
Caractérisation d'un calorimètre hadronique semi-digital pour ...
Upload
khangminh22Category
view
0download
0
DENIS GODIN
CARACTÉRISATION DES RÉCEPTEURS AcTIVÉS PAR PROTÉOLYSE DANS LES VAISSEAUX SANGUINS
Thèse présentée
à la Faculté des études supérieures de l'Université Laval
pour l'obtention du grade de Philosophiae Doctor (Ph-D.)
Département de pharmacologie FACULTÉ DE MÉDECINE
UNIVERSITÉ LAVAL QLJÉBEC
OCTOBRE 1997
O Denis Godin, 1997

National tibrary 1*1 of Canada Bibliothèque nationale du Canada
Acquisitions and Acquisitions et Bibliogaphic Services services bibliographiques 395 Wellington Street 395, nie Wellingtori Ottawa ON K1A O N 4 Ottawa ON K1A ON4 Canada Canada
The author has granted a non- exclusive licence allowing the National Library of Canada to reproduce, luan, distribute or sell copies of this thesis in microform, paper or electronic formats.
L'auteur a accordé une licence non exclusive permettant à la Bibliothèque nationale du Canada de reproduire, prêter, distnibuer ou vendre des copies de cette thèse sous la fome de microfiche/film, de reproduction sur papier ou sur format électronique.
The author retains ownership of the L'auteur conserve la propriété du copyright in this thesis. Neither the droit d'auteur qui protège cette thèse. thesis nor substantial extracts fiom it Ni la thèse ni des extraits substantiels may be printed or otherwise de celle-ci ne doivent être imprimés reproduced without the author's ou autrement reproduits sans son permission. autorisation.

Les récepteurs de la thrombine et PAR-2 forment la nouvelle famille des récepteurs
activés par protéolyse. De courts peptides correspondants à la nouvelle extrémité Na
temiinale suivant le clivage de chacun des récepteurs sont capables de reproduire la
plupart des effets médiés par ceux-ci. Nous avons effectué des études de structure-activité
dans le but d'obtenir des agonistes de bonne afnnité et résistants au métabolisme pouvant
être ensuite utilisés dans les essais biologiques. A l'aide d'une série d'inhibiteurs nous
avons comparé les voies de transduction UaraceUulaires impliquées dans les réponses
contractiles et mitogènes à la thrombine avec ceIles impliquées dans les réponses aux
peptides agonistes. Nous avons observé que l'effet contractile de la thrombine sur l'aorte
de laph est médié par la protéine kinase C (PKC) alors que la réponse au peptide agoniste
dépend en plus du calcium extracellulaire. Dans les cellules de muscle lisse en culture
(CML) la thrombine et les peptides agonistes augmentent la production de
phosphatidyiinositols mais seule la thrombine possède un effet rnitogene qui semble médié
par un signal tyrosine kinase. Dans le même modèle de l'aorte de lapin nous avons montré
que la relaxation a la trypsine et à SLIGRL-NHZ, impliquant le récepteur PAR-2, est
médiée par la production d'oxyde nitrique et constitue un essai spécifique pour ce
récepteur puisque ni la thrombine ni les agonistes synthétiques correspondants ne sont
capables d'induire une réponse relaxante dans ce rnodèk. Finalement nous montrons la
présence du récepteur PAR-2 dans les tissus vasculaires par hybridation Northern,
prouvant l'existence d'un récepteur distinct du récepteur de la thrombine.

RÉSUMÉ LONG
La découverte du récepteur de la thrombine a permis l'identification d'un
nouveau mécanisme d'activation des récepteurs couplés aux protéines G,
l'activation par protéolyse. Depuis, un deuxième récepteur activable par ce
mécanisme a été identifié, le récepteur PAR-2 (a second proteinase-activated
receptor »). De courts peptides, correspondants à la nouvelle extrémité N-
t e e a l e suivant le clivage du récepteur, sont capables de reproduire la plupart
des activités des protéases dans plusieurs modèles. Nous avons effectué des études
de structure-activité sur les peptides agonistes de chacun des récepteurs afin de
générer des agonistes de meilleure dfhité pour utilisation dans les différents
modèles biologiques. Nous avons caractérisé les voies de transduction
intracellulaires impliquées dans les réponses à la thrombine et aux agonistes
synthétiques dans le modèle de l'aorte de lapin isolée ainsi que dans les cellules de
muscle lisse en culture dérivées du même tissu. Nous avons observe que l'effet
contractile de la thrombine sur l'aorte de lapin est médié par la protéine kinase C
(PKC) alors que la réponse au peptide agoniste dépend du calcium extracellulaire
en plus du signal induit par cette kinase. Dans les .cellules de muscle lisse en
culture (Cm) la thrombine et les peptides agonistes augmentent la production de
ph~sphatidylinositols mais seule la dirombine possède un effet mitogene qui
semble médié par un signal tyrosine kinase. Dans le même modèle de l'aorte de
lapin nous avons montré que la relaxation à la trypsine et à SLIGRL-NH2,
impliquant le récepteur PAR-2, est médiée par la production d'oxyde nitrique et
constitue un essai spécifique pou. ce récepteur puisque ni la thrombine ni les
agonistes synthétiques correspondants ne sont capables d'induire une réponse
relaxante dans ce modèle. Finalement nous montrons la présence du récepteur
PAR-2 dans les tissus vasculaires par hybridation Northem, prouvant l'existence

d'un récepteur distinct du récepteur de fa thrombine. L'ensemble de ces résultats
suggère l'existence d'une sous-famille de récepteurs couplés aux protéines G : les
récepteurs activés par protéolyse. Leurs effets importants sur les différents types
de cellules vasculaires laissent présager que ceux-ci pourraient être appelés à jouer
un rôle physiologique primordid, entre-autres au niveau du maintien du tonus
vasculaire.

AVANT-PROPOS
En premier lieu je tiens à remercier le Dr. Guy Drapeau, mon directeur de
recherche, pour sa grande disponibilité et son soutien inconditionnel. Par le partage de ses
comaissances et plusieurs discussions enrichissantes il m'a permis de développer mon
autonomie en recherche et d'acquérir un bon jugement critique.
Je voudrais remercier également les Drs. Francis Rioux, François Marceau et
Dimtcho Batchvarov pour leur contribution à ma formation de chercheur par leur
enseignement et leurs précieux conseils.
Merci également à Nathalie Harvey et Johanne Boutillier pour leur aide très
précieuse dans le laboratoire, sans oublier les étudiants d'été qui ont égaiement joué un
rôle important à un moment ou l'autre de ce projet : Caroiine Beaulé, Geneviève
Lavergne, Julie Lemieux et Dominique Gauvin. Un merci tout spécial à tout le personnel
du Centre de recherche de l'Hôtel-Dieu de Québec, particulièrement à tous ceux et celles
qui ont généreusement donné de leur sang dans le cadre de ce projet.
Je suis reconnaissant envers la Fondation des maladies du coeur du Canada et le
Fond de la recherche en santé du Québec pour l'octroi de bourses de recherche dans le
cadre de mes études de doctorat.
Fuiaiement je témoigne toute ma gratitude à mon épouse Patncia ainsi qu'à toute
ma f W e pour leur support inconditionnel tout au long de mon cheminement.
Cette thèse est dédiée à Patricia et à ma f d e en signe de ma profonde
reconnaissance.

TABLE DES MATIÈRES
...................................... ..... .... RÉSUMÉ COURT ...W.,......... ..................... .......................... n
..................................................................................................................... RESUME LONG ............m
......................................................................................................... AVANT-PROPOS ................-.o.. V
LISTE DES FIGURES ...................................................................... .......~...................................... X
1.1 LES DIFF~RENTES C A T & ~ ~ E S DE ~CEPTEURS ............................................................................ 2
1.2 LA THROMBiNE : STRUCTURE ET FORMATiON ............................................... ,.. ........ 3
1.3 WCEPTEUR CLIVABLE DE LA THROMBINE ..................................................................................... 5
... ......*..-............ .........-................................................................................. 1.3.1 Structure ,.,, ,, 5
1.3.2 Mode d 'activation ....................~.~..................................................................................... 6
1.3.3 Peptides agonistes du récepteur de la thrombine .................................................................. 8
1.3 EFFETS DE TYPE HORMONAL DE LA THROMBINE ............................................................................ 9
1.5 RECEPTEWR PAR-2 ..................................................................................................................... 1 1
...*......... ......*.....*.............................*.......................*.............. 1.5.1 Clonage et expression.. ,. I I
1.5.2 Mode d 'activution et agonistes peptidiques ........................................................................ I I
.................................................................. 1.6 OEUECTIFS DE L ' ~ E .,................. ..,, ....,... ... 12
....................................................................................................... 2.0 MATEIUEL ET METHODES 15
2.1 SYNTHESE ET PURIFICATION DES PEPTIDES ................................................................................ 15
2.1.1 Synthèse ............................................................................................................................. 15
2.1.2 Purification ........................................................................................................................ 20
2.2 ANALYSE PAR HPLC ............................... ,., ........................................................................... 2 0
.......*....... ...........*............ 2.3 S T A B L ~ ~~XABOLIQUE DE N A T ~ N H ~ ET DE [SAR']NAT& .. 22
7 7 .................................................................................................................. 2.3.1 Plasma de lapin , ,

7 3 ........................................................................................ 2.3.2 Mesure de 1 'activité enzymatique ,,
2.4 MODÈLE DE L'AORTE DE LAPIN ISOLEE ....................................................................................... 23
2.5 PROT~COLES EXPÉEUMENTAUX ....................~............................................................................. 24
2.5.1 Eflet contractile de fa thrombine. de la trypsine et des agonistes des récepteurs de la
thrombine et PM-2.. ........................ ,., ...,., .............................................. 24
2-5-2 Relaxation de NATrNH2 et de [sur' J N A T ~ N H ~ dms l'huile ................... ... ............... 24
2.5.3 Mode d'action de la thrombine et de [s~#]NA T& . ... ..............~..................................... 25
2.5.4 E f i t des inhibiteurs me faboliques sur la régulation du récepteur de la thrombine ............. 26
2.5.5 Eflet relaxant de la trypsine et des agonistes PAR.2 .............. .... ..................................... 26 2.6 CELLüLES EN CULTURE DE MUSCLE LiSSE DÉRM~ES DE L'AORTE DE LAPIN (ch&) ........................ 27
............ ..........-.-......*........................................................................ 2.6.1 Mise en culture. ,,.. 27
2.6.2 Mesure de la production des phosphatidylinositois ............................................................. 28
2.6.3 Incorporation de la f~]fhyrnidine .................................... .... ............................................ 29
........................ 2.7 D~VELOPPEMENT D'UN ESSM DE LINSON POUR LE R~CEPTEUR DE LA THROMBINE 30 25 2.7. I Radiomarquage de la thrombine à l'iode radioactive ( 1) ............................................ 30
....................... 2.7.2 Essai de liaison pour le récepteur de lu thrombine et analyse de Scatchurd 31
2.8 EVALUATXON DES EFFFXS iN Wl7 DE NATeNfi ET DE [SAR']NAT@IHZ CHEZ LE LAPIN ANXWI%IE
32
................................ 2.9 n>ENTFiCATION DES ARN MESSAGERS CODANT POUR LE &CEPTEUR PAR-2 33
29.1 Prépara tton de la sonde PAR2 .......................................................................................... 33
39.1. I Isolation du fragment (sonde) par PCR ........................ .. .............................................. 33
2.9.2 Préparation de 1 'ARN messager de 1 'aorte et du coeur de lapin ................... .., ....,............ . 36
2- 9.3 Gel d'&V et tra&rt sur membrane de nylon .......................~........................................... 37
2- 9.4 Hybridation avec la sonde PM-2 ................................~...................................................... 37
2.10 ANALYSESTAT~STIQUE ................................ .......................................................................... 38
3.1 ÉTUDE DE STRUCTURE-A- DES PEPTIDES AWNISTES ET ~ ~ A G O M S T E S DU &CEPTEUR DE t~
THROMBiNE ........... .. ......................................................................................................................... 39
3 -2 MODE D'ACTION DE U ~ ~ O N I N E ET DES PEPT~DES NAT CHEZ t~ LAPIN ..................................... 46
3.2.1 Etudes de contractilité .......... . . ...... ..... ......................................................................... 46
3.2.2 Cellules de muscle lisse de l'aorte de lapin en culture ....................................................... SI
3.2.3 Eget des peptides NA T in vivo chez le lapin unesthésié ................................ ..,,,. ............. 57
3.3 EFFETS DES PEPTDES AGONISTES DU RÉCEPTEUR PAR-2 SUR L'AORTE DE LAPIN I S O ~ E ................ 60
3.4 ÉTUDE DE S T R U C T I J R E - A ~ ~ ~ ~ ~ ~ ~ ~ : DES P E m E S A0IUISTE.S DU RÉCEPTEUR PAR-2 ........................ 64
3.5 D~TECTION DE L'ARN MESSAGER CODANT POUR LE &CEPTEUR PAR-2 DANS L'AORTE DE LAPIN .... 67

4.1 STRUCTURE-ACTIVITÉ ET STJ~B& MÉTABOUQUE DES AGONISTES Eï ANTAWMSTES DU
RE~EPTEUR DE LA THROMBiNE ..................... ,..,. ..................... ,,. 69
4.2 ÉTUDE DU MODE D'AC~ON ET DE Ln D~ENSIBIWSATION DU &cEPTEUR DE LA THROMB IM ......... 73 .................. 4.2. J Modèle de l'aorte de lupin isolée ,, .....................................~............................. 73
............................... ......................... 4 2.2 Modèle de ceZitdes de muscle lisse en miiure ....,., 77
4.3 EFFETS Af VNO DES AGONISTES PEPïïûIQUES DU RÉCEPTEUR DE iA T?~ROMBINE ........................... 81
4.4 ÉTUDES iN VITRO DES EFFETs INDUITS PAR LE RÉCEFTEUR PAR-2 .................................................. 83
........................................................................................ APPENDICE A : CURRICULUM W A E 105
....... . APPENDICE B : ARTICLES SCIENTIFIQUES PWLIÉS .. .......... , ...,,..o....o..o.o............... 110

LISTE DES TABLEAUX
Tableau 1 Liste des peptides synthétisés pour les fins de l'étude
Tableau 2 Étude de structure-activité des peptides agonistes et antagonistes du récepteur de la thrombine
Tableau 3 Hydrolyse de m-M& et de [S~~'I'NAT~-NJ& par l'arninopeptidase M plasmatique
Tableau 4 Mode d'action de la thrombine et de [s~~']NAT&~& sur l'aorte de lapin isolée
Tableau 5 Production de phosphatidyhositols par les CML
Tableau 6 Étude de structure-activité des agonistes du récepteur PAR-2.

LISTE DES FIGURES
Figure 1 Facteurs impliqués dans la cascade de la coagulation
Figure 2 Modèle d'activation du récepteur de la thrombine
Figure 3 Effets de la thrombine sur différents types cellulaires
Figure 4 Synthèse en phase solide du peptide N A T m 2
Figure 5 Système HPLC et paramètres utilisés
Figure 6 Activité de la thrombine et des peptides agonistes sur l'aorte de lapin isolée
Figure 7 Courbes de relaxation dans l'huile de [S~~' ]NAT~-W et de NAT6-=
Figure 8 Effet de H-7 sur les courbes concentration-réponse à la thrombine et au [~ar'] NAT6-NH2 sur l'aorte de lapin isolée
Figure 9 Effet de la bréfeldine 4 de I'anisomycine et de la cyclohexhide sur l'activité contractile de la thrombine
Figure 10 Liaison de la 'u~-thrombine sur les CML de lapin
Figure 1 1 Liaison de la 125~-thrombine sur les CML de lapin, analyse de compétition
Figure 12 Effet de la thrombine et des peptides agonistes sur l'incorporation de la [3~thymidine dans les CML de lapin
Figure 13 Effet des inhibiteurs sur l'incorporation de la [3~]thyrnïdine induite par la thrombine chez les CML de lapin
Figure 14 Réponses hypotensives induites par l'injection I.V. de NAT6-= et de [sari] NAT6-NH2 chez le lapin anesthésié

Figure 15 Tracés des réponses hypotensives chez le lapin anesthésié
Figure 16 Tracés représentatifk de relaxation de l'aorte de lapin isolée suite à la stimulation par les agonistes des récepteurs de la thrombine et PAR-2 62
Figure 17 Effét contractile de la trypsine et de SLIGRL-NH2 sur I'aorte de lapin isolée
Figure 18 Effet de ARGGN02 sur la relaxation induite par la trypsine et SLIGRL-NH2 dans l'aorte de lapin isolée 65
Figure 19 Hybridation Northern pour le récepteur PAR-2 68
Figure 20 Modèle de la cinétique de ré-expression des récepteurs de la thrombine 78
Figure 2 1 Modèle de cascade des signaux de transduction activés suite à la stimulation du récepteur de la thrombine 82

XII
LISTE DES ABBREVIATIONS
Ac: acétyl Aca: acide aminocap roique ADN: acide désoxyribonucléique Ala (A): alanine ANOVA: analyse de variance Arg (R): arginine Arg-NOz: na-nitro-L-arginine ARN: acide ribonucléique Asn (N): asparagine Asp @): acide aspartique B-: capacité maximale de liaison Boc: tert-butyloxycarbonyl BOP: benzo t riazole- 1 -y l-oxy-tri~~(diméthy1amino)p hosphonim hexafluorop hosp hate BSA: albumine sérique bovine But: butyl CaC12: chlorure de calcium Ci: Curie Cha: cyclohexylalanine CH2Cl: chlorure de méthylène CML: cellules de musde lisse CO2: dioxyde de carbone CPM: coups par minute Cys (C): cystéine DAG: diacylglycérol d ATP : desoxyadénosine triphosphate dCTP : désoxyqtosine triphosphate dGTP : désoxyguanosine triphosphate DMSO: dirnéthyl suIfoxyde dTTP: désoxythymidine triphosphate ED: dose effectrice EDT A: ethy lènediaminetetraacét ate EGF: facteur de croissance des cellules endothéliaies Erb : erbstatine EtOH: éthanol FMOC : 9-fluorénylmét hyloxycarbonyl Glu (E): glutamine Gly (G): glycine HCI: acide chioridrique HEPES : N-[2- hydroxyéthyl] pipérazineeN y -[2-acide éthanesulfonique] Hg: mercure His (H): histidine

HPLC: chromatographie Liquide à haute presion (Hi& Pressure Liquid Chromatography) 1 iode IP : inositolp hosphate IPTG: isopropyl 0-D-thiogalactopyranoside 1 V. : intra-veineux KCI: chlorure de potassium Ko: constante de dissociation à l'équilibre du complexe ligand-récepteur KH~POI: phosphate de potassium monobasique KI: iodure de potassium KOH: hydrowde de potassium Leu (L): leucine LiC1: chlorure de lithium Lys (K): lysine MAP : pression artérielle moyenne MAPK: protéine kinase activée par la mitogénèse MBHA: 4-mét hylbenzhydry lamine MePhe: N-méthyl-phénylalanine MgCl*: chlorure de magnésium MgS04: sulfate de magnésium MOPS : acide 3-EN-morpholino]propane sulfonique NaC1: chlorure de sodium Na2C03: carbonate de sodium NaHC03: bicarbonate de sodium NaOAc: acétate de sodium NaOH: hydroxyde de sodium Na2S04: sulfate de sodium NAT: nouvelle extrémité amino-terminale (New Amino Terminus) NS : non-significatif 02: oxygène OBu' : O-butyl P: phosphore PAR-2: second récepteur activé par protéolyse (Proteolyse-activated Receptor-2) pb: paire de bases PB S : tampon phosphate/saline (Phosphate Buffered Saline) PCR: amplification en chaîne de la polymérase (Polymerase Chaine R e d o n ) PDGF: facteur de croissance dérivé des plaquettes PE: phényléphe PGE2: prostaglandine Ez PG12: prostaglandine b Phe (F): phénylalanine PIP : phosphatidylinositol PKC: protéine kinase C PLC: phospholipase C PMA: phorbol myrktate acétate PMC : 2,2,5,7,8-pentaméthykhroman-6-sulfonyl

PPACK: D-Phe-Pro-Arg-CH2CI Pro (P): prohe RPM: rotations par minute RT: récepteur de la thrombine Sar : saxo sine SDS: sodium dodécyl sulfate Ser (S): sérine SSBE: solution saline balancée de Earle SSC: solution saline-citrate SVF: s h m de veau foetal TBE: tampon TrisiBorateEDTA TFA: acide trifiuoroacétique TFMSA: acide trifluoromQhanesuIfonique T h . (T) : thrémine TK: tyrosine kinase tPA: facteur tissulaire d'activation du plasminogène Tris: tris~ydroxym&hyl)aminoéthane Trp (W): tryptophane trt: trityl Tyr (Y): tyrosine UV: ultra-vioIet Vai (V): vahe X-Gai: 5-bromo-4-chloro-3-indoly1-B-~-gda~topyraside


1 .O INTRODUCTION
1 . LES DIFFÉRENTES CATEGORIES DE RECEPTEURS
Les récepteurs membranaires peuvent être regroupés principalement en trois
catégories, dépendamment des mécanismes de transduction utilisés. Les récepteurs
liés aux canaux ioniques forment la première catégone. Leur activite est contrôlée
par des neurotransmetteurs. Ils sont impliqués majoritairement dans la
transmission synaptique rapide entre deux cellules électriquement excitables. Ce
type de signal est médié par une petite quantité de neurotransmetteurs qui ouvrent
ou ferment le canal ionique auquel ils se lient de façon transitoire. La perméabilité
ionique de la membrane cytoplasmique est modifiée provoquant un changement de
l'excitabilité de la cellule postsynaptique (Bemdge, 1985).
La deuxième catégorie est composée des récepteurs catalytiques. Lorsqu'ils
sont activés par leur ligand, ces récepteurs agissent directement comme une
enzyme. La quasi totalité de cette famille de récepteurs sont des protéines
transmembramikes possédant un domaine cytoplasmique ayant des propriétés
tyrosine kinase (Bemdge, 1985).
Les récepteurs couplés aux protéines G forment la troisième catégone. Ces
récepteurs activent ou inactivent indirectement un canal ionique ou une enzyme
liée à la membrane cytoplasmique. Les interactions entre le récepteur et le canal
ionique ou l'enzyme s'effectuent par l'entremise d'une troisième protéine, appelée
«protéine régulatrice liant le GTP » ou protéine G. Ces récepteurs liés aux
protéines G activent généralement une chaîne d'événements qui altère la

concentration d'une ou plusieurs molécules de signalisation intraceUulaire,
courm.ment appelées messagers ou médiateurs intracellulaires. Ces messagers
intracellulaires agissent à leur tour en altérant le comportement d'autres protéines
cibles à l'intérieur de la cellule. L'AMP cyclique, le calcium et les
phosphatidylinositols font partie des messagers intracellulaires les plus importants
(Snyder, 1985).
1.2 LA THROMBINE : STRUCTURE ET FORMATlON
La thrombine est une enzyme qui joue un rôle important dans la coagulation
du sang. Cette protéine est générée dans le plasma par le clivage de son précurseur,
la prothrombine, lorsque cette demière fait partie du complexe pro-thrombinase.
Les autres composantes de ce complexe, sont les facteurs X et V, le calcium et les
phospholipides membranaires (figure 1). La prothrombine est synthétisée par le
foie et est convertie en thrombine par clivage des liens peptidiques C-terminaux
des a r m e s 27 1 et 320. La région N-temiinale de la prothrombine est inactive et
ne semble pas avoir de rôle physiologique ou biologique. La thrombine comporte
une chaîne A (légère) et une chaîne B (lourde ou catalytique). Son poids est
d'environ 39 kDa comparativement à 7 1,6 kDa pour la prothrombine.
La thrombine est une protéine globulaire hautement structurée et de fome
ékpsoïde. Deux caractéristiques structurales sont particulièrement importantes a
l'activité de cette enzyme. En premier lieu une poche étroite s'étendant tout au
long de la molécule et contenant les trois acides amines catalytiques :
et sep5, constitue ie site actif de la molécule. Dans les années précédant le
clonage du récepteur de la thrombine, les principaux substrats naturels de cette

CASCADE ENZYMATIQUE 1
Figure 1 Représentation schématique des facteurs impliqués dans la cascade
enzymatique de la voie intrinsèque de Ia coagulation (Jobin, 1995).

enzyme reconnus à cette époque étaient les chaînes A et B du fibrinogène, mais
l'hydrolyse in vivo de plusieurs liens peptidiques de différentes protéines avait été
observée. Dans le cas des protéines de mammiferes le clivage par la thrombine
s'effectue à l'extrémité C-terminale du lien peptidique adjacent à un résidu
La deuxième partîcularité structurale impliquant l'interaction de la
thrombine avec ses substrats est l'exosite Liant les anions, également désigné
comme région basique de la molécule. Une série d'acides aminés (Arg6', kg7' et
sont localisés trés près de ~ r ~ " , et L~S'" dans la protéine repliée,
avec L~S", et L~S"' également situes à proximité. Ces résidus donnent
naissance a une région chargée positivement qui forme un site d'interaction avec
les régions acides du fibrinogène, de la fibrine, de la thrombomoduline, de
l'hirudine et du récepteur de la thrombine (revue par Stubbs et Bode, 1993).
1.3 RÉCEPTEUR CLlVABLE DE LA THROMBINE
1.3.1 Structure
Le récepteur clivable de la thrombine a jusqu'a ce jour été cloné et
séquencé chez plusieurs espèces : plaquettes humaines (Vu et cou., 1991a; Vu et
coll., 1991b), fibroblastes de poumons de hamster (Rasmussen et coll., 1991),
cellules de muscle lisse d'aorte de rat (Zhong et coll., 1992) et finalement Xenopus
laevis (Gerszten et cou., 1994). Le récepteur humain est composé de 425 acides
aminés et possède plusieurs caractéristiques structurales communes aux récepteurs
couplés aux protéines-G : sept domaines transmembranaires donnant naissance à

trois boucles extracellulaires et trois intracellulaires, une queue C-terminale
intracellulaire et finalement un long domaine N-terminal extracellulaire. Cette
dernière région comprend quelques caractéristiques structuraies essentielles pour
le bon fonctionnement du récepteur. Premièreme* un site de clivage pour la
thrombine bDPR/SFLL] situé entre les résidus 41 et 42 de la protéine humaine.
Deiurièmemenf au moins deux sites d'interaction avec la thrombine, le premier est
situé en Notemiinal du résidu &g4' et est responsable de la reconnaissance du site
de clivage, le second comprend les résidus 53 à 64 et lie l'exosite anionique de la
thrombine (Rydel et cd . , 1990).
1.3.2 Mode d'activation
La façon par laquelle le récepteur de la thrombine est activé Were de celle
des autres récepteurs notamment par l'implication d'une protéolyse enzymatique
Limitée. Un contact initial a tout d'abord Lieu entre l'exosite anionique de la
thrombine et la séquence complémentaire (KYEPF, résidus 50 à 55) du récepteur.
Dans un second temps le segment peptidique du récepteur, lorsque orienté
correctement, lie le site actif de l'enzyme. Le clivage du récepteur a lieu, générant
une nouvelle séquence N-terminale capable d'induire une réponse intracellulaire.
Cette activation surviendrait suite à I'interaction de 5 ou 6 acides aminés avec un
site de liaison situé probablement sur la deuxième boucle extracellulaire (acides
aminés 244-268) et/ou sur le domaine N-terminal (repliement de la chaîne
peptidique sur elle-même, acides aminés 76-93) (Gerszten et cou., 1994; Bahou et
coll., 1994). La figure 2 illustre le clivage de ce récepteur par la thrombine ainsi
que la stimulation par l'extrémité N-terminale nouvellement formée.

Figure 2 Modèle d'activation du récepteur de la thrombine (Vu et coll., 199 lb).

1.3.3 Peptides agonistes du récepteur de la thrombine
Une des preuves les plus pertinentes en faveur du mode d'activation
proposé pour le récepteur de la thrombine provient de l'observation que de courts
peptides identiques a la séquence située en position C-terminale du site de clivage
peuvent reproduire certains effets de la thrombine. Ces peptides sont capables
d'induire l'agrégation plaquettaire (Vu et COU., 1991a)' la libération de 5-
hyd romtamine (Sabo et COU., 1 992)' l'inhibition de 1' adénylyl-cyclase
(Vassallo et coll., 1992) et autres effets associés à la stimulation par la thrombine
de différents types cellulaires. Les concentrations requises de thrombine et de
peptides qûnistes du récepteur pour induire une réponse similaire sont néanmoins
très différentes. Ceci peut s'expliquer par la concentration de ligand libérée suite
au clivage par la thrombine ou bien par la conformation optimale adoptée par la
nouvelle extrémité N-teminale comparativement au peptide Libre en solution.
D'autres hypothèses avancées pour expliquer cette différence sont la possibilité
qu'il existerait plus d'un récepteur pour la thrombine et I'incapacité des peptides à
reproduire complètement les effets de la thrombine causés par un manque
d'induction de signaux provenant d'interactions, autres que la protéolyse, entre la
protéine intacte et le récepteur (Vouret-Craviari et COU., 1 993 ; Kinlough-Rathbone
et COU., 1993).
Il est connu depuis un certain temps que la thrombine peut produire des
réponses d'intensité variée dans plusieurs types cellulaires différents (Detwiler et
Feinman, 1973; Martin et coil., 1975). De quelle façon peut on obtenir des degrés
de réponse différents en présence d'un récepteur activé par une seule étape
protéolytique, alors qu'on s'attendrait à ce que tous les domaines N-terminaux des
récepteurs soient clivés, même en présence d'une faible quantité de thrombine?
L'hypothèse émise à ce jour suggère que la stimulation d'une molécule de

récepteur génère une certaine quantité de seconds messagers avant de devenir
inactif. Le degré de réponse cellulaire serait alors modulé par le nombre de
récepteurs activés (quantité fixe de seconds messagers) et la vitesse de dégradation
de ces mêmes messagers.
1.4 EFFETS DE TYPE HORMONAL DE LA THROMBINE
La thrombine possède une capacité étendue d'interaction avec une grande
variété de types cellulaires. En outre, dépendamment de la cible cellulaire, eue
peut induire rapidement toute une gamme de réponses physiologiques en plus des
événements reliés à la division cellulaire (Fenton, 1988; Berk et con., 1990;
Michel et cou., 1989; He et cou., 1991). Plusieurs de ces actions sont celles
précisément requises en situation inflammatoire, dans le remodelage tissulaire et
éventuellement dans la cicatrisation. Par exemple, la thrombine est impliquée au
niveau de la réponse inflammatoire. Elle provoque la chimiotaxie et l'adhésion des
cellules i.ammatoires (Bar-Shavit et cou., 1990; Hattori et cou., 1989). Elle
contribue à la contraction et au remodelage tissulaire en induisant différents
changements morphologiques chez les cellules endothéliales (Sago et Linuma,
1992) et les fibroblastes (Kolondey et Wysoherski, 1992). Finalement eile
contribue a la cicatrisation en stimulant la mitogénèse, soit directement (Glenn et
coll., 1980) et/ou par sa capacité à induire la sécrétion d'autres facteurs de
croissance (Garcia et COU., 1992). La thrombine joue un rôle central dans une
seconde cascade d'évènements suivant un dommage tissulaire; une cascade
d'effets post-coagulation ou cette enzyme agit, de concert avec d'autres molécules,
comme une homone et un facteur de croissance. Les interactions entre la
thrombine et les multiples types cellulaires sont illustrées a la figure 3.

Monocyte
&+j Plaquette
Figure 3 Effets de la thrombine sur Merents types cellulaires.

1.5.1 Clonage et expression
PAR-2 (Second Proteinase Activated Receptor) est le second membre de la
famille de récepteurs activés par protéolyse. I1 a été nommé ainsi car au moment
de sa découverte son agoniste naturel était encore inconnu. Il a tout d'abord été
identifié chez la souris (Nystedt et coll., 1994), cloné et séquencé chez la même
espèce (Nystedt et coll., 1995a) avant d'être caractérisé chez l'humain (Nystedt et
co l , 1995b; Bohm et c d . , 1996). Ce récepteur est fortement exprimé surtout au
niveau du pancréas et du tractus gastro-intestinal, notamment dans l'estomac.
1.5.2 Mode d'activation et agonistes peptidiques
Ce récepteur, comme le récepteur de la thrombine, est activé par protéolyse
Limitée de son extrémité N-terminale extracellulaire. L'enzyme ou le ligand
endogène responsable de l'activation de ce récepteur est inconnu à ce jour, par
contre nous savons qu'il peut être activé par de faibles concentrations de trypsine.
La nouvelle extrémité N-terminale générée par le clivage agit comme ligand
attaché et comme dans le cas du récepteur de la thrombine le peptide
correspondant à cette nouvelle extrémité, Ser-Leu-Iso-Gly-Arg-Leu (SLIGRL), est
capable de reproduire en bonne partie les effets de la trypsine. La découverte de ce
récepteur est récente de sorte qu'on ignore encore beaucoup sur la structure-
activité de cet agoniste peptidique ainsi que sur les effets cellulaires médiés par ce
récepteur.

1.6 OBJECTIFS DE L'ÉTUDE
La découverte du récepteur clivable de la thrombine et de son mode
d'activation particulier par la thrombine elle-même ainsi que par les peptides
agonistes se révèle être d'une grande importance. Au moment ou nous avons
entrepris nos travaux il y avait peu d'éléments rapportés dans la littérature à
propos de la structure et de l'action des agonistes du récepteur de la thrombine.
Par contre, d'autres groupes ont effectués en parallèle des travaux similaires aux
nôîres.
Dans un premier temps nous avons effectué une étude de structure-activité
dans le but de déterminer le nombre de résidus minimal nécessaire à l'activation
du récepteur. Par la suite nous nous sommes efforcés d'identilïer parmi ceux-ci les
acides aminés essentiels à l'induction d'une activité biologique. Comme ces
peptides sont sujet à l'hydrolyse par I'arninopeptidase M présente dans les
vaisseaux sanguins et les cellules musculaires lisses (Palmien et COU., 1989) nous
avons entrepris, pour pallier cet obstacle et éviter une interprétation erronée des
résultats obtenus dans les différents essais, la synthèse d'un peptide résistant à la
dégradation métabolique. Les réponses biologiques aux peptides synthétisés seront
évaluées in vitro dans le modèle de l'aorte de lapin isolée (mesure de la
contractilité) ainsi qu'in vivo chez le même animal (mesure de la pression artérielle
moyenne).
La transduction de signal qui suit la liaison de la thrombine ou des peptides
agonistes au ricepteur a très peu été étudiée à ce jour. Dans le but de mieux
caractériser les voies de transduction impliquées suite à l'activation du récepteur

par I'un ou l'autre des agonistes, nous évaluerons I'effet de différents inhibiteurs
sur la contraction de l'aorte de lapin induite par ces agonistes. Ces mêmes
inhibiteurs seront également utilisés pour identifier les seconds messagers produits
suite à la stimulation par la thrombine ou par les peptides agonistes, des cellules en
culture de muscle lisse dérivées de I'aorte de lapin.
L'activation du récepteur de la thrombine par protéolyse partielle nous a
amené à nous questionner sur la désensibilisation et la cinétique de réexpression
de ce récepteur suite à son clivage par l'enzyme. Pour ce faire nous utiliserons
différents inhibiteurs de la synthèse protéique ou du transport intracellulaire,
toujours dans le modèle de l'aorte de lapin isolée, afin de vérifier les réponses à la
thrombine suivant différents traitements avec chacun de ces agents.
La découverte tout récente du récepteur PAR-2 ouvre la porte à tout un
champ d'étude atin d'élucider son fonctionnement, le comparer avec le récepteur
de la thrombine et véfier sa présence au niveau du tissu .vasculaire. Il est
également important de vérifier la présence d'une activation croisée des deux
récepteurs par les agonistes synthétiques et leurs analogues. Une telle activation
fausserait l'interprétation des données obtenues avec les composés synthétiques.
Une étude de structure-activité de même nature que celle effectuée dans le cas du
récepteur de la thrombine sera entreprise, toujours dans le but d'identifier les
résidus essentiels à l'activation de ce second récepteur. L'essai de contractilité et
de relaxation de l'aorte de lapin isolée sera le modèle utilisé pour l'évaluation de
l'activité biologique des peptides issus de cette étude.
Finalement, dans le but de démontrer hors de tout doute la présence du
récepteur PAR-2 dans les vaisseaux sanguins, nous présentons une hybridation de

type Northern montrant la synthèse d'un ARN messager spécifique de ce récepteur
dans l'aorte de lapin.

2.1 SYNTHÈSE ET PURlFlCATlON DES PEPTIDES
2.1.1 Synthèse
Les peptides sont synthétisés manuellement par la méthodologie en phase
solide (Menifield, 1962). Les chaînes peptidiques sont assemblées sur des résines
Wang (alcool p-akoqhenzilique; Bachem, Tomance, CA) ou MBHA (4-
méthylbemhydrylamine; Bachem, Torrance, CA) en utilisant des acides aminés
protégés par le groupement 9-fluorényhéthyloxycarbonyl (FMOC; Atherton et
Sheppard, 1989). Les chaînes latérales fonctionnelles des résidus d'acides aminés
sont également protégées par les groupements indiqués entre parenthèses:
@@'MC), Asn(trt), ~ s p ( O ~ u ' ) , C y ~(trt), G~U(OBU~), Ly ~(Boc), Ser(But), ~hr(E3d)
et ~yr(~u' ) . Les acides aminés sont activés avec le réactif benzotriazole- 1 -yl-o-y-
tris-(diméthylamino) phosphonium hexafiuorophosphate (BOP; Castro et coll.,
1975). Les peptides sont clivés de la résine par traitement à l'acide
trifiuoroacetique (TFA; résines Wang) ou à l'acide trifluorométhanesulfonique
(TFMSA; résine MBHA). La figure 4 montre la synthèse en phase solide du
peptide NAT6-NH2. La liste des peptides synthétisés pour les h s de cette étude
est présentée au tableau 1.

I 5% DEA
Dipea 7 Hobt
, , 1 % Dipea
TFA Phénol Éthanedithiol Thioanisole Eau
Ser-Phe-Leu-Leu- Arg- Asn-NH, 49
Ser-Phe-Leu-Leu- Arg- Asn-NH,
Figure 4 Représentation schématique de la synthèse en phase solide du peptide
NAT6-NH2.

Tableau 1 Liste des peptides synthétisés pour les fins de cette étude.
Agonistes du récepteur de la thzombine :
eu'] NAT, -NH, Ser-Leu-Leu-Leu-Arg-Asn-NfS
[~ca'] NAT, -WC Aca-Phe-Leu-Leu-Arg-Asn-MI,
[ G ~ $ ~ ~ ( c & N H ) ~he'l NAT, -me Gly (CH2NH) Phe-Leu-Leu-Arg-Asn-NH,
Antagonistes du récepteur de la thrombine :
Butyraldéhyde A C&C&CH2C0 ( C H p H ) Leu-Leu-Arg-Asn-NH,
Butyraldéhyde B
Butyraldéhyde C
Cyclohexylmethyl

Prothrombine 367-380
Arg-Gly-Asp-Ala-Cys-Glu-Gly-Asp-Ser-
Gly-Gly-Pro-Phe-Val
Ala-Gly-Tyr-Lys-Pro-Asp-Glu-Gly-Lys-
Arg-Gly-Asp-Ala-Cys-Glu-Gly-Asp-Ser-
Gly-Gly-Pro-Phe-Val
Tyr-Pro-Pro-Trp-Asn-Lys-Asn-Phe-Thr-
Glu-Asn-Asp-Leu-Leu
Prothrombine 63-88
Retro NAT,-NH, (acides aminés D) asn-arg-leu-leu-phe-ser-NH,
Agonistes du recepteut PAR-2 :
Ser-Leu-Ile-Gly-Arg-Leu-NH,
Ser-Leu-Ile-Gly-Arg-Leu
Ala-Leu-Ile-Gly-Arg-Leu
Ser-Ala-Ile-Gly-Arg-Leu
Ser-Leu-Ala-Gly-kg-Leu
Ser-Leu-Ile-Ala-Arg-Leu
Ser-Leu-Ile-Gly-Ala-Leu
Ser-Leu-Ile-Gly-Arg-Ala
Sar-Leu-Ile-Gly-Arg-Leu

Sar-Phe-Ile-Gly-Arg-Leu
a. sarcosine ou N-méthyl-glycine
b, N-rnethyl-phénylalanine
c. acide aminocaproique
d. acétyl-glycine
e , lien amide réduit p s i

Les peptides bruts sont purifiés par chromatographie préparative (Drapeau
et Regoli, 1988) sur une colonne Michel-Miller 2.5 x 40 cm (Ace Glass, Vineland,
NJ) contenant une résine de type Cls 15-20 p (Vydac, Hesperia, CA) en phase
inverse. Le peptide brut est chargé sur une colonne préalablement équilibrée avec
une solution de 0.05% TFA/&O. L'élution du produit s'effectue à l'aide d'un
gradient de 5.50% acétonitrile/0.05% TFA/H20 et des &actions d'environ 10 ml
sont recueillies. La pureté des fiactions contenant le peptide est vérEée par
chromatographie liquide à haute performance (HPLC) analytique. Celles contenant
le produit pur sont rassemblées en une seule et lyophilisées. L'homogénéité du
produit final est contrôlée à nouveau par HPLC analytique.
2.2 ANALYSE PAR HPLC
L'analyse des peptides purs ou des échantillons provenant des études
métaboliques est effectuée en utilisant un système Waters équipé d'un intégrateur
(Waters 746) et d'un détecteur UV (Waters 486 ajusté à 214 nm). La séparation
des composés s'effectue sur une colonne Vydac CI1 10 p en phase inverse (3.9 x
300 mm) en utilisant un gradient linéaire de 5-65% acétonitde/0.05% TFA/H20 a
un débit de 2mVmin pendant 20 min. Les composantes de l'appareil ainsi que les
paramètres utilisés sont illustrés à la figure 5.

