
ERCC6L2 Mutations Link a Distinct Bone-Marrow-Failure Syndrome to DNA Repair and Mitochondrial...
11
ARTICLE ERCC6L2 Mutations Link a Distinct Bone-Marrow-Failure Syndrome to DNA Repair and Mitochondrial Function Hemanth Tummala, 1,6 Michael Kirwan, 1,6 Amanda J. Walne, 1 Upal Hossain, 1,5 Nicholas Jackson, 2 Corinne Pondarre, 3 Vincent Plagnol, 4 Tom Vulliamy, 1,7, * and Inderjeet Dokal 1,5,7 Exome sequencing was performed in three index cases with bone marrow failure and neurological dysfunction and whose parents are first-degree cousins. Homozygous truncating mutations were identified in ERCC6L2 in two of the individuals. Both of these mutations affect the subcellular localization and stability of ERCC6L2. We show here that knockdown of ERCC6L2 in human A549 cells sig- nificantly reduced their viability upon exposure to the DNA-damaging agents mitomycin C and Irofulven, but not etoposide and camptothecin, suggesting a role in nucleotide excision repair. ERCC6L2-knockdown cells also displayed H2AX phosphorylation, which significantly increased upon genotoxic stress, suggesting an early DNA-damage response. Intriguingly, ERCC6L2 was seen to translocate to the mitochondria and the nucleus in response to DNA damage, and ERCC6L2 knockdown induced intracellular reactive oxygen species (ROS). Treatment with the ROS scavenger N-acetyl cysteine attenuated the Irofulven-induced cytotoxicity in ERCC6L2-knock- down cells and abolished ERCCGL2 traffic to the mitochondria and nucleus in response to this DNA-damaging agent. Collectively, these observations identify a distinct bone-marrow-failure syndrome due to mutations in ERCC6L2, a gene implicated in DNA repair and mito- chondrial function. Introduction Bone-marrow-failure syndromes are a heterogeneous group of life-threatening disorders characterized by the inability of the bone marrow to make an adequate number of mature blood cells. 1,2 They can be variable in their severity and can affect either one or all of the hemato- poietic lineages. In some cases, individuals can be classified into recognized syndromes, such as Fanconi anemia (FA [MIM 227650]) 3 and dyskeratosis congenita (DC [MIM 305000]). 4 In these diseases, the categorization of affected individ- uals has been facilitated by the recognition of charac- teristic features, such as the diagnostic mucocutaneous symptoms of DC (abnormal skin pigmentation, nail dys- trophy, and leukoplakia). Alternatively, specific genetic and/or functional defects can be used in diagnosis, exem- plified by the characteristically increased chromosomal breakage observed in FA individuals. However, there are also cases where the bone marrow failure is associated with one or more extrahematopoietic abnormalities but does not fit into a recognized syndrome and the underly- ing genetic and functional basis is thus unknown. The recent availability of next-generation sequencing technology is making it possible to elucidate the genetic basis and pathophysiology of uncharacterized human diseases. From our repository of bone-marrow-failure cases, we chose three genetically uncharacterized index cases in whom to perform exome sequencing with the aim of iden- tifying variants in a common gene. They had trilineage (erythroid, myeloid, and megakaryocytic) bone marrow failure and came from consanguineous families; all three also had developmental delay characterized by learning disability, and two out of the three cases also had micro- cephaly (Table 1). We hypothesized that this approach would enrich for homozygous disease-causing mutations. Material and Methods Exome Sequencing Peripheral-blood samples were obtained with written consent under the approval of our local research ethics committee (London – City and East). DNA extracted from these samples was submitted to the Beijing Genomics Institute for exome sequencing. Ten micrograms of genomic DNA was supplied and, after passing quality control, was subjected to Agilent SureSelect library preparation and exome enrichment before being sequenced on the Illumina GAII system. Sequencing data were processed through the Illumina pipeline. Variants were called with the Genome Analysis Toolkit (GATK) v.2.7.4. All samples were called jointly with an additional data set of 1,005 locally sequenced exomes with unrelated conditions (University College London Exomes [UCL-ex] Consortium) after BAM file reduction as implemented by GATK using default options. We used the Illumina TruSeq target region (5 200 bp on each side) for variant calling. We followed the GATK best practices and implemented variant recalibration with separate models for SNPs and indels. We excluded read depth from our recalibration model owing to the large read-depth variability generated by the heterogeneous 1 Blizard Institute, Barts and The London School of Medicine and Dentistry, Queen Mary University of London, London E1 2AT, UK; 2 Department of Haematology, University Hospital, Coventry CV2 2DX, UK; 3 Institute of Pediatric Hematology and Oncology, Lyon I University, Lyon 69008, France; 4 University College London Genetics Institute, London WC1E 6BT, UK; 5 Barts Health NHS Trust, London E1 1BB, UK 6 These authors contributed equally to this work 7 These authors contributed equally to this work and are co-senior authors *Correspondence: [email protected] http://dx.doi.org/10.1016/j.ajhg.2014.01.007. Ó2014 by The American Society of Human Genetics. All rights reserved. 246 The American Journal of Human Genetics 94, 246–256, February 6, 2014
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of ERCC6L2 Mutations Link a Distinct Bone-Marrow-Failure Syndrome to DNA Repair and Mitochondrial...

ARTICLE
ERCC6L2 Mutations Link a DistinctBone-Marrow-Failure Syndrometo DNA Repair and Mitochondrial Function
Hemanth Tummala,1,6 Michael Kirwan,1,6 Amanda J. Walne,1 Upal Hossain,1,5 Nicholas Jackson,2
Corinne Pondarre,3 Vincent Plagnol,4 Tom Vulliamy,1,7,* and Inderjeet Dokal1,5,7
Exome sequencing was performed in three index cases with bone marrow failure and neurological dysfunction and whose parents are
first-degree cousins. Homozygous truncating mutations were identified in ERCC6L2 in two of the individuals. Both of these mutations
affect the subcellular localization and stability of ERCC6L2. We show here that knockdown of ERCC6L2 in human A549 cells sig-
nificantly reduced their viability upon exposure to the DNA-damaging agents mitomycin C and Irofulven, but not etoposide and
camptothecin, suggesting a role in nucleotide excision repair. ERCC6L2-knockdown cells also displayed H2AX phosphorylation, which
significantly increased upon genotoxic stress, suggesting an early DNA-damage response. Intriguingly, ERCC6L2 was seen to translocate
to the mitochondria and the nucleus in response to DNA damage, and ERCC6L2 knockdown induced intracellular reactive oxygen
species (ROS). Treatment with the ROS scavenger N-acetyl cysteine attenuated the Irofulven-induced cytotoxicity in ERCC6L2-knock-
down cells and abolished ERCCGL2 traffic to themitochondria and nucleus in response to this DNA-damaging agent. Collectively, these
observations identify a distinct bone-marrow-failure syndrome due tomutations in ERCC6L2, a gene implicated in DNA repair andmito-
chondrial function.
Introduction
Bone-marrow-failure syndromes are a heterogeneous
group of life-threatening disorders characterized by the
inability of the bone marrow to make an adequate number
of mature blood cells.1,2 They can be variable in their
severity and can affect either one or all of the hemato-
poietic lineages. In some cases, individuals can be classified
into recognized syndromes, such as Fanconi anemia (FA
[MIM 227650])3 and dyskeratosis congenita (DC [MIM
305000]).4
In these diseases, the categorization of affected individ-
uals has been facilitated by the recognition of charac-
teristic features, such as the diagnostic mucocutaneous
symptoms of DC (abnormal skin pigmentation, nail dys-
trophy, and leukoplakia). Alternatively, specific genetic
and/or functional defects can be used in diagnosis, exem-
plified by the characteristically increased chromosomal
breakage observed in FA individuals. However, there are
also cases where the bone marrow failure is associated
with one or more extrahematopoietic abnormalities but
does not fit into a recognized syndrome and the underly-
ing genetic and functional basis is thus unknown.
