 cellules pourrait correspondre à un comportement différent des
cellules destinées à contribuer au trophectoderme ou à la masse cellulaire interne. Il n'y a pour
l'instant pas de réponse à cette question, qui demanderait d'examiner la mise en place de
l'inactivation en parallèle à l'expression des marqueurs de lignage.
Le maintien de l'inactivation empreintée dans le trophectoderme est associé à l'acquisition d'un
mode de réplication asynchrone, en avance par rapport au chromosome X actif (Sugawara et al.,
1983). Dans des cellules d'origine trophoblastique possédant des propriétés de cellules souches
(cellules TS, pour trophoblastic stem cells), l'absence d'Eed, qui conduit à la perte de nombreuses
marques (complexes PRC2 et PRC1, H2Aub, H3K27me3, H4K20me1, macroH2A, mais aussi
recouvrement par Xist), n'a pas pour conséquence la réactivation du Xp. C'est également le cas dans
l'embryon à E7,5 pour les cellules de l'ectoderme extraembryonnaires, qui sont une source de
cellules TS. Par contre, Eed et les marques qui lui sont associées sont requis pour le maintien de
l'inactivation du Xp lorsque les mêmes cellules sont différenciées in vitro, ainsi que dans le cône
ectoplacentaire, plus différencié que l'ectoderme extraembryonnaire (Kalantry et al., 2006). Comme
l'association du complexe PRC2 (qui inclut Eed) avec le Xp est normalement perdue lors de la
différenciation des cellules trophoblastiques tant in vitro que in vivo (Plath et al., 2003; Silva et al.,
2003), la fonction de Eed serait plutôt dans l'établissement, en amont, d'une mémoire épigénétique
capable de perpétuer l'état inactif.
63

Le maintien de l'inactivation du Xp serait particulièrement critique lors de la différenciation des
structures extraembryonaires, entre E7,5 et E8,5. C'est à ce moment que l'absence d'Eed se révèle
léthale (Wang et al., 2001a). C'est également le cas lorsque Xist est muté sur le Xp (qui ne peut donc
pas être inactivé dans les tissus extraembryonnaires), mais pas lorsqu'il est muté sur le Xm, ce qui
traduit bien un défaut lié à l'inactivation empreintée et non à l'inactivation aléatoire (Marahrens et
al., 1997; Sado et al., 2001). Réciproquement, l'absence du gène Tsix sur le Xm a pour conséquence
l'inactivation ectopique de ce dernier, s'ajoutant chez les femelles à celle du Xp, et est léthale après
E9,5 (Lee, 2000; Sado et al., 2001). Dans ce cas, l'activation ectopique de Xist peut être expliquée
par l'absence de la répression qu'exerce normalement sur lui Tsix, sur laquelle nous reviendrons en
détail à partir du chapitre 2 de la seconde partie de l'introduction (p. 95).
Contrairement, à l'inactivation aléatoire, il n'y pas de méthylation des îlots CpG aux promoteurs
des gènes lors de l'inactivation empreintée. Cela pourrait correspondre à une nécessité moins grande
de stabiliser l'inactivation dans des tissus qui seront amenés à disparaître à la naissance. A l'appui de
cette idée, la réactivation du Xp a été observée dans des cellules du trophectoderme mural dès E4.5,
puis dans le cône ectoplacentaire à partir de E6 (Hadjantonakis et al., 2001), mais une telle
observation pourrait tout aussi bien être la conséquence de ploïdies aberrantes. De manière
sporadique, on observe également l'activité du Xp dans l'ectoderme extraembryonnaire à E7,5
(Sugimoto et al., 2000).
3.3.5 Inactivation empreintée dans l'endoderme primitif
L'inactivation dans l'endoderme primitif avait été décrite par le passé comme une deuxième
vague d'inactivation, vers 4,5 jours après la fécondation, après celle observée dans le trophoblaste et
précédent l'inactivation aléatoire observée dans l'épiblaste (Takagi et Sasaki, 1975; West et al.,
1977). S'il est désormais acquis que l'inactivation empreintée des cellules trophoblastiques est en
continuité avec l'inactivation mise en place dans la morula, il n'en va pas de même pour
l'endoderme primitif. En effet, ce dernier est dérivé de la masse cellulaire interne des blastocystes,
entre E3,5 et E4,5. Il n'est toujours pas clair si la détermination de l'identité de ces cellules précède
(modèle de sorting out, Chazaud et al., 2006) ou est consécutive à leur exposition à la cavité
blastocoelique (Berenika Pluza, communication personnelle). Autrement dit, le moment exact où
elles acquièrent une identité distincte des futures cellules embryonnaires est pour l'instant mal
connu. Mais la fenêtre temporelle durant laquelle cela a lieu correspond à la réactivation du Xp dans
les cellules de la masse cellulaire interne. Il n'est donc pas exclu qu'il y ait réactivation, puis
64

inactivation de novo, toujours sur un mode empreinté. Des expériences de culture ex vivo de masse
cellulaire interne semblent a priori exclure un tel scénario (Okamoto et al., 2004). Ainsi, après 24 h
de culture de masses cellulaires internes, prélevées à E3,5 et débarrassées du trophectoderme par
« chirurgie immune » (immunosurgery), les cellules qui sont sorties de la masse cellulaire interne,
dont on pense qu'elles ont une identité d'endoderme primitif, ont toute un domaine de d'ARN Xist,
suggérant qu'elles n'auraient pas réactivé leur Xp. Néanmoins, seule une véritable étude de lignage,
avec une cinétique précise, permettrait de confirmer une telle conclusion.
Lorsque l'endoderme primitif est formé, Eed, Enx1 et H3K27me3 y colocalisent avec le Xp
inactif (Mak et al., 2004). Ces marques sont cependant perdues dans l'endoderme viscéral (qui
dérive de l'endoderme primitif) (Kalantry et al., 2006) et lors de la dérivation de cellules souches
(cellules XEN, pour extra-embryonic endoderm) à partir de l'endoderme primitif (Kalantry et al.,
2006; Kunath et al., 2004). Contrairement aux lignages trophoblastiques, l'absence d'Eed n'a pas
d'effet sur l'inactivation du Xp dans l'endoderme primitif ou les cellules qui en dérivent (Kalantry et
al., 2006), ce qui pourrait s'expliquer par un apport de protéines maternelles encore suffisant lorsque
Eed serait requis pour l'établissement d'une mémoire épigénétique. Celle-ci devrait également
pouvoir être maintenue, dans les cellules XEN, en la quasi-absence d'enrichissement de H3K9me2
sur le Xp (qui est par ailleurs hypoacétylé sur H3K9 et hypométhylé sur H3K4) (Kunath et al.,
2005). Cela conduit à s'interroger sur l'existence éventuelle de facteurs additionnels contribuant à la
stabilité de l'inactivation empreintée dans l'endoderme extraembryonnaire.
3.3.6 Nature de l'empreinte parentale
Une question fondamentale lorsque l'on parle d'inactivation empreinté est de comprendre quelle
est l'empreinte conditionnant l'inactivation, de manière stéréotypée, du chromosome X paternel.
Théoriquement, deux cas de figures sont possibles. Soit l'empreinte vient du père et prédispose à
l'inactivation. Soit elle vient de la mère et elle rend réfractaire à l'inactivation.
L'existence d'une empreinte maternelle a été démontrée pour la première fois grâce à
l'observation de disomies uniparentales. En effet, lorsque la disomie est de type XmXmXp ou
XmXmY, les deux chromosomes X maternels demeurent actif, causant la mort des embryons en
milieu de gestation. Au contraire, des souris disomiques de forme XmXpY ont leur Xp inactif et
sont viables (Goto et Takagi, 1998; Goto et Takagi, 1999). Il existerait donc des marques empêchant
l'inactivation du Xm. Quand sont-elles acquises? Vraisemblablement lors de la maturation des
ovocytes. Cela a été révélé par des expériences consistant à générer des embryons par combinaison
65

d'un ovocyte arrêté en début de méiose et d'un ovocyte mature. Dans ce cas, en effet, seul le
chromosome X provenant de l'ovocyte arrêté est inactivé, tandis que le chromosome X provenant de
l'ovocyte mature est toujours actif (Tada et al., 2000). On peut se demander si la mise en place d'une
telle empreinte dépend uniquement de la maturation ovocytaire ou est également conditionnée par
la présence, avant la méiose, de deux chromosomes X. Un élément de réponse peut être apporté par
une étude récente qui s'est intéressée à la mise en place de l'empreinte conditionnant l'expression
monoallélique de gènes autosomiques dont l'expression est soumise à l'empreinte parentale
(Durcova-Hills et al., 2006b). Les auteurs y ont fait un usage élégant de la possibilité d'avoir des
femelles de génotype XY, lorsque le gène Sry porté par le chromosome Y est délété. Ils concluent,
pour le gène Peg3, dont l'expression est réprimée sur le chromosome d'origine maternelle, que
l'empreinte maternelle est réalisée de manière similaire lors de la maturation ovocytaire que la mère
soit de génotype XX ou XYΔSry. Les modalités de mise en place sur le Xm de l'empreinte révélée par
Goto et Takagi pourraient être testées de la même manière, en mettant à profit l'existence de souris
XYΔSry, qui sont heureusement fertiles. Il est probable qu'un telle empreinte agisse en contrôlant le
gène Xist, car des études par transgenèse ont permis de la circonscrire à une région de 210 kb
incluant ce dernier (Okamoto et al., 2005).
Existe-t-il, en miroir de l'empreinte maternelle, une empreinte paternelle qui prédéterminerait le
Xp à l'inactivation? A priori, une telle idée semble exclue car des souris aneuploïdes XpO sont
viables, ce qui suppose que le Xp ait pu être maintenu actif (Papaioannou et West, 1981). De même,
des androgénotes XpXp sont viables, au moins juqu'au stade E7,5 (Okamoto et al., 2000). Dans ces
derrniers, l'expression d'un transgène LacZ lié au chromosome X informe que l'inactivation est
devenue aléatoire dans tous les tissus, ce qui traduit le fait que les deux chromosomes X sont
épigénétiquement équivalents. Toutefois, une analyse par RNA-FISH révèle qu'une majorité des
cellules aux stades 4 à 16 cellules, et une proportion non négligeable d'entre elles (10 à 20 %) au
stade blastocyste, ont formé des domaines de Xist de manière aberrante (c'est-à-dire deux domaines
dans les cellules XX et un domaine dans les cellules XY). C'est encore le cas pour une minorité de
cellules à E6,5. Vu sous cet angle, il y aurait bien une empreinte paternelle conditionnant à
l'expression de Xist, mais pouvant être compensée au cours du développement.
Comment une telle empreinte est-elle acquise? Probablement durant la spermatogenèse.
Cependant, elle n'est pas directement liée à la MSCI, car un transgène de 210 kb contenant Xist,
inséré en simple copie sur un autosome, est capable de réaliser toutes les étapes de l'inactivation
empreintée jusqu'au stade blastocyste, sans pour autant faire partie de la vésicule sexuelle, ni même
faire l'objet de MSUC (Okamoto et al., 2005). Quelles autres possibilités avons nous? Il a été
66

suggéré que l'expression de Xist sur le Xp pourrait être la conséquence de la déméthylation de son
promoteur (comme c'est le cas pour une proportion importante du génome) durant la
spermatogenèse (Norris et al. 1994). Dans le même ordre d'idées, elle pourrait tirer partie du
remaniement complet de la chromatine paternelle lorsque les protamines, qui en permettent
l'empaquetage compact dans les spermatozoïdes, sont remplacés par les histones (Aoki et al., 1997;
van der Heijden et al., 2005).
Dans la même étude de Durcova-Hills présentée plus haut (Durcova-Hills et al., 2006b), les
auteurs se sont également intéressés au gène H19, dont seul l'allèle d'origine maternelle est
normalement exprimé (Tada et al., 1998). L'absence d'expression de l'allèle paternel résulte de la
méthylation, durant la spermatogénèse, d'un centre d'empreinte proche de son promoteur (cf
discussion générale, section 2.2.1, p. 229). Contrairement au gène Peg3 dont l'empreinte ne dépend
pas de la nature des chromosomes sexuels (voir plus haut), le centre d'empreinte de H19 est méthylé
différemment dans la lignée germinale de mâles XY et XX (obtenus grâce à l'expression d'un
transgène Sry). Mais cette méthylation diffère également lorsqu'elle est acquise dans la lignée
germinale de mâles XX et de femelles XX. Ainsi, à la fois le processus de spermatogenèse et la
nature des chromosomes sexuels pourraient influer sur la mise en place d'une empreinte paternelle
pour H19. A condition, toutefois, de considérer que la spermatogenèse se déroule de la même
manière dans les mâles XY et XX.
Malheureusement, la même expérience n'est pour l'instant pas réalisable concernant
l'inactivation du chromosome X car les mâles XX sont stériles et nous ne connaissons pas, dans le
cas de l'inactivation, de marque d'empreinte pouvant être observée avant la fécondation. Il serait par
contre intéressant de regarder comment se comporte le Xp lorsque le Xm n'a pas pu acquérir
d'empreinte, car provenant d'un ovocyte non mature.
Nous avons vu que dans les embryons androgénotes, l'expression aberrante de Xist était
compensée au cours du développement et ne conduisait pas à des défauts notables avant
l'implantation (Okamoto et al., 2000). De même, en l'absence de Xist sur le Xp, l'embryon se
développe correctement jusqu'au stade blastocyste (Marahrens et al., 1997). Cependant, l'expression
monoallélique de Xist dès le stade 2 cellules, ainsi que la cinétique d'apparition des autres marques
sur le Xi, suggèrent fortement que Xist est bien requis pour la mise en place de l'inactivation
empreintée, dans le cadre du développement normal de l'embryon (Huynh et Lee, 2003; Mak et al.,
2004; Okamoto et al., 2004). Cela n'a pourtant jamais été démontré en établissant, dans l'embryon
pré-implantatoire, les mêmes cinétiques d'apparition des marques spécifiques du Xi dans un
contexte mutant pour Xist. Dans le cas où Xist serait bel et bien requis, l'absence de défaut apparent
67

des embryons avant le stade blastocyste, lorsque Xist est muté sur le Xp, nous conduirait à supposer
que, jusqu'à ce stade, les embryons seraient peu sensibles au dosage des gènes exprimés à partir du
chromosome X, dont le poids pourrait être atténué par la contribution des ARN et protéines
maternelles.
3.3.7 Inactivation soumise à l'empreinte parentale chez l'homme
Chez l'homme, plusieurs arguments tendent à exclure l'existence d'une inactivation empreintée,
bien qu'aucun d'entre eux ne soit définitif.
Ainsi, contrairement à la souris, XIST y serait exprimé aussi bien dans les embryons mâles que
femelles du stade 2 cellules au stade blastocyste (Daniels et al., 1997; Ray et al., 1997). Cela
contredit a priori l'idée qu'une inactivation empreintée pourrait être mise en place à ces stades, à
moins qu'elle ne soit indépendante de XIST, ou que les transcrits détectés par RT-PCR ne
correspondent au gène TSIX, antisens à XIST.
D'autre part, les femmes de génotype XXX et les hommes de génotype XXY (syndrome de
Klinefelter) se développent à peu près correctement, bien que dans la majorité des cas le X
surnuméraire soit d'origine maternelle (MacDonald et al., 1994). A l'inverse, les souris XmXmXp
ou XmXmY meurent en milieu de gestation, ce que l'on attribue à leur incapacité à inactiver le Xm
surnuméraire dans les tissus extraembryonnaires, du fait de l'empreinte maternelle (Goto et Takagi,
1998; Goto et Takagi, 1999). Par conséquent, l'inactivation dans les tissus extraembryonnaires ne
semble pas dépendre, chez l'homme, d'une empreinte maternelle.
Enfin, plusieurs études ont été publiées qui concluent, selon les auteurs, à l'inactivation aléatoire
dans les cellules trophoblastiques du placenta (Looijenga et al., 1999; Migeon, 1998; Migeon et Do,
1979; Zeng et Yankowitz, 2003), ou à l'inactivation préférentielle du Xp dans ces mêmes cellules
(Goto et al., 1997; Harrison, 1989; Harrison et Warburton, 1986; Ropers et al., 1978). Les
divergences entre les différents travaux peuvent avoir plusieurs origines, parmi lesquelles l'âge des
tissus examinés, la pureté du matériel biologique et la méthode utilisée pour mesurer l'inactivation.
Ainsi, en utilisant une même méthode d'analyse, consistant à caractériser des isozymes de glucose-
6-phosphate dehydrogenase (G6PD) différentes sur les deux chromosomes X, les travaux les plus
anciens concluaient à l'inactivation préférentielle du Xp lorsque les cellules utilisées provenaient de
placentas prélevés à terme (Harrison, 1989; Harrison et Warburton, 1986; Ropers et al., 1978), et au
contraire à une inactivation aléatoire lorsqu'elles étaient dérivées de villi chorioniques obtenus vers
68

3 mois de gestation (Migeon et Do, 1979). Un deuxième test mesurant la méthylation au promoteur
du gène codant pour le récepteur humain des androgènes (HAR), en utilisant un polymorphisme de
site de restriction entre les deux chromosomes X, donne quant à lui des résultats a priori
indépendants de l'âge des tissus, mais également contradictoires. Ainsi, dans des cellules obtenues à
partir de placentas prélevés à terme, Looijenga et al. observent trois configurations – inactivation
aléatoire, inactivation préférentielle du Xp et inactivation préférentielle du Xm – de manière
variable, y compris entre échantillons provenant d'un même placenta (Looijenga et al., 1999).
D'autre part, Goto et al. concluent, à partir de cellules dérivées de villi chorioniques vers 3 mois de
gestation, à l'inactivation préférentielle du Xp (Goto et al., 1997), tandis que Zeng et al. mesurent
une inactivation aléatoire dans le même type d'échantillon, mais qui pourrait avoir été purifié de
manière plus efficace, de manière à éviter la contamination par des cellules maternelles (Zeng et
Yankowitz, 2003). L'observation d'une inactivation préférentielle du chromosome X paternel
pourrait donc résulter, selon les cas, d'une contamination maternelle, ou de la sélection secondaire,
au cours du développement, de cellules ayant maintenu leur Xm actif.
Néanmoins, l'analyse d'un plus grand nombre de gènes du chromosome X devrait être réalisée
pour exclure définitivement l'idée d'une inactivation empreintée dans les tissus extraembryonnaires
chez l'homme. A ce sujet, il peut être intéressant de rappeler que, dans les tissus somatiques
humains, un grand nombre de gènes échappent à l'inactivation, avec une variabilité importante selon
les individus (cf introduction, partie 1, section 2.5, p. 50) (Anderson et Brown, 1999; Carrel et
Willard, 2005). Il est possible qu'une telle variabilité soit retrouvée dans les tissus
extraembryonnaires, d'autant plus que l'inactivation y semble établie de manière moins stable que
dans les cellules somatiques (Migeon et al., 2005). Si tel était le cas, cela pourrait conduire à
relativiser les conclusions ne s'appuyant que sur l'expression de G6PD ou de HAR.
3.4 Inactivation aléatoire dans l'embryon
3.4.1 Mise en place
Nous avons déjà détaillé les étapes de l'inactivation aléatoire, bien documentées (quoiqu'avec de
nombreuses zones d'ombres) en mettant à profit le modèle des cellules ES. Nous ne reviendrons pas
là dessus. Il s'agit plutôt, ici, de situer l'inactivation aléatoire dans son contexte initial, celui du
développement embryonnaire.
69

L'analyse de l'expression de transgènes insérés sur le chromosome X, exprimant soit la β-
galactosidase, soit la GFP, a permis de situer la réalisation de l'inactivation aléatoire dans la plupart
des cellules entre E5,5 et E6,5 dans le premier cas (Sugimoto et al., 2000), et entre E6,0 et E6,75
dans le second (Hadjantonakis et al., 2001). Toutefois, à E10,5, il semblerait que 12 % des cellules
embryonnaires femelles aient leur deux chromosome X actifs d'après des marquages cytogénétique
(Webb et al., 1992). Cela conduit à une vision de la mise en place de l'inactivation plus étalée dans
le temps, mais qui pourrait être contrastée selon les gènes.
A E5,5 Xist a commencé à être exprimé à partir du Xm, et l'on observe à nouveau l'accumulation
de Eed et de H3K27me3 sur l'un des deux chromosomes X (Mak et al., 2004). L'inactivation
aléatoire commence donc un peu avant E5,5, mais on ne sait pas si sa cinétique est la même pour le
Xp et le Xm. On peut à ce sujet remarquer que le principal intérêt de la GFP, qui est de pouvoir
réaliser des observations in vivo, en temps réel, n'a pour l'heure pas été véritablement exploité, sans
doute du fait de la difficulté à cultiver des embryons aux stades qui nous intéressent le plus, vers
E5,5. Les progrès réalisés dans ce domaine offrent des perspectives excitantes, en particulier pour
examiner comment s'articulent inactivation du chromosome X et développement embryonnaire. Par
exemple, la mise en place de l'inactivation aléatoire étant, à l'échelle de l'embryon, concomitante au
déroulement de la gastrulation, il serait intéressant de savoir s'il y a ou non couplage entre l'une et
l'autre. Si tel était le cas, comme la gastrulation procède de l'arrière vers l'avant de l'embryon, nous
devrions assister, de manière coordonnée, à une vague d'inactivation que l'on pourrait visualiser par
l'extinction d'un transgène GFP sur le Xm. Remarquons qu'à ces stades, la comparaison des
cinétiques citées ci-dessus (Hadjantonakis et al., 2001; Mak et al., 2004) situe la demi-vie de la
GFP vraisemblablement aux environs de 12 h, ce qui serait suffisamment faible pour qu'une telle
observation soit réalisable.
Nous avons vu dans le cas des lignages trophoblastiques que la possession ou non de propriétes
de cellules souches était corrélée à des modalités différentes d'inactivation empreintée (cf
introduction, partie 1, section 3.3.4, p. 63). De la même manière, on peut supposer dans le cas de
l'inactivation aléatoires que les modalités de mise en place ou de maintien de l'inactivation diffèrent
en fonction des types ou lignages cellulaires. Un premier exemple en est donné par les cellules de la
notochorde, dans lesquelles l'activité des deux chromosomes X est observée jusqu'à E9,5 (Sugimoto
et al., 2000; Tam et al., 1994a). L'utilisation de marqueurs de lignage permettra peut-être de trouver
de nouveaux exemples du même type, dont on peut supposer qu'ils sont assez nombreux, compte-
tenu de l'étalement dans le temps de la mise en place de l'inactivation.
70

3.4.2 Compétence du chromosome X à être reprogrammé
Une autre manière de poser la question de la mise en place de l'inactivation au cours du
développement revient à s'interroger non plus sur le statut – actif ou inactif – des chromosomes X,
mais plutôt sur leur compétence ou non à être inactivé. On retrouve ici la notion de fenêtre de
compétence, discutée à la section 2.3.2 de la première partie de l'introduction (p. 45). L'observation
dans l'embryon des conséquences de l'expression ectopique d'un transgène inductible de Xist inséré
sur l'un des deux chromosomes X, par ailleurs dépourvu du gène Xist endogène, a permis de
positionner la perte d'une telle compétence entre E9,5 et E12,5 (Savarese et al., 2006). Cette
conclusion est tout au moins applicable à une population de cellules dont la disparition, consécutive
à l'inactivation des deux chromosomes X lorsque le transgène est exprimé, ne peut pas être
compensée dans l'embryon.
Dans l'individu adulte, la compétence à être inactivé réapparaît cependant dans certains types
cellulaire. C'est le cas des lymphocytes pré-B et pré-T, dans lesquels, bien que la compensation de
dose soit correctement maintenue, le Xa redevient compétent pour être inactivé en réponse à
l'expression en cis d'un transgène inductible de Xist (Savarese et al., 2006). C'est précisément à ce
moment de la maturation en lymphocytes B et T qu'on lieu des réarrangements aux loci Igh et
TCRβ, respectivement, aboutissant à la mise en place de l'exclusion allélique (Bassing et al., 2002;
Goldmit et al., 2005). L'acquisition d'une compétence nouvelle du chromosome X à être inactivé
pourrait donc n'être alors qu'une conséquence secondaire de la reprogrammation épigénétique
rendant possible de tels réarrangements.
Les expériences de transfert nucléaire se sont révélées être un autre moyen de tester la
compétence des chromosomes X à être reprogrammés lors du développement embryonnaire. Les
embryons créés par transfert du noyau de cellules somatiques dans des ovocytes connaissent une
inactivation aléatoire, témoignant de l'effacement des marques épigénétiques acquises auparavant
(Eggan et al., 2000; Nolen et al., 2005). Cet effacement a probablement lieu dans la masse
cellulaire interne des blastocystes, car la triméthylation de H3K27 associée au Xi de la cellule
donneuse est maintenue sans discontinuité avant ce stade (Bao et al., 2005). A l'inverse de
l'embryon strico sensus, dans le placenta, le Xi correspond en général au Xi de la cellule donneuse
(Eggan et al., 2000; Nolen et al., 2005). Il est tentant de penser que l'empreinte permettant cela
serait constituée, au moins en partie, par la triméthylation de H3K27. En effet, contrairement à
71

celle-ci, l'expression de Xist est initialement perdue, après le transfert nucléaire, puis réalisée de
manière aberrante à partir du stade 8 cellules, et n'est pas toujours corrélée à l'expression des gènes
du chromosome X (Nolen et al., 2005).
Nous avions avancé plus haut (cf introduction, partie 1, section 3.3.6, p. 65) que l'empreinte
parentale était vraisemblablement réalisée grâce à l'expression monoallélique de Xist. Si cela est
exact, alors l'empreinte mise en place lors d'un transfert nucléaire n'est pas équivalente à l'empreinte
parentale. Cependant, il n'est pas exclu que la reprogrammation des chromosomes X puisse être
réalisée de manière sensiblement différente après transfert de noyaux provenant d'autres types
cellulaires que ceux utilisés jusqu'à présent.
3.5 Réactivation du chromosome X inactif dans la lignée germinale
Au cours du développement normal de l'embryon, le Xi est réactivé dans les cellules germinales
primordiales (PGC, pour Primordial Germ Cells), à l'origine des gamètes. C'est d'ailleurs,
probablement, un prérequis pour pouvoir produire une empreinte maternelle durant la maturation
des ovocytes, sans entrer en conflit avec les marques déposées durant l'inactivation aléatoire,
également capables de produire une mémoire épigénétique.
Les PGC sont spécifiées à E7,25 dans le mésoderme extraembryonnaire (d'origine épiblastique)
(Ginsburg et al., 1990), avant de migrer dans l'endoderme du tube digestif postérieur à partie de
E8,5, pour finalement coloniser les gouttières génitales entre E10,5 et E11,5 (pour revue, McLaren,
2003). Les PGC provenant des mâles ou des femelles ont la même capacité à se différencier en
gamètes mâles ou femelles avant E13,5 (McLaren et Southee, 1997). C'est à ce stade que les PGC
femelles adoptent un comportement différent des PGC mâles, en entrant en prophase I de méiose.
Les cellules germinales mâles s'arrêteront quant à elles en mitose à E14,5 (McLaren, 1983).
Lorsqu'elles colonisent les gouttières génitales et cessent de migrer, les PGC connaissent
d'importants changements dans leur morphologie et leurs propriétés d'adhésion (De Felici et al.,
1992; Donovan et al., 1986; Garcia-Castro et al., 1997), commencent à perdre leur pluripotence et
leur capacité à proliférer (Matsui et al., 1992; McLaren, 1984; Resnick et al., 1992), et un nombre
important d'entre elles s'engagent dans l'apoptose (Coucouvanis et al., 1993; Wang et al., 1998). Un
faisceau d'observations converge pour situer la réactivation du Xi après la colonisation des
gouttières génitales, mais avant l'entrée en méiose, en se référant à l'expression du gène Hprt
72

(Monk et McLaren, 1981), d'un transgène LacZ (Tam et al., 1994b) et d'un transgène GFP (Chuva
de Susa Lopes, communication orale, 2006), tous situés sur le chromosome X, ainsi qu'à la
disparition des domaines de Xist (Nesterova et al., 2002).
Des résultats récents, cependant, suggèrent que la réactivation du Xi pourrait être initiée plus
tôt, avant la migration des PGC. Certains gènes du chromosome X commenceraient alors à être
exprimés de manière biallélique, tandis que l'accumulation de Xist sur le Xi serait progressivement
perdue (Sugimoto et Abe, communication orale, 2006). Ces observations sont en fait cohérentes
avec l'occurence d'importants remaniements épigénétiques intervenant dans la même fenêtre de
temps (Seki et al., 2005). Ainsi, à E8,0, le niveau global de méthylation de l'ADN et de H3K9me2
diminue, précédé par une perte transitoires des méthylase de l'ADN dans le noyau. A E8,5-E9,0, le
niveau global de H3K27me3 augmente fortement, probablement pour préserver la fonction
répressive associée aux deux marques précédentes, tout en permettant une plus grande plasticité
qu'avec H3K9me2. Enfin, lorsqu'a lieu la colonisation des gouttières génitales à E10,5, une
augmentation transitoire de la méthylation de H3K4 et de l'acétylation de H3K9, qui sont des
marques permissives à la transcription, est observée (Seki et al., 2005). A ce stade macroH2A1.2
n'est pas associé au Xi (Nesterova et al., 2002). Il semble donc que la réactivation progressive et
partielle du Xi puisse être reliée à la mise en place d'un état épigénétique global plus plastique, si ce
n'est permissif à la transcription. Cela pourrait contribuer à créer une sorte d'état métastable de
l'inactivation, qui ne basculerait complètement qu'après avoir reçu des signaux de différenciation
lors de l'entrée dans les futures gonades. Il serait intéressant de savoir, dans ce contexte, si la
réactivation du Xi procède en bloc, en remaniant en même temps la chromatine sur toute la
longueur du chromosome, ou si des gènes clés tels que Xist sont affectés en premier et dirigent le
processus.
3.6 Modèles cellulaires
Nous avions, dans le chapitre précédant, discuté de données relatives à l'épigénétique de
l'inactivation aléatoire, essentiellement obtenues en utilisant le modèle des cellules ES. Dans ce
chapitre, nous avons également été amené à évoquer des observations faites dans des modèles
cellulaires des tissus extraembryonnaires, en complément des données obtenues dans l'embryon. Il
peut être utile, à ce point de notre exposé, de faire un bref inventaire des modèles cellulaires
disponibles pour étudier ex vivo (ou in vitro, selon le point de vue), les différentes facettes de
l'inactivation:
73

– les cellules trophoblastiques souches (TS, pour trophoblastic stem) sont dérivées du
trophoblaste polaire des blastocystes à E3,5 ou de l'ectoderme extraembryonnaire à E6,5
(Tanaka et al., 1998); elles maintiennent l'inactivation empreintée acquise plus tôt durant le
développement, mais les modalité de ce maintien diffèrent selon qu'elles sont cultivées de
manière à préserver leurs caractéristiques de cellules souches ou que l'on induit leur
différenciation (cf introduction, partie 1, section 3.3.4, p. 63);
– les cellules souche extraembryonnaires endodermiques (XEN, pour extra-embryonary
endoderm) sont dérivées de la masse cellulaire interne de blastocystes à E3,5 et expriment à
la fois des marqueurs d'endoderme viscéral et d'endoderme pariétal (Kunath et al., 2005);
l'inactivation est réalisée de manière empreintée dans ces cellules, de manière sensiblement
différente des lignages trophoblastiques (cf introduction, partie 1, section 3.3.5, p. 64);
– les cellules souches embryonnaires (ES, pour embryonic stem) sont dérivées de la masse
cellulaire interne de blastocystes à E3,5 (Evans et Kaufman, 1981; Martin, 1981) et sont
probablement repésentatives des cellules de l'épiblaste vers E5,0; les deux chromosomes X
sont actifs lorsqu'elles sont cultivées de manière à préserver leur pluripotence, mais l'un d'eux
est inactivé lorsqu'on induit leur différenciation (cf intoduction, partie 1, chapitre 2, p. 23);
– les cellules germinales embryonnaires (EGC, pour embryonary germ cells) sont dérivées par
dé-différenciation des PGC prélevés entre E8,5 et E11,5 (Durcova-Hills et al., 2006a; Matsui
et al., 1992; Resnick et al., 1992) et leur profil d'expression génique est pratiquement
identique à celui des cellules ES (Mise, communication orale, 2006), leurs deux
chromosomes X étant par ailleurs actif (Stewart et al., 1994); il ne s'agit donc
vraisemblablement pas d'un modèle de choix pour étudier les PGC;
– les PGC dérivées in vitro à partir de cellules ES (Geijsen et al., 2004) et les cellules
germinales (GC, pour germline cells) mâles dérivées à partir de testicules néonatales
(Kanatsu-Shinohara et al., 2004) ont un profil d'expression génique plus proches de celui des
PGC in vivo (Mise, communication orale, 2006) et pourraient donc être de meilleurs modèles
cellulaires pour étudier la réactivation du Xi dans ces dernières.
Deux préoccupations constantes, auxquelles il est en général difficile de répondre, font jour
lorsqu'on fait appel à des modèles cellulaires pour mieux comprendre des processus
développementaux. Tout d'abord, les modèles cellulaires sont dérivés de lignages bien réels in vivo,
mais on ne sait pas à quel point le processus de dérivation en lui-même ou le maintien en culture
pendant des périodes prolongées les éloignent de leur lignage d'origine. L'exemple des EGC est
74

assez frappant, car elles ont plus de points en commun avec les cellules ES alors qu'elles dérivent de
PGC, mais à un moment où ces dernières acquièrent une certaine plasticité épigénétique (cf
introduction, partie 1, section 3.5, p. 72) permettant probablement leur dé-différenciation.
La deuxième difficulté vient de ce que le principal intérêt de ces modèles cellulaires tient à leur
capacité à récapituler in vitro certains processus développementaux. En général, cela a lieu en
différenciant les cellules, et le protocole employé pour cela n'est pertinent qu'au regard de la
question posée. Dans le cas des cellules ES, deux méthodes sont utilisés couramment, par de
nombreux laboratoires: la formation de corps embryonnaires (EB, pour embryoid bodies) et
l'induction par l'acide rétinoïque. Une étude comparative de ces deux méthodes au regard de la mise
en place de l'inactivation a été publiée (Farivar et al., 2004). En bref, les auteurs montrent que
l'inactivation de l'un des deux chromosomes X dans des cellules ES femelles est plus efficace dans
des EB, surtout lorsque ceux-ci sont réattachés puis laissés pousser en conditions adhérentes. Un
troisième mode de différenciation, qualifié de différenciation par dilution, consiste simplement à
laisser pousser les cellules ES ensemencées avec une faible densité cellulaire, dans un milieu ne
permettant pas de maintenir leur pluripotence, mais sans induction par une molécule particulière
(Morey et al., 2001). Une lecture attentive des protocoles mis en oeuvre par les différents
laboratoire révèle des différences de composition de milieu de culture ou de densité cellulaire, qui
ont des conséquences parfois importantes en fonction des lignées de cellules ES utilisées. Dans nos
mains, l'emploi de l'acide rétinoïque est relativement efficace (plus que pour Farivar et al.), avec en
moyenne 60 % des cellules ES femelles ayant effectivement inactivé l'un de leurs chromosomes X.
La différenciation par dilution l'est également, mais avec une variabilité plus grande selon les
expériences.
Toutefois, la proportion finale de cellules ayant inactivé un chromosome X n'est pas le seul
critère. Un système de différenciation pourrait par exemple permettre la prolifération préférentielle
d'un type cellulaire donné. A première vue, l'inactivation aléatoire est réalisée de manière assez
homogène (à quelques exceptions près) dans les différents lignages dérivés de l'épiblaste. Mais dans
le détail, personne ne le sait vraiment. Précisément, différencier des cellules ES vers des types
cellulaires bien déterminés serait un bon modèle pour évaluer cela. Une illustration de ce problème
est donnée par l'association du Xi à la modification d'histone H3K27me3. Lors de la différenciation
en EB de cellules ES, elle n'est observée que transitoirement (Plath et al., 2003). Au contraire,
H3K27me3 est généralement enrichi sur le Xi dans les tissus adultes, bien que cela ne soit pas
homogène entre les cellules d'une même population et que au moins un type cellulaire fasse
exception (Bao et al., 2005; Gilbert et al., 2003; Rougeulle et al., 2004). Il serait intéressant de
75

savoir si l'observation réalisée dans les cellules ES et tout à fait étrangère à ce qui a lieu in vivo,
auquel cas les cellules ES ne devraient pas être utilisées dans des différenciations de longue durée,
ou si elle est l'illustration du comportement habituel de certains types cellulaires.
A l'étude de l'initiation de l'inactivation correspond une autre exigence: faire en sorte que toutes
les cellules se comportent de la même manière au même moment, pour être en mesure d'observer
des évènements transitoires. Selon ce critère, la différenciation induite par l'acide rétinoïque est
idéale, car elle a lieu rapidement et de manière pratiquement synchrone, comme en témoigne la
perte rapide d'expression de marqueurs de cellules non différenciées (Vigneau et al., 2006). C'est
moins le cas pour les EB, car des cellules indifférenciées échappant à l'inactivation restent pendant
très longtemps présentes au centre de ces derniers (Farivar et al., 2004).
Les considérations exposées ici expliquent la préférence accordée, au cours du travail de thèse, à
la différenciation induite par l'acide rétinoïque. Nous allons bientôt voir pourquoi.
76

Partie 2 Le centre d'inactivation du chromosome X
Chapitre 1 Fonctions et caractérisation du centre d'inactivation
1.1 Comptage, choix et initiation en cis de l'inactivation
1.1.1 Comptage
Nous avons déjà introduit le concept de comptage à la section 1.3.1 de la première partie de
l'intoduction (p. 14). C'était en fait un élément constitutif de la définition de l'inactivation aléatoire
du chromosome X, telle qu'elle avait été proposée par Mary Lyon (Lyon, 1962). En bref, il s'agit de
dire que le nombre de chromosomes X actifs dépend à la fois du nombre total de chromosomes X et
du nombre d'autosomes, de telle sorte que l'un des deux chromosomes X soit inactif chez les
femelles (AA:XX), mais que l'unique chromosome X reste toujours actif chez les mâles (AA:XY).
Plutôt que de faire appel à un comptage des chromosomes X, on aurait pu imaginer que la
capacité à mettre en place l'inactivation était liée au sexe proprement dit. En d'autres termes, que les
femelles auraient été compétentes pour l'inactivation, mais pas les mâles. Plusieurs observations
indiquaient que cela était peu probable. Par exemple, le genotype XO correspond à des individus
femelles dont l'unique chromosome X est actif (Welshons et Russell, 1959). D'autre part, des
individus possédant un complément diploïde d'autosomes et un nombre variable de chromosomes X
(i.e. AA:XXY; AA:XXXX; AA:XXXXY) ont toujours, et indépendamment de leur sexe, un seul
chromosome X actif (Barr, 1963; Giannelli, 1963; Grumbach et al., 1963). Le nombre de
chromosomes X est donc déterminant, contrairement au sexe. Cela semble finalement assez
logique, car on sait désormais que le déterminisme sexuel est réalisé essentiellement en fonction de
l'expression du gène Sry lors de la formation des gonades (Lovell-Badge et Robertson, 1990;
Schmahl et al., 2000), c'est-à-dire bien après l'établissement de l'inactivation aléatoire. Donc, si
77

différence liée au sexe il y a, soit elle est rattachée à un mécanisme encore inconnu de déterminisme
sexuel précédent la mise en place de l'inactivation aléatoire, soit elle mesure en réalité le nombre de
chromosomes X, ce qui revient à parler de comptage.
Selon quelle règle chromosomes X et autosomes sont-il comptés? Dans des cellules somatiques
adultes tetraploïdes femelles (AAAA:XXXX), deux des chromosomes X sont inactivés. Par ailleurs,
pour un génotype AA:XXXX, trois chromosomes X sont inactifs dans chaque cellule. En tenant
également compte des exemples mentionnés au paragraphe précédent, la règle de comptage peut
donc être énoncée ainsi: nombre de X actif/nombre d'autosomes = 1/2 (Rastan, 1983). Autrement
dit, tous les chromosomes X sont inactivés, sauf un seul par complément diploïde d'autosomes. Les
bases moléculaires d'un tel comptage ne sont toujours pas connues, mais les travaux que nous allons
présenter dans les chapitres suivants, ainsi que le travail de thèse, ont permis d'en caractériser
certains acteurs. Un modèle minimal communément admis consiste à faire l'hypothèse d'un facteur
« bloquant » produit en quantité limitante par les autosomes: la quantité de ce facteur produite par
un complément diploïde d'autosomes serait juste suffisante pour empêcher l'inactivation d'un seul
chromosome X. Comme nous le discuterons plus loin, ce modèle n'est pas suffisant pour intégrer
certaines données expérimentales récentes, ce qui a conduit à le compléter, ou à proposer des
modèles alternatifs. En particulier, certaines observations, détaillées au chapitre 2 de la seconde
partie de l'introduction (p. 95), ont conduit à proposer l'existence d'un second facteur, dit de
« compétence », produit par chaque chromosome X (Lee, 2005) (fig. 12, p. 79). Dans ce modèle,
une dose de facteur de compétence, correspondant à la quantité produite par un seul chromosome X,
serait titrée par la quantité de facteur bloquant produite par un complément diploïde d'autosomes.
Comme dans le modèle à un seul facteur bloquant, le complexe ainsi formé par l'association d'une
dose de facteur de compétence à deux doses de facteurs bloquant empêcherait l'inactivation de se
produire sur un chromosome X par complément diploïde d'autosomes. Toutefois, pour qu'un
chromosome X puisse être inactivé, l'absence d'association avec le facteur bloquant ne serait pas
suffisante. Au contraire, la fixation d'une dose, non titrée, de facteur de compétence serait également
indispensable. Comme cette seconde dose serait présente dans les femelles (XX) mais pas dans les
mâles (XY), ces derniers ne devraient donc pas avoir la capacité de réaliser l'inactivation. Nous
montrerons plus loin (résultats, chapitre , p. 131) que les mâles ont bien cette capacité, ce qui limite
la nécessité d'un tel facteur bloquant.
Postuler l'existence d'un facteur bloquant, ou d'un facteur de compétence, soulève deux
interrogations. En premier lieu, sur la nature de ces facteurs. En second lieu, sur la nature de leur(s)
cible(s).
78

La nature de tels facteurs est problématique. Si l'on prend l'exemple du facteur bloquant, il ne
devrait en exister qu'une seule copie par complément diploïde d'autosome, pour pouvoir ne se fixer
que sur un seul chromosome X. Cependant, il est difficile d'imaginer qu'il s'agisse d'une molécule
unique, produite en un seul exemplaire, car dans ce cas, la production en serait vraisemblablement
aléatoire, il elle courrait des risques importants d'être dégradée avant d'atteindre sa cible. Pour
contourner ces écueils, un modèle de brisure de symétrie a été proposé récemment par Nicodemi et
Prisco (Nicodemi et Prisco, 2007) (fig. 13, p. 80). Selon ce modèle, le facteur bloquant n'aurait pas
d'existence propre mais serait représenté par un ensemble de molécules diffusibles produites par les
autosomes. L'attachement de ces molécules entre elles et au chromosome X devrait être possible et
énergétiquement favorable. Dans ce contexte, une rupture de symétrie a lieu spontanément et
conduit à la coalescence de l'ensemble des molécules sur un seul chromosome X, avec une
cinétique qui dépend de la concentration de ces molécules et de l'avantage énergétique procuré par
cette coalescence. Un tel modèle permet d'expliquer le comportement de cellules aneuploïdes, dans
la mesure où la concentration des molécules « bloquantes » varie avec le nombre d'autosomes
capables de les produire. Il intègre également un paramètre lié à l'affinité des molécules diffusibles
79
Fig. 12: Modèle à deux facteurs proposé par J. Lee (Lee, 2005)
(A) Dans le modèle à deux facteurs, le « comptage » représente la titration de facteurs autosomiques (facteurs A) et de facteurs liés aux chromosomes X (facteurs X) pour former un facteur bloquant (BF, pour blocking factor). Les facteurs X non titrés deviennent des facteurs des compétence (CF, pour competence factor). Les facteurs X pourraient être des facteurs diffusibles ou des éléments localisés en cis, sur le chromosome X. Le « choix » reflète la fixation du BF sur le futur Xa et la fixation du CF sur le futur Xi, de manière mutuellement exclusive. (B) En l'absence de sites de fixation pour le CF et le BF sur les deux chromosomes X (par exemple suite à la délétion d'un élément génomique régulant le comptage et le choix), quatre configurations sont obtenues dont deux seulement seraient viables. (C) Lorsque des sites de fixation des CF et BF sont insérés par transgenèse sur un autosome, ce dernier peut entrer en compétition avec les sites de fixation endogènes sur le chromosome X, et interférer ainsi avec le comptage et le choix.

80
Fig. 13: Modèle de brisure de symétrie proposé par M. Nicodemi (d'après Nicodemi et Prisco, 2007)
À gauche: Évolution du système de molécules bloquantes autour de deux chromosomes avec lesquels elles ont la même affinité, pour des énergies d'interaction des molécules entre elles de E0 = 2,4kT (à gauche) et E0 = 0 (à droite). La configuration à t = 0 est déterminée de manière aléatoire et est identique dans les deux cas. Lorsque E0 = 0, la diffusion des molécules suit un chemin aléatoire. Lorsque E0 = 2,4kT, les particules commencent à former de petits agrégats, puis finissent par se regrouper sur un seul chromosome, brisant ainsi la symétrie entre les deux chromosomes.À droite: La concentration moyenne des molécules bloquantes autour du chromosome de gauche, ρl, et du chromosome de droite, ρr, est donnée en fonction du temps sur une échelle logarithmique. Les cercles correspondent au cas où E0 = 2,4kT, et les carrés au cas où E0 = 0 (les symboles sont pleins pour ρl, et vides pour ρr). Si E0 = 0, la symétrie entre les deux chromosomes est préservée et ρl(t) = ρr (t). Quand E0 / kT = 2,4, la symétrie est cassée. Après une période transitoire, ρl s'accroît d'un degré de magnitude par rapport au point de départ et ρr → 0. Encart à droite: Diagramme de phase du système, calculé par simulations, est montré sur le plan énergie d'interactionconcentration (E0, c). La courbe décrit la limite de transition de phase. Encart à gauche: Temps τ mis pour assembler les molécules bloquantes en fonction de la distance L entre deux segments du chromosome X, dans un parallélépipède de dimensions Lx = l0, Ly = l0/2, et Lz = l0/2 (avec l0 = 64d0, Ex =E0 = 2,4kT, et c = 25 × 103). En particulier, le ratio (L)/ τ τ16 est montré, dans lequel τ16 est la valeur de référence pour L = 16d0, qui est la distance utilisée par les auteurs dans leur article. Le tracé a pour équation τ = a + bL².

