
Engineering of the synthetic metabolic pathway for ... - IS MUNI
177
MASARYK UNIVERSITY FACULTY OF SCIENCE LOSCHMIDT LABORATORIES DEPARTMENT OF EXPERIMENTAL BIOLOGY Engineering of the synthetic metabolic pathway for biodegradation of environmental pollutant Doctoral dissertation Pavel Dvořák Supervisors: Prof. Mgr. Jiří Damborský, Dr. Doc. RNDr. Zbyněk Prokop, Ph.D. BRNO 2014
-
Upload
khangminh22 -
Category
Documents
-
view
3 -
download
0
Transcript of Engineering of the synthetic metabolic pathway for ... - IS MUNI

MASARYK UNIVERSITY
FACULTY OF SCIENCE
LOSCHMIDT LABORATORIES
DEPARTMENT OF EXPERIMENTAL BIOLOGY
Engineering of the synthetic metabolic
pathway for biodegradation of
environmental pollutant
Doctoral dissertation
Pavel Dvořák
Supervisors:
Prof. Mgr. Jiří Damborský, Dr.
Doc. RNDr. Zbyněk Prokop, Ph.D.
BRNO 2014


Poděkování
Na tomto místě bych chtěl poděkovat svému školiteli Jiřímu Damborskému za
profesionální vedení, schopnost a odhodlání dělat kvalitní, nepodlézavou vědu, ochotu
setkávat se, diskutovat a radit, která není ani mezi školiteli postgraduálních studentů zcela
automatická, a konečně za jeho víru ve zdárné konce, která mě dovedla až k sepsání této
práce.
Velmi děkuji i Zbyňku Prokopovi, mému školiteli specialistovi, za jeho optimismus a cenné
rady a všem svým současným i minulým kolegům, kteří přispívali a přispívají k tomu, že
Loschmidtovy laboratoře jsou nejenom špičkovým vědeckým týmem, ale i příjemným
místem pro práci, kvůli kterému se člověk každé pondělní ráno rád přiměje vstát z postele.
Největší dík ale patří mé ženě Monice za její lásku, přátelství, toleranci a schopnost vracet
mě z badatelských výšin zpátky nohama na zem.

Bibliographic entry
Author: Mgr. Pavel Dvořák
Loschmidt Laboratories
Department of Experimental Biology
Faculty of Science
Masaryk University
Title of dissertation: Engineering of the synthetic metabolic pathway for
biodegradation of environmental pollutant
Study Programme: Biology
Field of study: Molecular and Cellular Biology
Supervisor: Prof. Mgr. Jiří Damborský, Dr.
Supervisor-specialist: doc. RNDr. Zbyněk Prokop, PhD.
Year of defence: 2014
Keywords: biocatalysis; biodegradation; kinetic modelling; metabolic
engineering; in vitro multi-enzyme reaction; synthetic
biology; 1,2,3-trichloropropane

Bibliografický záznam
Autor: Mgr. Pavel Dvořák
Loschmidtovy laboratoře
Ústav experimentální biologie
Přírodovědecká fakulta
Masarykova univerzita
Název disertace: Inženýrství syntetické metabolické dráhy pro biodegradaci
environmentálního polutantu
Studijní program: Biologie
Studijní obor: Molekulární a buněčná biologie
Školitel: Prof. Mgr. Jiří Damborský, Dr.
Školitel specialista: doc. RNDr. Zbyněk Prokop, PhD.
Rok obhajoby: 2014
Klíčová slova: biokatalýza; biodegradace; kinetické modelování;
metabolické inženýrství; in vitro multi-enzymová reakce;
syntetická biologie; 1,2,3-trichlorpropan


Scio me nihil scire.
(I know that I know nothing)
Socrates
Success consists of going from failure to failure without loss of enthusiasm.
Winston Churchill
© Pavel Dvořák, Masaryk University 2014


CONTENT
MOTIVATION 1
ABSTRACT 2
ABSTRAKT 4
INTRODUCTION 7
1. Emerging technologies for engineering of metabolic pathways 7
1.1 Metabolic engineering 9
1.1.1 Strategies and tools 10
1.2 Synthetic biology 17
1.2.1 Strategies and tools 18
1.3 Protein engineering 21
1.3.1 Strategies and tools 21
1.4 Perspectives 24
2. Engineering of biodegradation pathways 25
2.1 Strategies and tools 27
2.2 Perspectives 32
3. Engineering of the synthetic metabolic pathway for
biodegradation of 1,2,3 trichloropropane 34
3.1 Halogenated hydrocarbons and 1,2,3-trichloropropane 34
3.2 Overview of 1,2,3-trichloropropane pathway engineering 38
CONTRIBUTION TO THE RESULTS 47

CHAPTER 1
In vitro assembly and immobilization of the synthetic pathway for biodegradation of toxic recalcitrant pollutant 1,2,3- trichloropropane 49
SUPPLEMENTARY TABLES AND FIGURES 69
CHAPTER 2
Maximizing the efficiency of in vitro multi-enzyme process by
stoichiometry optimization 79
SUPPLEMENTARY TABLES AND FIGURES 93
CHAPTER 3
Computer-assisted engineering of the synthetic pathway for
biodegradation of 1,2,3-trichloropropane in heterologous host E. coli 99
SUPPLEMENTARY TABLES AND FIGURES 119
CHAPTER 4
Assembly of the synthetic pathway for biodegradation of
1,2,3-trichloropropane in Pseudomonas putida KT2440 CF1 131
SUPPLEMENTARY TABLES AND FIGURES 145
SUMMARY 147
REFERENCES 148
CURRICULUM VITAE 160
LIST OF PUBLICATIONS 162
LIST OF CONTRIBUTIONS AT CONFERENCES AND SYMPOSIA 163



Motivation
- 1 -
MOTIVATION
Nature possess a great potential to cope itself with many problems arising from
continuously increasing human activity on the Earth. Among others, this potential is
hidden in astonishing variability of metabolic pathways of living organisms. For example,
it is not completely rare phenomenon that bacterium can adapt its metabolic traits for
new substrate and break down a toxic polluting compound. However, in certain cases, the
evolution is not fast enough or ends in a deadlock. Such challenges can be possibly solved
by state-of-the-art tools of recently established scientific disciplines including metabolic
engineering, protein engineering and synthetic biology. Still, the huge complexity of
dynamic processes in living cell represents a major bottleneck for any attempts to
rationally engineer natural or synthetic metabolic pathways for the purpose of
biodegradation of environmental pollutants or biosynthesis of value added chemicals. The
complexity can be significantly reduced by reconstructing selected multi-enzyme
reactions in vitro or by assembling orthogonal metabolic modules within an organism.
Such model studies help us to understand the dynamic behavior of metabolic pathways
and the obtained knowledge can be step-by-step utilized for rational engineering of living
systems.
The objectives of the Ph.D. project and this Thesis:
1. Introduction to the fields of metabolic engineering and synthetic biology with special
attention devoted to the knowledge-based engineering of biodegradation pathways;
prologue to the selected model system - synthetic metabolic pathway for
biodegradation of anthropogenic pollutant 1,2,3-trichloropropane.
2. In vitro reconstruction and immobilization of the model pathway. Development of the
biodegradation process based on immobilized enzymes.
3. Detailed kinetic characterization of employed enzymes, development and validation of
kinetic model for the pathway in vitro.
4. Optimization of the pathway in vitro by employment of suitable engineered enzymes
and balancing of biocatalysts' stoichiometry using kinetic modelling.
5. Application of obtained knowledge and synthetic biology tools for rational engineering
of the pathway in heterologous host Escherichia coli and dissection of the pathway
bottlenecks in vivo.
6. Selection of suitable microbial host (chassis) for biodegradation of
1,2,3-trichloropropane and further evolution of the synthetic pathway.

Abstract
- 2 -
ABSTRACT
This Thesis describes the application of synthetic biology and metabolic engineering
approaches for rational redesign of metabolic pathway for biodegradation of important
environmental pollutant 1,2,3-trichloropropane (TCP). The emerging technologies for
engineering of biosynthetic and biodegradation pathways are described in the
Introduction of the Thesis and prologue to the origins of synthetic TCP route is provided.
The pathway consisting of three enzymes – haloalkane dehalogenase, haloalcohol
dehalogenase and epoxide hydrolase - from two different microorganisms can convert
toxic TCP into harmless product glycerol but suffers from several important bottlenecks.
The four studies described in the Results section of the Thesis aim at rational dissection of
these handicaps, optimization of pathway performance and construction of biocatalyst
utilizable for TCP removal from the contaminated sites.
Chapter 1 of the Results section describes reconstruction of the model pathway in in
vitro conditions and developing of a novel biotechnology for TCP transformation based on
immobilized enzymes. The efficiency of the pathway was enhanced by employment of
engineered haloalkane dehalogenase with improved activity toward TCP and the route
was immobilized in the form of purified enzymes or cell-free extracts. The performance of
the three-enzyme system was tested in batch and continuous operations. The study
provides the first available report on the use of an immobilized synthetic metabolic
pathway employing engineered enzyme for the biotransformation of the toxic industrial
waste into desirable commodity chemical.
Further improvement of the reaction efficiency can be achieved by tuning enzymes'
stoichiometry. Development of the workflow for maximizing the efficiency of in vitro
multi-enzyme process by stoichiometry optimization is addressed in Chapter 2. In this
study, we employed kinetic modelling to maximize the efficiency of a three-enzyme
system based on in vitro assembled TCP pathway. Mathematical modelling and one-pot
multi-enzyme laboratory experiments provided detailed insight into pathway dynamics,
enabled the selection of suitable engineered enzyme and afforded high yield of the final
product glycerol, while minimizing biocatalyst loadings. The study highlights the potential
of kinetic modelling for industrial biocatalysis and presents a broadly applicable strategy
for optimizing multi-enzyme processes.
Chapter 3 describes application of previously developed kinetic model for
computer-assisted engineering of TCP pathway in heterologous host Escherichia coli. We
assembled TCP route in the laboratory strain E. coli BL21 (DE3), and used it as an
orthogonal biological system for thorough investigation of pathway bottlenecks in vivo.
Variants of the pathway employing wild-type or engineered haloalkane dehalogenase
were designed using modified mathematical model. The E. coli recombinants with
optimized and non-optimized stoichiometry of pathway enzymes were constructed and
characterized in terms of their viability in presence of TCP and degradation efficiency. The
validated model was used to quantitatively describe the kinetic limitations of currently
available enzyme variants, and to predict improvements required for further pathway

Abstract
- 3 -
optimization. The study highlights the potential of rational engineering of microorganisms
for the degradation of toxic anthropogenic compounds.
Last chapter of the Results section, Chapter 4, is focused on transfer of TCP pathway
from laboratory strain E. coli BL21 (DE3) into robust heterologous host Pseudomonas
putida KT2440, equipped with repertoire of metabolic and physiological functions that
make it less susceptible to stress accompanying degradation of some problematic
compounds including chlorinated organic pollutants. We decided to implant the TCP
pathway into this host to verify its suitability for TCP biodegradation and further tuning of
the synthetic route by in vivo evolution. Synthetic operon encoding three enzymes from
TCP pathway was assembled and introduced into P. putida chromosome. Obtained
constructs will be further characterized and compared with their E. coli counterparts. The
concluding section of the chapter discusses future directions in engineering of the
synthetic metabolic pathway for biodegradation of TCP.
In summary, the Thesis presents an innovative concept for rational engineering of a
synthetic metabolic pathway for degradation of toxic recalcitrant compound based on
detailed understanding of studied system and exploitation of in vitro, in silico and in vivo
tools of synthetic biology and metabolic engineering.

Abstrakt
- 4 -
ABSTRAKT
Tato práce popisuje využití metod syntetické biologie a metabolického inženýrství k
racionálnímu redesignu metabolické dráhy pro biodegradaci důležitého
environmentálního polutantu 1,2,3-trichlorpropanu (TCP). V úvodu práce jsou čtenáři
přiblíženy nové technologie a přístupy pro inženýrství biosyntetických a biodegradačních
drah společně s původem a počátky studia syntetické TCP dráhy. Dráha, která se skládá ze
tří enzymů – halogenalkandehalogenasy, haloalkoholdehalogenasy a epoxidhydrolasy –
pocházejících ze dvou různých mikroorganismů, dokáže přeměnit toxický TCP na
neškodný produkt glycerol. Efektivita této přeměny je však negativně ovlivněna několika
významnými limitacemi dráhy. Čtyři studie, které jsou shrnuty ve výsledkové části této
práce, se snaží tato omezení popsat, racionálně optimalizovat fungování dráhy a připravit
biokatalyzátor, který by mohl být využit k dekontaminaci míst znečištěných TCP.
Kapitola 1 výsledkové části popisuje rekonstrukci modelové dráhy v podmínkách in
vitro a vývoj nové biotechnologie pro transformaci TCP založené na imobilizovaných
enzymech. Efektivita dráhy byla zvýšena zapojením halogenalkandehalogenasy s uměle
zvýšenou aktivitou s TCP a dráha byla imobilizována v podobě purifikovaných enzymů či
bezbuněčných extraktů. Výkon tříenzymového systému byl testován v třepaných lahvích i
v průtokovém bioreaktoru. Tato studie je jedinou doposud publikovanou prací popisující
využití imobilizované syntetické metabolické dráhy s enzymem upraveným proteinovým
inženýrstvím pro biotransformaci toxické průmyslové odpadní látky na žádanou
komoditní chemikálii.
Dalšího zvýšení efektivity reakce může být dosaženo úpravou stechiometrie enzymů
v dráze. Kapitola 2 popisuje vývoj pracovního postupu pro maximalizaci efektivity in vitro
multienzymového procesu pomocí optimalizace stechiometrie. V této studii jsme využili
kinetické modelování pro zlepšení efektivity tříenzymového systému založeného na in
vitro rekonstruované TCP dráze. Matematické modelování podpořené laboratorními
experimenty poskytlo detailní vhled do dynamického fungování dráhy, umožnilo výběr
vhodného enzymu upraveného proteinovým inženýrstvím a přispělo tak k získání
maximálního možného výtěžku finálního produktu glycerolu při využití minimálního
potřebného množství enzymů. Studie zdůrazňuje potenciál kinetického modelování pro
průmyslovou biokatalýzu a nabízí široce využitelnou strategii pro optimalizaci
multienzymových procesů.
Kapitola 3 se věnuje aplikaci dříve popsaného kinetického modelu v inženýrství TCP
dráhy v heterologním hostiteli, bakterii Escherichia coli. TCP dráha byla sestavena v
laboratorním kmeni E. coli BL21 (DE3) a tento ortogonální systém byl využit k důkladné
analýze limitací dráhy v podmínkách in vivo. Za využití modifikovaného matematického
modelu byly navrženy varianty dráhy s divokým typem halogenalkandehalogenasy i s
mutanty připravenými proteinovým inženýrstvím. Byly zkonstruovány rekombinantní
bakterie E. coli s optimalizovanou a neoptimalizovanou stechiometrií enzymů dráhy.
Konstrukty byly experimentálně charakterizovány z pohledu jejich schopnosti degradovat
TCP a přežít v jeho přítomnosti. Ověřený model byl použit ke kvantitativnímu popisu

Abstrakt
- 5 -
kinetických limitací v současnosti dostupných variant enzymů dráhy a k predikci dalších
kroků nutných ke zlepšení limitních parametrů. Tato studie zdůrazňuje potenciál
racionálního inženýrství mikroorganismů pro degradaci toxických látek antropogenního
původu.
Poslední kapitola výsledkové části, Kapitola 4, je zaměřena na popis přenosu TCP
dráhy z laboratorního kmene E. coli BL21 (DE3) do robustního hostitele, bakterie
Pseudomonas putida KT2440. Tato bakterie je přirozeně vybavena repertoárem
metabolických a fyziologických funkcí, které ji dělají méně náchylnou ke stresu
doprovázejícímu degradaci některých problematických látek včetně chlorovaných
organických polutantů. Rozhodli jsme se přenést TCP dráhu do tohoto hostitele, abychom
ověřili jeho použitelnost pro účely biodegradace TCP a dalšího vývoje dráhy pomocí in
vivo evoluce. Studie popisuje přípravu syntetického operonu kódujícího enzymy TCP
dráhy, jeho vnesení do chromosomu P. putida a základní charakterizaci vzniklých
konstruktů. Získané rekombinantní bakterie budou dále charakterizovány a srovnány s
konstrukty E. coli. V závěru kapitoly autor diskutuje možné budoucí směry dalšího
inženýrství syntetické metabolické dráhy pro biodegradaci TCP.
Tato práce tak prezentuje inovativní koncept racionálního inženýrství syntetické
metabolické dráhy pro degradaci toxického environmentálního polutantu, založený na
detailní znalosti studovaného biologického systému a využití in vitro, in silico a in vivo
nástrojů syntetické biologie a metabolického inženýrství.

- 6 -

Introduction
- 7 -
INTRODUCTION
1. Emerging technologies for engineering of metabolic
pathways
Metabolism is a network of life-sustaining biochemical reactions in living cells
catalyzed by enzymes. These reactions can be divided into two principal categories: (i)
catabolic, that break down organic matter and produce energy through cellular
respiration, and (ii) anabolic, that use energy to synthesize biomolecules. Series of
catabolic or anabolic enzymatic reactions are realized via metabolic pathways. The
metabolic pathways are closely interconnected with cell signalling pathways and, taking
into account the central dogma of molecular biology, also with information-storing
molecules DNA and RNA. Mutual dependences of these basic cell components and
principles of their orchestration for the purpose of cell maintenance, reproduction or
adaptation to the new conditions are still not fully understood [1]. The dynamic
complexity of a metabolism is encoded already in the word itself that is derived from
Greek μεταβολή [metabolē], which means a change. This complexity shelters yet
unexplored fortune and endless space of possible solutions for many problems of
mankind to come in 21st century, but is also an evident obstacle for our attempts to
domesticate microbes, animals or plants for biosynthesis of value-added chemicals or for
biodegradation of polluting compounds.
In contrast to their purely chemical counterparts, biocatalytic reactions can proceed
in moderate conditions, but with high rate and specificity. The humans have taken
advantage of the biochemical trails of living organisms in classical biotechnology for
thousands of years without the knowledge of molecular basis of cellular processes. Yeasts,
bacteria and fungi have been used in agriculture, food production and medicine. Recently,
the modern biotechnology adopted the principles of sustainable living and our effort to
decode and exploit the metabolic processes and the life itself has significantly increased.
In the last decades of 20th century, biologists and biochemists focused on study of complex
natural systems, e.g., bacterial cell, and tried to dissect them into pieces and understand
the structure and function of basic parts like nucleic acids and proteins [2–4]. At the same
time, they learned how to use some of the discovered biological components, e.g.,
restriction enzymes, DNA ligases or DNA polymerases, as tools for system manipulation
[5,6]. These initial discoveries in 1950s, 60s, and 70s gave rise to the recombinant DNA
technology and molecular biology.
Since that time, we have been moving from analytical age to a synthetic age. Now,
variety of parts and tools is available and the possibilities expanded together with
emerging fields of metabolic engineering, synthetic biology and protein engineering [7,8].
The toolkits of these three scientific disciplines are being combined to utilize and tailor
biocatalytic reactions both in vivo and in vitro (Figure 1). The increasing computational

Introduction
- 8 -
power and invention of chemical DNA synthesis allow the biologists and chemists to adopt
engineering principles and construct some biological parts like DNA molecules or
enzymes also de novo [9,10]. The recent corporate interest in metabolic engineering and
synthetic biology was highlighted by their placing among the TOP 10 emerging
technologies for 2012 selected by the World Economic Forum. These disciplines are also
embedded in the European Union call appeal for the Knowledge Based Bio-Economy
towards 2020. The detailed discussion on all three subjects would be behind the scope of
this text. Nevertheless, the fundamentals described in the following paragraphs will
provide sufficient introduction into the matter and explanation of important terms used in
the Results section of the Thesis.
Figure 1. Utilization of metabolic engineering, protein engineering and synthetic biology for tailoring metabolic pathways in vivo and in vitro. The toolkits and the levels of operation of three emerging technologies partially overlap. Synthetic biology targets predominantly nucleic acid level and manipulates the DNA and RNA parts in order to design controllable biological systems. Protein engineering aims to optimize stability and catalytic properties of crucial pathway enzymes. Metabolic engineering balances expression of pathway enzymes (blue, red and green circle), minimizes accumulation of intermediates (M1, M2), improves uptake of substrates (S) and maximizes formation of products (P) in the target pathway.

Introduction
- 9 -
1.1 Metabolic engineering
Metabolic engineering (ME) was firstly defined as a new scientific discipline by James
E. Bailey in 1991 as "...the improvement of cellular activities by manipulations of
enzymatic, transport, and regulatory functions of the cell with the use of recombinant
DNA technology." [11]. The first international conference on ME was held in 1996 in
Danvers, Massachusetts, USA, and in 1998 the journal Metabolic Engineering was
established. The field has evolved primarily to allow alternative biological production of
drugs, fuels, and biomaterials or biodegradation of anthropogenic pollutants by
engineered organisms. The effort is driven by the shortage of fossil reserves and
accumulation of toxic man-made chemicals in the environment. The ME has a great
potential for growth in following decades. Up to 20% of global chemistry market
(estimated at 2,292 billion US$) could by covered by biotechnological products by 2020
[12].
The ME is now understood as a practice of optimizing genetic and regulatory
processes within cells to: (i) improve the yield and productivity of native products
synthesized by organisms, (ii) extend the range of substrates or improve the uptake of
substrate, or (iii) establish production of products that are new to the host cell [7,13,14].
These goals can be achieved either by engineering of natural metabolic pathways present
in the host cell or synthetic routes assembled from enzymes (genes) originating from
different organisms. Metabolic engineers usually model these biocatalytic reactions using
mathematical apparatus, calculate a yield of useful products, and determine the
constraints for their production. Computational and experimental tools are applied to
overcome these constraints and establish cost effective process. Maximum yield or uptake
of desired substance must be balanced with the natural survival needs of the host cell.
Bacteria are widely accepted host organisms in ME. The most explored model
organism Escherichia coli is also the most frequently used host for molecular cloning and
heterologous expression of recombinant enzymes in ME [15]. Its major advantages are
sequenced genome (4.6 Mbp) and wide range of tools developed for its genetic
manipulation, rapid growth, fast doubling time (20-30 min) and high cell densities
achieved in simple synthetic media. E. coli BL21(DE3) (E. coli B F– dcm ompT hsdS(rB–,
mB–) gal λ(DE3)) is widely used strain for overexpression of recombinant proteins and
proved its utility also in many ME studies. The host is a lysogen of λDE3 and contains the
T7 RNA polymerase gene, under the control of the lacUV5 promoter, integrated into the
chromosome. IPTG is used to induce the expression of recombinant proteins cloned into
vectors downstream of a T7 RNA promoter and transformed into the BL21 (DE3) cells.
Other bacterial hosts utilized in academic or industrial laboratories are for example
Bacillus subtilis (production of food enzymes), Pseudomonas putida (biodegradation of
organic pollutants), Streptomyces (production of antibiotics) or cyanobacteria (biofuel
production). In certain cases, properties of procaryotic cells are not sufficient for
expression of target recombinant gene, e.g., due to the missing posttranslational

Introduction
- 10 -
modifications, and eucaryotic hosts like Saccharomyces cerevisiae (production of
bioethanol or terpenoids), fungi (production of antibiotics), mammalian or insect cells
(production of biopharmaceuticals) are employed. The selection of the suitable host
organism is the crucial step at the beginning of any ME project aimed at in vivo pathway
engineering. The engineering of microbial hosts will be discussed in the Introduction and
the Results parts of the Thesis.
1.1.1 Strategies and tools
Microorganisms have been employed for human benefit for thousands of years and
genetically manipulated for decades. Traditionally, species that naturally produced a
desired molecule were identified and then improved through classical strain engineering
on the basis of random mutagenesis induced by chemical (e.g., nitrosoguanidine) or
physical (e.g., UV irradiation) mutagens, application of mutator strains or adaptive
laboratory evolution in chemostat and subsequent screening for the best microbial
factories [7]. More recently, random gene knock-outs and overexpression through
transposon mutagenesis or whole-genome shuffling of selected parental strains with
beneficial properties were employed in a hunt for phenotypic improvement [16,17]. This
has been a very efficient strategy and has resulted in low-cost production of many
different chemicals including penicillin, citric acid, glutamate or lysine, to name a few
[18,19]. However, demanding screening and resulting variants with many undefined
mutations are disadvantageous characteristics of the classical approach.
The attempts to tune production strains rationally appeared with the advent of ME
and development of more sophisticated genetic engineering tools. The complexity of
solved problems made the field become highly interdisciplinary joining together
molecular biology, bioinformatics, microbiology, biochemistry, mathematics or system
biology and many omics disciplines, e.g., metabolomics, proteomics, genomics and
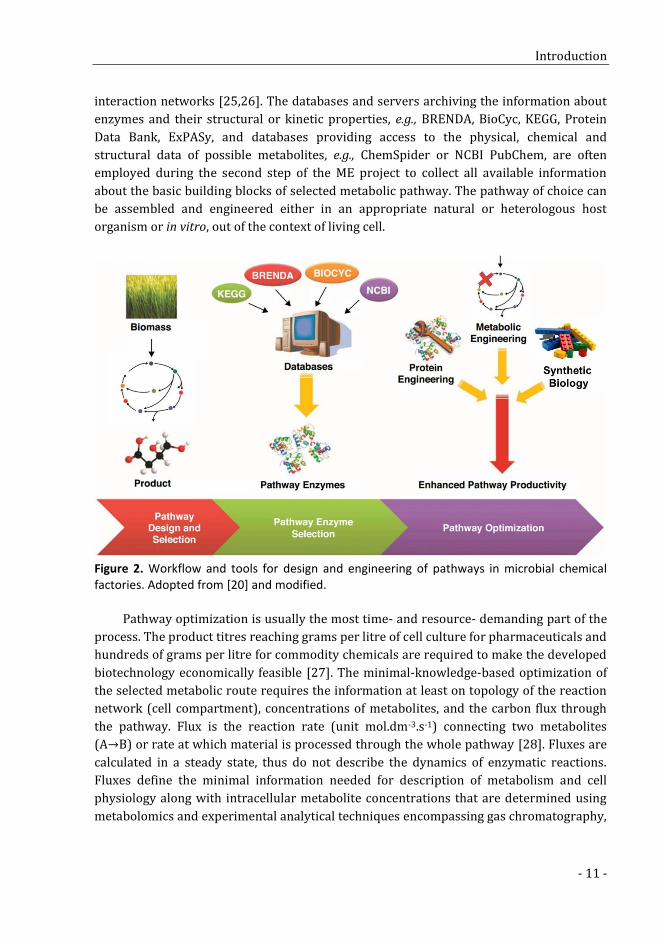
transcriptomics. Typical ME project now consists of three basic steps including: (i)
pathway selection and design, (ii) selection of suitable pathway enzymes, and (iii)
pathway optimization (Figure 2) [20]. Numerous computational and experimental tools
can be utilized to carry out these three steps. Thorough repertoires are listed in some
recent excellent reviews [20–23] and the following paragraphs will provide the overview
of only the most common tools.
Computational tools. The databases of anabolic and catabolic pathways like MetaCyc
or University of Minnesota Biocatalysis/Biodegradation Database can be used for
identification of pathway that can break down or produce a compound of interest. A more
advanced tool, Biochemical Network Integrated Computational Explorer (BNICE), predicts
also unknown pathways that are potentially chemically feasible, taking into account the
starting compound and/or product, the requested length of the pathway and the broader
reaction rules of the Enzyme Commission classification system [24]. Softwares like
CellDesigner or Cytoscape can visualize the defined reaction or metabolic-signalling
Introduction
- 11 -
interaction networks [25,26]. The databases and servers archiving the information about
enzymes and their structural or kinetic properties, e.g., BRENDA, BioCyc, KEGG, Protein
Data Bank, ExPASy, and databases providing access to the physical, chemical and
structural data of possible metabolites, e.g., ChemSpider or NCBI PubChem, are often
employed during the second step of the ME project to collect all available information
about the basic building blocks of selected metabolic pathway. The pathway of choice can
be assembled and engineered either in an appropriate natural or heterologous host
organism or in vitro, out of the context of living cell.
Figure 2. Workflow and tools for design and engineering of pathways in microbial chemical factories. Adopted from [20] and modified.
Pathway optimization is usually the most time- and resource- demanding part of the
process. The product titres reaching grams per litre of cell culture for pharmaceuticals and
hundreds of grams per litre for commodity chemicals are required to make the developed
biotechnology economically feasible [27]. The minimal-knowledge-based optimization of
the selected metabolic route requires the information at least on topology of the reaction
network (cell compartment), concentrations of metabolites, and the carbon flux through
the pathway. Flux is the reaction rate (unit mol.dm-3.s-1) connecting two metabolites
(A→B) or rate at which material is processed through the whole pathway [28]. Fluxes are
calculated in a steady state, thus do not describe the dynamics of enzymatic reactions.
Fluxes define the minimal information needed for description of metabolism and cell
physiology along with intracellular metabolite concentrations that are determined using
metabolomics and experimental analytical techniques encompassing gas chromatography,

Introduction
- 12 -
liquid chromatography, or capillary electrophoresis in combination with mass
spectrometry [29].
Mathematical models of metabolic networks play a central role in ME and pathway
optimization. Models are often deposited in standardized Systems Biology Markup
Language (SBML) in order to allow their sharing among diverse users. Modelling helps to
analyze the selected pathway (e.g., estimate thermodynamic feasibility) and identify
reactions that need to be modified to improve its performance. Flux Balance Analysis
(FBA) and Metabolic Flux Analysis (MFA) are two major techniques used for calculation
and modelling of intracellular fluxes [28,30]. FBA is a theoretical concept and represents a
direct application of linear programming to biological systems. It uses the stoichiometric
coefficients for each reaction in the system as the set of constraints for the optimization.
MFA determines intracellular fluxes from measurable rates of metabolite uptake and
secretion in growth medium. Substrates labelled with isotope 13C are used in chemostat
grown cultures to trace fluxes through a network. Eventually, the Cobra 2.0 toolbox for
Matlab, OptFlux or OptKnock platforms can implement constraint-based flux calculations
and suggest strategies based on knock-out or overexpression of target genes to optimize
metabolite production without compromising the cell growth [31–33]. Although, FBA and
MFA can deal with genome-scale metabolic models and do not require information on
enzyme kinetics, their output gives just steady-state approximation of dynamic reality in
living cell.
Kinetic modelling employed in the chapters 2 and 3 of the Thesis' Results section,
provides a good alternative to static flux calculations when reliable kinetic parameters of
pathway enzymes and some other input data, e.g., specific surface area of the cell,
permeability coefficient for substrate or approximate enzyme amount in the cell, are
available [15,34]. Kinetic constants can be obtained from enzyme databases or
determined experimentally with purified enzymes or cell-free extracts. One should keep in
mind that most of the kinetic constants deposited in databases are not standardized and
measured under in vivo-like conditions, which may be a limiting factor when applying
such data for kinetic modelling in in vivo systems [35,36]. Computational tools like
COPASI, Scientist, or E-Cell are eventually used to assemble the kinetic model of the
pathway in a form of kinetic and differential equations and simulate reaction time courses
for various conditions [37,38]. Once are the bottleneck reaction steps identified either by
kinetic modelling or flux analysis, experimental techniques are applied in order to target
corresponding gene(s) or regulatory mechanisms.
Experimental tools. Experimental tools for pathway optimizing are applied hand in
hand with the theoretical tools. They are mostly based on recombinant DNA technology
and their employment often generates transgenic organisms. Traditionally, engineering
input is conducted on the level of gene expression that affects the quantity of protein
molecules (Figure 3). Pathway optimizing often requires combination of gene deletions,
which may prevent the leakage of carbon to the unwanted by-products formed in
competing metabolic pathways, and balancing expression of several enzymes, preventing

Introduction
- 13 -
accumulation of intermediates potentially toxic to the host cell and streams the carbon
matter directly to the final product [39,40]. Expression of a target gene can be completely
nullified by knock-out techniques. Chromosomal gene disruption by phage λ Red
recombinase assisted homologous recombination is widely used procedure especially for
E. coli and other Gram-negative bacteria [41,42]. Engineering cuts at the level of gene
transcription and translation are utilized for fine-tuning of expression levels of pathway
components. Up and down regulation of gene expression can be achieved by: (i)
engineering synthetic promoter libraries, e.g., by random or targeted mutagenesis of
relevant promoter regions, (ii) engineering stability of mRNA molecules through tunable
intergenic regions with mRNA secondary structures or RNase cleavage sites, (iii) applying
RNA interference, or (iv) engineering ribosome binding sites [43–46]. Pathway
performance can be tuned also at the protein level through improved spatial organization
of enzymes and substrate channeling [47,48].
Assembly of new synthetic pathway in a heterologous host often requires parallel
overexpression of several genes. One popular approach, applied also in the Chapter 4 of
this Thesis' Results section, is to use the combinations of Novagen Duet plasmid vectors
(Merck Millipore, Germany) that carry compatible replication origins and independent
antibiotic markers. This allows effective propagation and maintenance of four plasmids
and simultaneous co-expression of up to eight genes in a single E. coli BL21(DE3) cell [49].
Duet vectors proved to be useful in ME studies focused on production of biodiesel,
heparosan, or pinocembrin [50–52]. Recently, Xu and co-workers modified Duet vectors
by introducing restriction sites for isocaudamer pairs AvrII, NheI, SpeI, and XbaI, allowing
rapid modular assembly of a number of regulatory elements (promoters, operators,
ribosome binding sites, and terminators) and multi-gene pathways [53]. Resulting
ePathBrick vectors provided a platform for optimizing pathway gene configuration and
gene order which was demonstrated by constructing 54 variants of flavonoid pathway
with three genes either in monocistronic, pseudo-operon or operon configuration within
one week. SEVA (Standard European Vector Architecture) plasmids with standardized
architecture (Figure 4) and nomenclature represent alternative modular vector system
tailored to allow cloning or expression of heterologous genes in P. putida and other Gram-
negative bacteria [54]. Various antibiotic markers, origins of replications, and cargos can
be combined in a single plasmid to fulfil the user's needs and rich collection of diverse
constructs is already available. The plasmids for the purpose of cloning, heterologous
expression, recombination or transposon mutagenesis were traditionally introduced into
the host by triparental mating, i.e. form of bacterial conjugation where a conjugative
plasmid present in one bacterial strain assists the transfer of a mobilizable plasmid
present in a second bacterial strain into a third bacterial strain [55]. Recently, faster and
convenient methods such as heat shock transformation or electroporation are frequently
used [56].

Introduction
- 14 -
Figure 3. Biomolecular targets on the genomic, transcriptional, translational and protein level (A) that can be engineered to adjust the amount and spatial organization of enzymes and improve functioning of metabolic pathways (B). Transcription machinery, enzyme promoters, ribosome binding sites (RBS), and translational machinery can be modified to adjust the expression of enzymes. Enzymes can be assembled using the protein scaffolds to optimize their spatial organization in a pathway and promote substrate channelling. Genome editing can be used to modulate host metabolism to improve flux through the target pathway. Schematic representation of two metabolic pathway in figure 3B shows the effect of increasing concentration of the second and third enzymes in the pathway on the titter of the product. Enzyme fluxes are represented by the size of the grey arrows and metabolite concentrations are represented by the size of the coloured circles. Adopted from [57] and modified.
Metabolic engineers have to keep in mind that the introduction of recombinant
plasmids and massive expression of heterologous genes can affect the fitness of the host
organism either by additional metabolic load or depletion of essential cofactors in redox
reactions [20,58]. Bypassing such troubles is possible by applying lower-copy-number
plasmids with weaker constitutive promoters and enzyme-mediated cofactor recycling
through overexpression of NAD+ kinase, transhydrogenases or dehydrogenases [59].
Additionally, expression of heterologous genes directly from host chromosome can be
beneficial for higher stability of desired genotype/phenotype and better host viability
[60]. Homologous recombination and site-specific or random transposition-based
techniques were developed to enable chromosomal insertion of single genes, gene clusters

Introduction
- 15 -
or whole synthetic operons [61–63]. Configuring pathway genes in the synthetic operon
further reduces metabolic burden. Expression of individual genes in an operon can be
tuned, e.g., by changing a gene order or multiplying the whole metabolic cassette in host
chromosome through increasing selection pressure in cell culture [64,65].
Figure 4. Overall organization of Standard European Vector Architecture (SEVA) plasmids. SEVA vectors are formed by three variable modules: a cargo (blue), a replication origin (green) and an antibiotic marker (magenta). Enzymes used to change the functional modules are shown with the same colour code. Modules are separated by three fixed regions (shared by all vectors) including the transcriptional terminators T0 and T1 and the origin oriT for broad host range transfer. Adopted from [54].
Design strategies. Since 1990's significant effort and funding have been directed
toward rational engineering of biotechnological processes. The process for production of
1,3-propanediol from glucose using a metabolically engineered E. coli K12 developed by
DuPont represents a textbook example of this approach [66]. 1,3-propanediol is a
commodity chemical used world-wide for the manufacturing of fabrics and range of
plastic-based materials. Achieving economically feasible yield of 135 g of 1,3-propanediol
per liter of cell culture required introduction and overexpression of six genes from three
different microorganisms, deletion of glycerol kinase and glycerol dehydrogenase genes
from competing pathway, and directed change in glucose uptake system of E. coli. The fact
that 575 scientists from DuPont and Genencor worked on this project continuously for 15
years illustrates the cost of viable biotechnology development and partially explains the
lack of similarly successful stories based on rational ME approach [15,67]. Thus far,
rational ME has generated interesting and sophisticated, but in many cases unproductive
demonstrations, due to the fact that effects of directed in vivo perturbations are still

Introduction
- 16 -
mostly unforeseen. Therefore, the field currently branches into more rapid strategies that
reduce the problematic complexity.
Multivariate modular metabolic engineering (MMME) is one of the solutions for
semi-rational design of cell factories. MMME approach divides multi-gene pathway of
choice into several metabolic modules based upon pathway bottleneck compounds and
enzyme turnover [68]. Expression of the modules is balanced via altering control elements
at the level of transcription (e.g,. promoter strength, gene copy number), translation (e.g.,
ribosome binding site), or enzyme properties (e.g., activity, inhibition). Eventually,
optimally balanced pathway is searched in reduced combinatorial space. The initial
implementation of this strategy enabled gram per liter production of taxadiene, precursor
of anticancer drug taxol, originally isolated from Taxus brevifolia Pacific yew tree, in E. coli
[69]. Treatment of a single patient would require harvesting dozens of full-grown trees or
complicated chemical synthesis of the compound consisting of up to 50 steps. Thus,
biosynthetic production provided reasonable alternative. The synthetic taxadiene
pathway was divided into upstream (8 genes) and downstream (2 genes) modules, which
were expressed separately from pairs of five selected plasmids with diverse copy numbers
and 3 different promoters. The screening of 32 constructed pathway variants resulted in
minimized accumulation of toxic intermediate and 15,000-fold improvement of taxadiene
production. The MMME strategy in combination with above mentioned Novagen Duet
vectors or ePathBrick vectors was successfully applied also for increasing flavonoids
production in E. coli or for achieving 8.6 gram per liter production of fatty acids in the
same host, respectively [52,70].
Despite the potential of utilizing microbial cell factories for biosynthesis or
biodegradation, possibly providing economic solutions to global challenges, the field of
ME still lacks a standards and universally applicable principles for strain optimization.
Nevertheless, the aim to engineer living systems on the rational basis remains the main
stream in the community. This desire made ME adopt strategies from another emerging
and closely related field of synthetic biology that has been recently established primarily
to understand the fundamentals of life and implement the knowledge for living system
manipulation.

Introduction
- 17 -
1.2 Synthetic biology
The origins of synthetic biology (SB) can be traced back to the completion of the
sequencing of human genome in 2001. Postgenomic era is characterized by
high-throughput sequencing and chemical synthesis of DNA (Figure 5). These
technological developments were followed by emergence of the companies that were able
not only read, but also write DNA and offer it as a commercial cost-effective product [14].
The age of SB was launched by constructing the first genetic counter and toggle switch in
E. coli. as well, because these innovations showed for the first time that artificial genetic
elements can be used for control of processes inside a microbe [71,72]. The competition
International Genetically Engineered Machine (iGEM) officially started in 2005 and since
that time has played an important role in the development of SB and dissemination of its
ideas among undergraduate and high school students [73]. In May 2010, an initial ascent
of SB culminated as a team of scientists led by Craig J. Venter synthesized Mycoplasma
mycoides genome de novo and introduced it into M. capricolum recipient cell [9].
The principal idea behind the SB is that any biological system can be regarded as a
combination of individual functional elements – not unlike those found in man-made
devices – and these parts can be combined in novel configurations to modify existing
properties or to create a new ones [74]. The overall aim of SB is to simplify biological
engineering by applying principles and designs adopted from electronic and computer
engineering in order to produce predictable and robust systems (e.g., genetic control
systems, metabolic pathways, chromosomes and cells) with novel functionalities that do
not exist in Nature [75]. This should be achieved through the assembly of
well-characterized, standardized, and reusable components.
Figure 5. Plot of price per base of DNA sequencing and synthesis. Adopted from www.synthesis.cc and modified.

Introduction
- 18 -
1.2.1 Strategies and tools
SB has been confronted with the need to precisely define its practice and to develop a
clear and generally applicable strategies from its very beginning. Synthetic biologists took
their lesson from the first decade of ME and realized that this was the only way how to
warrant and implement engineering principles into such a stochastic field as biology. The
awareness of biological complexity resulted in implementing Design-Build-Test-Analyze
(DBTA) cycle that was actually proposed for optimizing microbial production systems
already in 1991 by J.E. Bailey in his early description and vision of ME [11,75]. This
strategy can be linked to designing and building many variants of certain device or system
(e.g., genetic circuit, metabolic pathway, or whole microbial factory), selecting or
screening for variants with desired properties (e.g., the best response or the highest yield
of the product) and discarding the unproductive designs. The knowledge that is gained
from the initial rounds of DBTA cycle can be implemented in mathematical algorithms
used to improve future computer-aided designs. The more sophisticated software tools
for building biology are and the better characterized biological components we have, the
more requirement for the DBTA cycle will diminish and biological engineering will move
closer to the electronic and physical engineering disciplines [75].
There are several main areas of interest in SB that currently contribute to DBTA cycle
and the global collection of parts, devices, and basic knowledge of biological systems:
high-throughput DNA sequencing and synthesis [76]
standardization and registry of novel biological parts and devices [77]
mathematical modelling of biological systems and networks [78]
design of orthogonal systems [79]
minimal genomes and minimal cells/chasses [80]
synthetic genomes and whole-genome editing tools [81,82]
biosensors, synthetic genetic circuits and switches [83]
cell-free systems [67]
cell to cell communication [84]
de novo protein design [85]
Thorough description of all these topics is out of scope of this text and curious
readers are referred to the cited literature. The following paragraphs discuss the topics
relevant for experimental part of this Thesis.
DNA synthesis and parts standardization. Fast and affordable DNA synthesis
facilitated expansion of SB and represents the headstone for most of the research projects
including those focused on assembly of whole synthetic genomes [9,81]. Nowadays,
mostly microarray-based oligo synthesis is used to provide substrate (usually 5-50 oligos)

Introduction
- 19 -
for constructing larger synthetic fragments (usually 200-3000 bp) [76]. Scarless methods
including popular Gibson assembly, ligase cycling reaction or yeast recombination are
employed for combining sequence-verified gene-length fragments in even bigger
complexes [86,87]. Gene synthesis combined with parallel optimization of codons is very
powerful for improving expression of target genes in specific heterologous host [88].
Levels of proteins overexpressed in E. coli from codon-optimized genes can reach up to
30-40% of the total cellular protein. DNA synthesis also allows preparation and
modulation of genetic parts (promoters, ribosome binding sites, genes, terminators, etc.)
with verified sequences. Characterized parts that fulfil the defined standards in SB (e.g.,
BioBrick standard) are stored in parts registries and can be used for assembly of synthetic
genetic devices [77]. Precise definition of part property, e.g., of promoter strength or
plasmid copy number, for specific host organism and in specific conditions is crucial for
reliable input of mathematical modelling and successful design. This proved to be true
also in the Chapter 3 of the Thesis's Results section.
Mathematical modelling of biological systems and networks. The bottom-up
approach of SB and integration of DBTA cycle require close integration of experimental
and computational techniques. Similarly to ME, mathematical modelling is irreplaceable
for system design and optimization. The main tasks for SB computational tools include
collection and storage of literature data and datasheets from parts characterization,
implementation of parts in more complex modules, or model-based numerical simulations
of synthetic gene circuits and metabolic pathways [89]. Autodesk Cyborg
(www.autodeskresearch.com/projects/cyborg) and ClothoCAD (www.clothocad.org)
represent two ambitious projects that try to develop user-friendly modular software
platforms for SB data management and computer-aided design. Despite of the enormous
progress in computing capacity in recent years, the prediction accuracy of many
theoretical tools that deal with complex in vivo systems and stochastic biological
processes is still unsatisfactory. SB therefore develops several parallel strategies to
compensate for the complexity of a living cell.
Design of orthogonal systems. Orthogonal, or in other words parallel and
independent, systems are one of the key foundations in SB, making biology more
amenable to computational design. The ultimate goal for synthetic biologists is to develop
artificial genetic codes (e.g., quadruplet instead of triplet) and orthogonal transcription-
translation machineries that would allow assembly of novel metabolic pathways from
enzymes containing unnatural amino acids [79,90]. Such pathways could posses yet
unknown functions and synthesize variety of valuable compounds. Importantly,
orthogonal pathways or other modules should not interact with the natural counterparts
in the host cell. This allows better predictability of the component behaviour and
preceding of inhibitory cross-talk upon introduction of the pathway into an existing
metabolic network [91]. Also in the Chapter 3 of this Thesis, orthogonality of the studied
metabolic route in E. coli was achieved due to the fact that the reactions catalyzed by the
pathway enzymes were not naturally occurring in the heterologous host.

Introduction
- 20 -
Minimal genomes and minimal cells. SB aims to construct microbial chasses
(minimal host cells) either through lego-like de novo synthesis of viable cell or
systematical deletion of non-essential genes in the genomes of natural hosts. The latter
approach seems to be far more feasible at the moment. For example, transposon
mutagenesis-based procedure is used to produce The Keio collection of all viable E. coli
single-gene knock-outs to facilitate a systematic investigation of the regulation and
metabolism of E. coli [92]. The similar study in M. genitalium revealed that over 100 out of
482 genes were not essential for cell survival [80]. Although the function of many
essential genes is yet unknown the aim of the project is to create "M. laboratorium" chassis
with as few genes as possible. Similar attempts focus mostly on answering fundamental
questions of biology. But discarting non-essential functions in the widely used hosts like E.
coli, S. cerevisiae or P. putida can be very useful also for application purposes. It might
result in reduced complexity of the system and better efficiency of the biosynthetic or
biodegradation processes.
Engineering of cell-free systems. The major principle of cell-free synthetic biology is
that purified biomolecules or components in crude cell extracts, which can be better
monitored and modelled, substitute intact cells for constructing complex biomolecular
systems [67,93]. This approach overcomes current limitations of in vivo ME and SB, e.g.,
prospective toxicity of metabolites, interferences of multiple cell components, instability
of evolved genotypes, GMO restrictions, and provides high precision and freedom of
design. Cell-free systems can be applied for production of proteins (antibodies, vaccines,
biocatalysts) and diverse metabolites or construction of minimal cells from bottom up. In
vitro engineering is expanding also in the field of biocatalysis [29]. Metabolic pathways
reconstructed from purified enzymes or cell-free extracts allow verification of biocatalyst
functioning, determining kinetic parameters and evaluation of the network by kinetic
modelling [94-96]. In vitro multi-enzyme systems are intensively studied for their
potential in production of pharmaceuticals or biofuels. For example, Bujara and colleagues
utilized cell-free extract with endogenous glycolysis enzymes from genetically modified E.
coli for 10-step multi-enzyme synthesis of unnatural monosaccharides based on building
block dihydroxyacetone phosphate [97]. Del Campo and co-workers employed cell-free
system encompassing thirteen purified enzymes for conversion of abundant
monosacharide xylose into H2 approaching 100% of the theoretical yield [98]. Possible
disadvantages of in vitro metabolic networks are: (i) suboptimal efficiency due to the
lower enzyme concentration than in extremely dense cell cytoplasm and (ii) limited
recycling of the system [67]. These drawbacks can be solved by enzyme immobilization
and improved spatial organization via synthetic protein or DNA scaffolds that diminish
diffusion of pathway intermediates [48]. Alternatively, biocatalysts can be precipitated
and covalently interconnected in cross-linked enzyme aggregate (CLEA) particles [99].
This method was successfully applied for synthesis of toxic nucleotide analogues by
immobilized five-enzyme synthetic pathway with included ATP regeneration [100].
Similar strategy was adopted also for in vitro biodegradation of toxic environmental
pollutant described in the Chapter 1 of the Thesis.

