
Crystal structure of chondroitin AC lyase, a representative of a family of glycosaminoglycan...
13
Crystal Structure of Chondroitin AC Lyase, a Representative of a family of Glycosaminoglycan Degrading Enzymes James Fe ´ thie `re 1,3 , Bernhard Eggimann 2 and Miroslaw Cygler 1,3 * 1 Biotechnology Research Institute, NRC, 6100 Royalmount Avenue, Montre ´al Que ´bec, H4P 2R2, Canada 2 Ibex Technologies, 5485 Pare ´ Street, Montre ´al, Que ´bec H4P 2R2, Canada 3 Montre ´al Joint Centre for Structural Biology, Montreal Que ´bec, Canada Glycosaminoglycans (GAGs), highly sulfated polymers built of hexosa- mine-uronic acid disaccharide units, are major components of the extra- cellular matrix, mostly in the form of proteoglycans. They interact with a large array of proteins, in particular of the blood coagulation cascade. Degradation of GAGs in mammalian systems occurs by the action of GAG hydrolases. Bacteria express a large number of GAG-degrading lyases that break the hexosamine-uronic acid bond to create an unsatu- rated sugar ring. Flavobacterium heparinum produces at least five GAG lyases of different specificity. Chondroitin AC lyase (chondroitinase AC, 75 kDa) is highly active toward chondroitin 4-sulfate and chondroitin-6 sulfate. Its crystal structure has been determined to 1.9 A ˚ resolution. The enzyme is composed of two domains. The N-terminal domain of approxi- mately 300 residues contains mostly a-helices which form a doubly- layered horseshoe (a subset of the (a/a) 6 toroidal topology). The 370 residues long C-terminal domain is made of b-strands arranged in a four layered b-sheet sandwich, with the first two sheets having nine strands each. This fold is novel and has no counterpart in full among known structures. The sequence of chondroitinase AC shows low level of hom- ology to several hyaluronate lyases, which likely share its fold. The shape of the molecule, distribution of electrostatic potential, the pattern of con- servation of the amino acids and the results of mutagenesis of hyaluro- nate lyases, indicate that the enzymatic activity resides primarily within the N-terminal domain. The most likely candidate for the catalytic base is His225. Other residues involved in catalysis and/or substrate binding are Arg288, Arg292, Lys298 and Lys299. # 1999 Academic Press Keywords: crystal structure; glycosaminoglycan; chondroitin; chondroitinase; GAG lyase *Corresponding author Introduction Glycosaminoglycans (GAGs) are heterogeneous polymers built of repeating disaccharide units which are extensively N and O-sulfated. The disac- charide units of the polymers are composed of a hexosamine and a uronic acid linked by (1,3) or (1,4) linkages. They are usually covalently linked at the reducing end to core proteins (proteoglycans) through an O-glycosidic linkage to a serine residue. GAGs are divided into four main classes: chondroi- tin sulfate and dermatan sulfate, heparin and heparan sulfate, hyaluronic acid, and keratan sul- fate. Chondroitin sulfate and dermatan sulfate con- tain D-galactosamine linked to glucuronic or iduronic acid, respectively. Sulfation occurs to var- ious degrees at the 2 position of uronic acid and 4, 6 positions of galactosamine, and the amino group of galactosamine is acetylated (Iozzo, 1998). With the exception of hyaluronic acid, GAGs are synthesized as components of proteoglycans in the Golgi where extensive modifications to the repeat- ing disaccharides are made prior to subsequent secretion from the cell (Lidholt, 1997). Proteogly- cans remain either bound to the cell surface or become part of the extracellular matrix (ECM). Through their interactions with other ECM E-mail address of the corresponding author: [email protected] Abbreviations used: GAG, glycosaminoglycan; ECM, extracellular matrix; MAD, multiwavelength anomalous dispersion; MIRAS, multiple isomorphous replacement with anomalous scattering; FOM, figure of merit. Article No. jmbi.1999.2698 available online at http://www.idealibrary.com on J. Mol. Biol. (1999) 288, 635–647 0022-2836/99/190635–13 $30.00/0 # 1999 Academic Press
Transcript of Crystal structure of chondroitin AC lyase, a representative of a family of glycosaminoglycan...

Article No. jmbi.1999.2698 available online at http://www.idealibrary.com on J. Mol. Biol. (1999) 288, 635±647
Crystal Structure of Chondroitin AC Lyase, aRepresentative of a family of GlycosaminoglycanDegrading Enzymes
James FeÂthieÁre1,3, Bernhard Eggimann2 and Miroslaw Cygler1,3*
1Biotechnology ResearchInstitute, NRC, 6100Royalmount Avenue, MontreÂalQueÂbec, H4P 2R2, Canada2Ibex Technologies, 5485 PareÂStreet, MontreÂal, QueÂbecH4P 2R2, Canada3MontreÂal Joint Centre forStructural Biology, Montreal
QueÂbec, Canada*Corresponding author
through an O-glycosidic linkage
E-mail address of the [email protected]
Abbreviations used: GAG, glycosextracellular matrix; MAD, multiwadispersion; MIRAS, multiple isomorwith anomalous scattering; FOM, ®
0022-2836/99/190635±13 $30.00/0
Glycosaminoglycans (GAGs), highly sulfated polymers built of hexosa-mine-uronic acid disaccharide units, are major components of the extra-cellular matrix, mostly in the form of proteoglycans. They interact with alarge array of proteins, in particular of the blood coagulation cascade.Degradation of GAGs in mammalian systems occurs by the action ofGAG hydrolases. Bacteria express a large number of GAG-degradinglyases that break the hexosamine-uronic acid bond to create an unsatu-rated sugar ring. Flavobacterium heparinum produces at least ®ve GAGlyases of different speci®city. Chondroitin AC lyase (chondroitinase AC,75 kDa) is highly active toward chondroitin 4-sulfate and chondroitin-6sulfate. Its crystal structure has been determined to 1.9 AÊ resolution. Theenzyme is composed of two domains. The N-terminal domain of approxi-mately 300 residues contains mostly a-helices which form a doubly-layered horseshoe (a subset of the (a/a)6 toroidal topology). The �370residues long C-terminal domain is made of b-strands arranged in a fourlayered b-sheet sandwich, with the ®rst two sheets having nine strandseach. This fold is novel and has no counterpart in full among knownstructures. The sequence of chondroitinase AC shows low level of hom-ology to several hyaluronate lyases, which likely share its fold. The shapeof the molecule, distribution of electrostatic potential, the pattern of con-servation of the amino acids and the results of mutagenesis of hyaluro-nate lyases, indicate that the enzymatic activity resides primarily withinthe N-terminal domain. The most likely candidate for the catalytic base isHis225. Other residues involved in catalysis and/or substrate binding areArg288, Arg292, Lys298 and Lys299.
# 1999 Academic Press
Keywords: crystal structure; glycosaminoglycan; chondroitin;
chondroitinase; GAG lyaseIntroduction
Glycosaminoglycans (GAGs) are heterogeneouspolymers built of repeating disaccharide unitswhich are extensively N and O-sulfated. The disac-charide units of the polymers are composed of ahexosamine and a uronic acid linked by (1,3) or(1,4) linkages. They are usually covalently linked atthe reducing end to core proteins (proteoglycans)
to a serine residue.
ing author:
aminoglycan; ECM,velength anomalousphous replacementgure of merit.
