
Comparing inhibitory effect of tramadol on catalase of Pseudomonas aeruginosa and mouse liver

doi:10.1006/jmbi.2000.4094 available online at http://www.idealibrary.com on J. Mol. Biol. (2000) 302, 1193±1212
Crystal Structure of Mycobacterium tuberculosis6-Hydroxymethyl-7,8-dihydropteroate Synthase inComplex with Pterin Monophosphate: New Insight intothe Enzymatic Mechanism and Sulfa-drug Action
Arthur M. Baca1, Rachada Sirawaraporn2, Stewart Turley1,3,4,Worachart Sirawaraporn2 and Wim G. J. Hol1,3,4,5*
1Department of Bioengineeringand Biomolecular StructureCenter, University ofWashington, SeattleWA 98195, USA2Department of BiochemistryFaculty of Science, MahidolUniversity, Rama VI RoadBangkok, 10400, Thailand3Department of BiologicalStructure, University ofWashington, SeattleWA 98195, USA4Howard Hughes MedicalInstitute, University ofWashington, SeattleWA 98195, USA5Department of BiochemistryUniversity of WashingtonSeattle, WA 98195, USA
E-mail address of the [email protected]
Abbreviations used: MDR, multi-DHPS, 6-hydroxymethyl-7,8-dihydrpABA, para-aminobenzoic acid; PtP6-hydroxymethylpterin monophosp6-hydroxymethylpterin pyrophosph6-hydroxymethyl-7,8-dihydropterinEscherichia coli; Mtb, MycobacteriumStaphylococcus aureus; TB, tuberculo
0022-2836/00/051193±20 $35.00/0
The enzyme 6-hydroxymethyl-7,8-dihydropteroate synthase (DHPS) cata-lyzes the condensation of para-aminobenzoic acid (pABA) with 6-hydro-xymethyl-7,8-dihydropterin-pyrophosphate to form 6-hydroxymethyl-7,8-dihydropteroate and pyrophosphate. DHPS is essential for the de novosynthesis of folate in prokaryotes, lower eukaryotes, and in plants, but isabsent in mammals. Inhibition of this enzyme's activity by sulfonamideand sulfone drugs depletes the folate pool, resulting in growth inhibitionand cell death. Here, we report the 1.7 AÊ resolution crystal structure ofthe binary complex of 6-hydroxymethylpterin monophosphate (PtP) withDHPS from Mycobacterium tuberculosis (Mtb), a pathogen responsible forthe death of millions of human beings each year. Comparison to otherDHPS structures reveals that the M. tuberculosis DHPS structure is in aunique conformation in which loop 1 closes over the active site. The MtbDHPS structure hints at a mechanism in which both loops 1 and 2 playimportant roles in catalysis by shielding the active site from bulk solventand allowing pyrophosphoryl transfer to occur. A binding mode forpABA, sulfonamides and sulfones is suggested based on: (i) the new con-formation of the closed loop 1; (ii) the distribution of dapsone and sulfo-namide resistance mutations; (iii) the observed direction of the bondbetween the 6-methyl carbon atom and the bridging oxygen atom to thea-phosphate group in the Mtb DHPS:PtP binary complex; and (iv) theconformation of loop 2 in the Escherichia coli DHPS structure. Finally, theMtb DHPS structure reveals a highly conserved pterin binding pocketthat may be exploited for the design of novel antimycobacterial agents.
# 2000 Academic Press
Keywords: dihydropteroate synthase; Mycobacterium tuberculosis;sulfonamide; drug design; enzyme catalysis
*Corresponding authoring author:
drug-resistant;opteroate synthase;,hate; PtPP,ate; H2PtPP,pyrophosphate; Ec,tuberculosis; Sa,sis.
Introduction
Mycobacterium tuberculosis (Mtb), the causativeagent of human tuberculosis (TB), is responsiblefor the annual death of millions of human beingsand claims more lives than any other singleinfectious agent (Nakajima, 1993; Dolin et al.,1994; Snider et al., 1994; Rouhi, 1999). Almost one-third of the world's population is infected withM. tuberculosis and 10 % of these infected individ-uals will have active disease at some time in theirlife (Nunn & Kochi, 1993). The current treatmentprotocol for active TB includes at least six monthsof therapy with the ®rst line drugs of isoniazid,rifampin, pyrazinamide, and ethambutol (Bloom &
# 2000 Academic Press

1194 M. tuberculosis DHPS
Murrary, 1992; Snider & Roper, 1992; Bass et al.,1994). Multi-drug-resistant (MDR) tuberculosis hasrecently emerged, mainly because of the failure tocomplete the patient's treatment protocol. Becauseof the growing number of cases of MDR tuberculo-sis, the World Health Organization has declaredTB a global public health emergency (Nakajima,1993). It is clear that new antimycobacterial agentsneed to be developed.
Here, we have investigated the enzyme 6-hydro-xymethyl-7,8-dihydropteroate synthase (DHPS, EC2.5.1.15) from M. tuberculosis in order to explorethe possibilities this protein gives for the design ofanti-TB drugs. Diaminodiphenylsulfone or dapsone(Figure 1), a sulfone inhibitor of DHPS, has beenclinically used for decades to treat leprosy, whichis caused by the related organism Mycobacteriumleprae (Anand, 1996), and sulfamethoxazole(Figure 1) has been shown to have in vitro effectsagainst Mycobacterium avium and Mycobacteriumintracellulare (Raszka et al., 1994). M. tuberculosisDHPS is very similar to M. leprae DHPS (79 %sequence identity) and Mycobacterium avium DHPS(81 % sequence identity) (preliminary sequencedata from The Institute for Genomic Research). Thefact that sulfonamides and sulfones have beenused for clinical treatment of mycobacterial infec-tions underscores the observation that DHPS is apromising target for antimycobacterial drugs.
DHPS is essential for the de novo synthesis offolate in prokaryotes, in lower eukaryotes such asprotozoa and yeast, and in plants. DHPS is absentin mammals. This enzyme catalyzes the conden-sation of para-aminobenzoic acid (pABA) with6-hydroxymethyl-7,8-dihydropterin-pyrophosphate(H2PtPP) to form pyrophosphate and 6-hydroxy-methyl-7,8-dihydropteroate (Richey & Brown,1969; Shiota et al., 1969) (Figure 1). Dihydroptero-
ate is subsequently converted to the cofactor folatethrough the action of dihydrofolate synthase.
Because humans lack, while microorganismspossess DHPS, this enzyme has been longexploited as a selective drug target. Sulfa drugs,including sulfonamides and sulfones, were the ®rstsynthetic antimicrobial agents, being clinicallyused as early as 1933 and are still in use today(Wingard, 1991; Anand, 1996). As shown inFigure 1, sulfa drugs are structural analogues ofpABA, act as competitive antagonists of DHPS,and are effective inhibitors of folate biosynthesis(Woods, 1940; Brown, 1962; Shiota et al., 1964;Bock et al., 1974). Additionally, sulfa drugs can actas alternative substrates of DHPS, resulting in theformation of sulfa-pteroates that cannot be furtherconverted to folate (Roland et al., 1979; Swedberget al., 1979).
The sequences of several DHPS genes have beenreported from a variety of organisms. (LoÂpez et al.,1987; Kristiansen et al., 1990; Slock et al., 1990;Dallas et al., 1992; Volpe et al., 1992; Brooks et al.,1994; Triglia & Cowman, 1994; Kellam et al., 1995;Hampele et al., 1997; Pashley et al., 1997; ReÂbeilleÂet al., 1997; Swedberg et al., 1998; Gibreel & SkoÈ ld,1999; Nopponpunth et al., 1999). While DHPS is amonofunctional polypeptide in prokaryotes includ-ing Mycobacterial species (LoÂpez et al., 1987;Kristiansen et al., 1990; Slock et al., 1990; Dallaset al., 1992; Kellam et al., 1995; Hampele et al., 1997;Swedberg et al., 1998; Gibreel & SkoÈ ld, 1999;Nopponpunth et al., 1999), DHPS from plants andapicomplexan parasites is part of a bi-functionalenzyme (Brooks et al., 1994; Triglia & Cowman,1994; Pashley et al., 1997; ReÂbeille et al., 1997)with the preceding enzyme in the pathway6-hydroxymethyl-7,8-dihydropterin pyrophos-phokinase. DHPS in yeast is even part of a
Figure 1. Reaction and inhibitorsof DHPS. (a) Reaction catalyzed byDHPS; (b) oxidized substrate ana-logues used in crystallizationscreens with Mtb DHPS; (c) chemi-cal formulas of sulfa drugs,competitive antagonists of pABA;(d) two sulfa drugs used in thetreatment of Mycobacterial infec-tions.