The recent availability of next-generation sequencing
technology is making it possible to elucidate the genetic
basis and pathophysiology of uncharacterized human
diseases. From our repository of bone-marrow-failure cases,
we chose three genetically uncharacterized index cases in
whom to perform exome sequencing with the aim of iden-
1Blizard Institute, Barts and The London School of Medicine and Dentistry,
Haematology, University Hospital, Coventry CV2 2DX, UK; 3Institute of Ped4University College London Genetics Institute, London WC1E 6BT, UK; 5Bart6These authors contributed equally to this work7These authors contributed equally to this work and are co-senior authors
*Correspondence: [email protected]
http://dx.doi.org/10.1016/j.ajhg.2014.01.007. �2014 by The American Societ
246 The American Journal of Human Genetics 94, 246–256, February
tifying variants in a common gene. They had trilineage
(erythroid, myeloid, and megakaryocytic) bone marrow
failure and came from consanguineous families; all three
also had developmental delay characterized by learning
disability, and two out of the three cases also had micro-
cephaly (Table 1). We hypothesized that this approach
would enrich for homozygous disease-causing mutations.
Material and Methods
Exome SequencingPeripheral-blood samples were obtained with written consent
under the approval of our local research ethics committee
(London – City and East). DNA extracted from these samples
was submitted to the Beijing Genomics Institute for exome
sequencing. Ten micrograms of genomic DNA was supplied and,
after passing quality control, was subjected to Agilent SureSelect
library preparation and exome enrichment before being
sequenced on the Illumina GAII system. Sequencing data were
processed through the Illumina pipeline. Variants were called
with the Genome Analysis Toolkit (GATK) v.2.7.4. All samples
were called jointly with an additional data set of 1,005 locally
sequenced exomes with unrelated conditions (University College
London Exomes [UCL-ex] Consortium) after BAM file reduction
as implemented by GATK using default options. We used the
Illumina TruSeq target region (5 200 bp on each side) for variant
calling. We followed the GATK best practices and implemented
variant recalibration with separate models for SNPs and indels.
We excluded read depth from our recalibration model owing to
the large read-depth variability generated by the heterogeneous
Queen Mary University of London, London E1 2AT, UK; 2Department of
iatric Hematology and Oncology, Lyon I University, Lyon 69008, France;
s Health NHS Trust, London E1 1BB, UK
y of Human Genetics. All rights reserved.
6, 2014
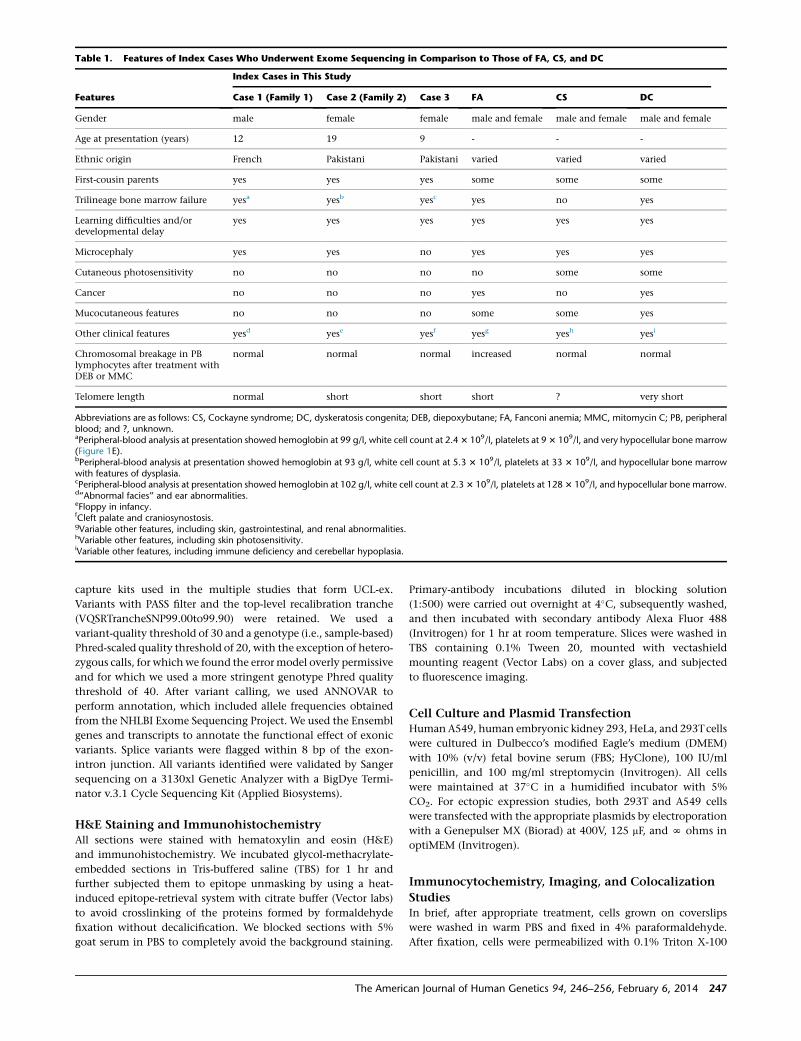
Table 1. Features of Index Cases Who Underwent Exome Sequencing in Comparison to Those of FA, CS, and DC
Features
Index Cases in This Study
Case 1 (Family 1) Case 2 (Family 2) Case 3 FA CS DC
Gender male female female male and female male and female male and female
Age at presentation (years) 12 19 9 - - -
Ethnic origin French Pakistani Pakistani varied varied varied
First-cousin parents yes yes yes some some some
Trilineage bone marrow failure yesa yesb yesc yes no yes
Learning difficulties and/ordevelopmental delay
yes yes yes yes yes yes
Microcephaly yes yes no yes yes yes
Cutaneous photosensitivity no no no no some some
Cancer no no no yes no yes
Mucocutaneous features no no no some some yes
Other clinical features yesd yese yesf yesg yesh yesi
Chromosomal breakage in PBlymphocytes after treatment withDEB or MMC
normal normal normal increased normal normal
Telomere length normal short short short ? very short
Abbreviations are as follows: CS, Cockayne syndrome; DC, dyskeratosis congenita; DEB, diepoxybutane; FA, Fanconi anemia; MMC, mitomycin C; PB, peripheralblood; and ?, unknown.aPeripheral-blood analysis at presentation showed hemoglobin at 99 g/l, white cell count at 2.43 109/l, platelets at 93 109/l, and very hypocellular bone marrow(Figure 1E).bPeripheral-blood analysis at presentation showed hemoglobin at 93 g/l, white cell count at 5.3 3 109/l, platelets at 33 3 109/l, and hypocellular bone marrowwith features of dysplasia.cPeripheral-blood analysis at presentation showed hemoglobin at 102 g/l, white cell count at 2.33 109/l, platelets at 1283 109/l, and hypocellular bone marrow.d‘‘Abnormal facies’’ and ear abnormalities.eFloppy in infancy.fCleft palate and craniosynostosis.gVariable other features, including skin, gastrointestinal, and renal abnormalities.hVariable other features, including skin photosensitivity.iVariable other features, including immune deficiency and cerebellar hypoplasia.
capture kits used in the multiple studies that form UCL-ex.
Variants with PASS filter and the top-level recalibration tranche
(VQSRTrancheSNP99.00to99.90) were retained. We used a
variant-quality threshold of 30 and a genotype (i.e., sample-based)
Phred-scaled quality threshold of 20, with the exception of hetero-
zygous calls, for whichwe found the errormodel overly permissive
and for which we used a more stringent genotype Phred quality
threshold of 40. After variant calling, we used ANNOVAR to
perform annotation, which included allele frequencies obtained
from the NHLBI Exome Sequencing Project. We used the Ensembl
genes and transcripts to annotate the functional effect of exonic
variants. Splice variants were flagged within 8 bp of the exon-
intron junction. All variants identified were validated by Sanger
sequencing on a 3130xl Genetic Analyzer with a BigDye Termi-
nator v.3.1 Cycle Sequencing Kit (Applied Biosystems).
H&E Staining and ImmunohistochemistryAll sections were stained with hematoxylin and eosin (H&E)
and immunohistochemistry. We incubated glycol-methacrylate-
embedded sections in Tris-buffered saline (TBS) for 1 hr and
further subjected them to epitope unmasking by using a heat-
induced epitope-retrieval system with citrate buffer (Vector labs)
to avoid crosslinking of the proteins formed by formaldehyde
fixation without decalicification. We blocked sections with 5%
goat serum in PBS to completely avoid the background staining.
The Americ
Primary-antibody incubations diluted in blocking solution
(1:500) were carried out overnight at 4�C, subsequently washed,
and then incubated with secondary antibody Alexa Fluor 488
(Invitrogen) for 1 hr at room temperature. Slices were washed in
TBS containing 0.1% Tween 20, mounted with vectashield
mounting reagent (Vector Labs) on a cover glass, and subjected
to fluorescence imaging.