SOLVANT A $0 + 0.05% TFA
Waters 5 10 I Waters 510 O CONTR~LEUR DE
Waters 680
Waters U6K I"""") I
COLONNE Cl, EN PHASE INVERSE ( Vydac IOp (3.9 x 300 mm) 1
DÉTECTEEURUV Waters 486 ajusté à 214 nm i
~ É G R A K U X 1 Waters 746 1 Programme: gradient linéaire 5.65% acétonitrile
Temps Débit %A %B
(min) (ml/min)
Figure 5 Représentation schématique du système HPLC et des paramètres utilisés

La résistance métabolique des peptides NAT6-N& et [ S ~ ~ ' W A T ~ - ~ est
évaluée en présence de plasma de lapin, source de l'enzyme aminopeptidase M.
2.3.1 Plasma de lapin
Le sang de lapins anesthésiés au pentobarbital de sodium (30 mgkg i-v.) est
recueilli dans des tubes héparinés à partir de l'artère carotide gauche. Les tubes
sont centrifugés à 450 x g pendant 15 minutes à température ambiante. Le plasma
est échantillonné en fiactions aliquotes et conservé a -20°C jusqu'à I'utilisation.
2.3.2 Mesure de Ifactivité enzymatique
Les peptides (50 PM) sont dissous dans une solution saline (pH 6.8) et
incubés pendant 5 min a 37°C avant l'addition de 15 p! de plasma de lapin
(volume total de 1000 pl). Les échantillons sont prélevés aux temps 0, 15, 30 et 60
min suivant le début de la période d'incubation et mélangés avec une solution de
T F m 0 à 10%. Les échantilIons sont ensuite centrifugés pendant 1 min à 10,000
x g avant l'analyse par HPLC. L'inhibition de l'aminopeptidase M est obtenue en
ajoutant un inhibiteur, I'amastatine (20 FM), dans les échantillons avant
l'incubation à 37°C. Pour l'analyse, 50 pl de chaque échantilion sont injectés dans
I'appareiI HPLC et le taux de dégradation métabolique est évalué à partir de la
diminution de la concentration du substrat peptidique (Drapeau et cou., 1993). Le
taux de dégradation est exprimé en substrat métabolisé (en m o l ) par minute par

ml de plasma. Ce taux est directement proportionnel au temps d'incubation ainsi
qu'à la quantité de plasma utilisée.
2.4 MODÈLE DE L'AORTE DE LAPIN ISOLÉE
L'aorte thoracique est isolée de lapins albinos de souche Nouvelle-Zélande
(1.5-2 kg; mâle ou femelle). Les animaux sont sacrinés par asphyxie dans une
chambre à CO2. L'aorte thoracique est prélevée et placée dans une solution de
Krebs fraîchement préparée et oxygénée (95% 05 5% COz). La composition de la
solution est la suivante (a): NaCl (117.5); KCl (4.7); =PO4 (1.2); MgS04
(1.18); CaC& (2.5); NaHC03 (25.0) et D-glucose (5.5). Le pH h a l de cette
solution se situe entre 7.35 et 7.40 (Marceau et COU., 1991). Une tige de métal de
faible diamètre est insérée délicatement dans la lumière du vaisseau afin de
faciliter la résection de l'excès de tissus adipeux et conjonctifs. L'aorte est ensuite
coupée en anneaux de 2 à 3 mm de longueur qui sont suspendus entre un support
métallique et une boucle de fii dans un bain à organe isolé de 5 ml contenant de
solution de Krebs oxygénée et maintenue à 37°C. Une tension basale de 2 g est
imposée aux tissus des le début de l'incubation in vitro et une période
d'équilibration variant entre 60 et 120 min, selon les protocoles, est allouée avant
l'enregistrement. Les changements isométriques dans la tension du tissu après
l'addition des agents pharmacologiques sont détectés par des transducteurs
isométriques (modèle 52-9545; Harvard Bioscience, South Natick, MA) reliés à
des enregisteurs graphiques (LKB 2210; Pharmacia, Baie-D'Urfé, QC) (Deblois et
COU., 1992).

2.5 PROTOCOLES EXPÉRIMENTAUX
2-5.1 €#et contractile de la thrombine, de la trypsine et des agonistes
des rdcepteurs de la thrombine et PAR2
L'effet contractile direct de la thrombine bovine p d é e (Thrombostat;
Parke-Davis), des peptides NAT, de la trypsine et des peptides agonistes du
récepteur PAR-2 est vé f ié par l'addition de concentrations croissantes
cumulatives dans le bain à organe isolé (courbes concentration-réponse
cumulatives) après une période d'équilibration des tissus de 60 min. Un maximum
de deux courbes, séparées par un temps d'attente de 90 m i . suivant le lavage des
tissus, sont obtenues avec chacun des tissus.
2.5.2 Relaxation de NA TrN& et de am^ T,NHz dans l'huile
La technique d'immersion dans l'huile (Kalsner et Nickerson, 1968; Regoli
et coll., 1974) est utilisée pour mesurer la vitesse de relaxation, dans l'huile
minerde, des anneaux d'aorte de lapin précontractés avec [ s ~ ~ ~ ] N A T ~ ~ ou
NAT6-NH2 en présence ou absence d'amastatine. Ces relaxations réflètent la
vitesse d'inactivation de ces peptides par les tissus. Les courbes de relaxation sont
obtenues en injectant des concentrations équi-actives de (4.4 x 104 M)
et de [ s ~ ~ ~ ] N A T ~ - w (1.4 x 1 0 ~ M) dans les bains où les anneaux d'aorte de
lapin sont suspendus dans la solution Krebs. Une fois le plateau de contraction
atteint, la solution Krebs est remplacée par de l'huile minérale. Les résultats sont
exprimés en pourcentage de relaxation en fonction du temps. Certaines

expériences sont réalisées en présence d'amastatine (20 FM), administrée dans les
bains 5 min avant I'agoniste.
2=5=3 Mode d'action de la thrombine et de [s~#]NAT&
Les tissus sont exposés à une concentration sous-maximale de [S~~']NAT~-
(40 CiM) après une période d'équilibration de 120 min. Les inhibiteurs sont
injectés dans Les bains 30 min avant chaque injection d'agoniste, des tissus
appariés servant de témoins sont utilisés pour évaluer l'effet des solvants. Les
mêmes tissus sont ensuite exposes aux temps 4h et 6h à la thrombine (60 nM) et
au KCl(20 mM) respectivement, pour les besoins de comparaison. Les inhibiteurs
à l'étude sont: la nifédipine, un bloqueur des canaux calciques de type L (Murad,
1990); le H-7, un inhibiteur des protéine kinases (Hidata et COU., 1984); le
diclofénac, un inhibiteur de la cyclooxygénase (Lewis et Fwst, 1987); l'erbsbtine,
un inhibiteur des tyrosine kinases (Imoto et coll., 1987) et D-Phe-Pro-Arg-CH2C1
(PPACK), un inhibiteur irréversible de l'activité catalytique de la thrombine
(Kettner et Shaw, 1979). Les concentrations des inhibiteurs utilisés sont
déterminées selon leur activité dans différents modèles in vitro (Levesque et coll.,
1993). Dans certaines experiences l'effet inhibiteur du H-7 est mieux documenté
en construisant des courbes concentration-effet sur une large échelle de
concentrations du peptide [s~.'JNAT~-NH, et de la thrombine. Un autre protocole
vise à déterminer la contribution de l'endothélium dans la réponse contractile
induite par chacun des agents. L'endothéiium est enlevé délicatement en frottant la
lumière du vaisseau avec une petite spatule et l'intensité du dommage intimai est
vérifiée par la perte de la réponse relaxante à l'acétylcholine (Furchgott et
Zawadzki, 1980). Finalement, un demier protocole évalue la contribution du
calcium extracellulaire dans la réponse contractile de [S~~']NAT~-W . de la

thrombine et du KCI. Ce protocole consiste à remplacer la solution régulière de
Krebs par une solution modifiée, exempte de calcium, 10 minutes avant chaque
ajout de stimulus contractile. Entre les stimulations les tissus sont incubés dans la
solution régulière de Krebs (avec calcium) afin d'éviter la dépletion des réservoirs
calciques intracellulaires.
2.5.4 Effet des inhibiteurs métaboliques sur la regulafion du
récepteur de la thrombine
Des courbes concentration-réponse cumulatives à la thrombine (2 nM à 60
nM) sont réalisées aux temps Ih, 3h et 6h suivant la période d'équilibration.
Différents inhibiteurs métaboliques sont utilisés a f h de vérifier leurs effets sur la
régulation du récepteur de la thrombine: la cycloheximide et I'anisomycine, des
inhibiteurs de la synthèse des protéines et la brefeldine A, un inhibiteur du
transport protéique entre le réticulum endoplasmique et l'appareil de Golgi. Les
inhibiteurs sont ajoutés dans les bains dès le début de la période d'incubation des
tissus et sont maintenus tout au long de l'étude. Des tissus appariés sont utilisés
dans chaque cas pour évaluer l'effet des solvants, s'il y a lieu.
2.5.5 Effet relaxant de la trypsine et des agonistes PAR-2
L'effet relaxant de la trypsine et des peptides agonistes du récepteur PAL2
est vérifié par l'addition de concentrations croissantes cumulatives (courbes
concentration-réponse cumulatives) sur les tissus préalablement précontractés à la
phényléphrine (PE; 0.5 PM). Une période d'équilibration des tissus de 60 min est
allouée avant la première injection de PE. Une fois le plateau atteint, une

concentration unique d'acétylcholine (100 nM) est testée a f i n de vérifier l'intégrité
fonctionnelle de l'endothélium. Après un second temps d'attente de 60 min un
maximum de deux courbes concentration-réponse, séparées par au moins 90 min,
sont obtenues avec chacun des tissus. Les résultats sont exprimés en pourcentage
de relaxation en fonction du plateau de PE.
2.6 CELLULES EN CULTURE DE MUSCLE LISSE DÉRIVÉES DE
L'AORTE DE LAPIN (CML)
2.6.1 Mise en culture
Plusieurs lignées distinctes de cellules de muscle lisse dérivées de l'aorte de
lapin sont obtenues à partir d'aortes thoraciques prélevées aseptiquement chez des
lapins fraîchement euthanasiés. L'aorte est placée dans une solution saline
balancée de Earle (SSBE). Le tissu est dégraissé, coupé en petits morceaux et
incubé pendant 60 min à 37OC dans une solution de collagénase (Type IA, 5
mg/ml) et d'élastase (Type III, 1 mg/ml) diluée dans le milieu de culture M-199.
Une fois la période d'incubation terminée, la suspension est centrifugée à 200 x g
pendant 5 min à température ambiante dans une cenûifbgeuse Sorvall RT6000D.
Le surnageant est ensuite décanté et le culot resuspendu dans le milieu de culture
M-199 supplémenté de 10% de sérum de veau foetal (SVF), 1% d'antibiotiques
(streptomycine 50 pg/d et pénicilline 50 U l d ) et 1% de L-glutamine (200 mM)
(M-199 complet). Les cellules en suspension et les fragments de tissu non-digéré
sont ensemencés dans des flacons de culture de 25 cm2 traités à la gélatine 0.2%
(gélatine 2%, Sigma; dans SSBE et 1% d'antibiotiques) pendant au moins 4 heures
afin d'assurer une bonne adhérence des cellules aux flacons. Les flacons sont
déposés dans un incubateur à 37°C à atmosphère contrôlée: 95% O2 et 5% COz.

Lors du repiquage les cellules sont détachées des flacons par exposition à une
solution de trypsine (O.OS%)/EDTA tétrasodique (0.53mM) à 37OC pendant 5 a 10
min. La suspension est recueillie et ensemencée à nouveau dans des flacons de 75
cm2 ou des plaques de 12 puits préalablement traitées à la gélatine. Les cellules
sont repiquées un maximum de 8 fois avant leur utilisation.
2.6.2 Mesure de la producfion des phosphatidylinosttols
Les cellules en culture (2.5 x 10' cellules / puit de 2.0 cm2) sont incubées
pendant 16 a 20 heures à 37OC dans 500 pl de M- 199 supplémenté de 20 pCi/ml
de myo-[3~inositol (Amersham, Oakville, ON). A la fin de la période
d'incorporation les puits sont lavés deux fois avec 500 pl de SSBE et les cellules
sont incubées a nouveau pendant 20 min dans du SSBE contenant 10 mM de LiCl.
Ensuite, les cellules sont stimulées avec la thrombine (10, 100 et 1000 nM) ou
NAT6-NH2 (10 et 100 FM) pendant exactement 5 min puis la réaction est arrêtée
en ajoutant 100 1 d'acide perchlorique 17.5% dans chacun des puits
(concentration finale de 5%). Les plaques sont incubées sur glace pendant 30 min
avant de recueillir le contenu des puits en grattant et en rinçant chacun des puits
avec 200 p1 d'acide perchlorique à 5%. (Balla et coll., 1988).
Les phosphatidylinositols sont extraits et séparés selon la méthode de
Guillemette et c d . (1989). Le contenu de chaque puit est centrifugé à 1800 x g
pendant 5 min a 4OC. Les culots, contenant les phospholipides, sont mélangés avec
10 ml de liquide à scintillation Ecolite + (ICN Biochemicals, Costa Mesa, CA).
Cette étape sert à quantifier l'incorporation du myo-[3H&nositol dans les
phosphoinositides. L'acide perchlorique contenu dans les surnageants est
neutralisé en additionnant 10 pl dYEDTA (100 mM) et 600 pi1 d'un mélange (1:l)

de 1,1,2-trïchlorotrifùoroéthane : tri-n-octylamine puis en agitant vigoureusement
pendant 30 sec @ownes et coii., 1986). Après centrifugation à 1800 x g pendant 1
min, la phase supérieure est recueillie et mélangée avec 20 pl de Tris-HC1 (pH 8.5)
et 5 pl de rouge phénol 3 mM. Cette solution est ensuite neutralisée avec du KOH
0.1 N, de façon a obtenir un pH final se situant entre 7 et 8 (indiqué par le rouge
phénol). Ces solutions, contenant les phosphatidylinositols, sont utilisées
immédiatement ou conservées à 4°C jusqu'à utilisation.
Les phosphatidylinositols sont charges sur des petites colomes contenant 1
ml de résine échangeuse d'anions (Dowex AG bX8, forme formatée, 200-400
mesh, #140-1450; Bio-Rad, Hercules, CA) (Bemdge et COU., 1983). On élue, par
étapes successives, le phosphatidylinositol (IP) avec 5 x 3 ml &O; l'inositol-4-
phosphate (Pl) avec 5 x 3 ml de formate d'ammonium 0.2 M dans l'acide
formique 0.1 M; I'inositol-4'5-bisphosphates (Ib) avec 5 x 3 ml de formate
d'ammonium 0.5 M dans l'acide forrnique 0.1 M; l'inositol- 1,4,5-triphosphates
(P3) avec 5 x 3 ml de formate d'ammonium 0.8 M dans l'acide formique 0.1 M et
l'inositol- 1,3,4,5-tétraphosphates (II$) avec 5 x 3 ml de formate d'ammonium 1 .O
M dans l'acide formique 0.1 M. La radioactivité de chaque fiaction recueillie est
ensuite quantifiée par scintillation liquide à l'aide d'un compteur à scintillation
12 15 Rack Beta 2 de LKB Wdac.
2.6.3 Incorporation de la f~lthymidine
La mesure de l'incorporation de la r3~thymidine est effectuée selon
Goldstein et WalI (1984) avec quelques modifications. Les cellules sont
ensemencées pour 24 heures dans du M-199 complet (avec sérum) dans des
plaques 12 puits (Costar, Nepean, ON) à une densité initiale de 30 000

ceilules/puit. Afin d'arrêter la croissance, on effectue 2 lavages avec du milieu M-
199 (sans s é m ) et on remplace le milieu de culture par du milieu pauvre en
sénun (M-199, 0.4% SVF, 1% giutamine et 1% antibiotiques) contenant du
diclofénac 0.5 PM. Après une période d'incubation de 72 heures les cellules sont
stimulées avec la thrombine, les peptides agonistes du récepteur de la thrombine,
le SVF ou d'autres agents pendant 24 heures. On ajoute ensuite 1 pCi de
[3Hlthymidine (Dupont, Missisauga, ON; 84 Ci/mmol) et on incube à nouveau
pour 24 heures. Les puits sont par la suite lavés deux fois avec une solution saline
(0.9% NaCl, pH 5.7) et les cellules sont détachées des puits par traitement avec
150 pl de la solution de trypsine (O.OS%)/EDTA tétrasodique (0.53 mM) jusqu'à
ce qu'elles n'adhèrent plus (approx. 20 min à température ambiante). Le contenu
des puits est placé dans des tubes de borosilicate de 12 x 75 mm auxquels on
ajoute 1 ml de Triton X-100 (1% dans la saline 0.9%) afin de perméabiliser les
membranes cellulaires. Le matériel intracellulaire est précipité par l'addition de 1
ml d'acide trichloroacétique 40% et on centrifuge à 2000 x g pendant 20 min à
4OC. Le surnageant est décanté et remplacé par 1 ml H20 distillée. Le culot est
resuspendu puis placé dans des flacons à scintillation auxquels on ajoute 10 ml
d7Ecolite + avant de procéder à la détermination du contenu en radioactivité.
2.7 DÉVELOPPEMENT D'UN ESSAI DE LIAISON POUR LE
RÉCEPTEUR DE LA THROMBINE
2.7.1 Radiomarquage de la thrombine ii l'iode radioactive (?)
La thrombine humaine purifiée (Calbiochem, La Jolla, CA) est marquée à
I''~'I selon une méthode initialement développée par Salacinski et coll. (198 1). Le
milieu de réaction est preparé en déposant une solution de 100 pg d'Iodogen

(Pierce, Rockford, IL) dans le chlorure de méthylène (CH2C1; 0.1 g/L) dans un
tube conique de 500 pl en polypropylène. L'évaporation du solvant engendre le
dépôt d'une couche d'environ 10 pg d710dogen sur la paroi du tube. Une solution
de thrombine, 150 U dans 50 pl de solution saline est placée dans le tube traité à
1'Iodogen en présence de 500 pCi de N ~ ' ~ ' I (Amersham, Oakville, ON) pendant 20
min. La réaction est arrêtée en ajoutant 150 pl d'une solution de 0.25 M KI f
Na2S04 préparée dans la saline. L'iode libre est ensuite séparé de la protéine
marquée a l'aide d'une colonne PD40 (Pharmacia, Baie d'Urfé, QC)
préalablement équilibrée avec de la saline contenant 0.1% d'albumine bovine.
2.7.2 Essai de liaison pour le récepteur de la thrombine et analyse de
Scatchard
Les cellules de muscle lisse sont ensemencées dans les plaques de 12 puits
(100 000 cellules 1 puit) pré-enduites de poly-L-lysine (50 ygM). Après 24 h
d'incubation à 37"C, 2 rinçages avec du milieu M-199 sans additif sont effectués,
suivis d'une période d'équilibration de 30 min dans le milieu d'essai. La
composition du milieu d'essai est la suivante: milieu M-199 auquel on ajoute 1%
d'albumine bovine puis 15 mM de tampon Hepes, le pH final de ce milieu est de
7.0. À la fin de la période d'équilibration le milieu est remplacé par du milieu
fiais. On ajoute dans les puits ~' '~~~-birornbine (0.3 nM à 300 nM) et un excès de
thrombine non-marquée (1 PM) dans une série de puits pour déterminer la liaison
non-spécifique (liaison à d'autres protéines ou au plastique). Après une période
d'incubation de 45 min, chaque puit est lavé rapidement 4 fois avec 2 ml de PBS
(O°C, pH 7.4). Les celIules sont lysées avec 1 ml de NaOH 0.1 M, la solution est
ensuite placée dans des tubes en polypropyléne 25 x 75 mm et la radioactivité est
quantifiée dans un compteur gamma Packard Cobra.

2.8 ÉVALUATION DES EFFETS IN VIVO DE NATs-NH2 ET DE
[SAR']NAT~-NH~ CHEZ LE LAPIN ANESTHÉSIÉ
Les lapins de souche Nouvelle-Zélande (1.5 - 2.5 kg, mâle ou femelle) sont
anesthésiés au pentobarbital de sodium (30 mgkg, I.V., ajusté individuellement).
De plus, la iidocaïne (2%) est utilisée comme anesthésique local aux sites
d'incision. La trachée est intubée et une assistance respiratoire est fournie par
I'intermédiaire d'une pompe respiratoire Harvard. Un cathéter de polyéthylène PE-
90 est inséré dans la carotide commune gauche, poussé jusqu'a l'aorte puis
connecté à une valve trois voies afin d'enregistrer le rythme cardiaque ainsi que la
pression sanguine à l'aide d'un transducteur de pression (Statham P23ID) connecté
à un polygraphe Grass (modèle 79). M n de prévenir la coagulation dans le
cathéter, chaque animal reçoit 500 U / kg d'héparine au début de l'expérience. Un
cathéter PE-50 est inséré dans la veine jugulaire droite pour les injections LV. des
peptides donnés en bolus de 0.3, 1.0 et 3.0 mgkg. Deux doses de chaque peptide
sont testées chez un même animal' I'inte~valie entre les doses est de 10 min ou
lorsque la pression sanguine se stabilise après la première injection. Dans certaines
expériences la veine jugulaire droite est utilisée pour l'infusion d'amastatuie à 70
pl / min (50 nmol I kg 1 min) à l'aide d'une pompe à seringue Sage (modèle 34 1A).
La veine jugulaire gauche est alors utilisée pour I'injection des peptides. La même
dose de peptide (1 mg / kg) est administrée deux fois chez le même animal; la
deuxième injection est donnée 12 min suivant le début de l'infusion continue de
I'amastatine qui se poursuit jusqu'au retour a la ligne de base. Chaque réponse
hypotensive est caractérisée quantitativement par la réponse maximale,
correspondant a la baisse de la pression sanguine moyenne (mm Hg).

2.9 lDENTlFlCAflON DES ARN
RÉCEPTEUR PAR-2
MESSAGERS
33
CODANT POUR LE
2.9. f Préparation de la sonde PAR-2
2.9.1.1 isolation du fragment (sonde) par PCR
Brièvement, la technique d'amplification élective in vitro ou PCR pour
Polymerase Chain Reaction est une méthode de synthèse enzymatique in vitro de
séquences définies d'ADN (Saiki et COU., 1985). La réaction utilise deux amorces
(oligonucléotides d'environ 15 à 30 bases) qui s'hybrident à chacun des brins de
I'ADN de part et d'autre de la région cible que l'on veut amplifier. L'élongation
des amorces est catalysée par l'ADN polymérase Taq (Saiki et COU., 1988) : une
polymérase résistante à la chaleur, isolée d'une bactérie thermophile appelée
Themus aquaticus. Une série répétitive de cycles (20 à 40) impliquant la
dénaturation de l'ADN gabanf l'hybridation des amorces et l'élongation des
amorces par la polymérase Taq résultent en une accumulation exponentielle du
fiagrnent cible d'ADN. Les extrémites du fragment sont d é f i e s par les régions 5'
de chacune des amorces. Étant donné que le produit de synthèse obtenu lors d'un
cycle donné peut servir de gabarit a son tour lors du prochain cycle, le nombre de
copies du fkagment cible doublera à chaque cycle.
La réaction d'amplincation se fait dans un volume total de 50 pl contenant
400 ng d'ADN, 5 pM de chacune des amorces, 200 FM de dATP, 200 pM de
dCTP, 200 pM de dGTP, 200 pM de dTTP, 20 mM de TrisoHC1 pH 8.4, 1.5 mM
MgC12 et 2.5 unites de polymérase Taq (Gibco BRL, Burlington, ON).

Les amorces utilisées sont les suivantes:
Les conditions de la réaction consistent en une dénaturation initiale de 3
min à 95OC suivie de 30 cycles de:
Dhturation, 1 min a 95°C;
Hybridation avec les amorces, 1 min à 60°C;
Extension des amorces, 1 min a 72OC;
puis d'une élongation h a l e de 10 min à 72°C.
20 pl de la réaction de PCR sont mélangés à 4 pi de bleu de chargement
(0.25% bleu de bromophénol; 0.25% xylène cyan01 FF; 15% Ficoll) puis chargés
sur un gel (1 1 x 14 cm) d'agarose 1% dans une solution de TBE 1X (100 mM Tris
pH 7.9; 83 mM acide borique; 1 mM EDTA). La migration s'effectue à 80 volts
dans le tampon TBE 1X jusqu'à ce que le bleu de bromophénol ait migré sur une
distance d'environ 10-12 cm-
La bande correspondant au fragment PCR amplifié est découpée et l'ADN
est séparé de l'agarose et des autres composantes du gel. La parcelle d'agarose
contenant le fiagrnient est placé dans un tube de 500 pl percé à sa base et
contenant une mince couche de laine de verre bien tassée. Ce tube est placé dans
un second tube de 1500 pl afin de recueillir l'éluat puis le tout est centnfigé 15
min à 12,000 RPM à température ambiante dans une centrifugeuse de table (IEC,
Costa Mesa, CA). On ajoute à l'éluat 1/10 de volume d'acétate de sodium 3 M @H

5.2) puis 3 volumes d'éthanol 95%. La solution est ensuite placée a -20°C pour
précipitation pendant au moins 1 heure. Le tout est centrifugé pendant 15 min à
10,000 x g à température ambiante, le culot d'ADN est ensuite lavé une fois à
l'éthanol 70% puis halement resuspendu dans 20 pl d'eau distillée stérile.
2.9.1.2 Clonage du fragment dans pCR 2.1
Le clonage du fiagrnent PCR s'effectue à I'aide d'une trousse de clonage
i< TA Cloning Kit )> (Invitrogen, San Diego, CA) selon la procédure recommandée
par le fabriquant (Mead et coll., 199 1; Clark, 1988). En bref, le fiagment est inséré
dans un vecteur de clonage pCR 2.1. Des cellules compétentes « One-Shot )) sont
utilisées pou. la transformation. Les clones positifs (colonies blanches) sont
identifiés par la méthode de sélection X-Gd / IPTG et repiques pour obtenir une
préparation de plasmides à plus grande échelle. Le fragment PCR est extrait du
vecteur par digestion à I'aide d'une endonucléase de restriction, EcoRI (Gibco
BRL, Burlington, ON). L'identité du f i m e n t est vérifiée par une réaction de
séquencage en utilisant la trousse a T7 Sequencing Kit >) (Phamacia, Baie d'Urfé,
QC) selon le protocoie établi par la compagnie (Sambrook et cou., 1989; Haltiner
et coll., 1985; Sanger et COU., 1977).
2.9.7.3 Marquage de la sonde PA R-2
Le marquage de la sonde s'effectue à l'aide d'une trousse << Oligolabelling
Kit » (Pharmacia, Baie dYUrfë, QC) en suivant les instructions du fournisseur
(Feinberg et Vogelstein, 1984). La sonde marquée avec le [~SPI~CTP
(Amersham, OakMlle, ON) est séparée des nucléotides libres et de la radioactivité
non-incorporée à l'aide d'une petite colonne Séphadex G-50 et ce en éluant avec
un tampon composé de 0.5 M NaCl, 50 mM Tris pH 7.5 et 10 mM EDTA.

L'activitk spécifique de la sonde est mesurée en méIangeant 1 p l de celle-ci avec 5
ml de liquide à scintillation Ecolite + dans un flacon à scintillation. Le contenu en
radioactivité est déterminé dans un compteur à scinflation 1215 Rack Beta 2 de
LKB Wallac.
2.9.2 Pléparation de I'ARN messager de l'aorte et du coeur de lapin
2.9.2.7 Extraction de IMRN total
L'extraction de 1'ARN total à partir des tissus de lapin s'effectue à l'aide du
réactif Trizol (Gibco BRL, Burlington, ON), un réactif commercial qui permet une
extraction rapide d7ARN de bonne qualité. L'extraction s 'effectue selon les
instructions qui accompagnent le réactif (Chomczynski et Sacchi, 1987; Ausubel et
coil., 1990). Brièvement, les tissus sont prélevés stérilement et placés
immédiatement dans de l'azote liquide pour inactiver les enzymes qui dégradent
I'ARN (RNases). Les tissus sont ensuite broyés dans un mortier à l'aide d'un
pilon, toujours dans l'azote liquide. On ajoute le Triml (1 ml par 100 mg de tissu)
lorsque les tissus sont bien homogénéisés. L'extraction se poursuit tel qu'indiqué
dans le feuillet d'instruction. Le rendement h a l se situe aux environs de 125 pg
d'ARN par gramme de tissu au départ.
2.9.2.2 Isolation de I'ARN messager à partir de I'ARN total
La trousse de préparation dYARN messager << polyA Spin mRNA Isolation
Kit » (New England Biolabs, Mississauga, ON) est utilisée pour obtenir I'ARN
polyA' (messager) à partir de I'ARN total isolé précédemment. L'e~chissement

s'effectue toujours selon les instructions fournies avec la trousse (Maniatis et COU.,
1989; Jacobson, 1987).
2.9.3 Gel d'ARN et transfert sur membrene de nylon
Les différents brins d'ARN messager isolés précédemment sont séparés sur
un gel d'agarose à 1.2% (11 x 14 cm) dans un tampon MOPS (MOPS 20 mM,
NaOAc 5 mM, EDTA disodique 1 mM) contenant 2% de formaldéhyde. Les
échantillons d7ARN messager (2.4 pg) sont mélangés selon une proportion 1: 1
avec du Bleu de chargement 2X pour ARN et déposés sur le gel. La migration
s'effectue à 100 V pendant 3 heures. Le gel est ensuite transféré sur une membrane
de nylon (Hybond N', Amersham, Oakville, ON). Le transfert est effectui dans
une solution SSC 20X (chlorure de sodium 3M, citrate de sodium 300 mM)
pendant 16 heures. Pour texminer I'ARN messager est fixe de façon covalente à la
membrane par exposition à un rayonnement ultra-violet pendant 10 min (5 min par
côté). La membrane est ensuite prête à être hybridée.
2.9.4 Hybridation avec la sonde PAR-2
La membrane est préhybridée pendant au moins 1 heure à 65°C' sous
agitation, dans une solution de préhybridation de façon à bloquer les sites non-
spécifiques. La sonde est ensuite ajoutée (1 x 106 CPM / ml de solution) et
l'incubation se poursuit pendant 16 heures, toujours sous agitation à 65°C.

La solution de préhybridation contient:
6X SSC;
5X Solution de Denhardt's (0.1% BSA, O. 1% Ficoll, O. 1% polyvinylpyrolidone);
0.5% SDS;
200 pghl d'Am de transfert de levure;
100 pg/ml ADN simple brin de spenne de saumon;
Eau stérile.
Après l'hybridation la membrane est lavée 2 fois dans une solution 2X de
SSC + 0.1% SDS à température ambiante pendant 15 min et 1 fois dans la même
solution pendant 45 min à 65°C.
La membrane est ensuite placée au contact d'un füm Kodak X-OMAT AR
en présence d'un écran intensifiant (Dupont, Mississauga, ON) pour l'analyse
autoradiographique.
2.1 0 ANALYSE STATISTIQUE
Les résultats sont exprimés par la moyenne k l'emeur-type. La différence
entre deux moyennes est déterminee au moyen du test t de Student pour
échantillons appariés ou non, selon le cas. Lorsque plusieurs moyennes sont
comparées avec un témoin, une analyse de variance (ANOVA) est effectuée suivie
du test de Dunnett pour les mesures paramétriques ou le test de hkall-Wallis
suivi du test de Mm-Wtney dans le cas des mesures non-paramébriques. Les
statistiques sont compilées à l'aide de deux programmes informatiques soit
Phiwiacological Calculations N (Tdarida et Murray, 1987) et << InStat 2.0 ».

3.1 ÉTUDE DE STRUCTURE-ACTIVITÉ DES PEPTIDES AGONISTES
ET ANTAGONISTES DU RÉCEPTEUR DE LA THROMBINE
Le récepteur de la thrombine est activé par de courts peptides
co~~espondants à l'extrémité N-terminale exposée lors du clivage du récepteur par
l'enzyme. Le peptide NATM ainsi qu'un m e n t plus court, NAT6, sont capables
de dupliquer en grande partie les effets produits par l'activation du récepteur
ciivable par la thrombine. Cependant ces peptides sont sensibles a l'action des
enzymes plasmatiques telle l'aminopeptidase M. Dans le but de développer des
agonistes de meilleure afnnité et métaboliquement stables, nous avons entrepris
une étude de structure-activité en utilisant le peptide NAT6 comme structure de
départ. L'activité des différents peptides a été mesurée en contractilté directe sur
l'aorte de lapin isolée. Dans le cas des peptides inactifs leur activité antagoniste a
également été vérifiée. Les résultats de cette étude apparaissent au tableau 2. Outre
le NAT6-NH2, un seul peptide a démontré une activité agoniste significative dans
cet essai : le [s~~']NAT~-NH~. Les peptides ayant subi d'autres modincations en
position 1 ou 2 se sont avérés très peu actifs voire inactifs, comme le démontrent
les peptides [ M ~ P ~ ~ ~ ] N A T ~ - N H ~ , ~ ~ U ~ J N A T ~ - N H ~ , [AC~']NAT~-NH~,
AC[G~~']NAT~-N&, [ ~ l ~ ' Y ( c H ~ N H ) P ~ ~ ~ ~ N A T ~ - N H ~ et [T~~']NAT~-NH~.
Différentes structures rapportées dans la littérature comme étant des antagonistes
du récepteur de la thrombine ont également été synthetisées et leur activité
antagoniste contre la thrombine ou NAT6-= a été védïée en utilisant le même
modèle : But(CH2NH)-Leu-Leu-Arg-Asn-NH2 (A), But(CH?NH)-Leu-Leu-Leu-
hg-Asn-NH2 (B), But(CHzNH)-Phe-Leu-Leu-Arg-Asn-NHz (C),

Tableau 2 Étude de structure-activité des peptides agonistes et antagonistes du
récepteur de la thrombine
Peptide
- -
Activité (EDSo) (@id) n
Agoniste Antagoniste
Butyraldéhyde A
Butyrddéhyde B
Butyraldéhyde C
Cyclohexylméthyl
P508-53 1
P5 17-53 1
Prothrombine 367-3 80
Rétro NATa-NH2

(cycIohexyIméthyI)2-N-Phe-Cha-Cba-Arg-Ly~-Pr~-Asn-Asp-Ly~-NH2, p5 17-53 O,
p508-532 et prothrombine 367-380. Aucune de ces structures n'a cependant
démontré d'activité dans notre modèle, plusieurs d'entre elles ont même démontré
un certain pouvoir agoniste (tableau 2).
L'incubation de NAT6-W avec le plasma de lapin, source
d'aminopeptidase, mène à une dégradation continue de ce peptide (tableau 3).
Nous avons montré que la dégradation métabolique de cet hexapeptide peut être
presque complètement abolie par l'inhibiteur d'aminopeptidases amastathe. Une
augmentation de la stabilité métabolique est observée lors de la substitution du
résidu Ser en position N-terminale par un résidu sarcosine dans la structure du
NAT6-NI&. Le produit résultant, le [s~~']NAT~-N&, est métaboliquement très
stable dans ce milieu (tableau 3).
L'activité biologique de la thrombine et des peptides NAT a été comparée à
l'aide du modèle de l'aorte de lapin isolée. Les courbes concentration-effet (figure
6) montrent que NAT&& est plus puissant que NATu pour induire une réponse
contractile du tissu. Lorsque comparé à NAT6-m, [S~.~']NAT&JH~ est un
agoniste plus puissant que la structure natureue. Par contre quand les tissus sont
traités avec NAT6- en présence d'amastatine la courbe concentration-effet de
ce peptide est déplacée à la gauche de celle de
d'amastatine la concentration requise pour obtenix
est d'environ 10 fois moindre qu'en l'absence de
[ s ~ ~ ' ] N A T ~ - ~ . En présence
une réponse contractile de 2 g
L'inhibiteur d'aminopepbdases.
Les réponses au peptide [s~~']NAT~-NH~ et à la thrombine ne sont pas modifiées
de façon significative en présence d'amastatine (résultats non montrés). Même si
la réponse B NATs-NH2 est potentialisée par l'amastatine, l'activité de ce peptide
par rapport à la thrombine demeure relativement basse. Les courbes concentration-

Tableau 3 Taux d'hydrolyse de NAT6-- et [S~~']NAT~-W par
I'aminopeptidase M plasmatique
PEPTIDE DÉGRADATION MÉTABOLIQUE
Plasma Plasma + amastatine
(nmoVrnin par ml) (nmoVmin par ml)
' Les vaieurs représentent la moyenne * !'erreur-type de 6 expériences.

0.001 0.01 0.1 1 10 100 Io00
[Agoniste] (pM)
Figure 6 Activité contractile de la thrombine, NATI4, [S~~'INAT~-N&, NAT6-
NH2 et NATs-NH2 + 20 pM amastathe sur l'aorte de lapin isolée.
Chaque point représente la moyenne l'erreur-type d'au moins 3
expériences.

effet complètes pour les peptides NAT ne sont pas disponibles, ces courbes
nécessitant de trop grandes quantités de produits.
Une autre démonstration du rôle important de la dégradation métabolique
par l'aminopeptidase M dans la modulation de la réponse de l ' a~r te de lapin au
peptide NAT6- est obtenue en utilisant la technique d'immersion dans l'huile.
Dans cette expérience la relaxation dans l'huile minérale des tissus préalablement
contractés dans une solution Krebs avec NAT6-NH2 ou [ s ~ ~ ' ] N A T ~ - ~ est
évaluée en fonction du temps (figure 7). Une série d'essais sont effectués en
présence ou absence d'amastatine. Les résultats présentés à la figure 7 montrent
qu'en l'absence d'amastatine la relaxation induite par [S~~']NAT~-W est
prolongée comparativement a celle induite par NAT6-=. Alon qu'une relaxation
complète à NAT&& est obtenue en 60 min, il faut 90 min pour obtenir le même
degré de relaxation avec [S~~']NAT~-W. En présence d'amastatine une nette
augmentation du temps de relaxation est observée avec NATs-N& alors que
seulement 60% de relaxation est atteint après une période de 90 min. Le temps de
relaxation induit par [S~~']NAT~-W n'est pas modifié en présence de l'inhibiteur
d'aminopeptidases. D'autres inhibiteurs tels l'inhibiteur de I'enzyme de
conversion de l'angiotensine captopril (5 pM) et l'inhibiteur d'endopeptidases
phosphoramidon (10 CIM) n'ont aucun effet sur le temps de relaxation à NATa-
NH2 (données non illustrées).

O 20 40 60
Temps (min)
Figure 7 Courbes de relaxation dans l'huile des anneaux d'aorte thoracique de
lapin exposées au [S~~']NAT~-NH~ (1.4 x 104 M), NATs-N& (4.4 x 104
M) et NAT6-NH2 (4.4 x lo4 M) en présence de 20 plbl d'amastatine.
Chaque point représente la moyenne k 1'eneu.r-type d'au moins 4
expériences.

