
Control aromaticity in the thermal decomposition of 2,5-dihydrofuran, 2,5-dihydrothiophene and...
15
Control aromaticity in the thermal decomposition of 2,5-dihydrofuran, 2,5-dihydrothiophene and 3-pyrroline: a kinetic and thermodynamic study via DFT Abolfazl Shiroudi • Ehsan Zahedi • Reza Zabihi Received: 8 August 2010 / Accepted: 6 October 2010 / Published online: 3 November 2010 Ó Akade ´miai Kiado ´, Budapest, Hungary 2010 Abstract A theoretical study of the thermal decomposition kinetics of 2,5-dihy- drofuran (1), 2,5-dihydrothiophene (2), and 3-pyrroline (3) has been carried out at the B3LYP/6-31??G**, B3PW91/6-31??G** and MPW1PW91/6-31??G** levels of theory. Our results show that the MPW1PW91/6-31??G** method is in good agreement with the available experimental values. The nucleus independent chemical shift (NICS) values of all reactants, TSs and products indicate that all studied structures are aromatic and the studied reactions are controlled kinetically and thermodynamically by the change of the aromaticity. Based on the optimized ground state geometries using the MPW1PW91/6-31??G** method, the natural bond orbital analysis (NBO) of donor–acceptor (bonding-antibonding) interactions revealed that by the increase of electronegativity of atom X O; S; NH ð Þ; LP e ðÞ X1 ! r C 2 H 6 resonance energies and also, the HOMO-LUMO energy-gaps in the ground state structures increase. The results also suggest that in compounds 1–3, the hydrogen elimination are controlled by LP(e) ? r* resonance energies. Keywords DFT Decomposition NBO NICS GIAO Introduction The kinetics of thermal decomposition of several compounds of the general type X.CH 2 .CH:CH.CH 2 have been previously studied using experimental techniques A. Shiroudi E. Zahedi (&) Chemistry Department, Islamic Azad University, Shahrood Branch, P.O.box 36155/133, Shahrood, Iran e-mail: [email protected]; [email protected] R. Zabihi Chemistry Department, Islamic Azad University, Karaj Branch, Tehran, Iran 123 Reac Kinet Mech Cat (2011) 102:21–35 DOI 10.1007/s11144-010-0254-3
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Control aromaticity in the thermal decomposition of 2,5-dihydrofuran, 2,5-dihydrothiophene and...

Control aromaticity in the thermal decompositionof 2,5-dihydrofuran, 2,5-dihydrothiophene and3-pyrroline: a kinetic and thermodynamicstudy via DFT
Abolfazl Shiroudi • Ehsan Zahedi • Reza Zabihi
Received: 8 August 2010 / Accepted: 6 October 2010 / Published online: 3 November 2010
� Akademiai Kiado, Budapest, Hungary 2010
Abstract A theoretical study of the thermal decomposition kinetics of 2,5-dihy-
drofuran (1), 2,5-dihydrothiophene (2), and 3-pyrroline (3) has been carried out at
the B3LYP/6-31??G**, B3PW91/6-31??G** and MPW1PW91/6-31??G**
levels of theory. Our results show that the MPW1PW91/6-31??G** method is in
good agreement with the available experimental values. The nucleus independent
chemical shift (NICS) values of all reactants, TSs and products indicate that all
studied structures are aromatic and the studied reactions are controlled kinetically
and thermodynamically by the change of the aromaticity. Based on the optimized
ground state geometries using the MPW1PW91/6-31??G** method, the natural
bond orbital analysis (NBO) of donor–acceptor (bonding-antibonding) interactions
revealed that by the increase of electronegativity of atom X O; S; NHð Þ; LP
eð ÞX1! r�C2�H6resonance energies and also, the HOMO-LUMO energy-gaps in the
ground state structures increase. The results also suggest that in compounds 1–3, the
hydrogen elimination are controlled by LP(e) ? r* resonance energies.
Keywords DFT � Decomposition � NBO � NICS � GIAO
Introduction
The kinetics of thermal decomposition of several compounds of the general type
X.CH2.CH:CH.CH2have been previously studied using experimental techniques
A. Shiroudi � E. Zahedi (&)
Chemistry Department, Islamic Azad University, Shahrood Branch,
P.O.box 36155/133, Shahrood, Iran
e-mail: [email protected]; [email protected]
R. Zabihi
Chemistry Department, Islamic Azad University, Karaj Branch, Tehran, Iran
123
Reac Kinet Mech Cat (2011) 102:21–35
DOI 10.1007/s11144-010-0254-3

[1–4]. It has been shown that elimination of molecular hydrogen occurs in the cases
when X is O, S, or NH. In the case of 2,5-dihydrofuran [2], it has been suggested that
the mechanism of the reaction involves elimination of hydrogen atoms from the
2- and 5-positions in the ring by means of six-central transition states (Fig. 1). The
kinetics of the gas-phase decomposition of 2,5-dihydrofuran (1), 2,5-dihydrothioph-
ene (2) and 3-pyrroline (3) have been experimentally studied [2–4]. As an
experimental result, Wellington and coworkers [2, 4] studied the decomposition of
2,5-dihydrofuran (1) between 342 and 409 �C and 2,5-dihydrothiophene (2).
Wellington [2] showed that the addition of nitric oxide, propylene or toluene had
no effect upon the rate decomposition of 2,5-dihydrofuran and the rate constant was
calculated to be 5.3 ± 0.1 9 1012 exp(-24408/T) s-1. Also, Wellington [4] studied
the decomposition of 2,5-dihydrothiophene and the rate constant has been obtained
as 1.66 9 1013 exp(-27579.3 ± 1761.5/T) s-1. Thomas et al. [3] indicated that the
rate constant of the decomposition of 3-pyrroline (3) in the temperature range of
317–358 �C is 1.9 ± 0.4 9 1012 exp(-22445.9 ± 121.3/T) s-1. Thomas showed
that the addition of nitric oxide had no effect on the rate of decomposition but the rate
of the decomposition increased in heterogeneous processes. The results prove the
reaction to be homogeneous, unimolecular and to obey a first-order rate law. The
main goal of this work is to understand the influence of the affective factors on
the transition state structures of the kinetics of dehydrogenation reactions 1–3
(Fig. 1), involving the formation of molecular hydrogen and related diene hetero-
cyclic compounds using theoretical methods and comparison with those obtained
experimental data. The purpose is to investigate the kinetic and thermodynamic
parameters, pre-exponential factors, and barrier heights of these reactions, and
consideration of the nature of the molecular mechanism of these decomposition
reactions using natural bond orbital (NBO) analysis. The stabilization energies (E2)
associated with LP(e) ? r* delocalizations in compounds 1–3 were systematically
and quantitatively investigated by NBO analysis. We interpreted the kinetic and
thermodynamic results with nucleus independent chemical shift (NICS). Density
functional theory (DFT) has been found to be successful for determining activation
barriers, molecular energies, molecular geometries, electronic and magnetic prop-
erties [5–11]. Also, NBO analysis based density functional theory (DFT) provided a
useful picture for bonding and dynamic behavior of the compounds [12].
