
Synthesis, optical properties and supramolecular order of π-conjugated...
10
Synthesis, optical properties and supramolecular order of p-conjugated 2,5-di(alcoxy)phenyleneethynylene oligomers Griselda Castruita a , Eduardo Arias a, * , Ivana Moggio a , Fátima Pérez a , Diana Medellín a , Román Torres a , Ronald Ziolo a , Amelia Olivas b , Emilia Giorgetti c , Maurizio Muniz-Miranda d a Centro de Investigación en Química Aplicada (CIQA), Boulevard Enrique Reyna 140, 25253 Saltillo, México b Centro de Ciencias de la Materia Condensada CCMC – UNAM, Carretera Tijuana-Ensenada Km 107, 22800 Ensenada, B. C., México c Istituto dei Sistemi Complessi ISC-CNR e INSTM Sezione di Firenze, Via Madonna del Piano 11, Sesto Fiorentino, 50019 Firenze, Italy d Dipartamento di Chimica, Universitá di Firenze, Sesto Fiorentino, Via della Lastruccia 3, Firenze, Italy article info Article history: Received 24 April 2009 Received in revised form 28 July 2009 Accepted 28 July 2009 Available online 3 August 2009 Keywords: Oligo(phenyleneethynylene)’s Supramolecular structure Liquid crystals Small angle X-ray diffraction Optical properties abstract Two series of 2,5-di(alkoxy)phenyleneethynylene oligomers were synthesized by Sonogashira–Heck cou- pling reaction. The chemical structure was corroborated by 1 H, 13 C, APT, DEPT-135 NMR, Raman, FTIR and MALDI-TOF mass spectrometry. The chemical structure of the molecules has been varied in order to study the effect on the physicochemical and optoelectronic properties of the different chain lengths of the lat- eral substituents (dodecanoxy and butoxy), of different terminal groups (H, Br and I), of different chain length (3, 5 and 7 repeat units in the main conjugated backbone). The thermal properties were analyzed by DSC, TGA and by temperature-dependent X-ray diffraction. The diffraction studies of the oligomers revealed a crystalline behavior for the butoxy series, while for the dodecanoxy series the X-ray patterns are consistent with a supramolecular organization formed of randomly distributed crystalline domains that exhibit a periodic structure at small angles, indicating the presence of a lamellar order. The optical properties can be modulated within a series by increasing the length of the conjugated oligomer chain. On the contrary, neither the length of the alkoxy substituents nor the terminal groups have effect on the shape of the absorption and emission spectra. Ó 2009 Elsevier B.V. All rights reserved. 1. Introduction The synthesis of families of phenyleneethynylene (PE) oligomers has been acquiring a growing interest for different research groups because oligomers with well defined length are soluble models that allow ascertaining a proper relationship of structure–properties face to their homologue polymers. Such correlations include the effect of p-electron delocalization on the optical, laser emission, photo and electroluminiscence properties [1]. Their hydrodynamic volume determined by size exclusion chromatography allows the construc- tion of calibration curves to calculate the actual molecular weight of the homologue polymers [2]. Furthermore, very particular supramo- lecular order is also exhibited in function of the oligomer length [3], which is a feature of importance for the development of devices that could emit polarized light [4] in electroluminescent diodes. Since PE oligomers have a potential application in optoelectronics nanotech- nology, new synthetic routes are also being explored to obtain them in few steps and in higher yields [2,5]. Furthermore, PEs have been designed to have many types of molecular architectures such as lin- ear, stared, hyperbranched and dendritic form [6]. The literature in this domain is enormous and several reviews have been reported [7]. Nonetheless, the comparison between the oligomers and poly- mer properties is mainly devoted to dialkyl or monoalkyl phenyle- neethynylenes, while very limited information is available for dialkoxy oligomers [7a]. In our laboratories we focused on dialkoxy phenyleneethynyl- ene oligomers with the aim of obtaining materials for light emit- ting diodes on the basis of the electron-donor effect of the ethers and on the promising results reported in literature for their homo- logue polymers [8]. In order to establish a correlation between the chemical structure of the molecules and their efficiency as emitting materials in LEDs, we report in this paper the synthesis of two ser- ies of PE oligomers bearing two-butoxy or two-dodecanoxy sub- stituents per phenyl ring. A study of their optical and structural properties was carried out by UV–vis, fluorescence spectroscopy, DSC, TGA and X-ray diffraction. The results are discussed in terms of the different chemical structures of the materials; in particular, we considered the different lengths of the lateral substituents, and of the main conjugated chains containing 3, 5 and 7 repeat units. We added different terminal groups to the oligomers (H, Br, I). On the basis of the work so far performed, the optoelectronic prop- erties of LEDs of all materials will be studied and reported in a forthcoming paper. 0022-2860/$ - see front matter Ó 2009 Elsevier B.V. All rights reserved. doi:10.1016/j.molstruc.2009.07.036 * Corresponding author. Tel.: +52 844 438 98 30; fax: +52 844 438 98 39. E-mail address: [email protected] (E. Arias). Journal of Molecular Structure 936 (2009) 177–186 Contents lists available at ScienceDirect Journal of Molecular Structure journal homepage: www.elsevier.com/locate/molstruc
-
Upload
guadalajara -
Category
Documents
-
view
0 -
download
0
Transcript of Synthesis, optical properties and supramolecular order of π-conjugated...

Journal of Molecular Structure 936 (2009) 177–186
Contents lists available at ScienceDirect
Journal of Molecular Structure
journal homepage: www.elsevier .com/ locate /molst ruc
Synthesis, optical properties and supramolecular order of p-conjugated2,5-di(alcoxy)phenyleneethynylene oligomers
Griselda Castruita a, Eduardo Arias a,*, Ivana Moggio a, Fátima Pérez a, Diana Medellín a, Román Torres a,Ronald Ziolo a, Amelia Olivas b, Emilia Giorgetti c, Maurizio Muniz-Miranda d
a Centro de Investigación en Química Aplicada (CIQA), Boulevard Enrique Reyna 140, 25253 Saltillo, Méxicob Centro de Ciencias de la Materia Condensada CCMC – UNAM, Carretera Tijuana-Ensenada Km 107, 22800 Ensenada, B. C., Méxicoc Istituto dei Sistemi Complessi ISC-CNR e INSTM Sezione di Firenze, Via Madonna del Piano 11, Sesto Fiorentino, 50019 Firenze, Italyd Dipartamento di Chimica, Universitá di Firenze, Sesto Fiorentino, Via della Lastruccia 3, Firenze, Italy
a r t i c l e i n f o
Article history:Received 24 April 2009Received in revised form 28 July 2009Accepted 28 July 2009Available online 3 August 2009
Keywords:Oligo(phenyleneethynylene)’sSupramolecular structureLiquid crystalsSmall angle X-ray diffractionOptical properties
0022-2860/$ - see front matter � 2009 Elsevier B.V. Adoi:10.1016/j.molstruc.2009.07.036
* Corresponding author. Tel.: +52 844 438 98 30; fE-mail address: [email protected] (E. Arias).
a b s t r a c t
Two series of 2,5-di(alkoxy)phenyleneethynylene oligomers were synthesized by Sonogashira–Heck cou-pling reaction. The chemical structure was corroborated by 1H, 13C, APT, DEPT-135 NMR, Raman, FTIR andMALDI-TOF mass spectrometry. The chemical structure of the molecules has been varied in order to studythe effect on the physicochemical and optoelectronic properties of the different chain lengths of the lat-eral substituents (dodecanoxy and butoxy), of different terminal groups (H, Br and I), of different chainlength (3, 5 and 7 repeat units in the main conjugated backbone). The thermal properties were analyzedby DSC, TGA and by temperature-dependent X-ray diffraction. The diffraction studies of the oligomersrevealed a crystalline behavior for the butoxy series, while for the dodecanoxy series the X-ray patternsare consistent with a supramolecular organization formed of randomly distributed crystalline domainsthat exhibit a periodic structure at small angles, indicating the presence of a lamellar order. The opticalproperties can be modulated within a series by increasing the length of the conjugated oligomer chain.On the contrary, neither the length of the alkoxy substituents nor the terminal groups have effect onthe shape of the absorption and emission spectra.