3.2 MODE D'ACTION DE LA THROMBINE ET DES PEPTIDES NAT
CHEZ LE LAPIN
3.2.7 Études de contacfilité
Une série d'experience a été réalisée afin de comparer le mode d'action de la
thrombine avec ceux du peptide [ s ~ ~ ' ] N A T ~ - ~ et du KCI, ce dernier étant
utilisé comme agoniste témoin. Des concentrations sous-maximales de chacun de
ces agonistes ont été choisies en se basant sur des expériences réalisées
précédemment (figure 6) (Levesque et COU., 1993). Ann d'éviter toute
tachyphylaxie, [S~~']NAT~-N& est toujours le premier agoniste injecté après la
période d'équilibration des tissus (2 h), suivi de la thrombine et du KCl en
conservant un intervalle de 2 h entre chaque injection d'agoniste. Des témoins
appariés sont utilisés pour évaluer l'effet des solvants, s'il y a lieu Les inhibiteurs
sont injectés 30 min avant l'enregistrement des réponses a chacun des agonistes.
Comme il est montré au tableau 4 le dommage intimal ne modifie pas la
réponse des tissus à ces agonistes alors qu'il bloque complètement la relaxation
induite par l'acétylcholine, utilisée comme témoin, sur des tissus précontractés à la
phényléphrine (données non illustrées). La nifédipine, un bloqueur des canaux
calciques dépendants du voltage, ne produit aucun effet sur la contraction induite
par la thrombine alors qu'une inhibition partielle de la contraction induite par
[S~~']NAT~-- est observée. Une inhibition prononcée de la réponse au KCl sur
les mêmes tissus prouve l'efficacité de cet inhibiteur dans cet essai. La contraction
induite par le KCI est lourdement aectée en présence d'une solution de Krebs
exempte de calcium, la réponse à [s~~']NAT~-N& est réduite d'environ 1/3 par
rapport au témoin, alors que celle de la thrombine n'est pas du tout affectée par

Tableau 4 Étude pharmacologique du mode d'action de la thrombine et de
[S~~'NAT~-N& sur l'aorte de lapin isolée
KCl
(20
Dommage intima1
Témoin
Nifédipine 100 nM
Solvant (EtOH)
Krebs sans calcium
Témoin
Diclofénac 0.5 pM
Solvant (Na2.C03)
Erbstatine anaiogue
Solvant (DMSO)
PPACK 1 pM
Soivant (saline)
- -- - - - -.
*** a Analyse statistique par le test t de Student : P < 0.05; P < 0.001.

l'absence de calcium. Le diclofénac, un inhibiteur de la cyclo-oxygénase des
acides gras, et l'erbstatine analogue, un inhibiteur de tyrosine kinases n'ont aucun
effet dépresseur sur la contraction induite par les trois agonistes présentés au
tableau 4. Un inhibiteur de l'activité catalytique de la thrombine, le PPACK,
prévient complètement la réponse contractile de l'aorte de lapin à la thrombine a
une concentration de 1 m. L'activité de [s~~']NAT~-NH, et du KC1 n'est pas
affectée par cet inhibiteur.
Suite à l'observation que des concentrations uniques de thrombine et de
[s~~']NAT~-N& sont affectées par I'inhibiteur de protéine kinases H-7, des
courbes concentration-effet complètes pour chacun de ces agonistes ont été
réalisées en présence de cet inhibiteur (figure 8). Comme nous pouvons l'observer
sur cette figure, le H-7 (10 CLM) réduit de façon signincative l'activité de toutes les
concentrations de thrombine et de [S~~']NAT~-NH~ utilisées. Des expériences
réalisées précédemment sur ces mêmes tissus ont révélé que le H-7 réduit la
contraction induite par le phorbol myristate acétate (PMA) (activateur de protéines
bases) alors qu'il affecte de façon très minimale la contraction au KCl (Levesque
et cou., 1993).
Un certain nombre d'expériences de désensibilisation du récepteur de la
thrombine ont été réalisées afin de vérifier la cinétique de réexpression des
récepteurs suivant une première stimulation. Pour ce faire 3 types d'agents
pharmacologiques ont été utilisés : la bréfeldine A, un inhibiteur du transport
protéique entre le réticulum endoplasmique et l'appareil de Golgi; l'anisomycine
et la cycloheximide, deux inhibiteurs de la synthèse protéique. Tel que montré à la
figure 9, les courbes concentration-réponse cumulatives à la thcombine effectuées
après une période d'équilibration d'une heure ne sont pas affectées par la présence
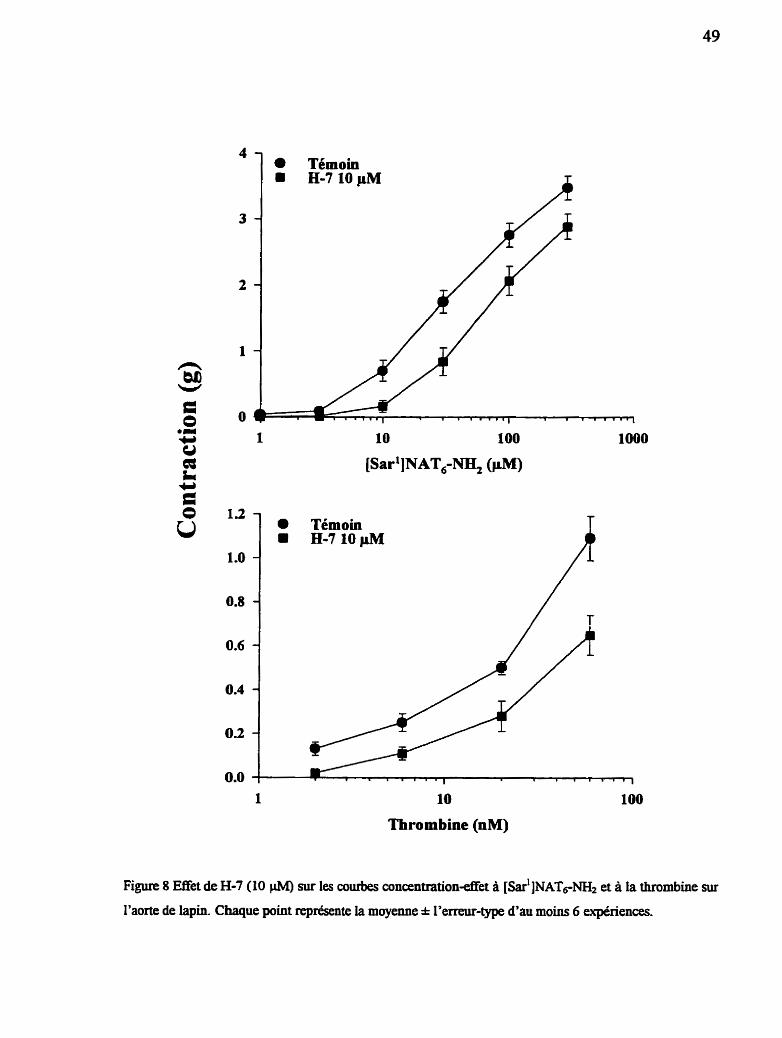
Témoin
Témoin EL7 10pM
10
Thrombine (nM)
Figure 8 Enet de H-7 (10 CiM) sur les courbes concentration4et A [ s ~ ~ ' ] N A T ~ N ~ & et à la thrombine sur
l'aorte de lapin. Chaque point représente la moyenae * l'erreur-type d'au moins 6 expériences.
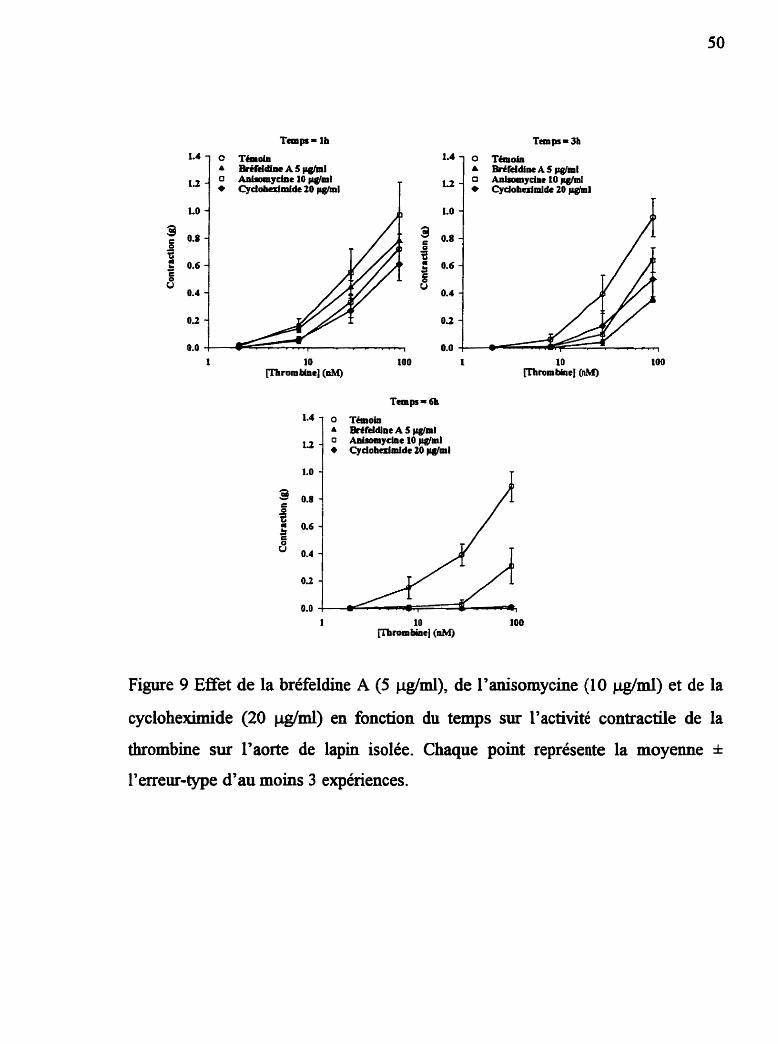
Figure 9 Effet de la bréfeldine A (5 @ml), de l'anisomycine (10 clg/ml) et de la
cycloheximide (20 &ml) en fonction du temps sur l'activité contractile de la
thrombine sur l'aorte de lapin isolée. Chaque point représente la moyenne * l'erreur-type d'au moins 3 expériences.

des inhibiteurs. Les courbes concentration-effet réalisées sur les mêmes tissus
après 3h d'équilibration montrent un effet certain mais peu prononcé des trois
substances inhibitrices. Finalement les courbes réalisées au temps 6h montrent que
les tissus mis en présence d'inhibiteurs ne répondent pas ou très peu à la
thrombine et ce par rapport aux tissus témoins, suggérant une population de
récepteurs diminuée, voire absente.
3.2.2 CelIuIes de muscle lisse de l'aorte de lapin en culture
3.2.2.1 Essai de liaison
La figure 10 montre les résultats de l'essai de liaison réalisé sur les cellules
en culture de muscle lisse de l'aorte de lapin en utilisant comme ligand la
thrombine marquée à l'iode 125. Le volet A de la figure représente la saturation du
récepteur avec les courbes de liaison totale, liaison spécifique et liaison non-
spécifique. La liaison spécifique est obtenue en soustrayant la liaison non-
spécifique, de la liaison totale, dans ce cas-ci la liaison spécifique est d'environ
30000 cpm. Le volet B de la figure représente l'analyse de Scatchard dérivée de la
courbe de saturation du volet A. Cette analyse démontre la présence de deux sites
de liaison pour la thrombine : un site de haute affinité de KD = 0.91 nM et un B,,
de 71.8 b o l / 106 cellules ainsi qu'un site de basse afsnté avec un KD = 3484 nM
et un B,, de 1005 f ino~ 106 cellules. L'expérience a été répétée en présence d'un
excès de thrombine non-marquée pendant la période d'équilibration avec le ligand.
Comme on peut le voir à la figure 11, la cinétique de liaison de la [L25~thrombine
est grandement affectée par cet excès de ligand ftoid. Le volet A de la figure 11
illustre la courbe de saturation du récepteur réalisée dans ces conditions et le volet
B représente l'analyse de Scatchard pour cet essai. On constate la disparition du

Liaison totale Liaison non-lpécifique
O 100 200 300 400 500 600
Lié (fmolel
Figure 10 Liaison de i25~-thr~rnbine sur les CML de lapin. (A) Courbe de saturation ternoin. (B)
Anaiyse de Scatchard.

'Ooo0 1 A Liaison totale 0 Liaison non-spéafique
100 150 200 250
[125~-t h rom bine] (nM)
O 100 200 300 400 500 600
Lié (fmole)
Figure 11 Liaison de lU~-thrombine sur les CML de lapin. (A) Courbe de saturation en présence de
1 phi de thrombine nonmarquée. (B) Analyse de Scatchard.

site de liaison à haute afnnité mais le site de basse afnnité demeure, le KI, de ce
site est de 1220 nM et son B, est de 985 f m o ~ 1 0 ~ cellules.
3.2.2.2 Production des phosphatidyl~nosfiols par les CML
Les cellules de muscle lisse en cuiture derivées de I'aorte de lapin
répondent positivement à la thrombine et au peptide NAT6-- en augmentant
leur production de phosphatidylinositols (IP) (tableau 5). Une augmentation
sigdicative de la production d'Pz et d91P3 est observée chez les cellules stimulées
par la thrombine. Même si l'on observe un effet concentration-dépendant avec la
thrombine, des valeurs sigmfïcatives sont obtenues seulement aux concentrations
de 10 et 1000 n M pour IP3 et 1000 nM dans le cas d'm. Le peptide NA'ï6-N&
augmente la production d'IP1, d'Pz et d'IP3; dans ces deux demiers cas
l'augmentation est concentration-dépendante et est significative. Le tableau 5
montre I'effet de concentrations croissantes de thrombine et de NAT6-NH2 sur la
production totale de phosphatidyIinositols.
3.2.2.3 Incorporation de la t~lthymidine par les CML
Le pouvoir qu'ont la thrombine et les peptides NATs-NH2 et [SU']NAT~-
a stimuler l'incorporation de la [3~thymidine chez les CML quiescentes a
également été testé. La thrombine stimule l'incorporation de la thymidine dans ces
cellules de façon concentration-dépendante (figure 12) avec un effet maximal
atteint a 10 nM. Contrairement a la thrombine, les peptides NAT6-m et
[S~~']NAT~-W ne provoquent pas d'incorporation même à des concentrations
dépassant les 100 W. Suite à cette observation, plusieurs inhibiteurs ont été

Tableau 5 Mesure des phosphatidylïnositols dans les cellules de muscle lisse de
l'aorte de lapin après 5 min d'exposition à diffirentes concentrations de thrombine
ou de NAT6-NHF
PHOSPHATIDYLINOSITOLS (c-p m . par puit)
S thdus IPr pz p 3 p4
Témoin 6995 * 1381 765 & 140 328 * 21 308 * 66
Thrombine 10 nM 8277 * 1743 929 * 97 437 * 32.' 273 * 24
Thrombine 100 nM 6038 * 1547 1377 * 371 509 * 79 236 * 39
Thrombine 1000 nM 1 1077 1552 3074 i 372" 1263 k 104"' 393 * 41
Analyse de varianceb NS P < 0.0001 P < 0.0001 P < 0.05
a Cellules provenant de 3 différentes lignées cellulaires au passage 4 ou 5. Les valeurs représentent
la moyenne * l'erreur-type de 9 expériences en tripikatas. Les résultats sont nonnalisés en c.p.m.
d71P par 100 000 c.p.rn. de myo-inositol associé aux eiiules.
Analyse de variance appliquée à chacune des colonnes (NS, non-significatif), suivie du test de
Mann-Whitney pour comparer chacun des groupes avec le groupe témoin : ' P < 0.05; " P < 0.01;
.*' P < 0.00 1

0.0 1 o. 1 1 10 100 Io00 1 0 m 1OOOOO
[Agoniste] (nM)
Figure 12 Effet de la thrombine, NAT6-NH2 et [S~~']NAT~-W sur
I'incorporation de la [3~thymidine dans les cellules en culture de
muscle lisse de l'aorte de lapin. La ligne pointillée indique le niveau
d'incorporation basal en présence de 0.4% de sérum de veau foetal.

utilisés afin d'élucider le mode d'activation de la synthèse d'ADN induite par la
thrombine chez les CML. Les résultats de cette étude apparaissent à la figure 13.
Des expériences récentes ont démontré que le diclofénac, un inhibiteur de la
cyclo-oxygénase des acides gras, est utile pour révéler l'incorporation de
thymidine provoquée par différents stimuli dans ce modèle (Levesque et COU.,
1993). Tel qu'illustré à la figure 13, le diclofénac (0.5 pM) révèle l'expression
maximale de l'incorporation de la thymidine chez les cellules stimulées par la
thrombine. En présence de diclofénac, la PG& et 17110prost (un analogue de PG12)
produisent une inhibition partielle de la synthèse d'ADN chez les cellules
stimulées par la thrombine (résultats non-montrés). La nifédipine provoque une
augmentation significative de l'incorporation de la thymidine autant chez le groupe
témoin que celui stimulé par la thrombine. Une légère augmentation est également
observée avec le PMA chez les cellules témoins alors que la Wortmannine, un
inhibiteur de la phospholipase D, ne produit aucune inhibition significative de
l'incorporation de thymidine chez les cellules témoins ou celles stimulées par la
thrombine. Lorsqu'utilisé a une concentration de 10 pM l'inhibiteur de la protéine
h a s e C, H-7, ne produit aucun effet autant chez le groupe témoin que chez celui
stimulé par la thrombine. Par contre à une concentration de 30 @A, cet inhibiteur
ramène l'effet de la thrombine à son niveau de base. Il en est de même pour
l'inhibiteur de tyrosine kinases : I'erbstatine analogue. Le H-7 utilisé a hautes
concentrations provoque également une diminution de I'incorporation chez le
groupe témoin, suggérant une certaine toxicité de ce produit chez ces cellules.
3.2.3 Effet des peptides NAT in vivo chez le lapin anesthésie
Lorsqu'injectés chez le lapin anesthésié, le NAT6-NH2 et le [S~~']NAT~-
N& produisent une réponse hypotensive qui est dose-dépendante et sensiblement
de même amplitude (figure 14). Cependa* à la plus haute dose injectée (3

Témoin ** ThmmbinelnM 1
Figure 13 Effet de différents inhibiteurs sur l'incorporation de la [3Hlthymidine
dans les cellules en culture de muscle lisse de l'aorte de lapin. Les
colonnes hachurées représentent I'incorporation basale (témoin); les
colonnes noires I'incorporation induite par stimulation avec la
thrombine (1 nM). PMA : phorbol myristate acétate; Erb : erbstatine.
Chaque barre représente la moyenne k l'erreur-type d'au moins 3
expériences. Analyse statistique par le test t de Student : P < 0.05; ** P ***
co.01; P co.001.

10"
Dose d'agoniste (molkg)
Figure 14 Réponses hypotensives induites par l'injection LV. de NAT6-NH2 O et
[ S ~ ~ ' W A T ~ - ~ (A). La diminution de la pression artérielle moyenne
(PAM) est exprimée comme étant la différence entre le niveau de base
et la réponse la plus grande obtenue suivant I'injection du peptide.
Chaque point montre la moyenne I l'erreur-type de 4 expéiences
réalisées chez des lapins difErents.

mgkg), l'effet hypotenseur de [s~~']NAT~-N& est signincativement plus
important ( P < 0.05; test t de Student) que celui induit par NAT6-MI2. Comme on
peut l'observer à la figure 15% l'injection de NAT6-MI2 produit une réponse
hypotensive rapide suivie d'un retour a la pression de base en moins d'une minute.
Pendant l'infusion d'amastatine chez le même animal, l'amplitude de la réaction
hypotensive est légèrement plus prononcée (figure 15b), et la durée de l'effet est
grandement augmentée prolongeant de façon signincative le retour B la pression
basale. Ce fait nous semble important.
3.3 EFFETS DES PEPTIDES AGONISTES DU RÉCEPTEUR PAR-2
SUR L'AORTE DE LAPIN ISOLÉE
Le récepteur PAR-2 est également activable par de courts peptides
correspondants à la nouvelle extrémité N-terminale dévoilée lors du clivage du
récepteur par la trypsine. La figure 16 montre les courbes concentration-réponse
contractiles réalisées chez l'aorte de lapin isolée avec la trypsine et le peptide
SLIGFtL-W. Les courbes à la thrombine et au NATs-NH2 sont représentées pour
£in de comparaison. Cette figure nous permet de constater que les récepteurs PAR-
2 et ceux de la thrombine réagissent de façon très semblable. Comparativement au
peptide SLIGRL-NH2, des concentrations environ 1000 fois plus petites sufnsent à
la trypsine pour agir, comme c'est le cas pour la thrombine et NAT6-NH2 au
niveau du récepteur de la thrombine (figure 16). Dans ce modèle, le peptide
SLIGRL-NH2 semble même plus puissant que NATd-NH2. Contrairement à la
thrombine et à NAT6-N&, la trypsine et SLIGRL-NH2 provoquent une relaxation
dépendante de l'endothélium dans l'aorte de lapin isolée et précontractée avec de
la phényléphrine. La figure 17 montre l'activité relaxante du peptide SLIGRL-NH2
(a) et de la trypsine @) ainsi que I'absence de réponse a NAT6-NH2 (c) et à la

B. Infusion d'amas~tine
n 1 min
Figure 15 Tracés représentatifs
lapin anesthésié des
de I'effet hypotenseur in vivo chez le
peptides agonistes du récepteur de la
thrombine. Ces tracés sont représentatifs de 4 expériences
réalisées chez des lapins différents.
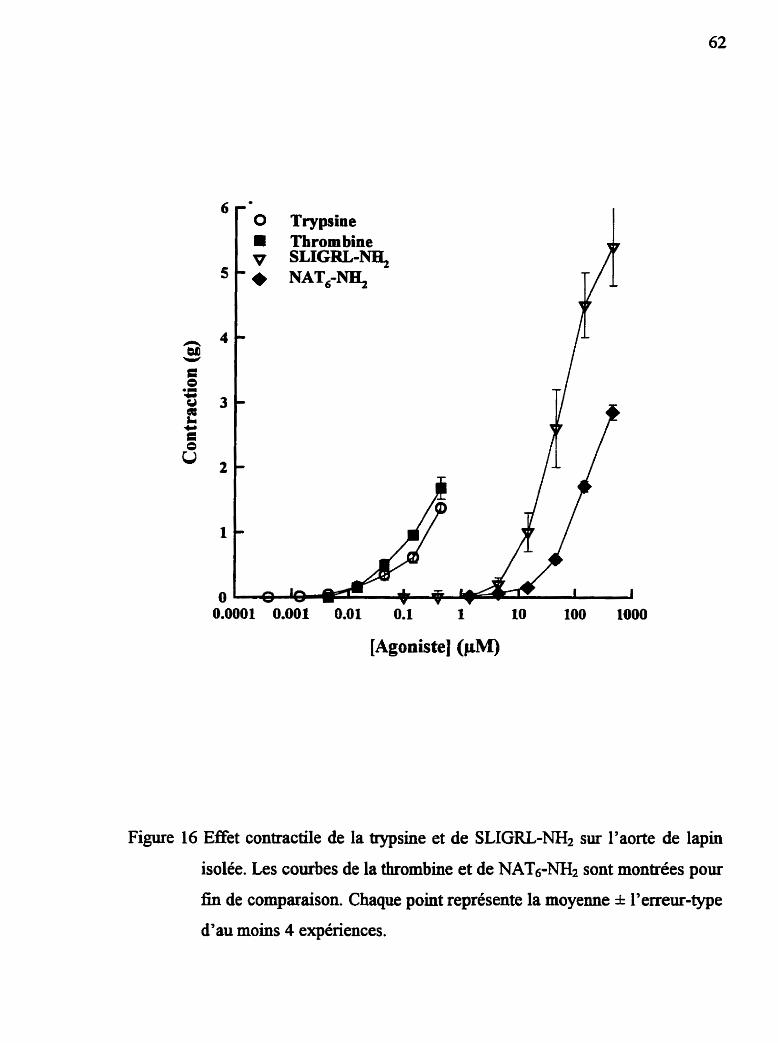
O Trypsine Thrombine
v SLIGRL-Na, ' NAT,-
[Agoniste] (PM)
Figure 16 Effet contractile de la trypsine et de SLIGRL-NH2 sur l'aorte de lapin
isolée. Les courbes de la thrombine et de NAT6-- sont montrées pour
£in de comparaison. Chaque point représente la moyenne l'erreur-type
d'au moins 4 expériences.
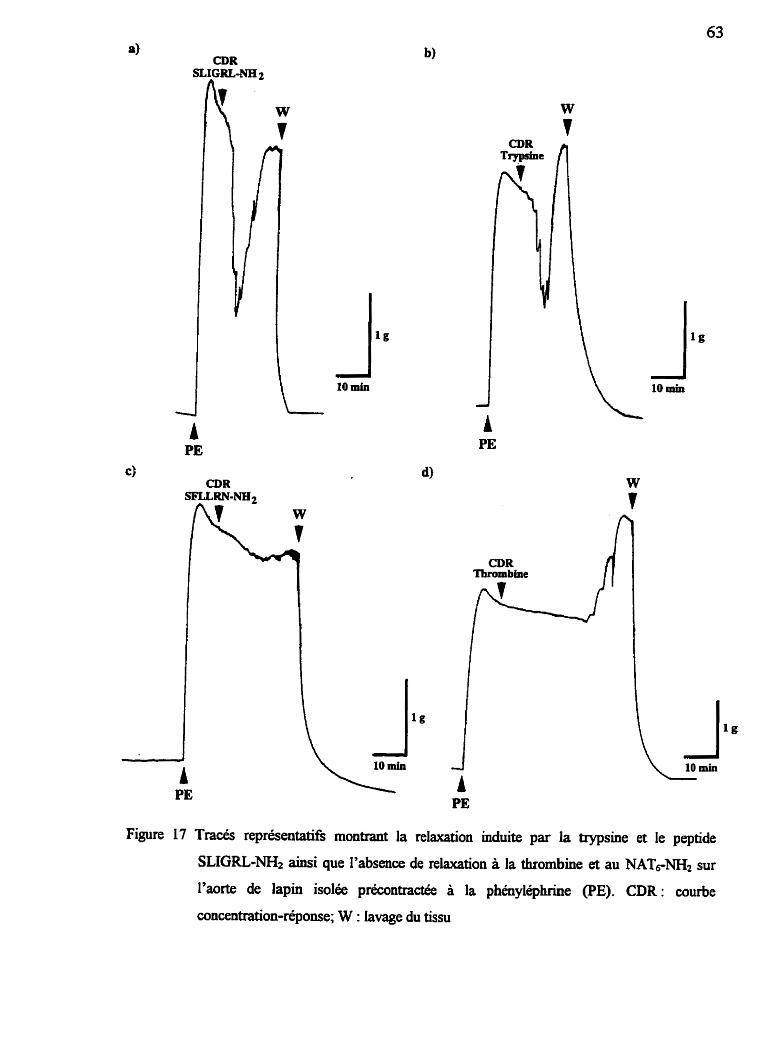
CDR
I l g 10 min
Figure 17 Tracés représentatifk montrant la relaxation induite par la trypsine et le peptide
SLIGRL-N& ainsi que I'absence de relaxation à la thrombine et au NATrNH2 sur
l'aorte de lapin isolée précontractée à la phényléphrine (PE). CDR: courbe
concentration-répoose; W : lavage du tissu

thrombine (d). Cette relaxation induite soit par la trypsine ou le peptide est
rapidement dominée par la réponse contractile des ceiiules de muscle lisse du
même vaisseau dès que les concentrations utilisées augmentent. La figure 18
représente graphiquement les réponses relaxantes à la trypsine et à SLIGRL-NH2;
l'inhibition par la nitro-L-arginine est également montrée, suggérant un mécanisme
d'action dépendant de la synthèse d'oxyde nitrique.
3.4 ÉTUDE DE STRUCTURE-ACTIVITE DES PEPTIDES AGONISTES
DU RÉCEPTEUR PAR-2
Une approche dinérente de celle utilisée dans le cas du récepteur de la
thrombine a été utilisée pour la synthèse d'agonistes peptidiques du récepteur
PAR-2. Le tableau 6 montre les différents peptides synthétisés lors de l'étude de
structure-activité. Le EDzov. calculé correspond à la concentration de peptide
nécessaire pour induire 20% de la relaxation maximale chez l'aorte de lapin pré-
contractée à la phényléphrine. Le premier volet de ce tableau montre l'hfiuence de
la substitution de chacun des résidus par un résidu alanine. Cette étude permet de
constater qu'il est difficile de substituer un résidu à l'intérieur de cette structure
sans perdre une grande partie de l'activité du peptide, à l'exception du résidu Arg
en position 5. Comme dans le cas des peptides agonistes du récepteur de la
thrombine, toute substitution des deux premiers acides aminés engendre des
peptides très peu actifs voire inactifs, tel que montré plus particulièrement dans le
second volet du tableau 6. Ces structures ont été synthétisées dans le but de
détecter toute réactivité croisée entre les récepteurs de la thrombine et PAR-2. Ces
séquences hybrides se sont cependant révélées inactives chez les deux récepteurs.

0.0001 0.001 0.01 0.1 1 10 100 Io00
[Agoniste] (pM)
Figure 18 Activité relaxante de la trypsine et de SLIGRL-NH2 su. I'aorte de lapin
isolée en présence ou non de N-G-nitro-L-arginine (Arg-NO2) (50 W. Chaque point représente la moyenne * l'erreur-type d'au moins 4
expériences.

Tableau 6 Étude de structure-activité des agonistes du récepteur PAR-2. Les
acides aminés soulignés indiquent les substitutions effectuées.
SLIGRL 9.67 * 0.43

3.5 DÉTECTION DE L'ARN MESSAGER CODANT POUR LE
RÉCEPTEUR PAR-2 DANS L'AORTE DE LAPIN
La présence du récepteur PAR-2 chez le lapin a été démontrée de façon
pharmacologique mais étant donné la possibilité de réactivité croisée avec le
récepteur de la thrombine, il s'avérait important de démontrer hors de tout doute la
présence de ce récepteur dans l'aorte de lapin. La figure 19 montre le transfert de
type Northern effectué avec de 1'ARN provenant de L'aorte et du coeur de lapin.
Un signal intense apparaît aux environs de 0.9 kb, longueur qui conespond à celle
de 1'- attendu. L'intensité plus grande du signal provenant du coeur est
possiblement due à une contamination par 1'ARN de cellules sanguines présentes
dans le coeur au moment de l'extraction.

Figure 19 Hybridation de type Northem utilisant une sonde spécifique pour le
récepteur PAR-2.

4.0 DISCUSSION
Les récepteurs activés par protéolyse forment une nouvelle classe de
récepteurs à sept domaines transmembmaires couplés aux protéines G. Ils sont
activés par clivage sélectif a un site particulier de leur extrémité N-terminale.
Jusqu'à maintenant deux récepteurs de cette famue ont été caractérisés, le
récepteur de la thrombine (Vu et cou., 1991) et le récepteur PAR-2 (Nystedt et
coll., 1994), un récepteur activable par la trypsine. L'expression de fortes densités
de chacun de ces récepteurs au niveau de l'endothélium vasculaire ainsi qu'au
niveau des cellules de muscle lisse vasculaire suggère que chacun de ces deux
récepteurs powait jouer un rôle important dans la régulation du tonus vasculaire.
4.1 STRUCTURE-ACTIVITÉ ET STABILITÉ MÉTABOLIQUE DES
AGONISTES ET ANTAGONISTES DU RÉCEPTEUR DE LA
THROMBINE
La découverte indiquant que le récepteur de la thrombine pouvait être activé
par de courts peptides correspondants à la nouvelle extrémité N-terminale dévoilée
par le clivage a initié toute une série d'études de structure-activité dans lesquelles
le peptide NAT14 a été utilisé comme structure de départ (Vassalo et COU., 1992;
Hui et COU., 1992; Scarborough et coll., 1992; Chao et cou., 1992). Ces études ont
principalement fait appel au modèle d'agrégation plaquettaire pour évaluer la
puissance des analogues ou fragments synthétisés. L'activité de NATl4 a
également été testée sur des tissus isolés tels l'aorte de rat et le muscle lisse
gastrique (Yang et cou., 1992) ainsi que l'aorte de lapin, où la thrombine agit de

façon synergique avec le facteur de croissance épidermique (Deblois et coll.,
1992). Nos résultats obtenus avec le modèle de l'aorte de lapin viennent confirmer
les expériences réalisées précédemment chez les plaquettes, à savoir que
l'hexapeptide NAT6- est un agoniste légèrement plus puissant que NATi4 et
que ce peptide peut senRr adéquatement de modèle pour le développement
d'agonistes ou d'antagonistes du récepteur de la thrombine.
La plupart des agonistes du récepteur de la thrombine décrits jusqu'à
maintenant sont des dérivés des peptides agonistes de ce récepteur. Des études de
structure-activité ont été entreprises afin de caractériser les éléments structuraux
requis pour obtenir une effet agoniste optimal. Certains changements ont
également été apportés à la structure peptidique dans l'espoir de développer un
antagoniste du récepteur. Nous avons démontré que l'effet agoniste optimal est
obtenu avec des peptides comportant 6 acides aminés. De plus, nos études ont
démontré que le groupement amide (MI2) à l'extrémité carboxy-terminale est
important pour obtenir un plein effet agoniste (résultats non-montrés). Le résidu
Ser N-texminal est également cmcial pour conserver un effet agoniste, le
remplacement de ce résidu par un groupement Aca, Ac-Gly ou Gly(CH2NH)
engendre des composés inactifs qui ne possèdent pas d'activité antagoniste. Ces
résultats sont en accord avec une étude récemment publiée par le groupe de
Natarajan et COU. (1995) qui montre que certaines substitutions, notamment avec
des résidus chargés, ne sont pas bien tolérées a cette position. D'autres études
menées par diffkrents groupes (Chao et cou., 1992; Scraborough et cou., 1992) ont
montré que lYacétyIation de ce résidu menait a un composé inactif. Notre étude
montre cependant que le résidu N-terminal peut tolérer un certain degré de
substitution, la N-méthylation est parfatement compatible avec l'activité
biologique de ce peptide. L'ajout d'un résidu Tyr en aval de la sérine modifie t r è s
peu le comportement agoniste du peptide NAT6-bJ& (tableau 2); ce composé a été

synthétisé dans le but d'être utilisé comme ligand dans d'éventuelles études de
liaison du récepteur de la thrombine avec les peptides agonistes.
Nous avons aussi tenté de remplacer le résidu Phe en position 2 par des
résidus contenant soit une chaîne latérale aromatique (MePhe) ou bien une chaîne
latérale hydrophobe (Leu). Ces substitutions, tout comme celles en position 1, ont
engendré des peptides ne possédant aucune activité agoniste ou antagoniste. La
seule substitution tolérée à cette position a été rapportée récemment dans la
littérature; il s'agit de la para-fluorophényldanine qui augmente de trois à cinq
fois le pouvoir agoniste de ce peptide (Nose et COU., 1993).
Plusieurs tentatives de développement d'antagonistes basées sur les études
de structure-activité ont été rapportées dans la littérature. Nous avons synthétisé
plusieurs de ces composés : But(CHzNH)-Phe-Leu-Leu-kg-Asn-N&
But(CH2NH)-Leu-Leu-Leu-Arg-Asn-N& et But(CH2NH)-Leu-Leu-Arg-Asn-,
(cyclohe~yhéth,y1)z-N-Phe-Cha-Cha-Arg-Lys-~o-Asn-Asp-Lys-~~
(Scarborough et cou., 1993)' RGDAI4 et RGDAu ainsi que le fiagrnent de la
prothrombine 367380 (tableau 2). Malgré ce qui a été publié, nous n'avons
observé aucune activité antagoniste dans notre modèle contrairement aux auteurs
de ces articles. Ceci est probablement dû au fait que ces antagonistes ont été testés
dans des modèles différents; il est plausible qu'ils se comportent Méremment
d'un modèle à l'autre. De plus, il n'existe pas a ce jour d'antagoniste pur du
récepteur de la thrombine rapporté dans la littérature.
D'autres études utilisant des tissus de lapins ont également révélé
l'importance des peptidases dans la modulation de l'activité biologique d'agonistes
et d'antagonistes synthétiques (Drapeau et cou., 1991, 1993). En prenant en
considération l'importance du résidu Ser N-terminal dans l'activité des peptides

NAT, nous avons
biologiques induites
examiné le rôle des aminopeptidases dans les réponses
par ces peptides. Les résultats indiquent que le plasma peut
dégrader NAT6-= par une activitk peptidase vraisemblablement associée à
l'aminopeptidase M, puisque l'activité de cette dernière est complètement inhibée
par de faibles concentrations d'amastatine. Cette observation est d'ailleurs
corroborée par le groupe de Coller et COU. (1992) qui ont montre que
l'aminopeptidase M était responsable du clivage du résidu Ser N-terminal d'un
fiagrnent N-terminal de 11 acides aminés dérivé de NATl4 (NATli). Les résidus
N-méthyl sont souvent utilisés pour fournir une protection contre les
aminopeptidases (Drapeau et cou., 1991); le remplacement du résidu N-terminal
Ser par une sarcosine dans la structure du NAT6* fournit une protection
complète contre l'activité de l'aminopeptidase plasmatique puisque [s~~']NAT~-
N& n'est pas dégradé dans les conditions où NATs-NH2 l'est complètement
(tableau 3).
L'aminopeptidase M, également présente au niveau des tissus vasculaires,
est capable d'innuencer les réponses biologiques au NAT6-NH2. Cette influence
est de nature quantitative plutôt que qualitative puisque la fome de la courbe
concentration-effet de NAT6-m2 est la même en présence ou absence
d'amastatine (figure 6) et elle est également de même nature que celle de NATl4 et
de [ s ~ ~ ' ~ A T ~ - N & . L'effet de cette arninopeptidase sur l'activité biologique des
peptides agonistes sur l'aorte de lapin est appréciable et montre I'importance de
pouvoir distinguer entre l'activité biologique et l'afnnité d'un peptide donné. Les
courbes concentration-effet de la figure 6 indiquent que NATo-NH2 possède une
mefleure aftùlt6 pour le récepteur que [S~~'JNAT~-W puisque dans les
conditions où le métabolisme n'est pas en cause la courbe concentration-effet de
NAT6-N& est déplacée à la gauche de celle de [s~~']NAT~-~&. Nous pouvons
donc affirmer que la substitution du résidu Ser par un résidu Sar a crée un produit

d'affinité légèrement diminuée dont la perte d'afnnté est compensée dans les
essais biologiques par une résistance métabolique augmentée.
Les expériences utilisant la technique d'immersion dans l'huile indiquent
que l'aminopeptidase est présente dans le micro-environnement immédiat du
récepteur et que le confinement de petites quantités d'agonistes dans la phase
liquide entourant le tissu mène à une relaxation graduelle qui peut être attribuée au
métabolisme de l'agoniste. Ceci est montré par la relaxation plus lente de la
contraction induite par [ s ~ ~ ' ] N A T ~ - ~ comparativement a la même expenence
réalisée avec NAT6--. Le temps de relaxation est maximal quand la contraction
est induite par le NAT6-= en présence d'amastatine (figure 7).
L'aminopeptidase M semble jouer un rôle important dans la dégradation de NAT6-
NH2 dans ce tissu puisque d'autres inhibiteurs de peptidases n'ont aucun effet sur
la relaxation de ces tissus.
4.2 ÉTUDE DU MODE D'ACTION ET DE LA DÉSENSIBILISATION DU
RECEPTEUR DE LA THROMBINE
4.2.1 Modele de l'aorte de lapin isolée
Nous avons étudié le mode d'action de la thrombine et d'un peptide
agoniste de ce récepteur (NAT6-N& ou [s~~']NAT~-w) dans le modèle de
l'aorte de lapin isolée (études de contractilité) ainsi que dans un modèle de cellules
de muscle lisse dérivées du même tissu (essais de liaison, production des
phosphatidylinositols et mitogénèse). Les réponses vasculaires à la thrombine
semblent vatier selon les espèces; certains lits vasculaires comme les artères

coronaires porcines
thrombine ou à un
et canines ainsi que les veines saphènes canines répondent à la
agoniste peptidique du récepteur clivable par une relaxation
dépendante de l'endothélium induite par le relâchement d'oxyde nihique ainsi que
par une contraction indépendante de ce même endothélium (Tesfamariam et coll.,
1993; Ku et Zaleski, 1993). La réponse contractile de l'aorte de lapin isolée à la
thrombine et à [S~~']NAT~-W est produite par le muscle lisse puisque la
résection de l'endothélium n'affecte pas la réponse à chacun des agonistes ou au
KC1 utilisé comme agoniste témoin (tableau 4).
Étant donné qu'il a déjà été démontré que la réponse contractile de l'aorte
de rat est dépendante du calcium extracellulaire, nous avons voulu vérifier si
l'aorte de lapin se comportait de la même façon. Une contraction soutenue est
observée avec la Uuombine en absence de calcium alors que la réponse au KCl,
(un stimulus dépolarisant dépendant du calcium extracellulaire) (Cauvin et coll.,
1983)' est inhibée d'environ 90% suite au retrait du calcium. Dans ces mêmes
conditions une inhibition modeste de la réponse à [ S ~ ~ ' ] N A T ~ ~ ! & est observée.
L'inhibition partielle de l'activité contractile de ce peptide suite au retrait du
calcium est du même ordre de grandeur que l'inhibition produite par la nifédipine,
un bloqueur des canaux calciques. Alors que la nifédipine n'affecte pas la réponse
contractile à la thrombine, elle abolit complètement la contraction induite par le
KCl, résultat qui vient confhner son rôle de bloqueur des canaux calciques
dépendants du voltage (Murad., 1990).
Il est important de mentionner que les réponses à la thrombine ainsi qu'aux
agonistes du récepteur clivable de l'aorte de lapin ou de rat Werent sur certains
aspects. Premieremenf la thrombine humaine ou de rat ne produit aucun effet sur
l'aorte de rat alors que cette demière répond bien aux peptides agonistes du
récepteur clivable (Antonacio et cou., 1993). En plus de montrer une relaxation

dépendante de l'endothélium, réponse qui n'est pas observée sur l'aorte de lapin,
les réponses contractiles de l'aorte de rat aux agonistes peptidiques dépendent
complètement du calcium extracellulaire. Dans les préparations d'aorte de lapin
l'effet de la thrombine est indépendant du calcium extracellulaire et la contraction
à [s~~']NAT~-= est seulement partiellement affectée par le retrait du calcium ou
le blocage des canaux calciques par la nifédipine. L'inhibition partielle de la
contraction induite par [S~~'INAT~-BJ&, provoquée par le retrait de calcium ou
l'addition de nifédipine dans le bain, laisse présager que l'activation du récepteur
par le peptide (qui ne nécessite ou provoque aucune protéolyse) dépend plus du
calcium extracellulaire que l'activation par clivage par la thrombine. L'inhibition
peut aussi être provoquée par l'activation d'un récepteur différent insensible à
l'activité protéolytique de la thrombine.
Les données obtenues avec les anneaux d'aorte de lapin suggèrent
I'implication de protéine kinases dans les réponses contractiles induites par la
thrombine ou le [s~~'JNAT~-NH~. La protéine kinase C est inhibée par le H-7
(Hidata et coll., 1984)' l'efficacité de ce dernier à réduire la réponse au PMA (un
activateur de la protéine kinase C) a déjà été démontrée (Levesque et COU., 1993).
Le H-7 déplace aussi les courbes concentration-réponse au [S~~']NAT~-W et à la
thrombine (figure 8). Par contre, l'utilisation de H-7 comme inhibiteur spécifique
de la protéine kinase C présente certaines limites, à savoir que cet inhibiteur peut
aussi inhiber d'autres protéine kinases, comme par exemple les protéine kinases
dépendantes des nucléotides cycliques (Hidata et coll., 1984). Alors qu'une
réponse contractile synergique de l'aorte de lapin à la thrombine et au EGF, qui
agit via un récepteur de type tyrosine kinase, a déjà été montrée (Deblois et coll.,
1992), l'inhibiteur de tyrosine kinases erbstatine ne bloque aucunement les
réponses à la thrombine et au peptide [S~~']NAT~-NH~. Comme il a déjà été décrit
précedement (Deblois et cd., 1992) PPACK, un inhibiteur du site catalytique de

la thrombine' bloque l'effet de cette demière mais n'a aucun effet sur la
contraction induite par [s~~'JNAT&IH~.
Le mode d'activation particulier du récepteur de la thrombine nous a amené
a nous interroger sur la cinétique de désensibilisation et de ré-expression de ce
récepteur suite au clivage par l'enzyme ou l'activation par les peptides agonistes.
Lors de certaines expériences nous avons pu observer la désensibilisation de
l'aorte de lapin à la thrombine suite a l'injection de plus de deux concentrations
massives de cette enzyme alors que plusieurs injections de peptides agonistes ne
menaient à aucune diminution de la réponse. Les courbes concentration-réponse
réalisées au temps 1 heure ne montrent aucune différence entre les tissus traités
avec la cycloheximide, l'anisomycine ou la bréfeldine A et les tissus témoins
(figure 9). Cette observation est normale puisque la population de réceptews est
déjà formée et exprimée à la surface de la cellule. Au temps 3 heures les
inhibiteurs ont un effet très mitigé sur les réponses à la thrombine, les différences
observées entre les groupes traités et le groupe témoin ne sont pas significatives.
La réponse quasi intacte peut s'expliquer par l'existence d'un ensemble de
récepteurs pré-fmés et entreposés en attente dans des vésicules. Lors de
l'activation des réceptews membranaires par la thrombine ces récepteurs seraient
transportés pour s'exprimer au niveau de la membrane. A partir de ce moment la
cellule est prête à être stimdée à nouveau par la thrombine. Ces observations ont
été également vérinées par Hein et colî. (1994) à l'aide d'anticorps spécifiques
pour le récepteur de la thrombine. A l'aide d'anticorps distincts (dirigés contre les
récepteurs intacts ou les récepteurs clivés) le groupe de Brass et cou. (1994) a
démontré que suite à l'activation par la thrombine, certains récepteurs sont
intemalisés puis, au lieu d'être dégradés comme la plupart des autres types de
récepteurs couplés aux protéines-G, sont ré-exprimes sous leur fome clivée. Ceci
implique qu'ils sont alors activables par les peptides agonistes mais pas par la

thrombine elle-même. La disparition de la réponse au temps 6 heures montre
l'effet des inhibiteurs sur la synthèse protéique et sur le transport des récepteurs
déjà synthétisés. L'ensemble de récepteurs pré-formés et entreposés est maintenant
épuisé, la synthèse protéique étant interrompue il n'y a plus de récepteurs intacts
exprimés à la surface de la cellule de muscle lisse. L'essai fonctionnel dans les
bains à organes isolés permet donc d'appuyer le modèle théorique des récepteurs
de réserve. L'illustration de ce modèle de cinétique d'expression des récepteurs de
la thrombine apparait à la figure 20.
4.2.2 Modèle de ce1Iuies de muscle lisse en culture
Les études de liaison effectuées avec la thrombine marquée sur les CML
montrent la présence de deux sites de liaison pour cette enzyme : un site de haute
affinité et l'autre de basse affinité. L'incubation des CML avec de la thrombine
non marquée pendant une heure avant l'ajout du ligand marqué induit la
disparition du site de haute affinit6, suggérant une forte désensibilisation de ce
récepteur. La siguification exacte de la présence d'un site de liaison de basse
affinité a la surface de ces cellules n'est pas très claire. Il s'agit possiblement de
récepteurs déjà clivés qui ne sont pas internalisés ou bien qui sont ré-exprimés
sous forme clivée. Cette observation a d'ailleurs déjà été faite dans un modèle
cellulaire différent (CHRF-288) par le groupe de Bras et coll. (1994). Les effets
de type hormonal de la thrombine sont vraisemblablement induits par le site de
haute afnnité puisque de très faibles concentrations de cette enzyme sont requises
pour produire des effets au niveau de la synthèse d'ADN ou de la production de
phosphatidylinositols (voir ci-dessous).