Cell Culture and Plasmid TransfectionHumanA549, human embryonic kidney 293, HeLa, and 293Tcells
were cultured in Dulbecco’s modified Eagle’s medium (DMEM)
with 10% (v/v) fetal bovine serum (FBS; HyClone), 100 IU/ml
penicillin, and 100 mg/ml streptomycin (Invitrogen). All cells
were maintained at 37�C in a humidified incubator with 5%
CO2. For ectopic expression studies, both 293T and A549 cells
were transfected with the appropriate plasmids by electroporation
with a Genepulser MX (Biorad) at 400V, 125 mF, and N ohms in
optiMEM (Invitrogen).
Immunocytochemistry, Imaging, and Colocalization
StudiesIn brief, after appropriate treatment, cells grown on coverslips
were washed in warm PBS and fixed in 4% paraformaldehyde.
After fixation, cells were permeabilized with 0.1% Triton X-100
an Journal of Human Genetics 94, 246–256, February 6, 2014 247

(TX100) in PBS, quenched in 50 mM NH4Cl, and blocked in 10%
goat serum and 1% BSA in PBS containing 0.05% TX100 for 1 hr.
Cells were incubated in the primary ERCC6L2 antibody (Abcam)
and the corresponding goat anti-rabbit secondary antibody conju-
gated to Alexa Fluor 488/568 (Invitrogen) in blocking solution for
1 hr separately. Cells were washed three times in PBS containing
0.05% TX100 between primary and secondary antibody incuba-
tions and mounted with vectashield containing DAPI (Vector
Labs). In cells expressing GFP-tagged wild-type (WT) and variant
forms of ERCC6L2, endoplasmic reticulum (ER) staining was
performed with mouse BiP antibody (Calbiochem). Autophagy
vacuoles were stained with LC3b antibody (Santa Cruz). For
lysosomes, A549 cells were stained with LysoTracker Red
DND-99 (Invitrogen) at a 100 nM/ml concentration; mito-
chondria were labeled with MitoTracker Orange CMTM Ros
(Invitrogen) at 37�C before fixation. Images were collected with
an LSM710 laser scanning confocal microscope (Olympus) under
relevant laser excitation, and the emitted signals were visualized
with ZEN software (Zeiss). For colocalization analysis, the back-
ground over noncellular regions was subtracted prior to determi-
nation of Pearson’s correlation coefficient (r2) in a minimum of
12 cells in each of three independent experiments. Image process-
ing was limited to contrast enhancement.
siRNA Transfection, Real-Time PCR, and
Immunoblotting DensitometryFor small interfering RNA (siRNA) studies, A549 cells were seeded
out in 6-well plates at 3.0 3 105 cells per well in antibiotic-free
DMEM containing 10% fetal calf serum (Invitrogen) and were
transfected the following day with Lipofectamine RNAiMAX
(Invitrogen). A pool of two siRNAs (SASI_Hs02_00305873 and
SASI_Hs01_00164316, Sigma-Aldrich) was used at a 30 nM final
concentration for ERCC6L2 knockdown, and a nontarget siRNA
(AllStars Negative Control siRNA, QIAGEN) was used as a negative
control. Mock-transfected cells were treated identically but
without any siRNA. Knockdown of ERCC6L2 expression was
confirmed by quantitative real-time PCR. RNA was extracted
with the RNeasy Mini Kit (QIAGEN), and cDNA was synthesized
with SuperScript II Reverse Transcriptase (Invitrogen). Quantita-
tive real-time PCR was performed with TaqMan probes
Hs02758991_g1 for ERCC6L2 and Hs00418541_m1 for GAPDH,
and reactions were run on an ABI 7500 thermal cycler (Applied
Biosystems). For immunoblotting, 30 mg of BSA-quantified protein
was loaded, transferred, and analyzed with a WesternBreeze
Chromogenic Kit (Invitrogen) and antibodies against ERCC6L2
(Abcam) and b-actin (Abcam), the latter of which was used as
a loading control. We performed semiquantitative analysis of
raw immunoblots by scanning the colorimetric blots at 600 dpi
resolution to TIFF format files, which we subjected to densitom-
etry analysis software (GelPro) to identify quantitative changes
in protein levels.
Cell Viability and ROS EvaluationAfter 48 hr exposure to ERCC6L2-siRNA-lipofection complexes
(SASI_Hs02_00305873 and SASI_Hs01_00164316, Sigma-Aldrich)
and nontarget siRNA (1027280, QIAGEN), medium was replaced
with DMEM containing 10% FBS supplemented with mitomycin
C (MMC), camptothecin (CPT), etoposide (ETP), and Irofulven
over a specified dose range in the presence or absence of N-acetyl
cysteine (NAC) (all purchased from Sigma-Aldrich). After further
48 hr incubation at 37�C, cells were washed in PBS, propidium
248 The American Journal of Human Genetics 94, 246–256, February
iodide was added to the cell suspension to a final concentration
of 5 mg/ml, and the cells were analyzed for staining on an LSRII
Flow Cytometer (Becton Dickinson). For the evaluation of basal
reactive oxygen species (ROS), cells were labeled with ROS marker
dihydroethidium (DHE, Invitrogen) at a 5 mM concentration, and
the fluorescence was measured by flow cytometry. Real-time
changes in ROS fluorescence upon Irofulven treatment were
measured at 37�C with standard 520 nm excitation and 610 nm
emission at 2 min intervals for a minimum of 60 min with the
use of a FLUROstar Optima plate reader (BMG Labtech). All read-
ings were normalized to the basal level (time ¼ 0). We calculated
statistical significance by comparing the linear regression of the
two curves with GraphPad Prism 5 software (GraphPad).
Subcellular FractionationSubcellular fractionation was performed according to a previously
described protocol5 with a slight modification. In brief, 1 3 106
DMSO-treated A549 cells and cells treated for 3 hr with MMC
(33 nM) or Irofulven (100 nM) were lysed in ice-cold HEPES
containing 5 mg/ml digitonin (Sigma Aldrich) and centrifuged at
3,000 rpm for extraction of cytoplasmic protein. Cell pellets
were resuspended in 1% Triton-X-114 in ice-cold PBS containing
a cocktail of protease and phosphatase inhibitors and centrifuged
at 14,000 rpm at 4�C for pelleting nuclei; the resulting superna-
tants consisting of membrane proteins were mixed in sample
buffer. The pellet-containing nuclei were briefly washed three
times in ice-cold radioimmunoprecipitation assay buffer contain-
ing a cocktail of protease and phosphatase inhibitors (Roche);
sonication followed twice for 10 s at 50% pulse. The final mixture
was shaken gently on ice for 15 min, and the protein supernatant
was obtained by centrifugation of lysates at 14,0003 g for 15 min.
Fractionated lysates were verified with antibodies against cyto-
plasmic GAPDH (Abcam), nuclear TATA binding protein (Abcam),
and the ER membrane protein BiP (Calbiochem) by immunoblot-
ting as described above.
Isolation of Mitochondrial Enriched LysatesExtracts enriched for mitochondrial proteins were obtained
according to a previously described protocol.6 In brief, after treat-
ment with MMC or Irofulven, cell pellets were resuspended in ten
packed cell volumes of buffer A (1 mM TrisHCl, pH 7.4, 0.13 M
NaCl, 5 mM KCl, and 7.5 mM MgCl2), pelleted, and resuspended
again in six packed cell volumes of homogenization buffer B
(10 mM Tris-HCl, pH 6.7, 10 mM KCl, 0.15 mM MgCl2, 1 mM
PMSF, and 1 mM DTT). Cells were homogenized on ice, and the
homogenate was transferred into a tube containing one packed
cell volume of 2 M sucrose solution and mixed gently. The cell
mixture was centrifuged at 1,2003 g for 5 min for pelleting unbro-
ken cells, nuclei, and large debris. The supernatant was transferred
to another tube and centrifuged at 7,000 3 g for 10 min for pellet-
ing mitochondria. The resulting supernatant was saved as soluble
cytoplasmic lysate 1. The mitochondrial pellet was resuspended in
three packed cell volumes of mitochondrial suspension buffer C
(10 mM TrisHCl, pH 6.7, 0.15 mM MgCl2, 0.25 M sucrose, 1 mM
PMSF, and 1 mM DTT) and centrifuged again at 9,500 3 g for
5 min for repelleting the mitochondria. The resulting supernatant
wasmixed with cytoplasmic lysate 1, obtained above. The pelleted
mitochondria were mixed with 13 sample buffer boiled at 95�Cfor 5 min and subjected to immunoblotting analysis. Mitochon-
drial fractionated lysates were verified with mitochondrial COXIV
(Abcam) by immunoblotting as described above.
