
Role of autophagy in cardiac fibroblast activation in ... - MSpace
206
Role of autophagy in cardiac fibroblast activation in cardiac fibrosis by Shivika Gupta A Thesis submitted to Faculty of Graduate Studies of The University of Manitoba in partial fulfilment of the requirements of the degree of Doctor of Philosophy Department of Physiology and Pathophysiology University of Manitoba Winnipeg Copyright ©2017 by Shivika Gupta
-
Upload
khangminh22 -
Category
Documents
-
view
3 -
download
0
Transcript of Role of autophagy in cardiac fibroblast activation in ... - MSpace

Role of autophagy in cardiac fibroblast
activation in cardiac fibrosis
by
Shivika Gupta
A Thesis submitted to Faculty of Graduate Studies of
The University of Manitoba
in partial fulfilment of the requirements of the degree of
Doctor of Philosophy
Department of Physiology and Pathophysiology
University of Manitoba
Winnipeg
Copyright ©2017 by Shivika Gupta

ii
Abstract
Following myocardial infarction (MI), initial cardiac remodeling or wound repair/healing is
beneficial and required for continued function. During the wound healing phase, cardiac
fibroblasts activate to become contractile and hypersynthetic myofibroblasts and contribute to
cardiac fibrosis. Chronic cardiac remodeling leads to the development of overt cardiac
hypertrophy and excessive extracellular matrix (ECM) deposition with attendant loss of
myocardial performance. There is no treatment that prevents or reverses fibrosis in the post-
MI heart. Induction of autophagy is linked to stress-induced ventricular remodeling in various
cardiac diseases and has not been well studied in fibroblast activation. As the plating of
primary cardiac fibroblasts on incompressible plastic substrate leads to activation of these
cells, we used this model system in primary rat cardiac fibroblasts to examine the relationship
between autophagy and fibroblast activation. 72 hours after plating of fibroblasts, we
observed the concomitant increase in expression of myofibroblast markers and induction of
autophagy, assessed via western blotting, immunofluorescence, and transmission electron
microscopy. Treatment with inhibitors of autophagy (BafilomycinA1 (Baf-A1), 3-
methyladenine (3MA), and chloroquine (CQ) decreased myofibroblast marker expression and
reduced myofibroblast contractility (assessed by collagen gel contraction assay). CQ
treatment suppressed fibroblast activation in unpassaged P0 cardiac fibroblasts. We used the
coronary artery ligation rat model of MI to further explore the effects of CQ administration in
development of cardiac fibrosis. At 4 and 8 weeks post-MI, there was no significant change
in ventricular function (assessed via echocardiography), however in isolated cardiac tissue
treated with CQ, we observed a decrease in myofibroblast marker expression. We also show
increased autophagy in the MI group animal. We also studied tissue fibrosis by comparing
the collagen accumulation between CQ treated and nontreated animals. Although CQ
treatment was associated with a decrease in -SMA expression in the 4 week post-MI group,

iii
there was no difference seen on collagen level among the same study groups. These data
support a causal link between autophagy and activation of cardiac fibroblast. This study
provides a new avenue for therapeutic options to suppress the activation of cardiac fibroblasts
and thereby reduce cardiac fibrosis.

iv
Acknowledgements
I would like to express gratitude to Dr. Ian Dixon for his support, guidance and
encouragement during the entire course of my Ph.D. training. I am grateful for his advice,
patience, and numerous opportunities he has provided me, which has helped me to become a
better researcher and more confident person. I would also like to thank my advisory
committee members including Drs. Andrew Halayko, Jeffrey Wigle, and Thomas Netticadan
for their valuable suggestions which helped to improve my research. Their patience and
efforts to enhance my critical thinking have allowed me to become a better graduate student.
I would specially like to extend my gratitude to Dr. Saeid Ghavami who has guided me
during the entire project with his expert knowledge in autophagy. I would like to thank the
CIHR, the Institute of Cardiovascular Sciences, the Wyrzykowski family and the Deacon
Foundation for supporting me. The financial support from these agencies has been critical to
my success.
I greatly appreciate our lab manager Sunil Rattan for his hands-on assistance in all the
experiments. Sunil has offered great advice professionally and personally during my training,
and has acted as both a teacher and a friend. His positive attitude and guidance was always
welcome. I am very grateful to my lab members Krista Filomeno, Dr. Matthew Zeglinski,
Morvarid Kavosh, and Natalie Landry for their support and comraderie. Scientific
discussions with all the labmates were always helpful. I would especially like to thank Krista
Filomeno, who has been an amazing friend and confidante. I would like to extent my
gratitude to the members of Kardami lab, Navid Kholini and Robert Fandrich for use and
instruction of equipment, and always providing their expertise when needed. I feel extremely
honored to be a part of the St. Boniface Hospital Albrechtsen Research Centre, and the
Institute of Cardiovascular Sciences. I would also like to give a special thank you to Dr.
Pawan Singal for his constant support and guidance to graduate students in the Institute. I

v
would also like to thank Dr. Singal’s lab members for sharing equipment, reagents, and
friendship, and to the office ICS staff (Mary Brown and Shweta Sharma), who are always
available to help with administrative issues and always have a smile on their faces. I also
extend my thanks to Drs. Peter Cattini (Head of the Department of Physiology and
Pathophysiology) and Janice Dodd.
Finally, I would like to thank my family and friends. Without their love and support I
would not have reached this stage in my life. I would like to thank my mother Dr. Sarita
Gupta and father Dr. Sharad Gupta for their endless support in every difficult situation and
motivating me during my program. I would also like to thank my friends Dr. Anurag
Sikarwar, Nisha Sengar, Dr. Biswajit Chaudhray, Triporna Lahiri, and Subhankar Ghosh here
in Winnipeg who were no less than a family. A very special thanks to Sikarwar and Ghosh
family – and their kids Prisha and Ronav who have given so much love and affection and
made me feel at home always. Also, I would like to thank my overseas childhood friends
Riddima, Ahsu and Vaishali whose precious friendship has given me strength and kept a
smile on my face throughout my life.
I would like to thank my husband Karan Sharma who has been a tremendous support
in this journey, without his unconditional love, patience and positive attitude it would not
have been possible to finish my PhD. Thank you for all the encouragement and motivational
Skype talks that have helped me to be a strong person.

vi
Table of Contents
Abstract ................................................................................................................................................... ii
Acknowledgements ................................................................................................................................ iv
List of Tables ......................................................................................................................................... ix
List of Figures ........................................................................................................................................ ix
List of Abbreviations ............................................................................................................................. xi
Introduction ............................................................................................................................................. 2
Cardiovascular Diseases and Epidemiology ....................................................................................... 2
Cardiovascular Disease and Cardiac Fibrosis............................................................................... 4
Myocardial Infarction and Wound Healing ........................................................................................ 5
Extracellular Matrix (ECM) .............................................................................................................. 10
Fibrillar Collagens ....................................................................................................................... 10
ED-A Fibronectin .......................................................................................................................... 11
Matricellular Proteins .................................................................................................................. 12
Cardiac Fibroblasts ........................................................................................................................... 13
Origins of Cardiac Fibroblasts: Development and Pathology ..................................................... 14
Cardiac Myofibroblasts ..................................................................................................................... 18
Activation of fibroblast to myofibroblasts – a continuum of phenotype .......................................... 18
Mechanical Stress ......................................................................................................................... 19
Transforming Growth Factor-β .................................................................................................... 20
Persistence of myofibroblasts in the heart following cardiac damage ......................................... 24
Animal Model of MI……………………………………………………………………………………………………………………24
Techniques to study cardiac structure and function………………………………………………………………….25
Cell death mechanisms ..................................................................................................................... 26
Apoptosis ....................................................................................................................................... 26
Necrosis......................................................................................................................................... 27
Autophagy ..................................................................................................................................... 27
Signaling pathways in regulation of autophagy ................................................................................ 33
PI3K-AKT/mTOR signaling .......................................................................................................... 33
Adenosine monophosphate kinase (AMPK) signaling .................................................................. 34
Hypoxia inducible factor-1α ......................................................................................................... 35
Autophagy in Development and Differentiation ............................................................................... 40

vii
Autophagy in Disease Pathogenesis ................................................................................................. 41
Autophagy in Cardiovascular Diseases ............................................................................................. 42
Autophagy: A therapeutic target in cardiovascular disease .............................................................. 46
Autophagy in phenoconversion or activation of fibroblasts ............................................................. 48
Rationale and Hypothesis…………………………………………………………………………………………………………….50
Rationale…………………………………………………………………………………………………………………………………….51
General Hypothesis ........................................................................................................................ …50
Specific Hypothesis 1. Mechanical stress induces autophagy and activation of cardiac fibroblasts in
vitro ................................................................................................................................................... 52
Objective 1.1: Determine the effect of mechanical stress on cardiac fibroblasts ............................. 52
Specific Hypothesis 2. Autophagy is necessary for mechanical stress-induced activation of cardiac
fibroblasts in vitro ............................................................................................................................. 52
Objective 2.1:Determine the Effect of Inhibition of Autophagy in Cardiac Fibroblasts .................. 52
Objective 2.2: Determine the effect of inhibition of autophagy on myofibroblast phenotype ........... 53
Objective 2.3: Determine the effect of inhibition of autophagy on myofibroblast function .............. 53
Objective 2.4: Determine the effect of inhibition of autophagy on myofibroblast function .............. 53
Specific Hypothesis 3.Inhibition of autophagy in vivo will attenuate cardiac fibrosis following
myocardial infarction ........................................................................................................................ 54
Objective 3.1: Determine the effect of inhibition of autophagy on cardiac remodeling ................... 54
Materials and Methods .......................................................................................................................... 55
Animal Ethics.................................................................................................................................... 55
Cell Isolation and Culture ................................................................................................................. 55
Treatment with Autophagy Inhibitors ........................................................................................... 56
Optimization of Autophagy Inhibitor Treatment by MTT Assay ................................................... 56
Protein Isolation from Adherent Cell Cultures ................................................................................. 57
Western Blot Analysis ...................................................................................................................... 58
Fluorescence Immunocytochemistry ................................................................................................ 59
Scratch Assay .................................................................................................................................... 59
Gel Contraction Assay ...................................................................................................................... 60
Transmission Electron Microscopy (TEM) ...................................................................................... 61
Experimental model of myocardial infarction .................................................................................. 61
Echocardiography ............................................................................................................................. 64
Pathological Analysis of Blood Serum ............................................................................................. 64
Protein Isolation from Whole Tissue ................................................................................................ 64

viii
Tissue Sectioning for Histology and Immunofluorescence .............................................................. 65
Masson’s Trichrome Staining ........................................................................................................... 65
Statistical analysis………………………………………………………………………………………………………………………..67
Chapter 1: Role of Autophagy in Fibroblast Activation In Vitro .......................................................... 68
Introduction ....................................................................................................................................... 68
Results ............................................................................................................................................... 70
Induction of Autophagy by Mechanical Stress .............................................................................. 70
Inhibition of Autophagy in Cardiac Fibroblasts ........................................................................... 72
Inhibition of Autophagy and Myofibroblast Features ................................................................... 78
Inhibition of Autophagy and Myofibroblast Function .................................................................. 86
Inhibition of Autophagy and Mitogen-Activated Protein Kinase (MAPK) Signaling ................... 93
Discussion ....................................................................................................................................... 101
Chapter 2: Autophagy and Cardiac Fibrosis In Vivo .......................................................................... 108
Introduction ..................................................................................................................................... 108
Results ............................................................................................................................................. 111
Fibroblast phenotype and inhibition of autophagy in the post-MI heart .................................... 111
Assessment of Biochemical Parameters for Cardiac, Liver and Kidney Damage ...................... 131
Functional Assessment Following Myocardial Infarction and Chloroquine Treatment ............ 135
General Discussion ............................................................................................................................. 148
Study Conclusions .............................................................................................................................. 152
Future Directions ................................................................................................................................ 153
Limitations of Study………………………………………………………………………………....156
References ........................................................................................................................................... 157

ix
List of Tables
Table 1: Types of CVD’s and their pathological features
Table 2: Autophagy proteins and their functions
Table 3: Optimized dosages for autophagy inhibitors
List of Figures
Figure 1: Phases of myocardial wound healing
Figure 2: Sources of cardiac fibroblasts
Figure 3: Activation of cardiac fibroblasts to myofibroblasts
Figure 4: Autophagy pathway and its modulators
Figure 5: Central hypothesis
Figure 6: Study Timeline
Figure 7: Temporal activation of autophagy and fibroblast in P0 adult rat cardiac fibroblasts
Figure 8: Baf-A1, CQ and 3MA treatment inhibits autophagy in P0 cardiac fibroblasts
Figure 9: Ultrastructure of rat ventricular cardiac fibroblasts
Figure 10: Inhibition of autophagy reduces expression of key cardiac myofibroblast markers
Figure 11: Immunofluorescence staining of P0 cardiac fibroblast
Figure 12: Effect of autophagy inhibition on cellular contractility of P0 cardiac fibroblasts
Figure 13: Scratch assay to assess cellular migration of cardiac fibroblasts in the presence and
absence of 7.5 nM Baf-A1, 5mM 3MA, 35 μM CQ
Figure 14: Effect of cell plating and CQ treatment on p38-MAPK phosphorylation in P0
primary cardiac fibroblasts
Figure 15: Effect of MAPK P-38 inhibition on primary rat cardiac fibroblast with and without
CQ treatment
Figure 16: Effect of MAPK inhibition on Baf-A1 treated and untreated P0 cardiac fibroblasts

x
Figure 17: Effect of inhibition of autophagy on primary cardiac fibroblast-to-myofibroblast
phenoconversion
Figure 18: LC-3β lipidation as a marker of autophagy in 4 week untreated and chloroquine-
treated post-MI rat hearts (control, myocardium remote to the scar and the infarct
scar).
Figure 19: α-SMA marker levels in 4 week untreated and chloroquine-treated post-MI rat
hearts.
Figure 20: Immunofluorescence staining of post-MI and sham-operated hearts at 4 weeks,
with and without chloroquine treatment
Figure 21: Masson’s trichrome staining for collagen content in 4 week post-infarct left
ventricles, with and without chloroquine treatment.
Figure 22: Hydroxyproline collagen quantification in 4 week post MI cardiac tissue.
Figure 23: LC-3β lipidation as a marker of autophagy autophagy in 8-week untreated and
chloroquine-treated rat hearts post-MI.
Figure24: α-SMA expression in 8-week post-MI hearts treated with chloroquine.
Figure 25: Immunofluorescence staining of 8 week post-MI hearts with and without
chloroquine
Figure 26: Biochemical parameters for cardiac, kidney and liver damage in 4 and 8 week
post-MI rat hearts with and without chloroquine treatment
Figure 27: Ejection Fraction and Fractional Shortening in Post-MI Rats
Figure 28: Heart Rate and Left Ventricular Posterior Wall Diameter in Post-MI rats

xi
List of Abbreviations
3MA 3-methyladenine
4EBP1 eIF4E-binding protein
ACTA2 Actin-2-α
ADP Adenosine diphosphate
AICAR 5-aminoimidazole-4-carboxamide ribonucleotide
AMP Adenosine monophosphate
AMPK 5' adenosine monophosphate-activated kinase
ANGII Angiotensin II
ANT Adenine nucleotide translocase
α-SMA α-Smooth Muscle Actin
AST/ALT Aspartate transaminase/alanine transaminase
ATP Adenosine triphosphate
Baf-A1 Bafilomycin A1
BAK Bcl-2-homologous antagonist/killer
BAX Bcl-2-like protein 4
Bcl-2 B-cell lymphoma 2
bHLH Basic helix-loop-helix
BMDC Bone marrow-derived cells
BNIP3 B-cell lymphoma 2/adenovirus E1B 19 kDa protein-interacting protein 3
BSA Bovine serum albumin

xii
CaMKK-b Calcium/calmodulin-dependent kinase kinase-b
CCN CTGF, Cyr61, nov proteins
CCN2/CTGF Connective Tissue Growth Factor
CD34 Cluster of differentiation 34
CD45 Cluster of differentiation 45/lymphocyte common antigen
CK/CK-MB Creatine kinase/creatine kinase-MB
CMA Chaperone-mediated autophagy
COPII Coat protein II
CQ Chloroquine
cTnI Cardiac troponin I
cTnT Cardiac troponin T
CVD Cardiovascular Disease
CyP-D Cyclophilin D
Cyr61 Cysteine rich protein 61
DEPTOR DEP domain-containing mTOR-interacting protein
DMEM Dulbecco's modified Eagle's medium
ECG Echocardiography
ECM Extracellular Matrix
ED-A FN ED-A Fibronectin
EF Ejection fraction
EGTA Ethylene glycol-bis(β-aminoethyl ether-N,N,N',N'-tetraacetic acid)

xiii
eIF4E Eukaryotic initiation factor 4E
EMT Epithelial to mesenchymal transition
EndMT Endothelial to mesenchymal transition
EPDC Epicardium-derived cells
FAK Focal adhesion kinase
FBS Fetal bovine serum
FGF Fibroblast growth factor
FIP200 Focal adhesion kinase family interactin protein of 200 kDa
FN Fibronectin
FoxO3 Forkhead box O3
FS Fractional shortening
GAGs Glucosaminoglycans
GAP GTPase-activating protein
G-CSF Granulocyte colony-stimulating factor
GF Growth factor(s)
GPCR G-protein coupled receptor
GTP Guanosine triphosphate
GβL G protein beta subunit-like
HDAC Histone deacetylase
HIF1α Hypoxia-inducible factor-1α
HR Heart rate
xiv
HRP Horseradish peroxidise
hsc70/hsc40 Heat shock cognate protein of 70/40 kDa
HSCs Hepatic stellate cells
IL Interleukin
IP3 Inositol triphosphate
I-Smad Inhibitory Smad
JNK c-Jun N-terminal kinase
kDa Kilodaltons
kPa Kilopascals
LAD Left anterior descending
LAMP2A Lysosome-associated membrane protein type 2A
LAP Latency-associated protein
LC3 Light chain variant 3
LDH Lactate dehydrogenase
LKB1 Liver kinase B1
LLC Large latent complex
LTBP Latent TGF-β binding protein
LV Left ventricular
LVPWd Left ventricular posterior wall diameter
MAP4K3 Mitogen-activated protein kinase kinase kinase kinase 3
Meox2 Mesenchyme homeobox 2
xv
MET Mesenchymal to epithelial transition
MI Myocardial Infarction
mLST8 mechanistic target of rapamycin complex subunit LST8
MMP Matrix metalloproteinase
mPTP Mitochondrial permeability transition pore
MRI Magnetic resonance imaging
mRNA Messenger ribonucleic acid
mTOR Mechanistic target of rapamycin
mTORC1/2 Mechanistic target of rapamycin complex ½
NFκB Nuclear factor kappa B
Nov Nephroblastoma overexpressed gene
OPN Osteopontin
P0 Passage 0 (unpassaged)
PBS Phosphate buffered saline
PDGF Platelet-derived Growth Factor
PE Phosphatidyl ethanolamine
PFA Paraformaldehyde
PGs Proteoglycans
PI3K Phosphatidylinositol 3 kinase
PIKK Phosphatidylinositol kinase-related kinase
PINK-1 PTEN-induced putative kinase 1

xvi
PRAS40 Proline-rich Akt substrate of 40 kDa
PRR5 Proline-rich protein 5
PTEN Phosphatase and tensin homolog
PVDF Polyvinylidene difluoride
Rab7 Ras-related GTP-binding protein 7
Rag Recombinant activating gene protein
Raptor Regulatory-associated protein of mTOR
REDD1 Regulated in development and DNA damage responses 1
Rheb Ras homolog enriched in brain
ROS Reactive Oxygen Species
R-Smad Receptor Smad
S100A4 S100 calcium-binding protein A4
S6K1 S6 kinase 1
SAHA Suberoylanilidehydroxamic acid
SDS-PAGE Sodium-dodecyl sulphate polyacrylamide gel electrophoresis
SLC Small latency complex
SMCs Smooth Muscle Cells
S-MEM Minimum essential media Spinner's modification
SMER Small molecule enhancer of rapamycin
Sox9 Sex-determining region Y-box 9
SPARC Secreted protein acidic and rich in cysteine

xvii
TBS Tris-buffered saline
Tbx5 T-box 5
Tcf21 Transcription factor 21
TEM Transmission electron microscopy
TGF-β Transforming Growth Factor-β
TLR Toll-like Receptor
TSP-1 Thrombospondin-1
TTE Transthoracic echocardiography
ULK1 Unc-51 like autophagy activating kinase 1
VDAC-1 Voltage-dependent anion-selective channel 1
Vps34 Vacuolar protein sorting 34
VSMCs Vascular Smooth Muscle Cells
Wk Week(s)
Zeb2 Zinc finger E-box binding homeobox 2

2
Introduction
Cardiovascular Diseases and Epidemiology
Cardiovascular disease (CVD) is a category of disease comprising the disorders related to
the heart and the vasculature, involving the circulatory system. CVDs can be described as
conditions resulting in compromised blood supply to and from the heart, causing
cardiovascular dysfunction. As per World Health Organization and Public Health Agency of
Canada guidelines, there are six major types of CVDs. The six categories of CVD are:
Table 1: Types of CVD’s and their pathological features:
No. Types Of CVD Pathological Features
1 Coronary heart disease Complete blockage of one or more coronary
arteries (referred to as MI) leading to tissue
necrosis.
Ischemic heart disease A partial blockage of one or more of the
coronary arteries leading to angina (chest
pain) and dyspnea (shortness of breath).
2 Cerebrovascular disease (stroke)
Blockage of the carotid artery or blood
vessels supplying blood to the brain due to a
hemorrhage (rupture of blood vessel) or
ischemia (blood clots), eventually leading to
stroke.
3 Peripheral vascular disease Reduced blood supply to extremities of the
body such as arms or limbs due to vascular
narrowing, closure, or spasm.

3
4 Heart failure
Inability of the heart to pump adequate
blood to the body due to chronic alcohol
consumption, myocardial infarction or
cardiomyopathy.
5 Rheumatic heart disease Bacterial infection in adults, affecting joints,
heart valves, endocardium (endocarditis), or
the pericardium (pericarditis).
6 Congenital heart disease An anatomical birth defect in the heart,
created during fetal cardiac development.
May involve defects of heart valves (such as
being too narrow or complete occlusion);
heart wall defects, such as impaired closure
of the inside walls of the atrium, which can
cause a reverse blood flow to the heart and
lungs; defects of myocardial muscle,
compromising efficiency; or improper
vessel connections, which may improperly
shunt blood travelling through the heart
chambers.
CVDs cause of death, globally representing 31% (17.5 million) of all deaths in 2012 [1].
Also on the global scale, out of 16 million deaths of people under the age of 70 due to non-
communicable disease, 37% are caused by CVDs. Among total CVD-related deaths,
approximately 43% are due to ischemic heart disease, 33% are due to stroke, and the other
26% are due to other causes (congestive heart failure, hypertension, rheumatic fever, etc.)

4
[1].Despite remarkable progress in treatment of heart disease, CVD is the leading cause of
death in over one-third of the country’s population [2]. Statistics Canada listed 1.3 million
Canadians living with heart disease in 2011, and around 14,000 died that year due to heart
attack [2].
The Canadian economy incurs $20.9 billion in annual costs due to heart disease and
stroke, even though studies show that 80% of premature heart disease and stroke can be
prevented by adopting a healthy lifestyle [3]. Incidence of cardiac fibrosis, in CVD is
associated with high mortality and there are no effective treatments for its prevention or
reversal post-MI [4].
Cardiovascular Disease and Cardiac Fibrosis
Cardiac fibrosis may be causal to heart failure in CVD [5] and is defined as replacement
and invasion of a healthy functional myocardium by excessive connective tissue if it can
interfere with systolic and diastolic function [6]. Among the six forms of CVD,
coronary/ischemic heart disease has been identified as the most frequent cause of cardiac
fibrosis. Coronary artery disease often leads to MI, which in turn stimulates cardiac fibrosis
[6] and eventual heart failure [5]. Other types of CVD, such as hypertension and diabetic
cardiomyopathy, have also been associated with development of cardiac fibrosis [7-9]. These
pathologies result in damage and loss of the myocardial tissue, ultimately resulting in heart
failure. Two types of cardiac fibrosis have been widely accepted to occur in heart [10, 11],
including:
1. Reactive interstitial fibrosis: This fibrosis is progressive leading to collagen
deposition in interstitium and is seen associated with hypertension, diabetes,
metabolic syndrome and in aging heart. It is an adaptive response to preserve

5
cardiac structure and function. There is a diffuse deposition of collagen
throughout the myocardium originating from the microvasculature.
2. Replacement fibrosis (also known as scarring fibrosis): This fibrosis is associated
with MI and cardiotoxicity, among other CVDs. It appears to replace myocytes
lost to necrosis following acute MI [12]. It is localized as in cases of ischemic
cardiomyopathy.
Myocardial Infarction and Wound Healing
In a normal, efficiently working heart there is a constant supply of oxygen and
nutrients provided mainly by the coronary circulation. Approximately 90% of heart attacks
are due to rupture of an atherosclerotic plaque in the coronary arteries, which causes
obstruction of blood supply to the heart via these arteries – also known as ischemia.
Prolonged ischemia leads to myocardial cell death, damage of the cardiac tissue, and loss of
cardiac function. MI can be diagnosed via electrocardiographic (ECG) findings, imaging
techniques such as magnetic resonance imaging (MRI), and by detecting elevated values of
biochemical markers of myocardial necrosis such as cardiac troponin T (cTnT) and I (cTnI)
and creatine kinase-MB (CK-MB) in the blood [13]. With time MI leads to global
pathological remodeling, leading to cardiac hypertrophy and ventricular arrhythmias.
Ultimately, this means that patients who survive acute MI are subsequently susceptible to the
development of heart failure, a leading cause of death [14].
After MI, the injured heart undergoes wound healing following the death of
cardiomyocytes[15]. Rapid necrosis of cardiomyocytes and myocardial tissue loss triggers
three overlapping phases of cardiac wound healing: (1) the inflammatory phase, (2) the cell
proliferation phase, and (3) the ECM synthesis and scar formation phase [16-18].

6
1) Inflammatory phase: This acute phase can last for hours or days. Tissue injury causes
release of reactive oxygen species (ROS) and intracellular proteins from the necrotic
cells in the infarcted area, and these products initiate an inflammatory cascade,
including activation of Toll-like receptors (TLRs), the transcription nuclear factor
kappa B (NF-κB), and the complement system [19, 20]. The first cell type to be
recruited after infarction are neutrophils, followed by monocytes [21]. Neutrophils
release inflammatory cytokines such as tumor necrosis factor-α (TNF-α) and
interleukins (IL-3, -6, and -8) that aid in recruiting monocytes, but which can also
lead to further damage and should be controlled [20]. The inflammatory response also
results in activation and release of MMPs (specifically MMP-2 and MMP-9) from
inflammatory cells and fibroblasts that help in ECM degradation [18, 22]. Activation
of the inflammatory phase cleans the dead and damaged tissue which is then followed
by release of TGF-β and IL-10 [23].Both of these factors inhibit inflammation and
induce infarct healing. Healing of the infarct begins with infiltration of monocytes and
lymphocytes[20].
2) Cell proliferation phase: Release of cytokines and growth factors such as TGF-β,
platelet-derived growth factor (PDGF) and fibroblast growth factor (FGF) induces the
proliferation and activation of cardiac fibroblasts to myofibroblasts, for which the
main role is to synthesize new ECM [24].The formation of new vessels to restore
tissue reperfusion (angiogenesis) is also an important feature of this phase [25]. The
closure of the infarct wound occurs via re-epithelization where epithelial epidermal
cells migrate inward to form the wound periphery. Angiogenesis and re-epithelization
help in the formation of granulation tissue, which is the newly formed connective
tissue with blood vessels [18]. Formation of new ECM provides structural integrity,

7
and accumulation of ECM components such as fibrillar collagens type I and III,
fibronectin, and glycoprotein signals the final phase of scar formation [26].
3) Extracellular matrix remodeling and scar maturation phase: This phase can continue
for weeks. A shift to matrix synthesis from degradation, due to activated
myofibroblasts in the infarct region, initiates scar formation [18]. With dynamic
changes in ECM structure, interactions between myofibroblasts and the ECM assist
with contracture of the infarct wound. Myofibroblasts form stress fibers containing
contractile proteins such as α-smooth muscle actin (α-SMA) that help in developing
tension needed for correct wound healing [27]. Once the injured tissue is repaired, a
process that can take weeks, resident myofibroblasts are removed via apoptosis [28].
However, this ideal scenario of wound healing might not occur in elderly patients
suffering from other cardiac and non-cardiac ailments. In adverse wound healing, the
myofibroblasts persist in the scar and distal, uninfarcted regions instead of undergoing
apoptosis, and continue to synthesis excess amounts of collagen and ECM proteins,
which lead to stiffening of the tissue, left ventricular dysfunction, and increased
chances of eventual heart failure [29, 30].

8
Figure 1: Phases of myocardial wound healing. Post-myocardial infarction (MI) the heart
undergoes overlapping wound healing phases for several days. Cardiomyocyte death occurs
for up to approximately 24 hours, followed by apoptosis or necrosis of the dead tissue,
continuing for up to 2 days post-MI. Thus begins an acute inflammatory phase which
continues for up to 5 days. The hallmark of this phase is recruitment of neutrophils followed
by monocytes and macrophages that phagocytose dead cells. Neutrophils release bioactive
factors such as cytokines, chemokines, and matrix metalloproteinases (MMPs), which help in
ECM degradation and removal of dead tissue. Macrophages recruited by the release of
inflammatory factors from dying myocardial tissue then engulf cellular debris via
phagocytosis. The next phase involves proliferation of cells that will initiate scar formation,
namely fibroblasts and myofibroblasts. The main cell types that initiate this phase are
endothelial cells, which release growth factors (GF), cytokines, and chemokines to induce
angiogenesis and endothelial to mesenchymal transition (EndMT). EndMT causes
proliferation and activation of cardiac fibroblasts to myofibroblasts, and initiates ECM
synthesis. This entire process occurs from 3-28 days. The final phase is scar formation and

9
maturation, which can continue for years. In this phase, activated myofibroblasts release
bioactive factors such as transforming growth factor- β (TGF-β), angiotensin II (ANGII) and
MMPs, which help in accumulation and maturation of ECM proteins. Myofibroblasts also
produce large amounts of ECM structural proteins (primarily fibrillar collagens) and contract,
which helps in wound contracture. In normal wound healing, once the scar tissue is formed at
the site of injury, myofibroblasts undergo apoptosis, halting the process of ECM deposition.