Introduction
- 21 -
Altogether, above mentioned SB tools and approaches help closing the enormous gap
between the cellular complexity and human desire for engineering. There is no doubt that
SB has revolutionized the field of ME. Amazing example of ME and SB consonance is a ten-
year project based on co-operation of University of California with company Amyris Inc.
resulting in commercial production of semi-synthetic artemisinine [75,101]. Artemisinine
is an important antimalarian drug precursor originally derived from plant Artemisia
annua. The goal of the project was to establish economically viable microbial production
of the precursor and substitute the fluctuating supply of plant-derived drug. The number
of design and experimental steps, e.g., balancing expression of 15 codon-optimized genes
from A. annua, selection of suitable S. cerevisiae chassis, chromosomal insertions, cell free
assays or gene knock-outs, stood between the initial idea and final production of 25 g per
litre of artemisinic acid converted to the artemisinine in the last chemical step.
Importantly, this case represents a precedent that supplied valuable information in DBTA
cycle and possibly simplified the development of similar processes for biotechnological
production of some other important pharmaceuticals like anticancer paclitaxel, analgesic
morphine, anti-HIV prostatin or antibiotic bleomycin [75]. ME and SB experts would
surely agree that another emerging discipline, protein engineering, will significantly
contribute to the success of these future enterprises.
1.3 Protein engineering
Basic building blocks of metabolic pathways are enzymes - powerful catalysts that
are able to increase reaction rates by up to seventeen orders of magnitude. Due to this
capacity, isolated enzymes are now widely utilized in molecular biology, food industry,
medicine, biodegradation or production of pharmaceuticals. However, only limited
number of enzymes naturally catalyzes the reactions that biotechnologists require and
which occur under conditions that are industrially acceptable and economically feasible.
Many efforts are currently devoted to the modification of enzyme properties to meet the
demands of bio-based industry. A scientific discipline dealing with designing and
constructing more powerful biocatalysts is called protein engineering (PE). Fine-tuning
enzyme characteristics through PE is desirable also for developing highly efficient systems
for ME. Specifically in strain engineering, enzyme variants with evolved kinetic properties
can replace traditional suboptimal approaches, based on overexpression of slow
biocatalysts, that can compromise system productivity and host viability through the
increased metabolic burden [8,58].
1.3.1 Strategies and tools
Three distinct strategies were developed in last two decades to allow the
construction and identification of mutant enzymes possessing desirable properties such
as increased activity, stability, modified specificity and selectivity or reduced

Introduction
- 22 -
substrate/product inhibition (Figure 6) [102,103]. The earliest approach was rational
design, which has been beneficial mostly in studying catalytic mechanism of enzymes and
recently also in knowledge-based modifications of enzymes' properties. Rational design
emphasizes the understanding of protein structure and amino acids interactions at the
beginning of the process. It exploits various computational tools and computer modeling
techniques, e.g., molecular docking, homology modelling or molecular dynamics
simulations, and site-directed mutagenesis for generation of targeted mutation(s)
resulting in single or several modified variants of enzyme [104]. State-of-the-art variant of
this strategy is de novo protein design which requires detailed understanding of the
desired catalytic meachanism and works with Rosetta software package [85,105].
Directed evolution as a newer approach has proven to be a powerful tool for the
improvement of enzymes [106,107]. Contribution of directed evolution is substantial
especially in particular cases, when neither the three-dimensional structure or homology
model nor the catalytic mechanism of the enzyme is known. Through the library
construction methods, based mostly on PCR protocol, directed evolution introduces
random mutations alongside the whole gene or recombines genes and generates huge
quantities of mutated sequences. Error-prone PCR or DNA-shuffling became widely used
methods of directed evolution [108,109]. Their randomness reminds the natural evolution
and can result in unexpected protein variants with improved functions. However, the
search for desired biocatalysts in huge mutant libraries is very demanding. The right
choice of high-throughput screening or selection method, usually based on colorimetric
assays, growth assays or fluorescence activated cell sorting (FACS), is crucial for
successful outcome of the directed evolution project.
Recent advances in enzyme engineering have used a combination of random
methods of directed evolution with elements of rational enzyme modification to
successfully by-pass certain limitations of both approaches. This focused directed
evolution, semi-rational approach or data-driven PE, targets multiple specific residues or
certain protein regions selected on the basis of prior structural and functional knowledge
[111,112]. The efficient choice of mutations likely to affect enzyme function is conducted
both experimentally and, on a much greater scale, computationally, using new powerful
algorithms and statistical tools, e.g. 3DM database, HotSpot Wizzard, FOLDX or ROSETTA
[104,113]. Focused directed evolution thus results in a smaller “smart“ libraries that are
easier to search through and more likely to yield positive results [114]. Particularly,
oligonucleotide-directed saturation mutagenesis proved to be useful for construction of
focused libraries [115].

Introduction
- 23 -
Figure 6. Comparison of the main protein engineering strategies. Adopted from [110] and modified.
Introduction of PE into the ME protocols is evident especially during the last few
years. Both rational design and directed evolution have successfully been used to engineer
diverse enzyme properties affecting the flux and thus the performance of the metabolic
pathways. Zhang and co-workers rationally engineered a promiscuous 2-keto-isovalerate
decarboxylase and 2-isopropylmalate synthase for preferred substrate specificity towards
a non-natural substrate [116]. Combination of mutated enzymes expanded E. coli
metabolism to produce unnatural alcohols as potential biofuels. In another recent report,
two enzymes from the diterpenoid biosynthetic pathway geranylgeranyl-diphosphate
synthase and levopimaradiene synthase were modified for levopimaradiene production in
E. coli [117]. Combination of rational approach based on homology modelling and
phylogenetic analysis in case of the first enzyme and random mutagenesis through error-
prone PCR of the second enzyme resulted in 2600-fold increase of levopimaradiene
biosynthesis.

Introduction
- 24 -
1.4 Perspectives
Synthesis of ME, SB, and PE will for sure be continuously intensified in following
years and will result in more powerful approaches for rapid improvement of metabolic
pathways and strain optimization. More experience and more information deposited in
DBTA cycle will allow better predictions and knowledge-based designs. SB devices like
biosensors and genetic switches will allow dynamic control of metabolic and signalling
pathways in engineered organisms [118]. It is expected that microfluidics, FACS and other
high-throughput techniques will revolutionize screening for new biocatalysts and
pathways or parts and strain characterization [119]. Besides that, phenomenon of natural
evolution and adaptation will draw more attention for possible fine tuning of rationally
designed microbial cell factories [120]. Importantly, progress in biological engineering
must go hand in hand with establishing regulatory frameworks and institutions that will
communicate the outcomes of the scientific projects to public and take care of biosafety,
ethics issues, and socio-economic aspects of conducted research.
Although the development in all three described areas has been exciting, our
understanding of cell biology remains unsatisfactory. This is obvious from limited
complexity and fitness of anthropogenic constructs [120]. ME and SB succeeded in
engineering microbes for biosynthesis of valuable chemicals in defined laboratory
conditions, but design of cells that will safely function in complex natural environments is
still a challenge. This is related especially to the engineering of metabolic pathways and
whole cells for biodegradation of toxic anthropogenic compounds and environmental
pollutants. Applications of the above described tools and approaches for this purpose will
be discussed in more detail in following chapters.

Introduction
- 25 -
2. Engineering of biodegradation pathways
The most of recent ME and SB applications are focused on production of
pharmaceuticals, commodity chemicals or biopolymers. The general interest in
biosynthesis is driven by unavoidable exhaustion of fossil resources and corresponding
economic aspects. Removal of environmental pollution caused by extensive activities of
consumer society represents another serious topic that deserves attention of biological
engineering. Recent pollution of soils, ground and surface waters constitutes a major
threat to the public health, not just in developing, but also in industrial countries including
EU states, China, and USA. In the Assessment of the European Environment Agency,
published in 2007, the EU estimated that there are approximately 250,000 known
contaminated sites in Europe that need to be cleaned up. Potentially polluting activities
occure in nearly 3,000,000 sites and the number of sites requiring remediation will
increase by 50% by 2025. Beside the medical and environmental consequences, this
situation signs considerable potential for growth of eco-industry focused on pollutant
removal and clean-up technologies.
Majority of the contaminants affecting the soil and water in Europe are organic
compounds such as mineral oil hydrocarbons, polyaromatic hydrocarbons, benzene
derivatives, and halogenated hydrocarbons (Figure 7). Many of organic polluting
compounds used in agriculture (e.g., pesticides dichlordifenyltrichlorethan, atrazine,
pentachlorophenol), industry (e.g., solvents as dichloroethane or dielectric fluids as
polychlorinated biphenyls) or military (e.g., explosives as trinitrotoluene) are of
anthropogenic origin and are called xenobiotics. Xenobiotics are the chemicals that have a
structure or a substituent on their structure that is not found in natural compounds. Some
of these compounds are considered as recalcitrant, in other words are not bioavailable or
not biologically degraded in the environment. Nevertheless, most of them are more or less
susceptible to biodegradation, i.e., biologically catalyzed reduction in their complexity
[121]. In case of organic compounds, biodegradation can sometimes lead to the complete
conversion of original compound to the inorganic products, i.e. mineralization.
Although plants and, to a lesser extent, animals may cause a number of changes in
structure of polluting chemicals, the major organisms causing the biological
transformations in soils, waters, and other environments are the microorganisms carrying
catabolic pathways. The biotransformations of organic pollutants catalyzed by microbial
metabolism are usually much faster than an abiotic degradation and depending on
microorganism can occur either in aerobic or anaerobic conditions. These factors together
with many advantageous properties mentioned in previous chapters made microbes and
especially bacteria favourable mean for bioremediation technologies, i.e., technologies
focused on removal of pollutants from contaminated sites in the environment either by
natural attenuation (little or no human action), biostimulation (addition of nutrients or
electron donors/acceptors to promote the growth of certain microbe) or bioaugmentation
(addition of natural or engineered microorganisms with the desired catalytic capabilities)

Introduction
- 26 -
[122]. Certain criteria formulated already 40 years ago by Dr. Martin Alexander must be
met for biodegradation to take place in the environment and bioremediation technology
to be seriously considered as a practical mean for treatment (Box 1) [121]. Some of the
early in situ or ex situ biotechnological processes really fulfilled these prerequisites and
resulted in successful local or world-wide applications of natural degraders in
anthropogenic operations. These include large-scale wastewater denitrification, removal
of uranium or application of 1,2-dichloroethane degrading bacterium for groundwater
treatment [123–125].
The uprise of recombinant DNA technology and ME in 1990s allowed transformation
of bioremediation from an empirical practice into an engineering science. The utlimate
goal of the new field was to engineer whole microbes, their biodegradation pathways and
corresponding enzymes toward directed in situ mineralization of desired pollutants. Such
"superbugs" were expected to provide economically feasible and environmentally friendly
alternative to costly conventional technologies for pollutants removal [126–128].
Furthermore, engineered bacteria could help solving the problem of persistent organic
pollutants (POPs) as dichlordifenyltrichlorethan (DDT), polychlorinated biphenyls (PCBs)
or dioxins that are resistant to natural biodegradation. The progress in design of
microorganisms and their metabolic pathways for the purpose of biodegradation and
bioremediation of organic pollutants achieved by emerging engineering technologies is
summarized in the following paragraphs.
Figure 7. Environmental contaminants affecting soil and groundwater in Europe (in %). Included are the data from Austria, Belgium, Czech Republic, Finland, Macedonia, Greece, Hungary, Italy, Luxembourg, Slovakia, Spain, and Sweden. Adopted from http://www.eea.europa.eu and modified.

Introduction
- 27 -
Box 1. The important criteria for practical feasibility of bioremediation technology formulated
by Martin Alexander [121].
1. A microorganism must exist that have the needed catalytic activity.
2. That microorganism must have the capacity to transform the compound at
reasonable rate and bring its concentration to the level that meets regulatory
standards.
3. The microorganism must not generate products that are toxic at the concentrations
likely to be achieved during the remediation.
4. The site must not contain concentrations of chemicals that are inhibitory to the
biodegrading species.
5. The target compound must be available to the microorganism.
6. Conditions at the site or in a bioreactor must be made conducive to the microbial
growth or activity.
7. The cost of the technology must be lower or, at worst, no more expensive than other
available technologies that can destroy the chemical.
2.1 Strategies and tools
Adaptive laboratory evolution and uprise of rational strain engineering. Early
attempts in biodegradation were limited to the isolation of microbes from contaminated
sites and studying natural degradation pathways in culturable bacteria [128]. Adaptive
laboratory evolution of bacteria carrying desirable trails was applied to speed up the rate
of pollutant's removal. This was achieved mostly by prolonged exposure of microbes to
the target chemical in chemostat or shaking flasks. The early experiment of Kellogg and
co-workers conducted in 1981 is an illustrative example of how the laboratory in vivo
evolution approach can be used to isolate microorganisms capable of utilizing persistent
compounds such as 2,4,5-trichlorophenoxyacetic acid, defoliating Agent Orange abused in
Vietnam War [129]. The same strategy was used also in more recent studies. For example,
faster degradation of widely used pesticide atrazine by Pseudomonas sp. ADP was
observed as a consequence of tandem duplications of atzB gene encoding the second
enzyme from atrazine pathway after 320 generations-long cultivation with pesticide as a
sole nitrogen source [130]. Similarly, amplification of tfdA gene encoding the first enzyme
in the pathway, 2,4-D dioxygenase, resulted in faster degradation of
2,4-dichlorophenoxyacetic acid, used as a sole carbon source during 1,000 generations-
long cultivation of Ralstonia sp. TFD41 [131]. These studies highlight the effect of tandem
gene duplication in the evolution of efficient catabolic pathways.
Very interesting strategy, which takes an advantage of adaptive laboratory evolution,
is genome shuffling. Genome shuffling has proven useful for engineering of multitrait
phenotypes that would be difficult to engineer rationally. Improvement of degradation of
anthropogenic pesticide pentachlorophenol (PCP) by Sphingobium chlorophenolicum
ATCC 39723 was addressed by this method, since the authors expected requirement of

Introduction
- 28 -
multiple mutations for the adaptations reducing the toxic effects of substrate and its
metabolites [132]. Three successive rounds of protoplast fusion and genome
recombinations of parents with improved phenotypes alternating with selection for
improved growth in the presence of PCP resulted in substantial improvements in both the
rate of PCP degradation and the concentration of PCP that could be tolerated. Analysis of
several improved strains proved that various combinations of mutations contributed to
the improved phenotypes. However, the mechanism of the adaptation remained
uncertain.
A patent and publication in Science by Chakrabarty and co-workers in 1981
represented a starting point of rational engineering for biodegradation and
bioremediation [129]. The work described preparation of recombinant P. putida strains
capable of breaking down crude oil by so called plasmid-assisted molecular breeding, i.e.,
dissemination of novel catabolic capabilities through directed bacterial conjugation and
plasmid transfer. Conjugation techniques allowing interspecies horizontal gene transfer
were utilized in many follow-uping studies [133]. Also Pseudomonads used by
Chakrabarty and co-workers remained popular hosts and model organisms for
biodegradation and bioremediation. The reason is that Psudomonads, encompassing
Gram-negative, aerobic, gamma-proteobacterial species, often possess remarkable
capacity to endure both endogenous and exogenous stresses [134]. Many Pseudomonads
are naturally resistant to high levels of solvents, probably due to the efficient efflux
pumps. Several environmental Pseudomonas strains were shown to mineralize aromatic
hydrocarbons and halogenated and nitro-organic compounds. In some P. putida strains
such as CSV86, aromatic compounds are even preffered substrates over glucose [135].
Namely P. putida KT2440 is generally regarded as safe organism, thus it is a convenient
model for laboratory experiments and suitable host for use in biotechnological processes.
Recruitment of new enzymes into existing catabolic pathways. The late 1980s and
early 1990s represented the golden era of biodegradation research with numerous
engineering attempts following the example of Chakrabarty and co-workers. The
persistence of many xenobiotics was in that time attributed mainly to the absence of
complete degradative pathways in a single organism [136,137]. Thus, the recruitment of
known complementary enzyme sequences by conjugative transfer of genes and so called
"patchwork assembly" of several existing natural pathways in a suitable host was believed
to generate a functioning synthetic routes allowing complete mineralization of target
persistent compounds such as PCBs, chlorinated benzoates or toluenes. Single genes or
catabolic gene clusters were heterologously expressed either from plasmids or
deliberately introduced into the chromosome of the host strain in a form of catabolic
cassette [133]. The system based on Tn5 minitransposon mediated delivery proved to be
very useful in studies aiming at stable expression of recruited enzymes from chromosome
of Gram-negative bacteria. The portfolio of plasmid delivery vectors utilizing Tn5-based
technology for random chromosomal insertions has expanded during the years [63,138].
Recently, also a Tn7-based broad-range bacterial cloning and expression vectors were

Introduction
- 29 -
prepared to allow site-specific implantation of metabolic modules at single attTn7 site of
various Gram-negative hosts [62].
Described tools and strategy were utilized for assembly of hybrid pathways in
various studies [139–144]. To name a few, Timmis and co-workers combined enzymes
from five different catabolic pathways of three distinct soil bacteria into functional route
for the degradation and mineralization of methylphenols and methylbenzoates [140].
Interestingly, the hybrid pathway was fully regulated and expressed only in response to
the presence of the substrate. In another work of the same group, a catabolic pathway for
alkylbenzoates carried on TOL plasmid of Pseudomonas was successfully restructured for
processing of the new substrate 4-ethylbenzoate [141]. The authors integrated the genes
from mutated bacteria selected for desired regulation of the pathway. More recently,
Tn5-based minitransposon system was utilized for transfer of three catabolic clusters
with eleven genes from three distinct bacteria to Cupriavidus necator H850 [143].
Engineered strain was the first designer bacterium to show aerobic growth on a wide
range of PCBs including two commercial pesticides Aroclor 1221 and Aroclor 1232.
Despite some success of "patchwork strategy" and engineering of "superbugs" with
extended substrate scope, this rather naive approach resulted also in many
disappointments. Almost textbook example is represented by engineered Pseudomonas
strains that failed to grow on 2-chlorotoluene as the only carbon source in well defined
laboratory conditions even though they possesed all genetic components presumed to be
necessary for substrate mineralization [145]. Similar failures can be explained by the fact
that the experimenters in the early and also in some recent studies underestimated or did
not have sufficient insight into important factors such as: (i) expression levels of
introduced heterologous enzymes and possible kinetic bottlenecks of assembled synthetic
routes, (ii) leakage of the side-products, (iii) thermodynamic feasibility of hybrid catabolic
network, (iv) complexity of regulatory networks, (v) or stress response and overall
physiology of the cell upon introduction of new metabolic modules and exposure to the
toxic substrates or intermediates [126–128]. Following paragraphs discuss selected
strategies that take listed factors into account and allow knowledge-based design of
strains with desired biodegradative capacities.
Engineering expression of enzymes in metabolic pathways. Tuning of individual
enzymes expression in natural or synthetic biodegradation pathways can help to
overcome or moderate the effect of unbalanced enzyme kinetics. One of the first studies
that dealt with the expression levels and activities of enzymes in the biodegradation
pathway was that of Mattozzi and co-workers [146]. The authors successfully designed a
P. putida strain that fully mineralized paraoxon, the worldwide known synthetic
phosphotriester insecticide, and used it as a sole source of carbon and phosphorus. In the
assembled catabolic pathway, one of the initial reaction intermediates diethyl phosphate
was degraded by serial action of three enzymes encoded by genes on synthetic operon.
The authors optimized the gene order in the operon and corresponding expression levels
according to the activities of individual enzymes measured in the cell lysates. In that way,

Introduction
- 30 -
they achieved faster paraoxon hydrolysis and utilization of diethyl phosphate. Several
studies reported the improvement of expression of dszB encoding 2-hydroxybiphenyl-2-
sulfinate sulfinolyase, the last rate limiting enzyme from the dibenzothiophene
biodesulfurization pathway. The overexpression of DszB in host cells and the performance
of the whole three-enzyme pathway was improved both by mutating the 5' untranslated
region of dszB [147] and by rearranging the gene order and removing the overlapping
structure in dsz operon [148]. The later work proved again that the positioning of gene in
the operon is an important determinant for its expression level. This phenomenon was
well described and explained in the recent study of Lim and co-wokers [64]. The
expression levels of pathway enzymes in host cell were addressed also in the work
described in the Chapters 3 and 4 of this Thesis.
Computer-aided design. Systems biology contributed significantly to the
rationalizing of the biodegradation field during the last few years. At the time of writing of
this Thesis NCBI public database lists 4647 sequences of bacterial genomes
(http://www.ncbi.nlm.nih.gov/genomes) from which many encode enzymes with yet
unknown biodegradative capacities. These huge pile of read genetic information together
with new powerful algorithms and computational tools that help scientists with mining of
relevant data allowed considerable progress in our understanding of dynamic interactions
that occur inside and in between the cells and the environments. Genome-scale metabolic
models and their interconnection with transcriptomic, proteomic, metabolomic, and flux
analyses under various growth and stress conditions completely changed our view on
popular biodegradative bacteria, such as P. putida [149,150]. Available genomic data
enabled also new insights into evolution of microbial metabolism toward biodegradation
of important xenobiotics [151].
Genome-based metabolic modelling can predict suitable targets for knock-out or
overexpression within the genome of a host organism. Thus, the desired task can be
sometimes achieved even without the need to incorporate heterologous genes [152]. One
of the modelling platforms utilizing genome-scale models, OptKnock, was found useful in
designing Geobacter sulfurreducens for higher respiration rates and electron transfer
[153]. Evolved strain could possibly enhance conversion of organic compounds to the
electricity in microbial fuel cells. The structure-activity relationship tool EcoliTox can
assess toxicity of selected substrates and metabolites throughout the metabolome of E.
coli [154]. Obtained information can be used for fine-tuning pathway expression in this
host.
Rich source of information on 219 biodegradation pathways is University of
Minnesota Biocatalysis/Biodegradation Database (UMBBD; http://eawag-bbd.ethz.ch).
Importantly, UMBBD can not only search through the existing metabolic routes, but allows
also predicting novel biotransformations leading from target xenobiotic substrate to the
unharmful molecule that can be metabolized by central catabolic pathways of a host cell.
The UMBBD Pathway Prediction System can highlight reactions that are likely to occur in
aerobic environment, but says nothing about the thermodynamic or kinetic feasibility of

Introduction
- 31 -
proposed pathways. Missing information on designer pathway thermodynamics can be
provided by the computational tool BNICE introduced in chapter 1.1.1 [24]. In theoretical
study of Finley and co-workers, metabolic flux analysis and genome-scale model of P.
putida implemented in BNICE were used to predict set of feasible pathways for
mineralization of 1,2,4-trichlorobenzene [155]. Although such theoretical predictions
should be experimentally verified, this pioneering work shows the possible way for those
who wish to utilize increasing power of computational modelling for design of novel
promissing catabolic routes. Unfortunately, majority of computational tools developed
nowadays for strain design are applied for improvement of the product yields rather then
biodegradation of recalcitrant pollutants [22].
SB approaches. Also SB, aiming at computer-aided designs and lego-like assemblies
of parts from diverse living organisms, has the potential to change the field of
biodegradation. SB approaches and devices such as toggle switches and genetic regulatory
circuits could be used for engineering bacterial consortia that would fulfil the demanding
biodegradation tasks better than individual microbes [156]. It is probable that
biodegradation in complex changeable environments will perform more reliably when
diverse metabolic and regulatory modules will be spread among members of consortium.
However, thus far the pioneering tests of consortial biodegradative capacities occured
only in defined laboratory conditions. For example, co-culture of engineered Escherichia
coli SD2 and Pseudomonas putida KT2440 was successfully employed for mineralizing
insecticide parathion in shaked flasks and in biofilm cultivation [157].
Development of novel regulatory systems for protein expression, cell-to-cell
communication and whole-cell biosensing that would allow parallel degradation and
seeking for toxic or explosive compounds in contaminated soils and waters represents
another challenging tasks for synthetic biologists [84,158,159]. SB-based strategies can be
also used to accelerate connection of target substrate with corresponding catalytic
activities. Aso and co-workers transplanted specialized membrane structures called
superchannels, allowing direct uptake of macromolecules from the environment into the
cytoplasm of Sphingomonas sp. A1, in the cell envelop of dioxin-degrading S. wittichii RW1
and achieve increased bioremediation capacity of this strain [160]. Opposite approach,
display of catabolic enzymes or whole pathways on the cell surface can increase efficiency
of substrate decomposition and diminish toxic effects of reactive metabolites on the
cytoplasmic structures [161].
Alternatively, the problem of metabolite toxicity could be addressed by engineering
biodegradative systems in vitro. This may encompass utilization of immobilized purified
pathway enzymes or crude cell extracs. Biodegradation field has not yet fully explored
benefits of this strategy. Predominantly, individual immobilized enzymes or whole-cell
biocatalysts are addopted in biotechnological processes aimed at contaminants removal
[162–164]. Rather exceptional study of Geuke and co-workers showed potential
application of in vitro engineering approch for biodegradation of technical
hexachlorocyclohexane (HCH) consisting of five isomers [165]. The authors used model

Introduction
- 32 -
system of two purified recombinant enzymes, dehydrochlorinase LinA and haloalkane
dehalogenase LinB, that are known to initiate biotransformation of prohibited insecticide
γ-HCH. They separately incubated HCH isomers with various ratios of LinA and LinB and
determined metabolic profiles of occuring sequential biotransformations. The analyses of
these profiles helped to judge the environmental fate of HCH isomers and proved that the
original HCH degradation pathway is optimized for γ-HCH, but not for other isomers. In
vitro approach proved to be useful for developing promissing biotechnology for
transformation of toxic anthropogenic pollutant into desirable commodity chemical
described in Chapter 1 of this Thesis.
Combination of ME and PE approaches for biodegradation. Significant
improvement of the biodegradation route performance can be achieved through
engineering activity or selectivity of the limiting enzymes. Activities of many enzymes that
act on anthropogenic compounds are derived from promiscuous activites that are
generally inefficient [166]. For example, Rhodococcus erythropolis N-acylhomoserine
lactone acylase hydrolyzes various lactones with values of kcat/Km up to 1.7x106 M-1.s-1 and
also unnatural substrate insecticide paraoxon, but with a significantly lower kcat/Km of 0.5
M-1.s-1 [167]. Promiscuous activities often result from substrate ambiguity, i.e., the ability
to bind and convert a molecule that resembles the normal substrate.
Numerous reports describe application of common PE methods such as epPCR, DNA
shuffling, site-directed or saturation mutagensis for engineering activity or selectivity of
individual catabolic enzymes [128,168]. However, the studies that report successful
implantation of constructed mutants in the context of the whole biodegradative pathway
and receiving host organims are not so common. Iwariky and co-workers successfully
applied Alcaligenes sp. KF711 harboring engineered monooxygenase P450CAM for
dehalogenation of pentachloroethane to trichloroethane under unoxic conditions. The
product was further degraded by hybrid dioxygenase in the presence of oxygen [169]. In
another study, promiscuous toluene orthomonooxygenase and epoxide hydrolase were
engineered using DNA-shuffling and saturation mutagenesis, respectively. Subsequent
employment of modified genes in recombinant E. coli with synthetic pathway allowed
faster aerobic degradation of chlorinated ethenes with lesser accummulation of
stress-inducing intermediates [170,171].
2.2 Perspectives
Numerous examples presented in the previous chapter clearly show that engineering
of microorganisms towards novel or improved biodegradative capacities remains a
challenge. The field of biodegradation still lacks success stories similar to those known
from the field of biosynthesis. Unfortunatelly, the high risk connected with engineering of
microbes for pollution removal and legislative barriers hindering their introduction to
practical applications let many scientific teams to change the topics of their research.

Introduction
- 33 -
Accordingly, application of ME and SB to biodegradation problems are rather rare in
current literature. This unsatisfactory situation can be attributed mainly to: (i) complexity
of biological systems and (ii) controversy on application of GMO in the field studies.
It is no doubt that the environmental concerns and regulatory constraints have
limited not just in situ applications of GMOs for bioremediation, but also the quality of
fundamental research and the overall progress in the field. The experiments of Lawrence
Wackett and his team on field-scale remediation of atrazine-contaminated soil with killed
recombinant E. coli expressing atrazine chlorohydrolase most probably represents the
only well documented case of GMO utilization for in situ bioremediation [172]. The
relaxation of restrictive policy is desirable because only the field-scale experiments can
lead to better understanding of designer organism behavior in real environments, where
both biotic and abiotic factors form the complex fluctuating bioremediation space [173].
Considering potential future in situ applications, some intelectual effort has already been
invested in development of technologies that would ensure the biosecurity and fate of
released GMOs. To avoid endangering the biodiversity and uncontrolled spreading of
foreign genes or recombinant microbes, the constructed degraders could be tracked
through bioluminiscence, fluorescence or watermarking of synthetic DNA or completely
discarded after performing the desired task by inducible suicidal systems [174–176].
Employment of immobile transgenic plants and their symbiotic bacteria for
phytobioremediation or application of GM microbes only within the boundaries of
well-defined decontamination zones or bioreactors could also reduce the risk of
unintenional dissemination [177]. Concerning the first limiting point, the only way towards microorganisms
performing well in a complex natural environment leads through deeper understanding of
fundamental principles of cell biology. The more intensive exploitation of the emerging
technologies introduced in this text should help with such a task. There is lots of space for
employment and further high-throughput data mining, functional identification and
experimental characterization of biological components under standardized conditions
[120]. Desirable is also wider utilizing of flux analysis and kinetic modelling [178],
inspecting biodegradation processes at a single cell, a single pathway, and a single
molecule levels [179], studying the formation and behavior of microbial consortia [156] or
development of more precise and reliable experimental tools for balancing of metabolic
networks and reduction of diverse stresses. Better synergy of PE and ME through
employment and evaluating of new enzyme variants in biodegradation pathways would
also be beneficial. The last but not least, the idea of DBTA cycle should be adopted by
biodegradation community, same as design of orthogonal and in vitro systems that might
help to reduce both problems of biological complexity and GMO controversy [67,75].
Enzymes with altered activity and enantioselectivity and some of the mentioned
novel ME and SB approaches have been recently used for engineering and understanding
of the synthetic metabolic pathway for biodegradation of anthropogenic pollutant
1,2,3-trichloropropane. The background and history of the pathway design, assembly, and
engineering is described in the following part of the Introduction.

Introduction
- 34 -
3. Engineering of the synthetic metabolic pathway for
biodegradation of 1,2,3-trichloropropane
The attempts to find natural degraders or engineer new microbes capable of efficient
removal of man-made halogenated hydrocarbons from the environment have continued
through the decades since 1980s until today. The engineering of the synthetic pathway for
aerobic utilization of 1,2,3-trichloropropane has been initiated already in 1989 and thus,
with its 25-years-long history, represents one of the most systematic efforts of that kind
reported so far. Before we come to the more detailed description of the pathway
engineering, the rationale on such endeavour will be provided in the following chapter.
3.1 Halogenated hydrocarbons and 1,2,3-trichloropropane
Many anthropogenic compounds mentioned in chapter 2.1 were halogenated
hydrocarbons. These chemicals had been considered as bearers of progress at the
beginning of their synthetic production in the first half of the last century. They were used
as insecticides (e.g., DDT, HCH), herbicides (e.g., atrazine), soil fumigants (e.g.,
1,2-dibromoethane) and plant growth regulators (e.g., halogenated benzoic acids).
Halogenated hydrocarbons protected millions of people from malaria and other wasting
diseases and enabled the Green Revolution in agriculture between 1940s and 1960s.
Manufacturing of reactive halogenated intermediates, such as epichlorohydrin or vinyl
chloride, supported a boom of chemical industry and introduced plastics into our lives.
However, the environmental-fate and toxicity studies triggered by the evidence provided
in famous book of Rachel Carson Silent Spring published in 1962, indicated that many of
these compounds can act as a persistent pollutants in the environment and can be harmful
to the ecosystems and human health [180]. Halogens became “not-to-trust elements“,
although it is now obvious that synthetic halogenated hydrocarbons have much more
structural analogues in the living nature than was ever supposed [181].
1,2,3-trichloropropane. Among other halogenated hydrocarbons, chlorinated
alkanes including 1,2-dichloroethane, 1,2-dibromoethane or 1,2,3-trichloropropane (TCP),
extensively used in agriculture or industry, represent common groundwater contaminants
in many developing and industrial countries [182]. Particularly TCP constitutes a hard nut
for remediation technologists due to its physicochemical properties, toxicity to living
organisms and environmental recalcitrance. TCP is colorless, volatile liquid with a sharp,
sweet odor, manufactured by major chemical companies such as Dow Chemical Company
or Shell. Recent report of Dow estimates worldwide annual production of TCP to be below
50,000 metric tons [183]. TCP is formed predominantly as a by-product during synthesis
of other important chemicals such as epichlorohydrin or cross-linking agent
hexafluoropropylene. It used to be present in soil fumigant 1,3-dichloropropene known
under trade name D-D and occured in past also in customer products, such as certain

Introduction
- 35 -
paint removers and cleaning agents. Mainly due to the improper waste management and
its incorporation in D-D, TCP became a widespread contaminant of soils, ground and
underground waters. Contamination was proved in several states in EU [184,185], USA
[186] and Asia [187]. In 2010, the US Environmental Protection Agency listed TCP as an
emerging contaminant. The EU Chemicals Agency considers TCP as a chemical of very high
concern because of its carcinogenic and toxic effects. TCP is anticipated human carcinogen
based on evidence of carcinogenicity in studies with experimental animals [188]. TCP
caused DNA damage, including formation of DNA adducts, and cell proliferation in rats
and mice and gene mutations or various chromosomal aberations were observed upon
exposure in bacteria, yeast, and mammalian cells in vitro [189,190]. Accidental ingestion
or inhalation of contaminated air resulted in acute intoxication and damage of liver in
humans [191]. Concentrations ranging from 0.1 to 74 μg.L-1 have been detected in
drinking water in Hawaii and California, two countries that continuously monitor TCP in
water sources [192]. State of Hawaii established maximal TCP level in a drinking water of
0.6 μg.L-1 (cca. 4.1 nM).
Abiotic transformations of TCP. The half-life of abiotic hydrolysis of TCP under
environmental conditions (25°C, pH=7) was estimated to be in the order of hundreds of
years [193]. Persistent nature and physicochemical properties of TCP listed in Table 1,
such as high density or limited water solubility (cca. 1.5 g.L-1), make a remediation of the
contaminated sites a difficult task [194]. Methods for in situ treatment of TCP-polluted
water, including vacuum extraction and subsequent chemical oxidation with ozone or
hydrogen peroxide, application of activated carbon or reduction with zero-valent zinc; are
under investigation [195]. However, pump and treat technologies and abiotic
transformations are expensive and relatively inefficient [194]. In vivo or in vitro
biotransformation, especially when coupled with the recycling of the biocatalyst, could
provide economical and efficient alternative for TCP removal.
Table 1. Basic physicochemical properties of metabolites from TCP pathway.a
chemical SMILES
molecular
weight density
boiling
point
melting
point logPa
(g.mol
-1) (g.ml
-1) (°C) (°C)
TCP C(C(CCl)Cl)Cl 147.4 1.39 156 -14 2.29
DCP C(C(CCl)Cl)O 129.0 1.36 182 NA 0.78
ECH C1C(O1)CCl 92.5 1.18 115-117 -57 0.35
CPD C(C(CCl)O)O 110.5 1.32 213 -40 -0.71
GDL C1C(O1)CO 74.1 1.12 61-62 -54 -0.93
GLY C(C(CO)O)O 92.1 1.25 182 18 -1.93
[a] Source of information: ChemSpider database http://www.chemspider.com/
[b] Calculated using ALOGPS 2.1 program [196].
Abbreviations: TCP, 1,2,3-trichloropropane; DCP, 2,3-dichloropropane-1-ol; ECH, epichlorohydrin;
CPD, 3-chloropropane-1,2-diol; GDL, glycidol; GLY, glycerol; NA, data not available.

Introduction
- 36 -
Biotic transformations of TCP. Biodegradation of TCP was observed both under
anaerobic and aerobic conditions. Recently, several Gram-negative bacterial strains from
genus Dehalogenimonas were isolated from contaminated groundwater and showed to
reductively dehalogenate TCP under anoxic conditions [197,198]. The major drawback of
this biotransformation is that it results in potentionally toxic products such as allyl
chloride or allyl alcohol and the mechanism of reductive dechlorination has not yet been
elucidated. In situ reductive dechlorination of contaminated groundwater can be
performed by direct injecting hydrogen-releasing compounds or hydrogen, which serves
as an electron donor [195]. However, this time-demanding strategy was applied only with
TCP concentrations below 1 mg.L-1. Anaerobic transformations might be more suitable for
in situ bioremediation than aerobic processes, due to the difficulty of supplying
homogeneous oxygen to the underground. Nevertheless, aerobic biodegradation
pathways are often studied, because they may lead to the complete mineralization of
target substrate and support growth of bacteria, which is highly desirable [128]. Aerobic
conversion of TCP was observed only in cometabolic mode, catalyzed by the methane
monooxygenase from the resting cells of methanotropic bacterium Methylosinus
trichosporium OB3b [199]. Calculations performed by Dolfing and Janssen proved that
complete mineralization of TCP by bacterial oxidative metabolism through lesser
chlorinated propanes and HCl formation is thermodynamically feasible [200]. Regrettably,
no organism capable of aerobic utilization of TCP as a source of carbon and energy for
growth has been isolated from polluted sites or enrichment cultures thus far. This can be
attributed to the: (i) relatively short presence of TCP in the environment and
corresponding lack of efficient dehalogenating enzymes and (ii) low adaptability potential
of bacteria toward this substrate [182]. Direct toxic effect of TCP or its reaction
intermediates or indirect incidence of reactive oxygen species (ROS), that can be formed
during aerobic biodegradation of chlorinated pollutants, may represent significant
handicap for evolution [201,202]. Due to the absence of natural degrader, TCP, together
with some other halogenated aliphatic hydrocarbons such as 1,2-dichloropropane or
trichloroethylene, was listed among extremely difficult targets for aerobic biodegradation
(Figure 8).

Introduction
- 37 -
Figure 8. Degradability of some aliphatic and aromatic compounds including those with
halogenated substituent(s). Adopted from [182].
Toxicity and oxidative stress. TCP toxicity mode of action on bacterial cells is not
fully understood. As can be deduced from studies on eukaryotes, TCP and its presumed
reaction intermediates, such as epoxides frequently produced by oxidative conversion of
chlorinated aliphatics, can form DNA adducts and induce genetic damage [200]. They can
also react with other biomacromolecules and cellular components, e.g., cell membranes.
Reactivity of TCP towards membrane structures could be promoted by high
hydrophobicity of the compound (Table 1). Saturation of unsaturated fatty acids by
halogenation or lipid peroxidation triggered by epoxides could cause change in membrane
fluidity, resulting in collapse of the electron transport chain and electrochemical potential
that maintains viability [203]. Electron transport chains are major sites of premature
electron leakage to oxygen. Disruption of respiratory chain could promote formation of
ROS, such as superoxide and hydrogen peroxide, and oxidative stress through further
damage of DNA and oxidation of lipids and proteins. More research is needed to validate
these hypothetical modes of action and will be an objective of future research in
Loschmidt Laboratories.

Introduction
- 38 -
It is evident that rapid enzymatic dehalogenation, forming harmless product
molecule acceptable in natural metabolic network of host organism, represents a critical
step for successful biodegradation of TCP. Selection of enzymes suitable for such task and
design of aerobic catabolic pathway for TCP mineralization are discussed in the following
chapter.
3.2 Overview of TCP pathway engineering
In 1989, van den Wijngaard and co-workers isolated from freshwater sediment
Gram-negative bacterial strain AD1 growing on epichlorohydrin (ECH) as the carbon and
energy source [204]. The bacterium, known recently as Agrobacterium radiobacter AD1,
was able of aerobic utilization of dihalogenated alcohols, such as 1,3-dichloropropan-1-ol,
via ECH, 3-chloropropane-1,2-diol (CPD), glycidol (GDL). Resulting reaction product
glycerol (GLY) enters the glycolysis upon phosphorylation and oxidation. Enzymes
expected to catalyze the reactions leading to GLY were epoxide hydrolase and haloalcohol
dehalogenase. The authors proposed TCP mineralization by addition of a single
dehalogenation step to the metabolic pathway of AD1.
Hydrolysis of a carbon-halogen bond in aliphatic halogenated alkanes and alkenes is
carried out by a spectrum of well characterized haloalkane dehalogenases (HLDs),
enzymes belonging to the α/β-hydrolase fold superfamily of proteins [205,206]. HLDs
have been reported to catalyze the first dehalogenation step in aerobic biodegradation
pathways of important halogenated pollutants, such as 1,2-dichloroethane,
1,2-dibromoethane and HCH [207]. Analysis of HLDs isolated during the last two decades
indicate that dehalogenation is the main function of these enzymes. Thus, mentioned
activities are probably not promiscuous and were rather evolved from activities toward
natural counterparts of halogenated xenobiotics [182]. First HLD capable of TCP
dehalogenation was characterized by Yokota and co-workers in 1987 (Table 2) [208]. The
authors described the enzyme from Corynebacterium sp. strain m15-3. (later renamed as
Rhodococcus sp. m15-3). Dehalogenase was later assigned DhaA and its sequence was
described by Kulakova and co-workers who isolated the gene from 1-chlorobutane
degrader Rhodococcus rhodochrous NCIMB13064 [209]. The enzyme converted prochiral
TCP into both enantiomers of 2,3-dichloropropan-1-ol (DCP) in almost equimolar ratio.
Importantly, dhaA gene was found to be quite widespread, occuring in several
geographically distinct Rhodococcus strains but never in bacteria possessing enzymes for
haloalcohol utilization [210]. Therefore, complementation and further evolution of these
functions within a single organism was unlikely, which may explain the absence of natural
TCP degrader. The attempts to obtain a recombinant bacterial biocatalyst by means of ME
and PE followed.

Introduction
- 39 -
ME and PE strategies applied to TCP project. The first recombinant strain capable
of TCP utilization was constructed in 1999 through heterologous expression of wild type
DhaA in A. radiobacter AD1 and assembly of the synthetic metabolic pathway (Figure 9)
[211]. Bosma and co-workers placed dehalogenase gene under the control of strong
constitutive promoter and introduced it into AD1 on a broad host range plasmid using
triparental mating. The resulting construct showed growth on 1,2,3-tribromopropane and
1,2-dibromo-3-chloropropane as a sole carbon sources in minimal medium, but not on
TCP. Only 0.7 mM (0.14 mmol ≌ 21 mg) of TCP was utilized by 200 mL cell culture of
starting OD450 of 0.07 within 25-day cultivation. This poor conversion of TCP was
attributed to the low activity of wild-type DhaA with the anthropogenic substrate TCP
(kcat/Km=40 s-1.M-1). The catalytic efficiency was more than two orders of magnitude lower
than that of dehalogenase DhlA from Xanthobacter autothropicus GJ10, the natural
degrader utilized in a groundwater purification plant treating xenobiotic pollutant
1,2-dichloroethane [125].
Bosma and co-workers published follow-up work describing improvement of DhaA
activity with TCP by PE in 2002. Although a crystal structure of DhaA has already been
available [212], they applied directed evolution, which was more frequently used for the
purpose of enzyme optimization in that time. One round of DNA shuffling and subsequent
epPCR resulted in variant DhaAM2 carying two substitutions (C176Y, Y273F) and 8-times
higher catalytic efficiency toward TCP [213]. Molecular modelling suggested that
introduced mutations minimized the occurence of unproductive binding mode of TCP in
the active site of DhaA. 200 mL culture of recombinant AD1 of starting OD450 of 0.14
constitutively expressing dhaAM2 gene under the strong dhla promoter on plasmid
utilized 3.6 mM (0.72 mmol ≌ 106 mg) of TCP within 10 days. Significant, but not
sustainable increase of optical density of the culture (final OD450 of 0.45) was observed.
This demontration represents a significant step towards viable TCP degrader, however,
the designed strain suffered from two limitations: (i) the dehalogenation of TCP by
DhaAM2 was not sufficiently fast to ensure rapid conversion of substrate that is toxic to
the cells already at the contration of 1 mM, (ii) cumulation of (S)-DCP caused by
enantioselectivity of haloalcohol dehalogenase.

Introduction
- 40 -
Table 2. Basic properties of the enzymes from TCP pathway.
enzyme
haloalkane
dehalogenase
DhaA
EC 3.8.1.5
haloalcohol
dehalogenase
HheC
EC 4.5.1.-
epoxide
hydrolase
EchA
EC 3.3.2.3
organism
Rhodococcus
rhodochrous
NCIMB13064
Agrobacterium
radiobacter AD1
Agrobacterium
radiobacter AD1
superfamily α/β-hydrolases
NAD(P)-binding
Rossmann-fold
domains
α/β-hydrolases
GenBank ID AF060871.1 AF397296.1 Y12804.1
residue
count/monomer 293 255 294
quarternary
structure
(PDB ID)
no
(1CQW)
homotetramer
(1ZMT)
no
(1EHY)
reaction
mechanism
nucleophilic
substitution
nucleophilic
substitution
nucleophilic
substitution
catalytic
residues
Glu130, His272,
Asp106
Ser132, Tyr145,
Arg149
Asp107, His275,
Asp246
substrate scope
broad specificity,
halogenated aliphatic
and cyclic compounds
broad specificity,
aliphatic and aromatic
vicinal haloalcohols
broad specificity,
1,2-epoxyalkanes,
styrene oxide
MW
(kDa)/monomer
pI/monomer
33.2
4.95
28.0
5.29
34.0
5.18
temp. optimum
(°C) 37 50 50
pH optimum 8.0-9.5 8.0-9.0 8.0-9.0
references [207,208] [214] [216]
Abbreviations: PDB, Protein Data Bank; MW, molecular weight; pI, isoelectric point.

Introduction
- 41 -
Accumulation of epoxides was not observed, but 10-times lower specific activity of
EchA with GDL than with ECH indicated a possible problem [214]. The flux of carbon and
energy through the synthetic pathway possesing these bottlenecks was obviously not
sufficient to provide enough GLY for sustainable growth, resulting in a failure of the
attempts to degrade TCP in a continuous packed-bed bioreactor inoculated with the
recombinant strain.
Cl
Cl Cl
Cl
Cl OHO
Cl
OH
OH
Cl
OH
O
OH
OH OH
1,2,3-trichloropropane (R,S)-2,3-dichloropropane-1-ol DCP
(R,S)-epichlorohydrin ECH
(R,S)-glycidol GDL
(R,S)-3-chloropropane-1,2-diol CPD
glycerol GLY
glycolysis
DhaA HheC
HheCEchA
EchA
TCP
glycerol kinase and glycerol-3-phosphate dehydrogenase
+H2O/-HCl -HCl
-HCl
+H2O
+H2O
Figure 9. Scheme of the synthetic metabolic pathway for aerobic degradation of 1,2,3-trichloropropane to the glycerol. At the turn of millenium, several studies appeared that were focused on deeper
characterization of two enzymes that form synthetic TCP pathway together with
haloalkane dehalogenase DhaA: haloalcohol dehalogenase HheC and epoxide hydrolase
EchA (Figure 9 and Table 2) [214–218]. Both enzymes were also engineered towards
improved activity, enantioselectivity, stability or broader substrate specificity, using
either rational design or directed evolution methods [219–223]. However, none of the
studies aimed at application of HheC or EchA in TCP biodegradation and no mutants with
properties optimized for this purpose were obtained. In contrast, DhaA and its activity
with difficult substrate TCP drew considerable attention. Using computational simulations
and quantum mechanical calculations, Banas and co-workers pointed out the significance
of access tunnels, connecting the bulk solvent with the burried active site of DhaA, for the
catalysis of TCP [224]. This finding was fundamental for further enhancement of DhaA
activity. In 2009, Pavlova and co-workers published a new mutant, DhaA31, with 32-times
improved turnover number and 26-times higher catalytic efficiency (kcat = 1.26 s-1; kcat/Km
= 1,050 s-1.M-1), when compared with wild type DhaA [225]. Applying computer-assisted

Introduction
- 42 -
design and site-directed and saturation mutageneses, three new substitutions (I135F,
V245F, L246I) were introduced into the main and slot tunnels of template DhaAM2.
Computational modelling and kinetic analyses revealed that new bulky residues protected
the active site from excess of water molecules, which otherwise disfavours nucleophilic
substitution. The enantioselectivity of DhaA31 remained unaltered.
Van Leeuwen and co-workers targeted the enantioselectivity of DhaA31 in order to
obtain the variants converting TCP predominantly into either (R)- or (S)-DCP [226]. Five
rounds of focused directed evolution employing saturation mutagenesis with restricted
codon sets provided mutant DhaA r5-90R with thirteen new amino acid substitutions and
improved (R) selectivity (e.e. = 90%) in comparison with DhaA31 (e.e. = 13%).
Unfortunately, mutagenesis affected the activity with TCP which dropped to the level of
DhaA (kcat/Km = 25 s-1.M-1). New DhaA variant could be potentially utilized in TCP pathway
to complement the function of (R)- selective HheC, because no haloalcohol dehalogenase
capable of efficient conversion of (S)-DCP has been isolated thus far.
The reseach group of Dick B. Janssen provided also alternative solution for
unmatching selectivity of DhaA and HheC. Arif and co-workers published a report on
isolation and characterization of novel P. putida strain MC4 that could utilize both
enantiomers of DCP as a sole carbon and energy source for growth [227]. The growth was
allowed by the lack of enantioselectivity of the initial oxidative
dehalogenase/dehydrogenase DppA that converted DCP to toxic 2-chloroacrolein and
2-chloroacrylic acid. Further degradation of 2-chloroacrylic acid in MC4 has not been
studied, but the pathway is supposed to continue through hydrolytic dechlorination to
yield pyruvate or reduction and further dehalogenation which could yield lactate. The
authors wanted to take advantage of the properties of the new strain for TCP
biodegradation and recently introduced dhaA31 gene with constitutive dhlA promoter into
the chromosome of MC4 using the transposon delivery system [228]. The resulting
recombinant bacterium P. putida MC4-5222 utilized 3.1 mM TCP in minimal medium
within 25 days of fed-batch cultivation. (volume of culture was not reported). The growth
of culture with slow growth rate (μ = 0.0012–0.008 h-1) resembling the one reported for
engineered strain AD1 with dhaAM2 expressed from plasmid was observed. Moreover, the
immobilized cells of MC4-5222 were tested for biodegradation of TCP in a laboratory scale
packed-bed reactor. The efficiency of TCP removal, supplied continuously at the
concentrations 0.30–0.33 mM, increased from 87 – 97% during 48 days of reactor
operation. The experiment proved good stability of chromosomal insertion and the
potential of cells to survive prolonged time interval in presence of TCP supplied as the
sole carbon source.
The engineered P. putida MC4-5222 represents a rare example of genetically
modified bacterium which showed growth on an important recalcitrant environmental
pollutant. Still, the recombinant cells were sensitive to the concentrations of TCP as low as
1 mM and further improvement of strain characteristics is desirable for a full scale
industrial application [228]. Adaptive laboratory evolution in the presence of chemical
mutagen ethyl methanesulfonate and TCP at the upper limit of tolerance were not

Introduction
- 43 -
successful. The authors attributed the limited fitness to the possible side reactions and
corresponding accumulation of reactive by-products, that might occur in metabolic
network of the strain MC4-5222 upon growth on TCP. However, these constraints are
difficult to trace back because pathway that converts DCP to the metabolites utilizable in
central metabolic routes is not well understood. The original TCP pathway encompassing
DhaA, HheC, and EchA, offers following advantages over new route from P. putida: (i) all
enzymes and metabolites of the pathway are known and characterized (Tables 1 and 2),
(ii) the enzymes do not need cofactors and were shown to be active under in vitro
conditions, and (iii) the pathway yields well defined utilizable product. These
characteristics proved to be useful in rational pathway engineering reported in the
Results part of this Thesis.
In this Thesis, the synthetic TCP pathway composed of DhaA, HheC and EchA
enzymes is used as a model system for knowledge-based engineering of an organism
degrading xenobiotic pollutant. A unique DBTA cycle encompassing following five steps
was used (Figure 10): (1) study of assembled TCP pathway with wild-type or engineered
enzymes both in soluble and immobilized forms in vitro, (2) determination of kinetic
parameters of individual enzymes and construction of a kinetic model describing the five-
step conversion of TCP to GLY, (3) application of the kinetic model for study of a dynamic
behaviour of in vitro three-enzyme system, (4) utilization of validated model for rational
design of the orthogonal pathway in laboratory strain E. coli, and (5) transfer of the most
promissing pathway variants into P. putida chassis. The data and new knowledge
generated within this project should provide deeper insight into this interesting model
system, should allow rational dissection of pathway bottlenecks and will hopefully lead to
the development of better biocatalyst for TCP biodegradation. Presented concepts and
tools should be applicable to engineering organisms degrading also other recalciltrant

Introduction
- 44 -
Figure 10. A knowledge-based strategy applied for rational engineering of a synthetic metabolic pathway for biodegradation of 1,2,3-trichloropropane. A) Metabolic pathway is initially reconstructed in vitro in a form of free or immobilized purified enzymes and its functioning is verified by measuring metabolite concentrations using available analytical techniques. Kinetic parameters of individual enzymes with their target substrates and reaction mechanisms are determined. B) Kinetic parameters and equations are utilized for development of a kinetic model. Model is experimentally validated with purified enzymes and used for dynamic in silico simulations of pathway performance under selected constraints. Productivity of the pathway is maximized through optimizing enzyme stoichiometry and incorporating available engineered enzymes. C) Synthetic pathway is assembled in a modular way as an orthogonal biological system in suitable heterologous host. Established mathematical model supplemented with relevant biological parameters is used to calculate all possible pathway variants within in vivo constraints. Only the best performing variants are constructed and experimentally characterized.