� 2009 Elsevier B.V. All rights reserved.
1. Introduction
The synthesis of families of phenyleneethynylene (PE) oligomershas been acquiring a growing interest for different research groupsbecause oligomers with well defined length are soluble models thatallow ascertaining a proper relationship of structure–properties faceto their homologue polymers. Such correlations include the effect ofp-electron delocalization on the optical, laser emission, photo andelectroluminiscence properties [1]. Their hydrodynamic volumedetermined by size exclusion chromatography allows the construc-tion of calibration curves to calculate the actual molecular weight ofthe homologue polymers [2]. Furthermore, very particular supramo-lecular order is also exhibited in function of the oligomer length [3],which is a feature of importance for the development of devices thatcould emit polarized light [4] in electroluminescent diodes. Since PEoligomers have a potential application in optoelectronics nanotech-nology, new synthetic routes are also being explored to obtain themin few steps and in higher yields [2,5]. Furthermore, PEs have beendesigned to have many types of molecular architectures such as lin-ear, stared, hyperbranched and dendritic form [6]. The literature in
ll rights reserved.
ax: +52 844 438 98 39.
this domain is enormous and several reviews have been reported[7]. Nonetheless, the comparison between the oligomers and poly-mer properties is mainly devoted to dialkyl or monoalkyl phenyle-neethynylenes, while very limited information is available fordialkoxy oligomers [7a].
In our laboratories we focused on dialkoxy phenyleneethynyl-ene oligomers with the aim of obtaining materials for light emit-ting diodes on the basis of the electron-donor effect of the ethersand on the promising results reported in literature for their homo-logue polymers [8]. In order to establish a correlation between thechemical structure of the molecules and their efficiency as emittingmaterials in LEDs, we report in this paper the synthesis of two ser-ies of PE oligomers bearing two-butoxy or two-dodecanoxy sub-stituents per phenyl ring. A study of their optical and structuralproperties was carried out by UV–vis, fluorescence spectroscopy,DSC, TGA and X-ray diffraction. The results are discussed in termsof the different chemical structures of the materials; in particular,we considered the different lengths of the lateral substituents, andof the main conjugated chains containing 3, 5 and 7 repeat units.We added different terminal groups to the oligomers (H, Br, I).On the basis of the work so far performed, the optoelectronic prop-erties of LEDs of all materials will be studied and reported in aforthcoming paper.

178 G. Castruita et al. / Journal of Molecular Structure 936 (2009) 177–186
2. Experimental
2.1. Synthesis
Experimental procedures and characterization of all compoundsare given in the Supplementary data section.
2.2. Physical measurements
NMR spectra were obtained at room temperature with a JeolEclipse spectrometer at 300 MHz for 1H and 75.4 MHz for 13C usingCDCl3 as solvent and internal reference. Thermogravimetric (TGA)and differential scanning calorimetry (DSC) analyses were carriedout on a DuPont 951 instrument under nitrogen at a heating rateof 10 �C/min. Small angle X-ray diffraction patterns were obtainedwith a Phillips X’PERT diffractometer equipped with a curvedgraphite monochromator using CuKa radiation at a wavelengthof 0.1542 nm. MALDI-TOF MS was performed either using a BrukerBIFLEX III. Spectra were recorded using the reflection mode inwhich samples were irradiated under high vacuum using a nitro-gen laser (wavelength 337 nm, 3 ns pulse); dithranol (Aldrich)
Scheme 1. Reagents and conditions: (a) BrAC12H25, NaOH, DMF, 120 �C, 24 h; (b) Br2, C16 h; (d) TBAF, THF, r.t., 0.5 h; (e) KIO3, I2, AcOH, H2SO4, H2O (92: 7: 1 vol.), 120 �C, 24[(C6H5)3P]2PdCl2, CuI, TMSA, Et3N, THF, 0 �C to r.t., 48 h; (i) TBAF, THF, r.t., 0.5 h.
Scheme 2. Reagents and conditions: (l) [(C6H5)3P]2PdCl2 (2.5% mol), CuI (1.5% mol), Et3NCuI, TMSA, Et3N, 80 �C, 16 h; (o) TBAF, THF, r.t., 0.5 h.
was used as the matrix, or in a Voyager DETM Pro (PerSeptive Biosys-tem, PE). Calibrated with oligothiophene standards in positive andnegative ion modes, reflector and linear and using a-cyano-4-hy-droxy-cinnamic acid or 2-[(2E)-3-(4-tert-butylphenyl)-2-methyl-prop-2-enylidene]malonitrile as the matrix. UV–vis spectra wererecorded on a Shimadzu 2401 UV spectrophotometer. Fluorescencespectra were obtained with a Perkin Elmer LS50B spectrofluorime-ter. Quantum yields in solution (UF.) were determined by adaptinga procedure reported in literature [9] and using as standard qui-nine sulfate in (0.1 M) H2SO4 (U = 0.54). The molecules where ex-cited next to the high energy absorption peak (310 nm) and at10 nm above the lower energy absorption peak. Three repetitionswere realized by using diluted solutions with absorbance at theexcitation wavelength between 0.027 and 0.1. The temperaturewas maintained at 25.5 ± 0.3 �C with a circulating water bath.MicroRaman spectra were collected using a Renishaw RM2000single grating spectrograph, coupled with a diode laser sourceemitting at 785 nm and sample irradiation was accomplishedusing a 50�microscope objective of a Leica Microscope DMLM. Ra-man light was filtered by a double holographic Notch filters systemand collected by an air-cooled CCD detector. The acquisition time
Cl4, 70 �C, 16 h; (c) [(C6H5)3P]2PdCl2 (2.5% mol), CuI (1.5% mol), TMSA, Et3N, 80 �C,h; (f) [(C6H5)3P]2PdCl2, CuI, TMSA, Et3N, 80 �C, 16 h; (g) TBAF, THF, r.t., 0.5 h; (h)
, 80 �C; (m) [(C6H5)3P]2PdCl2, CuI, Et3N, THF, 0 �C to r.t., 48 h; (n) [(C6H5)3P]2PdCl2,
G. Castruita et al. / Journal of Molecular Structure 936 (2009) 177–186 179
for each measurement was 10 s. All spectra were calibrated withrespect to a silicon wafer at 520 cm�1.