Figure 20 Modèle de la cinétique de ré-expression des récepteurs de la thrombine
(Brass et cou., 1994; Hein et cou., 1994).

Les CML de l'aorte de lapin placées en culture pendant quelques semaines
ont la capacité de répondre à la thrombine ainsi qu'aux peptides agonistes, NAT6-
NH2 dans le cas présenf en générant des phosphatidylinositols. L'augmentation de
la production de phosphatidylinositols après une exposition de 5 min à l'un ou
l'autre des agonistes, suggérant l'activation de la phospholipase C, est obtenue à
des concentrations comparables à celles utilisées pour l'obtention de réponses
contractiles sur les tissus isolés (tableau 5; figure 6). L'implication de la
phospholipase C suivant la stimulation du récepteur clivable par NAT6-NH2 est
cohérente avec la participation de la protéine kinase C, modulée par le
diacylglycérol, dans l'effet contractile induit par ce peptide et par la thrombine, tel
que mentionné précédemment lors de I'utilisation de l'inhibiteur de protéine
kinase H-7 (figure 8). Une augmentation signincative de la concentration de IP2 et
P3 survient avec les deux concentrations de NAT6-NH2 testées et une réponse
concentration-dépendante est observée. Une production concentration-dépendante
de phosphatidylinositols est également observée chez les cellules stimulées avec la
thrombine, quoique des niveaux signincatifs soient obtenus seulement pour IPs.
Encore une fois les concentrations nécessaires p o u obtenir cette réponse sont
semblables à celles utilisées lors des essais de contractilité de l'aorte de lapin
(figure 6). Des études effectuées par différents groupes ont montré l'implication de
la phospholipase C et de la protéine h a s e C suite à la stimulation du récepteur de
la thrombine chez les plaquettes (Brass et coll., 1986) et les cellules musculaires
lisses (Huang et Ives, 1989).
Même si la réponse à la thrombine ou à NATs-- des cellules en culture
de muscle lisse est similaire en ce qui a trait à la production des
phosphatidylinositols, seule la thrombine est capable d'induire la synthèse d'ADN
chez ces cellules (figure 12). NAT6-NH2 ou [s~~']NAT~-N& utilisés a des
concentrations variant jusqu'à 100 pM ne provoquent aucune augmentation

signincative de I'incorporation de [3~-thymidine alors qu'avec la thrombine une
courbe concentration-réponse complète est obtenue avec un seuil d'activation de
0.1 nM et un effet maximal obtenu à une concentration de 10 nM. Fait intéressant,
les concentrations de thrombine nécessaires pour produire l'incorporation de
thymidine sont beaucoup plus basses que celles nécessaires pour induire la
production de phosphatidylinositols ou la contraction de l'aorte de lapin (tableau
5; figure 6). D'autres résultats permettent de montrer que les sentiers de
signalisation intracellulaire impliqués dans la synthèse d'ADN chez les cellules de
muscle lisse de lapin Werent en partie de ceux qui induisent la réponse
contractile à la thrombine. La synthèse d'ADN est bloquée par I'erbstatine
analogue alors que cet inhibiteur de tyrosine kinases n'a aucun effet sur la réponse
contractile. H-7, a une concentration de 10 pM, est capable de déplacer la courbe
concentration-réponse contractde à la thrombine ou a [ S ~ ~ ' W A T ~ - N H ~ mais il
n'affecte aucunement la synthèse d'ADN. À 30 pM l'effet de H-7 sur
l'incorporation de thymidine est probablement un effet toxique puisque même les
cellules témoins sont affectées par ce traitement.
En tenant compte des Iimitations de notre approche pharmacologique, la
réponse contractile de l'aorte de lapin induite par la thrombine semble être induite
par l'activation de la PKC et cette réponse peut être reproduite avec les peptides
synthétiques. Par contre les réponses mitogènes induites par la thrombine, chez les
cellules de muscle lisse en culture, requièrent un signal tyrosine kinase qui n'est
pas généré par la stimulation du récepteur clivable avec les peptides agonistes. Ces
résultats sont en accord avec les résultats observés chez les fibroblastes de hamster
où les peptides agonistes du récepteur de la thrombine ne sont pas capables de
reproduire les effets mitogènes de la thrombine (Vouret-Craviari et coll., 1992).
L'inhibition par l'erbstatine de la réponse mitogène à la thrombine dans notre
modèle de cellules en culture indique qu'une tyrosine kinase est essentielle dans la

prolifération cellulaire7 du moins dans ce modèle. L'implication de tyrosine
kinases est également requise pour l'activité mitogène de la thrombine sur les
cellules de muscle lisse vasculaire de rats nouveaw-nés mais pas pour le
relâchement de calcium intracellulaire (Weiss et Nuccitelli, 1992). De plus,
d'autres études ont montré que les cellules de muscle lisse de I'aorte de rat
nécessitent une exposition prolong2e (18-21 heures) à la thrombine pour montrer
une réponse mitogène (Bachhuber et cou., 1995). Dans notre modèle les cellules
sont mises en présence des agonistes pendant plus de 24 heures et nous observons
effectivement une réponse à la thrombine. Dans le cas des peptides agonistes du
récepteur il y a lieu de se demander si le métabolisme de ces peptides ne serait pas
une des causes de leur incapacité à induire un effet mitogene chez ces cellules.
Une autre hypothèse concernant la façon dont le signal est généré par les cellules
de muscle lisse vasculaire, soit par l'implication d'un second récepteur ou par le
relâchement d'un facteur autocrine (Weiss et Maduri, 1993), reste à vérifier. La
figure 21 montre de façon schématique les voies de transduction induites par la
thrombine ou les peptides agonistes suite à la stimulation du récepteur.
4.3 EFFETS IN VIVO DES AGONISTES PEPTIDIQUES DU
RÉCEPTEUR DE LA THROMBINE
Les lapins anesthésiés et héparinés répondent de façon dose-dépendante aux
peptides NAT par une réaction hypotensive qui n'est pas entièrement caractérisée
à ce jour. L'augmentation modeste des réponses de [S~~']NAT~-W par rapport à
NAT6-- qui est observée lors de l'utilisation de hautes doses de ce peptide
(figure 14) est vraisemblablement la conséquence de deux paramètres observés

Site de + clivage Canal caleique de type L
Substrat r DAG p 3 1
ras î
i MAPa
I
i Facteurs nucIéaires
Synthèse d'ADN (mitogék) Noyau
Figure 21 Voies de transduction induites par la stimulation du récepteur de la
thrombine.

précédemment : la plus grande résistance au métabolisme et l'afnnité moindre de
[S~~']NAT~-W pour le récepteur clivable. Il semble évident maintenant que les
résultats observés in vitro sont reliées de près à l'activité in vivo de NAT6-=
puisque ~'amasbtine potentialise également la réponse hypotensive induite par ce
peptide chez le lapin anesthésié (figure 15b). La durée de la réponse est cependant
modifiée de façon plus importante que l'amplitude, une situation déjà observée
avec les peptides reliés à la bradykinine (Drapeau et COL, 1991). L'ensemble de
ces résultats in vivo permet de constater que les peptides agonistes peuvent être
utilisés dans ce modèle pour authentifier les effets vasculaires provenant de
l'activation du récepteur de la thrombine et ce sans provoquer de coagdation
disséminée.
4.4 ÉTUDES IN VITRO DES EFFETS INDUITS PAR LE RÉCEPTEUR
PAR-2
L'étude de structure-activité par la substitution de résidus alanine dans la
séquence du peptide agoniste du récepteur PAR-2, SLIGRL, nous a permis de
constater que ce peptide est beaucoup plus plus intolérant aux substitutions que
son équivalent chez le récepteur de la thrombine, NAT6-m. En effet, seule
l'arginine en position 5 ne semble pas être cruciale pour la préservation de
l'activité biologique de ce peptide. L'activité biologique a été mesurée dans le
modèle de relaxation de l'aorte de lapin isolée. Les autres substitutions effectuées
aux positions 1 à 4 et 6 ont produit des peptides de très faible activité (tableau 6):
La synthèse de peptides possédant un résidu sarcosine en position 1 a engendré des
composés inactifs dans le modèle de l'aorte de lapin isolée, autant en contractilité
qu'en relaxation. Cette substitution d'une sérine pour une sarcosine ne reproduit

aucunement les résultats observés avec le récepteur de la thrombine et l'agoniste
peptidique [s~~']NAT~-w. La figure 16 illustre les réponses contractiles à la
üypsine ainsi qu'aux peptides PAR-2' en utilisant comme modèles de comparaison
les courbes concentration-réponses à la thrombine et a NAT6-m. Cette figure
montre que les deux enzymes produisent leur effet contractile à des concentrations
environ 1000 fois moindres que celles nécessaires aux peptides pour produire le
même effet. Ceci peut être expliqué de façon waisemblable par une grande
différence d'-té entre les enzymes et les peptides face au récepteur. Les
enzymes possèdent plus d'un site de liaison ce qui explique la différence
d'&té. Le fait également que les peptides doivent se Lier à un récepteur non-
clivé (encombrement stérique) peut aussi intervenir dans l'*té de la liaison et
l'efficacité de ces agonistes.
La réponse relaxante de l'aorte de lapin isolée à la trypsine et aux peptides
agonistes du récepteur PAR-2 permet de bien distinguer les réponses induites par
chacun des récepteurs activés par protéolyse puisque ni la thrombine ni les
agonistes de ce récepteur n'induisent de relaxation dans ce même modèle (figure
17). Cette relaxation a la trypsine ou au peptide SLIGRL, dépendante de
l'endothélium vasculaire, est cependant surmontée très rapidement suite à leur
activité contractile sur les cellules de muscle lisse, tel que montré à la figure 17.
L'activité relaxante a été caractérisée davantage en utilisant un inhibiteur de
l'activité de la synthase de l'oxyde nitrique, la nitro-L-arginine. L'utilisation de cet
inhibiteur a permis de montrer l'implication de l'oxyde nitrique comme messager
dans la réponse relaxante à la trypsine ou aux peptides PAR-2 (figure 18).
Finalement, la présence du récepteur PAR-2 dans l'aorte de lapin a été
montrée par une hybridation de type Northem. Ce type d'hybridation permet de

détecter I'ARN messager codant pour une protéine en utilisant une sonde
spécfique pour cette protéine. Dans notre cas une sonde isolée par PCR à partir de
l'ADN ginornique humain a été utilisée pour hybrider I'ARN messager total
obtenu de l'aorte de lapin. Le signal obtenu est très intense et confirme
I'expression de ce récepteur dans ce tissu (figure 19). Pour fin de comparaisons la
même sonde a été hydridée en présence d'ARN total isolé du coeur du même lapin
et le signal est toujours présent, suggkrant donc encore une fois l'expression de
1'ARN messager codant pour le récepteur PAR-2 au niveau du coeur. La
différence dans l'intensité du signal entre le coeur et l'aorte peut s'expliquer par la
contamination du tissu cardiaque par des cellules sanguines présentes au moment
de l'isolation de I'ARN, d'où la production d'un ARN messager plus abondant et
la détection d'un signal plus intense. Des études réalisées en parallèle par
differents groupes ont montré la présence du récepteur PAR-2 dans plusieurs tissus
chez l'humain : le pancréas, les reins, le petit intesth, le coeur mais curieusement
pas dans l'aorte (Bohm et coll., 1996). Toutefois sa présence au niveau des tissus
vasculaires a été montrée chez l'humain par une autre équipe (Mirza et coll., 1996)
et chez la souris (Nystedt et coll., 1995a).

5. CONCLUSION
Cette étude nous a permis mieux de connaître le mécanisme d'activation des
récepteurs activés par protéolyse dans les vaisseaux sanguins, que ce soit
l'activation par les enzymes thrombine et trypsine ou l'activation par les peptides
agonistes de chacun de ces récepteurs. Nous en connaissons maintenant beaucoup
plus sur la façon dont ces récepteurs sont activés ainsi que les sentiers de
signalisation impliqués, comparativement à ce qui était connu au départ.
Les études de structure-activité pour les agonistes de chacun des récepteurs
ont permis de développer des structures de longueur minimale qui reproduisent en
bonne partie les effets induits par chacun des récepteurs. Dans le cas du récepteur
de la thrombine ces études ont mené au développement d'un peptide agoniste
résistant au métabolisme, un outil essentiel pour étudier les effets médiés par le
récepteur de la thrombine autant in vitro qu'in vivo.
L'étude des signaux de transduction induits par la stimulation du récepteur
de la thrombine par l'enzyme elle-même ainsi que par les peptides agonistes a
permis de constater que ceux-ci ne sont pas capables de reproduire tous les effets
médiés par la thrombine. La stimulation de la synthèse d'ADN par les cellules de
muscle lisse de l'aorte de lapin en culture par la thrombine et non par [S~~']NAT~-
N& constitue un bon exemple. Cette observation suggère l'existence de plus d'un
site de liaison ou d'un second signal de transduction nécessaire à l'initiation d'un
tel effet ou la stimulation d'un second récepteur différent du récepteur clivabie
dont il est question ici. Plusieurs études étayent cette dernière hypothèse (Connolly
et cou., 1996; Tay-Uyboco et cou., 1995; Jenkins et coll., 1995) et les travaux se
poursuivent a£h de localiser la source de ce deuxième signal. Jusqu'à récemment

la thèse d'un récepteur Mique pour la thrombine semblait la plus acceptable, le
manque d'effet des peptides agonistes serait plutôt relié a l'absence d'un signal de
transduction plutôt qu'a leur incapacité d'activer un second récepteur. Leur faible
taille ne leur permet possiblement pas d'adopter une conformation optimale pour
l'activation du récepteur comme le fait la thrombine. Cependant la clonage et la
caractérisation récente d'un troisième récepteur activé par protéolyse (PAR-3)' un
second récepteur pour la thrombine, pourrait mener B la solution du probleme.
Un rôle de régulation du tonus vasculaire par les agonistes est sous-entendu
par l'expression dense de ces récepteurs au niveau des différents types cellulaires
vasculaires, spécialement dans le cas du récepteur PAR-2 qui peut induire une
relaxation rapide, dépendante de l'endothélium, suivie d'une contraction modulée
par les cellules de muscle lisse. Les études se poursuivent toujours dans le but
d'identifier un agoniste endogène pour ce récepteur, agoniste qui pourrait jouer un
rôle important dans la régulation du tonus vasculaire.
Finalement, les essais in vivo utilisant les peptides agonistes du récepteur de
la thrombine ont démontré que ces agonistes synthétiques sont des outils utiles
pour étudier les actions de la thrombine dans un modèle où il est impossible
d'utiliser la thrombine elle-même dû au risque de coagulation disséminée.

6.0 PERSPECTIVES
Le récepteur de la thrombine grâce à sa capacité de stimuler différents types
cellulaires pourrait être appelé à jouer différents rôles. La resténose post-
angioplastie est causée en partie par ce récepteur. C'est un processus impIliquaflt
une forte croissance de la couche de cellules musculaires conséquemment au
délogement de la plaque athérosclérotique à l'aide d'un cathéter gonflable.
L'inhibition de la thrombine in vivo limite la réponse mitogene consécutive à un
dommage vasculaire dans un modèle de lapin hypercholesterolémique (McNamara
et cou., 1996). Cette étude appuie l'hypothèse de l'implication de la thrombine
dans cette réponse de croissance cellulaire. La thrombine pourrait également
contribuer à la réponse inflammatoire par stimulation des leucocytes et autres
cellules du système immunitaire.
La découverte d'antagonistes de chacun de ces récepteurs serait d'un intérêt
majeur pour la caractérisation des rôles et des modes de fonctionnement de chacun
d'eux. La découverte d'un antagoniste spécifique du récepteur clivable de la
thrombine serait également d'un très grand intérêt thérapeutique puisque les effets
mitogènes (et autres effets indésirables pendant la phase inflammatoire) seraient
contrôlés sans pour autant compromettre le phénomène de la coagulation maintenu
par le sentier thrombine-fibrinogène-fibrine.
En ce qui a trait au récepteur PAR-2 a peu près tout reste à faire. Il serait
intéressant d'étudier les effets de la trypsine et du peptide SLIGRL-NH2 dans le
modèle de cellules de muscle lisse de l'aorte de lapin en culture. La production
d'inositolphosphates et l'incorporation de [3~thymidine pourraient ensuite être

comparées avec les résultats obtenus en utilisant la thrombine et NATs-m. Étant
donné la relaxation dépendante de l'endothélium observée sur les tissus isolés il
serait pertinent d'étudier l'effet des agonistes PAR-2 sur des cellules endothéliales
en culture en utilisant les mêmes essais que dans le cas des cellules de muscle
lisse.
L'effet in vivo des agonistes PAR-2 n'a pas encore été évalué. La réponse
endothéliale observée sur l'aorte de lapin isolée laisse supposer un rôle important
de ce type de cellules dans la régulation du tonus vasculaire. L'effet du peptide
SLIGRL-NH2 sur la pression artkrielle moyenne pourrait être évalué à l'aide du
modèle de lapin utilisé dans le cas des agonistes peptidiques du récepteur de la
thrombine.
En demier lieu, la découverte récente du récepteur PAR-3 powait détenir
la réponse à plusieurs intérrogations soulevées lors de la présente étude. Il serait
intéressant d'étudier la présence et les effets de ce troisième récepteur activé par
protéolyse à l'aide des modèles in vitro et in vivo utilisés précédemment.

7.0 RÉFERENCES BIBLIOGRAPHIQUES
ANTONACCIO M.J., NORMANDIN D., SEFUIFINO R et MORELAND S.
(1993). Effects of thrombin and thrombin receptor activating peptides on rat aortic
vascular smooth muscle. J. Phannacol. Exp. Ther. 266 : 125-132.
ATHERTON E. et SHEPPARD RC. (1989). Solid phase peptide synthesis : a
practical approach. Ed. Rickwood D., Harnes B.D. IRL P~ess, M o r d
AUSUBEL F.M. Current protocols in molecdar biology, Vol. 2. Greene
hblishing Assoc. and Wiley-Interscience. New-York. p. A. 1.5.
BACHHUBER B.G., SAREMBOCK LJ., GIMPLE L.W., MCNAMARA C.A. et
OWENS G.K. (1995). Thrombin-induced mitogenesis in cultured aorîic smooth
muscle ceLls requires prolonged thrombin exposure. Am. J: Physiol. 268 : C 1141-
C 1 147.
BAHOU W.F., KUTOK J.L., WONG A., POTTER C.L. et COLLER %.S. (1994).
Identification of a novel thrombin receptor sequence required for activation-
dependent responses. Blood 84 : 4 195-4202.
BALLA T., BAUKAL A.J., GUILLEMETTE G. et CATT K.J. (1988). Multiple
pathways of inositol polyphosphate metabolism in angiotensin-stimulated adrenal
glomedosa ceus. J. Biol. Chem. 263 : 4083-409 1.

BAR-SHAW R, BENEZRA M., ELDOR A., HY-AM E., FENTON J.W. II.,
\KILNER G.D. et VLODAVSKY 1. (1990). Thrombin immobilized to extracellular
matrix is a potent mitogen for vascular smooth muscle cells : nonenzymatic mode
of action. Cell ReguI. I : 453-463.
BERK B.C., TAUBMAN M.C., CRAGOE E.J., FENTON J.W. II. et
GREINDLING K.K. (1990). Thrombin signal transduction mechanisms in rat
vascular smooth muscle cells. Calcium and protein kinase C-dependent and -
independent pathways. J Biol. Chem 265 : 17334-17340.
BERRIDGE M.J. (1985). The molecular basis of communication within the cell.
Sci. Am. 253 : 142-152.
BERRIDGE M.J., DAWSON RM.C., DOWNES C.P., HESLOP J.P. et IRVINE
RF. (1983). Changes in the levels of inositol phosphates after agonist-dependent
hydrolysis of membrane phosphoinositides. Biochem. J. 212 : 473-482.
BOHM S.K., WONG W., BROMME D., SMEEKENS S.P., ANDERSON D.C.,
O L L Y A., KAHN M., hiELKEN NA., COUGHLIN SR., PAYAN D.G. et
BUNNETT N.W. (1996). Molecular cloning, expression and potential hctions of
the huma, proteinase-activated receptor-2. Biochem. J. 314 : 100% 10 16.
BRASS L.F., PIZARRO S., AHUJA M., BELMONTE E., BLANCHARD N.,
STADEL J.M. et HOXIE LA. (1994). Changes in the structure and hct ion of the
human thrombin receptor during receptor activation, intemaIization, and recycling.
J. Biol. Chem. 269 : 2943-2952.

BRASS L.F., LAPOSATA M., BANGA H.S. et RITTIENHOUSE S.E. (1986).
Regdation of the phosphoinositide hydrolysis pathway in thrombin-stimdated
platelets by a pertussis tolan-sensitive guanine nucleotide-bindhg protein. J. Biol.
C h 261 : 1683846847,
CASTRO B., DORMOY J.R, EVIN G. et SELVE C. (1975). Réactifs de couplage
peptidique. IV. L'hexa£iuorophosphate de benzotriazo1yl-N-o~sdimethylamino
phosphonium (B.O.P.) Tetrahedron Len. 14 : 12 19-1222.
CAUVIN C., LOUTZENHISER R et BREEMEN C. (1983). Mechanisms of
calcium-mtagonist-induced vasodilation. Annu. Rev. Pharmacol. Toxzcol. 23 :
373-396.
CHAO B.H., KALKUNTE S., MAMGANORE J.M. et STONE S.R (1992).
Essential groups in synthetic agonist peptides for activation of the platelet
thrombin receptor. Biochemishy 31 : 6 175-6 178.
CHOMCZYNSKI P. et SACCHI N. (1987). Single-step method of RNA isolation
by acid guanidinium thiocyamte-phenol-chloroform extraction. Anal. Biochem.
162 : 156-159,
CLARK J.M. (1988). Novel non-templated nucleotide addition reactions catalyzed
by procaryotic and eucaryotic DNA polymerases. Nucleic Acids Res. 16 : 9677-
9686.

COLLER B.S., WARD P., CERUSO M., SCUDDER L.E., SPRINGER K.,
KUTOK J. et PRESTWICH G.D. (1992). Thrornbin receptor activating peptide :
importance of the N-terminal serine and its ionization state as judged by pH
dependence, nuclear magnetic resonance spectroscopy, and cleavage by
aminopeptidase M. Biochemishy 31 : 1 17 13-1 1720.
CONNOLLY A.J., ISHMARA H., KAHN M.L., FARESE Jr RV. et COUGHLIN
S.R (1996). Role of the thrombin receptor in development and evidence for a
second receptor. Nature 381 : 5 16-5 19.
DEBLOIS D., DRAPEAU G., PETITCLERC E. et MARCEAU F. (1992).
Synergism between the contractile effect of epidermal growth factor and that of
des-~r~~-brad~kinin or of a-thrombin in rabbit aortic rings. Br. J. Pharmacol. 105
: 959-967.
DETWILER T.C. et FEINlMAN RD. (1973). Kinetics of the thrombin-induced
release of adenosine triphosphate by platelets. Cornparison with release of
calcium. Biochernistry 12 : 282-289.
DOWNES C.P., HAWKINS P.T. et IRVINE RF. (1986). Inositol 1,3,4,5-
tetrakiphosphate and not phosphatidyhositol 3,4=biphosphate is the probable
precursor of inositol 1,3,4-trisphosphate in agonist-stimulated parotid gland.
Biochem. J. 258 : 501-506.
DRAPEAU G., AUDET R, LEVESQUE L., GODIN D. et MARCEAU F. (1993).
Development and in vivo evaluation of metabolically resistant antagonists of B1
receptors for klliins. J. Phannacol. Exp. Ther. 266 : 192-199.

DRAPEAU G., DEBLOIS D. et MARCEAU F. (199 1). Hypotensive effects of
~~s-des-~r~~-brad~kinin and metabolically protected agonists of BI receptors for
kinins. J. Pharmacol. Exp. Ther. 259 : 997-1003.
DRAPEAU G. et REGOLI D. (1988). Synthesis of bradykinin analogs. In :
Immunochemical Techniques, Part M : Chernotaxis and Infiammation, Methoh in
Enzymology, vol. 163. Ed. DiSabato G. Academic Press, New York. pp. 263-272.
FEINBERG A.P. et VOGELSTEIN B. (1984). A technique for radiolabeling DNA
restriction endonuclease fragments to high specïfïc actibity. Anal, Biochem 137 :
266-267.
FENTON J.W. II (1988). Regulation of thrombin generation and functions. Semin
Thromb. Hemostasis 14 : 234-240.
FURCHOTT, RF. et ZAWADZKI J.V. (1980). The obligatory role of
endothelial ceIis in the relaxation of artenal smooth muscle by acetylcholine.
Nature. 288 : 373-3 76.
GARCIA J.G.N., ASCHNER J.L. et MALIK A.B. (1992). Regulation of thrombin-
induced endothelial barrier dysfûnction and prostaglandin synthesis. In:
Thrombin. Ed. Berliner L.J. Plenum Press, New York. pp. 397-437.
GERSZTEN RE., CHEN J., ISHII M., ISHII K., WANG L., NANEVlCZ T.,
TURCK C.W., W T X H . et COUGHLIN S.R (1994). Specincity of the
thrombin receptor for agonist peptide is defined by it's extracellular surface.
Nature 368 : 648-65 1.

GLENN K., CARNEY D., FENTON J.W. II. et CUNNINGHAM D. (1980).
Thrombin active site regions required for fibroblast receptor binding and initiation
of cell division. J Biol. C h . 255 : 6609-6616.
GOLDSTEIN RH. et WALL M. (1984). Activation of protein formation and celî
division by bradykinin and des-kgg-bradykinin. J Biol. Chem. 259 : 9263-9268.
GUILLEMETTE G., LAMONTAGNE S., BOULAY G. et MOUILLAC B.
(1989). DifZerentiaI effects of hep& on inositol 1,4,5-triphosphate binding,
metabolism and calcium release activity in the bovine adrend cortex. Mol.
Phannacol. 35 : 339-344.
HALTINER M., KEMPE T. et TWAN R (1985). A novel strategy for constructïng
clustered point mutations. Nucl. Acids Res. 13 : 10 15- 1025.
HANSON S.R. et HARKER L.A. (1988). Intemption of the acute platelet-
dependent thrombosis by the synthetic antithrombin D-phenyl-alanyl-L-propyl-L-
arginyl chloromethylketone. Proc. Nad. Acud Sci. USA 85 : 3 184-3 188.
HARKER L.A. (1994). Strategies for inhibiting the effects of thrombin. Blood
Coagui. Fibrinolysis S(Supp1 1) : S-47-S-58.
HATTORI R, KAMILTON K.K., FUGATE RD., MCEVER R.P. et SIMS P.J.
(1989). Stimulated secretion of endothehl von Willebrand factor is accompanied
by rapid redistribution to the cell surface of intraceliular granule membrane protein
GMP-140. J: Biol. Chem. 264 : 7768-7771.

HE C.J., RONDEAU E., MEDCALF RL., LECAVE R, SCHLEUMNG W.D. et
SRAER J.D. (199 1). Thrombin increases proliferation and decreases fib~olytic
activity of kidney glomerular epithefiai ceas. J Cell. PhysioZ. 146 : 13 1-140.
HEIN L., ISHII K., COUGHLIN S.R et KOBILKA B.K. (1994). Intracellular
targeting and tra£ficking of thrombin receptors : a novel mechanism for
resensitlzation of a G protein-coupled receptor. J. Biol. Chem. 269 : 277 19-27726.
KIDATA H., INAGAKI M., KAWAMOTO M. et SASAKI Y. (1984).
Isoquinolinesnlfonarnides, novel and potent inhibitors of cyclic nucleotide
dependent protein kinases and protein kinase C. Biochemistry 23 : 5036-504 1.
HUANG C.L. et IVES H.E. (1989). Guanosine 5'-O-(3-thiotrisphosphate)
potentiates both thrombin and platelet-derived growth factor induced inositol
phosphate release in permeabilized vascular smooth muscle cells. J. Biol. Chem.
264 : 439 1-4397,
HUI KY., JAKUBOWSKI LA., WYSS V.L. et ANGLETON E.L. (1992).
Minimal sequence requirement of thrombin receptor agonist peptide. Biochem.
Biophys. Res. Commun 184 : 790-796.
MOTO M., UMEZAWA K., ISSHKI K., KUNMOTO S., SAWA T.,
TAKEUCHI T. et UMEZAWA H. (1987). Kinetic studies of tyrosine kinase
inhibition by erbstatin. J. Antibiot. 40 : 14 17-1473.

JACOBSON A. (1987). Methocls in Emymology. 152 : 254-257.
JANG 1-K., GULD H.K., ZISKZND A-A., LEWBACH RC., FALLON J.T. et
COLLEN D. (1990). Prevention of platelet-rich arterial thrombosis by selective
thrombin inhibition. Ciradation 81 : 2 19-225.
E N U N S A.L., HOWELLS G.L., SCOTT E., LE BONNIEC B.F., CURTIS M.A.
et STONE S.R (1995). The response to thrombin of human neutrophils : evidence
for two novel receptors. J Cell Science 108 : 3059-3066.
JOBIN F. (1995). L'hémostase nomde. In: L'Hémostme. Les Presses de
l'université Laval, Québec, Canada.
KALSNER S. et NICKERSON M. (1968). A method for the study of mechanism
of drug disposition in smooth muscle. C m J ~ s i o l . Phumacol. 46 : 7 19-730.
KETTNER C. et SHAW E. (1979). D-Phe-Pro-Arg-CH2C1 - A selective affinity
label for thrombin. Thromb. Res. 14 : 969-973.
KINLOUGH-RATHBONE RL., RAND M.L. et PACKHAM M.A. (1 993). Rabbit
and rat platelets do not respond to thrombin receptor peptides that activate human
platelets. Blood 82 : 103- 106.
KOLONDEY M.S. et WYSOLMERSKT RB. (1992). Isometric contraction by
fibroblasts and endotheliai cells in tissue culture : a quantitative study. J Cell.
Bzd. 177 : 73-82.

KU D.D. et ZALESKl J.K. (1993). Receptor mechanism of thrombin-induced
endothelitun-dependent and endothehum-independent coronary vascular effects in
dogs. J. Cardiovac. Phannacol. 22 : 609-6 16.
LEFKOVITS J., TOPOL E.J. (1994). Direct thrombin inhibitors in cardiovascular
medicine. CirmIation 90 : 1522- 1536.
LEVESQUE L., DRAPEAU G., GROSE J.H., IUOUX F. et MARCEAU F.
(1993). Vascnlar mode of action of kinin BI receptors and development of a
cellular mode1 for the investigation of these receptors. Br. J; Phmacol. 109 :
1254- 1262.
LEWIS A.J. et FURST D.W. (1987). Nonsteroidal Anti-innarnmato~y h g s :
Mechanism and Clinical Use. Marcel Dekker, New York.
MAMATIS T., FRITSCH E.F. et SAMBROOK L (1989). Molecular Cloning : A
Laboratory Manual. 2nd Ed Cold Spring Harbor. New-York. pp. 7.3.
MARCEAU F., PETITCLERC E., DEBLOIS D., PRADELLES P. et POUBELLE
P. (1991). Human interleukin-1 induces a rapid relaxation of the rabbit isolated
mesentexic artery. Br. J. Phannacol. 103 : 136791372.
MARTIN B.M., FEDJMAN RD. et DETWlLER T.C. (1975). Platelet stimulation
by thrombin and other proteases. Biochemzstry 14 : 1308-13 14.
MCNAMARA C.A., SAREMBOCK 1. J., BACHHUBER B.G., STOUFFER G.A.,
RAGOSTA M., BARRY W., GIMPLE L.W., POWERS E.R et OWENS G.K.
(1996). Thrombin and vascular smooth muscle cell proMeration : implications for
atherosclerosis and restenosis. Semin Thromb. Hem. 22 : 139-144.

MEAD D.A., PEY N.K, HERNSTADT C., MARCIL RA. et SMITH L.M.
(1991). A universal method for the direct cloning of PCR amplined nucleic acid.
Bio/Technology. 9 : 657663.
MERRIFIELD RB. (1962). Solid phase peptide synthesis. 1. The synthesis of a
tetrapeptide. J. Amer. Chem. Soc. 85 : 2 149.
MICHEL M.C., BRASS L.F., WILLIAMS A., BOKOCH G., LAMORTE V.J. et
MOTULSKY H.J. (1989). Alpha 2-adrenergic receptor stimulation mobilizes
intracellular Ca2+ in human eq&roleukemia cells. J: Biol. Chem. 264 : 4986-
4991.
MIRZA H., YATSULA V. et BAHOU W.F. (1996). The proteinase activated
receptor-2 (PAR-2) mediates mitogenie responses in human vascdar endothelid
ceus. Molecular characterization and evidence for functional coupling to the
thrombin receptor. J. Clin Invest. 97 : 1705- 1714.
MURAD F. (1990). Dnigs used for the treatment of angina : organic nitrates,
calcium-channel blockers and 8-adrenergic antagonists. In : The PhmacologicaZ
Bais of Therqpeutics. Ed. Gilman A.G., Rall T.W., Nies A.S. et Taylor P.
Pergamon Press, New York. pp. 764-783.
NATARAJAN S., RZEXINGER D., GAMBERDELLA M. et SEILER S. (1995).
« Tethered ligand >> derived penta peptide agonists of thrombin receptor : A study
of side chai. requirements for platelet activation Int. J. Pept. Protein Res. 45 :
145-151.

NOSE T., SHIMOHIGASHI Y., OHNO M., COSTA T., SHEMlZU N et OGINO
Y. (1993). Enhancement of thrombh receptor activation by thrombin receptor-
derived heptapeptide with para-fluorophenylalanine in place of phenylalanine.
Biochem. Biophys. Res. Commun 193 : 694-699.
NYSTEDT S., EMaSSON K., WAHLESDEDT C. et SUNDELIN J. (1994).
Molecular cloning of a potenbal proteinase activated receptor. Proc. Natl. Acad
SC^ UX4. 91 : 9208-9212.
NYSTEDT S., LARSSON Ao-K., ABERG H. et SUNDELIN J. (1995a). The
mouse proteinase-activated receptor-2 cDNA and gene. Molecular cloning and
functional expression. J. Bzol. Chem. 270 : 5950-5955.
NYSTEDT S., EMILSSON K., LARSSON A.K., STROMBECK B. et
SUNDELIN J. (1995b). Molecular cloning and actional expression of the gene
encoding the human proteinase-activated receptor 2. Eur. J. Biochem. 232 : 84-89.
PALMIERI F.E., BAUSBACK H.H. et WARD P.E. (1989). Metabolism of
vasoactive peptides by vascular endothelium and smooth muscle aminopeptidase
M. Biochem. Pharmacol. 38 : 173- 180,
RASMUSSEN U.B., VOURET-CRAVIARI V., JALLA S., SCHLESINGER V.,
PAGES G., PAVIRANI A., LECOCQ J.P., POUYSSEGUR J. et VAN
OBBERGHEN-SCHILLING E. (1991). cDNA cloning and expression of a
hamster alpha-thrombin receptor coupled to Ca2+ mobilization. FEBS Lett. 288 :
123-128.

REGûLI D., MOUX F., PARK W.K. et CHO1 C. (1974). Role of N-terminal
amino acid for the biological activity of angiotensin and inbibitory dogues. Can
J. Physol. Phannacol. 52 : 39-49.
RYDEL T.J., RAMCHANDRAN KG., TULINSKY A., BODE W., HUBER R,
ROITSCH C. et m 0 N II J.W. (1990). The structure of a complex of
recombinant hinidin and human alpha-thrombin. Science 249 : 277-280.
SABO T., GURWITZ D., MOTOLA L., BRODT P., BARAK R et ELHANATY
E. (1992). Structure-activity snidies of the thrombin receptor activating peptide.
Bzochern. Biophys. Res. Commun 188 : 604-610.
SAGO H. et LINUMA K. (1992). Cell shape change and cytosolic Ca2+ in human
umbelical-vein endothelial cells stimulated with thrombin. Thromb. Haemostaszs
67 : 33 1-334.
S A K I RK., SCHARF S., FALOONA F., MULLIS K.B., HORN G.T., ERLICH
H.A. et ARNHEM H. (1985). Enzymatic amplincation of B-globin genomic
sequences and restriction site analysis for diagnosis of sickle ceU anemia. Science
230 : 1350.
S A K I RK., GELFOND D.H., STOFFEL S., SCHARF S., HIGUCHi R, HORN
G.T., MULLIS K.B. et ERLICH H.A. (1988). Primer directed enzymatic
amplincation of DNA with a thennostable DNA polperase. Science 239 : 487-
491.

SALACINSKI P.R, McCLEAN C., SYKES J.E., CLEMENT-JONES V.V. et
LOWRY P.J. (1981). Iodination of proteins, glycoproteins and peptides using a
solid-phase oxidising agent, 1,3,4,6-tetrachloro-3,6-diphenyl glucoluril (Iodogen).
Anal. Biochem. 117 : 136-146,
SAMBROOK J., FRITSCH E.F. et MANlATlS T. (1989). Moleculur clonzng : A
laboruto y munual. Cold Spring Harbor Laboratory. Second Edition.
SANGER F., MCKLEN S. et COULSON A.R (1977). DNA sequencing with
chai.-terminating inhibitors. Proc. Natl. Sci. USA. 74 : 5463-5467.
SCARBOROUGH RM., NAUGTHON M.A., TENG W., HUNG D.T., ROSE J.,
VU T-K.H., WHEATON V.L, TURCK C.W. et COUGHLIN S.R (1992).
Tethered ligand agonist peptides : structural requirements for thrombin receptor
activation reveal mechanism of proteolytic unmasking of agonist hct ion. J. Biol.
Chem. 267 : 13146-13149.
SCARBOROUGH RM., TENG W., NAUGHTON M.A., ROSE J.W., ALVES
V., ARFSTEN A., RAMAKRISHNAN V. et BLACKHART B. (1993). Thrornbin
receptor antagonists derived fiom « tethered ligand » agonist peptides. 13'
Amencan Peptide Symposium, Edmonton, Canada : Abst. P632.
SC-CHER W.A., HERAN C.L., STEINBACHER T.E., SEILER S.M.,
MICHEL 1. et OGLETREE M.L. (1993). Cornparison of thrombin active site and
exosite inhibitors with hep& in models of arterial and venous tbrombosis J.
Phannacol. Exp. Ther. 267 : 1237-1242.