- 45 -

- 46 -

Contribution to the results
- 47 -
CONTRIBUTION TO THE RESULTS
The Results section of the Thesis consists of four chapters. Chapters 1 – 3 are based
on three original peer-reviewed articles. Chapter 4 is summarising data and results
under preparation for publication.
1. Dvorak, P., Bidmanova, S., Prokop, Z., and Damborsky, J. (2014) Immobilized
synthetic pathway for biodegradation of toxic recalcitrant pollutant
1,2,3-trichloropropane. Environ. Sci. Technol. 48, 6859-6866.
2. Dvorak, P., Kurumbang, N. P., Bendl, J., Brezovsky, J., Prokop, Z., and Damborsky,
J. (2014) Maximizing the efficiency of multi-enzyme process by stoichiometry
optimization. ChemBioChem DOI: 10.1002/cbic.201402265
3. Kurumbang, N. P.*, Dvorak, P.*, Bendl, J., Brezovsky, J., Prokop, Z., and
Damborsky, J. (2014) Computer-assisted engineering of the synthetic pathway for
biodegradation of a toxic persistent pollutant. ACS Synth. Biol. 3, 172-181.
(*shared first authors)
4. Dvorak, P., Nikel, P., de Lorenzo, V., Prokop, Z., Damborsky, J. (2014) Assembly of
the synthetic pathway for biodegradation of 1,2,3-trichloropropane in
Pseudomonas putida KT2440 CF1. (in preparation)
Author's contribution to the articles:
1. design of experiments, gene cloning, preparation of soluble and immobilized
enzymes and their characterization, development of analytical methods, assembly
of packed bed reactor, batch and continuous multi-enzyme reactions,
interpretation of data, writing of the manuscript
2. design of in vitro experiments, preparation of purified enzymes, determination of
steady-state kinetic parameters of enzymes, enatioselectivity measurements,
determination of inhibition constants, production of (S)-DCP by preparative
biocatalysis, batch multi-enzyme reactions, interpretation of data, writing of the
manuscript
3. design of in vitro and part of in vivo experiments, quantification of enzyme
content in cells by SDS-PAGE analysis and activity measurements, determination
of accumulation of TCP and its metabolites in host cells, degradation of TCP in
buffer by pre-induced resting cells, recovery of cells in LB medium, interpretation
of data, writing of the manuscript together with N. P. Kurumbang
4. design of experiments, construction of synthetic operon, cloning and
transformations, characterization of recombinant bacteria bearing synthetic
operon on plasmid and in chromosome, writing of the manuscript

- 48 -

- 49 -
CHAPTER 1
In vitro assembly and immobilization of
the synthetic pathway for biodegradation
of toxic recalcitrant pollutant
1,2,3-trichloropropane

Chapter 1
- 50 -
INTRODUCTION
1,2,3-trichloropropane (TCP) is an anthropogenic compound recently recognized as
an emerging contaminant of groundwater [186]. TCP is produced worldwide in quantities
reaching 50,000 metric tons annually and used by chemical companies as a solvent,
precursor of soil fumigants, and building block for synthesis of other chemicals, e.g.,
dichloropropene or polysulfone liquid polymers [183]. TCP is also formed as a by-product
during the synthesis of epichlorohydrin. Due to its massive production, TCP can often be
found at industrial and hazardous waste sites [229]. Recent incidents with drinking water
sources in California polluted by TCP emphasized the need to develop efficient
technologies for removing this toxic and carcinogenic compound from the environment.
Conventional remediation techniques are relatively inefficient, due to the physical
and chemical properties of the compound [230]. The exception is promissing reductive
conversion by zero-valent zinc [194]. In addition to abiotic transformations,
biotransformations are extensively studied for their ability to decontaminate sites
polluted with chemical contaminants [231]. Recently, isolated bacterial strains were found
to transform TCP under anaerobic conditions [197,198]. However, these
biotransformations often result in products toxic for surrounding environments and due
to the low efficiency can be applied only at limited (<1 mg.L-1) TCP concentrations [195].
No organism capable of aerobic TCP biodegradation has yet been isolated, probably due to
the anthropogenic nature of TCP and its recent introduction into the environment.
The absence of natural catabolic pathways for aerobic TCP utilization was addressed
by Bosma and co-workers [211,213]. The authors assembled a synthetic metabolic
pathway with the heterologous expression of haloalkane dehalogenase DhaA from
Rhodococcus sp. in the natural host Agrobacterium radiobacter AD1, capable of utilization
of haloalcohols. Engineered AD1 strain, expressing 5 times more active variant of
haloalkane dehalogenase, showed a slow growth on 1 mM TCP in minimal medium.
However, practical utility of this construct is limited by the toxicity of TCP for bacterial
cells above 1 mM concentration, isufficient activity of engineered haloalkane
dehalogenase, accumulation of toxic intermediates and legislative barriers on the use of
genetically modified microorganisms [213]. We recently addressed the problems of the
low catalytic efficiency of the first dehalogenation step and accumulation of intermediates
limiting the productivity of the pathway in vivo by combination of protein and metabolic
engineering [232].
In this study, we report the application of an immobilized enzymatic pathway for a
complete five-step degradation of TCP to the harmless product glycerol (GLY; Figure 1).
We immobilized engineered 32-times more active haloalkane dehalogenase DhaA31 from
Rhodococcus rhodochrous NCIMB 13064 [225], the wild-type haloalcohol dehalogenase
HheC and the wild-type epoxide hydrolase EchA from A. radiobacter AD1 into cross-linked
enzyme aggregates (CLEAs) and polyvinylalcohol (PVA) LentiKats lenses [233,234]. The
immobilized biocatalysts were used to convert TCP into the desirable commodity
chemical GLY in both batch and continuous systems. A comparison of the dynamic

Chapter 1
- 51 -
behavior of the complex multi-enzyme system in both soluble and immobilized forms is
provided. To the best of our knowledge, this is the first report on the use of an
immobilized synthetic metabolic pathway employing engineered enzyme for the
biotransformation of an environmental pollutant. The established immobilization strategy
is robust and suitable for scale-up. The developed biocatalyst is recyclable, resistant to
biodegradation, compatible with high input loads of TCP, and can operate under mild non-
sterile conditions. Moreover, the possibility to operate the process without the use of
genetically modified microorganisms makes this biotechnology suitable for environmental
applications.
Cl
Cl Cl
Cl
Cl OHO
Cl
OH
OH
Cl
OH
O
OH
OH OH
1,2,3-trichloropropane (R,S)-2,3-dichloropropane-1-ol DCP
(R,S)-epichlorohydrin ECH
(R,S)-glycidol GDL
(R,S)-3-chloropropane-1,2-diol CPD
glycerol GLY
(glycolysis)
DhaA HheC
HheCEchA
EchA
TCP
Figure 1. Scheme of the five-step synthetic metabolic pathway for biotransformation of 1,2,3-trichloropropane to glycerol. Used abbreviations of individual metabolites are shown. DhaA, haloalkane dehalogenase from Rhodococcus rhodochrous NCIMB 13064; HheC, haloalcohol dehalogenase; and EchA, epoxide hydrolase, both from Agrobacterium radiobacter AD1. The final product glycerol can be utilized in glycolysis when the pathway is assembled in vivo.

Chapter 1
- 52 -
MATERIAL AND METHODS
Reagents. TCP, 2,3-dichloropropane-1-ol (DCP), epichlorohydrin (ECH), 3-chloropropane-
1,2-diol (CPD), glycidol (GDL), and GLY standards were purchased from Sigma-Aldrich
(USA). All of the chemicals used in this study were of analytical grade. Bovine serum
albumin (BSA) for preparation of CLEAs was purchased from Sigma-Aldrich (USA). PVA
and polyethylene glycol of MW 1,000 were provided by LentiKat’s a.s. (Czech Republic).
The solution of the cross-linker dextran polyaldehyde (DPA) was prepared according to a
procedure described elsewhere [235]. A Free Glycerol Assay Kit was purchased from
BioVision (USA). The work with toxic compounds was conducted in a fume hood and with
protective equipment to minimize safety risks.
Gene synthesis and cloning. The nucleotide sequences of genes encoding wild-type
haloalcohol dehalogenase HheC and epoxide hydrolase EchA from Agrobacterium
radiobacter AD1 were downloaded from GenBank database (accession numbers
AF397296.1, Y12804.1, respectively). The tag sequence of six histidine codons was
attached downstream of the echA gene. Restriction sites for cloning into pET21b (NdeI,
BamHI) (Merck, Germany) and pET28b (NcoI, HindIII) were attached to the sequences of
the hheC and echA genes, respectively. Due to the creation of a NcoI restriction site, the
second codon of the echA gene (ACT) encoding threonine was substituted for a GCA codon
encoding alanine. Genes of HheC and EchA together with genes of the wild-type and
mutant haloalkane dehalogenase DhaA and DhaA31 were commercially synthesized
(Geneart, Germany). Sequences of all genes were optimized for expression in Escherichia
coli during synthesis. Synthetic genes were subcloned into the NdeI and BamHI restriction
sites of pET21b (dhaA, dhaA31 and hheC), and NcoI and HindIII restriction sites of pET28b
(echA). The resulting constructs pET21b-dhaA, pET21b-dhaA31, pET21b-hheC and
pET28b::echA were transformed into E. coli DH5α using the heat-shock method for
plasmid propagation.
Cultivation conditions and purification of enzymes. Competent cells of the E. coli
BL21(DE3) strain were transformed with pET21b-dhaA, pET21b-dhaA31, pET21b-hheC or
pET28b-echA using the heat shock method, and plated on LB agar plates with ampicillin
(100 µg.ml-1) or kanamycin (50 µg.ml-1). Plates were incubated overnight at 37°C. Single
colonies were used to inoculate 10 ml of LB medium with the respective antibiotic, and
cells were grown overnight at 37°C. Overnight cultures were used to inoculate 1 l of LB
medium with the respective antibiotic. Cells were cultivated at 37°C with shaking until an
OD600 of 0.4–0.6, after which expression was induced with 0.5 mM IPTG. Cells were then
cultivated overnight at 20°C. Biomass was harvested by centrifugation. Cells were washed
and resuspended in purification buffer A (20 mM K2HPO4 and KH2PO4, 0.5 M NaCl, 10 mM
imidazole, pH 7.5). 1 U of DNaseI (New England Biolabs, USA) per 1 ml of cell suspension
was added. Cells were disrupted by sonication using the ultrasonic processor Hielscher
UP200S (Teltow, Germany) with 0.3 s pulses and 85% amplitude. Cell lysate was

Chapter 1
- 53 -
centrifuged for 1 h at 21,000 g at 4°C, and the resulting cell-free extract was decanted.
DhaA, DhaA31 and EchA were purified using single-step nickel affinity chromatography.
Crude extract was applied to a 5 ml Ni-NTA Superflow column (Qiagen, Germany). The
column was attached to a BioLogic Duo Flow (Bio-Rad, USA). The buffer system consisted
of buffer A and buffer B (20 mM K2HPO4 and KH2PO4, 0.5 M NaCl, 500 mM imidazole, pH
7.5). Recombinant enzymes were eluted at 60% of buffer B during a two-step gradient
method: 0–10% in 5 column volumes and 10–60% of buffer B in 10 column volumes.
Fractions containing DhaA, DhaA31 or EchA were pooled and proteins were concentrated
using a stirred ultrafiltration cell (Millipore, USA). Enzymes were dialyzed against 50 mM
phosphate buffer (pH 7.5). HheC was purified using anion-exchange chromatography.
Crude extract was applied to a 35 ml glass Econo-Column (Bio-Rad, USA) packed with 25
ml of Q Sepharose Fast Flow (GE Healthcare, USA). The column was attached to a BioLogic
Duo Flow (Bio-Rad, USA). The buffer system consisted of buffer A (20 mM Tris-SO4, 1 mM
EDTA, 1 mM β-mercaptoethanol, pH 7.5) and buffer B (20 mM Tris-SO4, 1 mM EDTA, 1 mM
β-mercaptoethanol, 0.45 M (NH4)2SO4, pH 7.5 ). HheC was eluted at 20–25% of buffer B
during a two-step increasing linear gradient: 0–45% buffer B in 20 column volumes and
45– 100% buffer B in 5 column volumes. Fractions containing HheC were pooled and the
protein was concentrated. Enzyme was dialyzed against PD buffer (50 mM phosphate
buffer of pH 7.5 with 2 mM dithiothreitol). Concentrations of DhaA31, EchA and HheC
were determined by Bradford Reagent (Sigma-Aldrich, USA). Enzyme purity was judged
by SDS-PAGE analysis. Purified enzymes were stored for further use at 4°C.
Preparation of CLEAs. CLEAs of DhaA31 and EchA were prepared by dissolving 25 mg of
enzyme and 25 mg of BSA in 1 ml of 10 mM phosphate buffer (pH 7.5). The solution was
added to 9 ml of saturated ammonium sulfate (pH 8.0). After 1 h of incubation in an ice
bath with stirring, 1.3 ml of DPA was added and cross-linking occurred for another 1 h.
The resulting suspension was centrifuged at 4,000 g for 15 min at 4°C. The supernatant
was stored for the determination of residual enzymatic activity; CLEAs were re-suspended
in 20 ml of saturated sodium hydrogencarbonate and incubated with stirring in an ice
bath for 30 min. The suspension was centrifuged at 4,000 g for 15 min at 4°C and the
supernatant was removed. CLEAs were washed with a 50 mM phosphate buffer (pH 7.5)
and stored in 1 ml of this buffer at 4°C before further use.
Encapsulation of enzymes into PVA particles LentiKats. PVA (0.56 g) and polyethylene
glycol (0.34 g) were mixed with 3.7 ml of distilled water and heated at 98°C until PVA
dissolved completely. The liquid was cooled to 35°C, and 10 mg of free HheC in a PD buffer
(50 mM phosphate buffer of pH 7.5 with 2 mM dithiothreitol) or 1 g of CLEAs of EchA or
DhaA31 was added and mixed thoroughly. Small droplets (3-4 mm) of mixture were
dripped on plastic plates and incubated at 37°C until LentiKats lost 70% of their initial wet
weight. Dried LentiKats were soaked in 50 ml of 0.1 M sodium sulfate for 2 hrs to reswell.
HheC LentiKats were washed with a PD buffer and stored in the same buffer at 4°C.

Chapter 1
- 54 -
LentiKats of EchA and DhaA31 were washed with a 50 mM phosphate buffer (pH 7.5) and
stored at 4°C.
Preparation of combi-LentiKats from cell-free extracts. Cell-free extracts with DhaA31,
HheC or EchA were prepared from 200 ml of cell cultures using the same cultivation
conditions as described previously. Biomass was harvested by centrifugation. Cells were
washed and resuspended in 3 ml of 50 mM sodium phosphate buffer of pH 7.0. 1 U of
DNaseI (New England Biolabs, USA) per 1 ml of cell suspension was added. Cells were
disrupted by sonication using the ultrasonic processor Hielscher UP200S (Teltow,
Germany) with 0.3 s pulses and 85% amplitude. Cell lysates were centrifuged for 1 h at
21,000 g at 4°C, and the resulting cell-free extracts were decanted. Dithiothreitol was
added to the cell-free extract containing HheC to a final concentration of 2 mM. The total
protein concentration in the cell-free extracts was determined by Bradford Reagent.
Samples of cell-free extracts (5 µg of total protein) containing DhaA31, HheC or EchA were
loaded onto SDS-polyacrylamide gel. Gels were analyzed using a GS-800 Calibrated
Densitometer (Bio-Rad, USA). The amount of enzyme in the cell-free extract was
estimated according to a comparison of the trace density of its respective band with the
trace density of the standard sample band. CLEAs were prepared from cell-free extract
containing approximately 25 mg of DhaA31 or EchA. Combi-LentiKats were prepared by
mixing cell-free extract containing approximately 5 mg of HheC, CLEAs containing
approximately 5 mg of DhaA31 and aproximately 5 mg of EchA together with solubilized
PVA hydrogel with a mass ratio 1:4 (mixture of CLEAs and cell-free extract : PVA
hydrogel). Small droplets (3–4 mm) of mixture were dripped on plastic plates and
incubated at 37°C until combi-LentiKats lost 70% of their initial wet weight. Dried
combi-LentiKats were soaked in 50 ml of 0.1 M sodium sulfate for 2 hrs to re-swell.
Combi-LentiKats were washed with PD buffer and stored in the same buffer at 4°C.
Enzyme assays. Specific activities of soluble and immobilized DhaA31 with TCP and HheC
with DCP and CPD were assayed in 10 ml of a 50 mM Tris-SO4 buffer (pH 8.5) at 37°C with
a 10 mM substrate. The concentration of the reaction product (chloride ions) was
measured by the method of Iwasaki [236]. The specific activity of EchA was assayed by
following the substrate depletion in 10 μ of a 50 mM Tris-SO4 buffer (pH 8.5) at 37°C with
5 mM ECH or 10 mM GDL. Samples of the reaction mixture were mixed with acetone (1:1)
with internal standard hexanol and analyzed by gas chromatography (GC). The same GC
method was used for the quantitative analyses of all metabolites of the TCP pathway
except for GLY. The rates of abiotic conversions of all metabolites at selected time
intervals were negligible.
Storage stability of free and immobilized enzymes. Storage stability of free and
immobilized DhaA31, HheC and EchA was investigated at 4°C and in the case of free
enzymes, also at room temperature (22±2°C). Free and immobilized DhaA31 and EchA
were stored in 50 mM phosphate buffer (pH 7.5) without additives. Free and immobilized

Chapter 1
- 55 -
HheC was stored in PD buffer (pH 7.5; measurements at 4°C) or in 50 mM phosphate
buffer (pH 7.5) without additives (measurements at room temperature). The residual
activities of DhaA31, HheC and EchA stored at 4°C were measured with 10 mM TCP, DCP
and ECH, respectively, at certain time intervals using conditions described in the Enzyme
assays. The residual activities of DhaA31, HheC and EchA stored at 22±2°C were measured
with 10 mM TCP, DCP and ECH, respectively, in 10 ml of Tris-SO4 buffer of pH 8.2 at
22±2°C.
Confocal microscopy of immobilized enzymes. CLEAs of DhaA31 (200 µl) were
resuspended in 1 ml of carbonate/bicarbonate buffer (pH 9.5). The fluorescent dye
fluorescein 5(6)-isothiocyanate (FITC, Invitrogen, USA) was dissolved in dimethyl
sulfoxide (final concentration of 10 mg.ml-1). A solution of FITC (1 µl) was added to the
CLEAs, which were stained for 60 min at room temperature with shaking. After separation
by centrifugation at 4,024 g for 15 min at 4 °C, CLEAs were washed three times with
50 mM phosphate buffer (pH 7.5). EchA CLEAs (200 µl) were resuspended in 1 ml of
carbonate/bicarbonate buffer buffer (pH 8.3). The fluorescent dye pacific blue
succinimidyl ester (PBSE, Invitrogen, USA) was dissolved in dimethyl sulfoxide (final
concentration of 10 mg.ml-1). A solution of PBSE (5 µl) was added to the CLEAs, which
were stained for 60 min at room temperature with shaking. After separation by
centrifugation at 4,024 g for 15 min at 4 °C, CLEAs were washed three times with 50 mM
phosphate buffer (pH 7.5). The fluorescent dye X-rhodamine-5(6)-isothiocyanate
(5(6)-XRITC, Invitrogen, USA) was dissolved in dimethyl sulfoxide (final concentration of
1 mg.ml-1). A solution of 5(6)-XRITC (20 µl) was added to 200 µL of soluble HheC
(10 mg.ml-1) in carbonate/bicarbonate buffer (15 mM sodium carbonate, 30 mM sodium
bicarbonate, pH 9.5). HheC was stained for 60 min at room temperature with shaking.
Labelled enzyme was separated from unreacted dye using a disposable PD MiniTrap
column containing 2.1 ml of Sephadex G-25 medium (GE Healthcare, UK). All three
labelled enzymes (0.33 ml of solution of free HheC, 0.33 g of DhaA31 CLEAs and 0.33 g of
EchA CLEAs) were encapsulated in LentiKats by mixing together with 4 ml of PVA and PEG
solubilized in water. The resulting combi-LentiKats were washed with 200 ml of 50 mM
phosphate buffer (pH 7.5) and frozen in tissue freezing medium Jung at -26°C using a
Leica Cryocut 1800 Cryostat (Leica Microsystems, Germany). Samples were then
sectioned vertically and horizontally with a thickness of 5 μm and mounted on glass slides
with Mowiol 40-88 (Sigma-Aldrich, USA). Microscopy observations of labelled free HheC,
CLEAs of DhaA31 and EchA, and combi-LentiKats were performed with the Olympus IX81
imaging station FluoView500 (Olympus C&S Ltd., Czech Republic) using 10x, 20x and 40x
objectives. Images were analyzed using Olympus Fluoview (Olympus, Japan) and Imaris
(Bitplane, Switzerland).
Multi-enzyme conversion of TCP in batch system. The multi-enzyme conversion of TCP
to GLY was assayed in 15 ml of a 50 mM Tris-SO4 buffer (pH 8.5) in gas-tight glass vials
(Sigma-Aldrich, USA) incubated at 37°C. The reaction was initiated by adding 1 mg of

Chapter 1
- 56 -
DhaA31, HheC, and EchA in a soluble or immobilized form, into the reaction mixture with
5 mM TCP. The samples withdrawn from the reaction mixture (0.5 ml for soluble enzymes
or CLEAs and 0.1 ml for LentiKats) were mixed with acetone (1:1) containing hexanol as
an internal standard and analyzed by GC. Selected samples were analyzed by gas
chromatograph and mass spectrometer (GC-MS) (Agilent, USA) for identification of
metabolites otherwise routinely detected by GC. The concentration of GLY was
determined spectrophotometrically by the Free Glycerol Assay Kit. Samples of the
reaction mixture (0.1 ml) were heated at 95°C for 5 min, centrifuged at 18,000 g for 1 min,
diluted in an assay buffer and analyzed according to the manufacturer’s protocol.
Concentrations of GLY were calculated from absorbance at 570 nm. For evaluation of the
effects of pH and lower temperature, conversion of TCP was assayed in 25 ml glass vials
with a screw cap mininert valve (Sigma-Aldrich, USA) containing 15 ml of 50 mM sodium
phosphate buffer of pH 7.0 or 50 mM NaOH/glycine buffer of pH 10. Vials were incubated
in the water bath GLS Aqua Plus (Grant Instruments, UK) with shaking (200 rpm) at 20°C.
Recycling of immobilized enzymes in batch system. The reusability of DhaA31, EchA and
HheC immobilized in LentiKats was assayed in 25 ml glass vials with a screw cap mininert
valve (Sigma-Aldrich, USA) with 10 ml of 50 mM Tris-SO4 buffer (pH 8.5) and 1 mM TCP.
Each of ten cycles was started by the addition of LentiKats containing 1 mg of DhaA31,
2 mg of HheC, and 2 mg of EchA to the reaction mixture. After 100 min of incubation at
30°C, LentiKats were separated from the reaction mixture by decantation, washed in
25 ml of the reaction buffer and used for a new cycle. Samples of the reaction mixture
(0.5 ml) were collected at the beginning and end of each cycle, mixed with acetone (1:1)
containing an internal standard and analyzed by GC to determine the concentration of
TCP. In parallel, samples (0.1 ml) were taken for quantification of GLY under standard
conditions. The relative efficiency of the biocatalyst in each cycle was calculated from the
conversion of TCP to GLY, utilizing the conversion in the first cycle as 100%.
Multi-enzyme conversion of TCP in continuous system. Glass column 1 (28 cm in height,
1.5 cm internal diameter, 50 ml working volume) with 100 mg of DhaA31 immobilized in
47 g of wet LentiKats and glass column 2 (25 cm in height, 2.5 cm internal diameter,
100 ml working volume) with 100 mg of both HheC and EchA immobilized in 45 and 43 g
of LentiKats, respectively, were used for the removal of TCP in a ten-week continuous
operation of a packed bed reactor placed in fume hood at 22±2°C. A feed vessel contained
TCP dissolved under stirring in 1 l of distilled water buffered with 0.1 M Tris-SO4 (pH 8.2)
to a final theoretical concentration of 5 mM (week 1) or 10 mM (weeks 2-10). The
experimental concentrations of TCP, ranging from 2.25 to 7.97 mM, for evaluation of
system efficiency were determined in the input of column 1 (Figure 5 and Table S7 of
Supplementary tables and figures). The operation conditions for the packed bed reactor
are summarized in Table S2. Influent and effluent lines were constructed from
polytetrafluoroethylene tubing. Samples from the feed vessel, inlet of column 1 and
effluent vessels 1 and 2 were withdrawn periodically and analyzed by GC and the Free

Chapter 1
- 57 -
Glycerol Assay Kit. A new cycle of operation was started whenever the content of the feed
vessel was completely transferred to the effluent vessel 2. To evaluate possible leaching of
the immobilized enzymes from the reactor, samples from effluent vessel 2 were
withdrawn, concentrated 25 times using stirred ultrafiltration cells (Millipore, USA) and
analyzed by SDS-polyacrylamide gel electrophoresis (SDS-PAGE).
Operational conditions for GC and GC-MS analyses. A Gas Chromatograph 6890N with
flame ionization detector (Agilent Technologies, USA) and a capillary column ZB-FFAP
30 m x 0.25 mm x 0.25 µm (Phenomenex, USA) was used for routine analysis of samples
from multi-enzyme reactions. A Gas Chromatograph 7890A and Mass Spectrometer 5975C
MSD (Agilent Technologies, USA) equipped with a capillary column ZB-FFAP 30 m x
0.25 mm x 0.25 µm (Phenomenex, USA) was used for verification of the presence of
individual metabolites from the TCP pathway in selected samples from the multi-enzyme
reactions. Samples (2 μl) were injected into the GC with an inlet temperature of 250°C and
split ratio 20:1. The operational conditions for the column were: helium carrier gas with
an initial flow of 0.6 ml.min-1 for 1 min, followed by a flow gradient from 0.6 to
1.8 ml.min-1 (ramp 0.2 ml.min-1), temperature program set to give an initial column
temperature of 50°C for 1 min, followed by a temperature gradient from 50 to 220°C hold
for 2 min (ramp 25°C.min-1). The temperature of the detector was 250°C. MS scan speed
was 6.9 s-1. This method was used for all GC analyses. For that purpose, calibration curve
of 0 – 5 mM of TCP, DCP, ECH, CPD and GDL with internal standard hexan-1-ol was
prepared. Detection limits calculated using the software OriginPro v8 (OriginLab
Corporation, USA) were 3 μM, 5 μM, 6 μM, 186 μM and 22 μM for TCP, DCP, ECH, CPD and
GDL, respectively.

Chapter 1
- 58 -
RESULTS AND DISCUSSION
Multi-enzyme conversion of TCP using free enzymes.
We initially tested the ability of an in vitro assembled pathway to fully convert 5 mM
TCP to GLY in one-pot reaction at a time interval of 30 h. Purified DhaA, HheC, and EchA
were mixed in the mass ratio of 1:1:1 mg and incubated with TCP. Because the molecular
weights of DhaA, HheC, and EchA are similar (34, 29, and 35 kDa, respectively), the
proposed 1:1:1 ratio corresponded closely with the same molar ratio of enzymes. The pH
8.5 and temperature 37°C were selected to approach the reaction optima of all three
enzymes [209,216,237]. Using wild-type enzymes, TCP and the intermediates DCP and
GDL were degraded from 73% within 30 h of the reaction (Figure 2A and Table S3 in
Supplementary tables and figures). The percentage of degradation correlated well with
amount of GLY produced from TCP during the same time interval.
The time course of the reaction showed the major bottleneck of the pathway – slow
consumption of TCP by DhaA. Additionally, significant accumulations of two
intermediates, DCP and GDL, were observed. The accumulation of DCP was caused by the
high enantioselectivity (E≥100) of HheC [238]. Non-selective DhaA converts the prochiral
TCP into both enantiomers of DCP in an almost equimolar ratio. Since HheC prefers
(R)-DCP, (S)-DCP tends to accumulate. The specific activities of EchA with ECH and GDL
(29.5 µmol.min-1.mg-1 and 6.5 µmol.min-1.mg-1, respectively) in combination with the
previously reported kinetic parameters indicate that ECH is a much better substrate for
EchA than GDL [214]. Therefore, the substrate specificity of EchA is probably the main
cause of GDL accumulation during the multi-enzyme conversion of TCP.
To overcome the limitation of the first reaction step, the wild-type DhaA
(kcat = 0.04 s-1, kcat/Km = 40 s-1.M-1) was substitued with the mutant DhaA31 (kcat = 1.26 s-1,
kcat/Km = 1050 s-1.M-1), constructed in our laboratory using a computer-assisted design
[225]. The selectivity of DhaA31 with TCP remained unchanged. The benefit of engineered
DhaA31 in the multi-enzyme conversion of TCP was verified during the second
experiment with free enzymes. The time course of the reaction shows that TCP was
completely converted into its metabolites within the first 200 min of the reaction, and the
degradation of TCP, DCP, and GDL reached 99% of the theoretical maximum within 30 h of
the reaction (Figure 2B and Table S4). The conversion of TCP to GLY reached 95% of the
theoretical maximum. We conclude that complete in vitro biodegradation of TCP and its
biotransformation to the final product GLY is possible despite the suboptimal
stereochemistry and specificity in the pathway resulting in the accumulation of DCP and
GDL in the initial phase of reaction.

Chapter 1
- 59 -
Figure 2. Time courses of multi-enzyme conversions of 1,2,3-trichloropropane with free and immobilized enzymes. A) free purified enzymes DhaAwt, HheC, and EchA; B) free purified enzymes DhaA31, HheC, and EchA; C) immobilized purified enzymes DhaA31, HheC, and EchA; D) immobilized cell-free extracts containing DhaA31, HheC, and EchA. All reactions were performed using enzymes of mass ratio of 1:1:1 mg in 15 ml of reaction mixture. TCP, 1,2,3-trichloropropane; DCP, 2,3-dichloropropane-1-ol; ECH, epichlorohydrin; CPD, 3-chloropropane-1,2-diol; GDL, glycidol; GLY, glycerol. Each data point represents the mean value ± standard deviation from three independent experiments.
Immobilization of DhaA31, HheC and EchA.
Application of free enzymes in biotransformation processes is not practical due to
their complicated recycling, limited use in bioreactors, and low stability in harsh process
conditions such as elevated temperatures or the presence of organic co-solvents [162]. We therefore immobilized DhaA31, HheC, and EchA to avoid such limitations. Various
strategies for immobilization of individual biocatalysts are available [234]. Multi-enzyme
systems also benefit from immobilization, but development of joint immobilization
protocols for all employed catalysts is challenging. There is currently no protocol available
for immobilization of DhaA, HheC and EchA. Therefore, we tested enacapsulation of three
enzymes into PVA, which has proven its utility in immobilization of haloalkane
dehalogenase LinB, close relative of DhaA [240].

Chapter 1
- 60 -
The encapsulation of biocatalysts into PVA hydrogel is a widely used immobilization
technique [241,242]. Lens-shaped PVA hydrogel particles, known as LentiKats, are
promising matrices for biocatalysis due to the low cost, resistance to biodegradation and
good mechanical properties [234]. Their favorable geometry (thickness 200-400 μm)
allowes better mass transfer than spherical microbeads [243,244]. The biotechnology
based on LentiKats has already been used in large scale applications, e.g., synthesis of
beta-lactam antibiotics or denitrification in wastewater treatment. The size of LentiKats
allows easy separation from the reaction mixture and is suitable for application in packed
bed reactors, which can suffer from large pressure drops over the column when packed
with particles of inadequate size [245]. LentiKats provide a hydrophilic environment with
the pores sufficiently small to entrap whole cells, cross-linked enzymes, cross-linked
enzyme aggregates (CLEAs), or free enzymes of molecular weights higher than 80 kDa
[240,243,246].
In contrast to the HheC tetramer with a molecular weight of 116 kDa, smaller
monomeric molecules of DhaA31 and EchA are not suitable for direct encapsulation in
LentiKats. Therefore, aggregation and cross-linking of DhaA31 and EchA was carried out
to produce CLEA particles. Immobilization in CLEAs is straightforward technique which
has been used for many enzymes including hydrolases [99,233,247]. The protocols for
preparation of CLEAs from LinB and for miscroscopic monitoring of immobilized
biocatalyst were previsouly established in our laboratory [240,248]. Here we expand
these protocols also for immobilization of DhaA31 and EchA. To enable detailed
characterization of assembled pathway, immobilization was initially performed separately
for each of the three purified enzymes. CLEAs of DhaA31 and EchA were prepared by
mixing an enzyme solution with lyophilized bovine serum albumin, serving as a proteic
feeder [249]. The protein mixture was precipitated using ammonium sulfate, which is
widely used for preparing CLEAs due to its low cost and easy treatment [250]. The
suspension of aggregated enzymes was cross-linked using DPA, which had been shown to
have a less detrimental effect on enzymatic activity than the widely used glutaraldehyde
[251]. CLEAs and free HheC were labeled by fluorescent dyes to allow microscopic
monitoring of biocatalysts during the immobilization procedure (Figure 3). Free HheC and
wet CLEAs of DhaA31 and EchA were separately encapsulated in LentiKats in mass ratio
of 1:4 (enzyme or CLEAs: PVA gel).
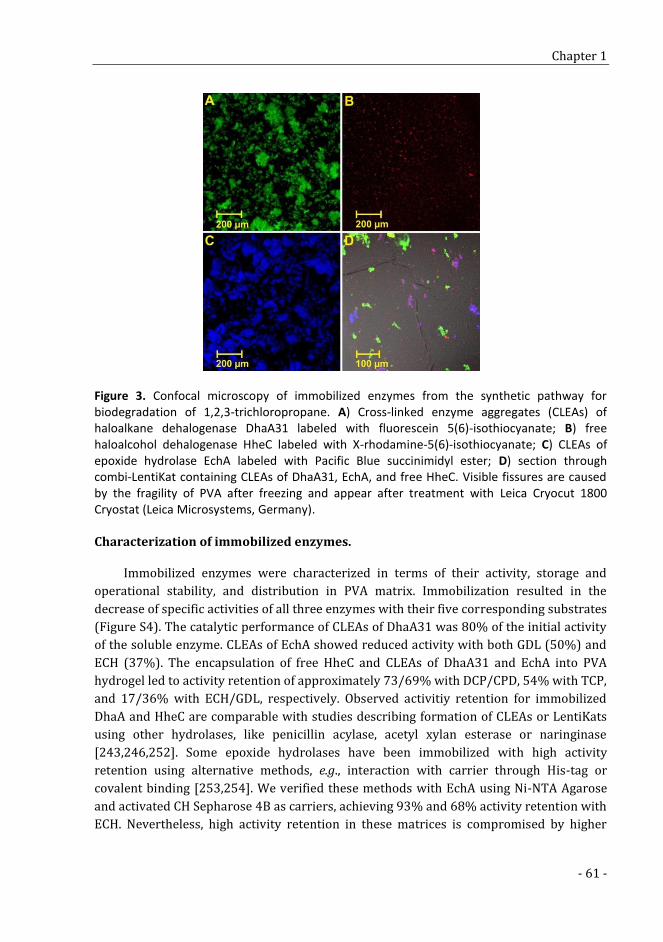
Chapter 1
- 61 -
Figure 3. Confocal microscopy of immobilized enzymes from the synthetic pathway for biodegradation of 1,2,3-trichloropropane. A) Cross-linked enzyme aggregates (CLEAs) of haloalkane dehalogenase DhaA31 labeled with fluorescein 5(6)-isothiocyanate; B) free haloalcohol dehalogenase HheC labeled with X-rhodamine-5(6)-isothiocyanate; C) CLEAs of epoxide hydrolase EchA labeled with Pacific Blue succinimidyl ester; D) section through combi-LentiKat containing CLEAs of DhaA31, EchA, and free HheC. Visible fissures are caused by the fragility of PVA after freezing and appear after treatment with Leica Cryocut 1800 Cryostat (Leica Microsystems, Germany). Characterization of immobilized enzymes.
Immobilized enzymes were characterized in terms of their activity, storage and
operational stability, and distribution in PVA matrix. Immobilization resulted in the
decrease of specific activities of all three enzymes with their five corresponding substrates
(Figure S4). The catalytic performance of CLEAs of DhaA31 was 80% of the initial activity
of the soluble enzyme. CLEAs of EchA showed reduced activity with both GDL (50%) and
ECH (37%). The encapsulation of free HheC and CLEAs of DhaA31 and EchA into PVA
hydrogel led to activity retention of approximately 73/69% with DCP/CPD, 54% with TCP,
and 17/36% with ECH/GDL, respectively. Observed activitiy retention for immobilized
DhaA and HheC are comparable with studies describing formation of CLEAs or LentiKats
using other hydrolases, like penicillin acylase, acetyl xylan esterase or naringinase
[243,246,252]. Some epoxide hydrolases have been immobilized with high activity
retention using alternative methods, e.g., interaction with carrier through His-tag or
covalent binding [253,254]. We verified these methods with EchA using Ni-NTA Agarose
and activated CH Sepharose 4B as carriers, achieving 93% and 68% activity retention with
ECH. Nevertheless, high activity retention in these matrices is compromised by higher

Chapter 1
- 62 -
price and lower mechanical stability, which makes them less suitable for full-scale
processes. The specific activity of soluble EchA with ECH (29.5 µmol.min-1.mg-1) is one
order of magnitude higher than the specific activity of DhaA31 with TCP (1.1 µmol.min-
1.mg-1) and HheC with DCP (1.6 µmol.min-1.mg-1) and CPD (3.0 µmol.min-1.mg-1). Despite
the reduction of the catalytic performance of EchA after immobilization, the pattern of
specific activities of three enzymes remained unchanged (Figure S1 in Supplementary
tables and figures). We therefore concluded that even 80% loss of the activity of EchA
towards ECH after immobilization in CLEAs and LentiKats would not result in the
accumulation of intermediates in the TCP pathway and the analogous immobilization
strategy was maintained also for this enzyme.
No enzymatic activities were detected in the supernatants obtained during the
preparation of CLEAs and LentiKats, besides low activity of HheC corresponding to
approximately 10% loss of enzyme during immobilization. Therefore, we expect that the
decreased activities of DhaA31, HheC and EchA are predominantly due to the partial
deactivation caused by aggregation, cross-linking, or encapsulation in PVA hydrogel,
rather than due to the leaching of enzymes during immobilization process.
Figure 4. Relative activities of free enzymes and enzymes immobilized in cross-linked enzyme aggregates (CLEAs) and LentiKats (LKs) with their corresponding substrates from the 1,2,3-trichloropropane pathway: TCP, 1,2,3-trichloropropane; DCP, 2,3-dichloropropane-1-ol; CPD, 3-chloropropane-1,2-diol; ECH, epichlorohydrin; GDL, glycidol. Colours indicate different enzymes: haloalkane dehalogenase DhaA31 (in green), haloalcohol dehalogenase HheC (red) and epoxide hydrolase EchA (blue). Error bars represent the standard deviation from three independent measurements. Specific activities of free enzyme with corresponding substrate used as the reference for 100% relative activity were: 1.09±0.02 μmol.min
-1.mg
-1 (DhaA31 with
TCP), 1.61±0.10 μmol.min-1
.mg-1
(HheC with DCP), 3.04±0.09 μmol.min-1
.mg-1
(HheC with CPD), 29.45±1.73 μmol.min
-1.mg
-1 (EchA with ECH), 6.46±0.64 μmol.min
-1.mg
-1 (EchA with GDL).

Chapter 1
- 63 -
The storage stability of LentiKats of DhaA31 and EchA in a phosphate buffer without
additives at 4°C was assayed during a three-month period and compared with the storage
stability of free enzymes (Figure S2). After this period, changes in the activity of
immobilized and free enzymes correlated for both DhaA31 and EchA. Free and
immobilized DhaA31 retained 93% and 86% of its initial activity with TCP, respectively.
EchA showed 121% and 122% of its initial activity in free and immobilized form,
respectively. The increase was statistically significant for free EchA (t-test, p<0.05).
Similar moderate increases in specific activity during storage were reported also for some
other enzymes [240,243].
HheC showed significantly lower storage stability than DhaA31 and EchA. Free HheC
stored in a phosphate buffer at 4°C lost all its activity with DCP within two months (data
not shown). Tang and co-workers previously suggested that inactivation of HheC is caused
by the monomerization of the enzyme and intramolecular disulfide bond formation under
oxidizing conditions [219]. Confirming this suggestion, the stability of HheC was
significantly improved by its storage in a buffer containing a reducing agent (Figure S2A).
Free and immobilized HheC retained 63% and 49% of its initial activity, respectively, after
two months of storage in the presence of 2 mM dithiothreitol. The lower activity of HheC
immobilized in LentiKats is due to partial leaching (Figure S2B). The storage stability of
free enzymes was tested also at room temperature (22±2°C), which was later applied
during the continuous removal of TCP in a packed bed reactor. In contrast to DhaA31 and
EchA, HheC showed better storage stability at room temperature than at 4°C (Figures S3).
The enzyme retained 70% of its initial activity after 10 weeks of storage at room
temperature in phosphate buffer without any additive.
Multi-enzyme conversion of TCP using immobilized enzymes.
Numerous studies describe the immobilization of individual enzymes or whole cells,
but reports on the effects of immobilization on the performance of complete biochemical
pathways are scarce [100]. For synthetic biodegradation pathways employing engineered
enzymes such reports are to the best of our knowledge missing. We applied TCP pathway
immobilized in LentiKats in a one-pot multi-enzyme reaction to study how the modified
activities of individual immobilized enzymes affected the time course of TCP
biodegradation. Immobilized enzymes were mixed in a ratio 1:1:1 mg and incubated with
5 mM TCP under the same conditions as had been used for soluble enzymes (Figure 2C
and Table S5). Despite the fact that activity of individual enzymes after immobilization
was reduced by 27–83%, the efficiency of the whole pathway was lower only about 11%
when compared to the conversion with free enzymes. The conversion of TCP to GLY after
30 h reached 84% and conversion of all intermediates 89% of the theoretical maximum.
We also studied the reusability of the immobilized TCP pathway in a batch system (Figure
S4). The immobilized pathway retained 77% of its initial efficiency of TCP conversion to
GLY after 10 successive cycles of batch operation.

Chapter 1
- 64 -
The application of purified enzymes immobilized individually provides better control
over the reaction by enabling the tuning of enzyme stoichiometry and by compensating
for the activity loss of individual enzymes. However, the purification and separate
treatment of all three enzymes increases the cost of the biocatalyst. We therefore
demonstrated that cell-free extracts obtained from E. coli BL21(DE3) cells expressing
DhaA31, HheC, or EchA can be utilized. For the preparation of the LentiKats, cell-free
extracts were mixed to provide a ratio of enzymes corresponding to 1:1:1. The
combi-LentiKats containing approximately 1 mg of each of the three enzymes were used
for the biodegradation of 5 mM TCP. The degradation of TCP and the intermediates DCP
and GDL reached 95% of the theoretical maximum within 30 h of reaction (Figure 2D,
Table S6). The overall reaction profile was very similar to the profile obtained with
purified immobilized enzymes (Figure 2C). The conversion of TCP to GLY reached 89% of
the theoretical maximum. Thus, the co-immobilization of enzymes and their improved
proximity resulted in conversion comparable by efficiency with free enzymes. The
proximity and homogeneous distribution of DhaA31, HheC, and EchA in combi-LentiKats
was verified by confocal microscopy (Figure 3D).
Multi-enzyme conversion of TCP using packed bed reactor.
The performance of immobilized pathway was also tested under continuous
operation using a packed bed reactor composed of two columns (Figure 5). Column 1 was
packed with LentiKats of DhaA31, column 2 with a mixture of LentiKats of HheC and EchA.
The total mass ratio of enzymes 1:1:1 corresponded to the previous batch experiments.
DhaA31 was separated from HheC and EchA in order to prevent the inhibition of the last
step in the pathway, the conversion of GLD to GLY by TCP (Ki = 0.21 mM). EchA was
combined with HheC in column 2 in order to prevent a reverse reaction and push the
reaction equilibrium during the conversion of DCP and CPD by HheC towards the product
[238]. The separation of enzymes into two columns also enabled better control over the
individual reaction steps and evaluation of their efficiency.
The packed bed reactor was operated at room temperature 22±2°C and pH 8.2 for 10
weeks under the conditions described in Table S1 in Supplementary tables and figures.
During that period, the reactor with an effective volume of 0.15 L processed 11 L of
contaminated water. Experimental concentrations of TCP for evaluating column 1
efficiency were determined in the input of the column 1 due to the leakage of TCP from the
pumping system before reaching the column (Table S7). The average leakage caused by
hydrophobic nature of TCP (log P = 2.24) and its tendency to penetrate through rubber
tubing of peristaltic pump was approximately 29%. The experimental concentrations of
TCP in the column 1 input for each of ten weeks of operation were 2.25, 3.03, 5.91, 6.54,
7.97, 6.31, 7.94, 6.65, 6.80, and 7.02 mM. The levels of residual TCP and produced DCP
were determined in the effluent of column 1. The levels of residual DCP and produced GDL
and GLY were determined in the effluent of column 2. These values were used to evaluate
the efficiency of both columns and the efficiency of GLY production from TCP (Figure 6).