3. Results and discussion
3.1. Monomer synthesis
The synthetic pathway used to obtain the required monomersfor oligomerization is depicted in Scheme 1. The chemical proce-dures were adapted and modified from the literature [10]; experi-mental procedures and characterization of compound 4–18 aregiven in the electronic Supplementary information [11]. The com-mercial 1,4-benzoquinone 1 was alkylated with 1-bromododecane(or 1-bromobutane) by the Williamson reaction, followed by the
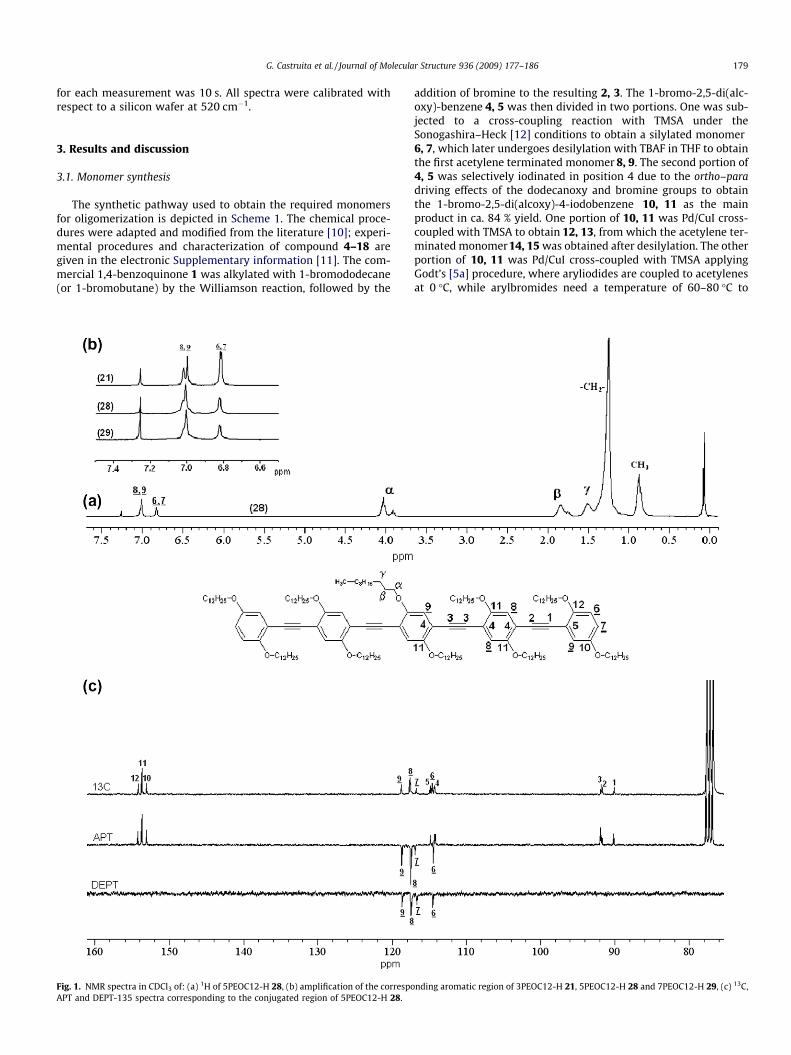
Fig. 1. NMR spectra in CDCl3 of: (a) 1H of 5PEOC12-H 28, (b) amplification of the correspoAPT and DEPT-135 spectra corresponding to the conjugated region of 5PEOC12-H 28.
addition of bromine to the resulting 2, 3. The 1-bromo-2,5-di(alc-oxy)-benzene 4, 5 was then divided in two portions. One was sub-jected to a cross-coupling reaction with TMSA under theSonogashira–Heck [12] conditions to obtain a silylated monomer6, 7, which later undergoes desilylation with TBAF in THF to obtainthe first acetylene terminated monomer 8, 9. The second portion of4, 5 was selectively iodinated in position 4 due to the ortho–paradriving effects of the dodecanoxy and bromine groups to obtainthe 1-bromo-2,5-di(alcoxy)-4-iodobenzene 10, 11 as the mainproduct in ca. 84 % yield. One portion of 10, 11 was Pd/CuI cross-coupled with TMSA to obtain 12, 13, from which the acetylene ter-minated monomer 14, 15 was obtained after desilylation. The otherportion of 10, 11 was Pd/CuI cross-coupled with TMSA applyingGodt’s [5a] procedure, where aryliodides are coupled to acetylenesat 0 �C, while arylbromides need a temperature of 60–80 �C to
nding aromatic region of 3PEOC12-H 21, 5PEOC12-H 28 and 7PEOC12-H 29, (c) 13C,

Fig. 2. Raman spectra of 3PEOC12-H 21, 5PEOC4-H 27, 5PEOC12-H 28, 7PEOC12-H29 and polymer I-nPEOC12. The insert figure shows the Infrared spectra ofmonomer 15 and 7PEOC12-H 29.
180 G. Castruita et al. / Journal of Molecular Structure 936 (2009) 177–186
effect the coupling reaction. Compounds 16, 17 was obtained in�80 % yield and its desilylation afforded the desired monomer18, 19.
3.2. Oligomer synthesis
The step by step synthetic route to obtain the oligomer series isoutlined in Scheme 2 either for two-butoxy chains per phenyl ringor two-dodecanoxy chains. We designed this route to avoid the useof triazenes as functional groups because of the difficulty to keeptheir purity as long as the oligomer length increases. However, itis worth mentioning that the divergent–convergent approach bythe use of the bifunctional acetylene–aryl–triazene monomer thatwe recently reported [5c] constitutes a more practical and easyroute to obtain oligomers larger than five repeated units. Monomer10, 11 was divided into three portions; one portion was Pd/CuIcross-coupled to 8, 9 to obtain the trimer hydrogen terminated20, 21. In the second portion only 11 was Pd/CuI cross-coupledto 9 at 0 �C in order to obtain a dimer terminated by a brominegroup 22, from which after being Pd/CuI cross-coupled to TMSA,23 was obtained and its desilylation afforded a dimer terminatedby an acetylene group 24. The third portion of 10, 11 undergoesPd/CuI cross-coupling with 14, 15 under Godt’s procedure to gen-erate a bromine terminated trimer 25, 26 in ca. 65 % yield; for thisparticular reaction, the internal temperature should not exceed0 �C for at least 4 h. Even with this control, the formation of highmolecular weight compounds could not be avoided because ofthe polymerization of 14, 15 with 10, 11. Therefore, 25 and 26 wereobtained pure only after being passed twice by silica gel chroma-tography and then by preparative GPC (Biorads, Bio-Beads SX1, tol-uene). The trimer 26 was also divided into two portions in order toform the pentamer 28 and the heptamer 29, while 25 was usedonly to form the pentamer 27. For the pentamers formation, 25(26) was Pd/CuI cross-coupled with 2.2 equivalents of 8 (9), whilefor the heptamer 29; 26 was cross-coupled with 2.2 equivalents ofdimer 24. Yield for oligomers 27, 28 and 29 was rather modest ofalmost 45, 46 and 39%, respectively. A large amount of dimer(52%) and tetramer (58%) having a diacetylene as the central group,from the homo-coupling of 8, 9 and 24 however, was isolatedamong the byproducts. The electron-donor character of the dodec-anoxy or butoxy chains appears to favor the oxidative Glaser [13]type homo-coupling of the symmetrical acetylenes rather thanthe Sonogashira–Heck cross-coupling reaction.