10
Extracellular Matrix (ECM)
Cardiac ECM forms the structural scaffold of the heart and is composed of non-
cellular matrix proteins. Cell types present in the ECM include fibroblasts, myocytes,
leukocytes, as well as smooth muscle and endothelial cells of the vasculature. The cardiac
ECM provides mechanical connections between cardiomyocytes, and so is the basis of the
structural and functional integrity of the myocardium [31, 32]. The dynamic nature of the
cardiac ECM means cellular components are under the influence of many cytokines, growth
factors, and hormones that coordinate turnover of matrix proteins and is in constant flux.
Under pathological triggers like ischemia, pressure overload, aging, and infection, the cardiac
ECM can undergo remodeling, primarily due to activation of MMPs [33]. While excessive
production of ECM causes fibrosis and ventricular dysfunction, inadequate ECM deposition
can lead to wall thinning and infarct expansion, resulting in clinical outcomes of aneurysm,
left ventricular (LV) dilatation, and cardiac rupture [34].
The matrix proteins comprising the non-cellular structural aspect of cardiac ECM are
fibrillar collagens, fibronectin, laminin, elastins, glycosaminoglycans (GAGs) and
proteoglycans (PGs). GAGs like hyaluronan, and PGs like syndecans and glypicans (cell
surface proteins) also facilitate with crosstalk between ECM proteins, growth factors, and
cytokines. Hyaluronan is the largest GAG in cardiac ECM and is elevated in MI [35], cardiac
hypertrophy [36] and myocarditis [37]. There are also the non-structural matricellularproteins
that include secreted protein acidic and rich in cysteine (SPARC), thrombospondin (TSP-1),
tenascin-C, osteopontin (OPN), periostin, proteolytic MMPs, and growth factors such as
connective tissue growth factor (CTGF), TGF-β, and PDGF [31].
Fibrillar Collagens
Fibrillar collagen types are the most abundant structural proteins in heart (specifically
collagen type I and III in the interstitium), along with the much less abundant but important

11
non-fibrillar collagen types IV, V, and VI [17]. Studies have shown on days 7 and 21 post-
MI, collagens I and III make up 30% and 60% of infarct scar area, respectively [34].Collagen
synthesis by fibroblasts and myofibroblasts, and other cell types such as vascular smooth
muscle cells (VSMCs) in the ECM, begins with pre-procollagens, which exit the endoplasmic
reticulum and enter the interstitial space. Hydroxylase enzyme processes pre-procollagens
into pro-collagens, transported via vesicles to the Golgi apparatus, where oligosaccharides are
added to form tropo-collagens. Tropo-collagens are linked by the membrane-bound enzyme
lysyl oxidase that acts on tropo-collagen lysine and hydroxylysine residues, resulting in the
formation of collagen fibers. Collagen turnover is critical in the heart, requiring a balance
between synthesis by myofibroblasts and degradation by collagenases (primarily MMP2 and
MMP9) [38]. This homeostasis of ECM is regulated via the TGF-β/Smad signaling pathways
in fibroblasts and myofibroblasts, discussed later [4]. Pathologically, circulating end terminal
peptides of pro-collagen in the serum are biomarkers for myocardial fibrosis, LV dysfunction
and risk of heart failure [34, 39]. Thus, collagen deposition and turnover plays a critical role
not only in scar formation but in overall cardiac remodeling and functioning of the heart post-
MI.
ED-A Fibronectin
Fibronectins (FN) area collection of proteins with a number of subtypes, and in heart
is an ECM glycoprotein expressed by a wide variety of cells and plays a critical role in cell
growth, adhesion, migration, and differentiation [40]. Fibronectins have been shown in
numerous studies to regulate the wound-healing process [41, 42]. Different splice variants
give rise to three modes of fibronectin (types I, II, and III) [43]. Fibronectin Type III
comprises 15 repeating highly conserved segments and has two variable exons extracellular
domain-A (ED-A) and -B (ED-B). The FN type III isoform containing the ED-A splice
variant is termed cellular FN and is present in the cardiac ECM [44]. ED-A FN production is

12
increased with conversion of fibroblasts to myofibroblasts, and is a key molecule involved in
fibrosis in various tissue types, including the lungs, liver, and heart [45]. Under the conditions
of tissue fibrosis, and fibroblast activation, ED-A FN is expressed in abundance by activated
fibroblasts, macrophages, and endothelial cells [32]. Several studies have demonstrated a
direct correlation between increased levels of ED-A FN and cell proliferation, increased cell
adhesion, and activation of fibroblasts to myofibroblasts [46, 47]. ED-A FN-deficient mice
show decreased left ventricular (LV) dysfunction post-MI as compared to wild type mice
[48], suggesting that ED-A FN is critical during developing cardiac fibrosis. Studies have
shown that in the post-MI heart, TGF-β induces myofibroblast secretion of ED-A FN [47, 49,
50]. The exact mechanism for this induction is not known, but Scleraxis, a basic helix-loop-
helix (bHLH) transcription factor, has been identified as the factor regulating TGF-β-induced
fibronectin expression [47, 51].
Matricellular Proteins
Matricellular proteins are the non-structural proteins of the ECM, expressed during
the embryonic development phase and absent in adulthood [52]. Studies have shown that
these proteins are re-expressed during tumor growth and tissue injury to help in matrix
remodeling and wound healing, through regulation of cell proliferation and migration [53].
For example, tenascin-C, important during embryogenesis, is re-expressed by the
mechanically stressed fibroblast specifically near the border zone of the infarct during post-
MI remodeling [54, 55]. Tenascin-C expression can be induced via growth factors such as
FGF and TGF-β, which are released during wound repair. Thrombospondins (TSP) are a
family of secreted glycoproteins that play key roles in cardiac pathologies. TSP-1and TSP-2
are shown to have a protective role in pressure overload (by activating TGF-β to induce ECM
synthesis) and post-MI (via inhibiting MMP2 activation) [31]. Osteopontin (OPN) is another
member of the matricellular proteins, which is up-regulated in macrophages following MI

13
and is important in reducing chamber dilatation, protecting from adverse cardiac remodeling
[56]. Levels of periostin are also increased following cardiac injury. Periostin plays a critical
role in the activation of fibroblasts and subsequent collagen deposition preventing infarct
rupture [31]. However, in a swine model of MI, recombinant periostin delivery to the
myocardium resulted in increased fibrosis [57], the role of this matricellular protein in cardiac
pathology remains unclear.The connective tissue growth factor, Cysteine rich protein 61
(Cyr61), and nephroblastoma overexpressed gene (nov) family of proteins, also known as
CCN proteins, have also been studied extensively in heart disease. Specifically, CTGF has
been elevated post-MI and in the pressure-overloaded heart, and is important in TGF-
signaling, angiogenesis, and myocyte survival [58, 59]. Matricellular proteins can serve as
important diagnostic markers for predicting the extent of adverse ECM remodeling following
myocardial injury. Namely, tenascin-C has been demonstrated as a useful marker for acute
myocarditis, post-MI remodeling, and heart failure; OPN levels serve as a useful marker of
LV dilatation; and serum CTGF correlates with plasma brain natriuretic peptide (BNP) and
TGF-β levels, thus serving as a useful marker for myocardial remodeling [60].
Cardiac Fibroblasts
The main cell types in the heart are cardiomyocytes, which make up 30% of the adult
rat heart, and non-myocytes, which make up the other 70%. Cardiac fibroblasts make up 70%
of this total of non-myocyte cells, which also includes endothelial cells and VSMCs [61-64].
Cardiac fibroblasts are long spindle-shaped cells lacking basement membrane and belonging
to mesenchymal origin [65]. The main function of cardiac fibroblasts is to maintain
homeostasis (synthesis and degradation) of ECM structural proteins. In the normal healthy
heart, cardiac fibroblasts maintain structural integrity through cell-cell and cell-ECM
interactions, ECM homeostasis, proliferation, and migration [62, 66]. Cardiac fibroblasts can
respond to chemical, mechanical, and electrical signals, which can transduce these signals

14
and produce stimulatory factors in response, as a means to signal to other cardiac cell types
like myocytes and endothelial cells to alter their function and affect ECM remodeling.
Fibroblasts secrete a variety of bioactive molecules such as growth factors TGF-B and PDGF,
cytokines such as IL-10, MMPs, and collagens [67]. Fibroblasts play an important role in
maintaining physiological function and affecting cellular function in pathological conditions
[68].
Origins of Cardiac Fibroblasts: Development and Pathology
Cardiac fibroblasts are a heterogeneous cell population as they can originate from
various cell sources under physiologic and pathologic conditions [65]. During embryonic
development, the proepicardial organ has been shown to be the source of the majority of
cardiac fibroblasts. Migratory proepicardial cells give rise to the embryonic epicardium [2].
These cells later undergo epithelial to mesenchymal transition (EMT) to form epicardial
derived cells (EPDCs) that invade atria and ventricle walls and differentiate into cardiac
fibroblasts. EPDC-originating fibroblasts eventually help to form the cardiac interstitium and
annulus fibrosis [69, 70]. EMT is regulated by numerous factors including growth factors
(FGFs, PDGF, and TGF-β) [71, 72], and transcription factors such as Sex-determining region
Y-box 9 (Sox9), T-box 5 (Tbx5), Thymosin β4, and transcription factor 21 (Tcf21) [71-73].
Tcf-21 has been reported to be an essential transcription factor determining that epicardial
cells will differentiate into cardiac fibroblasts [73]. FGF10 has been identified as another
important factor that plays a role in migration of cardiac fibroblast precursors into the
developing myocardium. Studies in FGF10-null mice show that the number of cardiac
fibroblasts in the heart was decreased, and the hearts were smaller in size [74]. Besides acting
as a source of cells of the cardiac interstitium, the endothelial cells in the atrioventricular
canal give rise to valvular fibroblasts, shown by some studies to occur through endothelial to

15
mesenchymal transition (EndMT) under regulation of various cytokines including PDGF,
TGF-β, and Wnt [75, 76].
Both processes (EMT and EndMT) are important drivers of embryonic cardiac
fibroblast maturation and heart development, but can also be activated during adult
cardiopathologies [76]. Proliferation and activation of cardiac fibroblasts is critical to wound
healing, and thus a substantial number of cardiac fibroblasts need to be recruited for proper
cardiac remodeling. Fate mapping studies have shown that EndMT is critical during fibrosis,
with up to 30% of fibroblasts in the post-MI heart originating from endothelial cells [77]. In
pathological scenarios, EMT and EndMT are efficient means for providing a source of
cardiac fibroblasts, which eventually differentiate into myofibroblasts [78]. Under
pathological conditions, profibrotic factors such as TGF-β and hypoxia activate EndMT [79].
Notch signaling has been found to activate the expression of TGF-β and activate EndMT
[80].
It was previously believed that resident fibroblasts were the primary cells contributing
to pathologic remodeling in the injured heart. This suggestion was supported by the
observation that cardiac fibroblasts respond to paracrine and autocrine release of bioactive
molecules such as TGF-β, FGF, and TNF-α released post-injury to initiate wound healing
[76]. In various studies of replacement fibrosis, collagen synthesis and matrix deposition was
initiated by resident fibroblasts at the site of injury [5, 11, 81, 82]. Studies in a pressure-
overload mouse model showed that inhibition of the profibrotic factor TGF-β attenuated
cardiac fibroblast proliferation, ECM production, and myofibroblast activation [82].
However, more recent studies on the origin of fibroblasts in cardiac pathologies have
revealed that the source is not limited to resident cardiac fibroblast, but that there is in fact a
heterogeneous population of cells contributing to cardiac fibroblasts in CVDs.

16
Bone marrow-derived cells (BMDCs) are another important source of fibroblasts in
cardiac pathology, with studies showing that almost 30% of cells in the infarcted area of post-
MI hearts are from a BMDC population in the first 14 days. Around 28 days post-MI, the
percentage of BMDCs in the infarct and border zone was reduced to 17%, with most being
myofibroblasts, as indicated by the expression of myofibroblast marker α-SMA [83]. The
timing of these findings suggests BMDCs play an important role during the early
inflammatory phase of myocardial wound healing. Monocytes also are present in the infarct
scar. Infarcted cardiac tissue shows the presence of cells which co-express monocytic and
myofibroblast markers (S100 calcium-binding protein A4 or S100A4; αSMA) [84].
Additionally, inhibition of monocyte recruitment decreases the cardiac fibroblast population
and myocardial remodeling after myocardial infarction [85].
Circulating fibrocytes are unique fibroblast progenitors of stem cell origin that
express phenotypic similarities with leukocytes. Fibrocytes co-expresses mesenchymal and
hematopoietic markers including lymphocyte common antigen or cluster of differentiation 45
(CD45), cluster of differentiation 34 (CD34), procollagen-1, and vimentin [86]. Cultured
fibrocytes, like myofibroblasts, express α-SMA, which is enhanced by TGF-β1 treatment.
Also like myofibroblasts, cultured fibrocytes exert contractile force which in the heart to aid
in cardiac wound contracture. Fibrocyte activation and trafficking during cardiac repair is a
significant source of myofibroblasts, which are critical to wound healing [87].
Perivascular cells or pericytes have also been studied as a putative source of cardiac
fibroblasts. These cells originate from the perivascular spaces in cardiac vessels [88].
Although pericytes were found to invade the injured kidney in a renal fibrosis model [89], the
role of pericytes in cardiac fibrosis is underexplored.

17
Figure 2: Sources of cardiac fibroblasts. During embryonic development, cardiac
fibroblasts originate via epithelial to mesenchymal transition (EMT)from the pro-epicardium,
or endothelial to mesenchymal transition (EndMT) from the endocardium. In the injured
adult heart, EMT and EndMT embryonic pathways are re-activated to recruit cardiac
fibroblasts for wound repair. In cardiac pathology, sources of fibroblasts also include bone
marrow-derived progenitor cells, monocytes, fibrocytes, and perivascular cells. These various
sources of cardiac fibroblasts also contribute to subsequent fibrosis as a source of
myofibroblast precursors.

18
Cardiac Myofibroblasts
Myofibroblasts were originally identified by Gabbiani et al. as a modified fibroblast
that contributes to growth, development, and tissue repair [27]. These cells share
characteristics with both fibroblasts and smooth muscle cells (SMCs) [27, 90]. Like
fibroblasts, myofibroblasts are a heterogeneous cell type, which can originate from resident
fibroblasts, SMCs, pericytes, and BMDCs (via EMT) [65]. They can produce copious
quantities of growth factors (such as TGF-β, PDGF, and CTGF) and other bioactive
molecules. Compared to fibroblasts, myofibroblast are larger, more motile, and
hypersynthetic for ECM proteins, specifically fibrillar collagens I and III [65].
Myofibroblasts produce stress fibers that coordinate wound contraction through incorporation
of contractile proteins such as α-SMA [91]. This property helps in activation of latent TGF-β,
increased ECM synthesis, collagen fibril reorganization, and development of tensile strength
during the wound healing process [92]. In the healthy heart, myofibroblast are usually absent,
except in valve leaflets. They only appear in the myocardium after cardiac injury in the
resulting scar, which is initially required for reparative fibrosis to prevent LV rupture.
However, their persistent presence and exaggerated fibrotic response is detrimental
eventually, leading to impaired cardiac function and eventual heart failure [93].
Activation of fibroblast to myofibroblasts – a continuum of phenotype
The distinction between cardiac fibroblasts and myofibroblasts is clear-cut with
respect to function, however, the question of how these cells activate in the case of cardiac
pathology has only recently been addressed in the literature. Fibroblast activation is regulated
through various factors, such as mechanical stress, growth factors (primarily TGF-β),
vasoactive peptides (e.g.: ANGII), and cytokines (e.g.: TNF-α)and injury [94]. Cardiac
fibroblasts maintain homeostasis of cardiac ECM, however, with their prolonged activation
mayeventually contribute to cardiac fibrosis [11]. In response to mechanical stress and

19
profibrotic factors, in vitro cardiac fibroblasts are converted to an intermediate phenotype
which is sometimes referred to as the proto-myofibroblast. Proto-myofibroblasts are
distinguished from fibroblasts by the de novo expression of α-SMA and increased expression
of ED-A FN [95]. ED-A FN is one of the ECM proteins expressed in proto-myofibroblasts
and marks the transition to a mature or super-mature myofibroblast [27]. Besides elevated
expression of these markers, mature myofibroblasts have increased focal adhesion proteins
such as vinculin, talin and paxilin [47, 96].
Mechanical Stress
In the healthy hearts, fibroblasts are buffered from exposure to severe mechanical
stress by a stable ECM network [97]. Under normal conditions, fibroblasts are present in an
environment where the external force is between 4 to 18kPa [98]. Due to the
mechanosensitive nature of cardiac fibroblasts, the first stimulus that activates a phenotypic
shift of fibroblast to myofibroblast is the change in mechanical environment following injury.
Architectural integrity of the tissue is lost and resident fibroblasts are then exposed to
constant mechanical stress. In the fibrosing heart, ECM stiffness is increased to around
100kPa [99]. Cardiac fibroblasts interact with the ECM through transmembrane proteins
called integrins, which translate these mechanical signals into the intracellular response,
including activation of profibrotic signaling cascades including the TGF-β/Smad and MAPK
pathways.
Constant mechanical stress and stiffened ECM results in mechanical activation of
latent TGF-β in the cardiac ECM, which induce a vicious cycle for secretion of TGF-β that
also stimulates further collagen production [100]. Mechanical tension has been shown to
induce ANGII production, which can further enhance TGF-βsignaling. ED-A FN expression
is also upregulated by increased mechanical tension [47]. McCulloch et al. examined the
effect of force-induced α-SMA expression in rat cardiac fibroblasts, and found that culture of

20
cardiac fibroblasts on rigid substrate increases cell tensile force and is associated with
increased α-SMA expression, both suggesting conversion to the myofibroblast phenotype. A
study on human burn scar biopsies showed that when scar tissue is subjected to mechanical
tension, a high number of α-SMA positive myofibroblasts are observed in the injury
site[101]. An in vivo study has also shown that mechanical tension in tissues is correlated
with increased expression of α-SMA [101]. Expression of myofibroblast markers α-SMA and
ED-A FN, and fibroblast activation to the profibrotic myofibroblast phenotype is
mechanically regulated [102, 103]. These changes results in excessive ECM deposition and
distorted architecture, which eventually causes pathological remodeling [5].
Transforming Growth Factor-β
Cardiac fibroblasts express various receptors on their cell surface which are
associated with fibrosis such as receptors for TGF-β, ANGII and PDGF. The ANGII type 1
receptor (AT-1) can induce increased mRNA and protein levels of TGF-β, and vice versa. It
has been shown in a study, that rat cardiac fibroblasts stimulated with TGF-β responded with
a significant increase in α-SMA and collagens type I and III, as compared to untreated
controls [104]. TGF-β, in an autocrine fashion increases the constitutive expression of α-
SMA stress fibres in the activating myofibroblast, and also upregulates its own mRNA and
protein expression [101]. Both mechanical tension and stimulation by TGF-β are necessary
for developing α-SMA positive myofibroblasts [105, 106]. Profibrotic TGF-β is the primary
growth factor involved in activation of myofibroblasts in various tissues including the
myocardium, and the development and maintenance of cardiac fibrosis.
Initially, TGF-β is released into the cardiac interstitium as an inactive dimeric form by
numerous cell types including cardiac fibroblasts and myofibroblasts. This inactive form,
latent TGF-β, is bound in the ECM until activated by mechanical stress and other profibrotic
factors. The TGF-βgene produces an N-terminal region called latency associated protein

21
(LAP) that non-covalently binds with TGF-β and forms a small latency complex (SLC). Once
released into the ECM the SLC forms a disulfide linkage with the large latent TGF-β-binding
protein (LTBP) to form a large latency complex (LLC). Further activation of the latent TGF-
β requires cleavage of LAP from this complex. Various factors can induce this cleavage such
as proteases (MMPs, TSP-1, and plasmin), acidic pH, release of ROS, or mechanical stress
[101, 107]. Myofibroblast contraction mediated by β1, β3, and αvβ5 integrin binding to LAP
can directly activate latent TGF-β1 from LLC in a protease-independent manner [106].
Once activated, TGF-β1 is released from the LLC into the ECM, where it binds to its
receptor, the serine threonine kinase cell surface homodimer TβRII [105]. TβRII then
undergoes autophosphorylation and activates TβRI forming a heterotetrameric complex
[108]. Intracellular receptor Smads (R-Smads) Smad2 and Smad 3 form a complex
(Smad2/3) that binds an intracellular phosphorylation site on TβRI. This initiates a cascade of
Smad-dependent signaling, resulting in formation of a downstream Smad3 and Co-Smad
4complex. The Smad3/4 complex translocates to the nucleus and upregulates the transcription
of fibrotic gene such as collagen I&III [109]. In the post-MI heart, there is a significant
increase in R-Smad2/3 phosphorylation coinciding with nuclear Smad accumulation and
increased collagen deposition, suggesting a critical role of TGF-β/Smad signaling in
transcriptional activation of profibrotic genes such as collagen and α-SMA.
Conversely a group of Smads inhibit gene expression called I-Smads, which includes
Smad6 and -7.I-Smad 7 inhibits TGF-β/Smad signaling by disrupting activation of R-Smads
by the TGF-β1 receptor [110]. The nuclear protein Ski, which appears to regulate anti-fibrotic
effects in fibroblast to myofibroblast conversion, is another negative regulator of TGF-β,
acting as an endogenous Smad inhibitor. Ski represses Smad-mediated TGF-β signaling by
inhibiting activation of profibrotic gene promoters by Co-Smad4 in the nucleus [111, 112].
However, Ski cannot act as a transcriptional inhibitor via direct binding to DNA, but requires

22
the presence of the Smad complex [112]. Ski is also assisted by other transcriptional regulator
proteins such as Meox1, Meox2, and Zinc finger E-box-binding homeobox 2 (Zeb2). Meox1
and Meox2 are homeodomain proteins required in development of murine skeletal muscle
[113]. Both are important in fibrosis. Meox2 can be down-regulated by physical stimuli, such
as mechanically damaged arteries [114]. Anti-fibrotic transcription factor Meox2 has been
found to inhibit profibrotic TGF-β effects, specifically by inhibiting EMT [115]. Zinc finger
E-box binding homeobox 2 (Zeb2), known for its role as a co-repressor in Smad-mediated
TGF-β signaling, has been repressed by nuclear Ski over-expression. Zeb2 repression of
mesenchyme homeobox 2 (Meox2) is relieved, resulting in upregulation of Meox2. Increased
Meox2 expression inhibits activation of profibrotic genes includingα-SMA, ED-A FN, and
collagens. However, in the injured heart, Ski is translocated to the cytosol, allowing Zeb2 to
down-regulate Meox2 – inducing expression of profibrotic genes and fibroblast to
myofibroblast phenotype conversion. This mechanism shows how Ski plays a role as
antifibrotic protein during ECM remodeling post-MI [116].

23
Figure 3: Activation of cardiac fibroblasts to myofibroblasts. In the healthy heart,
extracellular matrix (ECM) components are continuously turned over through synthesis and
degradation by interstitial cardiac fibroblasts. In cardiac fibrosis, myofibroblasts play an
important role in excessive synthesis and remodeling of the cardiac extracellular matrix in
response to mechanical stress or stimulation by profibrotic factors such as transforming
growth factor-β1 (TGF-β1) and angiotensin-II (ANGII). In these pathologies, adult
fibroblasts undergo a phenotypic transition to myofibroblastic cells, and thus acquire
contractile and hypersecretory characteristics. This transition is marked by increased α-SMA,
ED-A FN, and collagens type I and III.

24
Persistence of myofibroblasts in the heart following cardiac damage
Fibroblasts migrate to the infarct zone in post-MI hearts and undergo activation to
myofibroblasts and repopulate the infarct zone after the loss of cardiomyocytes [117].
Although myofibroblasts are necessary for wound healing after ischemic injury, their
persistent presence and excess production of collagen after proper wound healing leads to the
development of cardiac fibrosis, resulting in a stiff and non-compliant heart. Persistence of
myofibroblasts in the infarct and distal regions of the post-MI heart is purportedly caused by
a failure of these cells to undergo regulated cell death through apoptosis or other mechanisms
[118]. One potential cause of this failure of myofibroblasts to cease wound healing activities
is ongoing neuroendocrine dysregulation, potentially due to high wall stress resulting from
the presence of collagen-rich scar tissue. This environment may confer intermittent bouts of
ischemia, and therefore heart remains in a constant wound healing state, which contributes to
activated and persistent myofibroblasts in the myocardium [119].
Animal Models of MI:
Rat models have dominated research in cardiac biology due to many benefits over of
mice such as low cost, ease of handling and their larger size. In heart failure models,
myocardial injury is induced by various procedures: surgical, pharmacological, or
cauterization. The surgical method was first developed by Hans Selye and modified by
Pfeffer and coworkers, where they ligated the left coronary artery [120][121]. The procedure
requires performing a left thoracotomy on the anesthetized rat. Gentle pressure is applied on
the right side of the thorax and the heart is exteriorized. The left coronary artery is ligated
between the pulmonary artery outflow tract and the left atrium and the heart is returned to its
normal position. Finally the thorax is immediately sutured and closed [122]. Left coronary
artery ligation is the most common method used to induce acute myocardial damage in rat
and other animal models. Another widely used experimental model for induction of infarction

25
is achieved by administration of isoproterenol [123]. Isoproterenol is a beta-one adrenergic
receptor (B-AR) agonist and at high doses it induces cardiac myocyte necrosis and extensive
LV dilatation and hypertrophy [124]. The third method which is not used frequently due to
the limitations of reproducibility is the cauterization method. It consists of generating
overlapping burns in exposed rat hearts by applying a 2-mm-tipped soldering iron to the
epicardium of the left ventricle [125]. This where the animals show induction of transmural
injury in the left ventricle, with a low incidence of postoperative mortality [126].
Techniques used to study post-MI cardiac structure and function
Hemodynamic studies: The post–MI rat model is subjected to an invasive technique
where an atrial catheter is used to obtain systemic and LV blood pressure from
femoral and carotid arteries, respectively. Hemodynamic studies provide information
about systolic, diastolic and mean arterial pressure (MAP); heart rate (HR) as well as
central venous pressure (CVP) [127]. In post-MI rat model a reduction in load
dependent parameters of cardiac function (e.g. SV (stroke volumes), CO (Cardiac
Output), EF, dP/dt max/min) is observed at the end of 4 weeks, compared to sham-
operated control animals. These parameters provide an evidence of structural
changes in the myocardium such as scar thinning and myocardial hypertrophy at 4
weeks post MI [128].
Echocardiography: Echocardiography is one of the most useful diagnostic tools used
for assessing parameters of cardiac remodeling in small animal models of MI [129].
Along with the advantage of being non-invasive it is reproducible and gives accurate
estimations of the cardiac structure and function [130]. M-mode echocardiography is
widely used in laboratory experimental animals to measure the dimensions of cardiac
chambers and cardiac wall motion abnormalities [131]. It can also measure HR, SV,
LVESV, LVEDV, wall thickness (LVPWd), EF % and E/A ratio (ratio of peak

26
velocity flow in early diastole (the E wave) to peak velocity flow in late diastole
caused by atrial contraction the A wave ). M-mode echocardiography has a high
temporal and spatial resolution [132].
Magnetic resonance imaging (MRI): MRI is a very efficient and non-invasive
technique used to study the changes in left-ventricular geometry and function post-MI
[133] This technique can yield information on the myocardial function, left
ventricular mass and wall thickness in various animal models [134]. MRI also
provides valuable information about cardiac remodeling post-MI without the need to
sacrifice animals. Assessment of infarct size is an important parameter when studying
LV remodeling. This is possible with the MRI and delayed enhancement imaging
technique, where a contrasting agent such as gadolinium is administered and it allows
myocardial viability and infarct size to be determined [136]. Futhermore, the
advantages of MRI include its tomographic nature with high spatial and temporal
resolution and excellent soft tissue contrast [135].
Cell death mechanisms
Cell death mechanisms not only play an important role in homeostasis for
physiological function, but are also important in disease pathogenesis. The three main
mechanisms of cell death are apoptosis, necrosis, and autophagy. The primary mechanisms of
cell death are apoptosis and necrosis, which are distinctly regulated but share some
overlapping features.
Apoptosis
Apoptosis is characterized by cell shrinkage, cell fragmentation into membrane
enclosed apoptotic bodies, and phagocytosis by nearby cells, which acts as a clean-up
operation to prevent inflammation [137]. Apoptosis has been shown to be mediated by

27
intrinsic and extrinsic pathways and is dependent on caspases [138]. Cellular ATP levels are
maintained via continuous production and low energy expenditure.
Necrosis
Necrosis is characterized by loss of cell membrane integrity, cellular and organelle
swelling, and the onset of inflammation. It releases pro-inflammatory factors that activate the
inflammasome, further perpetuating the release of pro-inflammatory interleukins [139].
Unlike in apoptosis, adenosine triphosphate (ATP) levels and energy expenditure are
dramatically reduced in necrotic cells, due to severe mitochondrial damage [140].
Autophagy
Autophagy is a catabolic quality control mechanism for turnover of long-lived proteins
and organelles in bulk through lysosomal degradation. Autophagy occurs constitutively at
low levels in eukaryotic cells to perform housekeeping functions (eg, the destruction of
dysfunctional organelles). Breakthroughs in the field of autophagy research have come from
studies in yeast (Saccharomyces cerevisiae), including the discovery of over 32 autophagy-
related Atg genes [141]. Autophagy can be up-regulated by external stressors such as
starvation, oxidative stress, or hormonal imbalance, as well as in response to internal needs
such as removal of protein aggregates. This indicates that the process of autophagy is an
important survival mechanism [142]. To date three types of autophagy have been identified:
1. Chaperone-mediated autophagy (CMA): Chaperone molecules selectively target
cytosolic or membrane proteins containing the specific amino acid KFERQ motif for
lysosomal degradation. It is unique in its selectivity and direct shuttling of components
to the lysosome. In CMA, the chaperone heat shock cognate protein of 70 kDa (hsc70)
recognizes the specific KFERQ motif present on various proteins and facilitates their
interaction with lysosomes through lysosome-associated membrane protein type 2A