Fig. 1 Thermal decomposition of compounds 1–3 (Structures and numbering system)
22 A. Shiroudi et al.
123

Computational details
Initial estimates of the geometries of reactants and products were obtained by a
molecular-mechanics program PC-MODEL (88.0) [13] followed by full minimi-
zation using semi-empirical PM3 in the MOPAC 6.0 computer program [14, 15].
Energy minimum molecular geometries were located by minimizing the energy,
with respect to all geometric coordinates, without imposing any symmetry
constraints. The initial structures of the transition state geometries were obtained
using the optimized geometries of the equilibrium structures according to the
procedure of Dewar et al. (keyword SADDLE) [16]. Then, all initial geometries of
reactants, transition states and products were re-optimized using the DFT method at
B3LYP/6-31??G**, B3PW91/6-31??G** and MPW1PW91/6-31??G** levels
of theory by means of the GAUSSIAN 98 program package [17].
The nature of the stationary points for species has been fixed by means of the
number of imaginary frequencies. For minimum state structure, only real frequency
values and, in the transition states, only a single imaginary frequency value (with
negative sign) was accepted [18, 19]. The vibrational frequencies of minimum state
structures were calculated by the FREQ subroutine. The first order rate coefficient
k(T) was computed using the transition state theory (TST) [20, 21] and assuming
that the transmission coefficient is equal to 1, as expressed in the following relation:
kðTÞ ¼ kBT
h
� �exp �DG6¼
RT
� �ð1Þ
where DG= is the Gibbs free energy change between the reactant and its corre-
sponding transition structure, k and h are the Boltzmann and Planck constants,
respectively. DG= was calculated as follows:
DG 6¼ ¼ DH 6¼ � TDS 6¼ ð2Þ
and
DH 6¼ ¼ V 6¼ þ DZPVE þ DE Tð Þ ð3Þ
where V= is the potential energy barrier and DZPVE and DE(T) are the difference
of ZPVE and temperature corrections between the TS and the corresponding
reactant, respectively [22]. Comparison with experimental values shows better
agreement at the MPW1PW91/6-31??G** level of theory. A NICS study and
NBO [23] analysis were also performed using the MPW1PW91/6-31??G** level
of theory on the optimized structures.
Results and discussion
Kinetic and thermodynamic parameters
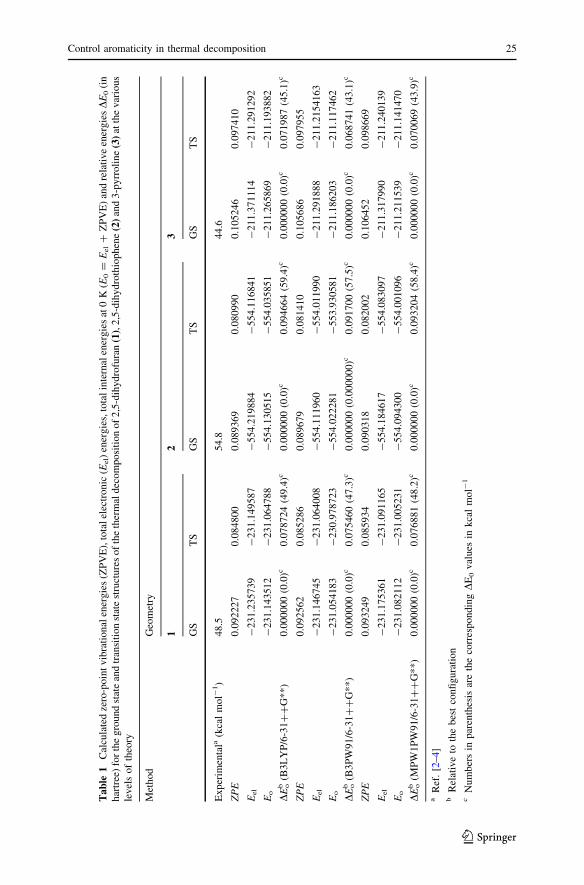
The zero-point vibrational energies (ZPVE), total electronic (Eel) energies and total
internal energies at 0 K (E0 = Eel ? ZPVE) of compounds 1–3, as calculated by
Control aromaticity in thermal decomposition 23
123

B3LYP/6-31??G**, B3PW91/6-31??G** and MPW1PW91/6-31??G** levels
of theory, are summarized in Table 1. Considering a comparison of activation
energies (DE0 is equal to activation energy in 0 K) from the DFT methods, the
MPW1PW91/6-31??G** results are in good agreement with the experimental data
[2–4] and show that the barrier height of the decomposition of compounds 1–3 is
48.24, 58.48 and 43.96 kcal mol-1. The effect of the electronegativity of the X
atom (O, S, NH) may be an important factor in this study. Oxygen is the most
electronegative of these atoms. This electronegativity may have the effect of
facilitating the formation of the transition state and hence the value of the adjusted
activation energy for the decomposition of 2,5-dihydrofuran is low, and is in
agreement with experimental studies [4]. In these reactions, the X atom and its
adjacent carbon atoms may become puckered out of the plane of the ring thus
bringing hydrogen atoms in the 2 and 5 positions close enough to form a six-centre
transition state. This would result in a loss of some vibrational freedom, forming a
more rigid structure [1]. The energy profiles for the thermal decompositions of
compounds 1–3 are depicted in Fig. 2.