SNYDER S.H. (1985). The molecular basis of communication between ceus. Sci.
Am 253 : 132-140.
STUBBS M.T. et BODE W. (1993). A player of many parts : the spotlight f d s on
thrombin's structure. Thromb. Res. 69 : 1-58.
TALLARIDA RJ. et MURRAY RB. (1987). Manual of pharmacologie
calcuIations with cornputer p r o g r m . Springer-Verlag New-York Inc. New-York.
TAY-UYBOCO J., POON MC., AHMAD S. et HOLLENBERG M.D. (1995).
Contractile actions of dirombin receptor-derived polypeptides in human umbelicai
and placental vascuiature : evidence for distinct receptor systems. Brit J.
Phannacol. 115 : 569-578.
TESFAMARZAM B., ALLEN G.T., NORMANDIN D. et ANTONACIO M.J.
(1993). Involvement o f the « tethered ligand » receptor in thrombin-induced
endothelium-mediated relaxations. Am. J. Physiol. 265 : H 1744-H 1749.
VASSAL0 RR, KIEBER-EMMONS T., CICHOWSKI K. et BRASS L.F.
(1992). Structure-hction relationships in the activation of platelet thrombin
receptors by receptor-derived peptides. J Biol. C h . 267 : 608 1-6085.
VOURET-CRAWARI V., VAN OBBERGHEN-SCHILLING E., SCIMECA J.C.,
VAN OBBERGHEN E. et POUYSSEGUR J. (1993). Daerential activation of
p44mapk (ERKI) by alpha-thrombin and thrombin-receptor peptide agonist.
Biochem. J. 289 : 209-214.

VOURET-CRAMARI V., VAN OBBERGHEN-SCHILLING E., RASMUSSEN
U.B., LECOCQ A. et POUYSSEGUR 3. (1992). Synthetic a-thrombin receptor
peptides activate G protein-coupled signalhg pathways but are unable to induce
mitogenesis. Mol. Biol. Ce12 3 : 95- 102.
VU T-K.H., HUNG D.T., WHEATON V.I. et COUGHLIN S.R (1991a).
Molecular cloning of a hctionnal thrombin receptor reveals a novel proteolytx
mechanism of receptor activation. Cell 64 : 1057-1068.
W T-K.H., WHEATON V.I., HUNG D.T., CHAM 1. et COUGHLIN S.R
(199 lb). Domains spec-g thrombin-receptor interaction. Nature 353 : 674-677.
WEISS RH. et MADURI M. (1993). The rnitogeniç effect of thrombin in vascular
smooth muscle cells is largely due to basic fibroblast growth factor. J. Biol. Chem
268 : 5724-5727.
WEISS RH. et NUCCITELLI R (1992). Inhibition of tyrosine phosphorylation
prevents tbrombin-induced mitogenesis, but not intracellular fÎee calcium release,
in vascular smooth muscle cells. J. Biol. Chem. 267 : 5608-5613.
YANG S.G., LANYYONU A., SAIFEDDINE M., MOORE G.J. et
HOLLENBERG M.D. (1992). Actions of thrombin and thrombin receptor peptides
analogues in gastric and aortic smooth muscle : development of bioassays for
structure-activity studies. Life Sci 51 : 1325-1332.
ZHONG C., HAYZER D.J., CORSON M.A. et RUNGE M.S. (1992). Molecular
cloning of the rat vascular smooth muscle thrombin receptor. Evidence for in vitro
regdation by basic fibroblast growth factor. J. Biol. Chem. 267 : 16975-16979.

APPENDICE A : CURRICULUM VITAE
Denis Godin, Ph.D.
Scolarité :
1994 - 1997 Doctorat en médecine expérimentale
Département de pharmacologie, Faculté de Médecine
Université Lavai, Ste-Foy, Québec, Canada
Mahise en médecine expérimentale
Département de pharmacologie, Faculté de Médecine
Université Laval, Ste-Foy, Québec, Canada
Baccalauréat en biochimie
Département de biochimie, Faculté des Sciences et de Génie
Université Laval, Ste-Foy, Québec, Canada
Bourses et distinctions :
1996 - 1997 Bourse d'étude de la Fondation des maladies du coeur du
Canada (renouvellement).
Bourse d'étude de la Fondation des maladies du coeur du
Canada.

Septembre 1996 Finaliste pour les meilleures présentations par fichage
IX International Vascular Biology Meeting, Seattle, WA.
Mai 1995
Mai 1994
Prix pour la meilleure présentation orale
Journée scientifique annuelie de l'Hôtel-Dieu de Québec
Prix pour la meilleure présentation orale
Journée scientifique annuelle de l'Hôtel-Dieu de Québec
1994 - 1996 Président de l'Association des Chercheurs et Chercheures
Étudiants en Médecine de l'université Laval.
Publications :
Membre du comité d'admission et de supervision du
programme de médecine expérimentale.
Représentant des étudiants du Centre de recherche de l'Hôtel-
Dieu de Québec.
Drapeau G., Brochu S., Godin D., Levesque L., Riowc F. et Marceau F. (1992).
Synthetic C5a receptor agonists : phannacology, metabolism and in vivo
cardiovascular and hematologic effects. Biochem. Phannacol. 45 : 1289-1299.

Drapeau G., Audet R, Levesque L., Godin D. et Marceau F. (1993). Development
and in vivo evaluation of metabolicdy resistant antagonists of BI receptors for
kinins. J. Pharmacol. Exp. Ther. 266 : 192-199.
Godin D., Marceau F., Beaulé C., Roux F. et Drapeau G. (1994). Aminopeptidase
modulation of pharmacological reçponses to synthetic thrombin receptor agonists.
Eur. J Pharmacol. 253 : 225-230.
Godin D., Roux F., Marceau F. et Drapeau G. (1995). Mode of action of
thrombin in the rabbit aorta. Br. J. Phunnacol. 115 : 903-908.
Wesley RB., Meng X.P., Godin D. et Galis Z.S. (1997). Extracellular rnatx-ix
modulates macrophage fiuictions characteristic to atherorna. Arterioscl. Thromb.
Vasc. Bzol. (Sous presse).
Godin D, Lemieux J., Harvey N. et Drapeau G. (1997). Second proteinase
activated receptor (PAR-2) characterization in the rabbit aorta. (En préparation).
Rélsumék de communication :
Drapeau G., Levesque L., Audet R, Godin D., Rioux F. et Marceau F.
Development and in vivo evaluation of metabolically resistant antagonists of BI
receptors for kinins. Experimental Biology 93, New Orleans, Louisiana, April
1993. FASEB J: 7 : A40, 1993.

Drapeau G., Godin Dm, Brochu S., Levesque L., Riom F. et Marceau F.
Pharmacological characterization of synthetic ligands for the anaphylatoxin C5a
receptor. Thirteenth Aumican Peptide Symposium, Edmonton, Alberta, June 20-
25, 1993.
Godin D , Marceau F., Brisson J., Brochu S., Rioux F. et Drapeau G. Synthèse et
pharmacologie de ligands du récepteur de l'anaphylatoxine C5a. Club de
recherches ciiniques du Québec, Pointe-au-Pic, Québec, Septembre 1993.
Médecine Sciences 9 (Suppl. 1) : 54, 1993.
Drapeau G., Harvey N., Audet R, Levesque L., Godin D, Rioux F. et Marceau F.
Structure-activity of peptides that bind to BI receptors for kinins : agonists,
antagonists, labeled ligands. Peptides and their antagonists in tissue injury.
Satellite Symposium of the XII' IUPHAR Congress, MontréaI, Québec, July 3 1 -
August 3, 1934. Can J. Physzol. Phannacol. 72 (Suppl. 2) : 21, 1994.
Gadin D, Rioux F., Marceau F. et Drapeau G. Intracellular pathways involved in
contractile and mitogenic responses of rabbit aortic tissues following thrombin
receptor activation. 1995 FASEB Summer Research Coderences, Copper
Mountain, Colorado, August 13-18, 1995.
Godin D., Harvey N., Rioux F., Marceau F. et Drapeau G. Voies intraceildaires
activées par la thrombine dans les vaisseaux sanguins. Club de recherches
cliniques du Québec, Bromont, Québec, Septembre 1995. Médecine Sciences 11
(Suppl. 2) : 42, 1995.

Godin D, Lemieux J., Riom F., Marceau F. et Drapeau G. Caractérisation du
récepteur PAR-2 dam les vaisseaux sanguins. 64' Congrés de 1'Acfas. Université
McGill, Montréal, Québec, Mai 1996.
Godin D, Batchvarov D., Lemieux J., Rioux F. et Drapeau G. Mechanism of the
contractile and relaxing effects induced by protease-activated receptors in the
rabbit aorta. IX International Vascular Biology Meeting, Seattle, Washington.
September 4-8 1996. J Vasc. Res. 33 (Suppl. 1) : 30, 1996.

APPENDICE B : ARTICLES SCIENTIFIQUES PUBLIES

BbalbnraJ P h m m d o g y . Vol. 6. No. 6. pp. IZSW2W. lm. Rmtcd iu Cirtu Britaia
SYNTHETIC CSa RECEPTOR AGONISTS
PHARMACOLOGY, METABOLJSM AND IN VIVO CARDIOVASCULAR AND HEMATOLOGIC EFFECïS
GUY DRAPMU,* Sm- BROCHU, DENIS GODIN, LUC LEVESQUE, FRANCIS Riom aad FRANCOIS MARCEAU
Ccatrc de recherche (Univcrsitd Laval), Hdtci-Dieu de Qudxc, -bec (Qutbac), Canada GIR 2J6
(Received 22 Septembcr 1992; acceptrd 19 Nooanbrr 1992)
Abstrict-Rccent investigations have producul novcl compounds that act on the rrccptor for ma- phylatoxin C5a. ïhcsc products are C-terminal analogues of C5a. some of which arc rndï fk î exteasively. We have rneasurcd the receptor affinities of such analogues in a binding asay on human oeutrophils (PMNs). We bave also charaderkd th& phannacological profiles in o h on the isolatcd rabbit portal vein and pdmonary artcry, on supcroxide rclcase by PMNs as wcU as in oiuu in the ancsthetized rabbit (acute hypotcnsive and acutmpcnic cffccm). The mctabolic &tancc of these anaiopes was aiso evaluatcd in the pr#cna of diffcrcnt pcptidascs. One of these wmpounds, MePhe-Lys-Pro-d=ba-Phe- D-Arg, behaved as an antagonist on the rcleasc of supcrortide by ncuuophils while excrting agonist activity in al1 othcr assays- fts partiai agonist statu was documcnted in a rcccptor down-regdation cxperimcnt on PMNs where iu activity was cornparcd with those of recombinant CS;a and of protamine which behavcs as a cornpetitive antagonist on thest œ h . Degradation s t u b iadicated that the discrepancy betwetn the affinity of certain analogues in vina and thcir potcncy in oiuo was probably tinked to thcir metabolic stability.
Wtthin the muItifunctionaI cornplement system, anaphylatoxin CSa pIays a prominent role as a pharmacologicaIly active fragment of CS that activates various leukocyte bctions (reviewed by Hugli [LI). Basophils and mast cells release histamine and other secondary mediators in rcsponse to C5a [l, 21. in phagocytic Icukocytes, namely neutrophils and monocytes/macrophages, several distinct acti- vation pathways are stimulated by C5a: there is a chernotactic and chemokineticresponse, an increased eicosanoid formation, a reIease of reactive radicals and of granule content [Il, an increased adherence to cultured endothelium [3] and a potentiation of the synthesis of intcrleukins 1 and 6 in monocytes [Ml. The receptor for C5a has been cloned and sequenced recently from human myeloid-like cell lines [7,8]. It bclongs to the rhodopsin suptrfamily of signaling proteins, consistent with the rok of a G protein in its signal transduction 191.
The in vivo pharmacology of CSa is characterized by two broad types of events. First, effects on circulating leukocytes are prominent and depend on
Comsponding author: Dr. Guy Drapeau, Centrc de recherche (Université Laval), Hdtel-Dieu de Qudbcc, 11 Cbtedu-Palais. Qudbec (Qudbec), Canada G1R 216- Tel. (4 18) 691 -5281 : FAX (4 18) 69 1-5439.
t Abbrcviations: ACE. angiotensin-converting enzyme; AmM. aminopeptidasc M: Ac-Phe, N-acetyl-phtnyl- alanine; BSA. bovine serum aibumin: Cha, cyclohexyl- alanine; CPN. carboxypeptidase N; DPN, 12-diphcnyl4 [3-(1-aaphthyl)-propyll-3 5-pyraxolidincdione; FMLP, N- lormyl-methiony I-lcucyl-phenyialanint; HBSS, Hanks* bal- anccd salt solution; MePhe, N-methyl-phcnylalanine; merge tpa. DL-mercaptomcth yl-3-guanidinoethylthiopr* panoic acid: and PMNs, potymorphonuclear Icukocytes.
the route of administration. Local administration (cg. intramchcal instillation, intradermal injection) promotes the migration of polymorphonudear lcdcocytes (PMNs)t from the blood into the tissues [IO, Il]. C5a, given by the intravenous route in the rabbit, produces an acute fa11 of the citculating aeutrophil count, interpreted as transient adhercna to the blood vesse1 wails, foiiowed by leukocytosis [12]. Recent evidence suggests that, under these circumstanlces, the bone marrow is actively delivering neutrophils to the blood, some of which are immature fonns, and that eicosanoids are not involved in the biphasic neutropenia/neutrophiüa respome [13,14]. The second type of Ut uivo response to C5a is dependent on secondarily relcased mediators and may affect many systems. Examples of this phenom- cnon are bronchospasm in guinea pigs and, in several spccies, a cornplex cardiovascuIar response that includw hypotension with puimonary hypertension and decreased cardiac output [12,15]. In the rabbit, the cardiovascular response is mainly dependent on the production of vasoconstrictor and vasodilator eicosanoids, such as thromboxane A2 and 6-keto- prostaglandin Ft,, and is independent of circulating, ncutrophils (12). ltsotated blood vesscls h m the rab- bit (l6.171. guinea pig [I8] and humans 119) exhibit in oitro mechanical responses to CSa that are not tachy- phytactic and are dependent mainly on eicosanoids; other mediaton such as histamine and nitric oxidc are possibly involved in minor components of the response [16181. There is significant evidence sug- gesting that responses to C5a in smooth muscle prep arations are ultimately exptained by the presenœ of tissue fonns of leukocytes (mast cells [l] or resident macrophages [39).

The puwerful activities of U a in vivo are regulated by arginine-carboxypeptidases found in blood serurn and also at the ceIl surface: the carboxypeptidases M and N p. 211. Othcr unstable arginine- carboxypepadasts are aisa formcd in plasma during coagulation [22,23 ] and may, in some instances, participate in C5a inactivation. =a-des-Arg, the resdting molccule, exhibits onIy a srnaii fraction of the potency of the partnt molenile in binding and functional assays [Il. There is evidcnce that CSa internalization by lcukocytes is foliowcd by extensive proteolysis [24]. ïndccd, the concentration of immunoreactive ma-des-Arg is very low in biological fiuids relative to the expected turn-over of CS and to Qa-des-Arg and C4ades-Arg concentrations [2Sî7].
The recent success of the synthesis of CSa receptor agonists modeled on the C-terminal rcgion of the molecule [ï8-311 has pemiaed a new approach to the study of U a actions and metabolic inactivation. Synthetic agouists with varying degrees of affinity and metabolic resistance have been testcd in cornparison to CSa in increasingly complex biological assays. Ultirnately, the complex integratcd car- diovascular and hematologic responses to ana- phyiatoxin CSa in the rabbit have been reproduced by the metaboiicaily resistant analogues with a potency approaching that of U a .
Indornethacin, amastatin. bestatin, captopril, salmon protamine (type X), N-forrnyl-methionyl- Ieucyl-phenylaianine (FMLP) and human recom- binant C5a from Escherichia coli (321 were purchased from Sigma (St. Louis. MO). DL-Mercaptomethyl- Iguanidinoethylthiopropanoic acid), (mergetpa) was purchased from Calbiochern (La Jolla, CA). 1.2 - Diphenyl - 4 - [3 - (1 - naphthyl) - propyl] - 3.5 - pyrazoIidinedione (DPN), a non-peptide FMLP antagonist [33,34]. was purchased from the Alfred Bader Chernid Library of Rare Chemicais (MI- waukee. W).
Synthesii of C-terminal analogues of CSa and HPLC anaiysk
CSa analogues were synthesued by solid phase rnethodology using equipment and methods pre- viously described [35.*1. HPLC analysis of pure peptides or samples from rnetabolic studies were performed using a Waters systern equipped with a 746 data module and a 486 UV detector set at 214 nm. Separations were achieved with a Vydac 10 pm (3.9 x 300 mm) reverse phase CIR column using a linear gradient of 5 4 5 % acetonitrile/O.OS% uifluoroacetic acid (TFA)/water at 2 ml/rnin over a pend of 20 min. For degradation studies, 50 pL of cach aliquot was injected and rates of peptide
' Drapeau G, Audct R. Levesque L, Godin D and Mamau F. Dcvciopmcnt and in vivo evatuation of mttabolically rcsistanr antagonists of BI rcccpton for kinins. Manuscript submittcd for publication.
metabotism were calcdated from the decrease of peptide substrate concentration.
The rnetabolic resistana of CSa analogues was evaluated in rabbit plasma as a potential source of arninoptptidase M (AmM) and carboxypeptidase N (CPN) activities, with a kidney mimvillar membrane preparation (neutrai endopeptidase 24. il activity), with purified angiotensin-convemng enzyme (A-) and in the prescnce of neutrophils. Rates of hydrolysis, evaluated by HPLC as described above, are expresscd as substrate consumption (in nmol) per minute per quantity of enzyme preparation (mg of protein, mL of plasma or number of ce&). Tntegration of peak areas and quantitation of peptide substrate were dculated automatically by the data module- Metabolites, arising from cleavage of substrate by enzyme preparations, were observed in every case, as peaks with retention times different from the peptide substrate, but were not identified, Rates of hydrolysis were directly proportional to the time of incubation and the amount of enzyme preparation -
Rabbit plarma. Blood was obtained from the carotid artery of anesthetized rabbits and drawn into heparinized tubes. The tubes were centrifuged at 500g for 15 min in a refrigerated table top centrifuge. The plasma was aliquoted and kept at -20" until used, For measurement of plasma activity. peptides ( S O M ) were placed in a Tris buffer (pH 7.5) containing 300 m M NaCl, O. 1 m M COCI: and 10 pM captopfil, The sarnple was incubated for 5 min at 37" before adding 1 S j L of plasma (total volume 1OOO pi.). Aliquots were taken at O, 15. 30 and 60 min. immersed in boiling water for 5min and centrifuged before KPLC analysis. Selective eval- uation of CPN or ArnM activity was obtained, respectively. by addingan AmMinhibitor (amastatin; IO 9M) or a CPN inhibitor (mergetpa: 10 PM) in the simple prior to incubation at 37".
Crude membrane preparation from rabbit kidneys. Kidneys were perfiued in situ with saline containing heparin (10 U/rnL) and semi-purified neutral endo- peptidase was prepared according to Fagny et al. (36). Briefly. the cortex was homogenized in a Hepes buffer (pH 7.5 with 10 mM MgC12). The homogenate was centrifuged at 1,500 g and the supernatant collected and centrifuged for 20 min at 15.000 g. The pellet was washed, rehomogenized in the Hepes buffer without MgClIV aliquoted and stored at -20". Protein concentration was measured by the method of Lomy etal. 1371. Peptides (50 jM) were incubated at 37" with the membrane preparation (final protein concentration 4.3 pg/mL) in a 50 mM Tris-HCi buffer, p H 7.6, containing bestatin (0.1 mM) and captopril (20 pM). The total incubation volume was lOOO pL and aliquots were withdrawn periodically, mixcd with 20.d of IO% TFA/water, and centrifugcd.
Puged ACE. Purified ACE (from rabbit lungs; Sigma) was dissolved in the same buffer used for plasma assays but without inhibitors or COCI?. The incubation conditions were also the sarne except for the use of 1 pL of ACE (final concentration 1 pg protein/rnL) as asource of enzyme instead of plasma.

Isoluted neunophih. Neutrophils were prepared from heparinized venous blood obtairied from healthy volunteers according to Bdyum [38]. Peptides (50 PM) were incubated with 6 x 1@ neuîmphils in Hanks' balanced salt solution (HBSS), totai voIume 200 .A. for 0, 15.30 and 60 min. At the end of the incubation the sample was centrifuged at 2000g for 1 min and 120 ,yL. of the supernatant was withdrawn and boiled for 5 min before HPLC anaiysis.
Binding to h m a n PMNs Human recombinant C5a (Sigma) was wI-labcled
using Iodogen (Pierce, Rockford, E). A solution of CSa. 10pg in 25 pL HBSS, was plaœd in a 1-mL Iodogen-coated tube: 500 pCi of NalZSI (Amersham, Oakville, Ontario) was added and the mixture alIowed to rcact for 15 min. The reaction was stopped by removing the mixture fiom the reaction tube and rnixing it with a solution of 0.25 M KI and NaSO, in HBSS. The free iodine was separated from the labeled protein using a PD-10 column (Pharmacia, Baie D'Urfé, Québec) equilibrated with HBSS buffer containing 0.1% (BSA) bovine serum albumin. The specific actïvity range was 220-570 Ci/ mmol.
For the binding assay. a suspension of PMNs (5 x IOb/mL) in HBSS containing 0.25% BSA was incubated for 60 min at O" with the appropriate concentration of ['"I]C5a. Total volume for binding assays was 200 uL. PMN-bound radioligand was then separated hom the unbound fiuid phase by centrifugation of a 1Oe.L sampIe of the ce11 suspension through a 3:7 (v/v) mixture of dioctyl phtha1ate:dibutyl phthalate (Aldrich Chemical Co., Milwaukee, WI) for 1 min at 10,000 g in a niicrofüge (Beckman, Fullerton, CA) [391. The tubes were then cut in half with scissors for evaluation of cell- bound [l31]C5a in the ce11 pellet-
Additional evidence of the agonist or antagonist behavior of CSe receptor ligands was obtained in the form of receptor dom-replation. Acute exposure to C5a at 37" tesults in an apparent los of binding sites. secondary to reuptor-ligand intemalization. without significant changes in the affinity of the residual receptors [40], In the present
midy. a suspeusion of PMNs (5 x 1O6/mL) in HBSS containing 0.25% BSA was incubatcd for 10 min at 37" in the presence of a mld ligand (CSa, a synthetic peptide, protamine or no ligand in controls) at a concentration proven to occupy a large fraction of the binding sites. Aftcr this incubation, the ceils were washed twiu-th ice-cofd HBSSBSA and the cclls were rcincubated as above at 0' to measure the binding of 2 nM [-IJCSa (non-specïfîc binding evaluated under each condition in the prcsence of a 100 nM concentration of cold CSa).
Supetoxide anion releae by newophilr Superoxide production was monitored as the
reduction of cytochrorne c for 5 min at 37" in tubes containing 2.5 x 10" neutrophils, 62.5 pM cytochrorne c (type Vf, Sigma) and U a or the desired anaiogue (total volume of 1 mL). The reactions were stopped by transferring the tubes to an ice-cold bath and the addition of superoxide dismutase (Iinalconcentration, 62.5 &nL) followed by centrifugation. The absorbanœ was read at 550 nrn and the amount of superoxide produced was caiculated by using an extinction coefficient of 21,000 M - ' ~ r n - ~ - Conditions tested included the direct analogue-induced superoxide release (possible agon- ist action) and the inhibitory effect of analogues or dmgs on C5a- or FMLP-induced effect (antagonist action).
Isolated blood uessek Rabbit vascular strips were prepared as described
1411 and were suspended in 5-rnL tissue baths containing Krebs solution [IS]. Their responses to agents were recorded using isometnc transducen (mode1 52-9545, Harvard Bioscience, South Natick. MA). The pulmonary artery and portal vein segments were cut helically and subjected to a baseline tension of 1 and OSg, respectiveIy. After an equilibration period of 1 hr, tissues were challenged with one of the chernotactic agents. The response to C5a is predominantly a relaxation. To record this type of response, tissues were always contracted with a submaximal concentration of phenylephrine (500 nM) for 1&30 min, which is the period required to reach a contraction plateau, before injecting the chernotactic peptides [17]. The effects of CSa or
Table 1 . Primaq structure of the C-terminal ponion of human and porcine C5a and of synthetic analoguesw
' Analogues 1 and II wcrc dcvclopcd fmm the porcine sequene and analogues III and IV from the human scqucncc. which are s h o w for cornparison. Substihrted rtsidues are undcrlined. Abbrcviations: Cha, cydohcxylalanine: Ac-Phc. N-aœtyi-phenylalanine; and McPhe, N-methyt- phcny lalaninc.

G. DRAPEAU n al.
Table 2. Rates of hydrdysis of analogues &IV by different enzyme prepacations'
Mctabotic degradation
' Vaiuts are the means 2 SEM of at least thret detcnninations; NS. not significant. Assays wcrc pcrformed at a finai substrate concentration of SOph4. Total incubation volume was 1 mL in which the following quantitics of enzyme pteparation wcre uscd for individuai asays: plasma. 15 &; ACE, t pg; kidncy extract. 4.3 pg of protein: and human neutrophiis, 6 x f cclis.
related peptides were tested up to four times at 9û- min intervals, with extensive washings between the tests. Results are expressed as the percentage of relaxation of the phenylephrine-induced plateau.
In vivo eualuation of CSa receptor agonisa
The hemodynamic and hematologic effects of CSa and analogues were evaiuated using New Zealand white rabbits of either sex (1.5 to 2.0 kg). The animals were anesthetized with sodium pentobarbital(30 mg/ kg. i.v., adjusted individually). Lidocaine (2%) was used in addition at sites of incision, The trachea was intu bated and ventilatory assistance was provided with a Harvard respiratory pump. A polyethylene catheter PE-90 was inserted into the right common carotid artery and pushed into the aorta for recording blood pressure with a pressure transducer (Statharn PUID) on a Grass polygraph (mode1 79). A Gfass tachograph (mode1 7PUC) was coupled electronically to the blood pressure recording system and provided a continuous recording of the heart rate. The carotid artery catheter was also used to obtain srna11 blood samples at specific tirnes for recording hematological responses to CSa- To avoid clotting in the catheter, the animals were given 400 U of heparin at the beginning of the expriment. A second PE-90 catheter was inserted into the Ieft externat jugular vein for intravenous injections. This Ut vivo mode1 has been used to perform a comparative evaiuation of CSa and of the new agonists synthesized with the following end points: acute blood pressure changes. acute neutropenia. A given animal was not injected more than twice with C5a or a related analogue, with a 45-min interval between doses; under these conditions. the response exhibits Iittle tachyphylaxis [12]. Each hypotensive episode was characterized quantitatively by the maximal response (fa11 of the mean blood pressure, mm Hg) and by the tirne course, evaluated by the period starting at the bolus injection and ending when half of the maximal hypotensive response had faded (tirne for half- recovery ) .
Arterial bIood sarnples were obtained 5 min before and 5 and 30min after CSa injection for a determination of the white blood cell counts (in a hemacytorneter after lysis of erythrocytes in 1% acetic acid) and a differential leukocyte count
(following Wright staining of blood smears). The results of the leukoqte counts are cxpressed as a percentage of the badine value (munt 5 min before C5a) for neutrophils and lymphocytes; other leukocyte types are infrequent in rabbit peripheral blood [42].
Results are expressed as means SEM. Student's r-test. one-way analysis of variance, Dunnett's test and linear regression were used in the fom of a software package (431 to analyze the data.
Peptide synthesis and rnetabolic studies The primary structures of the analogues syn-
thesized for this study are Iisted in Table 1. Anaiogue I has been reported by Ember er al. [28]; it is porcine CSa 60-74 with the T e and Phe6' substitutions. Peptide II is a shortened version of analogue 1 in which we have substituted an IIe residue for the N- terminai Gin? Analogue III is an extensively modified analogue of the C-terminal octapeptide of human C5a. Its structure is based on a compound (Ac-His-Lys-AspCha-Cha-Vala-Afa-Arg-OH) re- ported by Kawai et al. [30J, that we have modified slightly to include the N-terminal Phe6' for His substitution shown to improve affinity for the receptor [28,29]. Analogue IV is a hexapeptide introduced by MoIlison et al. (311 in a report where its activity on human neutrophils is described.
Analogues II-N were incubated with different enzyme preparations in order to examine their metabolic stability. These include plasma, in which ArnM and CPN activity were measured separately, purified ACE. a semi-purified neutral endopeptidase preparation and incubation with intact human neutrophils. Although the degradation of peptides was evaluated by decreasing substrate concentration, metabolite peaks, that increased as a hinction of tirne. were observed in parallel with substrate decrease. No attempt was made to identify these metabolites. Results presented in Table 2 show that in plasma. where selective CPN or AmM activities were examined, analogue 11 was metaboIized at comparable rates by either enzyme while the other

Fig. 1. Binding of [IYI]C5a to intact human PMNs. Tbe cxperiments wcre pcrformed shultancoudy with the =Us of a single donor. (A) Total (O), oon-spccific (x) and specific (A) binding in the 18 ph4 to 5.6 nbf range of radioligand. Non-spctific binding was estabiisbed in the presena of a 100 nM concentration of cold CSa ( 1 @d for the 5.6 aM wnccnnation Ievcl of [=I]CSa). (B) Scatchard plot analysis derived from the samt data and assuming a single class of binding sites. The correlation coefficient was 0.958. KD and B , values dcrived from the Scatchrd analysis wcrc 1.0 uM and 46.7 fmoI/l0' celis, rcspectively.
two analogues wcre cornpletely resistant. In fan at substrate concentrations of 50 pM, analogue ï I was degraded by ail preparations tested while, invenely, analogues III and IV were found resistant to al1 except for some degradation by neutrophils and marginal catabolism of andogue 111 by the kidncy extract. The rate for the catabolism of analogue II by ACE (500 nmol/min/mg protein) was almost ten times higher than the one obscrved with bradykinin (441, a peptide that also posseses a C-terminai Arg residue, whereas the rate for NEP degradation of this peptide was about half that obxrved with bradykinin analogues.' Enzymes involved in the degradation of the CSa analogues by intact neutrophils were not identified and, although pcaks probably attributable to metabolites wcre obscrvcd
' Drapeau G. Audet R. Lcvesque L, Godin D and Mamau F, Development and in vivo evaiuation of mttaboticaily rcsistant antagonists of B, reccpton for knins. Manuscript submitted for publication.
Fig. 2. Cornpetition for ["IICSa (2 nM) mcptor binding to intact PMNs by cold =a. synthctic anaiogucs. and the unrclatcd chernotactic peptide FMLP (A) and by protamine (B). Values are the means of criplicate dctenninations from cach one of three stparatc experimcnts. ï h e ordinate scale is the raiduai binding relative to the total binding without competing drug cstablishcd for cach donor. The non-
specific binding was subtraacd.
by HPLC anaiysis, pan of the decrease in substrate concentration may be due to uptake by neutrophils.
We compared the activitics of CSa and of its mimetic analogues in a well characteritcd model, the isolated hurnan neutmphil, to establish their relative potencies and agonist status. The binding of [iz'I]C5a to intact human PMNs was performed at CP and it was found to be saturable; the proportion of non-specific binding was relatively small (Fig. 1A). Scatchard plot analysis of the data (Fig. 18) rcvcalcd a nnglc dass of reccpton with KD and B,, values of. respectively 1.0 nM and 46.7 €moI/lP cells (about 28.000 reœpton per cc11 in this particular expenrncnt). These values are similar to those prcviously reported for the binding of purified CSa to intact human PMNs [a. 451. The abilities of CSa and its C-terminal analogues to displace the binding of [lBI]CSa at 00. were evaluated (Fig. 2). The unnlated chernotacric peptide, f-Met-Leu-Phe (FMLP). and protamine. a proposed CSa antagonist [W. were also tested. Ali the C-terminal analogues of C5a were at levt 1000-fold Iess potent than the recombinant cold protein to compte for binding. The order of potency was C5a > IV > III > II > 1. FMLP failed to compete for C5a binding sites.

Fig. 3. Superoxide anion relcase h m human PMNs stimuiated with C5a or with synthetic analogues. Tubes contained 2.5 x 10b neutrophils. ï h c control absorbane reading (no stimulus) was subtracted €rom the othcr rcadings. Values are the rncans z SEM of 3-9 deter- minations. The baxline releasc in cells incubatcd at 37'
without stimulus was 4.0 = 0.03 nmol/tube.
Peptide 1 exhibited a low relative potency and consequently was not systernatically studied in the other assays. Protamine concentration was expressed as weight because it is not necessarily a homo eneous protein: the icJo competition value against FzIlC5a was about 30 pg/mL. The abilities of C5a and of the synthetic fragments
to stimulate fiinaional responses after receptor occupancy were studied. CSa released superoxide anions from human PMNs at the relatively hi& concentration of 10 nM and above (Fig. 3). Analogue iI showed some activity at 100,uM. and peptide III was a significant releaser at 10 pM and above. The relative activities suggest an order of potency sirnilar to the receptor binding assay. with the exception of analogue N. which was found to be inactive as a superoxide anion releaser up to 100 pM. Claims of antagonist behavior for this peptide f311 prornpted us to test analogue IV as an antagonist of CSa since its receptor affinity had been shown to be significant (Fig. 2A). Analogue IV, at I0Op.M. suppressed completely and sefectiveiy the superoxide release from human PMNs stimulated with C5a. but not with FMLP (Fig. 4A). For cornparison. a non- peptide antagonist of FMLP, DPN [33.34] was shown to suppress superoxide release with the opposite specificity (Fig. 4B). The slight potentiation of CSa-induced superoxide release by DPN has been observed previously 1331, although it did not reach statistical significance . Protamine, at 100 pg/mL, behaved as a selective CSa antagonist (Fig. 4C).
ïhe agonist/antagonist s tatu of C5a receptor ligands was further assessed in dom-regulation expcriments. As shown in Fig. 5, the specific binding of [lZI]C5a (2nM) was variably depresscd by a preincubation with the mld form of the C5a receptor ligand under study. The concentration of the ligands used in the 10-min expenmental period at 37" was chosen on the bais of competition data prescnted in Fig. 2; a large proportion of the recepton was expected to be occupied in each case. C5a (10 nM)
Fig. 4. Selcctivc antagonim of chcmotaaic peptide- induccd supcroxide rcltase by peptide N (A). DPN (B) and protamint (C). The expcrimentai conditions wcrc as in Fig. 3. Values arc the means z SEM of 4 determinations in panel A, of 3-10 dttcnninations in pane1 B and of 6 dctcrminauons in panel C. Thc baselinc rclcasc in ceils incubated at 37" without stimulus was 2.3 I 0.3, 3.2 i 0.5 and 0.9 I 0.3 nmo!/nibe in experimcnts reportcd in panels A. B and C, respcctively (N = 4. 10, and 6, rcspcctivcly). DPN alone does not rclcase supcroxidc significantly [421,
nor did protamine (2.0 2 0.8 nmot/tube).
and analogue III (100 PM) desensitized extensively the human PMNs. as measured by the subsequent binding of [1z~]C5a (Fig. 5). Analogues II and N (100 and 30 @M. respectively) produced only a partial desensitization, whereas protamine (30 pg/mL) had no significant effect. as compared with controls.
Vwcuiar and in vivo Mects of CSa and sefected C- terminal analogues
C5a and the peptides II-IV were found to elicit biphasic mechanical response in rabbit isolated portal veins and pulmonary artenes preconnacted with phenyltphnne (Fig. 6): a srnall, brief and inconsistent contraction was followed by a more sustained relaxation that faded away over tirne. These reactions have been analyzed previously [16, 171. Only the relaxant response. expressed as a percentage of the

Co. II
Fig. 5. Down-mgulation of the spccific binding of ['ZsflCSa ta human P m s in cclls prctreatcd at 57" with coid ligands of the tc#pton for CSa. ïhc cch wcre cnpo~cd for 10 min to CSa (IO M. andogues Iï (100jM). iïl (100pM), or W (30 pM) or to rotamine (30 &IL), and thca washcd; 8 the bind'ng to [ IICSa (2nM) was estabLished at 00. as explaincd in the r e n . Coutrois wtre ptcincubatcd in HBSS- BSA b-cr oaly. The average value for contmls was 570 2 300 cpm, Values are the means * S E M of tcipiicate detcrmiaations in Ume separate cxperbncuts with diffcrent blood doaorr and arc cxprcssed as a petccntagc of the
contractde plateau, was used in the quantitative andysis of biological activity in the portal veins (Fig. 7A) and the pulmonary artenes (Kg. 7B). The ordcr of potency of the andogues was convenientiy assessed by comparing the mean amplitude observed at 0.1 to 1jM; this order was CSa > III N > TI in both tissues- Peptide TV behaved as an agonùt in these vascufar assays, possessing at least a large: fraction of the intnnsic activity of analogue m. The profiles of the relaxant response induced by the analagucs, notably the relaxation duration, were similar to the ones recorded with recombinant C5a m g . 6).
As reported previously [12], C5a clicited hypo- tensive responses in anesthetized rabbits (Table 3). CSa-induceci hypotension was not accompanied by asignificant change of hem rate (results not shown). Compared to the acute neutropenic effea (Table 4), an assay that was approximately one order of magnitude more sensitive, relativeiy large doses (1.7 nniol, Lv., and above) were nceded to produce hypotcnsive rtsponses. The acute neutropenic effect of C5a (recorded
5 min after i.v. boluses) was dose-related and ouiy transient, as it disappeared 30min after B a injection, except for the 5.6 nmol dose levei. There was only a partial recovery 30 min aftcr injection at this intense receptor stimulation Icvel. Lymphocyte
1-1
10 min Fig. 6. Rcpnsentative tracings of vascular responses to C5a (1 nM) and to analogues II1 and N (1 pM). C5a ar analogues wcn apptied in the bathing fidd of pnrontractcd rabbit vascular strips (PV, portal vein; PA. pulmonary artcry). Thcre were Wmin ptriods beiwetn applications of stimulants. The contraction was obtaincd with phcnylephrinc (PE, 500nM). Abxissa scde: time: orcfinate d e : isomeuic contraction. Ctoscd symbols refcr to the application of agents and open symbob to washout
of stimulants.

s i d a r in amplitude and in duration, doses of analogue II equivalent to 450 timcs that of C5a had to be given. S i d a rcsults were obtained with analogue IV at only 20 tirnes that of CSa. The neutropenic effects of peptides II, ï I I and iV 5 min afîer bolus injection indicated a relative activity of about 0.2, 2 and 16%. respeaively, compared to ma. Therefore, the order of potency for both the hypotensive and neutropenic effect was CSa> N mm,
1 O 00 000 POOO Manondu- DrSCUSSION
Recently, synthetic peptides that retained a Ggaificant fraction of the potcncy of C5a werc
407 I
synthesized by several groups [%31; present s-
! results]. Thcse analogues cxhibited the whole
ao- specmm of CSa effeas, such as ncutrophil chernotaxis, histamine telcase (281 and cytokine
in production in human mononuclcar cells [47]. The most active pe tides usually included a kcy substitution. Hi$ for Phe. which aras shown to incrcase the affinity of C-terminal fragments
j by several orders of magnitude [29]. Other
O modifications, consistinn rnainlv in tivdroohobic a9 t iO (W
~ a m t ~ w ~ t n t ~ ~ n '"OD substitutions, were designed to i k a s e ibe &nity 1301. Multivle substitutions with unnaturd amino
Fig. 7. Relaxant efiect of CSa and of analogues on the rabbit isolated portai vein (A) and on the rabbit isolatcd pulrnonary ancry (B). Values arc the mcans 2 SEM of 4-7
detcrminations.
counts were not affected significantiy by C5a (Table 4). The exvrirnental design excluded the study of the late neutrophil count rebund. The hypotensive and neutropenic effccts of
selected CSa synthetic peptides with varying degrees of metabolic resistance and affinity are shown in Tables 3 and 4. To obtain hypotensive responses
àcids. tht the rtsulting constraints on the peptide backbone, yielded peptide IV, claimed to antagonize some of the effects of C5a on human neutrophils [31]. The reIative metabolic resistanœ of these increasingiy modified peptides has not been reported. We have obxrved that some of these peptides, analogues III and IV, are quite resistant to peptidases present in blood or on ccU membranes such as A d , CPN, ACE and neutral endopeptidase 24.11. Al1 of these enzyme preparations have been shown to degrade analogue II, a relativeIy unmodified C- terminal andogue of CSa. The fragments generated by clcavage of analogue II by these peptidases were not identified. It is suspected, however, that CPN cleaves the C-terminal Arg residue to generate the
Tabte 3. Hypotcmivt cffccts and time for haü-rccovcry (ma) elicitcd by i.v. boluses of human anaphylatoxin C5a or synthetic analogues ia ancsthetizcd rabbits
Doses Bascline MAP AMAP TRso Peptide (x moI) N (mm Hg) (mm Hg) (w Ve hicle' C5a O. 17
0.56 1.7 5.6
II 850 W)(]
III 94 280 940
IV 12 35
120 --
O VehicIc for CSa: 0.1 mL of 0.25% BSA followcd by 0.3 m i saline. Results art mcans 2 SEM or the average of two determinations with values in parentheses. MAP = mean arttriai pressure.