GAGs are divided into four main classes: chondroi-tin sulfate and dermatan sulfate, heparin andheparan sulfate, hyaluronic acid, and keratan sul-fate. Chondroitin sulfate and dermatan sulfate con-tain D-galactosamine linked to glucuronic oriduronic acid, respectively. Sulfation occurs to var-ious degrees at the 2 position of uronic acid and 4,6 positions of galactosamine, and the amino groupof galactosamine is acetylated (Iozzo, 1998).
With the exception of hyaluronic acid, GAGs aresynthesized as components of proteoglycans in theGolgi where extensive modi®cations to the repeat-ing disaccharides are made prior to subsequentsecretion from the cell (Lidholt, 1997). Proteogly-cans remain either bound to the cell surface orbecome part of the extracellular matrix (ECM).
Through their interactions with other ECM# 1999 Academic Press

636 Crystal Structure of Chondroitinase AC
components they contribute to the general architec-ture and permeability properties of various connec-tive tissues such as basement membranes, cartilageand skin (Iozzo, 1998). A wide variety of proteinsthat bind to GAGs have been identi®ed (Hilemanet al., 1998). Among them are components of theblood coagulation cascade, basic and acidic ®bro-blast growth factors, plasma lipoproteins, ®bronec-tin, vitronectin, laminin, etc. It has also beenshown in recent years that a number of pathogensranging from viruses to protozoans interact speci®-cally with cell surface GAGs to recognize and bindto their target cells (Sawitzky, 1996).
Microorganisms are a major source of GAG-degrading enzymes (Sutherland, 1995), particularlyin the case of soil bacteria which may depend onconnective tissues in animal carcasses as a nutrientsource. Based on their catalytic mechanism GAG-degrading enzymes are divided into two distinctclasses: lyases and hydrolases. Cleavage of the hex-uronic acid-hexosamine bond always involves astandard glycosidase mechanism of either invert-ing or retaining type. On the other hand, cleavageof the hexosamine-hexuronic acid bond can occurthrough either a hydrolytic or a lyase mechanism,depending upon the enzyme. The lyase mechanisminvolves the removal of a relatively acidic C5 pro-ton of the uronic acid followed by the eliminationof a 4-linked hexosamine, resulting in the for-mation of an unsaturated C4-C5 bond on the hex-uronic acid moiety. Little is known about thecatalytic machinery of these enzymes and no three-dimensional structure of a GAG-degrading lyasehas been reported to date. The only polysaccharidelyase structures presently known are of pectateand pectin lyases (Yoder et al., 1993; Pickersgillet al., 1994; Mayans et al., 1997).
Bacterial GAG lyases fall into three categories:heparinases, chondroitinases, and hyaluronidasesand are somewhat better characterized biochemi-cally than the GAG hydrolases. Within each cat-egory there are endo- and exo- lyases. Severalbacterial GAG lyases are used routinely as tools inthe analysis of GAG composition and structure.However, only in recent years have such enzymesbeen cloned and sequenced (Su et al., 1996; Farrellet al., 1995; Canard et al., 1994; Hynes et al., 1995;Berry et al., 1994). Comparison of their amino acidsequences indicates the existence of several familiesof weakly homologous enzymes with no detectablehomology between the families, and raises thequestion of evolutionary links between theseenzymes.
Flavobacterium heparinum, when induced,expresses ®ve distinct and sequence-unrelatedGAG lyases: three heparinases and two chondroiti-nases. We have determined the ®rst crystal struc-ture of a GAG lyase, that of chondroitinase AC, ata resolution of 1.9 AÊ . This enzyme belongs to theendo-lyase family (Jandik et al., 1994). As the lackof homology suggested, its fold is different fromthat of pectin/pectate lyases. However, the
N-terminal domain, which most likely performsthe catalytic function, shows partial fold similarityto several glycosidases, enzymes that also breakdown carbohydrates but use a distinctly differentcatalytic mechanism. The shape of the domainsuggests that the chondroitin sulfate binding site islocated in a pronounced deep cleft within theN-terminal domain narrowing down substantiallythe list of potential catalytic residues.
Results
Chondroitinase AC (GenBank accession numberU27583 or gi1002525) is synthesized as a 700amino acid long polypeptide, including a 22 resi-due long signal peptide. The mature enzyme con-tains 678 residues and starts at Gln23. The ®rstthree N-terminal residues and the last one are dis-ordered and were not included in the model. Themolecule has approximate dimensions of45 AÊ � 50 AÊ � 95 AÊ and is composed of two dis-tinct domains with no cross-over between them(Figure 1(a)). The topology of chondroitinase AC isshown diagrammatically in Figure 1(b). TheN-terminal domain (residues 23-328), which com-prises approximately half of the protein, is almostexclusively a-helical. It is connected by a short lin-ker (residues 329-336) to the C-terminal half of themolecule (residues 337-699) built, with the excep-tion of one short a-helix, entirely of b-sheets.
The N-terminal aaa-helical domain
The N-terminal domain is composed primarilyof a-helices, 12 in total, ranging in length fromthree to four turns. Ten of the helices, a3 to a12,are arranged into pairs in the shape of a double-layered horseshoe, or an open toroid (Figure 2).The helices within each pair are consecutive in thelinear sequence and fold into hairpins. They aretilted by approximately 45 � to the plane of thehorseshoe. The inner layer of the horseshoe isformed by odd-numbered helices, all oriented thesame way, and the outer layer consists of the even-numbered helices, oriented opposite way.
Loops connecting a-helices within each hairpinare located on the same (top) side of the horseshoeand are from three to nine residues long. Loops con-necting the consecutive hairpins are located on theopposite, bottom side of the horseshoe, and varyconsiderably in length. The ®rst two connectionsare short, less than ten residues in length. The lasttwo are much longer, nearly 30 residues long. Thethird connecting loop (between a8 and a9) foldsinto a two-stranded antiparallel b-sheet, while thefourth one (between a10 and a11) forms an -loopwith short 310 helical turns in the middle.
Helices a1 and a2 do not participate in the pair-ing described above. Helix a1 is located on the topside of the horseshoe and closes off the central clefton one side. Helix a2 is roughly perpendicular toa1 and packs against helix a3 on the outer side ofthe horseshoe. This helix participates extensively in
the packing of protein molecules in the crystal.
Figure 1. (a) Ca trace of chon-droitinase AC displayed withMOLSCRIPT (Kraulis, 1991). TheC-terminal domain is shown inthick lines, residues His225,Arg288, Arg292, Lys298 andLys299 are displayed in full toshow their location relative to theC-terminal domain (see Figure 2).(b) Topology diagram of chondroi-tinase AC. a-Helices are displayedas cylinders and b-strands as thickarrows. Loops connecting strandswithin the same sheet or helicalhairpin are shown in black, thosebetween the sheets or hairpins ingray. The assignment of secondarystructures is as follows:a1 � 27-39, a2 � 46-56, a3 � 77-93,a4 � 103-119, a5 � 126-143, a6 �153-161, a7 � 174-189, a8 � 193-204, b1 � 215-217, b2 � 221-224,a9 � 233-249, a10 � 258-270, a11 �302-311, a12 � 316-327, b1 � 339-343, b2 � 348-353, b3 � 356-361,b4 � 387-391, b5 � 412-415, b6 �436-440, b7 � 445-453, b8 � 456-464, b9 � 469-478, b10 � 483-492, b11 � 497-499, b12 � 502-504, b13 � 508-514, b14 � 519-522,b15� 525-529, b16� 523-545, b17�557-571, b18� 576-584, a1 � 589-
593, b19� 601-606, b20 � 609-614,b21 � 619-624, b22 � 628-632, b23 �635-639, b24 � 643-648, b25 � 655-659, b26 � 667-674, b27 � 679-686,b28 � 696-699.