Table 1. Crystallographic data and re®nement statistics
A. Data collection statisticsSpace group P3221Unit cell dimensions (AÊ ) a � b � 63.1, c � 121.8Asymmetric unit Monomer, 29 kDaVm (AÊ 3/Da) 2.4Resolution limit (AÊ ) 1.7Observations 136,341Unique reflections 25,211Completeness overall
(outermost shell)0.805 (0.699)
Redundancy (outermost shell) 5.4 (4.1)Rmerge overall (outermost shell) 0.043 (0.177)
B. Refinement statisticsResolution range (AÊ ) 20.0-1.7R (%) 18.5Rfree (%) (5 % of data) 24.3Number of protein atoms 1848Number of PtP atoms 18Number of water molecules 244Average B of protein atoms (AÊ 2) 22.7Average B of PtP (AÊ 2) 16.6Average B of water (AÊ 2) 38.7rmsd from ideal stereochemistry
Bond lengths (AÊ ) 0.010Bond angles (deg.) 2.0Torsion angles (deg.) 15.6
Ramachandran parametersNon-glycine residues in most
favored region203 (94.4 %)
Non-glycine residues inadditionally allowed regiona
12 (5.6 %)
R, Rfree � �j jFoj ÿ jFcj j/� jFoj, where the working and freeR-factors are calculated using the working and free re¯ectionsets, respectively. The free re¯ections were held aside through-out the re®nement.
a As classi®ed by PROCHECK (Laskowski et al., 1993).
M. tuberculosis DHPS 1195
tri-functional enzyme, combining DHPS with thetwo preceding enzymes in the de novo folate bio-synthetic pathway: PPPK and 7,8-dihydroneopterinaldolase (Volpe et al., 1992). DHPS is reported tobe a homodimer in several prokaryotes, includingEscherichia coli, Staphylococcus aureus, Streptococcuspneumoniae, M. tuberculosis, and M. leprae (LoÂpezet al., 1987; Dallas et al., 1992; Hampele et al., 1997;Nopponpunth et al., 1999), whereas eukaryotic bi-functional DHPS is believed to be a dimer or tri-mer (Ferone, 1973; Walter & Konigk, 1980; Pashleyet al., 1997; ReÂbeille et al., 1997; Triglia et al., 1997).
Resistance to sulfonamides and sulfones is wide-spread in many pathogenic organisms, and inmany cases is associated with mutations in theDHPS gene (Huovinen et al., 1995; Vinnicombe &Derrick, 1999a,b). Other mechanisms of resistanceto sulfonamides and sulfones include the increasedproduction of pABA, gene ampli®cation of DHPS,an increased ability of the microorganism to usehost folate, and lastly, reduction of the microorgan-ism's cell permeability to sulfonamides (Anand,1996). Amongst bacteria, the most common modesof resistance appear to be increased production ofpABA and point mutations in DHPS (Anand,1996). With the advent of molecular cloning tech-niques, sequence analysis has revealed that singlemutations in the DHPS gene can confer resistanceto sulfonamides and sulfones. In M. leprae, resist-ance to dapsone is associated with two amino acidchanges in its DHPS (Kai et al., 1999).
Although there is a wealth of data on the phar-macological behavior of sulfa drugs, there areremarkably few biochemical or biophysical studiesthat have investigated the molecular mechanism ofthe immensely important sulfa drug target DHPS(Anand, 1996; Vinnicombe & Derrick, 1999b). Fewenzymatic studies of DHPS (Hampele et al., 1997;ReÂbeille et al., 1997; Vinnicombe & Derrick, 1999a)have appeared in the literature and the crystalstructures of only E. coli and S. aureus DHPS havebeen reported (Achari et al., 1997; Hampele et al.,1997). Here, we report the high-resolution structureof DHPS from M. tuberculosis in binary complexwith a pterin substrate analogue and discuss theimplications for the catalytic mechanism of thisclass of enzymes. We propose a possible bindingmode of pABA, sulfonamides, and sulfones, andrelate this to the distribution of known sulfona-mide/sulfone resistance mutations. Structural anal-ysis of M. tuberculosis DHPS reveals opportunitiesfor the design of novel selective antimycobacterialagents.
Results
The structure of M. tuberculosis DHPS
Quality of the structure
The re®ned 1.7 AÊ resolution structure ofM. tuberculosis (Mtb) DHPS in complex with6-hydroxymethylpterin monophosphate has a ®nal
R-factor of 18.5 % and an Rfree of 24.3 % (Table 1).Electron density is well de®ned for essentially allmain-chain and side-chain atoms of the 280residues in each subunit, except for: (i) the fourN-terminal residues Met1-Ala4; (ii) residuesGlu51-Val64 corresponding to the loop 2 regionconnecting strand b2 with helix a2; and (iii) thesix C-terminal residues Arg274-Gly280. Theprotein structure was evaluated by the programPROCHECK (Laskowski et al., 1993) for the stereo-chemical quality. The Ramachandran plot showsthat 94.4 % of the non-glycine and non-proline resi-dues are in the most favored region, and 5.6 % inthe additional allowed regions (Ramachandranet al., 1966). The overall average B-factor is 22.7 AÊ 2,with average B-factors of 19.5 AÊ 2 for main-chainatoms and 26.6 AÊ 2 for side-chains atoms (Table 1).
Structural characteristics of M. tuberculosis DHPS
Each subunit of Mtb DHPS adopts a ``TIM-barrel'' fold, with eight a-helices surrounding acentral barrel composed of eight parallel b-strandsof approximate dimensions 35 AÊ � 35 AÊ � 45 AÊ .The overall fold of Mtb DHPS is shown in Figure 2,and secondary structure elements are indicated in

Figure 2. Overall fold of the Mtb DHPS dimer. a-Helices are designated a1-a8, b-strands are labeled b1-b8, andconnecting loops 1-8 at the active site pole are indicated. 6-Hydroxymethylpterin monophosphate (PtP) is shown inpale blue. The inset shows the orientation of the barrels of the dimer. This Figure was produced with the programInsightII (Molecular Simulations, Inc.).
1196 M. tuberculosis DHPS
Figure 3. The a-b connections at the N-terminalpole of the a/b barrel are all composed of rela-tively short loops, no longer than seven amino acidresidues. In contrast, loops 1-8 at the C-terminalpole where the active site is located form moreextended connections and vary in length. Loops 3,4, 6, and 8 are compact, whereas loops 1, 2, 5, and7 form more extended structures consisting of 14,12, 18, and 14 residues, respectively. Loops 1, 2, 5,and 7 are likely to play a critical role during cataly-sis and in sulfa drug action, and will be discussedin greater detail below.
The crystal structure has one subunit per asym-metric unit with a dimer of Mtb DHPS generatedby the action of a crystallographic 2-fold. This is inagreement with gel ®ltration experiments, whichsuggest an apparent molecular mass of 56 kDa,approximately twice the 29 kDa predicted for thepolypeptide (Nopponpunth et al., 1999). Thedimerization interface is formed by subunit-sub-unit contacts mediated by a-helices a6, a7, a70, and
a8, and has a total buried surface area per mono-mer of �1500 AÊ 2 out of a total monomer surfacearea of 11,800 AÊ 2. (See Figure 2). All other subunit-subunit contacts in the crystal are much less exten-sive. Key residues in the dimer interface includeHis194, Trp270 and Ala273. These residues form ahydrophobic patch that composes about 12 % ofthe dimerization interface. These residues are nothighly conserved amongst all DHPS sequences, butare well conserved between M. tuberculosis,M. leprae, M. bovis, and M. avium DHPS sequences.
PtP Binding to M. tuberculosis DHPS
Mtb DHPS crystals could be grown only in thepresence of the oxidized pterin substrate analogueshown in Figure 1(b), 6-hydroxymethylpterinmonophosphate (PtP). PtP is more stable than pter-in pyrophosphate, since the monophosphate formis less prone to hydrolysis. Neither apo protein norDHPS complexed with PtPP resulted in crystals.

Figure 3. Structure-based DHPS sequence alignment. The DHPS sequences of the three bacterial species withreported three-dimensional structures are depicted (E. coli, Achari et al., 1997; S. aureus, Hampele et al., 1997; Mtb, thisstudy). b-Strands are yellow, loops blue, and a-helices green. Residues that were not observed in the DHPS structuresare indicated with lower-case letters. The program DSSP (Kabsch & Sander, 1983) was used for the assignment of sec-ondary structure elements. Residues absolutely conserved in 24 available DHPS sequences are indicated in red, resi-dues that are highly conserved in these DHPS sequences are indicated in pink. Blue dots indicate residues observedto interact with the pterin moiety of PtP, PtPP, or H2PtPP. Purple dots indicate residues interacting with the phos-phate or pyrophosphate moiety of PtP, PtPP, or H2PtPP. The pound symbols below Mtb Ser53 and Pro55 indicateresidues associated with dapsone resistance in the homologous M. leprae DHPS sequence (Kai et al., 1999), which is79 % identical with the Mtb DHPS sequence. This Figure was produced with the program IRIS Showcase (SiliconGraphics, Inc.).
M. tuberculosis DHPS 1197
Although pABA or sulfa drugs were present in thecrystallization buffer at a concentration of 3 mMalong with PtP, only PtP was observed in the MtbDHPS electron density map (Figure 4).
The phosphate moiety of PtP adopts a confor-mation in which O10, which bridges the phos-phorus atom with C9 attached to the pyrazinering, lies out of the ring plane with the C9-O10bond roughly perpendicular to the plane of thepterin moiety (Figure 4(a)), which is different from
the observed conformations of the a-phosphategroup in the E. coli DHPS:H2PtPP and S. aureusDHPS:PtPP binary complexes (Achari et al., 1997;Hampele et al., 1997), in which O10 and thea-phosphorus atom lie in the plane of the pterinring system (Figures 4(a) and 5). The signi®canceof this observation will be further discussed below.The value of the dihedral angle formed by N5, C6,C9 and O10 is 103.3 �. O10 hydrogen bonds toNH1 of the guanidinium group of Arg253. The
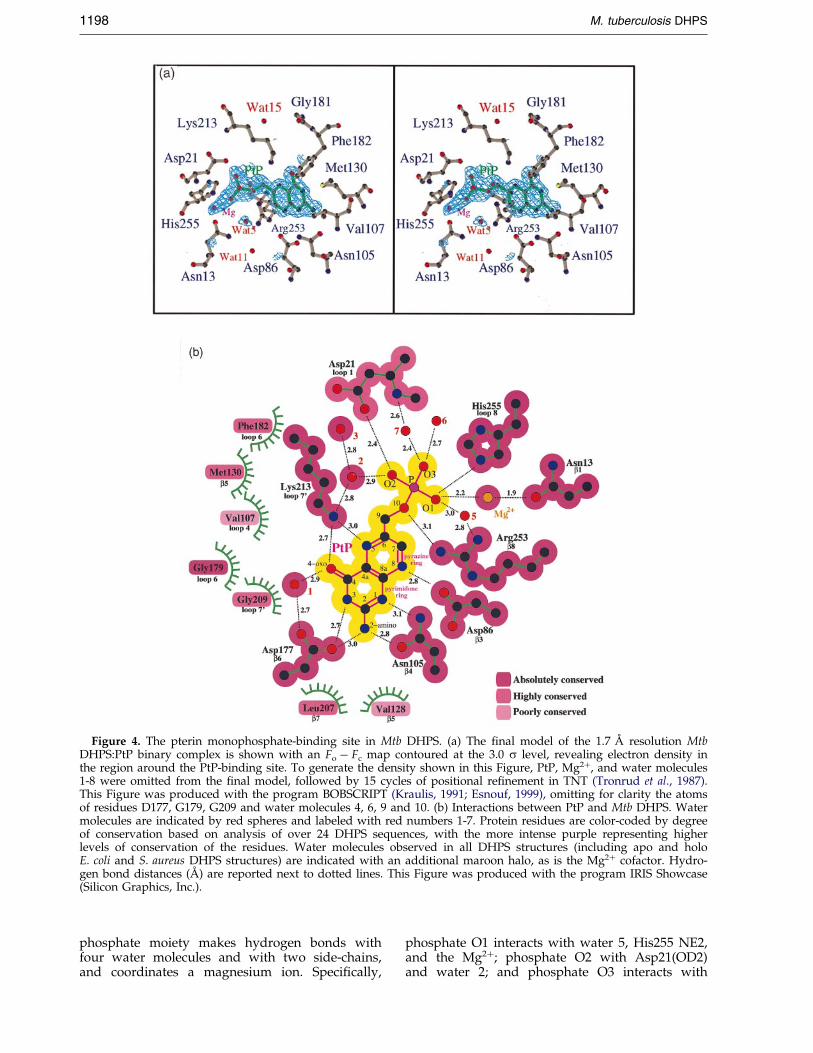
Figure 4. The pterin monophosphate-binding site in Mtb DHPS. (a) The ®nal model of the 1.7 AÊ resolution MtbDHPS:PtP binary complex is shown with an Fo ÿ Fc map contoured at the 3.0 s level, revealing electron density inthe region around the PtP-binding site. To generate the density shown in this Figure, PtP, Mg2�, and water molecules1-8 were omitted from the ®nal model, followed by 15 cycles of positional re®nement in TNT (Tronrud et al., 1987).This Figure was produced with the program BOBSCRIPT (Kraulis, 1991; Esnouf, 1999), omitting for clarity the atomsof residues D177, G179, G209 and water molecules 4, 6, 9 and 10. (b) Interactions between PtP and Mtb DHPS. Watermolecules are indicated by red spheres and labeled with red numbers 1-7. Protein residues are color-coded by degreeof conservation based on analysis of over 24 DHPS sequences, with the more intense purple representing higherlevels of conservation of the residues. Water molecules observed in all DHPS structures (including apo and holoE. coli and S. aureus DHPS structures) are indicated with an additional maroon halo, as is the Mg2� cofactor. Hydro-gen bond distances (AÊ ) are reported next to dotted lines. This Figure was produced with the program IRIS Showcase(Silicon Graphics, Inc.).
1198 M. tuberculosis DHPS
phosphate moiety makes hydrogen bonds withfour water molecules and with two side-chains,and coordinates a magnesium ion. Speci®cally,
phosphate O1 interacts with water 5, His255 NE2,and the Mg2�; phosphate O2 with Asp21(OD2)and water 2; and phosphate O3 interacts with