28
(LAMP2A)which acts as the receptor for the hsc70-target protein complex. Additional
chaperone proteins at the lysosomal membrane (Bag1, hip, hop, and hsc40) facilitate
unfolding of the target protein, and LAMP2A multimers form a translocation complex
that allows transport of the target protein to the lysosome [143]. CMA is increased with
elevated oxidative stress in the cell and causes degradation of oxidized proteins, acting
as an important adaptive cellular response to stress conditions [144].
2. Microautophaghy: Originally observed in yeast, microautophagy is a generally non-
selective process that involves cytosolic contents being directly internalised by the
lysosomal vacuolar membrane. It is especially important in cellular survival in
response to starvation, nitrogen deprivation, or rapamycin treatment. Unfortunately,
the molecular mechanisms that mediate microautophagy and its function in
mammalian cell physiology and pathology remains elusive [145].
3. Macro-autophagy –This is the most prevalent form of autophagy (hereafter simply
referred to as autophagy). In this process the whole organelle and misfolded proteins
are engulfed by a double membrane structure to form the autophagosome, which then
fuses with lysosomes to form autophagolysosomes [146, 147].
Autophagy is divided into four major steps. These are nucleation, elongation, closure, and
maturation, as outlined:
1. Nucleation: Also termed the initiation step or membrane isolation step, nucleation and
assembly of the initial phagophore (also called an isolation membrane) is initiated
usually by mechanistic target of rapamycin (mTOR) repression in response to nutrient
deprivation. One of the initial steps of nucleation involves activation of the class III
phosphatidylinositol 3-kinase (PI3K) vacuolar protein sorting 34 (Vps34). This
requires formation of a multiprotein complex composed of Vps34, a myristoylated
serine/threonine kinase p150, Atg14, and Beclin-1 [148]. Formation of this complex

29
induces formation of the phagophore [149, 150]. This step can be inhibited chemically
with 3-methyladenine [151].
2. Elongation: Involves the formation of a double-membrane vesicle structure through
two evolutionarily conserved (in both yeast and mammals) ubiquitin-like conjugation
systems. The first mechanism requires covalent conjugation of Atg12 to Atg5,
facilitated by E1-like Atg7 and E2-like Atg10, which is organized into a complex with
Atg16 (the Atg5-12-16 complex). The second mechanism involves
phosphatidylethanolamine (PE) conjugation with LC3 through protease action of
Atg4, Atg7, and E2-like Atg3. Proteolytic cleavage of the C-terminal region of LC3
by Atg4 converts it to LC-3I. LC-3I further conjugates with phosphatidyl
ethanolamine (PE) with the help of two ubiquitin-like modifier-activating (E-like)
enzymes Atg7 and Atg3. This lipid conjugation converts soluble LC3 to LC-3II (or
LC3-PE), the autophagy vesicle-associated form[152]. The fusion of this complex
with the membrane results in vesicle elongation.
3. Closure: Lipidated LC3 (eg,: LC-3II) and the associatedAtg5-12-16 complex
facilitatethe closure of the autophagosome and interact with an adapter protein p62.
p62 directly binds both poly- or mono-ubiquitinated proteins via its ubiquitin-
associated (UBA) domain [153, 154], and links the ubiquitinated cargos to the
autophagy machinery for autophagic degradation.LC3-II is the ‘gold standard’ used to
measure autophagy flux (i.e., the rate of formation of autophagosomes)[155-157].
4. Maturation: After phagosome formation, they undergo maturation through lysosomal
fusion forming autolysosomes. This final step of autophagy is governed by
translocation and fusion of autophagosomes with lysosomes. In yeast, maturation
occurs with the assistance of multiple proteins like SNAREs and UVRAG to form
autolysosomes [158]. In mammals, this process is not well understood, but involves

30
LAMP-2 and the GTP-binding protein Ras-related GTP-binding protein 7 (Rab7)
[159, 160]. Autolysosomes sequester damaged proteins and organelles, LC3 is
cleaved off by Atg4, recycled with other components, and subsequently degraded
such that its building blocks can be reused (see Figure 4) [161].
Apart from the role of Atg proteins, the cellular cytoskeletal system and secretory
pathways are also important in autophagy. Certain guanosine triphosphate (GTP) exchange
factors (such as Sec12 and Sec16,and two coatomer subunits of the coat protein II (COPII)
coat, Sec23 and Sec24) are needed for the biogenesis of autophagosomes [162]. Microtubules
are involved in autophagy in higher eukaryotes, as evidenced by inhibition of autophagosome
formation with the microtubule depolymerizing drug nocodazole in rat hepatocytes[163].In
mammalian cells, autophagosomes are formed at random locations in the cell and transported
toward the nucleus by the motor protein dynein, which requires the tubulin deacetylase
histone deacetylase 6 (HDAC6) for autophagic clearance of cellular components [164, 165].
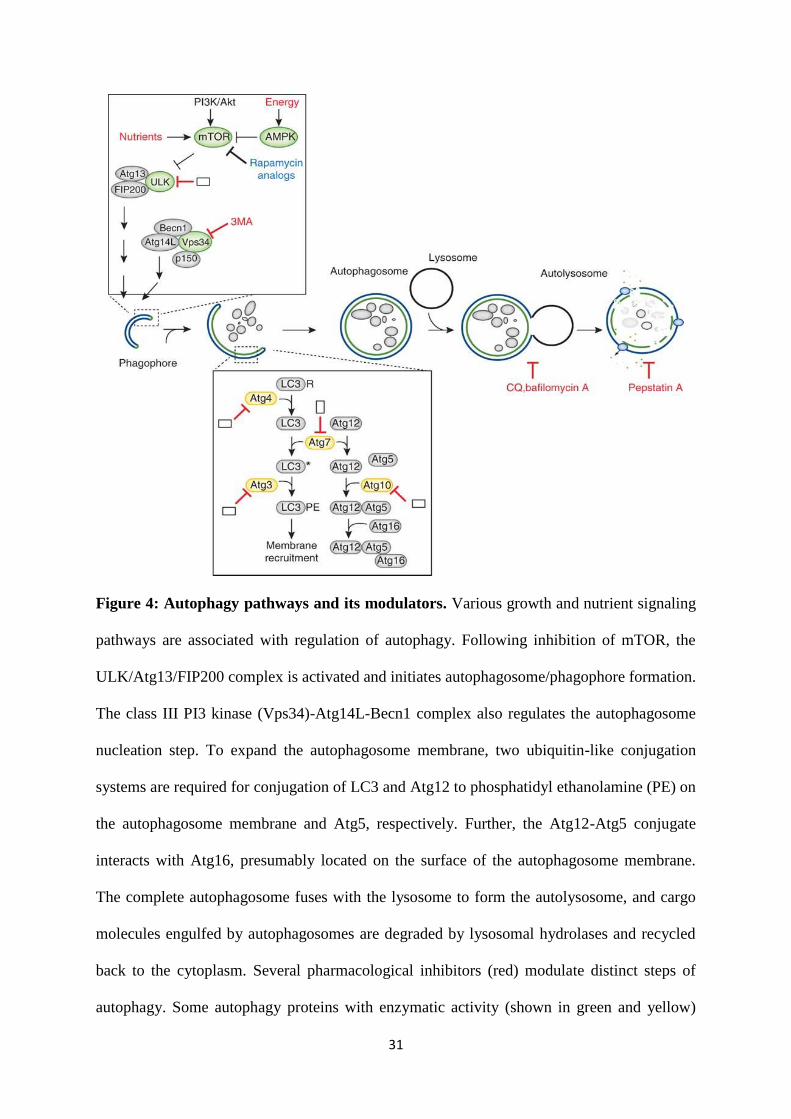
31
Figure 4: Autophagy pathways and its modulators. Various growth and nutrient signaling
pathways are associated with regulation of autophagy. Following inhibition of mTOR, the
ULK/Atg13/FIP200 complex is activated and initiates autophagosome/phagophore formation.
The class III PI3 kinase (Vps34)-Atg14L-Becn1 complex also regulates the autophagosome
nucleation step. To expand the autophagosome membrane, two ubiquitin-like conjugation
systems are required for conjugation of LC3 and Atg12 to phosphatidyl ethanolamine (PE) on
the autophagosome membrane and Atg5, respectively. Further, the Atg12-Atg5 conjugate
interacts with Atg16, presumably located on the surface of the autophagosome membrane.
The complete autophagosome fuses with the lysosome to form the autolysosome, and cargo
molecules engulfed by autophagosomes are degraded by lysosomal hydrolases and recycled
back to the cytoplasm. Several pharmacological inhibitors (red) modulate distinct steps of
autophagy. Some autophagy proteins with enzymatic activity (shown in green and yellow)

32
could be crucial target proteins for modulation of autophagy. Points where inhibitors could be
potentially developed are shown as blank boxes. CQ, chloroquine. Used with permission from
Heesun C et al., Nature Biotechnology, 2012.

33
Signaling pathways in regulation of autophagy
PI3K-AKT/mTOR signaling
Growth factors, G-protein coupled receptors (GPCR) and tyrosine kinase receptors on
the membrane activate the PI3K/Akt signaling pathway which induces cell growth,
differentiation and survival. PI3K/Akt signaling activates mTOR, which is a negative
regulator of autophagy and thus promotes survival [166]. mTOR is an evolutionarily
conserved serine/threonine protein kinase belonging to class I of the PI3K family that
regulates cell growth and metabolism as well as regulating autophagy [167-169]. Two
functionally distinct complexes are formed by mTOR: the autophagic regulator mTOR
complex 1 (mTORC1), and cell metabolism and cytoskeleton regulator mTOR complex 2
(mTORC2) [168]. In response to the nutrient environment, mTORC1 either down-regulates
autophagy (nutrient-rich environment) or upregulates autophagy (nutrient
deprivation).mTORC1 is composed of mTOR, regulatory-associated protein of mTOR
(Raptor), G protein beta subunit-like (GβL), and DEP domain-containing mTOR-interacting
protein (DEPTOR). This complex is directly inhibited byrapamycin [170]. During nutrient
deprivation, mTORC1 function is inhibited, allowing activation of autophagy through the
PI3K-adenosine monophosphate-activated protein kinase (AMPK)/mTOR axis [171, 172]. In
mammals, mTOR/Raptor-dependent signalling is mediated through amino acids via
activation of class III PI3K, while growth factors mediate this action through class I PI3K
[173, 174].
Amino acid activation acts downstream of phosphatases and endocytic trafficking
regulated by upstream mTORC1 kinase, Ste-20-related mitogen-activated protein kinase
kinasekinasekinase 3 (MAP4K3) and Vps34 [170, 175]. In addition, the GTPases
recombinant activating gene protein (Rag) and Ras homolog enriched in brain (Rheb) are also
important amino acid-dependent regulators of mTORC1. Amino acid-rich conditions favour

34
the active conformation of the Rag heterodimer (RagA/BGTP
-RagC/DGDP
) which interacts
with Raptor to translocate mTORC1 to the lysosome [176, 177]. The Rag heterodimer also
interacts with LAMP2 and Rheb, promoting further activation of the mTORC1 signaling
pathway and repression of autophagy. The tuberous sclerosis complex (TSC) proteins 1 and 2
(TSC1 and TSC2)serve as a checkpoint for autophagy by forming a TSC1/2 complex to
inhibit Rheb, decreasing mTORC1 activity and resulting in activation of autophagy [169,
171]. However, PI3K signaling will inhibit TSC1/2 by phosphorylation, allowing Rheb
activation, mTORC1 activity, and repression of the autophagy process [178].
The role of mTORC2, a complex comprising mTOR, GβL, rapamycin-insensitive
companion of mammalian target of rapamycin (RICTOR), and mSIN1, is less understood in
autophagy. Unlike mTORC1, mTORC2 is not directly inhibited by rapamycin.mTORC2acts
as a negative feedback activator and helps in inducing autophagy. mTORC2 is directed
towards Akt through action of the RICTOR subunit, and further to forkhead box O3 (FoxO3),
which is blocked by Akt [179, 180]. PI3K/pAKT does not regulate autophagy only via
mTORC-1; but through FoxO3 mediated transcriptional regulation of two autophagy related
genes LC3 and Bnip3 (a BH3-only protein), leading to the induction of autophagosome
formation [181].
Adenosine monophosphate kinase (AMPK) signaling
AMPK is an important regulator of autophagy. Under nutrient- and energy-deprived
conditions the ratio of adenosine monophosphate (AMP)to adenosine diphosphate (ADP)
increases, which activates AMPK viaphosphorylation by liver kinase B1 (LKB1) [182] and
calcium/calmodulin-dependent kinase kinase-b (CaMKK-b) [183]. Activated AMPK acts as
an upstream regulator of mTOR-1 and initiates autophagy by repressingTSC2 activity [184].
Activated AMPK phosphorylates elongation factor-2 kinase, balancing peptide elongation
and autophagy [185]. The unc-51 like autophagy activating kinase 1 (ULK1) complex

35
comprises Atg13, focal adhesion kinase (FAK) family interacting protein of 200 kDa
(FIP200) and Atg101 [186], which is phosphorylated by the AMPK/mTOR energy sensing
pathway and represses autophagy induction [187].
Hypoxia inducible factor-1α
Under conditions of hypoxia and oxidative stress the hypoxia-inducible factor-1α
(HIF1α) transcriptional factor accumulates in the nucleus and controls expression of various
genes that are important to metabolic adaptation [188]. Two important genes induced under
hypoxia are B-cell lymphoma-2/adenovirus E1B 19 kDa protein-interacting protein 3
(BNIP3) and BNIP3L. Protein products of there genes have been shown to increase
mitochondrial autophagy and thus reduce ROS production [189, 190]. BNIP3 and the protein
regulated in development and DNA damage responses 1 (REDD1) negatively regulate
mTORC1 activation through binding of the small GTPase Rheb and activation of the TSC
complex, respectively [191, 192]. Additionally, BNIP3 competes with Beclin-1 for B-cell
lymphoma2 (Bcl-2) binding, thus promoting autophagosome formation. The proteins
involved in autophagy and their functions are found in Table 1.
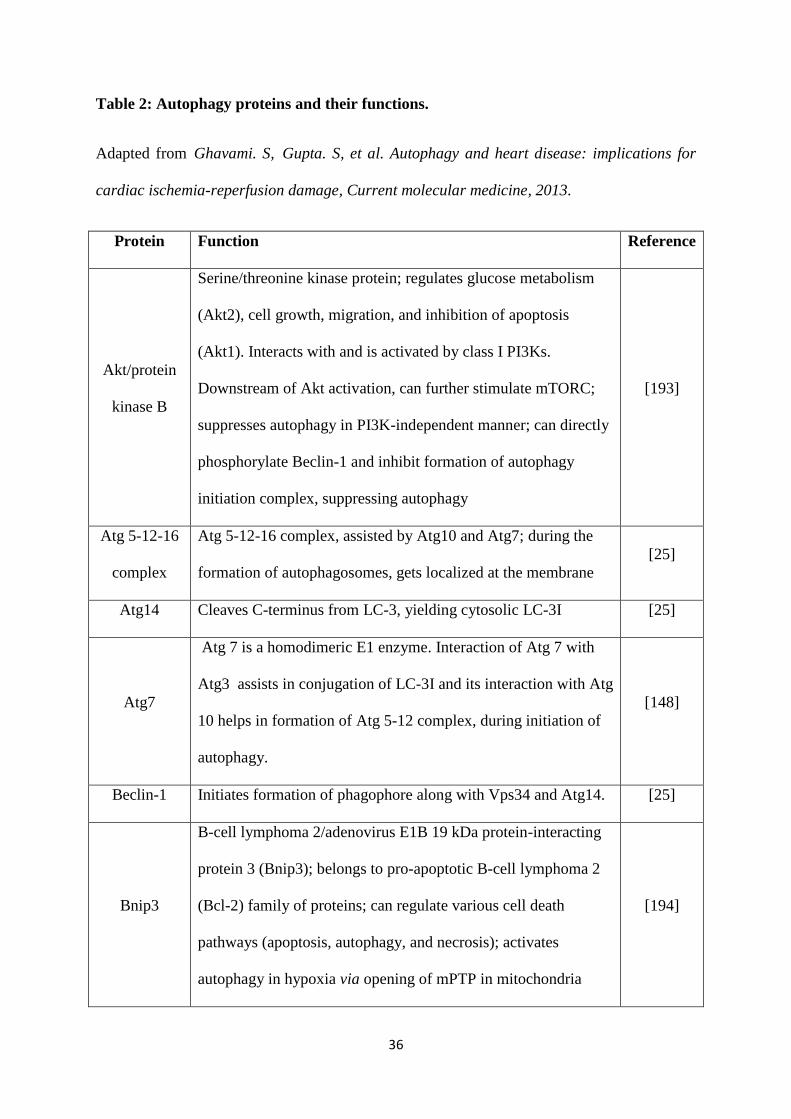
36
Table 2: Autophagy proteins and their functions.
Adapted from Ghavami. S, Gupta. S, et al. Autophagy and heart disease: implications for
cardiac ischemia-reperfusion damage, Current molecular medicine, 2013.
Protein Function Reference
Akt/protein
kinase B
Serine/threonine kinase protein; regulates glucose metabolism
(Akt2), cell growth, migration, and inhibition of apoptosis
(Akt1). Interacts with and is activated by class I PI3Ks.
Downstream of Akt activation, can further stimulate mTORC;
suppresses autophagy in PI3K-independent manner; can directly
phosphorylate Beclin-1 and inhibit formation of autophagy
initiation complex, suppressing autophagy
[193]
Atg 5-12-16
complex
Atg 5-12-16 complex, assisted by Atg10 and Atg7; during the
formation of autophagosomes, gets localized at the membrane
[25]
Atg14 Cleaves C-terminus from LC-3, yielding cytosolic LC-3I [25]
Atg7
Atg 7 is a homodimeric E1 enzyme. Interaction of Atg 7 with
Atg3 assists in conjugation of LC-3I and its interaction with Atg
10 helps in formation of Atg 5-12 complex, during initiation of
autophagy.
[148]
Beclin-1 Initiates formation of phagophore along with Vps34 and Atg14. [25]
Bnip3
B-cell lymphoma 2/adenovirus E1B 19 kDa protein-interacting
protein 3 (Bnip3); belongs to pro-apoptotic B-cell lymphoma 2
(Bcl-2) family of proteins; can regulate various cell death
pathways (apoptosis, autophagy, and necrosis); activates
autophagy in hypoxia via opening of mPTP in mitochondria
[194]
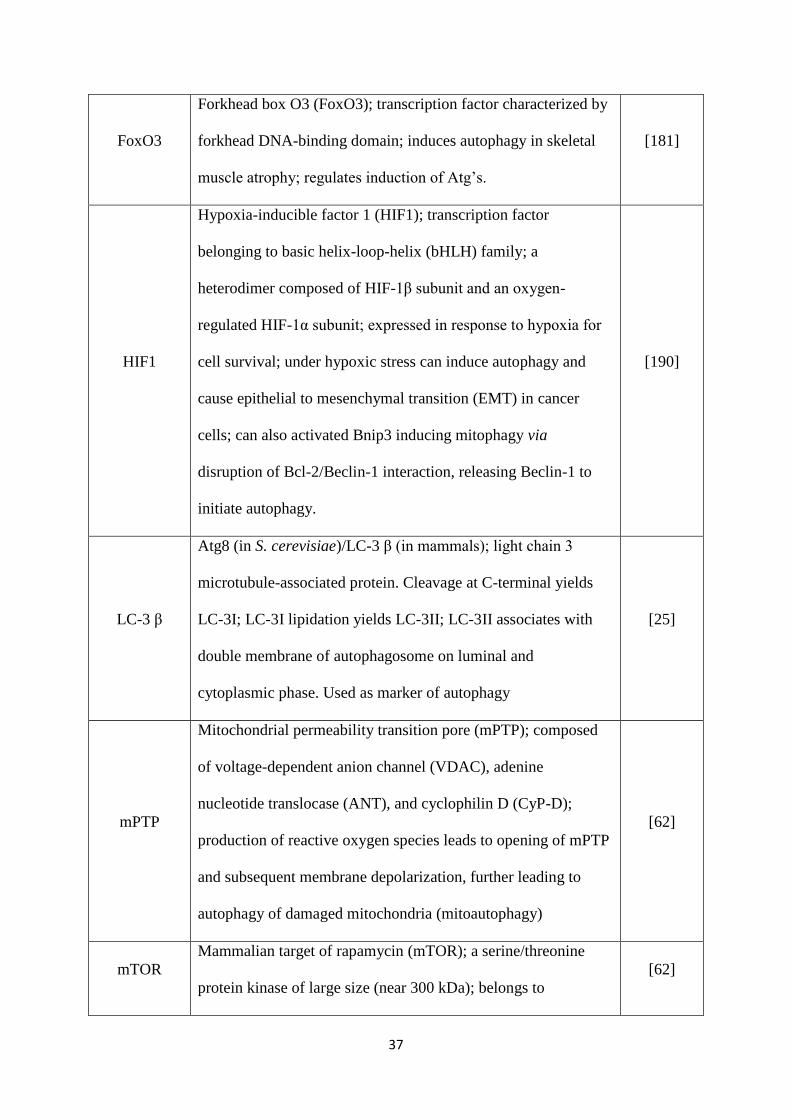
37
FoxO3
Forkhead box O3 (FoxO3); transcription factor characterized by
forkhead DNA-binding domain; induces autophagy in skeletal
muscle atrophy; regulates induction of Atg’s.
[181]
HIF1
Hypoxia-inducible factor 1 (HIF1); transcription factor
belonging to basic helix-loop-helix (bHLH) family; a
heterodimer composed of HIF-1β subunit and an oxygen-
regulated HIF-1α subunit; expressed in response to hypoxia for
cell survival; under hypoxic stress can induce autophagy and
cause epithelial to mesenchymal transition (EMT) in cancer
cells; can also activated Bnip3 inducing mitophagy via
disruption of Bcl-2/Beclin-1 interaction, releasing Beclin-1 to
initiate autophagy.
[190]
LC-3 β
Atg8 (in S. cerevisiae)/LC-3 β (in mammals); light chain 3
microtubule-associated protein. Cleavage at C-terminal yields
LC-3I; LC-3I lipidation yields LC-3II; LC-3II associates with
double membrane of autophagosome on luminal and
cytoplasmic phase. Used as marker of autophagy
[25]
mPTP
Mitochondrial permeability transition pore (mPTP); composed
of voltage-dependent anion channel (VDAC), adenine
nucleotide translocase (ANT), and cyclophilin D (CyP-D);
production of reactive oxygen species leads to opening of mPTP
and subsequent membrane depolarization, further leading to
autophagy of damaged mitochondria (mitoautophagy)
[62]
mTOR
Mammalian target of rapamycin (mTOR); a serine/threonine
protein kinase of large size (near 300 kDa); belongs to
[62]

38
phosphatidylinositol kinase-related kinase (PIKK) family.
Activity of mTOR is inhibited under nutrient starvation, a
critical step for autophagy induction in eukaryotes
mTORC1
mTOR complex 1; contains Raptor (KOG1 ortholog),
GβL/mLst8, proline-rich Akt substrate of 40 kDa (PRAS40) and
DEP domain-containing mTOR-interacting protein (DEPTOR);
key regulators of translation and ribosome biogenesis in yeast
and mammalian cells (respectively); responsible for autophagy
induction in response to starvation.
[62]
mTORC2
mTOR complex 2; contains Rictor (Avo3 ortholog), GβL/mLst8,
Sin1 (Avo1 ortholog), proline-rich protein 5 (PRR5/protor), and
DEPTOR. Rictor is a rapamycin insensitive companion of the
mTORC2 complex; mTORC2 is involved in regulating the
phosphorylation and activation of Akt/protein kinase B and
protein kinase C.
[62]
p62
Ubiquitin-binding scaffold protein; acts as cargo for localization
of ubiquitinated protein aggregates to autophagosomes for
degradation. Accumulates with inhibition of autophagy; used as
marker of autophagic flux.
[66]
PI3K
Signal transducer enzymes; phosphorylate the third hydroxyl
group of inositol ring of membrane-bound phosphatidylinositols.
Four major classes, based on structure: class I, II, III, and IV.
Class I substrates important for downstream Akt/mTOR
signaling
[195]
Raptor Regulatory associated protein of mTOR1 complex; an [180]
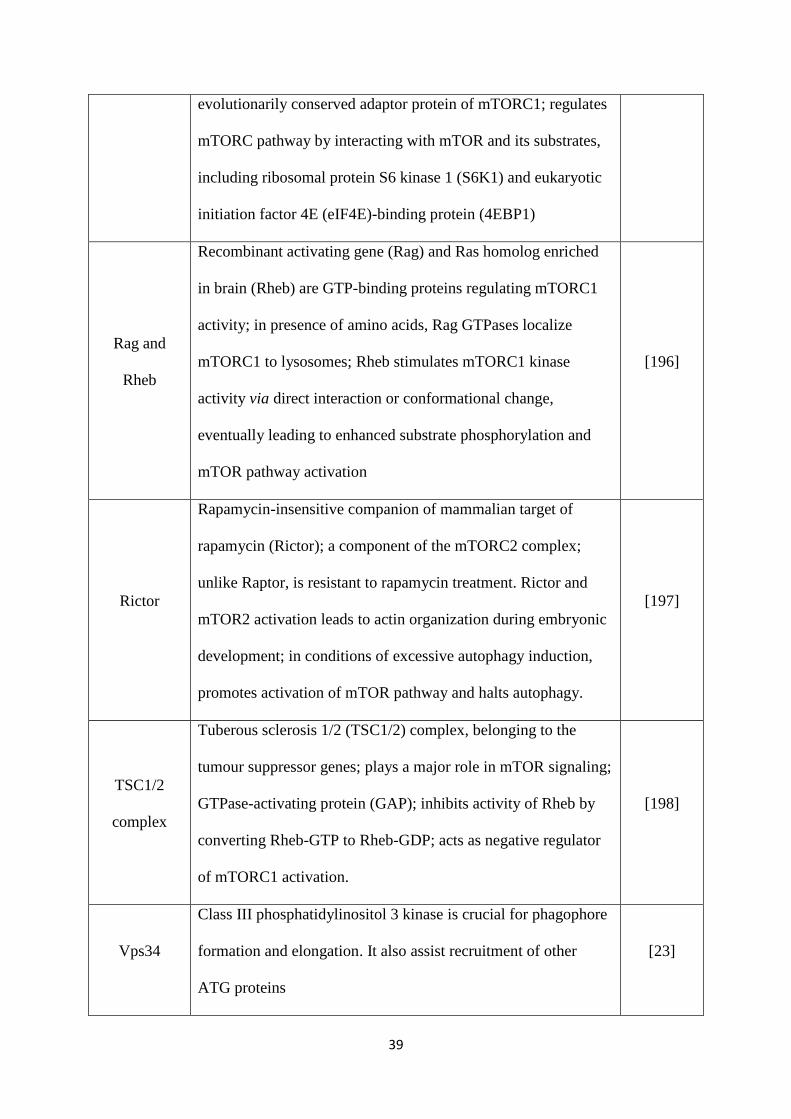
39
evolutionarily conserved adaptor protein of mTORC1; regulates
mTORC pathway by interacting with mTOR and its substrates,
including ribosomal protein S6 kinase 1 (S6K1) and eukaryotic
initiation factor 4E (eIF4E)-binding protein (4EBP1)
Rag and
Rheb
Recombinant activating gene (Rag) and Ras homolog enriched
in brain (Rheb) are GTP-binding proteins regulating mTORC1
activity; in presence of amino acids, Rag GTPases localize
mTORC1 to lysosomes; Rheb stimulates mTORC1 kinase
activity via direct interaction or conformational change,
eventually leading to enhanced substrate phosphorylation and
mTOR pathway activation
[196]
Rictor
Rapamycin-insensitive companion of mammalian target of
rapamycin (Rictor); a component of the mTORC2 complex;
unlike Raptor, is resistant to rapamycin treatment. Rictor and
mTOR2 activation leads to actin organization during embryonic
development; in conditions of excessive autophagy induction,
promotes activation of mTOR pathway and halts autophagy.
[197]
TSC1/2
complex
Tuberous sclerosis 1/2 (TSC1/2) complex, belonging to the
tumour suppressor genes; plays a major role in mTOR signaling;
GTPase-activating protein (GAP); inhibits activity of Rheb by
converting Rheb-GTP to Rheb-GDP; acts as negative regulator
of mTORC1 activation.
[198]
Vps34
Class III phosphatidylinositol 3 kinase is crucial for phagophore
formation and elongation. It also assist recruitment of other
ATG proteins
[23]