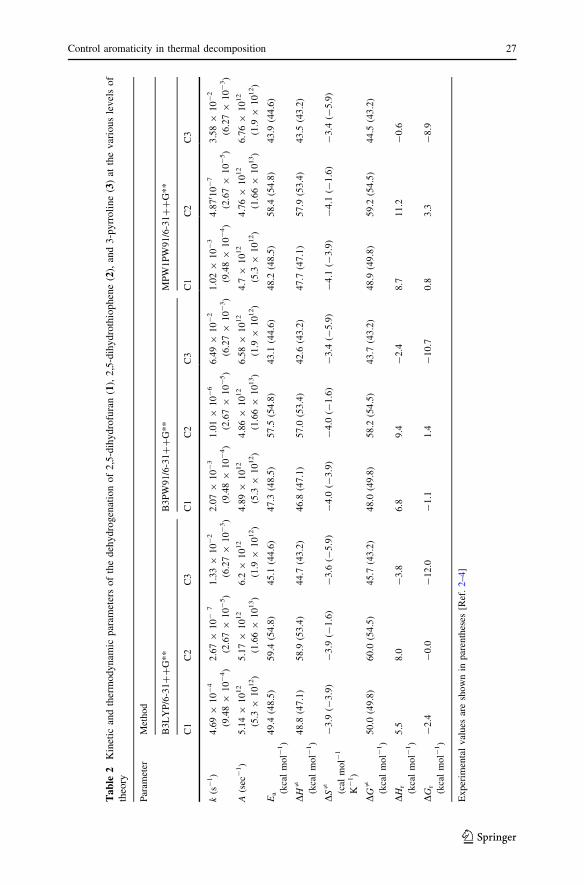
The comparative kinetic and thermodynamic parameters, i.e., enthalpies (DHr),
Gibbs free energies (DGr), enthalpies of activation (DH=), Gibbs activation free
energies (DG=), activation entropies (DS=), pre-exponential factors (A) and the
unimolecular rate coefficients (k) of the thermal decomposition of compounds 1–3are listed in Table 2. The thermal decomposition of compounds 1 and 2 are
endothermic processes. As can be seen, DS= values are relatively small, so that the
calculated DH= and DG= parameters are close to the DE0 values. According to the
experimental data, the decomposition rate constants at 400 �C for compounds 1–3are 9.48 9 10-4, 2.67 9 10-5 and 6.27 9 10-3 s-1 [2–4]; therefore the computed
values for compounds 1–3 at the MPW1PW91/6-31??G** level of theory are in
agreement with experimental data (1.02 9 10-3, 4.87 9 10-7, and 3.58 9
10-2 s-1). The MPW1PW91/6-31??G** level of theory shows that for compound
1, the rate constant, k1, can be represented as k1 = 4.7 9 1012 exp(-24280/T) s-1,
while rate constant for compound 2, k2, is 4.76 9 1012 exp(-29435/T) s-1 and for
compound 3, k3, is 6.76 9 1012 exp(-22128/T) s-1. Log A has been used to suggest
the type of transition state according to Benson [21, 24, 25]. The TS for the thermal
decomposition of compounds 1–3 is a cyclic six-membered structure, as suggested
by log A values from 12.67 to 12.83. The transition state geometric parameters
optimized at the MPW1PW91/6-31??G** theoretical level compared to the
reactants and are given in Table 3. The C2–H6 and C5–H11 bond distances increase,
which implies breaking of this bond (1.095–1.01–1.497–1.539 A) in the TSs. The
C2–C3 and C4–C5 bond distances reveal changes from single to double bond
character (1.498–1.508 to 1.429–1.438 A) in TSs. The H6–H11 bond is forming in
the TSs, and the C3–C4 bond distances reveal changes from single to double bond
character (1.33–1.333 to 1.367–1.372 A) in TSs [26]. The imaginary frequencies
characterized for the TS found for 2,5-dihydrofuran (1), 2,5-dihydrothiophene (2),
and 3-pyrroline (3) are 1590, 1622 and 1663 cm-1, respectively.
Hammond’s postulate can be interpreted in terms of the position of the transition
structure along the reaction coordinate, nT, as defined by Agmon [27]:
24 A. Shiroudi et al.
123
Ta
ble
1C
alcu
late
dze
ro-p
oin
tvib
rati
onal
ener
gie
s(Z
PV
E),
tota
lel
ectr
onic
(Eel)
ener
gie
s,to
tal
inte
rnal
ener
gie
sat
0K
(E0
=E
el
?Z
PV
E)
and
rela
tiv
een
erg
iesD
E0
(in
har
tree
)fo
rth
eg
rou
nd
stat
ean
dtr
ansi
tio
nst
ate
stru
ctu
res
of
the
ther
mal
dec
om
po
siti
on
of
2,5
-dih
yd
rofu
ran
(1),
2,5
-dih
yd
roth
iop
hen
e(2
)an
d3
-py
rro
lin
e(3
)at
the
var
ious
lev
els
of
theo
ry
Met
ho
dG
eom
etry
12
3
GS
TS
GS
TS
GS
TS
Ex
per
imen
tala
(kca
lm
ol-
1)
48
.55
4.8
44
.6
ZP
E0
.09
22
27
0.0
84
80
00
.089
36
90
.08
09
90
0.1
05
24
60
.097
41
0
Eel
-2
31
.23
573
9-
23
1.1
49
58
7-
55
4.2
19
88
4-
55
4.1
16
84
1-
21
1.3
71
11
4-
21
1.2
91
29
2
Eo
-2
31
.14
351
2-
23
1.0
64
78
8-
55
4.1
30
51
5-
55
4.0
35
85
1-
21
1.2
65
86
9-
21
1.1
93
88
2
DE
ob(B
3L
YP
/6-3
1?
?G
**
)0
.00
00
00
(0.0
)c0
.07
87
24
(49
.4)c
0.0
00
00
0(0
.0)c
0.0
94
66
4(5
9.4
)c0
.000
00
0(0
.0)c
0.0
71
98
7(4
5.1
)c
ZP
E0
.09
25
62
0.0
85
28
60
.089
67
90
.08
14
10
0.1
05
68
60
.097
95
5
Eel
-2
31
.14
674
5-
23
1.0
64
00
8-
55
4.1
11
96
0-
55
4.0
11
99
0-
21
1.2
91
88
8-
21
1.2
15
41
63
Eo
-2
31
.05
418
3-
23
0.9
78
72
3-
55
4.0
22
28
1-
55
3.9
30
58
1-
21
1.1
86
20
3-
21
1.1
17
46
2
DE
ob(B
3P
W9
1/6
-31?