à leur cible. Comme le suggèrent ses auteurs, le modèle de brisure de symétrie pourrait être
aisément étendu à un plus grand nombre de facteurs dont, en particulier, le facteur de compétence
proposé par J. Lee (Lee, 2005).
La recherche de cibles du facteur bloquant, ou plus généralement de facteurs de comptage, a
conduit à un important travail de dissection génétique d'une région particulière du chromosome X,
le centre d'inactivation du chromosome X (Xic chez la souris, et XIC chez l'homme, pour X-
chromosome inactivation center). Nous montrerons à la section 1.2 de la seconde partie de
l'introduction (p. 88) comment cette région, qui contrôle les différents aspects de l'initiation de
l'inactivation, a été caractérisée. Puis, à partir du chapitre 2 de la seconde partie de l'introduction
(p. 95), nous détaillerons l'étude fonctionnelle des éléments qui la constituent. La recherche de
cibles de facteurs de comptage peut être abordée sous deux angles. Une approche génétique définira
une telle cible comme un élément génomique, avec un rôle proéminent attribué à sa séquence et,
éventuellement, à sa position dans le génome. Une autre manière d'aborder cette question est de
considérer a priori que les facteurs de comptage ne se fixent pas à proprement parler à l'ADN mais
à la chromatine, et que les marques épigénétiques associées à cette dernière peuvent moduler son
affinité avec les facteurs de comptage. L'accent sera alors mis sur la caractérisation du profil
épigénétique particulier permettant leur fixation. Bien sûr, les deux approches ne sont pas
exclusives, mais au contraire s'enrichissent mutuellement. Les résultats obtenus durant cette thèse
en sont une illustration.
La cinétique avec laquelle est réalisé le comptage est un aspect important pour pouvoir en
comprendre les mécanismes. En particulier, il importe de savoir à quel moment le comptage est
réalisé, respectivement à l'initiation de l'inactivation. Comme il n'y a jamais eu de description de
cellule femelle sauvage AA:XX inactivant transitoirement ses deux chromosomes X, ni de cellule
mâle sauvage AA:XY inactivant transitoirement son unique chromosome X, il y a tout lieu de
penser que le comptage est un processus antérieur à l'initiation de l'inactivation. D'autre part, il peut
être qualifié de primaire car il n'implique pas la contre-sélection d'une population de cellules ayant
réalisé l'inactivation de manière aberrante. Toutefois, des expériences de fusion de cellules ES
apportent un éclairage différent. Ces expériences consistent à générer des cellules tétraploïdes de
génotype AAAA:XXXX, puis à induire leur différenciation in vitro et suivre la cinétique
d'inactivation de leurs chromosomes X. Initialement, toutes les configurations, de 0 à 4 Xi, sont
observées dans la population cellulaire en cours de différenciation et ce n'est que secondairement
que la configuration à 2 Xi, conforme à la règle de comptage, éclipse toutes les autres (Gribnau et
al, communication orale, 2006; Dubois et al, communication personnelle). Une telle observation
81

peut être expliquée de deux manières. Les cellules ne respectant par la règle de comptage pourraient
être contre-sélectionnées. Cela reviendrait à dire que le mécanisme habituel de comptage n'est pas
capable de gérer un génotype tetraploïde, et que les configurations aberrantes sont éliminées
secondairement par un processus que l'on pourrait qualifier de comptage secondaire. Une
explications alternative serait que le processus de comptage primaire puisse être réalisé tardivement
et conduise en particulier à la réversion de l'inactivation lorsqu'elle est réalisée en désaccord avec la
règle de comptage. Dans ce cas, cela signifierait que le mécanisme de comptage puisse être
opérationnel après la formation du domaine de Xist, ce qui pourrait être relié à la notion de fenêtre
d'initiation de l'inactivation proposée par A. Wutz (cf introduction, partie 1, section 2.3.2, p. 45)
(Wutz et Jaenisch, 2000). Il devrait être possible de discriminer entre ces deux possibilités –
comptage secondaire et comptage primaire tardif – en mesurant la mortalité cellulaire, qui devrait
être accrue dans le premier cas mais pas dans le second.
La notion de comptage a été essentiellement définie d'après l'observation de l'inactivation dans
les tissus somatiques. Mais peut-on parler de comptage dans le cas de l'inactivation empreintée? Les
conditions dans lesquelles est réalisée l'inactivation empreintée ont été développées à la section 3.3
de la première partie de l'introduction (p. 59). Nous avions présenté le cas des disomies
uniparentales de génotype XmXmXp ou XmXmY, pour lesquelles il semble que l'empreinte
maternelle prédomine sur un éventuel comptage, tous les Xm étant maintenus actifs. Au contraire,
dans des androgénotes XpXp, seulement 10 à 20 % des cellules ont un schéma d'inactivation
aberrant au stade blastocyste, ce qui implique que dans la majorité des cas un comptage correct
aurait pu être réalisé malgré l'empreinte paternelle. Il apparaît donc que le comptage ne serait
opérationnel que lorsque le devenir des chromosomes X n'est pas prédéterminé de manière forte par
des marques épigénétiques, ce qui est a priori le cas après la réactivation du Xp dans la masse
cellulaire interne des blastocystes.
La capacité à réaliser l'inactivation, et en particulier le comptage, de manière correcte, pourrait
donc être un bon indicateur de l'efficacité avec laquelle des cellule somatiques peuvent être
reprogrammées en cellules pluripotentes, ce que plusieurs techniques rendent désormais possibles.
Nous avions parlé à la section 3.4.2 de la première partie de l'introduction (p. 71) des expériences
de transfert nucléaire, dans lesquelles le Xi de la cellule donneuse est réactivé dans la masse
cellulaire interne des blastocystes, et participe correctement à l'inactivation aléatoire. Il est
également possible de reprogrammer directement des cellules somatiques différenciées en les
fusionnant avec des cellules ES ((Cowan et al., 2005; Do et Scholer, 2004; Tada et al., 2001);
Eggan.communication orale, 2006) ou par expression ectopique des gènes Oct3/4, Sox 2, c-Myc et
82

Klf4 (Maherali et al., 2007; Okita et al., 2007; Takahashi et Yamanaka, 2006; Wernig et al., 2007).
En particulier, lorsque des thymocytes XX sont fusionnés avec des cellules ES, leur Xi est réactivé
(Tada et al., 2001). Il serait intéressant de savoir si, lorsqu'elles sont différenciées in vitro, de telles
cellules chimériques peuvent réaliser l'inactivation de leurs chromosomes X en réalisant le
comptage de la même manière que des cellules ES ou que des cellules tétraploïdes obtenues par
fusion de cellules ES (voir plus haut). Dans des fibroblastes XX reprogrammés par l'expression
ectopique de Oct3/4, Sox 2, c-Myc et Klf4, le Xi est également réactivé et, lorsqu'elles sont
différenciées in vitro, de telles cellules, appelées iPS (pour induced pluripotent stem cells) semblent
réaliser l'inactivation aléatoire de la même manière que les cellules ES (Maherali et al., 2007).
Néanmoins, une cinétique plus précise de la mise en place de l'inactivation dans de telles cellules
reprogrammées devrait être réalisée pour déterminer si le comptage est primaire et antérieur à la
formation du domaine de Xist, comme dans les cellules ES, ou est réalisé de manière secondaire, ce
qui dénoterait un caractère partiel de la reprogrammation. Quoiqu'il en soit, aussi bien dans les
embryons obtenus par transfert nucléaire que dans les iPS différenciées in vitro, les deux
chromosomes X sont inactivés de manière équiprobable (Eggan et al., 2000; Maherali et al., 2007;
Nolen et al., 2005), indiquant que les marques précédemment associées au Xi ont été effacées (ce
type de données n'est pas disponible dans le cas des expériences de fusion). En d'autres termes, le
choix du chromosome X à inactiver a pu être réalisé de manière aléatoire. Cette notion de choix est
détaillée dans la section suivante.
1.1.2 Choix
Une fois déterminé que l'inactivation doit avoir lieu, et sur combien de chromosomes, il importe
de décider lesquels. C'est ce que nous qualifions de choix. La nécessité d'un choix pourrait paraître
superflue. En effet, si dans une cellule femelle un chromosome X initie, de manière aléatoire, le
processus d'inactivation et que le comptage fonctionne correctement, alors le second chromosome X
n'aura d'autre choix que de rester actif.
Précisément, la notion de choix s'est imposée car l'inactivation n'est pas toujours parfaitement
aléatoire. Au contraire, elle est souvent biaisée en faveur de l'un ou l'autre des deux chromosomes
X. Un tel biais peut être relié de manière directe à l'initiation de l'inactivation, auquel cas on parlera
de choix primaire. Il peut également apparaître secondairement, par contre-sélection de cellules
ayant l'un de leurs chromosomes X inactif. On parlera alors de choix secondaire. A l'inverse, il est
difficile d'utiliser la notion de choix concernant l'inactivation empreintée, car elle est réalisée de
83

manière stéréotypée, sans exception. Dès lors, toute altération du biais total en faveur de
l'inactivation du Xp sera plutôt attribuée à un défaut d'empreinte parentale, dont il est probable
qu'elle ne fasse pas appel aux mêmes mécanismes que le choix (cf introduction, partie 1,
section 3.3.6, p. 65).
L'observation d'un biais secondaire dans l'inactivation aléatoire correspond en général à la
présence d'un allèle muté sur l'un des deux chromosomes X. Dans ce cas, les cellules ayant l'allèle
muté sur le Xa seront généralement amenées à disparaître dans les tissus où la fonction du gène
correspondant est requise. Seules subsisteront alors, dans ces tissus, les cellules pour lesquelles
l'allèle fonctionnel de ce gène est sur le Xa, et ayant inactivé l'allèle muté. Une telle configuration
est observée dans de nombreuses maladies, notamment à l'origine de désordres mentaux, du fait de
l'enrichissement du chromosome X en gènes ayant une fonction dans le cerveau (pour revue, Franco
et Ballabio, 2006; Ropers, 2006). Notons cependant que le cas de figure opposé, où l'allèle sauvage
est retrouvé plus souvent sur le Xi, est également possible (Dobrovolny et al., 2005; Martinez et al.,
2005).
Le premier exemple de biais primaire dans le choix à avoir été décrit est associé aux allèles Xce
(pour X controlling element), chez la souris (Cattanach et Isaacson, 1967; Cattanach et Williams,
1972). Il en existe trois principaux: Xcea, dans les fonds génétiques 129Sv/Pas, C3H/HeJ, 101/H,
A/J, CBA/J et BALB/cByJ; Xceb, dans les fonds C57BL/6J, DBA/2J et JU/Ct; et Xcec dans le fond
CAST/Ei. Il est possible que d'autres allèles Xce, associés à d'autres fonds génétiques, viennent
compléter cette liste (Cattanach et al., 1969; Johnston et Cattanach, 1981; Simmler et al., 1993;
West et Chapman, 1978). Les allèles Xce sont ordonnés du plus « faible » (Xcea) au plus « fort »
(Xcec). Dans les individus hétérozygotes pour les allèles Xce, le chromosome porteur de l'allèle le
plus faible a le plus de chance d'être inactivé. Le biais le plus fort est observé dans les hétérozygotes
Xcea/Xcec, chez qui le chromosome porteur de l'allèle Xcea est inactivé dans ~75 % des cellules
(Plenge et al., 2000; de La Casa-Esperon et al., 2002). Au contraire, dans les individus de même
allèle Xce, le choix est essentiellement non biaisé (Krietsch et al., 1986; Plenge et al., 2000).
Cependant, il pourrait y avoir une source additionnelle de biais dans le choix primaire, tendant à
favoriser, faiblement, le chromosome X d'origine paternelle pour être inactivé (Bittner et al., 1997;
Chadwick et Willard, 2005; Forrester et Ansell, 1985; Fowlis et al., 1991; Plenge et al., 2000). Cela
pourrait suggérer que la réactivation du Xp dans le blastocyste n'efface pas la totalité des marques
épigénétiques acquises auparavant, dans le cadre de l'inactivation empreintée. La localisation du
locus Xce est un travail de longue haleine, toujours en cours. La région candidate est pour l'instant
84

centré sur Xist et son antisens Tsix (Chadwick et al., 2006), mais ces deux gènes n'en font pas partie
(Prissette et al., 2001; Simmler et al., 1993).
Le biais créé par les allèles Xce peut être altéré par des modificateurs autosomaux, dont trois
loci appelés Xiafs (pour X-inactivation autosomal factors) ont d'ores et déjà été identifiés à l'issu
d'un crible (Percec et al., 2002; Percec et al., 2003). Les allèles dominants ainsi caractérisés
semblent interagir spécifiquement avec les allèles Xce lors du choix primaire car 1) ils ne modifient
ni l'inactivation empreintée, ni l'expression de gènes autosomiques soumis à l'empreinte parentale;
2) ils n'ont pas d'effet sur le choix dans des homozygotes pour les allèles Xce; 3) ils altèrent le biais
lié à Xce dans tous les tissus testés; 4) leur effet a été observé peu après l'établissement de
l'inactivation aléatoire. Un important travail de caractérisation reste néanmoins à réaliser pour
déterminer quels sont les gènes associés aux loci Xiafs.
Il y a plusieurs manières d'envisager le processus de choix primaire. Il pourrait être réalisé une
fois pour toute, lors de la différenciation des cellules ES, peu avant le recouvrement du futur Xi par
Xist. Les deux chromosomes X seraient alors équivalents, du point de vue de l'inactivation, dans les
cellules ES indifférenciées. Dans cette optique, les modèles de comptage présentés à la section
précédente (Lee, 2005; Nicodemi et Prisco, 2007) pourraient également servir à expliquer le choix,
et un choix biaisé résulterait d'une affinité différentielle des deux chromosomes X pour les facteur
de comptage.
On peut également imaginer que l'inactivation serait initiée de manière stochastique, non
coordonnée entre les chromosomes. Dès lors qu'un chromosome X aurait initié l'inactivation, le
processus de comptage devrait intervenir pour empêcher l'inactivation du second chromosome X.
Le choix serait biaisé dans le cas ou les deux chromosomes X auraient une probabilité différente
d'initier l'inactivation en cis, par exemple parce qu'ils ne répondraient pas avec la même efficacité
aux signaux de différenciation la déclenchant.
Cependant, des observations récentes ont révélé que les chromosomes X alternent, dans les
cellules ES femelles, avant l'initiation de l'inactivation, entre deux états épigénétiques distincts qui
semblent prédéterminer leur futur statut actif ou inactif (Mlynarczyk-Evans et al., 2006).
Concrètement, ces états correspondent à l'observation par DNA-FISH sur des cellules fixées à la
paraformaldéhyde, pour différents gènes du chromosome X, d'un signal ponctuel simple sur un
allèle, et d'un signal dédoublé sur le second. Cette configuration « simplet-doublet » (SD) est
observée en phase S et n'est pas associée à une réplication asynchrone des deux chromosomes, d'où
sa dénomination de SIAR (SD signals that are Independent of Asynchronous DNA Replication)
(Mlynarczyk-Evans et al., 2006). Avant l'inactivation aléatoire, les cellules ES femelles alternent
85

entre les deux configurations SIAR possibles, les gènes d'un chromosome X donné étant
successivement détectés sous la forme d'un signal unique puis d'un doublet (Mlynarczyk-Evans et
al., 2006). Dans des fibroblastes femelles, ainsi que dans des cellules TS femelles, qui ont dans les
deux cas déjà inactivé l'un de leur chromosomes X, la configuration SIAR n'est plus observée.
D'autre part la fréquence avec laquelle un chromosome X est dans un état particulier (« simplet »
ou « doublet ») corrèle avec l'allèle Xce dont il est porteur (Mlynarczyk-Evans et al., 2006): la
détection d'un signal dédoublé est ainsi d'autant plus fréquente pour un chromosome que la
probabilité qu'il demeure actif est élevée. Une telle corrélation est également observée dans le cas
86
Fig. 14: Modèles de choix aléatoire (d'après Mlynarczyk-Evans et al., 2006)
(A) Les états SIAR décrits par MlynarczykEvans et al. suggèrent un modèle selon lequel, dans les cellules indifférenciées en réplication, les chromosomes X alternent de manière coordonnée entre un état de Xa présomptif (chromosome bleu clair) et un état de Xi présomptif (chromosome bleu foncé). Le devenir des chromosomes dépend de leur état au moment où un signal développemental initie l'inactivation. Le centre d'inactivation du chromosome X est également identifié par une configuration SIAR, mais opposée de celle du reste du chromosome X (MlynarczykEvans et al., 2006), d'où sa représentation par une bande de couleur différente de celle du chromosome qui le porte. (B) Modèle dans lequel les deux chromosomes X sont équivalents dans les cellules indifférenciées, avant qu'un signal développemental n'ait pour effet de marquer de manière différente les deux chromosomes X (croix grise), les désignant ainsi comme futur Xa ou Xi.

de mutants de Xist et Tsix, que nous présenterons plus loin (cf introduction, partie 2, chapitre 2,
p. 95), dans lesquels le choix est biaisé. Ces observations conduisent leurs auteurs à proposer un
modèle dans lequel choix et comptage opèrent à chaque instant dans une population de cellules ES
femelles indifférenciées, la proportion finale de cellules inactivant l'un ou l'autre de leurs
chromosomes X durant la différenciation étant prédéterminée par la proportion de cellules dans
chacune des configurations SIAR avant différenciation. Remarquons cependant que si la corrélation
entre SIAR et choix primaire est bien documentée, la causalité entre le premier et le second reste à
démontrer.
Quel que soit le modèle envisagé, la réalisation correcte du choix primaire suppose que
l'initiation de l'inactivation soit coordonnée entre les deux chromosomes. Une telle coordination
pourrait, selon les cas, relever du dispositif de comptage ou être directement liée aux mécanismes
de choix. Elle implique une communication entre les chromosomes X, qui pourrait être facilitée par
leur proximité dans le noyau. De fait, un rapprochement des chromosomes X est observé au cours
de la différenciation de cellules ES femelles, dans la période d'initiation du recouvrement du
chromosome X par Xist (Bacher et al., 2006; Xu et al., 2006). C'est-à-dire vers 1,5 à 2 jours de
différenciation induite par l'acide rétinoïque (Bacher et al., 2006) et entre 2 et 4 jours de
différenciation par la méthode des corps embryonnaires (Xu et al., 2006) (on remarquera
incidemment la différence de cinétique entre les deux méthodes, les cellules apparaissant moins
synchrones dans la seconde, comme mentionné à la section 3.6 de la première partie de
l'introduction, p. 73). Le modèle de Nicodemi et Prisco (cf section précédente) est celui qui décrit
le mieux l'avantage procuré par un tel rapprochement qui, en accroissant localement la
concentration des molécules « bloquantes », pourrait être l'élément déclencheur de la brisure de
symétrie parmi ces molécules.
Le rapprochement transitoire décrit dans les deux études (Bacher et al., 2006; Xu et al., 2006)
concerne plus précisément la région du chromosome X contrôlant le comptage, le choix et
l'initiation en cis de l'inactivation, appelée centre d'inactivation du chromosome X (Xic, pour X-
chromosome inactivation center), dont traitera la suite de ce texte. Nous reviendrons plus loin sur la
manière dont le rapprochement des Xic peut être altéré dans différentes lignées mutantes pour cette
région, qui sont également affectées pour le choix ou le comptage.
87

1.2 Caractérisation du centre d'inactivation du chromosome X
Le centre d'inactivation du chromosome X (Xic chez la souris, XIC chez l'homme) peut être
défini comme la région minimale capable de réaliser les fonctions de comptage, de choix et
d'initiation en cis de l'inactivation. Comme nous allons le voir, la définition de l'intervalle
génomique correspondant au centre d'inactivation est variable selon la méthode employée pour le
caractériser. Cela tient à des limitations pratiques, mais également conceptuelles, qui font que les
différentes approches sont avant tout complémentaires.
1.2.1 Définition cytogénétique
L'existence et la localisation du centre d'inactivation ont d'abord été déduites en examinant
l'inactivation du chromosome X dans des cellules porteuses de réarrangements chromosomiques
(Russell, 1983; Therman et al., 1983). Chez l'homme, sa taille a été estimée entre 680 et 1200 kb,
localisés sur la bande Xq13 du bras long du chromosome X (Brown, 1991; Lafreniere et al., 1993;
Lafreniere et Willard, 1993; Leppig et al., 1993; Willard, 1995; Willard, 1996). Chez la souris, la
région candidate définie de cette manière, bornée par la bande XD, est considérablement plus
étendue (Rastan, 1983; Rastan et Robertson, 1985). Néanmoins, la synténie entre les génomes
humain et murin a conduit à extrapoler à partir de l'homme une localisation plus précise du Xic chez
la souris (Heard et al., 1994). Dans les deux cas, le centre d'inactivation contient le gène Xist/XIST.
1.2.2 Définition par transgenèse
L'assomption que le Xic murin était synténique avec le XIC humain a pu être confirmée par des
expériences de transgenèse. Celles-ci avaient pour but d'évaluer l'aptitude d'un transgène à
reproduire les fonctions attribuées au Xic: comptage, choix et initiation en cis de l'inactivation.
Hypothèse étant faite que les mâles sont capables d'initier l'inactivation, le moyen le plus simple de
tester si un transgène participe au processus de comptage est de l'introduire sur un autosome dans
des cellules mâles. On s'attend alors à ce que le transgène soit « compté » comme un deuxième
chromosome X et que, par conséquent, l'inactivation puisse être initiée aussi bien à partir du
transgène que du Xic endogène.
88

Il a été montré que c'était bien le cas, lors de la différenciation de cellules ES mâles, lorsqu'un
YAC (Yeast Artificial Chromosome, chromosome artificiel de levure) de 450 kb incluant Xist était
inséré en multicopies sur un autosome (Lee et al., 1996; Lee et Jaenisch, 1997) (fig. 15, p. 89).
Trois configurations ont été observées par les auteurs: inactivation initiée à partir du transgène seul
(dans 53 à 76 % des cellules, selon les clones), à partir du transgène et du Xic endogène (35 à 44 %
des cellules) ou à partir du Xic endogène seul (0 à 2 %). Il semble donc que le transgène ne soit pas
compté comme un Xic ectopique unique. On peut avancer que cela est lié à son insertion en
multicopies. Chaque copie serait « comptée » une fois et pourrait initier l'inactivation
indépendamment des autres, ce qui donnerait un forte probabilité de voir l'autosome « décoré » par
l'ARN Xist.
La présence d'un centre d'inactivation ectopique n'a pas nécessairement pour conséquence de
conduire à l'inactivation chez les mâles. Un exemple en est donné chez l'homme où un individu
mâle porteur d'une duplication du XIC sur le chromosome X a pu été identifié (Schmidt et al.,
1991). Par conséquent, un seul XIC ou chromosome X a dû être compté, de manière à ce que
l'unique chromosome X reste actif. Mais on ne sait pas si un tel cas de figure pourrait être
généralisé, ou s'il fait plutôt figure d'exception car, précisément, si l'unique chromosome X avait été
inactivé, cela se serait traduit par une léthalité embryonnaire dont il est probable que les causes
n'auraient pas été détectées.
89
Fig. 15: Transgènes du Xic
Positionnement par rapport au Xic des transgènes mentionnés dans le texte. Les gènes présents dans le Xic sont représentés en gris, pour les gènes codants, et en couleur, pour les gènes non codants.

D'autre part, un YAC de 460 kb (Heard et al., 1996), très similaire au YAC de 450 kb (Lee et al.,
1996) présenté plus haut, ainsi qu'un YAC plus petit, long de 320 kb, récapitulent de la même
manière les fonctions du Xic lorsqu'ils sont insérés en multicopies sur un autosome dans des cellules
ES murines mâles, mais il en sont incapables lorsqu'ils sont insérés en simple copie (Heard et al.,
1996; Heard et al., 1997) (fig. 15, p. 89). Lorsqu'il est inséré en multicopie, le YAC de 460 kb se
rapproche transitoirement du Xic endogène au cours de la différenciation des cellules ES mâles in
vitro, à l'instar des deux Xic dans des cellules ES femelles (cf introduction, partie 1, section 1.1.2,
p. 83) (Bacher et al., 2006). Un tel rapprochement n'est pas observé lorsqu'il est inséré en simple
copie (Bacher et al., 2006), corrélant avec l'incapacité à reproduire les fonctions du Xic.
La différence de phénotype entre insertion en simple copie et en multicopies pourrait être reliée
à la difficulté de propager l'inactivation en cis sur des autosomes (cf introduction, partie 1,
section2.1, p. 32). La présence de plusieurs copies en tandem du Xic serait alors requise, soit pour
obtenir en cis un niveau d'expression de Xist plus élevé, soit pour reconstituer sur une région plus
étendue les propriétés du chromosomes X. Une explication alternative serait que les transgènes
utilisés ne représentent pas la totalité du Xic, mais que leur insertion en multicopies permettrait d'en
reconstituer de manière artificielle les propriétés. Une telle reconstitution semble plus naturelle
lorsqu'on ne pense pas à des séquences génomique particulières qui seraient absentes dans les
transgènes, mais au profil épigénétique qui leur est associé, lequel pourrait être généré à partir de
séquences distinctes. Nous reviendrons à la section 1.2.4 de la seconde partie de l'introduction
(p. 93) sur le profil épigénétique du Xic et, particulièrement, de la région en 5' de Xist, qui rend cette
dernière hypothèse vraisemblable.
Remarquons cependant que les mêmes transgènes de 460 kb ou des transgènes plus courts,
longs de 210 kb (fig. 15, p. 89), sont fonctionnels pour l'inactivation empreintée lorsqu'ils sont
insérés en simple copie : ils induisent l'inactivation empreintée de la même manière que le Xp
lorsqu'ils ont été transmis par la lignée germinale, mais pas lorsqu'ils proviennent de la lignée
germinale femelle (Okamoto et al., 2005). Ainsi, non seulement l'inactivation aléatoire mais
également l'inactivation empreintée sont régulés au niveau du Xic, mais selon des modalités
différentes que l'on pourrait rapprocher de la difficulté à parler de choix ou de comptage dans le cas
de l'inactivation .
La nécessité d'insérer des transgènes du Xic en multicopies pour récapituler le processus
d'inactivation aléatoire laisse penser que le principe même des expériences de transgenèses –
l'insertion sur un autosome – est potentiellement source d'artefact. Aussi, particulièrement lorsqu'on
s'intéresse au choix ou au comptage, ne doivent-elles être interprétées qu'avec prudence. Elles ont
90

néanmoins été poursuivies avec l'idée de délimiter plus précisément la région minimale pour ces
deux fonctions, qui pourrait être de 80 kb (πJL1, fig. 15, p. 89), incluant Xist et son antisens Tsix
(Lee et al., 1999), voire même de 35 kb, auquel cas elle est essentiellement constituée par le gène
Xist (Herzing et al., 1997). De manière comparable au transgène de 460 kb présenté plus haut, le
transgène de 80 kb (πJL1) inséré en multicopies sur un autosome se rapproche transitoirement du
Xic endogène au cours de la différenciation de cellules ES mâles (Xu et al., 2006). Par contre, dans
le cas du transgène de 35 kb (fig. 15, p. 89), il est probable que l'on atteigne les limites du système
par transgenèse pour définir les définir le Xic car, nous allons le voir, des travaux plus récent ont
montré que des éléments externes à ce transgène jouent un rôle essentiel tant pour le comptage que
pour le choix.
1.2.3 Composition du Xic
Pour contourner les limitations apparues dans les expériences de transgenèse, une approche
consiste à tester la fonction d'éléments du Xic candidats pour participer au comptage ou au choix, en
en réalisant la délétion au locus endogène. Une dissection génétique réalisée de la sorte implique de
mieux connaître ce qui compose le Xic, gènes ou potentiels éléments régulateurs.
Le séquençage, puis l'annotation de séquences synténiques du Xic, chez l'homme, la souris (fond
génétique 129Sv/Pas) et la vache, ont été réalisés il y a quelques années par Chureau et al. (Chureau
et al., 2002). Cette étude est, encore aujourd'hui, la seule à donner une vision synthétique de ce type
concernant le Xic, bien que d'autre génomes ou fonds génétiques (dans le cas de la souris) aient été
été séquencés depuis lors. Remarquons cependant que, lorsqu'elle a été réalisée, seule une partie du
Xic bovin avait été séquencée, en conséquence de quoi la comparaison des centres d'inactivation
murin et humain est plus détaillée qu'entre chacune de ces espèces et la vache.
Une telle approche comparative a permis d'identifier 11 gènes dans le Xic murin: Xpct, Cnbp2,
Ftx, Jpx, Xist, Tsx, Tsix, Chic1 (anciennement Brx), Cdx4, Ppnx et Nap1l2 (anciennement Bpx)2.
Certains étaient déjà connus: Xist (cf introduction, partie 1, section 2.1, p. 24) et Tsix (cf
introduction, partie 2, section 2.2, p. 98), mais aussi Xpct (Debrand et al., 1998), Tsx (Simmler et
al., 1996), Chic1 (Simmler et al., 1997), Cdx4 (Gamer et Wright, 1993) et Nap1l2 (Rogner et al.,
2000; Rougeulle et Avner, 1996). Parmi ces 11 gènes, 4 ne codent pas pour des protéines: Xpct, Xist,
2 Certains des noms donnés aux gènes ont une connotation particulière. Ainsi, Ftx fait référence à la Fête du Travail, Jpx au Jour de Pâques et Ppnx au Petit Papa Noël, toutes fêtes qui ont suivi la découverte de ces gènes, le x étant bien sûr pour le chromosome X.
91

Tsix, Ftx et Jpx. Par ailleurs, une transcription intergénique sens et antisens est détectée dans une
région, notée « région B », située 50 kb en amont de Xist (Chureau et al., 2002). La proportion
élevée de gènes non codant, ainsi que l'existence de la région B, ont pu être perçues, au moment où
a été réalisé cette étude, comme un trait particulier du Xic. C'est moins le cas aujourd'hui, dans la
mesure où des études à grande échelle ont montré que la majorité du gènome était transcrite, bien
que ne produisant pas de protéine (pour revue, Carninci, 2006; Frith et al., 2005; Mendes Soares et
Valcarcel, 2006). Les gènes identifiés chez la souris sont conservés chez l'homme, à l'exception de
Ppnx et celle, notoire, de Tsix, dont nous discuterons à la section 1.1 de la discussion (p. 215). Par
ailleurs, Tsx est exprimé chez la souris mais est un pseudogène chez l'homme.
La fonction de la plupart des gènes du Xic n'est pas connue. Ceux qui ont été le mieux
caractérisés sont bien entendu Xist et Tsix, dont nous avons déjà parlé, et que nous décrirons plus en
détail dans la suite du texte. Parmi les gènes du Xic codant pour des protéines, la plupart possèdent
des motifs conservés ou ont pu être rattachés à des familles protéiques. Ainsi, Cnbp2 code pour une
protéine à doigts de zinc (Chureau et al., 2002), Ppnx possède un motif de sécrétion (Chureau et al.,
2002), Xpct (X-linked PEST-containing transporter) appartient à la famille des transporteurs de
monocarboxylate (Lafrenière et al., 1994; Price et al., 1998), Chic1 à la famille CHIC (cysteine-
rich hydrophobic protein) (Simmler et al., 1997), Cdx4 à la famille caudal de gène à homéoboîte
(Gamer et Wright, 1993), et Nap1l2 à la famille Nap (Rogner et al., 2000; Rougeulle et Avner,
1996). Chez la souris, Cnbp2, Ppnx et Tsx sont exprimés spécifiquement dans les testicules. Ppnx
l'est également dans les cellules ES (Chureau et al., 2002). L'expression de Nap1l2 est spécifique
des neurones (Rogner et al., 2000; Rougeulle et Avner, 1996). Quant à Ftx et Jpx, leur expression
semble ubiquitaire (Chureau et al., 2002). Ainsi, l'expression des gènes du Xic est assez
représentative de la l'enrichissement du chromosomes X en gène exprimés dans la lignée germinale
ou dans le cerveau (cf introduction, partie 1, section 1.2, p. 12).
92
Fig. 16: Conservation du Xic entre la souris et l'homme (d'après Chureau et al., 2002)
Alignement du XIC humain avec le Xic murin montrant la conservation de la structure d'ensemble et de la plupart des gènes. Se référer au texte pour plus de détails.

L'ordre et l'orientation des gènes sont conservés entre la souris et l'homme, à l'exception de
Xpct, dont l'orientation est inversée entre les deux espèces. Toutefois, la région synténique humaine
est considérablement plus étendue (~2300 kb ) que chez la souris (714 kb séquencés). Dans les trois
espèces (souris, homme et vache), le centre d'inactivation est pauvre en dinucléotides GC, mais
enrichi en éléments répétés et en pseudogènes (22 chez la souris) (Chureau et al., 2002). Ces
derniers sont pour la plupart conservés entre les trois espèces, ce qui suggère qu'ils pourraient avoir
une fonction, par exemple dans la mise en place d'un profil épigénétique particulier. Ils serait à ce
titre intéressant de savoir s'ils sont exprimés au cours du développement embryonnaire. Parmi les
éléments répétés, ceux de type LINE sont enrichis dans les centres d'inactivation humain et bovin,
mais pas dans le centre d'inactivation murin. La distribution des éléments LINE ne supporte donc
que dans les deux premières espèces l'idée qu'ils joueraient un rôle dans la propagation de
l'inactivation en cis à partir du centre d'inactivation, envisagé comme un centre de nucléation (cf
introduction, partie 1, section 2.1, p. 32).
Il est possible qu'une partie des éléments de comptage ou de choix soit à chercher dans les
éléments conservés entre l'homme et la souris. A l'appui de cette idée, il a été montré que des
transgènes de YAC humains centrés sur XIST, de taille égale ou inférieure à 480 kb, étaient
reconnus comme des Xic lorsqu'il étaient insérés en multicopies sur un autosome dans des cellules
ES de souris mâles (Heard et al., 1999a; Migeon et al., 1999; Migeon et al., 2001b). Cependant,
contrairement aux transgènes de Xic murin, les transgènes humains sont « décorés » par un domaine
d'ARN XIST y compris dans des cellules ES non différenciées, et sont assez peu efficaces pour
conduire à l'inactivation ectopique de l'autosome hôte (Heard et al., 1999b; Migeon et al., 1999;
Migeon et al., 2001b). Cela suggère que des éléments de comptage sont reconnus de la même
manière dans les deux espèces, mais que l'interprétation de ce comptage en cis, au niveau du Xic,
serait régulée différemment, en particulier au cours du développement.
1.2.4 Epigénétique du Xic: propriétés de la région en 5' de Xist
La région en amont de Xist présente un profil épigénétique particulier. Elle est en effet enrichie
en marques répressives H3K9me2 et H3K27me3, détectées par immunofluorescence et ChIP, sur
340 kb en 5' de Xist. Cet enrichissement a été qualifié de hotspot de méthylation, et est observé dans
les cellules ES, mâles et femelles, indifférenciées et en début de différenciation (Heard et al., 2001;
Rougeulle et al., 2004). L'enzyme G9a est responsable du maintien de H3K9me2 au hotspot, mais
93

apparemment pas de sa déposition, qui serait antérieure à la dérivation des cellules ES (Rougeulle
et al., 2004). Un enrichissement en acétylation de H4 a également été observé dans cette région
dans les cellules ES femelles, mais pas dans les cellules ES mâles, ni dans des cellules ES femelles
hétérozygotes pour une délétion du premier exon de Xist (O'Neill et al., 1999). Il est dès lors tentant
d'imaginer qu'un telle acétylation différentielle pourrait participer au comptage. Le hotspot de
méthylation H3K9me2 et H3K27me3 a quant à lui été proposé pour intervenir comme centre de
nucléation pour l'inactivation du chromosome X, les marques H3K9me2 et H3K27me3 s'étendant à
l'ensemble du Xi en cours de différenciation (Heard et al., 2001; Rougeulle et al., 2004). Cette
hypothèse permettrait également d'expliquer pourquoi les transgènes du Xic insérés en simple copie
sur des autosomes sont incapables de récapituler l'inactivation (cf introduction, partie 2,
section 1.2.2, p. 88). En effet, ils sont tous tronqués dans la région du hotspot (Heard et al., 1999a;
Heard et al., 2001). Leur insertion en multicopies permettrait alors de reconstituer une region
suffisamment grande enrichie en H3K9me2 et H3K27me3, restituant les propriétés du hotspot
(Heard et al., 2001). Comme le rapprochement transitoire entre les Xic ectopiques et le Xic
endogène n'est observée que lorsque les premiers sont en multicopies, cela pourrait signifier que le
profil épigénétique associé au hotspot joue un rôle essentiel dans le rapprochement transitoire des
Xic (Bacher et al., 2006).
94
Fig. 17: Hotspots en 5' de Xist
Positionnement sur le Xic des hotspots de H3K9me2 et H3K27me3 (pointillés rouges) et d'acétylation de H4 (pointillés oranges). La limite à gauche du premier hotspot n'est pas connue. A titre indicatif, le transgène de 460 kb de Heard et al. est également positionné. Pour plus de détails, se référer au texte.

Chapitre 2 Dissection génétique du centre d'inactivation
2.1 Rôle de Xist dans le choix et le comptage
Nous avons vu à la section x que Xist initie le processus d'inactivation, à la mise en place de
laquelle il est indispensable. Cette nécessité de Xist est d'abord sur le chromosome supposé être
inactivé. Sans Xist il restera immanquablement actif (Marahrens et al., 1997; Marahrens et al.,
1998; Penny et al., 1996). Mais cela a-t-il à voir avec le comptage, le choix, ou tout simplement la
capacité à initier en cis le processus d'inactivation?
La première invalidation de Xist publiée consistait en la délétion de sa région promotrice et
d'une partie de son premier exon sur l'un des deux chromosomes X, porteur de l'allèle Xcea, dans
des cellules ES femelles hétérozygotes Xcea/Xceb (Penny et al., 1996) (XistΔProm-1, fig. 5, p. 27).
La conséquence en était de voir apparaître, lorsqu'elles étaient différenciées in vitro, deux
populations de cellules: avec leur chromosome X sauvage inactif, et avec leurs deux chromosomes
X actifs. De plus, la proportion de cellules ayant leurs deux chromosomes X actifs semblait
comparable à la proportion de cellules (de l'ordre de 65 %) qui auraient normalement inactivé le
chromosome X délété pour Xist, d'allèle Xcea (Penny et al., 1996). Dans des embryons chimériques
générés par l'aggrégation de ces cellules ES recombinées avec des morulas sauvages, la totalité des
cellules porteuses de la délétion de Xist ont leur chromosome X sauvage inactif à E10,5-12,5
(Penny et al., 1996). Tout se passe donc comme si comptage et choix primaires avaient fonctionné
correctement, mais que les cellules ayant choisi le chromosome X muté pour être inactivé, dans
l'impossibilité d'en réaliser l'inactivation, avaient conservé leurs deux chromosomes X actifs.
Comme le maintien de deux chromosomes X actifs dans des cellules différenciées est a priori non
viable, les cellules n'ayant pas inactivé le chromosome X sauvage auraient fini par mourir – ce qui
correspond à un mécanisme de comptage secondaire –, expliquant le biais total en faveur de
l'inactivation du chromosome X sauvage observé dans l'embryon. D'après Penny et al., Xist ne serait
donc impliqué ni dans le comptage, ni dans le choix primaires, mais uniquement dans l'initiation en
cis de l'inactivation.
95

Deux autres délétions de Xist, analysées in vivo, ont apporté des résultats contradictoires. La
première va du premier exon au cinquième exon et laisse en place P1, mais pas P2 (Marahrens et
al., 1997) (XistΔ1-5, fig. 5, p. 27). La seconde inclut la région promotrice dans sa totalité et se
poursuit jusqu'à l'exon 3 (Csankovszki et al., 1999) (Xist1lox, fig. 5, p. 27). Dans les deux cas,
l'inactivation ne peut pas être initiée en cis (Gribnau et al., 2005; Marahrens et al., 1997). A E7,5, le
chromosome X sauvage est inactivé dans toutes les cellules des embryons ayant hérité l'une ou
l'autre de ces mutations de leur mère (la transmission paternelle de délétions de Xist est léthale du
fait d'un défaut d'inactivation empreintée, cf introduction, partie 1, section 3.3, p. 59), de manière
tout à fait comparable à des embryons sauvages (Gribnau et al., 2005; Marahrens et al., 1998).
D'autre part, si ce biais total avait été réalisé de manière secondaire, on s'attendrait à observer la
mort d'environ la moitié des cellules de l'embryon entre E5,5, stade auquel est initiée l'inactivation
aléatoire, et E7,5, stade auquel la plupart des cellules ont leur chromosome X sauvage inactif. Cela
se traduirait vraisemblablement par un retard de développement embryonnaire ou une taille
inférieure des embryons portant la délétion, qui n'ont pas été observés à E7,5, E8, 5 ou E9,5 avec la
mutation de Marahrens et al. (Marahrens et al., 1998). On ne peut exclure que la disparition d'une
proportion importante de cellules ait été compensée par une prolifération plus importante des
cellules survivantes, mais le laps de temps très court (1 à 2 jours) durant lequel une telle
compensation aurait dû être réalisée permet d'en douter. C'est pourquoi les auteurs concluent que,
dans leurs mutants de Xist, le biais de choix favorisant l'inactivation du chromosome X sauvage est
réalisé de manière primaire (Gribnau et al., 2005; Marahrens et al., 1998). Ce sont des cellules ES
femelles porteuses de cette même délétion qui ont été utilisées par Mlynarczyk-Evans et al. pour
montrer que la configuration SIAR avait un caractère prédéterminant (cf introduction, partie 1,
section 1.1.2, p. 83). En effet, le signal dédoublé est dans ces cellules, avant différenciation,
toujours associé au chromosome X muté, qui est destiné à demeurer actif (Mlynarczyk-Evans et al.,
2006).
Les divergences entre le mutant de Penny et al., d'une part, et ceux de Marahrens et al. et
Csankovszki et al., d'autre part, pourraient résulter de modalités différentes de réalisation du choix
(ou du comptage) dans les cellules ES et dans l'embryon. Elles pourraient également être expliquées
par la délétion de régions différentes du gène Xist. En suivant ce raisonnement, la région délétée à la
fois par Marahrens et al. et Csankovszki et al., mais pas par Penny et al., qui s'étend du premier au
troisième exon de Xist, devrait contenir un élément requis pour le choix, fonctionnant de manière
96

indépendante de la région promotrice de Xist. Cette région pourrait être importante pour le choix en
tant qu'élément génomique, ou sous forme d'ARN, du fait de sa transcription en même temps que le
gène Tsix (cf introduction, partie 2, section 2.2, p. 98).
La délétion de l'exon 4 de Xist, pourtant fortement conservé entre l'homme et la souris, n'a quant
à elle aucun effet sur l'expression de Xist ou le déroulement correct de l'inactivation aléatoire
(Caparros et al., 2002).
Chez l'homme, il a été rapporté qu'une mutation rare, remplaçant une cytosine par une guanine
dans le promoteur de XIST, conduisait à l'inactivation préférentielle de l'allèle muté dans des
individus femelles (Plenge et al., 1997). Mais on ne sait pas si un tel biais est réalisé de manière
primaire ou secondaire. Il suggère toutefois que le niveau d'expression de XIST, qui pourrait être
altéré sur l'allèle porteur de cette mutation (Plenge et al., 1997), jouerait un rôle dans le choix.
Chez la souris, plusieurs mutations en 5' de Xist, qui conduisent de manière artéfactuelle à une
surexpression de ce dernier, ont également pour conséquence l'inactivation préférentielle de l'allèle
muté dans des embryons femelles (Nesterova et al., 2003; Newall et al., 2001). Il s'agit de délétions
se terminant 1 kb avant le promoteur de Xist, et s'étendant 2,5 kb (Δhs+neo, fig. 5, p. 27) (Nesterova
et al., 2003; Newall et al., 2001)) ou 8 kb (Δ5'+neo et Δ5'Δneo, fig. 5, p. 27) (Nesterova et al.,
2003) plus en amont, ainsi que de l'insertion, 1 kb en amont du promoteur de Xist, d'une cassette
néomycine sous contrôle du promoteur fort PGK (cassette PGKneo), transcrite antisens à Xist et
terminée par un signal de terminaison de transcription (SPA+neo, fig. 5, p. 27) (Nesterova et al.,
2003). Les mutants Δhs+neo et Δ5'+neo ont également inséré, en lieu et place de la région délétée,
une cassette PGKneo transcrite antisens à Xist, qui a été retirée dans le mutant Δ5'Δneo. Chacune
de ces quatre mutations ( Δhs+neo, Δ5'+neo, Δ5'Δneo et SPA+neo) conduit, dans des cellules ES
mâles indifférenciées, à une augmentation du niveau d'expression de Xist, associée à la formation
de domaines de Xist dans 10 à 15 % des cellules (Nesterova et al., 2003; Newall et al., 2001). Mais
on ne sait pas dans quelle mesure cela pourrait ou non conduire à l'inactivation ectopique de
l'unique chromosome X lors de la différenciation in vitro de ces cellules, et il n'est donc pas possible
de conclure sur un éventuel défaut de comptage. L'expression de Xist semble également augmentée
dans des embryons à E4,5 ayant hérité la mutation Δ5'+neo de leur mère (Nesterova et al., 2003).
La surexpression de Xist semble causée par l'activité bidirectionnelle du promoteur PGK dans les
mutants Δhs+neo, Δ5'+neo et SPA+neo), ainsi que par l'activité bidirectionnelle du gène Enox,
plus en amont, dans les mutants Δ5'+neo et Δ5'Δneo (Nesterova et al., 2003; Newall et al., 2001).
Le niveau d'expression ectopique de Xist dans les cellules ES mâles indifférenciées corrèle bien
97

avec le degré de biais en faveur de l'inactivation du chromosome muté observé dans les embryons
femelles. Un tel biais est réalisé dès E6,5 ce qui traduirait un défaut de choix primaire (Nesterova et
al., 2003).
En résumé, le niveau d'expression relatif de Xist sur chacun des chromosomes X influencerait
directement le choix primaire, mais uniquement lorsque le promoteur de Xist est présent ou que
l'expression de Xist peut être réalisée sur les deux chromosomes X (Penny et al., 1996). Dans ce
contexte, il est important de comprendre comment l'expression de Xist est régulée, particulièrement
avant l'initiation de l'inactivation, lorsque choix et comptage sont censés être réalisés. Comme nous
allons le voir, le gène Tsix est un élément essentiel pour cette régulation.
2.2 Le gène Tsix
L'existence d'une transcription antisens à Xist a été mise en évidence à la fin des années 1990
(Debrand et al., 1999; Lee et al., 1999; Mise et al., 1999). Le modèle prédominant était alors celui
d'une stabilisation des transcrits Xist en cours de différenciation, conduisant à leur accumulation au
niveau du chromosome X choisi pour être inactivé. D'emblée, le rôle d'une telle transcription
antisens dans la déstabilisation des transcrits Xist avant différenciation a été envisagé. Cela explique
l'intérêt immédiat qu'a suscité sa découverte, et l'important travail de caractérisation qui a suivi.
Cette transcription antisens à Xist fut associée à un gène, que l'on nomma Tsix (Lee et al., 1999),
dont l'unité de transcription majoritaire, longue de 40 kb, traverse le gène Xist, et dont l'ARN est
polyadénylé (Debrand et al., 1999; Lee et al., 1999). Des donnée convergentes en ce sens furent
obtenues par diverses méthodes, RNA-FISH brin spécifique (Lee et al., 1999), Northern blot
(Debrand et al., 1999), RT-PCR brin spécifique (Debrand et al., 1999; Lee et al., 1999; Mise et al.,
1999) et clonage de cDNA (Mise et al., 1999).
2.2.1 Variété des transcrits Tsix
Ce n'est pas d'un transcrit Tsix, mais de plusieurs transcrits dont on serait en droit de parler, d'un
point de vue strictement descriptif (fig. 18A, p. 100). Une première source de variété est dans le site
d'initiation de la transcription. L'origine du transcrit majoritaire a été positionnée, par primer
extension et 5' RACE (rapid amplification of cDNA ends), au sein d'un îlot CpG environ 15 kb en
aval du gène Xist. Deux sites d'initiation distants de 50 pb ont ainsi été identifiés (Lee et al., 1999).
98

L'îlot CpG en question est associé au minisatellite DXPas34 (Avner et al., 1998; Courtier et al.,
1995), sur lequel nous reviendrons en détail à la section 3.3 de la seconde partie de l'introduction
(p. 116).
Une transcription antisens à Xist, plus faible, peut être détectée en amont de ce premier site
d'initiation (Debrand et al., 1999), ce qui corrèle bien avec l'existence d'une transcription antisens
résiduelle lorsque l'îlot CpG mentionné plus haut est délété (Lee et Lu, 1999). La confirmation a été
apportée que cette observation traduisait l'existence d'un promoteur alternatif de Tsix, à l'origine
d'un transcrit minoritaire, environ 16 kb en amont du promoteur majoritaire (Sado et al., 2001). Ce
second promoteur a également été associé à un groupement de sites d'initiations identifiés par 5'
RACE, situés dans un îlot CpG (Ogawa et Lee, 2003). Un groupement de sites de transcription sens
et antisens a été caractérisé de la même manière au locus Xite, entre le promoteur majoritaire et le
promoteur minoritaire de Tsix (Ogawa et Lee, 2003; Stavropoulos et al., 2005). On ignore
cependant si la transcription initiée à cet endroit pourrait contribuer au transcrit Tsix tel que défini
plus haut, c'est-à-dire présentant un recouvrement de son unité de transcription avec celle du gène
Xist. Enfin, le minisatellite DXPas34 est également un lieu d'initiation de trancription sens et
antisens, (Cohen et al., 2007; Morey, 2003), avec la même interrogation quant à sa contribution aux
transcrits Tsix.
Plusieurs terminaisons de Tsix ont été caractérisées, créant une deuxième source de variété des
transcrits. La plupart d'entre elles sont localisées en 5' de Xist, à moins de 1,5 kb de son promoteur
(Debrand et al., 1999; Lee et al., 1999; Mise et al., 1999; Ogawa et Lee, 2003; Sado et al., 2001). Il
existe également un site de terminaison, mis en évidence par 3' RACE, peu après DXPas34 (Shibata
et Lee, 2003). De manière intéressante, un gradient d'abondance des transcrits antisens à Xist est
observé, avec environ dix fois plus de transcrits à proximité du promoteur majoritaire de Tsix qu'en
5' de Xist (Shibata et Lee, 2003). En admettant que ces transcrits soient tous générés aux mêmes
sites d'initiation, la terminaison des ARN Tsix pourrait donc être plus variable encore que ne le
laissaient supposer les variants de terminaison mentionnées ci-dessus.
L'épissage représente une troisième source de variété pour les transcrits Tsix. Les analyses
initiales par RT-PCR détectaient un transcrit antisens à toutes les positions testées le long du gène
Tsix, ce qui suggérait l'existence d'un transcrit non épissé (Debrand et al., 1999; Lee et al., 1999).
Différentes formes épissées de Tsix ont par la suite été caractérisées avec pour point commun,
lorsqu'elles ne sont pas terminées avant Xist, d'avoir une complémentarité limitée avec le transcrit
Xist, sur environ 1,9 kb au début de son premier exon (Sado et al., 2001; Shibata et Lee, 2003).
Cette région de complémentarité inclut la répétition A de Xist, requise pour l'extinction
99
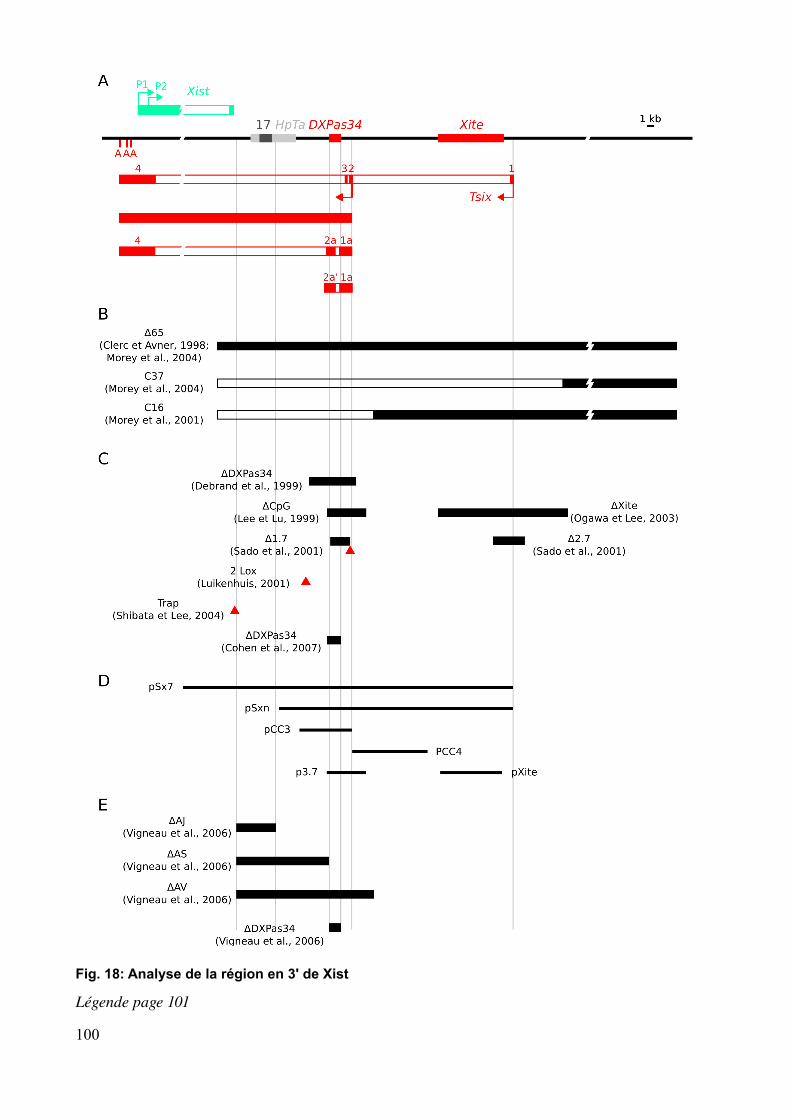
100
Fig. 18: Analyse de la région en 3' de Xist
Légende page 101

transcriptionnelle des gènes du chromosome X (Wutz et al., 2002). Les variations d'épissage sont
essentiellement au début de Tsix, associées soit à la transcription à partir du promoteur minoritaire
(Sado et al., 2001), soit à un épissage alternatif peu après le promoteur majoritaire, ayant pour
conséquence d'inclure ou non la séquence répétée DXPas34 dans le transcrit mature (Shibata et Lee,
2003). Les formes épissées (mesurées par RNAse Protection Assay) représentent environ 60 % des
transcrits totaux, avec apparemment une proportion comparable de formes incluant ou non
DXPas34 (Shibata et Lee, 2003).
En résumé, la représentation que nous nous faisons de Tsix est celle d'un ensemble de transcrits
issus d'un promoteur majoritaire, dont une proportion importante n'est pas épissée et dont les formes
épissées sont presque toujours complémentaires à l'ARN Xist, mais seulement sur une partie de son
premier exon. Il faut garder en mémoire que dans la plupart des cas, les variants de Tsix n'ont été
caractérisés que dans des cellules ES, mâles ou femelles. L'étude la plus complète réalisée à ce jour
dans l'embryon est celle de Sado et al., permettant de valider in vivo, dans le placenta, la présence
de certaines de ces formes (Sado et al., 2001).
101
Légende de la figure 18,p. 100: (A) Éléments génomiques en 3' de Xist et différents transcrits de Tsix connus. Le chromosome est figuré par une ligne continue noire. 17 désigne un ministallite formé par la répétition d'un motif de 17 pb (gris foncé). HpTa désigne une région d'homologie à un enhancer du récepteur des cellules préT (pTa, pour preT cell α) (en gris clair). DXPas34 (cf introduction, partie 2, section 3.3, p. 116) et Xite (cf introduction, partie 2, section 2.3.2.2, p. 108) sont figurés en rouge. Les transcrits Tsix sont représentés sous le Xic (en rouge), les rectangles pleins correspondant aux exons et les rectangles vides aux introns. Les différents sites de terminaison (pA) sont indiqués. (B) Délétion de 65 kb et complémentation de cette dernière en 3' de Xist. Les régions délétées sont figurées en rectangles noirs pleins, et les séquences réintroduites sous formes de rectangles noirs vides. (C) Mutations générées par d'autres laboratoire ciblant des éléments particuliers de la région de 65 kb en 3' de Xist. Les rectangles noirs pleins correspondent à des délétions, les triangles rouges à des signaux d'arrêt de transcription orientés de manière à piéger la transcription de Tsix. La mutation 2lox est celle présente dans les lignées Ma2L. (D) Transgènes en 3' de Xist inhibant le comptage et le choix selon (Lee, 2005). (E) Délétions en 3' de Xist générées durant la thèse (cf résultats, p. 131).