Figure 5. Geometry of the phosphate moiety in PtP. O10 bridges the pterin moiety with the phosphate or pyropho-sphate moieties of PtP and H2PtPP/PtPP, respectively. The PtP phosphate is not in the plane of the pterin ring sys-tem, whereas the a-phosphate moiety of H2PtPP and PtPP is in the plane of the pterin molecule. Mtb Arg253(corresponding to Ec Arg255 and Sa Arg239) is absolutely conserved in all DHPS sequences. Through its guanidiniumgroup, this arginine residue mediates hydrophobic interactions with one face of the pterin ring system by p-p stack-ing. Mtb Phe182 (corresponding to Ec Phe190 and Sa Phe172) and Mtb Met130 (corresponding to Ec Met 139 and SaMet128) are both highly conserved and mediate hydrophobic interactions with the opposite face of the pterin ringalong with Mtb Val107 (corresponding to Ec Ile 117 and Sa Gln105), which is not well conserved in DHPS sequences.
M. tuberculosis DHPS 1199
water molecules 6 and 7. In addition to interactingwith phosphate O1, the magnesium ion interactswith the OD1 of Asn13.
The pterin-binding pocket of DHPS is formed bythe side-chains of 12 residues (Figure 4) and uponbinding, the substrate analogue PtP buries 402 AÊ 2
out of a total of 418 AÊ 2 surface area. Each hydro-gen bond donor and acceptor of the pterin ringsystem is engaged in interactions with hydrogenbond donors or acceptors provided by DHPS side-chains. Polar interactions with the pterin moiety ofPtP are made by residues Asp86, Asn105, Asp177,Lys213, and a buried water molecule. The hydro-phobic parts of the pterin-binding pocket are pro-vided by Val107, Val128, Met130, Gly179, Phe182,Leu207, and Gly209 (Figure 4(b)).
Residue Arg253 serves a bifunctional role byproviding both hydrogen bonding and hydro-phobic interactions with PtP. Although this argi-nine residue does not hydrogen bond with anypterin ring atoms, its guanidinium NH2 hydrogenbonds to O10. The p-face of the guanidiniumgroup forms extensive hydrophobic interactionswith one face of the pterin ring system, most nota-bly with C8a, C7, C6 and C2. The possible import-ance of the p-p interactions of Arg253 with thepterin ring for the catalytic mechanism of DHPSwill be further discussed below.
Comparison of the M. tuberculosis, E. coli andS. aureus DHPS structures
Overall fold and different subunit orientations ofthe DHPSs
The overall TIM-barrel fold and dimerizationinterface of M. tuberculosis DHPS is similar to thatof E. coli (Ec) and S. aureus (Sa) DHPSs, which is inagreement with the fact that Mtb shares a 38 %sequence identity with both Ec and Sa DHPS's(Figure 3). The Mtb DHPS dimerization interface issimilar to that observed for the Sa and Ec DHPSdimers (Achari et al., 1997; Hampele et al., 1997). Inall three DHPS structures, the dimer interfaceinvolves helices a6, a7, a70, and a8, and buriesabout �1500 AÊ 2 per monomer. Individual residuescontributing to this dimer interface are not wellconserved amongst the three DHPSs.
Subunit A of the Ec DHPS dimer can be super-imposed onto subunit A of the Mtb DHPS dimerwith an rmsd of 0.78 AÊ for the a-carbon atoms of71 core residues. When subunit B of the Ec DHPSdimer is then compared to subunit B of the MtbDHPS dimer, the rmsd is 7.32 AÊ for the equivalentset of 71 core a-carbon atoms. This 7.32 AÊ r.m.s.d.re¯ects the fact that the two B-subunits are rotated17.9 � relative to each other. Subunit A of the Sa

Figure 6. Active site of three DHPS structures. (a) Residues interacting with the pyrophosphate moiety of H2PtPPin E. coli DHPS (Achari et al., 1997). E. coli DHPS loop 2 residues Thr62 and Arg63 interact with the a and b-phos-phate moieties, respectively, of H2PtPP. The b-phosphate-binding pocket is composed of His257, Asn22, and Ile20.Arg255 interacts with two of the b-phosphate oxygen atoms. (b) Residues interacting with the pyrophosphate moietyof PtPP in S. aureus DHPS (Hampele et al., 1997). S. aureus DHPS loop 2 residue Arg52 interacts with the a-phosphatemoiety of PtPP. The b-phosphate-binding pocket is composed of His241, Asn11, and Ile9. Arg239 is within hydrogenbonding distance of only one of the b-phosphate oxygen atoms. The other b-phosphate oxygen atom, which hydrogenbonds to the homologous Arg255 in the E. coli DHPS structure, interacts with a Mn2� in the S. aureus DHPS structure.(c) Residues interacting with the phosphate moiety of PtP in Mtb DHPS (this study). The phosphate interacts withHis255, Asn13, Asp21 (loop 1), and a Mg2�. The oxygen atom that connects the 6-methyl pterin with the phosphorusatom is within hydrogen bonding distance of the guanidinium group of Arg253, an arrangement quite different from
1200 M. tuberculosis DHPS

M. tuberculosis DHPS 1201
DHPS dimer can be superimposed onto the equiv-alent subunit of the Mtb DHPS dimer with anrmsd of 0.93 AÊ for the same set of 71 core a-carbonatoms. When the B subunits of the Sa and MtbDHPS dimers are then compared, the rmsd is6.90 AÊ for the equivalent set of a-carbon atoms,again re¯ecting that the two B subunits areoriented differently with respect to the A subunitby 16.5 �. When the A subunits of the Ec and SaDHPS dimers are then compared, the rmsd is0.90 AÊ for the equivalent set of a-carbon atoms,and their respective B subunits differ in orientationby 13.3 �. Finally, when the A subunits of Mtb, Ec,and Sa DHPS dimers are superimposed, it isobserved that there is a 17 � difference of in theangle for the axis of Mtb helix a8 compared to theEc and Sa helix a8. This helix is involved inthe dimerization interface in each of the DHPSstructures and its different orientation in the Mtbsubunit may explain the signi®cant differences inorganization of the three DHPS dimers.
The differences in subunit orientations in theMtb, Ec and Sa DHPS dimers and the orientationof Mtb helix a8 is likely to originate from the diver-gence in sequence of the amino acids contributingto the dimerization interface. An alternative possi-bility is that the differences in subunit orientationmay represent various states of the enzyme, whichis reported to have half-sites reactivity in otherspecies (Hampele et al., 1997; Vinnicombe &Derrick, 1999a). Communication across the dimerinterface by rotation of the subunits may result insubtle structural changes that could allow one sub-unit to be more or less active than the adjacentsubunit. The various apo and holo structures of Ecand Sa DHPS do not show changes in the dimerinterface upon ligand binding, but these studieswere all the result of soaking experiments, whichmight have masked rearrangements across thedimer interface. Clearly, further studies need to becarried out to establish how, and if, the dimer isdynamically changing its structure while theenzyme catalyzes the reaction.
Phosphate/pyrophosphate-binding pocket
Although the residues that form the pterinbinding pocket are highly conserved in sequenceand structure, the residues involved in forming thebinding site of the phosphate moiety are slightlydifferent between the Mtb, Ec, and Sa DHPS struc-tures, as shown in Figure 6. Interactions with thephosphate group of PtP in Mtb DHPS and with the
that in the E. coli and S. aureus DHPS structures. (d) Proposlyzed by DHPS. In the transition state, the partial positivemost likely assume a geometry in which this partial positivgated pterin ring system. In this geometry, O10 is perpendicuin the Mtb DHPS:PtP binary complex. The amino group of pphosphate. Residues that may be involved in catalysis areAsp21, Ser53, Arg54, Phe182, Lys213, Arg253 and His255. Tcase (Silicon Graphics, Inc.).
b-phosphate group of H2PtPP and PtPP in Ec andSa DHPS structures, respectively, are mediated byinteractions with Mtb Asn13 (corresponding to EcAsn22 and Sa Asn11), and with His255 (corre-sponding to Ec His257 and Sa His241). In the Mtbstructure, Asp21 of loop 1 interacts with the phos-phate group of PtP, but the corresponding homolo-gous residues Ec Asp30 and Sa Asp19 do notinteract with the pyrophosphate moiety, becauseMtb DHPS loop 1 is in a conformation distinctfrom that observed in either the Ec or Sa DHPSstructures, as further discussed below. Loop 2 resi-dues that interact with the pyrophosphate moietyof H2PtPP and PtPP in Ec and Sa DHPS structures(most notably Ec Thr62, Arg63, and Sa Arg52) can-not be compared to homologous residues in theMtb DHPS structure, since loop 2 was too ¯exibleto be observed in the latter structure.
Mtb Arg253 (corresponding to Ec Arg255 and SaArg239) contacts with one face of the pterin ringsystem via p-p interactions with the guanidiniumgroup in all three DHPS structures. As mentionedabove, in Mtb DHPS, the guanidinium group ofArg253 hydrogen bonds to O10, which bridges thepterin and phosphate moieties. Interactions of thisarginine in the Ec and Sa DHPS structures areslightly different, as its guanidinium group hydro-gen bonds with the two terminal oxygen atoms(Ob1 and Ob2) of the b-phosphate group of thepyrophosphate moiety in Ec DHPS:H2PtPP andwith terminal Ob1 of the b-phosphate group of thepyrophosphate moiety of Sa DHPS:Mn2�:PtPP(Figure 6(a) to (c)). The differences observedbetween the interactions of DHPS with the phos-phate and pyrophosphate moieties in the threestructures may represent snapshots of differentconformations the enzyme assumes during cataly-sis, as will be discussed further below.
Loop conformations
The loops on the active site containing theC-terminal pole of the b-barrel deserve specialattention, since they are likely to play a crucial anddynamic function during the catalytic action of theenzyme. The major differences between the differ-ent DHPS structures are in the conformations ofloop 1 (Mtb Val14-Asp27), loop 2 (Mtb Gly50-Asp61), loop 5 (Mtb His131-Val148), and loop 7(Mtb Leu220-Arg233). Loop 1 in the Mtb DHPSstructure adopts a conformation that is entirely dis-tinct from the conformations observed in the Ecand Sa DHPS structures (Figure 7(a)). In the Mtb
ed geometry of the transition state of the reaction cata-charge developed on the 6-methyl carbon atom woulde charge could be stabilized by the electron-rich conju-lar to the plane of the pterin, as observed approximately
ABA would attack C9 from a position opposite the pyro-indicated and include (Mtb numbering) Val11, Asn13,
his Figure was produced with the program IRIS Show-