Crystal Structure of Chondroitinase AC 637
Helices within the horseshoe contact each othermainly through non-polar side-chains in a fashiontypical for helix bundles. Several clusters of aro-matic side-chains are distributed throughout thedomain and participate in packing. Charged resi-dues are distributed mostly on the outside surfaceof the horseshoe and within the cleft. They formseveral salt bridges and contribute to the stabiliz-ation of this fold.
The C-terminal bbb-sheet domain
With the exception of a short a-helix, the b-sheetdomain is built entirely from b-strands which formfour sheets arranged in a nearly parallel fashion
and with only a small twist in each sheet (Figure 3).Viewed in the plane of the sheets and perpendicu-lar to individual strands, the domain is diamondshaped, with its geometric center located in-between sheet S1 and S2. Each sheet is built fromantiparallel strands. The ®rst two sheets are themost extensive, having nine strands each. In sheetS1 the ®fth strand is the shortest and divides thesheet into two halves. The N-terminal half providesa large fraction of the interface with the a-helicaldomain. In the second sheet, S2, the ®rst ®vestrands are the longest and pack against theN-terminal part of sheet S1. The remaining fourstrands are shorter and are tilted approximately30 � relative to the rest of the sheet (Figure 3). As a
result of this tilt and a shorter central strand within
Figure 3. Ribbon stereo drawing of the b-domain.Sheets S1 - S4 are displayed in different colors. The Ca2�
is shown as a yellow sphere. Loops within one sheet arecolored the same as the sheet, those that cross betweenthe sheets are colored yellow. The loops extending fromsheet S1 form one end of the diamond shape.
Figure 2. Ribbon stereo drawing of the a-helicaldomain showing the arrangement of a-helical hairpinsinto a horseshoe shape. Individual helices are inclined at�45 � angle to the horseshoe plane. Several highly con-served residues are shown in full, including His225,Arg288, Arg292, Lys298 and Lys299. Each hairpin ispainted in a different color. The ®rst two helices, a1 anda2, which are not part of the horseshoe arrangement areshown in light gray.
638 Crystal Structure of Chondroitinase AC
sheet S1, the centers of these two sheets are some-what farther apart than their remaining segments.Three water molecules are found buried betweenthe two sheets in this region, forming multiplehydrogen bonds to the side-chain and main-chainatoms. Sheet S3 contains six strands which lineagainst the ®ve longer strands of the second sheet.Finally, the last sheet, S4, has only four strands.The faces of the sheets that contact each other arelined with predominantly hydrophobic and aro-matic residues.
All connections between b-strands in sheetsS2, S3 and S4 are short, with the exception of acrossover from sheet S2 to sheet S3 which ismore extended and contains the only short a-helix found in this domain. On the other hand,the connections between strands in sheet S1, orloops which lead from sheet S1 to sheet S2, arelong and extend away from the sheet, formingthe second half of the diamond shape of thisdomain (Figure 3).
Domain interactions
The contacts between the N and C-terminaldomains extend over a large area, burying 3700 AÊ 2
of their surfaces (1800 AÊ 2 on the a-helical domainand 1900 AÊ 2 on the b-sheet domain) which corre-sponds to �15 % of the total molecular surface ofthe protein (24,700 AÊ 2). The a-helical domain con-tacts the C-terminal domain with the side of itshorseshoe shape, near one of the open ends(Figure 4). The surface of the a-helical domaininvolved in these interactions is convex and isformed by residues within the second half of thisdomain: the structured loop between helices a8and a9, the C-terminal part of helix a10 and the-loop between helices a10 and a11. This surface®ts into a depression formed by the N-terminal
half of sheet S1 and the extended loops of theC-terminal domain. Speci®cally, there are extensivecontacts involving strands b1 to b4 and all fourloops protruding on this side of the S1 sheet. Inaddition, there are also contacts with the mostextreme strand of sheet S3, formed by the C termi-nus of the protein (Figure 1). The cleft of the a-heli-cal domain is in the proximity of the C-terminaldomain and one end of the cleft is partially closedby the tips of the loops which extend from thesheet S1 (Figure 1). The central part of the interfaceis formed from hydrophobic and some unchargedpolar residues, with a notable exception of Asp282and Arg361. This aspartic acid is, however, hydro-gen bonded to the hydroxyl group of Tyr326 andto the NH group of the neighboring Asn284 whilethe arginine hydrogen bonds to the carbonyl ofAsn284 and to a water molecule. There are 11water molecules buried within the interface andalmost all hydrogen bonds within the interface arebridged through them. In addition, a His225...Glu371...Arg288...Glu376...Arg292 salt-bridgedcluster of charged side-chains at the edge of theinterface adds to the stability of this association.
Ca2� binding
In the course of structure determination it wasnoted that all heavy atom derivatives had a com-mon site. This site is located in the b-sheet domain,near the short middle strand b5 of sheet S1, on theside that is in contact with the a-helical domain. Astrong peak was observed in the difference electrondensity map at this position. The pentagonal bipyr-amidal coordination of oxygen atoms around thispeak indicated that this is likely a calcium ion. Theaverage distance from this site to the oxygenligands of 2.5 AÊ con®rms this assignment (Glusker,1991; McPhalen et al., 1991). The Ca2� coordinatesside-chains from the polypeptide chain immedi-ately preceding and following strand b5, tying theincoming and outgoing chains together (Figure 5).
Such a close approach of the two chains is pro-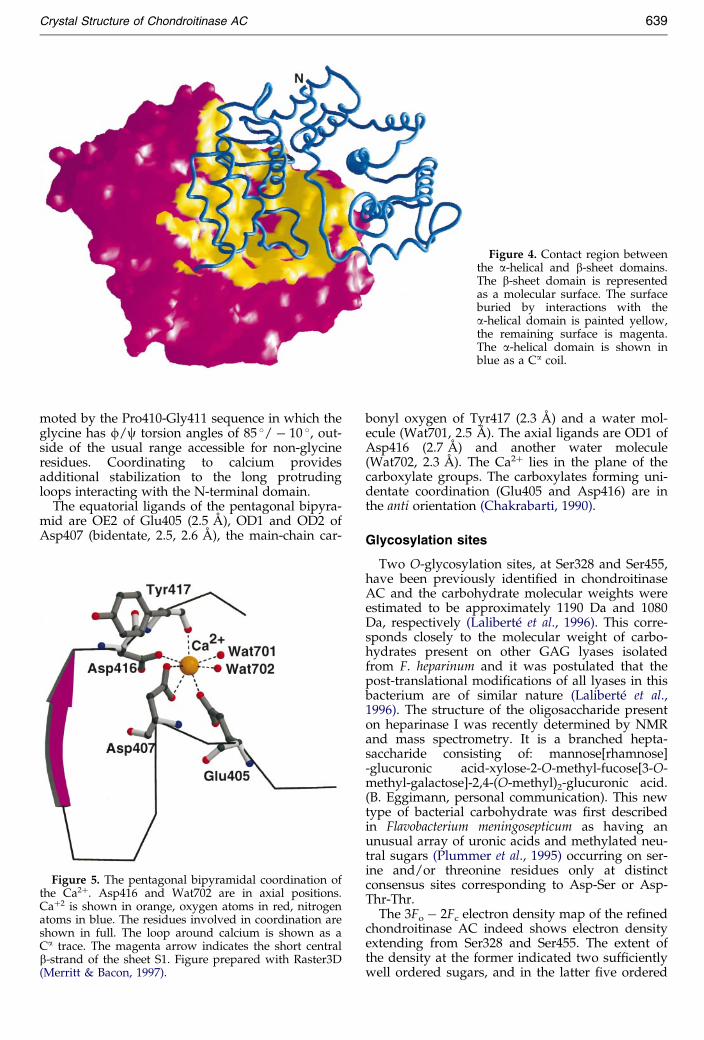
Figure 4. Contact region betweenthe a-helical and b-sheet domains.The b-sheet domain is representedas a molecular surface. The surfaceburied by interactions with thea-helical domain is painted yellow,the remaining surface is magenta.The a-helical domain is shown inblue as a Ca coil.
