
Defects in articular cartilage metabolism and early arthritis in fibroblast growth factor receptor 3...
36
Defects in articular cartilage metabolism and early arthritis in fibroblast growth factor receptor 3 deficient mice G. Valverde-Franco 1,3 , J.S. Binette 3 , W. Li 1,3 , H. Wang 1,3 , S. Chai 1,3 , F. Laflamme 4 , N. Tran- Khanh 5 , E. Quenneville 5 , T. Meijers 6 , A.R. Poole 2 , J.S. Mort 2 , M. D. Buschmann 5 and J.E. Henderson 1,3 . 1 J.T.N. Wong Laboratories for Mineralized Tissue Research, 2 Joint Diseases Laboratory, Shriner’s Hospital for Children and 3 Centre for Bone and Periodontal Research, McGill University, Montréal, Québec, Canada. Departments of 4 Engineering Physics and 5 Chemical Engineering, École Polytechnique, Montréal, Québec, Canada. 6 TNO Pharma, Utrechtseweg 48, The Netherlands Corresponding Author Janet E. Henderson PhD Associate Professor, Associate Dean Research Faculty of Medicine, McGill University J.T.N. Wong Laboratories for Mineralized Tissue Research Centre for Bone and Periodontal Research 740 Ave Dr Penfield, Room 2200 Montreal H3A 1A4 Tel: 514-398-3523 [email protected] © The Author 2006. Published by Oxford University Press. All rights reserved HMG Advance Access published April 19, 2006 by guest on May 3, 2014 http://hmg.oxfordjournals.org/ Downloaded from
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Defects in articular cartilage metabolism and early arthritis in fibroblast growth factor receptor 3...

Defects in articular cartilage metabolism and early arthritis in
fibroblast growth factor receptor 3 deficient mice
G. Valverde-Franco 1,3, J.S. Binette 3, W. Li 1,3, H. Wang 1,3, S. Chai 1,3, F. Laflamme 4, N. Tran-
Khanh 5, E. Quenneville 5, T. Meijers 6, A.R. Poole 2 , J.S. Mort 2, M. D. Buschmann 5 and
J.E. Henderson 1,3.
1 J.T.N. Wong Laboratories for Mineralized Tissue Research, 2 Joint Diseases Laboratory,
Shriner’s Hospital for Children and 3 Centre for Bone and Periodontal Research, McGill
University, Montréal, Québec, Canada. Departments of 4 Engineering Physics and 5 Chemical
Engineering, École Polytechnique, Montréal, Québec, Canada. 6 TNO Pharma, Utrechtseweg 48,
The Netherlands
Corresponding Author
Janet E. Henderson PhD
Associate Professor, Associate Dean Research
Faculty of Medicine, McGill University
J.T.N. Wong Laboratories for Mineralized Tissue Research
Centre for Bone and Periodontal Research
740 Ave Dr Penfield, Room 2200
Montreal H3A 1A4
Tel: 514-398-3523
© The Author 2006. Published by Oxford University Press. All rights reserved
HMG Advance Access published April 19, 2006 by guest on M
ay 3, 2014http://hm
g.oxfordjournals.org/D
ownloaded from

ABSTRACT
Fibroblast growth factor (FGF) receptor 3 has been identified as a key regulator of endochondral
bone development and of post natal bone metabolism through its action on growth plate
chondrocytes and osteoblasts respectively. It has also been shown to promote chondrogenesis
and cartilage production by cultured pre-chondrogenic cells in response to FGF18. In the current
studies we show that the absence of signalling through Fgfr3 in the joints of Fgfr3-/- mice leads to
premature cartilage degeneration and early arthritis. Degenerative changes in cartilage matrix
included excessive proteolysis of aggrecan core protein and type II collagen, as measured by
neoepitope immunoreactivity. These changes were accompanied by increased expression of
MMP13, type X collagen, cellular hypertrophy and loss of proteoglycan at the articular surface.
Using a novel micro-mechanical indentation protocol, it was shown that articular cartilage in the
humeral head of four month old Fgfr3-/- mice was less resistant to compressive force and less
stiff than that of littermate controls. These results identify Fgfr3 signaling as a potential target for
intervention in degenerative disorders of cartilage metabolism. by guest on May 3, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

INTRODUCTION
Hyaline cartilage is found in the developing growth plates and covering the articulating
surfaces of long bones in the adult skeleton (1,2). Growth plate cartilage becomes mineralised
and is partially resorbed to form a template on which trabecular bone of the primary spongiosa is
laid down during endochondral bone development (3). Articular cartilage remains un-
mineralised to form a resilient, low friction surface that acts to absorb the shock caused by high
impact mechanical loading. It is composed primarily of an avascular type II collagen and
proteoglycan (aggrecan) matrix that is secreted and maintained by relatively few chondrocytes in
the adult. Articular cartilage therefore has a very limited capacity for repair. Focal lesions caused
by trauma, infection or biochemical imbalance do not heal adequately and tend to progress
rapidly to osteoarthritis (4,5). This is accompanied by increased proteolysis of proteoglycan and
collagen leading to cartilage matrix degradation. Many lines of investigation are thus focused on
the development of improved technology for cartilage tissue engineering and assisted repair.
Assisted repair of focal defects in articular cartilage may be accomplished by ex-vivo
expansion of chondroprogenitor cells and subsequent transplantation in a scaffold with a source
of growth factors to promote differentiation and cartilage production (6-8). Growth factors that
have been used with some degree of success include bone morphogenic proteins (BMPs) and
transforming growth factor β (TGF β) (9-12), platelet-derived growth factor (PDGF) (13) and
fibroblast growth factors (FGFs) (14-17). In chondrocytes, FGFs signal through three of four
related receptors that undergo alternative splicing and are linked to the MEKK, phospholipase C
and STAT signaling pathways (18). Targeted mutations of the gene encoding FGF receptor
three (fgfr3) in mice resulted in developmental defects in endochondral bone that mimicked
those seen in humans carrying the equivalent activating point mutations (19-22). Mice deficient
by guest on May 3, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

in Fgfr3 signaling are phenotypically normal at birth but develop severe kyphosis, osteomalacia
and osteopenia by four months of age (23,24). Additional in vivo and in vitro work has shown
that these effects on growth plate cartilage and endochondral bone are mediated primarily
through FGF2 and FGF18 interactions with Fgfr3 (25-27). Furthermore, mRNA encoding
FGF18, FGFR2 and FGFR3 have been identified in articular cartilage explants removed from the
talus joints of adult human donors (28)
Previous work has therefore identified Fgfr3 signaling as a key regulator of chondrocyte
function in the developing growth plates and of osteoblast function in post natal bone. FGF2 has
also been identified as an important mitogen for articular chondrocytes ex vivo. The current
work was focused on defining the role of Fgfr3 signaling in post natal articular cartilage. The
shoulder and knee joints of Fgfr3+/+ and Fgfr3-/- mice were examined at four and nine months of
age for radiologic, histologic and molecular evidence of cartilage degradation. Compared with
their wild type littermates, Fgfr3-/- mice demonstrated narrowing of the humeral joint space and a
reduction in subchondral bone volume as early as four months of age. These changes were
accompanied by increased expression of type X collagen and increased cleavage of aggregan and
type II collagen, as evidenced by immunochemical staining with neo-epitope antibodies. These
results demonstrate an important role for FGFR3 in the maintenance of articular cartilage
integrity and identify the FGF-FGFR3 signaling pathway as a potential target for early
intervention in degenerative cartilage disease.
by guest on May 3, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