3.3. Chemical characterization
The chemical structure of all materials was confirmed by 1H, 13CNMR, Attached Proton Test (APT), Distortionless Enhancement byPolarization Transfer (DEPT-135) spectroscopies and by MALDI-TOF spectrometry. Fig. 1a shows the 1H NMR of the dodecanoxypentamer 28 as an example. In the aliphatic region, all oligomersand polymers show the same resonant peaks, but with differentintensities. The unresolved signal at d = 0.86 ppm is assigned tothe methyl protons of the dodecanoxy chains, the ethyls ACH2Aare centered at 1.24 ppm and the c-CH2AO protons appear at1.50 ppm; those of the b-CH2AO and a-CH2AO are centered at1.83 and 4.0 ppm, respectively. Two different aromatic proton sig-nals are identified in the oligomer series, Fig. 1b (inset): those sit-uated at each extremity of the oligomers, peaks (6 and 7) at6.81 ppm and the internals (8 and 9) at 7.07 ppm. In this figure,it can be seen that as the oligomer length increases, the internalsignal intensity also increases, while the externals always integratefor four protons. Assignment of each resonant peak to the corre-sponding carbon of the molecular structures by 13C NMR was morecomplicated because of overlapping signals. However, by takinginto account that the peak intensities are different as a function
of the degree of carbon substitution, as well as by applying othertechniques such as APT and DEPT-135, four type of carbons couldbe identified as seen in Fig. 1c: (i) the ethynylenes 1, 2 and 3 at89.89, 91.52 and 91.70 ppm, (ii) two ipso phenyleneethynylenes4 and 5 at 114.17 and 114.76 ppm, (iii) those belonging to themethines 6, 7, 8 and 9 at 114.53, 116.67, 117.52, 118.66 ppm and(iv) those substituted with the dodecanoxy 10, 11 and 12 at152.98, 153.62 and 154.11, respectively. The 13C APT spectra con-firmed the carbon assignment with all the quaternized carbonsas positive and the aromatic methines as negative. From this figure,it can be seen how hydrogen 7 of the terminated pentamer can beunequivocally identified. In the DEPT-135 experiment, only thearomatic carbons with an attached proton as is the case for themethines are visible as negative peaks, the rest of the ACH0A car-bons, including CDCl3, disappear.
The laser desorption/ionization time of flight (MALDI-TOF) massspectra confirmed the expected structure, for instance [11] for tri-mer 21, pentamer 28 and heptamer 29 a molecular ion [radical cat-ion] m/z of 1383.60, 2320.29 and 3259.50 were obtained, while therespective calculated mass for 21 was of C94H158O6: 1384.26, for 28of C158H262O10: 2321.76 and of C222H366O14: 3259.27. It was foundthat for these molecules a good signal/noise ratio can be obtainedby using dithranol or a-cyano-4-hydroxy-cinnamic acid or 2-[(2E)-3-(4-tert-butylphenyl)-2-methylprop-2-enylidene]malonitrile asmatrix and without to add cationized agents, such as Na, K or Li.
Fig. 2 shows the Raman spectra of 21, 28, 29 and I-nPEOC12that were obtained in thin films with a 785 nm excitation wave-length with the aim to avoid the strong fluorescence of these mate-rials in the visible region. Since the electronic properties of theoligomers are mainly related to the delocalization of the p-elec-trons through the (di-alcoxy)phenylAC„CA backbone, theAC„CA stretching group was analyzed by IR and Raman spectros-copy. The symmetry of these molecules, originates that the triplebond appears as a very intense peak in the Raman spectra becauseof the large polarizability change that is associated with this vibra-tion frequency. A shift to lower frequencies is observed going fromthe trimer 21 (m = 2205 cm�1), to the pentamer 28 (2200 cm�1) tothe heptamer 29 (2198 cm�1) and to the polymer I-nPEOC12(2195 cm�1). This shift is due to the low bond energy as a resultof the lowest electron density of the triple bond as far as the con-jugation increases, reaching maxima saturation energy for thepolymer. The delta of only 3 cm�1 between the heptamer and the

G. Castruita et al. / Journal of Molecular Structure 936 (2009) 177–186 181
polymer suggests that an effective conjugation could be reachedwith an oligomer having 9–10 repeat units. The lateral chain lengthas is expected has no influence in the AC„CA stretching frequen-cies, as an example in Fig. 2 is shown the spectrum of the butoxyhydrogen terminated pentamer 27 which shows a vibration fre-quency m of 2200 cm�1, similar to that of its homologue 28. In whatconcerns to the ACH = CHA vibration out of the plane of the alcoxyphenyl groups, the shift to lower frequencies is also dependant ofthe extend of conjugation being of 1598 cm�1 for 21, of1596 cm�1 for 28, of 1594 cm�1 for 29 and of 1594 cm�1 for I-nPEOC12. Contrary to what happened in the Raman spectra, inthe IR spectra, Fig. 2 insert, the corresponding AC„C-stretchingband becomes almost imperceptible, due to the lack of dipole mo-ment change, as can be seen for instance in the spectrum of the hep-tamer 29 at 2210 cm�1 and that is very close between each oligomer,in contrast to the that of the monomer 15 that shifts to 2110 cm�1.This former exhibits however a very intense „C-H stretching bandat 3280 cm�1 that disappears for the heptamer 29 as expected.
3.4. Electronic absorption and emission properties
In order to study the effect of the chain length of the lateral sub-stituents, chain length backbones and of different terminal groups(H, Br and I) on the optoelectronic properties of the oligomers, twoother oligomers were incorporated in the discussion; the dodecan-oxy iodine terminated trimer [5c,11] 30 and the butoxy bromineterminated pentamer [11] 31, with the following chemicalstructures:
C4H9O
OC4H9
C4H9O
OC4H9
C4H9O
OC4H9
C4H9O
OC4H9
C4H9O
OC4H9
Br
31
C12H25O
OC12H25
I
C12H25O
OC12H25
C12H25O
OC12H25
30
I Br
Additionally, the 2,5-di(butoxy) phenylenethynylene (Br-nPE-OC4) and the 2,5-di(dodecanoxy) phenylenethynylene (I-nPEOC12)[5b] polymers, homologues to the series, were also analyzed. Table 1collects all the optical data for the 2,5-di(alkoxy)phenyleneethynyl-ene oligomers studied in this work.
