
Functions and Mechanisms of Fibroblast Growth Factor (FGF ...

Original Article
Connective tissue growth factor—a novel mediator of angiotensinII-stimulated cardiac fibroblast activation in heart failure in rats
Mohammed Shakil Ahmed a,b, Erik Øie a,b, Leif Erik Vinge a,b, Arne Yndestad c, Geir ØysteinAndersen d, Yvonne Andersson a,b, Toril Attramadal a,b, Håvard Attramadal a,b,*
a MSD Cardiovascular Research Center, Rikshospitalet University Hospital, University of Oslo, Oslo N-0027, Norwayb Institute for Surgical Research, A3.1013, Rikshospitalet University Hospital, University of Oslo, Oslo N-0027, Norwayc Research Institute for Internal Medicine, Rikshospitalet University Hospital, University of Oslo, Oslo N-0027, Norway
d Department of Pharmacology, University of Oslo, Oslo N-0316, Norway
Received 19 September 2003; received in revised form 20 November 2003; accepted 4 December 2003
Abstract
The pathophysiologic mechanisms of myocardial remodeling in heart failure (HF) remain poorly understood. Using differential mRNAdisplay of myocardial tissue from rats with ischemic HF vs. controls we identified robust myocardial induction of the mRNA encodingconnective tissue growth factor (CTGF). The aim of this study was to investigate the sites of synthesis and the mechanisms of induction ofCTGF in failing myocardial tissue. The study demonstrates that myocardial expression of CTGF mRNA and protein is substantially elevatedin non-ischemic tissue from both the left and the right ventricles of rats with experimentally induced myocardial infarction (MI). The inductionof myocardial CTGF mRNA was shown to transcend from early post-infarction HF to chronic HF. In situ hybridization and immunohis-tochemical analysis of myocardial tissue sections demonstrated expression of CTGF confined to fibroblasts and endothelial cells ofnon-ischemic myocardial tissue. In subsequent experiments rats subjected to MI were randomized to treatment with the AT1 angiotensinreceptor antagonist losartan (12.5 mg/kg b.i.d. per os) or vehicle. Losartan attenuated ventricular hypertrophy, improved hemodynamics, andprevented the induction of myocardial CTGF mRNA observed in rats post-MI. To provide the cellular basis ofAng II-stimulated CTGF mRNAexpression, primary cultures of rat myocardial fibroblasts were stimulated with Ang II (10–7 M). Real-time reverse transcription–polymerasechain reaction and western blot analysis demonstrate that Ang II induces rapid, AT1 receptor-mediated elevations of CTGF mRNA and proteinin rat cardiac fibroblasts. Furthermore, CTGF was shown to stimulate fibroblast proliferation in vitro. In conclusion, this study demonstratesthat CTGF is a myocardial effector of Ang II-induced myocardial remodeling in HF mediated via AT1 receptors situated on cardiac fibroblasts.
© 2003 Elsevier Ltd. All rights reserved.
Keywords: Heart failure; Fibroblasts; Connective tissue growth factor; Angiotensin II
1. Introduction
Congestive heart failure (CHF) is associated with struc-tural alterations of myocardial tissue collectively termedmyocardial remodeling. The most characteristic features ofthese structural alterations include compensatory hypertro-phy of cardiac myocytes and myocardial fibrosis [1]. Thelatter involves both increased deposition of extracellular ma-trix (ECM) as well as altered composition of the ECM [2].Although such remodeling of myocardial tissue may initiallybe regarded as compensatory responses to increased wallstress and neurohormonal activation, the alterations ulti-
mately decrease left ventricular (LV) function and act asstrong predictors of mortality in patients with heart failure(HF). Despite considerable scientific data on the biochemicalcharacteristics of myocardial remodeling, the growth factorsinitiating these structural alterations of the heart still remainpoorly understood. We employed differential mRNA displayof myocardial tissue from rats with ischemic HF to identifyputative candidate genes in the mechanisms of myocardialremodeling. One of the genes that were strongly induced inthe failing hearts was the 38 kDa cysteine-rich immediateearly gene product, connective tissue growth factor (CTGF).Several features of CTGF prompted us to investigate themechanisms of induction of this growth factor in HF. CTGFis a member of the CCN (acronym of Cyr61/CEF-10,CTGF/Fisp-12, and Nov) family of growth factors [3]. These
* Corresponding author. Tel.: +47-23-073-520; fax: +47-23-073-530.E-mail address: [email protected] (H. Attramadal).
Journal of Molecular and Cellular Cardiology 36 (2004) 393–404
www.elsevier.com/locate/yjmcc
© 2003 Elsevier Ltd. All rights reserved.doi:10.1016/j.yjmcc.2003.12.004

growth factors are secreted, heparin-binding, and ECM-associated proteins involved in multiple cellular events in-cluding ECM production, cell adhesion, cell proliferation, orin some cell types apoptosis, as well as more complex bio-logical processes such as tumorigenesis, angiogenesis, andwound healing [4,5]. CTGF was found to be highly ex-pressed in fibroblasts and to trigger many of the cellularprocesses underlying fibrosis [6]. Indeed, several reportsprovide evidence implicating CTGF in the pathogenesis offibrotic disorders such as advanced atherosclerosis, sclero-derma, as well as liver- and kidney-fibrosis [7–10]. Theprofibrotic cytokine TGF-b has also been shown to increasethe mRNA levels of CTGF in fibroblasts in vitro [11]. Fur-thermore, TGF-b-stimulated secretion of collagen from fi-broblasts could be inhibited by neutralizing anti-CTGF anti-bodies [12]. Although, considerable evidence points to a roleof TGF-b in the pathophysiologic mechanisms of myocardialremodeling in HF, corresponding involvement of CTGF isunknown. In addition, certain G protein-coupled receptorshave also recently been shown to induce expression of CTGF[13]. Thus, the major aims of this study were (1) to elucidatethe cellular site(s) of synthesis of CTGF in failing myocardialtissue, and (2) to determine the mechanisms of induction ofCTGF mRNA in non-ischemic myocardial tissue in HF.Substantial evidence implicates the neurohormonal renin–angiotensin II (Ang II)-AT1 receptor axis in remodeling offailing myocardial tissue. However, to what extent CTGF isan autocrine/paracrine effector of Ang II was an issue thatremained to be resolved. In this study, investigations ofmyocardial tissue and isolated cardiac fibroblasts from ratswith ischemic HF provide strong evidence that CTGF is amediator of Ang II-stimulated myocardial remodeling.
2. Materials and methods
2.1. Animal preparation
We used the left coronary artery-ligated rat model of HFas previously described [14]. Briefly, male Wistar rats (z250g) were anesthetized with 1% isoflurane in 1/3 O2 and2/3 N2O and subjected to left thoracotomy with subsequentligation of the proximal portion of the left coronary artery.Except for ligation of the artery sham-operated rats under-went the same procedure. The animal experiments and hous-ing were in accordance with institutional guidelines andnational legislation conforming to The European Conventionfor The Protection of Vertebrate Animals Used for Experi-mental and Other Scientific Purposes of 18 March 1986.
2.2. Study protocol
The aim of the first series of experiments was to investi-gate regulation and cellular distribution of myocardial CTGFmRNA and protein during development of HF after myocar-dial infarction (MI). HF rats (n = 5 in each group) and
sham-operated rats (n = 4 in each group) were euthanised 2,7, or 42 d after induction of MI. The purpose of the secondseries of experiments was to investigate the effects of AT1
receptor antagonism on myocardial CTGF expression afterMI in rats. MI rats were randomized to treatment for 25 dwith the AT1 receptor antagonist losartan (Merck Sharp &Dohme, Whitehouse Station, USA; 12.5 mg kg–1 b.i.d. po;n = 6) or vehicle (water; n = 5) administered by gavage. Thetreatment protocol was initiated 3 d after induction of MI tominimize the possibility of a direct effect of losartan oninfarct size. Sham-operated rats that received no treatmentwere also included in the study (n = 5).