RESULTS
In previous work, signaling through Fgfr3 in chondrocytes was shown to play an important role
in the development and maintenance of growth plate cartilage. An extensive literature documents
show similarities in the regulation of growth plate and articular chondrocyte biology. We
therefore hypothesized that the congenital absence of Fgfr3 signaling would lead to altered
cartilage development and metabolism of cartilage in the joints of Fgfr3-/- mice.
Radiologic evidence of joint degeneration in FGFR3-/- mice
X-ray analysis of the knee (A, B, E, F left panel), shoulder (A, B, E, F right panel) and paws (C,
D, G, H) of Fgfr3+/+ and Fgfr3-/- mice at 4 months (A-D) and 12 months (E-H) showed
progressive joint deterioration (arrows), narrowing of the joint space (arrowheads) and soft tissue
swelling (asterisk) in Fgfr3-/- compared with Fgfr3+/+ mice (Fig 1). The micro-architecture of the
shoulder joints was assessed and quantified using micro CT. Three-dimensional rendering and
reconstruction of the scans from 4mo (Fig 2A-B) and 9mo (Fig 2C-D) mice showed progressive
collapse of the humeral neck, roughened bone surfaces, numerous osteophytes protruding into
the joint cavity and abnormal joint articulation in Fgfr3-/- mice compared with FGFR3+/+ mice.
These changes were accompanied by a narrowing of the joint space (Fig 2E) and a reduction in
sub-chondral bone volume in the Fgfr3-/- mice (Fig 2F).
Quantifiable, destructive changes in the joints of mice lacking FGFR3 signaling were
therefore evident as early as 4mo of age. To characterize these changes at the molecular level we
performed histological analyses using molecular markers of cartilage matrix degradation.
Premature cartilage degradation and type X collagen in the knees of FGFR3-/- mice
by guest on May 3, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

The knee joints of 4mo old mice (Fig 3A-D) stained with Safranin O (Fig 3A-B) appeared
similar except for an apparent increase in cell number and large lacunae and faint staining for
proteoglycan in the underlying cartilage layer in Fgfr3-/- compared with FGFR3+/+ mice. Faint
type X collagen immunoreactivity (Fig 3C-D), which is associated with chondrocyte
hypertrophy, was also apparent at the articular surface in Fgfr3-/- but not in the Fgfr3+/+ mice. By
9mo of age (Fig E-H), weak proteoglycan staining was also seen in the underlying mineralized
cartilage in both Fgfr3+/+ and Fgfr3-/- mice (Fig 3E-F). Fgfr3-/- mice also demonstrated a
significant reduction in proteoglycan at the articular surface, fibrillation and meniscal
hypertrophy, as well as an increase in type X collagen immunoreactivity (Fig 3G-H), Taken
together these changes suggested progression of the articular chondrocytes toward a terminally
differentiated phenotype, as seen in the transition zone of growth plate cartilage. It has been
proposed that these hypertrophic changes require a concomitant increase in the cleavage of
proteoglycan and type II collagen by locally active matrix metalloproteinases, particularly
MMP13 (29).
Increased cleavage of proteoglycan and type II collagen in the joints of Fgfr3-/- mice
The DIPEN antibody was used to distinguish fragments of aggregan generated by specific
cleavage of DIPEN341F342FGVG by MMP. The COL2-3/4Cs antibody was used to recognizes a
unique amino acid sequence neoepitope in the COOH-terminus of the large 3/4 fragment of the
a1(II) chain of type (II) collagen generated by cleavage by collagenase. Fig 4 shows a modest
increase in COL2-3/4Cs staining at 4mo in Fgfr3-/- joints (Fig 4D) and a significant increase in
both DIPEN (Fig 4F) and COL2-3/4Cs (Fig 4H) staining at 9mo compared with littermate
controls (Fig 4C, E, G). The increase in immunostaining in situ for type II collagen cleavage
by guest on May 3, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

products was corroborated by an increase in the circulating level of type II collagen metabolites
in Fgfr3-/- mice compared with age-matched controls (Table 1). In keeping with the reduction in
subchondral bone there was also an increase in circulating C-telopeptide, a breakdown product of
type I collagen, the major collagen present in bone
Increased MMP13 in the joints of Fgfr3-/- mice
Metalloproteinases (MMPs) 13 and 9 are the principal enzymes responsible for degradation of
hyaline cartilage in the growth plates and the articular joints. Using antisera raised against
MMP13 and MMP9 it was shown that MMP13 (Fig 5A-B, E-F) was increased in articular
cartilage of the humeral head at 4 and 9 months in Fgfr3-/- mice (Fig 5B, F) and at 9 months in
Fgfr3+/+ mice (Fig 5E). MMP9 immunoreactivity was not apparent in articular cartilage of either
genetic strain at any age tested (Fig 5C-D, G-H). In contrast, MMP was detected in bone marrow
cells, in trabecular bone and in growth plate chondrocytes (data not shown) in all mice at all
ages.
Altered biomechanical properties of articular cartilage in Fgfr3-/- mice
Cartilage stiffness and integrity are largely dependent on the presence of an intact cross-linked
network of collagen entrapping the highly charged proteoglycan, aggrecan, which in turn
effectively retains interstitial water. Biomechanical properties of cartilage are most accurately
assessed using isolated cylindrical disks (30). However, the small size of the mouse joint
precludes such an approach. We therefore developed a micro-indentation test that can be applied
to intact small joints, similar to a previously published method (31). The mean thickness of
articular cartilage at the head of the humerus, as shown in Fig 6, was 24.75 ± 6.93 in Fgfr3+/+
by guest on May 3, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

mice and 38.61 ± 7.65 microns in Fgfr3-/- mice (p <0.02). We then tested the resilience and
stiffness of articular cartilage on freshly dissected left humeral heads from 4-month old Fgfr3-/-
and Fgfr3+/+ mice. Although there was no detectable increase in aggregan cleavage at 4mo in
Fgfr3-/- mice, the loss of intact type II collagen resulted in a significant decrease in both cartilage
resilience (Fig 7A) and stiffness (Fig 7B). Similar, but more variable, changes were seen when
the right humerus was substituted for the left (data not shown).
by guest on May 3, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