40
Autophagy in Development and Differentiation
Autophagy is a dynamic process important in cell death and contributes to
development and differentiation [199]. During oocyte preimplantation, oocytes undergo
autophagy four hours post-fertilization in response to calcium oscillations wherein autophagy
is important for activating transcription and also for elimination of excess maternal proteins
[200, 201]. During the neonatal stage all tissues except the brain exhibit autophagy because
of starvation, as the maternal supply of nutrients from the placenta is ended. In this stage,
autophagy allows amino acids and other energy resources to be provided to the neonate
through activation of low energy-sensing AMPK and subsequent induction of various Atg
proteins [202]. The critical role of autophagy in development is evidenced by embryonic
lethality caused by Atg5 mutation and neonatal lethality in response to mutations of Atg3, 7,
9, and 16 [199]. Autophagy also plays an important role in cell differentiation, including
erythrocyte development, for removal of mitochondrial and other sub-cellular organelles
[203], in T and B lymphocyte differentiation [204], and in differentiation of adipocytes [205].
In terminally differentiated cells such as neurons and cardiac cells, autophagy processes
maintain cellular homeostasis. In the cardiovascular system, an optimal level of autophagy
has been found to be is critical within the following cell types- endothelial cells, vascular
smooth muscle cells, fibroblast, macrophages, and in cells of the myocardium. Excessive or
insufficient autophagy within these cell types can contribute to cardiac pathologies [206].
The zebrafish has been a model organism for most of the work demonstrating the role
of autophagy in cardiac development, with very few reports in the hearts of developing mice.
Studies in zebrafish demonstrated that morpholino and Atg mutations result in defective
cardiac and vascular development [207]. FGF is an important growth factor in cardiac
development which regulates cardiac stem cell differentiation to cardiomyocytes. In the

41
presence of FGF, autophagy is maintained at a low level whereas in its absence, autophagy is
highly induced, resulting in induction of cardiomyocyte differentiation. FGF prevents
premature differentiation of cardiac progenitor cells and regulates heart development during
embryogenesis [208, 209]. Autophagy in cardiac fibroblasts is induced either by serum
deprivation or β2 adrenergic stimulation, which can eliminate misfolded, trimeric forms of
type I pro-collagen produced in error by these cells [210, 211].
Autophagy in Disease Pathogenesis
Autophagy contributes to homeostasis and regulates various physiological processes
by mobilizing intracellular energy sources, degrading entire organelles such as mitochondria,
peroxisomes, ER and by preventing accumulation of altered or misfolded protein that might
otherwise be toxic and might trigger apoptosis [212]. It is the major lysosomal pathway for
the nonselective delivery of cytoplasmic components during periods of starvation or stress
[213, 214]. In certain other conditions autophagy can induce selective digestion of specific
organelles like mitochondria and peroxisomes [215]. There is evidence to support selective
autophagy Atg7-deficient mouse liver displays defects in peroxisome removal, and
chemically-induced endoplasmic reticulum stress leads to endoplasmic reticulum membrane
autophagy disease [216]. Dysregulated autophagy may contribute to the pathogenesis of
diseases [217]. Deficiencies in the autophagic pathway have been linked to several
neurodegenerative diseases such as Huntington’s, Parkinson’s and Alzheimer’s diseases
[218]. Autophagy has also shown to play role in cancer progression [219, 220] and infectious
diseases [221].
In most disease models and histopathological samples of diseased tissue, increased
autophagosome formation has been observed. However, this could be either because of
decreased removal or increased formation of autophagosomes, which relies on the complex
interplay of the various cellular regulators and activators of autophagy [222]. Defects in

42
autophagosome lysosome fusion and genetic or functional alterations causing impaired
delivery of autophagosomes to the lysosome are also implicated in disease pathogenesis. It is
thus possible that impaired autophagic clearance may contribute to the pathogenic
mechanism, for example in dynein mutations causing motor neuron diseases [165].
Accumulation of protein aggregates due to maladaptive autophagy is yet another player
initially correlated in neurodegenerative diseases, but later observed in other diseases as well
[223, 224]. Failure of autophagosomes fusion due to LAMP2 mutations or accumulation of
protein aggregates has also been observed in myodegenerative diseases [222].
Several studies in systemic and tissue-specific Atg mutations in mice have implicated
this protein in maladaptive autophagy causing cancer, and neurodegenerative diseases [225,
226]. Atg5- and Atg7-knockout mice display muscle cell deterioration and progressive
muscle weakness [227, 228]. Atg7-knockout mice also show reduced autophagy, resulting in
pancreatic β-cell degeneration and reduced insulin secretion [229, 230]. Reduced Beclin-1
expression in the brain is correlated with the increased occurrence of aging-related
neurodegeneration [231]. Atg mutations also produce gastrointestinal abnormalities similar to
those found in Crohn’s patients, including Paneth cell dysfunction and impaired granule
secretion [232, 233].
Autophagy in Cardiovascular Diseases
All the above literature signifies the role of autophagy in development, differentiation,
normal physiological processes, and in disease pathogenesis. The heart is critical in
maintaining homeostasis and thus the role of autophagy in cardiovascular pathology takes on
a modicum of importance. In the heart, autophagy serves a predominantly pro-survival
function by removing protein aggregates and damaged organelles under basal condition
[234]. The finding that Atg5 deficiency in murine hearts results in rapid LV dilatation and
contractile dysfunction underlines the important role autophagy plays in maintaining cellular

43
homeostasis in the myocardium [235]. Studies have demonstrated that constitutive autophagy
under basal conditions in the heart is an important mechanism for maintaining cardiomyocyte
size, overall myocardial structure, and proper morphology of mitochondria [236]. The heart
has a high rate of blood flow and contains more mitochondria compared to any other tissue of
the body, which implies a high rate of aerobic respiration for energy generation. Following
myocardial infarction, the heart will have compromised oxygen and nutrient availability due
to ischemia, resulting in upregulation of autophagy to ensure the availability of energy
substrates, recycling amino acids and free fatty acids for energy production [237]. In mice,
caloric restriction is also an inducer of autophagy post-MI, increasing ATP content and
reducing ventricular dilatation [238]. During cardiac ischemia and reperfusion, autophagy can
lead to cell death [239], and is frequently encountered in patients with coronary artery
disease, aortic vascular disease, and congestive heart failure. In models of MI, ischemia
reperfusion injury, and heart failure, adaptive and maladaptive autophagy appears to play an
important role in disease progression [240]. Reperfusion is the next step after long term
ischemia, an attempt to restore the availability of oxygen. However, reperfusion enhances
ROS generation, leading to the opening of the mitochondrial permeability transition pore
(mPTP) and permeabilization of the outer mitochondrial membrane by Bcl-2-like protein 4
(BAX)/Bcl-2-homologous antagonist/killer (BAK) (Bcl-2 pro-apoptotic proteins)[241, 242].
Opening of mPTP also allows an influx of solutes and water, disrupting the mitochondrial
proton gradient and causing depolarization of the mitochondrial membrane. As a result,
mitochondria are unable to generate ATP, and needed to be removed by autophagosomes in a
process known as a mitophagy, which prevents a potentially catastrophic loss of ATP.
Maintenance of ATP levels is one of the important survival mechanisms during ischemia
[241]. To protect from cell death, mitochondrial proteins are ubiquitinated by proteins
phosphatase and tensin homolog (PTEN)-induced putative kinase-1 (PINK-1) and the E3

44
ligase Perkin, allowing their interaction with the cargo protein p62. The complex of p62 and
ubiquitinated mitochondrial proteins is then sequestered by the autophagosome [243, 244].
Although these events prevent immediate cell death, they eventually cause
hypertrophic myocardial growth, increased afterload, and loss of contractile function,
resulting in heart failure. The magnitude of cardiomyocyte hypertrophy is correlated with
activation of autophagic flux. In mice, cardiomyocyte-specific restricted Beclin-1
expressionleads to increased autophagy, increased pathological remodeling, and early
mortality [245], which can be rescued by inducing haploinsufficiency of Beclin-1. Similarly,
HDAC inhibitors attenuate autophagy and blunt cardiomyocyte hypertrophy [246]. Treatment
of cultured neonatal cardiomyocytes with 3MA suppresses autophagy following ischemia
reperfusion and improves cell viability [247]. Similarly, in a rat model of doxorubicin-
induced cardiomyopathy, 3MA treatment is associated with decreased presence of autophagic
vacuoles and significant rescue of cardiac function [248]. The transcription factor GATA4, a
negative regulator of autophagy-related genes, has also been effective in inhibiting
doxorubicin-induced cardiomyopathy [249].
While this recent work indicates that inhibition of autophagy is detrimental in CVD,
and thus supports a maladaptive role of autophagy in disease progression, there are several
other observations where autophagy acts as a protective mechanism. Knockout of Atg5
induces ventricular enlargement and cardiac dysfunction, suggesting autophagy plays a
compensatory mechanism during heart failure. In ischemia, activation of AMPK and its
inhibition of mTOR induce autophagy. Inhibition of autophagy by overexpression of a
dominant negative AMPK, overexpression of Rheb, and overexpression of a dominant
negative form of glycogen synthase kinase 3β, or deletion of one allele of glycogen synthase
kinase-3β results in increased infarct size [250-252].

45
Although animal and cell culture studies provide evidence for both adaptive and
maladaptive roles of autophagy in the heart, most clinical data supports a maladaptive role of
autophagy in disease progression [253]. In two different studies examining explanted hearts
from patients undergoing partial ventriculectomy and transplantation, respectively,
researchers observed numerous autophagic vacuoles within degenerated cardiomyocytes,
signifying autophagy as a prominent source of cardiomyocyte death [254, 255]. Most studies
of autophagy in cardiac diseases have only examined its role in cardiomyocytes. In contrast,
the role of autophagy in fibroblasts, which play a crucial role in heart remodeling, have not
been evaluated much in this context. Recent evidence indicates autophagy may favor fibrosis
by promoting activation of fibroblasts. Our lab has previously demonstrated that increased
production of TGF-β is correlated with increased fibrogenesis and upregulation of autophagy
markers in cardiac myofibroblast in both a rat model of MI as well as human cardiac
myofibroblast derived from scar tissue [256]. Other groups have identified mTORC2
signalingas an inducer of production of the profibrotic growth factor CTGF as well as
fibroblast activation [257]. Aránguiz-Urroz et al. demonstrated that autophagy is involved in
ECM turnover via intracellular degradation of collagen by specific activation of β2
adrenergic receptors [210]. Another study found that increased autophagy correlates with
reduced fibrosis after transaortic constriction, a model of cardiac pressure overload [258].
Aside from its role in cardiomyocytes, there is evidence that autophagy regulates the vascular
smooth muscle cell (VSMC) phenotype, which possesses similar characteristics to
myofibroblasts. Increased autophagy promotes loss of the contractile phenotype and
increased VSMC proliferation, affecting vascular homeostasis [259]. Impaired autophagy
contributes to endothelial dysfunction, and increased production of ROS and inflammatory
cytokines [260]. Studies correlating autophagy in fibroblast and other cell types of
cardiovascular system in disease pathogenesis are rare in the literature and thus the

46
exploration and exploitation of autophagy in this context may provide novel therapeutic
interventions.
Autophagy: A therapeutic target in cardiovascular disease
The recent insights generated in the field of cardiovascular autophagy have raised the
prospect of targeting this cellular pathway for clinical applications. As basal levels of
autophagy help in cell survival and uncontrolled autophagy contributes to pathogenesis, it is
critical to understand the fine balance between these two distinct mechanisms if they are to be
manipulated for therapeutic gain [261]. There are several pharmacological activators and
inhibitors identified as potential regulators of cardiac autophagy [262]. Rapamycin is an
immune suppressive drug known to inhibit mTOR and activate autophagy [263]. Small
molecule enhancers of rapamycin (SMERs),which act independently of the mTOR pathway,
have also been identified [264].5-aminoimidazole-4-carboxamide ribonucleotide (AICAR),
an AMPK analogue which acts upstream of mTOR, stimulates autophagy [265]. Stress
inducers like brefeldin A, thapsigargin and tunicamycin increase the formation of autophagic
vesicles via expression of Beclin-1 and LC-3II, and have been shown to provide
cardioprotection [266]. Drugs targeting mTOR-independent pathways acting on myoinositol
phosphate metabolism have also been evaluated as inducers of autophagy through decreased
inositol triphosphate (IP3) receptor activity, including lithium chloride, sodium valproate,
xestospongin B, and carbamazepine [267, 268]. The FDA has approved two activators of
autophagy, minoxidil (a K+/ATP channel opener) and clonidine (a Gisignaling activator) for
use in humans [269]. Resveratrol is an antioxidant that enhances autophagy by activating
sirtuin, and AMPK is cardioprotective [270]. Metformin, a drug used in diabetes, stimulates
AMPK-dependent autophagy and helps in diabetic cardiomyopathy [271]. Cholesterol
lowering drugs including the various statins may reduce infarct size by activating mitophagy
[272].

47
Amongst the inhibitors of autophagy, 3MA is most well-known. It acts by inhibiting
autophagosome formation through interference of formation of the Vps34/Beclin-1 complex
[148]. It also acts as a PI3K inhibitor, as do two other drugs, wortmannin, and LY294002,
also found to inhibit autophagy [273]. As PI3K signalling is pleotropic in nature and involves
different classes of enzymes, some of which can activate autophagy, the context of its use is
of dire importance in determining its efficacy as a therapeutic. 3MA inhibits the activity of
p38 mitogen-activated protein kinase (MAPK) and c-Jun N-terminal kinase (JNK) as well
[274]. The histone deacetylase inhibitor trichostatin A inhibits autophagy and prevents or
reverses pathological cardiac remodeling by pressure-overload stress.
Suberoylanilidehydroxamic acid (SAHA), a different HDAC inhibitor, activates autophagy
and reduces cardiomyocyte death and infarct size after I/R [246, 275]. Similarly, granulocyte
colony-stimulating factor (G-CSF) treatment in hamsters with cardiomyopathy reverses
excessive activation of autophagy, which correlates with increased survival, improved cardiac
function, and reduced fibrosis [276]. So far, the most specific targets identified in level 3 are
inhibitors of v-ATPase (bafilomycin A1, concanamycinA). Bafilomycin A1 is a specific
inhibitor of v-ATPase in cells, and it inhibits the acidification of lysosomes and endosomes.
As early as 1998, bafilomycin A1 was reported to prevent maturation of autophagic vacuoles
by inhibiting the fusion between autophagosomes and lysosomes [277]. E64d and pepstatin A
are two autophagy inhibitors that function by suppressing lysosomal proteases. E64d is a
membrane-permeable inhibitor of cathepsins B, H, and L, whereas pepstatin A is an inhibitor
of cathepsins D and E.Lysosomotropic agents (able to penetrate the lysosome) such as
chloroquine (CQ) are also a valuable tool for blocking the late stage of autophagy [278]. As a
lysosomal lumen alkalizer (which includes chloroquine, hydroxychloroquine, NH4Cl, and
neutral red), it can also possesses anticancer properties. Lysosomotropic agents are the only
clinically relevant autophagy inhibitors widely used as anti-malarial and anti-rheumatoid

48
agents [279]. A newly developed dimeric form of chloroquine called Lys01 is a highly potent
autophagy inhibitor, ten-fold more potent that of CQ, that contains the spacer N,N-bis-(2-
aminoethyl)-methylamine as a connector between two chloroquine moieties [280]. A newer
water-soluble version called Lys05 is even more potent in its accumulation in the lysosome,
though at high doses has been associated with Paneth cell dysfunction in mice [281].
Sciarretta et al. have proposed several criteria for the selection of dugs targeting
autophagy [282]:
1. Activators or inhibitors of autophagy should be chosen based on the pathological
context.
2. The extent of autophagy modulation should be considered.
3. Autophagy is activated through different signaling mechanisms in different cardiac
diseases which should be understood.
4. Pharmacological modulators of autophagy may exert autophagy-independent functions,
which should be considered.
Targeting autophagy promises to be great therapeutic strategy to treat or prevent heart
diseases. To overcome the drawbacks of pharmacological modulators of autophagy, it is
necessary to explore all the underlying mechanisms in the contexts of CVD and the related
cellular processes involved in progressive diseases such as cardiac fibrosis.
Autophagy in activation of fibroblasts
Through secreting ECM components as well as autocrine and paracrine signaling
molecules that drive and maintain fibrosis, cardiomyocyte hypertrophy, and inflammation,
activated myofibroblasts are key drivers of cardiac fibrosis and cell death [283]. Recently, a
correlation has been established between dysregulation of autophagy and the development of
fibrosis in various organ systems, in the contexts of both up- and down-regulation of

49
autophagic processes [167]. In the liver, hepatic stellate cells were prevented from
differentiating into myofibroblast-like cells due to fibrosis-associated reduction in autophagy
[284, 285]. Autophagy was upregulated during the activation of quiescent hepatic stellate
cells (HSCs), leading to liberation of free fatty acids that undergo mitochondrial β oxidation.
In this mechanism, autophagy handles production of ATP to maintain the activated HSC
phenotype, which like myofibroblasts are hypersynthetic for ECM components and highly
proliferative, leading to hepatic fibrosis [285]. Fibrogenic kidney and lung cells have
demonstrated a reliance on autophagy in maintaining activated and fibrogenic phenotypes. In
a human embryonic fibroblast cell line, autophagy plays a critical role in activation to
fibroblasts[257]. In these cells, prolonged autophagy was associated with inhibition of
mTORC1 activation, and unanticipated augmentation of mTORC2 activation leading to
CTGF-dependent differentiation [257].The finding that inhibition of autophagy via PI3K with
LY294002, wortmannin, or 3MA prevented differentiation, as did inhibition through Atg7
silencing, indicates the critical role of autophagy in the fibroblast activation process.
However, the necessity of autophagy in activation of primary cardiac fibroblasts has not yet
been examined.

50
Rationale and Hypothesis
Rationale
The activation of cardiac fibroblasts to myofibroblasts is the hallmark event for
initiating cardiac fibrosis. As autophagy have been shown to participate in a number of
cardiac pathologies, including MI and is implicated in the induction of fibrogenesis, we
hypothesized (i) that autophagy is the causal for mechanical stress-induced activation of
cardiac fibroblasts to myofibroblasts in vitro and in vivo and (ii) that inhibition of autophagy
will abrogate the fibroblast activation in vitro, and (iii) that inhibition of autophagy in a rat
model of chronic MI will reduce the adverse cardiac remodeling of cardiac ECM in vivo.
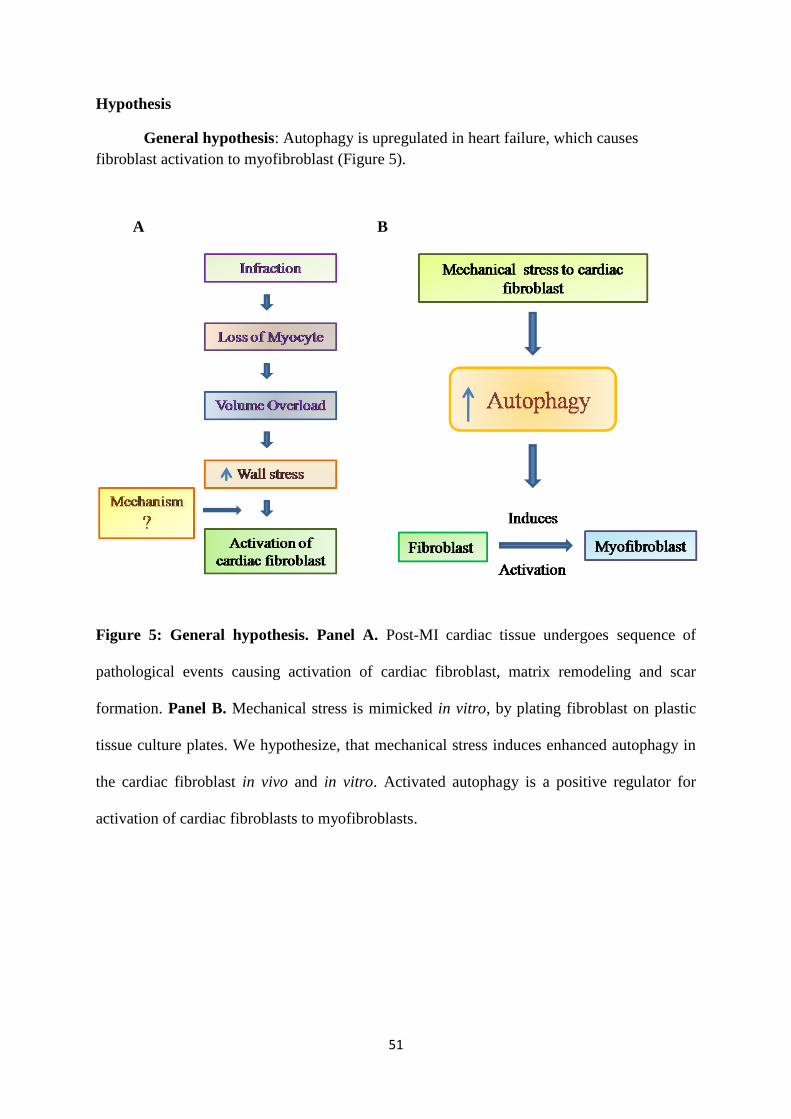
51
Hypothesis
General hypothesis: Autophagy is upregulated in heart failure, which causes
fibroblast activation to myofibroblast (Figure 5).
Figure 5: General hypothesis. Panel A. Post-MI cardiac tissue undergoes sequence of
pathological events causing activation of cardiac fibroblast, matrix remodeling and scar
formation. Panel B. Mechanical stress is mimicked in vitro, by plating fibroblast on plastic
tissue culture plates. We hypothesize, that mechanical stress induces enhanced autophagy in
the cardiac fibroblast in vivo and in vitro. Activated autophagy is a positive regulator for
activation of cardiac fibroblasts to myofibroblasts.
A B

52
Specific Hypothesis 1. Mechanical stress induces autophagy and activation of
cardiac fibroblasts in vitro
It has been established that mechanical stress both in vitro and in vivo induces
activation of cardiac fibroblasts to myofibroblasts [286], though this effect has not yet been
studied in freshly isolated P0 fibroblasts. Additionally, mechanical stress has been shown to
induce autophagy in cardiomyocytes [287], and is associated with the activation of
fibroblasts[257]. We hypothesized that mechanical stress concomitantly induces fibroblast
activation and induces autophagy in cardiac fibroblasts in vitro.
Objective 1.1: Determine the effect of mechanical stress on cardiac fibroblasts
Primary unpassaged (P0) primary rat cardiac fibroblasts were subjected to mechanical
stress by plating on non-compressible plastic tissue culture dishes. Time-dependent induction
of autophagy was examined by measuring levels of LC-3I and lipidated LC-3II at 24, 48, and
72 hours post-plating. Activation of cardiac fibroblasts to myofibroblasts was determined by
measuring protein expression of the myofibroblast marker α-SMA at 24, 48, and 72 hours.
Specific Hypothesis 2. Autophagy is necessary for mechanical stress-induced
activation of cardiac fibroblasts in vitro
Inhibition of autophagy has been shown to prevent myofibroblast conversion in
embryonic lung tissue fibroblasts [257], and TGF-β induced conversion of atrial fibroblasts
[256]. We hypothesized that inhibition of autophagic flux with the inhibitors Baf-A1, CQ,
and 3MA would prevent conversion of primary rat cardiac fibroblasts to myofibroblasts.
Objective 2.1:Determine the Effect of Inhibition of Autophagy in Cardiac Fibroblasts
To investigate the correlation of fibroblast activation of primary unpassaged P0 rat
cardiac fibroblasts and autophagy, we subjected these cells to mechanical stress by plating on
non-compressible substrate and treatment with inhibitors of autophagic flux Baf-A1, CQ, and

53
3MA at various concentrations. We then examined autophagic induction by measuring levels
of LC-3I, lipidated LC-3II, and the lysosomal cargo protein p62. Visualization of autophagy
inhibition in cardiac fibroblasts was achieved by immunofluorescence staining for LC3II, and
the presence and accumulation of autophagosomes, determined via transmission electron
microscopy (TEM).
Objective 2.2: Determine the effect of inhibition of autophagy on myofibroblast
phenotype
To determine the effect of autophagy inhibition on development of the myofibroblast
phenotype, we treated mechanically stressed fibroblasts with autophagy inhibitors for 48
hours and examined protein levels of key myofibroblast markers α-SMA and ED-A
Fibronectin, phosphorylation of p38 MAPK, and development of α-SMA stress fibers via
immunofluorescence.
Objective 2.3: Determine the effect of inhibition of autophagy on myofibroblast
function
To determine the effect of autophagy inhibition on developing myofibroblast function
in mechanically stressed cardiac fibroblasts, we treated cells with autophagy inhibitors for 48
hours and assayed cells for contractile ability (via collagen gel contraction assay) and
migratory ability (via scratch assay) at varying time-points.
Objective 2.4: Determine the effect of inhibition of autophagy on myofibroblast
function
As MAPK signaling has been shown to play an important role in fibroblast activation,
we determined the effect of mechanical stress on p38 MAPK phosphorylation levels at 24,
48, and 72 hours in the presence and absence of autophagy inhibitors.

54
Specific Hypothesis 3. Inhibition of autophagy in vivo will attenuate cardiac
fibrosis following myocardial infarction
There are numerous lines of evidence indicating that autophagy plays a role in cardiac
remodeling in the subacute stage post-injury in various animal models [288-290], though
studies are lacking in the later stages of cardiac remodeling. Thus, we hypothesized that
inhibition of autophagy in vivo will attenuate cardiac remodeling in post-MI rat hearts after 4
weeks.
Objective 3.1: Determine the effect of inhibition of autophagy on cardiac remodeling
To determine the effect of autophagy inhibition on cardiac remodeling, we utilized a
rat model of MI that employs permanent coronary ligation of the left anterior descending
(LAD) coronary artery and sham-operated control animals. Both groups were further
subdivided into those receiving daily treatment with CQ autophagy inhibitor and those
receiving control injections. We assessed post-MI function using serial echocardiography and
assessed remodeling via biochemical serum analysis and Masson’s trichrome staining for
collagen at 4 and 8 weeks post-MI.