Chapter 1
- 65 -
No leakage of intermediates was observed. Neither ECH nor CPD was detected in the
system.
The efficiencies of column 1 and 2 were higher than 95% during the first eight and
four weeks of operation, respectively (Figure 6 and Table S7). The efficiency of the overall
conversion of TCP to GLY decreased from values above 90% observed during the first four
weeks to values lower than 60% at the end of two months of operation. The decreased
reactor efficiency accompanied by an accumulation of unreacted TCP, DCP and GDL in
effluent vessels could be attributed to: (i) slow enzyme inactivation caused by oxidation of
HheC and thermal unfolding of DhaA31 and EchA and (ii) slow leaching of enzymes from
the reactor, which resulted in about 10% loss of HheC activity and about 5% loss of
DhaA31 and EchA activities during the operation (Figure 6B). In total, 65.5 mmol (cca.
9.7 g) of the 67.7 mmol of TCP that entered the packed bed reactor during 10 weeks of
operation were converted to intermediates (efficiency 97%), and 52.6 mmol of GLY were
produced (efficiency 78%).
Figure 5. Schematic overview and photograph of the packed bed reactor used for removing 1,2,3-trichloropropane from the water under continuous operation. The system consisted of two glass columns (C1 and C2) packed with immobilized biocatalysts, two peristaltic pumps (P1 and P2), and three glass vessels connected with polytetrafluoroethylene tubing. A feed vessel (FV) contained 1 L of 0.1 M Tris-SO4 with 5 mM (week 1) or 10 mM (week 2-10) of TCP. Effluent vessel 1 (EV1) was used to collect samples downstream of column 1 and as a feeding bottle for column 2; effluent vessel 2 (EV2) was used to collect final samples downstream of column 2. The site for determining the TCP concentration in the input of column 1 is indicated with a red arrow. The part of tubing with connector was unscrewed from the column and sample of water with TCP was withdrawn from the tubing downstream the pump 1.

Chapter 1
- 66 -
Figure 6. A) Efficiency of column 1 (filled) and column 2 (cross-hatched) of the packed bed reactor during the continuous biodegradation of 1,2,3-trichloropropane (TCP). Black diamonds with a solid line show the overall efficiency of TCP conversion to glycerol by immobilized biocatalysts in the two-step packed bed reactor. Efficiency was calculated from the concentration of TCP measured in the inlet of column 1 and the concentration of glycerol measured in effluent vessel 2. B) Residual concentrations of the metabolites from the synthetic pathway for biodegradation of TCP detected in effluent vessel 1 (TCP) and effluent vessel 2 (2,3-dichloropropane-1-ol, DCP; glycidol, GDL; glycerol, GLY) during 10 weeks of operating the packed bed reactor.

Chapter 1
- 67 -
CONCLUSIONS
Recent studies on bacterial utilization of 1,3-dichloroprop-1-ene and chlorinated
ethenes suggest that the biodegradation of chlorinated aliphatic pollutants is associated
with the accumulation of reactive intermediates and strong oxidative stress, representing
a significant barrier for the evolution of corresponding aerobic metabolic pathways in vivo
[170,202]. The absence of natural microorganisms carrying aerobic pathways for the
biodegradation of TCP and experiences gained during attempts to engineer
microorganisms growing on this toxic compound seem to support this view
[213,228,232]. Engineering synthetic pathway for converting toxic TCP to the harmless
GLY in vitro is an alternative approach, which does not suffer from the limitations of
engineered bacterium. It is now widely accepted that in vitro multi-enzyme systems
represent an emerging field of biocatalysis due to their simplicity, predictability, and
controllability [67,93]. This study shows that an in vitro assembly of natural or synthetic
enzymatic pathways can be a promising concept for the biodegradation of environmental
pollutants and can provide promising results especially when combined with protein
engineering techniques.
Developed biotechnology requires further validation before it can be scaled-up and
used in real conditions. The performance of the immobilized biocatalyst should be tested
in real water samples contaminated with TCP. Pretreatment of the contaminated water by
adjusting high salinity or eliminating possible enzyme-inhibiting constituents might be
necessary. Conversion of TCP to GLY was demonstrated to be efficient at 22±2°C and is
also possible in broad pH range 7-10 (Figure S5). Yet, buffering of the contaminated water
to pH close to the reaction optima of enzymes can be beneficial to achieve maximal
efficiency of the process. The favorable features of the presented biotechnology are: (i)
degradation of TCP using immobilized cell-free extracts instead of purified enzymes is
possible, (ii) material used for immobilization of enzymes is affordable, safe and
non-biodegradable, (iii) protocol for disposal of used LentiKats by burning is well
established, and (iv) the amount of enzymes necessary to degrade almost 10 g of TCP
during the operation of reactor can be obtained from less than 2 l of cell culture yet
without optimized cultivation conditions. An immobilized synthetic pathway works at TCP
concentrations that are close to the water solubility limit of TCP (10 mM), which is one
order of magnitude higher than concentrations tolerated by engineered microorganisms.
At the same time, the system has the potential to cope with significantly lower
concentrations of TCP. Kinetic parameters of DhaA31 for TCP (kcat = 1.26 s-1,
kcat/Km = 1050 s-1.M-1) are of the same order of magnitude as those of the haloalkane
dehalogenase DhlA for 1,2-dichloroethane (kcat = 3.3 s-1, kcat/Km = 6200 s-1.M-1), which has
already proven its utility in a groundwater purification plant treating micro- to nanomolar
concentrations of the pollutant [125].
The remaining bottlenecks of the pathway are: (i) lower activity of HheC with
non-preferred (S)-DCP, (ii) accumulation of GDL due to the substrate preference of EchA,
and (iii) gradual inactivation of HheC. These important, but not critical, limitations can be

Chapter 1
- 68 -
overcome by another round of protein engineering or modification of the immobilization
protocol. The flux of intermediates through the immobilized pathway can be further tuned
using kinetic modeling and optimization of enzyme stoichimetry, which is an objective of
the follow-up research described in Chapter 2 of this Thesis.

Chapter 1
- 69 -
SUPPLEMENTARY TABLES AND FIGURES Table S1. Nucleotide sequences of genes used in this study.[a] Synthetic gene dhaA with 6xHis tag, codon optimized for expression in E. coli
CATATGAGCGAAATTGGCACCGGTTTTCCGTTTGATCCGCATTATGTTGAAGTTCTGGGTGAACGTATGCATTATGTGGATGTTGGTCCGCGTGATGGTACACCGGTTCTGTTTCTGCATGGTAATCCGACCAGCAGCTATCTGTGGCGTAACATTATTCCGCATGTTGCACCGAGCCATCGTTGTATTGCACCGGATCTGATTGGTATGGGTAAAAGCGATAAACCTGATCTGGATTATTTCTTCGATGATCATGTGCGTTATCTGGATGCATTTATTGAAGCACTGGGTCTGGAAGAAGTTGTGCTGGTTATTCATGATTGGGGTAGCGCACTGGGTTTTCATTGGGCAAAACGTAATCCGGAACGTGTTAAAGGTATTGCCTGCATGGAATTTATTCGTCCGATTCCGACCTGGGATGAATGGCCTGAATTTGCACGTGAAACCTTTCAGGCATTTCGTACCGCAGATGTGGGTCGTGAACTGATTATTGATCAGAACGCATTTATCGAAGGTGCACTGCCGAAATGTGTTGTTCGTCCGCTGACCGAAGTTGAAATGGATCATTATCGTGAACCGTTTCTGAAACCGGTTGATCGCGAACCGCTGTGGCGTTTTCCGAATGAACTGCCGATTGCCGGTGAACCTGCAAATATTGTTGCACTGGTTGAAGCCTATATGAATTGGCTGCATCAGAGTCCGGTTCCGAAACTGCTGTTTTGGGGCACACCGGGTGTTCTGATTCCGCCTGCAGAAGCAGCACGTCTGGCAGAAAGCCTGCCGAATTGTAAAACCGTTGATATTGGTCCGGGTCTGCATTATCTGCAAGAAGATAATCCGGACCTGATCGGTAGTGAAATTGCACGTTGGCTGCCTGCACTGCATCATCATCACCATCATTAAGGATCC
Synthetic gene dhaA31 with 6xHis tag, codon optimized for expression in E. coli
CATATGTCCGAAATTGGCACCGGCTTCCCGTTTGATCCGCACTATGTTGAAGTTCTGGGCGAACGTATGCACTATGTTGATGTTGGTCCGCGTGATGGCACCCCGGTTCTGTTTCTGCACGGTAACCCGACGAGCTCTTATCTGTGGCGTAATATTATCCCGCATGTCGCCCCGAGTCACCGCTGCATTGCACCGGATCTGATCGGCATGGGTAAATCCGACAAACCGGATCTGGACTATTTCTTTGATGACCATGTCCGCTACCTGGATGCATTTATTGAAGCTCTGGGCCTGGAAGAAGTGGTTCTGGTGATCCATGACTGGGGCTCTGCGCTGGGTTTCCACTGGGCCAAACGTAATCCGGAACGCGTGAAAGGTATTGCGTGTATGGAATTTATCCGTCCGTTCCCGACCTGGGATGAATGGCCGGAATTTGCCCGCGAAACCTTTCAGGCGTTCCGTACGGCCGATGTTGGCCGCGAACTGATTATCGACCAAAACGCATTCATTGAAGGTGCTCTGCCGAAATATGTCGTGCGTCCGCTGACGGAAGTGGAAATGGATCATTACCGCGAACCGTTTCTGAAACCGGTTGACCGTGAACCGCTGTGGCGCTTCCCGAACGAACTGCCGATTGCAGGCGAACCGGCTAATATCGTTGCGCTGGTCGAAGCCTACATGAACTGGCTGCACCAGTCACCGGTGCCGAAACTGCTGTTTTGGGGCACCCCGGGTTTCATTATCCCGCCGGCAGAAGCAGCACGTCTGGCTGAATCGCTGCCGAATTGCAAAACGGTTGATATCGGCCCGGGTCTGCATTTTCTGCAAGAAGATAACCCGGACCTGATTGGCTCTGAAATTGCCCGCTGGCTGCCGGCTCTGCACCACCACCACCACCACTAAGGATCC
Synthetic gene dhaA90R with 6xHis tag, codon optimized for expression in E. coli
CATATGAGCGAAATTGGCACCGGTTTTCCGTTTGATCCGCATTATGTTGAAGTTCTGGGTGAACGTATGCATTATGTGGATGTTGGTCCGCGTGATGGTACACCGGTTCTGTTTCTGCATGGTAATCCGACCAGCAGCTATCTGTGGCGTAACATTATTCCGCATGTTGCACCGAGCCATCGTTGTATTGCACCGGATCTGATTGGTATGGGTAAAAGCGATAAACCTGATCTGGATTATTTCTTCGATGATCATGTGCGTTATCTGGATGCATTTATTGAAGCACTGGGTCTGGAAGAAGTTGTGCTGGTTATTCATGATTGGGGTAGCGCACTGGGTTTTCATTGGGCAAAACGTAATCCGGAACGTGTTAAAGGTATTGCCTGCATGGAATTTATTCGTCCGCTGACCACCTGGGATGAATGGCCTGAATTTGCACGTGAAACCTTTCAGGCATTTCGTACCGCAGATGTGGGTCGTGAACTGATTATTGATCAGAACATGTGGATTGAAGGTCTGATTCCGGCAGGCGTGATTCGCCCTCTGACCGAAGTTGAAATGGATCATTATCGTGAACCGTTTCTGAAACCGGTTGATCGCGAACCGCTGTGGCGTTTTCCGAATGAACTGCCGATTGCCGGTGAACCTGCAAATATTGTTGCACTGGTTGAAGCCTATATGAATTGGCTGCATCAGAGTCCGGTTCCGAAACTGCTGTTTTGGGGTAATCCGGGTTATCTGATTACACCGGCAGAAGCAGCACGTCTGGCAGAAAGCCTGCCGAATTGTAAAACCGTTGATATTGGTCCGGGTCTGCATTTTCTGCAAGAAGATAATCCGGACCTGATCGGTAGTGAAATTGCACGTTGGCTGCCTGCACTGCATCATCATCACCATCATTAAGGATCC
Wild-type gene hheC[b]
CATATGTCAACCGCAATTGTAACAAACGTTAAGCATTTTGGGGGAATGGGGTCTGCACTTCGTCTCTCGGAAGCAGGACATACAGTGGCTTGCCACGATGAAAGCTTCAAACAAAAGGACGAACTTGAAGCCTTTGCCGAAACCTATCCACAACTCAAACCAATGTCGGAACAAGAACCAGCGGAACTCATCGAGGCAGTTACCTCCGCTTATGGTCAAGTTGATGTACTTGTGAGCAACGACATATTCGCACCAGAGTTCCAACCCATAGATAAATACGCTGTAGAGGACTATCGCGGTGCGGTCGAGGCGCTACAAATTAGACCATTTGCACTGGTCAACGCCGTTGCAAGTCAAATGAAGAAGCGCAAAAGCGGACATATTATCTTTATTACCTCTGCAACGCCCTTCGGGCCTTGGAAGGAACTTTCTACCTACACGTCAGCCCGAGCAGGTGCATGCACCTTGGCAAATGCCCTTTCGAAGGAACTCGGTGAATACAACATTCCGGTGTTCGCAATAGGACCCAATTATCTTCACAGTGAAGATAGTCCCTACTTCTACCCCACAGAACCGTGGAAAACGAATCCAGAACACGTTGCCCATGTCAAAAAAGTCACT

Chapter 1
- 70 -
GCGCTCCAGCGGTTAGGTACACAGAAAGAATTGGGAGAACTCGTCGCGTTTCTCGCGTCTGGTAGTTGTGACTATCTGACCGGCCAGGTGTTCTGGTTGGCCGGCGGATTCCCAATGATCGAGCGTTGGCCTGGTATGCCCGAGTAGGGATCC
Synthetic gene echA with 6xHis tag, codon optimized for expression in E. coli
CCATGGCAATCCGTCGTCCTGAAGATTTCAAACACTATGAGGTCCAGCTGCCTGATGTTAAAATTCATTATGTCCGTGAGGGAGCCGGTCCGACACTGCTGCTGCTGCATGGTTGGCCTGGTTTTTGGTGGGAATGGTCGAAAGTCATCGGTCCACTGGCCGAGCACTATGATGTTATTGTGCCTGATCTGCGCGGTTTTGGTGATAGCGAGAAACCTGACCTGAACGATCTGAGCAAATATAGCCTGGATAAAGCCGCTGATGATCAAGCTGCCCTGCTGGATGCTCTGGGTATCGAAAAAGCCTATGTCGTGGGCCATGATTTTGCCGCTATTGTGCTGCACAAATTCATCCGTAAATATTCCGACCGTGTCATCAAAGCAGCCATCTTTGACCCGATTCAACCAGACTTTGGGCCGGTGTATTTTGGACTGGGCCATGTTCATGAAAGCTGGTATAGCCAGTTTCACCAACTGGACATGGCTGTTGAAGTGGTAGGCTCTTCACGTGAAGTGTGTAAAAAATATTTCAAACATTTCTTCGATCACTGGTCCTATCGTGACGAACTGCTGACAGAGGAGGAACTGGAAGTCCACGTGGACAATTGTATGAAACCGGATAATATCCACGGCGGGTTCAACTATTATCGTGCCAACATTCGTCCTGATGCTGCTCTGTGGACAGACCTGGATCATACCATGAGTGACCTGCCGGTTACTATGATTTGGGGTCTGGGCGACACATGTGTTCCTTATGCCCCACTGATTGAGTTTGTTCCGAAATATTATAGCAACTATACGATGGAAACCATCGAGGATTGTGGCCATTTTCTGATGGTGGAGAAACCGGAAATCGCCATCGACCGTATTAAAACGGCCTTCCGTCACCACCACCACCACCATTAAAAGCTT
[a] Restriction sites used for cloning are underlined and the 6xHis tag is shown in bold. [b] GenBank accession number AF397296.1.
Table S2. Operational conditions of the packed bed reactor. week of operation 1 1
a 2 3 4 5 6 7 8 9 10
operational period (h) 33 66 166 166 166 166 166 166 166 166 166
volumetric flow column 1 (ml.min
-1)
0.50 0.50 0.25 0.25 0.25 0.25 0.25 0.25 0.25 0.25 0.25
volumetric flow column 2 (ml.min
-1)
0.50 0.25 0.10 0.10 0.10 0.10 0.10 0.10 0.10 0.10 0.10
[a] Two cycles were conducted during the first week of operation to optimize running conditions.
Table S3. Concentrations of metabolites (mM) from the time course of the multi-enzyme conversion of 1,2,3-trichloropropane catalyzed by free purified DhaAwt, HheC and EchA. time (min)
TCP std. DCP std. ECH CPD GDL std. GLY std. sum
0 4.68 0.02 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 4.68
50 3.94 0.03 0.18 0.05 0.00 0.00 0.07 0.01 0.03 0.00 4.22
100 3.51 0.05 0.56 0.11 0.00 0.00 0.21 0.02 0.10 0.02 4.38
200 2.86 0.04 0.91 0.16 0.00 0.00 0.48 0.05 0.17 0.02 4.42
400 1.94 0.07 1.17 0.11 0.00 0.00 0.72 0.03 0.56 0.09 4.39
750 0.99 0.11 1.29 0.13 0.00 0.00 0.62 0.02 1.49 0.20 4.39
1260 0.34 0.10 1.12 0.16 0.00 0.00 0.43 0.01 2.60 0.21 4.49
1800 0.11 0.06 0.89 0.20 0.00 0.00 0.26 0.03 3.36 0.13 4.62
Abbreviations common for Tables S3-7: TCP, 1,2,3-trichloropropane; DCP, 2,3-dichloropropane-1-ol; ECH, epichlorohydrin; CPD, 3-chloropropane-1,2-diol; GDL, glycidol; GLY, glycerol; std., standard deviation. Each value represents the mean value ± standard deviation from three independent experiments.

Chapter 1
- 71 -
Table S4. Concentrations of metabolites (mM) from the time course of the multi-enzyme conversion of 1,2,3-trichloropropane catalyzed by free purified DhaA31, HheC and EchA. time (min)
TCP std. DCP std. ECH CPD GDL std. GLY std. sum
0 4.57 0.07 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 4.57
50 1.11 0.00 1.48 0.21 0.00 0.00 1.13 0.08 0.33 0.01 4.05
100 0.24 0.00 1.88 0.04 0.00 0.00 1.57 0.08 0.90 0.03 4.59
200 0.01 0.00 1.63 0.08 0.00 0.00 1.02 0.08 1.94 0.03 4.60
400 0.00 0.00 0.86 0.16 0.00 0.00 0.37 0.08 2.97 0.05 4.20
750 0.00 0.00 0.37 0.06 0.00 0.00 0.16 0.08 3.86 0.13 4.39
1260 0.00 0.00 0.11 0.01 0.00 0.00 0.03 0.01 4.20 0.02 4.34
1800 0.00 0.00 0.04 0.01 0.00 0.00 0.00 0.00 4.32 0.06 4.36
Table S5. Concentrations of metabolites (mM) from the time course of the multi-enzyme conversion of 1,2,3-trichloropropane catalyzed by immobilized purified DhaA31, HheC and EchA. time (min)
TCP std. DCP std. ECH CPD GDL std. GLY std. sum
0 4.71 0.27 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 4.71
50 1.86 0.19 1.14 0.03 0.00 0.00 0.33 0.05 0.11 0.04 3.44
100 0.89 0.06 1.54 0.18 0.00 0.00 0.88 0.05 0.28 0.02 3.59
200 0.13 0.01 1.54 0.16 0.00 0.00 1.46 0.15 1.06 0.14 4.19
400 0.00 0.00 1.30 0.12 0.00 0.00 1.04 0.15 2.05 0.06 4.39
700 0.00 0.00 1.02 0.16 0.00 0.00 0.53 0.04 2.77 0.16 4.32
1320 0.00 0.00 0.52 0.07 0.00 0.00 0.27 0.04 3.48 0.19 4.27
1800 0.00 0.00 0.34 0.07 0.00 0.00 0.16 0.03 3.94 0.07 4.44
Table S6. Concentrations of metabolites (mM) from the time course of the multi-enzyme conversion of 1,2,3-trichloropropane catalyzed by immobilized cell-free extracts with DhaA31, HheC and EchA. time (min)
TCP std. DCP std. ECH CPD GDL std. GLY std. sum
0 4.86 0.10 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 4.86
50 2.19 0.06 0.92 0.04 0.00 0.00 0.32 0.32 0.15 0.02 3.58
100 1.04 0.14 1.28 0.06 0.00 0.00 0.90 0.83 0.59 0.13 3.81
200 0.25 0.08 1.34 0.08 0.00 0.00 0.83 0.96 1.49 0.37 3.91
400 0.00 0.00 1.21 0.06 0.00 0.00 0.37 0.42 2.48 0.35 4.06
700 0.00 0.00 0.88 0.06 0.00 0.00 0.12 0.12 3.06 0.27 4.06
1320 0.00 0.00 0.47 0.02 0.00 0.00 0.00 0.00 4.14 0.24 4.61
1800 0.00 0.00 0.25 0.02 0.00 0.00 0.00 0.00 4.32 0.14 4.57

Chapter 1
- 72 -
Table S7. Concentrations of metabolites in a packed bed reactor and the calculated efficiencies of columns 1 and 2. weeks of operation
1 1 2 3 4 5 6 7 8 9 10
TCP in IC1 (mM)
2.25 3.03 5.91 6.54 7.33 7.97 6.31 7.94 6.65 6.80 7.02
TCP in EV1 (mM)
0.00 0.00 0.00 0.00 0.12 0.15 0.11 0.12 0.36 0.61 0.74
DCP in EV1 (mM)
2.12 2.89 5.55 6.67 6.64 7.45 5.78 8.03 6.52 6.14 7.52
DCP in EV2 (mM)
0.15 0.11 0.34 0.35 0.34 0.64 0.74 1.33 1.37 1.51 2.50
GDL in EV2 (mM)
0.00 0.00 0.00 0.00 0.00 0.17 0.26 0.47 0.52 0.64 1.09
GLY in EV2 (mM)
1.82 2.81 5.69 6.87 6.97 6.73 5.60 5.19 4.45 3.93 2.56
efficiency of C1 (%)
a
100 100 100 100 98 98 98 98 95 91 89
efficiency of C2 (%)
b
93 96 94 95 95 89 83 78 71 65 52
efficiency of GLY production (%)
c
81 93 96 100 95 84 89 65 67 58 36
Each number in mM represents a mean value from at least two technical replicates. Abbreviations: IC1, input of column 1; EV1, effluent vessel 1; EV2, effluent vessel 2; C1, column 1; C2, column 2.
[a] Calculated as:
[b] Calculated as:
[c] Calculated as:
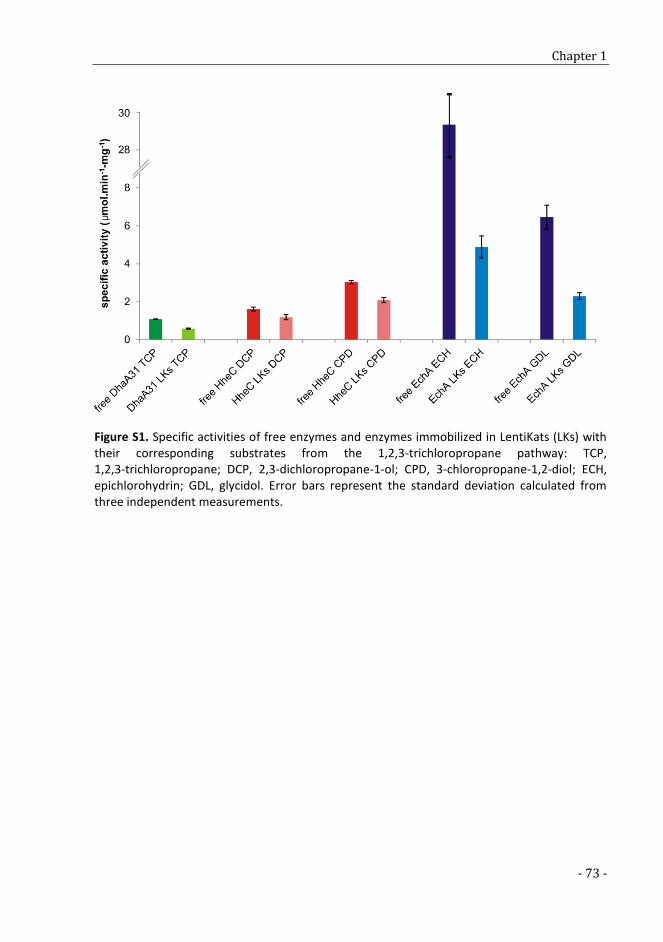
Chapter 1
- 73 -
Figure S1. Specific activities of free enzymes and enzymes immobilized in LentiKats (LKs) with their corresponding substrates from the 1,2,3-trichloropropane pathway: TCP, 1,2,3-trichloropropane; DCP, 2,3-dichloropropane-1-ol; CPD, 3-chloropropane-1,2-diol; ECH, epichlorohydrin; GDL, glycidol. Error bars represent the standard deviation calculated from three independent measurements.

Chapter 1
- 74 -
Figure S2. A) Storage stability of free and immobilized haloalkane dehalogenase DhaA31, haloalcohol dehalogenase HheC, and epoxide hydrolase EchA at 4°C measured during three-month interval. Activities measured at the beginning of storage, after 2 and 3 months of storage are diversified by fading colors. Free enzymes and LentiKats (LKs) were stored in 50 mM phosphate buffer of pH 7.5 without additives (DhaA31 and EchA), or with 2 mM dithiothreitol (HheC). Residual activities of DhaA31, HheC and EchA were measured with their corresponding substrates from the TCP pathway (1,2,3-trichloropropane, 2,3-dichloropropane-1-ol, and epichlorohydrin, respectively) at 37°C. A relative activity of 100% corresponds to the specific activity of: 1.13±0.03 µmol.min
-1.mg
-1 (free DhaA31); 0.58±0.03 µmol.min
-1.mg
-1 (LKs of
DhaA31); 1.61±0.20 µmol.min-1
.mg-1
(free HheC); 1.19±0.13 µmol.min-1
.mg-1
(LKs of HheC); 23.37±0.39 µmol.min
-1.mg
-1 (free EchA); 4.96±0.87 µmol.min
-1.mg
-1 (LKs of EchA). Error bars
represent the standard deviation calculated from three independent measurements. B) SDS-PAGE analysis of the buffer from storage of LKs of HheC, lane 1, standard sample of 1 µg of purified HheC; lane 2, buffer with fresh LKs; lane 3, 1 week of storage; lane 4, 2 weeks of storage; lane 5, 1 month of storage; lane 6, 2 months of storage; lane 7, 3 months of storage. The amount of leached enzyme after 3 months of storage was approximately 1.1 mg, which corresponds to 22% of the original amount of HheC immobilized in the LentiKats.

Chapter 1
- 75 -
Figure S3. Storage stability of free haloalkane dehalogenase DhaA31 (green columns), haloalcohol dehalogenase HheC (red columns), and epoxide hydrolase EchA (blue columns) at room temperature (22±2°C). Residual activities of DhaA31, HheC and EchA were measured with their corresponding substrates from the TCP pathway (1,2,3-trichloropropane, 2,3-dichloropropane-1-ol, and epichlorohydrin, respectively) at room temperature (22±2°C). Error bars represent the standard deviation from three independent measurements. Specific activities of fresh free enzyme with corresponding substrate used as the reference for 100% relative activity were: 0.44±0.04 μmol.min
-1.mg
-1 (DhaA31 with 1,2,3-trichloropropane),
0.29±0.02 μmol.min-1
.mg-1
(HheC with 2,3-dichloropropane-1-ol), and 14.11±0.07 μmol.min
-1.mg
-1 (EchA with epichlorohydrin).

Chapter 1
- 76 -
Figure S4. Recycling of the 1,2,3-trichloropropane pathway in LentiKats. The relative activity of the immobilized biocatalyst during the first cycle is 100% and corresponds to the production of 0.39 mM of glycerol from 1 mM of 1,2,3-trichloropropane during 100 minutes of reaction time in 50 mM Tris-SO4 buffer of pH 8.2 at 30°C. Error bars represent the standard deviation from three independent measurements.
Figure S5. Time courses of multi-enzyme conversions of 1,2,3-trichloropropane (TCP) with the pathway immobilized in LentiKats. A) reaction in 50 mM sodium phosphate buffer of pH 7.0; and B) 50 mM NaOH/glycine buffer of pH 10. All reactions were performed using enzymes with a mass ratio of 1:1:1 mg in 15 ml of reaction mixture at 20°C. DCP, 2,3-dichloropropane-1-ol; ECH, epichlorohydrin; CPD, 3-chloropropane-1,2-diol; GDL, glycidol; GLY, glycerol. Each data point represents the mean value ± standard deviation from three independent experiments.

- 77 -

- 78 -

- 79 -
CHAPTER 2
Maximizing the efficiency of in vitro
multi-enzyme process by stoichiometry
optimization

Chapter 2
- 80 -
INTRODUCTION
Multi-enzyme processes have great potential for the biosynthesis of fine and bulk
chemicals, bioremediation and biosensing [94,255,256]. Studies on two-enzyme systems
dominate the literature, but systems of three [95], four [257] and even twelve [258] or
thirteen [255] enzymes are known. Multi-enzyme systems are superior to single-enzyme
biocatalysis in that they can catalyze more complex synthetic schemes. In vitro
multi-enzyme networks enable simpler process control than analogous in vivo systems
and suffer less from reactant toxicity [29,259,260]. However, both system types often
exhibit reaction bottlenecks due to imbalanced enzyme properties.
Protein engineering is often used to improve enzymes’ catalytic properties and
stability [102,261]. Many engineered enzymes can be used in multi-enzyme processes but
methods for predicting their impact on productivity are lacking. Kinetic modelling is
essential for analyzing enzymatic reactions and can enable their rational optimization
[94,96,262,263]. However, there are only few accurate kinetic models of in vitro multi-
enzyme systems [95,96,255,257]. Available models rarely have experimental support and
their use in optimizing processes with engineered enzymes has not been adequately
explored.
The aim of this study was to develop a workflow for optimizing in vitro multi-enzyme
processes by using kinetic modelling to predict the effects of varying enzyme
stoichiometry and introducing available engineered enzymes. Our model system (Figure
1) was a synthetic metabolic pathway for the five-step biotransformation of toxic
industrial waste product 1,2,3-trichloropropane (TCP) into the commodity chemical
glycerol (GLY). This pathway has been previously assembled inside living cells
[211,213,232] and is based on haloalkane dehalogenase DhaA from Rhodococcus
rhodochroust NCIMB 13064 [209], haloalcohol dehalogenase HheC [216], and epoxide
hydrolase EchA from Agrobacterium radiobacter AD1 [237]. Three different haloalkane
dehalogenase variants were evaluated: (i) wild-type DhaA; previously constructed
mutants (ii) DhaA31with improved activity [225]; and (iii) DhaA90R with increased
enantioselectivity [226]. Kinetic models were built for pathways using each DhaA variant
and their enzyme stoichiometry was optimized under defined constraints.

Chapter 2
- 81 -
MATERIAL AND METHODS
Reagents. TCP, (R,S)-2,3-dichloropropane-1-ol (DCP), (R,S)-epichlorohydrin (ECH), (R,S)-
3-chloropropane-1,2-diol (CPD), (R,S)-glycidol (GDL), glycerol (GLY), acetone, ethyl
acetate, and hexanol were purchased from Sigma-Aldrich (USA). All chemicals used in this
study were of analytical grade. The Free Glycerol Assay Kit was purchased from BioVision
(USA).
Gene synthesis, cloning, cultivations and purification of enzymes. These procedures
were performed as described in Material and Methods section of Chapter 1.
Analytical techniques. Analytical techniques used in this study were described in
Material and Methods section of Chapter 1 of this Thesis.
Production of (S)-DCP by preparative kinetic resolution of (R,S)-DCP. Kinetics of HheC
with (S)-DCP was determined with the substrate prepared by kinetic resolution of
commercially available racemate using (R)-selective HheC. Kinetic resolution of (R,S)-DCP
was performed in 4 l of 50 mM Tris-SO4 buffer (pH 8.5) at 30°C using a 10 l screw cap
bottle. Substrate of purity ≥ 97% (Sigma Aldrich, USA) was added to a final concentration
of 22 mM. The enzymatic reaction was initiated by adding approximately 115 mg of HheC
as a crude extract and 125 mg of purified EchA. The excess of EchA prevented the reverse
reaction during the conversion of DCP and CPD by pushing the equilibrium toward the
product. The reaction was periodically monitored for 8 h and samples were analysed using
a Network GC System 6890N (Agilent Technologies, USA) equipped with a flame
ionisation detector and an Astec CHIRALDEX B-TA 30 m x 0.25 mm x 0.12 µm capillary
column (Sigma-Aldrich, USA). Samples (0.5 mL) were collected from the reaction mixture,
extracted with 1 ml of ethyl acetate, and dried with anhydrous sodium sulphate. Aliquots
(1 µl) were injected into the GC at an injector temperature of 200°C and a split ratio of
83:1. The operating column temperature was held at 100°C for 15 min. Helium was used
as the carrier gas at a continuous flow rate of 0.8 ml.min-1. The flame ionisation detector
was operated at 250°C. Reaction was terminated and unconverted (S) enantiomer was
extracted twice with 0.5 and 1 l of ethyl acetate. The organic phase was dried with sodium
sulphate. Ethyl acetate was removed at 40°C (water bath temperature) and 240 mbar and
then at 60°C (bath temperature) and 60 mbar using a Vacuum Rotavapor R-215 fitted
with a vacuum controller, vacuum pump and heating bath (Büchi, Switzerland). After the
evaporation of most of the ethyl acetate, the chemical composition of the residue was
determined by NMR after dissolution in CDCl3. NMR spectra were acquired using a 600
MHz AVANCE spectrometer (Bruker, Germany) with a double resonance TCI probe (1H, 13C). The resulting NMR spectra were compared to predicted NMR data calculated using
Advanced Chemistry Development Software v11.01 (ACD/Labs, Canada). The purity
(85%) of the synthesized (S)-DCP with residual ethyl acetate was determined by GC using
(R,S)-DCP as a reference material. The (S)-DCP was obtained obtained with e.e. ≥ 99%.

Chapter 2
- 82 -
Enzyme kinetics. Steady-state kinetic parameters for DhaA, DhaA31 and DhaA90R with
TCP and for HheC with (R,S)-DCP, (S)-DCP and (R,S)-CPD were determined in 25 ml micro-
flasks sealed with Mininert valves (Alltech, USA) in 10 ml of 50 mM Tris-SO4 buffer (pH
8.5) at 37°C. Because HheC is strongly (R)-selective, the kinetic constants for (R)-DCP,
which was commercially unavailable, were determined by measuring the initial velocity of
racemic DCP conversion. The initial velocity measurements were carried out at various
substrate concentrations that were verified by GC. The reaction was initiated by adding
the purified enzyme and terminated by mixing samples of the reaction mixture with 35%
(v/v) nitric acid. The concentration of the reaction product (chloride ions) was measured
using a Sunrise spectrophotometer (Tecan, Switzerland) at 460 nm after the addition of
mercuric thiocyanate and ferric ammonium sulfate. Specific activities were quantified by
measuring the slope of the initially-linear region in a plot of halide concentration against
time. The kinetic constants were calculated by non-linear fitting using Origin 7.5
(OriginLab Corporation, USA) or DynaFit 4 (BioKin, USA) based on the Michaelis-Menten
equation (1) where v is the reaction velocity, E0 is the enzyme concentration, kcat is the
turnover number, S is substrate concentration and Km is the Michaelis constant. Steady-
state kinetic parameters for EchA with (R,S)-ECH and (R,S)-GDL were determined by
monitoring the depletion of the substrate (initial concentration: 5 mM) in 10 ml of 50 mM
Tris-SO4 buffer (pH 8.5) at 37°C. The substrate concentration was verified by GC. The
reaction was initiated by adding the purified enzyme. Samples of the reaction mixture
(0.5 ml) were mixed with 0.5 ml of acetone containing the internal standard hexanol and
analyzed by GC. The numerical integration program Scientist 1.0 (MicroMath, USA) was
used to fit the ECH concentration progress curve to equation (1). The GDL concentration
progress curve was fitted to equation (2), where P is the concentration of the reaction
product and Ki is a product inhibition constant. The inhibition constant Kc was determined
by measuring the inhibitory effects of TCP at various concentrations (0, 0.5, 1.0 and
2.5 mM) on the conversion of 5 mM GDL by EchA in 15 ml of 50 mM Tris-SO4 buffer (pH
8.5) at 37°C (Figure S2 in Supplementary tables and figures). Experimental concentrations
of GDL and TCP were verified by GC. The numerical integration software Scientist 1.0
(MicroMath, USA) was used to fit the GDL concentration progress curves for each separate
inhibitor concentration to equation (3), where I is the inhibitor concentration. The
resulting expressions were compared to obtain a consensus value for the equilibrium
inhibition constant Kc.

Chapter 2
- 83 -
Enzyme enantioselectivity. The formation of the (R) and (S) enantiomers of DCP during
the conversion of TCP (2 mM) by DhaA, DhaA31 or DhaA90R was measured in 15 ml of
Tris-SO4 buffer (pH 8.5) at 37°C. Samples (0.5 ml) were taken periodically, mixed with
1 ml of ethyl acetate and vortexed for 30 s. The organic phase was then dried over sodium
sulphate and analysed by chiral GC as described previously.
Kinetic model development. The kinetic model of the TCP pathway was assembled based
on the reaction scheme shown in Figure 1 using the kinetic parameters listed in Table 1.
All five reaction steps in the multi-enzyme conversion of TCP exhibited Michaelis-Menten
kinetics. The conversion of the prochiral TCP into either (R)-DCP or (S)-DCP by DhaA,
DhaA31 or DhaA90R was described using equation (4). The rate constants kcat,TCP,(R)-DCP and
kcat,TCP,(S)-DCP were determined from the overall kcat for the appropriate DhaA variant with
TCP and the ratio of (R)- and (S)-DCP produced from the prochiral TCP by DhaA, DhaA31
or Dha90R. The formation of (R)-DCP and (S)-DCP and the subsequent conversion of both
enantiomers into (R,S)-ECH was described using equations (5) and (6). The remaining
reaction steps were assumed to be non-selective. The consecutive conversions of
(R,S)-ECH, (R,S)-CPD and (R,S)-GDL by HheC and EchA were described using equations
(7), (8) and (9), respectively. The final reaction step, i.e. the conversion of (R,S)-GDL into
the final product GLY by EchA, was described using Michaelis-Menten equations (9) and
(10). These expression feature the equilibrium constants Ki and Kc, which describe the
competitive inhibition caused by GLY and TCP, respectively.

Chapter 2
- 84 -
Optimizing the multi-enzyme conversion of TCP. The initial constraints used when
modelling the multi-enzyme conversion of TCP in order to verify the newly developed
kinetic model were as follows: the starting concentration of TCP was 2 mM and the initial
experimental concentration was as close to 2 mM as possible (Tables S1-S3); the
multi-enzyme reaction was allowed to proceed for 300 min; the concentrations of each of
the three enzymes (i.e. the DhaA variant, HheC and EchA) were 0.1 mg.ml-1; the total
enzyme loading in 10 ml of reaction mixture was 3 mg. Dynamic simulations of the multi-
enzyme system based on a series of Michaelis-Menten equations were performed using
code written in the Python 2.7 programming language. The equations were expressed in
differential form and integrated using Euler's method with a fixed step size of 0.18 s. The
effects of using different DhaA variants on the performance of the multi-enzyme process
were evaluated under the following constraints: at least 95% conversion of TCP into GLY
was to be achieved within 300 min while using the DhaA variant, HheC, and EchA in an
unoptimized mass ratio of 1:1:1 at a total enzyme loading of 3.0 mg. The optimization
algorithm increased the total enzyme loading in a stepwise fashion, using increments of
around 0.1 mg (i.e. increasing the loading of each individual enzyme by 0.066 mg in each
step) until the threshold of 95% conversion was surpassed. The enzyme stoichiometry in
each of three pathway variants was then optimized in order to identify the ratio of DhaA
variant, HheC and EchA that would yield 95% conversion of 2 mM TCP into GLY within
300 min while minimizing the combined loading of the three enzymes. The algorithm
searched for the most efficient ratio of three enzymes in the system starting with a total
enzyme loading of 3 mg. The total enzyme loading within the system was increased in
0.1 mg increments until the most efficient ratio surpassed 95% conversion. For each total
enzyme loading, 496 enzyme ratios were evaluated with step of 3% of a given total
amount.
Multi-enzyme conversion of TCP in batch experiments. The multi-enzyme conversion of
TCP into the final product GLY was assayed in 10 ml of 50 mM Tris-SO4 buffer (pH 8.5) in
25 ml Micro-flasks sealed with Mininert valves (Alltech, USA) and incubated in water bath
with shaking at 37°C. The reaction was initiated by adding a specific amount of purified
DhaA variant, HheC and EchA into the reaction mixture along with TCP at an initial
concentration of 2 mM. Samples were periodically taken from the reaction mixture, mixed
with acetone (1:1) containing hexanol as an internal standard, and analysed by GC to
determine the concentrations of TCP, DCP, ECH, CPD and GDL. Calibration curves for TCP,

Chapter 2
- 85 -
DCP, ECH, CPD and GDL concentrations of 0 – 5 mM were constructed to facilitate data
analysis. Selected samples were analysed by GC-MS to verify the identities of the
metabolites; in other cases, metabolites were identified based on flame ionisation
detection. The concentration of GLY in the reaction mixture was determined
spectrophotometrically using the Free Glycerol Assay Kit. Samples of the reaction mixture
(0.1 ml) were heated at 95°C for 5 min to terminate the enzymatic reaction and then
centrifuged at 18,000 g for 1 min, after which they were diluted in an assay buffer and
analysed according to the manufacturer's protocol. The GLY concentration was calculated
from the sample’s absorbance at 570 nm. A calibration curve for GLY concentrations of
10 – 80 µM was constructed by analyzing samples prepared from a 1 mM GLY standard
solution.
Figure 1. Synthetic pathway for the three-enzyme biotransformation of 1,2,3-trichloropropane. Five consecutive steps are catalyzed by the haloalkane dehalogenase DhaA, from Rhodococcus rhodochrous NCIMB 13064; the haloalcohol dehalogenase HheC and the epoxide hydrolase EchA from Agrobacterium radiobacter AD1. 1,2,3-trichloropropane (TCP) is converted via (R)-2,3-dichloropropane-1-ol ((R)-DCP) and (S)-2,3-dichloropropane-1-ol ((S)-DCP), epichlorohydrin (ECH), 3-chloropropane-1,2-diol (CPD), glycidol (GDL) to glycerol (GLY). Arrows in bold indicate key bottlenecks.

Chapter 2
- 86 -
RESULTS AND DISCUSSION
We initially prepared soluble enzymes with purities of ≥95% for DhaA and EchA and
≥85% for HheC. To validate the in vitro biotransformation of TCP into GLY, 1 mg each of
purified DhaA, HheC and EchA were mixed in 10 ml of Tris-SO4 buffer (pH 8.5) and
incubated with 2 mM TCP at 37°C for 300 min. The enzymes’ molecular weights are
similar (34.1, 29.3 and 36.5 kDa, respectively), so their mass ratio roughly equals their
molar ratio. A gas chromatography (GC) method for detecting and quantifying TCP and all
pathway intermediates in a single analysis was developed and used to monitor the
five-step process (Figure S1 in Supplementary tables and figures). The time course for the
conversion confirmed the pathway’s viability and revealed two major bottlenecks: (i) the
slow initial conversion of TCP to 2,3-dichloropropan-1-ol (DCP); and (ii) the mismatched
selectivity of DhaA and HheC, which caused (S)-DCP to accumulate (Figures 1 and 2).
Similar bottlenecks were identified in vivo [213,232].
We then investigated two DhaA mutants with properties tuned to address these
bottlenecks. The mutant DhaA31 was previously constructed in our laboratory using
computer-aided focused directed evolution of protein tunnels [225]. Its catalytic rate
towards TCP is 32 times that of wild-type DhaA. The mutant DhaAr5-90R (henceforth
“DhaA90R”) was obtained by van Leeuwen and co-workers via the focused directed
evolution of DhaA31. It has the same activity as wild-type DhaA but 7 times higher
selectivity for (R)-DCP formation [226]. Time courses for the three-enzyme conversion of
TCP using purified DhaA31 or DhaA90R were recorded under the conditions described for
the wild-type pathway. The differences between the resulting conversion profiles were
consistent with the kinetic properties of DhaA, DhaA31 and DhaA90R (Figure 2 and Tables
S1-S3 in Supplementary tables and figures).
Sixteen steady-state kinetic parameters were determined for each purified enzyme to
establish a kinetic model of the pathway (Table 1). All of the studied reactions exhibit
Michaelis-Menten (MM) kinetics. The conversion of glycidol (GDL) into GLY, was
described by a MM equation with two inhibition constants defining the inhibitory effects
of GLY (Ki = 1.00 mM) and TCP (Kc = 0.21 mM). These effects together with the substrate
preference of EchA for epichlorohydrin (ECH) caused GDL accumulation in vitro (Figure
2).

Chapter 2
- 87 -
Figure 2. Three-enzyme conversion of 1,2,3-trichloropropane (TCP) to glycerol (GLY) catalyzed by the pathway employing (a) wild-type DhaA, (b) engineered DhaA31 and (c) engineered DhaA90R, respectively. Experimental and simulated metabolite concentrations are indicated by symbols and solid lines, respectively. The reaction’s intermediates are 2,3-dichloropropane-1-ol (DCP), epichlorohydrin (ECH), 3-chloropropane-1,2-diol (CPD), and glycidol (GDL). The following parameters were constrained during the in vitro and in silico experiments: reaction volume (10 ml), initial TCP concentration (~2 mM; experimental concentrations are listed in Tables S1-S3), reaction time interval (300 min), the concentrations of each enzyme (DhaA variant, HheC and EchA; 0.1 mg.ml
-1), and the total loading of all three enzymes in the reaction
mixture (3 mg). Data points represent mean values from three independent measurements. Error bars are omitted for clarity; standard deviations are provided in Tables S1-S3 of Supplementary tables and figures.

Chapter 2
- 88 -
Table 1. Experimental steady-state kinetic parameters used in the kinetic model.
DhaA HheC
Km,TCP (mM) 1.01±0.08 Km,(R)-DCP (mM) 2.49±0.16
kcat,TCP,(R)-DCP (s-1
) 0.04a Km,(S)-DCP (mM) 3.33±0.51
kcat,TCP,(S)-DCP (s-1
) 0.03a Km,CPD (mM) 0.86±0.07
DhaA31 kcat,(R)-DCP (s-1
) 1.81±0.05
Km,TCP (mM) 1.79±0.09 kcat,(S)-DCP (s-1
) 0.08±0.00
kcat,TCP,(R)-DCP (s-1
) 0.58b kcat,CPD (s
-1) 2.38±0.06
kcat,TCP,(S)-DCP (s-1
) 0.47b EchA
DhaA90R Km,ECH (mM) 0.09±0.08
Km,TCP (mM) 12.56±2.99 Km,GDL (mM) 3.54±0.09
kcat,TCP,(R)-DCP (s-1
) 0.19c kcat,ECH (s
-1) 14.37±0.52
kcat,TCP,(S)-DCP (s-1
) 0.02c kcat,GDL (s
-1) 3.96±0.08
[a] Determined from the ratio of (R)- and (S)-DCP production as 56 % and 44 % of 0.07±0.00 s-1 for kcat,TCP,(R)-DCP and kcat,TCP,(S)-DCP, respectively (Material and Methods). [b] Determined as 55 % and 45 % of 1.05±0.02 s-1 for kcat,TCP,(R)-DCP and kcat,TCP,(S)-DCP, respectively (Material and Methods). [c] Determined as 90 % and 10 % of 0.21±0.03 s-1 for kcat,TCP,(R)-DCP and kcat,TCP,(S)-DCP, respectively (Material and Methods).
The kinetic model was validated against experimental data for the three-enzyme
conversion of 2 mM TCP (Figure 2). The two datasets were in good agreement – using
1:1:1 mixtures of the three enzymes with total enzyme loads of 3 mg, the predicted (and
measured) productivities of the DhaA, DhaA31 and DhaA90R pathways were 72% (62%),
85% (85%) and 45% (42%), respectively. No enzyme inactivation occurred, but the model
could be extended to include inactivation constants if necessary.
The model was then used to predict the DhaA, HheC and EchA loadings needed to
achieve 95% conversion of TCP to GLY under the chosen conditions by simulating
stepwise increases in enzyme loading within the reaction system until the productivity
goal was reached. At a fixed DhaA:HheC:EchA mass ratio of 1:1:1, the wild-type haloalkane
dehalogenase pathway reached the productivity goal using 2.4 mg of each enzyme, i.e. a
total enzyme load of 7.2 mg (Figure 3). The DhaA31 and DhaA90R pathways required
1.8 mg and 4 mg of each enzyme, respectively, (i.e. total enzyme loads of 5.4 or 12 mg) to
reach the goal. The differences in the modelled time courses and quantities of enzyme
required to achieve 95% conversion for the three pathway variants demonstrate the
profound effects of introducing engineered DhaA variants (Figure 3). Despite a

Chapter 2
- 89 -
pronounced accumulation of DCP and GDL during the initial 25 min, the DhaA31 pathway
was around 25% more efficient than the wild-type version. DhaA31 significantly
accelerated the conversion of TCP and thus accelerated the consumption of accumulated
GDL by suppressing TCP’s inhibitory effect. Conversely, DhaA90R reduced the system’s
efficiency despite effectively minimizing DCP accumulation. This selective but catalytically
inefficient mutant was thus not beneficial to the process.
We then evaluated the effects of enzyme stoichiometry on efficiency. An algorithm
was used to minimize the total enzyme load without sacrificing productivity. Optimal
enzyme mass ratios were calculated for each pathway. The modelled time courses for
individual reactions were similar in each case, but the optimized ratios and total enzyme
loadings differed significantly between pathways (Figure 3). Enzyme stoichiometry
optimization increased the efficiencies of the DhaA, DhaA31 and DhaA90R pathways and
reduced their total enzyme loadings required for >95% conversion by 21%, 41%, and
38%, respectively.
All of the optimization simulations were validated by testing the enzyme mass ratios
in vitro. The resulting data agreed very closely with the predictions (Figure 3). The
optimized DhaA90R pathway was around 10% less productive than predicted, possibly
because the Km of DhaA90R for TCP was underestimated due to the limited water
solubility of TCP (cca. 10 mM). In all other cases, the optimized systems achieved
productivities of 94% - 98% (Table S1-S3). Because the experimental time courses only
reflect optimal cases based on pre-defined constraints, we used the simulated data to
create 3D isoproductivity charts showing the effects of varying the loadings of the DhaA
variants, HheC and EchA (Figure 4). These charts show the limiting components for each
pathway and can be used to identify solutions with similar productivities.