6, 2014
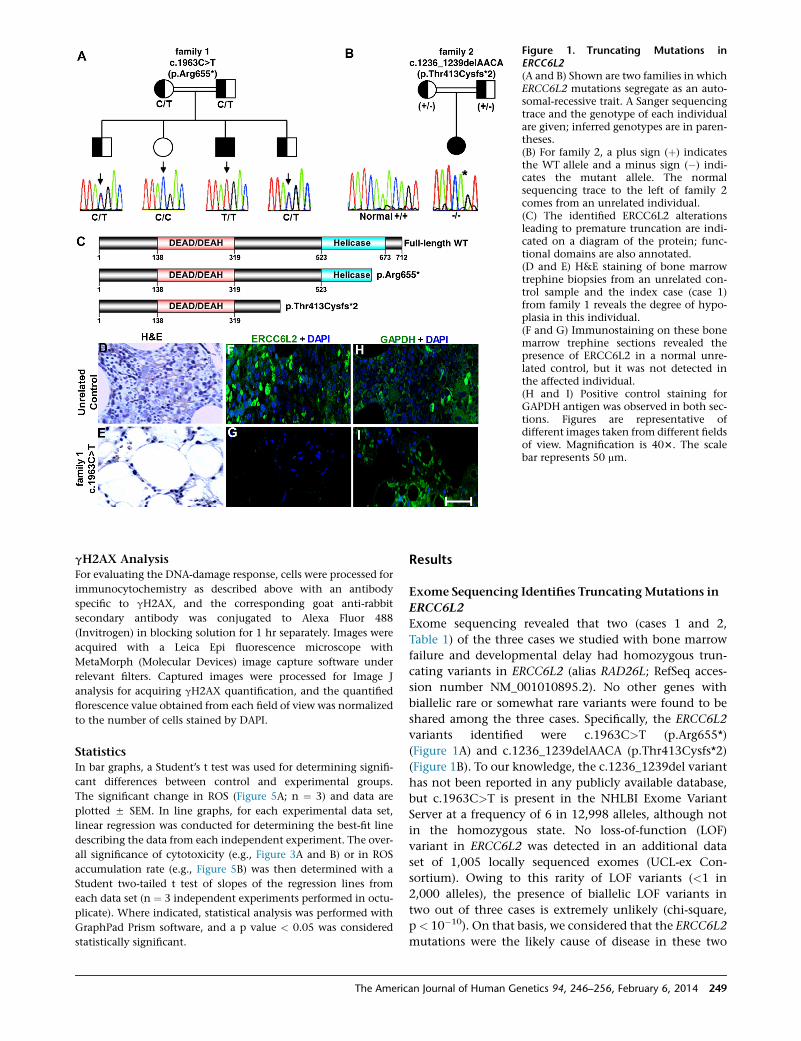
Figure 1. Truncating Mutations inERCC6L2(A and B) Shown are two families in whichERCC6L2 mutations segregate as an auto-somal-recessive trait. A Sanger sequencingtrace and the genotype of each individualare given; inferred genotypes are in paren-theses.(B) For family 2, a plus sign (þ) indicatesthe WT allele and a minus sign (�) indi-cates the mutant allele. The normalsequencing trace to the left of family 2comes from an unrelated individual.(C) The identified ERCC6L2 alterationsleading to premature truncation are indi-cated on a diagram of the protein; func-tional domains are also annotated.(D and E) H&E staining of bone marrowtrephine biopsies from an unrelated con-trol sample and the index case (case 1)from family 1 reveals the degree of hypo-plasia in this individual.(F and G) Immunostaining on these bonemarrow trephine sections revealed thepresence of ERCC6L2 in a normal unre-lated control, but it was not detected inthe affected individual.(H and I) Positive control staining forGAPDH antigen was observed in both sec-tions. Figures are representative ofdifferent images taken from different fieldsof view. Magnification is 403. The scalebar represents 50 mm.
gH2AX AnalysisFor evaluating the DNA-damage response, cells were processed for
immunocytochemistry as described above with an antibody
specific to gH2AX, and the corresponding goat anti-rabbit
secondary antibody was conjugated to Alexa Fluor 488
(Invitrogen) in blocking solution for 1 hr separately. Images were
acquired with a Leica Epi fluorescence microscope with
MetaMorph (Molecular Devices) image capture software under
relevant filters. Captured images were processed for Image J
analysis for acquiring gH2AX quantification, and the quantified
florescence value obtained from each field of view was normalized
to the number of cells stained by DAPI.
StatisticsIn bar graphs, a Student’s t test was used for determining signifi-
cant differences between control and experimental groups.
The significant change in ROS (Figure 5A; n ¼ 3) and data are
plotted 5 SEM. In line graphs, for each experimental data set,
linear regression was conducted for determining the best-fit line
describing the data from each independent experiment. The over-
all significance of cytotoxicity (e.g., Figure 3A and B) or in ROS
accumulation rate (e.g., Figure 5B) was then determined with a
Student two-tailed t test of slopes of the regression lines from
each data set (n ¼ 3 independent experiments performed in octu-
plicate). Where indicated, statistical analysis was performed with
GraphPad Prism software, and a p value < 0.05 was considered
statistically significant.
The Americ
Results
Exome Sequencing Identifies TruncatingMutations in
ERCC6L2
Exome sequencing revealed that two (cases 1 and 2,
Table 1) of the three cases we studied with bone marrow
failure and developmental delay had homozygous trun-
cating variants in ERCC6L2 (alias RAD26L; RefSeq acces-
sion number NM_001010895.2). No other genes with
biallelic rare or somewhat rare variants were found to be
shared among the three cases. Specifically, the ERCC6L2
variants identified were c.1963C>T (p.Arg655*)
(Figure 1A) and c.1236_1239delAACA (p.Thr413Cysfs*2)
(Figure 1B). To our knowledge, the c.1236_1239del variant
has not been reported in any publicly available database,
but c.1963C>T is present in the NHLBI Exome Variant
Server at a frequency of 6 in 12,998 alleles, although not
in the homozygous state. No loss-of-function (LOF)
variant in ERCC6L2 was detected in an additional data
set of 1,005 locally sequenced exomes (UCL-ex Con-
sortium). Owing to this rarity of LOF variants (<1 in
2,000 alleles), the presence of biallelic LOF variants in
two out of three cases is extremely unlikely (chi-square,
p< 10�10). On that basis, we considered that the ERCC6L2
mutations were the likely cause of disease in these two
an Journal of Human Genetics 94, 246–256, February 6, 2014 249

Figure 2. Truncating Mutations Affect the Localization and Degradation of ERCC6L2(A) Confocal-microscopy images demonstrate predominant cytoplasmic and nuclear localization of WT GFP-ERCC6L2 in A549 cells.(B and C) Both truncating variants of ERCC6L2 formed aggregates.(E–L) Colocalization in yellow was observed for both of the altered ERCC6L2 proteins with BiP antibody, which binds to the ER proteinBiP (E and F), LC3b antibody, which localizes to autophagic vacuoles (H and I), and LysoTracker, which stains lysosomes (K and L). Thecolocalization observed was not an artifact of cross-channel noise or bleed from a compliment channel. The Pearson correlationcoefficients for colocalization revealed r2 > 0.5 when measured for the entire cell with ZEN software (Zeiss).(D, G, and J) No colocalization was observed betweenWT ERCC6L2 and any of these organelle markers (Pearson correlation coefficientsr2 < 0.08).(M–O) Neither the WT nor altered forms of ERCC6L2 showed any colocalization with ubiquitin, as evidenced by the green stainingpattern (Pearson correlation coefficient r2 < 0.03). All panels are representative of images taken from different fields of view in threeseparate experiments. Images display DAPI (blue), GFP-tagged ERCC6L2 (green), and organelle-labeling markers (red). The scale barrepresents 30 mm.
individuals. No obvious disease-causing variant was
identified in the third case.