Figure 7. The different conformations of the conserved loop 1 of DHPS. (a) Superposition of the structures of E. coli(Achari et al., 1997), S. aureus (Hampele et al., 1997), and Mtb DHPS (this study) reveals that loop 1 can adopt differ-ent conformations. Loop 1 from E. coli (blue), S. aureus (yellow), and Mtb (green) are shown in the context of the restof the Mtb DHPS structure, shown in shades of red. The highly conserved Mtb Asp21 hydrogen bonds to PtP, but thehomologous aspartate residue in the E. coli and S. aureus loop 1 lies far from the active site. Distances between thea-carbon atoms of the homologous aspartate residues are indicated. This part of the Figure was produced with theprogram InsightII (Molecular Simulations, Inc.). (b) Multiple sequence alignment of DHPS loop 1 shows the highlevel of conservation of residues in this region. Conservation is indicated by shades of red, with bright red indicatingabsolute conservation and peach indicating low conservation. Positions with no detectable conservation arenot colored. Mtb Asp21 is in the highly conserved section of loop 1. This part of the Figure was produced with theprogram IRIS Showcase (Silicon Graphics, Inc.).
1202 M. tuberculosis DHPS
DHPS structure, this loop interacts with the PtPbinding site by closing down and ``capping'' theactive site (Figure 7(a)). In contrast, loop 1 is in anopen conformation in the Ec DHPS structures andis in an intermediate open conformation in mono-mer A of the Sa apo DHPS structure. When PtPPand Mn2� were soaked into the Sa apo crystals,loop 1 was not visible in monomer A, suggesting
that loop 1 became disordered upon substratebinding. Loop 1 was not visible in either the apo orholo structures of monomer B of Sa DHPS. In con-trast, loop 1 was visible and adopts roughly thesame conformation in both apo and holo Ec DHPSstructures, with its highly open conformationallowing substrates to be soaked into the active sitewithout resulting in disorder of loop 1. In each

M. tuberculosis DHPS 1203
case, the average B-factor of loop 1 is higher thanthe average B-factor for the whole protein, provid-ing further evidence for the ¯exibility of this loop.
It is intriguing that highly conserved residuesamongst all DHPSs are located on loop 1, includ-ing Mtb DHPS Asp21, which forms a hydrogenbond with the phosphate group of PtP(Figure 7(b)). In fact, Mtb DHPS Asp21 is highlyconserved among DHPS sequences, and is a gluta-mate only in the Helicobacter pylori DHPS sequence(Tomb et al., 1997). Although loop 1 exhibits a 70 %sequence identity between Mtb, Ec and Sa DHPSs,the homologous residue Ec DHPS Asp30 isapproximately 20 AÊ away from Mtb DHPS Asp21,and the homologous residue Sa DHPS Asp19 isapproximately 15 AÊ away from Mtb DHPS Asp21(Figure 7). Even though in all available structuresloop 1 is engaged in crystal contacts, this obser-vation makes it plausible that this highly con-served aspartate residue must travel relatively longdistances while loop 1 undergoes dramatic confor-mational changes to place its functional relevantresidues in the proper positions for catalysis. Thehigh level of sequence conservation, the relative¯exibility, and our observation of direct inter-actions with the active site suggest that loop 1 maybe important in the reaction mechanism of DHPS,as will be discussed below.
Electron density corresponding to loop 2 in theMtb DHPS structure could not be observed, indi-cating ¯exibility of this loop. In both apo and holoSa DHPS crystal structures, loop 2 was observed insubunit A but not in subunit B, suggesting ¯exi-bility of Sa loop 2 (Hampele et al., 1997). In con-trast, loop 2 could be observed in both apo andligand-soaked Ec DHPS crystal structures (Achariet al., 1997). In Mtb DHPS, loop 2 consists of 12residues, eight of which are highly conserved, andMtb Pro55 is absolutely conserved in wild-typeDHPS sequences. In Sa and Ec DHPSs, three ofthese residues are in contact with the pyropho-sphate of H2PtPP or PtPP (Figure 8(a) and (b)). Mtbloop 2 is discussed below in context with the pre-dicted binding mode of pABA and sulfa drugs.
Loop 5 in the Mtb DHPS structure, like loops 1and 2, adopts a conformation different from thecorresponding loop in E. coli and S. aureus(Figure 10). In contrast to loop 1, loop 5 residuesare poorly conserved and there is variation in thelength of this loop. Mtb DHPS loop 5 consists of 19residues versus 18 residues in E. coli, 11 residues inS. aureus, and ranges between 11 and 46 residuesin other species. Loop 5 in Mtb DHPS is stabilizedmainly by interactions with loop 6 and by a fewcontacts with loop 4. However, no interaction ofloop 5 with loop 2 could be observed, since loop 2was not visible in our electron density maps. Thisis in distinct contrast with what has been reportedfor the Ec and Sa DHPS structures. In the Ec DHPSstructures, loop 5 was observed to extensivelyinteract with loop 2, whereas in the Sa DHPSstructure, loop 5 interacts weakly with loop 2. It isinteresting that many of the amino acid differences
between the DHPS of M. tuberculosis and therelated organism M. leprae are localized withinloop 5. Although Mtb and M. leprae DHPS share anoverall sequence identity of 79 %, they share only42 % sequence identity in loop 5.
In summary, the loops of DHPS serve an import-ant functional role and, like the ¯uidic arms of anoctopus, can assume different conformations.Loops 1, 2, and 5 may be the most crucial loops forthe action of DHPS. The crystal structure of MtbDHPS reveals for the ®rst time direct evidence thatloop 1 may serve a key role in the catalytic actionof the enzyme. It is evident that further studies areneeded to fully elucidate the precise enzymatic roleof loop 1 residues, as well as of other putativeactive-site residues, while DHPS is carrying out thesynthesis of dihydropteroate.
Possible implications for catalysis
Even though our structure of Mtb DHPS in com-plex with PtP lacks the b-phosphate group of theactual substrate H2PtPP, our structure contains sev-eral interesting aspects that may help to shed lighton the mechanism of DHPS catalysis. This stems inparticular from the new conformation of loop 1and the out-of-plane orientation of the a-phosphategroup. Below we will look into the implications ofthese structural features for the catalytic mechan-ism combined with the possible enzymatic functionof several highly conserved residues in the DHPSenzyme family.
Three key phosphate-binding residues that maybe involved in facilitating pyrophosphoryl transferare visible in the Mtb DHPS:PtP binary complex(Figure 6(c)). The ®rst is the absolutely conservedMtb Asn13, which provides its OD1 to coordinatea magnesium ion, which then in turn is coordi-nated by O1 of the a-phosphate moiety of PtP. Inmonomer A of S. aureus Mn2�/PtPP-soaked DHPScrystals, a Mn2� was observed to be coordinatedby the OD1 of Sa Asn11, equivalent to Mtb Asn13,and by the terminal Ob2 of the b-phosphate groupof PtPP. The second key phosphate-binding resi-due is the highly conserved Mtb Asp21 of loop 1,which interacts directly with the terminal O2 of thephosphate moiety of PtP, with the a-phosphateoxygen atom most likely being protonated, sinceits pKa lies between 6.3 and 6.6 (Jencks &Regenstein, 1970). Ec DHPS Asp30 and Sa DHPSAsp19 are equivalent to Mtb Asp21, but in thesestructures these aspartate residues are far, i.e. up to20 AÊ , removed from the active site, in contrastwith the situation in Mtb DHPS (Figure 7). Thispoints to considerable rearrangements of loopsupon substrate binding and possibly during cataly-sis. The third absolutely conserved residue in thisregion is Mtb His255, which hydrogen bonds to theterminal O1 of the a-phosphate moiety of PtP andto the terminal Ob3 of the b-phosphate moiety ofH2PtPP and PtPP in the Ec and Sa DHPS struc-tures, respectively (Figure 6(a)-(c)). While MtbAsn13 and His255 have been discussed in connec-

1204 M. tuberculosis DHPS
tion with the Ec and Sa DHPS structures, the invol-vement of Asp21, provided by loop 1, is a newinsight obtained from the Mtb DHPS structure.
Mtb Arg253, a residue that is absolutely con-served in all reported DHPS sequences, is situ-ated at an intriguing position with respect toboth the pterin ring system and the pyropho-sphate moiety. It is engaged in p-p interactions
of its guanidinium group with the pyrazinering of the pterin. At the same time, the guani-dinium group of Mtb Arg253/Ec Arg255/SaArg239 interacts with the phosphate/pyropho-sphate moiety in each of the three DHPS struc-tures, but in each case in a different manner, asdiscussed above and shown in Figures 5 and 6.These differences may represent different snap-
Figure 8. Loops 2 and 5 ofDHPS. Superposition of the struc-tures of E. coli (Achari et al., 1997),S. aureus (Hampele et al., 1997), andMtb DHPS reveals that loops 2 and5 can adopt different confor-mations. Loops 2 and 5 from E. coli(blue), loops 2 and 5 from S. aureus(yellow), and loop 5 from Mtb(green) are shown in the context ofthe rest of the Mtb DHPS structure,depicted in shades of red. E. coliloop 5 interacts with E. coli loop 2,but S. aureus loop 5 does notinteract with S. aureus loop 2 asextensively. S. aureus loop 5 isseven amino acid residues shorterthan the corresponding E. coli loop5. Loop 2 was not observed inthe Mtb DHPS structure. ThisFigure was produced with theprogram InsightII (MolecularSimulations, Inc.).