Crystal Structure of Chondroitinase AC 639
moted by the Pro410-Gly411 sequence in which theglycine has f/c torsion angles of 85 �/ ÿ 10 �, out-side of the usual range accessible for non-glycineresidues. Coordinating to calcium providesadditional stabilization to the long protrudingloops interacting with the N-terminal domain.
The equatorial ligands of the pentagonal bipyra-mid are OE2 of Glu405 (2.5 AÊ ), OD1 and OD2 of
Asp407 (bidentate, 2.5, 2.6 AÊ ), the main-chain car-Figure 5. The pentagonal bipyramidal coordination ofthe Ca2�. Asp416 and Wat702 are in axial positions.Ca�2 is shown in orange, oxygen atoms in red, nitrogenatoms in blue. The residues involved in coordination areshown in full. The loop around calcium is shown as aCa trace. The magenta arrow indicates the short centralb-strand of the sheet S1. Figure prepared with Raster3D(Merritt & Bacon, 1997).
bonyl oxygen of Tyr417 (2.3 AÊ ) and a water mol-ecule (Wat701, 2.5 AÊ ). The axial ligands are OD1 ofAsp416 (2.7 AÊ ) and another water molecule(Wat702, 2.3 AÊ ). The Ca2� lies in the plane of thecarboxylate groups. The carboxylates forming uni-dentate coordination (Glu405 and Asp416) are inthe anti orientation (Chakrabarti, 1990).
Glycosylation sites
Two O-glycosylation sites, at Ser328 and Ser455,have been previously identi®ed in chondroitinaseAC and the carbohydrate molecular weights wereestimated to be approximately 1190 Da and 1080Da, respectively (Laliberte et al., 1996). This corre-sponds closely to the molecular weight of carbo-hydrates present on other GAG lyases isolatedfrom F. heparinum and it was postulated that thepost-translational modi®cations of all lyases in thisbacterium are of similar nature (Laliberte et al.,1996). The structure of the oligosaccharide presenton heparinase I was recently determined by NMRand mass spectrometry. It is a branched hepta-saccharide consisting of: mannose[rhamnose]-glucuronic acid-xylose-2-O-methyl-fucose[3-O-methyl-galactose]-2,4-(O-methyl)2-glucuronic acid.(B. Eggimann, personal communication). This newtype of bacterial carbohydrate was ®rst describedin Flavobacterium meningosepticum as having anunusual array of uronic acids and methylated neu-tral sugars (Plummer et al., 1995) occurring on ser-ine and/or threonine residues only at distinctconsensus sites corresponding to Asp-Ser or Asp-Thr-Thr.
The 3Fo ÿ 2Fc electron density map of the re®nedchondroitinase AC indeed shows electron densityextending from Ser328 and Ser455. The extent ofthe density at the former indicated two suf®ciently
well ordered sugars, and in the latter ®ve ordered
Figure 6. The electron density extending from theside-chain of Ser455. Modeled carbohydrate is shown inthick lines.
640 Crystal Structure of Chondroitinase AC
sugars. The remaining sugars are disordered in thecrystal. The shape of the electron density at bothsites corresponded well to the expected oligosac-charide structure (Figure 6). The well ordered sac-charide attached to Ser455 is involved in theinteractions with two arginine residues throughthe glucuronic acid. Its carboxylate group formssalt bridges with Arg415 and Arg421, which inturn ¯ank the Ca2�-coordinating residues Asp416and Tyr417. Additionally, a water molecule(Wat994) bridges one of the carboxylate oxygenatoms of glucuronic acid to the carboxylate ofAsp454, and hydrogen bonds to the ring oxygen ofthis sugar unit. Both carbohydrates extend into thesolvent channel between symmetry-related mol-ecules near the 4-fold axis, contacting mostly theside-chains from the parent molecule.
No speci®c role has been proposed for theseO-linked oligosaccharides. Their positioning in thestructure of chondroitinase AC and their speci®cinteraction with the protein, in the case of Ser455attached oligosaccharide, presents a possibility forsome role in protein folding, as in mammalian sys-tems.
Discussion
Chondroitin AC lyase is the second family ofpolysaccharide lyases for which three-dimensionalstructures have been determined. The previouslyinvestigated enzymes are those of pectin and pec-tate lyases which share a similar fold, a right-handed parallel b-helix (Yoder et al., 1993;Pickersgill et al., 1994; Mayans et al., 1997). Thisfold is likely common to other enzymes related tothe degradation of pectic substances from the plantcell wall (Henrissat et al., 1995). It is also rep-resented in glycosyl hydrolases but occurs rarely:out of 70 known families (Henrissat & Bairoch,1996; http://afmb.cnrs-mrs.fr/�pedro/CAZY/) ithas been found to date in only one (family 28,
rhamnogalacturonase, Petersen et al., 1997; polyga-lacturonase, Pickersgill et al., 1998). The fold ofchondroitin AC lyase is very different from that ofpectin or pectate lyases and represents a differentevolutionary solution to a similar chemical pro-blem. The molecule is divided into an a-helicaldomain and a b-sheet domain and, as we discussbelow, the catalytic residues are likely residingwithin the a-helical domain. The C-terminaldomain, although made of b-strands, forms a stackof b-sheets with no resemblance to the b-helix top-ology.
We have conducted an exhaustive searchthrough the available protein sequence databaseswith the program -BLAST (Altschul et al., 1997)and have used the program DALI (Holm &Sander, 1997) to search for fold similarity to otherproteins both at the level of individual domainsand at the level of the entire protein. The compari-son was made against all proteins deposited at theBrookhaven Protein Databank (Bernstein et al.,1977) as of September 1998.
Sequence homology
The search through sequence databases discov-ered several sequences with signi®cant homologyto chondroitinase AC. Sequences with the highestscores and which are homologous over the entirelength of chondroitinase AC correspond to severalbacterial hyaluronate lyases. Not only are hyaluro-nic acid and chondroitin or chondroitin sulfatesimilar in primary structure, but each substrate canbe acted upon by the other enzyme (Tam & Chan,1985; Lin et al., 1997; Gu et al., 1995).