Spthctic C5a reccptor agonists 1297
Table 4- Changes of poIyrnorphonucIear lcukocytc and lymphocyte counts in pcn'pheral blood of anesthcuttd rabbits at two diifercnt times (5 and 30 min) aftcr i.v. boluses of human recombinant C5a
or o f synthctic analogues - - - - - -
Polyrnorphonuclear Ieukocytcs L ~ m ~ h a c Y t a
Doses Responsc (Qo control) Rcspowe (95 control) Peptide ( X IO'q mol) N 5 min 30 min - 5 min 30 min
Vehicle' CSa 0.17
0.56 1.7 5.6
II 850 2500
IIX 94 280 940
N 12 35
120
Vehicle: 0.1 mL of 0.25% BSA followcd by 0.3 mL saiine- Rcsults are means 2 SEM or the average of two determinations with values in parentheses. Basdine (prc-injection) polymorphonudcar leukocytc and lymphocyte counts for all groups wcre. rcspcctively. 2464 220 and 1795 88 eus/& of blood (N = 56).
des-Arg rnetaboIite, ACE would Iikely libetate the C-terminal Gly-Arg dipeptide, while AmM would sequentiaily release the N-terminal residues. The relative importance of ACE and neutral-endo- peptidase as proteases involved in the regulation of CSa in vivo remains to be established. However these enzymes couid play a protective role against sub-clinical complement activation. It is of interest to note that clinica1 treatment of hypertension by ACE inhibitors is sometimes accompanied by side effects that are reminiscent of inflammatory processes.
b The competirion for [131!C5a receptor binding showed that analogue IV was the most potent one cxamined, followed by peptides III and II. In vitro biological assays generally confirmed this order of potency with some particular findings for peptide W. The latter behaved as a selective antagonist of CSa-induced superoxide release from human PMNs, suggesting that receptor binding by this hexapeptide is not followed by funcrional response.
The partial agonist status of analogue IV is also suggested by experiments designed to show Iigand- induced down-regulation (Fig. 5). C5a and anaiogue III were very effective to acutely dom-regulate C5a receptors at 37". a process presumably linked to Ligand-receptor complex interndization [a]. Howcver, the antagonist protamine, at a con- centration that ocmpies a large hction of the rece tors, failed to down-regulate the spccific binding P( of [l IlCsa. Only small dom-regulations have becn observed in cells pretreated with analogues II or N, possibly for different reasons. Analogue n exhibits a low affinity and is susceptible to degradation by human PMNs (Table 2), possibly resulting in a smaller effective receptor occupancy dunng the 10- min desensitization penod. Analogue IV, by opposition, has a much higher affinity and stabiiity
and was expected to occupy at 30pM at least as many receptors as analogue ILI in this series of experiments. The low down-regulation obsemd must be explained by a Iow, but measurable efficacy , supporting its intemediate status between a pure antagonist (protamine) and full agonists (CSa, analogue III). In their report Mollison et al. [31] showed that analogue TV is an antagonist of myeloperoxidase rtlease in human neutrophils whiie behaving as a partial agonist on the chernotactic responses observed in these celis.
The discrepant agonist-antagonist behavior of peptide IV may be explained by the hypothesis of an efficacy lower for this peptide than for CSa or the other peptides. The superoxide release assay is not a sensitive one for C5a relative to the degrce of receptor occupancy at the corresponding con- centrations (compare Fig. iA with Fig. 3). If a phannacologic cffect requires the stimulation of a very Iarge proportion of the receptors on each ceIl, Ligands with reduced efficacy are more likely to behave as antagonists (discussed by Leslie; [48]). In rabbit isolated vascular tissue, systerns that respond to subnanomolar concentrations of CSa, peptide IV behaved as an agonist, with possibly a weaker potency than expected from the binding data, relative to analogues III and II. This type of situation is expected for a low efficacy agonist if there is some "spare receptor" reserve in the rcsponding celis [48]. However, the different cxperirnental conditions in these two assays (Le. binding vs isolated tissues) and, possibly. the species barrier may also contribute to the potency difference. In uioo, peptide IV was a potent C5a mimetic. Thetefore, analogue IV does not appear to behave generally like an antagonist of CSa, but is possibly a "lead" compound towards ligand with lower efficacy that could be developed into antagonists. The discrepant agonist-antagonist

1298 G- DWU et al.
behavior of analogue IV dws not necessariIy mggtst the existence of multiple receptor types; binding data from Fig. I indicate a single type of sites in the ligand concentration range 18 pM to 5.6 nM.
The three C-teminal analogues of C5a examined for metabolic degradation by various peptidase sources exhibited wide differences of nisceptibility. However. multiple metabolic resistance did not result in a different time course for some of the CSa- induced effects. such as the relaxation of rabbit isolated vascular strips (Fig. 6) or the acute neutropenic effect following bolus injections in viuo (Table 3). The latter responses were n o longer detectable at the 3@min time point, except for extremely intense stimulation leveis. Mechanisms different from ligand metabotic disposition, such as receptor desensitization or Iigand diffusion and dilution in vivo. may account for the limitcd tirne course of these effects. The most striking effect of metabolic resistance in vivo was the gain in relative potency for analogues ïïï and IV, which approached the molar potency of C5a, despite a wide affinity gap between the analogues and the recombinant protein revealed by the in uiho assays. A good cornparison term for assessing the effect of metabolic resistance in vivo is the susceptible analogue II, which exhibited in oivo the typical wide gap of molar potency relative to C5a observed in uino.
The in vivo effect of the synthetic anaiogues can probably be explained entirely by their action on CSa recepron. Notably, their relative lack of effect on lymphocyte counts, a cet1 type which lacks recepton for C5a [49], supports such a proposition. Hypotension induced by analogue N was paraIIel to an increased central venous pressure (results not shown). sirnilar to CSa-induced hypotension [lt]. A full characterization of the rnechanisms by which these anaiogues ptoduced their effects will be possible when potent C5a receptor antagonists become avaiIable,
The present smdy suggests that the pharmacology of anaphylatoxin CSa can be approached successfully with the methodologies deveioped for much shorter peptides and with concepts developed from the study of conventional drugs. nie affinity, efficacy and metabolic disposition rate are three distinct par- ameters that govern the in vivo pharmacology of any CSa receptor ligand. The selective antagonist effect of protamine for CSa [46] has becn confirmed and studies are underway to define the minimal structure within this natural protein that binds the receptors for CSa.
Ackno wledgcmenrs-The technical hcl p of Ms. Daniciie Paré and fohanne Bouthillier is acknowledgcd. This work was supponed by a grant from the "Fonds de la recherche en Same du Québecw (FRSQ). GD. and F.M. arc Scholars of the FRSQ. S.B. is the rccipient of a summcr r#earch Studenahip awarded jointly by the Phamaccutical Manufaauren' Association of Canada and the Medica! Rcsearch Council of Canada.
1. Hugli TE. The stntctural buis for anaphylatoxins and chemotactic bnctions of C3a. C4a. and CSa. CRC Crit Reu Immunol 1: 321-366, 1981.
Johnson AR. Hugli TE and Müller-Eberhard HJ, RcIeasc of histamine fmm rat mast œIls by the complemcnt peptides Qa and CSa. immunology 28: 1067-1080. 1975. Tonnesen MG. SmedIy LA and Henson PM. Ncutrophil-endothclid ceII interactions. Modulation of neutrophif adhtsiveness induced by complcmcnt fragments CSa and CSa des Arg and formyl-mcthionyl- Ieucyl-phcnylalanine in o h . J Ciin Inutsr 74: 1581- 1592, 1983. Okusawa S, Dinarclio CA. Yanccy KB. Endrcs S. Lawley TJ. Frank MM, Burke JF and Geifand SA. U a induction of human intcrleukin 1. Synergisticcffcct with endotoxin or interferon-y. J fmmwiol 139 2635- W. 1981. Schindlcr R. Gclfand JA and DinarelIo CA, Recorn- binant C5a stimulates mmcription rathcr than transIation of interleukin-1 cïL-1) and tumor necrosis factor: Translational signal providcd by lipo- polysaccharide or IL-I itscff. Blood 76: 1631-1638, 1990. Scholz W. McCIurg MR. Cardenas Gf, Smith M, Noonan DJ, Hugli TE and Morgan EL, C5a-mcdiated relcasc of intcrlculrin-6 by human monocytes, Clin Immunol Immwiopathol SI: 297-307. 1990. Gerard NF and Gerard C. n i e chernotacsic reccptor for human CSa anaphylatoxin. Nonut 349: 614417, 1991. BouIay F. Mcry L. Tardif M, Brouchon L and Vignais P. Expression cloning of a reccptor for CSa anaphylatoxin on differcntiated HLdO cclls. Bio- chemistry JO: 2993-2999. 1991. Rollins TE, Siciliano S. Kobayashi S, CianciamIo DN, Bonilla-Argudo V, Collier K and Spnngcr MS, Purification of the active CSa reccptor from human poIymorphonuc1ea.r leukocytes as a receptor-G, complcx. Proc Nat1 Acad Sci USA 88: 971-975, 1991, Stimier NP. Hugli TE and Bloor CM, Pulmonary injury induccd by C3a and C5a anaphylatoxins. Am J Parhol 100: 327-348. 1980. Roxnbaum JT. HartiaIa KT, Webster RO. Howes EL and Goldstein IM, Antiinflamrnatory efka of endotoxin. Inhibition of rabbit pol ymorphonudear leukocyte rcsponsc to complement (CS)-denvcd peptides in viuo and in uirro. Am / Pathol 113: 291- 299. 1983. Lundbcrg C, Marceau F and Hugli TE. CSa-induud hcrnodynamic and htmatotogic changes in the rabbit: RoIe of cyclooxygcnasc producu and poly- morphonuclcar leukocytcs. Am J Pathol128: 47143. 1987. Kajita T and Hugli TE. CSa-induced neutrophilia. A pnmary humoral mechanism for temitment of neutrophils. Am J Pathal 137: 467477, 1990. Jagels MA and HugIi TE. Ncutrophii chemotactic factors promote lcukocytosis. A common mechanism for ceIIu1ar remitmcnt from bone marrow. J Immunoi 148: 111W128. 1992. Marceau F. Lundberg C and HugIi TE. Effccts of the anaphylatoxins on circulation. fmmunoptronnocology 14: 67-84. 1987. Hugli TE and Marceau F, Effects of the a a anaphylatoxin and its ttlationship to cyc1o-oxygcnasc metabolites in rabbit vaxular strips. Br J Phormacol w 7 2 ~ 7 3 3 , 1 9 8 ~ .
17. Petitclerc E and Mamau F, Relaxant cffcct of chemotactic peptides on rabbit v d a r strips: Evidencc for nitric oxide rclcase from a nonendothelial source. Blood VesscLr 28: 452463, 1991.
18. Marctau F and HugIi TE, Effect of U a and C5a anaphylatoxinson guinta pig blood vesseis.3 Phormacal Exp ïher Ub: 74S754, 1984.
19. Marceau F, deBIois D, Laplantc C. Petitclerc E,

Pelletier G. Grose JH and Hugli ïE, Contractile effcct of the chernotactic factors f-Met-Leu-Phe and C5a on the human isolatcd umbilical artery: RoIc of cydooxygcnase produas and tissue macrophages. C k R a 67: 105P-IWO. 1990.
20. Bokish VA and Müller-Eberhard KI, Anaphylatoxin inactivator of human plasma: Iu isolation and characicrizauon as a carbaxpcptidase- 3 Clin Inom 49: 2427-3336. 1970.
21. Skidgcl RA, Basic carboxypeptidascs: Rcgulaton of peptide hormonc actïvity. Trenàs PharmocoI Sri 9 2SeM4.1988.
22. Campbell W. Yonczu K. Shinohara T and Okada H. An arginine carhxypcptidase geacratcd dw-ng coagulation is diminished or absent in patients with rheumatoid anhritis. J tub Clin Invcrt 115: 61M12. 1990.
23. Hendeiks D, Wang W, Scharpé S. Lnrnrnaert MP and Van Sande hl, Purification and charaacrization of a new arginine carboxypeptida~ in h u ~ m serum. BiochUn Biophys A m 1034: 86-92. 1990.
24. Chenoweth DE and Hugli TE. Binding and degradation of C5a by hurnan neuuophils. 3 lmmunol1211: 1517. 1980.
25. Wagner JL and Hugli TE, Radioimmwoassay for anaphyiatoxins: A sensitive rncthad for determinhg compiement activation products in biologid fiuids. And Biochem î36: 7S88. 1983.
26. Hcideman M and Hugli TE. Anaphylatoxin gencration in multisystcrn organ failun. J T r a w 24: 1038-1043. 1984.
27. Hirnmetfarb J. Gcrard NP and Hakim RM. lntradiaiytic moduiation of granulocyte CSa reccpton. I Am Soc Nephrol2: ItCk926. 1991.
28. Ember JA. Sanderson SD, Taylor S, Kawahara M and Hugli TE. Biologic activity of synthctic analogues of CSa anaphylatoxin, J Immunal. in press.
29. Or YS. Clark RF. Lane B. Mol l in KW, Caner GW and Luly JR, Irnproverncnts in the minimum binding sequcncc of CSa: Examination of His-67. J Med Chem 35: U);?-4û4. 1992.
M. Kawai M. Quincy DA. Lane B. Mollison KW, Or YS. Luly IR and Caner GW. Strucnirefunction studics in a series of carboxyl-terminal octapeptide analogues of anaphylatoxin =a, 3 Mcd Chem 35: 22G223, 1992.
31. Mollison KW. Krause RA. Fcy TA, Miller L. Wicdcrnan FE. Kawai M and Lanc B. Hcxapeptidc analogs of CSa anaphylatoxin rcveal hctcrogenous ncutrophil agonism/anragonism. FASEB / 6: A2058, 1 =.
32. Mollison KW. Fey TA. Krause RA. Mandecki W, Fox IL and Carter GW, High-level CSa gcnc expression and recovcry of recombinant human C5a from &chcrichi& coli. Agents Actions 21: W 3 7 0 , 1981.
33. Levesque L. C.-Gaudreault R and Marceau F. The interaction of 35-pyrazolidinedione dmgs with reccpton for f-Met-Leu-Phe on hurnan ncutrophil
~e~kocyt~s : A study of ihc suu-acti~ity relation- sbip. Cm I PhyJiol PliormPurl69: 419-425, 1991-
34- Ltvcsqut L. C.-Gaudrcault R and Marceau F, Comparison of two dasscs of non-peptide d q p as antagonists of neutrophil reœpton for &Met-ku-Pk Pyrazolons and iodinatcd radiographie contras agents. Biuchern Pharmacol 43: 551.560. 1992.
35- Drapeau G and Rcgoli D, Syntbesis of bradykwn arialogs- In: Meihods in Entymology (Ed. DiSahto G). Vol- 163. pp. 26M72, Acadcmic Rcss. New York, 1988.
36- Fagny C, Miche1 A, &nard 1, Berkenboom G. Fontaine J and D&odt-Lanckmaa M. In o i m degradation of endothelin-l by endopeptidase 24.11 (enkcphaiinase), Pepndcr U: T I Z n 8 . 1991.
37. Lowry OH, Roscbmugh NJ, Farr AL and Randall RJ, Protein measurement with the Folin phtnol rcagcnt* J B b l Chm 193: 245-2'75.1951.
38. Bayum A. Isolation of leukocytcs h m huma blood. S d J CIUI Lmb Inuar 21 (Suppl9): %a, 196s.
39. Melewia FM. Plummer JM and Spiegclkrg HS, Comparison of the Fc rcccpton for I g E on human lympbocytcs and monocytes. I Immruiol 129: -569, 1983.
40. Huey R and Hugli TE. Charactcrizatioa of a #a rcccptor on human polymorphonuclear leukocytes ( P m . J Immwrol WJ: 2QU-t068.1985.
41. Laplante C. Trcmblay B and Matctau F, Reiaxaat cffcct of N-formyl-methioayl-lcucyt-pknylaianiac on rabbit vascular strips. J Phtzmucol Erp ïkr 24û: 77b 780. 1989.
42. Wcisbroth SH. 71ie Biology of the Labororory Rubbù. Academic Ptess. New York, 1974.
43. Taliarida RI and Murray Ml, Manuai of Phrmrrcologic Cai&tiolls with Comqurer Programs. Springer, New York, 1987.
44. Drapeau G. Chow A and Ward P. Metabolism of bradykinin analogs by angiotensin 1 converthg enzyme and cahxypeptidase N. Peptides 12: 631638, 1991.
45. Cbcnowcth DEand Hugli TE, Demonstrationof sptcific C5a rcccptor on intact human polymorphonuciear ieukocytes. Pmc Nad AC& Sci USA 75: 394S3947. 1978.
46. O k n UB. Selmcr J and Kahl JU. Complement CSa reccptor antagonism by protamine and poly-L-Arg on human Ieukocytes. Complemcnf 5: 153-162, 1988.
47. Morgan EL. Sandtnon S, Schoiz W. Noonan DJ, Weiglt WO and Hugli TE. !denfication and Ctiaractetllaaon of the effeffor region within human C5a responsiblc for stimulation uI IL4 synthcsis. J lmmunol 148: 3937-3942, 1992.
4û. Leslie FM, Metiiods uscd for the study of opioid reccptors. Phormocol Reo 39: K11-249, 1981.
49. Werfel T, Oppcrrnann M. Schulzt M. Ktieger G, Weber M and Go@ O, Binding of fluorescein-labeted anaphylatoxin C5a to human periphtrai btood, spleen, and bone marrow Icukocytcs. Blood 19: 152-160.1992.

Development and in Vivo Evaluation of Metabolically Resistant Antagonists of BI Receptors for Kinins'
GUY DRAPEAU, RiTCHIE AUDET, LUC LEVESQUE, DENIS GODIN and FRANÇûIS MARCEAU Cmtm de Recherche (vniv81Si18 Laval), Hdtel-Dieu de OuBbec, QuBbec, Canada
Acc%pW for pubkation January 1 t . 1993
ABsTRACt The & fe~~ptors for kinins are selectively stimulated by brady- kinin (BK) and L W K metabd'i that do not have the C- t m i M I arginine. des-ArgD-8~ and ~ysaes-w-~~, respec- tive&. 8, receptm mediate a definite subset of the carâbvas- cular effects of kinins in rmnd and septic animais. We have studii the metabdisrn of the best selective & antagmbt, Lys[leu"]des--@-BK, in order to support the ratiomi design of new antagonists mat have increased metabdic stabiïi. The affin@ of the new compounds was evaîuated uçing the p& scaie and was based on aie antagmism of the contractüe etfect of kinins in the rabbit isolatecl aortic preparation. Acetylathn of the a-amino group of the N-temiinai Lys residue provided an exce(- lent protection ftom the degradation by rabbit blood plasma This arid the inhibit effet3 of amastatin on the metaboliim of L y ~ [ ~ d e s - A & K indkated that aminopeptidas8 M (AmM) is the major route of inactivation for mis ciass of peptides in
plasma Varbus other rnodmc;itioris atforded a more or iess cbmplete resistance to purified angiotensin I amverthg eruyme (ACE). One analog, Ac-Lys[W6. Lerpldes-A@-BK, was f o u n d ~ t o t h e a b o v e - m e n W W e n z y m e s a n d toneutral emdopeptidase-24.11 extracted from rabbit Wney. This antag- oriist, aithaqh 100 times less poterit than the parent peptide ~ys[teu4]bes-~@-~~ on the rabbit a- preparation, was equi- potent in vivo against the hypotensive effect of a & agonist in IipopoSrsaccharide-treated rabbits, and, unlike the original am- pound, its effect was after the end of the infusion. Ac- Lys[MeAia6. Ld)lesArg%K does not antagonize & receptors either in vitro or in vivo. Camparative data ori other antagonists with varying degrees of potency and metabdi tesistance and ln vivo stud'i with peptidase inhibitors indiiite the major roles of neutraI endopeptidase-24.11 and of ACE as in vivo inactivation pathways for 6, antagonists.
AIthough the classification of receptors for %ininn still awaits the molecular cloning of al1 relevant reteptor m o l d e s , con- aiderable pharmacologie evidence supports the broad classifi- cation dong the Bi and Bz ~ p e s (Regoli and Bara&, 1980; Farmer and B m h , 19921. The Br receptor is selectively stim- ulated by kinin fragments of BK and Lys-BK without the C- terminal arginine residue (des- *-BK and Lys-des-Ar&'-BK, respectively). The development of cornpetitive, potent and se- lective entagonists (protoîypes [LeuS]des-w-BK and Lys[Leu8]des-@-BK) has consolidateci the pharmricologic identity of BI receptors (Regoli et d, 1977). Agonists and antagonists for Br receptors are conveniently assayeti on the rabbit aortic preparation, which exhibits a sustained and direct contractile response to kinins solely via this teceptor type (Regoli et d, 1977). The Bi receptors for Linins are usuaiiy not prominent enough
Rcccived for pubkation July '28.1992 ' Tbis w r k w u nipporrcd by gmnu Erom the Medicai iheareh Council of
C a r d a (gram MT-12217) and the -bec Hcart foundatioa. GD and FM are SchoLrr of the Fonda de la recherche en Santa du Quibec The "Centre & rucharche de 1'HÔtel-Dieu & Quibec" has nipponcd RA. and D.G. thmugi the r d of Studentrhips,
to account for the cardiovascular pharmacology of Linins, such as hypotension, vasodilation and increased vascular permeabil- ity. However, careful dpis hasi revealed that they acmunt for a significant part of the effect of lrinins in normal dogs (Toda et d, 1987; Müiler-Schweinitzer, 1988; Rhaieb et d, 1989; Lmtie et d, 1992) and that they CO-erist with & recep- tom, aRer an apparent up-regdation pmcess, in the cardiovas- cular system of rabbits or pigs that have b e n injected with bacteriai materials or that are esperîencing experimental ar- tbritis (Regoii et aL, 1981; Siebeck et ai, 1989; Farmer et al., 1991). Indeed, the contractiIe response to Bi receptor agonists is acquired in uitro in rabbit isolated vesseb in a process that is dependent on protein syuthesis and is ampIïfied by the cytokine interleukin- 1 (deBlois et d , 1988; deBlois et cd, 1991).
Peptidases play a key mie in channeling kinins toward Bi receptors and in terminating theu activity (Er* and Sloane, 1962; Proud et d, 1987; Erdoa, 1990). Carborypeptidase N (CPN; EC 3.4.17.3) and other argiaine ~(trboxypeptidsses in blood plasma or setum transfom BK and Lys-BK into BI agoni-. CPN has no aatinity for BI agonista or antagonists that do not possess a C-temird arginine residue (Drapeau et
ABBREVUtlONS: ACE, 8ngbtensin I mrerting enzyme; AmM, arninopeptüase M; BK, bradykuiii; CPN, carboxypeptidase N; LPS, lipopolysac- chadde: PAZ, -kg the coc~c~c~tration of antagoriist that reduces the effect of a double dose of agoriist to that of a single dase.
192

d, l99la) These enzyme activities are present in innnmmataiy exudatea (Proud et d, 1987). AmM (EC 3.4-Ils), also present in plnama and muciatm, hydrolyses Lys&-w-BK into des- Ar$-BK (Guimmes et aL, 1973; Proud et d, 1987; Sheikh and Kaplan, 19891, an agoni& of leseer affinity for the Bi receptors. ACE (EC 3.4.15.1) removes a dipeptide h m BK to yield an inactive metabolite-, ACE ah30 cleaws C-terminal tripeptidea h m Bt agonists or mtagonists, but at a alower rate and with lem affinty thrui for the removal of a dipeptide h m BK (Drapeau e t d, 1991a). Synthetic Bi receptor agoniete elicit hypotensive effects whose &nation is correlated with thek resistance to peptidaaes in Lipopolysaccharide (LPS)-treated rabbits (Drapeau et d, 1991b). In the present atudy we have synthesized a seriea of meta-
boiicaily pmtected Br nntagonista and evaluated theù in vwo pharmacologie activity against Sar [P-Phea]des-w-BK, a long-lsating BI receptor agoniat. Our data indiate that the in o h potency of Bi teceptor antagonists is related not oniy to antagoaist but also to their resiataace tio degradation by protmlytic enzymes. The ratiod design of tinin antago- niats thnt can be useà in physiopathologie investigations and therapeutics has to take these and other metabolic observations into consideration. HOE-140 is a very potent and sequence- reiated BK antagonist Wt is highiy selective for the Bz recep- tors and that erhibita excellent in vivo d i l i t y (Hock et cd, 1991; Wirth et d, 1991a). The dee-Ar$' form of this peptide, e chemicai untikely to be a spontaneously occurring metabolite, is a &ed BI-& antagonist of moderate affinity (Wuth et d, 1991b; Rhaleb et d, 1992). The= is evidence that the C- tenainal arginine of earlier & antagonists ia cleaved in biolog- icai systema to yield metabolites with some antaganist proper- ties on the Bi teceptan (Regoiï et d, 1986). Therefore selective, potent and stable BI-antagonists stiii have ta be produced.
Material and Methoâs
BI nntagoniiits were synttiesized by solid-phase methodology using either a manual synthesis apparatus or a semi-automatic peptide ayn- theaizer (Novasyn Cern, Aminotech, Ottawa, Canada). Peptide cbains wen mmbled on Wang or Novasyu KA mimi (Bachem, T o m c e , CA; Aminotech) h m Fmoc-pro- amino acids nctivattd with BOP reagtnt (Castro et ui, 1975). Peptides were cleaved h m the nain by trantment with trifluoruncetic acid (TFA). Crude aynthetic peptidcs m m purifid by prepnmtive chtomatogmphy as previoualy d w c r i (Drapeau and Regoü, 1988). The purity of the f ï~ .L products was mmmd by HPLC dtraIyaU, and manrr spectrometry nnaiyah (perfonned by Dr Micheal E v m , Université de Montréal) indicated the erpemcd molccular weighta.
HPLC Andyak HPLC U y s b of pure peptidea or ~ m p l e s from metabo1ic studies
war performed uahg a Waters aystcm equipped with a 746 data module end a 486 W dhtbCtor net at 214 am. Ssparatiom wen achieved with a Vydac 10p (3.9 x 300 mm) reverse-phase CU calumn ushg a ünear gradient of 5 to 65% aœtoaitrjie/TFA O.OS%/water at 2 ml/min wer a pend of 20 min. For degradation studiea, 50 of each aüquot wan injeckd, and ratta of peptide metabohm were cnîculated from the d t c r s n ~ of ptptide mbstrate concentration.
Rates of hydrolysl, waluatcd by HPLC, are expnsscd sll substxate conaumption (in nrnol) per minute per quantity of enzyme preparntion (mi of plasma or mg of protein). Integnition of peak areas and quanti- tation of peptide aubatrate were cdcuiattd automatidy by the data module. Metabotites nriainn h m clenvnm of the aubatrate bv enzvme
preparatioas were obsewed as peaka with retention tirna diffenn h m the peptide mibat3ate. but they were not identifid The rates O
hydiolysiil were dùectiy proportional to the time of incubation and u the amount of enzyme pnparation.
The metabolic rc~~istance of BI antagonists was waluatcd in the premnœ of rabbit plaama (AmM ectivity), with a kidney micmvillai membrane prepatation (enàopeptidsse activity) and 6 t h purifIed ACEa
W b i t piasma. BIood wm obtained frorn the carotid artery oi anesthetheci rabbits and drawn into h q m b h à tubes. The tubes were cenWbgd at 1500 rpm for 15 min in a refrigerated tabletop centrifuge. The p b waa aliquoted and kept at -20'C util use. For t&e rneamucment of plasma activity, peptides (50 were plac«i in a Tria buffer (pH 7.5 containhg 300 mM NaCI) and incubated 5 min nt 37'C kfore adding 15 pl of plnaml (total volume 1ûûû 4). AIîquots were taken at 0, 15, 30. and 60 min, bmemed in boiiing watcr for 5 min and œntrifuged More HPLC dyais.
Cmde membrane prepuation h m rabbit Iridneya Kidneys were perfus«l in d u with d i n e containing heparin (10 U/ml), and semi-purincd neutmi endopeptidnae was prepared acmrding to F o y et d (1991). Bnefly. the cortex was homogeaUcd in a Hepes M e r (pH 7.5 with IO m M MgC13. The hornoganate WJM con- at 1500 x g for 15 min, and the aupernatant waa coiiecîed aad centrifugcd 20 min nt 15,000 x g- The pellet was waahed, rthomogtnized in the H e p buffet without MgCh, niiquoted and stored at -20'C. Protein concen- tration was m d by the Lowry rnethd Peptides (50 &Q w e n incubateci at 37'C with the membrane pnpanition (6nai protein con- centration 4 3 &ml) in a 50-mM Tria HCI buffcr pH 7.6 containing bestatin (0.1 mM) and capbprii (20 m. Under these conditions, the nmnining lrininanc activity was almm compietely inbibited by thior- phan (25 a), thus esbblirhing its identity with neutmi endopeptidsrt- 24.11. Tbe total incubation voiume wes 1OOO d, and aliquots were periodically withdrawn at sequentid intemis, mired with 20 4 of 10% TFA/wattr, centrifugcd and d y s e d in the above-mentioned manner-
Purifhi ACE. Rvified ACE ( h m rabbit Iungs, Sigma, St. L,uuis. MO) wee diseoived in Trh M e r (pH 7.5) containhg 3ûû mM NnCl (final concentration of enzyme, 9 pg/xni). The kininase assay was carried out under the same conditions as those for pIasrna peptidases.
In VHm Evaluation of 81 Receptoi Antagonists Analogues of the potent a d aelective an-nist Lys -[LeaP]des-Aqf-
BK were evduated using rabbit aortic rings that wem preincubated for at lem 3.5 hr before starting the concentration-effm curve. Antago- niata were in contact with the tissues 30 min before constructing the concentmtion-effect a m e s in order ta reach gaod equiLibrium condi- tions. None of them was a BI agonist or partial agonist. Theu potency M anwniirte was e x p d using the pAt d e . pAz vdues were adculaa from the Schild segresaion; EDM for the contramile effkt of des -w-BK was cstabbhed in each tissue with or without the antug- oniat at neveral concentrations (as in Regoü et ui, 1977). The Schild plot parameten (PAZ, dope) and their mpective standard errors were calculateci h m the erptrimental pointa by a computer ptopam (Tai- hida and Murray, 1987). Some of the new antagoniau were chccked for selectivity by testing them a g a h t the contractile effect of BK in the rabbit hIated jugular vein erpoaed to captopri1 (1 a), a bioamay for & receptom for kiains (Regoü and Bara&, 1980).
New Zealaad white rabbits of either aex (1.b2.0 kg) were pmtnated with LPS (25 pg/kg iv.) 5 hr before anesthesia with sodium pentobar- bital (30 mg/ltg iv, adjusted individuaiiy). Lidocaine 2% was also uscd at the rites of incision. The trachea wss intubated, and ventilatory assistance wan providecl witk s Harvard respiratory pump. A polycth- ylene catheter PE-90 was i a s e d into the IeR common carotid artery &d pushed hta the .orta for tecording blood p-ure with a tranaducer (EM751, Elcomatic Ltd., G ~ R O W U.K.) on a Harvard

to the cathcter in order b intcmrpt che recording of the blood prwisure and to ihject kininn and other drugs intra-artarially through the same catheter. To avoid cIotting in the cathctcr, the irnimaLs were given 400 U of beparin at the kginning of the experiment. A second PE-50 catheter waa insertcd into the right carotid artery to infuse the antag- onist solutions or theîr saüne vehicle into the a o m at a rate of 70 pl/ min u s W a Sage syring pump (341A). This Uz uU>a mode1 hae been used to perform a compnrative evalua-
tion of the newly syntheziizd antaswist. Thne boluses (1 pg La) of the potent and long-acting Bt agonist Sar[D-Phe'ldes-Aqf-BK were givtn at 15-min i n t e d a Sach injection was f o U d by an hypoten- sive episode, initidy of 35 to 4û mm Hg amplitude and 6 min duration (Drapeau et d, 1991b). The teste!d Bt antagonist was infused at a constant rate starting 7 min before and continuing duriug the second episode to rnonimr the amplitude of the antagooist effect. The iafusion was mpped 15 min before the thinl episode. The third episode was recorded in order ta evaluate the persistence of Br receptor blockade a&r infusion.
A variation of the in uiuo protocol has k n u d to c o d m the role of specifïc peptidases in the inactivation of the prototype Bt antsgonisl, Lys-(Lcu8]Qa-AI$-BK. Agaiu, t h e boluses (1 rg La.) of the agonist Sar[D-Phe8]des-Ar&BK were given at 15 min internais. The 81 anmg- onist waa inhised at a constant rate that produced only a partial blockade of the agonist. The antagonist infusion was atarted 7 min before the second hypotensive episode and w u maintained untii the end of the experiment. A peptidase inhibitor was then adminiatemi starting 7 min More the thiitd episode to teat for potentiation of the blockade. The inhibitors selected were captoprii (1 mg/kg bolus), amas- min (infirsioa, 50 mnol/'g/min) and phosphoramidon (infusion, 25 pg/kg/min). The doses have been selected from relevant previous work ?.Üïamall animaia (Drapeau et d , 1991b; Matsumura et ui, 1990) to &bit ACE, nminopeptidase M and neutral enàapptidase-2di1, re- spectively.
LPS, extrricted h m E, wli serotype 0111B4, was h m Difco (Detroit, MI). Des-kg@-BK was putchased h m Bachem (Tomce, CA); Sar[D-Phen]des-Ar$-BK was synthesized as previowly deacnbed (Drapeau and Regoli, 1988). HOE LU) (D-Arg[H~a,Thi69-Tic7,0ifl]- BK) was a g i f t from Dr. K. Wh, (Hoechst AG, Frailkfurt. Getmany). Captoprü, thiarphau, bestatin, phosphoramidon and amastath were p u r c h d from Sigma (St. Louis, MO).
Each hmteneive episode was characterized quantitatively by the m&d response (faii of the mean b l d pressure, mm Hg) and by the time course, evaluated by the period suvring at the bolus injection and endhg when half of the maI;mlrl hypotensive response had fadeci (tirne for half-recovery). Results are erptessed as the means f standard emrs of the mean,
Design of metabolically reshtant BX antagonists. Lys[Leu8]des-Aqf-BK served as the reference antagbnist struc- ture for the synthesis of metabolically resistant B1 antagonists (table 1). The pharmacologic activity of BI antagonists as it was waluated on the isolated rabbit aorta and the results of degradation studiea are presented in table 2. Lys[hu8]des- Aqf-BK was a potent ant~gonist of BI receptors with a pA2 of 8.54 on the rabbit isolated aorta. As shown in figure 1, A and C, this antagonist was competitive and sunnountable when tested against the 8, agonist des-A&-BK (Schild plot dope close to unity). Lys[Leu8]dea-Ar&-BK was rapidly metabolized in rabbit plasma, and at the plasma dilution used in our assay, &gradation was due almoet entirely to Rminopeptidase activity because amastath (10 PM) reduced the plasma degradation of
this peptide to less than 5%. Lys[hua]des-Aqf-BK is a h cleaved by p d e d ACE and by endopeptidase activiw founc in a cru& microvillar membrane preparation of rabbit kidnej medulla ACE activity was campletely biocked by captopril(1C rM), whereas endopeptidase in the kidney preparation was extensively inhibited by thiorphan (25 a).
Acetylation of the a-cimino gmup of the N-terminal Lys residue of the basic structure produces an anaiog, Ac-Lys[ku8] des-ArgB-BK, thst was resistant to plesma Rminopeptidase activity but still susceptible to ACE and endopeptidase d m - dation. The d t s presented in table 2 show that N-terminai acetyiation is accompanied by a loss of antagonist activity of about 30% when compared with the parent compound Lys[Leua]des-A$-BK. Further extension of the structure of this antagoillstby insertion of an additional N-terminal Lys residue affords a compound, Ac-Lys-L,ys[Leud]des-w-BK, that is partidiy resistant to ACE metabdism. As an antagonist, however, this product is f -2 log unit weaker than Lys[Leub]des- Arg-BK.
Complete resistance to ACE metabolism was obtained when Sep was repbced by Aib or MeAla residues. Lntroduciug a p8eudopeptide bond between residues Phes and See afforded a partiai resistance to ACE. Because of their acetyleted N-ter- minal, these peptides are equally resistant ta plasma aminopep- tidase. Although the substitution of Se? with MeAla and Aib residues was made in order to obtain resistance to ACE, meta- bolic atudies with the kidney membrane preparation reveaied that Ac-Lys[MeAla6&u8]des-w-BK and Ac-Lys[Aibq Leualdes-ArgP-BK were also very resistant to endopeptidase activity. Under conditions where extensive degradation of Lys[LeuB]des-Aqf-BK occurred, no metabolism of the MeAla- or Aib-substituted antagonists was observed. As for Ac- Lys [P he5$(CH2NH) Ser',Leu8] -des-Ar8-BK, t his anaiog was degradecl by the kidney membrane pteparation.
The BL agonist Sar[D-Phe8]des-&$BK was also tested for degradation by the same sources of peptidases (table 2). It was found to be hi@y tesistant to al1 of them, including neutral endopep tidase. In terms of pharniacologic activity, the substitution of Aib
for Se9 was detrimental to antagonist activity (table 2). The other two structures, Ac-Lys[Phes$(CH2NH)Sef&u0]-des- ArgP-BK and Ac-Lys[MeAlas,Leu8]des-w-BK, were equally potent as Bi receptor antagonists, aithough they were about 100-fold les8 potent than the parent stmcture, Lys[bu8]des- ArgS-BK. Apart h m their reduced potency, the pharmacologic profiles of aii active analogues were those of typical and specific Bt antagonists, as shown in fig. lb to d for Ac-Lys[MeAla6,Leu8] des-Ar$-BK. The antagonism exerted by this peptide was fully competitive and surmountable, as seen by the Schild plot (fig. Ic); this analog a t IO4 M did not block BK activity on the & receptor as assayed on the jugular vein (fig. Id). Lys[Leu8]des- Arc-BK and ita metabolically resistant analog, Ac- Lys[Mehe,Leua]des-w-BK, also behaved as competitive antagoniats of the metabolically resistant Bi agonist Sar[D- Phe8]des-Arg-BK in the rabbit aortic preparation (pAz of 8.36 & 0.17 and 7.20 f 0.15, respectively).
In vivo pharmacological studies. The in uiuo activiw of the antagonists is presented in table 3. The hypotensive re- sponses to Sar[D-Phed]des-Arg9-BK before, during and after inh ion of each BI antagonist are expressed as the rnnrimal drop in b l d pressure after challenge by the agonist. In animais infused with the saiine vehicle of the antagonists, the BI agonist

Sar[D-Phe8]des-Aqf-BK produced hypotensive episodes of ap- pmIimatdy equal mgnitude, perhaps with some tachyphylaxis from the f k t to the third recording. The half-recovery times calculated for each of the hypotensive epiaodes were 374 f 20 sec, 380 I 64 sec and 369 f 39 aec, mpxtively (n = 6). Two infusion rates (5 and 25 rg/min) have been teated for each analog. At an infusion rate of 5 &min, O& one antegonist, Ac-Lys-Lys[Leu8]dea-Argg-BK produced a significant block of agonist activity, reducing the hypotensive response of Sar(D- Phe8]dea-w-BK by about two thirds. The effect of the other antagonists at this dose was not significant. At the -ion rate of 25 fig/min, all antagonisb in table 3, with the exception of Ac-Lys[Phe5$(CHzNH)Sef,Leu8)h-W-SK produceà aimilar and important reductions of the Bi agonist-induced hypotenaive response. The time of haif-recovery h m Sa@- Phe8]des-Ar#-BK-riiduced hypotension was not significantly infiuenceà by the antagoniste (data not shown); only the am- plitude was affected. The persistence of the antagonist effect wm measured by injecting Sar[D-Phe8]des-Arg-BK 15 min aftet the Ulfusion of each antagonist was etopped. Of aU ana- 1088, only Ac-Lya [MeAla6,Leu8]des-Ar$-BK showed a -- tant residual activity at this pst-infusion time period No residual activity was observed for the other antagoniab or for Ac-Lya(MeAlad&u4]des-ArgO-BK 30 min after ita infiision was etopped (data not shown).
An in u b protocol wae developed to coafirm the identity of the inactivation pathways for Bi autagonista uing the poten- tiation of the prototype antagonist by peptidase inhibitors (table 4). The design of t h experiment was baaed on the Bi agonist Sar [D-Phe8)des-Aqf'-BK utilized to reveal the BI recep- tor blockade. The in vivo effecta of Sar(D-Phe8]des-Arg-BK are not potentiateà by capt-priï or ~maatat in (Drapeau et d, 1991b), and this peptide is not metabolized tn a signifïcant extent by rabbit plasma, ACE or kidney extract (tabIe 2). The rnetaboiically susceptible antagoniat Lys[leua]dee-WBK
was infussd et a mugkdy effective rate (5 pg/ml) to bloch boluaes of the agoni& Captoprii and phosphoramidon siprnin- cantly potentiated the antagonïst, but not amastatin.
Figure 2 illustratea the apecificity of lcinin antagonists for Bi and Bz receptors in the cardiovascular system of the WS- pretreated anesthetid rabbits. Doses of BK snd of Sar[D- Phe8]des-Aqf-BK were appiied before, during and afhr idù- aion of Ac-Lys[MeAld,Leu8]des-ArgO-BK a BI receptor antag- oniat (fig. 2a). BK pduced a rapid and sharp decrease in blood pressure that was fiilly reversible in a relatively short period of time (figs. 2a and 2b). Sar[D-Phe6]des-Arg@-BK &O produced a bipkic hypotensive response as previoudy reported (Dra- peau et d, 1991b). The hypotensive activity of BK was not affecteci by the infusion of the Br antagonist, whereas the hypotemive resp0m-e to Sar[D-Phe6]des---BK was blocked The infusion of the & receptor antagonist HOE 140 (fig. 2b) led to a complete block of BK activity, wheteas that of Sa@ Phe8]des-Argg-BK wm not affected. The inhibitory effect of either antagonist persisM after its infusion was stoppeci.
Discussion
Lys[bu8]des-A$-BK is the most potent Bi antagonist aa egtabiished on the rabbit aorta (Regoii and B d , 1980). However, this peptide hm a ahort duration of action in uùm owing to ita rapid degradation (Marceau and Regoli, 1990). A series of modincations erere introduced in this structure to improve the metabolic stability toward sources of enzymes derived h m rabbit tisaues. The effect of the modincationa on the effinity (pA3 has been evduated. The potency in the in vivo blood pressure -y appeam to be a compler function of both the affinity and the metabolic resistance, ACE and AmM are two enzymes whose kinlliase activity ia
weU estabkhed (Er&, 1990; Proud et aL, 1987; Drapeau et al, 1991a). Bi agoniab with prolonged UI owo activity have ben

Fig. 1. In vilro p h m m k g y of two kinin & reœptor antagonists in rabbit Mood vessels. A, 8) C c m m t r a ~ e c t w e s for the contractile effect of debAfg@-BK on rabM aor<lc mgs in the .bumg a pnneros of various cmcmtratkns of the an(gonists. C) SchiId plot anaiysis of the data m pxds A end B. The & values interpoiated for L y s [ w N - B K and A c - L y s [ M e A i a e , ~ ~ - B K were 8.54 * 0.15 and 6.52 I 0.08, m m . Sbpes were 0.88 I 0.1 4 and -1 -02 0.08, . Dose ratio (DR) was detemiined by M i the EDso of des-A@-BK oôtained wim antagoriist by the €Dm of a mntroi cocIcBcItrath-et f f~without antagaika in th same tissue. 0) Lack of emct of Ac-Lys[MeAhe,Le~ des-Arg04~ (1 00 PM) on BKinduœd miac(ion of the rabbit isohted jugular vein. an effect mediatecl by & receptors. ln panils A, 8 and Do points are the means, and verticai bars, the S.E.M., of 2 to 8 deteminations.