?G
**
)0
.00
00
00
(0.0
)c0
.07
54
60
(47
.3)c
0.0
00
00
0(0
.00
00
00
)c0
.09
17
00
(57
.5)c
0.0
00
00
0(0
.0)c
0.0
68
74
1(4
3.1
)c
ZP
E0
.09
32
49
0.0
85
93
40
.090
31
80
.08
20
02
0.1
06
45
20
.098
66
9
Eel
-2
31
.17
536
1-
23
1.0
91
16
5-
55
4.1
84
61
7-
55
4.0
83
09
7-
21
1.3
17
99
0-
21
1.2
40
13
9
Eo
-2
31
.08
211
2-
23
1.0
05
23
1-
55
4.0
94
30
0-
55
4.0
01
09
6-
21
1.2
11
53
9-
21
1.1
41
47
0
DE
ob(M
PW
1P
W9
1/6
-31?
?G
**
)0
.00
00
00
(0.0
)c0
.07
68
81
(48
.2)c
0.0
00
00
0(0
.0)c
0.0
93
20
4(5
8.4
)c0
.000
00
0(0
.0)c
0.0
70
06
9(4
3.9
)c
aR
ef.
[2–4]
bR
elat
ive
toth
eb
est
con
fig
ura
tio
nc
Nu
mb
ers
inp
aren
thes
isar
eth
eco
rres
po
ndin
gD
E0
val
ues
ink
cal
mo
l-1
Control aromaticity in thermal decomposition 25
123

nT ¼1
2� ðDGr=DG 6¼Þ ð4Þ
The magnitudes of nT, indicate the degree of similarity between the transition
structure and the product. According to this equation, the position of the transition
state along the reaction coordinate is determined solely by DGr� (a thermodynamic
quantity) and DG= (a kinetic quantity). The values of nT for the thermal
decomposition of compounds 1–3, are 0.5042, 0.5144 and 0.4543. In fact the
transition state structures in the reactions 1 and 2 are more similar to the products
than to its reactants and the similarity between transition states and products
increases in the order of R2 [ R1 [ R3.
The nucleus independent chemical shift (NICS) study
In the NICS study, the gauge-invariant atomic orbital (GIAO) theory with the
MPW1PW91/6-31??G** method was applied to estimate the diamagnetic ring
current intensity on the optimized geometries of diene ring-heterocyclic reactants,
products and TSs in the gas phase. Magnetic properties of organic molecules arise
from the diamagnetic ring current of aromatic systems [28]. The nucleus
independent chemical shift (NICS) is defined as the negative value of the absolute
magnetic shielding in centers of rings or 1 A above or below the molecular plane
[29]. NICS at an empty point in space equals to zero and in principle does not
require reference molecules and calibrating (homodesmotic) equations for
Fig. 2 Energy profile for the unimolecular thermal decomposition of compounds 1–3
26 A. Shiroudi et al.
123
Tab
le2
Kin
etic
and
ther
modynam
icpar
amet
ers
of
the
deh
ydro
gen
atio
nof
2,5
-dih
ydro
fura
n(1
),2
,5-d
ihy
dro
thio
phen
e(2
),an
d3
-py
rro
lin
e(3
)at
the
var
ious
lev
els
of
theo
ry
Par
amet
erM
ethod
B3L
YP
/6-3
1?
?G
**
B3P
W91/6
-31
??
G**
MP
W1P
W91/6
-31
??
G**
C1
C2
C3
C1
C2
C3
C1
C2
C3
k(s
-1)
4.6
99
10
-4
(9.4
89
10
-4)
2.6
79
10
-7
(2.6
79
10
-5)
1.3
39
10
-2
(6.2
79
10
-3)
2.0
79
10
-3
(9.4
89
10
-4)
1.0
19
10
-6
(2.6
79
10
-5)
6.4
99
10
-2
(6.2
79
10
-3)
1.0
29
10
-3
(9.4
89
10
-4)
4.8
70 1
0-
7
(2.6
79
10
-5)
3.5
89
10
-2
(6.2
79
10
-3)
A(s
ec-
1)
5.1
49
10
12
(5.3
910
12)
5.1
79
10
12
(1.6
69
10
13)
6.2
910
12
(1.9
910
12)
4.8
99
10
12
(5.3
910
12)
4.8
69
10
12
(1.6
69
10
13)
6.5
89
10
12
(1.9
910
12)
4.7
910
12
(5.3
910
12)
4.7
69
10
12
(1.6
69
10
13)
6.7
69
10
12
(1.9
910
12)
Ea (k
cal
mol-
1)
49.4
(48.5
)59.4
(54.8
)45.1
(44.6
)47.3
(48.5
)57.5
(54.8
)43.1
(44.6
)48.2
(48.5
)58.4
(54.8
)43.9
(44.6
)
DH=
(kca
lm
ol-
1)
48.8
(47.1
)58.9
(53.4
)44.7
(43.2
)46.8
(47.1
)57.0
(53.4
)42.6
(43.2
)47.7
(47.1
)57.9
(53.4
)43.5
(43.2
)
DS= (cal
mol-
1
K-
1)
-3.9
(-3.9
)-
3.9
(-1.6
)-
3.6
(-5.9
)-
4.0
(-3.9
)-
4.0
(-1.6
)-
3.4
(-5.9
)-
4.1
(-3.9
)-
4.1
(-1.6
)-
3.4
(-5.9
)
DG=
(kca
lm
ol-
1)
50.0
(49.8
)60.0
(54.5
)45.7
(43.2
)48.0
(49.8
)58.2
(54.5
)43.7
(43.2
)48.9
(49.8
)59.2
(54.5
)44.5
(43.2
)
DH
r
(kca
lm
ol-
1)
5.5
8.0
-3.8
6.8
9.4
-2.4
8.7
11.2
-0.6
DG
r
(kca
lm
ol-
1)
-2.4
-0.0
-12.0
-1.1
1.4
-10.7
0.8
3.3
-8.9
Exper
imen
tal
val
ues
are
show
nin
par
enth
eses
[Ref
.2
–4
]
Control aromaticity in thermal decomposition 27
123

the evaluation of aromaticity. Negative values of NICS indicate a shielding effect
resulting from the induced diatropic ring current understood as aromaticity at a
specific point. On the contrary, its positive values are interpreted as the deshielded-
paratropic ring current and thus antiaromaticity. Schleyer and coworkers, based on
studies on an extensive set of heterocyclic compounds, proved that there are very
good linear correlations among the geometric, energetic, and magnetic properties
providing straightforward interpretation of the electronic structures and properties
of five-membered heterocycles with one heteroatom [30].