Several hyaluronate lyases show a low overalllevel of homology to chondroitinase AC extendingover the entire length of the enzymes (Figure 7).Hyaluronate lyases from Streptococcus agalactiae,Staphylococcus pneumoniae and S. aureus have 19-20 % identity to chondroitinase AC. A putativelyase from Streptomyces coelicolor shows 22 % iden-tity while hyaluronidase from Propionibacteriumacnes is 16 % identical. Other proteins display hom-ology to chondroitinase AC only within a limitedregion. Chondroitin lyase 2 from Bacteroides thetaio-taomicron shows discernable homology only start-ing from residue �210 of chondroitinase AC butextending through the rest of the protein at thelevel of 20 % identity. This range includes theC-terminal domain and the end of the N-terminaldomain, which is involved in contacts with the for-mer. Another protein with clear homology to chon-droitinase AC is a broad speci®city chondroitinaseABC from Proteus vulgaris, but most interestinglythe similarity extends only over the C-terminaldomains of both proteins. Chondroitinase ABC issigni®cantly larger than chondroitinase AC andhas a �600 residue long N-terminal domain. Sincethe N-terminal domain of chondroitinase AC ismost likely mainly responsible for substrate bind-ing and catalysis, the lack of homology betweentheir N-terminal domains while at the same time
having similarity between their C-terminal
Figure 7. Alignment of sequences of chondroitinase AC, several hyaluronidases and chondroitinase ABC. Putative active site residues His225, Arg288 and Arg292 aremarked with asterisks. The ®rst two are not conserved in lyase 2 and chondroitinase ABC which do not share homology with the N-terminal domain of chondroitinase ACand likely differ in the three-dimensional structure of their catalytic domains.Abbreviations: chon_AC, chondroitinase AC from F. heparinum (gi 1002525); S_agalact, hyaluronate lyase from S. agalactiae (gi 3287247); S_agal3502, hyaluronate lyase fromS. agalacticae strain 3502 (gi 595847); S_pneumoni, hyaluronidase from S. pneumoniae (gi 437705); S_aureus, hyaluronate lyase from S. aureus (gi 705406); S_coelic, putativesecreted lyase from S. coelicolor (gi 3355682); P_acnes, hyaluronidase from P. acnes (gi562086); lyase 2, lyase 2 from B. thetaiotaomicron (gi 895611); chon_ABC, chondroitinABC lyase from P. vulgaris (gi 1095454).

642 Crystal Structure of Chondroitinase AC
domains is somewhat surprising. It makes a strongcase for the importance of the C-terminal domainof chondroitinase AC for the overall function of theenzyme, although it is not clear what role it plays.
The BLAST search also indicated that there isweak homology of the region 45-205 of chondroiti-nase AC to some viral envelope proteins. This is aninteresting ®nding when considered that some ofthese proteins bind carbohydrates.
Fold similarities
Global comparison
The searches with the entire protein using theDALI algorithm detected no global similarities inthe sense that of 678 residues of chondroitinase ACno more than circa 200 (always within a singledomain) could be equivalenced to parts of otherproteins with an r.m.s. deviation of less than 5 AÊ .These results indicate that the overall topology/arrangement of domains of chondroitinase ACfrom F. heparinum is unique among proteins withknown three-dimensional structures. However, atthe level of the individual chondroitinase domainsinteresting similarities were found.
a-Helical domain
The main feature of this domain is the arrange-ment of ®ve hairpins of helices into a horseshoeshape which could be otherwise described as anincompletely closed toroid. Comparison with allthe proteins in the Protein Databank identi®ed sev-eral proteins that show similar toroidal topology ofa-helical hairpins. Among them are glucosylhydrolases from families 5, 8, 9 and 15 (see Davies& Henrissat, 1995), squalene-hopene cyclase(Wendt et al., 1997) and there is also a congruenceto the domains of farnesyltransferase (Park et al.,1997). The semblance of chondroitinase to polysac-charide hydrolases was rather unexpected since thecatalytic mechanisms of lyases and hydrolases aredifferent. Nevertheless, this observation pointstoward the possibility that the catalytic function ofthe chondroitinase resides in the a-helical domain.
The highest similarity score was to the C-term-inal domain of endoglucanase CelD (1clc, (a/a)6 ora/a toroid in SCOP classi®cation scheme) whichforms a toroid built of six a-helical hairpins. Out of303 Ca atoms of the chondroitinase domain 213atoms could be equivalenced to correspondingatoms in the endoglucanase with an r.m.s. devi-ation of 4.4 AÊ . The substrate binding site in CelD(PDB code 1gai) and other related glycosyl hydro-lases is located at the bottom of the toroid, wherethe inter-hairpin loops are located. The sixth hair-pin in these enzymes is made up by pairing theN-terminal helix with the C-terminal helix. Thishairpin is missing in chondroitinase AC and thatcreates a large cleft along the side of the horseshoe,
potentially enlarging the substrate binding area.A comparable degree of similarity was found tothe C-terminal domain of squalene-hopene cyclase:200 equivalences with 4.3 AÊ r.m.s. deviation. Thisdomain forms a very regular toroid made of sixhairpins. However, more interestingly, the N-term-inal domain of this cyclase has also a toroidalarrangement of helices but contains only foura-helical hairpins. The topology of the ®rst twohelices differs from others mentioned above asthey are not consecutive in the linear sequence.This domain superimposes though very well onthat of chondroitinase. The N-terminal domain ofcyclase contains even an equivalent for chondroiti-nase's helix a1, which runs near the center of thehorseshoe. Due to the differences in connectivity,the similarity to cyclase N-terminal domain wasnot detected by automated searches.
The third most signi®cant similarity was to thechain B of farnesyltransferase (Park et al., 1997).Here 202 Ca atoms were equivalenced with anr.m.s. deviation of 4.6 AÊ .
b-Sheet domain
The C-terminal domain consisting of a four-layered b-sheet has a partial structural homologyin one of the ®ve polypeptide chains of b-galactosi-dase from Escherichia coli (Jacobson et al., 1994). Of366 Ca atoms of the chondroitinase domain, 174are structurally homologous to the C-terminal seg-ment of chain A of b-galactosidase and superim-pose with a r.m.s. deviation of 3.8 AÊ . Thetopological similarity corresponds to sheets S1 andS2 of chondroitinase AC and encompasses all butthe last (in sequence) strand of sheet S2. This is thecentral strand of sheet S2 and, consequently,b-galactosidase has two side-by-side four-strandedb-sheets in place of a nine-stranded sheet of chon-droitinase AC. While the topology of connectionsand strand directions are the same in bothdomains, the loops differ signi®cantly in theirlengths and their conformations. It was shown forchain A of b-galactosidase that it folds indepen-dently of the rest of the molecule (Celada et al.,1974) and serves to position certain residues in theactive site located in the TIM barrel domain. Whilethe role of the b-sheet domain in chondroitinaseAC is not clear, its semblance to the b-galactosi-dase domain may suggest that the C-terminaldomain of chondroitinase AC plays a supportiverole in enzymatic activity.
The similarity to b-galacosidase does not extendbeyond the two large sheets. The assembly of thefour sheets into a single domain of chondroitinaseAC is thus topologically distinct and presents anew fold.
Substrate binding site
Although there is no direct evidence at this pointas to the location and identity of the active site ofchondroitinase AC, several lines of reasoning
suggest that the substrate binds to the N-terminal
Figure 8. Molecular surface rep-resentation of chondroitinase ACviewed from the side of the a-heli-cal domain (in the plane of thehorseshoe) onto the cleft. Thea-helical domain is on the right, theb-sheet domain is on the left.(a) The surface is colored by theelectrostatic potential calculatedwith GRASP (Nicholls et al., 1991).The top and the bottom of thea-helical domain outside of thecleft display positive potential. Theinternal surface of the cleft is lar-gely positive with a few smallstrongly negative areas. (b) Distri-bution of highly conserved residueson lyase surface. The N-terminal(a-helical) domain is coloredorange, the C-terminal domain incolored pale blue and the surfacecorresponding to highly conservedresidues is colored magenta. Thereis a high concentration of such resi-dues within the cleft.