DISCUSSION
Generalized destruction of articular cartilage, such as that seen in osteoarthritis, is a common
occurrence in people over the age of 60. In many instances it is believed to be a consequence of
prior joint injury that does not heal due to the limited capacity of cartilage for self repair. While
the signaling pathways linked to receptors for FGFs have been identified as potential targets for
intervention in assisted cartilage repair, little is known about their role in chondrogenesis and
cartilage metabolism. In previous work we demonstrated a critical role for Fgfr3 signaling in
cartilage production in vitro. In this study we have found that the absence of signaling through
Fgfr3 in chondrocytes in vivo leads to a similar degeneration of articular cartilage to that seen in
human OA. Significant differences in proteoglycan content, expression of MMP13 and type X
collagen and cleavage of aggrecan and type II collagen were seen as early as 4mo in Fgfr3-/-
mice compared with their littermate controls. This work identifies FGFR3 signaling as a critical
regulator of articular cartilage metabolism and a potential pathway for early intervention in
degenerative cartilage disease.
Reduction in joint space in Fgfr-/- mice
Under physiological circumstances the head of the humerus fits into a socket formed by the outer
edge of the scapula, where it is held in place by the rotator cuff. Micro CT analyses showed
misalignment of the humerus with the scapula, collapse of the humeral neck and an apparent
reduction in the joint space by 4mo of age in FGFR3-/- mice. The reduced space could not be
accounted for by a reduction in the thickness of articular cartilage in the Fgfr3-deficient mice, in
which the layer of uncalcified tissue was significantly thicker at 4mo of age. It did however
by guest on May 3, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

contain more hypertrophic chondrocytes and less matrix than that of the wild type mice, which
would have resulted in less resistance to compression when the joint was straightened for
imaging (see discussion of biomechanical properties). These defects were seen in association
with reduced sub-chondral bone volume and with the appearance of marginal osteophytes, a
typical feature of late OA (32-34). The fact that C-telopeptide was elevated in the serum of
Fgfr3-/- mice argues against focal bone loss due to inflammation, and for generalized bone loss as
a consequence of Fgfr3 deficiency (35-37). The later conjecture is supported by previous work
demonstrating increased MMP13, cortical thinning and osteopenia in 4mo old Fgfr3-/- mice
resulting from an intrinsic osteoblast defect (24).
Expression of type X collagen at the articular surface in Fgfr3-/- mice
The growth plates of long bones contain gradients of chondrocytes from committed chondro-
progenitor cells to fully differentiated hypertrophic cells that undergo apoptosis at the chondro-
osseous junction (3). The transition from immature matrix-producing cells to mature cells that
mineralize their surrounding matrix, is accompanied by a switch from primarily type II to type X
collagen. This transition is tightly regulated by coordinated signaling through growth factor
receptors, including PTH1R and FGFR3 (38-40). Expression of type X collagen in articular
cartilage is usually restricted to the layer of hypertrophic cells below the tidemark (41). Type X
collagen immunoreactivity was apparent in a few cells at the articular surface in 4mo old Fgfr3-/-
mice and greatly increased in 9mo mice deficient in Fgfr3 signaling. These results were
consistent with the decrease in safranin O-positive matrix and the increase in large lacunae at the
articular surface. These results support the hypothesis that Fgfr3 signaling is required to stabilize
the phenotype of articular chondrocytes and prevent progression to terminal differentiation.
by guest on May 3, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

Corroborating evidence comes from previous studies demonstrating Fgfr3 deficiency promoted
type X collagen expression and hypertrophic differentiation of growth plate chondrocytes
(23,40,42).
Increase in aggrecan and type II collagen cleavage in Fgfr3-/- mice
The tensile strength of articular cartilage is conferred by a healthy fibrillar network of type II
collagen that traps aggrecan complexes, which in turn confer stiffness. The initial cleavage of
type II collagen by endogenous collagenase/s generates a ¾ fragment that is recognized by the
site-specific antibody Col2-3/4Cshort (43). Similarly, cleavage of aggrecan by MMP is site-
specific between the G1 and G2 domains to generate a neo-epitope recognised by the VDIPEN
antibody (44). The increase in type II collagen and aggrecan cleavage products in the articular
cartilage of Fgfr3-/- mice resembled that seen in mice over-expressing MMP-13 (45). This
predicted increase in protease activity in the articular cartilage of Fgfr3-/- mice is consistent with
that shown previously in diaphyseal osteoblasts in 4mo old mice (24). FGF2 is a universal ligand
for all FGF receptors (46,47) and accumulates in the synovial fluid of arthritic joints, where it is
believed to stimulate MMP-13 (collagenase 3) production by articular chondrocytes (48,49).
Compensatory signaling by FGF2 interacting with FGFR1 or FGFR2, which are both up-
regulated in the absence of Fgfr3 (24,27), could have resulted in the observed increase in MMP-
13, which would result in increased type II collagen cleavage in the joints of Fgfr3-/- mice. The
fact that circulating levels of the cleavage products of both type II (3/4Cshort) and type I (CTX-1)
collagen were elevated in the circulation of Fgfr3-/- mice argues strongly for a generalized defect
in MMP-13 mediated collagen metabolism. This conjecture is supported by the observed
increase in MMP13 immunoreactivity in cartilage and bone over time. Consistent with the
by guest on May 3, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