The following general remarks can be made: (i) all the mole-cules absorb in the UV-blue and emit in the blue–green region; alight of relatively high energy. (ii) The electronic absorption spectraare characterized by an intense broad band with maximumwavelength ranging between 376 nm and 430 nm and a higher
Table 1Electronic absorption and emission properties of the 2,5-di(alkoxy)phenyleneethynylene m
Molecule kabs [nm] e[M�1 cm�1] � 104 kem [nm
3PEOC12-Br (26) 388 3.33 4173PEOC12-I (30) 384 9.34 4203PEOC12-H (21) 380 5.04 4115PEOC12-H (28) 412 6.36 4527PEOC12-H (29) 417 12.40 465I-nPEOC12 430 5.78*a 4763PEOC4-H (20) 379 6.42 4105PEOC4-H (27) 412 7.05 4513PEOC4-Br (25) 386 4.96 4175PEOC4-Br (31) 415 8.49 454Br-nPEOC4 420 3.39*b 470
* Based on the molecular weight of the repetitive unit. (Average aMw = 19,310; depolystyrene standards).
a estimated error: 10%.
energy absorption peak at around 300 nm. According to theoreticalcalculations on alcoxy substituted oligomers, the red shifted bandcan be associated with the electronic transition from HOMO toLUMO and the higher energy absorptions to those between lowerlying occupied molecular orbitals (HOMO-1, HOMO-2) to LUMO[14]. (iii) Contrary to the electronic absorption spectra, the fluores-cence ones are structured presenting a high energy intense peakand a shoulder at longer wavelength. This behavior has been pre-viously reported for other alcoxy phenyleneethynylenes andattributed to the fact that several rotamers are possible at theground state and each contributes optically in the UV–vis spectra.In the excited state, planarization of the geometry towards morequinoidal/cumulenic structures is promoted giving a more local-ized state [14]. (iv) In our molecules this effect can be further sup-ported by the Stokes’ shift values. These are in fact in the rangebetween Stokes losses for typical aromatic systems such as fluorine(around 1430 cm�1) and those of biphenyls, which assume a moreplanar conformation in the excited state (ca. 3310 cm�1) [15]. Asterm of comparison, Stokes shift for a phenyleneethynylene trimerand a pentamer bearing two hexyloxy lateral chains on the centralphenyl ring in dichloromethane was reported to be 2000 cm�1 and2200 cm�1, respectively [14a]. (v) The absorption spectra of all themolecules do not change with concentration and solvent type,excluding ground state interactions such as aggregation. Fig. 3ashows the spectra relative to 27 in chloroform as an example,while the spectra of 27 in toluene and DMF at different concentra-tion are included in the Supplementary data section. (vi) The fluo-rescence spectra recorded by exciting at different wavelength
exhibit the same shape (Fig. 3b shows the spectra relative to 27as example) and are practically independent on the solvent (seeSupplementary data section) suggesting that there is just one emit-ting state. Moreover, the excitation spectra recorded at the mainemission peak or at the shoulder (see Supplementary data section)are identical, indicating that both fluorescence bands are associ-ated with the same emitting state. (vii) The quantum yields in solu-tion (UF) are of medium value and in the typical range expected fordialkoxy substituted phenyleneethynylenes [7a]. The highestvalues are found for the pentamers. (viii) UF obtained by excitingat 310 nm is lower than that at the corresponding main absorption
aterials studied in this work in CHCl3.
] UF (kexc, nm) [%]a Stokes’ shift [cm�1] ES0–S1 [eV]
25 (310)66 (378) 1792 3.076 (310)14 (374) 2232 3.0538 (310)72 (370) 1985 3.0960 (310)84 (402) 2148 2.8121 (310)71 (407) 2475 2.7824 (310)37 (420) 2247 2.6819 (310)70 (369) 1995 3.0932 (310)74 (400) 2099 2.8134 (310)52 (376) 1868 3.0324 (310)83 (405) 2070 2.8012 (310)39 (410) 2533 2.71
gree of polymerization DP = 41. bMw = 10,587; DP = 43) determined by SEC using

Fig. 4. Normalized absorption and fluorescence (insert) spectra in CHCl3 ofmonomer 2 (dash-dotted line), 3PEOC4-H 20 (dotted lines), 3PEOC4-Br 25 (shortdash-dotted line), 5PEOC4-H 27 (short dashed lines), polymer Br-nPEOC4 (solidlines).
Fig. 3. (a) UV–vis spectra of 27 in chloroform at different concentrations (up to2 � 105 M). Inserted figure: UV–vis spectra of 27 in chloroform (solid line), toluene(dotted line) and DMF (dashed line). (b) Fluorescence spectra of 27 in chloroform atdifferent excitation wavelengths: 310 nm (trace a), 350 nm (trace b), 373 nm (tracec) and 400 nm (trace d).
182 G. Castruita et al. / Journal of Molecular Structure 936 (2009) 177–186
peak because the HOMO-1 or HOMO-2 ? LUMO electronic transi-tion occurs. This involves a higher energy than the HOMO–LUMOelectronic transition and correspondingly the internal conversionlosses are higher.
For sake of clarity, further discussion is presented by groups ofmolecules by studying the different parameters that can affectthem such as conjugated chain length, terminal groups and lateralchain length.
3.5. Effect of the conjugated chain length
The optical properties for the hydrogen terminated trimers 20,21, pentamers 27, 28, heptamer 29 and the homologue polymersBr-nPEOC4 and I-nPEOC12 show that the optical absorption andemission wavelengths increase with the oligomers length and thelimiting value is reached for the polymer, although the saturationof conjugation for the dodecanoxy series is nearly gained with anoligomer having 8–9 repeat units as the difference between thekmax of 28 and that of 29 is of only 5 nm and between 29 andthe homologue polymer is 13 nm. For the butoxy series, Fig. 4,the effective conjugation could be found with an oligomer having7–8 repeat units, given that the kmax between the pentamer andthe polymer is of only 7 nm. The corresponding molar absorptioncoefficient increases along the families in agreement with theoret-ical calculations on the oscillator strength of the HOMO–LUMO
electronic transition for alcoxy substituted phenyleneethynylenes[14b]. The emission wavelength also red shifts with the increasingconjugation and as a consequence, the energy of the first excitedstate decreases. The Stokes’ shift exhibits the same trend. In agree-ment with literature studies [14a], our molecules present anincreasing Stokes’ shift along with the number of repetitive unitsfor the same oligomer family, for instance passing from 21 to 28and to 29. This behavior evidences higher energy losses due tointernal conversion as long as the conjugation chain increases. Thisis consistent with theoretical studies [14a,14b], as the number ofpossible conformers for the ground state increases with the repet-itive units, the emitting state is always coplanar yielding to stron-ger changes in the geometry when passing from the ground to theexcited state. The fluorescence quantum yield increases with theincreasing repetitive units with the exception of the dodecanoxyheptamer 29. As the Stokes’ shift behavior is opposite, internallosses can not be the only factor to be involved in the fluorescencequantum yield. Other authors have reported that intersystemcrossing with triplet excited states formation is an important deac-tivation pathways for phenyleneethynylenes [16]. This effect cannot be discarded for our molecules and photophysical studies areconsidered to corroborate this possibility.
3.6. Effect of the terminal groups
A bathochromic shift of the absorption and emission maxima isobserved passing from the hydrogen terminated molecules to theirhalogenated homologues which can be ascribed to inductive elec-tronic effect of these termini groups. Bromine seems to impart amore aromatic character to the excited state as the Stokes’ shiftof bromine terminated oligomers is always lower than the H termi-nated homologue. However and in general UF for the halogenatedoligomers is lower than their corresponding hydrogen terminatedoligomers. As we have already mentioned, this could be due tothe triplet state formation and internal conversion losses. Sincethe Stokes’ shift trend is opposite to the fluorescence quantumyield, probably the internal conversion losses are less marked thanintersystem crossing. This latter is expected to be favored by thepresence of heavy atoms such as halogen terminal groups. How-ever, the investigation of long lived states by transient absorptionspectra and fluorescence decays studies is necessary to understandthis complex system.