Chapter 2
- 90 -
Figure 3. Optimization of three-enzyme conversion of 1,2,3-trichloropropane by kinetic modelling and employment of engineered enzyme variants. Calculated and measured results are indicated by solid lines and symbols, respectively. The following parameters were constrained: reaction volume (10 ml), initial TCP concentration (2 mM), and reaction duration (300 min). The initial optimization goal was 95% conversion of TCP into GLY within 300 min using a 1:1:1 ratio of wild-type enzymes. The calculated and experimentally verified total enzyme loading required to achieve this is 7.2 mg. The wild-type DhaA enzyme was then replaced with the mutants DhaA31 or DhaA90R to study the effect of their kinetics on the pathway (white arrows). Further optimization was achieved by tuning the enzyme ratios (grey arrows). Reductions in total enzyme loading achieved by employment of mutants or stoichiometry optimization alone and stoichiometry optimization together with mutants's effect are shown in circles and squares, respectively. Experimental concentrations of

Chapter 2
- 91 -
1,2,3-trichloropropane (TCP), 2,3-dichloropropane-1-ol (DCP), epichlorohydrin (ECH), 3-chloropropane-1,2-diol (CPD), glycidol (GDL) and glycerol (GLY) were determined by GC and using a glycerol assay kit. Data points represent mean values from three independent experiments. Error bars are omitted for clarity; standard deviations are provided in the Supporting Information (Tables S1-S3).
Figure 4. Isoproductivity color charts showing how biocatalyst concentration affects productivity in the three-enzyme conversion of 1,2,3-trichloropropane into glycerol. Applied constraints are described in the caption of Figure 3; the optimal solutions shown in that figure are indicated by white dots. Values on axes represent the loading of the corresponding enzyme in the reaction mixture. The black line indicates the threshold productivity of 95%.

Chapter 2
- 92 -
CONCLUSIONS
Our results demonstrate that modifying enzyme kinetic parameters and optimizing
enzyme stoichiometry both improved the efficiency of the studied multi-enzyme system.
However, the far simpler process of stoichiometry optimization had a greater impact than
introducing engineered enzymes. The optimized pathway using wild-type DhaA required
a similar total enzyme load to the non-optimized pathway using the engineered DhaA31
(5.7 mg versus 5.4 mg), showing that kinetic modelling alone can provide excellent
solutions in certain cases. Naturally, the best result was achieved with an optimized
pathway using engineered DhaA31: additive effects in this case reduced the catalyst load
required for 95% productivity by 56% relative to the unoptimized wild-type pathway
(Figure 3). This would be very economically beneficial in a large-scale industrial process.
In summary, we present an experimentally validated in silico optimization of a multi-
enzyme process by employing engineered enzyme variant and biocatalyst stoichiometry
tuning. Our workflow entails: (i) experimental verification of process viability, (ii)
determination of enzyme kinetics, (iii) identification of pathway bottlenecks and selection
of suitable engineered enzymes, (iv) development of a robust and accurate kinetic model,
(iv) experimental model validation, and (v) process optimization by in silico enzyme
stoichiometry modelling. Recent progress in efficient enzyme stabilization [264,265] and
ex-vivo cofactor regeneration [100,266] together with the development of integrated
analytical techniques [267], increases in computational power, and the growing
availability of kinetic data will enable the refinement of many useful biotransformations in
vitro and in silico. We believe that the workflow described herein and implemented in the
provided computer code represents a widely applicable strategy for rapid optimization of
multi-enzyme processes.

Chapter 2
- 93 -
SUPPLEMENTARY TABLES AND FIGURES Table S1. Concentrations of metabolites from time courses of 1,2,3-trichloropropane conversion catalyzed by wild-type DhaA, HheC and EchA.a
1 : 1 : 1 (SUM 3 mg)b; productivity 72 %
c
time (min)
TCP DCP ECH CPD GDL GLY SUM
0 2.03±0.06 0.00±0.00 0.00±0.00 0.00±0.00 0.00±0.00 0.00±0.00 2.03 10 1.80±0.01 0.08±0.02 0.00±0.00 0.00±0.00 0.00±0.00 n.d. n.d. 25 1.62±0.02 0.15±0.01 0.00±0.00 0.00±0.00 0.08±0.01 0.13±0.01 1.98 50 1.36±0.02 0.23±0.01 0.00±0.00 0.00±0.00 0.13±0.01 0.24±0.01 1.96 75 1.14±0.02 0.28±0.01 0.00±0.00 0.00±0.00 0.12±0.02 0.41±0.01 1.95
100 0.97±0.02 0.32±0.02 0.00±0.00 0.00±0.00 0.12±0.02 0.56±0.03 1.97 150 0.71±0.02 0.35±0.03 0.00±0.00 0.00±0.00 0.09±0.01 0.88±0.00 2.03 200 0.51±0.03 0.36±0.03 0.00±0.00 0.00±0.00 0.06±0.02 1.01±0.08 1.94 300 0.25±0.02 0.30±0.03 0.00±0.00 0.00±0.00 0.00±0.00 1.47±0.06 2.02
2.4 : 2.4 : 2.4 (SUM 7.2 mg)b; productivity 94 %
c
time (min)
TCP DCP ECH CPD GDL GLY SUM
0 1.95±0.06 0.00±0.00 0.00±0.00 0.00±0.00 0.00±0.00 0.00±0.00 1.95 10 1.54±0.06 0.09±0.01 0.00±0.00 0.00±0.00 0.06±0.01 n.d. n.d. 25 1.29±0.05 0.19±0.01 0.00±0.00 0.00±0.00 0.13±0.00 0.14±0.01 1.75 50 0.96±0.03 0.29±0.02 0.00±0.00 0.00±0.00 0.14±0.01 0.40±0.01 1.79 75 0.72±0.02 0.33±0.03 0.00±0.00 0.00±0.00 0.10±0.02 0.70±0.06 1.85
100 0.53±0.01 0.35±0.03 0.00±0.00 0.00±0.00 0.07±0.01 1.07±0.03 2.02 150 0.26±0.00 0.32±0.02 0.00±0.00 0.00±0.00 0.04±0.01 1.29±0.02 1.91 200 0.11±0.01 0.25±0.03 0.00±0.00 0.00±0.00 0.00±0.00 1.54±0.01 1.90 300 0.01±0.01 0.12±0.02 0.00±0.00 0.00±0.00 0.00±0.00 1.83±0.03 1.96
2.3 : 2.7 : 0.7 (SUM 5.7 mg)b; productivity 98 %
c
time (min)
TCP DCP ECH CPD GDL GLY SUM
0 1.95±0.11 0.00±0.00 0.00±0.00 0.00±0.00 0.00±0.00 0.00±0.00 1.95 10 1.62±0.10 0.08±0.02 0.00±0.00 0.00±0.00 0.06±0.00 n.d. n.d. 25 1.33±0.09 0.17±0.01 0.00±0.00 0.00±0.00 0.21±0.01 0.07±0.01 1.78 50 1.00±0.06 0.27±0.02 0.00±0.00 0.00±0.00 0.33±0.02 0.26±0.02 1.86 75 0.75±0.04 0.31±0.03 0.00±0.00 0.00±0.00 0.36±0.04 0.52±0.02 1.94
100 0.54±0.05 0.31±0.02 0.00±0.00 0.00±0.00 0.30±0.01 0.86±0.10 2.01 150 0.27±0.03 0.28±0.02 0.00±0.00 0.00±0.00 0.18±0.01 1.18±0.10 1.91 200 0.11±0.01 0.20±0.03 0.00±0.00 0.00±0.00 0.08±0.01 1.56±0.08 1.95 300 0.00±0.00 0.09±0.01 0.00±0.00 0.00±0.00 0.00±0.00 1.92±0.12 2.01
[a] Data represent mean values ± standard deviation calculated from three independent experiments. [b] Mass ratio of DhaA : HheC : EchA in mg of enzyme dissolved in 10 ml of reaction mixture. n.d., not determined
[c] Productivity was calculated using the formula:

Chapter 2
- 94 -
Table S2. Concentrations of metabolites from time courses of 1,2,3-trichloropropane conversion catalyzed by DhaA31, HheC and EchA.a
1 : 1 : 1 (SUM 3 mg)b; productivity 85 %
c
time (min)
TCP DCP ECH CPD GDL GLY SUM
0 2.09±0.10 0.00±0.00 0.00±0.00 0.00±0.00 0.00±0.00 0.00±0.00 2.09 5 1.18±0.03 0.46±0.02 0.00±0.00 0.00±0.00 0.00±0.00 n.d. n.d. 10 0.75±0.03 0.65±0.01 0.00±0.00 0.00±0.00 0.23±0.05 n.d. n.d. 15 0.48±0.03 0.75±0.01 0.00±0.00 0.00±0.00 0.43±0.04 n.d. n.d. 25 0.21±0.00 0.80±0.03 0.00±0.00 0.00±0.00 0.58±0.10 0.43±0.04 2.02 35 0.08±0.01 0.82±0.01 0.00±0.00 0.00±0.00 0.63±0.06 n.d. n.d. 50 0.02±0.02 0.78±0.02 0.00±0.00 0.00±0.00 0.37±0.05 0.87±0.05 2.04 75 0.00±0.00 0.69±0.01 0.00±0.00 0.00±0.00 0.00±0.00 1.19±0.05 1.88
100 0.00±0.00 0.63±0.02 0.00±0.00 0.00±0.00 0.00±0.00 1.37±0.18 2.00 150 0.00±0.00 0.49±0.01 0.00±0.00 0.00±0.00 0.00±0.00 1.44±0.00 1.93 200 0.00±0.00 0.38±0.01 0.00±0.00 0.00±0.00 0.00±0.00 1.53±0.09 1.91 300 0.00±0.00 0.24±0.00 0.00±0.00 0.00±0.00 0.00±0.00 1.78±0.09 2.02
1.8 : 1.8 : 1.8 (SUM 5.4 mg)b; productivity 96 %
c
time (min)
TCP DCP ECH CPD GDL GLY SUM
0 2.06±0.22 0.00±0.00 0.00±0.00 0.00±0.00 0.00±0.00 0.00±0.00 2.06 5 0.96±0.12 0.64±0.06 0.00±0.00 0.00±0.00 0.15±0.03 n.d. n.d. 10 0.45±0.06 0.79±0.06 0.00±0.00 0.00±0.00 0.45±0.09 n.d. n.d. 15 0.21±0.03 0.84±0.08 0.00±0.00 0.00±0.00 0.56±0.02 n.d. n.d. 25 0.05±0.02 0.79±0.08 0.00±0.00 0.00±0.00 0.53±0.02 0.67±0.02 2.04 35 0.01±0.01 0.77±0.06 0.00±0.00 0.00±0.00 0.35±0.01 n.d. n.d. 50 0.00±0.00 0.69±0.07 0.00±0.00 0.00±0.00 0.14±0.02 1.27±0.05 2.10 75 0.00±0.00 0.59±0.03 0.00±0.00 0.00±0.00 0.00±0.00 1.60±0.08 2.19
100 0.00±0.00 0.50±0.03 0.00±0.00 0.00±0.00 0.00±0.00 1.68±0.03 2.18 150 0.00±0.00 0.37±0.02 0.00±0.00 0.00±0.00 0.00±0.00 1.79±0.04 2.16 200 0.00±0.00 0.26±0.02 0.00±0.00 0.00±0.00 0.00±0.00 1.88±0.02 2.14 300 0.00±0.00 0.13±0.02 0.00±0.00 0.00±0.00 0.00±0.00 1.98±0.08 2.11
0.4 : 2.3 : 0.5 (SUM 3.2 mg)b; productivity 98 %
c
time (min)
TCP DCP ECH CPD GDL GLY SUM
0 2.02±0.12 0.00±0.00 0.00±0.00 0.00±0.00 0.00±0.00 0.00±0.00 2.02 10 1.39±0.11 0.27±0.02 0.00±0.00 0.00±0.00 0.16±0.01 n.d. n.d. 25 0.85±0.07 0.45±0.03 0.00±0.00 0.00±0.00 0.48±0.01 0.21±0.01 1.99 50 0.37±0.03 0.56±0.04 0.00±0.00 0.00±0.00 0.65±0.02 0.47±0.01 2.05 75 0.14±0.03 0.54±0.03 0.00±0.00 0.00±0.00 0.57±0.01 0.85±0.10 2.10
100 0.05±0.01 0.47±0.02 0.00±0.00 0.00±0.00 0.45±0.05 1.08±0.03 2.05 150 0.00±0.00 0.33±0.02 0.00±0.00 0.00±0.00 0.19±0.02 1.53±0.02 2.05 200 0.00±0.00 0.22±0.01 0.00±0.00 0.00±0.00 0.08±0.02 1.82±0.04 2.12 300 0.00±0.00 0.09±0.02 0.00±0.00 0.00±0.00 0.00±0.00 1.98±0.05 2.08
[a] Data represent mean values ± standard deviation calculated from three independent experiments. [b] Mass ratio of DhaA31 : HheC : EchA in mg of enzyme dissolved in 10 ml of reaction mixture. n.d., not determined
[c] Productivity was calculated using the formula:

Chapter 2
- 95 -
Table S3. Concentrations of metabolites from time courses of 1,2,3-trichloropropane conversion catalyzed by DhaA90R, HheC and EchA.[a]
1 : 1 : 1 (SUM 3 mg)b; productivity 42 %
c
time (min)
TCP DCP ECH CPD GDL GLY SUM
0 1.80±0.07 0.00±0.00 0.00±0.00 0.00±0.00 0.00±0.00 0.00±0.00 1.80 10 1.68±0.04 0.05±0.01 0.00±0.00 0.00±0.00 0.00±0.00 n.d. n.d. 25 1.60±0.04 0.05±0.00 0.00±0.00 0.00±0.00 0.00±0.00 0.10±0.01 1.75 50 1.51±0.01 0.05±0.01 0.00±0.00 0.00±0.00 0.00±0.00 0.16±0.01 1.82 75 1.45±0.01 0.05±0.01 0.00±0.00 0.00±0.00 0.00±0.00 0.25±0.00 1.75
100 1.35±0.01 0.06±0.01 0.00±0.00 0.00±0.00 0.00±0.00 0.31±0.02 1.72 150 1.22±0.02 0.06±0.01 0.00±0.00 0.00±0.00 0.00±0.00 0.45±0.00 1.73 200 1.13±0.03 0.06±0.01 0.00±0.00 0.00±0.00 0.00±0.00 0.53±0.06 1.72 300 0.91±0.02 0.06±0.00 0.00±0.00 0.00±0.00 0.00±0.00 0.75±0.03 1.72
4 : 4 : 4 (SUM 12 mg)b; productivity 85 %
c
time (min)
TCP DCP ECH CPD GDL GLY SUM
0 2.06±0.04 0.00±0.00 0.00±0.00 0.00±0.00 0.00±0.00 0.00±0.00 2.06 10 1.69±0.07 0.03±0.00 0.00±0.00 0.00±0.00 0.10±0.01 n.d. n.d. 25 1.40±0.02 0.05±0.02 0.00±0.00 0.00±0.00 0.13±0.05 0.21±0.01 1.79 50 1.27±0.09 0.04±0.01 0.00±0.00 0.00±0.00 0.13±0.02 0.51±0.01 1.95 75 1.06±0.08 0.06±0.01 0.00±0.00 0.00±0.00 0.09±0.04 0.82±0.04 2.03
100 0.88±0.04 0.04±0.01 0.00±0.00 0.00±0.00 0.05±0.05 0.97±0.04 1.94 150 0.67±0.03 0.04±0.01 0.00±0.00 0.00±0.00 0.00±0.00 1.39±0.10 2.10 200 0.44±0.04 0.04±0.02 0.00±0.00 0.00±0.00 0.00±0.00 1.56±0.03 2.04 300 0.28±0.02 0.01±0.01 0.00±0.00 0.00±0.00 0.00±0.00 1.75±0.06 2.04
5.2 : 1.5 : 0.8 (SUM 7.5 mg)b; productivity 88 %
c
time (min)
TCP DCP ECH CPD GDL GLY SUM
0 1.96±0.01 0.00±0.00 0.00±0.00 0.00±0.00 0.00±0.00 0.00±0.00 1.96 10 1.56±0.07 0.10±0.01 0.00±0.00 0.00±0.00 0.00±0.00 n.d. n.d. 25 1.30±0.05 0.10±0.01 0.00±0.00 0.00±0.00 0.24±0.05 0.09±0.02 1.73 50 1.00±0.04 0.10±0.00 0.00±0.00 0.00±0.00 0.47±0.04 0.32±0.02 1.89 75 0.83±0.02 0.10±0.01 0.00±0.00 0.00±0.00 0.50±0.04 0.62±0.02 2.05
100 0.66±0.03 0.09±0.01 0.00±0.00 0.00±0.00 0.34±0.05 0.84±0.03 1.93 150 0.46±0.03 0.09±0.01 0.00±0.00 0.00±0.00 0.17±0.02 1.31±0.09 2.03 200 0.32±0.01 0.08±0.03 0.00±0.00 0.00±0.00 0.09±0.02 1.56±0.07 2.05 300 0.19±0.01 0.06±0.01 0.00±0.00 0.00±0.00 0.00±0.00 1.72±0.10 1.97
[a] Data represent mean values ± standard deviation calculated from three independent experiments. [b] Mass ratio of DhaA31 : HheC : EchA in mg of enzyme dissolved in 10 ml of reaction mixture. n.d., not determined
[c] Productivity was calculated using the formula:
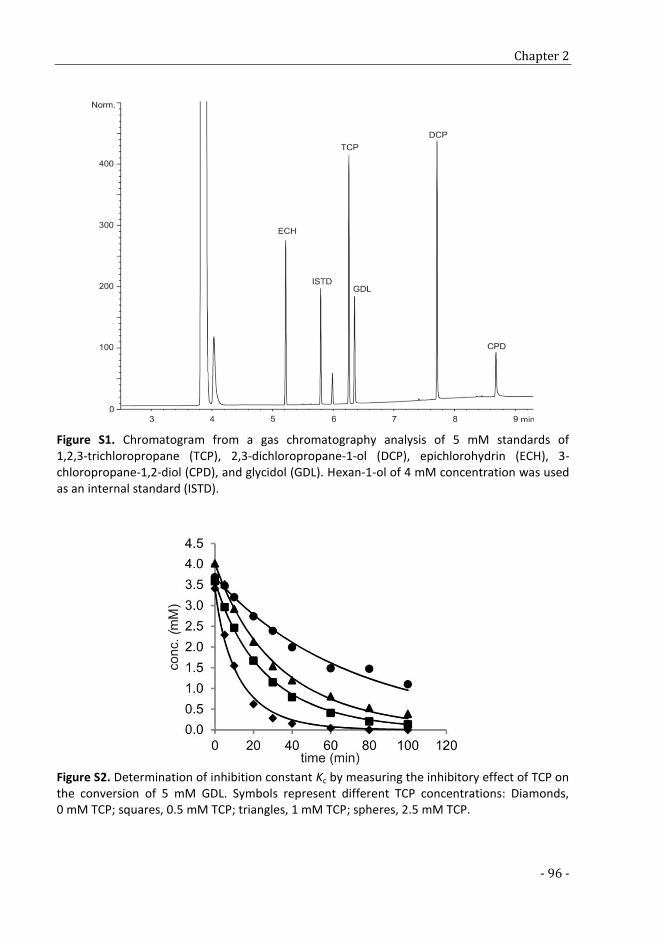
Chapter 2
- 96 -
Figure S1. Chromatogram from a gas chromatography analysis of 5 mM standards of 1,2,3-trichloropropane (TCP), 2,3-dichloropropane-1-ol (DCP), epichlorohydrin (ECH), 3-chloropropane-1,2-diol (CPD), and glycidol (GDL). Hexan-1-ol of 4 mM concentration was used as an internal standard (ISTD).
Figure S2. Determination of inhibition constant Kc by measuring the inhibitory effect of TCP on the conversion of 5 mM GDL. Symbols represent different TCP concentrations: Diamonds, 0 mM TCP; squares, 0.5 mM TCP; triangles, 1 mM TCP; spheres, 2.5 mM TCP.

- 97 -

- 98 -

- 99 -
CHAPTER 3
Computer-assisted engineering of the
synthetic pathway for biodegradation of
1,2,3-trichloropropane in heterologous
host Escherichia coli

Chapter 3
- 100 -
INTRODUCTION
Halogenated hydrocarbons, which are often anthropogenic compounds, are widely
used for agricultural, industrial and military purposes [268]. Once introduced into the
environment, these halogenated hydrocarbons often persist and cause a serious threat to
natural ecosystems and human health. Natural catabolic pathways for their mineralization
are inefficient or lacking, primarily due to the fact that most of the discussed chemicals
were present in the environment until last century. Engineering bacteria towards
enhanced biodegradation capacities has been intensively studied, because the
self-replicating whole-cell catalysts represent the cheapest option for bioremediation or
biotransformation processes. However only limited success has been achieved so far due
to the reasons described in previous two chapters of this Thesis [126,128]. One of the
most common failures is because of an imbalance in the expression or catalytic properties
of enzymes employed in synthetic routes. This factor often leads to an accumulation of
toxic intermediates, insufficient flux through the pathway, and limited fitness of the host
organism [145,146,269–271].
A number of approaches dealing with the unbalanced properties of enzymes in
engineered pathways have been reported in recent years. Improvement of pathway
performance can be achieved through the introduction of engineered enzyme variants
[116,117], kinetic modeling of the system [267,272] or modular optimization of the
pathway [69,70]. Although these examples come predominantly from the field of
biosynthesis of valuable chemicals, adoption of such synthetic biology tools holds
considerable promise for rational tuning of pathways for biodegradation of industrial
waste [231].
Example of halogenated hydrocarbon discussed here is a man-made
1,2,3-trichloropropane (TCP). TCP is an emerging toxic groundwater pollutant and
suspected carcinogen, which spreads to the environment mainly due to improper waste
management [186,195]. No naturally-occurring bacterial strain capable of aerobic
utilization of TCP has been isolated from nature thus far. However, TCP can be converted
to harmless glycerol (GLY) via previously described five-step catabolic pathway
assembled with haloalkane dehalogenase (DhaA) from Rhodococcus rhodochrous NCIMB
13064, haloalcohol dehalogenase (HheC) and epoxide hydrolase (EchA) from
Agrobacterium radiobacter AD1 [209,216,237]. The first recombinant strain partially
degrading TCP was constructed using heterologous expression of DhaA or an 8-times
more active mutant form of DhaAM2 in its natural host A. radiobacter AD1, possessing the
remainder of the pathway [211,213]. Although some increase in optical density of cultures
were observed in the latter case, the efficiency of TCP mineralization was insufficient for
supporting significant growth. It was proposed that the poor activity of haloalkane
dehalogenase towards TCP, and formation of toxic intermediates represent primary
bottlenecks of the pathway. Furthermore, equimolar production of the (S) and (R)
enantiomers of 2,3-dichloropropane-1-ol (DCP) from prochiral TCP by non-selective DhaA
and the resulting accumulation of (S)-DCP caused by high enantioselectivity of HheC

Chapter 3
- 101 -
towards (R)-DCP was observed and discussed as the factor limiting the flux of carbon
through the pathway. The effects of the described bottlenecks in the context of the whole
synthetic pathway have not been studied in detail.
Significant efforts have been invested in the past few years in engineering the first
enzyme of the TCP pathway; haloalkane dehalogenase (DhaA) [225,226,265,273].
Constructed mutants can possibly help overcome the previously mentioned limitations of
the TCP pathway. Mutant DhaA31, constructed in our laboratory using computer-assisted
directed evolution, showed 26-fold higher catalytic efficiency towards TCP than the wild
type enzyme (DhaAwt: kcat = 0.04 s-1, kcat/Km = 40 s-1 M-1; DhaA31: kcat = 1.26 s-1,
kcat/Km = 1050 s-1 M-1), while its enatioselectivity with the same substrate remained
unchanged [225]. Mutant r5-90R (referred here as DhaA90R), obtained after five rounds
of directed evolution of DhaA31 by van Leeuwen et al., converted prochiral TCP
predominantly into (R)-DCP (ee 90%), which is the preferred substrate for the selective
HheC [226]. DhaA90R possessed similar catalytic efficiency towards TCP as the wild-type
enzyme (kcat = 0.16 s-1; kcat/Km = 25 M-1 s-1). Thus, each of the mutants, DhaA31 and
DhaA90R, show improvement in one of the limiting factors of the TCP pathway.
After gaining a detailed knowledge about the performance and kinetics of the
synthetic TCP pathway in in vitro conditions, in this study, we carried out a systematic
analysis and rational optimization of the pathway also in heterologous host organism
Escherichia coli. Both engineered variants of DhaA were integrated to evaluate the effects
of increased activity and improved selectivity on the performance of the pathway. A
developed and in vitro verified kinetic model described in Chapter 2 of the Thesis was
supplied with new biological parameters and employed for rational selection of suitable
plasmid combinations and balancing the stoichiometry of the enzyme
DhaA/DhaA31/DhaA90R:HheC:EchA. The pathway was optimized towards faster removal
of toxic metabolites and higher production of GLY. Several E. coli constructs were
prepared based on the predictions and characterized for their viability and ability to
degrade TCP. Advantages and limitations of the engineered DhaA variants in the context of
the pathway assembled in vivo were analyzed and obtained information was used to
propose further steps for optimizing the pathway in heterologous host.

Chapter 3
- 102 -
MATERIAL AND METHODS
Chemicals, media, strains, and plasmids. 1,2,3-Trichloropropane (TCP),
2,3-dichloropropane-1-ol (DCP), epichlorohydrin (ECH), 3-chloropropane-1,2-diol (CPD),
glycidol (GDL) and glycerol (GLY) standards were purchased from Sigma-Aldrich (USA).
All chemicals used in this study were of analytical grade. All restriction enzymes and DNA
ligase were purchased from New England Biolabs (USA). A free Glycerol Assay Kit was
acquired from BioVision (USA). Luria Broth (LB) (Sigma Aldrich, USA) was used for
routine cultures. A synthetic mineral medium (SMM) containing 5.4 g of Na2HPO4.12H2O,
1.4 g of KH2PO4, 0.5 g of (NH4)2SO4, 0.2 g of MgSO4.7H2O, 2 ml of trace elements solution
and 1 ml of vitamin B1 (10 g.ml-1) per 1 l was used for selection experiments [274]. M9
minimal medium (Sigma Aldrich, USA) containing 0.2 g of MgSO4.7H2O and 2 ml of trace
elements per 1 l and 10 mM glucose were used for toxicity tests. Escherichia coli DH5α
(Life Technologies, USA) was used in cloning and plasmid propagation. E. coli BL21 (DE3)
(Life Technologies, USA) was used as a heterologous host for expression of the synthetic
pathway for the biodegradation of TCP. Plasmids pET21b, pACYCDuet-1, pETDuet-1,
pCDFDuet-1 (Merck Millipore, Germany) were used for subcloning and modular
construction of pathway variants.
Molecular techniques and culture conditions.The genes of the haloalcohol dehalogenase
encoding HheC and the epoxide hydrolase encoding EchA from Agrobacterium radiobacter
AD1, together with genes of the wild-type haloalkane dehalogenase from Rhodococcus
rhodochrous NCIMB 13064 encoding DhaAwt and the mutants DhaA90R and DhaA31 were
commercially synthesized (GeneArt, Germany). A tag sequence of six histidine codons was
attached downstream from the gene in all cases except for hheC. Sequences of all genes
except for hheC were codon-optimized for expression in E. coli during gene synthesis.
Synthetic genes were subcloned into the NdeI and BamHI restriction sites of pET21b. An
alternative NcoI restriction site was introduced at the beginning of the echA gene to enable
cloning into the first multiple cloning sites of Duet vectors. Upon introduction of the NcoI
site, the second codon of the echA gene, ACT encoding threonine, was substituted for GCA
encoding alanine. The constructs pET21b-dhaA, pET21b-dhaA31, pET21b-dhaA90R,
pET21b-hheC, and pET21b-echA were transformed into competent cells of E. coli DH5α
using the heat-shock method for plasmid propagation. Isolated plasmids were
transformed into E. coli BL21 (DE3) for evaluation of individual gene expression under
similar conditions. Subcloning of the genes coding for the synthetic pathway into Duet
plasmids is summarized in Table 1. E. coli BL21 (DE3) co-transformants, prepared by
modular combination of recombinant Duet vectors are summarized in Table 2. All plasmid
constructs were verified by sequencing (GATC, Germany).
Precultures of E. coli DH5α, BL21 (DE3) host cells, and co-transformants were
prepared by growth on LB medium with or without antibiotics at 37°C overnight. Final
concentrations of respective antibiotics (ampicillin 100 µg.ml-1, chloramphenicol
34 µg.ml-1, streptomycin 50 µg.ml-1) were used in the cultures with cells containing a

Chapter 3
- 103 -
single plasmid. Half concentrations of two relevant antibiotics were used in precultures,
and cultures of co-transformants. A standard protocol was developed for the cultivation of
the degraders. Precultures were used to inoculate fresh LB medium and cultures were
grown at 37°C until the cell density reached 1 at OD600. Expression of recombinant
enzymes was induced by 0.2 mM isopropyl-β-D-thiogalactopyranoside (IPTG) and
cultivation continued at 20°C. Cells were harvested during the late exponential phase by
centrifugation (5000 g, 15 min, 4°C), washed three times with sterile ice-cold SMM or M9
medium 50 mM sodium phosphate buffer pH 7.0, and used in further experiments.
Table 1. Plasmids used and recombinants constructed.
Duet vectorsa Origin Copy no. Recombinant
plasmids Restriction sites
pACYCDuet-1b P15A 10-12 pACYC-echA-hheC echA (NcoI/HindIII)
hheC (NdeI/KpnI) pCDFDuet-1
c CloDF13 20-40 pCDF-dhaAwt NdeI/KpnI
pCDF-dhaA31 NdeI/XhoI
pCDF-dhaA90R NdeI/KpnI pCDF-echA-hheC echA (NcoI/HindIII)
hheC (NdeI/KpnI) pETDuet-1
d ColE1 40 pETDuet-dhaAwt NdeI/KpnI
pETDuet-echA-hheC echA (NcoI/HindIII) hheC (NdeI/KpnI)
pETDuet-dhaA90R NdeI/KpnI
[a] Source – Merck Millipore [b] CmR, chloramphenicol resistance [c] SmR, streptomycin/spectinomycin resistance [d] ApR, ampicillin resistance.
Analytical techniques. A gas chromatograph 6890N with a flame ionisation detector
(GC-FID) and mass spectrometer (GC-MS) 5975C MSD (Agilent Technologies, USA), with
the capillary column ZB-FFAP 30 m x 0.25 mm x 0.25 µm (Phenomenex, USA) were used
for routine analysis and quantification of TCP and its metabolites. The separation method
used for both GC-FID and GC-MS used an inlet temperature of 250°C, split ratio 20:1,
helium carrier gas with an initial flow of 0.6 ml.min-1 for 1 min, followed by a flow
gradient of 0.2 ml.min-1 from 0.6 to 1.8 ml.min-1, and an oven temperature program set
initially to 50°C for 1 min, followed by a temperature gradient of 25°C.min-1 from 50 to
220°C with holding for 2 min. Different concentrations of these compounds were used to
prepare calibration curves for quantification.
Determination of toxicity of the substrate and intermediate metabolites. A growth test
in M9 medium containing 10 mM glucose was used to determine the toxicity of TCP and its
intermediates. The growth of E. coli cultures was evaluated at 37°C for 6 hours using 0.5,
1, 2, 4, 5, 10, 15 and 20 mM of TCP, DCP, ECH, CPD and GLD. To avoid the evaporation of
volatile compounds, cultivation was done in 25 ml glass vials with a screw cap mininert
valve (Sigma-Aldrich, USA). The concentration of TCP and intermediate compounds was

Chapter 3
- 104 -
monitored by GC. Samples (0.5 ml) were withdrawn and extracted with acetone (1:1),
containing hexan-1-ol as an internal standard and centrifuged for 2 min at 18,000 g. The
acetone extracts (2 μl) were injected directly into GC. E. coli cultures without the addition
of the tested compounds was used as a negative control. For growth monitoring, 1 ml
samples were withdrawn in 1 h intervals and measured at OD600. Toxicity data were fit
into polynomial equations from which the inhibitory concentration of individual
compounds (IC20 value) was determined.
Determination of minimal GLY concentration required for host cells growth. Single
colony of E. coli BL21 (DE3) was cultivated overnight in 10 ml of M9 medium containing
20 mM of GLY at 37°C. The preculture was washed two times with ice-cold sterile M9
medium to remove the residual GLY. Resuspension was concentrated to OD600 = 10. The
cultivation was started by addition of 100 l of washed preculture achieving the starting
OD600 0.1 in 5 ml of M9 medium with GLY concentrations varying from 0.01 to 2 mM. The
growth was monitored by measuring OD600 at 0, 3, 24 and 28 h time intervals. After 24 h of
cultivation, GLY was added to the cultures at the concentrations corresponding to the
starting ones. The growth curve with 1 and 2 mM GLY in M9 minimal medium was
determined once again to collect sufficient data between 3 h to 24 h. The seed culture of E.
coli BL21 (DE3) was prepared in M9 medium containing 20 mM of GLY at 37°C. Cell pellets
were collected, washed and concentrated to OD600 = 10. The cultivation started by
addition of 100 l of washed cells achieving the starting OD600 0.1 in 10 ml of M9
medium containing: (i) no carbon source as a negative control, (ii) 1 and 2 mM of GLY, and
(iii) 10 mM of glucose as a positive control. The growth was monitored by measuring
OD600 at 0, 3, 6, 9, 12 and 24 h after cultivation.
Determination of expression levels of the enzymes of the TCP pathway. The expression
levels of DhaAwt, DhaA31, DhaA90R, HheC and EchA were determined in E. coli BL21
(DE3) cells transformed with a pET21b vector and subcloned with corresponding genes,
as well as in the degraders prepared by standardized cultivation procedures. Washed cells
were resuspended in 10 ml of 50 mM sodium phosphate buffer and cell density adjusted
to 3.5 at OD600. 1 U of DNaseI per 1 ml of cell suspension was added. Cells were disrupted
with 5 cycles of sonication using a Hielscher UP200S (Teltow, Germany) ultrasonic
processor with 0.3 s pulses and an amplitude 85%. Each cycle consisted of 5 min
sonication followed by 5 min cooling at 4°C. The cell lysate was centrifuged for 1 h at
18,000 g at 4°C and the resulting cell-free extract (CFE) was decanted. The concentration
of total protein in CFEs was determined using Bradford reagent (Sigma Aldrich, USA).
Samples of CFE containing 5 μg of total protein were separated by sodium dodecyl sulfate
polyacrylamide gel electrophoresis (SDS-PAGE). CFE prepared from E. coli BL21 (DE3)
cells without plasmids were used as controls. For determination of the mass ratio, the
method was calibrated with standard samples containing 0.25, 0.5, 1, 1.5 and 2 μg of each
enzyme DhaA, EchA and HheC in purified form. Purification of DhaA, DhaA31, DhaA90R,
HheC and EchA was described in previous chapters of the Thesis. Gels were stained with

Chapter 3
- 105 -
Coomassie Brilliant Blue R-250 (Fluka, Switzerland) and analyzed using a GS-800
Calibrated Imaging Densitometer (Bio-Rad, USA). The amounts of DhaAwt, DhaA90R,
DhaA31, HheC and EchA in samples of CFE applied to the gel, and the corresponding
relative and mass ratios of the enzymes in the individual degraders were estimated from
trace densities of selected bands.
Verification of expression levels by activity measurements. The expression levels of
DhaAwt, DhaA31, HheC and EchA estimated from SDS-PAGE analysis of CFE samples using
deg31 and deg31-opt was verified by the measurement of individual enzyme activity in
CFE. The activity of DhaA, HheC and EchA was estimated using 10 mM TCP, 20 mM DCP
and 10 mM ECH, respectively. Substrates were dissolved in 10 ml of 50 mM Tris-SO4
buffer (pH 8.5) at 37°C. The reaction was initiated with the addition of CFE to a volume
corresponding to 1 mg of DhaA, 1 mg of HheC or 1 mg of EchA, as was estimated from the
expression levels analyzed by SDS-PAGE. CFE from E. coli BL21 (DE3) without plasmids
was used as a negative control. Samples (0.5 ml) of the reaction mixtures were withdrawn
at certain time intervals and mixed with a 0.5 ml solution of acetone in hexan-1-ol and
analyzed by GC. Specific activity was calculated as a decrease in substrate concentration in
μmol per minute per mg of the enzyme.
Mathematical modeling and optimization of the TCP pathway. The mathematical model
describing the TCP pathway was constructed using kinetic equations and parameters
obtained from in vitro enzyme kinetics with the purified enzymes and corresponding
substrates (TCP, DCP, ECH, CPD and GDL). All kinetic experiments were performed in
50 mM Tris-SO4 buffer at pH 8.5 and 37°C. Determination of kinetic parameters, and
construction of the model and its experimental verification for in vitro conditions is
described in Chapter 2 of this Thesis. Initial parameters for pathway optimization were:
starting concentration of TCP, 2 mM; time interval of multi-enzyme reaction, 300 min;
total mass of enzymes in the pathway, 3 mg. Allowed combinations of Duet vectors were
prepared using the following criteria: only two out of three employed Duet vector
derivatives (pACYC, pCDF and pETDuet) can be combined; either one or two genes coding
for enzymes from the TCP pathway can be expressed from one vector; only the echA gene
possesses the NcoI restriction site for subcloning into the first multiple cloning site of Duet
vectors, thus it can be expressed only from a vector with another subcloned gene and not
separately. The relative ratio of enzymes in a simulation was then determined by a
particular combination of plasmids carrying individual enzymes and a copy number of
individual plasmids (10 for pACYC, 20 for pCDF, and 40 for pETDuet). For all allowed
combinations of plasmids, dynamic simulations of the multi-enzyme system based on a
series of Michaelis-Menten equations were performed using the Python 2.7 programming
language. The differential form of equations was integrated using Euler's method with a
fixed step size of 0.05 min. The toxicity effect at individual times along degradation
profiles was calculated using previously obtained polynomial equations fitted to
experimental toxicity data and the actual concentration of each compound at a given time.

Chapter 3
- 106 -
The overall toxicity effect was calculated by integration of individual effects along the
profile.
Multi-enzyme reactions including variants of the DhaA enzyme with modified
catalytic properties were modeled using the following adjustments of the previously
described process. The total mass of enzymes was set to 0.1 mg, the time-interval of the
multi-enzyme reaction was set to 24 hours. A modification of the catalytic performance of
DhaA was as follows: KM was set to 1 mM, while the overall kcat was varied across the
range of 1 to 1000 min-1. The enantioselectivity of DhaA expressed as a ratio of catalytic
rate constants leading towards production of individual enantiomers of DCP was assigned
for values ranging from 1 to 200. To avoid the limitation of the relative ratio of enzymes in
the simulation by the employed plasmids, the mass of individual enzymes were assigned a
value from 0.0 to 0.1 mg with intervals of 0.0125 mg. The ratio resulting in the highest
end-point production of GLY was considered as representative of the optimized pathway.
Characterization of the degraders in minimal medium with TCP. Pre-induced cells of
the six constructed degraders (Table 2) were washed with sterile ice cold SMM medium
and resuspended in the same medium. A final concentration of 2 mM TCP was added to
15 ml of sterile SMM in 25 ml glass vials with a screw cap mininert valve (Sigma-Aldrich,
USA) and incubated at 30°C with shaking for 2 h to allow for complete dissolution.
Washed cells were adjusted to a final cell density 0.1 at OD600. Vials containing BL21 (DE3)
cells with and without TCP were used as controls. Flasks were continuously incubated for
5 days in a shaking incubator NB-205 (N-Biotek, South Korea) at 200 rpm and 30°C.
Samples for analysis of TCP and metabolites were withdrawn at 0, 6, 24, 48, 72, 96 and
120 h. Viability of cells at the beginning and end of their incubation with TCP was tested
by plating a sample of cell suspension diluted 2x104-times with ice-cold sterile SMM onto
LB agar plates. Plates were incubated overnight at 37°C and grown colonies were counted
using the Colony Picker CP7200 (Norgren Systems, USA).
Recovery of degraders in LB medium. Pre-induced cells of the six constructed degraders
were incubated at 37°C with shaking in 25 ml glass vials with a screw cap mininert valve
containing 10 ml of 50 mM sodium phosphate buffer (pH 7.0) with 3.5 mM TCP. The OD600
of cell suspensions was 3.5. BL21 (DE3) cells without plasmids were used as controls.
After 5 h of incubation, cell suspension samples (10 μl) were transferred into wells of a
96-well microtiter plate containing 100 μl of LB medium. The plate was incubated at 37°C
with shaking (900 rpm) in a shaking incubator (PST-100 HL Thermo Shaker, BIOSAN).
Cell growth was monitored by measuring OD600 at 30 min intervals using a microtiter-
plate reader (Tecan, Switzerland).
Degradation of TCP in buffer by pre-induced resting cells. Cell suspensions of pre-
induced deg31, deg31-opt and E. coli BL21 (DE3) without plasmids (negative control)
were diluted with sterile 50 mM sodium phosphate buffer to a final OD600 7. Glass vials
(25 ml) with a screw cap mininert valve containing 7.5 ml of the sterile 50 mM sodium

Chapter 3
- 107 -
phosphate buffer of pH 7.0 and 4 mM TCP were prepared separately and incubated for 1 h
at 37°C with shaking. The reaction was initiated by mixing 7.5 ml of the cell suspension
with 7.5 ml of buffer and dissolved TCP. The final concentration of TCP in 15 ml of the cell
suspension was 2 mM and the final theoretical OD600 was 3.5. The vials were incubated for
300 min at 30°C with shaking. Cell suspension samples (0.5 ml) were quenched in 0.5 ml
acetone with hexan-1-ol, vortexed and centrifuged at 18,000 g for 2 min. The
concentration of metabolites in the supernatant was analyzed using GC.
Detection of GLY produced from TCP degradation pathway. GLY concentration was
analysed by Free Glycerol Assay Kit (BioVision, USA) according to the manufacturer's
protocol. The pre-induced cells of deg31 and deg31-opt were prepared in LB medium. The
cells were washed two times with ice-cold buffer to remove residual LB. The cells with
final OD600 of 3.5 were incubated with 2 mM theoretical concentration of TCP for
300 minutes in 10 ml phosphate buffer of pH 7.0 supplied with 5 mM of glucose. The
concentration of TCP and DCP were monitored throughout the reaction course by GC.
Production of GLY at certain time points (0, 100, 200 and 300 min) was tested by Free
Glycerol Assay Kit. Samples of cell suspension (50 μl) were heated for 10 min at 95°C and
centrifuged at 18,000 g for 2 min. The supernatants were diluted and analysed using the
kit.
Determination of accumulation of TCP and metabolites in host cells. Accumulation of
TCP and intermediates in the cell biomass was evaluated after 3 h of incubation of cell
suspension of E. coli BL21 (DE3) with 2 mM of TCP, DCP or GDL. All experiments were
performed in 10 ml of 50 mM sodium phosphate buffer at 37°C using the cell density of
3.5 at OD600. After incubation, the cells were harvested by centrifugation at 4000 g for
15 min. Supernatant containing buffer was removed; cell pellet was resuspended in 0.5 ml
of acetone with hexan-1-ol, vortexed for 1 min and centrifuged. All the samples, i.e., initial
cell suspension, buffer and acetone supernatant, were analysed by GC. Proportional
concentrations of TCP, DCP and GDL in cell suspension, cell biomass and buffer were
calculated and compared.

Chapter 3
- 108 -
RESULTS AND DISCUSSION
The haloalkane dehalogenase DhaA, the haloalcohol dehalogenase HheC, and the
epoxide hydrolase EchA were transferred into a heterologous host, Escherichia coli BL21
(DE3), for systematic analysis and rational optimization of a synthetic metabolic pathway
for the biodegradation of TCP. The three enzymes and five metabolites of the pathway do
not naturally occur in E. coli. The possibility of interactions between original metabolism
of E. coli and the synthetic route (so called "metabolic cross-talk") is thus minimized,
which makes the TCP pathway in E. coli an optimal orthogonal system suitable for the
rational analysis of pathway bottlenecks [91].
Toxicity of TCP and intermediate metabolites for E. coli.
Both toxicity of metabolites and the flux of carbon through the pathway represent
the factors dictating the viability of the host organism. Toxicity of TCP and its metabolites
towards E. coli has not been reported. Thus, we tested the effects of various
concentrations of TCP, DCP, ECH, CPD and GDL on growth of the E. coli BL21 (DE3) culture
in mineral medium supplied with glucose. The concentration at which 20 percent of the
total cell population was inhibited (IC20) was calculated for each compound
(Supplementary Figure S1). Results revealed that TCP and epoxide ECH are the most toxic
among all the tested compounds with IC20 values of 1.35 and 1.41 mM, respectively
(Figure 1). The toxicity of both halogenated alcohols, out of which DCP tends to
accumulate in the pathway, was one order of magnitude lower. The second epoxide, GDL,
had an intermediate toxic effect with an IC20 of 6.25 mM. The analysis of TCP, DCP and GDL
accumulation in E. coli showed that TCP is the only compound which has the potential for
cellular accumulation due to its higher hydrophobicity (Supplementary Table S1). The
hydrophobicity and accumulation of the other metabolites was significantly lower due to
the introduced polar hydroxyl groups. These results suggest that concentrations of
metabolites from the TCP pathway detected in the supernatant of the buffer or medium
are a good representation of their intracellular concentrations.
Figure 1. Toxicity of TCP and intermediate metabolites for E. coli BL21 (DE3). Toxicity is expressed as inhibition concentration IC20.