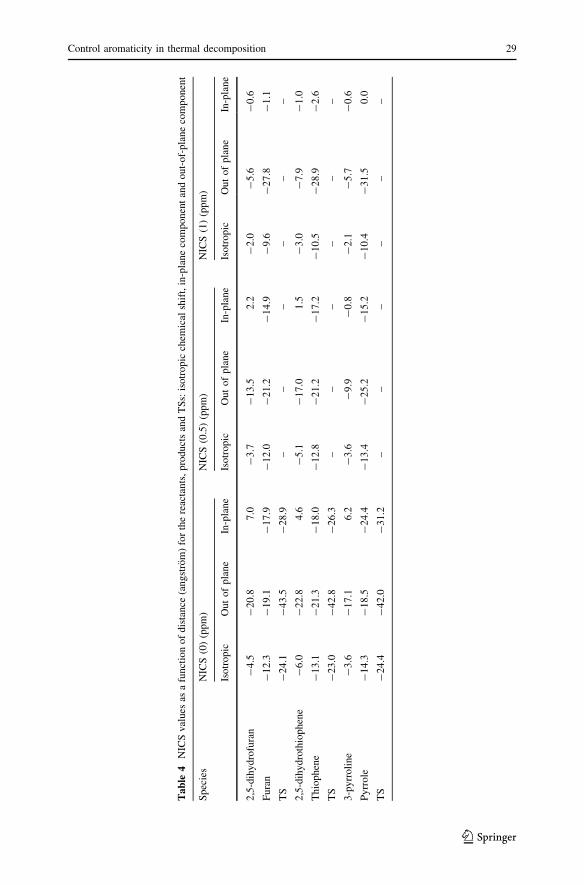
For all the reactants, TSs and products, the sets of points lying below and above
the rings’ geometric centers were used. Their locations correspond to distances from
0 to 1 A with 0.5 A steps. The NICS 0 A values calculated at the center of the ring
were influenced by r bonds, whereas the NICS 1 A values calculated at 1 A above
the plane were more affected by the p-system [31]. From Table 4, one may
conclude that all the studied reactants and products are aromatic. The structures
exhibit characteristic decreasing of the NICS values from the point located in the
geometric center of the ring to the 1 A above of it. The NICS values are the
isotropic chemical shifts of the respective bq’s, and the eigenvalues of the chemical
shift tensors were used to separate the isotropic NICS values into their in-plane and
out-of-plane components. Since the NICS values are distance-dependent, the
symbol ‘N@r’ will be used in this article to denote the NICS value (ppm) at distance
r(A) from the molecular plane. The magnitudes of the isotropic chemical shifts
for the products are very different; the minimal values are -12.3@0, -13.1@0 and
-14.3@0 for furan, thiophene and pyrrole. The strongest aromatic character in the
products was found for pyrrole, and the aromatic character is in the order
P3 [ P2 [ P1. Minimal values of the isotropic chemical shifts for the reactants are
-4.5@0, -6.0@0 and -3.6@0 for 2,5-dihydrofuran, 2,5-dihydrothiophene, and
3-pyrroline. The strongest aromatic character in the reactants was found for
2,5-dihydrothiophene, and the aromatic character is in the order: R2 [ R1 [ R3.
The transition states are not planar; therefore, the ring center in the C2–C3–C4–C5
Table 3 Structural parameters for the ground state and transition state structures of the thermal
decomposition of 2,5-dihydrofuran (1), 2,5-dihydrothiophene (2) and 3-pyrroline (3) at the MPW1PW91/
6-31??G** level of theory
State Compound
1 2 3
GS TS GS TS GS TS
Bond lengths (A)
r C2–H6 1.099 1.506 1.095 1.539 1.010 1.497
r C5–H11 1.099 1.506 1.095 1.539 1.010 1.497
r C2–C3 1.498 1.429 1.498 1.429 1.508 1.438
r C3–C4 1.330 1.371 1.331 1.372 1.333 1.367
d H6–H11 3.551 1.011 3.955 0.987 3.285 1.058
TS imaginary frequency (cm-1) 1590 1622 1663
Bond lengths are in angstroms (A)
28 A. Shiroudi et al.
123
Ta
ble
4N
ICS
val
ues
asa
fun
ctio
no
fd
ista
nce
(an
gst
rom
)fo
rth
ere
acta
nts
,pro
duct
san
dT
Ss:
isotr
opic
chem
ical
shif
t,in
-pla
ne
com
ponen
tan
dout-
of-
pla
ne
com
ponen
t
Sp
ecie
sN
ICS
(0)
(pp
m)
NIC
S(0
.5)
(pp
m)
NIC
S(1
)(p
pm
)
Iso
tro
pic
Ou
to
fp
lane
In-p
lane
Iso
trop
icO
ut
of
pla
ne
In-p
lane
Iso
trop
icO
ut
of
pla
ne
In-p
lan
e
2,5
-dih
yd
rofu
ran
-4
.5-
20
.87
.0-
3.7
-1
3.5
2.2
-2
.0-
5.6
-0
.6
Fu
ran
-1
2.3
-1
9.1
-1
7.9
-1
2.0
-2
1.2
-1
4.9
-9
.6-
27
.8-
1.1
TS
-2
4.1
-4
3.5
-2
8.9
––
––
––
2,5
-dih
yd
roth
iop
hen
e-
6.0
-2
2.8
4.6
-5
.1-
17
.01
.5-
3.0
-7
.9-
1.0
Th
iop
hen
e-
13
.1-
21
.3-
18
.0-
12
.8-
21
.2-
17
.2-
10
.5-
28
.9-
2.6
TS
-2
3.0
-4
2.8
-2
6.3
––
––
––
3-p
yrr
oli
ne
-3
.6-
17
.16
.2-
3.6
-9
.9-
0.8
-2
.1-
5.7
-0
.6
Py
rro
le-
14
.3-
18
.5-
24
.4-
13
.4-
25
.2-
15
.2-
10
.4-
31
.50
.0
TS
-2
4.4
-4
2.0
-3
1.2
––
––
––
Control aromaticity in thermal decomposition 29
123

plane is considered (Fig. 1). Minimal values of the isotropic chemical shifts for the
transition states are -24.1@0, -23.0@0 and -24.4@0 for 2,5-dihydrofuran,
2,5-dihydrothiophene, and 3-pyrroline. The strongest aromatic character in the
transition states was found for 3-pyrroline, and the aromatic character is in the order
TS3 [ TS1 [ TS2. The aromaticity on the ring center increased from reactants to
TSs and then decreases from TSs to products and the aromatic character of products
are higher than for reactants. The separation of the isotropic values into in-plane and
out-of plane contributions indicates that aromaticities of structures are controlled by
the out-of-plane component, with the exception of pyrrole, where it is in the ring
center. A quick look at the results reveals that the highest increases in the
aromaticity of ring along the dehydrogenation reaction (from reactant to TS and
from reactant to product) was found for 3-pyrroline, and the lowest increases was
found for 2,5-dihydrothiophene. These results indicated that the studied reactions
are controlled kinetically and thermodynamically by the changes of the aromaticity.