Bone Marrow of an Affected Individual Lacks
ERCC6L2
ERCC6L2 is located on chromosome 9 (9q 22.32) and spans
14 exons. Expasy prosite analysis of ERCC6L2 revealed an
N-terminal DEAH ATP-helicase domain and a catalytic
helicase C-terminal domain (Figure 1C). Pfam analysis
indicated that it belongs to the Snf2 family of helicase-
like proteins, which are involved in transcription regula-
tion, DNA repair, DNA recombination, DNA translocation,
and chromatin unwinding.7 ERCC6L2 is 712 amino acids
in length and has a predicted molecular weight of
81 kDa. Immunoblotting on human cell lines revealed a
72 kDa band (Figure S1A, available online), and the speci-
ficity of the antibody was verified by two siRNAs targeting
the ERCC6L2 sequence at two different regions. Both
siRNAs decreased the expression of mRNA, as analyzed
48 hr posttransfection by real-time PCR (Figure S1B), and
reduced protein levels, as detected by immunoblotting,
confirming the efficiency of this antibody in detecting
ERCC6L2 (Figure S1C). Immunostudies on A549 cells
250 The American Journal of Human Genetics 94, 246–256, February
revealed the presence of ERCC6L2 in both cytoplasmic
and nuclear compartments (Figures S1D–S1H).
H&E staining revealed that compared to those of an un-
related control, bone marrow trephine sections obtained
from the individual homozygous for the c.1963C>T
mutation were hypocellular (Figures 1D and 1E). Immuno-
histochemistry on these sections showed no detectable
staining for ERCC6L2 in the sample from the affected
individual, but it did show clear positive cells in the unre-
lated control (Figures 1F and 1G); both sections stained
positive for GAPDH (Figures 1H and 1I). These results indi-
cate that truncating mutations might have an impact on
ERCC6L2 stability.
Mislocalization andDegradation of ERCC6L2 Variants
To investigate the impact of truncating mutations on
ERCC6L2 stability, we expressed both N-terminal GFP-
fused WT and mutant plasmid cDNA constructs in human
cell lines. In human A549 cells,WTGFP-ERCC6L2 revealed
a continuous diffuse pattern throughout the cytoplasm
and nucleus (Figure 2A). In contrast, both truncated forms
of GFP-ERCC6L2 showed marked aggregate-like structures
(Figures 2B and 2C). The distinct aggregate pattern of the
6, 2014

Figure 3. ERCC6L2 Plays a Role in theDNA-Damage-Response Pathway(A and B) Compared to nontarget-siRNA-transfected cells, ERCC6L2-knockdowncells showed reduced survival after 48 hrtreatment with MMC and Irofulven in adose-dependent manner. Error bars repre-sent the SEM obtained from three inde-pendent experiments (one-way ANOVAwith Tukey’s test).(C and D) Compared to nontarget-siRNA-transfected cells, ERCC6L2-knockdowncells revealed gH2AX foci at basal level.(E and F) gH2AX foci were higher in num-ber in ERCC6L2-knockdown cells thanin nontarget-siRNA-transfected cells aftertreatment with Irofulven. Panels are repre-sentative of images taken from differentfields of view displaying gH2AX (green)and DAPI (blue). The scale bar represents5 mm.(G) The bar graph represents the quanti-fied gH2AX fluorescence value obtainedfrom each individual field of view andnormalized to the number of cells stainedby DAPI (n > 50) at each individualtime point. Error bars represent the SEM(Mann-Whitney test), derived from dataobtained from five fields of view at eachindividual time point from two indepen-dent experiments.
ERCC6L2 variants was not cell-type specific given that it
was also seen in 293T cells (Figure S2).
Further characterization of these variant ERCC6L2 aggre-
gates inA549cells revealed that theywere localized to theER
(Figures 2D–2F), autophagic vacuoles (Figures 2G–2I), and
lysosomes (Figures 2J–2L), as observed by costaining with
specific cell organelle markers. However, no colocalization
was observed with ubiquitin (Figures 2M–2O). Consistent
with the lack of ERCC6L2 staining in the affected individ-
ual’s bone marrow, these studies demonstrate that both
truncating alterations in ERCC6L2 impair its normal locali-
zation and thus result in aggregation in the ER and likely
degradation through ER-associated autophagy.8,9
ERCCL2 Knockdown Reduces Cell Viability after
Genotoxic Stress
Until now, the function of ERCC6L2 has not been defined
experimentally, but it has been shown to have a ubiquitous
expression profile (Figure S3). Among the Snf2 family of
proteins, it shares the greatest peptide sequence homology
with the chromatin-remodelling and DNA-repair proteins
ALC1, CHD1, CHD4, RAD54, ATRX, and ERCC6 (Figures
S4A and S4B). Mutations in ATRX (MIM 300032) have
been linked to X-linked mental retardation most often
accompanied by alpha-thalassemia (ATRX syndrome
[MIM 301040]),10 and mutations in ERCC6 (MIM
609413) have been shown to cause Cockayne syndrome
(CS [MIM 133540]), characterized clinically by dwarfism,
microcephaly, cachexia, and neurodegeneration.11–13
Functional studies on RAD54, ALC1, CHD1, and CHD4
in eukaryotic cells revealed their role in facilitating DNA
The Americ
repair through chromatin remodelling.14 Loss of ATRX
has been shown to cause genomic instability and altered
DNA-damage response.15 ERCC6 has also been shown to
play an important role in a subpathway of nucleotide
excision repair (NER), known as transcription-coupled
NER (TC-NER), and also in mitochondrial function.16–21
Considering the homology of these genes and the overlap
of the clinical features seen in our individuals (specifically
learning difficulties and microcephaly), we speculate that
ERCC6L2 might also play a role in the DNA-damage
response.
To test this, we attempted to mimic the truncating
mutations by using siRNA to knock down ERCC6L2
expression in human A549 cells. After successful knock-
down (Figure S1B), we exposed the cells to the following
clastogens: (1) MMC, a compound that causes interstrand
crosslinks that can be repaired by the FA pathway or NER,22
(2) Irofulven, a compound that specifically induces DNA
adducts recognized by TC-NER, but not global genome
NER,23 and (3) CPT and (4) ETP, which induce double-
stranded DNA breaks via inhibition of topoisomerase I
and II, respectively. After 48 hr exposure, cell viability
was assessed by flow cytometry. Compared to mock-trans-
fected cells or cells transfected with nontarget siRNA, cells
transfected with ERCC6L2 siRNA showed significantly
reduced viability after exposure to MMC (unpaired
Student’s two-tailed t tests, p < 0.01, Figure 3A) and
Irofulven (unpaired Student’s two-tailed t tests, p <
0.001, Figure 3B) in a dose-dependent manner. With CPT
or ETP, no difference was observed in cell survival between
cells transfected with ERCC6L2 siRNA and cells transfected
an Journal of Human Genetics 94, 246–256, February 6, 2014 251

with nontarget siRNA (Figure S5). The consistent increase
in MMC and Irofulven sensitivity suggests a role for
ERCC6L2 in the repair of interstrand crosslinks and specif-
ically in TC-NER of DNA damage. We note that there is
evidence suggesting a role for TC-NER in the repair of
MMC-induced interstrand crosslinks.24 Despite the fact
that ERCC6L2-knockdown cells also showed increased
susceptibility to ETP and, to a lesser extent, CPT, a similar
pattern was also observed in cells treated with nontarget
siRNA. These results prevent us from concluding that
ERCC6L2 also plays a role in DNA-repair pathways acti-
vated upon topoisomerase inhibition.
Knockdown of ERCC6L2 Induces a DNA-Damage
Response
Snf2 protein complexes have been shown to be involved
in the recruitment of gH2AX, a phosphorylated form of
histone 2A, to sites of DNA damage.25 Given the increased
sensitivity of ERCC6L2-knockdown cells to MMC and
Irofulven, we wanted to investigate the role of ERCC6L2
with respect to this marker of DNA damage. Immunostain-
ing with a gH2AX-specific antibody revealed the presence
of discrete foci in ERCC6L2-knockdown cells in the
absence of any genotoxic stress (Figures 3C and 3D).
Compared to nontarget-siRNA-transfected cells, cells
treated with Irofulven for 3 hr at a low concentration
(100 nM) showed a further significant increase in the level
of gH2AX (Figures 3E and 3F). The kinetics of the increase
in gH2AX was more pronounced in ERCC6L2-knockdown
cells than in nontarget-siRNA-transfected cells over time,
suggesting an early DNA-damage response (Figure 3G).