2.2.2 Expression de Tsix
2.2.2.1 Dans les cellules ES
Tsix est exprimé dans les cellules ES indifférenciées, mâles et femelles (Debrand et al., 1999;
Lee et al., 1999; Mise et al., 1999; Sado et al., 2001). Cela est vrai au locus endogène, mais
également à partir de transgènes du Xic insérés sur des autosomes dans des cellules ES mâles
(Debrand et al., 1999). Les transcrits Tsix sont plus nombreux que les transcrits Xist, d'environ un
facteur 100 au début du transcrit majoritaire de Tsix, et d'un facteur 10 au niveau de son dernier
exon (Shibata et Lee, 2003). Cependant, Xist est peu exprimé, à raison en moyenne de 10 copies par
cellule ES femelle et 4 copies par cellule mâle (Huynh et Lee, 2003). Il serait intéressant d'apprécier
dans quelle mesure le niveau relatif d'expression de Xist et de Tsix peut varier entre cellules d'une
même population. En particulier, on peut se demander si Xist est réellement exprimé dans toutes les
cellules ou si son niveau moyen d'expression traduit une forte expression dans quelques cellules
seulement. D'autre part, nous ne savons pas si, à un instant donné, l'expression de Xist et Tsix peut
avoir lieu à partir du même Xic, ou si l'un et l'autre sont exprimés de manière alternative.
L'évolution des techniques de marquage des ARN et d'imagerie devrait permettre d'apporter des
réponses précises à ces questions.
Dans les cellules ES mâles, comme dans les cellules ES femelles, l'expression de Tsix disparaît
en cours de différentiation. Cependant, lors de la différenciation de cellules ES femelles,
l'expression de Tsix est perdue plus rapidement sur le futur Xi que sur le futur Xa (Debrand et al.,
1999; Lee et al., 1999). En fonction des auteurs, l'extinction de la transcription antisens sur le futur
Xi précèderait toujours la formation d'un domaine d'ARN Xist (Lee et al., 1999) ou pourrait lui être
concomitante dans ~6 % des cellules différenciées (Debrand et al., 1999). Cette différence, minime,
pourrait être liée aux sondes employées pour le marquage par RNA-FISH de la transcription
antisens. Certaines des sondes utilisées par Lee et al. sont plus distantes du promoteur majoritaire
que celle utilisée par Debrand et al., centrée sur DXPas34. Compte-tenu du gradient d'abondance
des transcrits antisens mentionné plus haut, on pourrait alors imaginer une régulation différentielle
de la transcription à proximité du promoteur par rapport aux positions plus en aval,
complémentaires à Xist. Cela est cohérent avec l'observation que la transcription de Tsix à travers
102

Xist joue un rôle essentiel pour empêcher en cis la formation d'un domaine de Xist, rôle que n'aurait
pas la transcription antisens en 3' de Xist. Nous y reviendrons à partir de la section 2.3 de la seconde
partie de l'introduction (p. 104).
2.2.2.2 Dans l'embryon
Dans l'embryon, l'expression de Tsix est détectée par RNA-FISH, uniquement sur le Xm et dans
une faible proportion de cellules, aux stades 8 à 16 cellules (Mise et al., 1999). Elle est alors
associée à celle de Xist, à un niveau faible, sur le même chromosome, tandis qu'un domaine d'ARN
Xist « décore » le Xp. Cependant, Tsix n'est pas détecté par RT-PCR au stade 8 cellules (Sado et al.,
2001).
A E3,5 et E4,5 l'expression de Tsix est détectée par RT-PCR (Lee, 2000; Mise et al., 1999;
Sado et al., 2001) et par RNA-FISH, dans une proportion importante de cellules (Debrand et al.,
1999). C'est à ces stades que se différencient les trois premiers lignages – trophectoderme,
endoderme primitif et épiblaste – et que le Xp est réactivé dans les cellules de la masse cellulaire
interne. A E3,5, l'expression de Tsix est observée essentiellement à partir de l'allèle maternel (Lee,
2000; Sado et al., 2001). Néanmoins, dans les cellules où le Xp n'est plus « décoré » par l'ARN Xist
à E3,5-E4,5 l'expression de Tsix est toujours biallélique (Debrand et al., 1999).
Dans les tissus embryonnaires, l'expression de Tsix est détectée par RNA-FISH sur le Xa jusqu'à
E7,5, mais ne l'est déjà plus par RT-PCR à E5,5 (Mise et al., 1999). Cette extinction de Tsix
accompagne la mise en place de l'inactivation aléatoire, pour l'essentiel entre E5,5 et E7,5 (cf
introduction, partie 1, section 3.4.1, p. 69). Après E11,5 Tsix n'est plus détecté qu'à des niveaux
extrêmement faibles, que cela soit dans l'embryon ou dans des tissus adultes (Debrand et al., 1999;
Lee et al., 1999; Sado et al., 2001). Il serait intéressant cependant de déterminer si, après E7,5, cette
expression résiduelle de Tsix pourrait être spécifique de certains types cellulaires, par exemple dans
lesquels l'inactivation serait différée (comme c'est le cas dans la notochorde) ou faisant preuve d'une
certaine plasticité quand à l'inactivation du chromosome X (cf introduction, partie 1, sections 3.4.1
et 3.4.2, p. 69-71 ).
Au contraire de ce qui est observé dans les tissus embryonnaires, Tsix est exprimé fortement
dans le placenta à E11,5, mais uniquement depuis le Xm (Sado et al., 2001). Cette expression est
pratiquement perdue à E15,5 (Sado et al., 2001).
103

La détection de transcrits Tsix par RNA-FISH plus tôt (dès le stade 8 cellule) et plus tard (E7,5,
dans les tissus embryonnaires) que par RT-PCR est probablement liée à des différences de
sensibilité entre ces deux techniques, mais également à la manière dont elles ont été mises en
œuvre. En effet, l'expression de Tsix a été observée par RNA-FISH en utilisant une sonde
positionnée au début du transcrit majoritaire, c'est-à-dire là où les transcrits sont supposés être les
plus nombreux (cf inroduction, partie 2, section 2.2.2.1, p. 102). De plus, la détection du signal par
RNA-FISH dépend du niveau d'expression dans une cellule donnée, et non pas du niveau moyen
d'expression dans un population de cellules, ce qui est susceptible de conférer une meilleure
sensibilité lorsque l'expression est restreinte à certaines cellules. A l'inverse, dans les travaux cités,
lorsque l'expression de Tsix a été mesurée par RT-PCR, cela a été fait au moyen de couples
d'amorces positionnés sur le dernier exon de Tsix, où les transcrits sont supposés être moins
abondants, et en utilisant comme échantillon des ARN mélangés obtenus à partir de plusieurs
embryons. Ces observations soulignent la complémentarité des différentes techniques de détection
des ARN. Mais elles posent également, de nouveau, la question d'un rôle éventuel du gradient
d'abondance des transcrits Tsix au cours du développement ou, plus précisément, d'un rôle
spécifique de la région 5' de Tsix, centrée sur DXPas34, notamment lors du développement
embryonnaire précoce.
2.3 Fonctions de Tsix et dissection génétique en 3' de Xist
2.3.1 Implication de Tsix dans l'inactivation soumise à l'empreinte parentale
L'expression de Tsix dans l'embryon, décrite dans le paragraphe précédent, est symétrique de
celle de Xist à des stades ou dans des tissus où l'inactivation du chromosome X est empreintée.
Autrement dit, elle est observée toujours sur le Xm lorsque le Xp est inactivé de manière
empreintée (Lee, 2000; Sado et al., 2001). Par ailleurs, il existe plusieurs exemples de transcription
antisens impliquée dans la régulation de gènes autosomiques soumis à l'empreinte parentale (pour
revue, Reik et Walter, 2001). Dans ce contexte, on imagine aisément que Tsix puisse participer à la
régulation de l'inactivation empreintée. Cela n'est qu'en partie le cas, comme nous allons le voir.
Lorsqu'un allèle muté de Tsix, conduisant à l'absence ou quasi-absence d'expression de ce
dernier, est transmis par le Xp, le développement de l'embryon est normal et aucun défaut de
viabilité n'est mesuré (Lee, 2000; Sado et al., 2001). En revanche, lorsqu'il est transmis par le Xm,
104

une léthalité importante est observée dans les embryons mâles et femelles à E8,5, associée à une
dégénérescence des tissus extraembryonnaires (Lee, 2000; Sado et al., 2001). Comme Tsix est
exprimé spécifiquement sur le Xm dans le placenta, mais indifféremment sur le Xm ou le Xp dans
les lignages pour lesquels l'inactivation est aléatoire, il est logique de penser que l'effet observé est
la conséquence d'un défaut d'inactivation empreintée dans les tissus extraembryonnaires. Une telle
présomption est confortée par l'observation de l'expression ectopique de Xist sur le Xm dans les
blastocystes mâles et femelles à E3,5 (Lee, 2000). Celle-ci est à mettre en relation à E7,5 avec
l'observation de 8 à 21 % de cellules présentant, de manière anormale, soit leur unique chromosome
« décoré » par Xist chez les mâles, soit leurs deux chromosomes X « décorés » par Xist chez les
femelles (Sado et al., 2001). La désorganisation des tissus ne permettait cependant pas aux auteurs
d'être certain qu'une telle inactivation ectopique ne concernait que les lignages extraembryonnaire.
L'absence de lien entre la léthalité et d'éventuels défauts dans le tissus embryonnaires a cependant
été confirmé récemment par des expériences de tetraploïd rescue (« sauvetage par aggrégation de
cellules tétraploïdes » pourrait en être une traduction). Cette technique consiste à créer des
embryons en agrégeant des blastomères tétraploïdes, obtenus par fusion au stade 2 cellules, avec des
cellules ES diploïdes. Les premiers, dans ce cas de génotype sauvage, ne peuvent contribuer qu'aux
territoires extraembryonnaires, alors que la totalité de l'embryon est dérivé des secondes, qui sont
ici porteuses de l'allèle muté de Tsix sur le Xm. Les embryons crées de cette manière ne connaissent
pas de défaut de développement embryonnaire pouvant être attribué à l'absence de Tsix, confirmant
que la léthalité observée avait bien son origine dans les lignages extraembryonnaires (Ohhata et al.,
2006).
Nous avons distingué (cf introduction, partie 1, section 3.3, p. 59) plusieurs étapes dans
l'inactivation empreintée, notamment sa mise en place commençant dès le stade 4 cellules avec
l'apparition de domaines de Xist, et son maintien ou sa réversion, en fonction des lignages
cellulaires, au stade blastocyste. Nous l'avons vu, Tsix a un rôle dans la détermination du patron
correct d'inactivation empreintée dans les territoires extraembryonnaires. A quel moment cette
fonction intervient-elle? Il est peu probable que Tsix soit le facteur protecteur du Xm lors de la mise
en place de l'inactivation empreintée, car son expression est plus tardive, et n'est généralisée à la
plupart des cellules que dans le blastocyste. D'autre part, l'expression ectopique de Xist, en l'absence
de Tsix sur le Xm, est détectée à ce stade. Cela suggère que Tsix pourrait être requis dès la
formation des lignages extraembryonnaires dans les blastocystes, vers E3,5-E4,5.
105

Dans la mesure où Tsix réprime l'expression de Xist sur le Xm dans les tissus extraembryonaires,
on pourrait penser que, de manière similaire, l'expression de Tsix serait responsable de la
réactivation du Xp dans la masse cellulaire interne du blastocyste. Cela n'est apparement pas le cas
puisque, lorsque Tsix est muté sur le Xp, un transgène GFP porté par le Xp est correctement
exprimé à E6,5 (Kalantry et al., 2006). Mais il n'est pas certain que ce transgène ait au préalable été
inactivé de manière empreintée, avant la mise en place des lignages extraembryonnaires
(Hadjantonakis et al., 2001). De plus, dans les blastocystes à E3,5 et E4,5, l'expression de Tsix n'est
jamais détectée par RNA-FISH sur le chromosome X « décoré » par Xist (Debrand et al., 1999). Il
se pourrait donc que Tsix soit lui-même soumis à l'inactivation et que son expression ne puisse être
généralisée que lorsque deux conditions sont réunies: un signal développemental présent dans le
blastocyste et l'absence de « décoration » en cis par Xist.
La léthalité observée lors de la transmission par le Xm d'un allèle muté de Tsix présente de
fortes similitudes avec la transmission par le Xp d'un allèle muté de Xist (Marahrens et al., 1997).
De fait, ces deux mutations symétriques peuvent se complémenter, mais de manière partielle,
permettant un développment plus tardif, voire la naissance d'une faible proportion de souriceaux
viables (Sado et al., 2001). L'hypothèse la plus simple est d'imaginer que, dans ce cas, l'empreinte
serait « inversée », le Xp ne pouvant pas être inactivé, et le Xm l'étant plus ou moins
systématiquement. Le caractère partiel de cette complémentation fonctionnelle pourrait être
expliqué par une efficacité modérée de l'inactivation sur le Xm, du fait de l'empreinte maternellle,
ou par la nécessité de disposer de Xist et de Tsix selon des cinétiques sensiblement différentes au
cours du développement (Sado et al., 2001).
Il se trouve que la transmission maternelle de l'allèle mutant de Tsix généré par Sado et al. (Sado
et al., 2001) est léthale dans 98 % des cas, alors que 15 % des embryons mutants sont viables
lorsqu'il s'agit de l'allèle généré par Lee et al. (Lee et Lu, 1999). Dans le premier cas, il s'agit d'une
délétion (Δ1.7, fig. 18C, p. 100) centrée sur DXPas34, n'incluant pas le promoteur majoritaire de
Tsix, mais introduisant un site accepteur d'épissage et un signal de terminaison de transcription,
capables de piéger tous les transcrits provenant des promoteurs majoritaire et minoritaire. Dans le
second cas, il s'agit également d'une délétion (ΔCpG, fig. 18C, p. 100) centrée sur DXPas34, mais
incluant le promoteur majoritaire de Tsix sans piéger les transcrits provenant du promoteur
minoritaire. Une transcription résiduelle en aval de la délétion est d'ailleurs mesurée (Lee et Lu,
1999). Bien que la délétion du promoteur minoritaire ne conduise à aucun phénotype visible (Sado
et al., 2001), il est possible que la présence des transcrits originaires de ce dernier soit capable
d'atténuer le phénotype associé à l'absence du promoteur majoritaire.
106

2.3.2 Tsix et la région en 3' de Xist dans le choix
2.3.2.1 Rôle de Tsix dans le choix
Nous venons de voir que Tsix est requis pour empêcher en cis la formation d'un domaine de Xist
dans les cellules où l'inactivation est empreintée. Qu'en est-il pour l'inactivation aléatoire?
La première invalidation de Tsix publiée a consisté en la délétion de 3 kb, incluant le promoteur
de Tsix et DXPas34 (fig. 18C, p. 100), dans les transgène du Xic longs de 320 et 460 kb présentés
plus haut (fig. 15C, p. 89) (Debrand et al., 1999). Ces transgènes modifiés ont été insérés en
multicopie sur un autosome dans des cellules ES mâles. Lorsque ces cellules sont différenciées in
vitro, l'inactivation est initiée depuis les transgènes, mais jamais depuis le Xic endogène. Au
contraire, dans le cas de trangènes non modifiés, l'inactivation est initiée sur le chromosome X dans
7-10 % des cellules (Debrand et al., 1999). De telles observations pourraient traduire l'incapacité
des transgènes dans lesquels Tsix est invalidé à être pris en compte par le système de comptage,
contrairement aux transgènes non modifiés, ou révéler un biais total de choix en leur faveur pour
l'initiation de l'inactivation. Nous voyons que les expériences par transgenèse ne permettent pas de
discriminer entre ces deux possibilités. Par conséquent la détermination du rôle de Tsix dans le
choix, mais également, comme nous le verrons plus tard, dans le comptage, passe par son
invalidation au locus endogène.
Les mutations Δ1.7 et ΔCpG de Tsix décrites plus haut (fig. 18C, p. 100), lorsqu'elles sont
présentes à l'état hétérozygote, ont pour conséquence l'inactivation systématique du chromosome X
muté, aussi bien dans des cellules ES femelles que chez la souris (Lee, 2000; Lee et Lu, 1999; Sado
et al., 2001). Le chromosome X sauvage est toujours actif au cours de la différenciation de cellules
ES XΔCpGX (Lee et Lu, 1999), ce qui montre qu'il s'agit d'un défaut de choix primaire. Le même
biais de choix favorisant l'inactivation du chromosome X muté est observé dans des cellules ES
femelles où la transcription de Tsix est arrêtée avant de traverser le gène Xist par l'insertion d'un
accepteur d'épissage suivi d'un signal d'arrêt de transcription (2lox et Trap,fig. 18C, p. 100)
(Luikenhuis et al., 2001; Shibata et Lee, 2004). Il semblerait, ici aussi, que le biais observé
corresponde à un défaut de choix primaire (Shibata et Lee, 2004).
107

Dans l'une de ces lignées où les transcrits Tsix sont tronqués (2lox,fig. 18C, p. 100) le signal
dédoublé de la configuration SIAR (cf introduction, partie 2, section 1.1.2, p. 85) est toujours
associé, avant différenciation, au chromosome X sauvage, qui est destiné à demeurer actif. Cela
soutient, de même que le mutant de Xist évoqué plus haut, l'idée que les configurations SIAR
seraient prédéterminantes (Mlynarczyk-Evans et al., 2006).
De manière opposée au diverses invalidations de Tsix, lorsque l'expression de ce dernier est
artificiellement maintenue en cours de différenciation, l'allèle muté exprimant Tsix n'est jamais
choisi pour être inactivé (Luikenhuis et al., 2001; Stavropoulos et al., 2001). Cependant, il semble
s'agir dans ce cas d'un défaut de choix secondaire. L'expression ectopique de Tsix aurait alors pour
effet d'empêcher l'accumulation de Xist en cis, seulement après que le chromosome à inactiver ait
été choisi, avec pour conséquence l'absence d'inactivation dans la moitié des cellules (Stavropoulos
et al., 2001).
Notons que les expériences par perte de fonction de Tsix évaluent la fonction de Tsix dans la
fenêtre de temps où celui-ci est normalement exprimé. Au contraire des experiences par gain de
fonction, qui évaluent la fenêtre de temps durant laquelle le système est capable de réagir à
l'expression de Tsix. L'explication la plus simple permettant de résoudre l'opposition apparente entre
les résultats obtenus par perte et gain de fonction de Tsix consiste à faire l'hypothèse que ces deux
fenêtres ne sont pas exactement coïncidentes.
Lorsque, sur l'un des chromosomes X, dans des cellules ES femelles, la transcription de Tsix est
stoppée avant de traverser le gène Xist (Trap, fig. 18C, p. 100), l'insertion en 3' de Xist d'un
minigène exprimant une forme épissée (exons 2-3-4) de Tsix ne permet pas de restaurer le choix
(Shibata et Lee, 2004). Le choix requiert donc, ou bien l'expression d'autres formes de Tsix, épissées
ou non, ou bien la transcription en tant que telle de Tsix à travers le gène Xist.
2.3.2.2 Rôle de la région en 3' de Xist dans le choix
Tsix est-il seul responsable du choix dans le Xic? Il se pourrait que non. Lorsqu'une region de
65 kb, s'étendant du huitième et dernier exon de Xist à l'extrémité 3' du gène Chic1/Brx1, est délétée
sur l'un des deux chromosomes X dans des cellules ES femelles (XΔ65X), le chromosome X en
question est toujours choisi pour être inactivé lorsque les cellules sont différenciées in vitro
(fig. 18B, p. 100) (Clerc et Avner, 1998). Il a été montré que cela correspondait à un défaut de choix
primaire (Clerc et Avner, 1998). On remarquera que cette délétion inclut les deux promoteurs de
108

Tsix. Dès lors, son phénotype pourrait sembler assez banal, au vu des observations décrites dans la
section précédente. Pourtant, lorsque les premiers 16 kb de séquence en 3' de Xist, contenant le
promoteur majoritaire de Tsix, sont réinsérés dans cette délétion (cellules XΔ65+16kbX, fig. 18B,
p. 100), l'expression de Tsix est restaurée, mais pas le choix (Morey et al., 2001). Cela indique que
des éléments additionnels régulant le choix pourraient être présents dans la région non
complémentée.
Cette région contient le promoteur minoritaire de Tsix, mais celui-ci n'est pas requis pour le
choix (Sado et al., 2001). Elle contient également le locus Xite (Ogawa et Lee, 2003). Lorsque ce
dernier est délété, un biais dans le choix est observé, mais il est de très faible amplitude, et variable
selon les clones. L'absence de Xite ne suffirait donc pas à expliquer le biais total de choix dans les
cellules XΔ65+16kbX. Attardons-nous néanmoins sur les caractéristiques de Xite. Ce locus est associé à
l'initiation d'une transcription bidirectionnelle (Ogawa et Lee, 2003; Stavropoulos et al., 2005),
mais la continuation, vers le promoteur majoritaire de Tsix, des transcrits qui en sont issus n'est pas
requise pour la fonction de Xite dans le choix. Cette transcription est associée à la présence de sites
d'hypersensibilité à la DNAseI, tout deux variant de la même manière en fonction du fond génétique
et étant perdus en cours de différenciation (Ogawa et Lee, 2003). Pour ces raisons, Xite a été
proposé comme candidat pour le locus Xce, mais seule l'interversion de loci Xite entre fonds
génétiques d'allèles Xce différents permettrait de le démontrer. D'autre part, il est difficile de
déterminer si Xite est régulé activement en cours de différenciation, ou si la perte de son activité
transcriptionnelle n'est que la conséquence passive de l'inactivation du chromosome X.
Le rapprochement transitoire des Xic lors de la différenciation de cellules ES femelles, que nous
avons décrit précédemment (introduction, partie 2, section 1.1.2, p. 83), n'est pas observé dans les
cellules XΔ65X, qui inactivent systématiquement le chromosome porteur de la délétion de 65 kb
(Bacher et al., 2006). Mais il a lieu dans les cellules XΔ65+16kbX, dans lesquelles l'expression de Tsix
est restaurée, mais pas le choix (Bacher et al., 2006). D'autre part, ce rapprochement n'est pas altéré
dans les cellules XΔCpGX, bien que le choix soit fortement biaisé (Xu et al., 2006). Mais dans ce cas,
une transcription résiduelle provenant du promoteur minoritaire de Tsix est possible (Lee et Lu,
1999). Enfin, en l'absence de Xite, le rapprochement des deux Xic est seulement retardé, ce qui est
mis en relation par les auteurs avec le faible biais observé dans le choix (Xu et al., 2006).
Le rapprochement des Xic semble donc dépendre des éléments génomiques en 3' de Xist plus
que de la transcription de Tsix. Pour cette raison, il n'y a pas de corrélation stricte entre le choix et
ce rapprochement.
109

2.3.3 Tsix et la région en 3' de Xist dans le comptage
Un défaut de comptage peut être révélé, dans des cellules ES femelles différenciées, par la
présence de deux Xi ou de deux Xa, et dans des cellules ES mâles différenciées, par l'inactivation
de l'unique chromosome X, la règle de comptage (nombre de X actif / nombre d'autosomes = 1 / 2)
n'étant alors plus respectée. Nous allons voir dans quelles conditions ces configurations aberrantes
peuvent être observées.
2.3.3.1 Rôle de Tsix dans le comptage
Une approche parmi les plus directes pour tester la participation d'un gène ou d'un élément
génomique dans le comptage est d'en réaliser l'invalidation dans des cellules ES mâles. On s'attend,
si cet élément est requis pour le comptage, à observer l'inactivation ectopique de l'unique X lors de
la différenciation de ces cellules. Mais, nous en convenons, cela ne sera pas exactement la même
chose que de dire que cet élément est « compté », comme nous pouvions le faire pour les transgènes
du Xic insérés sur des autosomes (cf introduction, partie 2, section 1.2.2, p. 88).
Qu'en est-il des allèles mutants de Tsix dans les cellules mâles? Dans l'embryon, nous voyons
certes des domaines ectopiques, mais les analyses réalisées ne permettent pas de dire si une partie
des cellules concernées est ou non d'origine épiblastique (Lee, 2000; Sado et al., 2001). Des
éléments de réponse, contradictoires, sont par contre apportés par l'examen de cellules ES mâles
ayant un allèle muté de Tsix. Nos propres résultats, présentés plus loin, ont permis de lever certaines
de ces contradictions.
Deux lignées de cellules ES mâles dans lesquelles l'expression de Tsix est supprimée au locus
endogène ont été décrite par d'autres laboratoires comme conduisant à l'inactivation ectopique de
Tsix en cours de différenciation (Luikenhuis et al., 2001; Sado et al., 2001). Dans un cas, il s'agit de
cellules portant l'allèle muté Δ1.7 décrit plus haut (Sado et al., 2001) (fig. 18B, p. 100). Dans l'autre,
un accepteur d'épissage suivi d'un signal d'arrêt de transcription a été inséré dans la région 5' de Tsix
(Luikenhuis et al., 2001). Ces dernières cellules, qui sont désormais étudiées de manière intensive
dans notre laboratoire, ont pour nom Ma2L (fig. 18B, p. 100). En cours de différenciation induite
pas l'acide rétinoïque, des domaines ectopiques de Xist avaient été observés dans moins de 10 % des
cellules appartenant à l'une ou l'autre de ces deux lignées (Luikenhuis et al., 2001; Sado et al.,
2001), apparemment de manière transitoire (Sado et al., 2001).
110

D'après ces résultats, Tsix participerait donc bien à la voie de comptage. Néanmoins, compte-
tenu de la faible proportion de cellules dont le chromosome X est « décoré » par Xist, la totalité de
la voie de comptage ne semble pas dépendre de Tsix. Une autre interrogation fait jour: si
l'observation de domaines ectopiques de Xist est limitée dans le temps, cela traduit-il la mort des
cellules ayant inactivé leur unique X, c'est-à-dire un comptage secondaire, ou la réversion de toute
inactivation inappropriée, qui témoignerait d'un mécanisme de comptage primaire?
Au contraire des deux mutations précédentes, la délétion ΔCpG n'est pas associée à l'apparition
de domaines ectopiques de Xist au cours de la différenciation de cellules ES mâles (Lee et Lu,
1999). Cela a conduit J. Lee à proposer l'existence d'un modèle de comptage faisant appel à un
facteur bloquant et à un facteur de compétence, que nous avons détaillé plus haut (cf introduction,
partie 2, section 1.1.1, p. 78) (Lee, 2005; Lee et Lu, 1999). L'utilité d'un facteur de compétence
serait, pour partie, d'expliquer pourquoi les cellules possédant un unique Xic sont incapables
d'initier l'inactivation. Or, nous avons vu au paragraphe précédent que cela n'était pas toujours le
cas. Nous discuterons plus loin de ces différences entre lignées mutantes pour Tsix, en les
comparant à nos propres résultats (cf résultats, chapitre 1, p. 131).
Lorsque la délétion ΔCpG est présente à l'état homozygote, dans des souris femelles, elle
conduit à un surcroît de léthalité embryonnaire. Néanmoins, dans les souris qui survivent, le choix
est apparemment redevenu aléatoire et non biaisé. La meilleure explication en serait la mise en
place d'une inactivation chaotique dans l'embryon, suivie de la disparition des cellules présentant un
patron d'inactivation aberrant, ce qui révélerait en premier lieu un défaut de comptage (Lee, 2002).
Une telle inactivation chaotique a été confirmée dans des cellules ES homozygotes XΔCpG/XΔCpG qui,
lorsqu'elles sont différenciées, peuvent avoir deux Xi, un seul Xi, ou deux Xa (Lee, 2005).Dans ces
mêmes cellules, le rapprochement transitoire des Xic n'est pas observé en cours de différenciation,
alors qu'il n'est pas affecté lorsque la délétion ΔCpG est présente à l'état hétérozygote (Xu et al.,
2006). Cela pourrait suggérer un lien entre rapprochement des Xic et mécanisme de comptage,
plutôt qu'avec le mécanisme de choix. D'autre part, ces observation s'inscrivent dans le modèle à
deux facteurs proposé par J. Lee (fig. 12B, p. 79) (Lee, 2005).
2.3.3.2 Rôle de la région en 3' de Xist dans le comptage
La première délétion d'éléments du Xic conduisant à un défaut de comptage a été obtenue dès
1998 (Clerc et Avner, 1998), c'est-à-dire avant même la découverte de Tsix. Il s'agit de la même
délétion de 65 kb en 3' de Xist que nous avions présentée plus haut comme affectant le choix dans
111

des cellules ES femelles. Lorsque sont différenciées in vitro des cellules essentiellement XO
porteuses de cette délétion, leur chromosome X est recouvert par Xist dans une proportion
importante de cellules, mais de manière transitoire, la disparition des domaines d'ARN Xist étant
précédée par une importante mortalité cellulaire (Clerc et Avner, 1998). La confirmation a été
apportée plus tard que la même région était également requise pour empêcher l'inactivation de
l'unique chromosome X dans des cellules mâles (Morey et al., 2004). Ce phénotype a pu être
complémenté par la réinsertion, dans la délétion de 65 kb portée par des cellules XO, d'un fragment
d'ADN de 37 kb correspondant à la séquence immédiatement en 3' de Xist (Morey et al., 2004)
(C37, fig. 18B, p. 100). Par conséquent, cette région de 37 kb est candidate pour contenir des
éléments de comptage (Morey et al., 2004).
De tels éléments de comptage pourraient servir de numérateur en fixant les facteurs bloquants
ou de compétences. Par conséquent, s'ils sont présents en excès, ils devraient être capables de titrer
ces facteurs et d'inhiber ainsi l'inactivation dans des cellules ES femelles (fig. 12C, p. 79). C'est
précisément ce qui est observé lorsque différents transgènes (pSxn, pCC3, pCC4, p3.7 et pXite,
fig. 18D, p. 100) provenant de la région en 3' de Xist sont insérés en grand nombre de copies sur un
autosome dans des cellules ES femelles (Lee, 2005). En particulier, un transgène (p3.7) de 3,7 kb
correspondant à la région délétée dans les mutants ΔCpG, et un transgène du locus Xite (pXite), sont
chacun capable de réaliser ce phénotype. Des transgènes de plus grande taille (πJL2, πJL3 et pSx7,
fig. 18D, p. 100), contenant une partie ou la totalité du gène Xist, inhibent également l'inactivation
dans des cellules ES femelles, mais de manière partielle et variable selon les clones (Lee, 2005).
Aucun des transgènes mentionnés n'a, par contre, de phénotype observable dans des cellules ES
mâles (Lee, 2005).
De manière intéressante, dans des cellules ES femelles en cours de différenciation, les
transgènes pSx7, p3.7 et pXite se rapprochent transitoirement de l'un des Xic, et semblent inhiber
partiellement le rapprochement des Xic endogènes entre eux, (Xu et al., 2006). Un tel
rapprochement a également lieu entre le transgène pXite et le Xic endogène dans des cellules mâles,
mais il est dans ce cas sans conséquence (Xu et al., 2006). Il est tentant de penser que l'inhibition de
l'inactivation associée à ces transgènes serait la conséquence de leur interférence avec le
rapprochement des Xic, lequel serait, comme le suggèrent également les mutants XΔCpG/XΔCpG (cf
section précédente), essentiel au processus de comptage.
Le phénotype associé aux transgènes p3.7 et pXite est différent de celui observé lorsque les
mêmes éléments sont délétés de l'un des Xic endogènes dans des cellules ES femelles ( XΔCpGX et
XΔXiteX (cf section précédente). D'autre part, la délétion de Xite, de même que la délétion ΔCpG, ne
112

conduisent pas à un défaut de comptage dans les cellules ES mâles (Ogawa et Lee, 2003). Les
transgènes p3.7 et pXite ne semblent donc pas interférer seulement avec les séquences
correspondantes du Xic, par exemple pour la fixation de facteurs de comptage, mais pourraient plus
largement destructurer la région en 3' de Xist ou le Xic dans son ensemble, notamment en interférant
avec le rapprochement des Xic. Pour mieux comprendre les mécanismes d'action de ces transgènes,
il serait intéressant de savoir si le phénotype qui leur est associé pourrait être modulé en fonction de
leur nombre de copies. La comparaison de leurs profils transcriptionnels et chromatiniens avec ceux
des Xic endogènes pourrait également être informative.
113

114

Chapitre 3 Régulation de l'expression de Tsix et minisatellite
DXPas34
3.1 Facteurs généraux de transcription et marques chromatiniennes au
promoteur de Tsix
La transcription de Tsix est réalisée grâce à la polymérase II (Navarro et al., 2005). Sur le Xm
des cellules TS, ainsi que dans les cellules ES mâles ou femelles, où Tsix est exprimé, son
promoteur majoritaire est associé à la polymérase II et au facteur général de transcription TFIIB.
Les facteurs TFIIA, TFIIE, TFIIF et TFIIH ont également été observés au promoteur de Tsix dans
les cellules ES femelles indifférenciées (Navarro et al., 2005). Comme attendu, la machinerie de
transcription n'est pas recrutée au promoteur de Tsix sur le Xp des cellules TS, ni dans les
fibroblastes mâles ou femelles, où Tsix est éteint (Navarro et al., 2005).
L'association à la modification d'histone H3K27me3, considérée comme répressive pour la
transcription, est symétrique de l'association aux facteurs généraux de transcription: elle est
observée sur le Xp des cellules TS et dans les fibroblastes mâles ou femelles. Par contre,
l'association avec la marque H3K9me2, également répressive, n'est jamais observée.
L'acétylation des histones H3 et H4, en général associée à des promoteurs
transcriptionnellement actifs, est enrichie au promoteur majoritaire de Tsix dans les cellules ES
mâle, mais pas dans les fibroblastes mâles, ce qui corrèle avec l'expression de Tsix (Kimura et al.,
2002). La situation dans les cellules femelles n'est cependant pas connue. La modification
H3K4me2, également associée à l'activité transcriptionnelle, corrèle bien avec l'expression de Tsix
dans les cellules ES et sur le Xa des cellules TS. Mais, de manière inattendue, elle est également
enrichie au promoteur majoritaire de Tsix dans des fibroblastes mâles et femelles en l'absence
d'expression de ce dernier (Kimura et al., 2002; Navarro et al., 2005). Il est difficile d'associer cette
particularité à une quelconque fonction, mais il est tentant de penser qu'elle pourrait être liée aux
mécanismes de comptages ou de choix mis en oeuvre dans l'inactivation aléatoire (Navarro et al.,
2005).
115

3.2 Régulateurs transcriptionnels de Tsix
Des éléments régulant l'expression de Tsix ont été recherchés par Stavropoulos et al. au moyen
de tests luciférase, combinant le promoteur minimal de Tsix, obtenu à partir du promoteur
majoritaire, et des fragments d'ADN provenant d'une région s'étendant de 27 kb en amont à 4 kb en
aval de ce promoteur (Stavropoulos et al., 2005). Cette étude à permis de montrer que le promoteur
minimal était constitutivement actif, aussi bien dans des cellules mâles que femelles, différenciées
ou non. L'extinction en cours de différenciation, ainsi qu'un niveau plus fort d'expression avant
différenciation, sont obtenus en le combinant à des éléments enhancers mis en évidence au locus
Xite, d'une part, et dans une région centrée sur le promoteur minimal, d'autre part. Ce second
enhancer, qualifié de bipartite car scindé en deux au niveau du promoteur de Tsix, est constitué en
amont par une région de 4,9 kb, et en aval par une région de 4 kb incluant le minisatellite DXPas34
(Stavropoulos et al., 2005). Ce dernier est requis, mais pas suffisant, pour l'activité de l'enhancer
bipartite (Stavropoulos et al., 2005)
Xite a été par ailleurs positionné génétiquement en amont de Tsix (Ogawa et Lee, 2003).
Cependant, lorsqu'il est délété sur l'un des Xic dans des cellules femelles, l'expression de Tsix est
perdue plus rapidement sur l'allèle muté que sur l'allèle sauvage (Ogawa et Lee, 2003). Dans le
contexte du Xic, Xite permet donc de maintenir plus longtemps l'expression de Tsix en cis, alors que
les tests luciférase auraient suggéré un comportement opposé. Cela incite à une certaine retenue
dans l'interprétation des expériences reposant sur des tests luciférase, et à la poursuite de l'étude
détaillée du Xic au moyen de délétions ciblées dans le locus endogène, pour caractériser de manière
fiable les régulateurs transcriptionnels de Tsix.
DXPas34 est un bon candidat pour une telle approche. Nous présenterons les principales
caractéristiques de ce minisatellite à la section suivante, puis dans la section résultats, dans lequelle
nous détaillerons le phénotype associé à sa délétion.
3.3 Le minisatellite DXPas34
Nous avons déjà évoqué le minisatellite DXPas34 à quelques reprise, pour mentionner qu'il est
le lieu d'un épissage alternatif de Tsix (Shibata et Lee, 2003), et qu'il peut initier une transcription
bidirectionnelle, mise en évidence récemment (Cohen et al., 2007) (cf introduction, partie 2,
116

section 2.2, p. 98). De plus, dans les tests luciférase présentés à la section précédente, DXPas34 est
requis, mais pas suffisant, pour l'activité de l'enhancer bipartite, suggérant une fonction dans la
régulation transcriptionnelle de Tsix (Stavropoulos et al., 2005).
DXPas34 est formé par la répétition en tandem d'un motif de 34 pb, riche en CpG, s'étendant sur
1,2 kb. Il est associé à un îlot CpG qui contient le promoteur majoritaire de Tsix. La caractérisation
de DXPas34 a été faite initialement sur la base d'un profil de méthylation de l'ADN inhabituel, qui
semblait spécifique du Xa dans les cellules somatiques et les cellules ES différenciées (Avner et al.,
1998; Courtier et al., 1995; Prissette et al., 2001; Simmler et al., 1993).
Ainsi, l'ensemble constitué par DXPas34 et l'îlot CpG adjacent est globalement déméthylé au
niveau des CpG dans le sperme, les ovocytes, les embryons jusqu'au stade blastocyste, et les
cellules ES non différenciées (Prissette et al., 2001). Mais, dans les tissus embryonnaires et
extraembryonnaires à partir de E7,5, les CpG sont méthylés de manière plus importante chez les
mâles que les femelles (Prissette et al., 2001) Ils sont également méthylés de manière importante
lors de la différenciation de cellules ES mâles (Prissette et al., 2001). Cependant, le caractère
différentiel, entre les deux sexes, de cette méthylation apparaît moins prononcé dans les tissus
extraembryonnaires (Prissette et al., 2001). Prises dans leur ensemble, ces observations ont conduit
à associer au Xa la méthylation de DXPas34 et de la région promotrice de Tsix (Prissette et al.,
2001). Une telle conclusion est dans une certaine mesure paradoxale, car le chromosome X actif est
précisément celui qui est supposé avoir maintenu le plus longtemps l'expression de Tsix en cours de
différenciation (cf introduction, partie 2, section 2.2.2.1, p. 102), tandis que la méthylation de
l'ADN marque généralement les promoteurs inactifs. Cependant, il s'agit ici d'une marque tardive,
probablement postérieure aux évènements de comptage ou de choix. D'autre part, le degré de
méthylation dans les cellules somatiques varie en fonction du fond génétique (Avner et al., 1998;
Courtier et al., 1995; Prissette et al., 2001), mais indépendamment de l'allèle Xce (Prissette et al.,
2001). Il est donc difficile d'assigner une fonction à cette méthylation. Tout au plus peut-on
supposer qu'elle contribue à la stabilisation du patron d'inactivation, selon des mécanismes pour
l'instant inconnus.
Une telle méthylation différentielle présente néanmoins une certaine similitude avec les DMD
(Differentially Methylated Domains) associés aux gènes autosomiques dont l'expression est soumise
à l'empreinte parentale. Le centre d'empreinte des gènes H19 et Igf2 en est un exemple, parmi les
plus connus (pour revue, Wood et Oakey, 2006). Sa fonction est étroitement liée à son association
avec le facteur CTCF (CCCTC-binding factor), qui est elle-même abolie lorsque les CpG du DMD
sont méthylés. Cela a conduit à examiner la présence de CTCF au niveau de DXPas34.
117

De nombreux sites putatifs de fixation de CTCF sont prédits dans et à proximité de DXPas34
(fig. 19, p. 118) (Chao et al., 2002; Donohoe et al., 2007). La fixation de CTCF au niveau de
DXPas34 est effectivement observée in vivo par immunoprécipitation de chromatine (Chao et al.,
2002; Donohoe et al., 2007; Navarro, thèse de doctorat, 2006). Pour Navarro et al., CTCF est
associé à DXPas34 dans les cellules ES non différenciées, mais absent dans les fibroblastes, qu'il
s'agisse, dans les deux cas, de cellules mâles ou femelles (Navarro, thèse de doctorat, 2006). La
présence de CTCF dans des fibroblastes femelles est au contraire détectée par Chao et al. (Chao et
al., 2002). Enfin, Donohoe et al. observent, au cours de la différenciation de cellules ES mâles et
femelles, le maintien de CTCF à deux sites de fixation situés entre le promoteur de Tsix et
DXPas34, mais sa disparition à deux sites de fixation proches de DXPas34, en aval (par référence à
Tsix) de ce dernier (Donohoe et al., 2007).
Le comportement similaire observé dans les mâles et les femelles par Navarro et al. peut
paraître surprenant lorsqu'on sait que les CpG dans DXPas34 sont méthylés différentiellement, et
que la fixation de CTCF est sensible à la méthylation des CpG. Cependant, il semblerait que la
fixation de CTCF au niveau de DXPas34 soit moins sensible à la méthylation des cytosines
appartenant au motif CpG qu'à la méthylation des cytosines externes à ce motif (Chao et al., 2002).
D'autre part, la méthylation de certains sites putatifs de fixation de CTCF localisés à proximité de
DXPas34 révèle une certaine autonomie par rapport à la méthylation de l'ensemble des CpG de la
région. Elle est en effet réalisée à un niveau apparemment proche sur le Xa ou le Xi, dans des
cellules ES différenciées ou des cellules TS, mâles ou femelles. Ces sites sont également méthylés
dans le sperme, ce qui pourrait suggérer un rôle éventuel de DXPas34, via CTCF, dans l'acquisition
d'une empreinte paternelle (Boumil et al., 2006).
118
Fig. 19: Sites consensus de fixation de CTCF et YY1 dans et à proximité de DXPas34 (d'après Donohoe et al., 2007)
La deux délétions de DXPas34, générées par nous même (cf résultats, chapitre 1, p. 131) et par un autre laboratoire, sont représentées au dessus du locus DXPas34 (cf résultats, chapitre 1, p. 131).

La plupart des sites putatifs de fixation de CTCF dans DXPas34, et à proximité de celui-ci,
sont associés à des sites putatifs de fixation du facteur YY1 (fig. 19, p. 118) (Donohoe et al., 2007;
Kim et al., 2006). YY1 est un facteur de transcription à doigts de zinc de la famille Gli-Kruppel
(Kim et al., 2006). Il est l'orthologue de la protéine PHO, membre de la famille polycomb chez la
drosophile (Atchison et al., 2003). Comme CTCF, YY1 joue également un rôle essentiel dans la
régulation de certains loci soumis à l'empreinte parentale et est sensible à la méthylation de l'ADN
(Kim et al., 2006). Il est également très versatile quant à sa fonction, qui sert à activer ou réprimer
de nombreux gènes, selon le contexte (Dunn et Davie, 2003; Shi et al., 1997; Thomas et Seto,
1999).
La fixation de YY1 sur DXPas34 est observée dans les cellules ES indifférenciées mâles et
femelles (Donohoe et al., 2007; Navarro, thèse de doctorat, 2006). Elle est maintenue lors de la
différenciation de cellules femelles, alors que la fixation de CTCF a déjà été perdue sur certains
sites de fixation (Donohoe et al., 2007). Par contre, elle n'est plus détectée dans des fibroblastes
mâles ou femelles (Navarro, thèse de doctorat, 2006).
En l'absence de YY1 ou de CTCF dans des cellules ES mâles, l'expression de Tsix est fortement
diminuée et celle de Xist augmentée (Donohoe et al., 2007). Des expériences de co-
immunoprécipitation et des tests luciférase suggèrent que ces deux protéines pourraient collaborer
pour activer l'expression de Tsix (Donohoe et al., 2007). Toutefois, les cinétiques différentes de
perte de CTCF et de YY1 sur DXPas34 en cours de différenciation (Donohoe et al., 2007)
suggèrent que les fonctions de ces deux facteurs pourraient avoir un certain degré d'autonomie.
In vivo, l'invalidation de Ctcf par RNAi est léthale avant l'implantation. L'invalidation par
knock-out de Yy1 est léthale dans la période péri-implantatoire lorsqu'elle est réalisée sur les deux
allèles, mais ne conduit qu'à un retard modéré de développement à l'état hétérozygote (Donohoe et
al., 2007). Par contre, des blastocystes mâles hétérozygotes pour le knock-out de Yy1, cultivés in
vitro pendant 5-6 jours, témoignent d'une expression ectopique de Xist dans environ 30 % des
masses cellulaires internes différenciées et des excroissances trophoblastiques (trophoblast
outgrowth). Nous verrons plus loin qu'il est difficile d'attribuer ces défauts au seul rôle de DXPas34,
car CTCF et YY1 se fixent également au début du gène Xist. La mutagénèse ciblée des sites de
fixation de ces deux facteurs, plus facile à réaliser dans le cas du gène Xist, car les sites putatifs de
fixation sont moins nombreux, serait requise pour déterminer le rôle joué par CTCF et YY1
spécifiquement au niveau de DXPas34.
119

Enfin, sans que l'on sache si cela peut être relié à la présence de CTCF ou de YY1, des sites
d'hypersensibilité à la DNAse I, témoignant d'une chromatine « ouverte », accessible aux facteurs
de transcription, ont été caractérisés au niveau de DXPas34. Des sites d'hypersensibilité à la
DNAse I sont également présents au niveau de Xite et dans la région de l'enhancer bipartite en
amont du promoteur de Tsix (Boumil et al., 2006; Ogawa et Lee, 2003; Stavropoulos et al., 2005).
Une autre similarité entre DXPas34 et Xite est la capacité à initier une transcription bidirectionnelle
(Cohen et al., 2007; Ogawa et Lee, 2003; Stavropoulos et al., 2005). Exception faite de la
transcription initiée dans DXPas34, qui n'a été analysée que dans des cellules mâles, sites
d'hypersensibilité et transcription bidirectionnelle sont observés aussi bien dans des cellules ES
indifférenciées mâles que femelles, avant d'être perdus en cours de différenciation (Boumil et al.,
2006; Cohen et al., 2007; Ogawa et Lee, 2003; Stavropoulos et al., 2005). Il se pourrait que ces
similarités, particulièrement entre DXPas34 et Xite, traduisent une régulation globale de la région
en 3' de Xist, dont les modalités resteraient à découvrir.
120

Chapitre 4 Régulation de l'expression de Xist par Tsix
Dans tous les mutants de Tsix publiés à ce jour, l'absence d'expression de Tsix se traduit en cis
par une augmentation de l'expression de Xist dans les cellules ES indifférenciées, aussi bien mâles
que femelles (Lee et Lu, 1999; Luikenhuis et al., 2001; Morey et al., 2001; Sado et al., 2001; Sun et
al., 2006; Vigneau et al., 2006). Inversement, lorsque Xist est surexprimé de manière ectopique dans
des mutants de la région 5' de Xist, l'expression de Tsix n'est pas modifiée (Nesterova et al., 2003).
Tsix serait donc génétiquement en amont de Xist.
Par ailleurs, Tsix et Xist ont des effets opposés sur le choix primaire: l'invalidation de Tsix
conduit à l'initiation préférentielle de l'inactivation en cis, de la même manière que l'expression
ectopique de Xist, tandis que l'invalidation de Xist, tout au moins dans certains mutants, biaise le
choix primaire en faveur de l'inactivation du chromosome X en trans. Le choix du futur Xi semble
donc dépendre soit du rapport de la quantité des transcrits Xist et Tsix sur chaque chromosome, soit
du niveau d'expression de Xist, lui-même régulé par Tsix.
C'est de cette régulation de Xist par Tsix dont nous allons discuter à présent, en l'envisageant de
deux manières: aux niveaux post-transcriptionnel et transcriptionnel.
4.1 Régulation post-transcriptionnelle
Dans des cellules ES mâles indifférenciées mutantes pour Tsix (Ma2L et XΔ65Y), la quantité de
transcrits Xist est fortement augmentée mais aucun recrutement de TFIIB ou de PolII n'est observé à
son promoteur (Navarro et al., 2005). De plus, la transcription de Xist mesurée par run-on n'est pas
augmentée dans des cellules ES indifférenciées XΔCpGY (Sun et al., 2006). Tsix régule donc le
niveau d'expression de Xist de manière post-transcriptionnelle, tout au moins dans les cellules ES
mâles indifférenciées. D'autre part, lorsque dans des cellules ES femelles, Tsix est invalidé sur l'un
des chromosomes X, l'expression de Xist est augmentée spécifiquement à partir de ce chromosome
(Lee et Lu, 1999; Luikenhuis et al., 2001; Morey et al., 2001; Shibata et Lee, 2004). Par
conséquent, si l'on admet que, à l'instar des cellules mâles, Tsix régule Xist au niveau post-
121

transcriptionnel dans les cellules ES femelles indifférenciées, cette régulation doit avoir lieu avant
la diffusion éventuelle des transcrits Xist et Tsix dans le noyau, et devrait donc être réalisée dans un
laps de temps bref après la transcrtiption.
Un mode possible de régulation post-transcriptionnelle de Xist consisterait en la destabilisation
des transcrits Xist par Tsix. Cette idée avait initialement été avancée d'après l'observation d'une
stabilisation des transcrits Xist au cours de la différenciation de cellules ES, durant laquelle
l'expression de Tsix est perdue (Panning et al., 1997; Sheardown et al., 1997a). Néanmoins, cette
stabilisation avait été conclue à partir d'expériences antérieures à la découverte de Tsix, et a été
remise en cause récemment par des travaux tenant compte de ce dernier (Sun et al., 2006). Les
mêmes auteurs ont également évalué la stabilité de Xist dans des cellules ES mâles et femelles
mutantes pour Tsix (XΔCpGY et XΔCpGX, fig. 18, p. 100). Ils concluent que la stabilité de Xist n'est
modifiée ni au cours de la différenciation de cellules ES, ni par l'expression de Tsix (Sun et al.,
2006). La dégradation des ARN Xist par un mécanisme de type RNAi dépendant de Tsix semble
donc également peu probable, mais il n'est pas exclu qu'un mécanisme de ce type puisse conduire à
altérer la conformation de la chromatine au niveau de Xist, sans affecter la stabilité de ses transcrits.
Nous verrons à la section suivante qu'une modification de la chromatine dans la région promotrice
de Xist est effectivement observée dans les cellules où Tsix a été invalidé.
Il faut néanmoins nuancer l'observation de Sun et al. en remarquant que leur analyse de stabilité
a porté sur les transcrits totaux de Xist, mais n'a pas examiné ses formes épissées. Or, il a été montré
que l'abondance des transcrits Xist dans des cellules ES femelles différenciées pouvait être régulée
au moment de leur maturation par la voie NMD (non-sense RNA mediated decay). Plus
spécifiquement, la voie NMD est requise pour l'obtention de transcrits épissés, mais n'a pas d'effet
sur le niveau des transcrits non épissés de Xist (Ciaudo et al., 2006). La régulation de Xist par la
voie NMD n'est pas médiée par Tsix, dans la mesure où l'expression de ce dernier n'est pas affecté
lorsque sont interférés des éléments de la voie NMD (Ciaudo et al., 2006). Néanmoins, un rôle de
Tsix spécifiquement dans la stabilité ou l'obtention des formes épissées de Xist, indépendamment de
la voie NMD, reste à évaluer. Pour l'heure, dans les différentes lignées mutantes de Tsix,
l'augmentation consécutive du niveau d'expression de Xist a pu être observée, selon les études, en
mesurant la quantité des transcrits totaux ou épissés de Xist. Néanmoins, il est difficile de tirer des
conclusions quantitatives en comparant ces mesures faites dans des études ou avec des mutants
différents.
122

Tsix pourrait également réguler l'activité de Xist, en interagissant avec ses domaines
fonctionnels (par exemple la répétition A, cf p. 28) pour en inhiber la fonction. Une telle hypothèse
est envisageable grâce à la complémentarité des séquences de Xist et Tsix. Cependant, lorsque sur
l'un des chromosomes X, dans des cellules ES femelles, la transcription de Tsix est stoppée avant de
traverser le gène Xist (Trap, fig. 18, p. 100), et l'expression de Xist augmentée en conséquence sur
ce chromosome, l'insertion en 3' de Xist d'un minigène exprimant une forme épissée (exons 2-3-4)
de Tsix ne permet pas de restaurer un niveau normal d'expression de Xist (Shibata et Lee, 2004).
Plusieurs explications peuvent être avancées à cela: Tsix pourrait interagir avec l'ARN Xist grâce à
se forme non épissée, l'interaction entre les ARN Xist et Tsix pourrait dépendre de l'épissage de Tsix
ou de la transcription antisens à travers Xist, ou l'ARN produit par le minigène diffuserait trop
rapidement dans le noyau (par exemple parce qu'il est de petite taille ou qu'il ne serait pas pris en
charge par la machinerie d'épissage) pour pouvoir interagir avec l'ARN Xist. On voit donc qu'il est
difficile de dissocier ici la fonction de l'ARN Tsix de la manière dont il est produit ou modifié. Mais
s'il était prouvé que la machinerie de transcription ou de modification co-transcriptionnelle des
ARN joue un rôle dans la régulation post-transcriptionnelle de Xist par Tsix, cela expliquerait que
cette dernière soit circonscrite en cis dans les cellules femelles.
4.2 Régulation transcriptionnelle
Xist est transcrit à partir de deux promoteurs, P1 et P2. Dans les cellules somatiques
différenciées, environ 75 % des transcrits sont issus du second (Johnston et al., 1998). En moyenne,
10 copies d'ARN Xist sont présentes par cellule ES femelle, et 4 copies par cellule ES mâle, avant
différenciation (Sun et al., 2006). A cela correspond l'absence de détection de TFIIB et PolII par
ChIP au promoteur P1 (Navarro et al., 2005) et une transcription, mesurée par run-on, extrêmement
faible (Sun et al., 2006). Dans les cellules ayant inactivé l'un de leurs chromosomes X, l'expression
de Xist sur ce chromosome est régulée par le recrutement allélique de la machinerie de transcription.
Cela est observé aussi bien dans les cellules TS et XEN, dans lesquelles l'inactivation est
empreintée, que dans des fibroblastes ou dans des cellules ES différenciées, pour lesquels
l'inactivation est aléatoires (Navarro et al., 2005; Navarro et al., 2006). Le recrutement de cette
machinerie n'est pas réprimé par Tsix dans les cellules ES indifférenciées (Navarro et al., 2005),
mais elle l'est vraisemblablement en cours de différenciation (Navarro et al., 2006; Sado et al.,
2006). En effet, lorsque Tsix est tronqué dans des cellules ES mâles (lignée Ma2L: 2lox, fig. 18C,
p. 100), TFIIB est recruté sur P1, mais pas sur P2, en cours de différenciation, avec pour
123

conséquence la transcription de Xist mesurée grâce à la présence de PolII en élongation (Navarro et
al., 2006). Dans des embryons mâles, l'expression d'un allèle non fonctionnel de Xist, fusionné à la
GFP, est observée de manière ectopique à E13,5 lorsque Tsix a été invalidé en cis (cette
configuration a été baptisée Xdc3 par ses auteurs), mais dans ce cas, la plupart des ARN sont issus
de P2 (Sado et al., 2005). Cette différence pourrait correspondre à des cinétiques différentes
d'activation des promoteurs P1 et P2 en l'absence de Tsix, ou à un jeu complexe entre le recrutement
de la machinerie de transcription, d'une part, et l'initiation de la transcription, d'autre part,
permettant à P1 de recruter les facteurs requis au niveau de P2. Pour discriminer entre ces deux
possibilités, une quantification des transcrits entre P1 et P2 manque à l'étude de Navarro et al., et
une cinétique plus précoce au cours du développement devrait compléter l'analyse réalisée par Sado
et al.
L'absence de Tsix, si elle ne conduit pas à l'activation transcriptionnelle de Xist avant
différenciation, n'en a pas moins pour conséquence une modification de la chromatine au promoteur
de Xist, dont il a été proposé qu'elle « prédestinait » l'allèle ainsi ciblé a être surexprimé en cours de
différenciation (Navarro et al., 2006). En effet, en l'absence de transcription de Tsix à travers Xist,
dans des cellules ES mâles indifférenciées (lignée Ma2L), les marques de type euchromatique
H3K4me2, H3K4me3 et H3K9ac sont augmentées au niveau de P1 et P2, alors que sont diminuées
H3K9me3 et la méthylation des CpG, marques de type hétérochromatique (Navarro et al., 2006).
Les modifications H3K4me2, H3K4me3 et H3K9ac sont également présentes dans des fibroblastes
femelles, qui expriment Xist, mais pas dans des fibroblastes mâles, qui ne l'expriment pas (Navarro
et al., 2006). L'euchromatinisation de la région promotrice de Xist est, de manière similaire,
observée sur le chromosome Xdc dans des embryons mâles à E13,5. Elle se traduit dans ce cas par
l'hypométhylation de l'ADN, l'enrichissement en H3K4me2 et H4ac, et la diminution de H3K27me3
(Sado et al., 2005), qui sont normalement observés sur le Xi (McDonald et al., 1998; Navarro et al.,
2005; Sado et al., 2005; Sun et al., 2006). A l'inverse, le Xa est normalement caractérisé, dans la
région promotrice de Xist, par la méthylation de l'ADN et l'enrichissement en H3K27me3, ainsi que
l'absence des marques euchromatiques H3K4me2, H3K4me3, H3K9ac et H4ac (McDonald et al.,
1998; Navarro et al., 2005; Sado et al., 2005; Sun et al., 2006). D'autre part, sur le Xi ou le Xdc, la
conformation euchromatique dans la région promotrice de Xist est associée à la présence de sites
d'hypersensibilité à la DNAse I (Sado et al., 2005; Sheardown et al., 1997b). Enfin, mentionnons
3 Dans la configuration Xdc, l'invalidation de Tsix est réalisée grâce à la délétion Δ1.7 (fig. 18C, p. 100), et la mutation de Xist correspond à l'allèle XistGFP (fig. 5, p. 27).
124
que le facteur YY1 est associé au promoteur de Xist dans des cellules somatiques adultes,
spécifiquement chez les femelles, c'est-à-dire, probablement, spécifiquement sur le Xi (Kim et al.,
2006).
Pour que le mécanisme de choix primaire soit possible, il faut que la région promotrice de Xist
ait la capacité de basculer aussi bien vers une conformation euchromatique que hétérochromatique
en cours de différenciation. Une telle versatilité pourrait être facilitée par la combinaison
particulière de marques épigénétiques présentes avant différenciation. Dans les cellules ES femelles
indifférenciées, cette région est en effet enrichie en H3K4me2 et H3K9me3, mais appauvrie en
H3K4me3, H3K9ac et H3K27me3, une telle combinaison pouvant être interprétée comme un état
réprimé (par H3K9me3), mais potentiellement activable (grâce à H3K4me2) (Navarro et al., 2006).
L'ADN est par ailleurs globalement hypométhylé au niveau de la région promotrice de Xist dans les
embryons mâles et femelles jusqu'au stade blastocyste inclus (McDonald et al., 1998), ainsi que
dans les cellules ES femelles indifférenciées (Norris et al., 1994; Penny et al., 1996; Sado et al.,
125
Fig. 20: Régulation de la région promotrice de Xist par Tsix (d'après Navarro et al., 2006)
(A) Ratio des marques chromatiniennes mesurées par immunoprécipitation de chromatine (ChIP) dans les lignées Ma2L et Ma1L, les transcrits Tsix étant tronqués dans les premières mais pas dans les secondes. Les valeurs sont données sur une échelle logarithmique de base 2, et centrées sur le promoteur P1 de Xist (valeur 0 en abcisse). Les valeurs en abcisse sont en kb. Le promoteur P2 est à la position +1,5 kb. (B et C) Fixation de CTCF mesurée par ChIP aux mêmes positions génomiques, dans des cellules ES femelles sauvages (B) et dans des cellules ES mâles invalidées ou non pour Tsix (lignées Ma2L et Ma1L, respectivement) (C). Les auteurs proposent que la fixation de CTCF pourrait isoler la région promotrice de Xist du profil chromatinien observé dans le corps du gène Xist.