2.3. Hemodynamic measurements and tissue sampling
On the day of the experiments, the rats were anesthetizedwith gas anesthesia containing 1% isoflurane in 1/3 O2 and2/3 N2O. LV systolic pressure (LVSP) and LV end-diastolicpressure (LVEDP) were recorded by a 2F micromanometer-tipped catheter (model SPR-407, Millar Instruments, Hous-ton, TX, USA) inserted through the right carotid artery [14].After completion of the hemodynamic measurements, therats were euthanised by excision of the heart. The left andright ventricles were sectioned separating non-infarctedmyocardium from the infarcted area. Contamination of vi-able myocardial tissue with necrotic tissue was carefullyavoided.
Rat hearts were perfused and fixed in 4% paraformalde-hyde for 4 h (in situ hybridization; n = 3) or in Bouin’ssolution (2% paraformaldehyde and 0.2% picric acid inphosphate-buffered saline (PBS)) for 5 h (immunohis-tochemistry; n = 3).
2.4. Isolation of cardiac fibroblasts
Non-myocytes from hearts of HF and sham-operated ratswere obtained by differential centrifugation of cardiac cellsreleased after reverse Langendorf perfusion and enzymaticdigestion of the hearts as described previously [15]. Thehomogeneity of these primary isolates was assessed by im-munocytochemistry using anti-vimentin immunoreactivity(anti-porcine vimentin IgG1-kappa, clone V9, Zymed Labo-ratories, CA, USA) as fibroblast marker and anti-rat vonWillebrand factor immunoreactivity (purified polyclonalanti-rat von Willebrand factor, Cedarlane, Ont., Canada) asendothelial cell marker. These non-myocytes contained morethan 90% vimentin-positive cells, and less than 5% of thecells displayed anti-von Willbrand factor immunoreactivity.The cells from sham-operated rats were plated onto non-coated cell culture dishes, maintained and propagated inDulbecco’s modified Eagle’s medium (Life Technologies,Gaithersburg, MD, USA) supplemented with 10% fetal calfserum (Bio Whitaker) and 40 µg/ml garamycin in a humidi-fied atmosphere containing 5% CO2 at 37 °C. After the firstpassage >95% of the cells stained positive for vimentinand <1% displayed immunoreactivity against von Will-ebrand factor.
394 M.S. Ahmed et al. / Journal of Molecular and Cellular Cardiology 36 (2004) 393–404

2.5. Isolation of RNA and northern blot analysis
Total RNA was prepared from rat myocardial tissues byacid–phenol/chloroform extraction in the presence of chao-tropic salts (Trizol, Life Technologies, Gaithersburg, MD,USA) as described previously [15]. Northern blot analysiswas performed using a 404 bp fragment of the rat CTGFcDNA (nt 425–829; Accession No. AF120275). The northernblot was scanned by a laser phosphorimager and the autora-diographic signals were subjected to densitometric analysis(ImageQuant, Molecular Dynamics, Sunnyvale, CA, USA).The glyceraldehyde-3-phosphate dehydrogenase (GAPDH)cDNA probe was employed as a housekeeping gene (frag-ment of rat GAPDH cDNA; nt 458–994; GenBank, Acces-sion No. M17701).
2.6. Quantitative real-time RT-PCR analysis
Reverse transcription (RT) and polymerase chain reaction(PCR) of each sample were run in triplicates using the Taq-Man PCR Core Reagent Kit and the ABI Prism 7700 Se-quence Detector and software (Applied Biosystems, FosterCity, CA, USA) according to the manufacturer’s instructions.Sequence specific PCR primers and TaqMan probes weredesigned using the Primer Express software version 1.5 (Ap-plied Biosystems); see Table 1 for details. Each pair ofprimers was designed to span an intron to avoid amplificationof genomic DNA. SyBr Green assays (Table 1) were per-formed using 2 × SyBr Green Universal Master Mix (Ap-plied Biosystems) and 300 nM sense and anti-sense primers.The specificity of the SyBr Green assays was assessed bymelting point analysis. A standard curve was obtained byperforming amplifications of cDNA from serial dilutions ofmyocardial total RNA. For all specific mRNA amplifiedlinear inverse correlations were observed between amount ofRNA and CT values (number of cycles at threshold lines).
2.7. In situ hybridization
pBluescript SK+ containing the 404 bp CTGF cDNAfragment (nt 425–829) was linearized with either EcoRV orBamHI to synthesize digoxigenin-labeled anti-sense and
sense ribonucleotide probes using either T7 or T3 poly-merase, respectively. Paraffin-embedded (6 µm) sections ofrat hearts were deparaffinized in xylene, rehydrated, andtreated with 0.3% Triton X-100 in PBS for 15 min. Thesections were permeabilized with 10 µg/ml of proteinase K(Boehringer Mannheim, IN, USA), acetylated with 0.25%acetic anhydride, and subsequently dehydrated in ethanol.Prehybridization buffer (600 mM sodium chloride, 60 mMsodium nitrade; pH 7.0 (4×SCC) formamide, 1 × Denhardt’ssolution, 10% dextran, and 250 µg/ml sonicated salmonsperm DNA) was applied for at least 2 h at 42 °C. Subse-quently, digoxigenin-labeled riboprobe (5 ng/µl) was addedand hybridized overnight in a humidified chamber at 42 °C.After hybridization, the sections were rinsed, treated withRNase A, and finally washed in 0.1 × SSC (2 × 30 min at60 °C). After blocking, the sections were incubated withalkaline phosphatase-conjugated Fab fragment of sheep anti-digoxigenin IgG (1:500; Boehringer Mannheim) and positivesignals were detected by the NBT/BCIP color substrate sys-tem.
2.8. Generation and purification of anti-CTGF IgG
Rabbit anti-CTGF antiserum was prepared by immuniza-tion against a synthetic peptide (CFESLYYRKMYGDMA)from the carboxyl terminus of rat CTGF conjugated to key-hole limpet hemocyanin. Total IgG was purified by protein Aagarose chromatography. For affinity chromatography ofanti-CTGF IgG, the peptide (5 mg) was coupled to CNBr-activated sepharose (Amersham Biosciences). Total IgG wasincubated with the affinity matrix at 4 °C, and anti-CTGFIgG were eluted with 5 M MgCl2. The eluted antibodyfractions were dialyzed against TBS (0.05 M Tris; pH 7.4,0.15 M NaCl, and 0.02% sodium azide) and concentrated byultrafiltration. The concentration of purified anti-CTGF IgGwas 0.61 µg/µl.
2.9. Western blot analysis
Myocardial tissue or cells were homogenized and solubi-lized in buffer containing 3% Triton X-100, 300 mM NaCl,100 mM Tris; pH 7.3, 1 mM Na3VO4, 20 mM EDTA, 1%NP-40, 2 mM PMSF, and protease inhibitors (1 µg/ml each ofaprotinin, pepstatin, and leupeptin), denatured in Laemmlibuffer [16], separated on 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and fi-nally, transferred by electroblotting to a PVDF membrane.The membrane was blocked in PBS containing 0.05%Tween-20, 1% BSA, and 5% purified casein, and subse-quently incubated with purified anti-CTGF IgG (1:800) inantibody buffer (PBS containing 0.025% Tween-20, 0.5%BSA, and 2.5% purified casein). Finally, the membrane wasincubated with horseradish peroxidase conjugated anti-rabbit IgG (1:3000), and immunoreactive bands were visual-ized with the enhanced chemiluminescence detection system(ECL, Amersham Biosciences).
Table 1Oligonucleotide primers and probes used for real-time quantitative PCRanalysis of CTGF and RT1A mRNA levels
Oligonucleotide Sequence Accessionno.
Sense (CTGF) 5′-GGCTGGAGAAGCAGAGTCGT–3′ NM_022266Antisense(CTGF)
5′-GATGCACTTTTTGCCCTTCTTAA-3′
Probe (CTGF) 5′-ATGGTCAGGCCCTGTGAAGCTGACC-3′
Sense (AT1) a 5′-GTCATCCACCGAAATGTATACTTCAT-3′
X62295
Antisense (AT1)a
5′-TTGGTAAGGCCCAGCCCTAT-3′
a SyBr Green assay.
395M.S. Ahmed et al. / Journal of Molecular and Cellular Cardiology 36 (2004) 393–404

2.10. Immunohistochemistry
Immunohistochemical analysis of myocardial tissue sec-tions was performed as described previously [14] using thepurified rabbit anti-CTGF IgG described above and mono-clonal anti-porcine vimentin IgG1-kappa (V9, 1:100,Zymed). The avidin–biotin-peroxidase system (VectastainElite kit, Vector Laboratories, CA, USA) was used for signalamplification. Purified rabbit IgG (non-immune) or omissionof primary antibody was used as negative controls.