NS. NS. Pc.05
obtained by protecting them h m degradation by these two enzymes, the former enzyme sctivity being more important than the latter (Drapeau et d, 1991b). We therefore modifieci Lya[Leu8]des-A$-BK in otder to obtain BI antagonists with a similrir stability.
Acetylation of the a-rimino group of N-terminal midues protects peptides h m aminopeptidases. Ac-Lys[Leu8]des - Aqf-BK and ail other acetylated structures were effectively rea-t ta plasma AmM activity (table 2). Although Lys&eua] des-Aqf-BK rnetaboliem in piasma was almost entirrly attrib- utable to A d , protection h m this peptidase did not produce an antagunist with a prolonged activity in uwo. Ac-Lya[Led] des-k$-BK was appmximately equipotent to Lys[LetP]des- Arf-BK (table 3). An attempt to potentiate the latter unpm- tected antagonist with amastatin failed (table 4). ais0 indicating that the role of AmM is s m d compared with other activities.
Therefore, enzymes that are present on the vesse1 waiis and that are not abundant in plasma, such as ACE, could play an
important role in the degradation of des-Atg-kinins in uiuo. Subsequent modifications were designeci ta pmtect the BI an- tagoniats agaiust ACE. Dinerent approaches were pursued for this purpose, It bas been ahown that pmlonging Lys-BK with an extra N-terminai Lys residue (Lys-Lys-BK) affords at ieast partiai protection against ACE (Dorer et aL, 1974; Drapeau et aL, 1991a)- Ac-Lys-Lys[Leusldes-w-BK shows some resist- ance to ACE hydmlysis Ut oitto (table 2). This resistance may explah a gain of apparent in uwo activity relative to Lys[Leua] des-Aqf-BK, even though the affin;@ of AC-Lys-Lys[Leua]dea- Arf-BK for the BI receptor is 1 log unit Iess thRn that of Lys&eu8]des-&@-BK4 AC-Lys-Lys[Leu8]des-@-BK is the ody teated antagonist that simiificantly blocks the agonist SarfD-Phe8]des-A#BK at an infusion rate of 5 pg/min. This, and the potentiation of an unprotected BI antagonist by cap- topril (table 4). suggest the importance of ACE in the disposi- tion of BI antagonists in vivo. Aithough Ac-Lys-Lys&eu8]des- A d - B K is a potent antagonist in uko, it did not exhibit any prolonged activity.
Prevïoua investigations suggested that the Se9 tesidue was tolerant to substitution for the activity of L;nina on BI receptors (Regdi and Barabé, 1980). Substitution of Sep by MeAla or Aib residuea and modification of the Phes-Se? peptide bond were hvestigated in order to obtain ACE resistance. Indeed, ail these analogs exhibited partiai or complete ACE resist- ance, but their phanaacologic profiles differed consider- ably- Ac-Lys[Aib6,Leu8]&s-Arge-BK was inactive on the rabbit aorta (table 21, probably as a resuit of the important conformational constrainta irnposed by the introduction of auch a residue in a flexible peptide stmcture (Spatola, 1983). Accordingiy, this analog was not testeci ik vivo. The two other antagonists, Ac-Lys[MeAla6,Leu8]des-ArgP-BK and Ac- LyaEPhe6$(CH2M)SerB,Leu6]des-A$-BK, have several fea- tutes in common. Both were tesistant to AmM and ACE (t hough the resistance of Ac-Lys[f he6#(CH2NH)Se#&u8]des- AI$-BK to ACE was not complete), and they were equipotent Bt agonists as meanued on the rabbit aorta When tested in vivo, however, these analogs exhibited very different pharma- cologic profiles. No signifîcant antagonist activity was detecteà with Ac-Lys[P hes$(CH2NH)Ser6,Leu8] des -w-BK, but Ac-
Fig. 2 Hypotensive effect of BK and of Çarlû-Qheqckç-~-8K in LPS-pre- treated rabbits, A) Influence of a B1 m t o r antagoriist, Ac-Lys[MeAia6, -&g@-BK. 6) Influence of a Bz antagonist, HO€-140. Horizontal doted Unes represent aie periods of bitra-ar- tenalinfusiorisoftheantagonistsatüm Wited rates. The agoriists wece
tIOE I4û I p*m given as inMerial boluses at the in-
# dicateddoses.TheiastrwxKdingswere made 15 min after stopping the an* onistsinfusions.Eadicmeofthetrec- ings is tepresentative of 3 to 4 experi-
BK m t s .
BK S U ~ D . ~ - A ~ ~ ' - ~ K U s & n ~ o h * . a r 0.1 BI I sim-Piucw&.e~
5 min

Lys[MeAla6,k8]&-A$-BK waa quite potent Uz vivo, [email protected] icantly blocking agoniet activity a t an infusion rate of 25 pg/ min. IFLrthemore, this anaiog was the ody one to show any tesidual activity 15 min aRer the i n h i o n was stopped.
Comparing the in vivo activity of Ac-Lys[Phes# (CH2MI)Sef,Leud]des-w-BK and that of Ac-LysMeAlab, Leualdes-Ar#'-BK I d us to hypothesïze that another major earyme was involved in the degradation these peptides. 1n addition, phosphoramidon infusion potenthtecl the prototype Br autagonist infusion (table 4). Therefore, we Btudied the metabolism of aii these peptides by endopeptidase-24-11 (E.C. 3.4.24.11) found in a kidney extract (Fagny et d, 1991). Our results showed that Ac-Lys[Mefi8bu8ldes-w-BK and Ac- Lys[Aibd,Leu8]des-ArgD-BK were not metabolized by this en- zyme, whereas Ac-Lys [P he6$(CH~NH)&fl&u1]des-w-BK and ail other structures were bydrolysed by endopeptidase 24.11. The low afiïnity of Ac-Lys[PheS$(CH~NH)SeF&u8] h-Argg-BK, combined with its susceptibility to endopeptidase hydrolyaie, probably accounta for its low potency in viuo, whereas the endopep tidase reaiatance of Ac-Lys [MeAla6,Leu8] des-Argg-BK compensates for its iack of receptor affhity. Tbere may be more than one form of neutral endopeptidase in tissues, but they al1 cIeave the same bon& in bradykininr Glf-Phe5 and Pd-Phea (ZoIfaghari et d, 1989). In the BI antagonists that we have tested, the Pro7-Phe8 bond is probably immune to endopeptidase cleavage because Leu8 is the C-terminal sesidue. The GIY-PheS bond appears to be completely p r o W by the modification of the peptide backbone a t the Ievel of the sixth residue (substitution with Aib or MeAia), and the Gif-Phe6 bond is partinlly protected by the modification of the Phes-Se9 bond. These changes may alter enzyme Rfnnity, which is pos- tutateci to be dependent on multiple hydrogen bonds to-the peptide backbone, notably a t the Ievel of the S',-S'2 bond (Roques and Beaumont, 1990). Ac-Lys[MeAla6,Leu8]des-Arge-BK was found to be approxi-
mateiy equipotent to the prototype BI antagonist Lys[Leu8] des-Args-BK in uiuo, despite the fact that it exhibiteci a 10- fold decreased affinity in the rabbit aortic rings. The multiple metaboiic resistance of Ac-Lys[MeAla6,Leua]des-Arc-BK is probably related to th high in vivo potency. This compound retahed the key pharmacologie pmperties of the prototype king apparentiy cornpetitive and surmountable in vitro (fig. 1 Cl and being totdy selective for Bi receptors in vitm (fig. 1 D) and Ur uivo (fig. 2). Notably, it is inactive as an antago- nist of BK on the rabbit jugulas vein, whereas the des-Arg fragment of HOE-140 is a potent Bz receptor antagonist in this assay (pA2 of 8; Rhaleb et d, 19921- The structure Ac- Lys[MeAla6,Leu8]des-w-BK can be considered an interesting b i s for further improvements in stabiîîty and affinity. This study ilIustrates that peptide activity Ur vivo is W e d
to both receptor affinity and metabolic resistance. Acknodegmen t
The rachnicsl help o l Ms. Johnnne Bouthiiîier is acknmlcdpcd
Rahmncem CASTRO, B., DORMOY, J. R. M N , G. IWD Setm. CI Rcsctifr & cwpm
peptidique- W. L'htufluorophorpbate de berwtriau,lyl N-o.ytridimathyiam- in0 phorphoaium (B.0.P .). Teuahedron k 14: 1219-1!222,1975.
D ~ L O I S , D., B o ~ ~ ~ ~ L E R . J. AND m ~ e ~ t l , F.: Entct of gi\fc~c~rticOicis, monohes and growth famm on rite spontaneously dcwloping iarpomm of the &bit isotteâ aorta to des-Arp-bradybh Br. J. PhmacoL 83: 96%- 977, Issa
Dmtors, D., Bomnumt, J. AND Mmcmu. F.: P h eqmam to pmtein iyatherir inbibiton enhuicta tiaaue respoxue m des-At%-bradytinia: Pomiilt mie of intarleulia-1. Br. J. PhannicoL 103: 1057-lCi66,1991.
DOR~R. F. EL. RYAN. J. W. AND S m m , J- B i Hydmlysir of bradykinia and its hightr homol- by mgiotensin mnvertiog enqme. Biocbcm, J. 141: 915-93'7,1974.
DRAPMU, G, ~ O W , AND WARD, P. EL M s u b o w of bridykblhl r(lontu d . n u g o n U t r b y u i p o ~ a r i n I c o n ~ s n s ~ r m a a n d c u b o ~ N . Peptide% 12: 631-638.1991~
D~u~erru. G, &ors, D. AND Mmmau, PI Hypoteniivt c f 5 1 of Lyr-dcr- Ar+btidyLiPiadmnrboIicrllypmrcctcd y o a h of BI recepton for kiniaa. J. PharmacoL Exp. Tbr. 259: 997-1003,1901b.
DRAPEAU, G- AHD REGOU D.: Synthtsis of btrdykinin a d o g s In Immmochem- ical Tcchniqusr, Part M: ChcmotarU aad rnnimmitiau, Methoda in Enzy- mology, VOL 163, cd by G- DiSlbato, pp. 263-273. Academic Rua, New York, 1988
m, E. G. rsND SWAN& E & An snsymc in human bbod piasma tbat . . mctwaus bradykinin d icdkhu B i m . PhsrmiiEoL Il: 585-592.1962.
ERDOS. E. G i Som old ind mme new on kinin r n e t u b o b J. P-L 15: (-6) S-2W324.1990.
FAGNY, C., M~CHEL, A., ~~ONARD. LI B P ~ W ~ B O O M , G, FONT- J. AND D c s c n o r r r - h u c ~ u ~ ~ . MI In uitm dcgrrdition of endothciin-1 by endopep- cidue 24-11 (en.kephrrin.u). Peptides Ck 773-778.1991-
FABYER. S. G. AND BURCH, R MI Biochemical md molccular phannacaiogy of Linin mxptom. Ann. Rev. PhumacoL TWcaL 32: 511336.1992
FARMER, S. G., MC-, B. k, S. N- AND BURCH, R hk induction of vucular rmooth murlc bridykinin & receptors in uiPo during rntigen
wti Miom W 191-193.1991. Cvnua~es, J* A.. Bornes, D. R. -0. E S. AND PRADO, JI LI Kinin-
conwitiiy Minopcptidw fmm human aenrm. Biocbrm. PhumroL 22: 31 57-3l'IZ 1973-
Hom, F. J, W m , K., bus, U., Lm& W., GERHARDS, H. J., W m m , G., Ehwz, S., BUXPOHL, G, K ~ N I G . W., K ~ o u e , J. m SCHOWEEINS, B. A: Hœ 14û. a nta potent and long-Miag bradylinin-antagonise In uiao midiea. Br. J. PhumieoL 102: 769-773,1991.
L o f f n ~ M., mou, D., RHlueB, N. E. AND PLANTe, G. E: The d e of Bt- and &anin reeaptom in the rend nrbular and hemodynamic rcspomie to bmby- k h h Am. J. PhysioL 262: R72-R76.1992
Mmcmu. F. AND Recou, D. CI Kinin mxpton of the BI type and theu mmgoniiu la: BndylMin antago- Basic and ciinical -h, ed by R M. Burch, pp. 33-49, Mucc1 Dekker hc, New York, 1990.
~~AISUYURA, Y., HUMI, K., TAKAOKA. [H, AND MORULOTO. SU Phrnho&- don, a meullopmtaiouc Lihibitor, suppreaaea the hypcrtcnrivt effcct of big endothclin-1. Ru, J. Pharmacd f 6s: 103-106,1990.
MOueR-Sc- EI Changea in the venoua cornpliana by bndylMin and nngiotcnain II md ib signScaace for the wueubu effect of cycloqarïn A. Nauayn-Schmicd Arch. PhamiacoL 338: 69%703,1988
PROUD, D., B A U I I G ~ . C. K. NACL~RO, R bf. AND WARD. P. E.: meuboliam in h u ~ n n a d scmtions durhg erpcrimentally indud dergk rhinitia. J. ImmunoL 138: 428434.1987.
REGOLI. D. AND BARABé. J.: Pharmacology of bradykinin and relatrd k h h P-1. M. 32: 146,1980.
RECOU. D., Bmm& J. rn P m . W. K.: Rmptors for bradykinin in rabbit aortae. C a J. PhysioL Phannacol, 65: 853-867,1977.
REGOU. D.. DRAP~AU. G., ROYERO, P.. D~oN. S., RHALE& N. -EL, BARU& J., D ' O ~ s J u s r e . P. MD WARD. P.: Conversion of kioiar and their antago- ni.ts h m BI-nnptor amivators or blockers in isoiatd ves4s. Eur. J. Phar- muoL 121: 219-224,1986.
mou, D. C., M-u, F. AND LAY~CNE, J.: induction of B,-rrccpv,n for lllPiad in the mbbit by a bacterial lipopolysaccharide. Eut. J. PharmacoL 71: LE-L15.1981.
Rmua, N. -&.DION, S., BARABé. 3.. Roms~. N.. JUKIC, D., DRAP~AU. G. AND Rec01.1. DI &ceptors for k i n h in iroIatcd wM1 vessela of dop. Eut. J. PhumricoL 162: 419427.1989.
Rtiue8, N. -E., Co- F. AND W O U , D.: Nonselectivity of new bradykinin mt.gonira ~ D c BI nceptorn Litt Sci. 51: PL12SPL119.1992
Roques, P. MD Berrviuo~~, A: NeutmI endopeptib24.1L inhibitors: From adgeaics to hyperrtnrives? Trends PhatmacoL Sci 11: 245-249, Lm.
S ~ e a r ~ , 1, A. AND KAPLAN, A, P.: Mechinirm of digestion of bmdykinin aad I~lbradylllaio (Itallidio) in humun scnrm: Rote of cutioxypeptih, agio- tanain-coovercing enzyme and &termination of h d dtgmdntion producrs. Biochem, PhumrcaL 38: 993-1000,1989,
iw, WHAUEY, E. T., HO-N, fi-, WElFERT, J- AND M.: The hypotsnsive rrrponme to dem-A+bradyünin haeams duMg E. cdi septicsrnia in the pig. Adv. Exp, M d BioL 247b: 389493,1989.
SPAMLA. A P.: Peptide hckbone madifications A rtnrnirr-acrivity uralyaia of peptides containhg mide bond surrogates. Conformational constrainu, aad rakted backbone repiacementa In: ChemUvy and biochedmy of uniuo aci& peptides rnd proteina. vol. 7, pp. 267-357, Mual Dtkker ïnc., New York, 1983.
T-DA. R J. AND M ~ Y , R B.: Manual of phumacologic dcuiations with cornpuUr pmgmma (2nd edition). New York Sp~ger-Verhg New York. hc. 145 p., L987,
TODA. N., B u . K., AMSA. T. AND OKAMURA, T.: Heterogcneity in mechmima of bradykinin action in canine Uoiatcd b l d vamein. Eur. J. Pharmacoi. 136: 321-329.1987.
W m , K.. Brra~ont, G., STECHL, J., KNOLLE, J, H- S., S C H O ~ S , BU w - D - A r g [ H y p a , Tài'. D-Tic7, Oic']bdyünin ( d t i A r g ' O - ( H o e 1401) in r

potiat bdykiain BI mceptm auwniu Eur. J. PhinucoL 206: 217-218, Hu- iung maanbruis-bouad Mibii-exdOPW-ywd ~Y&I* of 1991b brdyItinin. eatyma 42: 16&173,1989.
WUtTH, K., HOC& P. J, ALôUS, U, Lnn. W., hPïMUUNN, H. G, A~ACNOB- m?otw, H, HENKE Sv Bnnw- G, KONXG, W., KNOLL& J. AND w rsgrint t ~ : GIly DtlPL(IP, PU, Airiruat R o h r of Phu- SCH~LKENS, B. k Hœ 140, i - potcnr .nd bng-aahg bridykinin-mmg- -*, Cam- & - & moral-Dicu & Quibcc, 11 C o t s d u - P U oaut: In viuo Iftdid.. Br. J. PhmmcoL 10% 7?4-717,1991s. Quibec. P.Q. Canada GïR 216.
Zou~c~ lu i r , R. Lrcna. D.. Bluc~zi, C. R.Cmmxo. P. C. AND BQIAL,F,JI

European Journal of Pharmacology
Aminopeptidase modulation of the pharniacological responses to synthetic thrombin receptor agonists
Denis Godin, Françuis Marceau, Caroline Beaulé, Francis Riou, Guy Drapeau * C m & ikhmhe ~Unwtrriréiutnai), Had-Diclc & Quiac, 11 Côte du Pahk, Québec (Quc'brc), Cimada GIR 2f6
(Reccived 12 August 1993; aceeptcd 25 Novcmber 1993)

ELSEVIER European Journal of Pharmacology 253 (1994) 23-230
Aminopeptidase modulation of the pharmacological responses to synthetic thrombin receptor agonists
Denis Godin, François Marceau, Caroline Beaulé, Francis Riow, Guy Drapeau * Centre & Rcchcrchc fUnir.emiiLarall. HÔreI-Dieu de Que'brc, III Côte du Palau. Que'bcc (Québecl, Camdrr GIR U6
(Rcceived 12 August 1993: acccpted 25 Novcmbtr 1993)
ïhrornbin is a contractile stimulus of isolated rabbit aortic rings and apparently produces its effects through the recently haracterized cleavable receptor. A synthetic hexapeptide, NAT6-m, (new amino terminus). was found to be the minimal active :ructure for full activation of this receptor. The N-termina1 Ser residue of NAT,-NH, is cruciai for biological activity. in this :udy we examined the metabolism of NAT6-NH2 in rabbit plasma, where it was rapidly d e p d e d by aminopeptidase M. In the resence of the aminopeptidase inhibitor amastatin, no metabolism was observed, On this basis a metabolically resistant nalogue. [Sar1]N~T6-m2. was designed. We compared the biological a W t y of thrombin. NAT,-NHI and [Sarf ]NAT~-NH, in ie abbit aorta and found that [Sar1lNAT6-NH2 was more potent than NAT,-NH,; however. in the presence of amastatin the mentration-effect curve for NAT,-NH, was shifted to the Ieft of that for [s~~')NAT~-NH,. The effects of [Sarl]N~T,-NH2 3d of thrornbin were not modified by the presence of the arninopeptidase inhiliitor. We also studied the effect of arnastatin on ie in vivo hypotensive response to NAT6-m2 and found that it was also influenced by aminopeptidase M inhibition. Our :sults show that aminopeptidase protection is important when evaiuating responses to synthetic agonists of the thrombin eavable receptor and that an in vivo model, the anesthesized and heparinized rabbit, may be weful for the development of gonists and antagonists of this receptor.
e). words: Thrombin; Thrombin receptor: Aminopeptidase M; Peptide; (Rabbit)
introduction
a-Thrombin is a known contractile stirnulus of the ibbit aortic preparation (Haver and Namm, 1984, id a cataiyticalIy active protease is required for this :tion. A cleavable receptor for a-thrombin from egakaryocyte-like ce11 lines has recentIy been doned id sequenced (Vu et al., 1991). This receptor is a ember of the rhodopsin superfamily of s ignahg Woteins. A new model of receptor activation was pro- ~sed in which a-thrombin acts on a typical cleavage :e located in the N-terminal region of the chah gosing a 'new amino terminus' (NAT) domain that :ts as a 'tethered' agonist of the receptor. Vu et al. 991) tested this model by showing that a synthetic tradecapeptide (NAT,,) corresponding to the NAT main is an agonist of the cellular receptor for a-
- krcspondiag author. Tel. (418) 691-5598, fax (418) 691-5439.
thrombin in humari plateiets and in Xenopus oocytes expressing the cloned receptor.
Thrombin is a potent modulator of bIood vesse1 tone and acts through both endothelium-dependent and -in- dependent mechanisms to either relax or contract vas- cuIar smooth muscle. NAT,, is reportedty a relaxing agent in the isolated rat aorta (Yang et al., 19921, but is a contractiie agonist in rabbit aortic preparations (De- blois et al., 19921, suggesting that the cleavable recep- tor mode1 may apply to these systems. The rabbit aortic model does not exhibit the high degree of tachyphy- laxis typicaI of the rat systern. ï h a t thrombin and NAT peptides are direct myotropic agents in the rabbit is further demonstrated by the rnitogenic effect of both types of agents in smooth muscle ceils derived Erom the aorta (Herbert et al., 1992).
Since the discover- of NAT peptides, several groups have show that NAT,, and other N-terminal frag- ments h i c the effect of thrnbin in several pharma- cologic systems, notably by inneasing phospholipase C

activity in cells (Troyer et al., 1992; Vouvret-CraviaR et al., 1992). A number of structure-activity studies sug- gest that the N-terminal hexapeptide Ser-Phe-Leu- Leu-Arg-Asn (NAT,) is the minimai active structure (Vassalo et al., 1992; Hui et al., 1992; Scarborough et al., 1992; Chao et al., 1992). These studies also stress the importance of the Ser-Phe residues in stirnulating the biological responses to NAT,; acetylating or remov- ing the Ser residue completely abolishes bioIogica1 activity as does interchanging Ser-Phe by Phe-Ser or replacing Phe by Aia.
In biological assays the N-terminal Ser residue may be susceptible to hydrolysis by aminopeptidases like aminopeptidase M, which is found in blood vesseIs, in smooth muscle cells (Palmieri et al., 1989) and in the circulation. The activity of this enzyme may lead to an underestimation of the affinity of susceptible peptides. Thus, in view of the importance of the N-terminal Ser residue for the activity of NAT peptides, we have examined the effect of arninopeptidase inhibition on the in vitro and in vivo phannacological responses to NAT,-NH,. We have also designed an amuiopepti- dase-resistant analogue, [Sar N NAT^-^ ,. and com- pared its activity to that of natural sequences.
2. Materiab and methods
2.1. Incubation of NAT& and [Sar ' / N A T ~ - N H ~ with plasma
The metabolic resistance of NAT6-NH2 and [ S ~ ~ ' ] N A T ~ - N H ~ was evaluated with rabbit plasma as a source of aminopeptidase M activity. Blood was ob- tained from the carotid artery of anesthetized rabbits and drawn into heparinized tubes. The tubes were centrifuged at 1500 rpm for 15 min in a refrigerated table top centrifuge. The plasma was aliquoted and kept at -20°C until use. For measurement of plasma activity, peptides (50 PM) were piaced in phosphate- buffered saline (pH 6.8). Each sample was incubated 5 min at 37°C before addition of 15 pi of plasma (tctàl volume 1000 pl). Aliquots were taken at 0, 15, 30 and 60 min, mixed with 20 p l of 10% trifluoroacetic acid/ water and centrifuged before HPLC (high performance liquid chromatography) aiialysis. Aminopeptidase inhi- bition was obtained by adding an aminopeptidase M inhibitor (amastatin; 20 PM) to the sarnple prior to incubation at 37°C. Rates of hydrolysis, evaluated by HPLC as previously described (Drapeau et al., 19931, were monitored as the decrease in the concentration of the substrate and are expressed as substrate consump- tion (in nmol) per min per ml of plasma. ïhese rates were directly proportional to the tirne of incubation and the amount of plasma.
The thoracic aorta was isolated fiom New Zealar White rabbits of either sex (15-2 kg). The vesseIs we cut into rings (2-3 mm width) and suspended betwee a metal hook and a thread Ioop under a basal tensic of 2 g in 5-ml organ chambers containing oxygenatt (95% 0,-5% CO,) and warmed (3PC) Krebs solutic (Detiiois et al., 1992). Their responses to agents wei isometncaiiy recorded as described previously (DebIo et al., 1992). At Ieast 1 h of equilibration was ailowec The direct contractile effecfs of purified bovine thon bin (Thrombostat, Parke-Davis, Scarborough. Canadi and of NAT peptides were assessed by adding a cumi Iative scaIe of concentrations (cumulative concentri tion-effect curve) to the bathing fluid. A maximum c two concentration-effect curves were obtained in eac tissue with a 9û-min interval between each. Contractil responses are expressed in grams.
An oii immersion technique (Kalsner and Nickel son, L968; Regoli et ai., 1974) was used to rneasure th rate of relaxation, in minera1 oil, of rabbit aorta ring precontracted with NAT6-m or [Sar1 ]NAï6-NH: These rates reflect the metaboIic disposition of th agonist by the tissue. ReIaxation curves were obtaine with equaliy effective doses of NAT6-m2 (1.4 X 10- M) and [ S a r i ] ~ ~ T 6 - W , (1.4 X 10-'M) on rabbit aort strips, suspended in 5-mi organ baths as describe above. The tissues were suspended in Krebs solutio and challenged with the agonist; after the contractio plateaus were reached the Krebs solution was replace( with mineral oil. The results are expressed as th1 percentage of relaxation as a function of time. In somi experiments the tissues were incubated with amastatii (20 gM) for 5 min before the agonist was added.
2.3. In ciro ecaluation of NATbNH2 and [ S a r l ] N ~ T , NH2
New Zealand rabbits of either sex (1.5-2.5 kg) werc anesthetized with sodium pentobarbital (30 mg/ kg i.v. adjusted individually). Lidocaine 2% was used in addi tion at sites of incision. The trachea was intubated am ventilatory assistance was provided with a Harvarc respiratory pump. A polyethylene catheter PE-90 wa inserted into the left comrnon carotid artery, pushec into the aorta and connected to a three-way valve ir order to record the cardiac rhythm and blwd pressurr wit h a pressure transducer (Stat ham Pî3ID) connectec to a Grass poIygraph (mode1 79). To avoid clotting ir, the catheter, the animais were given 500 U of heparin per kg of body weight, at the beginning of the experi. ment. A PE-50 catheter was inserted into the nghi jugular vein for i.v. injections of peptides given in bolu3 of 0.3, 1.0 and 3.0 mg/kg. Two doses of each peptide were tested in an animal at intervals of 10 min or when

the arterid pressure had stabiiized. For protocok re- quiring the infusion of amastatin, the rïght jugular vein was used to infuse amastatin at a rate of 70 pl/min (50 nmol/kg per min) by using a Sage syringe pump (mode1 341A). A carheter was placed in the left jugular vein for the injection of peptides. The same dose (1 mg/kg) of each peptide was injected twice in the animal; the second injection was given 12 min after a continuous infusion of amastatin was started that lasted untii recovery was complete. The hypotensive a c t ~ t y of each peptide is expressed as the maximum decrease of the mean arterial pressure (dMAP, in mm Hg uni&).
The tetradecapeptide NAT,, (H-Ser-Phe-Leu-Leu- Arg-Asn-Pro-Asn-Asp-Lys-Tyr-Glu-Pro-Phe-OH), NAT,NH, and [Sar ']NAT~-NH, were synthesized in our Iaboratory by using solid-phase rnethods as already descnied (Drapeau et al.. 1993). Amastatin, captopril and phosphoramidon were purchased £rom Sigma (St. Louis, MO).
When appropiate, the statistical significance of re- sults was assessed by using Student's t-test for un- paired data. A P value of 0.05 or less was considered significan t .
Incubation of NAT,-NH2 with rabbit plasma Ied to a continuous degradation of this product (Table 1). Metabolism of the natural hexapeptide sequence was ahos t completely abolished by the aminopeptidase inhibitor amastatin. Substituting the N-terminal Ser by a sarcosine residue in the structure of NAT6-W2 leads to a product, [ S ~ ~ ' ] N A T ~ - ~ ~ , that was stable under t hese conditions. The biological activity of thrombin and NAT peptides was evaluated on isolated
Tribie 1 Rate of hydrolysis of NAT6-NH: and [SarL]N~T6-NH, by plasma aminopcptidax M ' Peptide Metabolic degradation
Plasma Plasma + amastatin (nmol/min pcr ml) (nmol/min pcr ml)
NAT6-M 37 *4 2 & 2 [saï' ~ A T ~ - N H ~ 10.7 1.7 * 0.7
Assays were perfotmcd ai a final substratc concentration of 50 PM. Total incubation volume was 1 ml containning 15 P I of plasma. Amastatin concentration: 2û phfi a Values are the meansiS.EM. of at least threc detcminations.
Agonist Concentration (nM)
Fig. 1. Co~ccntration-effect cumcs for the connactife cffen of thrombin. NAT,,, [Sarx]N~T6-NH,. NAT6-NH, and NAT,-NH in the prcsencc of amastatin (20 PMI in rabbit aortic rings. Each point is the m e a n ~ S . E M . (vertical bars) of at least thrce individual determinations in rings dtrivcd h m diffcrcnt animais. Ordinate: tissue contraction uni& in grarns (g); abscissa: agonist concentration (nM).
rings from the rabbit aorta. The cumulative concentra- tion-effect curves (Fig. 1) indicate that NAT6-NH2 was more potent than NAT,, in inducing contraction of this tissue. When compared to NAT6-w2, [Sar '1- NA'ï6-mI was a more potent agonist than the naturai structure; however when the tissues were challenged with NAT6-m2 in the presence of amastatin, the concentration-effect curve was shifted to the left of the curve for [SarllNATcNH,. In the presence of amas- tatin the concentration required to obtain a contrac- tion of 2 g was about 10 times less than in the absence of the aminopeptidase inhibitor. The responses to [S~~']NAT~-NH, or to thrombin were not significantly modified in the presence of amastatin (data not shown). Aithough arnastatin potentiated the response to NAT,- NI,, the observed potency under these conditions was still IOW relative to that of thrombin. Full concentra- tion-effect curves for NAT peptides were not obtained because extreme concentrations wouId be required.
Further evidence for the role of aminopeptidase metabolism in the modulation of the response of the rabbit aorta to NAT,-NH, was obtained by using the oil immersion technique. In these experiments (Fig. 2) the relaxation in minerai oil of tissues previously con- tracted in Krebs solution with either NAT,-NH? or [SarL]N~T6-NH2 was evaiuated as a hinction of tirne. A series of tests were made in the absence and in the presence of amastatin. The results presented in Fig. 2 show that in the absence of amastatin the relaxation elicited by [Sar ']NAT,-NH , was more prolonged com- pared to that elicited by NAT,-NH2. Whereas near complete relaxation to NAT,-NH, was obtained in 60 min, a 90-min penod was needed for [Sar NAT^-NH,

Time (min)
Fig. 2. Relaxation curvcs obtaincd for I S a r 1 ] ~ ~ T 6 - M I 2 ( 1 . 4 ~ IO-~M). NAT6-m2 (4.4 x 10 -4M) and NAT'-ERIz (4.4 x IO-' Mlin the presence of amastatin (20 PM) in rabbit aonic rings. The oil immersion technique was used. Each point is the mean*S.E.M. (vertical bars) of ac lcast four determinations in ~ g s derived h m different animais. Ordinate: tissue relaxation in 8; abscissa: t h e (min after oil addition to the organ bath).
to reach the same degree of relaxation. In the presence of arnastatin a very marked increase in relaxation t h e was observed with NAT,-NH2 as only 60% relaxation was observed after a penod of 90 min. The relaxation tirne of the [Sar ']NAT,-NH ,-induced contraction was not modified in the presence of the aminopeptidase inhibitor. Other peptidase inhibitors such as the an- giotensin converting enryme inhibitor captopril(5 p M )
O J LO-' 10"
Dose of agonist (mol/kg)
Fig. 3. Hypotensive responscs to i.~. injections of NAT6-MI2 ( 4 . 0 ~ IO-'. 1.3 X IO-' and 4.0X L O - ~ mol/kg) and [S~ 'NAT~-NH, (4.1 x IO-', 1.4X IO-^ and 4.1 X IO-^ mol/kg) aiven in bolus (fmd voIumcs of 0.1 ml). Drop in mcan artcrial pressure (AMAP) is expresscd as the differcncc betwcen the baseline pressure and the lowest value obtained following injection of the peptide. Under the sûmc conditions, injection of 0.1 ml of peptide sohrent (saiine) was without cffcct. &ch point is the mcan~S.E.M. (vertical ban) of at lcast four determinations made in differcnt mimals. Ordinate: dc- crcasc of MAP in mm Hg units; abscissa: dose of agonist in moI/kg of animal weight.
B. Amasutin infusion
n t min
Fig. 4- Reprcscntative blmd pressure tracing showing the hypoten- ske cffect of a single dose (1 mg/ks) of NAT6-NH2; (A) control. (B) during the infusion of amastatin (50 nmol/kg per min). These results are represtntative of thrce expcriments in diffcrent ançsthetized rabbits i\rrow heads indicate the rime of peptide injection.
and the endopeptidase inhibitor phosphoramidon (10 pM) had no eEect on the relaxation time of NATGNH, (data not shown).
When NAT6-NH, and [SarL]N~TGNH, were in- jected in the anesthetized rabbit, they produced dose- dependent hypotensive episodes of similar amplitude although, at the highest dose injected (3 mg/kg), the hypotensive effect of (Sar1]N~TCNH, was significantly greater (P > 0.05; Student's t-test) than that of NAT6- NH2 (Fig. 3). As can be seen in Fig. 4a. injection of NAT6-NH, produced a rapid hypotensive episode wi th a return to baseline conditions in less than 1 min. During amastatin infusion in the sarne animal the amplitude of the hypotensive reaction was slightiy more pronounced but, more UnportantIy, amastatin infusion potentiated the duration of the effect (fig. 4b), Ieading to a prolonged recovery time.
4. Discussion
The discovery of the cleavable thrombin nceptor (Vu et al.. 1991) and of its activation by a synthetic tetradecapeptide, NAT,,, corresponding to the newly exposed N-terminus following thrombin cleavage of the receptor, has generated a senes of stmcnire-activ- ity studies in which NAT,, was used as the basic structure (Vassalo et al.. 1992; Hui et al.. 1992; Scar- borough et al., 1992; Chao et al., 1992). These studies

have maidy used platelet aggregation to assess the potency of the analogues or fragments that were syn- thesized. The actnnty of NAT,, has also been tested in isolated tissues such as the rat aorta and gastric smooth muscle (Yang et al.. 1991) as well as in the rabbit aorza, where thrombin activity sas synergistic with that of epidennal growth factor (Deblois et al., 1992). Our results in the rabbit aorta confirrn previous findings with platelets that the hexapeptide NAT6-NH2 is a slightly more potent agonist than NAT,, and that this hexapeptide is a suitable template for further analogue development.
Other studies in rabbit tissues have also s h o w the importance of peptidases in modulating the biotogical activity of synthetic peptide agonists and antagonists (Drapeau et al., 1991, 1993). Considering the impor- tance of the N-terminal Ser residue for NAT peptide activity, we examined the role of arninopeptidase activ- ity in the biotogicai response to these peptides. The present results indicate that plasma can degrade NAT,-NHT by peptidase activity most likely at- tn'butable to aminopeptidase M because of its full inhibition by retatively 101s- doses of amastatin. This is in accordance with obsen-ations made by Coller et al. (1992) that plasma aminopeptidase M cleaves the N- terminai Ser residue from an I l amino acid N-terminal fragment of NAT,, (NAT,, 1. N-Methyt residues are often used to provide protection from peptidases (Drapeau et al., 19911, and replacing the N-terminal Ser in NAT6-m2 by a sarcosine residue provides fui1 protection from plasma aminopeptidase activity as [Sar1]N~T6-MIZ was not metabolized in conditions where NAT,-NH2 was fully degraded (Table 1).
Aminopeptidase M is also present in vascular tissues and influences the biological response to NAT,-NH,. This influence is quantitative, not qualitative, since the shape of the concentration-effect cuve was the same in the presence and absence of amastatin (Fig. 1) and fo1Iows that of NAT,, and of [SarL]NAT6-MI,. The effect of aminopeptidase on the activity of peptide agonists in the rabbit aorta was appreciable and em- phasizes the importance of distinguishing between bio- logical potency and receptor affinity. The concentra- tion-effect curves indicate that NAT,-NH, has a higher affinity for the cleavabk thrombin receptor than [Sar ']NAT~-NH 2 , since in conditions where metabolism did not play a role, its concentration-effect curve was shifted from the right to the left of that for (Sar1]NAT6-Mi,. Thus it can be said that substitution of Ser by Sar leads to a product with a marginal loss of affinity that is more than compensated in biological assays by a gain in metabolic resistance.
Experiments using the oil immersion technique indi- cate that aminopeptidase M activity is present in the microenvironment of the receptor and that trapping of srnaIl quantities of agonist in the fluid phase irnmedi-
ately surrounding the tissue leads to a gradua1 relax- ation that is probably attniutable to metabolism of the agonist. This is shown by the slower relaxation of the [Sar ']NAT6-W ,induceci contraction as compared to that seen in e-xperiments invoIving NAT,-NH,. The relaxation time was maximal when the contraction was induced by NAT6-NH, in the presence of amastatin. Aminopeptidase M seerns to play a significant role in the disposition of NAT,-NH2 in this tissue since other peptidase inhibitors had no effect on the relaxation of these tissues.
Anesthetized and heparinized rabbits exibit a dose- dependent hypotensive response to NAT peptides that is not completely characterized. The modest increase in potency of [Sar1]NAT,-MI, compared to NAT6-NH2 observed at high doses of peptide in the in vivo assay (Fig. 3) probably reflects both the higher metabolic resistance and lower receptor affinity of the Sar-sub- stituted analogue. It seerns that the observations made in vitro may be related to the in vivo activity of NAT6- NH, since amastatin also potentiated the hypotensive actMty of this peptide in the anesthetked rabbit (Fig. 4). The duration of action was more potentiated than its amplitude. a situation previously observed with bradykinin-related peptides (Drapeau et al., 2991). Fi- nally, this study shows that the N-terminal amino group of NAT-related peptides can to1erate a certain degree of substitution. While previous studies have shown that acetylation of the N-terminal residue leads to inactive analogues (Chao et al., 1992; Scarborough et al., 19921, the present resu1ts indicate that N-methylation is com- patible with biological a c t ~ t y .
The relevance of the data presented here must also be considered in relation to the role of thrombin and of its cleavable receptor in normal physioiogy. It is likely that arninopeptidase activity exerts iittle effect on this system under normal conditions. This is reflected by the absence of effect of amastatin in potentiating the agonist effect of thrombin on the rabbit aorta. Thrombin activates the receptor by a cleavage that exposes a tethered agonist and it is uncertain whether aminopeptidase M has access to the newiy exposed terminus before it activates the receptor. AIso recent observations have shown that receptor intemalkation plays a key role in terrninating the actions initiated by thrombin (Hoxie et al., 1993). However, aminopepti- dase protection may be important in the developrnent of peptide-based antagonists of the thrombin receptor that will eventually be tested in vivo and in tissues where this enzyme is active. Our approach, which includes validation of new pharmacoIogica1 tools in vivo, may allow future applications in experimental pathoIogies in rabbits (for instance, dietary at herogene- sis: Cooke et al., 1992). Also, since cultured smooth muscle ceils derived from the rabbit aorta exhibit a rnitogenic response to either a-thrombin or NAT,,

(Herbert et al., 1992). protection frorn aminopeptidases may aIso be relevant to the development of agonists of this receptor for studies designed to explore its activity as a growth factor.
Supported by the Québec Heart and Stoke Foundation. GD and FM are Scholars of the 'Fonds de la Recherche en Santé du Québec'. The technical hclp of Mr- Ritchie Audct and Ms. Johanne Bouthillier is acknowledged.
6. References
C h a . Ba.. S. Kalkuntc. J.M. Maraganore and S.R Stone, 1992, Essential groups in synthetic agonist peptides for activation of the platelet thrornbin rcccptor, Biochemistry 31,6175.
Coller. B.S.. P. Ward. M. Ccniso. LE Scuddcr. K. Springcr, J. Kutok and GD. Prestwich. 1997 Thrombin rcceptor activating peptide: importance of the N-terminai serine and its ionkation state as judgcd by pH dependence. nuckar magnetic rcsonance spectroscopy, and clcavage by aminopeptidase M. Biochcmistry 31. 11713.
Cooke. J.P.. AH. Singer. P. Tsao. P. Zera and M E Bitlingham, 1992. Anriatherogtnic effects of L-arginine in the hypcrcboles- teroltmic rabbit J. Ciin. tnvest. 90. 1168.
Dcblois. D.. G. Drapeau, E- PetitcIerc and F. Marceau, 1992, Syner- gism bctwcen the contnctile cffcct of cpidcrmal growth factor and that of des-kgp-bradykinin or of a-thrombin in rabbit aonic rings. Br. J. Pharmacol. 105, 959.
Drapeau. G.. D. Debiois and F. Marceau, 1991. Hypotensive effccts of Lys-des-&g9-bradykinin and rnetabolically protectcd agonists of B, reccpton for kinins, I. Pharmacol. Exp. Thcr. 259,997.
Drapeau. G.. R. Audet. L. Levesque. D. Godin and F. Marceau. 1993. Dcveloprnent and in vivo ~ualuation of metabotically rais- tant aniagonists of B, recepton for kinins. S. Phannacol. Exp. Thcr. 266. 192-
Haver. V.M. and D.H. Namm, 19s . Characterimtion of the throm-
bin-induccd contraction of vascular smooth muscte, Blood Vts- sels 21, 53.
Herbert, f-M, 1. Lamarche and F. Da4 1% Induction of vascular smoath muscle aU growth by seiedivc activation of the thrombin rcceptor. Effcct of heparin, FEBS L c t ~ 3Of. 155.
Hoxk, J.& M. Ahuja, E. Belmonte. S. Pizarro, R Panon and LF. Brass, 1993, Intcrnalization and rccycling of activated thrombin rcccptors, J. BioL Chcm, 26û, 13756.
Hui, KY, J.A. Jakubowski, V.L Wyss and EL Angleton, 1991, Minimal sequene requinment of thrornbin receptor agonist p e p tide, Biochem Biophys. Rcs. Commun. 184, 790.
Kalsncr, S. and M- Nicktrson, 1968, A mcthod for the study of mcchanism of drug disposition in smooth muscle. Can. J. Physiol, Pharmacol, 36,719,
Palmieri. FE. EH- Bausback and P.E. Ward, 1989. Mctabolism of vasoactivc peptides by vascular endothclium and smooth muscle aminopeptidase M. Biochem. Pharmacoi. 38. 173.
Regoli, D.. F. Rioux, W.K Park and C Choi 1974. Role of N-tcrmi- na1 amino acid for the biological actMty of angiotensin and inhibitory analogues, Can. J- Physiol. Phannacol. 52, 39.
Scarborough, RM., M A Naughton, W. Teng, D.T. Hung, J. Rose, T.-KH. Vu, VJ. Wheaton. W. Turck and S.R Coughlin, 1991, Tcthcrcd ligand agonist peptides: structural rcquirernents for thrombin reuptor activation rtvcal mcchanism of pmtcolytic unmasking of agonist function. I. 0iol. Chem 267, 13146.
Troyer, D., R PadiIla, T. Smith, 1. Krtisberg and I.I.W. Glass, 1992, Stimulation of the thrombin receptor of human giomcnilar mesangid cc& by Ser-Phc-Leu-LÉu-Arg-Asn-Pro-Asn-AspLys- Tyr-Glu-Pro-Phe peptide. J. 6iol. Chem 267,20126.
Vassailo, KR, T. Kieber-Ernmons. K. Cichowski and LF. Brass. 1992, Structure-funcrion rclationships in the activation of plarelet thrombin reccpton by receptorderived peptides. J. Biol. Cbem. 267, 6081.
Vowret-Craviari. V.. E. Van Obbcrghtn-Schilling. U.B. Rasmussen and A Pavirani, 1992, Synthctic a-thrombin receptor peptides activate G proteinauplcd signalhg pathways but arc unablc to induce mitogenesis. Mol. Biol. Cell. 3.95.
Vu, T-KK., D.T. Hung, V.I. Whcaton and S.R. Coughlin, 1991. Molecuiar cloning of a functional thrombin rcccptor revcals a nwcl protcolytic mechanism of receptor activation, Cell 64. 1027.
Yang, S.G., A. Lanyyonu. M. Saifeddinc, GJ. Moore and M.D. Hohaberg, 1992 Actions of thrombin and thrombin receptor peptide analogues in gasvic and aortic smooth muscie: develop- ment of bioassays for structure-activity studies. Lift Sci. 51. 133.