55
Materials and Methods
Animal Ethics
Experimental protocols involving animals were reviewed and approved by the
University of Manitoba’s Animal Care Committee following the Canadian Council of Animal
Care Standards.
Cell Isolation and Culture
Cardiac fibroblasts were isolated by using retrograde Langendorff collagenase
perfusion, as previously described [291]. Juvenile male Sprague Dawley rats (150 to 200
grams) were anesthetized with a ketamine:xylazine cocktail (100 mg/kg ketamine; 10 mg/kg
xylazine) via intraperitoneal injection. Upon loss of limb reflexes, heparin (6 mg/kg) was
administered by intravenous injection in the femoral vein to prevent blood clotting. Hearts
were then excised and placed in a 100 mm cell culture dish containing 10 mL of Dulbecco’s
Modified Eagle’s Medium: Nutrient Mixture F12 (DMEM-F12; Gibco, USA) supplemented
with100 U/mL penicillin, 100 U/mL streptomycin and 1.0 μM ascorbic acid. The hearts were
cannulated to the apparatus from the aorta, and subjected to a 5-minute perfusion of DMEM-
F12 followed by a 6-minute perfusion with Minimum Essential Media Spinner’s
Modification (S-MEM; Gibco, USA) to halt myocardial contraction and promote cell
dissociation. Following S-MEM treatment, hearts were then perfused with DMEM-F12
supplemented with 0.1% (w/v) collagenase type II (Worthington Biochemical Corp., USA)
for 20 minutes to digest the tissue. The digested ventricles were then collected in a 100-mm
culture dish and digested via mincing and incubating in diluted collagenase (0.05% w/v) at
37°C for 10 min. The resulting tissue suspension was then neutralized by adding complete
growth media (DMEM-F12 supplemented with 10% fetal bovine serum/FBS, 100 U/mL
penicillin, 100 U/mL streptomycin, 1.0 μM ascorbic acid; 10% FBS DMEM-F12). The cells
were then filtered through a 40μm cell strainer (ThermoFisher Scientific, USA) to discard

56
undigested tissue; the suspension was collected in 50 mL conical tube and centrifuged at 750
x g for 7 minutes. After centrifugation, the cell pellets were re-suspended in 10 mL of
complete growth media. Lastly, each cell pellet was diluted to a final volume of 30 mL, and
10 mL aliquots were plated in 100mm dishes then incubated for 2 hours at 5% CO2, 37˚Cto
allow for cell adherence. Cell cultures were then washed twice with 1X phosphate-buffered
saline (PBS) and incubated with fresh 10% FBS DMEM-F12 media at 37˚C with 5% CO2
overnight. The next day, cultures were rinsed once with 1X PBS and fresh complete growth
media was added. Once the cultures reached 50-60% confluency they were used for
experimental assays. Unpassaged primary cells are henceforth referred to as P0 cardiac
fibroblasts.
Treatment with Autophagy Inhibitors
Three individual pharmacological inhibitors of autophagy were used in the study:
Bafilomycin A1 (Baf-A1), 3-Methyladenine (3MA), and Chloroquine (CQ) (all from Sigma,
USA).
Optimization of Autophagy Inhibitor Treatment by MTT Assay
The 3-(4, 5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide (MTT) assay
was used to determine the working concentrations of Baf-A1 and CQ and 3MA.
Approximately 5,000 isolated P0 cardiac fibroblasts were plated in 10% FBS DMEM-F12in
each well of a 96-well plate. For each autophagy inhibitor, a range of concentrations was
applied to a set of wells: 0.25 nM -1.0 nM for Baf-A1, 1.0 mM – 5.0 mM for 3MA, and 25
µM -100 µM for CQ. The assay was performed in triplicates for 24, 48 and 72 hours. After
each time point, 20 µL of MTT solution (5 mg/mL of MTT in PBS, filtered with 0.45 μM
filter) was added to each well and incubated at 37˚C for 3 hours. Further media was removed
and 200 µL of dimethyl sulfoxide (DMSO) was added and mixed via trituration. After
5minutes incubation at room temperature, relative cell proliferation was calculated by

57
measuring optical absorbance at a wavelength of 570 nm using a spectrophotometric plate
reader (Bio-Rad Laboratories, USA). The working concentrations for each autophagy
inhibitor are listed in Table 2.
Table 3: Optimized dosages for autophagy inhibitors. Dosages were determined via MTT
Assay in freshly isolated P0 primary rat cardiac fibroblasts.
Drug Name Optimized Working Concentrations
Bafilomycin-A1 0.5 nM, 0.75 nM
3-Methyladenine 1.0mM, 2.5 mM, 5.0 mM
Chloroquine 25 μM, 35 μM, 50 μM
Protein Isolation from Adherent Cell Cultures
Cells were treated with optimized doses of each drug for 48(for CQ and Baf-A1) or
72 (for 3-MA) hours. After incubation, the cells were rinsed once with 1X PBS, and
mechanically scraped on ice in 1.5 mL of cold PBS. The suspension was collected into 1.5
mL microcentrifuge tubes and centrifuged at 14000 xg for 5 minutes at 4oC. Pellets were re-
suspended in radioimmunoprecipitation assay buffer (RIPA), comprising 1% NP-40, 1.0
mMethylene glycol-bis (β-aminoethyl ether)-N,N,N',N'-tetraacetic acid (EGTA),
supplemented with phosphatase inhibitors (10 mMNaF, 1.0 mMNa3VO4) and protease
inhibitors (P8430 Protease Inhibitor Cocktail; Sigma, USA). Lysates were incubated on ice
for 1 hour, sonicated three times for 10 seconds each and centrifuged at 14,000 x g at 4oC for
15 minutes. The supernatant containing the isolated protein was transferred to fresh
microcentrifuge tubes.
Protein quantification was performed using the bicinchoninic acid method
[292].Samples were diluted 1:4 in RIPA buffer; then, 10 μL of each sample was loaded in

58
triplicate onto a 96-well plate. A set of solutions containing varying concentrations of bovine
serum albumin (BSA; Invitrogen, USA) standards was used to generate a standard curve. 200
μL of a solution of bicinchoninic acid and copper II sulphate (50:1 dilution v/v;
ThermoFisher Scientific, USA) was then added to each well and the plate was incubated at
37°C for 30 minutes. Protein concentrations were determined by measuring the absorbance of
the samples at 560 nm on an iMark microplate reader (Bio-Rad Laboratories, USA).
Western Blot Analysis
Protein samples underwent electrophoretic separation under reducing conditions using
one-dimensional 6-12% (non-gradient) sodium-dodecyl sulphate polyacrylamide gel
electrophoresis (SDS-PAGE). Lanes were loaded with 10-20 μg of protein, which were then
subjected to 150 V of electrical potential for 1.5 hours. Separated proteins were transferred to
polyvinylidene difluoride (PVDF) membranes at 300 mA for 75 minutes at 4°C. Membranes
were blocked in phosphate-buffered saline (PBS) with 0.2% Tween-20 (PBS-T) containing
10% (w/v) skim milk for 1.5 hours at room temperature. Primary antibodies were diluted in
either 3% skim milk in PBS or 5% bovine serum albumin (BSA) in Tris-buffered saline with
0.1% Tween-20 (TBS-T), as per the manufacturer’s instructions. The following primary
antibodies were used: α-SMA (1:5,000; Abcam, USA), ED-A FN (1:1,000; Millipore,
Canada), LC-3β (1:1,000; Sigma, USA), β-tubulin (1:5,000; Abcam, USA), eEF2 (1:1,000;
Cell Signaling, USA), p62 (1:1,000; Cell Signaling, USA), total-p38(1:,1000; Cell Signaling,
USA), and phospho-p38 (1:1,000; Cell Signaling, USA). Membranes were incubated with the
primary antibodies overnight at 4oC, with constant shaking. The following day, membranes
were washed three times with PBS-T, for 10 min each and then incubated with horseradish
peroxidase (HRP)-conjugated goat anti-mouse or anti-rabbit secondary antibodies (1:10,000;
Jackson Laboratories, USA) in PBS-T with 3% skim milk for 1.5 hours at room temperature,
with shaking. After 3-4 washes with PBS-T, protein bands were visualized using ECL or

59
ECL Pico (ThermoFisher Scientific, USA) per the manufacturer's instructions, and developed
on CL-Exposure X-ray film (Life Technologies, USA). Equal protein loading was confirmed
by probing for β-tubulin or eEF2. Films were scanned and digitized using a GS-8000
Densitometer (Bio-Rad Laboratories, USA) and quantified using Quantity One™software
(Bio-Rad Laboratories, USA).
Fluorescence Immunocytochemistry
Primary rat P0 cardiac fibroblasts were seeded onto 6-well tissue culture plates (~2.0
x 105 cells/well) containing glass cover slips and allowed to adhere overnight and then treated
with Baf-A1, CQ, or3MA for 48 hours. Cells were fixed with 4% (w/v) paraformaldehyde
(PFA) in PBS (pH 7.4) for 15 minutes at room temperature, followed by 15 minutes of
permeabilization with 0.1% (v/v) Triton-X 100 in PBS (Sigma, USA). Cells were washed
three times with 1X PBS and blocked for 90 minutes with 10% BSA at room temperature.
Cells were washed once with PBS and incubated overnight at 4oC with α-SMA and LC-3β
primary antibodies (1:500 in 1%(w/v) BSA in PBS). After washing 3 times for 5 minutes
each, cells were incubated with a fluorescent-tagged secondary antibody (Alexa Fluor® 555
anti-mouse or 488 anti-rabbit; Invitrogen; 1:700 in 1%(w/v) BSA in PBS for 90 minutes at
room temperature. Cover slips were mounted onto glass slides using Prolong® Gold antifade
reagent with 4’,6-Diamidino-2-phenylindole (DAPI; Molecular Probes, Life Technologies,
USA). Images were acquired using a Zeiss Axiovert 200M epifluorescence microscope and
analysed with AxioVision software (Carl Zeiss Canada, Canada).
Scratch Assay
P0 rat cardiac fibroblasts were used for cell scratch/wound healing assays. Silicone
inserts (Ibidi, USA) were placed in a 6-well culture dish. A cell suspension of 2 x 105 cells /
70 μL was prepared and added into each chamber of the inserts. The cells were allowed to
adhere and grow overnight in complete growth medium. The following day, the medium was

60
changed to low serum (2% FBS) for 24 hours and then treated with 3MA (5Mm), Baf-A1
(0.75 nM) and CQ (35 µM) for 48 hours. After 48 hours, inserts were removed and imaged
by light microscopy every 6hours until the control cells (untreated) reached 100%
confluency. Data was analyzed by comparing differences between wound width at 0, 12 and
24hours in control and treated groups using Wimasis Image Analysis software (Wimasis
Image Analysis, Germany).
Gel Contraction Assay
Collagen gels were prepared by mixing 7.0 mL of a cold collagen solution
(StemcellTechnologies, Canada) with 2.0 mL of 5X concentrated DMEM-F12 containing 100
U/mL penicillin, 100 U/mL streptomycin and 1.0 µM ascorbic acid(pH 7.4). The final
volume was adjusted to 10 mL using double distilled water (ddH2O). 600 µL of the solution
was added to each well of a 24-well plate and allowed to solidify overnight at 370C in a 5%
CO2 incubator. Approximately 2.0 x 104freshly-isolated P0 rat cardiac fibroblasts were plated
in each well with 10% FBS DMEM-F12 and were allowed to adhere for 2 hours. The cells
were rinsed twice with PBS followed by the addition of 10% FBS DMEM-F12. Cells were
allowed to grow for 24 hours in 10% FBS DMEM-F12, followed by 24 hours incubation in
low serum media (2% FBS DMEM-F12). Following serum deprivation, gels were released
using a circular cutting tool. Subsequently, the cells were divided into four groups: (i)
untreated control group, (ii) TGF-β-treated (10 ng/mL) positive control group, (iii) autophagy
inhibition withCQ (10, 25 and 35 μM), Baf-A1 (7.5 nM), or 3MA(2.5Mm), and (iv)
autophagy inhibition with TGF, CQ (10, 25 and 35 μM) + TGF-β1 (10 ng/mL), Baf-A1 (7.5
nM) + TGF-β1(10ng/mL), and 3-MA (2.5Mm)+ TGF-β1(10ng/mL) treatment groups. Images
were taken immediately post treatment (t=0 hours) as well as at 24 and 48 hours post-
treatment. Gels were analyzed using IDL Measure Gel software (University of Calgary,
Canada) to determine total gel surface area at each time point.

61
Transmission Electron Microscopy (TEM)
Cells were fixed in Karnovsky’s Fixative and the pellet was resuspended in 5%
sucrose 0.1M Sorensen’s buffer overnight at 4°C. Cells were pelleted and post-fixed with 1%
osmium tetroxide in 0.1 M Sorensen’s Phosphate Buffer for 2 hours, and dehydrated and
embedded in Embed 812 for sectioning. Semi-thin sections (1 μM) were cut from the blocks
and stained with toluidine blue for inspection, followed by sections of 200 nM which were
placed on copper grids for staining with uranyl acetate and counter staining with lead nitrate.
Imaging was done using a Philips CM10 electron microscope.
Experimental model of myocardial infarction
Adult male Sprague-Dawley rats of 250 – 300 grams body weight were used to create
an experimental model of myocardial infarction (MI). Our lab has extensive previous
experience with this technique[293].Animals were initially divided into two groups,
including(i) coronary ligated animals - animals underwent permanent surgical occlusion of
the left anterior descending coronary artery (LAD), and (ii) a sham-operated group wherein
animals underwent surgery, and the suture was introduced but was not tied, and thus we did
not occlude the LAD. Animals were then subdivided into timed groups, (i) 4-week and (ii) 8-
week post-MI, based on the time-point when the animals were to be sacrificed. To determine
the effect of autophagy inhibitory drug CQ in MI-induced animals, animals underwent
treatment with CQ (40mg/kg) for 3 weeks starting after week 1 post-surgery in the 4-week
model, and treatment for 2 weeks starting from week 6 in the 8-week model. There were 4
groups for each time-point; (i) sham; (ii) sham + CQ treatment (iii) MI, and (iv) MI+CQ
treatment, as is shown in Figure 6. Upon sacrifice, tissues were collected from distinct zones:
viable left ventricle (LV - border zone of infarct) and LV scar (infarct scar) from the ligated
(MI-induced) groups. Complete left ventricles were collected from the sham-operated groups.
Tissues for protein isolation were flash-frozen in liquid nitrogen and stored at-80oC until

62
further processing for various assays. Tissues required for histology and immunofluorescence
were frozen in Optimal Cutting Temperature (OCT) compound blocks and stored at -
80oC.Blood was also collected before and after CQ treatment, and serum was separated for
evaluation of toxicological parameters.
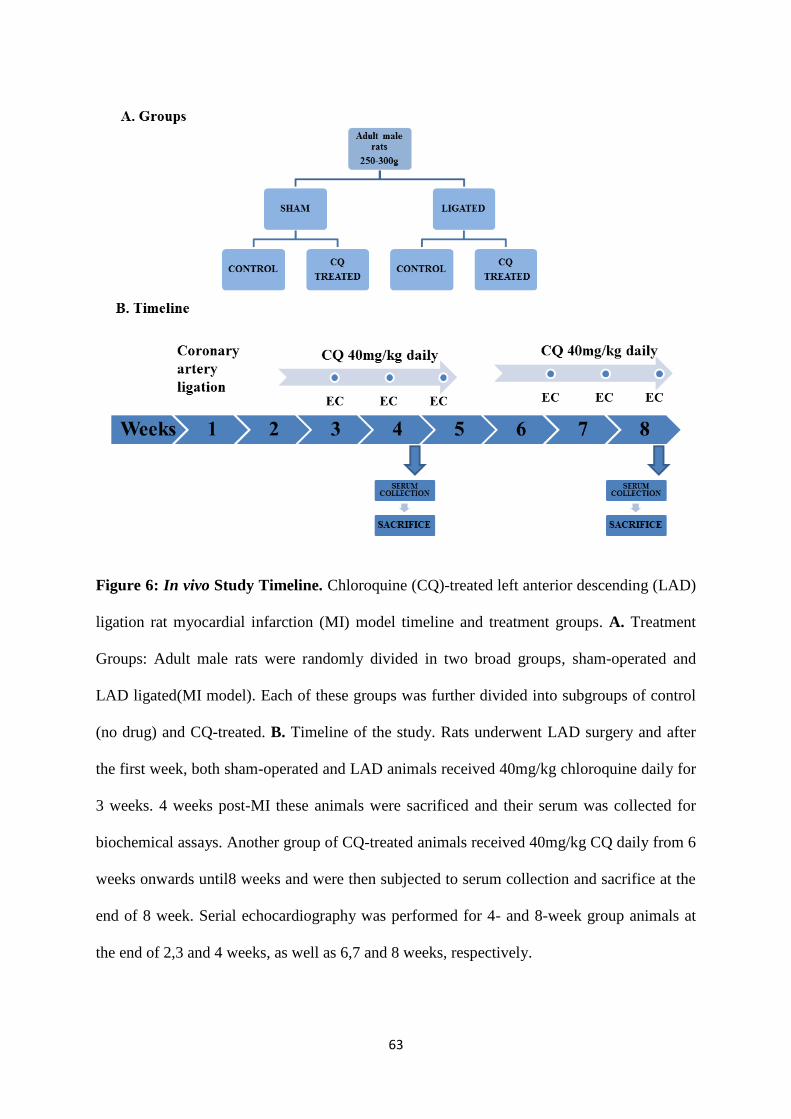
63
Figure 6: In vivo Study Timeline. Chloroquine (CQ)-treated left anterior descending (LAD)
ligation rat myocardial infarction (MI) model timeline and treatment groups. A. Treatment
Groups: Adult male rats were randomly divided in two broad groups, sham-operated and
LAD ligated(MI model). Each of these groups was further divided into subgroups of control
(no drug) and CQ-treated. B. Timeline of the study. Rats underwent LAD surgery and after
the first week, both sham-operated and LAD animals received 40mg/kg chloroquine daily for
3 weeks. 4 weeks post-MI these animals were sacrificed and their serum was collected for
biochemical assays. Another group of CQ-treated animals received 40mg/kg CQ daily from 6
weeks onwards until8 weeks and were then subjected to serum collection and sacrifice at the
end of 8 week. Serial echocardiography was performed for 4- and 8-week group animals at
the end of 2,3 and 4 weeks, as well as 6,7 and 8 weeks, respectively.

64
Echocardiography
Non-invasive transthoracic echocardiography (TTE) was performed serially on rats
post-surgery at 2, 3, and 4 weeks post-surgery in the 4-week MI-model and 5, 6,7, and 8
weeks post-surgery in the 8-week MI-model. To perform TTE, chest hairs of all the animals
were shaved with an electric razor after anesthesia with 3.0% (v/v) isoflurane gas. The
animals were maintained in this state with 2.0% isoflurane and TTE was performed using an
acoustic gel and 5.0 MHz probe (Vivid 7, GE Medical Systems, USA) in the parasternal short
axis view at the level of the papillary muscles to acquire M-mode images. EchoPAC PC
software (v.112, GE Medical Systems, USA) was used to analyze M-mode images.
Pathological Analysis of Blood Serum
Blood serum was collected from animals subject to LAD ligation or the sham
operation, immediately prior to sacrifice at 4 and 8 weeks, in order to determine effect of CQ
on liver, kidney, and cardiac tissues. Aspartate transaminase/alanine transaminase
(AST/ALT), creatinine, and lactate dehydrogenase (LDH) levels were tested for each organ,
respectively. 1.5 – 2mL of blood was collected in serum separator test tubes. Tubes
containing blood were centrifuged at 2,000 rpm for 15 minutes within one hour of blood
collection. The separated serum was then collected in individual 1.5 mL microcentrifuge
tubes for each test. The serum samples were sent to the pathology laboratory of St. Boniface
General Hospital (Winnipeg, MB, Canada) for analysis. LDH levels were assayed with 2
hours of sample collection, followed by the AST/ALT and creatinine determinations.
Protein Isolation from Whole Tissue
Cardiac tissues isolated from all four treatment group of animals, weighing 30-50mg
were stored at -80oC were used for protein isolation. Tissues were crushed using a motor and
pestle in liquid nitrogen, collected in centrifuge tubes, and mixed into SDS lysis buffer (10μL
for every 1.0 mg of tissue) containing 250 mmol/L Tris-HCl (pH 6.8), 4% SDS, 20% (v/v)

65
glycerol and1.0 mM ethylene glycol-bis (β-aminoethyl ether-N,N,N’,N’-tetraacetic) acid
(EGTA), plus phosphatase inhibitors (10 mMNaF, 1.0 mMNa3VO4) and protease inhibitors
(P8430 Protease Inhibitor Cocktail; Sigma, USA), followed by incubation on ice for one
hour. Samples were then sonicated three times, for 10 seconds each. Lysates were transferred
to QIAshredder columns (QIAgen, Germany) and centrifuged at 14,000xg at 4oC for 2
minutes. The supernatant collected in the bottom of the columns was used for protein content
determination (Smith’s assay, as described above) and Western blot analysis.
Tissue Sectioning for Histology and Immunofluorescence
Cardiac tissue excised from experimental MI-model rats at 4-weeks and 8-weeks
post-MI was stored at -80oCin OCT blocks. Tissue sections of were cut at a 5 μm thickness
using a microtome (Microm HM550 Cryostat; ThermoFisher Scientific). Sections were fixed
with 4% (w/v) PFA in PBS and used for immunofluorescence following the same protocol
used for fixed cells.
Masson’s Trichrome Staining
To determine the distribution of fibrous collagens within the myocardium from in vivo
preparations of hearts, we used Masson’s trichrome staining technique. Cardiac tissue
sections were obtained from frozen OCT blocks and mounted on glass slides. The sections
were blocked in Bouin’s fixative (5% (v/v) acetic acid, 9% (v/v) formaldehyde, 0.9% (w/v)
picric acid) at 60oC in a water bath. The sections were then washed 3 times with PBS to
remove the excess fixative and stained with Weigert’s iron hematoxylin working solution
(Hematoxylin, 95%alcohol, 29%ferric chloride and concentrated HCL) for 10 minutes at
room temperature. The excess solution was rinsed with water for 5 minutes. Next, the slides
were subjected to Biebrich scarlet-acid fuchsin solution 1% beibrich scarlet, 1% fuchsin acid
and glaial acetic acid) for 10-15 minutes. Then sections were washed with water and
subjected to phosphomolybdic-phosphotungstic acid for 10-15 minutes until the red staining

66
for collagens appeared. The slides were then directly stained in aniline blue for 10-15 minutes
and rinsed with water. Next, the slide preps were subjected to1% glacial acetic acid for 5
minutes and washed briefly for 5 minutes. Finally, slide preparations were dehydrated with
95% ethanol for5 minutes Sections were incubated with xylene twice(3 minutes each) and
mounted with coverslips using Permount™ mounting medium (Fisher Chemical, USA).
Images were acquired at 4X using light microscopy.
Hydroxyproline Assay
The Hydroxyproline assay is a semi-quantitative biochemical method for evaluating
tissue collagen content. This assay allows for the detection of hydoxyproline, common to
collagens and elastin, and because collagen content far exceeds elastin in heart tissue, has
been used as a measure of total collagen content in the tissue [294, 295]. The basic principle
of this assay is when oxidized hydroxyproline content in the tissue/serum samples react with
4 dimethylamino benzaldehyde (DMAB), producing a colored product which can be
quantitated by its absorbance at 560nm. We used the Sigma–Aldrich kit according to the
manufacturer’s instructions. We used 10 mg of the LV tissues (control and viable eg, remote
to the infarct scar) in our assays. The tissue was homogenized in 100µl water and subjected to
100 µl of concentrated HCL (12 M) at 120oC for 3 hours. Next the samples were carefully
collected and centrifuged at 13,000xg for 5 minutes in order to receive a clear supernatant.
Further, 5 -10µl of supernatant was added to a 96 well plate and left to evaporate in the oven
at 60 C until they were dry. Next we added 100µl of chloramine oxidation mixture and left
for 5 minutes so that it could oxidize the hydroxyproline. Later added 100 µl of DAMB and
incubated at 60o C for 90 minutes to give colored product. Finally we measured the
absorbance at 560nm and calculated the concentration of hydroxyproline content in our 4
study groups- sham, sham + CQ, MI and MI + CQ.

67
Statistical Analysis
Data are expressed as mean ± SEM. Each n represents a single animal, and all studies
were conducted with at least 3 individual animals in a given group. Statistical significance
between the groups was assessed by Student t-test or where multiple experimental groups are
compared one-way ANOVA followed by Student-Newman-Keuls post-hoc test using
SigmaPlot software (Systat Software, San Jose, CA, USA). A P-value < 0.05 was considered
statistically significant.

68
Chapter 1: Role of Autophagy in Fibroblast Activation In Vitro
Introduction
Autophagosomes have been observed in the myocardium in ischemia/reperfusion
models [17, 18], and at the cellular level, autophagy is involved in epithelial to mesenchymal
transition (EMT) as well as mesenchymal to epithelial transition (MET) [22, 23]. It has been
suggested that the nature of remodeling within cardiac tissue eg, whether it's adaptive or
maladaptive, depends upon the degree of autophagic induction [15]. Both EMT and MET
contribute to differentiation, wound healing and proliferation [23]. The link between cell
differentiation and autophagy has been demonstrated using inhibitors of autophagy.
Treatment with CQ or siRNA selective for ATG3-ATG7 suppresses starvation-induced
autophagy and subsequent EMT through TGF-β/Smad signaling in hepatocarcinoma cells
[22]. Our previous studies of human atrial fibroblasts indicate that the onset of autophagy and
cardiac fibrosis are tightly associated with one another. The use of inhibitors of autophagy
may be useful in revealing the role of autophagic pathways in pathological signaling
processes such as cardiac fibroblast activation to myofibroblasts [13, 16]. When isolated and
cultured in vitro, cardiac fibroblasts undergo rapid activation to myofibroblastic phenotype
which includes among other markers, increased expression of α-SMA [296]. Substrate
stiffness has been found to be a player in this activation [286, 297, 298]. Thus we employ
mechanical stress, in the form of a stiff plating substrate, to induce conversion of cardiac
fibroblasts to the myofibroblast phenotype, as previously published [102, 297, 299].
During autophagy in mammalian cells, autophagosomes engulf components in the
cytoplasm, including proteins and organelles. A soluble microtubule-associated protein
1A/1B-light chain 3 (LC3) is ubiquitously distributed in mammalian tissues as well as
cultured cells, and during engulfment of cytoplasmic contents by autophagosomes, its
cytosolic form (LC-3I) becomes simultaneously conjugated to phosphatidylethanolamine to

69
form LC3-phosphatidylethanolamine (LC-3II). LC-3II is recruited to autophagosomal
membranes, and is degraded alongside autophagosome contents when autophagosomes fuse
with lysosomes to form autolysosomes. Therefore, LC-3II turnover is an indicator of
autophagic activity, and immunodetection of lipidated (phosphatidylethanolamine-
conjugated) LC3 is used as a reliable method for monitoring induction of autophagy [300,
301]. In the final step where the lysosome fuses with autophagosomes, the digestive acidic
enzymes within this compartment degrade the cellular material. In order to successfully
complete autophagy, maintenance of the acidic pH (4.5 to 5) within the autophagolysosome
is very critical and is achieved by the action of membrane bound v-ATPases.
Pharmacological inhibitors such as Baf-A1and CQ interfere with membrane-bound v-ATPase
activity and neutralize the pH within the autophagolysosomes, and thus inhibit autophagy by
preventing autophagosome-lysosome fusion in the final step of the autophagic process [302,
303]. This mechanism is also referred to as autophagic flux inhibition, which results in
accumulation of cellular LC-3II [302]. Conversely, 3MA is a PI3K inhibitor that instead
targets hVPS34 and disrupts the initiation complex, inhibiting autophagy in its initial stages
[304]. We have used these drugs to study the effect of inhibition of autophagy and autophagic
flux in the activation of fibroblasts to hyperactive myofibroblasts. Additional measurement of
autophagic processes, such as levels of the cargo protein p62, which tags and escorts protein
aggregates to autophagosomes, can be measured as further examination of autophagic flux.
Lysosomal degradation of autophagosomes results in decreased p62 levels, and inhibition of
autophagy can induce accumulation of p62 [24].

70
Results
(Part of this work has been published in Oncotarget in October2016. This is an open-access
article distributed under the terms of the Creative Commons Attribution License.)
Title: Inhibition of autophagy inhibits the conversion of cardiac fibroblasts to cardiac
myofibroblasts.
Authors: Shivika S. Gupta, Matthew R. Zeglinski, Sunil G. Rattan, Natalie M. Landry, Saeid
Ghavami, Jeffrey T. Wigle, Thomas Klonisch, Andrew J. Halayko, Ian M.C. Dixon)
Contribution by SSG: Contributed to conceptualization of most of the experiments, carried
out and analyzed all experiments and wrote up 80% of the manuscript)
Induction of Autophagy by Mechanical Stress
Though it has been established that mechanical stress induces autophagy in
cardiomyocytes [287], this effect has not yet been studied in cardiac fibroblasts. Plating on
non-compressible tissue culture dishes induced time-dependent induction of autophagy in
freshly isolated unpassaged (P0) rat cardiac fibroblasts. Activation of cardiac fibroblasts to
myofibroblasts were confirmed by a significant increase in protein levels of α-SMA after 48
and 72 hours of plating, compared to 24 hour controls. Concomitantly, we observed a
significant 6-fold increase in lipidated LC-3II protein levels after as little as 48 hours after
plating, as compared to 24 hour controls. By 72 hours post-plating lipidated LC-3II still
maintained a 5.5-fold significant increase (P< 0.05) compared to the 24 hour control (Figure
7A and B).

71
Figure 7: Temporal activation of autophagy and fibroblast in P0 adult rat cardiac
fibroblasts. Panel A. There was a significant increase in the level of the autophagosome
marker, LC-3II at 48 and 72 hours post-plating on a non-compressible plastic substrate vs. 24
hours. Panel B. Western blot analysis for the myofibroblast marker α-SMA showed a
significant increase 48 and 72 hours after plating when compared to 24 hour controls. Data
are mean ± SEM (n = 3) (*P< 0.05 24 hours vs. 48 hours; #P< 0.05 24 hours vs. 72 hours;
φP< 0.05 48 vs. 72 hours).Significance was determined using one-way ANOVA followed by
a Student-Newman-Keuls post-hoc test.

72
Inhibition of Autophagy in Cardiac Fibroblasts
To determine the relationship between cardiac fibroblast activation and autophagy in
primary, unpassaged P0 rat cardiac fibroblasts, we treated cells with two inhibitors of
autophagic flux, bafilomycin-A1 (BafA1) and chloroquine (CQ), and with 3-methyladenine
(3MA), which inhibits autophagy initiation. Levels of LC-3I and lipidated LC-3II were
assayed via Western blotting, revealing a significant increase in LC-3II lipidation with Baf-
A1 and CQ treatment (P< 0.05) compared to controls (Figure 8A, B and C). No significant
change in lipidated LC-3II levels was observed with 3MA treatment. Autophagy inhibitors
have also been found to induce accumulation of p62 protein levels [24], and indeed, with CQ
treatment, we observed a 3.4-fold significant (P< 0.05) increase in p62 levels at 50 µM CQ as
compared to controls (Figure 8D).

73
#
A
B
C
#
B
A

74
Figure 8: Baf-A1, CQ and 3MA treatment inhibits autophagy in P0 cardiac fibroblasts.
Panel A and B 48 hours of Baf-A1 treatment (5.0 nM and 7.5 nM) showed no significant
change in LC-3I protein expression. However, there was a significant increase in the
expression levels of LC-3II with 7.5 nM Baf-A1 vs. untreated controls. Cells treated with CQ
(25 µM and 50 µM) for 48 hours demonstrated increased accumulation of LC-3II vs.
untreated controls. The 50 µM CQ treatment group was significantly increased vs. control
while the 25 µM group was trending upward but did not reach significance. Panel C.
Inhibition of autophagy in the 3MA treatment group showed no significant change in LC-3II
or LC-3I. Panel D. p62 protein levels were significantly increased following CQ (50 µM)
treatment vs. untreated control. Data are mean ± SEM (n=3-4) (#P< 0.05 control vs. 7.5 nM
Baf-A1 and 50 µM CQ). Significance was determined using one-way ANOVA followed by a
Student-Newman-Keuls post-hoc test.
D

75
We utilized transmission electron microscopy (TEM) to visualize autophagosomes in
cardiac fibroblasts treated with autophagic flux inhibition drugs (Baf-A1 and CQ).This
confirmed autophagosome presence and accumulation in both treatment groups compared to
controls (Figure 9A and B).
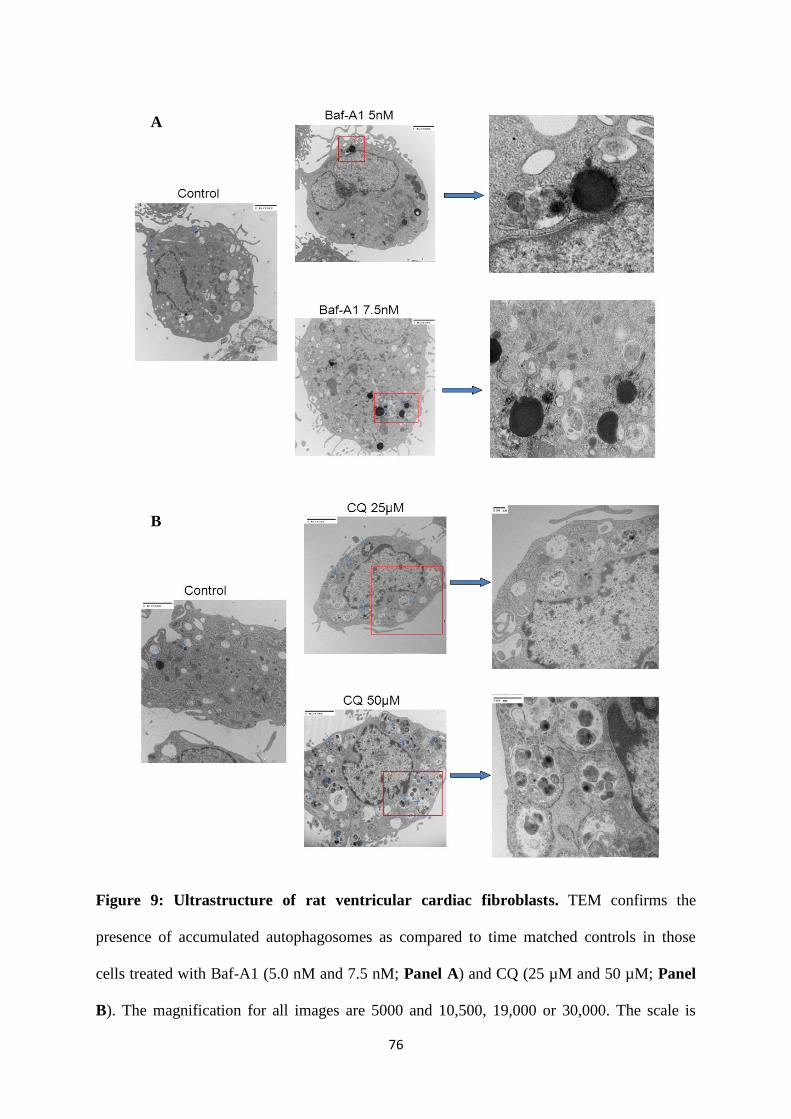
76
Figure 9: Ultrastructure of rat ventricular cardiac fibroblasts. TEM confirms the
presence of accumulated autophagosomes as compared to time matched controls in those
cells treated with Baf-A1 (5.0 nM and 7.5 nM; Panel A) and CQ (25 µM and 50 µM; Panel
B). The magnification for all images are 5000 and 10,500, 19,000 or 30,000. The scale is
A
B

77
shown on each image. Data are mean ± SEM (n=3-4) (#P< 0.05 control vs. 7.5 nM Baf-A1
and 50 µM CQ).