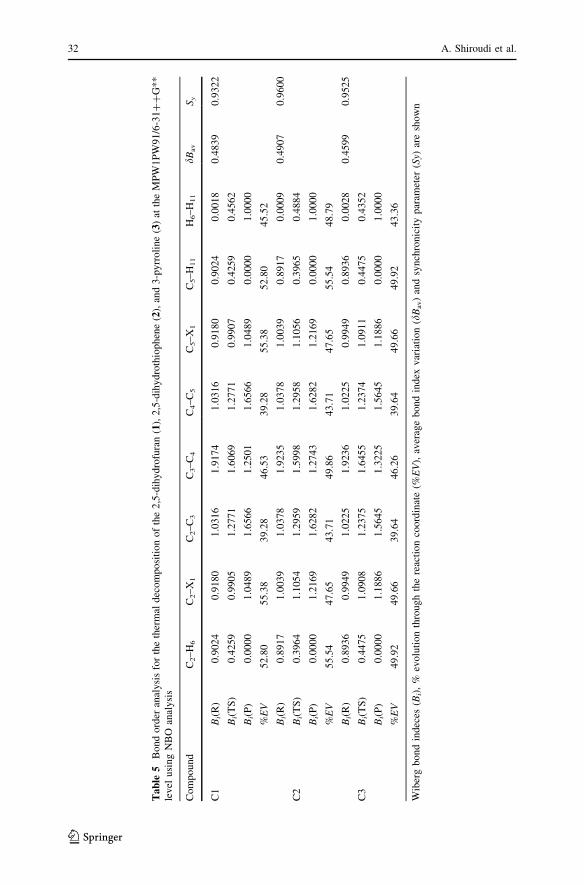
Bond order analysis
A more balanced measure of the extent of bond formation or bond breaking along a
reaction pathway is provided by the concept of bond order (B). This theoretical
technique has been used to investigate the molecular mechanism of chemical
reactions [32]. To follow the nature of the decomposition process, the Wiberg bond
indices have been computed by using the NBO [33, 34] analysis as implemented in
GAUSSIAN 98. There are several bond breaking/forming processes along the
fragmentation process and the global nature of the decomposition reaction can be
monitored by means of the synchronicity (Sy) concept [35], defined by the following
expression:
Sy ¼ 1�Pn
i¼1dBi�dBavj j
dBav
2n� 2ð5Þ
In this equation, n is the number of bonds directly involved in the reaction and the
relative variation of the bond index (dBi) for a bond i at the TS is given as a
percentage by:
dBi ¼BTS
i � BRi
BPi � BR
i
ð6Þ
where the superscripts R, TS, and P, refer to the reactant, transition state and
product. The evolution in bond change is calculated as:
%EV ¼ dBi � 100 ð7Þ
and average value is calculated from:
dBav ¼1
n
Xn
i¼1
dBi ð8Þ
Bond indices were calculated for those bonds changed in the reaction pathway, i.e.:
X1–C2, C2–C3, C3–C4, C4–C5, C5–X1, C2–H6, C5–H11 and H6–H11 bonds (Fig. 1);
30 A. Shiroudi et al.
123

all other bonds remain practically unaltered during the processes. The calculated
Wiberg indexes Bi for reactants, transition states and products for compounds 1–3,
enable us to examine the progress of the reactions (Table 5).
Reaction 1 leads to the cleavage of the C2–H6 and C5–H11 bonds through TS1 to
produce furan ? H2 located at 8.76 kcal mol-1 above the reactant at the
MPW1PW91/6-31??G** level. TS1 results from a simple elongation of the
breaking C2–H6 and C5–H11 bond lengths and the simultaneous shrinkage of
the H6–H11 distance as a result of the forming hydrogen molecule. The C2–H6 and
C5–H11 bonds are elongated by 1.506 A, and the forming H6–H11 bond is longer
than the equilibrium bond length in hydrogen molecule. Wiberg indeces show more
progress in the C2–X1 and C5–X1 bond breaking (about 55.38%) and less progress is
observed in the C2–C3 and C4–C5 single bond formation (39.28%). TS1 has an
imaginary frequency of 1590i cm-1 that indicates a well-defined transition state
geometry and the associated energy barrier located 48.24 kcal mol-1 above the
reactant at the MPW1PW91/6-31??G** level.
In reaction 2, the mechanism involves the simultaneous breaking of the C2–H6
and C5–H11 bonds and shrinkage of the H6–H11 distance through TS2 to produce
thiophene ? H2 located at 11.29 kcal mol-1 above the reactant at the MPW1PW91/
6-31??G** level. TS2 results from a simple elongation of the breaking C2–H6 and
C5–H11 bond lengths and the simultaneous shrinkage of the H6–H11 distance as a
result of the formation of a hydrogen molecule. The C2–H6 and C5–H11 bonds are
elongated by 1.539 A, and the forming H6–H11 bond is longer than the equilibrium
bond length in the hydrogen molecule. The changes of C2–H6 and C5–H11 bonds,
and the formation of the H6–H11 single bond are advanced considerably by 55.54,
55.54 and 48.79%. The structure of TS2 possesses an imaginary frequency at
1622i cm-1 and the associated energy barrier located 58.48 kcal mol-1 above the
reactant at the MPW1PW91/6-31??G** level.
Reaction 3 leads to the cleavage of the C2–H6 and C5–H11 bonds through TS3
to produce pyrrole ? H2 located at 0.68 kcal mol-1 below the reactant at the
MPW1PW91/6-31??G** level. TS3 results from a simple elongation of
the breaking C2–H6 and C5–H11 bond lengths and the simultaneous shrinkage of
the H6–H11 distance as a result of the forming H6–H11 bond. The C2–H6 and C5–H11
bonds are elongated by 1.497 A, and the forming H6–H11 bond is longer than the
equilibrium bond length in the hydrogen molecule. Moreover, the evolution in bond
changes for the breaking of C2–H6 and C5–H11 bonds is 49.92% in this reaction, and
the formation of the H6–H11 single bond is 43.36%. The imaginary frequency of
1663i cm-1 indicates a well-defined transition state geometry and the associated
energy barrier, located 43.96 kcal mol-1 above the reactant at the MPW1PW91/
6-31??G** level. The synchronicity values of the thermal decomposition of
compounds 1–3 are 0.93, 0.96 and 0.95, which revealed that the reaction pathways
can be described as concerted and slightly asynchronous.