Collectively, these results indicate that ERCC6L2 plays a
role in the DNA-damage-response pathway and that its
depletion sensitizes cells to genotoxic agents.
ERCC6L2 Traffics to the Mitochondria and Nucleus
after Genotoxic Stress
To further characterize the response of ERCC6L2 to
DNA-damaging agents, we performed immunocytochem-
istry on A549 cells. Compared to the normal diffuse
cytosolic and nuclear staining pattern seen in control
DMSO-treated cells (Figure S6A), a distinct perinuclear
accumulation of ERCC6L2 was seen in the cells treated
with both MMC and Irofulven (Figures S6B and S6C).
Immunoblotting on subcellular fractionated protein
lysates obtained from these cells revealed a dramatic
translocation of ERCC6L2 from cytosolic to membrane-
ous compartments and, to a lesser extent, toward the
nucleus after treatment with MMC or Irofulven for 3 hr
(Figures 4A and 4B).
ERCC6L2 has a distinct 13 amino acid N-terminal
sequence that harbors a putative mitochondrial targeting
sequence, as predicted by MitoProt software.26 This
prompted us to investigate extracts enriched with mito-
chondria obtained from A549 cells, and indeed these
showed an higher level of ERCC6L2 in mitochondria
than in the cytoplasm in cells treated with MMC and
252 The American Journal of Human Genetics 94, 246–256, February
Irofulven (Figures S6D and S6E). Immunocytochemistry
on cells labeled with MitoTracker and DAPI confirmed
the predominant localization of ERCC6L2 in the mito-
chondria (Figures 4C–4Q) and nucleus (Figures 4F, 4K,
and 4P) after MMC and Irofulven treatment. These results
are particularly intriguing in light of recent data indicating
that ERCC6 might act as a sensor of mtDNA damage19–21
and the central role that mitochondrial dysfunction plays
in the process of normal human aging.27
ERCC6L2 Knockdown Increases Intracellular ROS
Translocation of ERCC6L2 to themitochondria in response
to DNA-damaging agents led us to investigate its role in
mitochondrial function. We used ERCC6L2-knockdown
cells and nontarget-siRNA-transfected cells to quantify
intracellular ROS by using the ethidium-based probe DHE.
After successful knockdown, cells were stained with DHE
and subjected to flow cytometry. Compared to cells trans-
fected with nontarget siRNA, ERCC6L2-knockdown cells
showed a significant increase in intracellular ROS
(Figure 5A). To further investigate the changes in intracel-
lular ROS, we induced genotoxic stress with Irofulven
(100 nM) and monitored the change in intracellular ROS
over time. After treatmentwith Irofulven, ERCC6L2-knock-
down cells showed a significant increase (p < 0.0001,
comparing the linear regression of two curves) in the
change of intracellular ROS level over time in comparison
to nontarget transfected cells (Figure 5B). These results indi-
cate that depletion of ERCC6L2 results in increased ROS.
The Response of ERCC6L2 to Genotoxic Stress Is ROS
Dependent
To investigate a possible role for ROS in sensitizing
ERCC6L2-knockdown cells to genotoxic stress, we used
NAC, a well-known antioxidant that scavenges ROS.
ERCC6L2-knockdown cells and nontarget-siRNA-trans-
fected cells were treated with Irofulven at a cytotoxic
concentration (650 nM, refer to Figure 3B) in the presence
of NAC at two different concentrations (5 and 10 mM).
After 48 hr, flow cytometry analysis revealed that
compared to ERCC6L2-knockdown cells treated with
Irofulven alone, ERCC6L2-knockdown cells treated with
Irofulven and NAC had a significant (p < 0.0001) reduc-
tion of cell death in an NAC-dose-dependent manner
(Figure 5C). ERCC6L2-knockdown cells also stained nega-
tive for gH2AX upon treatment with Irofulven in the pres-
ence of NAC (data not shown). In addition, NAC inhibited
the Irofulven-induced translocation of ERCC6L2 to mito-
chondria (Figure 5D) and the nucleus (Figure S7). These
results suggest that ERCC6L2 traffic to both mitochondria
and nucleus is ROS dependent.
Discussion
In this study, we report on two homozygous truncating
ERCC6L2 mutations identified in individuals with
6, 2014
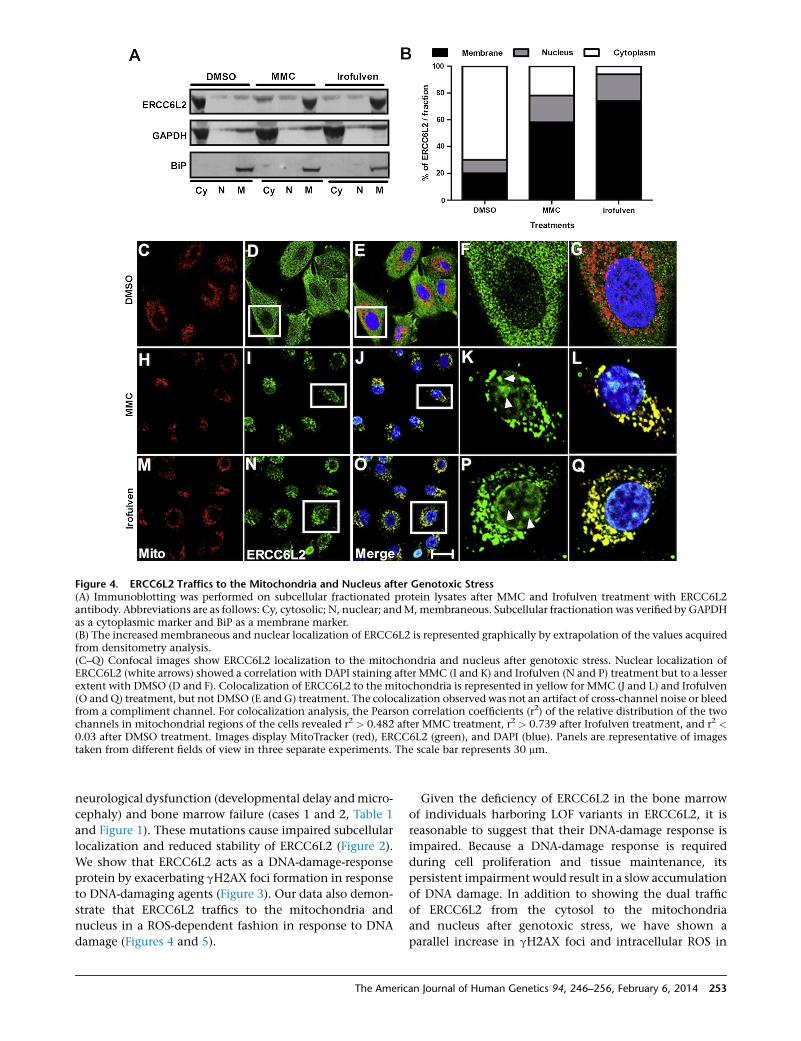
Figure 4. ERCC6L2 Traffics to the Mitochondria and Nucleus after Genotoxic Stress(A) Immunoblotting was performed on subcellular fractionated protein lysates after MMC and Irofulven treatment with ERCC6L2antibody. Abbreviations are as follows: Cy, cytosolic; N, nuclear; andM, membraneous. Subcellular fractionation was verified by GAPDHas a cytoplasmic marker and BiP as a membrane marker.(B) The increased membraneous and nuclear localization of ERCC6L2 is represented graphically by extrapolation of the values acquiredfrom densitometry analysis.(C–Q) Confocal images show ERCC6L2 localization to the mitochondria and nucleus after genotoxic stress. Nuclear localization ofERCC6L2 (white arrows) showed a correlation with DAPI staining after MMC (I and K) and Irofulven (N and P) treatment but to a lesserextent with DMSO (D and F). Colocalization of ERCC6L2 to the mitochondria is represented in yellow for MMC (J and L) and Irofulven(O and Q) treatment, but not DMSO (E and G) treatment. The colocalization observed was not an artifact of cross-channel noise or bleedfrom a compliment channel. For colocalization analysis, the Pearson correlation coefficients (r2) of the relative distribution of the twochannels in mitochondrial regions of the cells revealed r2 > 0.482 after MMC treatment, r2 > 0.739 after Irofulven treatment, and r2 <0.03 after DMSO treatment. Images display MitoTracker (red), ERCC6L2 (green), and DAPI (blue). Panels are representative of imagestaken from different fields of view in three separate experiments. The scale bar represents 30 mm.
neurological dysfunction (developmental delay andmicro-
cephaly) and bone marrow failure (cases 1 and 2, Table 1
and Figure 1). These mutations cause impaired subcellular
localization and reduced stability of ERCC6L2 (Figure 2).