published literature, MMP9 expression was apparent in growth plate but not in articular cartilage
(29, 45, 50). The increase in VDIPEN epitope is also consistent with upregulation of MMP-13
activity, although proteoglycan cleavage could also occur as a remodeling and repair response to
the loss of type II collagen integrity.
Impaired biomechanical properties of articular cartilage in Fgfr3-/- mice
The current literature indicates that the biomechanical properties and functional integrity of
articular cartilage deteriorates prior to the appearance of visible signs of surface erosion. Using
an arthroscopic indentation probe it was shown in a cohort of 50 patients with chronic anterior
cruciate ligament deficiency that cartilage stiffness decreased over time after injury and often
preceded the appearance of visible cartilage defects at the time of reconstructive surgery (51). In
other studies using focal indentation probes cartilage stiffness correlated with proteoglycan
content, as measured by gadolinium-enhanced magnetic resonance imaging but not with cartilage
thickness (52,53). Using a micro-mechanical indentation tester we showed a similar reduction in
cartilage stiffness in the shoulders of 4 month old Fgfr3-/- mice, in which the depth of articular
cartilage was in fact greater than that seen in Fgfr3+/+ mice. The loss of stiffness could be
attributed to increased degradation of type II collagen, demonstrated by in situ immunostaining
and ex vivo by Elisa assay of circulating levels of metabolites. Increased degradation of collagen
type II is in turn related to the increase in type X collagen hypertrophic cells seen at the articular
surface, which produce less proteoglycan than non-hypertrophic cells and occupy a larger
volume of the cartilage matrix. Further credence is given to this hypothesis by the observed
increase in type II collagen metabolism, demonstrated by in situ immunostaining and ex vivo by
Elisa assay of circulating levels of metabolites. This decrease in cartilage stiffness in association
by guest on May 3, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

with impaired type II collagen metabolism is reminiscent of that seen in 15 month old Col2a1+/-
mice (31), which synthesize approximately 50% less type II collagen than their wild type
littermates. Interestingly, these mice also exhibited a reduction in sub-chondral bone volume, as
seen in our own Fgfr3-/- mice at 4 months of age. The reduction in sub-chondral bone volume no
doubt reflects the defects in FGFR3-/- osteoblast function described previously (24) and most
probably led to the obvious deformation of the articulating surfaces seen at 9 months of age.
Another potential explanation for the loss of cartilage stiffness is joint mis-alignment, as shown
by X-ray and micro CT imaging, which could lead to reduced mobility. In fact it was shown that
11 weeks of joint immobilization in dogs resulted in a 42% decrease in cartilage stiffness (54).
However, the morphological defects in the skeletons of 4 month old Fgfr3-/- mice do not appear
to inhibit their activity or their access to food. The combination of joint misalignment and
reduced sub-chondral bone volume could however compromise the overall biomechanical
properties of diarthrodial joints and combined with premature cartilage degradation, could lead
to global joint deterioration. by guest on May 3, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

MATERIALS and METHODS
Mice
All mouse procedures were performed in accordance with McGill University guidelines, which
are set by the Canadian Council on Animal Care. Mice homozygous for targeted inactivation of
the gene encoding Fgfr3, and wild type littermates, were obtained from intercross of FGFR+/-
maintained on a C3H background as described (24). Cohorts of mice were anesthetized and
euthanised by exsanguination at two, four, six, nine and 12 months for analyses. Representative
data is shown for 4mo and 9mo old Fgfr3+/+ and Fgfr3-/- mice
Radiologic imaging
High resolution X-rays of the shoulder joints, knees, forepaws and hindpaws were obtained on
euthanised mice or the dissociated limbs using a Faxitron MX20, equipped with an FPX-2
Imaging system (Dalsa Medoptics, Waterloo, Ontario, Canada). Immediately after X-ray the left
shoulder was dissected free of soft tissue, fixed overnight in 4% paraformaldehyde, rinsed three
times with PBS and scanned on a Skyscan1072 microCT instrument (Skyscan, Antwerp,
Belgium). Image acquisition was performed at 100 kV and 98 µA with a 0.9° rotation between
frames to obtain two-dimensional images, which were used to generate three-dimensional
reconstructions. The joint space and subchondral bone volume were calculated with the three-
dimensional Creator software supplied with the instrument.
Histology, histochemistry and immunochemistry
by guest on May 3, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

All histology, histochemistry and immunohistochemistry procedures were performed essentially
as described previously (24,55). The right knee and shoulder joints were dissected free of soft
tissue, fixed for 16h in 4% paraformaldehyde at 4°C and rinsed three times in PBS. Bones were
decalcified in 4.13% EDTA for 14 days at 4°C, embedded in paraffin and 4 m sections cut on a
rotary microtome for staining with safranin-O and fast green to identify proteoglycan. Left
shoulder joints were embedded in LR white acrylic resin (London Resin Co. Ltd., London, UK)
and sections cut on an ultra microtome for staining with 0.2% toluidine blue for 1 minute to
identify mineralized and un-mineralized cartilage. Immunohistochemical analyses were
performed on 4µm sections of paraffin embedded tissue using the following primary antisera:
rabbit anti- C- terminal peptide Val-Asp-Ile-Pro-Glu-Asn of aggrecan G1 domain, (DIPEN
1: 800) (44); rabbit anti-peptide Gly-Pro-Hyp-Gly-Pro-Gln-Gly, which represents a type II
collagen primary cleavage site (COL 2 3/4C short peptide 1:200) (43); mouse anti-type X
collagen peptide Tyr-Asp-Pro-Arg-Thr-Gly-Ile-Phe-Thr (1:300) (56); rabbit anti-MMP9 peptide
(Sigma Saint Louis, MO, USA 1:200) and rabbit anti-MMP13 peptide ovalbumin conjugates
kindly provide by Dr. J.S. Mort (1:300).
Biomechanical testing
The left humeral heads of 4mo old Fgfr3+/+ and Fgfr3-/- mice were used for testing the
biomechanical properties of articular cartilage using a novel indentation test developed on a
Mach-1® micro-mechanical tester (BioSyntech Canada Inc., Laval, Canada). Samples were
dissected free of soft tissue and the distal metaphysis fixed with dental stone in a small plastic
container before dissecting the joint capsule to expose the articular surface. The humeral head
was immersed in PBS for 30 minutes at room temperature to equilibrate before controlled
by guest on May 3, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

compression was applied by an actuator with 0.1 µm displacement precision, which moved a
cylindrical flat tip indentor of 60 µm diameter positioned perpendicular to the articular surface.
Ramp compressions at 5µm/s were applied until a 2g load was reached at two different positions
on each humeral head and the force response captured by a load cell with 7 mg precision. The
point of contact was determined by visual inspection of the loading curve and cartilage stiffness
was assessed by calculating load-displacement slopes in the displacement intervals of 2.5 to 5.5
m (S1), 5.5 to 8.5 m (S2) and 8.5 to 11.5 μm (S3). The results were analyzed by MANOVA,
using the general linear model with Fgfr3+/+ vs Fgfr3-/- as a categorical predictor and
displacement as a continuous predictor.
Biochemical analyses
A commercial ELISA assay was used to determine circulating levels of the type I collagen
degradation product CTX-I (Mouse C-telopeptide, Nordic Bioscience Diagnostics, USA).
Circulating type II collagen cleavage products were measured with the DELFIA® ELISA assay (
Perkin Elmer, Wellesley, Ma. USA) that uses a monoclonal C2C antibody raised against the
carboxyl-terminal 3/4 fragment of the degraded 1 (II) chain. This assay uses a time-resolved
immunofluorometric technique that greatly reduces background fluorescence (57).
by guest on May 3, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