Natural bond orbital analysis
Delocalization of electron density between the filled (bonding or lone pair) Lewis
type NBOs and the empty (antibonding and Rydberg) non-Lewis NBOs leads to
Control aromaticity in thermal decomposition 31
123
Tab
le5
Bo
nd
ord
eran
alysi
sfo
rth
eth
erm
ald
eco
mp
osi
tio
no
fth
e2
,5-d
ihy
dro
fura
n(1
),2
,5-d
ihy
dro
thio
ph
ene
(2),
and
3-p
yrr
oli
ne
(3)
atth
eM
PW
1P
W9
1/6
-31?
?G
**
lev
elu
sin
gN
BO
anal
ysi
s
Com
po
und
C2–
H6
C2–
X1
C2–
C3
C3–
C4
C4–
C5
C5–
X1
C5–
H11
H6–
H11
dBav
Sy
C1
Bi(
R)
0.9
02
40
.918
01
.031
61
.917
41
.03
16
0.9
18
00
.902
40
.001
80
.483
90
.932
2
Bi(
TS
)0
.425
90
.990
51
.277
11
.606
91
.27
71
0.9
90
70
.425
90
.456
2
Bi(
P)
0.0
00
01
.048
91
.656
61
.250
11
.65
66
1.0
48
90
.000
01
.000
0
%E
V5
2.8
05
5.3
83
9.2
84
6.5
33
9.2
85
5.3
85
2.8
04
5.5
2
Bi(
R)
0.8
91
71
.003
91
.037
81
.923
51
.03
78
1.0
03
90
.891
70
.000
90
.490
70
.960
0
C2
Bi(
TS
)0
.396
41
.105
41
.295
91
.599
81
.29
58
1.1
05
60
.396
50
.488
4
Bi(
P)
0.0
00
01
.216
91
.628
21
.274
31
.62
82
1.2
16
90
.000
01
.000
0
%E
V5
5.5
44
7.6
54
3.7
14
9.8
64
3.7
14
7.6
55
5.5
44
8.7
9
Bi(
R)
0.8
93
60
.994
91
.022
51
.923
61
.02
25
0.9
94
90
.893
60
.002
80
.459
90
.952
5
C3
Bi(
TS
)0
.447
51
.090
81
.237
51
.645
51
.23
74
1.0
91
10
.447
50
.435
2
Bi(
P)
0.0
00
01
.188
61
.564
51
.322
51
.56
45
1.1
88
60
.000
01
.000
0
%E
V4
9.9
24
9.6
63
9.6
44
6.2
63
9.6
44
9.6
64
9.9
24
3.3
6
Wib
erg
bo
nd
indec
es(B
i),
%ev
olu
tion
thro
ugh
the
reac
tion
coord
inat
e(%
EV
),av
erag
ebond
index
var
iati
on
(dB
av)
and
syn
chro
nic
ity
par
amet
er(S
y)ar
esh
ow
n
32 A. Shiroudi et al.
123

transfer of occupancy from the localized NBOs of the idealized Lewis structure into
the empty non-Lewis orbitals (and thus, departure from the idealized Lewis
structure description). It is referred to as ‘delocalization’ correction to the zeroth-
order natural Lewis structure to a stabilizing donor–acceptor interaction. The
energies of these interactions can be estimated by the second order perturbation
theory [36, 37]. For each donor NBO (i) and acceptor NBO (j), the stabilization
energy (E2) associated with i ? j delocalization, is explicitly estimated by the
following equation [37, 38]:
E2 ¼ DEij ¼ qi½F2ði;jÞ=ðei � ejÞ� ð9Þ
where qi is the donor orbital occupancy, ei and ej, are diagonal elements (orbital
energies) and F(i,j) is the off-diagonal NBO Fock matrix elements.
Based on the optimized ground state geometries using the MPW1PW91/6-
31??G** method, the NBO analysis of donor–acceptor interactions revealed that
the stabilization energies associated with the electronic delocalization from non-
bonding Lone-Pair orbitals LP eð ÞX1
� �to r�C1�H2
antibonding orbitals of compounds
1–3. The LP eð ÞX1! r�C2�H6resonance energies for compounds 1–3 are 5.32, 3.10
and 4.14 kcal mol-1, while the LP(e)x1 occupancies in compounds 1–3 are 1.93418,
1.95132 and 1.93516 (Table 6). Also, the LP eð ÞX1! r�C2�H6delocalizations could
fairly explain the decrease of occupancies of LP(e)x1 nonbonding orbitals in the
rings of compounds 1–3 (1 \ 3 \ 2). The results revealed that by the increase of
electronegativity of atom X (O, S, N), LP eð ÞX1! r�C2�H6resonance energies
increase and the HOMO-LUMO energy-gaps in the ground state structures increase.
The results also suggest that in compounds 1–3, the hydrogen elimination are
controlled by LP(e) ? r* resonance energies.
Table 6 NBO calculated resonance energies (E2) (kcal mol-1), based on the MPW1PW91/6-31??G**
level calculated geometries, for LP eð ÞX1! r�C2�H6delocalization, and rC2�H6
bonding and r�C2�H6anti-
bonding orbitals occupancies, HOMO energies, LUMO energies and HOMO-LUMO gaps in the 2,5-
dihydrofuran (1), 2,5-dihydrothiophene (2), and 3-pyrroline (3)
MPW1PW91/6-31??G**
1 2 3
Occupancies
rC2�H61.97819 1.97960 1.97522
r�C2�H60.02751 0.02194 0.02838
LP(e)91 1.93418 1.95132 1.93516
Stabilization energy (E2)
LP eð ÞX1! r�C2�H65.32 3.10 4.14
HOMO,LUMO energies (a.u.)