2.11. Purification of recombinant CTGF and cellproliferation assay
Recombinant CTGF was purified as described [17].Briefly, rat CTGF cDNA encoding the entire open readingframe was cloned into the mammalian expression vectorpRK-5 and transiently transfected into COS-7 cells. Serum-free medium was collected 48 h post-transfection and sub-jected to cation-exchange chromatography. To assess thehomogeneity and integrity of purified recombinant CTGF,400 ng of protein eluate was subjected to SDS-PAGE andsubsequent analysis of Coomassie blue staining or westernblot analysis, respectively.
To determine the mitogenic activity of CTGF, purifiedrecombinant CTGF was added to quiescent Rat-2 fibroblasts(ATCC, USA) and incubated for 19 h followed by addition of1 µCi/ml of [3H]-thymidine to pulse the cells for additional3 h. The cells were subsequently washed four times withice-cold PBS, incubated twice (30 min each time on ice) withice-cold 5% trichloroacetic acid, briefly washed once withice-cold ethanol, and finally lysed with lysis buffer contain-ing 0.1 M NaOH and 0.1% SDS. Incorporated [3H]-thymidine was determined by liquid scintillation spectrom-etry.
2.12. Statistical analysis
All values are given as mean ± S.E. unless otherwiseindicated. Between-group variations were assessed by two-tailed unpaired t-test. For multiple comparions ANOVA wasperformed and post-hoc analysis with Bonferroni’s test wasemployed whenever stated in figure legends. P values <0.05were considered to be statistically significant.
3. Results
3.1. Hemodynamic measurements
Hemodynamic measurements recorded at day 7 after liga-tion of the left coronary artery revealed substantial LV dys-function. As shown in Table 2, LVSP was significantly lowerand LVEDP was significantly higher in the HF rats as com-pared to the sham-operated rats.
3.2. Myocardial expression of CTGF mRNA during HF
The time course of CTGF mRNA expression in the non-ischemic LV and in the right ventricle after induction of MI is
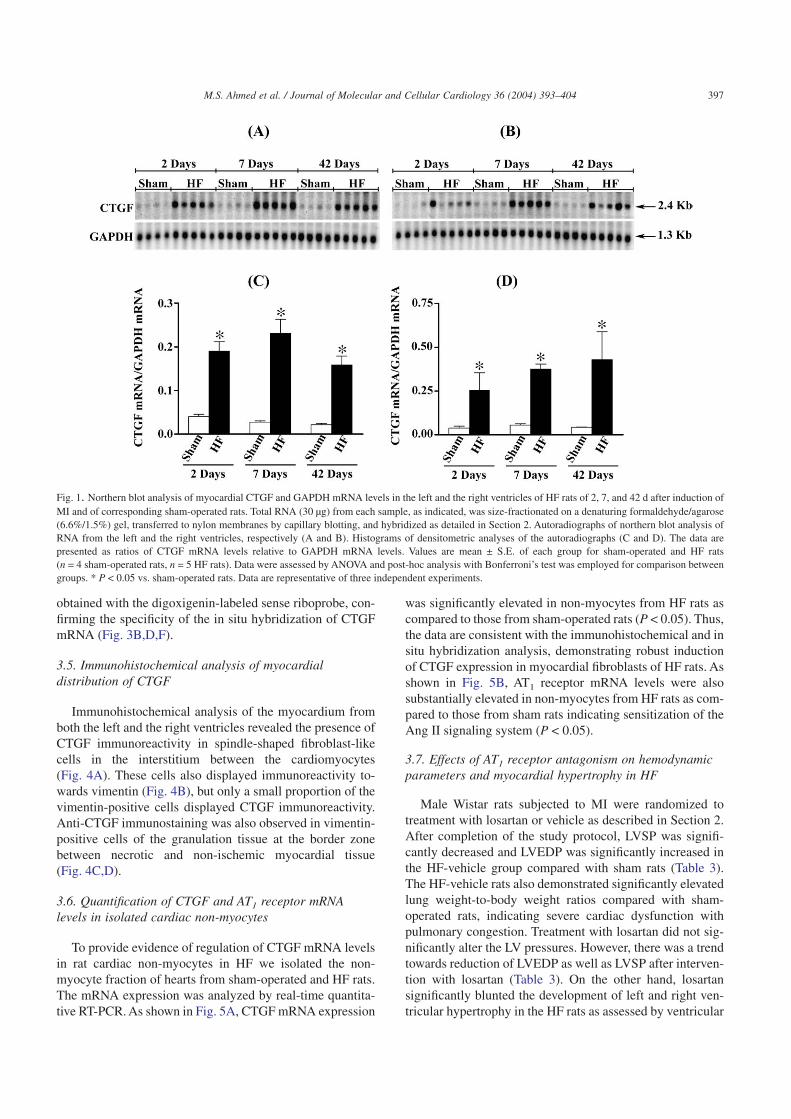
illustrated in Fig. 1. Northern blot analysis demonstrated thatCTGF mRNA levels were low but consistently detectable inthe left and right ventricles of sham-operated rats. Dramaticinduction of CTGF mRNA expression was observed in bothleft (panels A and C) and right (panels B and D) ventricles ofHF rats at 2, 7, and 42 d after MI compared to the sham-operated rats. In the LV, 5-fold increase (P < 0.05) of CTGFmRNA levels was seen already 2 d after MI, reaching peaklevels 7 d after MI (8-fold increase, P < 0.05). Forty-two daysafter MI, LV CTGF mRNA levels remained persistentlyelevated compared to the sham group (7-fold increase;P < 0.05). In the right ventricle, myocardial CTGF mRNAlevels in the HF rats were 8-fold above the levels seen in thesham rats at days 2 and 7 after MI (P < 0.05) and were furtherelevated 42 d after induction of MI (10-fold above levels insham group; P < 0.05).
3.3. Myocardial CTGF protein levels in HF
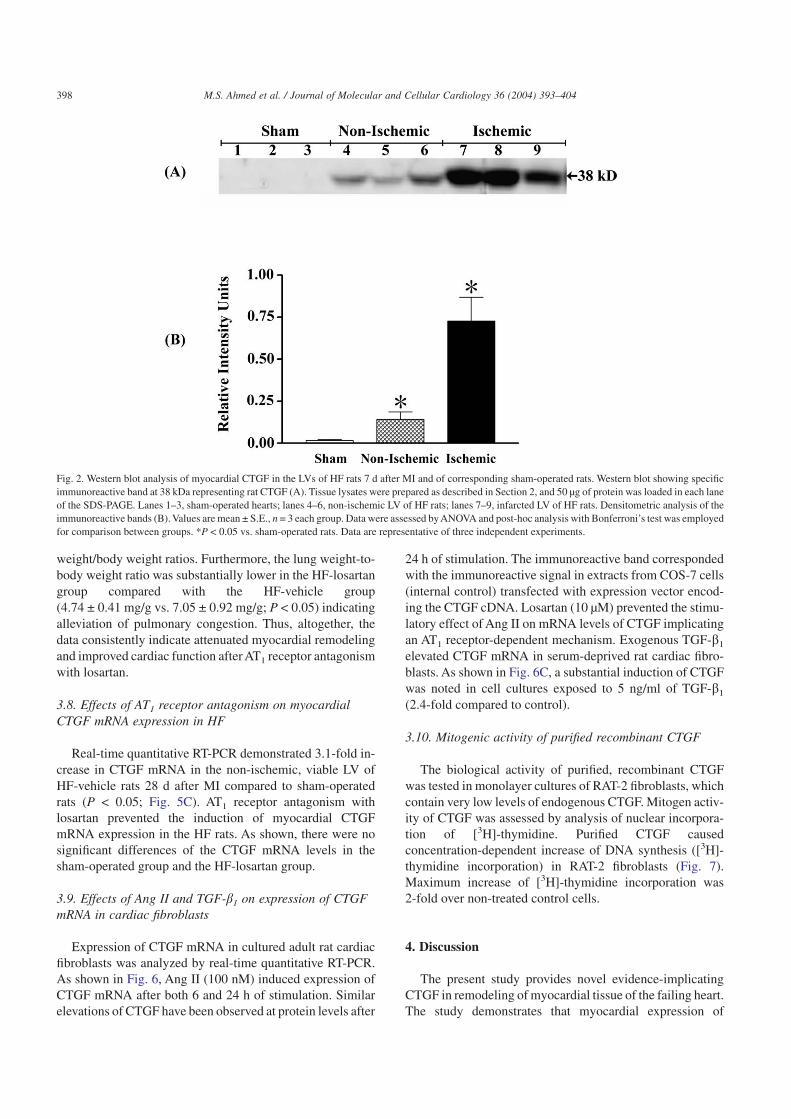
Western blot analysis of myocardial tissue extracts fromsham rats revealed immunoreactivity corresponding to theexpected mass of CTGF (38 kDa). The immunoreactive bandcorresponded with the immunoreactive signal in extractsfrom COS-7 cells transfected with expression vector encod-ing rat CTGF cDNA. Increased intensity of this band wasobserved in extracts from both ischemic and non-ischemicregions of hearts from HF rats compared to sham-operatedrats (Fig. 2). Highest expression levels were observed in theischemic zone (40-fold the levels in the sham group,P < 0.05). However, substantial induction of CTGF was seenin non-ischemic LV tissue (8-fold the levels in the shamgroup; P < 0.05).