Mode of action of thrombin in the rabbit aorta
Denis Godin, Francis Rioux, François Marceau & 'Guy Drapeau
Centre de recherche (Université Laval), Hôtel-Dieu de Québec, I l Côte du Palais, Québec (Québec), Canada GlR 256
1 Thrombin is a vasoactive proteasc that eticits the contraction of the rabbit aorta by activating a G- protein coupled receptor through cleavagc of its N-ccnninal extracellular domain. Synthetic peptides corrcsponding to the ncwly exposcd N-terminus, foiiowing thrombin cleavage, have ben show to rcproducc some of the activitics of thrombin in the rabbit aorta. 2 IntraaUular pathways involvcd in the cont rade responst of the rabbit aorta to thrombin and synthetic peptides wert uramincd by use of a series of inhibitors. A similar method was appticd to characterk the mitogcnic effect of thrombin on cultured srnooth musde aiis (SMCs) deriveci from the samc tissue. 3 Resuits from tbis study indicatc that the contractile rcsponst of the rabbit aorta to thrornbin is depeudent on the activation of protein kinase C (PKC) and independent of extracellular calcium. The contractiie responst to thrombin can bc M y rcproduccd by peptide agonists relatcd to the N-terminai rcccptor sequena. Howwer, subtie secm to exist betwccn the mechanism of the contrade efkct of thrombin and of the synthctic peptides, as both PKC activation and cxtraceliuiar calcium were found to participate in the contractile effcct of the synthetic peptides. 4 In culturcd SMCs, bo& kombin and the synthetic peptides in& inositol phosphate nunover; howcver, ouiy ttvombin clicited a mitogcnic efftct, which occurs at thrombin concentrations weU below thox nedeci to increase inont01 phosphate turnover signiûcantly. Activation of a tyrosine kinase pathway is involvcd in the mitogcnic effkct of thrombin on aortic SMCç. 5 AItogether these results suggest the existence of subtIe ciifferences berneen the mode of action of thrornbin and of synthetic peptides relatai to the N-terminal thrombin rectptor sequenct, in the rabbit aorta.
Keywords: Thrombin; thrombin cleavabIe rcccptor; smooth muscle cck; rabbit aorta
Introduction
a-Thrombin is a key enzyme involved in blood coagulation and although its activity on soluble substrates in the circulation has been txtcnsively studied, thrornbin also exhibits hormone-like activity in many ceU types (for rcview see Fenton, 1988 and Coughh et al., 1992). The cloning of a receptor mcdiating some of these activities has revealed a ncw cIass of G-protcin coupled receptors that are activated by iimited prottolysis of the N-terminal extraceiiuiar domain (Vu er al., 1991); a second receptor that is activated in a similar way has reccntiy k e n describeci (Nystedt et al., 1994). Thcse receptors c m bc acti- vated by synthetic peptides, as short as fivc or six amino acids, such as Ser-Phe-ku-Leu-Grg-hsn-NH2 (NAToNH3 for the thrombin r a p t o r (Hui et al.. 1992): corresponding to the newly generatcd aminoterminus rcccptor sequenœ foiiowing prote01 ytic cleavage.
Thrombin is a moduiator of vesse1 tone that can act by either endotheliurn-dqendent or -independent mechanism and the relaxation or contraction induced by thrombin can be re- produced by synthetic agonists of the cleavable reccptor. In the intact rat aona peptide agonists are reiaxing agents (Yang er ut., 1992); however, contractile rtsponses arc obscrved when the endothelium is removed (Antonaccio er al., 1993)- Stimu- lation of the dog coronary artdes by an agonist of the thrornbin cleavablc reccptor also produces an cndothelium- dependent rdaxation that is transfomed into a contractile response in the absena of cndothetium (Ku & Zaieski, 1993). In the rabbit aorta thrornbin. NAT6-NH2 and an arnino- peptidasc rcsistant analogue, [SarLpAT6-NH2 (Godin et al., I994), produce a dosedependent contraction suggesting that the cleavable reccptor is involved in this activity.
Thrombin has also bcen found to exen rnitogenic activity in several ce11 types (Fenton, 1988). However, intraceiluiar signals
' Author for comspdndcncc.
generated by peptide agonists of the cleavable receptor secm insuffiCient to inducc mitosis in hamster fibroblasts (Vourct- Cravian et al., 1992). In an effort to characterize f d e r the second messengers involved in the contractile rcsponse of the rabbit aorta, foiiowing stimulation by cither thrornbin or agonist peptides, we have evaluated the effect of differcnt in- hibitors in the presence of these contractile agents. We have ako used these inhibitors to characteritc second mcsscngers activateci following sllmulation by thrombin or agonist pep- tides of cuitured vascular smooth muscle cek (SMCs) dcrivcd from the rabbit aorta. Our results reveal a difference in the way contractile responses are obtained through activation by thrombin or peptide agonists and chat a profirative response of SMCs derived from the aorta is obtained only by stimula- tion with thrombin.
Methads
The thoracîc aorta was isolatcd from New Zealand White rabbits of either sex (1.5 - 2 kg). n i e vessels were cut into rings (2 -3 mm width), suspended between a metal hook and a thread loop under a basal tension of 2 g in 5 ml organ chambers containing oxygenated (95% O2:5% CO3 and warmed (37°C) Krebs solution (Marceau et al., 1991). Their respoases to agents were recorded isornetricaily as describcd prcviously (Bouthillicr et al., 1987).
The mechaaism of the contractile efféct of the thrornbin rtceptor agonist, [Sar1]N~~&H2 was anaiysed by applying a submaximal concentration (40 g ~ ) of this peptide to the tis- sues after an cquilibration period of 2 h. The concentration- effect relationship for this agent can bc found elsewhcrc (Godin et ut., 1994). ïnhibitory drugs were applicd 30 min bcfore cach agonist and paircd controt tissues wcre used to

evaluate the effect, if any, of the drug vehicle. Thc sarne tissues were aiso chaiitngtd at 4 and 6 h by thrombin (60 n ~ ) and KCl (20 mM), respectively, for purposes of cornparison- The drugs used in this study wert dedipine, an L-type Ca2' channe1 blockcr (Murad, 1990), H-7, a prottin kirme inhibitor (Hidata et al., 1984), diclofenac. a cydoloxygenase inhibitor (Lewis & Funt, 1987). erbstatin, a tyrosine kinase inhibitor (Imoto et al., 1987) and ~Phe-Pro-Arg-CHzC1 (PPACK), an irrevcrsible inhibitor of the catalytic activity of tbrombin (Kettner h Shaw, 1979). The concentrations of the dmgs wcre choscn according to their reporteci activity ia various in vitro systems (Lcvcsque er al., 1993). The effet of the kinase in- hibitor, H-7, was fuRhcr documentcd over a widt range of concentrations of the thrombin rtccptor agonists; cumuiative concentrationeffect curves for [Sa.r"ATmz and thrombin were constructeci, In order to evaluate the contribution of the endothelhm in the contractile response to each agent, the cndothelium was carefdy removcd by rubbing with a metal spatuIa, The tissues wcrc controiled by asscssing the loss of the relaxant eKect of acctylcholine (Furchgott & Zawadzki, 1980). We also replacd the Krcbs solution by a cafcium-frct form to evaluate the contribution of extracellular calcium to the con- tractile efféct of each agent, The Krcbs solution was rcplaced by the calcium-fixe fom 10 min More each contractile sti- mulation and the tissue preparation was maintaineci in normal Krebs solution betwecn tests to avoid depletion of intraceDuIar Ca" pools,
Cell culture
Several distinct lines of rabbit SMCs were estabiished from rabbit aorta aseptically rcmoved from frtshiy k i k i animais, The tissues (approximate weight 200 mg) were incubateci at 37°C for 1.5 h in medium 199 (Gibco) supplemented with 10% foetal bovine serum (FBS; Gibco), antibiotics (strcptomycin, 50 pg ml-', and penicillin 50 u ml-': G'bco), bacteriai col- iagenase (type IA, 3 mg ml-') and elastase (type III, 1 mg ml"); al1 enzymes were obtained from Sigma (St-Louis, MO, U.S.A.), Dispersed cciis and panIy digested tissue frag- ments were centrifuged and resuspended in medium 199 sup- ptcmcnted with 10% FBS and antibio tics. This suspension was seeded directly in 25 cm' flasks coated with 0.2% gelatin. Cells were passaged on _pelatincoated surfaces at confiuency with a bnef trypsin (O.OS%)IEDTA (0.53 ma0 treatment. The ce1ls were studied at passages 3 to 7 only.
r3Hj-t hymidine incorporation
Cells, in Medium 199 containing IO% FBS, were plated at an initial density of 3 x IO4 cells per well in gelatin-coatcd 12-weil disposable plastic plates (Costar). The ceils were growth-ar-
resteci, by incubation for 72 h in medium 199 containhg 0.4% FBS, beforc bting cxposcd to thrombin, NATcNH2, [Sari]- NAT&&, FBS or to various drugs for 4û h. Enhanad thymidine incorporation was assesseci after a 24 h uposure ta rHj-thymidine (Dupont, Mississauga, Ont, Canada; O. 1 pCi ml"; specific activity 84 Ci mmol"). The ulls wcre then washed twicc with saline, harvested with trypsin (O.OS%)/ EDTA (0.53 a!) and coiiccted in borosiricate tcst tub. The w e k were rinsed once with 1 ml Triton X-LOO 1 %/saline and nucltic acids prccipitated with 1 ml trichioroaatic acid 40%; water. Aftcr a 20 min antrifugation at 2,700 g, the supcr- natant was withdrawn and the pellet rcsuspnided in I ml distiiitd water, placcd into scintillation vials containing 10 ml Ecolite + scintiIlation 0uid (ICN Biomcdicals, Irvine CA, USSA-) and counted in a iiquid scintiiiation counter (Beckman LS 8000).
Inositol phosphate production
Labelling and stimulation of rabbit aorta SMCs wcre catried out according to B a h et al. (1988) with some modifications. Bricfly, cek in medium 199 supplemcntcd with 10% FBS and antibiotics, wcre &cd at a dcnsity of 2.5 x l@ in 2 cm2 weiis and culturd for 24 h. The ails wcre then washed thrce times with serum-frce medium. The incorporation medium (500 pi) consistai of medium 199 containing 20 $Ci mi-' of myo-rw- inositol (Amcrsham, spedc activity 82 Ci mol''), was ad- ded to the wells for a labelhg ptriod of 16-20 h, After in- cubation at 37°C in a humidifieci atmosphere containhg 5% CO3 each well was washed twice with Earle's baland salt solution (EBSS). The ails were f e c r incubated for 20 min in EBSS with 10 r a ~ LiCl then challengeci with thrombin or NAT&& at différent conltnuations for exactly 5 min- Sti- mulation was terminated by the addition of perchlonc acid (final concentration 5%).
Inositol phosphates were extracted and separated as pre- viously describeci (Guiliernette et a/., 1989). At the end of the experirnént, the ceils were kept on ice for 30 min and thm scraped from the weiis and transferred into tubes, The weUs wcre rinsed with additional perchloric acid (5% W/V) and the resdting media was cmtrifuged at 10,000 g for 5 min. Per- chioric acid was extracted from the supernatant with a 1 : 1 mixture of Freon and tri-n-octylamine (Domes et al., 1986; reagents from Sigma). After aeutraiization, the sampIes were applied to anion exchange nsin (Dowex AG 1-X8 formate, 200-400 mesh, Bio-Rad No. 140-1450) columns (1 ml bed volume) for the separation of the inositol phosphates accord- ing to Berridge er ai. (1983). The inositol phosphates were sequtntially eluted with 3 x 5 mi of distilied water ( f a in- ositol), 3 x 5 ml of 0.2 M ammonium formate/O.l M formic acid (inositot monophosphatcs, PI), 3 x 5 mi of 0.5 M am-
Table 1 Pharmacological analysis of the mode of action O€' thrombin and the thrombin rcccpcor agonist [Sar1W~Ts-NH2 in the rabbit aortic preparation
Intima1 damage Conuol Nifidipine 100 nM Vchicle (Ethanol) Calcium-frcc Krebs Control Diclofenac 0.5 pM Vchictc (h'a2C03) Erbstatin analogue Vchick (DMSO) PPACK 1 p M Vchicle (saline)
'Values in each trcated group w m cornparcci to conuols from the same ani& by StudentSs r tcst for paircd samplw: 'Pc0.05; -P<0.01; o**P<O.OO1.

monium formatt[O. 1 M formic acid (imositol biphosphatcs, Pd, 3 x 5 ml of 0.8 M ammonium [email protected] M formic acid (ïmositol trisphosphtes, iP3) and 3 x 5 ml of 1.0 M ammonium formatc/0-1 M fonnic acid (ïmositol tetrakisphosphatcs, P.). Thc f-ons wert collecteci, mixeci with Ecolitt + scintillation fluid and counted for radioactivity in a liquid scintiUation counter.
Reagents
The foiiowing dnigs wert purchascd from Sigma (St-Louis MO, U.S.A.): nifedipine, di cl of^^^^^^:, PPACK, PGE2 (pros-
Figure 1 Effect of H-7 (V 10 pbd) on tht cumulative dose-tespouse curve for [ s ~ ~ ' ~ A T ~ - N H ~ (a) and thrombin (b). The rcsuIts an cxpressed in g of dcvcloped conuaction. Values arc the mean* s.c.mcan of at Iast 6 dcterminations. Statistical anaiysis by Studcat's t test for p a i d samplcs: * P < 0.05; **P<O.OOI (a control respo-1.
Tabk 2 InositoI phosphates mcasurcmcnts in rabbit aortic smooth thrombin or NATrNHIa
taglandin Ez) and PMA (phorbol 12-myristate 1Eacctatc). Er bs tatin analogue and H-7 (1 45-isoquholintsulp hony1)-2- mtthylpipcrazine) wcrt pufchased from Biomol Res. Lab. Inc. (Plymouth Meeting PA, U.S.A.) Wottmannin was from Cal- biochem (San Diego, CA, U.S.A.), iloprost fiom &rkx Ca- nacia Inc. (Lachine QC, Canada) and Thrombostat (bovine thrombin) from Parke-Davis (Scarborough ON, Canada). NAT&& and [Sar1]NATm2 wert synthcsized in our ia- boratory by soiid phase peptide synthesis methods. Thcir homogeneity was assessed by high perfomanœ liquid chro- matography and thcir identity con6nned by mas spcctro- metry.
A series of ~periments using cabbit aortic rings was carricd out to compare the mode of action of thrombin with an ami- nopeptidase rcsistant agonist of the thrombin clcavable re- ceptor, [Sax1JNAT~NH2 (Godin et al., 1994), and KC1 uscd as a control agonist. Submaximal concentrations of these ago- nists were choscn bascd on prcvious cxperiments (Godin et d., 1994, Lcvesqut et al., 1993). In ordm to avoid any tachyphy- laxis, [Sar1JNATsNEIr was always the 6rst agoaist injccted aBcr an quiiiiration period of 2 h, foUowd by thrombin and KCI succcssively with an interval of 2 h bctween agonists. Paired controls were uscd to evaiuate the effect of solvents. AU inbibitory drugs were applied 30 min bcfore rccording the nt- sponse to a c h agonist.
As shown in Table 1, intima1 damage did not modify the response to these agonists while it completely blocked the re- iaxation induccd by acetylcholine, used as a control, of tissues pre-contracted with phenyltphrine (data not shown). Niedi- pine, the voltage-operated calcium channel blocker, was in- active against the thrombin-induccd contraction of the rabbit aorta whcreas a partial inhibition of the contraction induccd by [Sart]NATGNH2 was observai. Extensive inhibition of the dcpoIariring agent KCI occurrcd in the same tissues, thus providing an internai control for the efkctiveness of the drug in our assay. Tbe contraction induced by KCI was profoundly afftcted in the presence of a Krebs sotution devoid of calcium while the contraction induced by [Sar1]NATcNHt was reduced to 213 of the control and the efféct of thromfiin was left un- changed by wkium ternoval.
Didofenac, an inhibitor of Fany acid cyclo-oxygenase, and the erbstatin analogue, an inhibitor of tyrosine kinases, did not have any depressaat effect on the contraction inducai by the three agonists prcsentcd in Table 1. An inhibitor of the m a - lytic activity of tbrombin, PPACK, completely prevented the
muscle celfs 5 min after txposure to diffcrcnt conceutrations of
Srnooth muscle ccüs Irom 3 dinerat œU Lines were used at parias 4 or 5 (values arc the means 2 s.c.mcan of 9 expriments composed of tripiicatc &tenninations). The d t s have betn normaliotd as c.p.m. of IP per 100,000 c.p.m. of the rcmaining œU-associateci myo- inositol in each ccU weU. bOnc-way analysk of variance wu pdormed on a c h calumn (NS, not sisnificant). Mann-Whitney's test was then applied to compare the effcct of cach traitment with the contra1 cclls: *P<O,OS; **P<O.OI; ***P<0.001.

contraction of the rabbit aorta by thrombin at a concentration of 1 m. The activitics of the peptide agonist and of KCi wcre not jnflucnccd by this inhibitor.
Foliowing the observation that singie doses of thrombin or [Sar1mATrNH2 wert afF& by the protein kinase inhibitor H-7, frill cumulative concentration-eff' curvts for t h e agoaists wcrc establishd in the prcsenœ of this inhibitor. As shown in Figure 1, H-7 (10 FM) signiscantiy depresscd aiI concentrations of thrombin and [Sar1JNATrNH2- Prcvious cxpMmcnts on the same tissue have dso shown that H-7 significaatiy reduced the contraction to PMA while it afkcted ody minimafly contractions induccd by KCl (Levtsque et al., t 993).
Cellular mode1 of vasculut effecrs
Evidence was obtained that cultured SMCs dcrived from the rabbit aorta rcspond to thrombin and NAT6-NH2 by inducing
Figure 2 Efftct of thrombin (O), ? iAf6-m2 (9) and [Sari]- NAT6-NH2 (V) on the synthcsis of DNA in cuitund cabbit wiscular smooth muscle ceiis. Data prrscntcd arc the mc2us1s.c. of stveral txpcfimcnts involving viplicatc detcrmiaations. Dashcd line ttprc- scnts basa1 incorporation (0.49.0 F BS).
Table 3 [.'~-thymidine incorporation by rabbit aortic nnooth muscle ceilsa
Contra1 Diclofcnac 0.5 pM +- PGEt 0.3 pM + noprost 0.3 ph4 +- Niftdipine 0.1 pM + Wortmannin
IO-' M + PMA 1 pg mr' +Ha7 10 pM + H-7 30 p M + Erbstatin analogue
10 pM ANOVA
'Values arc the mean is.c.mean of at least 12 detcrmina- tions from 4 separate expcrimeats with differcnt a l 1 l ins at passages 4 and 5 (NT: not testeci). One-way analysis of variana was pcrformd on each column. Mann-Whitney's test was then applied in cach colwnn to compare the cffect of each utaunent with tbc badine or the thrombin- stimulated thymidine incorporation mpcctively (fint Linc; *P< 0.05; 8mP<0.01; ~mmPcO.ûûl). ï b c effcct of thrornbin treamcnt alone within cach rrcated group was assesseci by Student's t test and was significant (P<0.0001) for ail groups cxcept H-7 (30 PM) and etbstatin analogue-acatcd groups.
inositol phosphate (IP) turnover (Table 2). Significant inctc8scs in the production of Ifi and IP3 wcrc obsmrcd in œk stimulattd with thrombin. Although adose-dependent cffcct is observcd for thrombin, signiscant values wtrc obtaintd only at concentra- tions of 10 and 1 0 n~ for IP3 formation and 1000 nM for IP2. NATcNHz increased IP,, and ïP3 formation with a sig- nificant dose-dependent relation for both Pz and PI. Throm- bin, NATrPUi and [Sarl]NAT,&& wert also testcd for thcir abiiity to inducc the incorporation of [3HJ-thymidine in quies- cent ccüs of the samc origin that werc maintahcd in 0.4% FBS. lhrombin stimuiatcd the incorporation of thymidine in thesc ceils in a dose-rtIatod manner (Figure 2). with maximum in- corporation attained a t a dose of 10 n~ while NAT&& and [Sarl)NATsNH~ at doses in exces of la) p~ failed to produœ any significant thymidine incorporation in these ails. Several inhibitors werc testcd for their abiiity to modulate the thrombin- indu& DNA synthcsis into rabbit culturcci aortic SMCs (Table 3). Prcvious experiments had shown that diclofenac, an inhibitor of fatty acid c y c l o - o x y g ~ , was useful in rcvcaiing agonist- induccd thymidine incorporation in this mode1 (Lcvcsqut et al., 1993). As shown in Table 3 diclofenac (0.5 p ~ ) revealed max- imum expression of thymidine incorporation foliowing stimu- lation with thrombin. Ln the pr#ence of diclofenac, PGEz and iloprost (a PGIz analogue) produccâ a partial inhibition ofDNA synthcsis in uils stimuiatcd by thrombin. Nicdipine productd a sisnifiant incrcase of thymidine incorporation in both control and thrombin-stimulated ah. A siight incnase was also ob- served with P M A while wortmannin, a phospholipase D in- hibitor, did not produœ any signilïcant inhibition of thrombin- stimdated thymidine incorporation. At a dose of 10 p~ the PKC inhibitor. H-7, was without effcct on either control or thrombin-stimulated ab, howevcr, H-7, at a higher dose (30 p ~ ) and the erbstatin analogue (a tyrosine kinase inhibitor) reduced the effkct of thrombin to baseline values. At the samt dose, H-7 also reductd DNA synthesis in control cclIs.
Discussion
The comparative mode of action of thrombin and a synthetic agonist of the cleavable receptor for thrombin, either NATr NH2 or [SarL]XATCNH2 was investigated using a fresh rabbit aortic prcparation (contractility assay) and cdtured SMCs denved from the same tissue (IP turnover, mitosis). Vascuiar responses to thrornbin secm to v a y according to species; some vascular beds. such as pig and dog coronary artcries or dog saphcnous vtins respond to thrornbin, or an agonist peptide of the cleavable reccptor, with an endotheliumdependeat re- laxation that is mediateci by the release of nitric oxide and a endo thehum-indendent contraction (Tesfamaiam es al.. 1993; Ku & Zaleski. 1993). The contractile rcsponsc of the rabbit isolattd aona to thrombin and [Sar1]NAT6-NH2 is mediattd by smooth muscle since mechanical removal of the cn- dothetium did not a î k t the rcsponse to either agonisu or to KCI w d as a control agonist (Table 1).
As the contractile rcsponse of tbc rat aorta has bcen shown to be dependent on extracelluiar calcium, we investigatcd whether the rabbit aorta operates in a similar manner. A sus- tained contraction was observeci with thrombin in the absence of cxtraœUuIar calcium in the bathing tiuid, whik the con- traction to KCl, a depolarking stimulus dependent on extra- cellular Ca2' (Cauvin et al., 1983), was inhibiteci by almost 90% on calcium removal. In the sanie conditions a modest inhibition of the contraction to the agonist peptide, [Sar'] NAT6-NHZ was observed. ï h c partial inhibition of the con- tractile activïty of this peptide by calcium removal was also in the same order as that produœd by the calcium chamel biocker nifedipine. While nifedipinc had no effect on the con- traction to thrombin, it abolished the KC1-induced contrac- tion, which is consistent with the inhibitory effect of this drug on the voltage-operatcd calcium channel (Murad, 1990). It is worth noting that the responses to thrombin and agonists of the cleavable rcccptor of the rabbit and rat aorta dincr in

several tcspects- Fvst of aiI, human or rat thrombin have no efftct on the rat aorta, which on the othcr hand rcsponds to synthctic agoaists of the thrombin cleavable rtccptor (Anto- naccio et al., 1993). Aside from showing an endotheliumde- pendent relaxation, which is not observai in the rabbit aorta, contractile responscs of the rat aorta to peptide agonists rdy on extracclidar Cz?' while in the rabbit prcparation the effect of thrombin is independent of extracciiular Ca2+ and the contraction to [Sar1NATcNH: is only partially aEectcd by Ca2' removal or blockade of Caz- channek by nifdpine. The partial inhibition of [Sar'l-NAT~Ni~inductd contractile re- sponses by CaZ' nmoval or nifcdipine in the rabbit aorta may indicate that tcctptor activation by the peptide agonis& which does not involve protcolysis, may bc more dependent on ex- ti dcclluiar Caf' than activation foiiowing cleavage of the se- ccptor by thrombin, It may aiso be due to the activation of another rcccptor that is insensitive to the protcolytk action of thrombin. A second G-protein couplcd rtccptor that is pro- teoliticafly activated has rccently been cloned from a mouse genornic Iibrary (Nystcdt et al., 1994). Although the en- dogenous protcase that phsumably activates this reçtptor has not betn ideucificd, this rtctptot is insemitive to thrombin but rnay be activated by trypsin. Like the thrombin rroeptor, it is ais0 activatcd b y a synthetic huapeptide, Ser-Leu-Re-Gly- Arg-Leu correspondhg to the ncwly cxposed N-terminus fol- lowing prottoiytic cleavagc. Aiso structure-activity studits on rat and guinea-pig tissues using a stries of peptide analogues suggcst the existence of thrombin raxptor subtypes for pep tides related to NATsNHr (Koiienberg et al., 1993).
Data obtained in rabbit aortic ring also suggest the in- volvement of protein kbasc(s) in the contractions provoked by thrornbin or [Sar1JNATGhiH2. Protein kinase C is in- hibited by H-7 (Hidata er al., 1984), and accordingly has been shown to rcduce the contractite effcct of PMA, a direct ac- tivator of protein kinase C, in aortic rings (Levesque et d, 1993). H-7, in the samc assay, also shiftcd the concentration- rcsponse curvc to both [Sartw.4T&I'H2 and thrombin (Fig- ure 1). However, the use of H-7 as a protein kinase C in- hibitor has some limitations o ~ i n g to other inhibitory effects of this drug, as for example, on cyclic nucleotidedepcndcnt protein kinases (Hidata et al.. 1984). White the contractile response of the rabbit aorta to thrombin has b e n s h o w to act synergistically with chat of EGF the receptor for which is a tyrosine kinase (Deblois et al.. 1992), erbstatin, an inhibitor of this type of receptor did not block the rcsponses of any of the agonists in Table 1. As pre\%ousIy observed (Deblois et al., 1992), PPACK. in accordancc with its inhibition of the cataiytic site of thrombin, did block the contractile eEcct of the latter while it had no effect on [SarL]NAT6-NHz-induccd contraction. SMCs denved from the rabbit aorta and cultured ovcr
wecks retaineci the capacity to respond to bot& the agonist of the cleavable receptor. in this case NAT,&&, and to thrombin by the generation of phosphoinositides, The in- creased IP concentration after 5 min exposure to either ago- nist, evidence of the activation of phospholipase C, was obtained at concentrations comparable to those used for the contractile response of isolated tissues (ïable 1; Godin et al., 1994). ï h e activation of phospholipase C following stimula-
References
tion of the cIcavablc m t o r by NAT** is consistent with the participation of diacylglyccrol-nguiatcd protcin kinase C in the contractile effcct of this peptide and of thrombin as r e f d to above with the use of the protcin kinase inhibitor H-7 (Figure 1). Significant increascs in iP2 and IP3 occurrçd at bath concentrations of NAT6-NHt testcd and a gradcd rc- sponse is okrved. A dose-dependent iP production was also observeci in cclls stimulateci with thrombin although sipificant only for IP3. Hcre also the doses uscd to obtain this ceponse are si& to those ncctssary to contract the rabbit aorta (Godin et al., 1994).
AIthough the responsc of c u i t d rabbit SMCs to throm- bin and NATGNHr is similar, with regard to IP breakdown, only thrombin could induce DNA synthesis in these œils (Figure 2). NATrNE12 or [Sar1jNAT6-NH2 at doses up to 100 phf did not provoke any significant [3Hj-thyrnidine in- corporation whercas with thrombin, a full concentration-efftct curve was obtaintd with a threshold dose as mail as 0.1 n~ whiie a EU cffect was obtatined at 10 n ~ . Intenstingly the concentrations of thrombin needed to produce the incorpora- tion of thymidine secm well bclow those nceded to inducc IP breakdown or contraction of the isolatcd aorta (ïable 2; Godin et al., 1994). Thme are also other indications that the intraceiluiar pathways involvcd in DNA synthesis in rabbit SMCs are at Icast partly dinerent from those mediating the contractile rcsponse to thrombin. DNA synthesis was blockd by the trbstatin analogue while this tyrosine kinase inhibitor had no effect on the contractile response. H-7, at a con- azntration of IO PM, can shift the contractile dose-rcsponse m e to both [SartjNATd-NHz and thrombin but does not afllect DNA synthesis. At 30 pM the effect of H-7 on thymidine incorporation is probably of a toxic nature as control cc& were also affected.
Within the limitations of our pharrnacoIogica1 approach, the contracüc response of the rabbit aorta to thrombin s#ms to be mediated by the activation of PKC and can bc rc- produced with synthetic agonists, However mitogcnic rc- sponses to thrombin, of cultured vascular SMCs derived from this tissue, nquire a tyrosine kinase signal that is not gentratcd by stimulation of the cleavabie teccptor with peptide agonists. Thesc [indings agree with obsemations in hamster fibroblasts where peptide agonists of the thrombin receptor do not rc- produce the mitogenic efféct of thrombin (Vouet-Craviari et al., 1993). The inhibition of the mitogenic efféct of thrombin in our cultured cells by erbstatin indicates that a tyrosine kinase signai is essential For ce11 proliferation in this modef. Tyrosine kinase activity is also required for the mitogcnic activity of thrombin on neonatal rat vascu1.u SMCs but is not needed for intraœUular catcium relcase (Weiss & Nuccitelli, 1992). How this signal is generated in rabbit vascuIar SMCs whether through another type of receptor or through the autocrine release of a growth factor (Weiss & Maduri, 1993) rernains to be established,
Supportcd by the Medical Research CounciI of Canada (grant MT- 12570) and by the Qui& Hcart Foundation, F.M. and G.D. are SchoIan of the -Fonds de la recherche en Santé du Québec' and D.G. is a ncipient of a Studentship from the samc apncy.
AMONACCIO. MJ.. NORMANDIS. D., SERAFINO, R. & MORE- BERRIDGE. MJ.. DAWSON. R.M.C.. DUWNES. C.P.. HESLOP. S.P. & LAND. S. (1993). EKecu of thrombin and thrombin rcceptor IRVINE. R.F. (1983). Changes in the lcvels o f inositol phosphates activriting peptides on rat aortic vascular smooth miisclc. J. afttr agonist-dependent hydrolysis of membrane phosphoino- Pharmacol. EXP. Ther.. 266, 125 - 132. sitides. Biochem. J.. 212, 473 -482.
BALM. T., BAUKAL. A.J., GUILLE1\1ETTE, G. k C A R . Kf. (1988). BOüTHILLIER. J.. DEBLOIS. D. k MARCEAU. F. (1987). Studics on Multiple pathways of inositol polyphosphate metabolism in the induction of pharm;icological rcsponses to d e s - ~ r ~ ~ - angiotcnsin-sùmulated adrcnal gomenilosa cclls. 3. Biol. Chem., bradykinin in rilro and in vivo. Br. J. Pharmacol.. 92,257 - 264. 263,4083 - 409 1.

CAUWN, C., LOUTZEMnSER. R & v m BREEMEN. C- (1983). Mccùankms of calcium-aatagonist-induccd vasodiiation, A m n n r r Rcv. Phmmacol. Toxicol., 23, 373 - 396.
COUGHLM. S.R. T-KH., VU. D-T, HUNG, & WHEATON, V.I. (1992). Charactcrization of the cIoned platelet thrombin nccptor: issues and oppomnitics. J. C l h Invest., 89,351 -355.
DEBLOIS. D.. DRAPEAU. G.. PEïITCLERC, E & MARCEAU. F. (1992). Syacrgism bctween the contractile effect of epidmnal growth factor and that of des-A$-bradykinin or of a-thrombin in rabbit aortic rings. Br. 3. Pharmacol., 105,939-967.
DOWNES. C.P.. HAWKINS, P.T. & IRVINE. RF. (1986). hositoi 1,3,4,5-tetraiuphosphate and not phosphatidylinositol 3,4bi- phosphate is the probable pttcunor of inositol 1.3.4-trispho-
csphare in agonist-stimulatcd parotid gland, Biochm. J., 258, 50 1 - 506.
FENTON rr. S.W. (1988). Regdation of thrombiu gcncration and function. Semin. Thromb- Hemosr., 14, 234-240.
FURCHGO'IT. RF. & ZAWADZKI. LV. (1980). The obligatory rote of endothelia1 cclls in the rela~ation of anerial srnooth muscle by autylchotine. Nasure, 288,373 -r 376.
GODIN, D.. MARCEAU, F.. BMULE, C., RIOUX, F. & DRAPEAU. G. (1994). Aminopeptidase moddaùon of the pharrnacoiogical responscs to synthetic thrombin antagonists. Ew. 3. Pharmacol., 2S3.225 -DO.
GUILLEMETTE, G., LA%fONTAGNE, S., BOLXAY. G. & MOUILLAC, 0. (1989). Differcntial e£fccts of hepatin on inositol 1,4,5- triphosphate binding, rnetabotism and calcium ~ I e a s c actin'ty in the bovine adrcnal cortex. Mol. Pharmacol., 35, 339 -344.
HIDATA. H.. INAGAKI. M., KAWAMOTO. S. & SASAKI. Y. (1984). Isoquinolinesulfonamr*d~~. novel and potent inhibitors of cydic nucleotide dependent protein kinases and protcin kinase C. Bioehemktry, 23, 5036 - 503 1.
HOLLENBERG, M.D.. ADEBAYO. A. LAAWONU, M.S. & MOORE, GJ. (1993). Role of the amino- and carboxyl-terminai domains of thrombin rcceptor-derived polypeptides in biological activity in vascuIar endothelium and gastric smooth muscle: cvidence for reccptor subtypes. Moi. Pharmacol., 43,921 -930.
HUI, K.Y., JAKUBOWSKI, I.A.. W S S . V.L. & ANGLETON, E.L. (1992). Minimal sequencc requinment of thrombin rcceptor agonist peptide. Biochem. Biophyx Res. Commun., 184,790 -796.
IMOTO, M., UMEZAWA K., ISSHIKI. K.. K W O T O . S.. SAWk T., TAKEUCHI. T. & UMEUWA. H. (1987). Kinetic studies of tyrosine kinase inhibition by crbstau'n. 3. Antibior., 40, 1417- 1473.
KETïNTSR. C. & SHAW. E. (1 979). D-Phc-Pro-ArgCHzCf - A selectivc affinity label for thrombin. Thromb, Res., 14,969-973.
KU. D.D. B ZALESKI. J.K. (1993). Rcccptor mtchanisrn of thrombin- induccd cndothtlium-dependent and tndothelium-independent coronan vascuiar e ffccts in dogs. 1. Cardiovasc. Pharmacol., 22, 609 - 6 16.
LEVESQtlE, L. DRAPEAU. G, GROSE S-H, RIOUX. F. h MARCEAU, F. (t993). VascuIar mode of action of Linin BI ruxpton and development of a cellular mode1 for the investigation of these cecepton. Br- 3, Pharmacol,, 109, 1254 - 1262
LEWIS, AJ. & FURST. D.W. (1987). ETonsreroidoI Anri-hj?umnasory Drugs: Mechanisms d Cfhicul Use, Ntw-York Marcel Dekker-
MARCEAU. F.. PEïKCLERC, E.. DEBLOIS, D., PRADELLES. P. & POCIBULE, P.E. (1991). Human interlcukin-1 indu- a rapid rrlaxtion of the rabbit isofated mesentcric artery. Br. 3- Pha)7~eol., 103, 1367- 1372,
MURAD, F. (1990)- Dmgs used for the trcatment of angina- organic nitrates, cialcium-channtl blockers and &adtcnergic antagonists. In The Pharmacobgicd Busir of ThetapeuticS. cd. Gilman, A.G., Rall, T.W., Nies, A.S. Bt Taylor, P. pp. 764 - 783. New-York: Pergamon Press.
NYSïEDT, S., EMILSSON, K. WAHLESTEDT, C. & SUNDELIEI, 1. (1994). MoIccular cloning of a potcntial protcinase activattd rcceptor. Proc. NatL R e d . Sci. U.S.A., 91,9208 -9212.
TESFAMARIAM, B.. ALLEN. G-T-. NORMANDIN. D. dt ANTONAC- CIO, MJ. (1993). Involvement of the 'tethercd ligand' rcœptor in thrombin-induced endotheiium-mcdiated rdaxations. Am. J. Physiol., 265, H 1 744 - 9.
VOURETSRAVIARI, V., VMJ OBBERGHEN-SCHILLING, E, RAS- MUSSEN, U.B., LïCOCQ, A. & POL'YSSEGUR. J. (1992). Synthetic z-thrombin nceptor peptides activate G protein-couplcd signal- ing pathways but are unable to indue mitogenesis. Mot. Biol. Cell, 3,95 - 102.
W. TX-H., HUNG, D.T., WEiEATM. VJ, & COUGHLM. S.R. (1991). Motecular cloaing of a functional thrombin reçcptor ccwals a novel proteolytic mcchanism of mcptor activation. Celt* aI, LOS7 - 1068.
WEISS, RH. & MADURI. M. (1993). The mitogcaic effect of thrombin in vascular smooth muscle cells is largely due to basic fibroblast growth factor. 3. Biol. Chem.. 268, 5724 - 5727.
WEISS, RH. & NUCClTELLt R (1992). Inhibition of tyrosine phosphorylation prcvents thrombin-induced mitogenesis, but not intracc~lulat frct caIciurn release, in vascular ssnooth muscle ~ 1 1 s . J. Biol. Chem., 267, 5608 -561 3.
YANG. S.G.. LANIYONU. A., SAIFEDDiNE. M.. MOORE, GJ. & HOLLENBERG, M.D. (1992). Actions of thrombin and thrombin reccptor peptide analogues in gasu-ic and aonic smooth muscle: dtve1opment of bioassays for sutu=tw-activity studies. Li& Sci., Sl,f3tS - 1332.
(Received Febtuary 22. 1995 Aceepred Murch 27.1995)

IMAGE EVALUATION TEST TARGET (QA-3)
Copyright © 2022 FDOKUMEN




