78
Inhibition of Autophagy and Myofibroblast Features
The concomitant increase in myofibroblast marker levels and lipidated LC- II lead us
to question whether inhibition of autophagy may affect the process of activation in P0 cardiac
fibroblasts. Using Western blot analysis, we determined that all three autophagic inhibitors
significantly reduced (P< 0.05) protein levels of myofibroblast markers α-SMA and ED-A
FN (Figure 10A, B and C). Treatment with 50 µM CQ completely abolished expression of α-
SMA and ED-A FN.
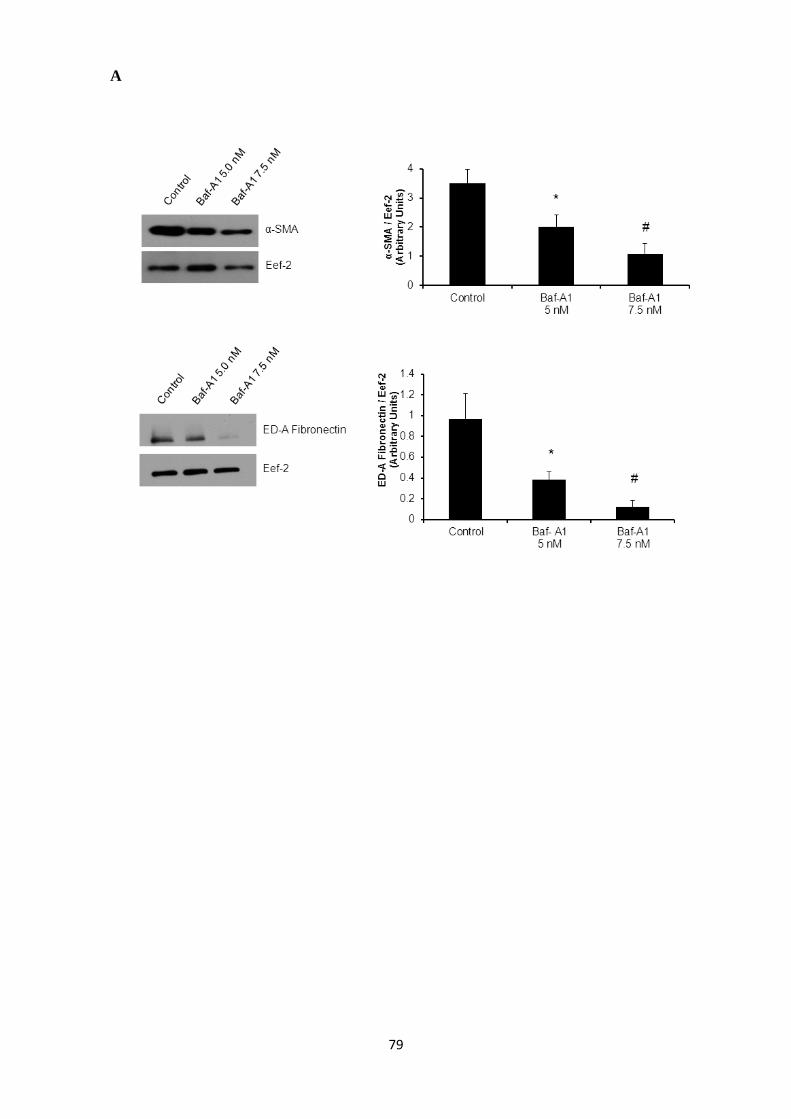
79
A
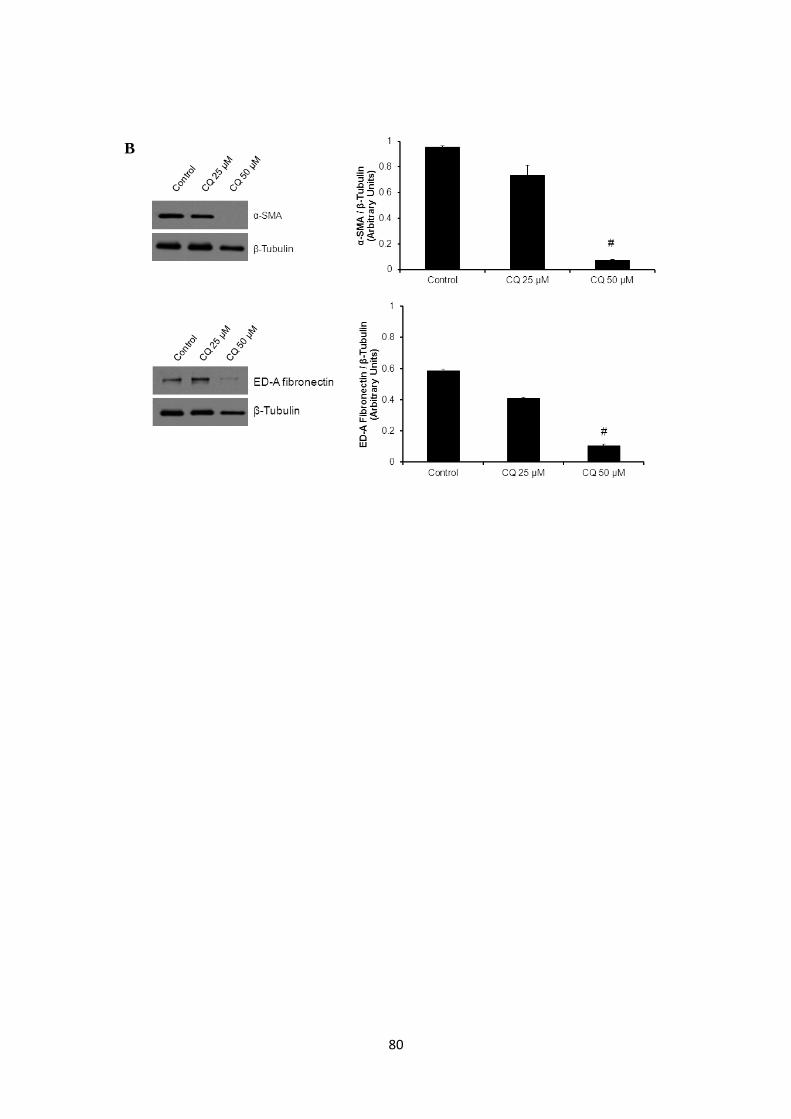
80
B

81
Figure 10: Inhibition of autophagy reduces expression of key cardiac myofibroblast
markers. Western blot analysis of P0 cardiac fibroblasts treated with autophagy inhibitors
show decreased expression levels for ED-A FN and α-SMA. Panel A. 48 hours of Baf-A1
(5.0 nM and 7.5 nM) treatment resulted in a significant reduction in α-SMA (40 % and 70%)
and ED-A FN (35% and 50%) levels respectively. Panel B. 50 µM CQ treatment for 48
hours shows a significant decrease in α-SMA and in ED-A FN levels to near undetectable
levels. Panel C. Treatment groups of 1 mM, 2.5 mM and 5 mM 3MA for 72 hours show
decreased expression levels of α-SMA and ED-A FNvs. controls. Data are expressed as mean
± SEM (n=3-4) (*P< 0.05 control vs. 5.0 nM Baf-A1, 2.5 mM 3MA; #P< 0.05 control vs. 7.5
nM Baf-A1, 5 mM 3MA, 50 µM CQ). Significance was determined using one-way ANOVA
followed by a Student-Newman-Keuls post-hoc test.
C

82
Further examination of autophagic flux via immunofluorescence staining for α-SMA
revealed a decrease in α-SMA-positive stress fibre formation (a key feature of the
myofibroblast) following treatment with all three inhibitors, as well as increased punctate
LC-3β staining, compared to controls (Figure 11A, B and C).

83
A
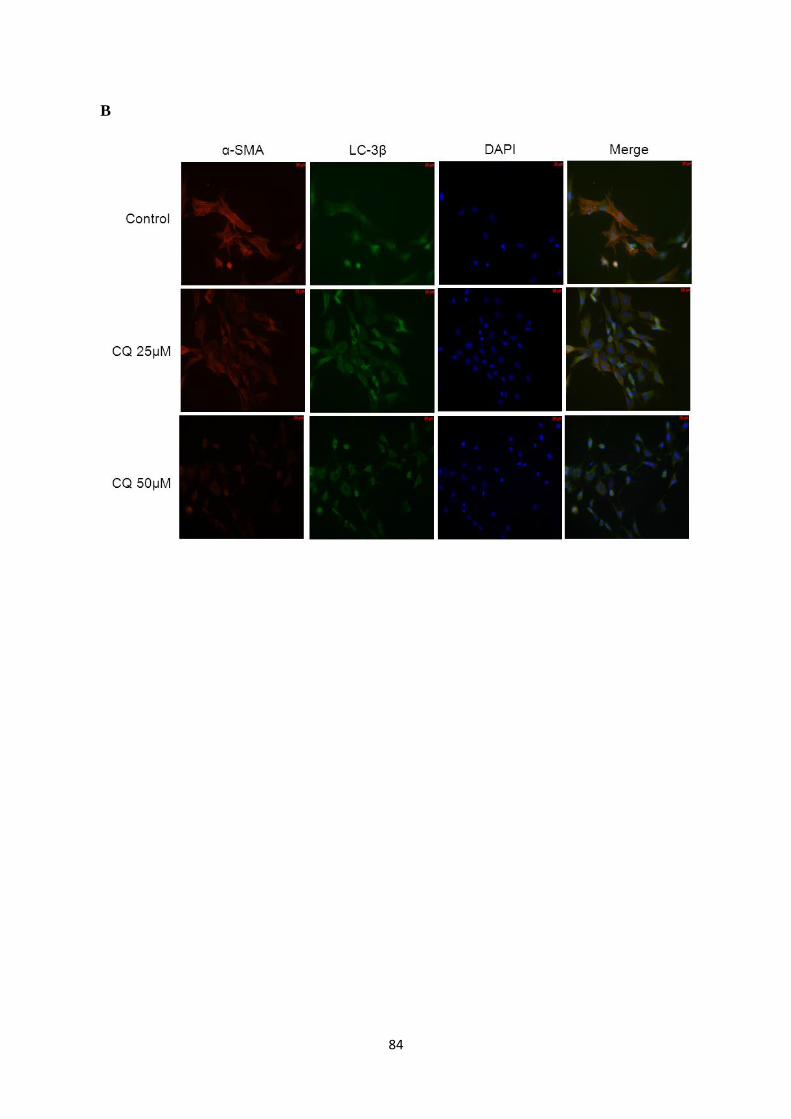
84
B
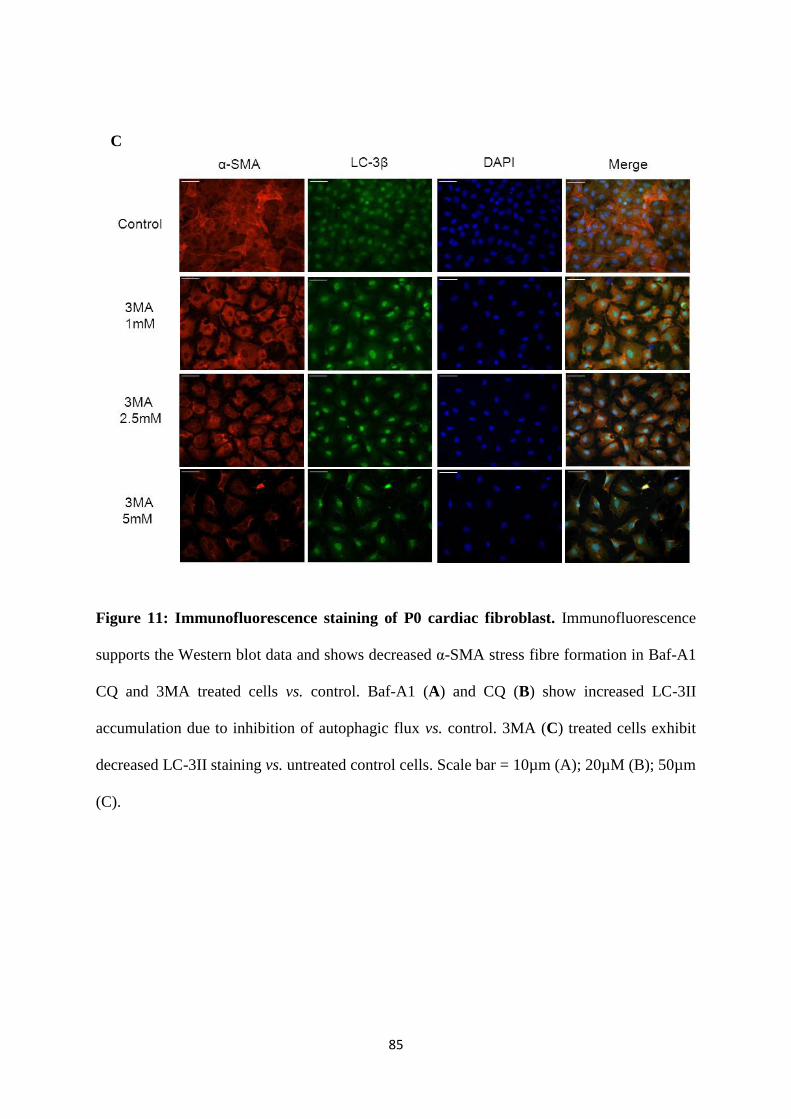
85
Figure 11: Immunofluorescence staining of P0 cardiac fibroblast. Immunofluorescence
supports the Western blot data and shows decreased α-SMA stress fibre formation in Baf-A1
CQ and 3MA treated cells vs. control. Baf-A1 (A) and CQ (B) show increased LC-3II
accumulation due to inhibition of autophagic flux vs. control. 3MA (C) treated cells exhibit
decreased LC-3II staining vs. untreated control cells. Scale bar = 10µm (A); 20µM (B); 50µm
(C).
C

86
Inhibition of Autophagy and Myofibroblast Function
We sought to further examine the effect of autophagy inhibition on not only
myofibroblast morphology, but also myofibroblast function – namely contractility and
motility, both of which are increased in the wound healing response and subsequent
activation of fibroblasts to myofibroblasts. To assess contractility, we used a collagen gel-
based contraction assay to determine surface contractility of cells as previously described by
our group and others, utilizing TGF-β1 as a potent inducer of contraction [50, 305]. Treatment
with Baf-A1 (7.5 nM) and CQ (25 and 35 µM), but not 3MA, significantly (P< 0.05)
inhibited TGF-β1-induced contraction after 72 hours (Figures12A, B and C), as compared to
TGF-β1 stimulated positive controls.

87
A
B
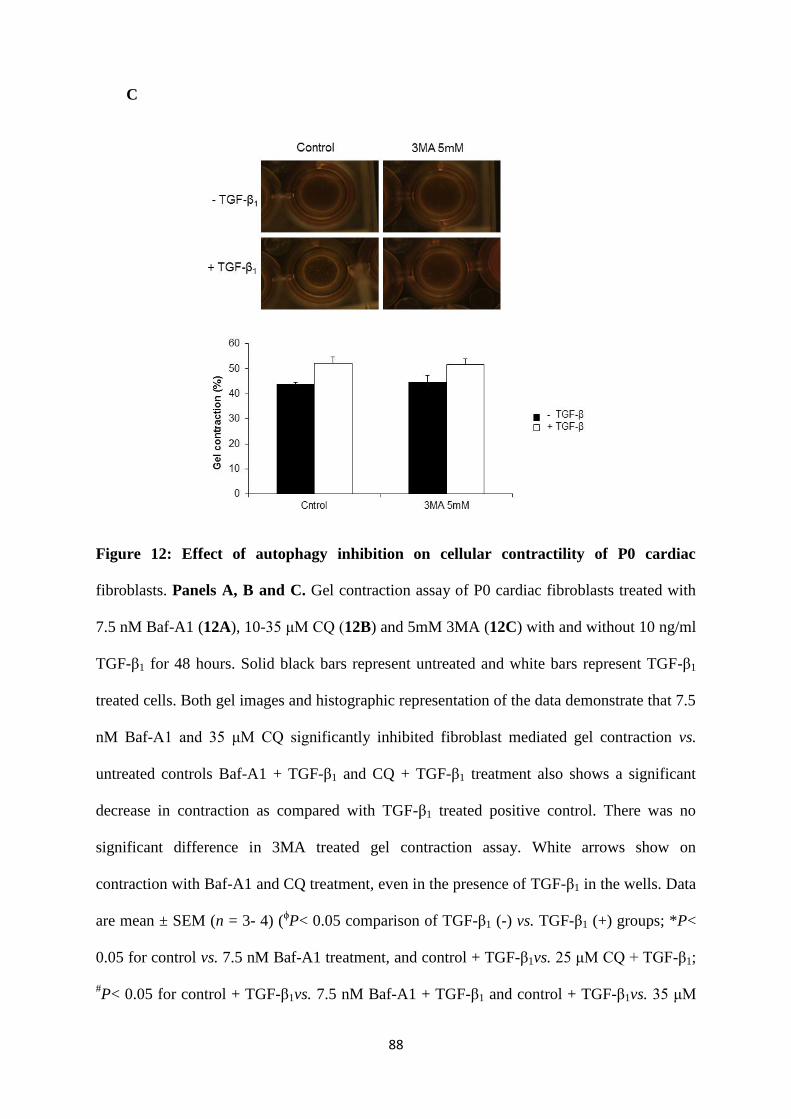
88
Figure 12: Effect of autophagy inhibition on cellular contractility of P0 cardiac
fibroblasts. Panels A, B and C. Gel contraction assay of P0 cardiac fibroblasts treated with
7.5 nM Baf-A1 (12A), 10-35 μM CQ (12B) and 5mM 3MA (12C) with and without 10 ng/ml
TGF-β1 for 48 hours. Solid black bars represent untreated and white bars represent TGF-β1
treated cells. Both gel images and histographic representation of the data demonstrate that 7.5
nM Baf-A1 and 35 μM CQ significantly inhibited fibroblast mediated gel contraction vs.
untreated controls Baf-A1 + TGF-β1 and CQ + TGF-β1 treatment also shows a significant
decrease in contraction as compared with TGF-β1 treated positive control. There was no
significant difference in 3MA treated gel contraction assay. White arrows show on
contraction with Baf-A1 and CQ treatment, even in the presence of TGF-β1 in the wells. Data
are mean ± SEM (n = 3- 4) (ϕP< 0.05 comparison of TGF-β1 (-) vs. TGF-β1 (+) groups; *P<
0.05 for control vs. 7.5 nM Baf-A1 treatment, and control + TGF-β1vs. 25 μM CQ + TGF-β1;
#P< 0.05 for control + TGF-β1vs. 7.5 nM Baf-A1 + TGF-β1 and control + TGF-β1vs. 35 μM
C
C

89
CQ + TGF-β1). Significance was determined using one-way ANOVA followed by a Student-
Newman-Keuls post-hoc test.

90
We used a traditional scratch assay to determine the relationship between autophagy
and cardiac fibroblast migration. These assays were performed in the presence and absence of
autophagic inhibitors and migration measured via time required for fibroblasts to fill the
empty wounded area (eg, migrate). Compared to time 0 hour controls, by 12 hours there was
reduced fibroblast migration with inhibition of autophagy 3MA, Baf-A1 and CQ; Figure 13).
After 24 hours, CQ continued to inhibitfibroblast migration, compared to untreated controls
which had fully covered the denuded area. With 35 µM CQ treatment, an approximate 3-fold
and 6-fold reductions in cell migration were observed after 12 and 24 hours, respectively.
Although Baf-A1 treatment appeared to reduce cell migration at the 12 hour time-point via
visual observation, there was not a statistically significant difference in migration. Treatment
with 3MA did not significantly alter cardiac fibroblast migration, even after 24 hours (Figure
13).
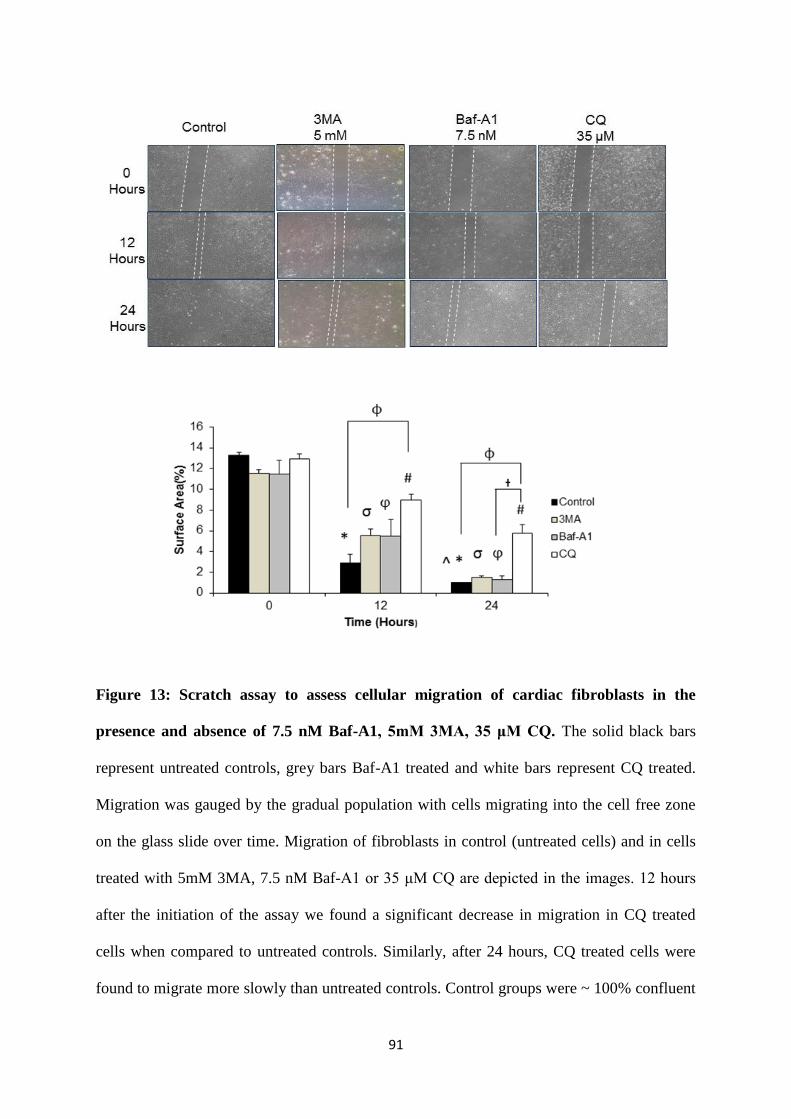
91
Figure 13: Scratch assay to assess cellular migration of cardiac fibroblasts in the
presence and absence of 7.5 nM Baf-A1, 5mM 3MA, 35 μM CQ. The solid black bars
represent untreated controls, grey bars Baf-A1 treated and white bars represent CQ treated.
Migration was gauged by the gradual population with cells migrating into the cell free zone
on the glass slide over time. Migration of fibroblasts in control (untreated cells) and in cells
treated with 5mM 3MA, 7.5 nM Baf-A1 or 35 μM CQ are depicted in the images. 12 hours
after the initiation of the assay we found a significant decrease in migration in CQ treated
cells when compared to untreated controls. Similarly, after 24 hours, CQ treated cells were
found to migrate more slowly than untreated controls. Control groups were ~ 100% confluent

92
at 24 hours. (*P< 0.05 for 0 hours vs. 12 hours and 24 hours in untreated controls; σP< 0.05
for 0 hoursvs. 12 hours and 24 hours in 5 mM 3MA treated groups;φP< 0.05 for 0 hours vs.
12 hours and 24 hours in 7.5 nM Baf-A1 treated groups;#P< 0.05 for 0 hours vs. 12 and 24
hours in 35 μM CQ treatment groups; †P< 0.05 for Baf-A1 vs. CQ 35 μM at 24 hours;
ϕP<0.05 for untreated control vs. CQ 35 μM, treated groups within the same time point). Data
are mean ± SEM (n = 3- 4). Significance was determined using one-way ANOVA followed
by a Student-Newman-Keuls post-hoc test.

93
Inhibition of Autophagy and Mitogen-Activated Protein Kinase (MAPK) Signaling
MAPK signaling is implicated in the acquisition of the myofibroblast phenotype, eg
the activation of cardiac fibroblasts. Our preliminary study of the effects of MAPK signaling
on myofibroblast phenotype included Western analysis of P42/44 (ERK) and p38 expression
in both basal and phosphorylated forms. When we compared plating duration (24h, 48h and
72h) among groups, we found no changes in ERK phosphorylation. We also used Western
blotting to test for phosphorylation (eg, activation) of p38 MAPK in unpassaged P0 cardiac
fibroblasts over 72 hours (Figure 14). At both 48 and 72 post-plating, we observed a
significant (P< 0.05) reduction of 80-90% in the ratio of phospho-p38: total p38 levels
(Figure 14A) compared to 24 hour controls. Inhibition of autophagic flux with CQ (50 µM)
resulted in a 12-fold increase in phospho-p38: total p38 levels after 48 hours, comparable to
unpassaged P0 fibroblasts 24 hours post-plating (Figure 14B).