M. tuberculosis DHPS 1205
shots of the interactions that this completelyconserved arginine residue engages in duringcatalysis.
Another absolutely conserved residue, MtbLys213, provides an e-amino group that hydrogenbonds with the 4-oxocarbonyl O4 and the N5 ofthe pyrazine ring of PtP in our Mtb DHPS:PtPbinary complex. Similar interactions occur in the Ecand Sa DHPS structures (Achari et al., 1997;Hampele et al., 1997). Together with the positivelycharged guanidinium group of Mtb Arg253, theamino group of Lys213 is the second positivecharge interacting with the ``C6-end'' of the pyra-zine ring, i.e., the N5, C6, C7 region, with possibleelectron-withdrawing consequences for the C6-C9bond to be attacked by the incoming amino groupof the second substrate, pABA.
Another completely conserved functional groupis the hydroxyl side-chain of Mtb Ser53 from loop2, which is equivalent to Ec Thr62 and Sa Thr51. Inthe E. coli enzyme, the hydroxyl group of thisresidue interacts with terminal oxygen atoms ofthe a-phosphate moiety of H2PtPP, while its back-bone amide group is engaged in hydrogen bondformation with this phosphate moiety. In SaDHPS, both the hydroxyl group and backboneamide group of Thr51 engage in hydrogen bondsto the OE1 of Sa Gln 105 (equivalent to MtbVal107), which packs against one side of the pterinface. Even though loop 2, and hence Mtb Ser53, isnot visible in our electron density maps of MtbDHPS, it is likely that at some stage during cataly-sis Mtb Ser53 interacts with the a-phosphatemoiety of its H2PtPP substrate.
The mobility of loop 2 (Figure 8) has other intri-guing aspects. Loop 2 residue Arg63 of Ec DHPS(corresponding to Mtb Arg54), hydrogen bonds tothe terminal Ob3 of the b-phosphate moiety ofH2PtPP, but extends from loop 2 to occupyroughly the space where residues Ser20 and Asp21of loop 1 reside in our Mtb DHPS structure. Loop 2of Ec and Sa DHPSs differ slightly from each other,with the Ca position of Sa Arg52 (corresponding toboth Mtb Arg55 and Ec Arg63) residing 4 AÊ fromthe equivalent position in the Ec structure(Figure 8). Unlike Ec Arg63, the equivalent SaArg52 does not occupy space where residues arefound in the closed conformation of Mtb loop 1. SaArg52 replaces Ec Thr62 as the hydrogen bondingpartner with Oa1 of the a-phosphate moiety ofPtPP. Because (i) loop 2 is in slightly different con-formations in the Ec and Sa DHPS structures(Figure 8); (ii) Ec Arg63 and Sa Arg52 interact withdifferent parts of the pyrophosphate moiety in theEc and Sa DHPS structures; and (iii) the equivalentMtb Arg54 is invisible in the Mtb DHPS structure,it is quite possible that loop 2 undergoes dynamicmotion during catalysis such that this highly con-served arginine residue hydrogen bonds to differ-ent parts of the pyrophosphate during substratebinding along the reaction pathway. In summary,Mtb Ser53 and Arg54 may hydrogen bond tothe pyrophosphate moiety of the H2PtPP during
catalysis and serve to facilitate pyrophosphoryltransfer.
The a-phosphate position of PtP in the MtbDHPS structure is particularly interesting. Theobserved position of the phosphate moiety in PtPplaces this oxygen atom roughly perpendicular tothe plane of the pterin ring system. This is in con-trast with the observed conformations of thea-phosphate moiety in the Ec DHPS:H2PtPP and SaDHPS:PtPP binary complexes, in which the a-phos-phate group lies in the plane of the pterin ring sys-tem, as can be seen in Figure 5. The b-phosphategroup of the pyrophosphate moiety of H2PtPP canbe modeled into the Mtb DHPS:PtP structure and®ts within the b-phosphate-binding site consistingof Mtb Asn13, His255, and Val11. (The homologousresidues in E. coli and S. aureus create similarb-phosphate binding sites in the Ec DHPS:H2PtPPand Sa DHPS:PtPP:Mn2� structures). Hence, theout-of-plane oxygen atom for PtP seen with MtbDHPS may be closer to the transition state than theobserved conformation of H2PtPP or PtPP.
Further support for the possibility that the pos-ition of the a-phosphate group in the MtbDHPS:PtP structure is relevant for the actual reac-tion between pABA and H2PtPP stems from thedirection of the C9-O10 bond. It is most likely thatthe nucleophilic attack by the amino nitrogen atomof pABA occurs from a direction approximately inline with the C9-O10 bond, but obviously with thenitrogen atom on the side of C9 opposite from thatwhere O10 is observed. In our structure, such anincoming position for pABA can be modeled asshown in Figure 6(d). In this model, pABA inter-acts with the highly conserved Phe182 as well aswith the highly conserved loop 2 residues Ser53and Arg54. Phe182 is conserved in all DHPSsequences except in Toxoplasma gondii DHPS, inwhich the corresponding residue is isoleucine(Pashley et al., 1997). Interestingly, the bindingmode of pABA proposed from this protein:sub-strate complex is in agreement with the location ofthe sulfa-drug-resistant mutations of residues inother species equivalent to Mtb Ser53 and MtbPro55, as discussed below. In a mechanism consist-ent with our hypothesized pABA binding site,the positive charges of the absolutely conservedArg253 and Lys214 next to the pyrazine ring sys-tem would be suitably positioned to enhance thepolarization of the C6-C9 bond, promoting a par-tial positive charge on C9 and thereby facilitatingthe reaction with the pABA amino nitrogen atom.
In summary, the available information suggestskey catalytic functions for seven residues; Asn13,Asp21, Ser53, Arg54, Phe182, Lys213, Arg253 andHis255 (Figures 4(b) and 6(d)).
Predicted binding mode of pABA, dapsone,and sulfonamides
Although pABA and various sulfa drugs werepresent in the crystallization buffers and cryopro-tectants at a concentration of 3 mM, these ligands

1206 M. tuberculosis DHPS
were not observed in the Mtb DHPS structure. Thebinding mode of pABA has not been reported forany DHPS. In fact, only the structure of Ec DHPSsoaked with H2PtOH and sulfanilamide has pro-vided a position of the simplest sulfonamidebound to DHPS (Achari et al., 1997). When the MtbDHPS:PtP binary complex is compared to the EcDHPS:H2PtOH:sulfanilamide ternary complex(PDB code 1ajz.pdb; Achari et al., 1997), it appearsthat the closed loop 1 conformation of Mtb DHPSoccupies the space where sulfanilamide wasobserved to bind in the homologous Ec DHPSdrug-soaked crystal structure. As mentioned ear-lier, since many residues of loop 1 are highly con-served, it is quite likely that the position of loop 1in the Mtb DHPS structure is of catalytic relevance,in particular since the absolutely conserved Asp21makes a direct interaction with O2 of the PtP phos-phate moiety (Figure 4(b)). Hence, we investigatedwhether we could predict an alternative bindingmode for pABA and sulfa drugs to DHPS.
Loop 2 was not observed in the Mtb DHPS struc-ture, but was observed in both the Ec and SaDHPS structures to contribute highly conservedresidues to the interaction with the pyrophosphatemoiety of H2PtPP and PtPP, respectively. There-fore, it can be expected that loop 2 plays animportant role in DHPS catalysis. Dapsone resist-ance has been attributed to mutations in the dhpsgene of M. leprae (Kai et al., 1999). The reporteddapsone-resistance mutations, M. leprae Thr53Ala/Ile and Pro55Leu correspond to Mtb loop 2 resi-dues Ser53 and Pro55. Because M. leprae and MtbDHPSs share a 75 % sequence identity in loop 2,it is most likely that this loop assumes similarfunctions and conformations in both enzymes.Therefore, M. leprae dapsone-resistant mutationsmay provide information that can be used to pre-dict a binding mode for dapsone in the Mtb DHPScrystal structure.
In order to explore this possibility, the coordi-nates of the residues corresponding to E. coli loop2 (Ec residues Glu60 to Ser70) were grafted ontothe Mtb DHPS structure and the four non-identicalE. coli side-chains were changed into the corre-sponding Mtb residues. In the Mtb DHPS model soobtained, the positions of the two dapsone-resist-ant mutations in loop 2 lie very close to the activesite. Additionally, there is only one solvent-accessi-ble channel to the active site, since Mtb DHPS loop1 is in a closed conformation. The most likely bind-ing site for pABA, dapsone, and sulfonamides islocated within this channel. Residues contributingto the formation of this channel include Mtb loop 2residues Ser53, Arg54, and Pro55; loop 5 residuesHis141 and Pro143; loop 6 residues Gly181,Phe182, Lys184, and Thr185; and loop 70 residuesLys213 and Arg214. Two of these residues, Phe182and Lys213, interact with the pterin moiety. Ofthese channel residues, the pABA-binding site ismost likely lined by Ser53, Arg54, Pro55, Phe182,and Lys213. When this channel is closed by thebinding of a pABA, dapsone, or sulfonamide mol-
ecule, the active site becomes shielded from bulksolvent water, which agrees well with the need toprevent water serving as the nucleophile thatwould hydrolyze the activated substrate, yielding6-hydroxymethyl-7,8-dihydropterin. Shielding ofthe active site during magnesium-dependent pyro-phosphoryl transfer from bulk water by the closureof ¯exible loops has been observed crystallographi-cally in the PRTase enzymes (HeÂroux et al., 1999;Shi et al., 1999). The modeled binding mode of dap-sone spans across the tip of loop 2, and places dap-sone close to the resistance mutations (Figure 9)with Arg54 and Pro55 approximately 3 AÊ from thephenyl ring containing the amino group deepest inthe binding site, Ser53 �6 AÊ from this aminogroup, and Gly181 and Ala183 about 3-5 AÊ fromthe other phenyl group of the symmetric(Figure 1(d)) dapsone molecule.
The position of the modeled pABA/dapsone/sulfonamides in the Mtb DHPS structure places theaniline ring of these ligands orthogonal to the pos-ition of sulfanilamide observed in the E. coliDHPS:H2PtOH:sulfanilamide structure (Achariet al., 1997). The modeled position may re¯ect thebinding mode after closure of loop 1, which wasopen in the E. coli structure. The binding mode ofsulfanilamide reported in the E. coli DHPS complex(Achari et al., 1997) allows little room for the het-erocyclic substitutions at the sulfonamide positionthat are common in many sulfa drugs, whereasthere is space for these substitutions in the channelthat we predict to be the sulfa-drug-binding site.
The modeled binding mode of pABA/dapsone/sulfonamides in the Mtb DHPS structure is closerthan sulfanilamide in the Ec DHPS structure to thedistribution of sulfonamide-resistant mutationsobserved from a variety of species, which havebeen found to lie mainly on the loop regions ofDHPS. As can be seen in Figure 10 and in Table 2,many of the mutations lie on loop 2 between MtbSer53 and Asp61. Interestingly, mutations occur inthree organisms at positions equivalent to MtbSer53 and Pro55, the residues that we used to pre-dict the binding mode of dapsone. Mutations occurat positions corresponding to Mtb Phe19 andSer20, both located on loop 1. In fact, a singlemutation in E. coli and N. meningitidis DHPSs cor-responding to the Mtb Phe19 position can convertthe enzyme from being sulfonamide-sensitive tosulfonamide-resistant (Dallas et al., 1992; FermeÂret al., 1995; FermeÂr & Swedberg, 1997). The pre-sence of a dominant sulfa drug-resistant mutationat a position corresponding to Phe19 supports ourhypothesis that loop 1 plays an important role inDHPS catalysis by placing residue Asp21 at theactive site. Mtb Phe19 packs against the loop 2grafted from E. coli, within 5 AÊ of the absolutelyconserved Pro55, another position that, whenmutated, confers sulfonamide-resistance in severaldifferent species. Therefore, one can imagine thatthe equivalent Phe19Ile (E. coli) and Phe19Leu(N. meningitides) mutations may indirectly modu-late the dynamic behavior of loop 2 through