1996; Sun et al., 2006). Mais il semble méthylé dans les cellules ES mâles, tout au moins pour un
petit nombre de positions testées (Norris et al., 1994; Sun et al., 2006). Cette méthylation semble
dépendre de l'association des ARN Tsix avec le méthylase Dnmt3a (Sun et al., 2006).
Une autre étude (Sun et al., 2006) a néanmoins des conclusions différentes de celle de Navarro
et al. (Navarro et al., 2006), concernant la régulation par Tsix de la chromatine au promoteur de
Xist. Leurs auteurs n'y observent en effet pas de modification des marques H3K4me2, H3K27me3
et H4ac dans des cellules ES femelles indifférenciées XΔCpGX, qui n'expriment pas Tsix sur l'un de
leur chromosome X. D'autre part, dans des cellules ES mâles XΔCpGY, ils ne mesurent pas de
diminution de la méthylation de l'ADN à proximité du promoteur de Xist, aussi bien lorsque les
cellules sont indifférenciées qu'en cours de différenciation. Dans les cellules ES femelles
indifférenciées XΔCpGX, la méthylation de l'ADN au promoteur de Xist ne semble pas non plus
altérée sur le chromosome porteur de la délétion, mais après différenciation, elle est observée, de
manière attendue, presque exclusivement sur le chromosome X sauvage, qui dans ces lignées n'est
jamais inactivé. Ces différences avec les profils présentés par Navarro et al. appellent une remarque,
concernant la résolution des deux études. Ainsi, Sun et al. n'ont testé qu'une seule position dans la
région promotrice de Xist, contrairement à Navarro et al., qui ont réalisé pour chacune des
modifications un profil incluant de nombreuses positions et permettant de définir précisément les
limites, de part et d'autre des promoteurs P1 et P2, des modifications qu'ils observent. Précisément,
les observations de Sun et al. ne sont pas en désaccord avec les mesures réalisées par Navarro et al.
aux mêmes positions, qui sont à la marge de la région euchromatinisée en l'absence de Tsix
(Navarro, 2006; Navarro et al., 2006).
Cette région est à peu près celle où les séquences des transcrits épissés de Tsix et de Xist sont
complémentaires (Navarro et al., 2006). Il est donc tentant de penser que le mécanisme permettant
cette euchromatinisation mette en jeu une interaction directe des transcrits Xist et Tsix, dont nous
avons discuté à la section précédente. On peut également imaginer qu'il utiliserait la terminaison de
Tsix ou l'épissage de son dernier exon pour définir les bornes de la région euchromatique.
Cependant, Xist est correctement réprimé, et sa région promotrice est correctement euchromatinisée,
à E13,5, dans des mutants de Tsix déficients pour l'épissage du dernier exon, ce qui exclut un rôle
du mécanisme d'épissage (Sado et al., 2006). Quant à tester la fonction des transcrits Tsix, cela
pourrait être fait en regardant si l'expression d'un minigène de Tsix, en l'absence de transcription
antisens à travers Xist (mutant décrit p. 108) (Shibata et Lee, 2004), conduit à l'euchromatinisation
de la région promotrice de Xist dans des cellules ES indifférenciées.
126

D'autre part, l'absence d'activation transcriptionnelle de Xist malgré l'euchromatinisation de sa
région promotrice, dans les cellules ES indifférenciées mutées pour Tsix, suggère l'existence d'un
répresseur transcriptionnel de Xist, perdu en cours de différenciation (Navarro et al., 2005) (fig. 21,
p. 128). On peut également imaginer que, bien que le promoteur de Xist soit euchromatinisé, la
chromatine associée au reste du locus Xist ne soit pas compétente pour permettre l'expression de ce
gène à un fort niveau. Des remaniements de nature épigénétique devraient alors avoir lieu en cours
de différenciation pour permettre cette expression. Comme nous le verrons à la section suivante,
puis au chapitre 3 de la section résultats (p. 165), Tsix joue aussi un rôle important dans la
détermination du profil épigénétique d'ensemble du locus Xist/Tsix.
4.3 Remodelage de la chromatine associé à l'expression de Tsix
Le corps du gène Xist est associé aux marques H3K4me2 et H3K9me3, et à l'absence des
marques H3K4me3, H3K9ac et H3K27me3 (Navarro et al., 2006). Lorsque Tsix est tronqué (lignée
Ma2L) ou absent (Δ65, fig. 18B, p. 100), la marque H3K4me2 y est pratiquement perdue (Morey
et al., 2004; Navarro et al., 2005). Mais elle est restaurée en même temps que l'expression de Tsix,
après avoir ôté le signal d'arrêt de transcription dans Ma2L, ou complémenté la délétion de 65 kb de
manière à restaurer l'expression de Tsix (C16, fig. 18B, p. 100) (Morey et al., 2004; Navarro et al.,
2005). L'absence de Tsix conduit également à l'apparition de H3K27me3 dans le corps du gène Xist,
qui sera développée dans l'article 2 (p. 165), mais n'a pas d'effet sur les autres marques citées plus
haut. Des observations similaires ont été réalisées avec la délétion ΔCpG (fig. 18C, p. 100), qui a
pour conséquence l'enrichissement en H3K27me3 et la perte de H3K4me2 et d'acétylation de H4
dans le corps du gène Xist en cis de la délétion, dans des cellules ES femelles (Sun et al., 2006).
Perte de H3K4me2 et apparition de H3K27me3 correspondent à l'acquisition de caractères
hétéchromatiques. Or, nous avons dit (cf section précédente) qu'en l'absence de Tsix, la région
promotrice de Xist était euchromatinisée. Comment concilier de telles observations, apparemment
contradictoires? En premier lieu, il est raisonnable de penser que ces deux fonctions de Tsix doivent
être protégées, ou isolées, l'une de l'autre. A ce titre, il est intéressant de rappeler que
l'euchromatinisation de la région promotrice de Xist est bien circonscrite: elle commence peu en
amont de P1 et s'achève au niveau de P2. Il a été montré que cette région était délimitée par deux
pics de fixation de CTCF dans les cellules ES indifférenciées, que Tsix soit ou non tronqué, ainsi
que dans des fibroblastes femelles (Navarro et al., 2006)4. Comme CTCF est connu pour pouvoir
4L'amplitude des ces pics peut néanmoins varier selon que Tsix est exprimé ou non. Voir le profil fig. 20, p. 125 dans
127

réaliser une fonction insulatrice, y compris sur le chromosome X (Filippova et al., 2005) ou vis-à-
vis de H3K9me3 (Cho et al., 2005), il s'agit d'un bon candidat pour délimiter spatialement les
fonctions de Tsix au promoteur et dans le corps de Xist. Par ailleurs, des observations suggèrent
qu'une fonction de CTCF au promoteur de Xist pourrait être conservée chez l'homme (Pugacheva et
al., 2005).
Il a été proposé que la déposition de H3K4me2 dans le corps de Xist pourrait contribuer à
effacer les marques d'empreinte et à rendre les deux allèles de Xist épigénétiquement équivalent, au
moment de la réactivation du Xp dans la masse cellulaire interne des blastocystes (Navarro et al.,
les lignées Ma2L, qui est très proche de celui observé dans des fibroblastes femelles (Navarro et al., 2006).
128
Fig. 21: Modèle de reprogrammation des chromosomes X proposé par Navarro et al. (Navarro et al., 2005)
Légende page 129

2005) (fig. 21, p. 128). Dans la mesure où l'expression de Tsix est perdue en cours de
différenciation, la régulation de H3K4me2 et H3K27me3 par Tsix pourrait également jouer un rôle
essentiel lors de la mise en place de l'inactivation aléatoire. Tester cette hypothèse requiert de mieux
comprendre comment Tsix peut influer sur de telles marques, et quelle est leur cinétique au cours du
développement. Nous développerons cette question chapitre 3 de la section résultats (p. 165).
129
Légende de la figure 21, p. 128: Les stades développementaux sont représentés à gauche, et la région Xist/Tsix sur les deux chromomosomes X à droite, en visàvis. L'intensité du trait de la flèche illustre dans chaque figure le niveau d'expression. (A) Dans les embryons préimplantoires, l'expression de Xist sur l'allèle paternel et de Tsix sur l'allèle maternel est réalisée grâce à la fixation différentielle sur leur promoteur des facteurs généraux de transcription, ainsi que de modifications d'histones telles que H3K4me2. Il s'agit ici d'une extrapolation à partir de modèles cellulaires des tissus extraembryonnaires, réalisant l'inactivation sur un mode empreinté également observé dans les embryons préimplantatoires. (B) Au moment de l'implantation, un répresseur pour l'instant inconnu (figuré par un point d'interrogation) réprimerait le recrutement de la machinerie de transcription sur les promoteurs de Xist, spécifiquement dans la masse cellulaire interne des blastocystes. Parallèlement, l'expression biallélique de Tsix effacerait les profils de modification d'histones en induisant le recrutement d'un fort niveau de H3K4me2 (flèches roses), ce qui aurait pour conséquence de rendre les deux allèles de Xist épigénétiquement équivalents. (C) Lorsque l'inactivation aléatoire est initiée, la répression de Tsix sur l'un des chromosomes X aurait pour conséquence un recrutement de H3K4me2 (et plus généralement une euchromatinisation) au niveau du promoteur de Xist, tandis que la disparition du facteur répresseur autoriserait le recrutement de la machinerie de transcription. La conséquence en serait un fort niveau d'expression monoallélique de Xist. (D) Dans l'épiblaste des embryons femelles, d'autres marques épigénétiques permettraient de consolider l'expression monoallélique de Xist, et l'extinction biallélique de Tsix.

130

Résultats
Chapitre 1 Tsix et le minisatellite DXPas34 jouent un rôle
essentiel dans le comptage
Une région candidate pour contenir des éléments nécessaires au processus de comptage, longue
de 37 kb, a été caractérisée en 3' de Xist par Morey et al. (Morey et al., 2004). Les expériences
ayant permis cette caractérisation ont été décrites à la section 2.3.3.2 de la seconde partie de
l'introduction (p. 111). Cette région contient, en particulier, les éléments suivant:
– une région d'homologie à un enhancer du gène pTa (pre-T cell receptor α) dans les cellules
pre-T (Reizis et Leder, 1999);
– la répétition en tandem, sur environ 2,7 kb, d'un motif de 17 pb, partageant en deux la région
d'homologie mentionnée avant;
– le minisatellite DXPas34;
– le promoteur majoritaire de Tsix;
– le locus Xite;
– le promoteur minoritaire de Tsix.
Nous avons entrepris de tester la fonction des quatre premiers éléments de cette liste, au moyen
de trois délétions ciblées dans le Xic, de taille croissante en partant de la fin du gène Xist, ainsi que
par une délétion précise du minisatellite DXPas34. Toutes ces délétions ont été réalisées par
recombinaison homologue dans des cellules ES mâles, de manière à déterminer si les éléments
ciblés jouent un rôle dans le processus de comptage, mais aussi à pouvoir en tester plus facilement
la participation éventuelle à la régulation de l'expression de Tsix.
Lorsque nous avons entrepris ce travail, différentes invalidations de Tsix donnaient des résultats
contradictoires quant à son rôle dans le processus de comptage (cf introduction, partie 2,
section 2.3.3.1, p. 110), et il était généralement admis qu'il n'y participait pas. D'autre part, plusieurs
délétions incluant DXPas34 avaient été réalisées, mais généralement avec l'objectif d'étudier le rôle
131

de Tsix, et par conséquent, soit en délétaient le promoteur, soit inséraient un signal d'arrêt de
transcription pour Tsix (fig. 18, p. 100). Enfin, les connaissances concernant DXPas34 étaient plus
limitées qu'elles ne le sont aujourd'hui. Nous connaissions le profil de méthylation différentielle de
ces CpG (Avner et al., 1998; Courtier et al., 1995; Prissette et al., 2001; Simmler et al., 1993) ainsi
que, mais de manière moins précise qu'aujourd'hui, sa capacité à fixer CTCF (Chao et al., 2002).
Enfin, certaines données, non publiées, suggéraient que DXPas34 lui-même était capable d'initier
une transcription antisens à Xist (Morey, 2003).
Les résultats présentés dans l'article qui suit on permis de montrer de le rôle essentiel joué par
Tsix pour empêcher l'inactivation d'être initiée dans les cellules mâles, ainsi que la fonction de
DXPas34 dans la régulation transcriptionnelle de Tsix.
1.1 Article 1: An essential role for the DXPas34 tandem repeat and Tsix
transcription in the counting process of X chromosome inactivation
132

133

134

135

136

137

138

Fig. 5. Targeting strategy used for the deletion of DXPas34 and Southern blot analysis of the
resulting ES cell lines. (A) The homologous recombination construct is depicted underneath the map
of the region. Probe HF (empty box) and restriction sites relevant for Southern blot analysis are
indicated (B, Bsu36I, BE, BstEII, P, PshAI). Primers d34f and d34r were used for PCR screening of
the neomycin-resistant clones. Primers dneof and dneor were used to screen for the removal of the
selection cassette after cre expression. The nomenclature of the various cell lines corresponding to
the mutations is indicated on the right. (B) Southern blot analysis of the Neo34#1 and 34#1
clones. Probes, restriction enzymes, and cell lines are indicated. A second independent clone
(34#2) was controlled in the same way and showed similar profile. The absence of integration of
the homologous recombination construct elsewhere in the genome was further checked by DNA-
FISH (not shown).
Fig. 6. Artifactual effects of the pgk1-neo selection cassette can partially mask the phenotype
associated with the targeted deletion of the DXPas34 tandem repeat. Location of the PCR assays
used in A and B is described in Fig. 1C. (A) Real-time quantitative random RT-PCR analysis of Xist
and Tsix normalized to Arpo in undifferentiated ES cells. Both the increase in Xist levels and the
decrease in Tsix levels, which are detected in the 34#2 ES cell line, are masked when the pgk1-neo
cassette is left in place as in the Neo34#2 ES cell line. Columns and error bars represent the means
and standard deviations of the 2-CT ratios of Xist/Tsix versus Arpo (Xist/Tsix, n = 3; Arpo, n = 3).
Cycle thresholds for RT reactions lacking reverse transcriptase were systematically nine cycles
greater than RT+. Similar results and conclusions were drawn when using the 34#1 and Neo34#1
paired ES cell lines. (B) Real-time quantitative random RT-PCR analysis of Xist and Tsix
normalized to Arpo in differentiated ES cells. The induction of Xist expression observed in the
34#2 ES cell line upon differentiation is reduced in the Neo34#2 ES cell line. Higher levels of
Tsix RNA were found in the Neo34#2 ES cell line when compared with the 34#2 ES cell line
during differentiation.
Fig. 7. Quantitative RT-PCR (qRT-PCR) triplicate measurements of the levels of spliced Tsix
transcript normalized to the Arpo reporter in ES cells differentiated by using retinoic acid. The
kinetics of reduction of Tsix levels during differentiation are similar in the CK35 and 34#1 ES cell
lines for positions X9 and Te3 (and for positions Te23 and Te1; see Fig. 2c).
139

Fig. 8. Strategy for targeting nested deletions to within Tsix and Southern blot analysis of the
resulting ES cell lines. (A) General protocol. A first round of homologous recombination
(insertional vectors were used) targets, upstream from the region to be deleted, a loxP site associated
with a transcribed hygromycin gene (H), a puromycin thymidine-kinase (PTK) ORF, and a FRT
site. A second round of homologous recombination is then used to target, downstream of the region
to be deleted, a cassette bearing the neomycin gene (Neo) a loxP and a FRT site. loxP site-specific
recombination mediated by cre recombinase expression generates the expected deletion and
simultaneously reconstitutes a functional PTK gene allowing for puromycin selection of the deleted
cells. The PTK gene is removed by expression of the Flp recombinase. (B) Map of the Xist/Tsix
region featuring the probes (empty boxes) used in the Southern blot analysis and relevant restriction
sites (K, KpnI; A, ApaI). The AJ, AS, and AV nested deletions generated from the CK35 ES
cell line are indicated below the map. (C) Southern blot analysis of the AJ#1, AS#1, and AV#1
ES cell lines. Independent clones AJ#2, AS#2, and AV#2 gave identical results. Probes,
restriction enzymes, and cell lines are indicated.
Fig. 9. (A) RNA-FISH pattern (green) in DAPI-stained (blue) nuclei using probe 510 showing a
signal similar to wild-type in the AJ#1 and a collection of faint and dispersed nuclear dots in the
Ma2L ES cell line. The digital imaging treatment fully respects the aspect and the intensity range
observed visually under the microscope by S.V. and P.C. (B) Ectopic Xist RNA accumulation in
differentiated male ES cells results in silencing of the MeCP2 gene. RNA-FISH patterns for Xist
(green) and X-linked gene MeCP2 (red) in DAPI-stained (blue) nuclei of the AV#1 ES cell line
differentiated for 3 days. (Left) Shown are nuclei carrying a domain of Xist RNA accumulation.
(Right) Shown are nuclei lacking a domain of Xist RNA accumulation. In each category, the
percentage of nuclei where MeCP2 signal was detected or not detected is indicated. Failure to detect
a signal for MECP2 is significantly higher in cells presenting a Xist domain than in cells lacking a
Xist domain. This suggests that MECP2 is silenced in 46% of cells of the former category.
Table 1. Oligonucleotides used in this work
Primers used for RT-PCR and chromatin immunoprecipitation (ChIP) analysis
PCR Forward primer Reverse primer
Name Sequence Name SequenceOct3/4 Oct31f ccccaatgccgtgaagttg Oct31r tcagcagcttggcaaactgtt
X3 XistUp3 gcttcacaaggtaaccaacca XistLo3 tgcacagtcaagcaggagtt
X9 XistUp9 tgaccagtacctcgcaagttc XistLo9 ctaagagcacctggctccac
140
Xgr X129 ggttctctctccagaagctaggaaag Xgr1 gctactcacttgtgtattatatggcatga
Xcr X129 ggttctctctccagaagctaggaaag Xcr1 tggtagatggcattgtgtattatatgg
Xe7 XistEx7Up gccatcctccctacctcagaa XistEx7Lo cctgacattgttttccccctaa
X6 XistUp6 tagctgcaatccctttctgc XistLo6 aatgtgggtgttggaaatcaa
cTa cTsixUpA ggagcctaaacctgtctgtc cTsixLoA gtgtgtcatagctcaagagg
Tpb TpbUp tcaatgaaacaacaccttaccttctag TpbLo acattggaggaagaaactgaggtt
Te3 Te3f cgcatagctggcaagcattt Te3r gggacagcggaagagatgg
Te23 Te23f cgcatagctggcaagcattt Te23r aagaagagcgtgatagccagct
X11 XistUp11 gcgcttgcaggtacttttg XistLo14 aagagccttaggtcccgcc
Tpg TpgUp tggagctctttcatgttcttcctt TpgLo atgaatgggcttcttgaatttctact
Upt1 Upt1f aacaggcagaaactggctgaag Upt1r tgaccccactgctcccct
Te1 Te1f tcagtttgagtacagacaccaggc Te1r gacagagtgaaaatccggaagttg
Hprt prom.
HprtUp1 ggcagcgtttctgagcca HprtLo1 aaagcagtgaggtaagcccaac
Arpo prom.
ArpoUp2 ccaataggcatggacgacgt ArpoLo2 cccgcgtgtgccttttatag
Arpo ArpoUp5 tccagaggcaccattgaaatt ArpoLo5 tcgctggctcccacctt
Primers used for RTPCR and chromatin immunoprecipitation (ChIP) analysis
PCR Forward primer Reverse primer
Name Sequence Name Sequence
d34 d34f taagccattagtccgggaacg d34r tcgaccaggcaagctgcta
X18N X18Nf1 aagtgtgtagcgcttgcaggt X18Nr1 gcgcctcaagagccttagg
dneo dneof tttgccttgcccgcc dneor ggtgcccgaagtccttgtt
pblnr pblnrf1 ccatatccgggtgcggtag pblnr1 ttccgcgatatcacttccatg
sl slf1 aaagctagcttacccacgccaggcttattg slr1 tgccgcataacggaggaat
sr srf1 cttcagcctcagacaaacgtgac srr1 aaacaattgggttgaaaatgagggctataccatg
vl vlf1 aaagctagcgcatgtgtactatatcacccccattc
vlr1 ccctttggttgctgttttatatttttc
vr vrf1 ctatacaatatgagtgccattataacatcaca vrr1 aaacaattgtcatactcataaactcacagcacctatga
3X 3X1 caatgaatgggccagaaaagc 3X2 gccagcaccccttattccttac
141

Supporting Text.
Construction of the recombination vectors, selection, and screening procedures. List of the
oligonucleotides used in this work (see Table 1).
Targeted deletion of DXPas34
pBln6534 was produced through recombineering in pBln65 to exchange the DXPas34
sequence (coordinates of the deleted sequence in AJ421479.1/GI:21425583:139060-140335) with a
PGK/Tn5 neomycin-polyA floxed cassette. The entire structure of pBln6534 was extensively
analyzed by restriction digestion, and sequences surrounding the DXPas34 deletion were
sequenced. pBln65 has been previously generated by gap-repair cloning of a 65-kb sequence
(coordinates in AJ421479.1/GI:21425583:127005-192614) from the fully sequenced bacterial
artificial chromosome (BAC) 399K20 into the NotI linearized pBln vector (DNA and structure
available on request), which contains a pBelo origin and an ampicillin resistance gene. pBln6534
was linearized by using ISceI and electroporated in the CK35 ES cell line (Xcell Gene Pulser; Bio-
Rad). After selection with 320 g/ml G418 (Invitrogen), resistant clones were screened by PCR
(d34f/d34r and x18f/x18r). Three positive clones of 270 were checked for normal karyotype and
analyzed by Southern blot. Clones neo34#1 and 34#2 were lipofected (Lipofectamin 2000;
Invitrogen) with the cre-expression plasmid pOG231 to excise the selection cassette, resulting in the
34#1 and 34#2 ES cell lines, which were further analyzed by Southern blot.
AJ, AS, and AV nested targeted deletions
Two recipient plasmids were constructed. pHLPTKFm contains a pMC-hygromycin-polyA
gene, separated from a puromycin-thymidine-kinase fusion ORF by a loxP site (the PTK ORF was
obtained from plasmid pTKPuroTKpA8, a gift from Dr. S. Tajbakhsh, Institut Pasteur, Paris).
pFLNCNM contains a neomycin gene carrying a loxP site between the pMC promoter and the ATG
for neo. Additional information on these plasmids is available on request. Mouse genomic
sequences were introduced into pHLPTKFm and pFLNCNM by gap repair recombineering using
the 399K20 BAC as donor. Coordinates of mouse genomic sequences (as in
AJ421479.1/GI:21425583) in the homologous recombination vectors were as follows: pHLPTKFm-
Aa (119539-129547), pFLNCNM-Jt (133808-143718), pFLNCNM-St (139040-150182), and
pFLNCNM-Vt (143275-154066). The structure of these plasmids was extensively verified by
restriction digestion. A first round of homologous recombination was performed by electroporating
the CK35 ES cell line with linearized plasmid pHLPTK-Aa. Transfected cells were selected by
142

using 180 g/ml hygromycin (Roche Applied Science, Indianapolis), PCR-screened, and clones
35Aa16 and Aa1112 were used for the next round of homologous recombination. Plasmids
pFLNCNM-Jt, pFLNCNM-St, and pFLNCNM-Vt were independently electroporated into the
35Aa16 and Aa1112 ES cell lines to generate, respectively, the AJ, AS, and AV deletions. After
selection with 320 g/ml G418, resistant clones from each individual plate were pooled separately,
and pools were independently lipofected with pOG231 and selected with 1 g/ml puromycin
(Sigma). Independent clones from each pool were extended and lipofected with the pCAGGS-FLPe
plasmid (Gene Bridge, Dresden, Germany), selection with Gancyclovir 1.2 g/ml was applied, and
resistant clones were PCR screened (3 × 1/3 × 2) for deleted clones with a single FRT site left in
place. Southern blot analysis was performed by using the Csl, AP, and VL probes.
143
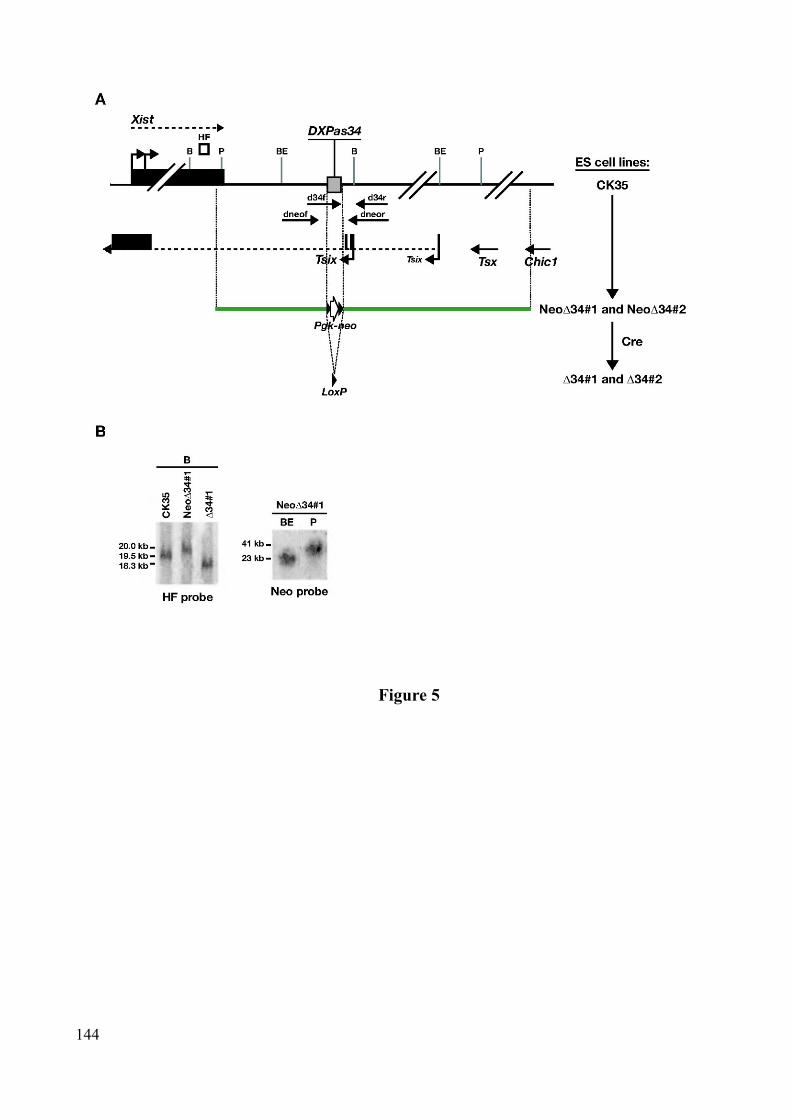
144
Figure 5

145
Figure 6
Figure 7

146
Figure 8
Figure 9

1.2 Tsix est un élément essentiel de la voie de comptage dans les cellules
ES mâles
Les résultats présentés dans l'article 1 ont démontré que, dans des cellules ES mâles, 1) le
minisatellite DXPas34 est un activateur transcriptionnel de Tsix; 2) l'expression de Tsix est requise
pour empêcher l'inactivation de l'unique chromosome X et est par conséquent un élément essentiel
de la voie de comptage; 3) la région comprise entre la fin de Xist et DXPas34 n'est pas nécessaire
pour le comptage ou l'expression correcte de Tsix.
Parmi les lignées de cellules ES mâles étudiées, les mutations NeoΔPas34, ΔDXPas34, ΔAV et
Ma2L constituent une série allélique pour le niveau d'expression de Tsix avant différenciation:
l'expression de Tsix est diminuée de moitié dans les cellules NeoΔPas34, de 90 % dans les cellules
ΔDXPas34, et est quasiment nulle dans les cellules ΔAV et Ma2L. Dans ces cellules, le niveau
d'expression de Xist est corrélé de manière inverse à celui de Tsix. De plus, le niveau d'expression
de Tsix avant différenciation corrèle avec la proportion de cellules qui inactivent leur unique
chromosome X en cours de différenciation. Cette proportion est maximale pour les cellules ΔAV et
Ma2L, dans lesquelles Tsix n'est pas du tout exprimé. Incidemment, l'inactivation est initiée dans la
lignée Ma2L de manière plus efficace que ce qui avait précédemment publié (Luikenhuis et al.,
2001), soulignant probablement l'importance des conditions de différenciation lorsqu'on s'intéresse
à l'inactivation du chromosome X (cf introduction, partie 1, section 3.6, p. 73).
1.2.1 Stabilité de l'inactivation dans les cellules mâles
Bien que les cellules mâles mutées pour Tsix initient de manière efficace l'inactivation de leur
unique chromosome X, la proportion de cellules avec un Xi décroît après avoir atteint un maximum
vers 3 à 4 jours de différenciation. Après 12 jours, seulement 8 % des cellules porteuses de la
délétion ΔAV ont encore un domaine d'ARN Xist (données non publiées). Une première explication
à cette décroissance serait que l'inactivation ait été réalisée de manière instable et l'on assisterait
alors à sa réversion. Une seconde possibilité serait la mort des cellules différenciées qui auraient
inactivé de manière stable leur unique chromosome X.
L'instabilité de l'inactivation dans les cellules mâles pourrait être conditionnée par un profil
épigénétique particulier de son chromosome X, comparativement aux cellules femelles. Un
enrichissement de certaines marques épigénétiques (hyperacétylation de H2A, H2B, H3 et H4,
147

hyperméthylation de H3K4 et hypométhylation de H3K9) a en effet été observé dans des cellules
ES femelles, de manière équivalente sur leurs deux chromosomes X, mais pas dans des cellules ES
mâles, au niveau de quatre gènes de ménage liés au chromosome X (G6pdx, Hprt, Pgk1 et Rps4)
(O'Neill et al., 2003). Cependant, cet enrichissement est maintenu plus longtemps sur le Xa que sur
le Xi lors de la différenciation de cellules ES femelles (O'Neill et al., 2003). Il semblerait donc
paradoxal que l'absence de ces marques – qui plus est, de type euchromatique – puisse contribuer à
destabiliser l'inactivation dans les cellules mâles. D'autre part, de telles marques n'ont été mesurées
que sur un petit nombre de gènes et pour une ou deux positions seulement dans chacun d'entre eux.
Les technologies de type ChIP on Chip rendent aujourd'hui possible leur caractérisation à plus
grande échelle, et de manière plus détaillée, ce qui permettrait de savoir dans quelle mesure les
profils épigénétiques des chromosomes X diffèrent entre les cellules ES mâles et femelles et, le cas
échéant, si ils pourraient être modifiées dans des lignées mutantes pour le choix ou le comptage.
Pour vérifier la seconde hypothèse – celle d'une inactivation stable accompagnée de mort
cellulaire, en l'absence d'expression de Tsix dans des cellules mâles – il faudrait pouvoir mesurer un
surcroît de mortalité consécutif à l'inactivation ectopique. Mais cela n'est pas trivial car, lors de la
différenciation des cellules ES, une importante mortalité est normalement observée. Nous ne
sommes ainsi pas parvenu à mesurer un surcroît significatif de mortalité dans des expériences de
mesure de la mortalité globale par un marquage au bleu trypan, sans qu'il nous soit pour autant
possible de conclure (donnée non publié). Nous avons tenté une approche alternative, qui consistait
à mesurer un surcroît de mortalité spécifiquement dans les cellules ayant inactivé leur chromosome
X. Il s'agissait dans ce cas de combiner un marquage d'apoptose au moyen d'un test Annexin à un
marquage des domaines d'ARN Xist par RNA-FISH. La combinaison de ces marquages, aux
exigeances contradictoires, demande une mise au point importante. Aussi nos premiers résultats, qui
suggéreaient un surcroît d'apoptose dans les cellules possédant un domaine de Xist, n'ont-ils pas pu
être reproduits (donnée non publiée), et nous avons décidé de ne pas poursuivre cette approche.
On ne sait pas si l'absence de Tsix conduit, comme dans les cellules ES, à initier de manière
ectopique l'inactivation de l'unique chromosome X dans des embryons mâles. Il est probable que
cela soit le cas, car un allèle non fonctionnel de Xist est surexprimé de manière durable dans des
embryons mâles n'exprimant pas Tsix (chromosome Xdc, cf note p. 124) (Sado et al., 2005).
Néanmoins, l'expression ectopique de Xist, si elle a lieu, ne semble pas pénaliser la survie
d'embryons mutés pour Tsix, lorsque la léthalité liée au défaut d'inactivation empreintée est évitée
par tetraploid rescue (Ohhata et al., 2006). Par conséquent, ou bien l'expression ectopique de Xist
148

ne conduit pas à l'inactivation stable de l'unique chromosome X, ou bien ce dernier est inactivé de
manière stable dans une proportion de cellules suffisamment faible pour que leur disparition
n'entraîne pas de léthalité embryonnaire.
La capacité des lignées ΔAV et Ma2L à initier l'inactivation de manière ectopique contraste avec
l'absence d'inactivation dans les cellules XΔCpGY générées par J. Lee (Lee et Lu, 1999). Cela pourrait
être expliqué par l'existence d'une transcription antisens à Xist résiduelle dans ces dernières (Lee et
al., 1999). Cette transcription résiduelle pourrait être originaire du promoteur minoritaire de Tsix,
mais dans ce cas, il est surprenant que nous ne l'observions pas dans les mutants ΔAV, et il est peu
probable qu'elle soit plus élevée que celle mesurée dans les mutants ΔDXPas34 qui, pourtant,
initient l'inactivation. Une autre explication à l'absence d'inactivation dans les cellules XΔCpGY
pourrait être la présence d'un cassette de sélection exprimant la néomycine sous contrôle du
promoteur PGK, laissée en place dans la délétion ΔCpG telle qu'elle a été publiée originellement
(Lee et Lu, 1999). En effet, il a été montré qu'une telle cassette de sélection pouvait être à l'origine
d'une transcription ectopique, en particulier au niveau des gènes Xist (Nesterova et al., 2003;
Newall et al., 2001) et Tsix (Cohen et al., 2007; Vigneau et al., 2006). Cependant, il nous a été
confirmé que la délétion ΔCpG produisait le même phénotype après excision de la cassette
néomycine (Lee, communication personnelle). Enfin, il est possible que le mode de différenciation
en corps embryonnaires (cf p. 75), qui a été utilisé pour l'analyse des cellules XΔCpGY, ne permette
pas de détecter l'inactivation ectopique dans des cellules mâles, dont nous avons vu qu'elle était
réalisée de manière transitoire. Lors d'une différenciation en corps embryonnaires, les cellules
semblent en effet moins synchrones que lors d'une différenciation induite par l'acide rétinoïque, et
leur différenciation est étalée sur une période de temps plus longue, ce qui est peu propice à la
détection d'un événement transitoire, qui se retrouverait ainsi « dilué ». Dans nos mains, lorsque les
cellules ΔAV et Ma2L sont différenciées en corps embryonnaires qui, après avoir été formés en
suspension, poursuivent leur différenciation en conditions adhérentes, nous observons à la
périphérie de ces corps embryonnaires réattachés une frange de cellules possédant un domaine
ectopique d'ARN Xist (donnée non publié). Au contraire, de tels domaines ne sont pratiquement
jamais observés dans les cellules du corps embryonnaire lui-même (donnée non publié). Cela
pourrait s'expliquer par la manière dont les cellules se différencient à partir de corps embryonnaires
réattachés. En effet, les cellules différenciées sortent des corps embryonnaires et se répartissent en
monocouche autour d'eux, avant que les corps embryonnaires ne finissent par disparaître, après une
culture prolongée. Les cellules ΔAV et Ma2L possédant un domaine ectopique de Xist semblent
donc se répartir sur un « front de différenciation », et reproduire ainsi le phénotype observé lors de
149

leur différenciation induite par l'acide rétinoïque. Néanmoins, à un instant donné, la proportion
totale de ces cellules est faible, si l'on comptabilise également les cellules indifférenciées présentes
dans le corps embryonnaires, et les cellules sorties depuis plus longtemps, qui auraient perdu leur
domaine de Xist.
L'absence d'inactivation dans les cellules XΔCpGY avait conduit J. Lee à proposer l'existence d'un
facteur de compétence, pour expliquer l'incapacité des cellules mâles à inactiver leur chromosome
X. Un modèle à deux facteurs en avait été déduit (cf introduction, partie 1, section 1.1.1, p. 78). Au
vu de nos résultats, un tel modèle devrait être réévalué. Nous en discuterons au chapitre 3 de la
discussion, p. 235.
1.2.2 Niveau d'expression de Xist et formation de domaines d'ARN Xist
Dans les cellules ΔAV et Ma2L indifférenciées, la quantité d'ARN Xist mesurée par RT-PCR
quantitative est augmentée d'un facteur 50 par rapport à des cellules sauvages. De ce fait, elle est
comparable à celle observée dans les cellules ΔDXPas34 en cours de différenciation. Cependant,
contrairement aux cellules ΔDXPas34 différenciées, l'augmentation des transcrits Xist dans les
cellules ΔAV et Ma2L indifférenciées n'a pas pour conséquence la formation de domaines d'ARN
Xist. Le signal obtenu en RNA-FISH pour Xist ressemble plutôt à des éclats dispersés. Or, dans les
cellules ES indifférenciées, le territoire chromosomique présente à peu près le même degré de
compaction qu'en cours de différenciation (Chaumeil et al., 2006). Par conséquent, la cause de ces
éclats est plutôt à chercher dans l'incapacité des ARN Xist à être correctement localisés. Cela
pourrait dépendre de la structure primaire de ces ARN (variants d'initiation, de terminaison,
d'épissage...), de leur structure secondaire (par exemple, structuration en tige-boucle de certaines
répétitions), de la présence ou de l'absence de facteurs avec lesquels ils interagiraient, ou des
propriétés de la chromatine sur le chromosome X, en particulier au niveau d'éventuels centres de
nucléation (cf introduction, partie 2, section 1.2.4, p. 93). A ce sujet, on peut remarquer qu'un
transgène inductible de Xist, localisé sur le chromosome X hors du Xic est capable d'initier
l'inactivation dans des cellules ES indifférenciées (Wutz et Jaenisch, 2000). Cela pourrait être relié
au contexte chromosomique différent dans lequel sont situés le transgène et le gène Xist endogène
ou, plus prosaïquement, on pourrait penser que le niveau absolu des transcrits Xist serait plus élevé
dans les lignées transgéniques de Wutz et al. que dans les cellules ΔAV et Ma2L, et échapperait
ainsi aux systèmes de contrôle présents dans les cellules indifférenciées.
150

Par ailleurs, l'expression de Xist augmente encore en cours de différenciation dans les cellules
ΔAV et Ma2L, bien que les transcrits Tsix sont déjà absents dans les cellules indifférenciées.
L'augmentation de l'expression de Xist est en réalité observée en cours de différenciation quel que
soit le niveau initial d'expression de Tsix, y compris dans les cellules ES mâles sauvages. Cela
suggère que l'expression de Xist serait régulée, pendant la différenciation, indépendamment de Tsix,
ce qui pourrait être relié, en cours de différenciation, à la perte d'un répresseur transcriptionnel,
proposée par Navarro et al. (cf p. 128) (Navarro et al., 2005).
1.3 DXPas34 régule l'expression de Tsix
1.3.1 DXPas34 et autres éléments régulant l'expression de Tsix
En en réalisant la délétion ciblée dans des cellules ES mâles, nous avons pu démontrer le rôle
essentiel joué par le minisatellite DXPas34 comme activateur transcriptionnel de Tsix. En l'absence
de DXPas34, l'expression de Tsix est réduite d'environ 90 %. Il est vraisemblable que le caractère
hypomorphe du phénotype des cellules ΔDXPas34, comparativement à des mutant supprimant la
totalité de l'expression de Tsix (i.e. ΔAV et Ma2L), réside dans l'expression résiduelle de 10 % des
transcrits Tsix. Cette expression est perdue en cours de différenciation avec une cinétique
comparable à l'extinction de l'expression de Tsix observée dans les cellules sauvages.
L'extinction transcriptionnelle de Tsix en cours de différenciation pourrait correspondre à
l'existence de régulateurs additionnels de Tsix, dont l'activité serait elle-même modifiée en cours de
différenciation. Nous avions présenté, à la section 3.2 de la seconde partie de l'introduction (p. 116)
la caractérisation au moyen de tests luciférases de deux enhancers de Tsix, l'un d'eux contenant
DXPas34 et le second correspondant à Xite (Stavropoulos et al., 2005). Au moins deux
caractéristiques communes à ces enhancers sont perdues en cours de différenciation, l'initiation
d'une transcription bidirectionnelle (dans DXPas34 et Xite) et la présence de sites d'hypersensibilité
à la DNAseI (dans DXPas34, en amont du promoteur majoritaire de Tsix et dans Xite). Le
comportement similaire de Xite et de DXPas34 pourrait expliquer que, lorsque DXPas34 est délété,
la cinétique d'extinction de Tsix ne soit pas modifiée, à condition d'admettre que Xite soit le
principal responsable de l'expression résiduelle de Tsix. Il serait intéressant de le vérifier avec des
mutants dans lesquels auraient été délétés à la fois DXPas34 et Xite. De plus, en cours de
différenciation, la fixation de CTCF est perdue au niveau de DXPas34 avec une cinétique
151

comparable à l'extinction de Tsix (la perte de YY1 est plus tardive) (cf introduction, partie 2,
section 3.3, p. 116 ), ce qui pourrait rendre compte de la perte de l'activité activatrice de DXPas34.
Pour cette même raison, il pourrait être judicieux de rechercher la présence de CTCF au niveau de
Xite.
1.3.2 Comparaison avec un second mutant de DXPas34
Une seconde délétion de DXPas34 a été publiée récemment (Cohen et al., 2007). Elle diffère de
celle que nous avons généré en étant moins longue de 50 pb vers le promoteur de Tsix et plus
étendue de 300 pb du côté opposé (fig. 19, p. 118).
Cette délétion affecte le niveau d'expression de Tsix dans des cellules ES mâles un peu
différemment de la notre: la baisse du niveau d'expression de Tsix en 5' est un peu moins importante
que celle que nous observons (14 % d'expression résiduelle contre 10 %), mais elle est plus
marquée dans la région 3', complémentaire à Xist (4 % d'expression résiduelle contre 10 %). Il est
possible que les différences de taille et de position entre les deux délétions en soient responsables.
Cela pourrait correspondre à la présence d'un élément régulateur ou d'un promoteur additionnel, qui
ne serait délété que dans la délétion de Cohen et al. Cette idée est confortée par des tests luciférase
réalisés par Cohen et al., consistant à placer en amont d'un rapporteur luciférase dénué de
promoteur, soit le minisatellite DXPas34 (c'est-à-dire, pratiquement, la région correspondant à notre
délétion ΔDXPas34), soit la région correspondant à leur délétion ΔDXPas34, plus étendue de
300 pb en aval (dans le sens de la transcription de Tsix). Dans les deux cas, l'activité luciférase
mesure une transcription antisens (par référence à Xist) initiée dans ces régions, mais celle-ci est
plus importante lorsque sont présentes les 300 pb additionnelles (Cohen et al., 2007). Une
transcription sens, beaucoup plus faible, est également mesurée, mais dans ce second cas
uniquement. Ces observations suggèrent donc la présence en aval de DXPas34 d'un promoteur ou
d'un élément régulant la transcription initiée dans DXPas34.
Une explication alternative aux différences de niveau d'expression de Tsix dans les deux lignées
ΔDXPas34 pourrait correspondre à une destabilisation des transcrits Tsix consécutive à la délétion
de Cohen et al. Dans ce cas, la région de 300 pb déjà mentionnée pourrait être essentielle à la
stabilisation des transcrits Tsix. Cette stabilisation pourrait intervenir au niveau des transcrits
primaires ou épissés. On peut également imaginer que l'épissage de certaines isoformes de Tsix
152

pourrait être altéré dans le mutant de Cohen et al., mais pas dans le notre. A ce sujet, il est
intéressant de rappeler que l'un des variants d'épissages de Tsix est épissé entre ses exons 3a et 4 peu
en aval de la délétion de Cohen et al. (fig. 18, p. 100).
Le profil d'expression de Tsix dans les cellules mâles ΔDXPas34 générées par Cohen et al. est
tout à fait similaire, quantitativement et qualitativement, à celui observé dans les cellules ES mâles
ΔCpG (Cohen et al., 2007). D'autre part, dans les deux cas, leur phénotype a été analysé en les
différenciant par la méthode des corps embryonnaires. Aussi n'est-il pas étonnant que, comme dans
les cellules XΔCpGY, Cohen et al. n'observent pas d'inactivation ectopique en cours de
différenciation. Les raisons permettant d'expliquer une telle différence avec nos résultats ont déjà
été discutées p. 149 concernant la délétion ΔCpG, et sont tout à fait applicables à nos délétions
ΔDXPas34 respectives.
Dans des cellules ES femelles dont l'un des chromosomes X porte la délétion ΔDXPas34,
Cohen et al. observent un biais de choix en faveur de l'inactivation de l'allèle muté. Cela est
consistant avec la réduction observée en cis du niveau d'expression de Tsix, et l'augmentation
consécutive du niveau d'expression de Xist (Cohen et al., 2007). Néanmoins, le biais est légèrement
moins prononcé dans les cellules XΔDXPas34X que dans les cellules XΔCpGX. Comme la variation du
niveau d'expression de Tsix n'est pas très différente dans les deux cas, cela pourrait traduire une
sensibilité très importante au niveau d'expression de Tsix, ou un rôle de la région promotrice distinct
de l'expression de Tsix.
De manière inattendue, Cohen et al. observent, tardivement au cours de la différenciation des
cellules ES XΔDXPas34X, une augmentation de faible amplitude du niveau d'expression de Tsix
spécifiquement à partir de l'allèle muté (Cohen et al., 2007). Cela les conduit à proposer une
fonction ambivalente pour DXPas34, celle d'un activateur de l'expression de Tsix dans les cellules
non différenciées ou en début de différenciation, et celle d'un répresseur de Tsix dans les cellules
différenciées. Cette ambivalence pourrait être mise en relation avec le changement des propriétés de
DXPas34 en cours de différenciation, notamment la méthylation de ses îlots CpG et la perte, selon
des cinétiques différentes, de CTCF et YY1. Il serait également important de regarder si une
dérépression similaire de Tsix, lorsque DXPas34 est délété, peut être observée dans l'embryon.
Nous n'avons pas encore obtenu de cellules ES femelles portant la délétion de DXPas34, et
permettant d'adresser son rôle dans le choix, comme l'on fait Cohen et al. Par contre, des souris
recombinantes porteuses de cette délétion ont été générées. Nous présentons les résultats
préliminaires obtenus avec ces lignées à la section suivante, en les comparant avec les souris
recombinante obtenues par Cohen et al. à partir de leur propre délétion de DXPas34.
153