3.4. In situ detection of myocardial CTGF mRNA
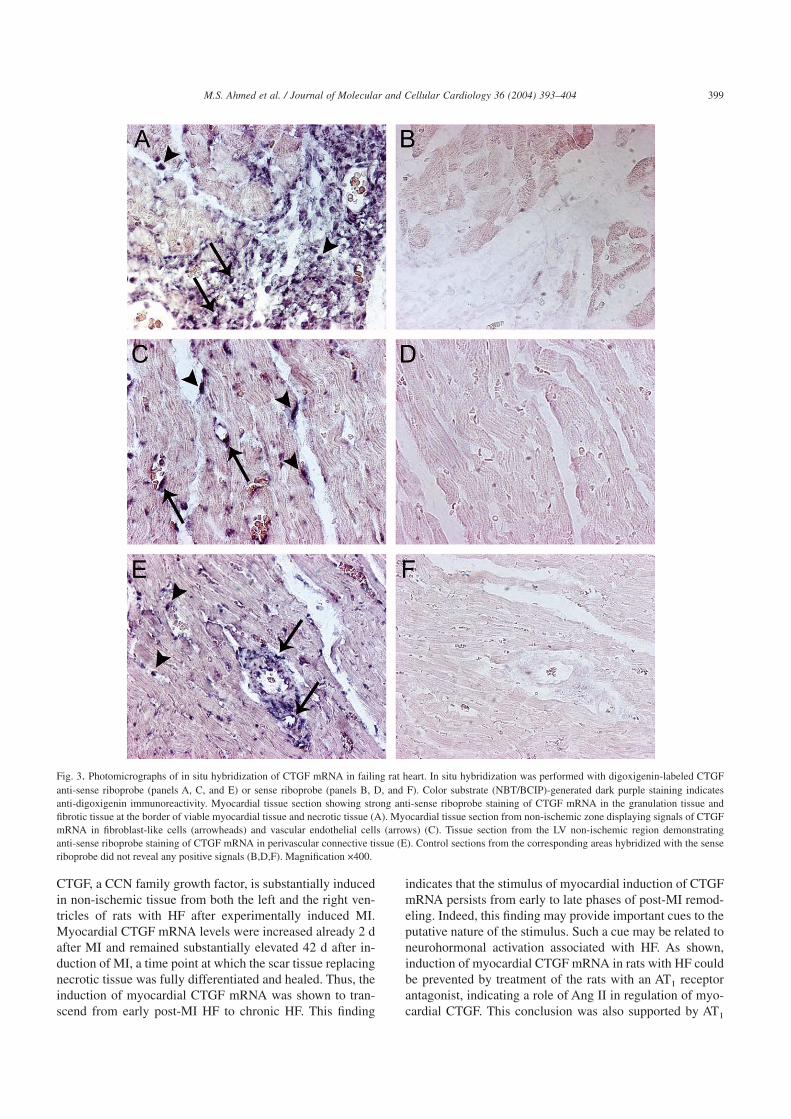
Fig. 3 demonstrates in situ hybridization of a digoxigenin-labeled CTGF anti-sense riboprobe in sections of myocardialtissue 7 d after MI. The most intense staining of CTGFmRNA was observed in the granulation tissue and differen-tiating scar tissue in the transition zone between necrotic andviable myocardium (Fig. 3A). In the granulation tissue,CTGF mRNA was identified in fibroblast-like cells and en-dothelial cells. However, CTGF mRNA expression was alsoseen in myocardial tissue distal to the ischemic zone, pre-dominantly in fibroblast-like cells between the cardiomyo-cytes and in vascular endothelial cells (Fig. 3C). In thisrespect, robust signals for CTGF mRNA were observed inperivascular connective tissue (Fig. 3E). No signals were
Table 2Hemodynamic measurements after induction of MI or sham operation
LVSP (mmHg) LVEDP (mmHg)Sham (n = 12) 118 ± 3 4 ± 1HF 2 d post-MI (n = 5) 99 ± 5 * 22 ± 1 *HF 7 d post-MI (n = 5) 105 ± 4 * 25 ± 2 *HF 42 d post-MI (n = 5) 96 ± 4 * 26 ± 3 *
Values are mean ± S.E. * P < 0.05 vs. sham.
396 M.S. Ahmed et al. / Journal of Molecular and Cellular Cardiology 36 (2004) 393–404
obtained with the digoxigenin-labeled sense riboprobe, con-firming the specificity of the in situ hybridization of CTGFmRNA (Fig. 3B,D,F).
3.5. Immunohistochemical analysis of myocardialdistribution of CTGF
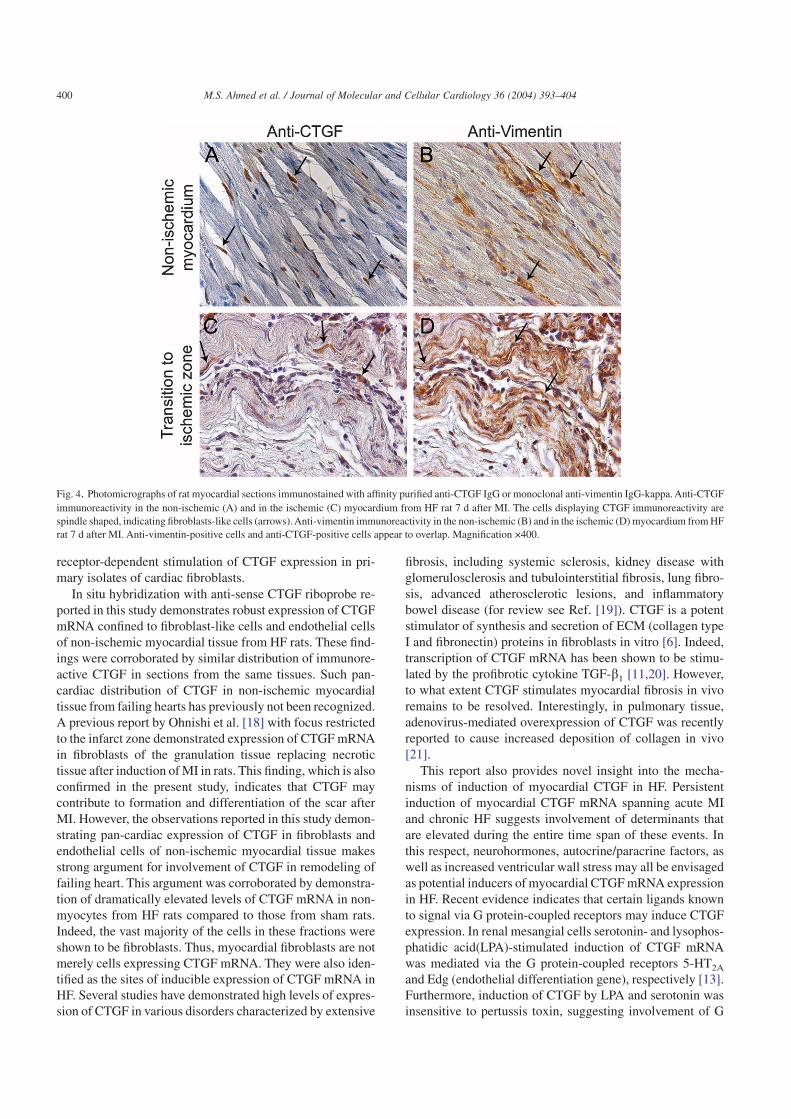
Immunohistochemical analysis of the myocardium fromboth the left and the right ventricles revealed the presence ofCTGF immunoreactivity in spindle-shaped fibroblast-likecells in the interstitium between the cardiomyocytes(Fig. 4A). These cells also displayed immunoreactivity to-wards vimentin (Fig. 4B), but only a small proportion of thevimentin-positive cells displayed CTGF immunoreactivity.Anti-CTGF immunostaining was also observed in vimentin-positive cells of the granulation tissue at the border zonebetween necrotic and non-ischemic myocardial tissue(Fig. 4C,D).