Figure 9. Modeled binding site of dapsone. (a) The Mtb DHPS structure has loop 2 (purple) grafted from the E. colistructure. Mtb loop 1 and the grafted loop 2 close over the active site and shield it from bulk solvent. Dapsone(green) can be modeled into the only remaining solvent-accessible channel to the PtP (yellow) site. (b) Residues con-tributing to the pABA/sulfa drug binding site come from loop 1 (grey), loop 2 (purple), loop 5 (blue), loop 6 (pink),and loop 7 (orange). Loop 2 was taken from the homologous E. coli DHPS structure (Achari et al., 1997) and graftedonto the Mtb DHPS structure (in red). Loop 2 residues Thr53 and Pro55, shown in purple, are mutated in the hom-ologous M. leprae DHPS in response to dapsone (Kai et al., 1999). Residues that may contribute to dapsone bindingand that may form a general pABA/sulfa drug binding region include (Mtb numbering) loop 2 residues Ser53,Arg54, and Pro55; loop 5 residues His141 and Pro143; and loop 6 residues Gly181, Phe182, Lys184, and Thr185; andloop 7 residues Lys213 and Arg214. The 4-amino group of dapsone (in green) is in line for nucleophilic attack of the6-methyl carbon atom of PtP (in yellow). This Figure was produced with the program InsightII (Molecular Simu-lations, Inc.).
M. tuberculosis DHPS 1207
interactions with Pro55, altering the pABA-bindingpocket so as to better exclude sulfonamides. Thiswas not observed before, since the position of thecorresponding phenylalanine residue (Ec Phe28) inthe E. coli structure (Achari et al., 1997) is 20 AÊ
away from the residue corresponding to Mtb Pro55(Ec Pro64), due to the fact that Ec loop 1 is not in aconformation that places loop 1 residues nearthe active site, as does our Mtb DHPS structure.Finally, DHPS mutations at positions equivalentto Mtb Gly181 and Ala183, located on loop 6,confer resistance to sulfonamides in N. menin-gitidis and Plasmodium falciparum, respectively.Mutagenesis studies at the equivalent positions ofthe N. meningitidis DHPS enzyme show thatN. meningitidis Gly194Cys or duplication ofSer193-Gly194, both part of loop 6, can result in adrug-resistant enzyme (FermeÂr et al., 1995; FermeÂr& Swedberg, 1997). Interestingly, this regionGly181-Thr185 of loop 6 contributes several resi-dues to our predicted dapsone-binding site(Figures 9 and 10).
The position of the modeled pABA/dapsone/sulfonamides in the Mtb DHPS structure derivedfrom mutational data is consistent with a putativetransition state geometry depicted in Figure 6(d),in which the 4-amino group of the attacking
nucleophile displaces the pyrophosphate from theopposite side of the 6-methyl carbon atom. Theabsolutely conserved residues Ser211 and Arg212,whose function was previously de®ned as forminga pABA/sulfonamide-binding site based on theE. coli:H2PtOH:sulfanilamide ternary complex(Achari et al., 1997), can now be re-evaluated inlight of our Mtb DHPS structure. The side-chain ofMtb Ser211 makes, in our structure, a hydrogenbond with ND1 of the imidazole ring of His255,which then allows the NE2 of this absolutely con-served histidine to form a hydrogen bond to anoxygen atom of the modeled b-phosphate moietyof H2PtPP (Figure 6(a)-(c)). Arg212 hydrogenbonds via its NH1 to the backbone carbonyl groupof Asp21 (loop 1) (Figure 6(c)). This interactionwith Arg212 was not observed in either the Ec orSa DHPS structures because, as described above,the highly conserved Asp21 was far from the activesite (Achari et al., 1997; Hampele et al., 1997). Ofcourse, great ¯exibility of loops 1 and 2 is observedin the DHPS structures reported to date, so that itis likely that the substrate pABA and the largevariety of sulfonamide and sulfone inhibitorsmay very well each have subtle or signi®cantdifferences in position upon binding DHPS. Yet theavailable evidence points to a general binding site

Table 2. Sulfonamide and dapsone-resistance mutations observed in DHPS from six organisms
Organisms Mutation observedCorresponding
Mtb residue Structure References
E. coli Phe28Leu, Ile Phe19 Loop 1 Dallas et al. (1992); FermeÂr &Swedberg (1997)
N. meningitidis Phe31Leu Phe19 Loop 1 FermeÂr et al. (1995); FermeÂr &Swedberg (1997)
P. carinii Phe23Leu Ser20 Loop 1 Lane et al. (1997)M. leprae Thr53Ile,Ala Ser53 Loop 2 Kai et al. (1999)P. carinii Thr55Ala Ser53 Loop 2 Lane et al. (1997); Kazanjian
et al. (1998); Mei et al. (1998)P. falciparum Ser436Ala, Phe Ser53 Loop 2 Wang et al. (1997)P. falciparum Ala437Gly Arg54 Loop 2 Wang et al. (1997)S. pneumoniae Arg58-Pro59 duplication Arg54-Pro55 Loop 2 Padaycachee & Klugman (1999)M. leprae Pro55Leu Pro55 Loop 2 Kai et al. (1999)P. carinii Pro57Ser Pro55 Loop 2 Lane et al. (1997); Kazanjian
et al. (1998); Mei et al. (1998)E. coli Pro64Ser Pro55 Loop 2 Vedantam et al. (1998)S. pneumoniae Arg insertion after Gly60 Gly56 Loop 2 Padaycachee & Klugman (1999)S. pneumoniae Ser61 duplication Ala57 Loop 2 Maskell et al. (1997)P. carinii His60Asp Thr58 Loop 2 Lane et al. (1997)S. pneumoniae Ile66-Glu67 duplication Val60-Asp61 Loop 2 LoÂpez et al. (1987)P. carinii Ile111Thr Val107 Loop 4 Lane et al. (1997)P. falciparum Lys540Glu Trp132 Loop 5 Wang et al. (1997)N. meningitidis Gly194Cys, Ser193-Gly194
duplicationGly181 Loop 6 FermeÂr et al. (1995); FermeÂr &
Swedberg (1997)P. falciparum Ala581Gly Ala183 Loop 6 Wang et al. (1997)P. falciparum Ala613Ser, Thr Gly217 a70 Wang et al. (1997)P. carinii Val248Gly Ala245 a7 Lane et al. (1997)
Figure 10. Distribution of sulfa-drug-resistance mutations in DHPS. Mutations that confer sulfa drug-resistance toDHPS from six different species (Table 2) have been mapped onto the Mtb DHPS structure with ``loop 2`` graftedfrom E. coli DHPS. The equivalent position in the Mtb DHPS structure corresponding to where a mutation has beenobserved is indicated with a sphere at its Ca, and the sphere is color-coded by species; N. meningitidis blue, S. pneumo-niae green, P. falciparum cyan, P. carinii orange. Positions that are mutated in several species, equivalent to Mtb DHPSresidues 19 (loop 1), 53 and 55 (loop 2), are indicated by large red spheres, and are close to the active site and puta-tive dapsone binding region. This Figure was produced with the program InsightII (Molecular Simulations, Inc.).
1208 M. tuberculosis DHPS

M. tuberculosis DHPS 1209
for at least pABA and dapsone (Figures 9 and 10),which ®ts well into a picture of a likely catalyticmechanism (Figure 6(d)).
Structural insights for the design of novelantimycobacterial agents
The highly conserved pterin-binding pocket maybe exploited for the design of novel DHPS inhibi-tors. Sulfa drugs are generally substituted on thesulfonamide position, as shown in Figure 1(c).Extension of a sulfa drug into the pterin pocketwould mean modi®cation of the sulfonamide orsulfone drug molecule in a position opposite towhere substitutions have traditionally been made.This may result in a new set of chemically diversemolecules that may serve as lead compounds forantimycobacterial drug design. Additionally, thehighly conserved nature of the pterin-bindingpocket may render it less prone to drug-resistantmutations, while the hydrophobic nature of manyresidues lining this pocket may create opportu-nities to add hydrophobic substitutions to novelinhibitors. This could be highly bene®cial for themembrane translocation steps of orally availableanti-tuberculosis agents. Obviously, one has toavoid binding of such inhibitors to pterin andfolate binding sites of key human proteins. Theabsolutely conserved Arg253, however, may func-tion as a distinctive feature of the pterin-bindingpocket of DHPS. Transition state inhibitors againstDHPS may also serve as powerful leads for novelantimycobacterial agents, since the reaction cata-lyzed by DHPS is not present in humans. In sum-mary, the crystal structure of Mtb DHPS presentsexciting opportunities to be explored for the designof new antimycobacterial agents.
Materials and Methods
Synthesis of pterin substrate analogues
The pterin substrate analogues used in this studyare shown in Figure 1(b). 6-Hydroxymethylpterinpyrophosphate (PtPP) and 6-hydroxymethylpterin mono-phosphate (PtP) were synthesized from 6-hydoxy-methylpterin (Sigma) and pyrophosphoric acid (Aldrich)adapting the protocol of Shiota et al. (1964) and thenseparated using HPLC (Hewlett Packard 1100 SeriesAutosampler, Vydac C18 reverse phase column). Theidentities of PtPP and PtP were con®rmed with MALDImass spectrometry.
Co-crystallization of DHPS with pterin monophosphate
Recombinant Mtb DHPS was overexpressed in E. coliand puri®ed as described (Nopponpunth et al., 1999).The Mtb DHPS protein sample was at a concentration of14 mg/ml in a buffer containing 50 mM potassium phos-phate (pH 7.0), 1 mM DTT, and 20 % (v/v) glycerol.When screening for crystallization conditions of binaryor ternary DHPS complexes, the pterin derivatives (PtPor PtPP shown in Figure 1(b)) were added to the proteinsolution to a ®nal concentration of 3 mM and pABA orvarious sulfa drugs were added to a ®nal concentration
of 3 mM. 7,8-Oxidized versions of pterins were used incrystallization experiments, since pterin binding toDHPS is relatively independent of the reduction state ofthe pterin ring, as shown by the ability of S. aureusDHPS to bind an oxidized pterin substrate (Hampeleet al., 1997) and by the similar Ki values of folate(15 mM), dihydrofolate (15 mM), and tetrahydrofolate(8 mM in S. pneumoniae) (Vinnicombe & Derrick, 1999a).Despite extensive screening, crystals of apo-DHPS,binary complex PtPP:DHPS, or ternary complex PtPP:(pABA or sulfa drug):DHPS could not be grown. MtbDHPS crystals were obtained only when PtP waspresent. Initial crystallization conditions, which weresubsequently optimized, were found using the CrystalScreen Kit I from Hampton Research. Crystallization wascarried out using the sitting drop vapor diffusion meth-od in which 1 ml of protein solution was added to 1 ml ofreservoir and equilibrated against 0.5 ml of reservoir sol-ution in a sealed chamber at 25 �C. The reservoir solutioncontained 100 mM sodium acetate (pH 4.6), 200 mMammonium acetate, and 30 % (w/v) PEG 4000. Bipyra-midal shaped crystals grew within one to two days withdimensions of 0.30 mm � 0.20 mm � 0.20 mm. The crys-tals used in data collection were grown in the presenceof 3 mM PtP and 3 mM pABA or sulfa drugs.
Data collection and processing
Crystals were ¯ash-frozen in a liquid nitrogen streamusing 15 % glycerol added to the reservoir solution as acryoprotectant. The pH of the cryoprotectant was 5.8. Adata set of the DHPS:PtP binary complex was collectedat 140 K to 1.7 AÊ resolution using a Rigaku RU 200 rotat-ing anode X-ray generator (CuKa; l 1.54 AÊ ) and a RaxisIIc area detector. This data set was processed usingDENZO and scaled with SCALEPACK (Otwinowski &Minor, 1997). The crystal was trigonal with space groupP3221, cell dimensions a � b � 62.9 AÊ , c � 121.4 AÊ .
Structure determination and refinement
The structure of Mtb DHPS:PtP was solved by molecu-lar replacement methods with the AMoRe program(Navaza, 1994) using data from 8 to 3.5 AÊ . The searchmodel was an apo version of S. aureus DHPS monomerA (PDB code 1ad1.pdb; Hampele et al., 1997), with non-identical residues compared to Mtb DHPS (percentageidentity � 37.4) replaced by alanine. The top solutionbefore rigid-body re®nement yielded a correlation coef®-cient of 23.5 %, and an R-factor of 52.5 %. The asym-metric unit contains a monomer of 28.8 kDa, but acrystallographic 2-fold axis generates a dimer.
Mtb DHPS was re®ned with the software packagesCNS (Pannu & Read, 1996; Adams et al., 1997; BruÈ ngeret al., 1998) and TNT (Tronrud et al., 1987) using datafrom 20 to 1.7 AÊ . Iterative model building was doneemploying the program O (Jones et al., 1991) using sa-weighted 2Fo ÿ Fc and Fo ÿ Fc electron density maps(Read, 1986). All re®nement procedures were corrobo-rated by analysis of the free R-factor (BruÈ nger, 1992),with 5 % of the observed re¯ections belonging to the testset. Loops 1, 2, 5, and 7, b-strand 1 and a-helix 8 wereremoved from the starting model, since initial electrondensity maps in these areas could not be well inter-preted. Several rounds of simulated annealing, alternatedwith manual model building, were performed. Duringthe course of model building, loops 1, 5, and 7, b-strand1 and a-helix 8 were added to the model. A total of 244