ACKNOWLEDGEMENTS
This work was supported by grants to JEH from the Canadian Institutes of Health Research
(CIHR), the Arthritis Society of Canada (TAS) and the Canadian Arthritis Network Centres of
Excellence, and to MDB from CIHR. GVF was supported by a studentship from the Fonds de la
recherche en santé du Québec (FRSQ) and SC, FL and NT-K by training awards from the CIHR
Strategic Training Program in Skeletal Health Research. JEH is a Chercheur Boursier Senior of
the FRSQ and MDB holds a Canada Research Chair in Cartilage Tissue Engineering. The
authors acknowledge the expert technical assistance of Ailian Li (J.T.N. Wong Laboratories) and
Miren Gratton (Centre for Bone and Periodontal Research).
by guest on May 3, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

REFERENCES
1. Poole, A.R. (1993) Cartilage in health and disease. In McCarty, D.J. and Koopman, W.J.
(eds), Arthritis and allied conditions: a textbook of rheumatology. Lea & Febiger,
Philadelphia, pp. 279-333.
2. Poole, A.R., Laverty S. and Mwale F. (2000) Endochondral bone formation and
development in the axial and appendicular skeleton. In Henderson, J.E. and Goltzman, D.
(eds), The osteoporosis primer. Cambridge University Press, Cambridge UK, pp. 3-17.
3. Kronenberg, H. (2003) Developmental regulation of the growth plate. Nature, 423, 332-
336.
4. Hunziker, E.B. (2002) Articular cartilage repair: basic science and clinical progress. A
review of the current status and prospects. Osteoarthritis Cart., 10, 432-463.
5. Solchaga, L.A., Goldberg V.M. and Caplan A.I. (2001) Experimental models of cartilage
repair: cartilage regeneration using principles of tissue engineering. Clin. Orthop. Rel.
Res., 391S, S161-S170.
6. Korblin, M. and Estrov Z. (2003) Adult stem cells for tissue repair: a new theraputic
concept. N. Engl. J. Med., 349, 570-582.
7. Dowthwaite, G.P., Bishop J.C., Redman S.N., Khan I.M., Rooney P., Evans D.J.R.,
Haughton L., Bayram Z., Boyer S., Thomson B., Wolfe M.S. and Archer C.W. (2003)
The surface of articular cartilage contains a progenitor cell population. J. Cell. Sci., 117,
889-897.
by guest on May 3, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

8. Ochi, M., Adachi N., Nobutu H., Yanada S., Ito Y. and Agung M. (2004) Articular
cartilage repair using tissue engineering technique-novel apporach with minimally
invasive procedure. Artificial Organs, 28, 28-32.
9. Reddi, A.H. (2003) Cartilage morphogenetic proteins: role in joint development,
homoeostasis, and regeneration. Ann. Rheum. Dis., 62 S2, 73-78.
10. Issack, P.S. and DiCesare P.E. (2003) Recent advances toward the clinical application of
bone morphogenetic proteins in bone and cartilage repair. Am. J. Orthop., 32, 429-436.
11. Li, W.J., Tuli R., Okafor C., Derfoul A., Danielson K.G., Hall D.J. and Tuan R.S. (2005)
A three-dimensional nanofibrous scaffold for cartilage tissue engineering using human
mesenchymal stem cells. Biomaterials, 26, 599-609.
12. Sekiya, I., Larson B.L., Vuoristo J.T., Reger R.L. and Prockop D.J. (2005) Comparison
of effect of BMP-2, -4, and -6 on in vitro cartilage formation of human adult stem cells
from bone marrow stroma. Cell. Tiss. Res., 320, 269-276.
13. Barbero, A., Grogan S., Schafer D., Heberer M., Mainil-Varlet P. and Martin I. (2004)
Age related changes in human articular chondrocyte yield, proliferation and post-
expansion chondrogenic capacity. Osteoarthritis Cart., 12, 476-484.
14. Henson, F.M., EA E.A.B. and Davies M.E. (2005) Promotion of the intrinsic damage-
repair response in articular cartilage by fibroblastic growth factor-2. Osteoarthritis Cart.,
13.
15. Madry, H., Emkey G., Zurakowski D. and Trippel S.B. (2004) Overexpression of human
fibroblast growth factor 2 stimulates cell proliferation in an ex vivo model of articular
chondrocyte transplantation. J. Gene Med., 6, 238-245.
by guest on May 3, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

16. Stevens, M.M., Marini R.P., Martin I., R R.L. and V P.S. (2004) FGF-2 enhances TGF-
beta1-induced periosteal chondrogenesis. J. Orth. Res., 22, 1114-1119.
17. Veilleux, N. and Spector M. (2005) Effects of FGF-2 and IGF-1 on adult canine articular
chondrocytes in type II collagen-glycosaminoglycan scaffolds in vitro. Osteoarthritis
Cart., 13, 278-286.
18. Ornitz, D.M. and Marie P.J. (2002) FGF signaling pathways in endochondral and
intramembranous bone development and human genetic disease. Genes Dev., 16, 1446-
1465.
19. Naski, M.C., Colvin J., Coffin J. and Ornitz D.M. (1998) Repression of hedgehog
signalling and BMP4 expression in growth plate cartilage by fibroblast growth factor
receptor 3. Development, 125, 4977-4988.
20. Li, C., Chen L., Iwata T., Kitagawa M., Fu X.-Y. and Deng C.-X. (1999) A Lys644Glu
substitution in fibroblast growth factor receptor 3 (FGFR3) causes dwarfism in mice by
activation of STATs and Ink4 cell cycle inhibitors. Hum. Mol. Genet., 8, 35-44.
21. Bellus, G.A., Bamshad M.J., Przylepa K.A., Dorst J., Lee R.R., Hurko O., Jabs E.W.,
Curry C.J.R., Wilcox W.R., Lachman R.S., Rimoin D.L. and Francomano C.A. (1999)
Severe achondroplasia with developmental delay and acanthosis nigricans (SADDAN).
Am. J. Med. Genet., 85, 53-65.
22. Iwata, T., Li C.-L., Deng C.-X. and Francomano C. (2001) Highly activated FGFR3 with
the K644M mutation causes prolonged survival in severe dwarf mice. Hum. Mol. Genet.,
10, 1255-1264.
by guest on May 3, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