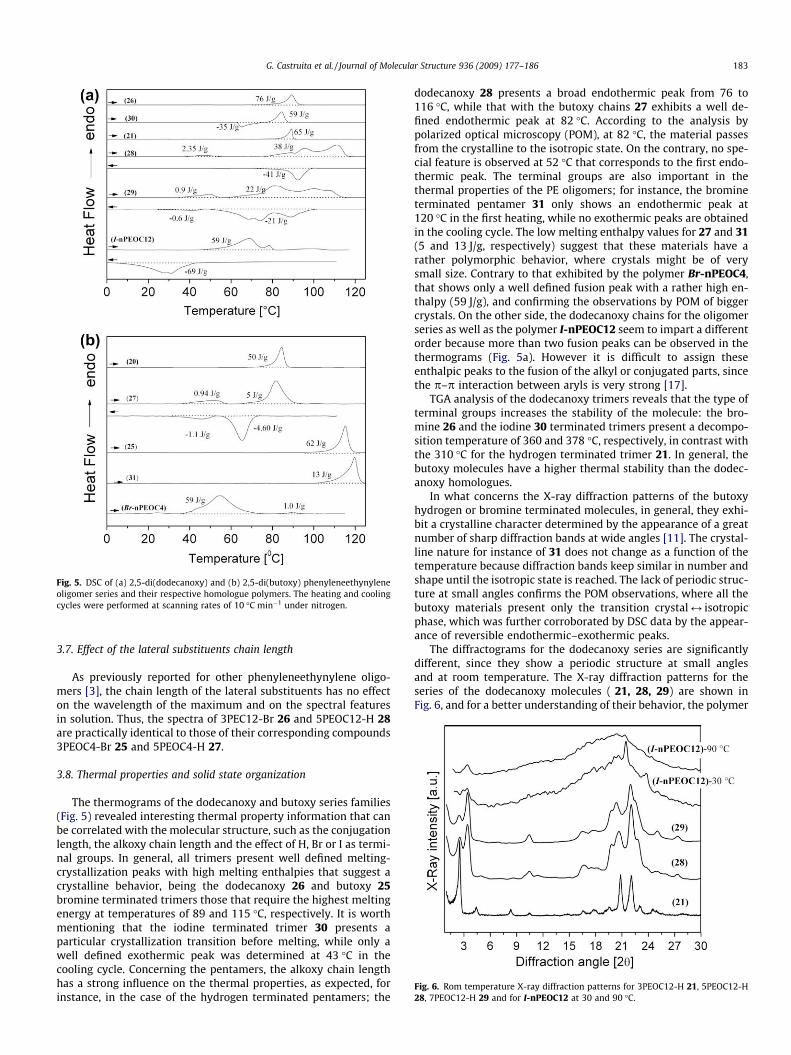
Fig. 5. DSC of (a) 2,5-di(dodecanoxy) and (b) 2,5-di(butoxy) phenyleneethynyleneoligomer series and their respective homologue polymers. The heating and coolingcycles were performed at scanning rates of 10 �C min�1 under nitrogen.
Fig. 6. Rom temperature X-ray diffraction patterns for 3PEOC12-H 21, 5PEOC12-H28, 7PEOC12-H 29 and for I-nPEOC12 at 30 and 90 �C.
G. Castruita et al. / Journal of Molecular Structure 936 (2009) 177–186 183
3.7. Effect of the lateral substituents chain length
As previously reported for other phenyleneethynylene oligo-mers [3], the chain length of the lateral substituents has no effecton the wavelength of the maximum and on the spectral featuresin solution. Thus, the spectra of 3PEC12-Br 26 and 5PEOC12-H 28are practically identical to those of their corresponding compounds3PEOC4-Br 25 and 5PEOC4-H 27.
3.8. Thermal properties and solid state organization
The thermograms of the dodecanoxy and butoxy series families(Fig. 5) revealed interesting thermal property information that canbe correlated with the molecular structure, such as the conjugationlength, the alkoxy chain length and the effect of H, Br or I as termi-nal groups. In general, all trimers present well defined melting-crystallization peaks with high melting enthalpies that suggest acrystalline behavior, being the dodecanoxy 26 and butoxy 25bromine terminated trimers those that require the highest meltingenergy at temperatures of 89 and 115 �C, respectively. It is worthmentioning that the iodine terminated trimer 30 presents aparticular crystallization transition before melting, while only awell defined exothermic peak was determined at 43 �C in thecooling cycle. Concerning the pentamers, the alkoxy chain lengthhas a strong influence on the thermal properties, as expected, forinstance, in the case of the hydrogen terminated pentamers; the
dodecanoxy 28 presents a broad endothermic peak from 76 to116 �C, while that with the butoxy chains 27 exhibits a well de-fined endothermic peak at 82 �C. According to the analysis bypolarized optical microscopy (POM), at 82 �C, the material passesfrom the crystalline to the isotropic state. On the contrary, no spe-cial feature is observed at 52 �C that corresponds to the first endo-thermic peak. The terminal groups are also important in thethermal properties of the PE oligomers; for instance, the bromineterminated pentamer 31 only shows an endothermic peak at120 �C in the first heating, while no exothermic peaks are obtainedin the cooling cycle. The low melting enthalpy values for 27 and 31(5 and 13 J/g, respectively) suggest that these materials have arather polymorphic behavior, where crystals might be of verysmall size. Contrary to that exhibited by the polymer Br-nPEOC4,that shows only a well defined fusion peak with a rather high en-thalpy (59 J/g), and confirming the observations by POM of biggercrystals. On the other side, the dodecanoxy chains for the oligomerseries as well as the polymer I-nPEOC12 seem to impart a differentorder because more than two fusion peaks can be observed in thethermograms (Fig. 5a). However it is difficult to assign theseenthalpic peaks to the fusion of the alkyl or conjugated parts, sincethe p–p interaction between aryls is very strong [17].
TGA analysis of the dodecanoxy trimers reveals that the type ofterminal groups increases the stability of the molecule: the bro-mine 26 and the iodine 30 terminated trimers present a decompo-sition temperature of 360 and 378 �C, respectively, in contrast withthe 310 �C for the hydrogen terminated trimer 21. In general, thebutoxy molecules have a higher thermal stability than the dodec-anoxy homologues.
In what concerns the X-ray diffraction patterns of the butoxyhydrogen or bromine terminated molecules, in general, they exhi-bit a crystalline character determined by the appearance of a greatnumber of sharp diffraction bands at wide angles [11]. The crystal-line nature for instance of 31 does not change as a function of thetemperature because diffraction bands keep similar in number andshape until the isotropic state is reached. The lack of periodic struc-ture at small angles confirms the POM observations, where all thebutoxy materials present only the transition crystal M isotropicphase, which was further corroborated by DSC data by the appear-ance of reversible endothermic–exothermic peaks.
The diffractograms for the dodecanoxy series are significantlydifferent, since they show a periodic structure at small anglesand at room temperature. The X-ray diffraction patterns for theseries of the dodecanoxy molecules ( 21, 28, 29) are shown inFig. 6, and for a better understanding of their behavior, the polymer

Fig. 7. X-ray diffraction patterns at different temperatures for: (a) 5PEOC12-H 28and (b) 7PEOC12-H 29.