154

Chapitre 2 Resultats préliminaires sur la fonction de DXPas34
dans l'embryon
Deux souris mâles chimériques porteuses de la délétion ΔDXPas34 ont été générées en
collaboration avec le Centre d'Ingénierie Génétique Murine, dirigé par Francina Langa-Vives, à
partir des deux clones indépendants de cellules ES mâles ΔDXPas34, dont le phénotype a été
détaillé plus haut. Les cellules ES sont de fond génétique 129S2/SvPas (nous abrégerons par 129) et
le premier mâle chimérique obtenu a été croisé dans un premier temps avec des femelles de fond
génétique C57BL/6N (nous abrégerons par B6). Les données chiffrées que nous allons présenter
correspondent à la descendance de ces premiers croisements, sur trois générations. Nous ne ferons
qu'évoquer le résultat des croisements planifiés plus tardivement, pour lesquels les effectifs sont en
cours d'accroissement.
2.1 Transmission maternelle de la délétion ΔDXPas34 et rôle de
DXPas34 dans l'inactivation empreintée
Dans les cellules ES mâles ΔDXPas34, l'expression de Tsix est fortement diminuée. Nous nous
attendions par conséquent à observer dans la souris un phénotype similaire à celui des mutants de
Tsix, éventuellement atténué par le caractère hypomorphe de la mutation ΔDXPas34. L'effet le plus
évident aurait alors été un déficit de transmission maternelle de la mutation associé à un défaut
d'inactivation empreintée. En F2, nous n'observons cependant pas de déficit associé à la
transmission maternelle de l'allèle ΔDXPas34, qui est réalisée dans des proportions mendeliennes
(F2, fig. 22, p. 156). En F3, après croisement en retour avec des mâles de fond génétique 129 (F3,
fig. 22, p. 156), un déficit lié à la transmission maternelle de la délétion est cependant observé dans
les femelles (χ², p = 0,016), mais la transmission de l'allèle muté est au contraire favorisé dans les
mâles (χ², p = 0,028) (fig. 22, p. 156). Un autre croisement (F4*, fig. 22, p. 156), sans dilution
supplémentaire du fond génétique B6, ne reproduit pas de manière significative ces écarts à une
transmission mendélienne. Cependant, la transmission préférentielle de l'allèle ΔDXPas34 est
également observée, quoique de manière peu significative (χ², p = 0,061), dans la descendance du
155

156
Fig. 22: Transmission de la délétion ΔDXPas34
Croisements et effectifs obtenus à chaque génération à partir du premier mâle chimérique obtenu. Les fonds génétiques sont précisés (B6:C57BL/6N; 129:129Sv/Pas). Se référer au texte pour plus de détails.

croisement XΔDXPas34X x XΔDXPas34Y (F3*, fig. 22, p. 156). Dans tous les cas, aucun déficit de
descendance mâle ou femelle ne peut être mesuré de manière significative. En combinant les
effectifs obtenus pour les trois premiers croisements (F2 + F3 + F4*, fig. 22, p. 156), le déficit en
femelles ΔDXPas34 n'est pas statistiquement significatif (χ², p = 0,36), contrairement au biais en
faveur des mâles ΔDXPas34 (χ², p = 0,02). Là encore, aucune différence significative entre les
effectifs mâles et femelles n'est mesurée (χ², p = 0,94).
Comme ces croisements ont été réalisées avec des souris de fonds génétiques mélangés 129 et
B6, et que nous observons un biais de transmission de la mutation dans les mâles et les femelles en
F3, après croisement en retour sur le fond génétique 129, mais pas en F2, nous pouvions supposer
que le fond génétique B6 contribuerait à atténuer le phénotype de la délétion ΔDXPas34. C'est
pourquoi nous avons commencé les mêmes croisements (F2 et F3) à partir du second mâle
chimérique, mais en fond pur 129 (en utilisant la descendance de ce mâle croisé avec une femelle
129). Les premiers résultats, préliminaires, semblent correspondre à une transmission mendélienne,
et ne reproduisent pas de biais à l'encontre de la transmission dans les femelles, ou en faveur de la
transmission de la délétion dans les mâles (travail en cours).
Une seconde explication à la transmission non mendelienne de la délétion en F3 mais pas en F2,
dans le fond mélangé B6,129, pourrait être l'origine parentale de la délétion transmise par la mère.
Ainsi, les femelles croisées pour obtenir la descendance F2 ont hérité la mutation de leur père (le
mâle chimérique), tandis que les femelles utilisées pour générer la descendance F3 ont hérité la
mutation de leur mère. L'absence d'écart significatif à une transmission mendelienne à la génération
F4*, issue du croisement de femelles ayant hérité la mutation de leur père, pourrait soutenir cette
idée. Néanmoins, chez les mâles, la transmission de la mutation semble favorisée, bien que de
manière non significative (p = 0,12), ce qui correspond à une configuration plus proche de celle
observée en F3 que de celle observée en F2. Des effectifs plus importants en F4* seraient donc
probablement nécessaires pour pouvoir conclure. D'autre part, l'observation d'un excès, quoique peu
significatif (χ², p = 0,061), de mâles portant la délétion ΔDXPas34 dans la descendance du
croisement XΔDXPas34X x XΔDXPas34Y (F3*, fig. 22, p. 156) pourrait aller dans le sens d'un effet lié à
l'origine parentale de la mutation transmise par la mère. Lorsque seront déterminées les conditions
permettant de reproduire le biais en faveur de la transmission de la délétion dans les mâles, il sera
important de déterminer à quel moment il apparaît, pour pouvoir en comprendre la signification
biologique.
157

Un second type d'expériences a été entrepris pour adresser un effet éventuel de la délétion
ΔDXPas34 sur l'inactivation empreintée. Des blastocystes de génotype XΔDXPas34Y et XΔDXPas34X, de
fond génétique mélangé B6,129 ou de fond pur 129, ont été prélévés à E3,5 et cultivés ex vivo
pendant 4 jours. Dans ce modèle, les cellules trophoblastiques s'étalent pour former un halo (ou
trophoblast outgrowth) autour de la masse cellulaire interne. Après marquage par RNA-FISH, un
domaine ectopique d'ARN Xist est observé sur le Xm dans environ 10 % des cellules
trophoblastiques pour les cultures de blastocystes XΔDXPas34X, et dans 5 à 11 % des cellules
trophoblastiques pour les cultures de bastocystes XΔDXPas34Y (Molaro et Clerc, travail en cours). Des
domaines ectopiques ne sont par contre pas observés dans des blastocystes sauvages cultivés de la
même manière. Malgré l'expression ectopique de Xist dans une proportion de cellules, il ne semble
pourtant pas y avoir de défaut visible d'attachement des blastocystes, ni de prolifération des cellules
trophoblastiques, ce qui est en accord avec l'absence de déficit de transmission maternelle de la
délétion, dans la plupart des croisements présentés plus haut. L'absence d'effet significatif de
l'expression ectopique de Xist sur le Xm, pourrait être dû au faible nombre de cellules concernées,
dont la mort liée à l'inactivation du Xm serait compensée au cours du développement, où à
l'inefficacité des domaines ectopiques à conduire à l'inactivation du Xm. Un moyen d'évaluer cette
seconde hypothèse serait de tester, sur le Xm, la présence de marques épigénétiques associées
chromosome X inactif, en parallèle à la détection du domaine d'ARN Xist.
2.2 DXPas34 et le comptage dans l'embryon
Dans des embryons femelles, l'invalidation de Tsix sur les deux chromosomes X au moyen de la
délétion ΔCpG a pour conséquence un surcroît de léthalité embryonnaire, s'ajoutant à l'effet de la
transmission maternelle de la délétion. Ce surcroît de mortalité est expliqué par l'inactivation
chaotique des chromosomes X, qui correspond à un défaut de comptage (cf p. 111). Si tel était le
cas pour la délétion ΔDXPas34, nous devrions observer un déficit de femelles homozygotes dans la
descendance de croisements XΔDXPas34X x XΔDXPas34Y. Un tel déficit n'est pas observé avec notre
délétion ΔDXPas34 (χ², p=0,28) (F3*, fig. 22, p. 156). De plus, elle peut être maintenue à l'état
homozygote sans difficulté apparente. Il ne semble donc pas y avoir de défaut de comptage résultant
en une inactivation chaotique dans les femelles.
Cependant, lorsque des blastocystes de génotype XΔDXPas34Y sont prélévés à E3,5 et cultivés in
vitro pendant 4 jours, des domaines ectopiques de Xist sont observés dans 3 à 15 % des cellules de
la masse cellulaire interne, selon les blastocystes, ce qui n'est pas le cas dans des cultures de
158

blastocystes sauvages (Molaro et Clerc, travail en cours). Cette proportion est variable, mais en
général plus faible que ce qui avait été observé dans les cellules ES en cours de différenciation. Cela
pourrait résulter d'un impact de la délétion ΔDXPas34 sur le comptage qui serait plus modéré dans
l'embryon que dans les cellules ES, ou de la plus grande difficulté à visualiser l'inactivation
ectopique, probablement transitoire (cf p. 147), dans le modèle ex vivo de culture de blastocystes
comparativement aux cellules ES en différenciation. Il serait d'autre part important de confirmer, en
utilisant des marqueurs de lignage, l'appartenance de ces cellules au lignage épiblastique, et non à
l'endoderme extraembryonnaire ou au lignage trophoblastique.
2.3 DXPas34 et le choix dans l'embryon
Dans la mesure où tous les mutants de Tsix connus conduisent à un biais de choix (cf
introduction, partie 2, section 2.3.2.1, p. 107), et que DXPas34 régule l'expression de Tsix, il est
logique de penser que la délétion de DXPas34 sur l'un des deux chromosomes X dans des cellules
femelles puisse également être à l'origine d'un biais de choix. Nous avons indiqué plus haut qu'une
seconde délétion de DXPas34 avait cet effet dans des cellules ES femelles (cf résultats,
section 1.3.2, p. 152) (Cohen et al., 2007).
Pour évaluer l'existence d'un biais de choix lié à la délétion ΔDXPas34, nous avons croisé des
femelles XΔDXPas34XΔDXPas34 (fond mélangé B6,129) avec des mâles sauvage 129.Pgk1a, de manière à
disposer d'un polymorphisme permettant de détecter l'expression de Xist par RT-PCR à partir de
chacun des allèles. Les mâles 129.Pgk1a sont essentiellement de fond génétique 129 à l'exception
de la région du Xic, héritée du fond génétique C3H.Pgk1a et portant l'allèle Xcec (Courtier et al.,
1995). Les femelles XΔDXPas34XΔDXPas34 utilisées pour ce croisement ont normalement hérité l'allèle
Xcea correspondant au fond génétique 1295. Dans des embryons femelles aux stades E9,5 et E10,5
issus de ce croisement, l'expression de Xist est réalisée à hauteur de 64 % à partir de l'allèle muté.
En admettant que la proportion des transcrits issus du chromosome XΔDXPas34 est représentative de la
proportion de cellules ayant inactivé ce chromosome, le biais observé n'est pas supérieur à celui
attendu dans des embryons sauvages de mêmes allèles Xce. En effet, dans des souris sauvages
hétérozygotes Xcea/Xcec, le chromosome X portant l'allèle Xcea est inactivé dans, en moyenne, 75-
5 Il n'est cependant pas exclu qu'elles aient acquis à sa place l'allèle Xceb, après recombinaison avec le chromosome X de fond génétique B6 apporté en F1. Mais comme l'allèle Xce a été positionné dans le Xic, à proximité de DXPas34, la probabilité d'un tel événement de recombinaison est faible. Il sera néanmoins important de valider les résultats présentés dans un fond génétique pur.
159

80 % des cellules (Chadwick et Willard, 2005)6. La variabilité importante que nous observons entre
individus pour le choix du chromosome X inactif est également en accord avec celle observée dans
des embryons sauvages (Chadwick et Willard, 2005). Ces observations indiquent donc que la
présence à l'état hétérozygote de notre délétion ΔDXPas34 ne conduit pas à biaiser le choix du
chromosome X inactif. L'analyse complémentaire d'un nombre plus grand d'embryons est en cours,
et conforte pour l'instant les résultats présentés ici.
2.4 Discussion et comparaison avec la délétion ΔDXPas34 générée par
Cohen et al.
Une seconde délétion de DXPas34 (Cohen et al., 2007), introduite plus haut (résultats,
section 1.3.2, p. 152), a un phénotype plus similaire à celui observé pour les différents mutants de
6 Dans une configuration Xceb/Xcec, le chromosome portant l'allèle Xceb est inactif dans 7075 % des cellules (Chadwick et Willard, 2005)
160
Fig. 23: Expression de Xist à partir de l'allèle ΔDXPas34 dans des embryons femelles XΔDXPas34XΔDXPas34
L'expression des allèles maternels et paternels de Xist a été mesuré par RTPCR quantitative en temps réel avec des amorces alléliques pour les fonds génétiques 129 et 129.Pgk1a. Le rapport de l'expression de l'allèle maternel et de l'allèle paternel est donné en % sur l'axe des ordonnées. Les embryons présentés sont tous des embryons femelles de génotype X DXPas34Δ X, prélevés aux stades indiqués. Le croisement ayant permis de les obtenir est indiqué en haut de la figure. Les rapports mesurés ne sont pas significativement distincts de ceux attendus dans des embryons femelles sauvages hétérozygotes Xcea/Xcec, dans lesquels l'allèle Xcea est apporté par une mère de fond génétique 129 et l'allèle Xcec par un père de fond génétique 129.Pgk1a.

Tsix et, en particulier, la délétion ΔCpG. Elle se traduit par un défaut d'inactivation empreintée,
comme en témoignent la léthalité embryonnaire de 73 % des mâles et 91 % des femelles ayant
hérité la mutation de leur mère, d'une part, et un défaut d'attachement et de prolifération des cellules
trophoblastiques lorsque des embryons XΔDXPas34X ou XΔDXPas34Y sont cultivés in vitro, d'autre part
(Cohen et al., 2007). De plus, dans des culture de blastocystes de génotype XΔDXPas34X et XΔDXPas34Y,
Cohen et al. mesurent par RT-PCR, dans les cellules trophoblastiques, l'expression de Xist à partir
du Xm, lorsque celui-ci est porteur de la délétion ΔDXPas34 (Cohen et al., 2007). La léthalité
causée par la transmission maternelle de cette délétion est certes élevée, mais plus faible que pour la
délétion ΔCpG (Cohen et al., 2007; Lee, 2000). La transmission paternelle de la délétion ΔDXPas34
ne semble quant à elle pas causer de léthalité.
D'autre part, Cohen et al. n'ont pas pu obtenir de descendance homozygote pour leur délétion
ΔDXPas34, ce qui est cohérent avec la proximité de son phénotype et de celui de la délétion ΔCpG.
Néanmoins, les auteurs ne donnant pas de donnée chiffrée à ce sujet, il est impossible de savoir si,
dans les deux cas, la pénétrance du phénotype est la même. Enfin, ils vérifient, dans des fibroblastes
dérivés de souris femelles, l'existence d'un biais de choix en faveur de l'inactivation du chromosome
X portant la délétion ΔDXPas34, qu'ils ont également mis en évidence dans des cellules ES femelle
(cf résultats, section 1.3.2, p. 152) (Cohen et al., 2007). Mentionnons cependant un point important
sur lequel Cohen et al. restent évasifs, qui consiste à savoir s'ils ont, ou non, dans leurs souris,
excisé la cassette PGKneo utilisée pour sélectionner la délétion de DXPas34. Or, nous l'avons déjà
vu, cette cassette est potentiellement source d'artefact (cf p. 149). Nous leur ferons néanmoins crédit
sur ce point, en admettant qu'ils ont pu observer ou reproduire l'essentiel de leurs phénotypes après
excision de cette cassette.
Ainsi, bien que nos deux délétions conduisent à une diminution importante de l'expression de
Tsix dans les cellules ES mâles, les phénotypes qui leur sont associés dans l'embryon sont très
différents. Pourtant, dans les deux cas, la délétion de DXPas34 a pour effet l'expression ectopique
de Xist sur le Xm dans des excroissances trophoblastiques (trophoblast outgrowth) mâles et
femelles, ainsi que, tout au moins pour notre délétion, dans des cellules embryonnaire mâles. Cela
suggère que, de la même manière que dans les cellules ES, Tsix serait régulé par DXPas34 dans
l'embryon, aussi bien dans les lignages embryonnaires qu'extraembryonnaires. Les différences
entre nos mutants pourraient alors être expliquées par le degré de diminution de l'expression de Tsix.
A ce sujet, nous avions déjà évoqué (p. 152) les différences entre les deux délétions de DXPas34
concernant le niveau d'expression de Tsix dans la région de complémentarité avec Xist, mesuré dans
les cellules ES mâles. De ce point de vue, la lignée de Cohen et al. est essentiellement amorphe,
161

tandis que la notre est hypomorphe, avec 10 % d'expression résiduelle antisens à Xist. Il serait
intéressant de voir si de telles différences sont retrouvées dans l'embryon. Nous avions par ailleurs
proposé que cette différence pourrait avoir son origine dans la délétion par Cohen et al. d'une région
de 300 pb, qui n'est pas concernée par notre délétion. Au vu de la similarité du phénotype observé
par Cohen et al. avec celui décrit pour la mutation ΔCpG, il est intéressant de mentionner ici que
dans les deux cas, la région de 300 pb a été délétée.
Il est raisonnable d'imaginer qu'un telle différence d'expression de Tsix en 3' (bien que
l'expression en 5' soit affectée de manière comparable à la notre) conduirait, dans les mutants de
Cohen et al., à initier l'inactivation sur le Xm dans une proportion plus importante de cellules
trophoblastiques, dont la disparition ne pourrait pas être compensée au cours du développement. Il
serait donc intéressant de pouvoir corréler, dans nos deux mutants, la proportion de cellules
trophoblastiques ayant un domaine ectopique de Xist et la variation dans ces cellules des niveaux
d'expression de Xist et de Tsix, à partir du Xm. Dans le cas de Tsix, il sera important, notamment,
d'en mesurer les niveaux d'expression en 5' et en 3'. Pour l'instant, nos résultats ne permettent pas
une telle comparaison, car Cohen et al. ont évalué l'expression ectopique de Xist par RT-PCR,
contrairement à nous, qui avons observé la présence de domaines d'ARN Xist sur le Xm par RNA-
FISH.
Le même type de comparaison serait utile dans les tissus embryonnaires, en particulier pour
comprendre l'absence de biais de choix dans nos mutants. Cette absence de biais est d 'autant plus
surprenante que nous avons nous-même montré que le niveau d'expression de Xist, avant et après
différenciation, était inversement corrélé à celui de Tsix avant différenciation (cf article 1, p. 131), et
qu'il est admis d'autre part que le niveau d'expression relatif de Xist sur chacun des chromosomes X
détermine la proportion de cellules dans lesquelles l'un ou l'autre de ces chromosomes est inactivé
(cf introduction, partie 2, section 2.1, p. 95). Ils se pourrait donc que, dans les embryons femelles
porteurs de notre délétion de DXPas34, l'expression de Tsix ne soit pas affectée comme dans les
mâles, une alternative étant que l'expression résiduelle de Tsix serait suffisante pour réaliser
correctement le choix. Cela correspondrait alors à un effet de seuil, dont il serait utile de
comprendre les fondements, en analysant les différents aspects (transcriptionnels, post-
transcriptionnels) de l'initiation de l'inactivation dans des cellules femelles XΔDXPas34X. Dans tous les
cas, une analyse détaillée sera facilitée par la dérivation de cellules ES femelles, qui est en cours.
La similarité de niveau d'expression de Tsix en 3' entre les mutants ΔDXPas34 de Cohen et al.
et les mutants ΔCpG pourrait expliquer, dans les deux cas, la léthalité des souris homozygotes pour
ces délétions. Néanmoins, il faudrait vérifier dans les premiers que cela correspond, comme dans les
162

seconds, à la mise en place d'une inactivation chaotique. Lorsque la délétion ΔCpG est présente à
l'état hétérozygote, cela a également pour conséquence de ne plus permettre le rapprochement
transitoire des Xic. Néanmoins, nous ne savons pas si cela peut être relié directement au phénotype
d'inactivation chaotique. L'examen du rapprochement des Xic dans des cellules ES femelles portant
l'une ou l'autre des délétions de DXPas34 apporterait certainement des éléments de réponse à cette
interrogation. Elle permettrait peut-être, également, de mieux comprendre l'importance respective
pour le rapprochement des Xic de l'expression de Tsix, en 5' et en 3', et de l'élément génomique
DXPas34 en tant que tel.
163

164

Chapitre 3 Rôle de Tsix dans la régulation épigénétique du Xic
L'invalidation de Tsix a deux conséquences sur la configuration de la chromatine dans le Xic. Au
niveau du promoteur de Xist, elle se traduit par l'enrichissement en marques de type euchromatique
et la perte de marques de type hétérochromatique, dont il a été proposé que cela prédéterminait
l'activation transcriptionnelle de Xist et l'initiation de l'inactivation en cis, qui ont lieu en cours de
différenciation (cf introduction, partie 2, section 4.2, p. 123). Au niveau du corps du gène Xist, on
assiste au contraire à la perte de H3K4me2 (une marque euchromatique) et l'enrichissement en
H3K27me3 (une marque hétérochromatique) (cf introduction, partie 2, section 4.3, p. 127). Ces
remarques suscitent de nombreuses interrogations, dont deux ont été adressées dans l'article qui suit.
Tout d'abord, comment Tsix peut-il contrôler la configuration de la chromatine dans le corps du
gène Xist? De manière intéressante, un enrichissement important en H3K27me3 est mesuré en 5' de
Xist, dans une région qualifiée de hotspot de méthylation, dont il a été fait l'hypothèse qu'elle
servirait de centre de nucléation pour la propagation de l'inactivation en cis (cf introduction,
partie 2, section 1.2.4, p. 93).L'enrichissement en H3K27me3 observé dans les mutants de Tsix
pourrait-il alors être la conséquence d'une extension du hotspot?
D'autre part, on peut s'interroger sur le lien entre le niveau d'expression de Tsix avant
différenciation, et le devenir du chromosome X en cis en cours de différenciation. En particulier, le
rôle de la configuration de la chromatine au promoteur de Xist, qui dépend elle-même de
l'expression de Tsix, est à préciser. Si cette configuration était réellement prédéterminante, on
s'attendrait à ce qu'elle ne soit pas la même dans des lignées mutantes exprimant différents niveaux
de Tsix et initiant l'inactivation de manière ectopique avec des fréquences variables. Pour tester cela,
la série allélique constituée par les lignées de cellules ES mâles Ma2L, ΔAV et ΔDXPas34
représente un outil de choix.
Les deux interrogations exposées ici nous ont donc conduit à examiner le profil épigénétique du
Xic dans ces lignées mutantes pour Tsix, ainsi que dans des cellules sauvages, en incluant une
perspective développementale.
165

3.1 Article 2: Tsix is a boundary element controlling H3K27 tri-
methylation at the X-inactivation center prior to X-inactivation
166

Tsix is a Boundary Element Controlling H3K27 Tri-methylation at the X-inactivation
Center Prior to X-inactivation
Pablo Navarro, Sébastien Vigneau, Corinne Chureau, Philip Avner, Philippe Clerc, and
Claire Rougeulle.
Unité de Génétique Moléculaire Murine, URA 2578, Institut Pasteur
75724, Paris Cedex 15, France
Corresponding Author: Claire Rougeulle,
e-mail [email protected], phone +33.1.45.68.86.53, fax +33.1.45.68.86.56
167

168

169

170

171

172

173

174

175

176

177

178

179

180

181

182

183

184

185

186

187

188

189

190

191

192

193

194

195

196

197

198

199

200

201
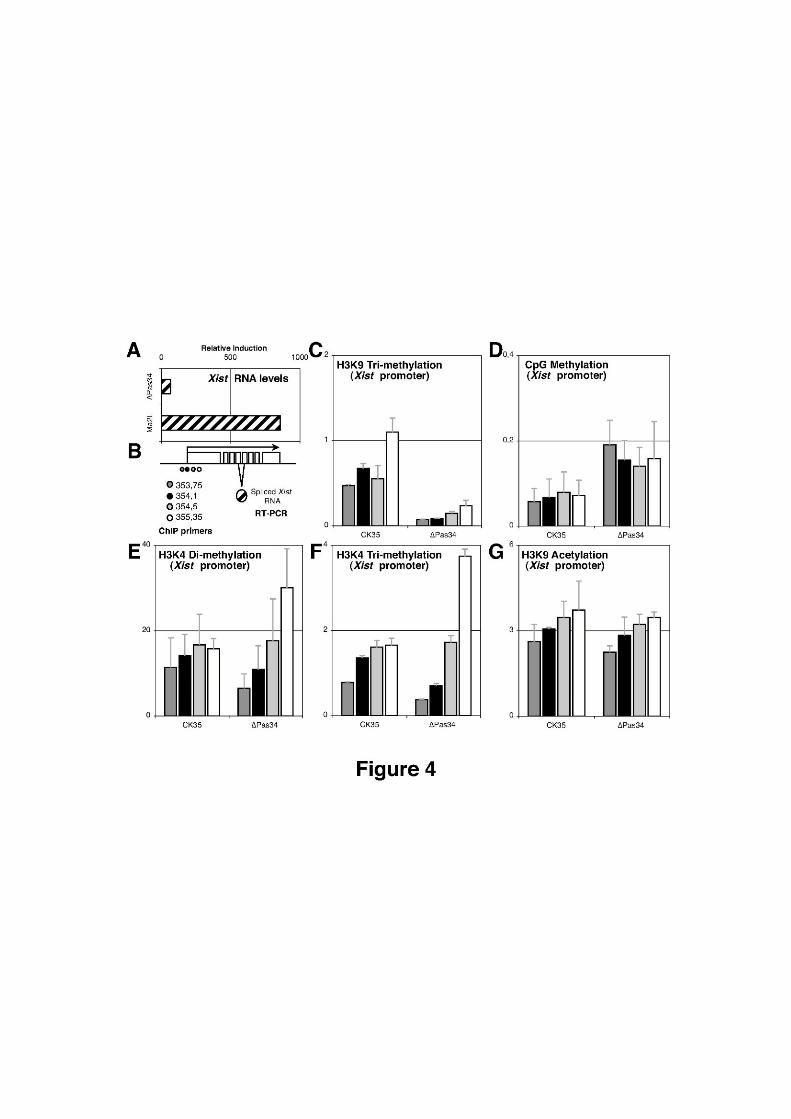
202

203

204

205
206
207

208

209

3.2 Principales conclusions
Comment Tsix régule-t-il les propriétés de la chromatine au niveau du Xic? Comment ces
propriétés évoluent-elles au cours du développement, et quelles sont leurs fonctions? Voici deux
interrogations auxquelles l'article précédent a tenté d'apporter des éléments de réponse. Rappelons-
en les principales conclusions.
Tout d'abord, l'arrêt de transcription de Tsix, marqué par l'accumulation de PolII, délimite une
région pauvre en marque répressive H3K27me3 au locus Xist/Tsix, comparé à l'enrichissement en
H3K27me3 observé au hotspot de méthylation en 5' de Xist. Dans les différents mutants de Tsix
examinés (Ma2L, ΔAV et ΔDXPas34), l'enrichissement en H3K27me3 mesuré dans le corps du
gène Xist est en continuité avec le hotspot, ce qui suggère qu'il pourrait résulter de la propagation en
cis de la méthylation H3K27me3 à partir de ce dernier.
L'enrichissement en H3K27me3 au locus Xist/Tsix, consécutif à l'invalidation de Tsix, est
observé dans les cellules ES mâles indifférenciées, mais a disparu après 4 jours de différenciation.
La même disparition de H3K27me3 est mesurée au hotspot, dans des cellules sauvages ou mutées
pour Tsix. Dans des fibroblastes, l'enrichissement en H3K27me3 corrèle par contre avec la présence
d'un Xi, dans la mesure où il est observé dans les fibroblastes femelles mais pas dans les
fibroblastes mâles. Cela dénote le caractère transitoire, sur le Xi, de la perte de H3K27me3 au
niveau du Xic.
D'autre part, dans les mutants de Tsix étudiés, la fréquence d'inactivation ectopique en cours de
différenciation est inversement corrélée, non seulement avec le niveau d'expression de Tsix, mais
également avec le degré d'euchromatinisation du promoteur de Xist, mesurés dans les cellules
indifférenciées. Au contraire, les mêmes modifications de la chromatine sont observées dans le
corps du gène Xist pour les trois lignées ΔAV, Ma2L et ΔDXPas34, malgré un niveau variable
d'expression de Tsix.
Enfin, de manière inattendue, Tsix semble exercer une régulation de H3K27me3 et du niveau
d'expression des gènes dans une région étendue d'au moins 300 kb en 5' de Xist.
210

3.3 Bivalence des fonctions de Tsix dans le Xic
L'extension du domaine de H3K27me3 dans les mutants de Tsix peut être interprétée de deux
manière Elle pourrait correspondre à la propagation en cis de l'enrichissement observé dans le
hotspot, auquel cas l'expression de Tsix créerait une barrière à cette propagation. Elle pourrait
également ne pas dépendre hotspot, mais être conditionnée plutôt par les propriétés de l'ensemble
du locus Xist/Tsix, selon que Tsix est exprimé ou non.
La corrélation entre accumulation de PolII et délimitation du territoire enrichi en H3K27me3
semble privilégier la première hypothèse., et désigner l'accumulation de PolII pour une fonction
d'élément barrière. Cette corrélation est parfaite si l'on se réfère aux cellules ES sauvages et aux
mutants Ma2L, dans lesquels Tsix est tronqué dans sa partie 5'. Dans les mutants ΔDXPas34,
H3K27me3 est enrichi jusqu'à la région promotrice de Tsix, dans laquelle une accumulation faible
de PolII continue à être observée, malgré la perte de DXPas34 (Vigneau et al., 2006). L'expression
résiduelle de Tsix, de l'ordre de 10 % de celle observée dans les cellules sauvages, ne freine donc
pas l'extension du domaine d'enrichissement en H3K27me3, qui ne s'arrêterait que lorsque
l'accumulation de PolII serait suffisamment importante. Deux modes d'action permettant à
l'accumulation de PolII d'avoir une fonction délément barrière ont été discutés dans l'article: cette
accumulation pourrait conduire au recrutement d'enzymes s'opposant à H3K27me3 (par exemple
des déméthylases), ou provoquer une discontinuité dans l'enchaînement des nucléosomes, qui serait
préjudiciable à la propagation en cis de H3K27me3.
Cependant, si l'extension du domaine enrichi en H3K27me3 ne dépend que de l'accumulation de
PolII, il est surprenant qu'en l'absence du promoteur majoritaire de Tsix, dans les mutants ΔAV, elle
ne se poursuive pas au-delà de la délétion. Cela pourrait être lié à l'accumulation ectopique de PolII
à la fin du transcrit minoritaire de Tsix, pourtant exprimé à un niveau inférieur à l'expression
résiduelle du transcrit majoritaire dans les mutants ΔDXPas34 (Vigneau et al., 2006). Une
explication alternative consiste à faire l'hypothèse d'un antagonisme entre la déposition de
H3K4me2, associée à l'expression de Tsix (Morey et al., 2004; Navarro et al., 2005; Navarro et al.,
2006), et l'accumulation de H3K27me3. Dans ce cas, l'accumulation de H3K27me3 ne serait qu'un
effet indirect de l'absence de H3K4me2. Cela serait cohérent avec le fait que H3K4me2 est
également fortement diminué dans les mutants ΔDXPas34. Mais cette explication supposerait que
H3K4me2 soit également enrichi en amont du promoteur majoritaire de Tsix, malgré le faible
niveau d'expression du transcrit minoritaire. Cela n'a pas été évalué de manière extensive mais, pour
une position testée en amont du promoteur majoritaire de Tsix, le niveau de H3K4me2 serait a
211

priori suffisant pour restreindre l'extension de H3K27me3, si un tel antagonisme était avéré
(Navarro et al., 2006). De manière générale, la configuration d'ensemble de la chromatine, et non
pas seulement H3K4me2, pourrait contrarier l'enrichissement en H3K27me3. Remarquons qu'une
telle explication peut tout aussi bien s'inscrire dans l'un ou l'autre des modèles mentionnés plus haut,
à savoir la propagation en cis de H3K27me3 à partir du hotspot, ou son recrutement
indépendamment de ce dernier.
Contrairement à l'enrichissement en H3K27me3 dans le corps du gène Xist, qui est comparable
dans les trois mutants de Tsix étudiés, les réorganisation de la chromatine dans la région promotrice
de Xist est différente dans la lignée ΔDXPas34, hypomorphe pour Tsix, et dans les lignées ΔAV et
Ma2L, essentiellement amorphes. Ainsi, au promoteur de Xist, seule la marque hétérochromatique
H3K9me3 diminue de la même manière dans les trois lignées, alors que l'enrichissement en
marques euchromatiques H3K4me2, H3K4me3, H3K9ac n'est observée de manière importante, et
sur l'ensemble de la région promotrice de Xist, que dans les mutants amorphes pour Tsix. La
méthylation des CpG augmente quant à elle dans les cellules ΔDXPas34, alors qu'elle diminue dans
les cellules ΔAV et Ma2L. Au vu de ces observations, on pourrait penser que la marque H3K9me3
au promoteur de Xist est plus sensible à la transcription antisens que les autres modifications
d'histones, H3K4me2, H3K4me3 et H3K9ac. Il est en revanche plus difficile d'expliquer
l'augmentation de la méthylation des CpG dans les mutants ΔDXPas34. On peut supposer qu'il
s'agirait là d'un effet secondaire de la « lecture » de la nouvelle combinaison de modifications
d'histones au promoteur de Xist. Cela suggère en tout cas que la méthylation de l'ADN et la
modification H3K9me3 ne seraient pas, à ce locus, dépendant l'un de l'autre, contrairement à ce qui
est généralement observé pour ces deux marques (Ikegami et al., 2007; Wu et al., 2007). Surtout,
les modification de la chromatine observées dans les cellules ΔDXPas34 illustrent l'autonomie des
profils chromatiniens dans le corps du gène Xist et dans sa région promotrice. Nous avons déjà
évoqué que cela pourrait être lié à la délimitation de cette région promotrice par des pics de fixation
de CTCF, qui pourraient avoir une fonction insulatrice (cf introduction, partie 2, section 4.3,. 127).
Quel qu'en soit le mécanisme sous-jacent, le niveau d'expression de Tsix et le degré
d'euchromatinisation de la région promotrice de Xist dans les cellules ES mâles indifférenciées
corrèlent avec le niveau d'expression de Xist et la fréquence d'inactivation ectopique observés après
différenciation. Pour relier les effets de l'invalidation de Tsix avant différenciation à ceux observés
en cours de différenciation, il est possible d'imaginer le scénario suivant. Dans les cellules
indifférenciées, l'expression de Tsix permet la mise en place au locus Xist/Tsix d'une chromatine qui
serait permissive pour l'expression de Xist, mais Tsix maintient simultanément le promoteur de Xist
212

dans un état réprimé, bien que facilement activable (cf introduction, partie 2, section 4.2, p. 123).
L'enrichissement en H3K27me3 au locus Xist/Tsix observé en l'absence de Tsix pourrait faire l'office
d'un répresseur de la transcription de Xist, régulé dynamiquement, et représenter ainsi une sécurité
supplémentaire pour empêcher l'inactivation d'être initiée dans les cellules indifférenciées. Elle
expliquerait alors l'absence d'activation transcriptionnelle de Xist, malgré l'euchromatinisation de
son promoteur et aurait donc la même fonction que le répresseur transcriptionnel proposé
précédemment par Navarro et al. (cf p. 128) (Navarro et al., 2005). Cette fonction justifierait
également que H3K27me3 soit amené à disparaître en cours de différenciation, au moins
transitoirement, durant la phase d'initiation de l'inactivation. On peut également imaginer que la
configuration chromatinienne associée à l'enrichissement en H3K27me3 s'oppose, dans les cellules
indifférenciées à la localisation des transcrits Xist sur le Xic, ce dernier ne pouvant dès lors pas
servir de centre de nucléation pour la formation d'un domaine d'ARN Xist. Après la phase
d'initiation de l'inactivation, l'enrichissement en H3K27me3 dans le Xic accompagnerait la mise en
place des marques hétérochromatiques sur le Xi, ce qui expliquerait qu'il ne soit pas observé dans
les fibroblastes mâles. Il serait intéressant d'établir une cinétique précise de la perte, puis de la
restauration de H3K27me3, de manière à déterminer dans quelle mesure ces évènements sont
associés à l'initiation ou à la consolidation de l'inactivation.
3.4 Régulation globale du Xic par Tsix
De manière inattendue, l'absence d'expression de Tsix dans les cellules indifférenciées mâles
Ma2L et ΔAV a pour conséquence une augmentation de H3K27me3 et une diminution de
l'expression des gènes (essentiellement Xpct et Cnbp2, dans une moindre mesure Ftx et Jpx) dans
l'ensemble de la région du hotspot, soit environ 300 kb. Cette situation rappelle le rôle joué,
notamment, par les ARN Kcnqot1 et Air dans la régulation de domaines de gènes soumis à
l'empreinte parentale. Nous discuterons plus en détail à la section 2.2.1 de la discussion (p. 229) des
rapprochements qui peuvent être fait entre ces deux gènes et Tsix, ainsi que, plus généralement, des
points communs entre le fonctionnement du Xic et d'autres exemples d'expression monoallélique.
Il est important de noter, cependant, que nous ne savons pas si la régulation par Tsix de la région
en 5' de Xist est ou non médiée par Xist. Il serait intéressant de le tester au moyen de lignées
mutantes, sur le même chromosome, pour Xist et Tsix. La configuration Xdc générée par Sado et al.
(cf note p. 124) pourrait être utilisée dans ce but. L'idée que Xist servirait d'intermédiaire pour cette
régulation est cohérentes avec l'hypothèse que le hotspot interviendrait en tant que centre de
213

nucléation pour l'initiation de l'inactivation. En effet, en l'absence de Tsix, le niveau des transcrits
Xist est augmenté ce qui, malgré les réserves déjà exprimées quant à sa capacité à être localisé
correctement (cf p. 150), pourrait initier au niveau du hotspot la mise en place de l'inactivation. Un
tel effet devrait être limité dans le temps car nous avons montré que la méthylation H3K27me3
était ensuite perdue au hotspot, quoique de manière transitoire. Cette période de temps durant
laquelle la fonction du hotspot comme centre de nucléation dépendrait de H3K27me3 pourrait
correspondre à la fenêtre d'initiation définie par Wutz et al. (cf p. 45). L'information véhiculée par
H3K27me3 pourrait ensuite être relayée au hotspot par d'autres marques (en particulier H3K9me2,
qui y est enrichie et n'est pas altéré dans des mutants de Tsix), et sur le reste du chromosome par la
propogation en cis de l'inactivation.
L'observation d'un lien entre expression de Tsix et configuration épigénétique en 5' de Xist
pourrait permettre également de mieux articuler entre elles certaines des observations de Bacher et
al. concernant le rapprochement transitoire des Xic en cours de différenciation (Bacher et al., 2006).
En effet, ce rapprochement dépend de Tsix (cf p. 109), et il peut être observé entre le Xic endogène
et des transgènes du Xic uniquement lorsque ceux-ci sont insérés en multicopie (cf p. 93). Il a été
proposé que cela correspondait à la reconstitution d'un hotspot synthétique dans les transgènes en
multicopies et que le hostpot serait requis pour le rapprochement des Xic (Bacher et al., 2006). Dans
ce cas, le rôle joué par Tsix dans le rapprochement des Xic pourrait être médié par le hotspot. Cette
hypothèse est d'autant plus séduisante que l'on sait que la mobilité à l'intérieur du volume nucléaire
peut être régulée par la structure chromatinienne et l'activité transcriptionnelle (Branco et Pombo,
2006). Il serait donc intéressant de vérifier une telle fonction pour le hotspot en en réalisant la
délétion ciblée dans des cellules femelles.
214

Discussion générale
Chapitre 1 Conservation de Tsix et de DXPas34
Globalement, la fonction du gène Xist et les marques épigénétiques associées au Xi semblent
bien conservées parmi les mammifères, et notamment entre l'homme et la souris, qui ont été les plus
étudiés (cf introduction, partie 1, chapitre 2, p. 23). Cela semble moins le cas lorsqu'on s'intéresse à
la régulation développementale de l'expression de Xist. Sont là pour en témoigner, chez l'homme,
l'absence d'expression empreintée de Xist avant l'implantation et, probablement, dans les tissus
extraembryonnaires (cf introduction, partie 1, section 3.3.7, p. 68) (Daniels et al., 1997; Ray et al.,
1997), ainsi que l'inaptitude d'un transgène du XIC humain à réguler correctement l'expression de
XIST lorsqu'il est inséré dans des cellules ES murines (cf p. 93) (Migeon et al., 2001b).
Or, nous avons montré que, chez la souris, Tsix et DXPas34 étaient deux éléments requis pour
que l'inactivation soit mise en place de manière appropriée. Qu'en est-il chez les autres espèces de
mammifères et, en particulier chez l'homme?
1.1 Conservation du gène Tsix
La comparaison de séquences synténiques entre l'homme, la souris et la vache a montré un
faible degré de conservation du gène Tsix (Chureau et al., 2002). L'expression de TSIX, est
cependant observée chez l'homme (Migeon et al., 2001a), et une transcription antisens à Xist est
détectée chez la vache (Farazmand et al., 2004). Dans les deux espèces, l'inactivation est aléatoire
dans les cellules somatiques. Il semble qu'elle le soit également dans les tissus extraembryonnaires
chez l'homme (cf introduction, partie 1, section 3.3.7, p. 68), mais que l'inactivation soit empreintée
dans ces derniers chez la vache (Dindot et al., 2004; Xue et al., 2002).
Chez l'homme, plusieurs isoformes de TSIX ont été caractérisées, qui sont apparemment toutes
terminées avant le premier exon de XIST (fig. 24B, p. 216) (Chow et al., 2003; Migeon et al.,
2001a). Une telle conclusion souffre néanmoins plusieurs réserves. Tout d'abord, elle résulte
215

216
Fig. 24: Conservation de Tsix chez l'homme
Légende page 217

d'observations par RT-PCR et RNA-FISH, réalisées avec une résolution insuffisante pour exclure
l'existence d'exons de petite taille, notamment au niveau du premier exon de XIST. D'autre part, elle
correspond à l'expression de TSIX dans des cellules ES murines, à partir d'un transgène du XIC
humain (Migeon et al., 2001a), dans des fibroblastes dérivés de cellules germinales primordiales
(désignés comme human embryoid body derived, EBD) (Migeon et al., 2001a), ou dans des
carcinomes embryonnaires humains, dérivés de tumeurs de la lignée germinale mâle (Chow et al.,
2003). Or, il n'est pas certain que ces systèmes soient représentatifs de la fonction que TSIX pourrait
avoir, au cours du développement, dans l'établissement de l'inactivation aléatoire. Il est d'ailleurs
intéressant de constater que, chez la souris, il existe un gradient d'abondance des transcrits Tsix de 5'
en 3' (cf introduction, partie 2, section 2.2.2.1, p. 102) (Shibata et Lee, 2003), et que la détection des
transcrits Tsix en 5' et en 3' ne coïncide pas toujours au cours du développement embryonnaire (cf
introduction, partie 2, section 2.2.2.2, p. 103). Cela pourrait également être le cas chez l'homme, et
une analyse plus systématique devrait être faite pour pouvoir affirmer que les transcrits TSIX n'ont
pas de complémentarité avec la région 5' de XIST.
L'expression de XIST et de TSIX a été analysée par RNA-FISH et RT-PCR dans plusieurs
lignées cellulaires, dont des cultures de villi chrorioniques (cellules trophoblastiques du placenta)
collectés à la naissance, des fibroblastes foetaux (obtenus entre 5 et 8 semaines de gestations) et des
fibroblastes de peau de nouveaux-nés, d'enfants de différents âges et de jeunes adultes (Migeon et
al., 2002). Dans tous les cas, l'expression de XIST n'est observée, comme attendu, que chez les
femelles, et correspond à l'inactivation de l'un des deux chromosomes X. Mais de manière
surprenante, l'expression de TSIX est également spécifique des femelles et est encore détectée dans
217
Légende de la figure 24, p. 216: (A) Carte du locus Xist/Tsix chez la souris. (B) Carte du locus XIST/TSIX chez l'homme représentant les différentes isoformes de TSIX. La transcription sens représentée en pointillés vert a été observée dans une lignées de cellules rénales adultes femelles (Chow et al., 2003). Les types cellulaires dans lesquels ont été identifiés les transcrits TSIX (en rouge) sont précisés. Les pointillés rouges figurent une incertitude quant à l'origine du transcrit dans les cellules EBD (pour embryoid body derived, fibroblastes dérivés de cellules germinales primordiales) (Migeon et al., 2001a). La transcription antisens à XIST est discontinue dans des carcinomes embryonnaires dérivés de tumeurs de cellules germinales mâles, mais on ne sait pas si cela correspond à une forme épissée de TSIX ou à deux transcrits distincts (Chow et al., 2003). Les origines de transcription de TSIX ont été localisées précisément par 5'RACE pour des formes exprimées à partir d'un transgène du XIC humain inséré sur un autosome dans des cellules ES murines mâles (Migeon et al., 2001a). Elles sont toutes situées dans un intervalle de 1 kb et révèlent l'existence d'un premier exon alternatif, (C) Dotplot figurant les trois blocs de séquences conservés en 3' de Xist/XIST entre l'homme et la souris (Lee et al., 1999)

certains individus jusqu'à l'âge de 8 ans. De plus, elle est réalisée à partir du Xi, et l'ARN TSIX
semble former un domaine inclus dans le domaine d'ARN XIST (Migeon et al., 2002). L'expression
coïncidente de XIST et TSIX à partir du même chromosome, et l'existence d'un domaine d'ARN
TSIX, sont également observée dans des cellules EBD (Migeon et al., 2002), ainsi qu'à partir de
transgènes du XIC insérés sur un autosome dans des cellules somatiques humaines (Chow et al.,
2003), et dans des cellules ES murines mâles (cf p. 93) (Migeon et al., 2002). Seuls les carcinomes
embryonnaires témoignent d'une expression différente, dans la mesure où XIST n'y est détecté par
RNA-FISH que dans 10% des cellules sous la forme d'un signal ponctuel. L'expression de TSIX,
faiblement détectée par RT-PCR, n'a cependant pas été regardée dans ces cellules par RNA-FISH
(Chow et al., 2003).
Dans tous les cas, lorsque TSIX est exprimé, l'expression au niveau du dernier exon de XIST,
détectée par RT-PCR quantitative brin spécifique, coïncide avec l'expression antisens en 3' de XIST
détectée par RT-PCR, brin spécifique (Chow et al., 2003) ou non (Migeon et al., 2001a; Migeon et
al., 2002). De plus, dans des cellules somatiques femelles adultes, Chow et al. observent une
transcription sens (mais pas de transcription antisens) en 3' du site normal de terminaison de XIST.
Une telle transcription sens n'est cependant pas détectée par les mêmes auteurs dans des cellules
somatiques humaines exprimant XIST à partir d'un transgène du XIC (Chow et al., 2003). Enfin, les
marquages par RNA-FISH semblent avoir été réalisés dans tous les cas avec des sondes double brin
(Chow et al., 2003; Migeon et al., 2002)7, dans les régions a priori non chevauchantes des deux
gènes. Aussi, les données sont-elles convaincantes pour dire que TSIX et XIST sont coexprimés à
partir du même chromosome chez l'homme (d'après les RT-PCR brin spécifiques et l'absence de
marquage par RNA-FISH de TSIX sur le Xa), mais il n'est pas exclu que les domaines d'ARN TSIX
observés par Migeon et al. (Migeon et al., 2002) correspondent à une fraction des transcrits XIST
dont la transcription aurait été poursuivie au-delà du site normal de terminaison.
Quoiqu'il en soit, l'expression de TSIX ne semble être en cis un obstacle ni à l'expression, ni à
l'accumulation de XIST. En particulier, la coexpression de XIST et TSIX à partir du même
chromosome dans le placenta est cohérente avec l'absence probable d'inactivation empreintée dans
les tissus extraembryonnaires humains (cf introduction, partie 1, section 3.3.7, p. 68). Cependant,
les cellules dans lesquelles l'expression de TSIX a été étudiée sont toutes dérivées de tissus,
embryonnaires et extraembryonnaires, obtenus postérieurement à la mise en place de l'inactivation.
7 La position exacte de la sonde en 3' de XIST et la méthode utilisée pour son marquage ne sont en réalité pas précisés par Chow et al. (Chow et al., 2003), qui n'ont fait une analyse par RNAFISH de la transcription de TSIX que dans des cellules somatiques l'exprimant à partir d'un transgène du XIC. Pour les cellules analysées par Migeon et al. (Migeon et al., 2002), les sondes utilisées sont décrites explicitement comme n'étant pas brin spécifique.
218