3.6. Quantification of CTGF and AT1 receptor mRNAlevels in isolated cardiac non-myocytes
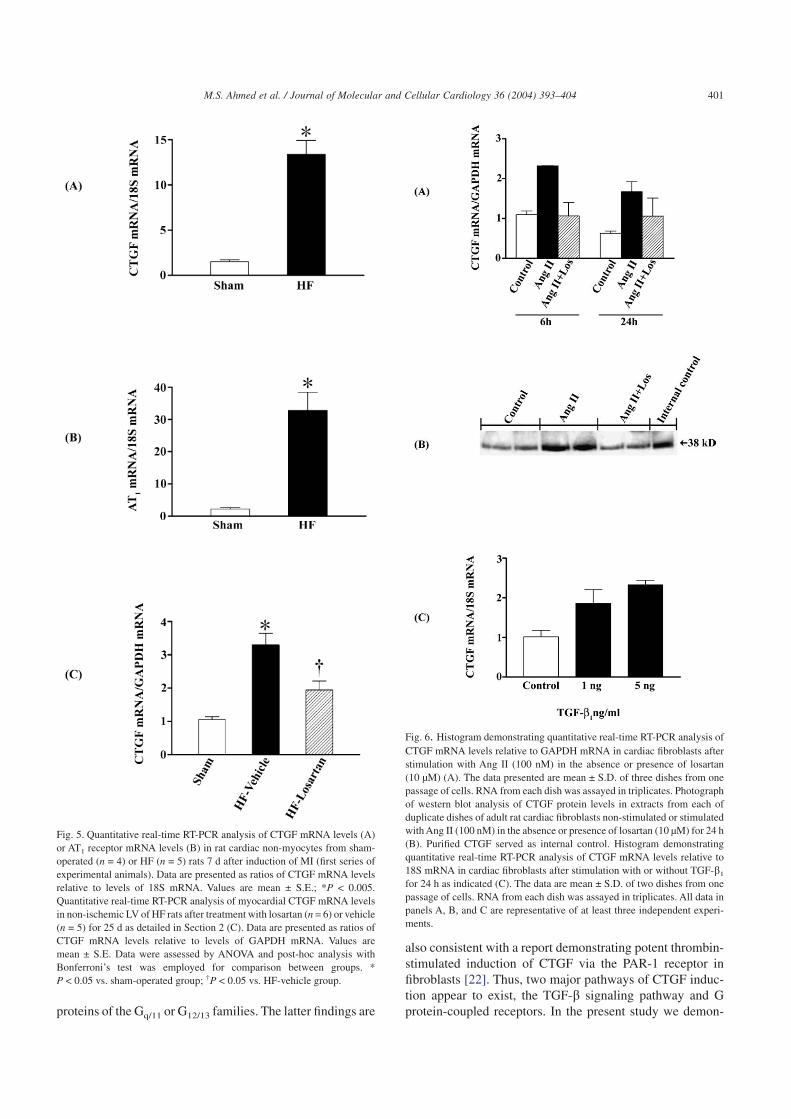
To provide evidence of regulation of CTGF mRNA levelsin rat cardiac non-myocytes in HF we isolated the non-myocyte fraction of hearts from sham-operated and HF rats.The mRNA expression was analyzed by real-time quantita-tive RT-PCR. As shown in Fig. 5A, CTGF mRNA expression
was significantly elevated in non-myocytes from HF rats ascompared to those from sham-operated rats (P < 0.05). Thus,the data are consistent with the immunohistochemical and insitu hybridization analysis, demonstrating robust inductionof CTGF expression in myocardial fibroblasts of HF rats. Asshown in Fig. 5B, AT1 receptor mRNA levels were alsosubstantially elevated in non-myocytes from HF rats as com-pared to those from sham rats indicating sensitization of theAng II signaling system (P < 0.05).
3.7. Effects of AT1 receptor antagonism on hemodynamicparameters and myocardial hypertrophy in HF
Male Wistar rats subjected to MI were randomized totreatment with losartan or vehicle as described in Section 2.After completion of the study protocol, LVSP was signifi-cantly decreased and LVEDP was significantly increased inthe HF-vehicle group compared with sham rats (Table 3).The HF-vehicle rats also demonstrated significantly elevatedlung weight-to-body weight ratios compared with sham-operated rats, indicating severe cardiac dysfunction withpulmonary congestion. Treatment with losartan did not sig-nificantly alter the LV pressures. However, there was a trendtowards reduction of LVEDP as well as LVSP after interven-tion with losartan (Table 3). On the other hand, losartansignificantly blunted the development of left and right ven-tricular hypertrophy in the HF rats as assessed by ventricular
Fig. 1. Northern blot analysis of myocardial CTGF and GAPDH mRNA levels in the left and the right ventricles of HF rats of 2, 7, and 42 d after induction ofMI and of corresponding sham-operated rats. Total RNA (30 µg) from each sample, as indicated, was size-fractionated on a denaturing formaldehyde/agarose(6.6%/1.5%) gel, transferred to nylon membranes by capillary blotting, and hybridized as detailed in Section 2. Autoradiographs of northern blot analysis ofRNA from the left and the right ventricles, respectively (A and B). Histograms of densitometric analyses of the autoradiographs (C and D). The data arepresented as ratios of CTGF mRNA levels relative to GAPDH mRNA levels. Values are mean ± S.E. of each group for sham-operated and HF rats(n = 4 sham-operated rats, n = 5 HF rats). Data were assessed by ANOVA and post-hoc analysis with Bonferroni’s test was employed for comparison betweengroups. * P < 0.05 vs. sham-operated rats. Data are representative of three independent experiments.
397M.S. Ahmed et al. / Journal of Molecular and Cellular Cardiology 36 (2004) 393–404
weight/body weight ratios. Furthermore, the lung weight-to-body weight ratio was substantially lower in the HF-losartangroup compared with the HF-vehicle group(4.74 ± 0.41 mg/g vs. 7.05 ± 0.92 mg/g; P < 0.05) indicatingalleviation of pulmonary congestion. Thus, altogether, thedata consistently indicate attenuated myocardial remodelingand improved cardiac function after AT1 receptor antagonismwith losartan.
3.8. Effects of AT1 receptor antagonism on myocardialCTGF mRNA expression in HF
Real-time quantitative RT-PCR demonstrated 3.1-fold in-crease in CTGF mRNA in the non-ischemic, viable LV ofHF-vehicle rats 28 d after MI compared to sham-operatedrats (P < 0.05; Fig. 5C). AT1 receptor antagonism withlosartan prevented the induction of myocardial CTGFmRNA expression in the HF rats. As shown, there were nosignificant differences of the CTGF mRNA levels in thesham-operated group and the HF-losartan group.
3.9. Effects of Ang II and TGF-b1 on expression of CTGFmRNA in cardiac fibroblasts
Expression of CTGF mRNA in cultured adult rat cardiacfibroblasts was analyzed by real-time quantitative RT-PCR.As shown in Fig. 6, Ang II (100 nM) induced expression ofCTGF mRNA after both 6 and 24 h of stimulation. Similarelevations of CTGF have been observed at protein levels after
24 h of stimulation. The immunoreactive band correspondedwith the immunoreactive signal in extracts from COS-7 cells(internal control) transfected with expression vector encod-ing the CTGF cDNA. Losartan (10 µM) prevented the stimu-latory effect of Ang II on mRNA levels of CTGF implicatingan AT1 receptor-dependent mechanism. Exogenous TGF-b1
elevated CTGF mRNA in serum-deprived rat cardiac fibro-blasts. As shown in Fig. 6C, a substantial induction of CTGFwas noted in cell cultures exposed to 5 ng/ml of TGF-b1
(2.4-fold compared to control).