Chapter 3
- 109 -
Minimal GLY requirement for the growth of E. coli.
Aerobic mineralization of TCP should provide sufficient energy for sustainable
growth based on thermodynamic calculations [200]. however, TCP cannot be used at high
concentration during the cultivation due to its toxicity towards the host cells. Thus, only a
limited amount of GLY can be achieved in a culture medium from the degradation of TCP.
GLY is one of the suitable carbon sources that can be utilized by E. coli for energy
generation and cellular biosynthesis. Paliy et al reported the effect of different minimal
media and gluconeogenic carbon sources on the growth of E. coli BL21 and showed that
the strain grew reasonably well in mineral medium supplied with 2 g.l-1 GLY (~21.7 mM)
[275]. Paalme et al. also observed the growth rate of E coli K-12 on various carbon sources
at a concentration of 2 g.l-1 and reported the growth rate varying from 0.22 h-1 on acetate
to 0.77 h-1 on glucose with casamino acids [276]. The growth rate on GLY was 0.32 h-1. In
this study, we determined the minimal concentration of GLY necessary for observable
growth of E. coli BL21 (DE3) cell culture of OD600 0.1 in the mineral medium spiked with
GLY ranging from 0.01 mM to 2 mM (0.922 mg.l-1 to 184.3 mg.l-1). 1mM and 2 mM
concentration of GLY provided carbon and energy for significant increase in OD600 during
the first 3 h of cultivation, but were not sufficient to support the growth continuously for
24 h (Supplementary Figure S2). Repeated feeding of GLY at similar concentrations to the
cultures after 24 h resulted significant increase in OD600 at the concentration ≥ 1 mM.
Construction of TCP degraders by the assembly of an engineered pathway in E. coli.
The genes coding for enzyme of TCP pathway including two mutant variants of dhaA
- dhaA90R and dhaA31 - were commercially synthesized and codon-optimized for
expression in E. coli except for hheC which showed good expression in E. coli even without
optimization. Initially, all genes were subcloned separately into NdeI and BamHI
restriction sites of pET21b vector to compare the expression of individual enzymes under
the T7 promoter. SDS-PAGE analysis of cell-free extracts (CFE) obtained from pre-induced
cells prepared with standardized cultivation protocol showed comparable expression of
all enzymes in E. coli BL21 (DE3) (Supplementary Figure S3). Expression of enzymes
represented by the densities of corresponding bands was related to the expression of
DhaAwt which was defined as 1.0. The expression of the other enzymes was 1.3, 0.8, 1.3,
and 1.3 for DhaA90R, DhaA31, HheC, and EchA, respectively. To allow subcloning of echA
into the first multiple cloning site of Duet vectors, NcoI restriction site was introduced at
the start of the gene altering its second codon ACT (threonine) for GCA (alanine). This
single amino acid substitution had no effect either on enzyme expression or its activity.
The TCP pathway was assembled in two modules, separating expression of the first
enzyme (DhaA) from expression of the second and third enzymes (HheC and EchA). The
two major bottlenecks of the pathway: (i) poor activity of the first enzyme with TCP and
(ii) formation of toxic intermediates, were thus dissected. Modular assembly of the
pathway was carried out using combinations of two out of three Duet vectors: pACYC,

Chapter 3
- 110 -
pCDF and pETDuet. The Duet vector system has already proven its utility in modular
engineering of biosynthetic multi-enzyme pathways [50–53]. Individual Duet vectors can
be combined in a single host cell due to the different origins of the replication and
antibiotic resistance marker (Table 1) [49]. The modular expression strength can be
calculated using the promoter strength and plasmid copy number [69,70]. The expression
levels of subcloned genes can be estimated from the copy numbers of combined plasmids,
since all Duet vectors contain the same T7 promoter and the differences in expression of
individual genes from the TCP pathway under this promoter were found to be negligible
(Supplementary Figure S3).
The starting variants of the TCP pathway were constructed by subcloning dhaAwt,
dhaA90R or dhaA31 into the second multiple cloning site of pCDF, and echA and hheC into
the first and second multiple cloning sites of pETDuet, respectively. This combination
should provide an almost equal ratio of subcloned enzymes based on their reported copy
numbers. The combination of plasmids, consisting of the first enzyme (DhaA) in pCDF, and
the second and third (HheC and EchA) enzymes in pETDuet, was the same in all three
variants. Plasmid pairs were co-transformed into E. coli, resulting in three constructs
denoted degWT, deg90R, and deg31 (Table 2). Degraders were cultivated in LB medium
using a standardized cultivation protocol. An approximate estimation of expression
profiles obtained from SDS-PAGE analysis of CFE showed that the relative ratio of
enzymes in the three degraders was close to 0.2:0.4:0.4 (Supplementary Figure S4).
Table 2. Theoretical and experimental (in bold) ratios of enzymes in the constructed degraders. Degrader Plasmid combinations Theoretical ratio
Experimental ratio DhaA:HheC:EchA
Sum of enzymes
a
degWT pCDF-dhaAwt + pETDuet-echA-hheC 0.20 : 0.40 : 0.40 0.28 : 0.38 : 0.34 1.00
degWT-opt pETDuet-dhaAwt + pCDF-echA-hheC 0.50 : 0.25 : 0.25 0.56 : 0.25 : 0.19 0.94
deg90R pCDF-dhaA90R + pETDuet-echA-hheC 0.20 : 0.40 : 0.40 0.16 : 0.40 : 0.44 1.03
deg90R-opt pETDuet-dhaA90R + pACYC-echA-hheC 0.67 : 0.16 : 0.17 0.70 : 0.12 : 0.18 0.95
deg31 pCDF-dhaA31 + pETDuet-echA-hheC 0.20 : 0.40 : 0.40 0.12 : 0.42 : 0.46 1.06
deg31-opt pCDF-dhaA31 + pACYC-echA-hheC 0:50 : 0.25 : 0.25 0.60 : 0.16 : 0.24 0.74
[a] The sum of the three enzymes in each degrader was calculated from the density of corresponding bands on SDS-PAGE gels and compared to the degWT value (set as 1.00).
Optimization of the ratio of the three enzymes in the TCP pathway was conducted
using an extended version of the previously developed kinetic model. The model is based
on the Michaelis-Menten steady-state kinetic parameters of DhaA variants, HheC, and
EchA, determined from their corresponding substrates in vitro. The concentration of each
enzyme is kept constant at 0.1 mg.ml-1. New constraints defining the toxicity of individual

Chapter 3
- 111 -
metabolites and copy numbers of the used plasmids determining the expression level of
enzymes were introduced for optimization of the pathway in vivo. The model was used to
calculate all possible combinations of the two plasmids within the defined constraints
(Supplementary Tables S2-S4). Calculated time courses of the modeled multi-enzyme
conversion of 2 mM TCP at a time interval of 300 min were visualized using Gnuplot.
Twelve resulting combinations for each of the pathway variants and the corresponding
twelve relative expression ratios were ranked according to: (i) the efficiency of GLY
production and (ii) the level of the overall toxicity in the system, assuming additivity of
the metabolites’ toxic effects. Plasmid combinations used in the non-optimized degraders
degWT, deg90R and deg31 (pCDF-dhaA variant+pETDuet-echA-hheC) were ranked lower
compared to highly ranked degraders with optimized expression levels.
A single combination of plasmids (pETDuet-dhaA90R+pACYC-echA-hheC) showed
the best rank, leading to the highest GLY production and the lowest toxicity for the
degrader employing the DhaA90R variant (Supplementary Table S3). Variants with
DhaAwt and DhaA31 showed three and four equally good combinations, respectively,
from which one combination was selected for experimental construction (Supplementary
Tables S2 and S4). The plasmid combination pETDuet-dhaAwt+pCDF-echA-hheC was
selected for degWT-opt and pCDF-dhaA31+pACYC-echA-hheC for deg31-opt (Table 2).
Comparison of the calculated values representing GLY production and overall toxicity
showed that both degWT-opt and deg90R-opt produced 2-fold more GLY and less toxic
metabolites, in comparison to degWT and deg90R (Supplementary Tables S2 and S3).
Calculated values for plasmid combinations bearing catalytically efficient DhaA31 plus
HheC and EchA, showed that this biochemical pathway cannot be effectively optimized for
GLY production within the defined constraints (Supplementary Table S4), and deg31-opt
was expected to provide improvement only in terms of a lower level of toxic metabolites.
Experimental characterization of the TCP degraders.
The resting cells of six degraders were experimentally characterized in terms of
their: (i) expression profile, (ii) viability, and (iii) degradation capacity. The degraders
were cultivated in LB medium using a standardized cultivation protocol with heterologous
protein expression induced by IPTG. The degraders with over-expressed enzymes of the
TCP pathway were harvested and disintegrated using sonication. Equal amounts of CFEs
were loaded on SDS-PAGE and the expression profiles of six degraders were analyzed by
densitometry (Figure 2). Relative ratios of DhaA variants, HheC and EchA were calculated
from band densities and compared with the theoretical relative ratios predicted by
modeling (Table 2). An excellent agreement between theoretical and experimental values
was observed. The relative sum of the three enzymes from the TCP pathway was
calculated as a sum of the band densities, and revealed that degraders with an optimized
ratio contained equal or lower total amounts of the enzymes than non-optimized ones.

Chapter 3
- 112 -
Figure 2. SDS-PAGE analysis of the expression profiles of the six constructed degraders. M: protein marker (14.4, 18.4, 25, 35, 45, 66.2, and 116 kDa), 1: degWT, 2: degWT-opt, 3: deg90R, 4: deg90R-opt, 5: deg31, 6: deg31-opt and Std: standard sample with purified DhaAwt, HheC and EchA containing 0.25 μg of each enzyme. The theoretical molecular weights of the DhaA variants, HheC and Echa are 34, 29 and 35 kDa, respectively.
The viability and ability to degrade TCP was tested with resting cells of the degraders
resuspended in SMM medium with a final OD600 of 0.1. Cells were incubated at 30°C with
2 mM TCP and no additional carbon source. Time course of TCP and DCP concentrations
was monitored by GC analysis over 120 h (Figures 3A-C). No significant accumulation of
other metabolites of the TCP pathway was observed. Viabilities of the six degraders and
host with and without TCP as controls are presented in Figure 3D. No GLY was detected in
cell cultures suggesting its utilization by the cells. The production of GLY via the TCP
pathway was confirmed in parallel by incubation of induced cells of deg31 and deg31-opt
in the presence of 2 mM TCP and 5 mM glucose (Supplementary Figure S5). Theoretical
concentrations of produced GLY in cultures without glucose and relative toxicity for each
degrader were calculated from recorded time course concentrations of TCP and DCP
(Figures 3E and F).
The averaged data from the three independent experiments revealed that all three
degraders with rationally optimized ratios of pathway enzymes degraded TCP faster than
non-optimized constructs. Experimental time courses of TCP and DCP concentrations
correlated with the relative ratios of enzymes in the degraders. Differences in profiles
corresponded to the catalytic parameters of the DhaA variant and the relative ratio of
haloalkane dehalogenase with respect to the other two associated enzymes (Figure 3A-C
and Table 2). Deg90R and deg90R-opt with selective DhaA90R showed minimal
accumulation of DCP, but their degradation potential was limited by a low catalytic
efficiency with TCP. Deg31 and deg31-opt, both containing DhaA31, the non-selective
variant with improved catalytic efficiency towards TCP, showed the fastest conversion of

Chapter 3
- 113 -
TCP, but also the most significant accumulation of DCP. These two effects were even more
pronounced in the deg31-opt with higher expression of DhaA31.
Viability reflects the physiological state of the cells heterologously expressing the
synthetic pathway after their exposure to toxic TCP (Figure 3D). DegWT-opt showed a
small, but statistically significant difference in cell viability compared to degWT,
suggesting that optimization of the expression levels provided benefits to the cells. The
difference in viability of deg90R and deg90R-opt was statistically insignificant – the low
production of GLY was not balanced by a higher selectivity of DhaA90R and reduced
accumulation of DCP. The most striking result was the superior viability of both degraders
containing the DhaA31 enzyme, connected to a higher production of GLY and/or reduced
toxicity compared to the degraders expressing DhaAwt and DhaA90R. Deg31-opt removes
the toxic metabolites faster than deg31, but the effect of the reduced toxicity on the
viability of this degrader is not visible, suggesting that both degraders surpassed some
threshold level beyond which toxicity does not play a role and only the amount of
produced GLY is the key factor determining cell viability. Further improvement in viability
should be achieved by a more efficient production of GLY. Approximately 1 mM
concentration of GLY produced within 5 days by the best variants, deg31 and deg31-opt, is
probably not sufficient to provide energy for growth and compensation for the oxidative
stress induced during the aerobic mineralization of toxic chlorinated substrates [202].
The ability of the degraders to recover from short-term (300 min) exposure to a
higher concentration (3.5 mM) of TCP was tested (Supplementary Figure S6). Resulting
growth curves corresponded well with the viabilities of degraders calculated from plating
after 120 h incubation with a lower concentration of TCP. Only deg31 and deg31-opt
recovered faster than the host BL21 (DE3) without the synthetic pathway (control) and
with comparable growth. Degraders like degWT and deg90R, with a non-optimized ratio
of pathway enzymes, and deg90R-opt, with an optimized ratio but poor conversion of TCP,
recovered slower than the control. Only degWT-opt showed comparable growth with host
cells without the introduced pathway, again confirming the benefits of pathway
optimization. The data confirm that the constructs containing DhaA31 do not have an
increased metabolic burden [277].
Identification of pathway bottlenecks.
Two constructs carrying DhaA31 and showing the highest viability were selected for
further characterization. Pre-induced cells of elevated cell densities (OD600 3.5) were used
to secure complete degradation of 2 mM TCP during a shorter incubation time (300 min).
The mass ratio of enzymes in both degraders was estimated by SDS-PAGE and verified
with measurements of catalytic activities in CFE (Supplementary Table S5). Obtained data
were used for determination of the enzyme concentrations and applied to calculations of
the degradation profiles of deg31 and deg31-opt. In both cases the experimental profiles
showed a good agreement with predictions (Figure 4). Accumulation of DCP and GDL was
observed for both constructs. A more pronounced accumulation of DCP was observed for

Chapter 3
- 114 -
deg31-opt, with faster removal of TCP and a corresponding lower toxicity of the system.
The accumulated DCP was analyzed by chiral GC and found to be mainly composed of the
(S)-enantiomer. Theoretical conversions of TCP to GLY at the end of incubation calculated
from experimental concentrations of TCP at time 0 min and concentrations of the
accumulated metabolites at 300 min was similar for both deg31 and deg31-opt (75% and
69%). This is in agreement with the data previously collected over 120 h for conversion of
2 mM TCP, and suggests that the degraders containing DhaA31 cannot be further
optimized for better production of GLY without further engineering of DhaA properties
(Supplementary Table S4).

Chapter 3
- 115 -
Figure 3. Degradation profiles and viability of the six degraders. (A-C) Degradation profiles of the degraders containing DhaAwt, DhaA90R and DhaA31, respectively. The diamonds and squares refer to concentrations of TCP and DCP of non-optimized (red) and optimized (blue) variants. D) Viability of the host and degraders after 120 h incubation in SMM medium with TCP, relative to the viability at the beginning of the experiment. E) GLY production calculated from concentrations of TCP and DCP detected at the end of incubation. F) Overall toxicity of metabolites to degraders presented in arbitrary units (AU). Standard deviations were obtained from three independent experiments.
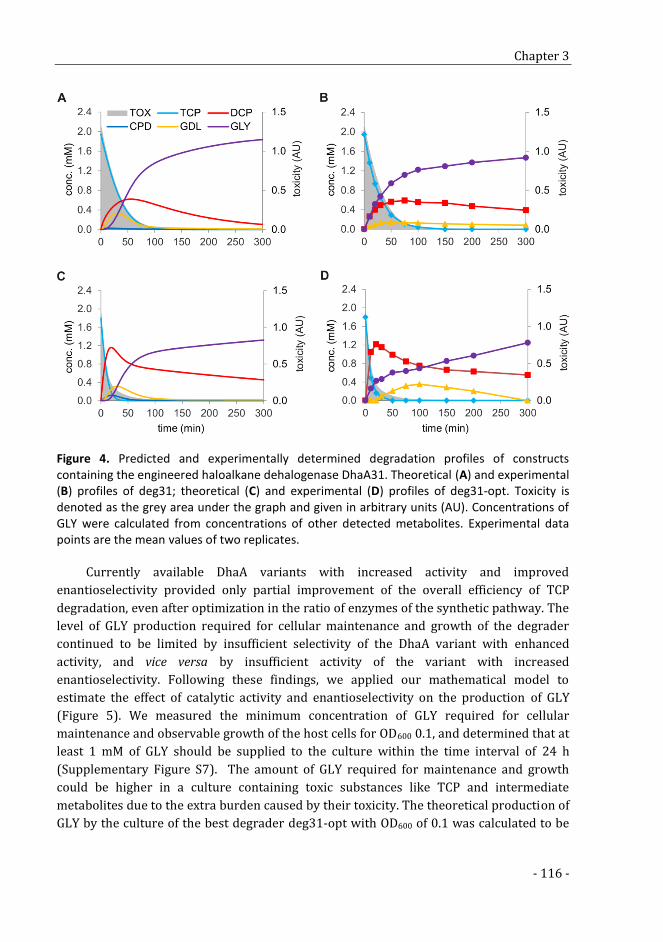
Chapter 3
- 116 -
Figure 4. Predicted and experimentally determined degradation profiles of constructs containing the engineered haloalkane dehalogenase DhaA31. Theoretical (A) and experimental (B) profiles of deg31; theoretical (C) and experimental (D) profiles of deg31-opt. Toxicity is denoted as the grey area under the graph and given in arbitrary units (AU). Concentrations of GLY were calculated from concentrations of other detected metabolites. Experimental data points are the mean values of two replicates. Currently available DhaA variants with increased activity and improved
enantioselectivity provided only partial improvement of the overall efficiency of TCP
degradation, even after optimization in the ratio of enzymes of the synthetic pathway. The
level of GLY production required for cellular maintenance and growth of the degrader
continued to be limited by insufficient selectivity of the DhaA variant with enhanced
activity, and vice versa by insufficient activity of the variant with increased
enantioselectivity. Following these findings, we applied our mathematical model to
estimate the effect of catalytic activity and enantioselectivity on the production of GLY
(Figure 5). We measured the minimum concentration of GLY required for cellular
maintenance and observable growth of the host cells for OD600 0.1, and determined that at
least 1 mM of GLY should be supplied to the culture within the time interval of 24 h
(Supplementary Figure S7). The amount of GLY required for maintenance and growth
could be higher in a culture containing toxic substances like TCP and intermediate
metabolites due to the extra burden caused by their toxicity. The theoretical production of
GLY by the culture of the best degrader deg31-opt with OD600 of 0.1 was calculated to be

Chapter 3
- 117 -
0.6 mM per 24 h, suggesting that DhaA31 does not possess the required catalytic
properties. To increase GLY production to 1 mM per 24 h, the model suggested the need
for further improvement of the catalytic efficiency of DhaA31 by 4-fold, or improvement of
the enantioselectivity by 20-fold. Alternatively, the employment of a DhaA variant with
combined improvement of catalytic efficiency by 1.2-fold and enantioselectivity by 10-fold
in favor of production of (R)-DCP would produce 1 mM GLY within 24 h. A DhaA variant
possessing the properties enabling the highest possible pathway efficiency (1.6 mM GLY
per 24 h) should have 4-fold improvement in catalytic efficiency and 20-fold higher
enantioselectivity compared to DhaA31.
Figure 5. Hypersurface plot describing the effect of catalytic efficiency (kcat/Km) and enantioselectivity (E-value) of DhaA on the production of GLY. The positions of the three variants of DhaA are indicated by red dots. The substrate TCP is supplied at 2 mM. The kinetic constants of DhaAwt, DhaAR90 and DhaA31 were experimentally determined and differ slightly from data reported in the literature.

Chapter 3
- 118 -
CONCLUSIONS
The toxicity of the substrate/metabolites and insufficient carbon/energy flow can
limit viability of the host organism and represent possible bottlenecks in the TCP
degradation pathway. The most toxic substances within the pathway are TCP
(IC20 = 1.35 mM) and ECH (IC20 = 1.41 mM). Other intermediates of the pathway are
significantly less toxic. Since accumulation of ECH inside the cells does not take place, the
hydrophobic substrate TCP represents the single most important toxic substance of the
pathway. A concentration of GLY ≥ 1 mM supports cellular maintenance and growth of E.
coli BL21 (DE3) with an initial OD600 0.1.
A kinetic model was employed to improve the TCP degradation pathway using
engineered enzyme variants and balanced enzyme ratios. Predictions were evaluated in
terms of minimized toxicity of intermediates and maximized production of GLY, and
showed excellent correspondence with experimental data. Expression levels of the
enzymes can also be reliably predicted from a combination of plasmids with different
copy numbers. A statistically significant increase in the viability of degWT-opt compared
to degWT demonstrates that optimization of the enzyme ratio is a useful approach for
improvement of overall pathway performance, and that the kinetic model employing in
vitro measured kinetic parameters of individual enzymes is applicable for rational
pathway engineering also in suitable heterologous host organism. The model can be used
for the design of constructs with optimized expression levels and prediction of minimum
requirements for catalyst properties.
Toxicity of the substrate does not limit viability of the cells employing the highly
active variant of the first enzyme, haloalkane dehalogenase DhaA31, in the pathway. This
is due to rapid conversion of the highly toxic TCP to the less toxic DCP. However, the
growth and viability of the cells are limited by the insufficient production of GLY.
Production of GLY can be improved by further engineering the first enzyme of the
pathway towards a higher catalytic efficiency (kcat/Km >2300 s-1M-1) or higher
enantioselectivy (E-value >20), or by a combination of both properties (kcat/Km >700 s-1M-1
and E-value >10). Removal of the second enzyme selectivity of the pathway, haloalcohol
dehalogenase HheC, represents another option for improvement of the carbon/energy
flow. Construction of a new generation of enzyme catalysts for degradation of TCP is
currently on-going in our laboratory. In parallel, further options for adaptive laboratory
evolution of the whole synthetic pathway in alternative heterologous host are studied in
order to obtain efficient, self-reproducing, and stress-resistant biocatalyst suitable for
biotechnological applications. These options are outlined in the last chapter of the Thesis.

Chapter 3
- 119 -
SUPPLEMENTARY TABLES AND FIGURES
Table S1. Accumulation of selected metabolites of TCP pathway inside the E. coli cells. concentration (mM) in factor
logPa cell suspension buffer cell biomass biomass/buffer
TCP 2.29 2.18 ± 0.15 2.17 ± 0.15 10.30 ± 1.39 4.7
DCP 0.78 2.27 ± 0.09 2.25 ± 0.10 2.23 ± 0.19 1.0
GDL -0.93 2.24 ± 0.09 2.20 ± 0.10 1.53 ± 0.51 0.7
[a] Calculated using ALOGPS 2.1 program [196].

Ch
apte
r 3
- 1
20
-
Ta
ble
S2
. Pre
dic
tio
n o
f G
LY
pro
du
ctio
n a
nd
to
xici
ty e
xpo
sure
fo
r tw
elv
e p
oss
ible
co
nst
ruct
s u
sin
g se
lect
ed c
om
bin
atio
ns
of
pla
smid
s an
d
Dh
aAw
t en
zym
e.
Exp
erim
enta
lly
p
rep
ared
in
itia
l (r
ed)
and
o
pti
miz
ed
(gre
en)
con
stru
cts
are
hig
hli
ghte
d.
pla
sm
id 1
pla
sm
id 2
norm
aliz
ed r
atio
DhaA
: H
heC
: E
ch
A
convers
ion
to G
LY
rank
GL
Y
toxic
ity
rank
toxic
ity
avera
ge
rank
pD
UE
T-d
ha
A
pC
DF
-hhe
C-e
chA
0.5
0
0.2
5
0.2
5
70.9
%
1
14.0
0
2
1
pD
UE
T-d
ha
A
pA
CY
C-h
heC
-ech
A
0.6
7
0.1
7
0.1
7
69.0
%
2
11.1
2
1
1
pC
DF
-dha
A
pA
CY
C-h
heC
-ech
A
0.5
0
0.2
5
0.2
5
70.9
%
1
14.0
0
2
1
pA
CY
C-h
heC
pD
UE
T-d
ha
A-e
chA
0.4
4
0.1
1
0.4
4
59.9
%
4
15.3
2
3
2
pC
DF
-hhe
C
pD
UE
T-d
ha
A-e
chA
0.4
0
0.2
0
0.4
0
63.4
%
3
16.6
2
4
2
pA
CY
C-h
heC
pC
DF
-dha
A-e
ch
A
0.4
0
0.2
0
0.4
0
63.4
%
3
16.6
2
4
2
pD
UE
T-h
heC
pC
DF
-dha
A-e
ch
A
0.2
5
0.5
0
0.2
5
52.9
%
5
23.1
9
5
3
pC
DF
-hhe
C
pA
CY
C-d
haA
-ech
A
0.2
5
0.5
0
0.2
5
52.9
%
5
23.1
9
5
3
pC
DF
-dha
A
pD
UE
T-h
heC
-ech
A
0.2
0
0.4
0
0.4
0
44.4
%
6
25.7
1
6
4
pA
CY
C-d
haA
pC
DF
-hhe
C-e
chA
0.2
0
0.4
0
0.4
0
44.4
%
6
25.7
1
6
4
pD
UE
T-h
heC
pA
CY
C-d
haA
-ech
A
0.1
7
0.6
7
0.1
7
34.3
%
7
28.2
1
7
5
pA
CY
C-d
haA
pD
UE
T-h
heC
-ech
A
0.1
1
0.4
4
0.4
4
26.5
%
8
31.5
4
8
6

Ch
apte
r 3
- 1
21
-
Ta
ble
S3
. Pre
dic
tio
n o
f G
LY
pro
du
ctio
n a
nd
to
xici
ty e
xpo
sure
fo
r tw
elv
e p
oss
ible
co
nst
ruct
s u
sin
g se
lect
ed c
om
bin
atio
ns
of
pla
smid
s an
d
Dh
aA9
0R
en
zym
e.
Exp
erim
enta
lly
p
rep
ared
in
itia
l (r
ed)
and
o
pti
miz
ed
(gre
en)
con
stru
cts
are
hig
hli
ghte
d.
pla
sm
id 1
pla
sm
id 2
norm
aliz
ed r
atio
DhaA
: H
heC
: E
ch
A
convers
ion
to G
LY
rank
GL
Y
toxic
ity
rank
toxic
ity
avera
ge
rank
pD
UE
T-d
ha
A
pA
CY
C-h
heC
-ech
A
0.6
7
0.1
6
0.1
7
62.9
%
1
20.4
2
1
1
pD
UE
T-d
ha
A
pC
DF
-hhe
C-e
chA
0.5
0
0.2
5
0.2
5
56.8
%
2
23.3
1
2
2
pC
DF
-dha
A
pA
CY
C-h
heC
-ech
A
0.5
0
0.2
5
0.2
5
56.8
%
2
23.3
1
2
2
pA
CY
C-h
heC
pD
UE
T-d
ha
A-e
chA
0.4
4
0.1
1
0.4
4
52.2
%
3
24.4
5
3
3
pC
DF
-hhe
C
pD
UE
T-d
ha
A-e
chA
0.4
0
0.2
0
0.4
0
50.3
%
4
25.5
0
4
4
pA
CY
C-h
heC
pC
DF
-dha
A-e
ch
A
0.4
0
0.2
0
0.4
0
50.3
%
4
25.5
0
4
4
pD
UE
T-h
heC
pC
DF
-dha
A-e
ch
A
0.2
5
0.5
0
0.2
5
34.1
%
5
30.1
5
5
5
pC
DF
-hhe
C
pA
CY
C-d
haA
-ech
A
0.2
5
0.5
0
0.2
5
34.1
%
5
30.1
5
5
5
pC
DF
-dha
A
pD
UE
T-h
heC
-ech
A
0.2
0
0.4
0
0.4
0
30.0
%
6
31.7
3
6
6
pA
CY
C-d
haA
pC
DF
-hhe
C-e
chA
0.2
0
0.4
0
0.4
0
30.0
%
6
31.7
3
6
6
pD
UE
T-h
heC
pA
CY
C-d
haA
-ech
A
0.1
7
0.6
7
0.1
7
21.2
%
7
33.2
9
7
7
pA
CY
C-d
haA
pD
UE
T-h
heC
-ech
A
0.1
1
0.4
4
0.4
4
18.0
%
8
35.2
6
8
8
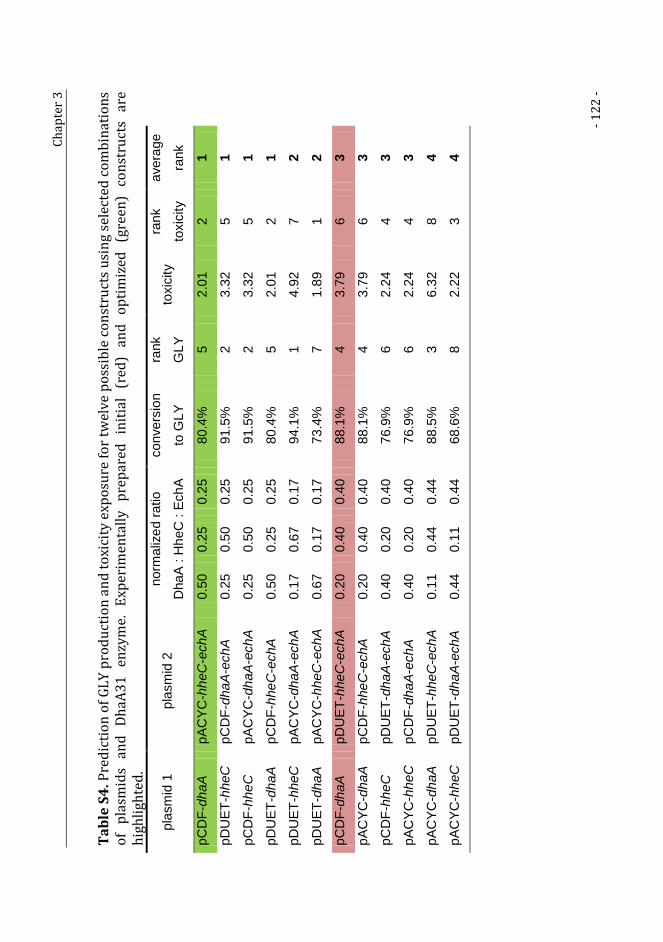
Ch
apte
r 3
- 1
22
-
Ta
ble
S4
. Pre
dic
tio
n o
f G
LY
pro
du
ctio
n a
nd
to
xici
ty e
xpo
sure
fo
r tw
elv
e p
oss
ible
co
nst
ruct
s u
sin
g se
lect
ed c
om
bin
atio
ns
of
pla
smid
s an
d
Dh
aA3
1
enzy
me.
E
xper
imen
tall
y
pre
par
ed
init
ial
(red
) an
d
op
tim
ized
(g
reen
) co
nst
ruct
s ar
e h
igh
ligh
ted
.
pla
sm
id 1
pla
sm
id 2
norm
aliz
ed r
atio
DhaA
: H
heC
: E
ch
A
convers
ion
to G
LY
rank
GL
Y
toxic
ity
rank
toxic
ity
avera
ge
rank
pC
DF
-dha
A
pA
CY
C-h
heC
-ech
A
0.5
0
0.2
5
0.2
5
80.4
%
5
2.0
1
2
1
pD
UE
T-h
heC
pC
DF
-dha
A-e
ch
A
0.2
5
0.5
0
0.2
5
91.5
%
2
3.3
2
5
1
pC
DF
-hhe
C
pA
CY
C-d
haA
-ech
A
0.2
5
0.5
0
0.2
5
91.5
%
2
3.3
2
5
1
pD
UE
T-d
ha
A
pC
DF
-hhe
C-e
chA
0.5
0
0.2
5
0.2
5
80.4
%
5
2.0
1
2
1
pD
UE
T-h
heC
pA
CY
C-d
haA
-ech
A
0.1
7
0.6
7
0.1
7
94.1
%
1
4.9
2
7
2
pD
UE
T-d
ha
A
pA
CY
C-h
heC
-ech
A
0.6
7
0.1
7
0.1
7
73.4
%
7
1.8
9
1
2
pC
DF
-dha
A
pD
UE
T-h
heC
-ech
A
0.2
0
0.4
0
0.4
0
88.1
%
4
3.7
9
6
3
pA
CY
C-d
haA
pC
DF
-hhe
C-e
chA
0.2
0
0.4
0
0.4
0
88.1
%
4
3.7
9
6
3
pC
DF
-hhe
C
pD
UE
T-d
ha
A-e
chA
0.4
0
0.2
0
0.4
0
76.9
%
6
2.2
4
4
3
pA
CY
C-h
heC
pC
DF
-dha
A-e
ch
A
0.4
0
0.2
0
0.4
0
76.9
%
6
2.2
4
4
3
pA
CY
C-d
haA
pD
UE
T-h
heC
-ech
A
0.1
1
0.4
4
0.4
4
88.5
%
3
6.3
2
8
4
pA
CY
C-h
heC
pD
UE
T-d
ha
A-e
chA
0.4
4
0.1
1
0.4
4
68.6
%
8
2.2
2
3
4

Chapter 3
- 123 -
Table S5. Determination of relative and mass ratio of enzymes of TCP pathway in two selected degraders deg31 and deg31-opt. deg31 DhaA31 HheC EchA sum
relative ratioa 0.15 0.38 0.47 1
mass ratio (mg)a 0.62 ± 0.09 1.50 ±0.22 1.90 ± 0.23 4.02 ± 0.53
activity in CFE (μmol.min-1
.mg-1
) 1.27 ± 0.03 1.45 ± 0.02 31.17 ± 2.01
activity of ctrl. (μmol.min-1
.mg-1
)b 1.47 ± 0.14 1.22 ± 0.08 33.35 ± 2.76
activity in CFE/control (%) 86 119 93
corrected mass ratio (mg) 0.53 1.79 1.77 4.09
deg31-opt DhaA31 HheC EchA sum
relative ratioa 0.64 0.17 0.19 1
mass ratio (mg)a 2.20 ± 0.10 0.56 ± 0.07 0.66 ± 0.08 3.43 ± 0.16
activity in CFE (μmol.min-1
.mg-1
) 1.12 ± 0.06 1.04 ± 0.07 32.55 ± 0.92
activity of ctrl. (μmol.min-1
.mg-1
)b 1.47 ± 0.14 1.22 ± 0.08 33.35 ± 2.76
activity in CFE/control (%) 76 85 98
corrected mass ratio (mg) 1.67 0.48 0.65 2.8
[a] Mass ratio and sum of enzymes in mg corresponds to the amount of enzymes in CFE obtained from 10 ml of cell suspension of OD600 of 3.5 by sonication. Ratios were calculated from band densities on SDS-polyacrylamide gel. [b] Control activities were determined with purified enzymes.

Chapter 3
- 124 -
Figure S1. Polynomial equations describing the dependence of E. coli growth on concentration of individual metabolites: A) TCP, B) DCP, C) ECH, D) CPD, and E) GLD.
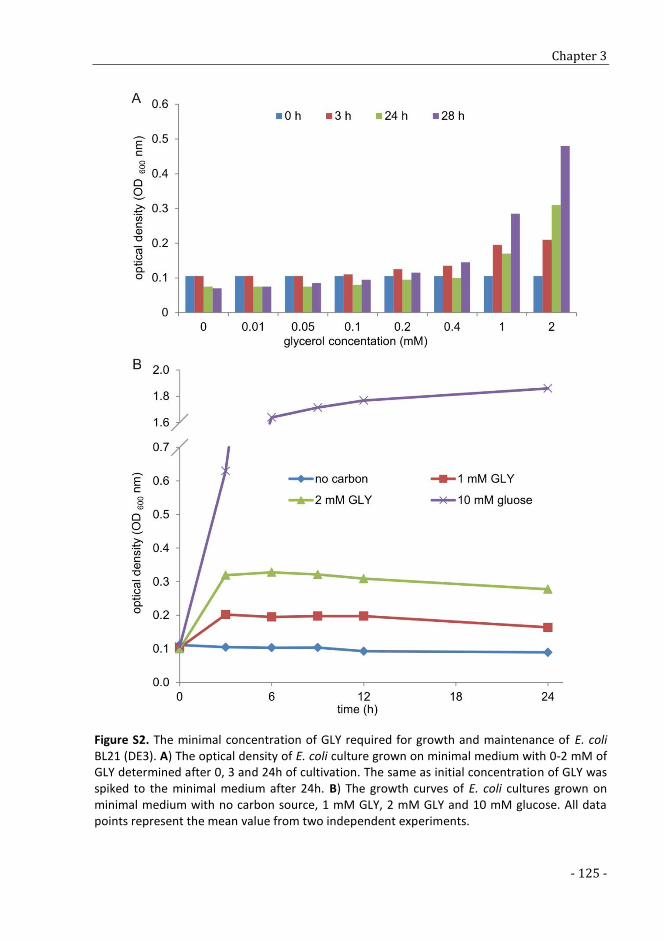
Chapter 3
- 125 -
Figure S2. The minimal concentration of GLY required for growth and maintenance of E. coli BL21 (DE3). A) The optical density of E. coli culture grown on minimal medium with 0-2 mM of GLY determined after 0, 3 and 24h of cultivation. The same as initial concentration of GLY was spiked to the minimal medium after 24h. B) The growth curves of E. coli cultures grown on minimal medium with no carbon source, 1 mM GLY, 2 mM GLY and 10 mM glucose. All data points represent the mean value from two independent experiments.

Chapter 3
- 126 -
Figure S3. Comparison of the expression of DhaA variants, HheC and EchA from plasmid pET21b. 5 μg of protein were loaded on the gel. Expression of DhaAwt represented by the band density was set as 1.0, expression of the other enzymes related to this value was 1.3 for DhaA90R, 0.8 for DhaA31, 1.3 for HheC and 1.3 for EchA. M, protein marker (14.4, 18.4, 25, 35, 45, 66.2, 116 kDa).
Figure S4. Comparison of the expression profiles of unoptimized degraders. M: protein marker (units in kDa), 1: detWT, 2: deg90R, 3: deg31 and Std: standard sample with 0.25 μg of purified DhaAwt, HheC and EchA.

Chapter 3
- 127 -
Figure S5. Verification of GLY production during whole-cell conversion of TCP with deg31-opt (A) and deg31 (B) of cell density corresponding to the OD600 of 3.5. Glucose (5 mM) was added to the minimal medium as a preferred carbon source. Decrease in concentration of GLY in later phase of reaction was most likely caused by early depletion of glucose. Chiral analysis of 2,3-dichloropropane-1-ol accumulated during conversion of TCP by deg31-opt (C).
Figure S6. Recovery of degraders in LB medium after exposure to 3.5 mM TCP for 300 min. The data represent the mean value from two independent experiments.

Chapter 3
- 128 -
Figure S7. Prediction of DhaA variants optimized for the production of GLY using the engineered TCP pathway in the host cells of OD600 0.1 incubated with 2 mM TCP as a sole carbon and energy source. The existing variants are in red, variants with improved production of GLY (1 mM within 24 h period) are in yellow and optimal variant is in green.

- 129 -

- 130 -

- 131 -
CHAPTER 4
Assembly of the synthetic pathway for
biodegradation of 1,2,3-trichloropropane
in Pseudomonas putida KT2440 CF1

Chapter 4
- 132 -
INTRODUCTION
The performance of the synthetic pathway for biodegradation of
1,2,3-trichloropropane (TCP) was previously tested and optimized in laboratory strain
Escherichia coli BL21 (DE3). This strain is routinely used for overexpression of
recombinant proteins and often serves as a model organism also for metabolic
engineering or synthetic biology studies [15]. However, applicability of E. coli degraders in
adaptive laboratory evolution or continuous biodegradation process is limited due to: (i)
instability of plasmid-based expression system in prolonged cultivations, (ii) sensitivity of
E. coli to the toxic effect of TCP, and (iii) strong overexpression of heterologous genes from
T7 promoter, which may result in extensive metabolic burden for the host cell
[58,60,277,278]. Therefore, we decided to transfer TCP pathway into the host organism
and expression system that could be better suited for further evolution of the synthetic
route and potential biotechnological applications.
Pseudomonas putida KT2440 is a saprophytic non-pathogenic soil bacterium with
ability to naturally mineralize aromatic hydrocarbons (e.g., toluene or xylene), chloro- and
nitro- organic compounds including some pesticides, herbicides and even explosive
chemicals [134]. Recent studies revealed that its alternative variant of Embden-Meyerhof-
Parnas pathway (archetypal glycolysis) known as Entner-Doudoroff pathway empowers
P. putida KT2440 with higher tolerance to the oxidative stress which often accompanies
aerobic bacterial utilization of chlorinated pollutants [202,279]. The metabolic versatility
and robustness made P. putida a host of choice for several projects focused on engineering
of biodegradation pathways [141,145,146]. Increasing popularity of P. putida as a host
organism is accompanied by rising number of genetic tools developed for the purpose of
strain engineering. SEVA plasmids with standardized architecture and nomenclature
represent state-of-the-art vector system with regulatory elements tailored to allow
cloning or expression of heterologous genes in P. putida and other Gram-negative bacteria
[54]. They provide unique variability in terms of antibiotic markers, origins of
replications, and cargos that can be combined in a single plasmid. The pSEVA vectors were
recently modified to allow also scarless deletions of genes or implementation of
regulatory and metabolic modules in a chromosome of Gram-negative bacteria through
specialized mini-transposon-based delivery systems [138,42]. Some of these tools were
used to engineer strain KT2440 Cell Factory 1 (CF1) in a way that a prophages and genes
encoding proteins of flagellum were deleted [280,281]. These modifications provided P.
putida KT2440 with increased energy charge and genomic stability, making it a suitable
chassis for heterologous expression of recombinant enzymes and synthetic metabolic
pathways.
The aim of this study was to transplant the most efficient variant of synthetic TCP
pathway from E. coli into P. putida KT2440 CF1 and express genes for haloalkane
dehalogenase DhaA31, haloalcohol dehalogenase HheC, and epoxide hydrolase EchA from
suitable SEVA vector and from P. putida chromosome. The degrading capacity of obtained

Chapter 4
- 133 -
P. putida constructs was compared with one of two most promissing E. coli degraders,
deg31 described in the Chapter 3. The conducted experiments represent a good starting
point for a broader study focused on comparison of E. coli and P. putida degraders and
development of a robust microbial cell factory for biodegradation of TCP.

Chapter 4
- 134 -
MATERIAL AND METHODS
Chemicals, media, strains, and plasmids. TCP, DCP, ECH, CPD, GDL, and GLY standards
were purchased from Sigma-Aldrich (USA). All chemicals used in this study were of
analytical grade. All restriction enzymes were purchased from New England Biolabs
(USA). Phusion High-Fidelity DNA Polymerase was from New England Biolabs (USA) and
GoTag DNA Polymerase was from Promega (USA). T4 DNA ligase and dNTPs were from
Roche Applied Science (USA). Oligonucleotides were synthesized by Sigma Aldrich (USA).
NucleoSpin Gel and PCR Clean-up kit was purchased from Macherey-Nagel (Germany). A
free Glycerol Assay Kit was acquired from BioVision (USA). Luria Broth (LB) (Sigma
Aldrich, USA) was used for routine cultures. M9 minimal medium (Sigma Aldrich, USA)
containing 2 mM MgSO4.7H2O and 2 ml of trace elements per 1 l and 10 mM glucose were
used for toxicity tests [274]. E. coli DH5α (Life Technologies, USA) or CC118λpir was used
in cloning and plasmid propagation. E. coli BL21 (DE3) (Life Technologies, USA) or P.
putida KT2440 CF1 [281] were used as a heterologous hosts and plasmids pCDF and
pETDuet (Merck Millipore, Germany) or pSEVA238 and pBEXK [54,138] were used for
expression of the synthetic pathway for the biodegradation of TCP. Properties of
employed strains and plasmids are listed in Table 1.
Determination of toxicity of TCP for P. putida. A growth test in M9 medium containing
10 mM glucose was used to determine the toxicity of TCP for P. putida KT2440 CF1. The
experiment was carried out as described for E. coli in Material and Methods section of
Chapter 3 of this Thesis with minor modifications. The growth of P. putida cultures was
evaluated at 30°C for 6 hours using 1, 2, 4, and 6 mM of TCP. The concentration of TCP was
monitored by GC. To minimize the evaporation of TCP, cultivation was done in 100 ml
Erlenmeyer flasks with penetrable Suba Seal rubber septa (Sigma-Aldrich, USA).
PCR amplification of dhaA31, hheC and echA from pET21b and pET28b. The genes
dhaA31, hheC, and echA were amplified from the template plasmid constructs
pET21b-dhaA31, pET21b-hheC and pET28b-echA together with the ribosome binding site
(RBS) and C-terminal 6xHis tag (DhaA31 and EchA) by PCR using oligonucleotide primers
dhaA31-F/R, hheC-F/R, and echA-F/R listed in Table 2. To amplify genes, approximately
2 ng of template DNA was mixed in 50 μl reaction mixture containing Phusion HF Buffer
with MgCl2, 3% DMSO, dNTPs of final concentration 0.2 mM each, 0.5 μM of each primer,
and 1 U of Phusion High-Fidelity DNA Polymerase. PCR cycling conditions were as follows:
after an initial DNA denaturation step of 3 min at 94°C, the reaction mixtures were
subjected to 30 cycles consisting of 10 s at 98°C, 30 s at 57°C and 30 s at 72°C. Additional
5 min annealing step at 72°C followed. PCR products were verified by agarose
electrophoresis and purified from gel using NucleoSpin Gel and PCR Clean-up.
Construction of synthetic TCP operon. Restriction analysis of dhaA31, hheC and echA was
conducted using the on-line tool NEBcutter V2.0 (http://tools.neb.com/NEBcutter2/, New

Chapter 4
- 135 -
England Biolabs, USA). Purified PCR products were double-digested with KpnI/BamHI
(dhaA31His), BamHI/XbaI (hheC) and XbaI/PstI (echAHis) and subcloned in a step-wise
manner into the polylinker of vector pSEVA238 digested with corresponding restriction
enzymes (Figure 1). Ligation mixtures were transformed into the competent E. coli DH5α
using heat shock method. Clones were checked for plasmids with inserts by colony PCR
using 1 U of GoTag DNA Polymerase or by restriction analysis. Primers p470, PS2, TCPo1
and TCPo2 listed in Table 2 were used for sequencing of the final constructs including
synthetic TCP operon (Secugen, Spain). Verified plasmid construct pSEVA238-dhaA31-
hheC-echA (signed here as pSEVA238-TCPop) was double digested with KpnI and PstI and
TCP operon was subcloned also into the pBEXK vector (Figure 1) cut with the same
restrictases. Gene cloning techniques and other molecular biology procedures followed
standard protocols [56] or manufacturer's instructions.
Transformation of plasmid constructs into electrocompetent P. putida KT2440 CF1.
Constructs pSEVA238-TCPop, pBEXK-TCPop, and empty pSEVA238 were transformed into
competent P. putida KT2440 CF1 by electroporation. Electrocompetent cells were
prepared as described by Choi et al. by washing the cell biomass twice with 0.3 M sucrose
[282]. Plasmid DNA was added to a 100 μl aliquot of the cell suspension. The mixture was
transferred to a 0.2-cm gap width cuvette and electroporated using the program EC2 of a
MicroPulserTM apparatus (Bio-Rad Laboratories, USA), which corresponds to a single
pulse of 2.5 kV (field strength 12.5 kV.cm-1) and a time constant of ca. 5 ms. Following
electropulsing, 1 ml of LB medium was added quickly to the cells and incubated for 90 min
at 30°C. Adequate dilutions of the resulting culture were then plated onto LB agar plates
with Km (50 μg.ml-1) and plates were incubated overnight at 30°C. Transformants were
checked for pSEVA238-TCPop by colony PCR and restriction analysis and verified clones
were used for expression tests. Single colonies of transformants after electroporation of
pBEXK-TCPop were checked for the loss of the plasmid marker ApR and chromosomal
insertions were verified also by colony PCR using the primers cFRT-Ab-F and cKm-R.
Test of expression of enzymes from TCP pathway in P. putida KT2440 CF1. Single
colonies of KT2440 CF1 transformants were used for inoculation of overnight culture in
LB medium containing Km. Cell were grown at 30°C with shaking (180 rpm). 200 μl of
overnight culture was used to inoculate 20 ml of LB medium with Km. Cultures were
grown at 30°C with shaking (180 rpm) until OD600 of about 0.6. Expression of recombinant
enzymes was induced with 5 mM 3-methylbenzoate (0.5 M stock in milliQ water was
prepared by dissolution of 3-methylbenzoate in presence of sodium hydroxide). Induction
continued overnight (13 – 14 h) at 20°C with shaking (180 RPM). Cells were pelleted by
centrifugation at 4,000 g at 4°C. Cell biomass was resuspended in 50 mM sodium
phosphate buffer (pH 7.0) and cells were disrupted by sonication. Cell lysates were
centrifuged at 21,000 g for 45 min at 4°C. Resulting cell-free extracts (CFE) were decanted
and samples of CFE and cell pellets were loaded on 15 % sodium dodecyl sulfate
polyacrylamide gel. After SDS-PAGE, gels were stained with Coomassie Brilliant Blue

Chapter 4
- 136 -
R-250 (Fluka, Switzerland) and analyzed using a GS-800 Calibrated Imaging Densitometer
(Bio-Rad, USA).
Degradation of TCP in buffer by pre-induced resting cells. Cell suspensions of
pre-induced deg31, pre-induced P. putida KT2440 CF1 with pSEVA238-TCPop or empty
pSEVA238 (negative control) and three randomly picked clones of P. putida KT2440 CF1
with TCP operon in chromosome were diluted with sterile 50 mM sodium phosphate
buffer to a final OD600 7.0. Glass vials (25 ml) with a screw cap mininert valve containing
7.5 ml of the sterile 50 mM sodium phosphate buffer of pH 7.0 and 4 mM TCP were
prepared separately and incubated for 1 h at 37°C with shaking. The reaction was initiated
by mixing 7.5 ml of the cell suspension with 7.5 ml of buffer and dissolved TCP. The final
concentration of TCP in 15 ml of the cell suspension was 2 mM and the final theoretical
OD600 was 3.5. The vials were incubated for 300 min at 30°C with shaking. Cell suspension
samples (0.5 ml) were quenched in 0.5 ml acetone with hexan-1-ol, vortexed and
centrifuged at 18,000 g for 2 min. The concentration of metabolites in the supernatant was
analyzed using GC and the production of GLY was monitored by Glycerol Assay Kit as
described in Material and Method sections of previous chapters of this Thesis. The
stability of TCP degradation phenotype of P. putida constructs with synthetic operon
inserted in chromosome was verified by twenty rounds of serial plating of clones on LB
agar plates without antibiotics and repeated determination of TCP degradation in buffer
with pre-induced cells.
Determination of growth curve of E. coli BL21 (DE3) and P. putida KT2440 CF1 on GLY.
The overnight cultures of E. coli BL21 (DE3) and P. putida KT2440 CF1 grown in LB
medium were washed by ice cold M9 minimal medium with 2 mM MgSO4.7H2O and trace
elements and used for inoculation of 20 ml of the same medium but with 0.2% GLY to the
final OD600 of 0.1. The growth curves of cultures grown with shaking (180 rpm) at 30°C
were determined by measuring optical density at 600 nm for 30 h.
Analytical techniques. Analytical techniques used in this study were described in
Material and Methods section of Chapter 3 of this Thesis.