184 G. Castruita et al. / Journal of Molecular Structure 936 (2009) 177–186
I-nPEOC12 was also added to the discussion. From this figure, thetrimer 3PEOC12-H 21 shows four high order reflections at2h = 2.49, 4.39, 8.33 and 10.47�, with a ratio of ca. 1:2:3:4 that isindicative of a lamellar system and representative of a smecticmesophase with a layer spacing d = 35.4 Å. Well defined intensepeaks are observed for d = 4.26 and 4.03 Å and at higher angles abroad peak with d = 3.62 Å is also observed. The diffractogram ofthis trimer does not change with temperature [11], indicating thatthe nature of the mesophase is maintained up to the isotropic melt.
We performed molecular simulations for the conformation ofthe oligomers. They were optimized from the equilibrium geome-tries through Density Functional Theory based on the hybrid B3LYPfunction and using the 3-21G� and 6-31G(d) basis. They revealedthat trimer 21 has an extended conformation lengthL = 36.87 ± 0.086 Å from aliphatic to aliphatic chain and with a con-jugated backbone length of l = 18.7 Å. This value suggests that alkylchains are slightly tilted (16�) with respect to the normal; cosu = d/L. From Fig. 6, we also note that 5PEOC12-H 28, 7PEOC12-H 29 andthe I-nPOC12 polymer: (i) both oligomers show two high orderpeaks at 2h = 2.49 and 3.43�. The peak at 2.49� (as the correspond-ing trimer) decreases in intensity from the trimer to the pentamerand to the heptamer, while the reflection peak centered at 3.43�,that appeared from the pentamer becomes more intense for theheptamer. The corresponding interlamellar spacing for these an-gles is 25.7 and 35.4 Å, respectively. These values do not matchwith the molecular dimensions that were calculated to be36.8 � 31.0 Å for the pentamer 28 and 36.8 � 45.8 Å for the hepta-mer 29. Therefore, a tilt angle of the alkyl chains of 16 and 46�must be considered also for these oligomers, Table 2. (ii) The poly-mer exhibits one broad first order peak at 3.11� at 30 �C;d = 28.41 Å that becomes more defined as a function of the temper-ature, but not as sharp as those of the oligomers, with a very smallreduction of the interlamellar spacing d = 26.6 Å at 90 �C and a cal-culated tilted angle of 40� (Le Moigne [10a] reports a d = 29.5 and24.5 Å at 30 and 80 �C for a similar polymer but bromine termi-nated). As a whole, these results suggest that trimer 21 adopts apreferential molecular order where the alkyl chains are tilted at16� as mentioned above and illustrated in the model of Fig. 8a.The pentamer 28 and the heptamer 29 present rather the coexis-tence of at least two supramolecular ordered domains, where thealkyl chains are tilted of 16 and 46�. These domains are orientedin all directions as is sketched in Fig. 8b. For example, the micro-graph of Fig. 8c, corresponding to the heptamer 29 observed
Table 2Experimental calculated diffraction spacing d(100) at a given 2 angle and in function of theextended conformation from aliphatic to aliphatic chain and l corresponds to the conjuga
Molecule N [�C] 2h [�] d(100)[Å]
3PEOC12-H (21) 30 2.49 35.4860 2.51 35.2075 2.53 34.9285 2.53 34.92
5PEOC12-H (28) 30 2.49, 3.43 35.48, 2580 2.53, 3.39 34.92, 2690 2.55, 3.33 34.64, 26
100 2.55, 3.23 34.64, 27110 2.57, 3.17 34.38, 27
7PEOC12-H (29) 30 2.49, 3.43 35.48, 2580 2.57, 3.41 34.38, 2595 3.21, 3.61 27.52, 24
100 3.67 24.34110 3.71 23.81
I-nPEOC12 30 3.11 28.4160 3.32 26.6175 3.32 26.6190 3.32 26.61
between crossed polarizers at 95 �C shows a crystalline phase,where crystals are randomly distributed in all directions. Furtherinvestigations of the other materials by POM and X-ray diffractionwere performed with temperatures corresponding to the phase re-gions seen by DSC. They revealed that the trimer 21 [11], the pen-tamer 28 (Fig. 7a) and the heptamer 29 (Fig. 7b) show the same
temperature for the dodecanoxy series. L is the theoretical molecular length on theted backbone length. The molecular tilt angle was calculated from: cos = d(100)/L.
L [Å] l [Å] Tilt angle u [�]
36.87 ± 0.086 18.7 16171919
.76 36.87 ± 0.086 31.0 16, 46
.06 19, 45
.53 20, 44
.35 20, 42
.87 21, 41
.76 36.87 ± 0.086 45.8 16, 46
.91 21, 45
.47 42, 484950
36.87 ± 0.086 — 40444444

Fig. 8. (a) Possible molecular ordering deduced from X-ray diffraction powder for the 2,5-di(dodecanoxy) series; three dimensional view I, a side view II and a top view III.The different molecular net parameters such as l, L, , c and d are collected in Table 2. (b) Sketch that schematically represents the formation of small crystals randomlydistributed and oriented during melting. (c) POM micrographs captured during the second cooling cycle of 7PEOC12-H 29 and showing the crystals formation from melting at95 �C and at (d) 80 �C.
G. Castruita et al. / Journal of Molecular Structure 936 (2009) 177–186 185
behavior: a crystalline–isotropic–crystalline transition. In otherwords, the materials practically crystallize from the melt as seenin the photo in Fig. 8c and d, taken at 95 and 80 �C in the coolingcycle of the heptamer 29 and adopting a lamellar order withoutevidence of any of the known textures reported in literature. Athigher angles, it is evident that only the polymer presents a diffuseband centered at aprox. 4.4 Å, Fig. 6, while the other moleculespresent a series of half broad bands covering the range between2h = 16.6 � and 2h = 23.0 � with spacing d = 5.3, 4.48, 4.4, 4.0 and3.86 Å. This reflection bands are associated with the order of thedodecanoxy side chains, being the average distance between twochains c = 4.0 Å, Fig. 8a. In addition, it is worth noting the presenceof a very diffuse scattering at 2h = 25�, d = 3.6 Å (d from Fig. 8a)indicating the existence of p–p interactions between phenyl ringsof the conjugated backbone as previously found for other arylenee-thynylenes [18]. After analyzing this series of 2,5-di(dodecanoxy)phenyleneethynylene molecules, we conclude that the term ‘‘sani-
dic” proposed by Bunz [7a,19] for other similar PPEs polymers canalso be applied to these oligomers that match well with brickshape and because of their board-like phases.
4. Conclusions
Two families of 2,5-di(alkoxy)phenyleneethynylene oligomerswere synthesized by the Sonogashira–Heck reaction. The chemicalstructure of the molecules was varied in order to study the effecton the optical and physicochemical properties of the materials ofthe following parameters: (a) chain length of the conjugated back-bone in a series of 2,5-di(dodecanoxy) phenyleneethynylene oligo-mers and polymer, (b) chain length of the lateral ether substituents(dodecanoxy/butoxy) and (c) terminal groups (hydrogen, bromineand iodine terminated dodecanoxy trimers). A bathochromic shiftwas observed when passing from the trimer to the pentamer and

186 G. Castruita et al. / Journal of Molecular Structure 936 (2009) 177–186
in the dodecanoxy series when passing to the heptamer and thento the polymer according to the increasing conjugation. A similarbehavior was observed in the Raman spectra, where the AC„CAstretching vibration moves to lower frequencies as a result of theincrease in the p-electron delocalization of the conjugated back-bone. The type of terminal group substituted in the trimers has agreat effect in the quantum yields in solution following the orderH > Br > I. The pentamers are the molecules with the highest quan-tum yields in solution. The chain length of the lateral substituentsor the terminal groups does not affect the shape of the absorptionand emission spectra, and all the molecules emit in the blue–greenregion. The X-ray diffraction studies of the oligomers reveal, ingeneral, a crystalline behavior for the butoxy series, while for thedodecanoxy series the X-ray patterns are consistent with a supra-molecular organization, that gives rise to randomly distributedcrystalline domains exhibiting a periodic structure at small angles,and indicating the presence of lamellar order. These oligomers,which match with brick shape and present board-like phases,could be classified as ‘‘sanidic”, as proposed for other PPEs.