Il serait donc possible que TSIX serve à réguler XIST avant ou pendant la mise en place de
l'inactivation, éventuellement par l'intermédiaire de transcrits plus longs, se poursuivant à travers le
premier exon de XIST. On ne sait pas précisément quand cette mise en place pourrait avoir lieu. La
détection de XIST par RT-PCR, du stade 2 cellules au stade blastocyste, aussi bien dans les
embryons mâles que femelles (Daniels et al., 1997; Ray et al., 1997), semble indiquer que
l'inactivation ne pourrait initiée qu'à partir du stade blastocyste. Néanmoins, il est tout a fait possible
que l'expression détectée dans ces travaux soit celle de TSIX, qui était alors inconnu.
Compte-tenu des difficultés à étudier le développement embryonnaire précoce chez l'homme, il
est probable que l'utilisation de lignées de cellules souches embryonnaires humaines (hES, pour
human embryonic stem) permettra d'améliorer notre compréhension de l'inactivation dans notre
espèce. Mais la représentativité de ces cellules par rapport à la masse cellulaire interne des
blastocystes se pose avec encore plus d'acuité que chez la souris, dans la mesure où l'étude
comparative dans les blastocystes est plus difficile, et où les cellules hES qui ont été dérivées
jusqu'à présent ont des profils d'inactivation variables (Hoffman et al., 2005). Dans la plupart des
cellules hES femelles caractérisées, qui expriment des marqueurs de cellules souches, l'un des
chromosomes X est déjà inactivé, bien que son association à MACROH2A1, qui est une marque
tardive du Xi, ne soit acquise que lorsque ces cellules sont différenciées (Hoffman et al., 2005). Il
serait important de savoir s'il s'agit là d'un trait particulier lié au processus de dérivation des cellules
hES, où si l'inactivation pourrait être mise en place plus précocement chez l'homme que chez la
souris. Enfin, il serait intéressant de connaître, lors de l'embryogenèse précoce et dans les cellules
hES, l'expression du gène TSIX, mais également la structure des transcrits TSIX, et en particulier
leur degré de complémentarité à XIST.
Chez la vache, les données concernant l'expression antisens à Xist sont nettement plus
préliminaires. On ne peut d'ailleurs pas parler de gène Tsix, dans la mesure où la transcription
antisens n'a été analysée, par RT-PCR brin spécifique, qu'à une seule position dans le premier exon
du gène Xist (Farazmand et al., 2004). Elle est détectée, de même que l'expression de Xist, dans
plusieurs organes embryonnaires chez les femelles. Mais seule l'expression antisens est observée
dans les mêmes organes chez les mâles (Farazmand et al., 2004). Des transcrits antisens sont
également présents chez l'adulte dans les organes génitaux (ovaires, oviductes et testicules), et dans
les muscles des mâles (Farazmand et al., 2004). Le gène Tsix, s'il existait, pourrait donc être soit
exprimé de manière ubiquitaire, soit spécifiquement sur le Xa, auquel cas il pourrait être requis pour
219

s'opposer en cis à l'expression de Xist, notamment en interagissant avec son premier exon. Si un
profil similaire était observé dans le placenta, cela pourrait expliquer que l'inactivation y soit
empreintée chez la vache.
Chez le campagnol, l'expression de Tsix est en accord avec un tel profil. Tsix y est en effet
exprimé à partir du Xa dans les tissus adultes mâles et femelles, où l'inactivation est aléatoire, ainsi
que dans les tissus extraembryonnaires, pour lesquels l'inactivation est empreintée (Shevchenko et
al, communication orale). Cela suggère, là encore, que Tsix pourrait être requis pour réprimer en cis
l'expression de Xist, aussi bien dans les tissus embryonnaires qu'extraembryonnaires.
Au final, il apparaît que Tsix pourrait avoir une fonction dans la répression de Xist chez d'autres
mammifères que la souris, l'homme faisant, peut-être, exception. Néanmoins, l'expression de Tsix,
qui est éteinte rapidement après la mise en place de l'inactivation chez la souris, ne l'est pas dans les
autres espèces étudiées. Cela pourrait tenir à la présence d'éléments régulateurs de Tsix distincts
entre ces espèces, et avoir des implications quant à la manière dont y sont réalisés les processus de
choix et de comptage lors de l'inactivation aléatoire.
1.2 Conservation des éléments régulant l'expression de Tsix
La région 5' de Tsix est peu conservée entre la souris, l'homme et la vache, à l'exception de trois
blocs de séquence en 3' de Xist/XIST, conservés entre l'homme et la souris (fig. 24C, p. 216)
(Chureau et al., 2002; Lee et al., 1999). Parmi ceux-ci, seul le premier bloc, qui correspond à la fin
de Xist, est également conservé chez le vache (Chureau et al., 2002). Le troisième bloc est proche
du promoteur majoritaire de Tsix chez la souris, et de l'origine du transcrit TSIX caractérisé par
Migeon et al. (Migeon et al., 2001a). Cependant, l'îlot CpG associé à la région promotrice de Tsix
chez la souris n'est pas conservé chez l'homme et la vache (Chureau et al., 2002; Migeon et al.,
2001a). De manière similaire, le minisatellite DXPas34 est peu conservé chez les mammifères
(Chureau et al., 2002; Cohen et al., 2007).
Le degré de conservation le plus important pour DXPas34 est observé chez deux rongeurs, le rat
(fig. 25, p. 223) (Cohen et al., 2007) et le campagnol (Shevchenko et al., communication orale,
2006). Le motif répété de 34 pb, définissant classiquement DXPas34 chez la souris et appelé ici A1,
est très dégénéré chez le rat (où il est appelé motif A), chez qui il a perdu la séquence consensus de
fixation de CTCF mais conservé celle pour YY1. Il y est également présent en plus faible nombre
de copies (13 copies, contre 29 chez la souris dans le fond génétique 129) (Cohen et al., 2007).
Chez la souris, un second motif, A2, homologue du motif A trouvé chez le rat, a été identifié, mais il
220

ne possède pas de consensus pour la fixation de CTCF. Plus en amont de DXPas34, un troisième
motif de 31 pb est répété 35 fois chez le rat, mais seules 6 copies fortement dégénérées en sont
présentes chez la souris. Cependant, dans les deux espèces, ce motif possède le consensus de
fixation de CTCF. Chez l'homme, seul le premier groupe de motifs (A) est présent, mais à raison
seulement de 7 exemplaires répartis sur 3 kb et, bien que leur séquence soit très dégénérée, ils ont
conservé le consensus pour la fixation de CTCF (Cohen et al., 2007; Kim et al., 2006).
Chez la souris même, un polymorphisme du nombre de répétition du motif de 34 pb est observé
entre fonds génétiques. Il a été monté que ce polymorphisme était indépendant du locus Xce
(Prissette et al., 2001). Mais il pourrait ne pas être sans incidence sur la fonction de DXPas34, ce
qu'il serait intéressant d'évaluer expérimentalement, en modifiant le nombre de répétitions dans un
fond génétique donné.
Le locus Xite semble également peu conservé parmi les mammifères (Chureau et al., 2002;
Ogawa et Lee, 2003), y compris chez le campagnol (Shevchenko et al., communication orale,
2006). Nous avons décrit précédemment comment les propriétés de DXPas34 et Xite étaient
régulées au cours de la différenciation de cellules ES murines, de manière coïncidente avec
l'extinction transcriptionnelle de Tsix (cf introduction, partie 2, sections 3.3, p. 116). Cela
correspond en particulier, pour les deux loci, à la disparition de sites d'hypersensibilité à la DNAseI
et de la capacité à initier une transcription bidirectionnelle. En cours de différenciation, DXPas34
est également hétérochromatinisé et perd son association avec les facteurs CTCF et YY1. D'autre
part, des tests luciférase suggèrent qu'en l'absence de régions additionnelles telles que Xite ou un
enhancer bipartite incluant DXPas34, le promoteur de Tsix est constitutionnellement actif dans les
cellules différenciées (cf introduction, partie 2, section 3.2, p. 116). La faible conservation de
DXPas34 et Xite chez l'homme et la vache, mais également, bien que dans une moindre mesure
pour DXPas34, chez le campagnol, pourrait donc expliquer que dans ces trois espèces, l'expression
de Tsix ne soit pas réprimée après la mise en place de l'inactivation (cf section précédente).
De manière intéressante, il a été proposé que DXPas34 aurait dérivé, chez les euthériens, d'un
rétroélément de la famille ERVL (endogenous retrovirus) (Cohen et al., 2007). Les rétrotransposons
fonctionnant comme des unité autonomes, susceptibles d'apporter un un plusieurs promoteurs,
enhancers, ou insulateurs (Gerasimova et Corces, 1996; Willoughby et al., 2000), il est possible
que certaines de ces fonctions aient été recrutées pour la régulation du gène Tsix. Par la suite,
DXPas34 aurait pu acquérir une fonction spécifiquement chez la souris ou les rongeurs, grâce à
l'amplification de certains motifs, tandis qu'une fonction différente provenant de l'ancêtre de
DXPas34 aurait été retenue chez l'homme.
221

Inversement, on peut penser que certaines des fonctions de l'ancêtre de DXPas34 aient été
préservées dans différentes espèces, malgré le faible degré de conservation de séquence. L'absence
de corrélation stricte entre conservation de séquence et conservation de la fonction d'éléments
régulateurs est bien illustrée par la difficulté à caractériser des motifs nucléotidiques communs aux
différents sites d'hypersensibilité à la DNAse I et permettant d'en prédire l'existence (Noble et al.,
2005). Au contraire, une approche prenant comme critère la structure locale de l'ADN parvient à
faire cette prédiction de manière efficace (Greenbaum et al., 2007). Par ailleurs, la caractérisation à
l'échelle du génome des profils transcriptionnels, de modications d'histones et de réplication de
l'ADN, contribue désormais à faire émerger la notion de domaines ou territoires fonctionnels
(Thurman et al., 2007), qui ne dépendent qu'indirectement de la séquence de l'ADN. L'appartenance
d'un locus à un domaine fonctionnel pourrait ainsi être préservée au cours de l'évolution malgré la
divergence de sa séquence nucléotidique.
Cela pourrait contribuer à expliquer le faible degré de conservation généralement observé pour
les sites de fixation des facteurs de transcription, aussi bien entre différentes espèces de drosophile
(Dermitzakis et al., 2003; Emberly et al., 2003; Richards et al., 2005) qu'entre l'homme et la souris
(Dermitzakis et Clark, 2002). La redondance fonctionnelle entre sites de fixation d'un même
élément régulateur pourrait également être une source d'évolution rapide de sa séquence. C'est par
exemple le cas chez la drosophile pour un des enhancers (S2E, pour stripe 2 enhancer) du gène
even-skipped (Ludwig et al., 2000; Ludwig et al., 2005). Le patron d'expression associé à cet
enhancer est remarquablement bien conservé entre différentes espèces de drosophiles, bien que sa
structure, et les nombreux sites de fixation de facteurs de transcription qui lui sont associés aient
divergé de manière importante. Les auteurs démontrent que cette conservation de fonction résulte
de la co-évolution des moitié 5' et 3' de l'enhancer, ce qui aurait été a priori impossible en l'absence
de redondance fonctionnelle des éléments qui le composent. On peut imaginer un scénario similaire
dans le cas de DXPas34, dont la structure a évolué rapidement, y compris entre espèces
phylogénétiquement proches, telles que la souris et le rat. Le grand nombre et la redondance
fonctionnelle des sites de fixation de CTCF et YY1 pourrait ainsi être en partie responsable de la
rapidité de cette évolution.
Une fonction de DXPas34 aurait également pu être maintenue chez l'homme, dans la mesure où
certains sites consensus de fixation de CTCF ont été conservés dans les séquences homologues au
motif A de DXPas34. Si cette fonction est liée à la régulation transcriptionnelle de Tsix, on doit
imaginer qu'elle correspond à un moment particulier du développement, où la fonction de Tsix serait
222

requise de manière similaire dans les différentes espèces. Cela pourrait être lors de la mise en place
de l'inactivation, pour laquelle nous disposons, pour l'instant, de peu d'informations en dehors de la
souris.
Enfin, il est bon de mentionner que, y compris si la mise en place de l'inactivation est très
différente chez l'homme et la souris, cela n'enlève en rien l'intérêt de poursuivre une étude détaillée
de l'inactivation chez cette dernière. En guise de boutade, on pourrait tout aussi bien se demander si
l'homme est un bon modèle pour étudier la souris: la réponse serait probablement négative. Plus
sérieusement, l'inactivation du chromosome X, et particulièrement sa régulation au niveau du Xic,
sont un excellent modèle pour comprendre des mécanismes épigénétiques de portée générale. Dans
le chapitre suivant, nous allons précisément discuter certains parallèles qui peuvent être faits entre
ce que nous savons du fonctionnement du Xic et de la régulation d'autres régions où l'expression des
gènes est monoallélique.
223
Fig 25: Conservation de DXPas34 (d'après Cohen et al., 2007)
(A) Dot plot entre la souris et le rat pour DXPas34. Le minisatellite DXPas34 tel qu'il est défini classiquement correspond à la répétition du motif A1 chez la souris. Des détails sont donnés dans le texte pour les différents motifs (A1, B2, A, B). (B) Alignement des motifs A et B retrouvés ches la souris, le rat et l'homme.

224

Chapitre 2 Le Xic, cas particulier ou paradigme?
2.1 ARN non codants et expression monoallélique
Récemment, des études à grande échelle ont montré que la plus grande partie du génome des
mammifère était transcrite (Carninci, 2006; Frith et al., 2005; Mendes Soares et Valcarcel, 2006).
La plupart des transcrits non annotés ainsi révélés sont probablement des ARN non codants, un
grand nombre d'entre eux possédant des transcrits antisens. Dans la majorité des cas, trancription
sens et antisens sont coordonnées, les gènes étant coexprimés et/ou leur niveau d'expression étant
inversement corrélé (Chen et al., 2005; Katayama et al., 2005). De fait, tant chez les eucaryotes que
les procaryotes, la transcription antisens est utilisée pour réguler l'expression de nombreux gènes
(Ogawa et Lee, 2002; Terryn et Rouze, 2000; Wagner et Simons, 1994). Le couple Xist/Tsix semble
s'insérer dans ce schéma, particulièrement si l'on se réfère à l'expression coordonnées de ces deux
gènes au cours du développement. En particulier, Xist et Tsix sont tous les deux exprimés dans les
tissus extraembryonnaires et dans la masse cellulaire interne des blastocystes. En revanche,
l'expression de Tsix est perdue dans les tissus embryonnaires. Il serait intéressant de savoir s'il s'agit
là d'un trait particulier au couple Xist/Tsix, ou si la coexpression, dans une même cellule, de gènes
sens et antisens est plus répandue dans les lignages extraembryonnaires et dans les cellules
indifférenciées, ou possédent des propriétés de cellule souche. La généralisation programmée des
études à haute résolution du transcriptome apportera certainement une réponse.
Une autre particularité des gènes Xist et Tsix est le caractère généralement monoallélique de leur
expression, la seule exception notable étant dans la masse cellulaire interne des blastocystes ou dans
les cellules ES qui en sont dérivées. Inactivation du chromosome X mise à part, les deux dispositifs
d'expression monoalléliques les plus étudiés sont l'exclusion allélique des gènes codant pour les
immunoglobulines, et l'expression soumise à l'empreinte parentale. A ce jour, environ 70 gènes dont
l'expression est soumise à l'empreinte parentale ont été caractérisés chez les mammifères (par
commodité de language, nous parlerons d'expression « empreintée », comme nous en avons pris
l'habitude concernant l'inactivation du chromosome X). Ils sont en général organisés en groupes de
gènes corégulés au sein d'un même territoire génomique. La plupart de ces groupes contiennent au
225

moins un ARN non-codant exprimé de manière empreintée (pour revue, Yang et Kuroda, 2007).
Pour deux de ces ARN, Air et Kcnq1ot1, il a été démontré, en en réalisant une forme tronquée, que
leur transcription régulait l'expression empreintée d'un gène antisens (Igf2r et Kcnq,
respectivement), mais également de gènes plus distants appartenant au même territoire (Mancini-
Dinardo et al., 2006; Sleutels et al., 2002). Cela rappelle bien évidemment la régulation de
l'expression de Xist par Tsix, ainsi que la régulation globale que semble exercer Tsix sur la région en
5' de Xist (cf résultats, section 3.4, p. 213). Cependant, de même que pour Tsix, on ne sait pas si le
fait de transcrire ou le transcrit en tant que tel réalisent ces fonctions.
Comme Tsix vis-à-vis de Xist, l'ARN non codant Air contrôle la configuration de la chromatine
au promoteur du gène antisens Igf2r (Yamasaki et al., 2005). Ainsi, dans des cultures primaires de
neurones, Air n'est pas exprimé et l'expression de Igf2r est biallélique, ce qui corrèle au promoteur
de ce dernier avec l'hypométhylation de l'ADN, l'acétylation des histones H3 et H4 et la
diméthylation de H3K4. A l'inverse, dans des fibroblastes et dans des cellules gliales en culture, seul
l'allèle paternel de Air et l'allèle maternel de Igf2r sont exprimés avec, dans ce cas, entre les allèles
paternels et maternels, un enrichissement différentiel des marques chromatiniennes au promoteur de
Igf2r (Yamasaki et al., 2005). L'expression biallélique de Igf2r semble acquise lors de la maturation
neuronale, car elle n'est pas observée dans les progéniteurs neuronaux et les cellules gliales in vivo.
Elle ne serait donc pas associée à des propriétés de cellules souches. Inversement, l'expression
biallélique de Tsix, et le faible niveau d'expression de Xist, observés dans les cellules ES et dans la
masse cellulaire interne des blastocyste, laissent penser qu'ils pourraient dépendre de facteurs
servant à maintenir un état pluripotent, qui sont perdus en cours de différenciation.
De manière similaire à Xist, l'allèle paternel de Kcnq1ot1 est exprimé à partir du stade 2
cellules, tandis que l'allèle maternel est réprimé (Lewis et al., 2006). Néanmoins, contrairement à
Xist, son expression demeure par la suite empreintée. La transcription de Kcnq1ot1 est requise pour
la répression en cis de gènes dont l'expression est empreintée soit de manière ubiquitaire, soit
spécifiquement dans les tissus extraembryonnaires (Mancini-Dinardo et al., 2006). Cette répression
semble médiée par le complexe Eed-Ezh2, et associée à l'enrichissement en marques répressives
H3K27me3 et H3K9me2, qui n'est apparemment pas restreint aux régions promotrices (Lewis et
al., 2004; Umlauf et al., 2004). La situation pourrait donc être plus similaire à celle observée dans le
Xic au niveau du hotspot, ou d'une manière générale sur le Xi.
De manière intéressante, dans un système épisomal où la région promotrice de Kcnq1ot1, qui
correspond au centre d'empreinte KvDMR1, est flanquée de gènes rapporteurs, ces derniers sont
réprimés avec d'autant plus d'efficacité que la transcription initiée dans KvDMR1 se poursuit sur une
226

grande distance (Kanduri et al., 2006; Thakur et al., 2004). Cette répression est associée en
l'enrichissement en marque chromatiques répressives, initialement aux positions où la transcription
originaire de KvDMR1 traverse un gène rapporteur antisens, puis sur l'ensemble de l'épisome
(Kanduri et al., 2006). Bien que le système utilisé ici soit très artificiel, il illustre l'importance que
pourrait avoir la taille des unités de transcription de couples de gènes sens/antisens pour la
régulation génique ou le contrôle du profil épigénétique dans les régions adjacentes. Cela est à
rapprocher de la grande taille de l'unité de transcription Xist/Tsix (53 kb), mais également des
transcrits Air (108 kb) (Lyle et al., 2000) et Kcnq1ot1 (54 kb) (Engemann et al., 2000).
L'expression de Tsix régule la configuration chromatinienne non seulement au promoteur de
Xist mais également, de manière opposée, sur l'ensemble de l'unité de transcription de Xist/Tsix. Il a
été proposé que ce remodelage permettrait de rendre épigénétiquement équivalents les deux Xic
dans la masse cellulaire interne des blastocystes, où Tsix est exprimé bialléliquement, de façon à
permettre le choix aléatoire du chromosome à inactiver (Navarro et al., 2005). Cette hypothèse a
pour corollaire que l'expression de Tsix et l'acquisition concomitantes de certaines marques
épigénétiques (enrichissement en H3K4me2 et appauvrissement en H3K27me3) correspondent à un
état de la chromatine compétent pour pouvoir initier ultérieurement l'inactivation. On peut faire le
parallèle avec ce qui est observé avant la recombinaison intrachromosomique VH-DJH précédent
l'exclusion allélique, au locus codant pour la chaîne lourde des immunoglobulines (pour revue,
Bassing et al., 2002; Johnson et al., 2003). Chez la souris, ce locus comprend plusieurs centaines de
segments de gène dits « variables » (variable, VH), 16 segments dits de « diversité » (diversity, DH)
et 4 segments de « jonction » (joining, JH) (Chevillard et al., 2002) qui sont recombinés lors de la
maturation des précurseurs des lymphocytes B pour donner un unique gène IgH fonctionnel.
Lorsque ce dernier est exprimé sur l'un des chromosomes, la recombinaison est inhibée au locus
IgH, ce qui a pour conséquence l'exclusion allélique, assurant qu'un seule type d'anticorps soit
produit dans un même clone de lymphocyte B. Avant la recombinaison VH-DJH, une transcription
antisens génique et intergénique est mise en place au locus VH (Bolland et al., 2004), qui devient
hyperacétylé sur les histones H3 et H4, mais de manière confinée aux gènes et à leur promoteur
(Chowdhury et Sen, 2001; Johnson et al., 2003). Transcription antisens et hyperacétylation étant
perdus après la recombinaison (Bolland et al., 2004; Chowdhury et Sen, 2001; Johnson et al.,
2003), elles pourraient être requises précisément pour permettre l'accessibilité du locus à la
machinerie de recombinaison, en conférant à chaque segment une compétence égale à être
recombiné. Les mécanismes permettant d'acquérir une telle compétence épigénétique pourraient
227

être assez généraux, et être également à l'origine de la déstabilisation de l'inactivation du
chromosome X observée lors de la maturation des lymphocytes B (cf introduction, partie 1,
section 3.4.2, p. 71).
Tsix est capable de modifier la chromatine de manière opposée sur son unité de transcription et
au promoteur de Xist vraisemblablement grâce à l'isolement de ce dernier par deux pics de fixation
de CTCF (Navarro et al., 2006). Au moins un autre exemple de ce type existe dans la littérature, au
locus DM1 (Cho et al., 2005). Une répétition CTG y est en effet hétérochromatinisée (methylation
H3K9 et recrutement de HP1γ) sous l'effet d'une transcription antisens la traversant, alors qu'elle est
insérée dans une région plus large caractérisée par un enrichissement en méthylation de H3K4. La
restriction de l'hétérochromatinisation au niveau de la répétition corrèle bien avec la fixation de
CTCF de part et d'autre: lorsque cette fixation est perdue à la suite d'une expansion de la répétition,
les marques hétérochromatiques s'étendent en cis (Cho et al., 2005). De manière interessante, le
positionnement phasé des nucléosomes semble jouer un rôle important pour la configuration
épigénétique de ce locus (Filippova et al., 2001), ce qui n'a pas encore été évalué au locus Xist/Tsix.
Les quelques exemples mentionnés dans les paragraphes précédents illustrent que la plupart des
mécanismes mis en jeu au Xic sont également utilisés à d'autres loci. La spécificité du Xic réside
donc, plutôt, dans la combinaison particulière de ces mécanismes et la manière dont ils sont régulés
au cours du développement. Si on la compare à l'expression de gènes soumis à l'empreinte
parentale, une des particularités les plus éloquentes de l'inactivation du chromosome X est la
possibilité de choisir, de manière aléatoire, l'un des deux chromosomes X pour être inactivé.
L'existence d'un mécanisme de comptage en est le corollaire car, sans lui, des schémas
d'inactivation aberrants, léthaux, seraient possibles. Le minisatellite DXPas34, ou une région un peu
plus grande l'incluant, jouent un rôle essentiel dans ces deux processus, choix et comptage, ainsi
que dans la réalisation correcte de l'inactivation empreintée dans les tissus extraembryonnaires. Or,
DXPas34 présente des similarités évidentes avec les centres d'empreinte (ICR, pour imprinting
control regions), contrôlant l'expression de groupe de gènes soumis à l'empreinte parentale. A ce
titre, percevoir les similitudes et les différences de DXPas34 avec les ICR peut être informatif.
C'est ce dont nous discutons à la section suivante.
228

2.2 Centres d'empreinte et répétitions en tandem
2.2.1 Comparaison entre DXPas34 et les centres d'empreinte
Les ICR sont les lieux d'acquisition de marques différentielles (ou empreinte) dans la lignée
germinale mâle et femelle (Li et al., 2004; Lucifero et al., 2004). Cela correspond en général à la
présence de DMD (differentially methylated domains) qui controlent l'expression des gènes en cis
(pour revue, Verona et al., 2003). L'empreinte primaire serait définie par la méthylation
différentielle au niveau des DMD. Il se pourrait donc que les modifications d'histones
n'interviennent qu'ultérieurement pour propager en cis ou consolider l'information véhiculée par la
méthylation de l'ADN. Cependant, on doit garder à l'esprit que la rareté du matériel biologique
correspondant à la lignée germinale ou aux stades préimplantatoires n'a pas permis d'évaluer
correctement le rôle des marques épigénétiques autres que la méthylation de l'ADN au niveau des
ICR. Les progrès technologiques en ce domaine donnent quelques espoirs que cela pourra bientôt
être fait (O'Neill et al., 2006).
L'un des ICR les mieux caractérisés, et présentant le plus de similitudes avec DXPas34, est celui
régulant l'expression des gènes Igf2 et H19 (fig. 26, p. 230) (Thorvaldsen et al., 1998). Il consiste en
un DMD riche en GC, localisé de -2 à -4 kb par rapport au site d'initiation de la transcription de
H19. Ce DMD est nécessaire pour permettre l'expression mutuellement exclusive de H19 et Igf2,
respectivement à partir du chromosome maternel et paternel (Thorvaldsen et al., 1998; Tremblay et
al., 1997). Sur le chromosome maternel, le DMD est hypométhylé et fixe CTCF au niveau de quatre
répétions d'un motif de 21 pb riche en GC (Bell et Felsenfeld, 2000; Hark et al., 2000; Stadnick et
al., 1999). La présence de CTCF est associée à une fonction insulatrice empêchant l'interaction du
promoteur de Igf2 avec des enhancers situés plus en aval (Schoenherr et al., 2003; Szabo et al.,
2004). Elle semble également requise pour protéger la région de la méthylation au cours de
l'ovogenèse et du développement embryonnaire, et pour initier en cis l'expression de H19 (Engel et
al., 2006; Fedoriw et al., 2004). Sur le chromosome paternel, le DMD est hyperméthylé durant la
spermatogenèse (Davis et al., 2000; Tremblay et al., 1997). La méthylation est propagée en cis avec
pour conséquence de réprimer l'expression de H19. Elle s'oppose également à la fixation de CTCF
et à la formation d'un insulateur, ce qui permet au promoteur de Igf2 d'interagir avec ses enhancers
(Bell et Felsenfeld, 2000; Hark et al., 2000).
229

Au locus H19/Igf2, la fixation de CTCF sur le DMD réalise donc au moins deux fonctions:
activatrice pour H19 et insulatrice entre Igf2 et ses enhancers. Il est possible que la fixation de
CTCF au niveau de DXPas34 puisse être également ambivalente quant à ses fonctions. Il a été
montré que DXPas34 se comportait comme un enhancer de Tsix (cf résultats, chapitre 1, p. 131). La
fixation de CTCF sur DXPas34 dans les cellules ES pourrait contribuer à cette fonction soit
directement, soit en isolant Tsix d'un répresseur, qui reste à identifier. Il a également été proposé que
la fixation de CTCF sur DXPas34 pourrait isoler le promoteur de Xist d'un enhancer plus en aval
(Chao et al., 2002). Une telle fonction est cependant difficile à distinguer du rôle joué par DXPas34
comme régulateur de l'expression de Tsix. On peut néanmoins observer que les lignées ΔAV et
Ma2L, dans lesquelles Tsix est invalidé, sont identiques pour ce qui est de l'expression de Xist ou du
profil chromatinien au locus Xist/Tsix, bien que dans un cas (ΔAV) DXPas34 ait été délété, et que
dans le second (Ma2L), il soit toujours présent. Par conséquent, si l'enhancer proposé par Chao et
al. existait, son rôle serait limité par rapport à celui de Tsix, ou il serait incapable de fonctionner en
l'absence de Tsix. On peut également envisager qu'il ne soit pas actif chez les mâles ou qu'il aurait
230
Fig. 26: Fonction insulatrice du DMD au locus H19/Igf2
Sur l'allèle d'origine paternelle (en haut), le DMD (en rouge) est méthylé (sucettes noires), avec pour conséquences d'empêcher la fixation de CTCF, rendant ainsi accessible au gène Igf2 un enhancer localisé plus en aval, ce qui permet l'expression de Igf2. La méthylation est également propagée en cis sur le promoteur du gène H19, qui n'est pas exprimé. Sur l'allèle d'origine maternelle (en bas), CTCF se fixe au DMD, qui n'est pas méthylé, et crée un élément insulateur entre le gène Igf2 et ses enhancers, empêchant l'expression de Igf2. Le gène H19, dont le promoteur n'est pas méthylé est quant à lui exprimé.

été délété dans ΔAV, auquel cas il serait localisé à proximité du promoteur de Tsix.
La fonction insulatrice réalisée par la fixation de CTCF à l'ICR de H19 semble correspondre à la
mise en place de structures chromatiniennes complexes, incluant la formation de boucles de
chromatine séquestrant spatialement Igf2 de ses enhancer (Kurukuti et al., 2006; Murrell et al.,
2004). En particulier, une interaction privilégiée, vraisemblablement médiée par CTCF, est établie
sur le chromosome maternel entre l'ICR et un DMD situé en 3' du gène Igf2 (Kurukuti et al., 2006;
Murrell et al., 2004). Il semblerait également que l'ICR de H19 interagisse à plus grande échelle
avec des loci distincts, sur le même chromosome et sur d'autres chromosomes, et serait ainsi partie
prenante d'un réseau de régulation génique ou épigénétique structuré spatialement (Ling et al.,
2006; Zhao et al., 2006). On est tenté de penser que le même type d'interaction impliquant CTCF
pourrait avoir lieu entre DXPas34 et la région promotrice de Xist, voire entre les deux pics de
fixation de CTCF délimitant cette dernière. De telles interactions pourraient avoir lieu en cis, mais
aussi en trans, et jouer de la sorte un rôle dans le choix. On peut également imaginer que le Xic
interagisse de la même manière avec des régions distantes du chromosome X qui serviraient de
centre relais pour la mise en place de l'inactivation, ou au contraire de manière à isoler spatialement
les régions échappant à l'inactivation, dans la délimitation desquelles CTCF semble jouer un rôle (cf
introduction, partie 1, section 2.5, p. 50). Les technologies permettant de mesurer de telles
interactions n'en sont qu'à leurs débuts et la possibilité de disposer de bons contrôles revêt de ce fait
une importance cruciale (pour revue, Dekker, 2006). A ce titre, l'existence d'un grand nombre de
délétions générées dans le Xic représente un avantage certain pour l'analyse de la conformation
tridimensionnelle de cette région, car elle permet d'exclure plus facilement un grand nombre
d'interaction faussement positive.
La fixation de YY1 est également observée au niveau de DXPas34 (Donohoe et al., 2007; Kim
et al., 2006; Navarro, thèse de doctorat, 2006), trait qu'il partage avec le promoteur du gène Xist, et
les ICR associés aux gènes empreintés Peg3 et Nespas (Kim et al., 2006). Comme pour CTCF, la
fixation de YY1 peut dans certains cas permettre la formation d'un insulateur. Mais une telle
fonction n'est pas reproduite, lors de tests in vitro, par les sites de fixation de YY1 présents au
niveau de DXPas34 ou du promoteur de Xist, contrairement à ceux présents à l'ICR du gène Peg3
(Kim et al., 2006).
CTCF et YY1 sont tous les deux sensibles à la méthylation de l'ADN pour leur fixation (Bell et
Felsenfeld, 2000; Hark et al., 2000; Kanduri et al., 2000; Kim et al., 2003). Or, DXPas34 est
méthylé différenciellement mais, contrairement au DMD du locus H19/Igf2, pas dans les gamètes,
et pas avant la différenciation des lignages embryonnaires et extraembryonnaires. Certains sites de
231

fixation de CTCF à proximité de DXPas34 sont néanmoins méthylés dans le sperme, mais perdent
cette marque après la fécondation (Boumil et al., 2006). Ils ne seront méthylés de nouveau que dans
les tissus embryonnaires et extraembryonnaires différenciés, de manière identique sur les deux
allèles (Boumil et al., 2006) (cf introduction, partie 2, section 3.3, p. 116). De plus, la fixation de
CTCF et YY1, qui est observée dans les cellules ES au niveau ou à proximité de DXPas34, semble
être réalisée de la même manière sur les deux allèles, et est perdue en cours de différenciation aussi
bien chez les mâles que chez les femelles (Boumil et al., 2006; Navarro, thèse de doctorat, 2006).
On pourrait néanmoins imaginer que certains des sites de fixation de CTCF à proximité de
DXPas34 puissent, à travers leur méthylation, véhiculer une empreinte paternelle qui contribuerait
à la mise en place de l'inactivation empreintée immédiatement après la fécondation. Mais
l'information associée à cette empreinte devrait ensuite être transmise sous une autre forme, ou
l'inactivation empreintée se maintenir de manière autonome. Une telle empreinte serait
indépendante de Tsix, puisque l'expression de ce dernier est postérieure à celle de Xist et à la mise
en place de l'inactivation empreintée. D'autre part, si la méthylation différentielle de DXPas34 joue
un rôle dans l'inactivation aléatoire en modulant la fixation de CTCF, cela ne peut être que
transitoirement, en cours de différenciation, où l'on peut imaginer qu'elle serait mise en place avec
des cinétiques différentes sur les deux allèles. Par conséquent, la méthylation au niveau de
DXPas34 est utilisée (si tant est qu'elle le soit) très différemment de ce que l'on observe à l'ICR du
232
Fig. 27: Modèle de régulation du locus H19/Igf2 (d'après Kurukuti et al., 2006)
Modèle montrant les interactions mesurées par 3C en cis sur le locus H19 dans des foies néonataux de souris. Le modèle fait l'hypothèse de la relocalisation d'une partie du locus incluant H19 dans un centre de chromatine active (ACH, pour active chromatin hub). L'exclusion du ACH serait contrôlée par l'ICR et deux autres éléments, MAR3 (matrix attachment region 3) et DMR1 (differentially methylated region 1), réunis en un complexe par CTCF. Une boucle contenant le gène Igf2 serait ainsi formée. HSS, DNase1 hypersensitive region; IGS, intergenic sequence region; en10, enhancerconserved sequence (enhancer mésodermal de H19); en4, endodermal H19 enhancer.

locus H19/Igf2, ou pour les gènes empreintés en général, et semble s'être adaptée aux particularités
de l'inactivation du chromosome X: réversibilité de l'inactivation au stade blastocyste, et choix
aléatoire du chromosome X à inactiver.
2.2.2 Fonction de la répétition en tandem au niveau de DXPas34
Il a été montré récemment que DXPas34 était transcrit bidirectionellement (Cohen et al., 2007).
Cela ouvre la possibilité que des siRNA puissent être générés dans ce minisatellite et contribuer à sa
régulation. Dans la mesure où DXPas34 est constitué par la répétition en tandem d'un motif de
34 pb, il est tentant d'imaginer un mécanisme similaire à celui proposé par Robert Martiensenn
(Martienssen, 2003) pour la régulation de l'hétérochromatine centromérique: une activité RdRP
(RNA dependant RNA Polymerase) pourrait être initiée sur chacune des répétitions à partir du lot de
siRNA déjà présents, par conséquent sans réduction en taille de l'ARN double brin produit à chaque
cycle. Si l'existence d'une activité RdRP était avérée chez les mammifères, la présence de
répétitions en tandem dans DXPas34 pourrait donc être un élément essentiel pour pouvoir amplifier
rapidement une activité RNAi. Comme la transcription bidirectionnelle est perdue dans les cellules
différenciées, elle serait donc amenée à jouer un rôle dans les cellules non différenciées ou en cours
de différenciation, précisément dans la fenêtre de temps où l'inactivation est mise en place,
l'expression de Tsix est perdue et le choix du futur Xi est supposé s'accomplir. La capacité de
l'activité RNAi à pouvoir être amplifiée rapidement pourrait alors être déterminante. Elle pourrait
contribuer à la disparition rapide des transcrits Tsix, soit directement en les ciblant pour être
dégradés, soit en induisant le recrutement de marques hétérochromatiques au niveau de DXPas34,
avec pour conséquence d'inhiber sa fonction d'activateur transcriptionnel de Tsix.
La recherche de siRNA originaires de DXPas34 est donc une perspective intéressante, et devrait
probablement être entreprise au cours d'une cinétique précise de différenciation, dans des cellules
femelles et mâles, c'est-à-dire réalisant ou non l'inactivation.
233

234

Chapitre 3 Choix et comptage: une distinction illusoire?
Nous avons, dans une large partie de ce texte, cherché à présenter de manière claire les notions
de comptage et choix, et à en préciser les mécanismes. Ces deux notions sont utiles d'abord parce
qu'elles permettent de mettre un nom sur des phénotypes bien distincts: l'inactivation d'un nombre
aberrant de chromosomes X, dans le premier cas, et l'inactivation préférentielle de l'un des deux
chromosomes X, dans le second. Néanmoins, on ne peut considérer le comptage et le choix comme
véritablement différents que si ils font appels à des mécanismes distincts, moléculairement ou
temporellement. Ce constat est particulièrement d'actualité dans les cellules femelles, où les deux
mécanismes sont requis pour que l'inactivation soit mise en place de manière correcte.
Or nous avons vu que les mêmes acteurs pouvaient jouer un rôle dans le comptage et le choix.
Cela est en particulier le cas pour Tsix et, d'une manière générale, pour la région en 3' de Xist. En
l'absence de l'un ou l'autre, le choix est biaisé dans les cellules femelles, et l'inactivation est initiée
dans les cellules mâles. Mais pour un élément tel que Xite, l'absence d'inactivation dans les mâles
lorsqu'il est délété (Ogawa et Lee, 2003) ne signifie pas l'absence de fonction dans le comptage,
puisque des transgènes de Xite insérés en grand nombre de copie sont capables d'interférer avec le
mécanisme de comptage dans les cellules femelles (Lee, 2005). De même, la délétion ΔCpG n'a-t-
elle d'effet sur le comptage que dans les cellules femelles, lorsqu'elle est présente à l'état
homozygote (Lee, 2002; Lee, 2005), et des transgènes reprenant la séquence délétée dans ΔCpG
produisent dans des cellules femelles le même effet que des transgènes de Xite (Lee, 2005) (cf
introduction, partie 2, section 2.3.3.2, p. 111). Deux interprétations sont possibles à ces résultats.
La première revient à considérer que le comportement de cellules mâles et femelles est
fondamentalement différent vis-à-vis de l'inactivation. Les mâles seraient ainsi incapables d'initier
l'inactivation. Une raison possible en serait qu'il leur manquerait un facteur de compétence pour
l'inactivation, ce qui correspond au modèle à deux facteurs proposé par J. Lee (cf p. 78) (Lee,
2005). Or, ce modèle a été élaboré en particulier pour expliquer pourquoi la délétion ΔCpG ne
conduisait pas à initier l'inactivation dans les mâles (Lee et Lu, 1999). Pour cette raison précise, le
modèle à deux facteurs ne permet pas d'expliquer pourquoi, dans plusieurs autres mutants de Tsix,
l'inactivation est initiée dans des cellules mâles (cf introduction, partie 2, section 2.3.3.1, p. 110, et
résultats, chapitre 1, p. 131).
235

Il importe donc de considérer choix et comptage sous un angle différent. Deux contributions
récentes, l'une expérimentale, l'autre théorique, sont à ce titre particulièrement intéressantes. Tout
d'abord, l'observation d'un rapprochement transitoire des Xic en cours de différenciation (Bacher et
al., 2006; Xu et al., 2006). Les mêmes transgènes qui interfèrent avec le comptage dans des cellules
femelles interfèrent avec ce rapprochement en se substituant à l'un des Xic (Xu et al., 2006). D'autre
part, des transgènes du Xic qui ne sont pas capables de se rapprocher d'un Xic endogène ne sont pas
non plus capables de récapituler l'initiation de l'inactivation (Bacher et al., 2006). Le processus de
rapprochement transitoire des Xic est donc tout désigné pour contribuer de manière essentielle au
comptage. Par ailleurs, le choix requiert en théorie que les Xic puissent, à un moment donné,
communiquer entre eux. Leur rapprochement pourrait y contribuer de manière essentielle, mais une
lignée mutante (XΔ65+16X) montre qu'il n'est pas suffisant (Bacher et al., 2006).
Ainsi donc, un modèle qui permettrait de concilier choix et comptage avec le rapprochement
transitoire des Xic serait du plus grand intérêt. Le modèle qui intègre aujourd'hui le mieux la
nécessité d'un rapprochement des Xic pour ces deux processus est le modèle de brisure de symétrie
proposé par Nicodemi et Prisco (Nicodemi et Prisco, 2007), que nous avions présenté à la section
p. 79. Dans les cellules femelles, la brisure permettant l'agrégation de toutes les molécules
bloquantes sur un seul chromosome ne serait possible que si la concentration locale de ces
molécules est suffisamment importante, ce qui pourrait être favorisé par le rapprochement des deux
Xic, qui en serait l'évènement déclencheur. Dans les cellules mâles, l'agrégation aurait quand même
lieu, car il n'y aurait pas la compétition d'un second chromosome X. Compte-tenu du rôle essentiel
joué par Tsix dans le comptage, et notamment de l'initiation de l'inactivation dans la lignée Ma2L en
l'absence de toute délétion d'élément génomique, il faudrait considérer que l'agrégation des
molécules bloquantes sur un Xic dépend avant tout du profil épigénétique mis en place par Tsix. En
l'absence de Tsix, le Xic ne serait pas « vu » par ces molécules qui, si il existe, iraient s'agréger sur le
second Xic.
En adoptant le modèle de brisure de symétrie, nous faisons donc disparaître la distinction entre
choix et comptage. Mais nous faisons apparaître une nouvelle distinction, qui correspond aux
paramètres de ce modèle que sont la concentration en molécules bloquantes, le gain énergétique
procuré par l'association de ces molécules entre elles, et le gain énergétique procuré par leur
association avec le chromosome X. Du point de vue du chromosome X, le dernier paramètre
dépendrait du profil épigénétique contrôlé par Tsix., et le premier du rapprochement transitoire des
Xic.
236

Malgré tout, le modèle de brisure de symétrie paraît en opposition avec le modèle d'alternance
des chromosomes X entre états épigénétiques prédéterminants avancé par Mlycnarzyk-Evans et al.
(Mlynarczyk-Evans et al., 2006) (cf p. 85). Un moyen de concilier ces deux modèles serait de
considérer que la brisure de symétrie est instable, et réalisée de nouveau à chaque division cellulaire
dans les cellules indifférenciées, et qu'elle ne deviendrait irréversible qu'en cours de différenciation.
Une alternative serait que les états épigénétiques (SIAR) mis en évidence par Mlycnarzyk-Evans et
al. correspondraient à la variation au cours des divisions cellulaires de l'un des paramètres du
modèle de Nicodemi et Prisco, qui pourrait être l'affinité du chromosome X avec les molécules
bloquantes.
237

238

Chapitre 4 Perspectives du travail de thèse
La continuation logique du travail de thèse suit deux directions: poursuivre la caractérisation de
la fonction de DXPas34 in vivo et mieux comprendre de quelle manière DXPas34 régule
l'expression de Tsix, essentiellement à travers une étude in vitro. Les questions en suspens ont déjà
été discutées au fil du texte. Il s'agit donc dans ce bref chapitre plutôt de faire le point sur celles qui
devront être traitées en priorité.
Dans les cellules ES mâles, nous avons montré que DXPas34 était un activateur transcriptionnel
de Tsix. Dans l'embryon, il semble que la délétion de DXPas34 n'ait cependant d'effet ni sur
l'inactivation empreintée, ni sur le choix, bien que Tsix soit essentiel à l'un et à l'autre.
On pourrait comprendre l'absence d'effet sur l'empreinte par le caractère hypomorphe de notre
délétion: elle n'affecterait qu'un petit nombre de cellules dont la disparition pourrait être compensée
au cours du développement. L'absence de biais de choix est a priori plus surprenante car, d'une part,
nous avons montré dans des cellules ES mâles une corrélation inverse entre le niveau d'expression
de Tsix et de Xist (cf chapitre 1, p. 131), et d'autre part, il a été montré que le niveau d'expression
relatif de Xist sur chacun des chromosomes X déterminait la proportion de cellules dans lesquelles
l'un ou l'autre de ces chromosomes est inactivé (cf introduction, partie 2, section 2.1, p. 95).
Autrement dit, on s'attendrait à obtenir une réponse graduelle à une altération de l'expression de
Tsix, plutôt qu'une réponse de type tout ou rien, comme lorsqu'il s'agit de la survie d'embryons.
On peut bien entendu penser que, pour ce qui est du choix, la variabilité interindividuelle aurait
pu masquer un biais de faible amplitude causé par l'absence de DXPas34 sur l'un des allèles. D'où
l'intérêt d'examiner un plus grand nombre d'embryons, ce qui est en cours. Deux autres explications
pourraient être avancées: l'expression résiduelle de Tsix serait suffisante pour réaliser correctement
le choix, ou DXPas34 ne serait pas, dans les embryons femelles, un activateur transcriptionnel de
Tsix aussi important qu'il l'est dans les cellules ES mâles. De tels arguments conviendraient tout
aussi bien pour expliquer l'absence de léthalité liée à un défaut d'inactivation empreintée.
Pour discriminer entre ces deux possibilités, il faudrait quantifier l'expression de Tsix dans les
différents tissus embryonnaires, ou dans des modèles cellulaires que nous devrions dériver: cellules
ES femelles pour étudier le choix, et cellules TS ou XEN pour l'inactivation empreintée. La
239

génération de cellules ES femelles permettrait en outre d'examiner le positionnement des Xic en
cours de différenciation, et en particulier leur éventuel rapprochement, dans un contexte mutant
pour DXPas34.
Nous avons mentionné qu'une seconde délétion de DXPas34 (Cohen et al., 2007), légèrement
plus étendue, a un phénotype plus comparable à celui observé en l'absence de Tsix, dans la mesure
où elle conduit à un défaut d'inactivation empreintée et de choix. D'autre part, l'expression de Tsix
semble plus sévèrement altérée en 3' par la délétion de Cohen et al. que par la notre. Cela pourrait
suggérer l'existence d'un site d'initiation de transcription ou d'un élément régulant l'expression de
Tsix dans une région de 300 pb différenciant les deux délétions de DXPas34. L'initiation de
transcrits dans cette région devrait pouvoir être facilement mise en évidence. En revanche,
l'identification d'un régulateur additionnel de Tsix requerrait d'en réaliser la délétion ciblée.
CTCF et YY1 sont deux excellents candidats pour expliquer la régulation de Tsix par DXPas34
(cf introduction, partie 2, section 3.3, p. 116). Néanmoins, il est difficile d'en adresser le rôle
spécifiquement au niveau de DXPas34, bien que d'autres laboratoires s'y soient employés (Donohoe
et al., 2007). En effet, CTCF et YY1 sont également fixés au début du gène Xist et il est délicat de
discriminer, dans des mutants de CTCF ou YY1 si l'effet que l'on observe résulte d'une dérégulation
de Xist ou de Tsix. Par conséquent, l'approche à retenir consisterait à muter de manière ciblée les
sites de fixation de CTCF et YY1, ce qui est plus facile au niveau de Xist, car dans le cas de
DXPas34, cela reviendrait à muter pratiquement chacun des motifs répétés. Par ailleurs, une
approche de type 3C pourrait être utilisée pour mesurer l'interaction éventuelle de DXPas34 avec la
région promotrice de Xist, via la fixation de CTCF.
Nous avons envisagé une autre perspective intéressante (cf discussion, section 2.2.2, p. 233), qui
serait la production de siRNA au niveau de DXPas34, rendue possible par l'existence d'une
transcription bidirectionnelle dans ce dernier. De tels siRNA devront être recherchés, avec une
attention particulière portée à la période de mise en place de l'inactivation.
D'une manière générale, la délétion de DXPas34 conduit à un phénotype hypomorphe, comparé
à celui résultant de l'absence totale de Tsix. Cela est vrai pour les deux délétions de DXPas34, celle
générée par Cohen et al. et la notre, et il se pourrait que les différences entre ces deux délétions
aient pour cause un degré différent d'altération du niveau d'expression de Tsix, combiné à un effet de
seuil pour en observer les effets, particulièrement in vivo. Si ce scenario était conforté par la
quantification des niveaux d'expression de Tsix in vivo, les lignées mutantes pour DXPas34,
pourraient être utilisées pour tester le rôle d'autres facteurs – par exemple des facteurs de
240

modification de la chromatine – dans l'initiation de l'inactivation. On peut en effet supposer qu'en
l'absence de DXPas34, le seuil de détection d'autres mutations au phénotype également hypomorphe
serait abaissé, ce qui en faciliterait la caractérisation.
Pour finir, une question centrale en biologie est de savoir comment la variation continue d'un
paramètre peut conduire à basculer entre deux états radicalement différents. Cela est généralement
interprété en terme d'effet de seuil, mais le support moléculaire d'un tel seuil est généralement mal
compris. L'inactivation du chromosome X, et particulièrement sa régulation au niveau du Xic,
offrent un cadre idéal pour aborder cette question du point de vue des mécanismes épigénétiques.
241

242

Annexe: Nouvelle publiée dans Médecine/Science
243

244

245

246

Bibliographie
Adler, DA., West, JD. and Chapman, VM.. (1977) Expression of alphagalactosidase in preimplantation mouse embryos Nature 267, 838839.Agelopoulos, M. and Thanos, D.. (2006) Epigenetic determination of a cellspecific gene expression program by atf2 and the histone variant macroh2a EMBO J 25, 48434853.Aitken, RJ. and Marshall Graves, JA.. (2002) The future of sex Nature 415, 963.Alekseyenko, AA., Larschan, E., Lai, WR., Park, PJ. and Kuroda, MI.. (2006) Highresolution chipchip analysis reveals that the drosophila msl complex selectively identifies active genes on the male x chromosome. Genes Dev 20, 848857.Allen, E., Horvath, S., Tong, F., Kraft, P., Spiteri, E., Riggs, AD. and Marahrens, Y.. (2003) High concentrations of long interspersed nuclear element sequence distinguish monoallelically expressed genes Proc Natl Acad Sci U S A 100, 99409945.Anderson, CL. and Brown, CJ.. (1999) Polymorphic xchromosome inactivation of the human timp1 gene Am J Hum Genet 65, 699708.Angelov, D., Molla, A., Perche, P., Hans, F., Cote, J., Khochbin, S., Bouvet, P. and Dimitrov, S.. (2003) The histone variant macroh2a interferes with transcription factor binding and swi/snf nucleosome remodeling Mol Cell 11, 10331041.Aoki, F., Worrad, DM. and Schultz, RM.. (1997) Regulation of transcriptional activity during the first and second cell cycles in the preimplantation mouse embryo Dev Biol 181, 296307.Atchison, L., Ghias, A., Wilkinson, F., Bonini, N. and Atchison, ML.. (2003) Transcription factor yy1 functions as a pcg protein in vivo EMBO J 22, 13471358.Austin, CR. and Amoroso, EC.. (1957) Sex chromatin in early cat embryos Exp Cell Res 13, 419421.Avner, P., Prissette, M., Arnaud, D., Courtier, B., Cecchi, C. and Heard, E.. (1998) Molecular correlates of the murine xce locus Genet Res 72, 217224.Ayoub, N., Richler, C. and Wahrman, J.. (1997) Xist rna is associated with the transcriptionally inactive xy body in mammalian male meiosis Chromosoma 106, 110.Baarends, WM., Hoogerbrugge, JW., Roest, HP., Ooms, M., Vreeburg, J., Hoeijmakers, JH. and Grootegoed, JA.. (1999) Histone ubiquitination and chromatin remodeling in mouse spermatogenesis. Dev Biol 207, 322333.Baarends, WM., Wassenaar, E., van der Laan, R., Hoogerbrugge, J., SleddensLinkels, E., Hoeijmakers, JHJ., de Boer, P. and Grootegoed, JA.. (2005) Silencing of unpaired chromatin and histone h2a ubiquitination in mammalian meiosis Mol Cell Biol 25, 10411053.Bacher, CP., Guggiari, M., Brors, B., Augui, S., Clerc, P., Avner, P., Eils, R. and Heard, E.. (2006) Transient colocalization of xinactivation centres accompanies the initiation of x inactivation
247

Nat Cell Biol 8, 293299.Bai, X., Alekseyenko, AA. and Kuroda, MI.. (2004) Sequencespecific targeting of msl complex regulates transcription of the rox rna genes EMBO J 23, 28532861.Bailey, JA., Carrel, L., Chakravarti, A. and Eichler, EE.. (2000) Molecular evidence for a relationship between line1 elements and x chromosome inactivation: the lyon repeat hypothesis Proc Natl Acad Sci U S A 97, 66346639.Bannister, AJ., Zegerman, P., Partridge, JF., Miska, EA., Thomas, JO., Allshire, RC. and Kouzarides, T.. (2001) Selective recognition of methylated lysine 9 on histone h3 by the hp1 chromo domain Nature 410, 120124.Bao, S., Miyoshi, N., Okamoto, I., Jenuwein, T., Heard, E. and Azim Surani, M.. (2005) Initiation of epigenetic reprogramming of the x chromosome in somatic nuclei transplanted to a mouse oocyte EMBO Rep 6, 748754.Barchi, M., Mahadevaiah, S., Di Giacomo, M., Baudat, F., de Rooij, DG., Burgoyne, PS., Jasin, M. and Keeney, S.. (2005) Surveillance of different recombination defects in mouse spermatocytes yields distinct responses despite elimination at an identical developmental stage Mol Cell Biol 25, 72037215.Barr, ML.. (1963) Some properties of the sex chromosomes and their bearing on normal and abnormal development. Second International Conference on Congenital Malformations , .Barr, ML. and Bertram, EG.. (1949) A morphological distinction between neurones of the male and female, and the behavior of the nucleolar satellite during accelerated nucleoprotein synthesis Nature 163, 676677.Barton, DE., David, FN. and Merrington, M.. (1964) The positions of the sex chromosomes in the human cell in mitosis. Ann Hum Genet 28, 123128.Bassing, CH., Swat, W. and Alt, FW.. (2002) The mechanism and regulation of chromosomal v(d)j recombination Cell 109 Suppl, S4555.Beletskii, A., Hong, YK., Pehrson, J., Egholm, M. and Strauss, WM.. (2001) Pna interference mapping demonstrates functional domains in the noncoding rna xist Proc Natl Acad Sci U S A 98, 92159220.Bell, AC. and Felsenfeld, G.. (2000) Methylation of a ctcfdependent boundary controls imprinted expression of the igf2 gene Nature 405, 482485.Bellani, MA., Romanienko, PJ., Cairatti, DA. and CameriniOtero, RD.. (2005) Spo11 is required for sexbody formation, and spo11 heterozygosity rescues the prophase arrest of atm/ spermatocytes J Cell Sci 118, 32333245.Belmont, AS., Bignone, F. and Ts'o, PO.. (1986) The relative intranuclear positions of barr bodies in xxx nontransformed human fibroblasts. Exp Cell Res 165, 165179.Bernstein, BE., Kamal, M., LindbladToh, K., Bekiranov, S., Bailey, DK., Huebert, DJ., McMahon, S., Karlsson, EK., Kulbokas, EJ3., Gingeras, TR., Schreiber, SL. et al.. (2005) Genomic maps and comparative analysis of histone modifications in human and mouse Cell 120, 169181.
248