94
Figure 14: Effect of cell plating and CQ treatment on p38-MAPK phosphorylation in P0
primary cardiac fibroblasts. Panel A. Phosphorylation of p38-MAPK was detected at 24, 48
and 72 hours after plating of P0 cardiac fibroblasts onto a non-compressible plastic substrate.
Total p38-MAPK was also analysed. Phospho-p38 (P-p38) expression was significantly
decreased after 48 and 72 hours of culture when compared to 24 hour controls. Panel B.
Western blot images indicate an increase in phosphorylation of p38-MAPK in the presence of
50 µM CQ treated for 48 hours, as compared to untreated controls. Lane protein loading was
normalized using β-tubulin. Data are mean ± SEM (n=3) (*P< 0.05 applies to the
comparisons of 24 hours plated cells vs. 48 hours plating; #P< 0.05 applies to the comparison
of 24 hours plating vs. the 72 hours plated group; φP< 0.05 for control vs. 50 µM CQ).
Significance was determined using one-way ANOVA followed by a Student-Newman-Keuls
post-hoc test.
B
A

95
To further investigate the role of MAPK signaling in autophagy and induction of
myofibroblast phenotype in cardiac fibroblasts, we utilized a p38 MAPK inhibitor SB203580,
both in the presence and absence of autophagic inhibitors. Pre-treatment with 10 µM
SB203580 show no change in p38 phosphorylation, as compared to control.(Figure 15A).
Combination treatment with both SB203580 + CQ induced a significant increase in p38
phosphorylation, although this increase did not differ significantly from that of CQ alone
(Figure 15A). To examine the effect of MAPK and autophagic inhibition, we examined their
effects on autophagic flux via Western blotting for lipidated LC-3β II levels. Treatment with
SB230580 both in the absence and presence of CQ had no effect on lipidated LC-3β II levels,
though CQ treatment alone showed a significant increase in lipidated LC-3β II (P < 0.05;
Figure15B). Similar effects were observed when SB203580 was used in the presence and
absence of 7.5 nM Baf-A1 (Figure 16A and B).
We then examined the effects of SB203580 in the presence and absence of autophagy
inhibitors on the on fibroblast activation within P0 cardiac fibroblasts. Treatment with
SB203580 alone did not significantly affect protein levels of the myofibroblast markers α-
SMA and ED-A FN compared to untreated controls (Figure 15C and D). Co-treatment with
SB203580 + CQ significantly (P < 0.05) reduced myofibroblast marker protein levels
compared to both untreated and SB203580 alone-treated groups (Figure 15C and D). No
additive effects were observed with SB203580 in conjunction with CQ treatment, as the
addition of SB203580 to CQ inhibition did not result in any significant difference in the
amount of decrease of α-SMA and ED-A FN. Again, similar results were observed with
SB203580 and Baf-A1 co-treatment (Figure 16C and D). Between the two inhibitors of
autophagic flux, CQ had a more pronounced effect on increasing p38 phosphorylation and
repressing the myofibroblast phenotype.
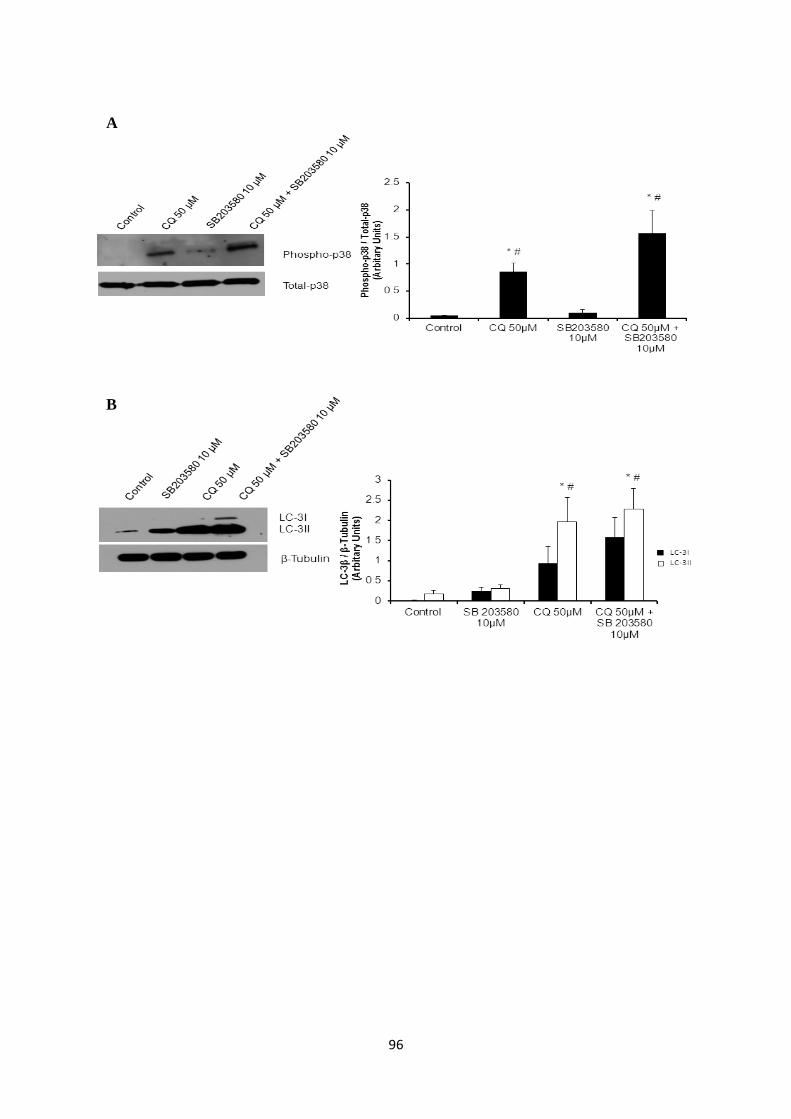
96
A
B
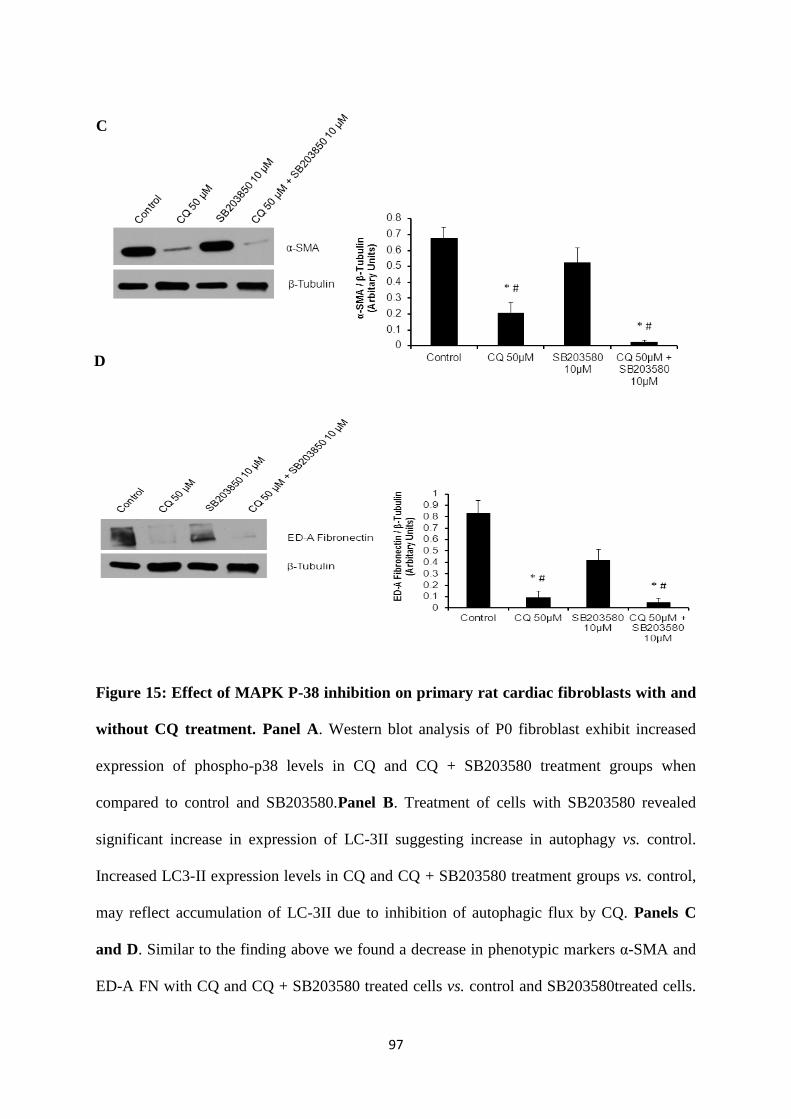
97
Figure 15: Effect of MAPK P-38 inhibition on primary rat cardiac fibroblasts with and
without CQ treatment. Panel A. Western blot analysis of P0 fibroblast exhibit increased
expression of phospho-p38 levels in CQ and CQ + SB203580 treatment groups when
compared to control and SB203580.Panel B. Treatment of cells with SB203580 revealed
significant increase in expression of LC-3II suggesting increase in autophagy vs. control.
Increased LC3-II expression levels in CQ and CQ + SB203580 treatment groups vs. control,
may reflect accumulation of LC-3II due to inhibition of autophagic flux by CQ. Panels C
and D. Similar to the finding above we found a decrease in phenotypic markers α-SMA and
ED-A FN with CQ and CQ + SB203580 treated cells vs. control and SB203580treated cells.
C
D

98
SB203580 treatment was associated with similar expression of both phenotypic markers
when compared to control. Data are expressed as mean ± SEM and representative of 3
individual animals and experiments (eg, n=3) (*P<0.05 control vs.50µM CQ or CQ +
SB203580; #P<0.05 SB203580vs.50µM CQ or CQ + SB203580). Significance was
determined using one-way ANOVA followed by a Student-Newman-Keuls post-hoc test.

99
A
B
C

100
Figure 16: Effect of MAPK inhibition on Baf-A1 treated and untreated P0 cardiac
fibroblasts. Panel A. Phospho-p38 levels show significant increase in Baf-A1 and Baf-A1 +
SB203580 treated cells vs control and SB203580 treated cells. Panel B. Autophagy marker
LC-3II exhibit increased expression in SB203580 treated vs. control group. Baf-A1 treatment
in presence and absence of SB203580 show accumulation of LC-3II suggesting inhibition of
autophagic flux compared to control. Panels C and D. Phenotypic markers α-SMA and ED-
A fibronectin show significant decrease in expression pattern when compared to control and
SB203580. Data are expressed as mean ± SEM and representative of 3 individual animals
and experiments (eg, n=3) (*P<0.05 control vs.7.5nM Baf-A1 or CQ + SB203580; #P<0.05
SB203580vs.7.5 nM Baf-A1 or Baf-A1 + SB203580). Significance was determined using
one-way ANOVA followed by a Student-Newman-Keuls post-hoc test.
D

101
Discussion
Wound healing and tissue fibrogenesis are processes requiring the activation and
migration of fibroblasts, as well as the hypersynthetic and contractile abilities of
myofibroblasts. Fibroblast activation to myofibroblast is a critical hallmark of both processes,
of which TGF-β has been shown to be a major driver [306]. TGF-β signaling in fibrosis
occurs both through a canonical Smad-dependent pathway as well as TGF-β - linked p38-
MAPK signaling [307]. Recently, our group demonstrated that autophagy may regulate TGF-
β1-induced fibrogenesis in primary human atrial myofibroblasts [256].
Autophagy in eukaryotes is associated with enhanced longevity [308], as well as
cytoprotection in fibroblasts. Embryonic mouse fibroblasts overexpressing the pro-
autophagic ATG5 gene were found to better resist cellular stress [309]. Additionally,
autophagy is downregulated in fibrotic lesions from various tissues including the lung and
liver [310, 311]. Autophagy has been extensively examined in the context of cardiomyocyte
function and death in response to pathological stimuli, especially in myocardial remodeling.
Cardiomyocyte loss (both apoptotic and non-apoptotic) are crucial in cardiac remodeling and
the development of heart failure, and have been demonstrated in association with oxidative
stress and increased autophagy in ischemia/reperfusion and pressure overload cardiac models
[312, 313]. In both models, inhibition of pro-autophagic Beclin-1 ameliorated adverse cardiac
remodeling. In fact, autophagic inhibition has been shown to alleviate fibroproliferative
events in numerous tissues [314]. However, these studies only describe the autophagic
response before and after disease development, while the current results target the importance
of autophagic flux in the fibroblast activation in freshly isolated, unpassaged P0 rat cardiac
fibroblasts. Studies such as these are rare within the literature due to the technical hurdles
associated with experimentation involving quiescent P0 cells, and thus the need for the
current study is rationalized.

102
We utilized an established and highly effective method of inducing fibroblast
activation, that is, plating on hard substrates [4, 14, 27, 33]. Even in primary rat hepatic
stellate cells from TGF-β-null mice, culture on stiff substrates induces the myofibroblast
phenotype [315]. In turn, long-term mechanical strain in skin and heart tissues further
potentiates scarring and fibrosis [297, 316]. Numerous studies have demonstrated rapid
induction of the myofibroblast phenotype in primary fibroblasts plated on non-compressible
tissue culture substrates [296, 305, 317]. Within 16 - 48 hours of plating, our primary
unpassaged P0 rat cardiac fibroblasts exhibited significant increases in protein levels of key
myofibroblast markers α-SMA and ED-A FN, as well as increased presence of α-SMA stress
fibers, an effect that persisted to 72 hours post-plating. The combination of these findings
provides a strong body of evidence supporting the recapitulation of mechanical stress-
induced fibroblast activation in our P0 cardiac fibroblasts. This stiffness-induced fibrobast
activation was accompanied by increased expression of lipidated LC-3β II at both 48 and 72
hours post-plating. This agrees with previously published data from our lab which pointed to
a causal effect of TGF-β1 stimulation in induction of autophagy and cell activation. Cellular
activation is closely linked to increased ECM synthesis in both post-coronary artery bypass
graft (CABG) human atrial and post-MI rat ventricular cardiac tissues [318]. Additionally,
studies in hepatocytes and hepatic carcinoma cell metastasis have demonstrated a
requirement for autophagy in hepatocyte phenotype plasticity and TGF-β1 stimulated EMT
[319].
As autophagy has been described by our lab [256] and others [301] as a necessary
cellular process in fibrotic tissues, we sought to elucidate associated mechanisms in cardiac
fibroblasts activation to myofibroblasts. Western blotting for accumulated p62 and lipidated
LC-3β II levels confirmed successful inhibition of autophagy with all three inhibitors (Baf-
A1, CQ, and 3MA). Via TEM, we confirmed autophagic inhibition through the observation

103
of autophagosome accumulation. A novel finding of this work is the prevention of cardiac
fibroblast activation to myofibroblasts with autophagic inhibition, as demonstrated by
decreased Western blot protein levels of the markers α-SMA and ED-A FN in comparison to
untreated control plated on stiff substrates. Moreover, we determined that CQ was the most
effective of the three drugs in inhibiting activation of cardiac fibroblasts. This is in agreement
with a study by He et al. [320], which showed CQ treatment of hepatic stellate cells in vivo in
a liver fibrosis model was able to diminish α-SMA expression and cellular organization.
However, no studies to date have demonstrated the role of autophagy in repressing cardiac
fibroblast activation, as well as myofibroblast function (specifically cell contraction and
migration), though there is data for other cell types [227, 321, 322]. For example, ATG5
knockout cardiomyocytes exhibit decreased contractile force, and loss of ATG7 in skeletal
muscle induces myocyte loss and reduce force production [227]. Our finding that inhibition
of autophagic flux with either, Baf-A1 and CQ abrogates cardiac myofibroblast contraction
agrees with these findings. In terms of migration, the effects of autophagy have been
examined in HeLa and other transformed cell types [301, 323]. In HeLa cells, ATG3, -5, or -
7 knockdown promoted increased migration compared to competent cells [301]. In MDA-
MB-231 breast cancer cells, autophagic activation with trifluoperazine treatment inhibited
cell migration [323]. These reports contrast with our present findings, in which we find
inhibition of autophagic flux with either Baf-A1 or CQ inhibited migration of cardiac
fibroblasts over 24 hours. One potential reason for this difference in findings may be that
genetic inhibition of ATG gene expression does not specifically target autophagic flux. This
discrepancy may also be the result of vastly different cellular models (our primary,
unpassaged cells vs. immortalized/cancer cell lines). On the other hand, a study in MGC803
gastric cancer cells showed that CQ treatment inhibited cell migration in a dose-dependent
manner [324], which agrees with our findings. It is important to note that CQ possesses non-

104
specific inhibitory actions on Toll-like receptor 9 (TLR9), a key factor in cell migration [324]
– thus, although the decreased migratory response to CQ treatment in cardiac fibroblasts was
not anticipated, it may be at least partially attributed to non-specific effects of CQ on TLR9
function. It is interesting to note that 3MA treatment did not significantly affect
myofibroblast functions (contractility and migration). This may be due to the different
inhibitory mode of 3MA, which instead of targeting autophagic flux, blocks formation of
autophagosomes via inhibition of PI3K. Thus, it may be that certain phases of the autophagic
process are more critical for the process of cardiac fibroblasts activation than others.
Nonetheless, inhibition of autophagic flux with either Baf-A1 or CQ prevented not only
morphological phenoconversion features, but also critical myofibroblast functions –
suggesting autophagy is an important process in development of the cardiac myofibroblast.
Concurrent with autophagy induction and activation of the myofibroblast phenotype
(48 hours post-plating), we also observed a significant decrease in phospho-p38: total p38
ratio. Inhibition of autophagy with CQ resulted in an increased expression level of phospho-
p38: total p38 similar to that observed in untreated P0 cardiac fibroblasts at 24 hours after
plating. This data show an inverse relationship between phospho-p38 and markers of both
fibroblast activation and autophagy. Therefore to establish a causal relationship for p38
activation we sought to determine the effect of inhibitors of autophagy in the presence and
absence of an inhibitor of p38 MAPK activity and assess both markers of myobiroblast. To
examine the effect of p38 phosphorylation and autophagic flux on fibroblast activation we
treated cells with SB203580 alone as well as a co-treated CQ or Baf-A1and SB203580for 48
hours. As expected we found that SB203580 treatment alone, which means phospo-p38
inhibition, did not alter the expression of α- SMA and ED-A FN. Next we found co-treatment
of P0 primary fibroblasts resulted in a reduction of the myofibroblast phenotype compared to
controls. We anticipated there should have been a higher degree of expression of α-SMA and

105
ED-A FN in the co-treatment groups when compared to CQ and Baf-A1 treatment alone.
However, there was no difference in the level of reduction between CQ or Baf-A1 treated and
CQ or Baf-A1 + SB203580 treated. Which could be due to use of two pharmacological
inhibitors used together. Possibly the Phospho-p38 inhibitor could not work along in
combination with CQ or Baf-A1. Thus to explore the effects of MAPK and autophagy
inhibition simultaneously a genetic KO of p38 along with CQ or Baf-A1 should be tested.
Although phosphorylation of p38 MAPK is clearly related to inhibition of autophagy, the
functional significance of this event remains unknown. In a rat model of hepatic
ischemia/reperfusion injury, Fang et al. [48] also observed a significant increase in p38
phosphorylation with CQ treatment as soon as 6 hours and up to 48 hours after treatment,
which supports our current findings.
A schematic of these findings in primary unpassaged P0 rat cardiac fibroblasts –
plating on stiff substrates, fibroblast activation, the induction of autophagy, and the effect of
inhibition – is shown in Figure 16. It can be concluded that mechanical stress induces both
fibroblast activation and autophagy in cardiac fibroblasts, and inhibition of autophagy blocks
autophagolysosomes formation and results in significant repression of the cardiac
myofibroblast phenotype, findings that support our previous work [305].
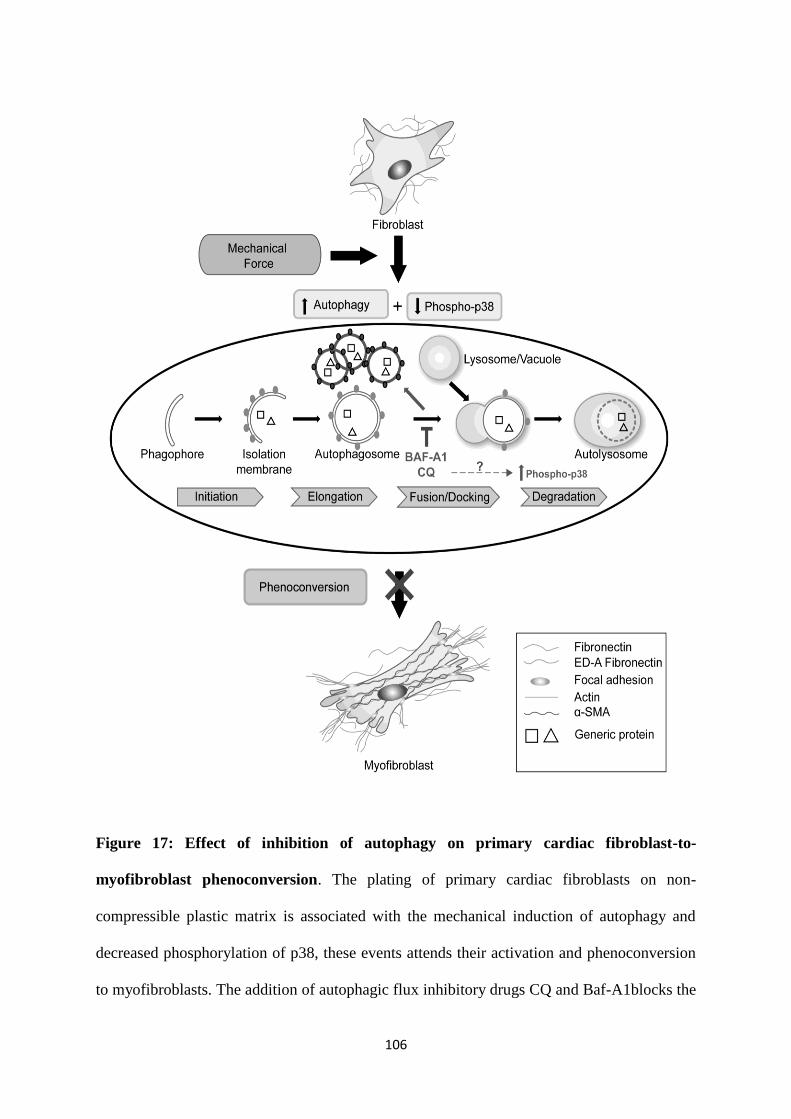
106
Figure 17: Effect of inhibition of autophagy on primary cardiac fibroblast-to-
myofibroblast phenoconversion. The plating of primary cardiac fibroblasts on non-
compressible plastic matrix is associated with the mechanical induction of autophagy and
decreased phosphorylation of p38, these events attends their activation and phenoconversion
to myofibroblasts. The addition of autophagic flux inhibitory drugs CQ and Baf-A1blocks the

107
fusion of the autophagosome and lysosome, a key component of autophagy, concomitantly
inducing p38 phosphorylation. The mechanism of inducing p38 phosphorylation by CQ and
Baf-A1 is currently unknown. Comprehensively, autophagic flux inhibition significantly
suppresses the myofibroblast phenotype.

108
Chapter 2: Autophagy and Cardiac Fibrosis In Vivo
Introduction
Myocardial remodeling is a key event after myocardial infarction (MI). One of the
important hallmarks of cardiac remodeling is the activation of cardiac fibroblasts to
hypersecretory myofibroblasts [101]. Fibroblasts comprise the largest cell population in the
myocardium, arising from various sources, and are principally involved in producing
structural proteins such as fibrillar collagens, which form much of the cardiac extracellular
matrix (ECM) [62]. Cardiac injuries (such as MI) trigger fibroblast activation to
myofibroblasts [119]. After MI, the heart undergoes wound healing wherein fibrillar collagen
deposition contributes to the infarct scar [65]. While this may be viewed as a beneficial event
in the short-term post-MI heart, chronic uninhibited and inappropriate scarring often occurs
post-MI and the cells responsible go on to infiltrate the non-infarcted tissue. Reduced
integrity and elasticity of cardiac tissue results in thickening and stiffening of the remnant or
surviving myocardium, which is also referred to as cardiac fibrosis [325]. Cardiac
myofibroblasts play a key role not only in reparative fibrosis post-MI, but also in
hypertrophy-induced remodeling and fibrosis. Phenoconversion to myofibroblasts is
promoted by growth factors such as transforming growth factor beta (TGF-β), platelet-
derived growth factor (PDGF), and connective tissue growth factor (CTGF), vasoactive
peptides such as angiotensin II (ANGII), as well as a variety of other profibrotic cytokines
released by cardiomyocytes and non-myocyte cells in the injured heart [94]. Physical changes
in the ECM resulting from cardiac injury doubly promote remodeling by allowing the release
and activation of latent TGF-β as well as exerting mechanical tension that has also been
shown to promote fibroblast activation [326, 327]. This activation of fibroblasts shifts the
balance to favour ECM synthesis, leading to ECM thickening and contraction, increasing
synthesis and accumulation of fibrotic depositions that can replace the myocytes and/or

109
interrupt the myocyte–myocyte interactions in the myocardium [328, 329]. Enhanced
stiffness of the myocardium impedes ventricular contraction and relaxation as well as
electrical signaling between cardiomyocyte, leading to distorted architecture, impaired
systolic and/or diastolic function of the heart, and arrhythmias [330].
Recent evidence from our lab suggests that autophagy favours fibrosis by enhancing
activation of cardiac fibroblasts to myofibroblasts [256]. Autophagy is associated with
acquisition of markers of fibroblast activation including increased protein levels of
ACTA2/α-SMA (actin-α-2/α-smooth muscle actin), enhanced expression of collagens, and
the formation of stress fibres, which impart contractility for proper wound contracture [256,
288]. This work demonstrated that autophagic fibroblasts also have increased expression and
secretion of CTGF. Inhibition of autophagy using mTOR inhibitor, class I phosphoinositide
3-kinase and class III phosphatidylinositol 3-kinase (PI3K) inhibitors, or through ATG7
silencing, prevented fibroblast activation and attenuated LV remodeling in a mouse model of
MI. In spite of these findings, the mediators linking autophagy to fibroblast activation and
fibrosis still remains largely undefined.
In Chapter One, we demonstrated a link between autophagy and fibroblast activation
using an in vitro mechanical stress model of rat cardiac fibroblast activation (eg, plating on a
noncompressible matrix). One of the critical components for autophagy is the proper
functioning of the lysosome and/or promoting its fusion with autophagosomes. Thus, we
hypothesized that the autophagic insult observed during MI could be attenuated using
chloroquine, which is a lysosomal lumen alkalizer that blocks the autophagy progress by
impairing lysosome formation. Chloroquine and its analog hydroxychloroquine are the only
clinically relevant autophagy inhibitors that are widely used as anti-cancer, anti-malarial and
anti-rheumatoid agents [331]. Recently, Ohm et al. (2014) demonstrated that chloroquine can

110
inhibit TLR9-mediated inflammatory responses in murine cardiac fibroblasts. Of interest,
altered TLR 9 signaling has pathophysiological significance both in various cardiovascular
disorders and cardiac remodeling [332].
As we established the efficacy of chloroquine as an anti-fibrotic agent in our in vitro
model of cardiac fibroblast activation to myofibroblasts, we sought to determine the effects of
chloroquine at the preclinical level using in vivo rat model of MI induced via permanent
ligation of the left anterior descending artery (LAD). Interruption of blood flow via LAD
ligation leads to ischemia, mimicking MI and leads to the development of a fibrotic scar
tissue. This surgical procedure imitates the pathobiological and pathophysiological aspects
occurring in MI and thus is apt for the study of protective effect of chloroquine in post-MI
cardiac remodeling.
Methodology
As described in the Materials and Methods section above, we utilized permanent
surgical occlusion of the LAD in rats as an experimental model of MI, and sham-operated
controls underwent surgery without permanent occlusion of the LAD. Timed myocardial
infarction (MI) and sham treatment groups received 40 mg/kg/day injected chloroquine
(CQ) either for 3 weeks (from week 1 in the 4-week model) or 2 weeks (from week 6 in
the 8-week model) (Figure 6). Histology was used to assess autophagy and cardiac fibrosis
parameters, as described in detail in the Materials and Methods section. Echocardiography
was performed at 4 or 8 weeks to evaluate functional parameters such as ejection fraction
(EF%), fractional shortening (FS%), heart rate (HR), and left ventricular posterior wall
thickness diameter (LVPWd). All rats with MI were examined by an echocardiographist to
confirm the results.

111
Results
Fibroblast phenotype and inhibition of autophagy in the post-MI heart
We utilized Western blotting to determine changes in protein levels of the autophagy
markers LC-3I and LC-3II, with β-tubulin as a control for total protein loading. At 4 weeks
post-MI, although we observed a trend towards increased accumulation of the autophagy
marker LC-3I in untreated (no drugs; ND) viable and scar regions of the heart, the differences
were not significant (Figure 18A). No notable changes in LC-3II accumulation were observed
in untreated hearts post-MI. However, in CQ-treated hearts, there was a modest (but non-
significant) trend towards increased LC-3I in the viable region and a significant (P ≤ 0.05)
increase in LC-3I in the scar region, compared to sham-operated CQ-treated controls (Figure
18A). We observed a trend towards an increase in LC-3II in viable and scar regions of post-
MI CQ-treated hearts, though this change was not significant (Figure 18A). When comparing
ND and CQ-treated groups, we observed that CQ treatment caused increased accumulation of
LC-3I in post-MI viable and scar regions. In summary these results show increased
autophagy in the infarct scar vs control. CQ treatment in scar region led to significant LC-3II
accumulation which is associated with inhibition of autophagy.
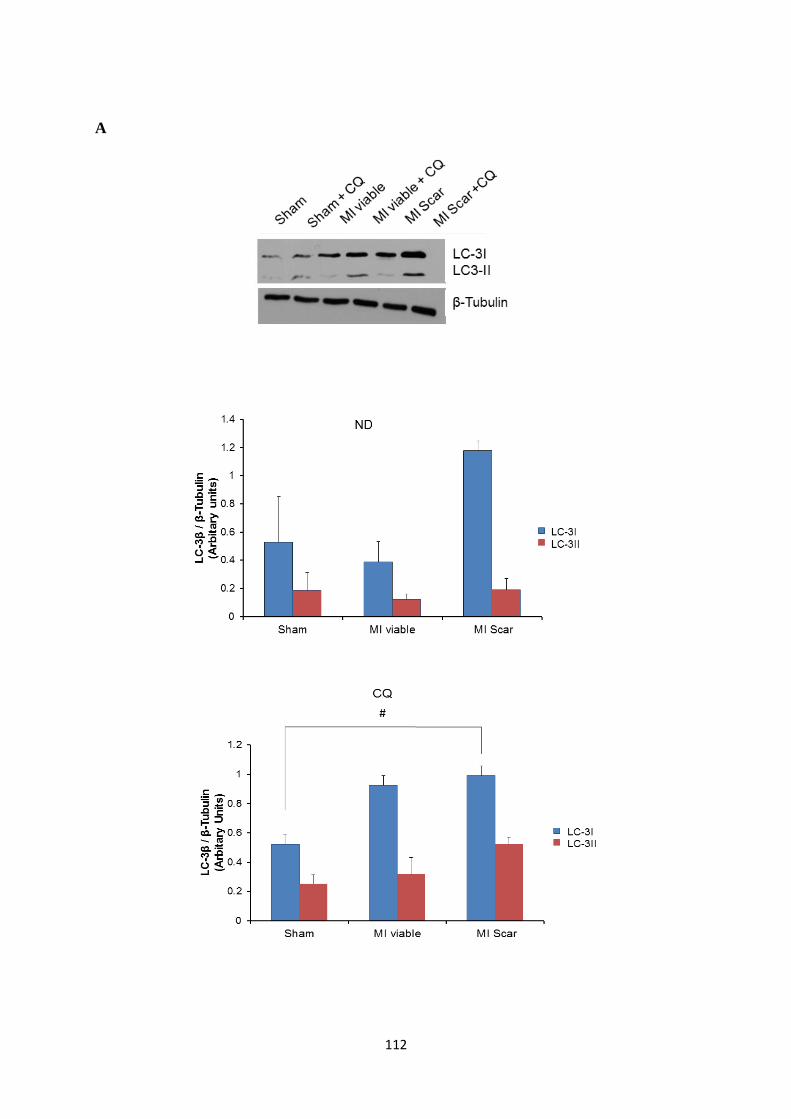
112
A
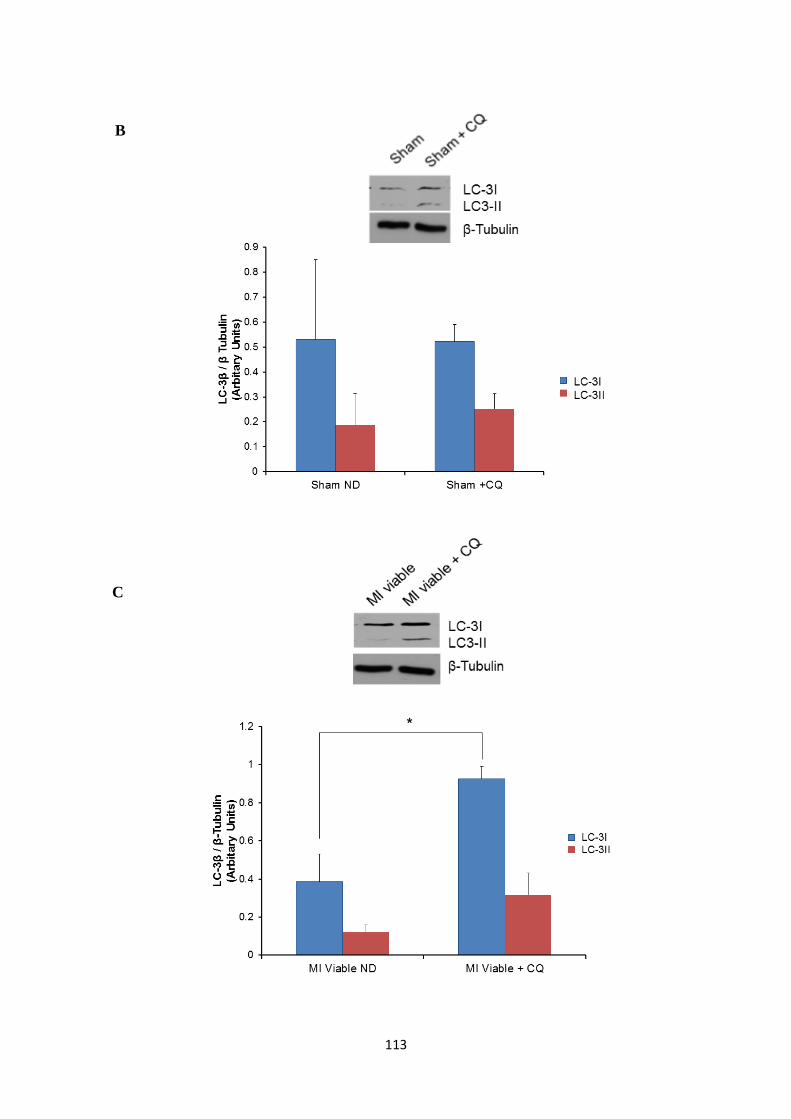
113
B
C
B
C

114
Figure 18: LC-3 lipidation as a marker of autophagy in 4 week untreated and
chloroquine-treated post-MI rat hearts (control, myocardium remote to the scar and the
infarct scar). Panel A. Western blot for autophagy markers LC-3I and LC-3II in untreated
(no drugs; ND) and chloroquine (CQ)-treated hearts. Levels of LC-3I trended toward increase
in the post-MI scar region of untreated (no drug; ND) hearts. No change in LC-3II levels was
observed between untreated sham-operated and MI hearts. CQ treatment (40mg/kg-1
/day for 2
weeks beginning at 1 week post-MI) induced significantly higher levels of LC-3I in post-MI
scar regions, but not viable regions. CQ treatment produced a trend towards increased levels
of LC-3II in post-MI viable and scar regions, though levels did not differ significantly. ND:
no drugs, #compared to sham-operated, P < 0.05.Significance was determined using one-way
ANOVA followed by a Student-Newman-Keuls post-hoc test.
D

115
Panel B. Western blot for LC-3I and LC-3II in sham-operated untreated vs. CQ-treated
hearts. There was no significant difference in autophagy marker levels in sham-operated
hearts in untreated vs. CQ-treated groups. Panel C. Western blot for LC-3I and LC-3II in the
viable region of post-MI hearts, comparing untreated vs. CQ-treated groups. There was a
significant increase in LC-3I expression in the viable region of post-MI hearts treated with
CQ, compared to untreated hearts(*P<0.05 compared to no drugs; ND).Significance was
determined using student-t test. No changes were observed in LC-3II levels with CQ
treatment. Panel D. Western blot for LC-3I and LC-3II in the scar region of post-MI hearts,
comparing untreated vs. CQ-treated groups. No changes were observed in LC-3I levels for
untreated and CQ-treated hearts. There was a significant increase in LC-3II expression in the
scar region of post-MI hearts treated with CQ, compared to untreated scar controls. (#P<
0.05compared to no drugs, data is mean ± SEM, n=3-6 distinct cardiac samples per
group).Significance was determined using student-t test.