5. Supplementary data
Experimental procedures, characterization of compounds, MAL-DI-TOF MS spectra, UV–vis spectra of 27 in toluene and DMF at dif-ferent concentration, Small Angle X-ray (SAX) diffraction patternsof butoxy oligomers, SAX at different temperatures for 21, and1H, HETCOR, APT, 13C NMR spectra of selected oligomers are given.
Acknowledgments
This work was supported by CONACYT through project U51504-R and by the Collaboration program CONACYT-CNR (México–Italy).We thank Dr. Oscar E. Contreras and E. Aparicio (CCMC-UNAM) forassistance in the collection of X-ray data.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, inthe online version, at doi:10.1016/j.molstruc.2009.07.036.
References
[1] (a) T. Maillou, J. Le Moigne, V. Dumarcher, L. Rocha, B. Geffroy, J.M. Nunzi, Adv.Mater. 14 (2002) 1297;(b) G.B. Cunningham, Y. Li, S. Liu, K. Schanze, J. Phys. Chem. B 107 (2003)12569.
[2] D.L. Person, J.M. Tour, J. Org. Chem. 62 (1997) 1376.[3] H. Li, D.R. Powell, R.K. Hayashi, R. West, Macromolecules 31 (1998) 52.[4] E. Arias, J.C. Arnault, T. Maillou, I. Moggio, D. Guillon, J. Le Moigne, B. Geffroy,
Synth. Met. 127 (2002) 229.[5] (a) U. Ziener, A. Godt, J. Org. Chem. 62 (1996) 6137;
(b) H. Kukula, S. Veit, A. Godt, Eur. J. Chem. 277 (1999);(c) N. González-Rojano, E. Arias-Marín, D. Navarro-Rodríguez, S. Weidner,Synlett 8 (2005) 1259.
[6] (a) R. Metivier, R. Amengual, I. Leray, V. Michelet, J.P. Genet, Org. Lett. 6 (2004)739;(b) J.S. Moore, Z. Xu, Macromolecules 24 (1991) 5893;(c) R. Wu, J.S. Schumm, D.L. Pearson, J.M. Tour, J. Org. Chem. 61 (1996) 6906.
[7] (a) U.H.F. Bunz, Chem. Rev. 100 (2000) 1605;(b) M. Gholami, R.R. Tykwinski, Chem. Rev. 106 (2006) 4997;(c) J.S. Schumm, D. Pearson, J.M. Tour, Angew. Chem. Int. Ed. Engl. 33 (1994)1360;(d) K. Müllen, G. Wegner, Electronic Materials: The Oligomer Approach, Wiley-VCH, Weinheim, 1998.
[8] (a) J.S. Yang, T.M. Swager, J. Am. Chem. Soc. 120 (1998) 11864;(b) Y. Pang, J. Li, B. Hu, F.E. Karasz, Macromolecules 31 (1998) 6730;(c) M. Hirohata, K. Tada, T. Kawei, M. Onoda, K. Yoshino, Synth. Met. 85 (1997)1273.
[9] A.T.R. Williams, S.A. Winfield, J.N. Miller, Analyst 108 (1983) 1067.[10] (a) M. Moroni, J. Le Moigne, T.A. Pham, J.Y. Bigot, Macromolecules 30 (1997)
1964;(b) C. Weder, M.S. Wrighton, Macromolecules 29 (1996) 5157;(c) P. Wautelet, M. Moroni, L. Oswald, J. Le Moigne, A. Pham, J.Y. Bigot,Macromolecules 29 (1996) 446;(d) P. Bharathi, U. Patel, T. Kawaguchi, D.J. Pesak, J.S. Moore, Macromolecules28 (1995) 5955;(e) M. Moroni, J. Le Moigne, S. Luzzati, Macromolecules 27 (1994) 562.
[11] See electronic supplementary information.[12] (a) H.A. Dieck, R.F. Heck, J. Organomet. Chem. 93 (1975) 259;
(b) R.F. Heck, Palladium Reagent in Organic Syntheses, Academic Press, NewYork, 1990;(c) C. Amatore, A. Lutand, A. Suarez, J. Am. Chem. Soc. 115 (1993) 9531.
[13] C. Glaser, Ann. Chem. Pharm. 137 (1870) 154.[14] (a) P.V. James, P.K. Sudeep, C.H. Zurres, K.G. Thomas, J. Phys. Chem. A 110
(2006) 4329;(b) L.T. Liu, D. Yaron, M.I. Sluch, M.A. Berg, J. Phys. Chem. B 110 (2006) 18844;(c) N. Li, K. Jia, S. Wang, A. Xia, J. Phys. Chem. A 111 (2007) 9393;(d) Y. Matsunaga, K. Takeshi, T. Akasaka, A.R. Ramesh, P.V. James, K.G. Thomas,P.V. Kamat, J. Phys. Chem. B 112 (2008) 14539.
[15] I.B. Berlman, Handbook of Fluorescence Spectra of Aromatic Molecules, seconded., Academic Press, London, New York, 1971.
[16] (a) P.K. Sudeep, P.V. James, K.G. Thomas, P.V. Kamat, J. Phys. Chem. A 110(2006) 5642;(b) J.E. Rogers, T.M. Cooper, P.A. Fleitz, D.J. Glass, D.G. McLean, J. Phys. Chem. A106 (2002) 10108.
[17] H. Barrientos, E. Arias, I. Moggio, J. Romero, O. Rodríguez, E. Giorgetti, T. DelRosso, Synth. Met. 147 (2004) 267.
[18] (a) E. Arias-Marin, J. Le Moigne, T. Maillou, D. Guillon, I. Moggio, B. Geffroy,Macromolecules 36 (2003) 3570;(b) E. Arias Marin, J.C. Arnault, D. Guillon, T. Maillou, J. Le Moigne, B. Geffroy,J.M. Nunzi, Langmuir 16 (2000) 4309.
[19] (a) L. Kloppenburg, D. Jones, J.B. Claridge, H-C. Loye, U.H.F. Bunz,Macromolecules 32 (1999) 4460;(b) J.N. Wilson, P.M. Windscheif, U. Evans, M.L. Myrick, U.H.F. Bunz,Macromolecules 35 (2002) 8681.











![Thermal, oxidative and radiation stability of polyimides III. Polyimides based on N-[3-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)phenyl]acetamide and different diamines](https://static.fdokumen.com/doc/165x107/63448d5903a48733920af0ae/thermal-oxidative-and-radiation-stability-of-polyimides-iii-polyimides-based-on.jpg)