Bittner, RE., Popoff, I., Shorny, S., Hoger, H. and Wachtler, F.. (1997) Dystrophin expression in heterozygous mdx/+ mice indicates imprinting of x chromosome inactivation by parentoforigin, tissue, strain and positiondependent factors Anat Embryol (Berl) 195, 175182.Blewitt, ME., Vickaryous, NK., Paldi, A., Koseki, H. and Whitelaw, E.. (2006) Dynamic reprogramming of dna methylation at an epigenetically sensitive allele in mice PLoS Genet 2, e49.Boggs, BA., Cheung, P., Heard, E., Spector, DL., Chinault, AC. and Allis, CD.. (2002) Differentially methylated forms of histone h3 show unique association patterns with inactive human x chromosomes Nat Genet 30, 7376.Boggs, BA., Connors, B., Sobel, RE., Chinault, AC. and Allis, CD.. (1996) Reduced levels of histone h3 acetylation on the inactive x chromosome in human females Chromosoma 105, 303309.Bolland, DJ., Wood, AL., Johnston, CM., Bunting, SF., Morgan, G., Chakalova, L., Fraser, PJ. and Corcoran, AE.. (2004) Antisense intergenic transcription in v(d)j recombination Nat Immunol 5, 630637.Borden, J. and Manuelidis, L.. (1988) Movement of the x chromosome in epilepsy. Science 242, 16871691.Borsani, G., Tonlorenzi, R., Simmler, MC., Dandolo, L., Arnaud, D., Capra, V., Grompe, M., Pizzuti, A., Muzny, D. and Lawrence, C.. (1991) Characterization of a murine gene expressed from the inactive x chromosome Nature 351, 325329.Bortoluzzi, S., Rampoldi, L., Simionati, B., Zimbello, R., Barbon, A., d'Alessi, F., Tiso, N., Pallavicini, A., Toppo, S., Cannata, N., Valle, G. et al.. (1998) A comprehensive, highresolution genomic transcript map of human skeletal muscle Genome Res 8, 817825.Boumil, RM., Ogawa, Y., Sun, BK., Huynh, KD. and Lee, JT.. (2006) Differential methylation of xite and ctcf sites in tsix mirrors the pattern of xinactivation choice in mice Mol Cell Biol 26, 21092117.Bourgeois, CA., Laquerriere, F., Hemon, D., Hubert, J. and Bouteille, M.. (1985) New data on the insitu position of the inactive x chromosome in the interphase nucleus of human fibroblasts. Hum Genet 69, 122129.Branco, MR. and Pombo, A.. (2006) Intermingling of chromosome territories in interphase suggests role in translocations and transcriptiondependent associations PLoS Biol 4, e138.Brockdorff, N., Ashworth, A., Kay, GF., McCabe, VM., Norris, DP., Cooper, PJ., Swift, S. and Rastan, S.. (1992) The product of the mouse xist gene is a 15 kb inactive xspecific transcript containing no conserved orf and located in the nucleus Cell 71, 515526.Brown, CJ. and Greally, JM.. (2003) A stain upon the silence: genes escaping x inactivation Trends Genet 19, 432438.Brown, CJ., Ballabio, A., Rupert, JL., Lafreniere, RG., Grompe, M., Tonlorenzi, R. and Willard, HF.. (1991) A gene from the region of the human x inactivation centre is expressed exclusively from the inactive x chromosome Nature 349, 3844.Brown, CJ., Hendrich, BD., Rupert, JL., Lafreniere, RG., Xing, Y., Lawrence, J. and Willard, HF.. (1992) The human xist gene: analysis of a 17 kb inactive xspecific rna that contains conserved
249

repeats and is highly localized within the nucleus Cell 71, 527542.Brown, SD.. (1991) Xist and the mapping of the x chromosome inactivation centre Bioessays 13, 607612.Buzin, CH., Mann, JR. and SingerSam, J.. (1994) Quantitative rtpcr assays show xist rna levels are low in mouse female adult tissue, embryos and embryoid bodies Development 120, 35293536.Cao, R. and Zhang, Y.. (2004) The functions of e(z)/ezh2mediated methylation of lysine 27 in histone h3 Curr Opin Genet Dev 14, 155164.Caparros, M., Alexiou, M., Webster, Z. and Brockdorff, N.. (2002) Functional analysis of the highly conserved exon iv of xist rna Cytogenet Genome Res 99, 99105.Carninci, P.. (2006) Tagging mammalian transcription complexity Trends Genet 22, 501510.Carrel, L. and Willard, HF.. (2005) Xinactivation profile reveals extensive variability in xlinked gene expression in females Nature 434, 400404.Carrel, L., Cottle, AA., Goglin, KC. and Willard, HF.. (1999) A firstgeneration xinactivation profile of the human x chromosome Proc Natl Acad Sci U S A 96, 1444014444.Carrel, L., Park, C., Tyekucheva, S., Dunn, J., Chiaromonte, F. and Makova, KD.. (2006) Genomic environment predicts expression patterns on the human inactive x chromosome. PLoS Genet 2, e151.Cattanach, BM. and Isaacson, JH.. (1967) Controlling elements in the mouse x chromosome Genetics 57, 331346.Cattanach, BM. and Williams, CE.. (1972) Evidence of nonrandom x chromosome activity in the mouse Genet Res 19, 229240.Cattanach, BM., Pollard, CE. and Perez, JN.. (1969) Controlling elements in the mouse xchromosome. i. interaction with the xlinked genes Genet Res 14, 223235.Chadwick, BP.. (2007) Variation in xi chromatin organization and correlation of the h3k27me3 chromatin territories to transcribed sequences by microarray analysis Chromosoma 116, 147157.Chadwick, BP. and Lane, TF.. (2005) Brca1 associates with the inactive x chromosome in late sphase, coupled with transient h2ax phosphorylation Chromosoma 114, 432439.Chadwick, BP. and Willard, HF.. (2001) Histone h2a variants and the inactive x chromosome: identification of a second macroh2a variant Hum Mol Genet 10, 11011113.Chadwick, BP. and Willard, HF.. (2003) Setting the stage. eedenx1 leaves an epigenetic signature on the inactive x chromosome Dev Cell 4, 445447.Chadwick, LH. and Willard, HF.. (2005) Genetic and parentoforigin influences on x chromosome choice in xce heterozygous mice Mamm Genome 16, 691699.Chadwick, LH., Pertz, LM., Broman, KW., Bartolomei, MS. and Willard, HF.. (2006) Genetic control of x chromosome inactivation in mice: definition of the xce candidate interval Genetics 173, 21032110.Chambeyron, S. and Bickmore, WA.. (2004) Chromatin decondensation and nuclear reorganization of the hoxb locus upon induction of transcription Genes Dev 18, 11191130.Chao, W., Huynh, KD., Spencer, RJ., Davidow, LS. and Lee, JT.. (2002) Ctcf, a candidate trans
250

acting factor for xinactivation choice Science 295, 345347.Charlesworth, B.. (1996) The evolution of chromosomal sex determination and dosage compensation Curr Biol 6, 149162.Charlesworth, D., Charlesworth, B. and Marais, G.. (2005) Steps in the evolution of heteromorphic sex chromosomes Heredity 95, 118128.Chaumeil, J., Le Baccon, P., Wutz, A. and Heard, E.. (2006) A novel role for xist rna in the formation of a repressive nuclear compartment into which genes are recruited when silenced Genes Dev 20, 22232237.Chaumeil, J., Okamoto, I., Guggiari, M. and Heard, E.. (2002) Integrated kinetics of x chromosome inactivation in differentiating embryonic stem cells Cytogenet Genome Res 99, 7584.Chazaud, C., Yamanaka, Y., Pawson, T. and Rossant, J.. (2006) Early lineage segregation between epiblast and primitive endoderm in mouse blastocysts through the grb2mapk pathway. Dev Cell 10, 615624.Chen, J., Sun, M., Hurst, LD., Carmichael, GG. and Rowley, JD.. (2005) Genomewide analysis of coordinate expression and evolution of human cisencoded senseantisense transcripts Trends Genet 21, 326329.Cheng, MK. and Disteche, CM.. (2006) A balancing act between the x chromosome and the autosomes. J Biol 5, 2.Chevillard, C., Ozaki, J., Herring, CD. and Riblet, R.. (2002) A threemegabase yeast artificial chromosome contig spanning the c57bl mouse igh locus J Immunol 168, 56595666.Cho, DH., Thienes, CP., Mahoney, SE., Analau, E., Filippova, GN. and Tapscott, SJ.. (2005) Antisense transcription and heterochromatin at the dm1 ctg repeats are constrained by ctcf Mol Cell 20, 483489.Chow, JC., Hall, LL., Clemson, CM., Lawrence, JB. and Brown, CJ.. (2003) Characterization of expression at the human xist locus in somatic, embryonal carcinoma, and transgenic cell lines. Genomics 82, 309322.Chow, JC., Yen, Z., Ziesche, SM. and Brown, CJ.. (2005) Silencing of the mammalian x chromosome Annu Rev Genomics Hum Genet 6, 6992.Chowdhury, D. and Sen, R.. (2001) Stepwise activation of the immunoglobulin mu heavy chain gene locus EMBO J 20, 63946403.Chu, DS., Dawes, HE., Lieb, JD., Chan, RC., Kuo, AF. and Meyer, BJ.. (2002) A molecular link between genespecific and chromosomewide transcriptional repression Genes Dev 16, 796805.Chureau, C., Prissette, M., Bourdet, A., Barbe, V., Cattolico, L., Jones, L., Eggen, A., Avner, P. and Duret, L.. (2002) Comparative sequence analysis of the xinactivation center region in mouse, human, and bovine Genome Res 12, 894908.Ciaudo, C., Bourdet, A., CohenTannoudji, M., Dietz, HC., Rougeulle, C. and Avner, P.. (2006) Nuclear mrna degradation pathway(s) are implicated in xist regulation and x chromosome inactivation PLoS Genet 2, e94.Ciavatta, D., Kalantry, S., Magnuson, T. and Smithies, O.. (2006) A dna insulator prevents
251

repression of a targeted xlinked transgene but not its random or imprinted x inactivation Proc Natl Acad Sci U S A 103, 99589963.Clemson, CM., Chow, JC., Brown, CJ. and Lawrence, JB.. (1998) Stabilization and localization of xist rna are controlled by separate mechanisms and are not sufficient for x inactivation J Cell Biol 142, 1323.Clemson, CM., Hall, LL., Byron, M., McNeil, J. and Lawrence, JB.. (2006) The x chromosome is organized into a generich outer rim and an internal core containing silenced nongenic sequences Proc Natl Acad Sci U S A 103, 76887693.Clemson, CM., McNeil, JA., Willard, HF. and Lawrence, JB.. (1996) Xist rna paints the inactive x chromosome at interphase: evidence for a novel rna involved in nuclear/chromosome structure J Cell Biol 132, 259275.Clerc, P. and Avner, P.. (1998) Role of the region 3' to xist exon 6 in the counting process of xchromosome inactivation Nat Genet 19, 249253.Cohen, DE., Davidow, LS., Erwin, JA., Xu, N., Warshawsky, D. and Lee, JT.. (2007) The dxpas34 repeat regulates random and imprinted x inactivation. Dev Cell 12, 5771.Cohen, SM., Brylawski, BP., CordeiroStone, M. and Kaufman, DG.. (2003) Same origins of dna replication function on the active and inactive human x chromosomes J Cell Biochem 88, 923931.Collins, N., Poot, RA., Kukimoto, I., GarcíaJiménez, C., Dellaire, G. and VargaWeisz, PD.. (2002) An acf1iswi chromatinremodeling complex is required for dna replication through heterochromatin. Nat Genet 32, 627632.Cooper, P., Keer, JT., McCabe, VM., Hamvas, RM., Brown, SD., Rastan, S. and Brockdorff, N.. (1993) Physical mapping of 2000 kb of the mouse x chromosome in the vicinity of the xist locus Genomics 15, 570575.Costanzi, C. and Pehrson, JR.. (1998) Histone macroh2a1 is concentrated in the inactive x chromosome of female mammals Nature 393, 599601.Costanzi, C., Stein, P., Worrad, DM., Schultz, RM. and Pehrson, JR.. (2000) Histone macroh2a1 is concentrated in the inactive x chromosome of female preimplantation mouse embryos Development 127, 22832289.Cote, J., Quinn, J., Workman, JL. and Peterson, CL.. (1994) Stimulation of gal4 derivative binding to nucleosomal dna by the yeast swi/snf complex Science 265, 5360.Coucouvanis, EC., Sherwood, SW., CarswellCrumpton, C., Spack, EG. and Jones, PP.. (1993) Evidence that the mechanism of prenatal germ cell death in the mouse is apoptosis. Exp Cell Res 209, 238247.Courtier, B., Heard, E. and Avner, P.. (1995) Xce haplotypes show modified methylation in a region of the active x chromosome lying 3' to xist Proc Natl Acad Sci U S A 92, 35313535.Cowan, CA., Atienza, J., Melton, DA. and Eggan, K.. (2005) Nuclear reprogramming of somatic cells after fusion with human embryonic stem cells Science 309, 13691373.Cremer, T. and Cremer, C.. (2001) Chromosome territories, nuclear architecture and gene
252

regulation in mammalian cells Nat Rev Genet 2, 292301.Csankovszki, G., McDonel, P. and Meyer, BJ.. (2004) Recruitment and spreading of the c. elegans dosage compensation complex along x chromosomes Science 303, 11821185.Csankovszki, G., Nagy, A. and Jaenisch, R.. (2001) Synergism of xist rna, dna methylation, and histone hypoacetylation in maintaining x chromosome inactivation J Cell Biol 153, 773784.Csankovszki, G., Panning, B., Bates, B., Pehrson, JR. and Jaenisch, R.. (1999) Conditional deletion of xist disrupts histone macroh2a localization but not maintenance of x inactivation Nat Genet 22, 323324.Daniels, R., Zuccotti, M., Kinis, T., Serhal, P. and Monk, M.. (1997) Xist expression in human oocytes and preimplantation embryos Am J Hum Genet 61, 3339.Davis, TL., Yang, GJ., McCarrey, JR. and Bartolomei, MS.. (2000) The h19 methylation imprint is erased and reestablished differentially on the parental alleles during male germ cell development Hum Mol Genet 9, 28852894.De Felici, M., Dolci, S. and Pesce, M.. (1992) Cellular and molecular aspects of mouse primordial germ cell migration and proliferation in culture Int J Dev Biol 36, 205213.de La CasaEsperon, E., LoredoOsti, JC., PardoManuel de Villena, F., Briscoe, TL., Malette, JM., Vaughan, JE., Morgan, K. and Sapienza, C.. (2002) X chromosome effect on maternal recombination and meiotic drive in the mouse Genetics 161, 16511659.de la Cruz, X., Lois, S., SanchezMolina, S. and MartinezBalbas, MA.. (2005) Do protein motifs read the histone code? Bioessays 27, 164175.de Napoles, M., Mermoud, JE., Wakao, R., Tang, YA., Endoh, M., Appanah, R., Nesterova, TB., Silva, J., Otte, AP., Vidal, M., Koseki, H. et al.. (2004) Polycomb group proteins ring1a/b link ubiquitylation of histone h2a to heritable gene silencing and x inactivation Dev Cell 7, 663676.Debrand, E., Chureau, C., Arnaud, D., Avner, P. and Heard, E.. (1999) Functional analysis of the dxpas34 locus, a 3' regulator of xist expression Mol Cell Biol 19, 85138525.Debrand, E., Heard, E. and Avner, P.. (1998) Cloning and localization of the murine xpct gene: evidence for complex rearrangements during the evolution of the region around the xist gene. Genomics 48, 296303.Dekker, J.. (2006) The three 'c' s of chromosome conformation capture: controls, controls, controls Nat Methods 3, 1721.Dermitzakis, ET. and Clark, AG.. (2002) Evolution of transcription factor binding sites in mammalian gene regulatory regions: conservation and turnover. Mol Biol Evol 19, 11141121.Dermitzakis, ET., Bergman, CM. and Clark, AG.. (2003) Tracing the evolutionary history of drosophila regulatory regions with models that identify transcription factor binding sites. Mol Biol Evol 20, 703714.DiazPerez, S., Ouyang, Y., Perez, V., Cisneros, R., Regelson, M. and Marahrens, Y.. (2005) The element(s) at the nontranscribed xist locus of the active x chromosome controls chromosomal replication timing in the mouse Genetics 171, 663672.DiazPerez, SV., Ferguson, DO., Wang, C., Csankovszki, G., Wang, C., Tsai, S., Dutta, D.,
253

Perez, V., Kim, S., Eller, CD., Salstrom, J. et al.. (2006) A deletion at the mouse xist gene exposes transeffects that alter the heterochromatin of the inactive x chromosome and the replication time and dna stability of both x chromosomes Genetics 174, 11151133.Dietzel, S., Schiebel, K., Little, G., Edelmann, P., Rappold, GA., Eils, R., Cremer, C. and Cremer, T.. (1999) The 3d positioning of ant2 and ant3 genes within female x chromosome territories correlates with gene activity Exp Cell Res 252, 363375.Dindot, SV., Kent, KC., Evers, B., Loskutoff, N., Womack, J. and Piedrahita, JA.. (2004) Conservation of genomic imprinting at the xist, igf2, and gtl2 loci in the bovine. Mamm Genome 15, 966974.Disteche, CM., Filippova, GN. and Tsuchiya, KD.. (2002) Escape from x inactivation. Cytogenet Genome Res 99, 3643.Do, JT. and Scholer, HR.. (2004) Nuclei of embryonic stem cells reprogram somatic cells Stem Cells 22, 941949.Dobrovolny, R., Dvorakova, L., Ledvinova, J., Magage, S., Bultas, J., Lubanda, JC., Poupetova, H., Elleder, M., Karetova, D. and Hrebicek, M.. (2005) Recurrence of fabry disease as a result of paternal germline mosaicism for alphagalactosidase a gene mutation Am J Med Genet A 134, 8487.Donohoe, ME., Zhang, L., Xu, N., Shi, Y. and Lee, JT.. (2007) Identification of a ctcf cofactor, yy1, for the x chromosome binary switch Mol Cell 25, 4356.Donovan, PJ., Stott, D., Cairns, LA., Heasman, J. and Wylie, CC.. (1986) Migratory and postmigratory mouse primordial germ cells behave differently in culture Cell 44, 831838.Dunn, KL. and Davie, JR.. (2003) The many roles of the transcriptional regulator ctcf Biochem Cell Biol 81, 161167.DurcovaHills, G., Adams, IR., Barton, SC., Surani, MA. and McLaren, A.. (2006a) The role of exogenous fibroblast growth factor2 on the reprogramming of primordial germ cells into pluripotent stem cells Stem Cells 24, 14411449.DurcovaHills, G., Hajkova, P., Sullivan, S., Barton, S., Surani, MA. and McLaren, A.. (2006b) Influence of sex chromosome constitution on the genomic imprinting of germ cells Proc Natl Acad Sci U S A 103, 1118411188.Duret, L., Chureau, C., Samain, S., Weissenbach, J. and Avner, P.. (2006) The xist rna gene evolved in eutherians by pseudogenization of a proteincoding gene Science 312, 16531655.Duthie, SM., Nesterova, TB., Formstone, EJ., Keohane, AM., Turner, BM., Zakian, SM. and Brockdorff, N.. (1999) Xist rna exhibits a banded localization on the inactive x chromosome and is excluded from autosomal material in cis Hum Mol Genet 8, 195204.Dyer, KA., Canfield, TK. and Gartler, SM.. (1989) Molecular cytological differentiation of active from inactive x domains in interphase: implications for x chromosome inactivation. Cytogenet Cell Genet 50, 116120.Eggan, K., Akutsu, H., Hochedlinger, K., Rideout, W3., Yanagimachi, R. and Jaenisch, R.. (2000) Xchromosome inactivation in cloned mouse embryos Science 290, 15781581.
254

Eils, R., Dietzel, S., Bertin, E., Schröck, E., Speicher, MR., Ried, T., RobertNicoud, M., Cremer, C. and Cremer, T.. (1996) Threedimensional reconstruction of painted human interphase chromosomes: active and inactive x chromosome territories have similar volumes but differ in shape and surface structure. J Cell Biol 135, 14271440.Emberly, E., Rajewsky, N. and Siggia, ED.. (2003) Conservation of regulatory elements between two species of drosophila. BMC Bioinformatics 4, 57.Emerson, JJ., Kaessmann, H., Betran, E. and Long, M.. (2004) Extensive gene traffic on the mammalian x chromosome Science 303, 537540.Engel, N., Thorvaldsen, JL. and Bartolomei, MS.. (2006) Ctcf binding sites promote transcription initiation and prevent dna methylation on the maternal allele at the imprinted h19/igf2 locus Hum Mol Genet 15, 29452954.Engemann, S., Strödicke, M., Paulsen, M., Franck, O., Reinhardt, R., Lane, N., Reik, W. and Walter, J.. (2000) Sequence and functional comparison in the beckwithwiedemann region: implications for a novel imprinting centre and extended imprinting. Hum Mol Genet 9, 26912706.Epstein, CJ., Smith, S., Travis, B. and Tucker, G.. (1978a) Both x chromosomes function before visible xchromosome inactivation in female mouse embryos Nature 274, 500503.Epstein, CJ., Travis, B., Tucker, G. and Smith, S.. (1978b) The direct demonstration of an xchromosome dosage effect prior to inactivation Basic Life Sci 12, 261267.Evans, HJ., Ford, CE., Lyon, MF. and Gray, J.. (1965) Dna replication and genetic expression in female mice with morphologically distinguishable x chromosomes Nature 206, 900903.Evans, MJ. and Kaufman, MH.. (1981) Establishment in culture of pluripotential cells from mouse embryos Nature 292, 154156.Fackelmayer, FO.. (2005) A stable proteinaceous structure in the territory of inactive x chromosomes J Biol Chem 280, 17201723.Falconer, DS.. (1953) [total sexlinkage in the house mouse.] Z Indukt Abstamm Vererbungsl 85, 210219.Fang, J., Chen, T., Chadwick, B., Li, E. and Zhang, Y.. (2004) Ring1bmediated h2a ubiquitination associates with inactive x chromosomes and is involved in initiation of x inactivation J Biol Chem 279, 5281252815.Farazmand, A., Basrur, PK., Stranzinger, G., Graphodatskaya, D., Reyes, ER. and King, WA.. (2004) Expression of xist sense and antisense in bovine fetal organs and cell cultures. Chromosome Res 12, 275283.Farivar, S., Yamaguchi, S., Sugimoto, M. and Takagi, N.. (2004) Xchromosome inactivation in differentiating mouse embryonic stem cells carrying xlinked gfp and lacz transgenes Int J Dev Biol 48, 629635.Fedoriw, AM., Stein, P., Svoboda, P., Schultz, RM. and Bartolomei, MS.. (2004) Transgenic rnai reveals essential function for ctcf in h19 gene imprinting Science 303, 238240.FernandezCapetillo, O., Mahadevaiah, SK., Celeste, A., Romanienko, PJ., CameriniOtero, RD., Bonner, WM., Manova, K., Burgoyne, P. and Nussenzweig, A.. (2003) H2ax is required for
255

chromatin remodeling and inactivation of sex chromosomes in male mouse meiosis Dev Cell 4, 497508.Filippova, GN., Cheng, MK., Moore, JM., Truong, J., Hu, YJ., Nguyen, DK., Tsuchiya, KD. and Disteche, CM.. (2005) Boundaries between chromosomal domains of x inactivation and escape bind ctcf and lack cpg methylation during early development Dev Cell 8, 3142.Filippova, GN., Thienes, CP., Penn, BH., Cho, DH., Hu, YJ., Moore, JM., Klesert, TR., Lobanenkov, VV. and Tapscott, SJ.. (2001) Ctcfbinding sites flank ctg/cag repeats and form a methylationsensitive insulator at the dm1 locus. Nat Genet 28, 335343.Fischle, W., Wang, Y., Jacobs, SA., Kim, Y., Allis, CD. and Khorasanizadeh, S.. (2003) Molecular basis for the discrimination of repressive methyllysine marks in histone h3 by polycomb and hp1 chromodomains Genes Dev 17, 18701881.Forrester, LM. and Ansell, JD.. (1985) Parental influences on x chromosome expression Genet Res 45, 95100.Fowlis, DJ., Ansell, JD. and Micklem, HS.. (1991) Further evidence for the importance of parental source of the xce allele in x chromosome inactivation Genet Res 58, 6365.Franco, B. and Ballabio, A.. (2006) Xinactivation and human disease: xlinked dominant malelethal disorders Curr Opin Genet Dev 16, 254259.Fraser, AS., Sobey, CC., Spicer, CC.. (1953) Mottled, a sexmodified lethal in the house mouse J Genet 51, 217.Frith, MC., Pheasant, M. and Mattick, JS.. (2005) The amazing complexity of the human transcriptome Eur J Hum Genet 13, 894897.Galton, M. and Holt, SF.. (1965) Asynchronous replication of the mouse sex chromosomes Exp Cell Res 37, 111116.Gamer, LW. and Wright, CV.. (1993) Murine cdx4 bears striking similarities to the drosophila caudal gene in its homeodomain sequence and early expression pattern. Mech Dev 43, 7181.Ganesan, S., Richardson, AL., Wang, ZC., Iglehart, JD., Miron, A., Feunteun, J., Silver, D. and Livingston, DM.. (2005) Abnormalities of the inactive x chromosome are a common feature of brca1 mutant and sporadic basallike breast cancer. Cold Spring Harb Symp Quant Biol 70, 9397.Ganesan, S., Silver, DP., Drapkin, R., Greenberg, R., Feunteun, J. and Livingston, DM.. (2004) Association of brca1 with the inactive x chromosome and xist rna. Philos Trans R Soc Lond B Biol Sci 359, 123128.Ganesan, S., Silver, DP., Greenberg, RA., Avni, D., Drapkin, R., Miron, A., Mok, SC., Randrianarison, V., Brodie, S., Salstrom, J., Rasmussen, TP. et al.. (2002) Brca1 supports xist rna concentration on the inactive x chromosome Cell 111, 393405.GarciaCastro, MI., Anderson, R., Heasman, J. and Wylie, C.. (1997) Interactions between germ cells and extracellular matrix glycoproteins during migration and gonad assembly in the mouse embryo J Cell Biol 138, 471480.Gartler, SM. and Riggs, AD.. (1983) Mammalian xchromosome inactivation Annu Rev Genet 17, 155190.
256

Geijsen, N., Horoschak, M., Kim, K., Gribnau, J., Eggan, K. and Daley, GQ.. (2004) Derivation of embryonic germ cells and male gametes from embryonic stem cells Nature 427, 148154.Gerasimova, TI. and Corces, VG.. (1996) Boundary and insulator elements in chromosomes. Curr Opin Genet Dev 6, 185192.Giannelli, F.. (1963) The pattern of xchromosome deoxyribonucleic acid synthesis in two women with abnormal sexchromosome complements Lancet 1, 863865.Gilbert, DM.. (2002) Replication timing and transcriptional control: beyond cause and effect Curr Opin Cell Biol 14, 377383.Gilbert, N., Boyle, S., Sutherland, H., de Las Heras, J., Allan, J., Jenuwein, T. and Bickmore, WA.. (2003) Formation of facultative heterochromatin in the absence of hp1 EMBO J 22, 55405550.Gilbert, SL. and Sharp, PA.. (1999) Promoterspecific hypoacetylation of xinactivated genes Proc Natl Acad Sci U S A 96, 1382513830.Gilfillan, GD., Straub, T., de Wit, E., Greil, F., Lamm, R., van Steensel, B. and Becker, PB.. (2006) Chromosomewide genespecific targeting of the drosophila dosage compensation complex Genes Dev 20, 858870.Ginsburg, M., Snow, MH. and McLaren, A.. (1990) Primordial germ cells in the mouse embryo during gastrulation Development 110, 521528.Goldmit, M., Ji, Y., Skok, J., Roldan, E., Jung, S., Cedar, H. and Bergman, Y.. (2005) Epigenetic ontogeny of the igk locus during b cell development Nat Immunol 6, 198203.Gomez, M. and Brockdorff, N.. (2004) Heterochromatin on the inactive x chromosome delays replication timing without affecting origin usage Proc Natl Acad Sci U S A 101, 69236928.Goodfellow, PJ., Mondello, C., Darling, SM., Pym, B., Little, P. and Goodfellow, PN.. (1988) Absence of methylation of a cpgrich region at the 5' end of the mic2 gene on the active x, the inactive x, and the y chromosome Proc Natl Acad Sci U S A 85, 56055609.Goto, T., Wright, E. and Monk, M.. (1997) Paternal xchromosome inactivation in human trophoblastic cells Mol Hum Reprod 3, 7780.Goto, Y. and Takagi, N.. (1998) Tetraploid embryos rescue embryonic lethality caused by an additional maternally inherited x chromosome in the mouse Development 125, 33533363.Goto, Y. and Takagi, N.. (1999) [control of x chromosome inactivation in mammals] Tanpakushitsu Kakusan Koso 44, 16821690.Goto, Y., Gomez, M., Brockdorff, N. and Feil, R.. (2002) Differential patterns of histone methylation and acetylation distinguish active and repressed alleles at xlinked genes Cytogenet Genome Res 99, 6674.Graves, JA.. (1982) 5azacytidineinduced reexpression of alleles on the inactive x chromosome in a hybrid mouse cell line. Exp Cell Res 141, 99105.Graves, JAM.. (2006) Sex chromosome specialization and degeneration in mammals Cell 124, 901914.Graves, JAM., Gecz, J. and Hameister, H.. (2002) Evolution of the human xa smart and sexy
257

chromosome that controls speciation and development Cytogenet Genome Res 99, 141145.Greaves, IK., Rangasamy, D., Devoy, M., Marshall Graves, JA. and Tremethick, DJ.. (2006) The x and y chromosomes assemble into h2a.zcontaining [corrected] facultative heterochromatin [corrected] following meiosis Mol Cell Biol 26, 53945405.Greenbaum, JA., Parker, SCJ. and Tullius, TD.. (2007) Detection of dna structural motifs in functional genomic elements. Genome Res 17, 940946.Gribnau, J., Luikenhuis, S., Hochedlinger, K., Monkhorst, K. and Jaenisch, R.. (2005) X chromosome choice occurs independently of asynchronous replication timing J Cell Biol 168, 365373.Grumbach, MM., Morishima, A. and Taylor, JH.. (1963) Human sex chromosome abnormalities in relation to dna replication and heterochromatinization Proc Natl Acad Sci U S A 49, 581589.Grutzner, F. and Graves, JAM.. (2004) A platypus' eye view of the mammalian genome Curr Opin Genet Dev 14, 642649.Grutzner, F., Rens, W., TsendAyush, E., ElMogharbel, N., O'Brien, PCM., Jones, RC., FergusonSmith, MA. and Marshall Graves, JA.. (2004) In the platypus a meiotic chain of ten sex chromosomes shares genes with the bird z and mammal x chromosomes Nature 432, 913917.Gudmundsdottir, K. and Ashworth, A.. (2006) The roles of brca1 and brca2 and associated proteins in the maintenance of genomic stability. Oncogene 25, 58645874.Gupta, V., Parisi, M., Sturgill, D., Nuttall, R., Doctolero, M., Dudko, OK., Malley, JD., Eastman, PS. and Oliver, B.. (2006) Global analysis of xchromosome dosage compensation J Biol 5, 3.Hadjantonakis, AK., Cox, LL., Tam, PP. and Nagy, A.. (2001) An xlinked gfp transgene reveals unexpected paternal xchromosome activity in trophoblastic giant cells of the mouse placenta Genesis 29, 133140.Hadjantonakis, AK., Gertsenstein, M., Ikawa, M., Okabe, M. and Nagy, A.. (1998) Noninvasive sexing of preimplantation stage mammalian embryos Nat Genet 19, 220222.Hall, LL., Clemson, CM., Byron, M., Wydner, K. and Lawrence, JB.. (2002) Unbalanced x;autosome translocations provide evidence for sequence specificity in the association of xist rna with chromatin Hum Mol Genet 11, 31573165.Hamada, FN., Park, PJ., Gordadze, PR. and Kuroda, MI.. (2005) Global regulation of x chromosomal genes by the msl complex in drosophila melanogaster Genes Dev 19, 22892294.Hark, AT., Schoenherr, CJ., Katz, DJ., Ingram, RS., Levorse, JM. and Tilghman, SM.. (2000) Ctcf mediates methylationsensitive enhancerblocking activity at the h19/igf2 locus Nature 405, 486489.Harrison, KB.. (1989) Xchromosome inactivation in the human cytotrophoblast Cytogenet Cell Genet 52, 3741.Harrison, KB. and Warburton, D.. (1986) Preferential xchromosome activity in human female placental tissues. Cytogenet Cell Genet 41, 163168.Heard, E.. (2005) Delving into the diversity of facultative heterochromatin: the epigenetics of the
258

inactive x chromosome Curr Opin Genet Dev 15, 482489.Heard, E. and Disteche, CM.. (2006) Dosage compensation in mammals: finetuning the expression of the x chromosome. Genes Dev 20, 18481867.Heard, E., Avner, P. and Rothstein, R.. (1994) Creation of a deletion series of mouse yacs covering a 500 kb region around xist Nucleic Acids Res 22, 18301837.Heard, E., Clerc, P. and Avner, P.. (1997) Xchromosome inactivation in mammals Annu Rev Genet 31, 571610.Heard, E., Kress, C., Mongelard, F., Courtier, B., Rougeulle, C., Ashworth, A., Vourc'h, C., Babinet, C. and Avner, P.. (1996) Transgenic mice carrying an xistcontaining yac Hum Mol Genet 5, 441450.Heard, E., Mongelard, F., Arnaud, D. and Avner, P.. (1999a) Xist yeast artificial chromosome transgenes function as xinactivation centers only in multicopy arrays and not as single copies Mol Cell Biol 19, 31563166.Heard, E., Mongelard, F., Arnaud, D., Chureau, C., Vourc'h, C. and Avner, P.. (1999b) Human xist yeast artificial chromosome transgenes show partial x inactivation center function in mouse embryonic stem cells Proc Natl Acad Sci U S A 96, 68416846.Heard, E., Rougeulle, C., Arnaud, D., Avner, P., Allis, CD. and Spector, DL.. (2001) Methylation of histone h3 at lys9 is an early mark on the x chromosome during x inactivation Cell 107, 727738.Helbig, R. and Fackelmayer, FO.. (2003) Scaffold attachment factor a (safa) is concentrated in inactive x chromosome territories through its rgg domain Chromosoma 112, 173182.Hendriksen, PJ.. (1999) Do x and y spermatozoa differ in proteins? Theriogenology 52, 12951307.Hendriksen, PJ., Hoogerbrugge, JW., Themmen, AP., Koken, MH., Hoeijmakers, JH., Oostra, BA., van der Lende, T. and Grootegoed, JA.. (1995) Postmeiotic transcription of x and y chromosomal genes during spermatogenesis in the mouse Dev Biol 170, 730733.HernandezMunoz, I., Lund, AH., van der Stoop, P., Boutsma, E., Muijrers, I., Verhoeven, E., Nusinow, DA., Panning, B., Marahrens, Y. and van Lohuizen, M.. (2005) Stable x chromosome inactivation involves the prc1 polycomb complex and requires histone macroh2a1 and the cullin3/spop ubiquitin e3 ligase Proc Natl Acad Sci U S A 102, 76357640.Herzing, LB., Romer, JT., Horn, JM. and Ashworth, A.. (1997) Xist has properties of the xchromosome inactivation centre Nature 386, 272275.Hoffman, LM., Hall, L., Batten, JL., Young, H., Pardasani, D., Baetge, EE., Lawrence, J. and Carpenter, MK.. (2005) Xinactivation status varies in human embryonic stem cell lines. Stem Cells 23, 14681478.Hong, YK., Ontiveros, SD. and Strauss, WM.. (2000) A revision of the human xist gene organization and structural comparison with mouse xist Mamm Genome 11, 220224.Hong, YK., Ontiveros, SD., Chen, C. and Strauss, WM.. (1999) A new structure for the murine xist gene and its relationship to chromosome choice/counting during xchromosome inactivation Proc Natl Acad Sci U S A 96, 68296834.
259

HoyerFender, S., Costanzi, C. and Pehrson, JR.. (2000) Histone macroh2a1.2 is concentrated in the xybody by the early pachytene stage of spermatogenesis Exp Cell Res 258, 254260.Hurst, LD.. (2001) Evolutionary genomics. sex and the x Nature 411, 149150.Huynh, KD. and Lee, JT.. (2003) Inheritance of a preinactivated paternal x chromosome in early mouse embryos Nature 426, 857862.Ikegami, K., Iwatani, M., Suzuki, M., Tachibana, M., Shinkai, Y., Tanaka, S., Greally, JM., Yagi, S., Hattori, N. and Shiota, K.. (2007) Genomewide and locusspecific dna hypomethylation in g9a deficient mouse embryonic stem cells. Genes Cells 12, 111.Izban, MG. and Luse, DS.. (1992) Factorstimulated rna polymerase ii transcribes at physiological elongation rates on naked dna but very poorly on chromatin templates J Biol Chem 267, 1364713655.Jablonka, E. and Lamb, MJ.. (1988) Meiotic pairing constraints and the activity of sex chromosomes J Theor Biol 133, 2336.Jacobs, SA., Taverna, SD., Zhang, Y., Briggs, SD., Li, J., Eissenberg, JC., Allis, CD. and Khorasanizadeh, S.. (2001) Specificity of the hp1 chromo domain for the methylated nterminus of histone h3 EMBO J 20, 52325241.Jegalian, K. and Page, DC.. (1998) A proposed path by which genes common to mammalian x and y chromosomes evolve to become x inactivated Nature 394, 776780.Jenuwein, T. and Allis, CD.. (2001) Translating the histone code Science 293, 10741080.Jeppesen, P. and Turner, BM.. (1993) The inactive x chromosome in female mammals is distinguished by a lack of histone h4 acetylation, a cytogenetic marker for gene expression Cell 74, 281289.Johnson, K., AngelinDuclos, C., Park, S. and Calame, KL.. (2003) Changes in histone acetylation are associated with differences in accessibility of v(h) gene segments to vdj recombination during bcell ontogeny and development Mol Cell Biol 23, 24382450.Johnston, CM., Nesterova, TB., Formstone, EJ., Newall, AE., Duthie, SM., Sheardown, SA. and Brockdorff, N.. (1998) Developmentally regulated xist promoter switch mediates initiation of x inactivation Cell 94, 809817.Johnston, PG. and Cattanach, BM.. (1981) Controlling elements in the mouse. iv. evidence of nonrandom xinactivation Genet Res 37, 151160.Jurka, J., Kohany, O., Pavlicek, A., Kapitonov, VV. and Jurka, MV.. (2004) Duplication, coclustering, and selection of human alu retrotransposons Proc Natl Acad Sci U S A 101, 12681272.Kalantry, S. and Magnuson, T.. (2006) The polycomb group protein eed is dispensable for the initiation of random xchromosome inactivation PLoS Genet 2, e66.Kalantry, S., Mills, KC., Yee, D., Otte, AP., Panning, B. and Magnuson, T.. (2006) The polycomb group protein eed protects the inactive xchromosome from differentiationinduced reactivation Nat Cell Biol 8, 195202.KanatsuShinohara, M., Inoue, K., Lee, J., Yoshimoto, M., Ogonuki, N., Miki, H., Baba, S., Kato, T., Kazuki, Y., Toyokuni, S., Toyoshima, M. et al.. (2004) Generation of pluripotent stem
260

cells from neonatal mouse testis Cell 119, 10011012.Kanduri, C., Pant, V., Loukinov, D., Pugacheva, E., Qi, CF., Wolffe, A., Ohlsson, R. and Lobanenkov, VV.. (2000) Functional association of ctcf with the insulator upstream of the h19 gene is parent of originspecific and methylationsensitive. Curr Biol 10, 853856.Kanduri, C., Thakur, N. and Pandey, RR.. (2006) The length of the transcript encoded from the kcnq1ot1 antisense promoter determines the degree of silencing. EMBO J 25, 20962106.Karachentsev, D., Sarma, K., Reinberg, D. and Steward, R.. (2005) Prset7dependent methylation of histone h4 lys 20 functions in repression of gene expression and is essential for mitosis Genes Dev 19, 431435.Katayama, S., Tomaru, Y., Kasukawa, T., Waki, K., Nakanishi, M., Nakamura, M., Nishida, H., Yap, CC., Suzuki, M., Kawai, J., Suzuki, H. et al.. (2005) Antisense transcription in the mammalian transcriptome Science 309, 15641566.Kay, GF., Barton, SC., Surani, MA. and Rastan, S.. (1994) Imprinting and x chromosome counting mechanisms determine xist expression in early mouse development Cell 77, 639650.Kay, GF., Penny, GD., Patel, D., Ashworth, A., Brockdorff, N. and Rastan, S.. (1993) Expression of xist during mouse development suggests a role in the initiation of x chromosome inactivation Cell 72, 171182.Keohane, AM., Barlow, AL., Waters, J., Bourn, D. and Turner, BM.. (1999) H4 acetylation, xist rna and replication timing are coincident and define x;autosome boundaries in two abnormal x chromosomes Hum Mol Genet 8, 377383.Keohane, AM., O'neill, LP., Belyaev, ND., Lavender, JS. and Turner, BM.. (1996) Xinactivation and histone h4 acetylation in embryonic stem cells Dev Biol 180, 618630.Khalil, AM., Boyar, FZ. and Driscoll, DJ.. (2004) Dynamic histone modifications mark sex chromosome inactivation and reactivation during mammalian spermatogenesis Proc Natl Acad Sci U S A 101, 1658316587.Khil, PP., Smirnova, NA., Romanienko, PJ. and CameriniOtero, RD.. (2004) The mouse x chromosome is enriched for sexbiased genes not subject to selection by meiotic sex chromosome inactivation Nat Genet 36, 642646.Kim, J., Kollhoff, A., Bergmann, A. and Stubbs, L.. (2003) Methylationsensitive binding of transcription factor yy1 to an insulator sequence within the paternally expressed imprinted gene, peg3. Hum Mol Genet 12, 233245.Kim, JD., Hinz, AK., Bergmann, A., Huang, JM., Ovcharenko, I., Stubbs, L. and Kim, J.. (2006) Identification of clustered yy1 binding sites in imprinting control regions Genome Res 16, 901911.Kimura, H., Tada, M., Hatano, S., Yamazaki, M., Nakatsuji, N. and Tada, T.. (2002) Chromatin reprogramming of male somatic cellderived xist and tsix in es hybrid cells Cytogenet Genome Res 99, 106114.Knezetic, JA. and Luse, DS.. (1986) The presence of nucleosomes on a dna template prevents initiation by rna polymerase ii in vitro Cell 45, 95104.
261

Ko, MS., Threat, TA., Wang, X., Horton, JH., Cui, Y., Wang, X., Pryor, E., Paris, J., WellsSmith, J., Kitchen, JR., Rowe, LB. et al.. (1998) Genomewide mapping of unselected transcripts from extraembryonic tissue of 7.5day mouse embryos reveals enrichment in the tcomplex and underrepresentation on the x chromosome Hum Mol Genet 7, 19671978.KofmanAlfaro, S. and Chandley, AC.. (1970) Meiosis in the male mouse. an autoradiographic investigation Chromosoma 31, 404420.Kohlmaier, A., Savarese, F., Lachner, M., Martens, J., Jenuwein, T. and Wutz, A.. (2004) A chromosomal memory triggered by xist regulates histone methylation in x inactivation PLoS Biol 2, E171.Kratzer, PG. and Gartler, SM.. (1978) Hgprt activity changes in preimplantation mouse embryos Nature 274, 503504.Kraus, WL., Manning, ET. and Kadonaga, JT.. (1999) Biochemical analysis of distinct activation functions in p300 that enhance transcription initiation with chromatin templates Mol Cell Biol 19, 81238135.Krietsch, WK., Fehlau, M., Renner, P., Bucher, T. and Fundele, R.. (1986) Expression of xlinked phosphoglycerate kinase in early mouse embryos homozygous at the xce locus Differentiation 31, 5054.Kumar, S. and Subramanian, S.. (2002) Mutation rates in mammalian genomes Proc Natl Acad Sci U S A 99, 803808.Kunath, T., Arnaud, D., Uy, GD., Okamoto, I., Chureau, C., Yamanaka, Y., Heard, E., Gardner, RL., Avner, P. and Rossant, J.. (2005) Imprinted xinactivation in extraembryonic endoderm cell lines from mouse blastocysts Development 132, 16491661.Kunath, T., Strumpf, D. and Rossant, J.. (2004) Early trophoblast determination and stem cell maintenance in the mousea review Placenta 25 Suppl A, S328.Kurukuti, S., Tiwari, VK., Tavoosidana, G., Pugacheva, E., Murrell, A., Zhao, Z., Lobanenkov, V., Reik, W. and Ohlsson, R.. (2006) Ctcf binding at the h19 imprinting control region mediates maternally inherited higherorder chromatin conformation to restrict enhancer access to igf2 Proc Natl Acad Sci U S A 103, 1068410689.Kurz, A., Lampel, S., Nickolenko, JE., Bradl, J., Benner, A., Zirbel, RM., Cremer, T. and Lichter, P.. (1996) Active and inactive genes localize preferentially in the periphery of chromosome territories J Cell Biol 135, 11951205.Lachner, M., O'Carroll, D., Rea, S., Mechtler, K. and Jenuwein, T.. (2001) Methylation of histone h3 lysine 9 creates a binding site for hp1 proteins Nature 410, 116120.Lafreniere, RG. and Willard, HF.. (1993) Pulsedfield map of xq13 in the region of the human x inactivation center Genomics 17, 502506.Lafreniere, RG., Brown, CJ., Rider, S., Chelly, J., TaillonMiller, P., Chinault, AC., Monaco, AP. and Willard, HF.. (1993) 2.6 mb yac contig of the human x inactivation center region in xq13: physical linkage of the rps4x, phka1, xist and dxs128e genes Hum Mol Genet 2, 11051115.Lafrenière, RG., Carrel, L. and Willard, HF.. (1994) A novel transmembrane transporter encoded
262

by the xpct gene in xq13.2. Hum Mol Genet 3, 11331139.Lahn, BT. and Page, DC.. (1999a) Retroposition of autosomal mrna yielded testisspecific gene family on human y chromosome Nat Genet 21, 429433.Lahn, BT. and Page, DC.. (1999b) Four evolutionary strata on the human x chromosome Science 286, 964967.Latham, KE. and Rambhatla, L.. (1995) Expression of xlinked genes in androgenetic, gynogenetic, and normal mouse preimplantation embryos Dev Genet 17, 212222.Lebon, JM., Tam, PP., SingerSam, J., Riggs, AD. and Tan, SS.. (1995) Mouse endogenous xlinked genes do not show lineagespecific delayed inactivation during development Genet Res 65, 223227.Lee, JT.. (2000) Disruption of imprinted x inactivation by parentoforigin effects at tsix Cell 103, 1727.Lee, JT.. (2002) Homozygous tsix mutant mice reveal a sexratio distortion and revert to random xinactivation. Nat Genet 32, 195200.Lee, JT.. (2005) Regulation of xchromosome counting by tsix and xite sequences Science 309, 768771.Lee, JT. and Jaenisch, R.. (1997) Longrange cis effects of ectopic xinactivation centres on a mouse autosome Nature 386, 275279.Lee, JT. and Lu, N.. (1999) Targeted mutagenesis of tsix leads to nonrandom x inactivation Cell 99, 4757.Lee, JT., Davidow, LS. and Warshawsky, D.. (1999) Tsix, a gene antisense to xist at the xinactivation centre Nat Genet 21, 400404.Lee, JT., Strauss, WM., Dausman, JA. and Jaenisch, R.. (1996) A 450 kb transgene displays properties of the mammalian xinactivation center Cell 86, 8394.Leppig, KA., Brown, CJ., Bressler, SL., Gustashaw, K., Pagon, RA., Willard, HF. and Disteche, CM.. (1993) Mapping of the distal boundary of the xinactivation center in a rearranged x chromosome from a female expressing xist Hum Mol Genet 2, 883887.Lewis, A., Green, K., Dawson, C., Redrup, L., Huynh, KD., Lee, JT., Hemberger, M. and Reik, W.. (2006) Epigenetic dynamics of the kcnq1 imprinted domain in the early embryo. Development 133, 42034210.Lewis, A., Mitsuya, K., Umlauf, D., Smith, P., Dean, W., Walter, J., Higgins, M., Feil, R. and Reik, W.. (2004) Imprinting on distal chromosome 7 in the placenta involves repressive histone methylation independent of dna methylation. Nat Genet 36, 12911295.Li, J., LeesMurdock, DJ., Xu, G. and Walsh, CP.. (2004) Timing of establishment of paternal methylation imprints in the mouse Genomics 84, 952960.Lifschytz, E. and Lindsley, DL.. (1972) The role of xchromosome inactivation during spermatogenesis (drosophilaallocyclychromosome evolutionmale sterilitydosage compensation) Proc Natl Acad Sci U S A 69, 182186.Lindsley, DL., Sandler, L., Baker, BS., Carpenter, AT., Denell, RE., Hall, JC., Jacobs, PA.,
263