23. Colvin, J.S., Bohne B.A., Harding G.W., McEwen D.G. and Ornitz D.M. (1996) Skeletal
overgrowth and deafness in mice lacking fibroblast growth factor receptor 3. Nature
Genet., 12, 390-397.
24. Valverde-Franco, G., Liu H., Davidson D., Chai S., Carvajal H.V., Goltzman D., Ornitz
D.M. and Henderson J.E. (2004) Defective bone mineralization and osteopenia in young
adult FGFR3-/- mice. Hum. Mol. Genet., 13, 271-284.
25. Liu, Z., Xu J., Colvin J.S. and Ornitz D.M. (2002) Coordination of chondrogenesis and
osteogenesis by fibroblast growth factor 18. Genes Dev., 16, 859-869.
26. Ohbayashi, N., Shibayama M., Kurotaki Y., Imanishi M., Fujimori T., Itoh N. and
Takada S. (2002) FGF18 is required for normal cell proliferation and differentiation
during osteogenesis and chondrogenesis. Genes Dev., 16, 870-879.
27. Davidson, D., Blanc A., Filion D., Wang H., Plut P., Pfeffer G., Buschmann M.D. and
Henderson J.E. (2005) FGF18 signals through FGFR3 to promote chondrogenesis . J.
Biol. Chem., 280, 20509-20515.
28. Ellsworth, J.L., Berry J., Bukowski T., Claus J., Feldhaus A., Holderman S., Holdren
M.S., Lum K.D., Moore E.E., Raymond F., Ren H.P., Shea P., Sprecher C., Storey H.,
Thompson D.L., Waggie K., Yao L., Fernandest R.J., Eyre D.R. and Hughes S.D. (2002)
Fibroblast growth factor-18 is a trophic factor for mature chondrocytes and their
progenitors. Osteoarthritis Cart, 10, 308-320.
29. D'Angelo, M., Yan Z., Nooreyazdan M., Pacifici M., Sarment D.S., Billings P.C. and
Leboy P.S. (2000) MMP-13 is induced during chondrocyte hypertrophy. J. Cell.
Biochem., 77, 678-693.
by guest on May 3, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

30. Fortin, M., Soulhat J., Sirazi-Adl A., Hunziker E.B. and Buschmann M.D. (2000)
Unconfined compression of articular cartilage: nonlinear behaviour and comparison with
a fibril-reinforced biphasic model. J. Biomech. Eng., 122, 189-195.
31. Hyttinen, M.M., Toyras J., Lapvetelainen T., Lindblom J., Prockop D.J., Li S.W., Arita
M., Jurvelin J.S. and Helminen H.J. (2001) Inactivation of one allele of the type II
collagen gene alters the collagen network in murine articular cartilage and makes
cartilage softer. Ann. Rheum. Dis., 60, 262-268.
32. Hunter, D.J., Zhang Y., Niu J., Tu X., Amin S., Goggins J., Lavalley M., Guermazi A.,
Gale D. and Felson D.T. (2005) Structural factors associated with malalignment in knee
osteoarthritis: the Boston osteoarthritis knee study. J. Rheumatol., 32, 2192-2199.
33. Scharstuhl, A., Glansbeek H.L., Beuningen H.M.v., Vitters E.L., Kraan P.M.v.d. and
Berg W.B.v.d. (2002) Inhibition of endogenous TGFb during experimental osteoarthritis
prevents osteophyte formation and impairs cartilage repair. J. Immunol., 169, 507-514.
34. Zhang, Y.W., Su Y., Lanning N., Swiatek P.J., Bronson R.T., Sigler R., Martin R.W. and
vandeWoude G.F. (2005) Targeted disruption of Mig-6 in the mouse genome leads to
early onset degenerative joint disease. Proc. Natl. Acad. Sci. (USA), 102, 11740-11745.
35. Walsh, N.C. and Gravallese E.M. (2004) Bone loss in inflammatory arthritis:
mechanisms and treatment strategies. Curr. Opin. Rheum., 16, 419.27.
36. Lajeunesse, D. and Reboul P. (2003) Subchondral bone in osteoarthritis: a biologic link
with articular cartilage leading to abnormal remodeling. Curr. Opin. Rheum., 15, 628-
633.
37. Garnero, P. and Delmas P.D. (2004) Noninvasive techniques for assessing skeletal
changes in inflammatory arthritis: bone biomarkers. Curr. Opin. Rheum., 16, 428-434.
by guest on May 3, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

38. Vortkamp, A., Lee K., Lanske B., Segre G.V., Kronenberg H.M. and Tabin C.J. (1996)
Regulation of the rate of cartilage differentiation by Indian Hedgehog and PTH-related
protein. Science, 273, 613-622.
39. Kobayashi, T., Chung U., Schipani E., Starbuck M., Karsenty G., Katagiri T., Goad D.,
Lanske B. and Kronenberg H. (2002) PTHrP and indian hedgehog control differentiation
of growth plate chondrocytes at multiple steps. Development, 129, 2977-2986.
40. Amizuka, N., Davidson D., Liu H., Valverde-Franco G., Chai S., Sasaki T., Ozawa H.,
Hammond V., Ornitz D.M., Goltzman D. and Henderson J.E. (2003) Signaling by
fibroblast growth factor receptor 3 (FGFR3) and parathyroid hormone related protein
(PTHrP) coordinate cartilage and bone development. Bone, 34, 13-25.
41. Poole, A.R., Kojima T., Yasuda T., Mwale F., Kobayashi M. and Laverty S. (2001)
Cartilage biology: composition and structure of articular cartilage. Clin. Orthop. Rel.
Res., 391S, S26-S33.
42. Deng, C., Wynshaw-Boris A., Zhou F., Kuo A. and Leder P. (1996) Fibroblast growth
factor receptor 3 is a negative regulator of bone growth. Cell, 84, 911-921.
43. Billinhurst, R.C., Dahlberg L., Ionescu M., Reiner A., Bourne R., Rorabeck C., Mitchell
P., Hambor J., Diekmann O., Tschesche H., Chen J., vanWart H. and Poole A.R. (1997)
Enhanced cleavage of type II collagen by collagenases in osteoarthritic articular cartilage.
J. Clin. Invest., 99, 1534-1545.
44. Hughes, C.E., Caterson B., Fosang A.J., Roughly P.J. and Mort J.S. (1995) Monoclonal
antibodies that specifically recognize neoepitope sequences generated by 'aggrecanase'
and matrix metalloproteinase cleavage of aggrecan: application to catabolism in situ and
in vitro. Biochem. J., 305, 799-804.
by guest on May 3, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