We show that ERCC6L2 acts as a DNA-damage-response
protein by exacerbating gH2AX foci formation in response
to DNA-damaging agents (Figure 3). Our data also demon-
strate that ERCC6L2 traffics to the mitochondria and
nucleus in a ROS-dependent fashion in response to DNA
damage (Figures 4 and 5).
The Americ
Given the deficiency of ERCC6L2 in the bone marrow
of individuals harboring LOF variants in ERCC6L2, it is
reasonable to suggest that their DNA-damage response is
impaired. Because a DNA-damage response is required
during cell proliferation and tissue maintenance, its
persistent impairment would result in a slow accumulation
of DNA damage. In addition to showing the dual traffic
of ERCC6L2 from the cytosol to the mitochondria
and nucleus after genotoxic stress, we have shown a
parallel increase in gH2AX foci and intracellular ROS in
an Journal of Human Genetics 94, 246–256, February 6, 2014 253

Figure 5. The Relationship among ERCC6L2, ROS, and Genotoxic Stress(A) Compared to cells transfected with nontarget siRNA, A549 cells transfected with ERCC6L2 siRNA showed an increase in intracellularROS levels. Error bars represent the SEM of three independent experiments performed in triplicate (**p < 0.01).(B) Changes in ROS levels upon Irofulven stimulation were monitored over time. Error bars represent the SEM of eight readings in threeindependent experiments.(C) Analysis with fluorescence-activated cell sorting revealed reduced cytotoxicity of Irofulven in ERCC6L2-knockdown cells in the pres-ence of NAC in a concentration-dependent manner. Error bars represent the SEM from three independent experiments.(D) Immunocytochemistry on A549 cells treated with Irofulven (650 nM) in the presence of NAC (10 mM). Images display MitoTracker(red), ERCC6L2 (green), and DAPI (blue). Panels are representative of images taken from different fields of view in three separate exper-iments. The scale bar represents 30 mm.
ERCC6L2-knockdown cells. Increasing ROS levels leading
to alteration in cellular homeostasis is considered to be of
major pathological significance in neurodegenerative dis-
orders28 and bone marrow failure.29
There is also evidence that the increase in ROS levels
and incompetent DNA-damage repair contributes to the
pathogenesis of bone-marrow-failure syndromes.30 For
example, although FA is primarily characterized by chro-
mosome instability, developmental abnormalities, and
cancer susceptibility, it also shows reduced mitochondrial
function and increased ROS production.31 CS and the
functionally related xeroderma pigmentosum (XP [MIM
278700, 610651, 278720, 278730, 278740, 278760, and
278780]) are also caused by an inability to repair DNA
damage, particularly UV-induced DNA crosslinking32 and
increased ROS production.32,33 Cells from CS individuals
and CS mouse models have also shown increased ROS
activity and accumulation of damaged mitochondria as
a result of defective mitochondrial clearance by auto-
phagy.19 Recently, studies on DC lymphoblasts have
revealed elevated levels of ROS and a pronounced DNA-
damage response leading to a proliferative defect and
apoptosis.34,35 It is also known that increased ROS levels
impair the self-renewal capacity of hematopoietic progen-
itor cells in bone marrow.31 Taking all this together, we
propose that an increase in ROS and the accumulation of
254 The American Journal of Human Genetics 94, 246–256, February
DNA damage are the underlying causes of the disease
observed in our affected individuals.
Although some features overlap with previously
described syndromes, the individuals reported in this study
also have significant differences (Table 1). For instance, FA
is characterized by abnormal chromosomal breakage, but
analysis of peripheral-blood lymphocytes in our affected
individuals did not identify such defects. The increased
sensitivity to MMC in FA is also much more pronounced
than that observed in our ERCC6L2-knockdown assays
(Figure 3A) and the primary cells of these individuals (Table
1). Equally, bone marrow failure is not a reported feature of
CS, although neurological abnormalities are present
in most cases.36 XP, on the other hand, is primarily charac-
terized by hypersensitivity to sunlight and subsequent
development of carcinomas, and only some cases present
with neurologic disease.36 Interestingly, a recent study re-
ported a variant in ERCC4 (XPF) in a CS individual with
overlapping features of XP (severe photosensitivity and
abnormal skin pigmentation) and FA (abnormal chromo-
somal breakage).37 Hoyeraal Hreidarsson syndrome (HHS
[MIM 300240]) is characterized by neurological dysfunc-
tion, microcephaly, and bonemarrow failure and is consid-
ered to be a severe form of DC,38 a telomere maintenance
disorder,39,40 which itself commonlymanifests withmuco-
cutaneous abnormalities and bone marrow failure.4 Our
6, 2014

individuals lacked the mucocutaneous features, immuno-
deficiency, and cerebellar hypoplasia typically seen in DC
and HHS and, in comparison to age-matched controls,
had variable telomere lengths when measured by multi-
plex PCR41 (Table 1 and Figure S8). It is also notable that
exome sequencing did not identify variations in any of
the known genes associated with bone marrow failure in
these individuals.
Because there is no precise match between all the fea-
tures in the cases reported here and other genetically
characterized disorders, we propose the presently described
disorder as a distinct bone-marrow-failure syndrome. We
also believe that our study highlights the combined roles
of intracellular ROS and DNA damage in driving bone
marrow failure.
Supplemental Data
Supplemental Data include eight figures and can be found with
this article online at http://www.cell.com/AJHG.
Acknowledgments
We would like to thank the families and clinicians who contrib-
uted to this research, as well as Martine French for providing
bone marrow slides. This work was funded by TheWellcome Trust
and Medical Research Council. We would also like to thank Gary
Warnes and Ann Wheeler for support and technical assistance in
fluorescence-activated cell sorting and imaging.
Received: November 13, 2013
Accepted: January 10, 2014
Published: February 6, 2014
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://www.1000genomes.org
ANNOVAR, www.openbioinformatics.org/annovar
BioGPS, http://www.biogps.org
dbSNP, http://www.ncbi.nlm.nih.gov/SNP/
GATK, http://www.broadinstitute.org/gatk/
MitoProt, http://ihg.gsf.de/ihg/mitoprot.html
NHLBI Exome Sequencing Project (ESP) Exome Variant Server,
http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.
omim.org
Pfam, http://pfam.sanger.ac.uk
Prosite, http://www.expasy.ch/Prosite
References
1. Young, N.S., Bacigalupo, A., and Marsh, J.C. (2010). Aplastic
anemia: pathophysiology and treatment. Biol. Blood Marrow
Transplant. 16 (1 Suppl), S119–S125.
2. Dokal, I., and Vulliamy, T. (2010). Inherited bone marrow fail-
ure syndromes. Haematologica 95, 1236–1240.
3. Kottemann, M.C., and Smogorzewska, A. (2013). Fanconi
anaemia and the repair of Watson and Crick DNA crosslinks.
Nature 493, 356–363.
The Americ
4. Walne, A.J., and Dokal, I. (2009). Advances in the understand-
ing of dyskeratosis congenita. Br. J. Haematol. 145, 164–172.
5. Holden, P., and Horton, W.A. (2009). Crude subcellular frac-
tionation of cultured mammalian cell lines. BMC Res. Notes
2, 243.
6. Kluck, R.M., Bossy-Wetzel, E., Green, D.R., and Newmeyer,
D.D. (1997). The release of cytochrome c from mitochondria:
a primary site for Bcl-2 regulation of apoptosis. Science 275,
1132–1136.
7. Flaus, A., Martin, D.M., Barton, G.J., and Owen-Hughes, T.
(2006). Identification of multiple distinct Snf2 subfamilies
with conserved structural motifs. Nucleic Acids Res. 34,
2887–2905.
8. Yorimitsu, T., Nair, U., Yang, Z., and Klionsky, D.J. (2006).
Endoplasmic reticulum stress triggers autophagy. J. Biol.
Chem. 281, 30299–30304.