Crystal Structure of Chondroitinase AC 643
domain. The ®rst, and visually most compelling,indication comes from the shape of the molecule.The cleft along the side of the N-terminal domainis large enough to contain a long polysaccharidechain and, with some rearrangement of the longloop between helices a2 and a3, could easily bindthe middle of the GAG chain. The presence of sev-eral basic side-chains within the cleft, His225,Arg288, Arg292, Lys298 and Lys299, and othersnear the top and bottom rims, creates a positivepotential surface on the inside and near the ends ofthe cleft (Figure 8(a)), as expected for the bindingsite of the highly negatively charged chondroitinsulfate substrate. Interestingly, in the middle of thecleft there is a patch of negative potential createdby the presence of Asp178. This aspartate is con-served in all homologous lyases (Figure 7) andmost likely plays a role in the catalysis.
The second clue comes from the comparisonwith other enzymes with toroidal helical topology
for which structures of complexes with substrateanalogs are known, namely glucoamylase (Aleshinet al., 1996) and squalene-hopene cyclase (Wendtet al., 1997). When their structures (N-terminaldomain in the case of squalene-hopene cyclase) aresuperimposed on the a-helical domain of chondroi-tinase AC, the substrate analogs in the twoenzymes are found in roughly the same location inthe end of the cleft near the C-terminal domain.Such a location of the substrate binding site wouldindicate that the b-sheet domain plays an activerole in substrate binding, an observation supportedby the function of the b-sheet domain of b-galacto-sidase (Jacobson et al., 1994).
Sequence alignment with hyaluronate lyases pro-vides an even more convincing argument (Figures 7and 8(b)). Early work on the mechanism of hyaluro-nate lyases implied a role for a histidine in theactivity of the enzyme (Greiling et al., 1975), andmore recently His479 of hyaluronate lyase fromgroup B streptococci (S. agalactiae) has been ident-
i®ed as essential for the enzymatic activity (Lin et al.,
644 Crystal Structure of Chondroitinase AC
1997). This histidine occurs within a stretch of wellconserved residues and corresponds to His225 inchondroitinase AC (Figure 7). At the end of thisstretch is a conserved sequence GR288XXSR292,with the two arginine residues lining the cleft andpossibly involved in chondroitin binding (Figures 1and 2). Several aromatic residues within the cleft,Trp126, Trp127, Tyr234, are also well conserved(Figure 8(b)). They are well suited for stackingagainst the hexose rings of the substrate. Trypto-phan residues have been shown to play a role insubstrate binding in pectate lyase from Fusariummoniliformae (Rao et al., 1996). This is also in agree-ment with earlier biochemical data stressing theimportance of hydrophobic interactions in theenzyme-substrate complex formation for chondroi-tinase AC (Hiyama & Okada, 1977). In addition tothese aromatic residues, several hydrophobic side-chains are conserved in this cleft and may contrib-ute to this hydrophobic surface. This potential sub-strate binding site is complemented by Trp427 fromthe C-terminal domain, which comes from the loopthat participates in coordinating the Ca2�.
Catalytic mechanism and active site residues
Most lyases require a free carboxylate group atthe C5 position of the uronic acid of a GAG poly-mer for ef®cient enzymatic cleavage (Linhardt et al.,1986; Ernst et al., 1995). This electron withdrawinggroup enhances the acidity of the proton at pos-ition C5. In the case of chondroitin A and C, theleaving groups (on C4 and C5) are in the unfavor-able axial-equatorial relationship, which is also incommon with hyaluronan. A three step mechanismof the lyase action was proposed several years ago(Gacesa, 1987). The ®rst step consists of the neu-tralization of the carboxylate anion on the glucuro-nic acid, which provides resonance stabilization of
the enolate anion intermediate. Next, a generalTable 1. Data collection, phasing, and re®nement statistics
Native Pb
Cell (a � b, c) (AÊ ) 87.2, 193.3 87.6, 193.4 87Resolution (AÊ ) 1.9 1.9 2.3No. unique refl. 56,348 56,329 27Completeness (%) 94.6(78.8)b 93.6(62.6) 79Multiplicity 3 2.5 3.5I/s(I) overall 12.3 17.4 23I/s(I) > 5 (%) 81 78.7 92Rmerge (%)c 5.7(17.5) 6.4(34) 4.9Riso (%) - 10 7.3FOMd - 0.27 0.3
RefinementResolution (AÊ ) Atoms�solvent R-factorf Rf
g
6-1.9 5437�532 19.6 26
a Wavelength for Tb[MAD] 1.616 1.648 1.649.b Completeness in the highest resolution shells is given in parenthc Rmerge � � (jIi ÿ hIij)/� (Ii); where hIi is the average intensity of td Overall FOM as de®ned in the PHASES package.e Overall FOM as de®ned in MLPhare.f R-factor � � j(jFoj ÿ jFcj)j/� (jFoj).g Rfree; 10 % of the data was set aside for calculation of a cross-vali
base-catalyzed abstraction of the C5 proton takesplace with the change of hybridization of C5.Finally, the third step involves the elimination ofthe 4-O-glycosidic bond. The role of the generalbase is likely played by a histidine as it has beenshown that a chemical modi®cation of histidineresidues (Greiling et al., 1975) or site-directed modi-®cation of a speci®c histidine in hyaluronate lyaseshad a profound effect on activity (Lin et al., 1997).Histidine has also been implicated in heparin lyaseactivity (Godavarti et al., 1996).
Based on sequence homology with hyaluronatelyases and on a role in catalysis of His479 of hya-luronate lyase from S. agalactiae (above), we postu-late that the corresponding His225 ofchondroitinase AC plays the role of a general base.It would abstract the C5 proton from glucuronicacid resulting in the formation of an unsaturatedC4-C5 bond with a subsequent cleavage of theglycosidic bond followed by the elimination of the4-linked hexosamine. The two arginine residues,Arg288 and Arg292, are well positioned for theinteraction with the carboxylic group on glucuronicacid or with one of the sulfate groups and arelikely important for catalysis. We are now in theprocess of mutating these residues.
Materials and Methods
Crystallization
The chondroitinase AC from F. heparinum was homo-logously overexpressed in the original strain and puri-®ed as described in a previously established protocol(FeÂthieÁre et al., 1998). The puri®ed enzyme was crystal-lized using 15-17 % (w/v) poly(ethylene glycol) mono-methyl ester 2000 as a precipitating agent with 80 mMammonium acetate buffer (pH 6.5), and 400 mM sodiumacetate. The crystals belong to tetragonal system withspace group P43212 and cell dimensions a � b � 87.2 AÊ ,
c � 193.3 AÊ . There is one molecule in the asymmetricTb La Tb[MAD]a
.5, 194.1 87.5, 193.8 87.2, 193.32.3 2.6
,293 23,926 21,636.5(31.2) 69.7(24.0) 89.5(76.1)
3.5 4.5.1 19.2 15.5.9 96.5 84.8(9.1) 5.3(9.4) 9.2(29.6)
6.1 -3 0.34 0.45e
ree rmsd bonds (AÊ ) rmsd angle (�).0 0.008 1.5
esis.he re¯ection, and Ii is the intensity of the Ith observation.
dated R-factor.

Crystal Structure of Chondroitinase AC 645
unit. Diffraction data were collected at cryogenic tem-perature at the Cornell High Energy Synchrotron Source(CHESS) on beamlines A1 and F2. The crystals were¯ash-frozen in the mother liquor supplemented with25 % (v/v) glycerol. The diffraction of native crystalsextended to 1.9 AÊ resolution. All collected data were pro-cessed with the Denzo/scalepack package (Otwinowski& Minor, 1997). Table 1 shows data collection statistics.