Chapter 4
- 137 -
Table 1. Bacterial strains and plasmids used in this study
Strain or plasmid Relevant characteristicsa Source or reference
Escherichia coli
Dh5α Cloning host; Φ80lacZΔM15 recA1 endA1 gyrA96
thi-1 hsdR17(rK−mK
+) supE44 relA1
deoR Δ(lacZYA-argF)U169
Life Technologies, USA
CC118λpir Cloning host; araD139 Δ(ara-leu)7697 ΔlacX74
galE galK phoA20 thi-1 rpsE
rpoB(RifR) argE(Am) recA1 λpir lysogen
[55]
BL21 (DE3) Expression host; F– ompT gal dcm lon hsdSB(rB
-
mB-) λ(DE3 [lacI lacUV5-T7 gene 1 ind1 sam7
nin5])
Life Technologies, USA
Pseudomonas putida
KT2440 Cell Factory 1
(CF1)
Engineered KT2440 derived from P. putida mt-2
cured of the TOL plasmid pWW0;
Δprophages1,2,3,4 ΔTn7 ΔendA1 ΔendA2
ΔhsdRMS Δflagella ΔTn4652
[281]
Plasmids
pCDF Expression vector; CDF(CloDF13) T7lac SmR Merck Millipore, Germany
pETDuet Expression vector; ColE1(pBR322) T7lac ApR Merck Millipore, Germany
pSEVA238b Expression vector; oriV(pBBR1) xylS Pm aphA
KmR
[54]
pBEXK Mini-Tn5 delivery vector derived from
pBAM1; tnpA oriV(R6Kγ) oriT(RK2) xylS Pm bla
FRT-aphA-FRT ApR Km
R
[138]
[a] Antibiotic markers: Ap, ampicillin; Sm, streptomycin; Km, kanamycin. [b] Plasmids belonging to the Standard European Vector Architecture (SEVA) collection.
Table 2. Nucleotide sequences of primers used in this study. dhaA31-F
1 5' TCGGTACCCTTTAAGaaggagATATACATATGTCCGAAATTG 3'
dhaA31-R2 5' AATGGATCCTTAGTGGTGGTGGTGG 3'
hheC-F2 5' TTCGGATCCCTTTAAGaaggagATATACATATGTCAACCGCA 3'
hheC-R3 5' TCTCTAGATTACTCGGGCATACCAGGCCA 3'
echA-F3 5' TCTCTAGACTTTAAGaaggagATATACCATGGCAATC 3'
echA-R4 5' TCCTGCAGTTAATGGTGGTGGTGGTGGTG 3'
p470 5' GGTTTGATAGGGATAAGTCCAG 3'
PS2 5' GCGGCAACCGAGCGTTC 3'
TCPo1 5' GAACGAACTGCCGATTGCAG 3'
TCPo2 5' CGAATCCAGAACACGTTGC 3'
cFRT-F 5' CTTGCAATGGTACAATCCTCC 3'
cKm-R 5' CGGAATGCTATGCAGACG 3'
Introduced restriction sites are underlined: 1KpnI; 2 BamHI; 3XbaI; 4PstI. Sequence of original ribosome binding site from pET vector is represented by small letters.

Chapter 4
- 138 -
Figure 1. Schematic representation of the Tn5-based insertional vector pBEX for regulated expression of heterologous genes in Gram-negative bacteria. The functional elements of the plasmid backbone include an antibiotic resistance marker (bla encoding ß-lactamase), a hyperactive transposase (tnpA), a conditional vegetative origin of replication (oriR6Kγ), an origin of transfer region (oriT), and two mosaic elements (ME-O and ME-I). The expression modules span edited copy of the P. putida xylS gene together with the inducible Pm promoter. Promoter is in front of a multiple cloning site followed by strong T0 terminator from phage λ. Expression module is transferred into the chromosome of recipient bacterium along with an removable cassette consisting of the antibiotic resistance marker (aphA-encoded aminoglycoside 3'-phosphotransferase conferring kanamycin resistance) bracketed by FRT sequences. Adopted from [138] and modified.

Chapter 4
- 139 -
RESULTS AND DISCUSSION
The response of P. putida KT2440 CF1 to the toxic effect of TCP was initially tested
and compared with E. coli BL21 (DE3). TCP was previously showed to be the most toxic
compound from all the metabolites in TCP pathway. Effect of various concentrations of
TCP on growth of the P. putida KT2440 CF1 culture in mineral medium supplied with
glucose was determined using the protocol described for E. coli BL21 (DE3) in Chapter 3
of the Thesis. The concentration of TCP at which the growth of total cell population was
reduced by 20% (IC20) was calculated (Supplementary Figure S1). The IC20 value for P.
putida KT2440 CF1 (1.32 mM) was almost identical to the value previously published for
E. coli BL21 (DE3) (1.35 mM). The result shows that TCP is equally detrimental for both
bacterial hosts suggesting the same toxicity mode of action.
The TCP pathway genes were then subcloned from pET to SEVA vector system to
allow transfer of the pathway from E. coli to P. putida. Genes dhaA31, hheC and echA with
their RBSs were amplified from plasmid constructs pET21b-dhaA31, pET21b-hheC, and
pET28b-echA by PCR (Figure 2) and new restriction sites for subcloning into the
polylinker of SEVA vectors were introduced upstream and downstream of the genes.
Restriction sites were selected with care to allow serial subcloning of all three genes in the
polylinker and construction of a synthetic operon, which could be easily transferred in
between various SEVA vectors.
Plasmid pSEVA238 was used for the assembly of synthetic operon (Figure 3). The
plasmid carries inducible Pm/xylS promoter from P. putida TOL plasmid pWW0,
kanamycin resistance marker, and the origin of replication from pBBR1 that confers the
vectors with medium-copy number (30 – 40 molecules in a single P. putida cell). The
genes coding for enzymes of TCP pathway were subcloned into pSEVA238 in step-wise
manner resulting in constructs pSEVA238-hheC, pSEVA238-hheC-echA and pSEVA238-
TCPop. New constructs were verified by colony PCR, restriction analysis (Figure 2) and
sequencing. The plasmid constructs were transformed into the electrocompetent P. putida
CF1 cells and the expression of individual genes of TCP pathway after induction with 5
mM 3-methylbenzoate was evaluated by SDS-PAGE analysis of CFEs obtained from
disrupted cell biomass (Figure 4A). Interestingly, analysis revealed that the relative ratio
of DhaA31, HheC and EchA expressed from pSEVA238-TCPop in P. putida under selected
conditions (0.11:0.49:0.40) was very similar to the ratio achieved previously in E. coli
deg31 after induction with 0.2 mM IPTG (0.12:0.42:0.46). The expression of DhaA31 in P.
putida was significantly lower than the expression of HheC and EchA although the front
position of dhaA31 in the synthetic operon and long transcription distance from the
beginning of the gene to the end of the operon predetermined it rather for higher
expression [64,283]. The relative amount of all three enzymes in total soluble protein of P.
putida was about 38% while in E. coli deg31 the content of DhaA31, HheC, and EchA in cell
had reached 50% of the total soluble protein.

Chapter 4
- 140 -
Figure 2. Electrophoretic analysis of products of PCR amplification of dhaA31 (935 bp), hheC (801 bp), and echA (937 bp) from pET plasmids (gel on left) and restriction analysis of pSEVA238-TCPop (7744 bp) cut with KpnI and PstI. Length of synthetic TCP operon (bottom band) is 2685 bp. The 500 bp molecular ruler (Bio-Rad, USA) was used for analyses.
Figure 3. Map of plasmid construct pSEVA238-TCPop with synthetic operon encoding three enzymes from pathway for biodegradation of TCP. Operon is expressed from inducible xylS-Pm promoter and transcription is terminated at single rho-independent transcriptional terminator T0 from phageλ. Each gene in operon possesses its own ribosome binding site (RBS). The primers used for sequencing are shown at their annealing sites (in blue).

Chapter 4
- 141 -
The TCP operon was subsequently subcloned also into the suicide pBEXK plasmid
with minitransposon system for delivery of selected cargo into the chromosome of host
bacterium (Figure 1). The synthetic operon was integrated into the chromosome of P.
putida KT2440 CF1 as described by Nikel and de Lorenzo to ensure genetic stability of
engineered strain even in prolonged cultivations [138]. The integration in six selected
clones was verified by parallel plating on LB agar plates with kanamycin and ampicillin
and by colony PCR with insert- and plasmid backbone-specific primers. Expression of
individual genes of TCP pathway in E. coli deg31, P. putida CF1 pSEVA238-TCPop and one
of the clones of P. putida CF1 with TCP operon was compared after induction with 0.2 mM
IPTG (E. coli) or 5 mM 3-methylbenzoate (P. putida; Figure 4B). SDS-PAGE analysis
showed that the relative ratio of DhaA31, HheC and EchA expressed from operon in
chromosome of P. putida remained unchanged when compared with expression from
pSEVA238. Also the relative amount of all three enzymes in total soluble protein of P.
putida (35%) was similar to the value determined for recombinant strain expressing
enzymes from plasmid.
Figure 4. A) SDS-PAGE analysis of expression of DhaA31 (34 kDa), HheC (29 kDa) and EchA (35 kDa) in Pseudomonas putida CF1. M, protein marker (6.9, 21, 29, 35.8, 56.2, 101, 125, 210 kDa); 1, cell-free extract (CFE) of CF1 with empty plasmid pSEVA238; 2, CFE of CF1 with pSEVA238-hheC; 3, CFE of CF1 with pSEVA238-hheC-echA; 4, CFE of CF1 with pSEVA238-TCPop. B) Comparison of expression of individual enzymes in: 1, Escherichia coli deg31; 2, P. putida CF1 pSEVA238-TCPop; 3, P. putida CF1 with synthetic TCP operon introduced into the chromosome. Preinduced cells of P. putida KT2440 with pSEVA238-TCPop and six selected clones
with TCP operon in the chromosome were incubated with 2 mM TCP in buffer for 300 min
and the degradation profiles of P. putida degraders were determined and compared with
reaction time course collected for E. coli deg31 (Figure 5). The degradation profile of P.
putida carrying pSEVA238-TCPop was very similar to the profile of deg31. This result

Chapter 4
- 142 -
corresponded well with the similarity of relative ratios of DhaA31, HheC, and EchA in E.
coli and P. putida constructs. The theoretical conversion of TCP to GLY reached 68% in
both constructs. Interestingly, the TCP degradation profile of six P. putida clones with
single copy of synthetic operon in chromosome (Figure 5C – average data) was
comparable with the profile of cells expressing TCP pathway from medium copy plasmid.
Nevertheless, conversion of TCP varied in between individual constructs with operon
integrated in chromosome and was in some degree slower, reaching theoretical
conversion of TCP to GLY of 61% on average. Slight variations in profiles of individual
clones can be attributed to the different sites of integration, because the context of
integration position might influence the level of expression [138]. Specification of
integration sites of six selected P. putida constructs by arbitrary primed PCR and
sequencing is in progress. Stability of phenotype of six constructs with chromosomal
insertion was verified by repeated determination of ability to produce GLY from TCP after
12 successive transfers of cells grown for 12 h in liquid LB medium without TCP or
antibiotic (data not shown).
Although there was not much variation in degradation profiles of E. coli and P. putida
recombinants, their potential to utilize produced GLY differed substantially (Figure 5D).
While deg31 utilized all of generated GLY within 300 min interval both P. putida
constructs showed continuous accumulation of GLY. Our observation is in agreement with
recent study of Nikel and co-workers showing that P. putida KT2440 grown on GLY has
considerably long lag phase, which may last up to 18 h [284]. Both E. coli and P. putida are
genetically equipped with the functions required for GLY metabolism. The prolonged lag
phase of P. putida can be attributed to the differences in central carbon metabolism and
rearrangements of the whole metabolic network that is necessary to start growth on GLY
[284].
On the other hand, Nikel and co-workers claim that the growth of P. putida on GLY as
a sole carbon source eventually results in a specific metabolic mode of bacterium that is
characterized by: (i) remarkably low level of physiological stress, when compared with
growth on other carbon sources, and (ii) very efficient conversion of substrate into
biomass [284]. We verified the latter by recording the growth curve of P. putida KT2440
CF1 in minimal medium with 0.2% GLY (Supplementary Figure S2). Compared to E. coli
BL21 (DE3), the P. putida culture showed longer lag phase but achieved higher optical
density at the end of exponential phase of growth. Our future research will focus on
comparison of the levels of physiological stress accompanying the degradation of TCP and
utilization of produced GLY in E. coli deg31 and P. putida constructs by viability testing
and flow cytometry measurements with cells labelled by stress-specific fluorophores.
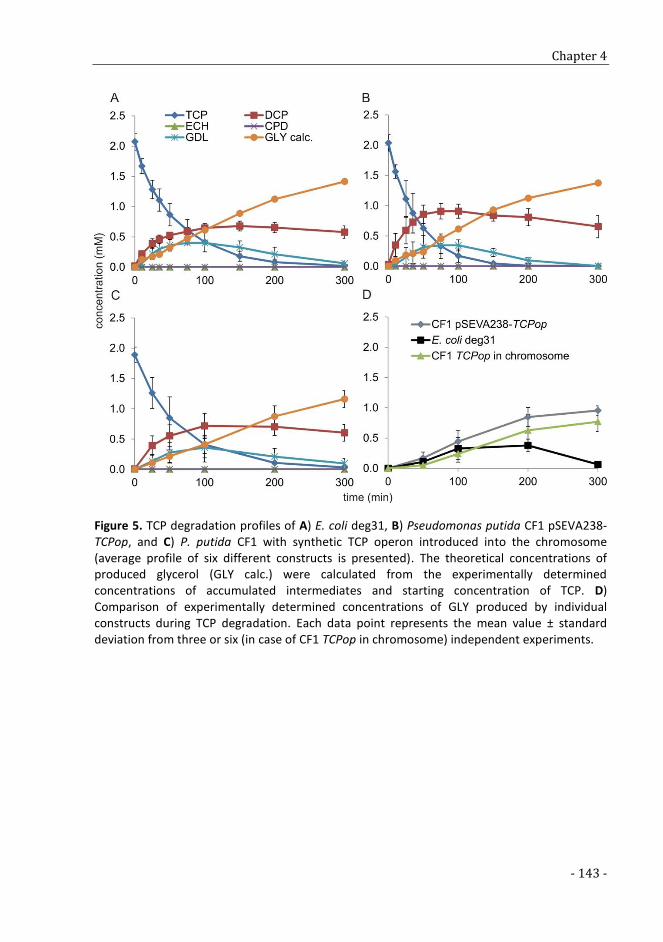
Chapter 4
- 143 -
Figure 5. TCP degradation profiles of A) E. coli deg31, B) Pseudomonas putida CF1 pSEVA238-TCPop, and C) P. putida CF1 with synthetic TCP operon introduced into the chromosome (average profile of six different constructs is presented). The theoretical concentrations of produced glycerol (GLY calc.) were calculated from the experimentally determined concentrations of accumulated intermediates and starting concentration of TCP. D) Comparison of experimentally determined concentrations of GLY produced by individual constructs during TCP degradation. Each data point represents the mean value ± standard deviation from three or six (in case of CF1 TCPop in chromosome) independent experiments.

Chapter 4
- 144 -
CONCLUSIONS AND OUTLOOK Genes encoding enzymes from syntetic TCP pathway were successfully subcloned
from pET to SEVA vector system with inducible Pm/xylS promoter, which enabled
expression of TCP pathway in P. putida KT2440 CF1. The strain showed similar respons to
the toxicity of TCP as E. coli BL21 (DE3). Synthetic operon consisting of dhaA31, hheC, and
echA genes was assembled in pSEVA238 and subcloned into plasmid vector pBEXK, which
was utilized for implantation of the construct into the chromosome of KT2440 CF1. P.
putida recombinants with ratio of enzymes and TCP degradation profiles resembling
those of E. coli deg31 were obtained. These recombinants will be used for thorough
comparative study of two different microbial strains carrying TCP pathway of same
productivity and for further in vivo evolution of TCP pathway.
We expect that shortening of the lag phase by additional engineering interventions
targeting GLY metabolism will make P. putida attractive host for biodegradation of TCP. As
was demonstrated in several bacterial species, shorter lag phase on GLY can be reached
e.g., by deleting glpR represor that regulates expression of glp gene cluster for GLY
metabolism [285,286]. On the other hand, certain applications can take advantage from
GLY accumulating in KT2440 CF1 strain during growth on preffered co-substrate like
glucose or succinate. In such cases, GLY can be streamed in alternative biosynthetic
pathways for production of some value-added compounds, e.g., 1,3-propanediol, citric
acid, or bioplastics like polyhydroxyalkanoates [287]. Thus, toxic industrial waste TCP
could be converted into useful chemicals using complex biochemical networks of whole-
cell biocatalyst that cannot be, with recent state of knowledge, reconstructed in vitro.
Alternatively, accumulated GLY as a direct product of TCP conversion could be
detected and quantified using available enzymatic kits with specific fluorescence probes.
Such system could be employed in fluorescence activated cell sorting (FACS) of clones
resulting from adaptive laboratory evolution of P. putida recombinants incubated with
TCP or clones containing variants of synthetic TCP operon randomized by error-prone
PCR, oligonucleotide-directed randomization or some other mutant library generating
methods [119,288].
We believe, that newly constructed bacterial recombinants and proposed methods
will be benefitial for evolution of more efficient TCP pathway lacking the bottlenecks
described in previous chapters of the Thesis, namely, unbalanced enantioselectivity of
DhaA31 and HheC resulting in accumulation of (S)-DCP or unfavourable substrate
preference of EchA causing accumulation of GDL. In our future research, we aim to further
study the effects of TCP on the host cell physiology, to remove discussed bottlenecks and
to gain bacterial degrader capable of sustainable growth on TCP as the sole carbon and
energy source.

Chapter 4
- 145 -
SUPPLEMENTARY FIGURES
Figure S1. Linear dependence of inhibition of Pseudomonas putida KT2440 CF1 growth on concentration of TCP. Data points represent a mean value from three independent experiments ± standard deviation.
Figure S2. Growth curves of Escherichia coli BL21 (DE3) and Pseudomonas putida KT2440 CF1 obtained during cultivation in minimal medium with 0.2% glycerol. The data points represent a mean value from duplicate measurement.

- 146 -

Summary
- 147 -
SUMMARY
Recent chemical pollution of soil, ground water and surface water constitutes a
major threat to the ecosystems and the public health. Reports of renowned environment
protection agencies predicate that the situation will become worse and the number of
sites requiring remediation will further increase. Biotransformations using either natural
or recombinant microorganisms are extensively studied for their ability to decontaminate
polluted sites. However, rational engineering of microorganisms towards novel or
improved biodegradative capacities performed in the complex environments remains a
challenge. Broader employment of cutting edge techniques and approaches of emerging
biological engineering disciplines can help with both better understanding of studied
biological systems and exploitation of obtained knowledge in practical applications.
This Thesis summarizes the work in which synthetic biology and metabolic
engineering approaches were applied for study and rational redesign of metabolic
pathway for biodegradation of important environmental pollutant TCP. We first
reconstructed the pathway under in vitro conditions and developed a novel biotechnology
for TCP transformation based on immobilized enzymes. Then, we improved the efficiency
of the three-enzyme system with help of engineered biocatalyst and kinetic model using
the data obtained from detailed characterization of individual enzymes. We have
successfully assembled the pathway in heterologous host E. coli BL21 (DE3) and showed
that the same kinetic model can be applied also for tuning of the in vivo orthogonal system.
Eventually, one of the most promising pathway variants was transferred to the new host,
P. putida KT2440 CF1, whose capacity to become a suitable chassis for further evolution of
TCP route is under investigation.
The Thesis presents an innovative concept for rational engineering of a synthetic
metabolic pathway for degradation of toxic recalcitrant compounds. This concept is based
on detailed understanding of studied system and exploitation of in vitro, in silico and in
vivo tools of synthetic biology and metabolic engineering. Conducted research generated a
deeper insight into a studied model system, allowed rational dissection of pathway
bottlenecks, resulted in development of new in vitro biocatalysts applicable for TCP
biodegradation and will hopefully lead to the design of better microbial degrader capable
of sustainable growth on TCP. The developed approaches and molecular tools should be
generally applicable also to other environmental pollutants.

References
- 148 -
REFERENCES
(1) De Lorenzo, V. From the selfish gene to selfish metabolism: revisiting the central dogma. BioEssays
News Rev. Mol. Cell. Dev. Biol. 2014, 36, 226–235. (2) Watson, J. D.; Crick, F. H. The structure of DNA. Cold Spring Harb. Symp. Quant. Biol. 1953, 18, 123–
131. (3) Rich, A. Recent studies on the structure of ribonucleic acid. Prog. Neurobiol. 1956, 1, 114–121. (4) Pauling, L.; Corey, R. B.; Branson, H. R. The structure of proteins; two hydrogen-bonded helical
configurations of the polypeptide chain. Proc. Natl. Acad. Sci. U. S. A. 1951, 37, 205–211. (5) Arber, W.; Linn, S. DNA modification and restriction. Annu. Rev. Biochem. 1969, 38, 467–500. (6) Meselson, M.; Yuan, R. DNA restriction enzyme from E. coli. Nature 1968, 217, 1110–1114. (7) Nielsen, J.; Fussenegger, M.; Keasling, J.; Lee, S. Y.; Liao, J. C.; Prather, K.; Palsson, B. Engineering
synergy in biotechnology. Nat. Chem. Biol. 2014, 10, 319–322. (8) Pirie, C. M.; De Mey, M.; Jones Prather, K. L.; Ajikumar, P. K. Integrating the protein and metabolic
engineering toolkits for next-generation chemical biosynthesis. ACS Chem. Biol. 2013, 8, 662–672. (9) Gibson, D. G.; Glass, J. I.; Lartigue, C.; Noskov, V. N.; Chuang, R.-Y.; Algire, M. A.; Benders, G. A.;
Montague, M. G.; Ma, L.; Moodie, M. M.; et al. Creation of a bacterial cell controlled by a chemically synthesized genome. Science 2010, 329, 52–56.
(10) Jiang, L.; Althoff, E. A.; Clemente, F. R.; Doyle, L.; Röthlisberger, D.; Zanghellini, A.; Gallaher, J. L.; Betker, J. L.; Tanaka, F.; Barbas, C. F.; et al. De novo computational design of retro-aldol enzymes. Science 2008, 319, 1387–1391.
(11) Bailey, J. E. Toward a science of metabolic engineering. Science 1991, 252, 1668–1675. (12) Ghisalba, O.; Meyer, H.-P.; Wohlgemuth, R.; Flickinger, M. C. Industrial Biotransformation. In
Encyclopedia of Industrial Biotechnology; John Wiley & Sons, Inc., 2009. (13) Yang, Y.-T.; Bennett, G. N.; San, K.-Y. Genetic and metabolic engineering. Electron. J. Biotechnol. 1998, 1,
134–141. (14) Stephanopoulos, G. Synthetic biology and metabolic engineering. ACS Synth. Biol. 2012, 1, 514–525. (15) Smolke, C. The Metabolic Pathway Engineering Handbook: Fundamentals; 1 edition.; CRC Press: Boca
Raton, 2009. (16) Alexeyev, M. F.; Shokolenko, I. N.; Croughan, T. P. New mini-Tn5 derivatives for insertion mutagenesis
and genetic engineering in gram-negative bacteria. Can. J. Microbiol. 1995, 41, 1053–1055. (17) Zhang, Y.-X.; Perry, K.; Vinci, V. A.; Powell, K.; Stemmer, W. P. C.; del Cardayré, S. B. Genome shuffling
leads to rapid phenotypic improvement in bacteria. Nature 2002, 415, 644–646. (18) Thykaer, J.; Nielsen, J. Metabolic engineering of beta-lactam production. Metab. Eng. 2003, 5, 56–69. (19) De Graaf, A. A.; Eggeling, L.; Sahm, H. Metabolic engineering for L-lysine production by
Corynebacterium glutamicum. Adv. Biochem. Eng. Biotechnol. 2001, 73, 9–29. (20) Dhamankar, H.; Prather, K. L. J. Microbial chemical factories: recent advances in pathway engineering
for synthesis of value added chemicals. Curr. Opin. Struct. Biol. 2011, 21, 488–494. (21) Copeland, W. B.; Bartley, B. A.; Chandran, D.; Galdzicki, M.; Kim, K. H.; Sleight, S. C.; Maranas, C. D.;
Sauro, H. M. Computational tools for metabolic engineering. Metab. Eng. 2012, 14, 270–280. (22) Medema, M. H.; van Raaphorst, R.; Takano, E.; Breitling, R. Computational tools for the synthetic
design of biochemical pathways. Nat. Rev. Microbiol. 2012, 10, 191–202. (23) Santos, C. N. S.; Stephanopoulos, G. Combinatorial engineering of microbes for optimizing cellular
phenotype. Curr. Opin. Chem. Biol. 2008, 12, 168–176. (24) Hatzimanikatis, V.; Li, C.; Ionita, J. A.; Henry, C. S.; Jankowski, M. D.; Broadbelt, L. J. Exploring the
diversity of complex metabolic networks. Bioinforma. Oxf. Engl. 2005, 21, 1603–1609. (25) Funahashi, A.; Matsuoka, Y.; Jouraku, A.; Morohashi, M.; Kikuchi, N.; Kitano, H. CellDesigner 3.5: A
Versatile Modeling Tool for Biochemical Networks. Proc. IEEE 2008, 96, 1254–1265. (26) Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N. S.; Wang, J. T.; Ramage, D.; Amin, N.; Schwikowski, B.;
Ideker, T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504.
(27) Tang, W. L.; Zhao, H. Industrial biotechnology: tools and applications. Biotechnol. J. 2009, 4, 1725–1739.
(28) Stephanopoulos, G. Metabolic fluxes and metabolic engineering. Metab. Eng. 1999, 1, 1–11. (29) Meyer, A.; Pellaux, R.; Panke, S. Bioengineering novel in vitro metabolic pathways using synthetic
biology. Curr. Opin. Microbiol. 2007, 10, 246–253.

References
- 149 -
(30) Becker, J.; Klopprogge, C.; Herold, A.; Zelder, O.; Bolten, C. J.; Wittmann, C. Metabolic flux engineering of L-lysine production in Corynebacterium glutamicum--over expression and modification of G6P dehydrogenase. J. Biotechnol. 2007, 132, 99–109.
(31) Schellenberger, J.; Que, R.; Fleming, R. M. T.; Thiele, I.; Orth, J. D.; Feist, A. M.; Zielinski, D. C.; Bordbar, A.; Lewis, N. E.; Rahmanian, S.; et al. Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox v2.0. Nat. Protoc. 2011, 6, 1290–1307.
(32) Rocha, I.; Maia, P.; Evangelista, P.; Vilaça, P.; Soares, S.; Pinto, J. P.; Nielsen, J.; Patil, K. R.; Ferreira, E. C.; Rocha, M. OptFlux: an open-source software platform for in silico metabolic engineering. BMC Syst. Biol. 2010, 4, 45.
(33) Burgard, A. P.; Pharkya, P.; Maranas, C. D. Optknock: a bilevel programming framework for identifying gene knockout strategies for microbial strain optimization. Biotechnol. Bioeng. 2003, 84, 647–657.
(34) Parachin, N. S.; Bergdahl, B.; van Niel, E. W. J.; Gorwa-Grauslund, M. F. Kinetic modelling reveals current limitations in the production of ethanol from xylose by recombinant Saccharomyces cerevisiae. Metab. Eng. 2011, 13, 508–517.
(35) Costa, R. S.; Machado, D.; Rocha, I.; Ferreira, E. C. Critical perspective on the consequences of the limited availability of kinetic data in metabolic dynamic modelling. IET Syst. Biol. 2011, 5, 157–163.
(36) Van Eunen, K.; Kiewiet, J. A. L.; Westerhoff, H. V.; Bakker, B. M. Testing biochemistry revisited: how in vivo metabolism can be understood from in vitro enzyme kinetics. PLoS Comput. Biol. 2012, 8, e1002483.
(37) Mendes, P.; Hoops, S.; Sahle, S.; Gauges, R.; Dada, J.; Kummer, U. Computational modeling of biochemical networks using COPASI. Methods Mol. Biol. Clifton NJ 2009, 500, 17–59.
(38) Tomita, null; Hashimoto, null; Takahashi, null; Shimizu, null; Matsuzaki, null; Miyoshi, null; Saito, null; Tanida, null; Yugi, null; Venter, null; et al. E-CELL: Software Environment for Whole Cell Simulation. Genome Inform. Workshop Genome Inform. 1997, 8, 147–155.
(39) Ma, S. M.; Garcia, D. E.; Redding-Johanson, A. M.; Friedland, G. D.; Chan, R.; Batth, T. S.; Haliburton, J. R.; Chivian, D.; Keasling, J. D.; Petzold, C. J.; et al. Optimization of a heterologous mevalonate pathway through the use of variant HMG-CoA reductases. Metab. Eng. 2011, 13, 588–597.
(40) Koffas, M. A. G.; Jung, G. Y.; Stephanopoulos, G. Engineering metabolism and product formation in Corynebacterium glutamicum by coordinated gene overexpression. Metab. Eng. 2003, 5, 32–41.
(41) Datsenko, K. A.; Wanner, B. L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 6640–6645.
(42) Martínez-García, E.; de Lorenzo, V. Transposon-based and plasmid-based genetic tools for editing genomes of gram-negative bacteria. Methods Mol. Biol. Clifton NJ 2012, 813, 267–283.
(43) Blazeck, J.; Alper, H. S. Promoter engineering: recent advances in controlling transcription at the most fundamental level. Biotechnol. J. 2013, 8, 46–58.
(44) Pfleger, B. F.; Pitera, D. J.; Smolke, C. D.; Keasling, J. D. Combinatorial engineering of intergenic regions in operons tunes expression of multiple genes. Nat. Biotechnol. 2006, 24, 1027–1032.
(45) Hebert, C. G.; Valdes, J. J.; Bentley, W. E. Beyond silencing--engineering applications of RNA interference and antisense technology for altering cellular phenotype. Curr. Opin. Biotechnol. 2008, 19, 500–505.
(46) Salis, H. M.; Mirsky, E. A.; Voigt, C. A. Automated design of synthetic ribosome binding sites to control protein expression. Nat. Biotechnol. 2009, 27, 946–950.
(47) Dueber, J. E.; Wu, G. C.; Malmirchegini, G. R.; Moon, T. S.; Petzold, C. J.; Ullal, A. V.; Prather, K. L. J.; Keasling, J. D. Synthetic protein scaffolds provide modular control over metabolic flux. Nat. Biotechnol. 2009, 27, 753–759.
(48) You, C.; Myung, S.; Zhang, Y.-H. P. Facilitated substrate channeling in a self-assembled trifunctional enzyme complex. Angew. Chem. Int. Ed Engl. 2012, 51, 8787–8790.
(49) Tolia, N. H.; Joshua-Tor, L. Strategies for protein coexpression in Escherichia coli. Nat. Methods 2006, 3, 55–64.
(50) Nawabi, P.; Bauer, S.; Kyrpides, N.; Lykidis, A. Engineering Escherichia coli for biodiesel production utilizing a bacterial fatty acid methyltransferase. Appl. Environ. Microbiol. 2011, 77, 8052–8061.
(51) Zhang, C.; Liu, L.; Teng, L.; Chen, J.; Liu, J.; Li, J.; Du, G.; Chen, J. Metabolic engineering of Escherichia coli BL21 for biosynthesis of heparosan, a bioengineered heparin precursor. Metab. Eng. 2012, 14, 521–527.
(52) Wu, J.; Du, G.; Zhou, J.; Chen, J. Metabolic engineering of Escherichia coli for (2S)-pinocembrin production from glucose by a modular metabolic strategy. Metab. Eng. 2013, 16, 48–55.
(53) Xu, P.; Vansiri, A.; Bhan, N.; Koffas, M. A. G. ePathBrick: a synthetic biology platform for engineering metabolic pathways in E. coli. ACS Synth. Biol. 2012, 1, 256–266.
(54) Silva-Rocha, R.; Martínez-García, E.; Calles, B.; Chavarría, M.; Arce-Rodríguez, A.; de Las Heras, A.; Páez-Espino, A. D.; Durante-Rodríguez, G.; Kim, J.; Nikel, P. I.; et al. The Standard European Vector

References
- 150 -
Architecture (SEVA): a coherent platform for the analysis and deployment of complex prokaryotic phenotypes. Nucleic Acids Res. 2013, 41, D666–D675.
(55) Herrero, M.; de Lorenzo, V.; Timmis, K. N. Transposon vectors containing non-antibiotic resistance selection markers for cloning and stable chromosomal insertion of foreign genes in gram-negative bacteria. J. Bacteriol. 1990, 172, 6557–6567.
(56) Green, M. R. Molecular Cloning: A Laboratory Manual (Fourth Edition): Three-volume set; 4th edition.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, N.Y, 2012.
(57) Boyle, P. M.; Silver, P. A. Parts plus pipes: synthetic biology approaches to metabolic engineering. Metab. Eng. 2012, 14, 223–232.
(58) Glick, B. R. Metabolic load and heterologous gene expression. Biotechnol. Adv. 1995, 13, 247–261. (59) San, K.-Y.; Bennett, G. N.; Berríos-Rivera, S. J.; Vadali, R. V.; Yang, Y.-T.; Horton, E.; Rudolph, F. B.;
Sariyar, B.; Blackwood, K. Metabolic engineering through cofactor manipulation and its effects on metabolic flux redistribution in Escherichia coli. Metab. Eng. 2002, 4, 182–192.
(60) Santos, C. N. S.; Yoshikuni, Y. Engineering complex biological systems in bacteria through recombinase-assisted genome engineering. Nat. Protoc. 2014, 9, 1320–1336.
(61) St-Pierre, F.; Cui, L.; Priest, D. G.; Endy, D.; Dodd, I. B.; Shearwin, K. E. One-step cloning and chromosomal integration of DNA. ACS Synth. Biol. 2013, 2, 537–541.
(62) Choi, K.-H.; Gaynor, J. B.; White, K. G.; Lopez, C.; Bosio, C. M.; Karkhoff-Schweizer, R. R.; Schweizer, H. P. A Tn7-based broad-range bacterial cloning and expression system. Nat. Methods 2005, 2, 443–448.
(63) Loeschcke, A.; Markert, A.; Wilhelm, S.; Wirtz, A.; Rosenau, F.; Jaeger, K.-E.; Drepper, T. TREX: a universal tool for the transfer and expression of biosynthetic pathways in bacteria. ACS Synth. Biol. 2013, 2, 22–33.
(64) Lim, H. N.; Lee, Y.; Hussein, R. Fundamental relationship between operon organization and gene expression. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 10626–10631.
(65) Tyo, K. E. J.; Ajikumar, P. K.; Stephanopoulos, G. Stabilized gene duplication enables long-term selection-free heterologous pathway expression. Nat. Biotechnol. 2009, 27, 760–765.
(66) Nakamura, C. E.; Whited, G. M. Metabolic engineering for the microbial production of 1,3-propanediol. Curr. Opin. Biotechnol. 2003, 14, 454–459.
(67) Hodgman, C. E.; Jewett, M. C. Cell-free synthetic biology: thinking outside the cell. Metab. Eng. 2012, 14, 261–269.
(68) Biggs, B. W.; De Paepe, B.; Santos, C. N. S.; De Mey, M.; Kumaran Ajikumar, P. Multivariate modular metabolic engineering for pathway and strain optimization. Curr. Opin. Biotechnol. 2014, 29C, 156–162.
(69) Ajikumar, P. K.; Xiao, W.-H.; Tyo, K. E. J.; Wang, Y.; Simeon, F.; Leonard, E.; Mucha, O.; Phon, T. H.; Pfeifer, B.; Stephanopoulos, G. Isoprenoid pathway optimization for Taxol precursor overproduction in Escherichia coli. Science 2010, 330, 70–74.
(70) Xu, P.; Gu, Q.; Wang, W.; Wong, L.; Bower, A. G. W.; Collins, C. H.; Koffas, M. A. G. Modular optimization of multi-gene pathways for fatty acids production in E. coli. Nat. Commun. 2013, 4, 1409.
(71) Elowitz, M. B.; Leibler, S. A synthetic oscillatory network of transcriptional regulators. Nature 2000, 403, 335–338.
(72) Gardner, T. S.; Cantor, C. R.; Collins, J. J. Construction of a genetic toggle switch in Escherichia coli. Nature 2000, 403, 339–342.
(73) Vilanova, C.; Porcar, M. iGEM 2.0--refoundations for engineering biology. Nat. Biotechnol. 2014, 32, 420–424.
(74) De Lorenzo, V.; Danchin, A. Synthetic biology: discovering new worlds and new words. EMBO Rep. 2008, 9, 822–827.
(75) Paddon, C. J.; Keasling, J. D. Semi-synthetic artemisinin: a model for the use of synthetic biology in pharmaceutical development. Nat. Rev. Microbiol. 2014, 12, 355–367.
(76) Kosuri, S.; Church, G. M. Large-scale de novo DNA synthesis: technologies and applications. Nat. Methods 2014, 11, 499–507.
(77) Røkke, G.; Korvald, E.; Pahr, J.; Oyås, O.; Lale, R. BioBrick assembly standards and techniques and associated software tools. Methods Mol. Biol. Clifton NJ 2014, 1116, 1–24.
(78) Daniel, R.; Rubens, J. R.; Sarpeshkar, R.; Lu, T. K. Synthetic analog computation in living cells. Nature 2013, 497, 619–623.
(79) An, W.; Chin, J. W. Synthesis of orthogonal transcription-translation networks. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 8477–8482.
(80) Glass, J. I.; Assad-Garcia, N.; Alperovich, N.; Yooseph, S.; Lewis, M. R.; Maruf, M.; Hutchison, C. A.; Smith, H. O.; Venter, J. C. Essential genes of a minimal bacterium. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 425–430.

References
- 151 -
(81) Dymond, J. S.; Richardson, S. M.; Coombes, C. E.; Babatz, T.; Muller, H.; Annaluru, N.; Blake, W. J.; Schwerzmann, J. W.; Dai, J.; Lindstrom, D. L.; et al. Synthetic chromosome arms function in yeast and generate phenotypic diversity by design. Nature 2011, 477, 471–476.
(82) Cheng, J. K.; Alper, H. S. The genome editing toolbox: a spectrum of approaches for targeted modification. Curr. Opin. Biotechnol. 2014, 30C, 87–94.
(83) Nakamura, H.; Shimomura-Shimizu, M.; Karube, I. Development of microbial sensors and their application. Adv. Biochem. Eng. Biotechnol. 2008, 109, 351–394.
(84) Silva-Rocha, R.; de Lorenzo, V. Engineering multicellular logic in bacteria with metabolic wires. ACS Synth. Biol. 2014, 3, 204–209.
(85) Siegel, J. B.; Zanghellini, A.; Lovick, H. M.; Kiss, G.; Lambert, A. R.; St Clair, J. L.; Gallaher, J. L.; Hilvert, D.; Gelb, M. H.; Stoddard, B. L.; et al. Computational design of an enzyme catalyst for a stereoselective bimolecular Diels-Alder reaction. Science 2010, 329, 309–313.
(86) Gibson, D. G.; Young, L.; Chuang, R.-Y.; Venter, J. C.; Hutchison, C. A.; Smith, H. O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345.
(87) Kok, S. de; Stanton, L. H.; Slaby, T.; Durot, M.; Holmes, V. F.; Patel, K. G.; Platt, D.; Shapland, E. B.; Serber, Z.; Dean, J.; et al. Rapid and Reliable DNA Assembly via Ligase Cycling Reaction. ACS Synth. Biol. 2014, 3, 97–106.
(88) Gustafsson, C.; Minshull, J.; Govindarajan, S.; Ness, J.; Villalobos, A.; Welch, M. Engineering genes for predictable protein expression. Protein Expr. Purif. 2012, 83, 37–46.
(89) Freemont, P. S.; Kitney, R. I. Synthetic Biology - A Primer; 1 edition.; World Scientific Publishing: Singapore ; London, 2012.
(90) Wang, K.; Schmied, W. H.; Chin, J. W. Reprogramming the genetic code: from triplet to quadruplet codes. Angew. Chem. Int. Ed Engl. 2012, 51, 2288–2297.
(91) Kim, J.; Copley, S. D. Inhibitory cross-talk upon introduction of a new metabolic pathway into an existing metabolic network. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, E2856–E2864.
(92) Long, C. P.; Antoniewicz, M. R. Metabolic flux analysis of Escherichia coli knockouts: lessons from the Keio collection and future outlook. Curr. Opin. Biotechnol. 2014, 28C, 127–133.
(93) Hold, C.; Panke, S. Towards the engineering of in vitro systems. J. R. Soc. Interface R. Soc. 2009, 6 Suppl 4, S507–S521.
(94) Santacoloma, P. A.; Sin, G.; Gernaey, K. V.; Woodley, J. M. Multienzyme-Catalyzed Processes: Next-Generation Biocatalysis. Org. Process Res. Dev. 2011, 15, 203–212.
(95) Ishii, N.; Suga, Y.; Hagiya, A.; Watanabe, H.; Mori, H.; Yoshino, M.; Tomita, M. Dynamic simulation of an in vitro multi-enzyme system. FEBS Lett. 2007, 581, 413–420.
(96) Van Hecke, W.; Bhagwat, A.; Ludwig, R.; Dewulf, J.; Haltrich, D.; Van Langenhove, H. Kinetic modeling of a bi-enzymatic system for efficient conversion of lactose to lactobionic acid. Biotechnol. Bioeng. 2009, 102, 1475–1482.
(97) Bujara, M.; Schümperli, M.; Billerbeck, S.; Heinemann, M.; Panke, S. Exploiting cell-free systems: Implementation and debugging of a system of biotransformations. Biotechnol. Bioeng. 2010, 106, 376–389.
(98) Martín del Campo, J. S.; Rollin, J.; Myung, S.; Chun, Y.; Chandrayan, S.; Patiño, R.; Adams, M. W. W.; Zhang, Y.-H. P. High-yield production of dihydrogen from xylose by using a synthetic enzyme cascade in a cell-free system. Angew. Chem. Int. Ed Engl. 2013, 52, 4587–4590.
(99) Sheldon, R. A. Cross-linked enzyme aggregates (CLEAs): stable and recyclable biocatalysts. Biochem. Soc. Trans. 2007, 35, 1583–1587.
(100) Scism, R. A.; Bachmann, B. O. Five-component cascade synthesis of nucleotide analogues in an engineered self-immobilized enzyme aggregate. Chembiochem Eur. J. Chem. Biol. 2010, 11, 67–70.
(101) Paddon, C. J.; Westfall, P. J.; Pitera, D. J.; Benjamin, K.; Fisher, K.; McPhee, D.; Leavell, M. D.; Tai, A.; Main, A.; Eng, D.; et al. High-level semi-synthetic production of the potent antimalarial artemisinin. Nature 2013, 496, 528–532.
(102) Bornscheuer, U. T.; Huisman, G. W.; Kazlauskas, R. J.; Lutz, S.; Moore, J. C.; Robins, K. Engineering the third wave of biocatalysis. Nature 2012, 485, 185–194.
(103) Davids, T.; Schmidt, M.; Böttcher, D.; Bornscheuer, U. T. Strategies for the discovery and engineering of enzymes for biocatalysis. Curr. Opin. Chem. Biol. 2013, 17, 215–220.
(104) Damborsky, J.; Brezovsky, J. Computational tools for designing and engineering enzymes. Curr. Opin. Chem. Biol. 2014, 19, 8–16.
(105) Röthlisberger, D.; Khersonsky, O.; Wollacott, A. M.; Jiang, L.; DeChancie, J.; Betker, J.; Gallaher, J. L.; Althoff, E. A.; Zanghellini, A.; Dym, O.; et al. Kemp elimination catalysts by computational enzyme design. Nature 2008, 453, 190–195.
(106) Brakmann, S. Discovery of superior enzymes by directed molecular evolution. Chembiochem Eur. J. Chem. Biol. 2001, 2, 865–871.

References
- 152 -
(107) Neylon, C. Chemical and biochemical strategies for the randomization of protein encoding DNA sequences: library construction methods for directed evolution. Nucleic Acids Res. 2004, 32, 1448–1459.
(108) Cadwell, R. C.; Joyce, G. F. Randomization of genes by PCR mutagenesis. PCR Methods Appl. 1992, 2, 28–33.
(109) Stemmer, W. P. Rapid evolution of a protein in vitro by DNA shuffling. Nature 1994, 370, 389–391. (110) Eriksen, D. T.; Lian, J.; Zhao, H. Protein design for pathway engineering. J. Struct. Biol. 2014, 185, 234–
242. (111) Hidalgo, A.; Schliessmann, A.; Molina, R.; Hermoso, J.; Bornscheuer, U. T. A one-pot, simple
methodology for cassette randomisation and recombination for focused directed evolution. Protein Eng. Des. Sel. PEDS 2008, 21, 567–576.
(112) Chaparro-Riggers, J. F.; Polizzi, K. M.; Bommarius, A. S. Better library design: data-driven protein engineering. Biotechnol. J. 2007, 2, 180–191.
(113) Pavelka, A.; Chovancova, E.; Damborsky, J. HotSpot Wizard: a web server for identification of hot spots in protein engineering. Nucleic Acids Res. 2009, 37, W376–W383.
(114) Jochens, H.; Bornscheuer, U. T. Natural diversity to guide focused directed evolution. Chembiochem Eur. J. Chem. Biol. 2010, 11, 1861–1866.
(115) Reetz, M. T.; Carballeira, J. D. Iterative saturation mutagenesis (ISM) for rapid directed evolution of functional enzymes. Nat. Protoc. 2007, 2, 891–903.
(116) Zhang, K.; Li, H.; Cho, K. M.; Liao, J. C. Expanding metabolism for total biosynthesis of the nonnatural amino acid L-homoalanine. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 6234–6239.
(117) Leonard, E.; Ajikumar, P. K.; Thayer, K.; Xiao, W.-H.; Mo, J. D.; Tidor, B.; Stephanopoulos, G.; Prather, K. L. J. Combining metabolic and protein engineering of a terpenoid biosynthetic pathway for overproduction and selectivity control. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 13654–13659.
(118) Hwang, I. Y.; Tan, M. H.; Koh, E.; Ho, C. L.; Poh, C. L.; Chang, M. W. Reprogramming microbes to be pathogen-seeking killers. ACS Synth. Biol. 2014, 3, 228–237.
(119) Wang, B. L.; Ghaderi, A.; Zhou, H.; Agresti, J.; Weitz, D. A.; Fink, G. R.; Stephanopoulos, G. Microfluidic high-throughput culturing of single cells for selection based on extracellular metabolite production or consumption. Nat. Biotechnol. 2014, 32, 473–478.
(120) Church, G. M.; Elowitz, M. B.; Smolke, C. D.; Voigt, C. A.; Weiss, R. Realizing the potential of synthetic biology. Nat. Rev. Mol. Cell Biol. 2014, 15, 289–294.
(121) Alexander, M. Biodegradation and Bioremediation, Second Edition; 2 edition.; Academic Press: San Diego, 1999.
(122) El Fantroussi, S.; Agathos, S. N. Is bioaugmentation a feasible strategy for pollutant removal and site remediation? Curr. Opin. Microbiol. 2005, 8, 268–275.
(123) Francis, C. W.; Mankin, J. B. High nitrate denitrification in continuous flow-stirred reactors. Water Res. 1977, 11, 289–294.
(124) Lovley, D. R.; Phillips, E. J. P.; Gorby, Y. A.; Landa, E. R. Microbial reduction of uranium. Nature 1991, 350, 413–416.
(125) Stucki, G.; Thueer, M. Experiences of a Large-Scale Application of 1,2-Dichloroethane Degrading Microorganisms for Groundwater Treatment. Environ. Sci. Technol. 1995, 29, 2339–2345.
(126) De Lorenzo, V. Recombinant bacteria for environmental release: what went wrong and what we have learnt from it. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2009, 15 Suppl 1, 63–65.
(127) Ramos, J.-L.; Marqués, S.; van Dillewijn, P.; Espinosa-Urgel, M.; Segura, A.; Duque, E.; Krell, T.; Ramos-González, M.-I.; Bursakov, S.; Roca, A.; et al. Laboratory research aimed at closing the gaps in microbial bioremediation. Trends Biotechnol. 2011, 29, 641–647.
(128) Wittich, R.-M.; Dillewijn, P. van; Ramos*, J.-L. Rational Construction of Bacterial Strains with New/Improved Catabolic Capabilities for the Efficient Breakdown of Environmental Pollutants. In Handbook of Hydrocarbon and Lipid Microbiology; Timmis, K. N., Ed.; Springer Berlin Heidelberg, 2010; pp. 1247–1254.
(129) Kellogg, S. T.; Chatterjee, D. K.; Chakrabarty, A. M. Plasmid-assisted molecular breeding: new technique for enhanced biodegradation of persistent toxic chemicals. Science 1981, 214, 1133–1135.
(130) Devers, M.; Rouard, N.; Martin-Laurent, F. Fitness drift of an atrazine-degrading population under atrazine selection pressure. Environ. Microbiol. 2008, 10, 676–684.
(131) Nakatsu, C. H.; Korona, R.; Lenski, R. E.; de Bruijn, F. J.; Marsh, T. L.; Forney, L. J. Parallel and divergent genotypic evolution in experimental populations of Ralstonia sp. J. Bacteriol. 1998, 180, 4325–4331.
(132) Dai, M.; Copley, S. D. Genome shuffling improves degradation of the anthropogenic pesticide pentachlorophenol by Sphingobium chlorophenolicum ATCC 39723. Appl. Environ. Microbiol. 2004, 70, 2391–2397.