1210 M. tuberculosis DHPS
water molecules were added with the CNS program andcarefully checked for proper hydrogen bonding geome-try. Pterin monophosphate was also included in themodel, using input topology and parameter ®les gener-ated by the program XPLODE (Kleywegt & Jones, 1997).In the ®nal stages of re®nement, a magnesium ion wasincluded, which was 1.93 AÊ from the OD1 of Asn13 and2.17 AÊ from O1 of the PtP phosphate. The ®nal model (R18.5 %, Rfree 24.3 %) includes Mtb DHPS residues 5 to 50and 65 to 274, PtP, Mg2�, and 244 water molecules. Asummary of crystallographic information, data proces-sing and re®nement statistics is given in Table 1.
Protein Data Bank accession numbers
The atomic coordinates for the re®ned structure ofM. tuberculosis DHPS in complex with PtP have beendeposited in the RCSB Protein Data Bank, accession code1eye.pdb.
Acknowledgments
Sequence data of M. avium DHPS were obtained fromThe Institute for Genomic Research website (http://www.tigr.org). Sequencing of M. avium was accom-plished with the support of NIAID. We thank DrsMichael Feese, Stephen Suresh, Francis Athappilly,Christophe Verlinde and other members of the Biomole-cular Structure Center for assistance, advice, and discus-sion. We are grateful to Drs Erkang Fan and Feng Hongfor assistance regarding the synthesis of the substrateanalogue. This research was supported, in part, bythe National Science and Technology DevelopmentAgency, Thailand. A.M.B. is supported by the WhitakerFoundation. W.G.J.H. is grateful to the Murdock Charita-ble Trust for a major equipment grant to the Biomolecu-lar Structure Center.
References
Achari, A., Somers, D. O., Champness, J. N., Bryant,P. K., Rosemond, J. & Stammers, D. K. (1997). Crys-tal structure of the anti-bacterial sulfonamide drugtarget dihydropteroate synthase. Nature Struct. Biol.4, 490-497.
Adams, P. D., Pannu, N. S., Read, R. J. & BruÈ nger, A. T.(1997). Cross-validated maximum likelihoodenhances crystallographic simulated annealingre®nement. Proc. Natl Acad. Sci. USA, 94, 5018-5023.
Anand, N. (1996). Sulfonamides and sulfones. InBurger's Medicinal Chemistry and Drug Discovery(Wolff, M. E., ed.), pp. 527-573, John Wiley & Sons,New York.
Bass, J. B. L. S. F., Hopewell, P. C., O'Brien, R., Jacobs,R. F., Ruben, F., Dixie, E., Snider, J. & Thornton, G.(1994). Treatment of tuberculosis and tuberculosisinfection in adults and children. Am. J. Respir. Crit.Care Med. 149, 1359-1374.
Bloom, B. R. & Murrary, C. J. L. (1992). Tuberculosis:commentary on a reemergent killer. Science, 257,1055-1064.
Bock, L., Miller, G. H., Schaper, K.-J. & Seydel, J. K.(1974). Sulfonamide structure-activity relationshipsin a cell-free system. 2. Proof for the formation of asulfonamide-containing folate analog. J. Med. Chem.17, 23-28.
Brooks, D. R., Wang, P., Read, M., Watkins, W. M.,Sims, P. F. & Hyde, J. E. (1994). Sequence variationof the hydroxymethyldihydropterin pyrophosphoki-nase: dihydropteroate synthase gene in lines of thehuman malaria parasite, Plasmodium falciparum,with differing resistance to sulfadoxine. Eur. J.Biochem. 224, 397-405.
Brown, G. M. (1962). The biosynthesis of folic acid. II.Inhibition by sulfonamides. J. Biol. Chem. 237, 536-540.
BruÈ nger, A. T. (1992). The R-value: a novel statisticalquantity for assessing the accuracy of crystal struc-tures. Nature, 355, 472-475.
BruÈ nger, A. T., Adams, P. D., Clore, G. M., DeLano,W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S.,Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J.,Rice, L. M., Simonson, T. & Warren, G. L. (1998).Crystallography & NMR system: a new softwaresuite for macromolecular structure determination.Acta Crystallog. sect. D, 54, 905-921.
Dallas, W. S., Gowen, J. E., Ray, P. H., Cox, M. J. &Dev, I. K. (1992). Cloning, sequencing and enhancedexpression of the dihydropteroate synthase gene ofEscherichia coli MC4100. J. Bacteriol. 174, 5961-5970.
Dolin, P. J., Raviglione, M. D. & Kochi, A. (1994). Globaltuberculosis incidence and mortality during 1990-2000. Bull. World Health Org. 72, 213-220.
Esnouf, R. M. (1999). Further additions to MolScriptversion 1. 4, including reading and contouring ofelectron-density maps. Acta Crystallog. sect. D, 55,938-940.
FermeÂr, C. & Swedberg, G. (1997). Adaptation to sulfo-namide resistance in Neisseria meningitidis may haverequired compensatory changes to retain enzymefunction: kinetic analysis of dihydropteroatesynthases from N. meningitidis expressed in aknockout mutant of Escherichia coli. J. Bacteriol. 179,831-837.
FermeÂr, C., Kristiansen, B. E., Skold, O. & Swedberg, G.(1995). Sulfonamide resistance in Neisseria meningiti-dis as de®ned by site-directed mutagenesis couldhave its origin in other species. J. Bacteriol. 177,4669-4675.
Ferone, R. (1973). The enzymic synthesis of dihydrop-teroate and dihydrofolate by Plasmodium berghei.J. Protozool. 20, 459-464.
Gibreel, A. & SkoÈ ld, O. (1999). Sulfonamide resistance inclinical isolates of Campylobacter jejuni: mutationalchanges in the chromosomal dihydropteroatesynthase. Antimicrob. Agents Chemother. 43, 2156-2160.
Hampele, I. C., D'Arcy, A., Dale, G. E., Kostrewa, D.,Nielsen, J., Oefner, C., Page, M. G., Schonfeld, H. J.,Stuber, D. & Then, R. L. (1997). Structure and func-tion of the dihydropteroate synthase from Staphylo-coccus aureus. J. Mol. Biol. 268, 21-31.
HeÂroux, A. E. L. W., Ross, L. J., Davis, R. L. & Borhani,D. W. (1999). Crystal structure of Toxoplasma gondiihypoxanthine-guanine phosphoribosyltransferasewith XMP, pyrophosphate, and two Mg2� ionsbound: insights into the catalytic mechanism.Biochemistry, 38, 14495-14506.
Huovinen, P., Sundstrom, L., Swedberg, G. & Skold, O.(1995). Trimethoprim and sulfonamide resistance.Antimicrob. Agents Chemother. 39, 279-289.
Jencks, W. P. & Regenstein, J. (1970). Ionization con-stants of acids and bases. In Handbook of Biochemis-try - Selected Data for Molecular Biology (Sober, H. A.,