Structure determination
Sequence comparison of chondroitinase AC to pro-teins deposited in the Brookhaven Protein Databank(Bernstein et al., 1977) revealed no homologous proteinwith a known structure. Therefore a search for heavyatom derivatives was undertaken, and conventionalMIRAS and MAD (Hendrickson, 1991) methods wereused to solve the enzyme's structure. Several derivativeswere identi®ed. Compounds containing lead, terbiumand lanthanum bound at a common single site. InitialMIRAS phases were obtained from these derivatives andsupplemented with phases from a terbium MAD exper-iment (Table 1). The MAD phases were estimated withMLPhare (CCP4, 1998) and yielded a ®gure-of-merit(FOM) of 0.46 for data between 40 and 2.6 AÊ . Thesephases were improved by density modi®cation with DM(CCP4, 1998) and yielded a ®nal overall FOM for the ter-bium MAD data of 0.6. An electron density map calcu-lated to 2.6 AÊ was readily interpretable. Clear electrondensity was present for most of the polypeptide chain.Further improvement of the map was expected fromcombining these terbium MAD-based phases with theMIRAS phases from other derivatives This was doneusing program SIGMAA (Read, 1986). The resulting mapwas only marginally improved (FOM of 0.65 for 2.6 AÊ
data) as compared to the solvent ¯attened MAD map,likely due to common heavy-atom sites. Model buildingproceeded with the program O (Jones et al., 1991), andover 90 % of the residues were included in the initialmodel. No density was present for the ®rst three aminoacids at the N terminus, and there are two sections inthe b-sheet domain that show some disorder (477-482and 568-577). These segments were positioned approxi-mately and given initially partial occupancies.
Refinement
Re®nement was carried with the X-Plor program(BruÈ nger, 1993) with re¯ections in the 6-1.9 AÊ resolutionrange and I > 2s(I). Approximately 10 % randomly cho-sen re¯ections were used for cross-validation. The initialstructure was subjected to rigid body minimizationwhich yielded an initial R-factor of 0.48 The ®rst cycle ofre®nement which consisted of minimization, torsionangle dynamics, and individual B-factor re®nement,yielded an R-factor of 0.282 and Rfree of 0.332. Analysisof the spatial distribution of re¯ection intensities indi-cated a small degree of diffraction anisotropy: scatteringin the c* direction was stronger than in the two otherdirections. The overall anisotropic B-factor was deter-mined with X-Plor, and appropriate corrections appliedto Fobs. Subsequent cycles of re®nement were followedby manual rebuilding during which solvent molecules,carbohydrates and the Ca2� were added stepwise.
The re®nement converged, with the R-factor of 0.196and Rfree of 0.260. All pertinent data are given in Table 1.The ®nal model includes residues 26-699, 532 water mol-
ecules, seven sugar units of O-linked carbohydrates andone Ca2�. Inspection of the structure with the programPROCHECK (Laskowski et al., 1993) showed more then90 % of the non-proline and non-glycine residues in themost favorable regions.
Data Bank accession numbers
The coordinates are deposited with the BrookhavenProtein Databank, accession code 1CB8; structure factorshave accession code R1CB8SF.
Acknowledgements
We thank Drs A. Matte and J.D. Schrag for readingthe manuscript and their helpful comments and Dr S.Raymond for help in producing the Figures. J.F. was apost-doctoral fellow from the Medical Research Councilof Canada during the course of this work, and is now atMax Planck Institute - Med Res. Jahnstrasse 29, Heidel-berg, 69120, Germany.
References
Aleshin, A. E., Stoffer, B., Firsov, L. M., Svensson, B. &Honzatko, R. B. (1996). Crystallographic complexesof glucoamylase with maltooligosaccharide analogs:relationship of stereochemical distortions at thenonreducing end to the catalytic mechanism. Bio-chemistry, 35, 8319-8328.
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J.,Zhang, Z., Miller, W. & Lipman, D. J. (1997).Gapped BLAST and PSI-BLAST: a new generationof protein database search programs. Nucl. AcidsRes. 25, 3389-3402.
Bernstein, F. C., Koetzle, T. F., Williams, G. J., Meyer,E. E. J., Brice, M. D., Rodgers, J. R., Kennard, O.,Shimanouchi, T. & Tasumi, M. (1977). The ProteinData Bank: a computer-based archival ®le formacromolecular structures. J. Mol. Biol. 112, 535-542.
Berry, A. M., Lock, R. A., Thomas, S. M., Rajan, D. P.,Hansman, D. & Paton, J. C. (1994). Cloning andnucleotide sequence of the Streptococcus pneumoniaehyaluronidase gene and puri®cation of the enzymefrom recombinant Escherichia coli. Infect. Immun. 62,1101-1108.
BruÈ nger, A. T. (1993). X-Plor Version 3.1, Yale Univer-sity.
Canard, B., Garnier, T., Saint-Joanis, B. & Cole, S. T.(1994). Molecular genetic analysis of the nagH geneencoding a hyaluronidase of Clostridium perfringens.Mol. Gen. Genet. 243, 215-224.
CCP4, (1998). Collaborative Computational ProjectNumber 4 (1994). The CCP4 suite: programs forprotein crystallography. Acta Crystallog. sect. D, 50,760-763.
Celada, F., Ullmann, A. & Monod, J. (1974). Animmunological study of complementary fragmentsof beta-galactosidase. Biochemistry, 13, 5543-5547.
Chakrabarti, P. (1990). Interaction of metal ions withcarboxylic and carboxamide groups in protein struc-tures. Protein Eng. 4, 49-56.
Davies, G. & Henrissat, B. (1995). Structures and mech-anisms of glycosyl hydrolases. Structure, 3, 853-859.
Ernst, S., Langer, R., Cooney, C. L. & Sasisekharan, R.(1995). Enzymatic degradation of glycosaminogly-
cans. Crit. Rev. Biochem. Mol. Biol. 30, 387-444.
646 Crystal Structure of Chondroitinase AC
Farrell, A. M., Taylor, D. & Holland, K. T. (1995). Clon-ing, nucleotide sequence determination andexpression of the Staphylococcus aureus hyaluronatelyase gene. FEMS Microbiol. Letters, 130, 81-85.
FeÂthieÁre, J., Shilton, B. H., Li, Y., Allaire, M., Laliberte,M., Eggimann, B. & Cygler, M. (1998). Crystalliza-tion and preliminary analysis of chondroitinase ACfrom Flavobacterium heparinum. Acta Crystallog. sect.D, 54, 279-280.
Gacesa, P. (1987). Alginate-modifying enzymes. A pro-posed uni®ed mechanism of action for the lyasesand epimerases. FEBS Letters, 212, 199-202.
Glusker, J. P. (1991). Structural aspects of metal ligand-ing to functional groups in proteins. Advan. ProteinChem. 42, 1-76.
Godavarti, R., Cooney, C. L., Langer, R. & Sasisekharan,R. (1996). Heparinase I from Flavobacterium hepari-num. Identi®cation of a critical histidine residueessential for catalysis as probed by chemical modi®-cation and site-directed mutagenesis. Biochemistry,35, 6846-6852.
Greiling, H., Stuhlsatz, H. W., Eberhard, T. & Eberhard,A. (1975). Studies on the mechanism of hyaluronatelyase action. Connect. Tissue Res. 3, 135-139.
Gu, K., Linhardt, R. J., Laliberte, M. & Zimmermann, J.(1995). Puri®cation, characterization and speci®cityof chondroitin lyases and glycuronidase from Flavo-bacterium heparinum. Biochem. J. 312, 569-577.
Hendrickson, W. A. (1991). Determination of macromol-ecular structures from anomalous diffraction of syn-chrotron radiation. Science, 254, 51-58.