HOMO -0.24885 -0.22859 -0.23773
LUMO -0.00260 -0.00507 -0.00494
HOMO–LUMO gap 0.24625 0.22352 0.23279
Control aromaticity in thermal decomposition 33
123

Conclusion
The decomposition of the 2,5-dihydrofuran (1), 2,5-dihydrothiophene (2), and 3-
pyrroline (3) has been studied at the B3LYP/6-31??G**, B3PW91/6-31??G**
and MPW1PW91/6-31??G** levels of theory. The MPW1PW91/6-31??G**
level of theory gives activation energies very close to the experimental values and
the decomposition barrier height of compound 3 is the lowest. The values of nT for
the decomposition compounds 1–3, are 0.5042, 0.5144 and 0.4543; therefore, the
similarity between transition states and products increase with respect to reactions in
the order of R2 [ R1 [ R3. In the NICS study, separation of the isotropic values
into in-plane and out-of-plane contributions indicates that aromaticities of products
are controlled by the out-of-plane component, with the exception of the pyrrole in
the ring centre. The strongest aromatic character in the products and TSs was found
for pyrrole and the studied reactions are controlled kinetically and thermodynam-
ically by change of the aromaticity. The NBO analysis revealed that the activation
energies for hydrogen elimination in compounds 1–3 are controlled by LP eð ÞX1
� �!
r�C2�H6resonance energies. Also, NBO results show that the synchronicity values of
decomposition reaction of compounds 1–3 are about 0.93–0.96; therefore, these
reactions are concerted and slightly asynchronous.
Acknowledgment The authors would like to thank Dr D. Tahmassebi for his help on the operation of
the Gaussian 98 programs in this study.
References
1. James TL, Wellington CA (1968) J Chem Soc (A) 2398–2400
2. Wellington CA, Walters WD (1961) J Am Chem Soc 83:4888–4891
3. Thomas AL, Wellington CA (1969) J Chem Soc (A) 2895–2897
4. Wellington CA, James TL, Thomas AC (1969) J Chem Soc (A) 2897–2900
5. Becke AD (1993) J Chem Phys 98:5648–5652
6. Lee C, Yang W, Parr RG (1988) Phys Rev B 37:785–789
7. Hehre WJ, Radom L, Schleyer PVR, Pople JA (1986) Ab initio molecular orbital theory. Wiley,
New York
8. Nori-Shargh D, Shiroudi A, Oliaey AR, Deyhimi F (2007) J Mol Struct (THEOCHEM) 824:1–7
9. Mucsi Z, Chass GA, Csizmadia IG (2009) J Phys Chem B 113:10308–10314
10. Mucsi Z, Viskolcz B, Csizmadia IG (2007) J Phys Chem A 111:1123–1132
11. Mucsi Z, Kortvelyesi T, Viskolcz B, Csizmadia IG, Novak T, Keglevich G (2007) Eur J Org Chem
2007:1759–1767
12. Nori-Shargh D, Roohi F, Deyhimi F, Naeem-Abyaneh R (2006) J Mol Struct (THEOCHEM)
763:21–28
13. Serena Software, Box 3076, Bloomington, IN, USA
14. Stewart JJP (1990) J Comput Aided Mol Des 4:1
15. Stewart JJP, QCPE 581, Department of Chemistry, Indiana University, Bloomington, IN, USA
16. Dewar MJS, Heally EF, Stewart JJP (1984) J Chem Soc Faraday Trans2 80: 227–234
17. Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Zakrazewski VG,
Montgomery JA, Startmann RE Jr, Burant JC, Dapprich S, Millam JM, Daniels AD, Kudin KN,
Strain MC, Farkas O, Tomasi J, Barone V, Cossi M, Cammi R, Mennucci B, Adamo C, Clifford S,
Ochterski J, Petersson GA, Ayala PY, Cui Q, Morokuma K, Malik DK, Rabuck AD, Raghavachar K,
Foresman JB, Cioslowski J, Ortiz JV, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I,
Gomperts R, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanyakkara A, Gonzalez C,
34 A. Shiroudi et al.
123

Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Andres JL, Head-Gordon M, Replogle
ES and Pople JA. GAUSSIAN 98 (Revision A.3) Gaussian Inc. Pittsburgh, PA, USA (1998)
18. Ermer O (1975) Tetrahedron 31:1849–1854
19. McIver-Jr JW (1974) Acc Chem Res 7:72–77
20. Benson SW (1960) The foundations of chemical kinetics. McGraw-Hill, New York
21. Glasstone KJ, Laidler KJ, Eyring H (1941) The theory of rate processes. McGraw-Hill, New York
22. Serrano AJ, Lorono M, Cordova T, Chuchani G (2008) J Mol Strcut (THEOCHEM) 859:69–72
23. Glendening ED, Reed AE, Carpenter JE, Weinhold F, NBO version 3.1
24. O’Neal HE, Benson SW (1967) J Phys Chem 71:2903–2921
25. Benson SW (1960) Thermochemical kinetics. Wiley, McGraw-Hill, New York
26. Nori-Shargh D, Shiroudi A, Naeem-Abyaneh R, Nozari M (2005) Phosphorus, Sulfur Silicon
180:435–442
27. Chuang CH, Lien MH (2004) Eur J Org Chem 2004:1432–1443
28. Cysewski P (2005) J Mol Struct (THEOCHEM) 714:29–34
29. Schleyer PVR, Maerker C, Dransfeld A, Jiao H, van Eikema Hommes NJR (1996) J Am Chem Soc
118:6317–6318
30. Schleyer PVR, Jiao H, Goldfuss B, Freeman PK (1995) Angew Chem Int Ed Engl 34:337–340
31. Nigam S, Majumder C, Kulshreshtha SK (2006) Angew Chem Sci 118:575–578
32. Lendvay G (1989) J Phys Chem 93:4422–4429
33. Reed AE, Curtiss LA, Weinhold F (1988) Chem Rev 88:899–926
34. Reed AE, Weinstock RB, Weinhold F (1985) J Chem Phys 83:735–746
35. Moyano A, Pericals MA, Valentini E (1989) J Org Chem 54:573–582
36. Badenhoop JK, Weinhold F (1999) Int J Quantum Chem 72:269–280
37. Zahedi E, Aghaie M, Zare K (2009) J Mol Struct (THEOCHEM) 905:101–105
38. Carpenter JE, Weinhold F (1988) J Mol Struct (THEOCHEM) 169:41–62
Control aromaticity in thermal decomposition 35
123
















![Aromaticity of the planar hetero[8]circulenes and their doubly charged ions: NICS and GIMIC characterization](https://static.fdokumen.com/doc/165x107/6335e0f002a8c1a4ec01f590/aromaticity-of-the-planar-hetero8circulenes-and-their-doubly-charged-ions-nics.jpg)