45. Neuhold, L.A., Killar L., Zhao W., Sung M.L.A., Warner L., Kulik J., Turner J., Wu W.,
Billinghurst C., Meijers T., Poole A.R., Babij P. and DeGennaro L.J. (2001) Postnatal
expression in hyaline cartilage of constitutively active human collagenase-3 (MMP-13)
induces osteoarthritis in mice. J. Clin. Invest , 107, 35-44.
46. Ornitz, D.M. and Leder P. (1992) Ligand specificity and heparin dependence of fibroblast
growth factor receptors 1 and 3. J. Biol. Chem., 267, 16305-16311.
47. Shimoaka, T., Ogasawara T., Yonamine A., Chikazu D., Kawano H., Nakamura K., Itoh
N. and Kawaguchi H. (2002) Regulation of osteoblast, chondrocyte and osteoclast
functions by fibroblast growth factor (FGF) 18 in comparison with FGF2 and FGF10. J.
Biol. Chem., 277, 7493-7500.
48. Orito, K., Koshino T. and Saito T. (2003) Fibroblast growth factor 2 in synovial fluid
from an osteoarthritic knee with cartilage regeneration. J. Orth. Sci., 8, 294-300.
49. Wang, X., Manner P.A., Horner A., Shum L., Tuan R.S. and Nuckolls G.H. (2004)
Regulation of MMP-13 expression by RUNX2 and FGF2 in osteoarthritic cartilage.
Osteoarthritis Cart., 12, 963-973.
50. Colnot, C., Thompson X., Miclau T., Werb Z. and Helms J.A. (2003) Altered fracture
repair in the absence of MMP9. Development, 130, 4123-4133.
51. Vasara, A.L., Jurvelin J.S., Peterson L. and Kiviranta I. (2005) Arthroscopic cartilage
indentation and cartilage lesions of anterior cruciate ligament-deficient knees. Am. J.
Sports Med., 33, 408-414.
52. Niederauer, G.G., Niederauer G.M., Cullen L.C., Athanasiou K.A., Thomas J.B. and
Niederauer M.Q. (2004) Correlation of cartilage stiffness to thickness and level of
degeneration using a handheld indentation probe. Ann. Biomed. Eng., 32, 352-359.
by guest on May 3, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

53. Samosky, J.T., Burstein D., Grimson W.E., Howe R., Martin S. and Gray M.L. (2005)
Spatially-localized correlation of dGEMRIC-measured GAG distribution and mechanical
stiffness in the human tibial plateau. J. Orth. Res., 23, 93-101.
54. Jurvelin, J., Kiviranta I., Tammi M. and Helminen J.H. (1986) Softening of canine
articular cartilage after immobilization of the knee joint. Clin. Orth. Rel. Res., 207, 246-
252.
55. Tran-Khan, N., Hoemann C.D., McKee M.D., Henderson J.E. and Buschmann M.D.
(2005) Aged bovine chondrocytes display a diminshed capacity to produce a collagen-
rich, mechanically functional cartilage extracellular matrix. J. Orth. Res., 23, 1354-1362.
56. vanderKraan, P.M., Vitters E.L., Meijers T.H., Poole A.R. and vandenBerg W.B. (1998)
Collagen type I antisense and collagen type IIA messenger RNA is expressed in adult
murine articular cartilage. Osteoarthritis Cart., 6, 417-426.
57. Poole, A.R., Ionescu M., Fitzcharles M.A. and Billinghurst R.C. (2004) The assessment
of cartilage degradation in vivo: development of an immunoassay for the measurement in
body fluids of type II collagen cleaved by collagenases. J. Immunol. Methods, 294, 145-
153.
by guest on May 3, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

FIGURE LEGEND
Fig 1 X-ray analysis of joints at 4 and 12 months in Fgfr3+/+ and Fgfr3-/- mice
High resolution Faxitron x-rays of the knees (A, B, E, F left), shoulder joints (A, B, E, F right)
and paws (C, D, G, H) of Fgfr3+/+ and Fgfr3-/- mice at 4 months (A-D) and 9 months (E-H) of
age. Arrows and arrowheads show progressive joint destruction and asterisks show soft tissue
swelling in the Fgfr3-/- mice . Images captured at x2 magnification and are representative of 8-10
mice of each genotype at each age.
Fig 2 Quantitative microCT analysis of the shoulder joints of Fgfr+/+ and Fgfr-/- mice
Representative three dimensional reconstructions (A-D left hand panels) and mid-sagital two
dimensional sections (A-D right hand panels) of the shoulder joints of Fgfr3+/+ and Fgfr3-/- mice
at 4 months (A, B) and 9 months (C, D) of age were captured on a Skyscan®1072 instrument.
Significant deformation and mis-alignment of the joint was seen at 4 months and 9 months in
Fgfr3-/- mice, in association with a reduction in the joint space (F) and significant thinning of the
sub-chondral bone plate (G). Fgfr3+/+ (white bars) and Fgfr3-/- (black bars). Results in panels E
and F are expressed as the means ±SD of 3-5 measurements made on 6-8 mice of each genotype
at each age. * Significantly different from Fgfr3+/+ * p< 0.01
Fig 3 Histochemical and immunochemical staining for proteoglycan and type X collagen
The knee joints of Fgfr+/+ and Fgfr3-/- mice at 4 months (A-D) and 9 months (E-H) of age were
decalcified, embedded in paraffin and stained with Safranin O to identify proteoglycan (A, B, E,
F) or immunostained to identify type X collagen (C, D, G, H). Articular cartilage in the distal
femur and proximal tibia in Fgfr3-/- mice showed an increase in cell number and large lacunae at
by guest on May 3, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