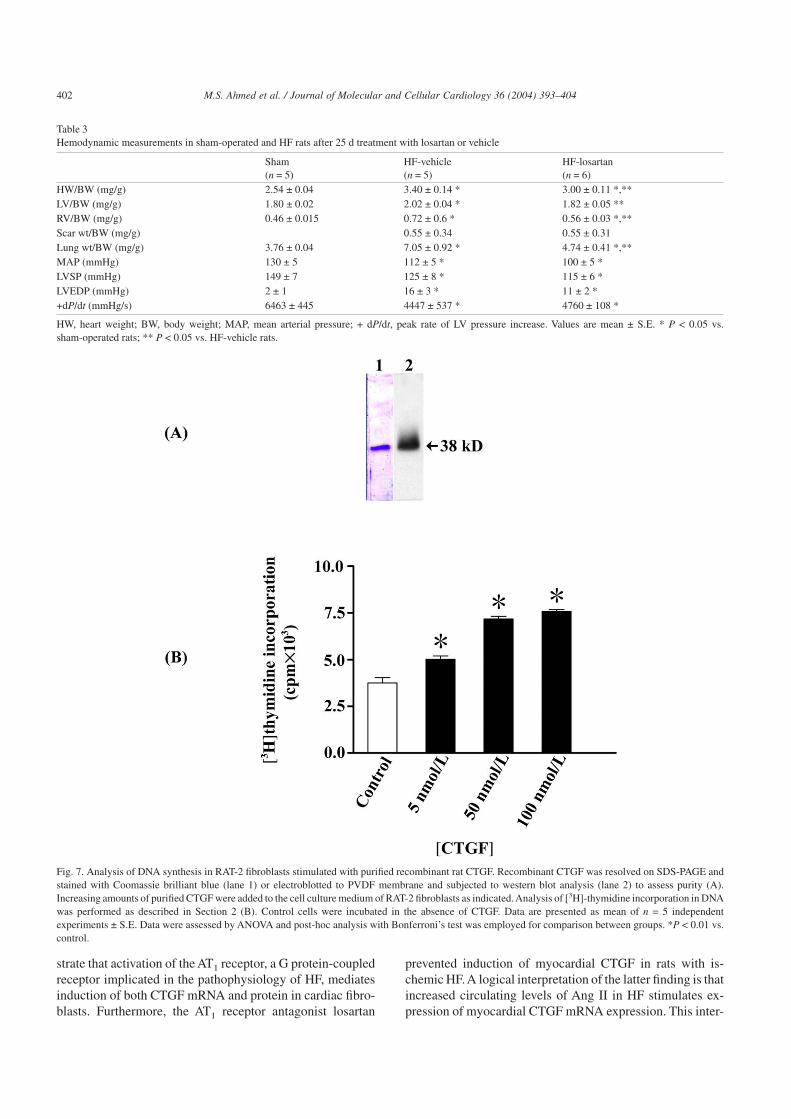
3.10. Mitogenic activity of purified recombinant CTGF
The biological activity of purified, recombinant CTGFwas tested in monolayer cultures of RAT-2 fibroblasts, whichcontain very low levels of endogenous CTGF. Mitogen activ-ity of CTGF was assessed by analysis of nuclear incorpora-tion of [3H]-thymidine. Purified CTGF causedconcentration-dependent increase of DNA synthesis ([3H]-thymidine incorporation) in RAT-2 fibroblasts (Fig. 7).Maximum increase of [3H]-thymidine incorporation was2-fold over non-treated control cells.
4. Discussion
The present study provides novel evidence-implicatingCTGF in remodeling of myocardial tissue of the failing heart.The study demonstrates that myocardial expression of
Fig. 2. Western blot analysis of myocardial CTGF in the LVs of HF rats 7 d after MI and of corresponding sham-operated rats. Western blot showing specificimmunoreactive band at 38 kDa representing rat CTGF (A). Tissue lysates were prepared as described in Section 2, and 50 µg of protein was loaded in each laneof the SDS-PAGE. Lanes 1–3, sham-operated hearts; lanes 4–6, non-ischemic LV of HF rats; lanes 7–9, infarcted LV of HF rats. Densitometric analysis of theimmunoreactive bands (B). Values are mean ± S.E., n = 3 each group. Data were assessed by ANOVA and post-hoc analysis with Bonferroni’s test was employedfor comparison between groups. *P < 0.05 vs. sham-operated rats. Data are representative of three independent experiments.
398 M.S. Ahmed et al. / Journal of Molecular and Cellular Cardiology 36 (2004) 393–404
CTGF, a CCN family growth factor, is substantially inducedin non-ischemic tissue from both the left and the right ven-tricles of rats with HF after experimentally induced MI.Myocardial CTGF mRNA levels were increased already 2 dafter MI and remained substantially elevated 42 d after in-duction of MI, a time point at which the scar tissue replacingnecrotic tissue was fully differentiated and healed. Thus, theinduction of myocardial CTGF mRNA was shown to tran-scend from early post-MI HF to chronic HF. This finding
indicates that the stimulus of myocardial induction of CTGFmRNA persists from early to late phases of post-MI remod-eling. Indeed, this finding may provide important cues to theputative nature of the stimulus. Such a cue may be related toneurohormonal activation associated with HF. As shown,induction of myocardial CTGF mRNA in rats with HF couldbe prevented by treatment of the rats with an AT1 receptorantagonist, indicating a role of Ang II in regulation of myo-cardial CTGF. This conclusion was also supported by AT1
Fig. 3. Photomicrographs of in situ hybridization of CTGF mRNA in failing rat heart. In situ hybridization was performed with digoxigenin-labeled CTGFanti-sense riboprobe (panels A, C, and E) or sense riboprobe (panels B, D, and F). Color substrate (NBT/BCIP)-generated dark purple staining indicatesanti-digoxigenin immunoreactivity. Myocardial tissue section showing strong anti-sense riboprobe staining of CTGF mRNA in the granulation tissue andfibrotic tissue at the border of viable myocardial tissue and necrotic tissue (A). Myocardial tissue section from non-ischemic zone displaying signals of CTGFmRNA in fibroblast-like cells (arrowheads) and vascular endothelial cells (arrows) (C). Tissue section from the LV non-ischemic region demonstratinganti-sense riboprobe staining of CTGF mRNA in perivascular connective tissue (E). Control sections from the corresponding areas hybridized with the senseriboprobe did not reveal any positive signals (B,D,F). Magnification ×400.
399M.S. Ahmed et al. / Journal of Molecular and Cellular Cardiology 36 (2004) 393–404
receptor-dependent stimulation of CTGF expression in pri-mary isolates of cardiac fibroblasts.
In situ hybridization with anti-sense CTGF riboprobe re-ported in this study demonstrates robust expression of CTGFmRNA confined to fibroblast-like cells and endothelial cellsof non-ischemic myocardial tissue from HF rats. These find-ings were corroborated by similar distribution of immunore-active CTGF in sections from the same tissues. Such pan-cardiac distribution of CTGF in non-ischemic myocardialtissue from failing hearts has previously not been recognized.A previous report by Ohnishi et al. [18] with focus restrictedto the infarct zone demonstrated expression of CTGF mRNAin fibroblasts of the granulation tissue replacing necrotictissue after induction of MI in rats. This finding, which is alsoconfirmed in the present study, indicates that CTGF maycontribute to formation and differentiation of the scar afterMI. However, the observations reported in this study demon-strating pan-cardiac expression of CTGF in fibroblasts andendothelial cells of non-ischemic myocardial tissue makesstrong argument for involvement of CTGF in remodeling offailing heart. This argument was corroborated by demonstra-tion of dramatically elevated levels of CTGF mRNA in non-myocytes from HF rats compared to those from sham rats.Indeed, the vast majority of the cells in these fractions wereshown to be fibroblasts. Thus, myocardial fibroblasts are notmerely cells expressing CTGF mRNA. They were also iden-tified as the sites of inducible expression of CTGF mRNA inHF. Several studies have demonstrated high levels of expres-sion of CTGF in various disorders characterized by extensive
fibrosis, including systemic sclerosis, kidney disease withglomerulosclerosis and tubulointerstitial fibrosis, lung fibro-sis, advanced atherosclerotic lesions, and inflammatorybowel disease (for review see Ref. [19]). CTGF is a potentstimulator of synthesis and secretion of ECM (collagen typeI and fibronectin) proteins in fibroblasts in vitro [6]. Indeed,transcription of CTGF mRNA has been shown to be stimu-lated by the profibrotic cytokine TGF-b1 [11,20]. However,to what extent CTGF stimulates myocardial fibrosis in vivoremains to be resolved. Interestingly, in pulmonary tissue,adenovirus-mediated overexpression of CTGF was recentlyreported to cause increased deposition of collagen in vivo[21].