M. tuberculosis DHPS 1211
ed.), pp. J187-J226, The Chemical Rubber Co.,Cleveland.
Jones, T. A., Zou, J. Y., Cowan, S. W. & Kjeldgaard, M.(1991). Improved methods for building proteinmodels in electron density maps and the location oferrors in these models. Acta Crystallog. sect. A, 47,110-119.
Kabsch, W. & Sander, C. (1983). Dictionary of protein sec-ondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers, 22,2577-2637.
Kai, M., Matsuoka, M., Nakata, N., Maeda, S., Gidoh,M., Maeda, Y., Hashimoto, K., Kobayashi, K. &Kashiwabara, Y. (1999). Diaminodiphenylsulfoneresistance of Mycobacterium leprae due to mutationsin the dihydropteroate synthase gene. FEMS Micro-biol. Letters, 177, 231-235.
Kazanjian, P., Locke, A. B., Hossler, P. A., Lane, B. R.,Bartlett, M. S., Smith, J. W., Cannon, M. &Meshnick, S. R. (1998). Pneumocystis cariniimutations associated with sulfa and sulfone pro-phylaxis failures in AIDS patients. AIDS, 12, 873-878.
Kellam, P., Dallas, W. S., Ballantine, S. P. & Delves, C. J.(1995). Functional cloning of the dihydropteroatesynthase gene of Staphylococcus haemolyticus. FEMSMicrobiol. Letters, 134, 165-169.
Kleywegt, G. J. & Jones, T. A. (1997). Model-buildingand re®nement practice. Methods Enzymol. 277, 208-230.
Kraulis, P. J. (1991). MOLSCRIPT: a program to produceboth detailed and schematic plots of protein struc-tures. J. Appl. Crystallog. 24, 946-950.
Kristiansen, B. E., Radstrom, P., Jenkins, A., Ask, E.,Facinelli, G. & Skold, O. (1990). Cloning and charac-terization of a DNA fragment that confers sulfon-amide resistance in a serogroup B, serotype 15strain of Neisseria meningitidis. Antimicrob. AgentsChemother. 34, 2277-2279.
Lane, B. R., Ast, J. C., Hossler, P. A., Mindell, D. P.,Bartlett, M. S., Smith, J. W. & Meshnick, S. R.(1997). Dihydropteroate synthase polymorphisms inPneumocystis carinii. J. Infect. Dis. 175, 482-485.
Laskowski, R. A., MacArthur, M. W., Moss, D. S. &Thornton, J. M. (1993). PROCHECK: a program tocheck the stereochemical quality of protein struc-tures. J. Appl. Crystallog. 26, 283-291.
LoÂpez, P., Espinosa, M., Greenberg, B. & Lacks, S. A.(1987). Sulfonamide resistance in Streptococcus pneu-moniae: DNA sequence of the gene encoding dihy-dropteroate synthase and characterization of theenzyme. J. Bacteriol. 169, 4320-4326.
Maskell, J. P., Sefton, A. M. & Hall, L. M. (1997). Mech-anism of sulfonamide resistance in clinical isolatesof Streptococcus pneumoniae. Antimicrob. AgentsChemother. 41, 2121-2126.
Mei, Q., Gurunathan, S., Masur, H. & Kovacs, J. A.(1998). Failure of co-trimoxazole in Pneumocystis car-inii infection and mutations in dihydropteroatesynthase gene. Lancet, 351, 1631-1632.
Nakajima, H. (1993). Tuberculosis: a global emergency.World Health, 46, 3.
Navaza, J. (1994). AMoRe: an automated package formolecular replacement. Acta Crystallog. sect. A, 50,157-163.
Nopponpunth, V., Sirawaraporn, W., Greene, P. J. &Santi, D. V. (1999). Cloning and expression ofMycobacterium tuberculosis and Mycobacterium leprae
dihydropteroate synthase in Escherichia coli.J. Bacteriol. 181, 6814-6821.
Nunn, P. & Kochi, A. (1993). A deadly duo - TB andAIDS. World Health, 46, 7-8.
Otwinowski, Z. & Minor, W. (1997). Processing of X-raydiffraction data collected in oscillation mode.Methods Enzymol. 276, 307-326.
Padaycachee, T. & Klugman, K. P. (1999). Novel expan-sions of the gene encoding dihydropteroatesynthase in trimethoprim-sulfamethoxazole-resistantStreptococcus pneumoniae. Antimicrob. Agents Chemo-ther. 43, 2225-2230.
Pannu, N. S. & Read, R. J. (1996). Improved structurere®nement through maximum likelihood. Acta Crys-tallog. sect. A, 52, 659-668.
Pashley, T. V., Volpe, F., Pudney, M., Hyde, J. E., Sims,P. F. G. & Delves, C. J. (1997). Isolation and molecularcharacterization of the bifunctional hydroxymethyl-dihydropterin pyrophosphokinase-dihydropteroatesynthase gene from Toxoplasma gondii. Mol. Biochem.Parasitol. 86, 37-47.
Ramachandran, G. N., Venkatchalam, C. M. & Krimm,S. (1966). Stereochemical criteria for polypeptideand protein chain conformations 3. Helical andhydrogen-bonded polypeptide chains. Biophys. J. 6,849-872.
Raszka, W. V. J., Skillman, L. P. & McEvoy, P. L. (1994).In vitro susceptibility of clinical isolates of Mycobac-terium avium and M. intracellulare to folate antagon-ists. Diagn. Microbiol. Infect. Dis. 18, 201-204.
Read, R. J. (1986). Improved Fourier coef®cients formaps using phases from partial structures witherrors. Acta Crystallog. sect. A, 42, 140-149.
ReÂbeilleÂ, F., Macherel, D., Mouillon, J.-M., Garin, J. &Douce, R. (1997). Folate biosynthesis in higherplants: puri®cation and molecular cloning of abifunctional 6-hydroxymethyl-7,8-dihydropterinpyrophosphokinase/7,8-dihydropteroate synthaselocalized in mitochondria. EMBO J. 16, 947-957.
Richey, D. P. & Brown, G. M. (1969). The biosynthesisof folic acid: puri®cation and properties of theenzymes required for the formation of dihydro-pteroic acid. J. Biol. Chem. 244, 1582-1592.
Roland, S., Ferone, R., Harvey, R. J., Styles, V. L. &Morrison, R. W. (1979). The characteristics and sig-ni®cance of sulfonamides as substrates for Escheri-chia coli dihydropteroate synthase. J. Biol. Chem. 254,10337-10345.
Rouhi, A. M. (1999). Tuberculosis: a tough adversary.Chem. Eng. News, 77, 52-69.
Shi, W., Li, C. M., Tyler, P. C., Furneaux, R. H.,Grubmeyer, C., Schramm, V. L. & Almo, S. C.(1999). The 2.0 AÊ structure of human hypoxanthine-guanine phosphoribosyltransferase in complex witha transition-state analog inhibitor. Nature Struct.Biol. 6, 588-593.
Shiota, T., Disraely, M. N. & McCann, M. P. (1964). Theenzymatic synthesis of folate-like compounds fromhydroxymethyldihydropteridine pyrophosphate.J. Biol. Chem. 239, 2259-2266.
Shiota, T., Baugh, C. M., Jackson, R. & Dillard, R.(1969). The enzymatic synthesis of hydroxymethyl-dihydropteridine pyrophosphate and dihydrofolate.Biochemistry, 8, 5022-5028.
Slock, J., Stahly, D. P., Han, C. Y., Six, E. W. &Crawford, I. P. (1990). An apparent Bacillus subtilisfolic acid biosynthetic operon containing pab,an amphibolic trpG gene, a third gene requiredfor synthesis of para-aminobenzoic acid, and the

1212 M. tuberculosis DHPS
dihydropteroate synthase gene. J. Bacteriol. 172,7211-7226.
Snider, D. E. J. & Roper, W. L. (1992). The new tubercu-losis. N. Engl. J. Med. 326, 703-705.
Snider, D. E. J., Raviglione, M. & Kochi, A. (1994).Tuberculosis: pathogenesis, protection and control.In Tuberculosis: Pathogenesis, Protection and Control(Bloom, B. R., ed.), pp. 2-11, Am. Soc. Microbiol.,Washington, DC.
Swedberg, G., Castensson, S. & Skold, O. (1979). Charac-terization of mutationally altered dihydropteroatesynthase and its ability to form a sulfonamide-con-taining dihydrofolate analog. J. Bacteriol. 137, 129-136.
Swedberg, G., Ringertz, S. & Skold, O. (1998). Sulfona-mide resistance in Streptococcus pyogenes is associ-ated with differences in the amino acid sequenceof its chromosomal dihydropteroate synthase. Anti-microb. Agents Chemother. 42, 1062-1067.
Tomb, J. F., White, O., Kerlavage, A. R., Clayton, R. A.,Sutton, G. G., Fleischmann, R. D., Ketchum, K. A.,Klenk, H. P., Gill, S., Dougherty, B. A., Nelson, K.,Quackenbush, J., Zhou, L., Kirkness, E. F. &Peterson, S., et al. (1997). The complete genomesequence of the gastric pathogen Helicobacter pylori.Nature, 388, 539-547.
Triglia, T. & Cowman, A. F. (1994). Primary structureand expression of the dihydropteroate synthetasegene of Plasmodium falciparum. Proc. Natl Acad. Sci.USA, 91, 7149-7153.
Triglia, T., Menting, J. G., Wilson, C. & Cowman, A. F.(1997). Mutations in dihydropteroate synthase areresponsible for sulfone and sulfonamide resistancein Plasmodium falciparum. Proc. Natl Acad. Sci. USA,94, 13944-13949.
Tronrud, D. E., Holden, H. M. & Matthews, B. W.(1987). An ef®cient general-purpose least-squares
re®nement program for macromolecular structures.Acta Crystallog. sect. A, 43, 489-501.
Vedantam, G., Guay, G. G., Austria, N. E., Doktor, S. Z.& Nichols, B. P. (1998). Characterization ofmutations contributing to sulfathiazole resistance inEscherichia coli. Antimicrob. Agents Chemother. 42, 88-93.
Vinnicombe, H. G. & Derrick, J. P. (1999a). Dihydrop-teroate synthase from Streptococcus pneumoniae:characterization of substrate binding order and sul-fonamide inhibition. Biochem. Biophys. Res. Commun.258, 752-757.
Vinnicombe, H. G. & Derrick, J. P. (1999b). Dihydrop-teroate synthase: an old drug target revisited.Enzyme Catalysis. Struct. Dynam. Chem. 278, 53-58.
Volpe, F., Dyer, M., Scaife, J. G., Darby, G., Stammers,D. K. & Delves, C. J. (1992). The multifunctionalfolic acid synthesis fas gene of Pneumocystis cariniiappears to encode dihydropteroate synthase andhydroxymethyldihydropterin pyrophosphokinase.Gene, 112, 213-218.
Walter, R. D. & Konigk, E. (1980). 7,8-Dihydropteroate-synthesizing enzyme from Plasmodium chaboudi.Methods Enzymol. 66, 564-570.
Wang, P., Lee, C. S., Bayoumi, R., Djimde, A., Doumbo,O., Swedberg, G., Dao, L. D., Mshinda, H., Tanner,M., Watkins, W. M., Sims, P. F. & Hyde, J. E.(1997). Resistance to antifolates in Plasmodium falci-parum monitored by sequence analysis of dihydrop-teroate synthetase and dihydrofolate reductasealleles in a large number of ®eld samples of diverseorigins. Mol. Biochem. Parasitol. 89, 161-177.
Wingard, L. B. (1991). Human Pharmacology: Molecular toClinical, Wolfe Medical Publications Ltd., London.
Woods, D. D. (1940). The relation of p-aminobenzoicacid to the mechanism of the action of sulphanila-mide. Br. J. Exp. Pathol. 21, 74-90.
Edited by I. Wilson
(Received 12 May 2000; received in revised form 1 August 2000; accepted 1 August 2000)
Copyright © 2022 FDOKUMEN




