4 months (B) and a reduction in cell number and proteoglycan content at 9 months (F). Type X
collagen immunoreactivity was seen in the articular cartilage of Fgfr3-/- mice at 4 and 9 months
(D, H) but not in that of Fgfr3+/+ mice (C, G). Results are representative of those obtained from 5
mice of each genotype at each age. Magnification at source x20.
Fig 4 Immunochemical staining for neoepitopes of aggrecan and type II collagen
The knee joints of Fgfr3+/+ and Fgfr3-/- mice at 4 months (A-D) and 9 months (E-H)) of age were
decalcified, embedded in paraffin and stained with DIPEN, to identify aggrecan cleaved by
MMPs (A, B, E, F) or with an antibody that recognizes the ¾ C terminal fragment of type II
collagen cleaved by MMP13 (C, D, G, H). In Fgfr3-/- mice, aggrecan cleavage was increased at 9
months (F) compared with Fgfr3+/+ (E) mice. Type II collagen cleavage was significantly
increased at 4 months (D) and 9 months (H) compared with wild type control (C and G
respectively). Results are representative of those from 5 mice of each genotype at each age.
Magnification at source x20
Fig 5 Immunochemical staining for MMP13 and MMP9 in Fgfr3+/+ and Fgfr3-/- mice
The humerus of Fgfr+/+ and Fgfr3-/- mice at 4 months (A-D) and 9 months (E-H) of age were
decalcified, embedded in paraffin and immunostained with antisera that recognises MMP13 (A-
B, E-F) or MMP9 (C-D, G-H). Whereas MMP13 expression increased over time in both genetic
strains the staining was far more intense in Fgfr3-/- mice. Little difference was seen in MMP9
expression between Fgfr+/+ and Fgfr3-/.
Fig 6 Identification of un-calcified cartilage in the humerus of Fgfr3+/+ and Fgfr3-/- mice
by guest on May 3, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

Specimens were prepared in the same manner as for Fig 5 and stained with Safranin O (A-B, E-
F) or Toluidine blue (C-D, G-H) to measure the depth of un-mineralized cartilage at the articular
surface. While the depth of unmineralized cartilage appeared greater in Fgfr3-/- mice compared
with Fgfr+/+, there was significantly less subchondral bone in the mutant mice at both ages.
Fig 7 Biomechanical properties of articular cartilage in Fgfr3+/+ and Fgfr3-/- mice
The freshly dissected heads of humeri from 4 month old Fgfr3+/+ and Fgfr3-/- mice were
subjected to loading by a flat tip indentor applied by an actuator moving at 5 m/s with a 0.1 µm
displacement precision (Mach-1® micro-mechanical tester). The load was measured
continuously and is shown at displacements of 4 m, 7 m and 10 m (A), while cartilage
stiffness was approximated as slopes of load vs displacement in the displacement intervals of 2.5
to 5.5 m (S1), 5.5 to 8.5 m (S2) and 8.5 to 11.5 μm (S3) (B). The response to loading (A) and
resulting stiffness (B) were significantly decreased in the Fgfr3-/- mice (hatched line) compared
with Fgfr3+/+ mice (solid line), as determined by MANOVA with Fgfr+/+ / Fgfr3-/- and
displacement as the predictors. The results are representative of 25 measurements made on n=13
FGFR3+/+ mice and 24 measurements on n=15 FGFR3-/- mice.
by guest on May 3, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from
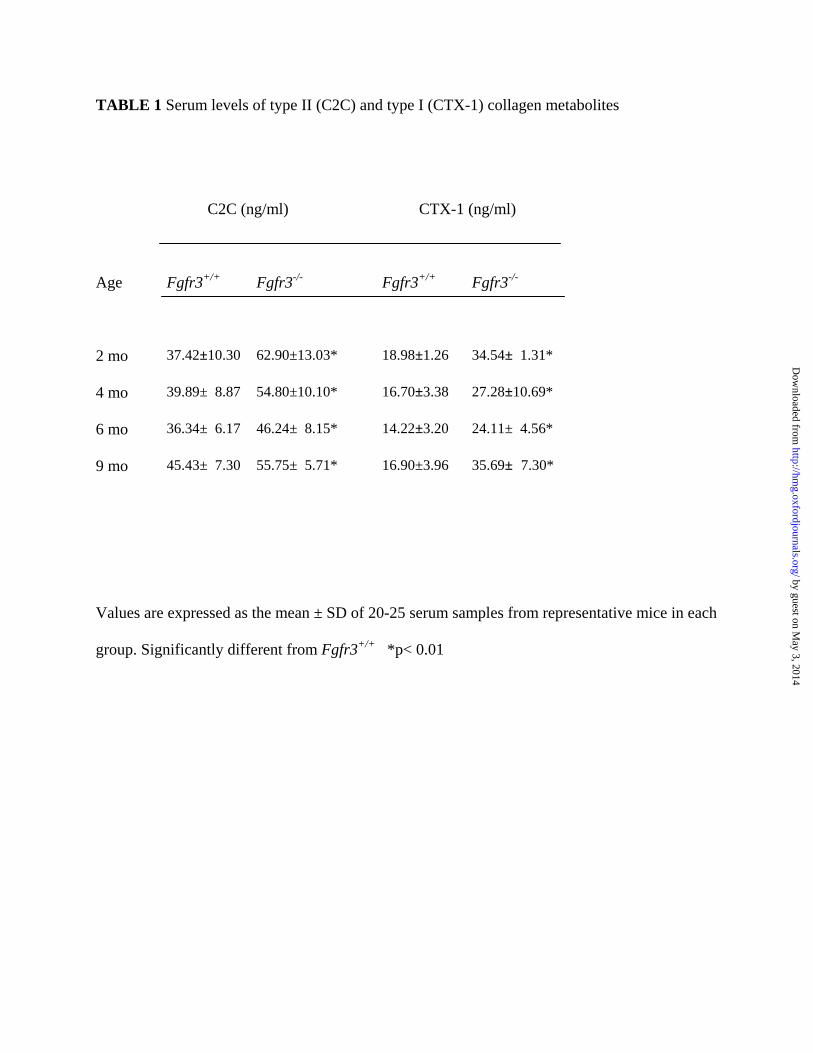
TABLE 1 Serum levels of type II (C2C) and type I (CTX-1) collagen metabolites
C2C (ng/ml) CTX-1 (ng/ml)
Age
Fgfr3+/+ Fgfr3-/- Fgfr3+/+ Fgfr3-/-
2 mo 37.42±10.30 62.90±13.03* 18.98±1.26 34.54± 1.31*
4 mo 39.89± 8.87 54.80±10.10* 16.70±3.38 27.28±10.69*
6 mo 36.34± 6.17 46.24± 8.15* 14.22±3.20 24.11± 4.56*
9 mo 45.43± 7.30 55.75± 5.71* 16.90±3.96 35.69± 7.30*
Values are expressed as the mean ± SD of 20-25 serum samples from representative mice in each
group. Significantly different from Fgfr3+/+ *p< 0.01
by guest on May 3, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

by guest on May 3, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

by guest on May 3, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

by guest on May 3, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

by guest on May 3, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

by guest on May 3, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

by guest on May 3, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from

by guest on May 3, 2014
http://hmg.oxfordjournals.org/
Dow
nloaded from




