This report also provides novel insight into the mecha-nisms of induction of myocardial CTGF in HF. Persistentinduction of myocardial CTGF mRNA spanning acute MIand chronic HF suggests involvement of determinants thatare elevated during the entire time span of these events. Inthis respect, neurohormones, autocrine/paracrine factors, aswell as increased ventricular wall stress may all be envisagedas potential inducers of myocardial CTGF mRNA expressionin HF. Recent evidence indicates that certain ligands knownto signal via G protein-coupled receptors may induce CTGFexpression. In renal mesangial cells serotonin- and lysophos-phatidic acid(LPA)-stimulated induction of CTGF mRNAwas mediated via the G protein-coupled receptors 5-HT2A
and Edg (endothelial differentiation gene), respectively [13].Furthermore, induction of CTGF by LPA and serotonin wasinsensitive to pertussis toxin, suggesting involvement of G
Fig. 4. Photomicrographs of rat myocardial sections immunostained with affinity purified anti-CTGF IgG or monoclonal anti-vimentin IgG-kappa. Anti-CTGFimmunoreactivity in the non-ischemic (A) and in the ischemic (C) myocardium from HF rat 7 d after MI. The cells displaying CTGF immunoreactivity arespindle shaped, indicating fibroblasts-like cells (arrows). Anti-vimentin immunoreactivity in the non-ischemic (B) and in the ischemic (D) myocardium from HFrat 7 d after MI. Anti-vimentin-positive cells and anti-CTGF-positive cells appear to overlap. Magnification ×400.
400 M.S. Ahmed et al. / Journal of Molecular and Cellular Cardiology 36 (2004) 393–404
proteins of the Gq/11 or G12/13 families. The latter findings are
also consistent with a report demonstrating potent thrombin-stimulated induction of CTGF via the PAR-1 receptor infibroblasts [22]. Thus, two major pathways of CTGF induc-tion appear to exist, the TGF-b signaling pathway and Gprotein-coupled receptors. In the present study we demon-
Fig. 5. Quantitative real-time RT-PCR analysis of CTGF mRNA levels (A)or AT1 receptor mRNA levels (B) in rat cardiac non-myocytes from sham-operated (n = 4) or HF (n = 5) rats 7 d after induction of MI (first series ofexperimental animals). Data are presented as ratios of CTGF mRNA levelsrelative to levels of 18S mRNA. Values are mean ± S.E.; *P < 0.005.Quantitative real-time RT-PCR analysis of myocardial CTGF mRNA levelsin non-ischemic LV of HF rats after treatment with losartan (n = 6) or vehicle(n = 5) for 25 d as detailed in Section 2 (C). Data are presented as ratios ofCTGF mRNA levels relative to levels of GAPDH mRNA. Values aremean ± S.E. Data were assessed by ANOVA and post-hoc analysis withBonferroni’s test was employed for comparison between groups. *P < 0.05 vs. sham-operated group; †P < 0.05 vs. HF-vehicle group.
Fig. 6. Histogram demonstrating quantitative real-time RT-PCR analysis ofCTGF mRNA levels relative to GAPDH mRNA in cardiac fibroblasts afterstimulation with Ang II (100 nM) in the absence or presence of losartan(10 µM) (A). The data presented are mean ± S.D. of three dishes from onepassage of cells. RNA from each dish was assayed in triplicates. Photographof western blot analysis of CTGF protein levels in extracts from each ofduplicate dishes of adult rat cardiac fibroblasts non-stimulated or stimulatedwith Ang II (100 nM) in the absence or presence of losartan (10 µM) for 24 h(B). Purified CTGF served as internal control. Histogram demonstratingquantitative real-time RT-PCR analysis of CTGF mRNA levels relative to18S mRNA in cardiac fibroblasts after stimulation with or without TGF-b1
for 24 h as indicated (C). The data are mean ± S.D. of two dishes from onepassage of cells. RNA from each dish was assayed in triplicates. All data inpanels A, B, and C are representative of at least three independent experi-ments.
401M.S. Ahmed et al. / Journal of Molecular and Cellular Cardiology 36 (2004) 393–404
strate that activation of the AT1 receptor, a G protein-coupledreceptor implicated in the pathophysiology of HF, mediatesinduction of both CTGF mRNA and protein in cardiac fibro-blasts. Furthermore, the AT1 receptor antagonist losartan
prevented induction of myocardial CTGF in rats with is-chemic HF.A logical interpretation of the latter finding is thatincreased circulating levels of Ang II in HF stimulates ex-pression of myocardial CTGF mRNA expression. This inter-
Table 3Hemodynamic measurements in sham-operated and HF rats after 25 d treatment with losartan or vehicle
Sham(n = 5)
HF-vehicle(n = 5)
HF-losartan(n = 6)
HW/BW (mg/g) 2.54 ± 0.04 3.40 ± 0.14 * 3.00 ± 0.11 *,**LV/BW (mg/g) 1.80 ± 0.02 2.02 ± 0.04 * 1.82 ± 0.05 **RV/BW (mg/g) 0.46 ± 0.015 0.72 ± 0.6 * 0.56 ± 0.03 *,**Scar wt/BW (mg/g) 0.55 ± 0.34 0.55 ± 0.31Lung wt/BW (mg/g) 3.76 ± 0.04 7.05 ± 0.92 * 4.74 ± 0.41 *,**MAP (mmHg) 130 ± 5 112 ± 5 * 100 ± 5 *LVSP (mmHg) 149 ± 7 125 ± 8 * 115 ± 6 *LVEDP (mmHg) 2 ± 1 16 ± 3 * 11 ± 2 *+dP/dt (mmHg/s) 6463 ± 445 4447 ± 537 * 4760 ± 108 *
HW, heart weight; BW, body weight; MAP, mean arterial pressure; + dP/dt, peak rate of LV pressure increase. Values are mean ± S.E. * P < 0.05 vs.sham-operated rats; ** P < 0.05 vs. HF-vehicle rats.
Fig. 7. Analysis of DNA synthesis in RAT-2 fibroblasts stimulated with purified recombinant rat CTGF. Recombinant CTGF was resolved on SDS-PAGE andstained with Coomassie brilliant blue (lane 1) or electroblotted to PVDF membrane and subjected to western blot analysis (lane 2) to assess purity (A).Increasing amounts of purified CTGF were added to the cell culture medium of RAT-2 fibroblasts as indicated.Analysis of [3H]-thymidine incorporation in DNAwas performed as described in Section 2 (B). Control cells were incubated in the absence of CTGF. Data are presented as mean of n = 5 independentexperiments ± S.E. Data were assessed by ANOVA and post-hoc analysis with Bonferroni’s test was employed for comparison between groups. *P < 0.01 vs.control.
402 M.S. Ahmed et al. / Journal of Molecular and Cellular Cardiology 36 (2004) 393–404

pretation is supported by a recent report where transgenicrats with elevated circulating levels of Ang II were found toexpress increased levels of myocardial CTGF mRNA andprotein [23]. Previous studies with Ang-converting enzymeinhibitors or AT1 receptor antagonists in experimental mod-els of HF have shown that these interventions exert salutaryeffects on progression of the disease [24,25]. A well-recognized target of Ang II antagonism in HF is related tofibroblast activation and deposition of ECM proteins in theheart. The present study provides compelling evidence thatthe factor targeted by AT1 receptors antagonism may indeedbe fibroblast-generated CTGF.