Henrissat, B. & Bairoch, A. (1996). Updating thesequence-based classi®cation of glycosyl hydrolases.Biochem. J. 316, 695-696.
Henrissat, B., Heffron, S. E., Yoder, M. D., Lietzke, S. E.& Jurnak, F. (1995). Functional implications of struc-ture-based sequence alignment of proteins in theextracellular pectate lyase superfamily. Plant Physiol.107, 963-976.
Hileman, R. E., Fromm, J. R., Weiler, J. M. & Linhardt,R. J. (1998). Glycosaminoglycan-protein interactions:de®nition of consensus sites in glycosaminoglycanbinding proteins. Bioessays, 20, 156-167.
Hiyama, K. & Okada, S. (1977). Action of chondroiti-nases. III. Ionic strength effects and kinetics in theaction of chondroitinase AC. J. Biochem. (Tokyo), 82,429-436.
Holm, L. & Sander, C. (1997). Dali/FSSP classi®cation ofthree-dimensional protein folds. Nucl. Acids Res. 25,231-234.
Hynes, W. L., Hancock, L. & Ferretti, J. J. (1995). Anal-ysis of a second bacteriophage hyaluronidase genefrom Streptococcus pyogenes: evidence for a thirdhyaluronidase involved in extracellular enzymaticactivity. Infect. Immun. 63, 3015-3020.
Iozzo, R. V. (1998). Matrix proteoglycans: from molecu-lar design to cellular function. Annu. Rev. Biochem.67:609-52, 609-652.
Jacobson, R. H., Zhang, X. J., DuBose, R. F. & Matthews,B. W. (1994). Three-dimensional structure of beta-galactosidase from E. coli. Nature, 369, 761-766.
Jandik, K. A., Gu, K. & Linhardt, R. J. (1994). Actionpattern of polysaccharide lyases on glycosaminogly-cans. Glycobiology, 4, 289-296.
Jones, T. A., Zhou, J. Y., Cowan, S. W. & Kjeldgaard, M.(1991). Improved methods for binding proteinmodels in electron density maps and the location oferrors in these models. Acta Crystallog. sect. A, 47,
110-119.Kraulis, P. J. (1991). MOLSCRIPT: a program to produceboth detailed and schematic plots of protein struc-tures. J. Appl. Crystallog. 24, 946-951.
LaliberteÂ, M., Eggimann, B., Zimmerman, J., Huang, L.& van Halbeek, H. (1996). Determination of the gly-cosylation sites of glycosaminoglycan lyases fromFlavobacterium heparinum. Protein Sci. 5, (suppl. 1),435-s.
Laskowski, R. A., MacArthur, M. W., Moss, D. S. &Thornton, J. M. (1993). PROCHECK: a program tocheck the stereochemical quality of protein struc-tures. J. Appl. Crystallog. 26, 283-291.
Lidholt, K. (1997). Biosynthesis of glycosaminoglycansin mammalian cells and in bacteria. Biochem. Soc.Trans. 25, 866-870.
Lin, B., Averett, W. F. & Pritchard, D. G. (1997). Identi®-cation of a histidine residue essential for enzymaticactivity of group B streptococcal hyaluronate lyase.Biochem. Biophys. Res. Commun. 231, 379-382.
Linhardt, R. J., Galliher, P. M. & Cooney, C. L. (1986).Polysaccharide lyases. Appl. Biochem. Biotechnol. 12,135-176.
Mayans, O., Scott, M., Connerton, I., Gravesen, T.,Benen, J., Visser, J., Pickersgill, R. & Jenkins, J.(1997). Two crystal structures of pectin lyase Afrom Aspergillus reveal a pH driven conformationalchange and striking divergence in the substrate-binding clefts of pectin and pectate lyases. Structure,5, 677-689.
McPhalen, C. A., Strynadka, N. C. & James, M. N.(1991). Calcium-binding sites in proteins: a struc-tural perspective. Advan. Protein Chem. 42, 77-144.
Merritt, E. A. & Bacon, D. J. (1997). Raster3D: photorea-listic molecular graphics. Methods Enzymol. 277, 505-524.
Nicholls, A., Sharp, K. A. & Honig, B. (1991). Proteinfolding and association: insights from the interfacialand thermodynamic properties of hydrocarbons.Proteins: Struct. Funct. Genet. 11, 281-296.
Otwinowski, Z. & Minor, W. (1997). Processing of X-raydiffraction data collected in oscillation mode.Methods Enzymol. 276, 307-326.
Park, H. W., Boduluri, S. R., Moomaw, J. F., Casey, P. J.& Beese, L. S. (1997). Crystal structure of proteinfarnesyltransferase at 2.25 Angstrom resolution.Science, 275, 1800-1804.
Petersen, T. N., Kauppinen, S. & Larsen, S. (1997). Thecrystal structure of rhamnogalacturonase A fromAspergillus aculeatus: a right-handed parallel betahelix. Structure, 5, 533-544.
Pickersgill, R., Smith, D., Worboys, K. & Jenkins, J.(1998). Crystal structure of polygalacturonase fromErwinia carotovora ssp. carotovora. J. Biol. Chem. 273,24660-24664.
Pickersgill, R. W., Jenkins, J., Harris, G., Nasser, W. &Robert-Baudouy, J. (1994). The structure of Bacillussubtilis pectate lyase in complex with calcium.Nature Struct. Biol. 1, 717-723.
Plummer, T. H. J., Tarentino, A. L. & Hauer, C. R.(1995). Novel, speci®c O-glycosylation of secretedFlavobacterium meningosepticum proteins. Asp-Serand Asp-Thr-Thr consensus sites. J. Biol. Chem. 270,13192-13196.
Rao, M. N., Kembhavi, A. A. & Pant, A. (1996). Role oflysine, tryptophan and calcium in the beta-elimin-ation activity of a low-molecular-mass pectate lyase
from Fusarium moniliformae. Biochem. J. 319, 159-164.
Crystal Structure of Chondroitinase AC 647
Read, R. J. (1986). Improved Fourier coef®cients formaps using phases from partial structures witherrors. Acta Crystallog. sect. A, 42, 140-149.
Sawitzky, D. (1996). Protein-glycosaminoglycan inter-actions: infectiological aspects. Med. Microbiol.Immunol. (Berlin), 184, 155-161.
Su, H., Blain, F., Musil, R. A., Zimmermann, J. J., Gu, K.& Bennett, D. C. (1996). Isolation and expression inEscherichia coli of hepB and hepC, genes coding forthe glycosaminoglycan-degrading enzymes hepari-nase II and heparinase III, respectively, from Flavo-bacterium heparinum. Appl. Environ. Microbiol. 62,
2723-2734.(Received 16 December 1998; received i
Sutherland, I. W. (1995). Polysaccharide lyases. FEMSMicrobiol. Rev. 16, 323-347.
Tam, Y. C. & Chan, E. C. (1985). Puri®cation andcharacterization of hyaluronidase from oral Peptos-treptococcus species. Infect. Immun. 47, 508-513.
Wendt, K. U., Poralla, K. & Schulz, G. E. (1997). Struc-ture and function of a squalene cyclase. Science, 277,1811-1815.
Yoder, M. D., Keen, N. T. & Jurnak, F. (1993). Newdomain motif: the structure of pectate lyase C, asecreted plant virulence factor. Science, 260, 1503-
1507.Edited by D. Rees
n revised form 9 March 1999; accepted 10 March 1999)




