9. Ogata, M., Hino, S., Saito, A., Morikawa, K., Kondo, S., Kane-
moto, S., Murakami, T., Taniguchi, M., Tanii, I., Yoshinaga,
K., et al. (2006). Autophagy is activated for cell survival after
endoplasmic reticulum stress. Mol. Cell. Biol. 26, 9220–9231.
10. Ratnakumar, K., and Bernstein, E. (2013). ATRX: the case of a
peculiar chromatin remodeler. Epigenetics 8, 3–9.
11. Mallery, D.L., Tanganelli, B., Colella, S., Steingrimsdottir, H.,
van Gool, A.J., Troelstra, C., Stefanini, M., and Lehmann,
A.R. (1998). Molecular analysis of mutations in the CSB
(ERCC6) gene in patients with Cockayne syndrome. Am. J.
Hum. Genet. 62, 77–85.
12. Laugel, V., Dalloz, C., Durand, M., Sauvanaud, F., Kristensen,
U., Vincent, M.C., Pasquier, L., Odent, S., Cormier-Daire, V.,
Gener, B., et al. (2010). Mutation update for the CSB/ERCC6
and CSA/ERCC8 genes involved in Cockayne syndrome.
Hum. Mutat. 31, 113–126.
13. Weidenheim, K.M., Dickson, D.W., and Rapin, I. (2009).
Neuropathology of Cockayne syndrome: Evidence for im-
paired development, premature aging, and neurodegenera-
tion. Mech. Ageing Dev. 130, 619–636.
14. Lans, H., Marteijn, J.A., and Vermeulen, W. (2012). ATP-
dependent chromatin remodeling in the DNA-damage
response. Epigenetics Chromatin 5, 4–18.
15. Lovejoy, C.A., Li, W., Reisenweber, S., Thongthip, S., Bruno, J.,
de Lange, T., De, S., Petrini, J.H., Sung, P.A., Jasin, M., et al.;
ALT Starr Cancer Consortium (2012). Loss of ATRX, genome
instability, and an altered DNA damage response are hall-
marks of the alternative lengthening of telomeres pathway.
PLoS Genet. 8, e1002772.
16. Velez-Cruz, R., and Egly, J.M. (2013). Cockayne syndrome
group B (CSB) protein: at the crossroads of transcriptional
networks. Mech. Ageing Dev. 134, 234–242.
17. Licht, C.L., Stevnsner, T., and Bohr, V.A. (2003). Cockayne
syndrome group B cellular and biochemical functions. Am.
J. Hum. Genet. 73, 1217–1239.
18. Aamann, M.D., Sorensen, M.M., Hvitby, C., Berquist, B.R.,
Muftuoglu, M., Tian, J., de Souza-Pinto, N.C., Scheibye-Knud-
sen, M., Wilson, D.M., 3rd, Stevnsner, T., and Bohr, V.A.
(2010). Cockayne syndrome group B protein promotes mito-
chondrial DNA stability by supporting the DNA repair associ-
ation with the mitochondrial membrane. FASEB J. 24, 2334–
2346.
19. Scheibye-Knudsen, M., Ramamoorthy, M., Sykora, P.,
Maynard, S., Lin, P.C., Minor, R.K., Wilson, D.M., 3rd, Cooper,
M., Spencer, R., de Cabo, R., et al. (2012). Cockayne syndrome
group B protein prevents the accumulation of damaged
an Journal of Human Genetics 94, 246–256, February 6, 2014 255

mitochondria by promoting mitochondrial autophagy. J. Exp.
Med. 209, 855–869.
20. Berquist, B.R., Canugovi, C., Sykora, P., Wilson, D.M., 3rd, and
Bohr, V.A. (2012). Human Cockayne syndrome B protein
reciprocally communicates with mitochondrial proteins and
promotes transcriptional elongation. Nucleic Acids Res. 40,
8392–8405.
21. Kamenisch, Y., and Berneburg, M. (2013). Mitochondrial
CSA and CSB: protein interactions and protection from
ageing associated DNA mutations. Mech. Ageing Dev. 134,
270–274.
22. Iyer, V.N., and Szybalski, W. (1963). A molecular mechanism
of mitomycin action: linking of complimentary strands.
Proc. Natl. Acad. Sci. USA 50, 355–362.
23. Koeppel, F., Poindessous, V., Lazar, V., Raymond, E., Sarasin,
A., and Larsen, A.K. (2004). Irofulven cytotoxicity depends
on transcription-coupled nucleotide excision repair and is
correlated with XPG expression in solid tumor cells. Clin.
Cancer Res. 10, 5604–5613.
24. Zheng, H., Wang, X., Warren, A.J., Legerski, R.J., Nairn, R.S.,
Hamilton, J.W., and Li, L. (2003). Nucleotide excision repair-
and polymerase eta-mediated error-prone removal of mito-
mycin C interstrand cross-links. Mol. Cell. Biol. 23, 754–761.
25. Ryan, D.P., andOwen-Hughes, T. (2011). Snf2-family proteins:
chromatin remodellers for any occasion. Curr. Opin. Chem.
Biol. 15, 649–656.
26. Claros, M.G., and Vincens, P. (1996). Computational method
to predict mitochondrially imported proteins and their target-
ing sequences. Eur. J. Biochem. 241, 779–786.
27. Sahin, E., and DePinho, R.A. (2012). Axis of ageing: telomeres,
p53 and mitochondria. Nat. Rev. Mol. Cell Biol. 13, 397–404.
28. Trushina, E., and McMurray, C.T. (2007). Oxidative stress and
mitochondrial dysfunction in neurodegenerative diseases.
Neuroscience 145, 1233–1248.
29. Rossi, D.J., Jamieson, C.H., and Weissman, I.L. (2008). Stems
cells and the pathways to aging and cancer. Cell 132, 681–696.
30. Pang, Q. (2011). HSCs: stressing out over ROS. Blood 118,
2932–2934.
256 The American Journal of Human Genetics 94, 246–256, February
31. Kumari, U., Ya Jun, W., Huat Bay, B., and Lyakhovich, A.
(2014). Evidence of mitochondrial dysfunction and impaired
ROS detoxifying machinery in Fanconi Anemia cells. Onco-
gene 33, 165–172.
32. Kraemer, K.H., Patronas, N.J., Schiffmann, R., Brooks, B.P.,
Tamura, D., and DiGiovanna, J.J. (2007). Xeroderma pigmen-
tosum, trichothiodystrophy and Cockayne syndrome: a
complex genotype-phenotype relationship. Neuroscience
145, 1388–1396.
33. Hayashi, M. (2008). Roles of oxidative stress in xeroderma
pigmentosum. Adv. Exp. Med. Biol. 637, 120–127.
34. Pereboeva, L., Westin, E., Patel, T., Flaniken, I., Lamb, L., Klin-
gelhutz, A., and Goldman, F. (2013). DNA damage responses
and oxidative stress in dyskeratosis congenita. PLoS ONE 8,
e76473.
35. Kirwan, M., Beswick, R., Walne, A.J., Hossain, U., Casimir, C.,
Vulliamy, T., and Dokal, I. (2011). Dyskeratosis congenita and
the DNA damage response. Br. J. Haematol. 153, 634–643.
36. Brooks, P.J. (2013). Blinded by the UV light: how the focus on
transcription-coupled NER has distracted from understanding
the mechanisms of Cockayne syndrome neurologic disease.
DNA Repair (Amst.) 12, 656–671.
37. Kashiyama, K., Nakazawa, Y., Pilz, D.T., Guo, C., Shimada, M.,
Sasaki, K., Fawcett, H., Wing, J.F., Lewin, S.O., Carr, L., et al.
(2013). Malfunction of nuclease ERCC1-XPF results in diverse
clinical manifestations and causes Cockayne syndrome, xero-
derma pigmentosum, and Fanconi anemia. Am. J. Hum.
Genet. 92, 807–819.
38. Dokal, I. (2011). Dyskeratosis congenita. Hematology (Am Soc
Hematol Educ Program) 2011, 480–486.
39. Shtessel, L., and Ahmed, S. (2011). Telomere dysfunction in
human bone marrow failure syndromes. Nucleus 2, 24–29.
40. Savage, S.A., and Bertuch, A.A. (2010). The genetics and clin-
ical manifestations of telomere biology disorders. Genet.
Med. 12, 753–764.
41. Cawthon, R.M. (2009). Telomere length measurement by a
novel monochrome multiplex quantitative PCR method.
Nucleic Acids Res. 37, e21.
6, 2014




