It could be argued, however, that prevention of myocardialinduction of CTGF mRNA by losartan, as reported in thisstudy, might not be directly attributable to antagonism of AT1
receptors on cardiac fibroblasts. The increased load on theheart during HF with increased wall stress provides anotherputative mechanism of induction of myocardial CTGFmRNA. Physical effectors, such as tension, cyclic stretch,and static pressure, all induce transcription of CTGF infibroblasts in vitro [26]. In the present study, minor reduc-tions of LVSP and LVEDP were observed in the HF-losartangroup as compared to the HF-vehicle group. Thus, the inhibi-tory effects of losartan on myocardial CTGF mRNA expres-sion could be secondary to reductions of both preload andafterload with corresponding reductions in wall stress. Al-though wall stress may impact on myocardial CTGF mRNAexpression, investigations of isolated cardiac fibroblasts inthis study unequivocally demonstrate that Ang II stimulatesCTGF expression. Indeed, cardiac fibroblasts are recognizedtarget cells of Ang II and express AT1 receptors [27]. Further-more, this study presents evidence that AT1 receptor expres-sion in cardiac fibroblasts increased in HF suggesting sensi-tization of the Ang II signaling system in these cells in HF.
In conclusion, the present study establishes CTGF as anovel growth factor and putative player in the pathophysi-ological mechanisms of myocardial remodeling. Further-more, the data provide compelling evidence of Ang II-mediated induction of CTGF, a multifunctional secretedgrowth factor in cardiac fibroblasts in HF. We also demon-strate that CTGF may be targeted by AT1 receptor antago-nism, i.e. an established intervention in HF with documentedefficacy on pathologic myocardial remodeling. However, thespecific roles of CTGF in cardiac remodeling and fibrosis areimportant issues to be resolved in future studies.
Acknowledgements
This study was supported by grants from the NationalResearch Council, the Norwegian Council for Cardiovascu-lar Research, and the MSD-Medinnova Research Fund.
References
[1] Pfeffer MA, Braunwald E. Ventricular remodeling after myocardialinfarction. Experimental observations and clinical implications. Cir-culation 1990;81:1161–72.
[2] Sun Y, Zhang JQ, Zhang J, Lamparter S. Cardiac remodeling byfibrous tissue after infarction in rats. J Lab Clin Med 2000;135:316–23.
[3] Bork P. The modular architecture of a new family of growth regulatorsrelated to connective tissue growth factor. FEBS Lett 1993;327:125–30.
[4] Shimo T, Nakanishi T, Nishida T, et al. Connective tissue growthfactor induces the proliferation, migration, and tube formation ofvascular endothelial cells in vitro, and angiogenesis in vivo. J Bio-chem (Tokyo) 1999;126:137–45.
[5] Hishikawa K, Oemar BS, Tanner FC, Nakaki T, Luscher TF, Fujii T.Connective tissue growth factor induces apoptosis in human breastcancer cell line MCF-7. J Biol Chem 1999;274:37461–6.
[6] Frazier K, Williams S, Kothapalli D, Klapper H, Grotendorst GR.Stimulation of fibroblast cell growth, matrix production, and granula-tion tissue formation by connective tissue growth factor. J InvestDermatol 1996;107:404–11.
[7] Paradis V, Dargere D, Vidaud M, et al. Expression of connective tissuegrowth factor in experimental rat and human liver fibrosis. Hepatol-ogy 1999;30:968–76.
[8] Clarkson MR, Gupta S, Murphy M, Martin F, Godson C, Brady HR.Connective tissue growth factor: a potential stimulus for glomerulo-sclerosis and tubulointerstitial fibrosis in progressive renal disease.Curr Opin Nephrol Hypertens 1999;8:543–8.
[9] Oemar BS, Werner A, Garnier JM, et al. Human connective tissuegrowth factor is expressed in advanced atherosclerotic lesions. Circu-lation 1997;95:831–9.
[10] Igarashi A, Nashiro K, Kikuchi K, et al. Connective tissue growthfactor gene expression in tissue sections from localized scleroderma,keloid, and other fibrotic skin disorders. J Invest Dermatol 1996;106:729–33.
[11] Grotendorst GR, Okochi H, Hayashi N. A novel transforming growthfactor beta response element controls the expression of the connectivetissue growth factor gene. Cell Growth Differ 1996;7:469–80.
[12] Duncan MR, Frazier KS, Abramson S, et al. Connective tissue growthfactor mediates transforming growth factor beta-induced collagensynthesis: down-regulation by cAMP. FASEB J 1999;13:1774–86.
[13] Hahn A, Heusinger-Ribeiro J, Lanz T, Zenkel S, Goppelt-Struebe M.Induction of connective tissue growth factor by activation of heptahe-lical receptors. Modulation by Rho proteins and the actin cytoskel-eton. J Biol Chem 2000;275:37429–35.
[14] Øie E, Vinge LE, Tønnessen T, et al. Transient, isopeptide-specificinduction of myocardial endothelin-1 mRNA in congestive heartfailure in rats. Am J Physiol 1997;273:H1727–36.
[15] Vinge LE, Øie E, Andersson Y, Grøgaard HK, Andersen G, Attra-madal H. Myocardial distribution and regulation of GRK and beta-arrestin isoforms in congestive heart failure in rats. Am J PhysiolHeart Circ Physiol 2001;281:H2490–9.
[16] Laemmli UK. Cleavage of structural proteins during the assembly ofthe head of bacteriophage T4. Nature 1970;227:680–5.
[17] Kireeva ML, Latinkic BV, Kolesnikova TV, et al. Cyr61 and Fisp12are both ECM-associated signaling molecules: activities, metabolism,and localization during development. Exp Cell Res 1997;233:63–77.
[18] Ohnishi H, Oka T, Kusachi S, et al. Increased expression of connectivetissue growth factor in the infarct zone of experimentally inducedmyocardial infarction in rats. J Mol Cell Cardiol 1998;30:2411–22.
[19] Blom IE, Goldschmeding R, Leask A. Gene regulation of connectivetissue growth factor: new targets for anti-fibrotic therapy? Matrix Biol2002;21:473–82.
[20] Chen MM, Lam A, Abraham JA, Schreiner GF, Joly AH. CTGFexpression is induced by TGF- beta in cardiac fibroblasts and cardiacmyocytes: a potential role in heart fibrosis. J Mol Cell Cardiol 2000;32:1805–19.
403M.S. Ahmed et al. / Journal of Molecular and Cellular Cardiology 36 (2004) 393–404

[21] Bonniaud P, Margetts PJ, Kolb M, et al. Adenoviral gene transfer ofconnective tissue growth factor in the lung induces transient fibrosis.Am J Respir Crit Care Med 2003;168:770–8.
[22] Chambers RC, Leoni P, Blanc-Brude OP, Wembridge DE, Laurent GJ.Thrombin is a potent inducer of connective tissue growth factorproduction via proteolytic activation of protease-activated receptor-1.J Biol Chem 2000;275:35584–91.
[23] Finckenberg P, Inkinen K, Ahonen J, et al. Angiotensin II inducesconnective tissue growth factor gene expression via calcineurin-dependent pathways. Am J Pathol 2003;163:355–66.
[24] De Carvalho Frimm C, Sun Y, Weber KT. Angiotensin II receptorblockade and myocardial fibrosis of the infarcted rat heart. J Lab ClinMed 1997;129:439–46.
[25] Varo N, Etayo JC, Zalba G, et al. Losartan inhibits the post-transcriptional synthesis of collagen type I and reverses left ventricu-lar fibrosis in spontaneously hypertensive rats. J Hypertens 1999;17:107–14.
[26] Hishikawa K, Oemar BS, Nakaki T. Static pressure regulates connec-tive tissue growth factor expression in human mesangial cells. J BiolChem 2001;276:16797–803.
[27] Lijnen PJ, Petrov VV, Fagard RH. Angiotensin II-induced stimulationof collagen secretion and production in cardiac fibroblasts is mediatedvia angiotensin II subtype 1 receptors. J Renin Angiotensin Aldoster-one Syst 2001;2:117–22.
404 M.S. Ahmed et al. / Journal of Molecular and Cellular Cardiology 36 (2004) 393–404
Copyright © 2022 FDOKUMEN




