Miklos, GL., Davis, BK., Gethmann, RC., Hardy, RW. et al.. (1972) Segmental aneuploidy and the genetic gross structure of the drosophila genome Genetics 71, 157184.Ling, JQ., Li, T., Hu, JF., Vu, TH., Chen, HL., Qiu, XW., Cherry, AM. and Hoffman, AR.. (2006) Ctcf mediates interchromosomal colocalization between igf2/h19 and wsb1/nf1 Science 312, 269272.Lingenfelter, PA., Adler, DA., Poslinski, D., Thomas, S., Elliott, RW., Chapman, VM. and Disteche, CM.. (1998) Escape from x inactivation of smcx is preceded by silencing during mouse development Nat Genet 18, 212213.Looijenga, LH., Gillis, AJ., Verkerk, AJ., van Putten, WL. and Oosterhuis, JW.. (1999) Heterogeneous x inactivation in trophoblastic cells of human fullterm female placentas. Am J Hum Genet 64, 14451452.LovellBadge, R. and Robertson, E.. (1990) Xy female mice resulting from a heritable mutation in the primary testisdetermining gene, tdy Development 109, 635646.Lucchesi, JC., Kelly, WG. and Panning, B.. (2005) Chromatin remodeling in dosage compensation Annu Rev Genet 39, 615651.Lucifero, D., Mann, MRW., Bartolomei, MS. and Trasler, JM.. (2004) Genespecific timing and epigenetic memory in oocyte imprinting Hum Mol Genet 13, 839849.Ludwig, MZ., Bergman, C., Patel, NH. and Kreitman, M.. (2000) Evidence for stabilizing selection in a eukaryotic enhancer element. Nature 403, 564567.Ludwig, MZ., Palsson, A., Alekseeva, E., Bergman, CM., Nathan, J. and Kreitman, M.. (2005) Functional evolution of a cisregulatory module. PLoS Biol 3, e93.Luger, K., Mader, AW., Richmond, RK., Sargent, DF. and Richmond, TJ.. (1997) Crystal structure of the nucleosome core particle at 2.8 a resolution Nature 389, 251260.Luikenhuis, S., Wutz, A. and Jaenisch, R.. (2001) Antisense transcription through the xist locus mediates tsix function in embryonic stem cells Mol Cell Biol 21, 85128520.Lyle, R., Watanabe, D., te Vruchte, D., Lerchner, W., Smrzka, OW., Wutz, A., Schageman, J., Hahner, L., Davies, C. and Barlow, DP.. (2000) The imprinted antisense rna at the igf2r locus overlaps but does not imprint mas1. Nat Genet 25, 1921.Lyon, MF.. (1961) Gene action in the xchromosome of the mouse (mus musculus l.) Nature 190, 372373.Lyon, MF.. (1962) Sex chromatin and gene action in the mammalian xchromosome Am J Hum Genet 14, 135148.Lyon, MF.. (1963) Lyonisation of the x chromosome Lancet 2, 11201121.Lyon, MF.. (1998) Xchromosome inactivation: a repeat hypothesis Cytogenet Cell Genet 80, 133137.Lyon, MF.. (2003) The lyon and the line hypothesis. Semin Cell Dev Biol 14, 313318.Ma, M. and Strauss, WM.. (2005) Analysis of the xist rna isoforms suggests two distinctly different forms of regulation Mamm Genome 16, 391404.MacDonald, M., Hassold, T., Harvey, J., Wang, LH., Morton, NE. and Jacobs, P.. (1994) The
264

origin of 47,xxy and 47,xxx aneuploidy: heterogeneous mechanisms and role of aberrant recombination Hum Mol Genet 3, 13651371.Mahadevaiah, SK., Turner, JM., Baudat, F., Rogakou, EP., de Boer, P., BlancoRodriguez, J., Jasin, M., Keeney, S., Bonner, WM. and Burgoyne, PS.. (2001) Recombinational dna doublestrand breaks in mice precede synapsis Nat Genet 27, 271276.Maherali, N., Sridharan, R., Xie, W., Utikal, J., Eminli, S., Arnold, K., Stadtfeld, M., Yachechko, R., Tchieu, J., Jaenisch, R., Plath, K. et al.. (2007) Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution Cell Stem Cell 1, 5570.Mahy, NL., Perry, PE. and Bickmore, WA.. (2002a) Gene density and transcription influence the localization of chromatin outside of chromosome territories detectable by fish J Cell Biol 159, 753763.Mahy, NL., Perry, PE., Gilchrist, S., Baldock, RA. and Bickmore, WA.. (2002b) Spatial organization of active and inactive genes and noncoding dna within chromosome territories J Cell Biol 157, 579589.Mak, W., Baxter, J., Silva, J., Newall, AE., Otte, AP. and Brockdorff, N.. (2002) Mitotically stable association of polycomb group proteins eed and enx1 with the inactive x chromosome in trophoblast stem cells. Curr Biol 12, 10161020.Mak, W., Nesterova, TB., de Napoles, M., Appanah, R., Yamanaka, S., Otte, AP. and Brockdorff, N.. (2004) Reactivation of the paternal x chromosome in early mouse embryos Science 303, 666669.Makalowski, W. and Boguski, MS.. (1998) Evolutionary parameters of the transcribed mammalian genome: an analysis of 2,820 orthologous rodent and human sequences. Proc Natl Acad Sci U S A 95, 94079412.ManciniDinardo, D., Steele, SJS., Levorse, JM., Ingram, RS. and Tilghman, SM.. (2006) Elongation of the kcnq1ot1 transcript is required for genomic imprinting of neighboring genes Genes Dev 20, 12681282.Marahrens, Y., Loring, J. and Jaenisch, R.. (1998) Role of the xist gene in x chromosome choosing Cell 92, 657664.Marahrens, Y., Panning, B., Dausman, J., Strauss, W. and Jaenisch, R.. (1997) Xistdeficient mice are defective in dosage compensation but not spermatogenesis Genes Dev 11, 156166.Margueron, R., Trojer, P. and Reinberg, D.. (2005) The key to development: interpreting the histone code? Curr Opin Genet Dev 15, 163176.Martienssen, RA.. (2003) Maintenance of heterochromatin by rna interference of tandem repeats Nat Genet 35, 213214.Martin, GR.. (1981) Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells Proc Natl Acad Sci U S A 78, 76347638.Martinez, R., BonillaHenao, V., Jimenez, A., Lucas, M., Vega, C., Ramos, I., Sobrino, F. and Pintado, E.. (2005) Skewed x inactivation of the normal allele in fully mutated female carriers determines the levels of fmrp in blood and the fragile x phenotype Mol Diagn 9, 157162.
265

Matsui, J., Goto, Y. and Takagi, N.. (2001) Control of xist expression for imprinted and random x chromosome inactivation in mice Hum Mol Genet 10, 13931401.Matsui, Y., Zsebo, K. and Hogan, BL.. (1992) Derivation of pluripotential embryonic stem cells from murine primordial germ cells in culture Cell 70, 841847.McCarrey, JR. and Dilworth, DD.. (1992) Expression of xist in mouse germ cells correlates with xchromosome inactivation Nat Genet 2, 200203.McCarrey, JR., Dilworth, DD. and Sharp, RM.. (1992) Semiquantitative analysis of xlinked gene expression during spermatogenesis in the mouse: ethidiumbromide staining of rtpcr products Genet Anal Tech Appl 9, 117123.McDonald, LE., Paterson, CA. and Kay, GF.. (1998) Bisulfite genomic sequencingderived methylation profile of the xist gene throughout early mouse development. Genomics 54, 379386.McDonel, P., Jans, J., Peterson, BK. and Meyer, BJ.. (2006) Clustered dna motifs mark x chromosomes for repression by a dosage compensation complex Nature 444, 614618.McKee, BD. and Handel, MA.. (1993) Sex chromosomes, recombination, and chromatin conformation Chromosoma 102, 7180.McLaren, A.. (1983) Primordial germ cells in mice Bibl Anat 24, 5966.McLaren, A.. (1984) Meiosis and differentiation of mouse germ cells Symp Soc Exp Biol 38, 723.McLaren, A.. (2003) Primordial germ cells in the mouse Dev Biol 262, 115.McLaren, A. and Southee, D.. (1997) Entry of mouse embryonic germ cells into meiosis Dev Biol 187, 107113.McNeil, JA., Smith, KP., Hall, LL. and Lawrence, JB.. (2006) Word frequency analysis reveals enrichment of dinucleotide repeats on the human x chromosome and [gata]n in the x escape region. Genome Res 16, 477484.Memili, E., Hong, YK., Kim, DH., Ontiveros, SD. and Strauss, WM.. (2001) Murine xist rna isoforms are different at their 3' ends: a role for differential polyadenylation. Gene 266, 131137.Mendes Soares, LM. and Valcarcel, J.. (2006) The expanding transcriptome: the genome as the 'book of sand' EMBO J 25, 923931.Mermoud, JE., Costanzi, C., Pehrson, JR. and Brockdorff, N.. (1999) Histone macroh2a1.2 relocates to the inactive x chromosome after initiation and propagation of xinactivation J Cell Biol 147, 13991408.Mermoud, JE., Popova, B., Peters, AHFM., Jenuwein, T. and Brockdorff, N.. (2002) Histone h3 lysine 9 methylation occurs rapidly at the onset of random x chromosome inactivation Curr Biol 12, 247251.MetzlerGuillemain, C., Luciani, J., Depetris, D., Guichaoua, MR. and Mattei, MG.. (2003) Hp1beta and hp1gamma, but not hp1alpha, decorate the entire xy body during human male meiosis Chromosome Res 11, 7381.Meyer, BJ., McDonel, P., Csankovszki, G. and Ralston, E.. (2004) Sex and xchromosomewide repression in caenorhabditis elegans Cold Spring Harb Symp Quant Biol 69, 7179.Migeon, BR.. (1998) Nonrandom x chromosome inactivation in mammalian cells. Cytogenet Cell
266

Genet 80, 142148.Migeon, BR. and Do, TT.. (1979) In search of nonrandom x inactivation: studies of fetal membranes heterozygous for glucose6phosphate dehydrogenase Am J Hum Genet 31, 581585.Migeon, BR., Axelman, J. and Jeppesen, P.. (2005) Differential x reactivation in human placental cells: implications for reversal of x inactivation. Am J Hum Genet 77, 355364.Migeon, BR., Chowdhury, AK., Dunston, JA. and McIntosh, I.. (2001a) Identification of tsix, encoding an rna antisense to human xist, reveals differences from its murine counterpart: implications for x inactivation Am J Hum Genet 69, 951960.Migeon, BR., Kazi, E., HaisleyRoyster, C., Hu, J., Reeves, R., Call, L., Lawler, A., Moore, CS., Morrison, H. and Jeppesen, P.. (1999) Human x inactivation center induces random x chromosome inactivation in male transgenic mice Genomics 59, 113121.Migeon, BR., Lee, CH., Chowdhury, AK. and Carpenter, H.. (2002) Species differences in tsix/tsix reveal the roles of these genes in xchromosome inactivation. Am J Hum Genet 71, 286293.Migeon, BR., Winter, H., Kazi, E., Chowdhury, AK., Hughes, A., HaisleyRoyster, C., Morrison, H. and Jeppesen, P.. (2001b) Lowcopynumber human transgene is recognized as an x inactivation center in mouse es cells, but fails to induce cisinactivation in chimeric mice Genomics 71, 156162.Min, J., Zhang, Y. and Xu, R.. (2003) Structural basis for specific binding of polycomb chromodomain to histone h3 methylated at lys 27 Genes Dev 17, 18231828.Mise, N., Goto, Y., Nakajima, N. and Takagi, N.. (1999) Molecular cloning of antisense transcripts of the mouse xist gene Biochem Biophys Res Commun 258, 537541.MlynarczykEvans, S., RoyceTolland, M., Alexander, MK., Andersen, AA., Kalantry, S., Gribnau, J. and Panning, B.. (2006) X chromosomes alternate between two states prior to random xinactivation PLoS Biol 4, e159.Moen, PTJ., Johnson, CV., Byron, M., Shopland, LS., de la Serna, IL., Imbalzano, AN. and Lawrence, JB.. (2004) Repositioning of musclespecific genes relative to the periphery of sc35 domains during skeletal myogenesis Mol Biol Cell 15, 197206.Mohandas, T., Sparkes, RS. and Shapiro, LJ.. (1981) Reactivation of an inactive human x chromosome: evidence for x inactivation by dna methylation. Science 211, 393396.Monesi, V.. (1965) Differential rate of ribonucleic acid synthesis in the autosomes and sex chromosomes during male meiosis in the mouse Chromosoma 17, 1121.Monk, M. and Harper, MI.. (1979) Sequential x chromosome inactivation coupled with cellular differentiation in early mouse embryos Nature 281, 311313.Monk, M. and McLaren, A.. (1981) Xchromosome activity in foetal germ cells of the mouse J Embryol Exp Morphol 63, 7584.Morey, C.. (2003) Caractérisation du rôle de la région en aval du gène xist lors de l'inactivation du chromosome x murin par mutagenèse ciblée dans les cellules es Thèse de Doctorat , .Morey, C., Arnaud, D., Avner, P. and Clerc, P.. (2001) Tsixmediated repression of xist
267

accumulation is not sufficient for normal random x inactivation Hum Mol Genet 10, 14031411.Morey, C., Navarro, P., Debrand, E., Avner, P., Rougeulle, C. and Clerc, P.. (2004) The region 3' to xist mediates x chromosome counting and h3 lys4 dimethylation within the xist gene EMBO J 23, 594604.Motzkus, D., Singh, PB. and HoyerFender, S.. (1999) M31, a murine homolog of drosophila hp1, is concentrated in the xy body during spermatogenesis Cytogenet Cell Genet 86, 8388.Murrell, A., Heeson, S. and Reik, W.. (2004) Interaction between differentially methylated regions partitions the imprinted genes igf2 and h19 into parentspecific chromatin loops Nat Genet 36, 889893.Namekawa, SH., Park, PJ., Zhang, L., Shima, JE., McCarrey, JR., Griswold, MD. and Lee, JT.. (2006) Postmeiotic sex chromatin in the male germline of mice Curr Biol 16, 660667.Namekawa, SH., Vandeberg, JL., McCarrey, JR. and Lee, JT.. (2007) Sex chromosome silencing in the marsupial male germ line. Proc Natl Acad Sci U S A 104, 97309735.Nanda, I., Shan, Z., Schartl, M., Burt, DW., Koehler, M., Nothwang, H., Grutzner, F., Paton, IR., Windsor, D., Dunn, I., Engel, W. et al.. (1999) 300 million years of conserved synteny between chicken z and human chromosome 9 Nat Genet 21, 258259.Navarro, P.. (2006) Mécanismes transcriptionnels et chromatiniens de la régulation de xist par son antisens tsix These de Doctorat.Navarro, P., Page, DR., Avner, P. and Rougeulle, C.. (2006) Tsixmediated epigenetic switch of a ctcfflanked region of the xist promoter determines the xist transcription program Genes Dev 20, 27872792.Navarro, P., Pichard, S., Ciaudo, C., Avner, P. and Rougeulle, C.. (2005) Tsix transcription across the xist gene alters chromatin conformation without affecting xist transcription: implications for xchromosome inactivation Genes Dev 19, 14741484.Nesterova, TB., Barton, SC., Surani, MA. and Brockdorff, N.. (2001a) Loss of xist imprinting in diploid parthenogenetic preimplantation embryos Dev Biol 235, 343350.Nesterova, TB., Johnston, CM., Appanah, R., Newall, AET., Godwin, J., Alexiou, M. and Brockdorff, N.. (2003) Skewing x chromosome choice by modulating sense transcription across the xist locus Genes Dev 17, 21772190.Nesterova, TB., Mermoud, JE., Hilton, K., Pehrson, J., Surani, MA., McLaren, A. and Brockdorff, N.. (2002) Xist expression and macroh2a1.2 localisation in mouse primordial and pluripotent embryonic germ cells Differentiation 69, 216225.Nesterova, TB., Slobodyanyuk, SY., Elisaphenko, EA., Shevchenko, AI., Johnston, C., Pavlova, ME., Rogozin, IB., Kolesnikov, NN., Brockdorff, N. and Zakian, SM.. (2001b) Characterization of the genomic xist locus in rodents reveals conservation of overall gene structure and tandem repeats but rapid evolution of unique sequence Genome Res 11, 833849.Newall, AE., Duthie, S., Formstone, E., Nesterova, T., Alexiou, M., Johnston, C., Caparros, ML. and Brockdorff, N.. (2001) Primary nonrandom x inactivation associated with disruption of xist promoter regulation Hum Mol Genet 10, 581589.
268

Nguyen, DK. and Disteche, CM.. (2006) Dosage compensation of the active x chromosome in mammals Nat Genet 38, 4753.Nicodemi, M. and Prisco, A.. (2007) Symmetrybreaking model for xchromosome inactivation Phys Rev Lett 98, 108104.Noble, WS., Kuehn, S., Thurman, R., Yu, M. and Stamatoyannopoulos, J.. (2005) Predicting the in vivo signature of human gene regulatory sequences. Bioinformatics 21 Suppl 1, i33843.Nogami, M., Nogami, O., Kagotani, K., Okumura, M., Taguchi, H., Ikemura, T. and Okumura, K.. (2000) Intranuclear arrangement of human chromosome 12 correlates to largescale replication domains Chromosoma 108, 514522.Nolen, LD., Gao, S., Han, Z., Mann, MRW., Gie Chung, Y., Otte, AP., Bartolomei, MS. and Latham, KE.. (2005) X chromosome reactivation and regulation in cloned embryos Dev Biol 279, 525540.Norris, DP., Brockdorff, N. and Rastan, S.. (1991) Methylation status of cpgrich islands on active and inactive mouse x chromosomes Mamm Genome 1, 7883.Norris, DP., Patel, D., Kay, GF., Penny, GD., Brockdorff, N., Sheardown, SA. and Rastan, S.. (1994) Evidence that random and imprinted xist expression is controlled by preemptive methylation Cell 77, 4151.Nusinow, DA. and Panning, B.. (2005) Recognition and modification of sex chromosomes Curr Opin Genet Dev 15, 206213.Nusinow, DA., HernandezMunoz, I., Fazzio, TG., Shah, GM., Kraus, WL. and Panning, B.. (2007) Poly(adpribose) polymerase 1 is inhibited by a histone h2a variant, macroh2a, and contributes to silencing of the inactive x chromosome J Biol Chem 282, 1285112859.O'Neill, LP., Keohane, AM., Lavender, JS., McCabe, V., Heard, E., Avner, P., Brockdorff, N. and Turner, BM.. (1999) A developmental switch in h4 acetylation upstream of xist plays a role in x chromosome inactivation. EMBO J 18, 28972907.O'Neill, LP., Randall, TE., Lavender, J., Spotswood, HT., Lee, JT. and Turner, BM.. (2003) Xlinked genes in female embryonic stem cells carry an epigenetic mark prior to the onset of x inactivation Hum Mol Genet 12, 17831790.O'Neill, LP., VerMilyea, MD. and Turner, BM.. (2006) Epigenetic characterization of the early embryo with a chromatin immunoprecipitation protocol applicable to small cell populations. Nat Genet 38, 835841.Odartchenko, N. and Pavillard, M.. (1970) Late dna replication in male mouse meiotic chromosomes Science 167, 11331134.Ogawa, Y. and Lee, JT.. (2002) Antisense regulation in x inactivation and autosomal imprinting Cytogenet Genome Res 99, 5965.Ogawa, Y. and Lee, JT.. (2003) Xite, xinactivation intergenic transcription elements that regulate the probability of choice Mol Cell 11, 731743.Ohhata, T., Hoki, Y., Sasaki, H. and Sado, T.. (2006) Tsixdeficient x chromosome does not undergo inactivation in the embryonic lineage in males: implications for tsixindependent silencing
269

of xist Cytogenet Genome Res 113, 345349.Ohhata, T., Tachibana, M., Tada, M., Tada, T., Sasaki, H., Shinkai, Y. and Sado, T.. (2004) Xinactivation is stably maintained in mouse embryos deficient for histone methyl transferase g9a Genesis 40, 151156.Ohno, S.. (1967) Sex chromosomes and sexlinked genes Monographs on endocrinology 1, .Ohno, S. and Hauschka, TS.. (1960) Allocycly of the xchromosome in tumors and normal tissues Cancer Res 20, 541545.Ohno, S., Becak, W. and Becak, ML.. (1964) Xautosome ratio and the behavior pattern of individual xchromosomes in placental mammals Chromosoma 15, 1430.Ohno, S., Kaplan, WD. and Kinosita, R.. (1959) Formation of the sex chromatin by a single xchromosome in liver cells of rattus norvegicus Exp Cell Res 18, 415418.Okamoto, I., Arnaud, D., Le Baccon, P., Otte, AP., Disteche, CM., Avner, P. and Heard, E.. (2005) Evidence for de novo imprinted xchromosome inactivation independent of meiotic inactivation in mice Nature 438, 369373.Okamoto, I., Otte, AP., Allis, CD., Reinberg, D. and Heard, E.. (2004) Epigenetic dynamics of imprinted x inactivation during early mouse development Science 303, 644649.Okamoto, I., Tan, S. and Takagi, N.. (2000) Xchromosome inactivation in xx androgenetic mouse embryos surviving implantation Development 127, 41374145.Okita, K., Ichisaka, T. and Yamanaka, S.. (2007) Generation of germlinecompetent induced pluripotent stem cells. Nature , .Osborne, CS., Chakalova, L., Brown, KE., Carter, D., Horton, A., Debrand, E., Goyenechea, B., Mitchell, JA., Lopes, S., Reik, W. and Fraser, P.. (2004) Active genes dynamically colocalize to shared sites of ongoing transcription Nat Genet 36, 10651071.Ouyang, Y., Salstrom, J., DiazPerez, S., Nahas, S., Matsuno, Y., Dawson, D., Teitell, MA., Horvath, S., Riggs, AD., Gatti, RA. and Marahrens, Y.. (2005) Inhibition of atm and/or atr disrupts gene silencing on the inactive x chromosome Biochem Biophys Res Commun 337, 875880.Pageau, GJ. and Lawrence, JB.. (2006) Brca1 foci in normal sphase nuclei are linked to interphase centromeres and replication of pericentric heterochromatin. J Cell Biol 175, 693701.Pageau, GJ., Hall, LL. and Lawrence, JB.. (2007) Brca1 does not paint the inactive x to localize xist rna but may contribute to broad changes in cancer that impact xist and xi heterochromatin J Cell Biochem 100, 835850.Panning, B., Dausman, J. and Jaenisch, R.. (1997) X chromosome inactivation is mediated by xist rna stabilization Cell 90, 907916.Papaioannou, VE. and West, JD.. (1981) Relationship between the parental origin of the x chromosomes, embryonic cell lineage and x chromosome expression in mice Genet Res 37, 183197.Parada, L. and Misteli, T.. (2002) Chromosome positioning in the interphase nucleus Trends Cell Biol 12, 425432.PARK, WW.. (1957) The occurrence of sex chromatin in early human and macaque embryos J Anat 91, 369373.
270

Penny, GD., Kay, GF., Sheardown, SA., Rastan, S. and Brockdorff, N.. (1996) Requirement for xist in x chromosome inactivation Nature 379, 131137.Percec, I., Plenge, RM., Nadeau, JH., Bartolomei, MS. and Willard, HF.. (2002) Autosomal dominant mutations affecting x inactivation choice in the mouse Science 296, 11361139.Percec, I., Thorvaldsen, JL., Plenge, RM., Krapp, CJ., Nadeau, JH., Willard, HF. and Bartolomei, MS.. (2003) An nethylnnitrosourea mutagenesis screen for epigenetic mutations in the mouse. Genetics 164, 14811494.Peters, AHFM., Mermoud, JE., O'Carroll, D., Pagani, M., Schweizer, D., Brockdorff, N. and Jenuwein, T.. (2002) Histone h3 lysine 9 methylation is an epigenetic imprint of facultative heterochromatin Nat Genet 30, 7780.Plath, K., Fang, J., MlynarczykEvans, SK., Cao, R., Worringer, KA., Wang, H., de la Cruz, CC., Otte, AP., Panning, B. and Zhang, Y.. (2003) Role of histone h3 lysine 27 methylation in x inactivation Science 300, 131135.Plath, K., Talbot, D., Hamer, KM., Otte, AP., Yang, TP., Jaenisch, R. and Panning, B.. (2004) Developmentally regulated alterations in polycomb repressive complex 1 proteins on the inactive x chromosome J Cell Biol 167, 10251035.Plenge, RM., Hendrich, BD., Schwartz, C., Arena, JF., Naumova, A., Sapienza, C., Winter, RM. and Willard, HF.. (1997) A promoter mutation in the xist gene in two unrelated families with skewed xchromosome inactivation. Nat Genet 17, 353356.Plenge, RM., Percec, I., Nadeau, JH. and Willard, HF.. (2000) Expressionbased assay of an xlinked gene to examine effects of the xcontrolling element (xce) locus Mamm Genome 11, 405408.Pokholok, DK., Harbison, CT., Levine, S., Cole, M., Hannett, NM., Lee, TI., Bell, GW., Walker, K., Rolfe, PA., Herbolsheimer, E., Zeitlinger, J. et al.. (2005) Genomewide map of nucleosome acetylation and methylation in yeast Cell 122, 517527.Popova, BC., Tada, T., Takagi, N., Brockdorff, N. and Nesterova, TB.. (2006) Attenuated spread of xinactivation in an x;autosome translocation Proc Natl Acad Sci U S A 103, 77067711.Price, NT., Jackson, VN. and Halestrap, AP.. (1998) Cloning and sequencing of four new mammalian monocarboxylate transporter (mct) homologues confirms the existence of a transporter family with an ancient past Biochem J 329 ( Pt 2), 321328.Priest, JH., Heady, JE. and Priest, RE.. (1967) Delayed onset of replication of human x chromosomes J Cell Biol 35, 483487.Prissette, M., ElMaarri, O., Arnaud, D., Walter, J. and Avner, P.. (2001) Methylation profiles of dxpas34 during the onset of xinactivation Hum Mol Genet 10, 3138.Pugacheva, EM., Tiwari, VK., Abdullaev, Z., Vostrov, AA., Flanagan, PT., Quitschke, WW., Loukinov, DI., Ohlsson, R. and Lobanenkov, VV.. (2005) Familial cases of point mutations in the xist promoter reveal a correlation between ctcf binding and preemptive choices of x chromosome inactivation Hum Mol Genet 14, 953965.Rasmussen, TP., Wutz, AP., Pehrson, JR. and Jaenisch, RR.. (2001) Expression of xist rna is sufficient to initiate macrochromatin body formation Chromosoma 110, 411420.
271

Rastan, S.. (1983) Nonrandom xchromosome inactivation in mouse xautosome translocation embryoslocation of the inactivation centre J Embryol Exp Morphol 78, 122.Rastan, S. and Robertson, EJ.. (1985) Xchromosome deletions in embryoderived (ek) cell lines associated with lack of xchromosome inactivation J Embryol Exp Morphol 90, 379388.Rattner, BP. and Meller, VH.. (2004) Drosophila malespecific lethal 2 protein controls sexspecific expression of the rox genes Genetics 166, 18251832.Ray, PF., Winston, RM. and Handyside, AH.. (1997) Xist expression from the maternal x chromosome in human male preimplantation embryos at the blastocyst stage Hum Mol Genet 6, 13231327.Reik, W. and Walter, J.. (2001) Genomic imprinting: parental influence on the genome Nat Rev Genet 2, 2132.Reizis, B. and Leder, P.. (1999) Expression of the mouse pret cell receptor alpha gene is controlled by an upstream region containing a transcriptional enhancer J Exp Med 189, 16691678.Resnick, JL., Bixler, LS., Cheng, L. and Donovan, PJ.. (1992) Longterm proliferation of mouse primordial germ cells in culture Nature 359, 550551.Rice, WR.. (1987) Genetic hitchhiking and the evolution of reduced genetic activity of the y sex chromosome Genetics 116, 161167.Rice, WR.. (1996) Sexually antagonistic male adaptation triggered by experimental arrest of female evolution Nature 381, 232234.Richards, S., Liu, Y., Bettencourt, BR., Hradecky, P., Letovsky, S., Nielsen, R., Thornton, K., Hubisz, MJ., Chen, R., Meisel, RP., Couronne, O. et al.. (2005) Comparative genome sequencing of drosophila pseudoobscura: chromosomal, gene, and ciselement evolution. Genome Res 15, 118.Richardson, AL., Wang, ZC., De Nicolo, A., Lu, X., Brown, M., Miron, A., Liao, X., Iglehart, JD., Livingston, DM. and Ganesan, S.. (2006) X chromosomal abnormalities in basallike human breast cancer. Cancer Cell 9, 121132.Rogers, RS., Inselman, A., Handel, MA. and Matunis, MJ.. (2004) Sumo modified proteins localize to the xy body of pachytene spermatocytes Chromosoma 113, 233243.Rogner, UC., Spyropoulos, DD., Le Novère, N., Changeux, JP. and Avner, P.. (2000) Control of neurulation by the nucleosome assembly protein1like 2. Nat Genet 25, 431435.Ropers, H.. (2006) Xlinked mental retardation: many genes for a complex disorder Curr Opin Genet Dev 16, 260269.Ropers, HH., Wolff, G. and Hitzeroth, HW.. (1978) Preferential x inactivation in human placenta membranes: is the paternal x inactive in early embryonic development of female mammals? Hum Genet 43, 265273.Ross, MT., Grafham, DV., Coffey, AJ., Scherer, S., McLay, K., Muzny, D., Platzer, M., Howell, GR., Burrows, C., Bird, CP., Frankish, A. et al.. (2005) The dna sequence of the human x chromosome Nature 434, 325337.Rougeulle, C. and Avner, P.. (1996) Cloning and characterization of a murine brain specific gene bpx and its human homologue lying within the xic candidate region. Hum Mol Genet 5, 4149.
272

Rougeulle, C., Chaumeil, J., Sarma, K., Allis, CD., Reinberg, D., Avner, P. and Heard, E.. (2004) Differential histone h3 lys9 and lys27 methylation profiles on the x chromosome Mol Cell Biol 24, 54755484.Rugarli, EI., Adler, DA., Borsani, G., Tsuchiya, K., Franco, B., Hauge, X., Disteche, C., Chapman, V. and Ballabio, A.. (1995) Different chromosomal localization of the clcn4 gene in mus spretus and c57bl/6j mice Nat Genet 10, 466471.Russell, LB.. (1983) Xautosome translocations in the mouse: their characterization and use as tools to investigate gene inactivation and gene action , .Russell, LB. and Bangham, JW.. (1961) Variegatedtype position effects in the mouse Genetics 46, 509525.Russell, LB., Bangham, J.. (1959) Genetics 44, 532.Sado, T., Fenner, MH., Tan, SS., Tam, P., Shioda, T. and Li, E.. (2000) X inactivation in the mouse embryo deficient for dnmt1: distinct effect of hypomethylation on imprinted and random x inactivation Dev Biol 225, 294303.Sado, T., Hoki, Y. and Sasaki, H.. (2005) Tsix silences xist through modification of chromatin structure Dev Cell 9, 159165.Sado, T., Hoki, Y. and Sasaki, H.. (2006) Tsix defective in splicing is competent to establish xist silencing Development 133, 49254931.Sado, T., Tada, T. and Takagi, N.. (1996) Mosaic methylation of xist gene before chromosome inactivation in undifferentiated female mouse embryonic stem and embryonic germ cells. Dev Dyn 205, 421434.Sado, T., Wang, Z., Sasaki, H. and Li, E.. (2001) Regulation of imprinted xchromosome inactivation in mice by tsix Development 128, 12751286.Savarese, F., Flahndorfer, K., Jaenisch, R., Busslinger, M. and Wutz, A.. (2006) Hematopoietic precursor cells transiently reestablish permissiveness for x inactivation Mol Cell Biol 26, 71677177.Schartl, M.. (2004) Sex chromosome evolution in nonmammalian vertebrates Curr Opin Genet Dev 14, 634641.Scheuermann, MO., Tajbakhsh, J., Kurz, A., Saracoglu, K., Eils, R. and Lichter, P.. (2004) Topology of genes and nontranscribed sequences in human interphase nuclei Exp Cell Res 301, 266279.Schimenti, J.. (2005) Synapsis or silence Nat Genet 37, 1113.Schmahl, J., Eicher, EM., Washburn, LL. and Capel, B.. (2000) Sry induces cell proliferation in the mouse gonad Development 127, 6573.Schmidt, M., Du Sart, D., Kalitsis, P., Fraser, N., Leversha, M., Voullaire, L., Foster, D., Davies, J., Hills, L. and Petrovic, V.. (1991) X chromosome inactivation in fibroblasts of mentally retarded female carriers of the fragile site xq27.3: application of the probe m27 beta to evaluate x inactivation status Am J Med Genet 38, 411415.Schoenherr, CJ., Levorse, JM. and Tilghman, SM.. (2003) Ctcf maintains differential methylation at the igf2/h19 locus Nat Genet 33, 6669.
273

Schubeler, D., MacAlpine, DM., Scalzo, D., Wirbelauer, C., Kooperberg, C., van Leeuwen, F., Gottschling, DE., O'Neill, LP., Turner, BM., Delrow, J., Bell, SP. et al.. (2004) The histone modification pattern of active genes revealed through genomewide chromatin analysis of a higher eukaryote Genes Dev 18, 12631271.Seki, Y., Hayashi, K., Itoh, K., Mizugaki, M., Saitou, M. and Matsui, Y.. (2005) Extensive and orderly reprogramming of genomewide chromatin modifications associated with specification and early development of germ cells in mice Dev Biol 278, 440458.Shao, CS. and Takagi, N.. (1991) Karyotypes and x chromosome inactivation in segregants of a murine xautosome translocation, t(x;4)37h Jpn J Genet 66, 433447.Sharman, GB.. (1971) Late dna replication in the paternally derived x chromosome of female kangaroos Nature 230, 231232.Sharp, AJ., Spotswood, HT., Robinson, DO., Turner, BM. and Jacobs, PA.. (2002) Molecular and cytogenetic analysis of the spreading of x inactivation in x;autosome translocations Hum Mol Genet 11, 31453156.Sheardown, SA., Duthie, SM., Johnston, CM., Newall, AE., Formstone, EJ., Arkell, RM., Nesterova, TB., Alghisi, GC., Rastan, S. and Brockdorff, N.. (1997a) Stabilization of xist rna mediates initiation of x chromosome inactivation Cell 91, 99107.Sheardown, SA., Newall, AE., Norris, DP., Rastan, S. and Brockdorff, N.. (1997b) Regulatory elements in the minimal promoter region of the mouse xist gene Gene 203, 159168.Shetty, S., Griffin, DK. and Graves, JA.. (1999) Comparative painting reveals strong chromosome homology over 80 million years of bird evolution Chromosome Res 7, 289295.Shi, Y., Lee, JS. and Galvin, KM.. (1997) Everything you have ever wanted to know about yin yang 1..... Biochim Biophys Acta 1332, F4966.Shibata, S. and Lee, JT.. (2003) Characterization and quantitation of differential tsix transcripts: implications for tsix function Hum Mol Genet 12, 125136.Shibata, S. and Lee, JT.. (2004) Tsix transcription versus rnabased mechanisms in xist repression and epigenetic choice. Curr Biol 14, 17471754.Silva, J., Mak, W., Zvetkova, I., Appanah, R., Nesterova, TB., Webster, Z., Peters, AHFM., Jenuwein, T., Otte, AP. and Brockdorff, N.. (2003) Establishment of histone h3 methylation on the inactive x chromosome requires transient recruitment of eedenx1 polycomb group complexes Dev Cell 4, 481495.Silver, DP., Dimitrov, SD., Feunteun, J., Gelman, R., Drapkin, R., Lu, SD., Shestakova, E., Velmurugan, S., Denunzio, N., Dragomir, S., Mar, J. et al.. (2007) Further evidence for brca1 communication with the inactive x chromosome Cell 128, 9911002.Simmler, MC., Cattanach, BM., Rasberry, C., Rougeulle, C. and Avner, P.. (1993) Mapping the murine xce locus with (ca)n repeats Mamm Genome 4, 523530.Simmler, MC., Cunningham, DB., Clerc, P., Vermat, T., Caudron, B., Cruaud, C., Pawlak, A., Szpirer, C., Weissenbach, J., Claverie, JM. and Avner, P.. (1996) A 94 kb genomic sequence 3' to the murine xist gene reveals an at rich region containing a new testis specific gene tsx Hum Mol
274

Genet 5, 17131726.Simmler, MC., Heard, E., Rougeulle, C., Cruaud, C., Weissenbach, J. and Avner, P.. (1997) Localization and expression analysis of a novel conserved brain expressed transcript, brx/brx, lying within the xic/xic candidate region. Mamm Genome 8, 760766.Simpson, AJG., Caballero, OL., Jungbluth, A., Chen, Y. and Old, LJ.. (2005) Cancer/testis antigens, gametogenesis and cancer. Nat Rev Cancer 5, 615625.SingerSam, J., Chapman, V., LeBon, JM. and Riggs, AD.. (1992) Parental imprinting studied by allelespecific primer extension after pcr: paternal x chromosomelinked genes are transcribed prior to preferential paternal x chromosome inactivation Proc Natl Acad Sci U S A 89, 1046910473.Singh, ND., Davis, JC. and Petrov, DA.. (2005) Xlinked genes evolve higher codon bias in drosophila and caenorhabditis Genetics 171, 145155.Sirchia, SM., Ramoscelli, L., Grati, FR., Barbera, F., Coradini, D., Rossella, F., Porta, G., Lesma, E., Ruggeri, A., Radice, P., Simoni, G. et al.. (2005) Loss of the inactive x chromosome and replication of the active x in brca1defective and wildtype breast cancer cells. Cancer Res 65, 21392146.Sleutels, F., Zwart, R. and Barlow, DP.. (2002) The noncoding air rna is required for silencing autosomal imprinted genes Nature 415, 810813.Smith, KP., Byron, M., Clemson, CM. and Lawrence, JB.. (2004) Ubiquitinated proteins including uh2a on the human and mouse inactive x chromosome: enrichment in gene rich bands Chromosoma 113, 324335.Solari, AJ.. (1974) The behavior of the xy pair in mammals Int Rev Cytol 38, 273317.Stadnick, MP., Pieracci, FM., Cranston, MJ., Taksel, E., Thorvaldsen, JL. and Bartolomei, MS.. (1999) Role of a 461bp grich repetitive element in h19 transgene imprinting Dev Genes Evol 209, 239248.Stavropoulos, N., Lu, N. and Lee, JT.. (2001) A functional role for tsix transcription in blocking xist rna accumulation but not in xchromosome choice Proc Natl Acad Sci U S A 98, 1023210237.Stavropoulos, N., Rowntree, RK. and Lee, JT.. (2005) Identification of developmentally specific enhancers for tsix in the regulation of x chromosome inactivation Mol Cell Biol 25, 27572769.Stewart, CL., Gadi, I. and Bhatt, H.. (1994) Stem cells from primordial germ cells can reenter the germ line Dev Biol 161, 626628.Strahl, BD. and Allis, CD.. (2000) The language of covalent histone modifications Nature 403, 4145.Straub, T., Gilfillan, GD., Maier, VK. and Becker, PB.. (2005) The drosophila msl complex activates the transcription of target genes Genes Dev 19, 22842288.Sugawara, O., Takagi, N. and Sasaki, M.. (1983) Allocyclic early replicating x chromosome in mice: genetic inactivity and shift into a late replicator in early embrogenesis Chromosoma 88, 133138.Sugawara, O., Takagi, N. and Sasaki, M.. (1985) Correlation between xchromosome inactivation and cell differentiation in female preimplantation mouse embryos Cytogenet Cell Genet 39, 210219.
275

Sugimoto, M., Tan, SS. and Takagi, N.. (2000) X chromosome inactivation revealed by the xlinked lacz transgene activity in periimplantation mouse embryos Int J Dev Biol 44, 177182.Sun, BK., Deaton, AM. and Lee, JT.. (2006) A transient heterochromatic state in xist preempts x inactivation choice without rna stabilization Mol Cell 21, 617628.Szabo, PE., Tang, SE., Silva, FJ., Tsark, WMK. and Mann, JR.. (2004) Role of ctcf binding sites in the igf2/h19 imprinting control region Mol Cell Biol 24, 47914800.Tada, M., Takahama, Y., Abe, K., Nakatsuji, N. and Tada, T.. (2001) Nuclear reprogramming of somatic cells by in vitro hybridization with es cells. Curr Biol 11, 15531558.Tada, T., Obata, Y., Tada, M., Goto, Y., Nakatsuji, N., Tan, S., Kono, T. and Takagi, N.. (2000) Imprint switching for nonrandom xchromosome inactivation during mouse oocyte growth Development 127, 31013105.Tada, T., Tada, M., Hilton, K., Barton, SC., Sado, T., Takagi, N. and Surani, MA.. (1998) Epigenotype switching of imprintable loci in embryonic germ cells Dev Genes Evol 207, 551561.Takagi, N. and Sasaki, M.. (1975) Preferential inactivation of the paternally derived x chromosome in the extraembryonic membranes of the mouse Nature 256, 640642.Takagi, N., Sugawara, O. and Sasaki, M.. (1982) Regional and temporal changes in the pattern of xchromosome replication during the early postimplantation development of the female mouse Chromosoma 85, 275286.Takahashi, K. and Yamanaka, S.. (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663676.Tam, PP., Williams, EA. and Tan, SS.. (1994a) Expression of an xlinked hmglacz transgene in mouse embryos: implication of chromosomal imprinting and lineagespecific xchromosome activity Dev Genet 15, 491503.Tam, PP., Zhou, SX. and Tan, SS.. (1994b) Xchromosome activity of the mouse primordial germ cells revealed by the expression of an xlinked lacz transgene Development 120, 29252932.Tanaka, S., Kunath, T., Hadjantonakis, AK., Nagy, A. and Rossant, J.. (1998) Promotion of trophoblast stem cell proliferation by fgf4 Science 282, 20722075.Taylor, JH.. (1968) Rates of chain growth and units of replication in dna of mammalian chromosomes J Mol Biol 31, 579594.Taylor, JH. and Miner, P.. (1968) Units of dna replication in mammalian chromosomes Cancer Res 28, 18101814.Terryn, N. and Rouze, P.. (2000) The sense of naturally transcribed antisense rnas in plants Trends Plant Sci 5, 394396.Thakur, N., Tiwari, VK., Thomassin, H., Pandey, RR., Kanduri, M., Göndör, A., Grange, T., Ohlsson, R. and Kanduri, C.. (2004) An antisense rna regulates the bidirectional silencing property of the kcnq1 imprinting control region. Mol Cell Biol 24, 78557862.Therman, E., Sarto, GE. and Buchler, DA.. (1983) The structure and origin of giant nuclei in human cancer cells Cancer Genet Cytogenet 9, 918.Thomas, MJ. and Seto, E.. (1999) Unlocking the mechanisms of transcription factor yy1: are
276

chromatin modifying enzymes the key? Gene 236, 197208.Thorvaldsen, JL., Duran, KL. and Bartolomei, MS.. (1998) Deletion of the h19 differentially methylated domain results in loss of imprinted expression of h19 and igf2 Genes Dev 12, 36933702.Thurman, RE., Day, N., Noble, WS. and Stamatoyannopoulos, JA.. (2007) Identification of higherorder functional domains in the human encode regions. Genome Res 17, 917927.Tremblay, KD., Duran, KL. and Bartolomei, MS.. (1997) A 5' 2kilobasepair region of the imprinted mouse h19 gene exhibits exclusive paternal methylation throughout development Mol Cell Biol 17, 43224329.Tsuchiya, KD., Greally, JM., Yi, Y., Noel, KP., Truong, J. and Disteche, CM.. (2004) Comparative sequence and xinactivation analyses of a domain of escape in human xp11.2 and the conserved segment in mouse Genome Res 14, 12751284.Turner, BM.. (1993) Decoding the nucleosome Cell 75, 58.Turner, JMA.. (2005) Sex chromosomes make their mark Chromosoma 114, 300306.Turner, JMA.. (2007) Meiotic sex chromosome inactivation Development 134, 18231831.Turner, JMA., Aprelikova, O., Xu, X., Wang, R., Kim, S., Chandramouli, GVR., Barrett, JC., Burgoyne, PS. and Deng, C.. (2004) Brca1, histone h2ax phosphorylation, and male meiotic sex chromosome inactivation. Curr Biol 14, 21352142.Turner, JMA., Mahadevaiah, SK., Elliott, DJ., Garchon, H., Pehrson, JR., Jaenisch, R. and Burgoyne, PS.. (2002) Meiotic sex chromosome inactivation in male mice with targeted disruptions of xist. J Cell Sci 115, 40974105.Turner, JMA., Mahadevaiah, SK., Ellis, PJI., Mitchell, MJ. and Burgoyne, PS.. (2006) Pachytene asynapsis drives meiotic sex chromosome inactivation and leads to substantial postmeiotic repression in spermatids Dev Cell 10, 521529.Umlauf, D., Goto, Y., Cao, R., Cerqueira, F., Wagschal, A., Zhang, Y. and Feil, R.. (2004) Imprinting along the kcnq1 domain on mouse chromosome 7 involves repressive histone methylation and recruitment of polycomb group complexes. Nat Genet 36, 12961300.Vallender, EJ. and Lahn, BT.. (2004) How mammalian sex chromosomes acquired their peculiar gene content Bioessays 26, 159169.van der Heijden, GW., Dieker, JW., Derijck, AAHA., Muller, S., Berden, JHM., Braat, DDM., van der Vlag, J. and de Boer, P.. (2005) Asymmetry in histone h3 variants and lysine methylation between paternal and maternal chromatin of the early mouse zygote Mech Dev 122, 10081022.Verona, RI., Mann, MRW. and Bartolomei, MS.. (2003) Genomic imprinting: intricacies of epigenetic regulation in clusters Annu Rev Cell Dev Biol 19, 237259.Vigneau, S., Augui, S., Navarro, P., Avner, P. and Clerc, P.. (2006) An essential role for the dxpas34 tandem repeat and tsix transcription in the counting process of x chromosome inactivation Proc Natl Acad Sci U S A 103, 73907395.Vigodner, M. and Morris, PL.. (2005) Testicular expression of small ubiquitinrelated modifier1 (sumo1) supports multiple roles in spermatogenesis: silencing of sex chromosomes in
277

spermatocytes, spermatid microtubule nucleation, and nuclear reshaping Dev Biol 282, 480492.VincentSalomon, A., GanemElbaz, C., Manié, E., Raynal, V., SastreGarau, X., StoppaLyonnet, D., Stern, M. and Heard, E.. (2007) X inactivespecific transcript rna coating and genetic instability of the x chromosome in brca1 breast tumors. Cancer Res 67, 51345140.Volpi, EV., Chevret, E., Jones, T., Vatcheva, R., Williamson, J., Beck, S., Campbell, RD., Goldsworthy, M., Powis, SH., Ragoussis, J., Trowsdale, J. et al.. (2000) Largescale chromatin organization of the major histocompatibility complex and other regions of human chromosome 6 and its response to interferon in interphase nuclei J Cell Sci 113 ( Pt 9), 15651576.Wagner, EG. and Simons, RW.. (1994) Antisense rna control in bacteria, phages, and plasmids Annu Rev Microbiol 48, 713742.Wakefield, MJ., Keohane, AM., Turner, BM. and Graves, JA.. (1997) Histone underacetylation is an ancient component of mammalian x chromosome inactivation Proc Natl Acad Sci U S A 94, 96659668.Wang, J., Mager, J., Chen, Y., Schneider, E., Cross, JC., Nagy, A. and Magnuson, T.. (2001a) Imprinted x inactivation maintained by a mouse polycomb group gene Nat Genet 28, 371375.Wang, PJ., McCarrey, JR., Yang, F. and Page, DC.. (2001b) An abundance of xlinked genes expressed in spermatogonia Nat Genet 27, 422426.Wang, PJ., Page, DC. and McCarrey, JR.. (2005) Differential expression of sexlinked and autosomal germcellspecific genes during spermatogenesis in the mouse Hum Mol Genet 14, 29112918.Wang, RA., Nakane, PK. and Koji, T.. (1998) Autonomous cell death of mouse male germ cells during fetal and postnatal period Biol Reprod 58, 12501256.Warburton, PE., Giordano, J., Cheung, F., Gelfand, Y. and Benson, G.. (2004) Inverted repeat structure of the human genome: the xchromosome contains a preponderance of large, highly homologous inverted repeats that contain testes genes. Genome Res 14, 18611869.Watson, JM., Spencer, JA., Riggs, AD. and Graves, JA.. (1990) The x chromosome of monotremes shares a highly conserved region with the eutherian and marsupial x chromosomes despite the absence of x chromosome inactivation Proc Natl Acad Sci U S A 87, 71257129.Watson, JM., Spencer, JA., Riggs, AD. and Graves, JA.. (1991) Sex chromosome evolution: platypus gene mapping suggests that part of the human x chromosome was originally autosomal Proc Natl Acad Sci U S A 88, 1125611260.Webb, S., de Vries, TJ. and Kaufman, MH.. (1992) The differential staining pattern of the x chromosome in the embryonic and extraembryonic tissues of postimplantation homozygous tetraploid mouse embryos. Genet Res 59, 205214.Welshons, WJ. and Russell, LB.. (1959) The ychromosome as the bearer of male determining factors in the mouse Proc Natl Acad Sci U S A 45, 560566.Wernig, M., Meissner, A., Foreman, R., Brambrink, T., Ku, M., Hochedlinger, K., Bernstein, BE. and Jaenisch, R.. (2007) In vitro reprogramming of fibroblasts into a pluripotent escelllike state. Nature , .
278

West, JD. and Chapman, VM.. (1978) Variation for x chromosome expression in mice detected by electrophoresis of phosphoglycerate kinase Genet Res 32, 91102.West, JD., Frels, WI., Chapman, VM. and Papaioannou, VE.. (1977) Preferential expression of the maternally derived x chromosome in the mouse yolk sac Cell 12, 873882.White, WM., Willard, HF., Van Dyke, DL. and Wolff, DJ.. (1998) The spreading of x inactivation into autosomal material of an x;autosome translocation: evidence for a difference between autosomal and xchromosomal dna Am J Hum Genet 63, 2028.Willard, HF.. (1995) The sex chromosomes and x chromosome inactivation The Metabolic and Molecular Bases of Inherited Disease , .Willard, HF.. (1996) X chromosome inactivation, xist, and pursuit of the xinactivation center Cell 86, 57.Williams, RRE.. (2003) Transcription and the territory: the ins and outs of gene positioning Trends Genet 19, 298302.Willoughby, DA., Vilalta, A. and Oshima, RG.. (2000) An alu element from the k18 gene confers positionindependent expression in transgenic mice. J Biol Chem 275, 759768.Wood, AJ. and Oakey, RJ.. (2006) Genomic imprinting in mammals: emerging themes and established theories PLoS Genet 2, e147.Wu, J., Wang, S., Potter, D., Liu, JC., Smith, LT., Wu, Y., Huang, TH. and Plass, C.. (2007) Diverse histone modifications on histone 3 lysine 9 and their relation to dna methylation in specifying gene silencing. BMC Genomics 8, 131.Wutz, A. and Jaenisch, R.. (2000) A shift from reversible to irreversible x inactivation is triggered during es cell differentiation Mol Cell 5, 695705.Wutz, A., Rasmussen, TP. and Jaenisch, R.. (2002) Chromosomal silencing and localization are mediated by different domains of xist rna Nat Genet 30, 167174.Xiao, C., Sharp, JA., Kawahara, M., Davalos, AR., Difilippantonio, MJ., Hu, Y., Li, W., Cao, L., Buetow, K., Ried, T., Chadwick, BP. et al.. (2007) The xist noncoding rna functions independently of brca1 in x inactivation Cell 128, 977989.Xu, N., Tsai, C. and Lee, JT.. (2006) Transient homologous chromosome pairing marks the onset of x inactivation Science 311, 11491152.Xue, F., Tian, XC., Du, F., Kubota, C., Taneja, M., Dinnyes, A., Dai, Y., Levine, H., Pereira, LV. and Yang, X.. (2002) Aberrant patterns of x chromosome inactivation in bovine clones. Nat Genet 31, 216220.Yamasaki, Y., Kayashima, T., Soejima, H., Kinoshita, A., Yoshiura, K., Matsumoto, N., Ohta, T., Urano, T., Masuzaki, H., Ishimaru, T., Mukai, T. et al.. (2005) Neuronspecific relaxation of igf2r imprinting is associated with neuronspecific histone modifications and lack of its antisense transcript air Hum Mol Genet 14, 25112520.Yang, PK. and Kuroda, MI.. (2007) Noncoding rnas and intranuclear positioning in monoallelic gene expression Cell 128, 777786.Yoshida, M., Horinouchi, S. and Beppu, T.. (1995) Trichostatin a and trapoxin: novel chemical
279

probes for the role of histone acetylation in chromatin structure and function. Bioessays 17, 423430.Zechner, U., Wilda, M., KehrerSawatzki, H., Vogel, W., Fundele, R. and Hameister, H.. (2001) A high density of xlinked genes for general cognitive ability: a runaway process shaping human evolution? Trends Genet 17, 697701.Zeng, S. and Yankowitz, J.. (2003) Xinactivation patterns in human embryonic and extraembryonic tissues. Placenta 24, 270275.Zhang, L., Huynh, KD. and Lee, JT.. (2007) Perinucleolar targeting of the inactive x during s phase: evidence for a role in the maintenance of silencing. Cell 129, 693706.Zhao, Z., Tavoosidana, G., Sjolinder, M., Gondor, A., Mariano, P., Wang, S., Kanduri, C., Lezcano, M., Sandhu, KS., Singh, U., Pant, V. et al.. (2006) Circular chromosome conformation capture (4c) uncovers extensive networks of epigenetically regulated intra and interchromosomal interactions Nat Genet 38, 13411347.Zirbel, RM., Mathieu, UR., Kurz, A., Cremer, T. and Lichter, P.. (1993) Evidence for a nuclear compartment of transcription and splicing located at chromosome domain boundaries Chromosome Res 1, 93106.Zlatanova, J. and van Holde, K.. (1998) Linker histones versus hmg1/2: a struggle for dominance? Bioessays 20, 584588.Zuccotti, M., Boiani, M., Ponce, R., Guizzardi, S., Scandroglio, R., Garagna, S. and Redi, CA.. (2002) Mouse xist expression begins at zygotic genome activation and is timed by a zygotic clock Mol Reprod Dev 61, 1420.
280















![Assemblages diagnostiques du syndrome X fragile: gène, symptômes et biosocialité [Diagnostic assemblages of Fragile X Syndrome: genes, symptoms and biosociality]](https://static.fdokumen.com/doc/165x107/6315aa0a3ed465f0570bab1c/assemblages-diagnostiques-du-syndrome-x-fragile-gene-symptomes-et-biosocialite.jpg)