References
- 153 -
(133) De Lorenzo, V.; Timmis, K. N. Analysis and construction of stable phenotypes in gram-negative bacteria with Tn5- and Tn10-derived minitransposons. Methods Enzymol. 1994, 235, 386–405.
(134) Nikel, P. I.; Martínez-García, E.; de Lorenzo, V. Biotechnological domestication of pseudomonads using synthetic biology. Nat. Rev. Microbiol. 2014, 12, 368–379.
(135) Shrivastava, R.; Basu, B.; Godbole, A.; Mathew, M. K.; Apte, S. K.; Phale, P. S. Repression of the glucose-inducible outer-membrane protein OprB during utilization of aromatic compounds and organic acids in Pseudomonas putida CSV86. Microbiol. Read. Engl. 2011, 157, 1531–1540.
(136) Reineke, W.; Knackmuss, H. J. Microbial metabolism of haloaromatics: isolation and properties of a chlorobenzene-degrading bacterium. Appl. Environ. Microbiol. 1984, 47, 395–402.
(137) Brenner, V.; Arensdorf, J. J.; Focht, D. D. Genetic construction of PCB degraders. Biodegradation 1994, 5, 359–377.
(138) Nikel, P. I.; de Lorenzo, V. Implantation of unmarked regulatory and metabolic modules in Gram-negative bacteria with specialised mini-transposon delivery vectors. J. Biotechnol. 2013, 163, 143–154.
(139) Lehrbach, P. R.; Zeyer, J.; Reineke, W.; Knackmuss, H. J.; Timmis, K. N. Enzyme recruitment in vitro: use of cloned genes to extend the range of haloaromatics degraded by Pseudomonas sp. strain B13. J. Bacteriol. 1984, 158, 1025–1032.
(140) Rojo, F.; Pieper, D. H.; Engesser, K. H.; Knackmuss, H. J.; Timmis, K. N. Assemblage of ortho cleavage route for simultaneous degradation of chloro- and methylaromatics. Science 1987, 238, 1395–1398.
(141) Ramos, J. L.; Wasserfallen, A.; Rose, K.; Timmis, K. N. Redesigning metabolic routes: manipulation of TOL plasmid pathway for catabolism of alkylbenzoates. Science 1987, 235, 593–596.
(142) Jiang, J.; Zhang, R.; Li, R.; Gu, J.-D.; Li, S. Simultaneous biodegradation of methyl parathion and carbofuran by a genetically engineered microorganism constructed by mini-Tn5 transposon. Biodegradation 2007, 18, 403–412.
(143) Wittich, R.-M.; Wolff, P. Growth of the genetically engineered strain Cupriavidus necator RW112 with chlorobenzoates and technical chlorobiphenyls. Microbiol. Read. Engl. 2007, 153, 186–195.
(144) Walker, A. W.; Keasling, J. D. Metabolic engineering of Pseudomonas putida for the utilization of parathion as a carbon and energy source. Biotechnol. Bioeng. 2002, 78, 715–721.
(145) Haro, M. A.; de Lorenzo, V. Metabolic engineering of bacteria for environmental applications: construction of Pseudomonas strains for biodegradation of 2-chlorotoluene. J. Biotechnol. 2001, 85, 103–113.
(146) De la Peña Mattozzi, M.; Tehara, S. K.; Hong, T.; Keasling, J. D. Mineralization of paraoxon and its use as a sole C and P source by a rationally designed catabolic pathway in Pseudomonas putida. Appl. Environ. Microbiol. 2006, 72, 6699–6706.
(147) Reichmuth, D. S.; Blanch, H. W.; Keasling, J. D. Dibenzothiophene biodesulfurization pathway improvement using diagnostic GFP fusions. Biotechnol. Bioeng. 2004, 88, 94–99.
(148) Li, G.-Q.; Ma, T.; Li, S.-S.; Li, H.; Liang, F.-L.; Liu, R.-L. Improvement of dibenzothiophene desulfurization activity by removing the gene overlap in the dsz operon. Biosci. Biotechnol. Biochem. 2007, 71, 849–854.
(149) Sohn, S. B.; Kim, T. Y.; Park, J. M.; Lee, S. Y. In silico genome-scale metabolic analysis of Pseudomonas putida KT2440 for polyhydroxyalkanoate synthesis, degradation of aromatics and anaerobic survival. Biotechnol. J. 2010, 5, 739–750.
(150) Kim, J.; Oliveros, J. C.; Nikel, P. I.; de Lorenzo, V.; Silva-Rocha, R. Transcriptomic fingerprinting of Pseudomonas putida under alternative physiological regimes. Environ. Microbiol. Rep. 2013, 5, 883–891.
(151) Shapir, N.; Mongodin, E. F.; Sadowsky, M. J.; Daugherty, S. C.; Nelson, K. E.; Wackett, L. P. Evolution of catabolic pathways: Genomic insights into microbial s-triazine metabolism. J. Bacteriol. 2007, 189, 674–682.
(152) Poblete-Castro, I.; Binger, D.; Rodrigues, A.; Becker, J.; Martins Dos Santos, V. A. P.; Wittmann, C. In-silico-driven metabolic engineering of Pseudomonas putida for enhanced production of poly-hydroxyalkanoates. Metab. Eng. 2013, 15, 113–123.
(153) Izallalen, M.; Mahadevan, R.; Burgard, A.; Postier, B.; Didonato, R.; Sun, J.; Schilling, C. H.; Lovley, D. R. Geobacter sulfurreducens strain engineered for increased rates of respiration. Metab. Eng. 2008, 10, 267–275.
(154) Planson, A.-G.; Carbonell, P.; Paillard, E.; Pollet, N.; Faulon, J.-L. Compound toxicity screening and structure-activity relationship modeling in Escherichia coli. Biotechnol. Bioeng. 2012, 109, 846–850.
(155) Finley, S. D.; Broadbelt, L. J.; Hatzimanikatis, V. In silico feasibility of novel biodegradation pathways for 1,2,4-trichlorobenzene. BMC Syst. Biol. 2010, 4, 7.
(156) Brenner, K.; You, L.; Arnold, F. H. Engineering microbial consortia: a new frontier in synthetic biology. Trends Biotechnol. 2008, 26, 483–489.

References
- 154 -
(157) Gilbert, E. S.; Walker, A. W.; Keasling, J. D. A constructed microbial consortium for biodegradation of the organophosphorus insecticide parathion. Appl. Microbiol. Biotechnol. 2003, 61, 77–81.
(158) Phelan, V. V.; Liu, W.-T.; Pogliano, K.; Dorrestein, P. C. Microbial metabolic exchange--the chemotype-to-phenotype link. Nat. Chem. Biol. 2012, 8, 26–35.
(159) Sinha, J.; Reyes, S. J.; Gallivan, J. P. Reprogramming bacteria to seek and destroy an herbicide. Nat. Chem. Biol. 2010, 6, 464–470.
(160) Aso, Y.; Miyamoto, Y.; Harada, K. M.; Momma, K.; Kawai, S.; Hashimoto, W.; Mikami, B.; Murata, K. Engineered membrane superchannel improves bioremediation potential of dioxin-degrading bacteria. Nat. Biotechnol. 2006, 24, 188–189.
(161) Zhang, H.; Li, Q.; Ye, T.; Zhang, Z.; Li, L. Optimization of the whole-cell catalytic activity of recombinant Escherichia coli cells with surface-immobilized organophosphorus hydrolase. J. Environ. Biol. Acad. Environ. Biol. India 2013, 34, 315–319.
(162) Ba, S.; Arsenault, A.; Hassani, T.; Jones, J. P.; Cabana, H. Laccase immobilization and insolubilization: from fundamentals to applications for the elimination of emerging contaminants in wastewater treatment. Crit. Rev. Biotechnol. 2013, 33, 404–418.
(163) Zhao, H.-P.; Ontiveros-Valencia, A.; Tang, Y.; Kim, B. O.; Ilhan, Z. E.; Krajmalnik-Brown, R.; Rittmann, B. Using a two-stage hydrogen-based membrane biofilm reactor (MBfR) to achieve complete perchlorate reduction in the presence of nitrate and sulfate. Environ. Sci. Technol. 2013, 47, 1565–1572.
(164) Tong, H.-W.; Mutlu, B. R.; Wackett, L. P.; Aksan, A. Manufacturing of bioreactive nanofibers for bioremediation. Biotechnol. Bioeng. 2014, 111, 1483–1493.
(165) Geueke, B.; Garg, N.; Ghosh, S.; Fleischmann, T.; Holliger, C.; Lal, R.; Kohler, H.-P. E. Metabolomics of hexachlorocyclohexane (HCH) transformation: ratio of LinA to LinB determines metabolic fate of HCH isomers. Environ. Microbiol. 2013, 15, 1040–1049.
(166) Khersonsky, O.; Tawfik, D. S. Enzyme promiscuity: a mechanistic and evolutionary perspective. Annu. Rev. Biochem. 2010, 79, 471–505.
(167) Afriat, L.; Roodveldt, C.; Manco, G.; Tawfik, D. S. The latent promiscuity of newly identified microbial lactonases is linked to a recently diverged phosphotriesterase. Biochemistry (Mosc.) 2006, 45, 13677–13686.
(168) Parales, R. E.; Ditty, J. L. Laboratory evolution of catabolic enzymes and pathways. Curr. Opin. Biotechnol. 2005, 16, 315–325.
(169) Iwakiri, R.; Yoshihira, K.; Ngadiman, null; Futagami, T.; Goto, M.; Furukawa, K. Total degradation of pentachloroethane by an engineered Alcaligenes strain expressing a modified camphor monooxygenase and a hybrid dioxygenase. Biosci. Biotechnol. Biochem. 2004, 68, 1353–1356.
(170) Rui, L.; Kwon, Y. M.; Reardon, K. F.; Wood, T. K. Metabolic pathway engineering to enhance aerobic degradation of chlorinated ethenes and to reduce their toxicity by cloning a novel glutathione S-transferase, an evolved toluene o-monooxygenase, and gamma-glutamylcysteine synthetase. Environ. Microbiol. 2004, 6, 491–500.
(171) Lee, J.; Cao, L.; Ow, S. Y.; Barrios-Llerena, M. E.; Chen, W.; Wood, T. K.; Wright, P. C. Proteome changes after metabolic engineering to enhance aerobic mineralization of cis-1,2-dichloroethylene. J. Proteome Res. 2006, 5, 1388–1397.
(172) Strong, L. C.; McTavish, H.; Sadowsky, M. J.; Wackett, L. P. Field-scale remediation of atrazine-contaminated soil using recombinant Escherichia coli expressing atrazine chlorohydrolase. Environ. Microbiol. 2000, 2, 91–98.
(173) De Lorenzo, V. Systems biology approaches to bioremediation. Curr. Opin. Biotechnol. 2008, 19, 579–589.
(174) Liu, X.; Germaine, K. J.; Ryan, D.; Dowling, D. N. Genetically modified Pseudomonas biosensing biodegraders to detect PCB and chlorobenzoate bioavailability and biodegradation in contaminated soils. Bioeng. Bugs 2010, 1, 198–206.
(175) Liss, M.; Daubert, D.; Brunner, K.; Kliche, K.; Hammes, U.; Leiherer, A.; Wagner, R. Embedding permanent watermarks in synthetic genes. PloS One 2012, 7, e42465.
(176) Paul, D.; Pandey, G.; Jain, R. K. Suicidal genetically engineered microorganisms for bioremediation: need and perspectives. BioEssays News Rev. Mol. Cell. Dev. Biol. 2005, 27, 563–573.
(177) Aken, B. V.; Correa, P. A.; Schnoor, J. L. Phytoremediation of polychlorinated biphenyls: new trends and promises. Environ. Sci. Technol. 2010, 44, 2767–2776.
(178) Chakrabarti, A.; Miskovic, L.; Soh, K. C.; Hatzimanikatis, V. Towards kinetic modeling of genome-scale metabolic networks without sacrificing stoichiometric, thermodynamic and physiological constraints. Biotechnol. J. 2013, 8, 1043–1057.
(179) Nikel, P. I.; Silva-Rocha, R.; Benedetti, I.; de Lorenzo, V. The private life of environmental bacteria: pollutant biodegradation at the single cell level. Environ. Microbiol. 2014, 16, 628–642.

References
- 155 -
(180) Carson, R.; Lear, L.; Wilson, E. O. Silent Spring; Anniversary edition.; Houghton Mifflin Company: Boston, 2002.
(181) Oberg, G. The natural chlorine cycle--fitting the scattered pieces. Appl. Microbiol. Biotechnol. 2002, 58, 565–581.
(182) Janssen, D. B.; Dinkla, I. J. T.; Poelarends, G. J.; Terpstra, P. Bacterial degradation of xenobiotic compounds: evolution and distribution of novel enzyme activities. Environ. Microbiol. 2005, 7, 1868–1882.
(183) The Dow Chemical Company. Product Safety Assessment: 1,2,3-Trichloropropane; The Dow Chemical Company, 2008.
(184) Miermans, C. J.; van der Velde, L. E.; Frintrop, P. C. Analysis of volatile organic compounds, using the purge and trap injector coupled to a gas chromatograph/ion-trap mass spectrometer: review of the results in Dutch surface water of the Rhine, Meuse, Northern Delta Area and Westerscheldt, over the period 1992-1997. Chemosphere 2000, 40, 39–48.
(185) Liska, I.; Barceló, D.; Grasserbauer, M. Strategy for the screening of organic pollutants in a river basin — an overview of the Nitra river monitoring programme. TrAC Trends Anal. Chem. 1996, 15, 326–334.
(186) U.S. Environmental Protection Agency. Emerging Contaminant – 1,2,3-Trichloropropane (TCP); U.S. Environmental Protection Agency, 2009.
(187) Yamamoto, K.; Fukushima, M.; Kakutani, N.; Kuroda, K. Volatile organic compounds in urban rivers and their estuaries in Osaka, Japan. Environ. Pollut. Barking Essex 1987 1997, 95, 135–143.
(188) U.S. Environmental Protection Agency. Toxicological Review of 1,2,3-Trichloropropane; CAS No. 96-18-4; U.S. Environmental Protection Agency: Washington, DC, 2009.
(189) La, D. K.; Schoonhoven, R.; Ito, N.; Swenberg, J. A. The effects of exposure route on DNA adduct formation and cellular proliferation by 1,2,3-trichloropropane. Toxicol. Appl. Pharmacol. 1996, 140, 108–114.
(190) Tafazoli, M.; Kirsch-Volders, M. In vitro mutagenicity and genotoxicity study of 1,2-dichloroethylene, 1,1,2-trichloroethane, 1,3-dichloropropane, 1,2,3-trichloropropane and 1,1,3-trichloropropene, using the micronucleus test and the alkaline single cell gel electrophoresis technique (comet assay) in human lymphocytes. Mutat. Res. 1996, 371, 185–202.
(191) Liu, P.; Liang, Y.-G.; Meng, Q.-Y.; Zhang, C.-G.; Wang, H.-C.; Zhang, X.-G.; Li, G.; Liu, Z.-Y.; He, Y.-Z. Successful therapy with hemoperfusion and plasma exchange in acute 1,2,3-trichloropropane poisoning. Hum. Exp. Toxicol. 2012, 31, 523–527.
(192) Tardiff, R. G.; Carson, M. L. Derivation of a reference dose and drinking water equivalent level for 1,2,3-trichloropropane. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2010, 48, 1488–1510.
(193) Pagan, M.; Cooper, W. J.; Joens, J. A. Kinetic studies of the homogeneous abiotic reactions of several chlorinated aliphatic compounds in aqueous solution. Appl. Geochem. 1998, 13, 779–785.
(194) Salter-Blanc, A. J.; Tratnyek, P. G. Effects of solution chemistry on the dechlorination of 1,2,3-trichloropropane by zero-valent zinc. Environ. Sci. Technol. 2011, 45, 4073–4079.
(195) Samin, G.; Janssen, D. B. Transformation and biodegradation of 1,2,3-trichloropropane (TCP). Environ. Sci. Pollut. Res. Int. 2012, 19, 3067–3078.
(196) Tetko, I. V.; Tanchuk, V. Y. Application of associative neural networks for prediction of lipophilicity in ALOGPS 2.1 program. J. Chem. Inf. Comput. Sci. 2002, 42, 1136–1145.
(197) Yan, J.; Rash, B. A.; Rainey, F. A.; Moe, W. M. Isolation of novel bacteria within the Chloroflexi capable of reductive dechlorination of 1,2,3-trichloropropane. Environ. Microbiol. 2009, 11, 833–843.
(198) Bowman, K. S.; Nobre, M. F.; da Costa, M. S.; Rainey, F. A.; Moe, W. M. Dehalogenimonas alkenigignens sp. nov., a chlorinated-alkane-dehalogenating bacterium isolated from groundwater. Int. J. Syst. Evol. Microbiol. 2013, 63, 1492–1498.
(199) Bosma, T.; Janssen, D. B. Conversion of chlorinated propanes by Methylosinus trichosporium OB3b expressing soluble methane monooxygenase. Appl. Microbiol. Biotechnol. 1998, 50, 105–112.
(200) Dolfing, J.; Janssen, D. B. Estimates of Gibbs free energies of formation of chlorinated aliphatic compounds. Biodegradation 1994, 5, 21–28.
(201) Van Hylckama Vlieg, J. E.; Poelarends, G. J.; Mars, A. E.; Janssen, D. B. Detoxification of reactive intermediates during microbial metabolism of halogenated compounds. Curr. Opin. Microbiol. 2000, 3, 257–262.
(202) Nikel, P. I.; Pérez-Pantoja, D.; de Lorenzo, V. Why are chlorinated pollutants so difficult to degrade aerobically? Redox stress limits 1,3-dichloroprop-1-ene metabolism by Pseudomonas pavonaceae. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2013, 368, 20120377.
(203) Crebelli, R.; Andreoli, C.; Carere, A.; Conti, L.; Crochi, B.; Cotta-Ramusino, M.; Benigni, R. Toxicology of halogenated aliphatic hydrocarbons: structural and molecular determinants for the disturbance of chromosome segregation and the induction of lipid peroxidation. Chem. Biol. Interact. 1995, 98, 113–129.

References
- 156 -
(204) Wijngaard, A. J. V. D.; Janssen, D. B.; Witholt, B. Degradation of Epichlorohydrin and Halohydrins by Bacterial Cultures Isolated from Freshwater Sediment. J. Gen. Microbiol. 1989, 135, 2199–2208.
(205) Janssen, D. B. Evolving haloalkane dehalogenases. Curr. Opin. Chem. Biol. 2004, 8, 150–159. (206) Koudelakova, T.; Bidmanova, S.; Dvorak, P.; Pavelka, A.; Chaloupkova, R.; Prokop, Z.; Damborsky, J.
Haloalkane dehalogenases: biotechnological applications. Biotechnol. J. 2013, 8, 32–45. (207) Koudelakova, T.; Chovancova, E.; Brezovsky, J.; Monincova, M.; Fortova, A.; Jarkovsky, J.; Damborsky, J.
Substrate specificity of haloalkane dehalogenases. Biochem. J. 2011, 435, 345–354. (208) Yokota, T.; Omori, T.; Kodama, T. Purification and properties of haloalkane dehalogenase from
Corynebacterium sp. strain m15-3. J. Bacteriol. 1987, 169, 4049–4054. (209) Kulakova, A. N.; Larkin, M. J.; Kulakov, L. A. The plasmid-located haloalkane dehalogenase gene from
Rhodococcus rhodochrous NCIMB 13064. Microbiol. Read. Engl. 1997, 143 ( Pt 1), 109–115. (210) Poelarends, G. J.; Zandstra, M.; Bosma, T.; Kulakov, L. A.; Larkin, M. J.; Marchesi, J. R.; Weightman, A. J.;
Janssen, D. B. Haloalkane-utilizing Rhodococcus strains isolated from geographically distinct locations possess a highly conserved gene cluster encoding haloalkane catabolism. J. Bacteriol. 2000, 182, 2725–2731.
(211) Bosma, T.; Kruizinga, E.; de Bruin, E. J.; Poelarends, G. J.; Janssen, D. B. Utilization of trihalogenated propanes by Agrobacterium radiobacter AD1 through heterologous expression of the haloalkane dehalogenase from Rhodococcus sp. strain M15-3. Appl. Environ. Microbiol. 1999, 65, 4575–4581.
(212) Newman, J.; Peat, T. S.; Richard, R.; Kan, L.; Swanson, P. E.; Affholter, J. A.; Holmes, I. H.; Schindler, J. F.; Unkefer, C. J.; Terwilliger, T. C. Haloalkane dehalogenases: structure of a Rhodococcus enzyme. Biochemistry (Mosc.) 1999, 38, 16105–16114.
(213) Bosma, T.; Damborský, J.; Stucki, G.; Janssen, D. B. Biodegradation of 1,2,3-trichloropropane through directed evolution and heterologous expression of a haloalkane dehalogenase gene. Appl. Environ. Microbiol. 2002, 68, 3582–3587.
(214) Rink, R.; Fennema, M.; Smids, M.; Dehmel, U.; Janssen, D. B. Primary structure and catalytic mechanism of the epoxide hydrolase from Agrobacterium radiobacter AD1. J. Biol. Chem. 1997, 272, 14650–14657.
(215) Nardini, M.; Ridder, I. S.; Rozeboom, H. J.; Kalk, K. H.; Rink, R.; Janssen, D. B.; Dijkstra, B. W. The x-ray structure of epoxide hydrolase from Agrobacterium radiobacter AD1. An enzyme to detoxify harmful epoxides. J. Biol. Chem. 1999, 274, 14579–14586.
(216) Van Hylckama Vlieg, J. E.; Tang, L.; Lutje Spelberg, J. H.; Smilda, T.; Poelarends, G. J.; Bosma, T.; van Merode, A. E.; Fraaije, M. W.; Janssen, D. B. Halohydrin dehalogenases are structurally and mechanistically related to short-chain dehydrogenases/reductases. J. Bacteriol. 2001, 183, 5058–5066.
(217) De Jong, R. M.; Tiesinga, J. J. W.; Rozeboom, H. J.; Kalk, K. H.; Tang, L.; Janssen, D. B.; Dijkstra, B. W. Structure and mechanism of a bacterial haloalcohol dehalogenase: a new variation of the short-chain dehydrogenase/reductase fold without an NAD(P)H binding site. EMBO J. 2003, 22, 4933–4944.
(218) Tang, L.; Lutje Spelberg, J. H.; Fraaije, M. W.; Janssen, D. B. Kinetic mechanism and enantioselectivity of halohydrin dehalogenase from Agrobacterium radiobacter. Biochemistry (Mosc.) 2003, 42, 5378–5386.
(219) Tang, L.; van Hylckama Vlieg, J. E. T.; Lutje Spelberg, J. H.; Fraaije, M. W.; Janssen, D. B. Improved stability of halohydrin dehalogenase from Agrobacterium radiobacter AD1 by replacement of cysteine residues. Enzyme Microb. Technol. 2002, 30, 251–258.
(220) Tang, L.; Torres Pazmiño, D. E.; Fraaije, M. W.; de Jong, R. M.; Dijkstra, B. W.; Janssen, D. B. Improved catalytic properties of halohydrin dehalogenase by modification of the halide-binding site. Biochemistry (Mosc.) 2005, 44, 6609–6618.
(221) Rui, L.; Cao, L.; Chen, W.; Reardon, K. F.; Wood, T. K. Active site engineering of the epoxide hydrolase from Agrobacterium radiobacter AD1 to enhance aerobic mineralization of cis-1,2-dichloroethylene in cells expressing an evolved toluene ortho-monooxygenase. J. Biol. Chem. 2004, 279, 46810–46817.
(222) Rui, L.; Cao, L.; Chen, W.; Reardon, K. F.; Wood, T. K. Protein engineering of epoxide hydrolase from Agrobacterium radiobacter AD1 for enhanced activity and enantioselective production of (R)-1-phenylethane-1,2-diol. Appl. Environ. Microbiol. 2005, 71, 3995–4003.
(223) Fox, R. J.; Davis, S. C.; Mundorff, E. C.; Newman, L. M.; Gavrilovic, V.; Ma, S. K.; Chung, L. M.; Ching, C.; Tam, S.; Muley, S.; et al. Improving catalytic function by ProSAR-driven enzyme evolution. Nat. Biotechnol. 2007, 25, 338–344.
(224) Banás, P.; Otyepka, M.; Jerábek, P.; Petrek, M.; Damborský, J. Mechanism of enhanced conversion of 1,2,3-trichloropropane by mutant haloalkane dehalogenase revealed by molecular modeling. J. Comput. Aided Mol. Des. 2006, 20, 375–383.
(225) Pavlova, M.; Klvana, M.; Prokop, Z.; Chaloupkova, R.; Banas, P.; Otyepka, M.; Wade, R. C.; Tsuda, M.; Nagata, Y.; Damborsky, J. Redesigning dehalogenase access tunnels as a strategy for degrading an anthropogenic substrate. Nat. Chem. Biol. 2009, 5, 727–733.

References
- 157 -
(226) Van Leeuwen, J. G. E.; Wijma, H. J.; Floor, R. J.; van der Laan, J.-M.; Janssen, D. B. Directed evolution strategies for enantiocomplementary haloalkane dehalogenases: from chemical waste to enantiopure building blocks. Chembiochem Eur. J. Chem. Biol. 2012, 13, 137–148.
(227) Arif, M. I.; Samin, G.; van Leeuwen, J. G. E.; Oppentocht, J.; Janssen, D. B. Novel dehalogenase mechanism for 2,3-dichloro-1-propanol utilization in Pseudomonas putida strain MC4. Appl. Environ. Microbiol. 2012, 78, 6128–6136.
(228) Samin, G.; Pavlova, M.; Arif, M. I.; Postema, C. P.; Damborsky, J.; Janssen, D. B. A Pseudomonas putida Strain Genetically Engineered for 1,2,3-Trichloropropane Bioremediation. Appl. Environ. Microbiol. 2014, 80, 5467–5476.
(229) (IRIS), U. E. O. N. I. R. I. S. 1,2,3-Trichloropropane (CASRN 96-18-4) | IRIS | US EPA http://www.epa.gov/iris/subst/0200.htm (accessed Aug 15, 2014).
(230) Sarathy, V.; Salter, A. J.; Nurmi, J. T.; O’Brien Johnson, G.; Johnson, R. L.; Tratnyek, P. G. Degradation of 1,2,3-trichloropropane (TCP): hydrolysis, elimination, and reduction by iron and zinc. Environ. Sci. Technol. 2010, 44, 787–793.
(231) Copley, S. D. Evolution of efficient pathways for degradation of anthropogenic chemicals. Nat. Chem. Biol. 2009, 5, 559–566.
(232) Kurumbang, N. P.; Dvorak, P.; Bendl, J.; Brezovsky, J.; Prokop, Z.; Damborsky, J. Computer-assisted engineering of the synthetic pathway for biodegradation of a toxic persistent pollutant. ACS Synth. Biol. 2014, 3, 172–181.
(233) Cao, L.; van Rantwijk, F.; Sheldon, R. A. Cross-linked enzyme aggregates: a simple and effective method for the immobilization of penicillin acylase. Org. Lett. 2000, 2, 1361–1364.
(234) Immobilization of Enzymes and Cells; Guisan, J. M., Ed.; Methods in BiotechnologyTM; Humana Press: Totowa, NJ, 2006; Vol. 22.
(235) Drobchenko, S. N.; Isaeva-Ivanova, L. S.; Kleiner, A. R.; Lomakin, A. V.; Kolker, A. R.; Noskin, V. A. An investigation of the structure of periodate-oxidised dextran. Carbohydr. Res. 1993, 241, 189–199.
(236) Iwaji Iwasaki, S. U. New Colorimetric Determination of Chloride using Mercuric Thiocyanate and Ferric Ion. Bull. Chem. Soc. Jpn. - BULL CHEM SOC JPN 1952, 25, 226–226.
(237) Jacobs, M. H.; Van den Wijngaard, A. J.; Pentenga, M.; Janssen, D. B. Characterization of the epoxide hydrolase from an epichlorohydrin-degrading Pseudomonas sp. Eur. J. Biochem. FEBS 1991, 202, 1217–1222.
(238) Spelberg, L.; Harald, J. Enantioselective biocatalytic conversions of epoxides, University of Groningen, 2003.
(239) Ba, S.; Arsenault, A.; Hassani, T.; Jones, J. P.; Cabana, H. Laccase immobilization and insolubilization: from fundamentals to applications for the elimination of emerging contaminants in wastewater treatment. Crit. Rev. Biotechnol. 2013, 33, 404–418.
(240) Bidmanova, S.; Damborsky, J.; Prokop, Z. Immobilization of Haloalkane dehalogenase LinB from Sphingobium japonicum UT26 for Biotechnological Applications. J. Biocatal. Biotransformation 2013, 02.
(241) Jurchescu, I.-M.; Hamann, J.; Zhou, X.; Ortmann, T.; Kuenz, A.; Prüße, U.; Lang, S. Enhanced 2,3-butanediol production in fed-batch cultures of free and immobilized Bacillus licheniformis DSM 8785. Appl. Microbiol. Biotechnol. 2013, 97, 6715–6723.
(242) Cárdenas-Fernández, M.; Neto, W.; López, C.; Álvaro, G.; Tufvesson, P.; Woodley, J. M. Immobilization of Escherichia coli containing ω-transaminase activity in LentiKats®. Biotechnol. Prog. 2012, 28, 693–698.
(243) Nunes, M. A. P.; Fernandes, P. C. B.; Ribeiro, M. H. L. High-affinity water-soluble system for efficient naringinase immobilization in polyvinyl alcohol-dimethyl sulfoxide lens-shaped particles. J. Mol. Recognit. JMR 2012, 25, 580–594.
(244) Czichocki, G.; Dautzenberg, H.; Capan, E.; Vorlop, K.-D. New and effective entrapment of polyelectrolyte-enzyme-complexes in LentiKats. Biotechnol. Lett. 2001, 23, 1303–1307.
(245) Sheldon, R. A. Characteristic features and biotechnological applications of cross-linked enzyme aggregates (CLEAs). Appl. Microbiol. Biotechnol. 2011, 92, 467–477.
(246) Wilson, L.; Illanes, A.; Pessela, B. C. C.; Abian, O.; Fernández-Lafuente, R.; Guisán, J. M. Encapsulation of crosslinked penicillin G acylase aggregates in lentikats: evaluation of a novel biocatalyst in organic media. Biotechnol. Bioeng. 2004, 86, 558–562.
(247) Tran, D. N.; Balkus, K. J. Perspective of Recent Progress in Immobilization of Enzymes. ACS Catal. 2011, 1, 956–968.
(248) Bidmanova, S.; Hrdlickova, E.; Jaros, J.; Ilkovics, L.; Hampl, A.; Damborsky, J.; Prokop, Z. Microscopic monitoring provides information on structure and properties during biocatalyst immobilization. Biotechnol. J. 2014, 9, 852–860.

References
- 158 -
(249) Shah, S.; Sharma, A.; Gupta, M. N. Preparation of cross-linked enzyme aggregates by using bovine serum albumin as a proteic feeder. Anal. Biochem. 2006, 351, 207–213.
(250) Schoevaart, R.; Wolbers, M. W.; Golubovic, M.; Ottens, M.; Kieboom, A. P. G.; van Rantwijk, F.; van der Wielen, L. a. M.; Sheldon, R. A. Preparation, optimization, and structures of cross-linked enzyme aggregates (CLEAs). Biotechnol. Bioeng. 2004, 87, 754–762.
(251) Mateo, C.; Palomo, J. M.; van Langen, L. M.; van Rantwijk, F.; Sheldon, R. A. A new, mild cross-linking methodology to prepare cross-linked enzyme aggregates. Biotechnol. Bioeng. 2004, 86, 273–276.
(252) Montoro-García, S.; Gil-Ortiz, F.; Navarro-Fernández, J.; Rubio, V.; García-Carmona, F.; Sánchez-Ferrer, A. Improved cross-linked enzyme aggregates for the production of desacetyl beta-lactam antibiotics intermediates. Bioresour. Technol. 2010, 101, 331–336.
(253) Choi, S. H.; Kim, H. S.; Lee, I. S.; Lee, E. Y. Functional expression and magnetic nanoparticle-based Immobilization of a protein-engineered marine fish epoxide hydrolase of Mugil cephalus for enantioselective hydrolysis of racemic styrene oxide. Biotechnol. Lett. 2010, 32, 1685–1691.
(254) Yildirim, D.; Tükel, S. S.; Alagöz, D.; Alptekin, O. Preparative-scale kinetic resolution of racemic styrene oxide by immobilized epoxide hydrolase. Enzyme Microb. Technol. 2011, 49, 555–559.
(255) Ardao, I.; Zeng, A.-P. In silico evaluation of a complex multi-enzymatic system using one-pot and modular approaches: Application to the high-yield production of hydrogen from a synthetic metabolic pathway. Chem. Eng. Sci. 2013, 87, 183–193.
(256) Schoffelen, S.; van Hest, J. C. M. Chemical approaches for the construction of multi-enzyme reaction systems. Curr. Opin. Struct. Biol. 2013, 23, 613–621.
(257) Findrik, Z.; Vasić-Racki, D. Biotransformation of D-methionine into L-methionine in the cascade of four enzymes. Biotechnol. Bioeng. 2007, 98, 956–967.
(258) Itoh, A.; Ohashi, Y.; Soga, T.; Mori, H.; Nishioka, T.; Tomita, M. Application of capillary electrophoresis-mass spectrometry to synthetic in vitro glycolysis studies. Electrophoresis 2004, 25, 1996–2002.
(259) Lopez-Gallego, F.; Schmidt-Dannert, C. Multi-enzymatic synthesis. Curr. Opin. Chem. Biol. 2010, 14, 174–183.
(260) Billerbeck, S.; Härle, J.; Panke, S. The good of two worlds: increasing complexity in cell-free systems. Curr. Opin. Biotechnol. 2013, 24, 1037–1043.
(261) Bommarius, A. S.; Blum, J. K.; Abrahamson, M. J. Status of protein engineering for biocatalysts: how to design an industrially useful biocatalyst. Curr. Opin. Chem. Biol. 2011, 15, 194–200.
(262) Blackmond, D. G. Reaction progress kinetic analysis: a powerful methodology for mechanistic studies of complex catalytic reactions. Angew. Chem. Int. Ed Engl. 2005, 44, 4302–4320.
(263) Xue, R.; Woodley, J. M. Process technology for multi-enzymatic reaction systems. Bioresour. Technol. 2012, 115, 183–195.
(264) Reetz, M. T.; Carballeira, J. D.; Vogel, A. Iterative saturation mutagenesis on the basis of B factors as a strategy for increasing protein thermostability. Angew. Chem. Int. Ed Engl. 2006, 45, 7745–7751.
(265) Koudelakova, T.; Chaloupkova, R.; Brezovsky, J.; Prokop, Z.; Sebestova, E.; Hesseler, M.; Khabiri, M.; Plevaka, M.; Kulik, D.; Kuta Smatanova, I.; et al. Engineering enzyme stability and resistance to an organic cosolvent by modification of residues in the access tunnel. Angew. Chem. Int. Ed Engl. 2013, 52, 1959–1963.
(266) Lauterbach, L.; Lenz, O.; Vincent, K. A. H₂-driven cofactor regeneration with NAD(P)+-reducing hydrogenases. FEBS J. 2013, 280, 3058–3068.
(267) Bujara, M.; Schümperli, M.; Pellaux, R.; Heinemann, M.; Panke, S. Optimization of a blueprint for in vitro glycolysis by metabolic real-time analysis. Nat. Chem. Biol. 2011, 7, 271–277.
(268) Bhatt, P.; Kumar, M. S.; Mudliar, S.; Chakrabarti, T. Biodegradation of Chlorinated Compounds—A Review. Crit. Rev. Environ. Sci. Technol. 2007, 37, 165–198.
(269) Cámara, B.; Herrera, C.; González, M.; Couve, E.; Hofer, B.; Seeger, M. From PCBs to highly toxic metabolites by the biphenyl pathway. Environ. Microbiol. 2004, 6, 842–850.
(270) Pollmann, K.; Wray, V.; Pieper, D. H. Chloromethylmuconolactones as critical metabolites in the degradation of chloromethylcatechols: recalcitrance of 2-chlorotoluene. J. Bacteriol. 2005, 187, 2332–2340.
(271) Martínez, P.; Agulló, L.; Hernández, M.; Seeger, M. Chlorobenzoate inhibits growth and induces stress proteins in the PCB-degrading bacterium Burkholderia xenovorans LB400. Arch. Microbiol. 2007, 188, 289–297.
(272) Li, R.-D.; Li, Y.-Y.; Lu, L.-Y.; Ren, C.; Li, Y.-X.; Liu, L. An improved kinetic model for the acetone-butanol-ethanol pathway of Clostridium acetobutylicum and model-based perturbation analysis. BMC Syst. Biol. 2011, 5 Suppl 1, S12.
(273) Gray, K. A.; Richardson, T. H.; Kretz, K.; Short, J. M.; Bartnek, F.; Knowles, R.; Kan, L.; Swanson, P. E.; Robertson, D. E. Rapid Evolution of Reversible Denaturation and Elevated Melting Temperature in a Microbial Haloalkane Dehalogenase. Adv. Synth. Catal. 2001, 343, 607–617.

References
- 159 -
(274) Assis, H. M. S.; Sallis, P. J.; Bull, A. T.; Hardman, D. J. Biochemical Characterization of a Haloalcohol Dehalogenase from Arthrobacter erithii H10a. Enzyme Microb. Technol. 1998, 22, 568–574.
(275) Paliy, O.; Gunasekera, T. S. Growth of E. coli BL21 in minimal media with different gluconeogenic carbon sources and salt contents. Appl. Microbiol. Biotechnol. 2007, 73, 1169–1172.
(276) Paalme, T.; Elken, R.; Kahru, A.; Vanatalu, K.; Vilu, R. The growth rate control in Escherichia coli at near to maximum growth rates: the A-stat approach. Antonie Van Leeuwenhoek 1997, 71, 217–230.
(277) Diaz Ricci, J. C.; Hernández, M. E. Plasmid effects on Escherichia coli metabolism. Crit. Rev. Biotechnol. 2000, 20, 79–108.
(278) Ramchuran, S. O.; Holst, O.; Karlsson, E. N. Effect of postinduction nutrient feed composition and use of lactose as inducer during production of thermostable xylanase in Escherichia coli glucose-limited fed-batch cultivations. J. Biosci. Bioeng. 2005, 99, 477–484.
(279) Chavarría, M.; Nikel, P. I.; Pérez-Pantoja, D.; de Lorenzo, V. The Entner-Doudoroff pathway empowers Pseudomonas putida KT2440 with a high tolerance to oxidative stress. Environ. Microbiol. 2013, 15, 1772–1785.
(280) Martínez-García, E.; Jatsenko, T.; Kivisaar, M.; de Lorenzo, V. Freeing Pseudomonas putida KT2440 of its proviral load strengthens endurance to environmental stresses. Environ. Microbiol. 2014.
(281) Martínez-García, E.; Nikel, P. I.; Chavarría, M.; de Lorenzo, V. The metabolic cost of flagellar motion in Pseudomonas putida KT2440. Environ. Microbiol. 2014, 16, 291–303.
(282) Choi, K.-H.; Kumar, A.; Schweizer, H. P. A 10-min method for preparation of highly electrocompetent Pseudomonas aeruginosa cells: application for DNA fragment transfer between chromosomes and plasmid transformation. J. Microbiol. Methods 2006, 64, 391–397.
(283) Chizzolini, F.; Forlin, M.; Cecchi, D.; Mansy, S. S. Gene position more strongly influences cell-free protein expression from operons than T7 transcriptional promoter strength. ACS Synth. Biol. 2014, 3, 363–371.
(284) Nikel, P. I.; Kim, J.; de Lorenzo, V. Metabolic and regulatory rearrangements underlying glycerol metabolism in Pseudomonas putida KT2440. Environ. Microbiol. 2014, 16, 239–254.
(285) Schweizer, H. P.; Po, C. Regulation of glycerol metabolism in Pseudomonas aeruginosa: characterization of the glpR repressor gene. J. Bacteriol. 1996, 178, 5215–5221.
(286) Escapa, I. F.; del Cerro, C.; García, J. L.; Prieto, M. A. The role of GlpR repressor in Pseudomonas putida KT2440 growth and PHA production from glycerol. Environ. Microbiol. 2013, 15, 93–110.
(287) Yang, F.; Hanna, M. A.; Sun, R. Value-added uses for crude glycerol--a byproduct of biodiesel production. Biotechnol. Biofuels 2012, 5, 13.
(288) Wang, H. H.; Isaacs, F. J.; Carr, P. A.; Sun, Z. Z.; Xu, G.; Forest, C. R.; Church, G. M. Programming cells by multiplex genome engineering and accelerated evolution. Nature 2009, 460, 894–898.

Curriculum vitae
- 160 -
CURRICULUM VITAE
Name: Pavel Dvořák
Birth date: August 9, 1983
Place of birth: Kroměříž (Czech Republic)
Nationality: Czech
Affiliation:
Loschmidt Laboratories, Department of Experimental Biology, Faculty of Science, Masaryk
University, Kamenice 5/A13, 625 00 Brno, Czech Republic; phone: +420 602 377 908; FAX:
+420-5-49496302; e-mail: [email protected]
Education
2009 – now: Ph.D. student, Molecular and Cell Biology, Faculty of Science,
Masaryk University
2009: MSc. in Genetics and Molecular Biology, Faculty of Science, Masaryk
University (passed with honours)
2007: Bc. in Genetics and Molecular Biology, Faculty of Science, Masaryk
University (passed with honours)
Employment
2013-now: research assistant, Research Centre for Toxic Compounds in the
Environment RECETOX, Faculty of Science, Masaryk University
2011-now: Ph.D. student, International Clinical Research Center (FNUSA-ICRC),
Brno, Czech Republic
2009-2011: research assistant, Mendel's Centre for Education in Biology,
Biomedicine and Bioinformatics, Department of Biology, Medical Faculty,
Masaryk University
Research interests
biocatalysis; rational design and directed evolution of enzymes; mutagenesis,
screening and selection techniques; synthetic biology and metabolic engineering
of bacteria
Teaching activities
Loschmidt Laboratories Summer School of Protein Engineering; supervisor,
mentor and lecturer (lecture Molecular biology in protein engineering)
Molecular Biotechnology, lectures on Metabolic Engineering (in Czech),
Molecular Biotechnology Practice (in Czech), lecturer

Curriculum vitae
- 161 -
Academic stays
9 – 11/2013: Research stay at the Centro Nacional de Biotecnología, Madrid,
Spain, group of prof. Víctor de Lorenzo (contact person), techniques of genetic
engineering applied for metabolic engineering of bacteria.
12/2010: Research stay at the Wissenschafts Zentrum Straubing, Technische
Universität München (Germany), group of dr. Volker Sieber (contact person dr.
Jan K. Gutterl), robotic screening of mutant libraries generated by directed
evolution of industrially relevant enzymes.
International practical courses
6/2014: International Synthetic and Systems Biology Summer School, Taormina,
Sicily, Italy
11/2012: Advanced Course Metabolomics for Microbial Systems Biology, BSDL-
EDU, TU Delft, Delft, Netherlands
9/2010: Protein Engineering - Rational Design & Directed Evolution, Dechema
Summer School, Institute of Biochemistry, Greifswald University, Germany
Awards
2014: Award of the Dean of the Faculty of Science, Masaryk University for
exemplary representation of the faculty in abroad.
2014: The Sigma-Aldrich award of Gerty T. and Carl. F. Cori for the best young
scientist presentation, Interdisciplinary Meeting of Young Biologists, Biochemists,
and Chemists, Milovy, Czech Republic
2010: Award of the Dean of the Faculty of Science, Masaryk University for
organisation of The Student Scientific Conference on GMO Research
2009: Award of the Dean of the Faculty of Science, Masaryk University for
excellent students of master studies
2008: Award of the Czech Society for Biochemistry and Molecular Biology for the
best presentation at XII. Meeting of Biochemists and Molecular Biologists, Section
of Young Investigators, Brno, Czech Republic
Research projects
2010-2013: Specific Research, Category b) Support of specific research projects
focused on organization of student scientific conferences. Co-founder of student
club Biomania and organizer of The International Student Scientific Conference
on Biotechnology and Biomedicine (www.biomania.cz/conference-2013)
2008-2009: Masaryk University Rector's Program for Support of Creative
Activities of Students. Part A, Support of Excellent Diploma Theses. Semi-rational
design and construction of haloalkane dehalogenases for biotechnological
applications; 20081431A0006; principal investigator.

List of publications
- 162 -
LIST OF PUBLICATIONS
Dvorak, P., Bidmanova, S., Prokop, Z., Damborsky, J., 2014: Immobilized Synthetic
Pathway for Biodegradation of Toxic Recalcitrant Pollutant 1,2,3-Trichloropropane.
Environmental Science and Technology. 48, 6859-6866. (ACS Editors' Choice May 24 2014)
Dvorak, P., Kurumbang, N.P., Bendl, J., Brezovsky, J., Prokop, Z., Damborsky, J., 2014:
Maximizing the Efficiency of Multi-enzyme Processes by Stoichiometry Optimization.
ChemBioChem, DOI: 10.1002/cbic.201402265.
Kurumbang, N.P.*, Dvorak, P.*, Bendl, J., Brezovsky, J., Prokop, Z., Damborsky, J., 2014:
Computer-Assisted Engineering of the Synthetic Pathway for Biodegradation of a Toxic
Persistent Pollutant. ACS Synthetic Biology. 3: 172-181. (*shared first author)
Koudelakova, T., Bidmanova, T., Dvorak, P., Pavelka, A., Chaloupkova, R., Prokop, Z.,
Damborsky, J., 2013: Haloalkane Dehalogenases: Biotechnological Applications.
Biotechnology Journal. 8: 32-45.
Klvana, M., Pavlova, M., Koudelakova, T., Chaloupkova, R., Dvorak, P., Prokop, Z.,
Stsiapanava, A., Kuty, M., Kuta-Smatanova, I., Dohnalek, J., Kulhanek, P., Wade, R.C.,
Damborsky, J., 2009: Pathways and Mechanisms for Product Release in the Engineered
Haloalkane Dehalogenases Explored using Classical and Random Acceleration Molecular
Dynamics Simulations. Journal of Molecular Biology. 392(5): 1339-1356.

List of contributions at conferences and symposia
- 163 -
LIST OF CONTRIBUTIONS AT CONFERENCES AND SYMPOSIA
Dvorak, P., Kurumbang, N.P., Bendl, J., Brezovsky, J., Prokop, Z., Damborsky, J., Computer-
Assisted Engineering of Synthetic Pathway for Biodegradation of Anthropogenic Pollutant
Under In Vitro and In Vivo Conditions. International Synthetic and Systems Biology Summer
School, 15.-19.6.2014. Taormina, Italy (poster)
Dvorak, P., Kurumbang, N.P., Bendl, J., Brezovsky, J., Prokop, Z., Damborsky, J.,
Engineering of Metabolic Pathway for Biodegradation of Anthropogenic Pollutant Using
Methods of Synthetic Biology (in Czech). Interdisciplinary Meeting of Young Biologists,
Biochemists, and Chemists, 13.-16.5.2014. Milovy, Czech Republic (oral presentation)
Dvorak, P., Kurumbang, N.P., Bendl, J., Brezovsky, J., Prokop, Z., Damborsky, J., In Vitro and
In Silico Engineering of Multi-enzyme Reactions. Biotrans, 21.-25.7.2013. Manchester, UK
(oral presentation)
Dvorak, P., Kurumbang, N.P., Bendl, J., Brezovsky, J., Prokop, Z., Damborsky, J., Rational
Engineering of Synthetic Biodegradation Pathway. Symposium Consistent Bioprocess
Development, 1.3.2013. Technische Universität Berlin, Germany (invited lecture)
Dvorak, P., Prokop, Z., Bednar, D., Brezovsky, J., Bidmanova, S., Damborsky, J., In Vitro
Protein and Metabolic Engineering of Biodegradation Pathway. Biotrans, 2.-6.10.2011,
Giardini Naxos, Italy (oral presentation)
Dvorak, P., Pavlova, M., Klvana, M., Brezovsky, J., Chaloupkova, R., Volfova, I., Damborsky,
J., Increasing Activity of Bacterial Biocatalyst with Toxic Anthropogenic Substrate Using
Methods of Protein Engineering. 25th International Symposium on Microscale
Bioseparations, 21.3.-25.3.2010, Prague, Czech Republic (poster)
Dvorak, P., Pavlova, M., Klvana, M., Brezovsky, J., Chaloupkova, R., Prokop, Z., Damborsky,
J., Increasing the Activity of Haloalkane Dehalogenase DhaA with Anthropogenic Substrate
1,2-Dichloroethane Using Methods of Focused Directed Evolution. XX. Enzyme Egineering,
20.-24.9.2009, Gröningen, Netherlands (poster)
Dvorak, P., Pavlova, M., Klvana, M., Chaloupkova, R., Prokop, Z., Nagata, Y., Uhlirova, R.,
Brezovsky, J., Damborsky, J., Semi-rational Engineering of Haloalkane Dehalogenase DhaA
Towards Improved Activity with 1,2-Dichloroethane. XII. Meeting of Biochemists and
Molecular Biologists, Section of Young Investigators, 6.-7.2.2008, Brno, Czech Republic (oral
presentation)

- 164 -

- 165 -




