116
To determine the effect of MI and CQ treatment on myofibroblast presence in rat
hearts at 4 weeks, we performed Western blotting analysis for the key myofibroblast marker
α-SMA. In untreated hearts, MI was associated with increased α-SMA protein levels in viable
tissue, though this difference was not statistically significant. However, MI is linked to a
marked and significant (P< 0.05) increase in α-SMA levels in scar tissue vs controls (Figure
19). These results indicate an increase in cardiac fibroblast activation at 4 weeks post-MI in
the infarct scar of these rats. CQ treatment was effective in significantly (P< 0.05) reducing
α-SMA expression in both the viable and scar regions of post-MI hearts, compared to
untreated controls for these regions (Figure 19).

117
Figure 19: -SMA marker levels in 4 week untreated and chloroquine-treated post-MI
rat hearts.Western blot analysis of α-SMA (marker of activated fibroblasts)from left
ventricular tissue lysates of sham-operated and MI rat hearts, either untreated or treated with
chloroquine (CQ; 40mg/kg-1
/day for 2 weeks beginning at 1 week post-MI). α-SMA
expression in untreated and CQ-treated sham hearts was low. In scar regions of post-MI
hearts, α-SMA was significantly increased, compared to viable post-MI and sham-operated
hearts. Treatment with CQ significantly reduced α-SMA levels in these regions, as compared
to the same regions with no drug (ND). (*compared to sham ND; #compared to MI viable
ND; фcompared to MI viable ND;
ccompared to MI scar ND; P< 0.05 for all comparisons).

118
Data is mean ± SEM (n=3-6 distinct cardiac samples per group). ND: No drugs. Significance
was determined using one-way ANOVA followed by a Student-Newman-Keuls post-hoc test.

119
To complement these findings in left ventricular lysates, we used immunostaining to
examine autophagy (via LC-3β accumulation) and fibroblast activation (α-SMA staining) in
cardiac tissues at 4 weeks in sham-operated and MI groups with and without CQ treatment.
Immunofluorescence staining for LC-3β staining was increased in post-MI hearts with and
without CQ treatment (Figure 20). We also observed an increase in α-SMA staining in post-
MI hearts, compared to sham-operated controls, which was apparently diminished with CQ
treatment post-MI. These results confirm that elevated autophagy accompanies accumulation
of α-SMA untreated infarct scar. CQ treatment was associated with reduced expression of α-
SMA in the left ventricle at 4 weeks post-MI. Thus treatment with CQ in 4 week post-MI rat
hearts is associated with reduction of fibroblast activation when compared to the untreated
MI rat hearts.
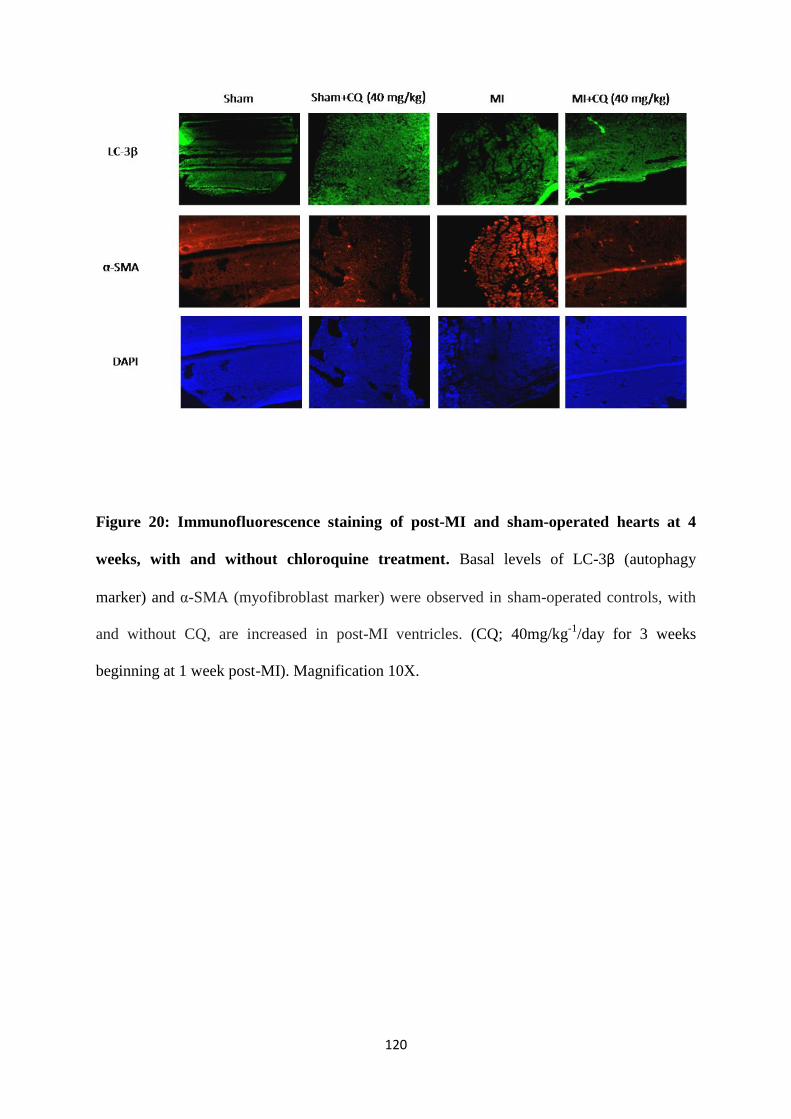
120
Figure 20: Immunofluorescence staining of post-MI and sham-operated hearts at 4
weeks, with and without chloroquine treatment. Basal levels of LC-3β (autophagy
marker) and α-SMA (myofibroblast marker) were observed in sham-operated controls, with
and without CQ, are increased in post-MI ventricles. (CQ; 40mg/kg-1
/day for 3 weeks
beginning at 1 week post-MI). Magnification 10X.

121
As the reduction ofα-SMA expression in CQ treated 4 week-post MI rats was
significantly reduced, it is reasonable to explore the concomitant effect of CQ treatment on
collagen accumulation in infarcted myocardium using Masson’s trichrome imaging and
hydroxyproline assay. While increased collagen content was present in the 4 week post MI
rat heart sections vs. controls, we found that CQ treatment of these animals did not affect
collagen levels post-MI

122
Figure 21: Masson’s trichrome staining for collagen content in 4 week post-infarct left
ventricles, with and without chloroquine treatment. Blue staining in control and
experimental sections above reveals collagen deposition. At 4 weeks after infarction, collagen
staining is increased in intensity and density of staining, and is unchanged with CQ treatment
vs. untreated controls. Magnification 4X.

123
To quantify and validate our Masson’s trichorme stain experiements, we assayed
hydroxyproline content across various groups. The helical structure of collagen is due to the
post-translational hydroxylation of the prolines present in the collagen. Thus presence of
hydroxyproline is restricted to fibrillar collagens and elastin. Due to the low content of elastin
proteins, the precedent in the literature is to use hydroxyproline content to infer myocardial
fibrillar collagen content [294]. Our results with this second assay paralleled those noted
inMasson’s staining experiments. That is, we found increased collagen accumulation found in
4 week post-MI untreated animal vs control. There was no significant reduction found in our
CQ treated MI groups vs untreated MI groups. This set of data revealed that the CQ treatment
is linked to deactivation of fibroblasts, but not with any change in collagen accumulation.

124
Figure 22: Hydroxyproline collagen quantification in 4 week post MI cardiac tissue:
Histographical representation of the hydroxyproline content in 4 week post MI rat hearts
indicate increased collagen content in the MI untreated groups when compared to untreated
sham. We observed no significant difference in myocardial hydroxyproline content of the
MI+ CQ treated samples vs. control. Values are expressed as mean of 4 -6 per group. (*
P<0.05 experimental vs. controls). Significance was determined using one-way ANOVA
followed by a Student-Newman-Keuls post-hoc test.
*

125
Similar to in the 4-week study group, we assessed expression for autophagy and
myofibroblast markers in 8-week study group hearts post-MI for untreated and CQ-treated
animal tissue lysates. Western blot analysis for LC-3I and LC-3II show a trend (but not
significant) toward increased expression of LC-3II in MI scar tissue, compared to MI viable
and sham-operated, in untreated hearts (Figure 23). CQ-treated groups show a similar trend
(non-significant) to increased expression of both autophagy markers (Figure 23). Our analysis
of α-SMA expression showed similar results. In untreated hearts, α-SMA levels were
significantly increased in the MI scar, but not in the viable region, compared to sham-
operated controls (Figure 24). Treatment with CQ appeared to show a trend (non significant)
towards decreased α-SMA levels in the MI scar region (Figure 24). Immunofluorescence
staining for 8-week study group hearts post-MI with and without CQ also support the
Western blot data, showing some degree of decrease in α-SMA staining with CQ treatment
post-MI (Figure 25).
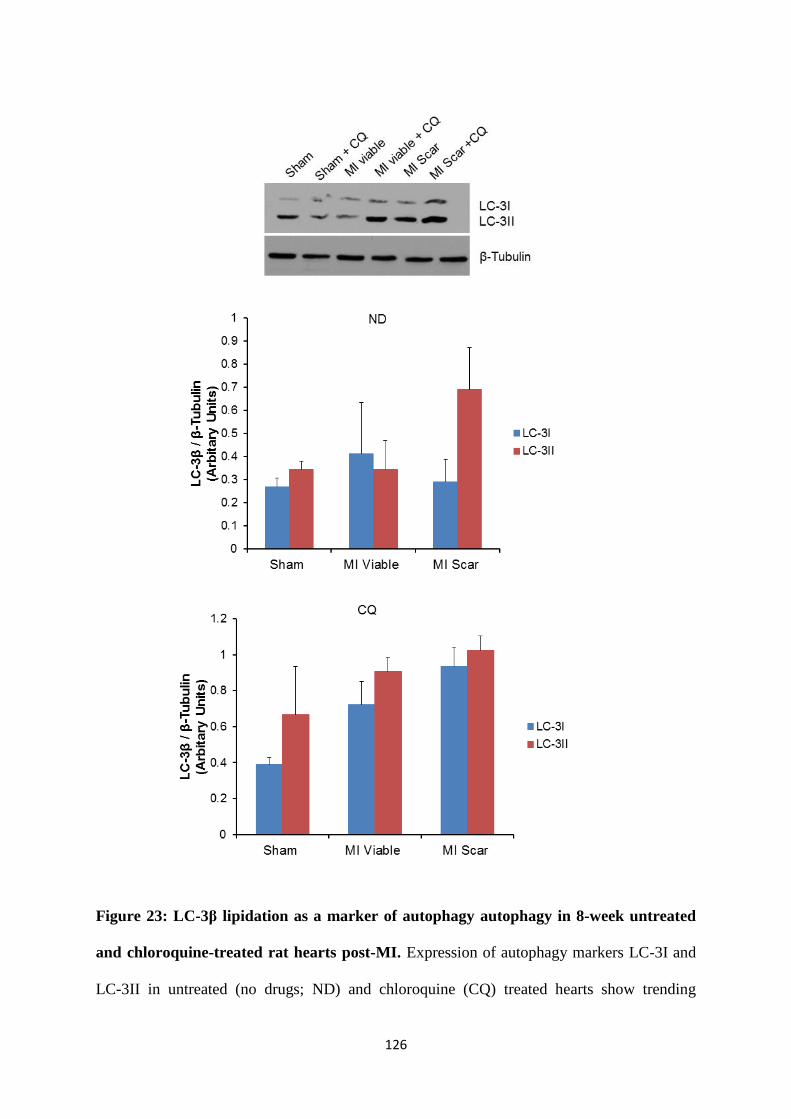
126
Figure 23: LC-3β lipidation as a marker of autophagy autophagy in 8-week untreated
and chloroquine-treated rat hearts post-MI. Expression of autophagy markers LC-3I and
LC-3II in untreated (no drugs; ND) and chloroquine (CQ) treated hearts show trending

127
increase in LC-3II levels in the post-MI scar region of untreated (no drug; ND) heart and CQ
treated heats when compared to sham-operated treated and untreated groups respectively.
However this did not reach significance. Data are expressed as mean ± SEM (n=3 - 4). ND:
No drugs.

128
Figure 24: α-SMA expression in 8-week post-MI hearts treated with chloroquine. α-
SMA expression was assessed in CQ treated and untreated post-MI hearts after at end of 8
week post-MI. We saw a significant increase in α-SMA expression in the infarct scaring the
non-treated groups when compared to sham-operated ND group (*compared to sham ND). In
contrast, no significant similar increase ofα-SMA expression was observed in the MI+ Scar
CQ treated vs. the untreated group of same region. We interpret this finding to show that
autophagy is suppressed in the CQ treated infarct scar samples. Data is the mean ± SEM of
n=3-4 hearts). ND: No drugs. Significance was determined using one-way ANOVA followed
by a Student-Newman-Keuls post-hoc test.

129

130
Figure 25: Immunofluorescence staining of 8 week post-MI hearts with and without
chloroquine. LC-3β (autophagy marker) and α-SMA (myofibroblast marker) were observed
in sham-operated controls, with and without CQ, with increase in post-MI ventricles. (CQ;
40mg/kg/day for 3 weeks beginning at 5 week post-MI). Mgnification 10X.

131
Assessment of Biochemical Parameters for Cardiac, Liver and Kidney Damage
The enzymes routinely measured clinically to diagnose and monitor myocardial
infarction include lactate dehydrogenase (LDH) in the early stages following MI. Increased
LDH long-term may indicate damage to various tissues, including the heart, liver, kidney,
muscle, lungs, and erythrocytes [333, 334]. To diagnose organ damage, biochemical assays
are used to measure serum levels of creatinine for kidney damage [335], and aspartate amino
transferase (AST)and aspartate transaminase (ALT) for liver damage [336]. There was no
difference in the LDH or creatinine levels in any of the groups at 4 and 8 week post-MI when
compared to sham-operated controls, suggesting that the drug has no effects of widespread
tissue or kidney-specific damage. We observed a trend towards elevation in AST and ALT
levels at 4 and 8 weeks in the CQ treated MI groups (Figure 26), indicating a possibility of
low-level sustained damage to the liver following CQ treatment post-MI.
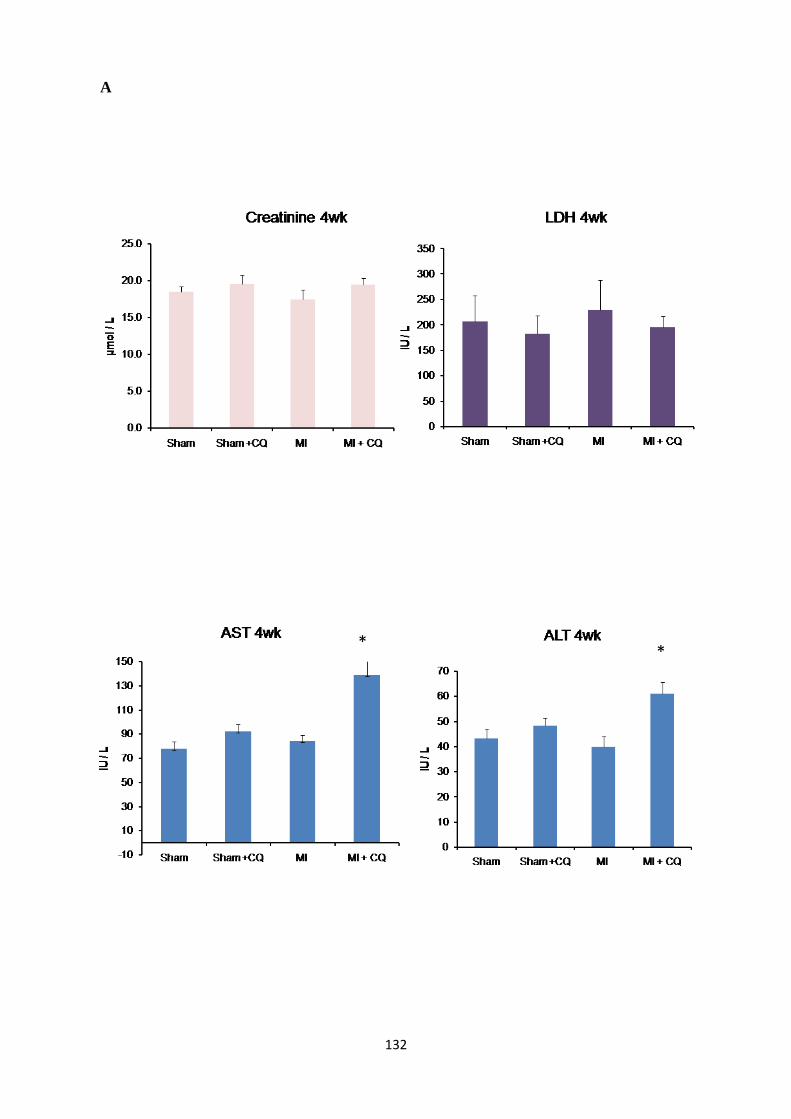
132
A
* *

133
B

134
Figure 26: Biochemical parameters for cardiac, kidney and liver damage in 4 and 8
week post-MI rat hearts with and without chloroquine treatment. Panel A. At 4 weeks,
no significant differences were observed in serum markers for wide spread tissue damage
(lactate dehydrogenase; LDH) nor damage to the kidney (creatinine). There was a significant
increase in serum marker of liver (aspartate amino transferase (AST) and aspartate
transaminase (ALT).(* P< 0.05 MI vs. MI + CQ). Significance was determined using one-
way ANOVA followed by a Student-Newman-Keuls post-hoc test. Panel B. At 8 weeks,
there are still no significant increases in these damage markers, though again there is a trend
towards increased AST and ALT in the CQ-treated post-MI group. (CQ; 40 mg/kg-
1/day).Data expressed as mean ± SEM (n=4-6).

135
Functional Assessment Following Myocardial Infarction and Chloroquine Treatment
To determine whether CQ treatment of post-MI animals translated to improvements in
cardiac functional changes, we assessed cardiac functional parameters using
echocardiography. We confirmed via biochemical assay that our 40mg/kg-1
/day CQ drug
regime did not invoke tissue damage (Figure 26).Using non-invasive transthoracic
echocardiography (TTE), we obtained the M-mode echocardiography images and evaluated
the standard functional parameters of cardiac function including injection fraction (EF),
fractional shortening (FS), left ventricular posterior wall diameter (LVWPd) and heart rate
(HR). All the parameters were monitored in the second, third and fourth week in the 4-week
study group, and during the fifth, sixth and eighth week in the 8-week study group. In the 8-
week study group, we observed significant reductions in ejection fraction to approximately
60% in post-MI animals, compared to sham-operated controls, remaining consistent
throughout the 2-4 week period. Treatment with CQ was not associated with any change in
the ejection fraction in infarcted animals, which remained significantly lower than in sham-
operated + CQ animals and at similar levels as untreated post-MI groups, around the 60%
mark (Figure 27A). We also observed significantly decreased fractional shortening,
indicating reduced contractility, from~50% in sham-operated-operated controls (with and
without CQ) to less than 30% in post-MI animals, either with or without CQ treatment
(Figure 27A). In the 8-week study group, we observed similar results, with ejection fraction
being reduced by about 30% in post-MI animals with or without CQ. Similarly, fractional
shortening was reduced to less than 30% in all post-MI animals, again regardless of CQ
treatment (Figure 28B). Heart rate and LVPWd were highly variable in the 4-week treatment
group, but there were no significant differences between any of the treatment or control
groups (Figure 28A). Similarly, we found no difference in these parameters in the 8-week
study group (Figure 28B).
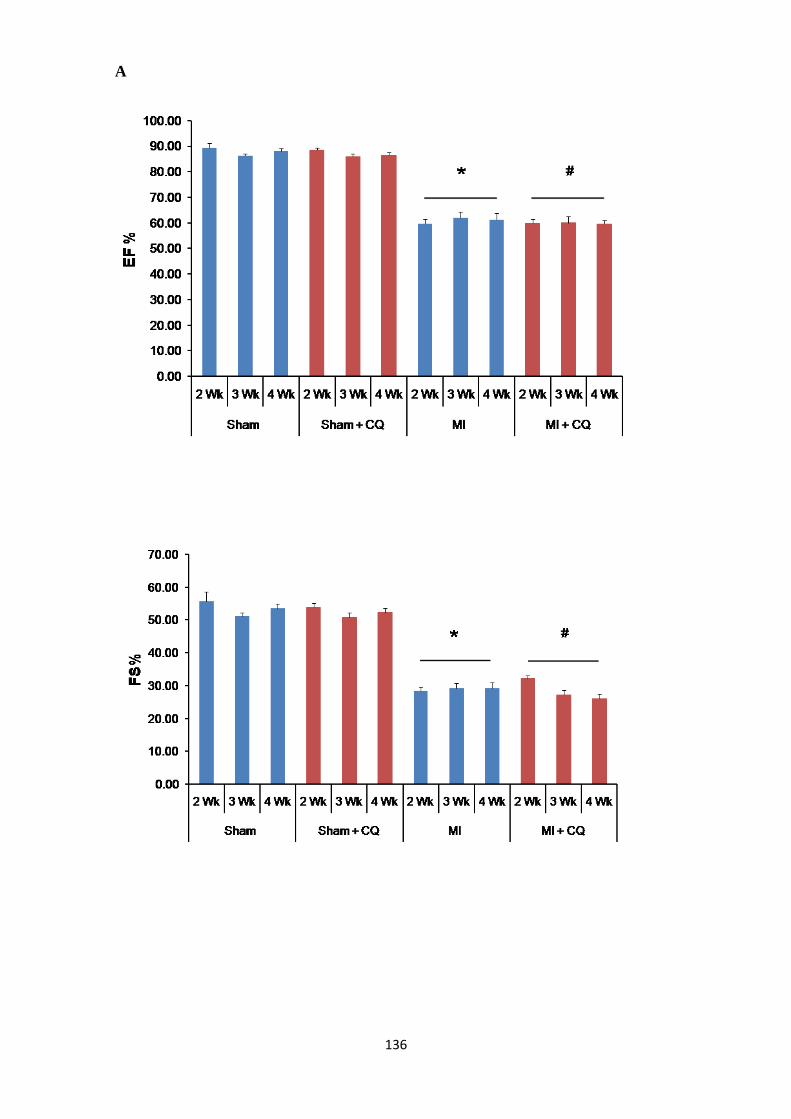
136
A
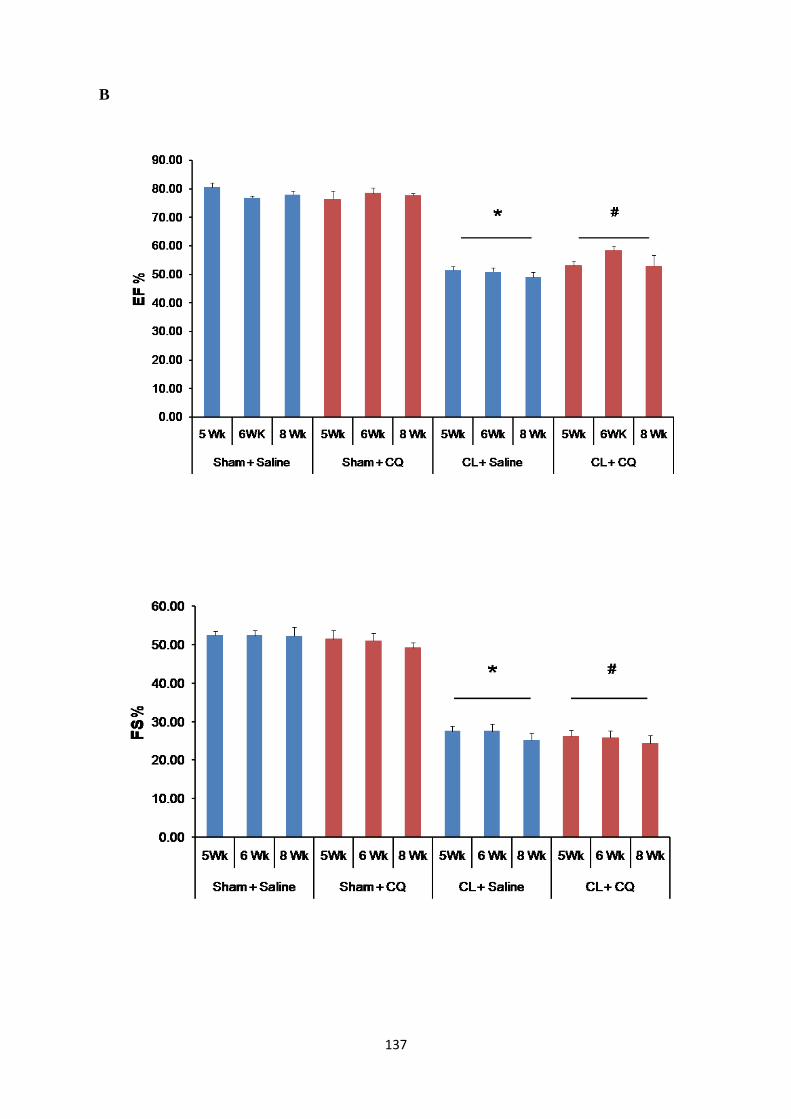
137
B

138
Figure 27: Ejection Fraction and Fractional Shortening in Post-MI Rats. M-Mode
transthoracic echocardiography (TTE) was performed at 2, 3 and 4 weeks (for 4-week study
groups) or 5, 6, and 8 weeks (for 8-week study groups) after the LAD- ligation surgery to
assess ejection fraction (EF) and fractional shortening (FS). Panel A. In the 4 week study
group, EF and FS were both significantly reduced following MI, with or without CQ
treatment. Panel B. In the 8 week study group, EF and FS were significantly reduced post-MI
regardless of CQ treatment. No progressive decrease in function was observed over the
duration of TTE examinations. (*compared to sham-operated; #compared to sham-operated +
CQ; P < 0.05 for all comparisons). Data is mean ± SEM (n=4-6). Significance was
determined using one-way ANOVA followed by a Student-Newman-Keuls post-hoc test.

139
A
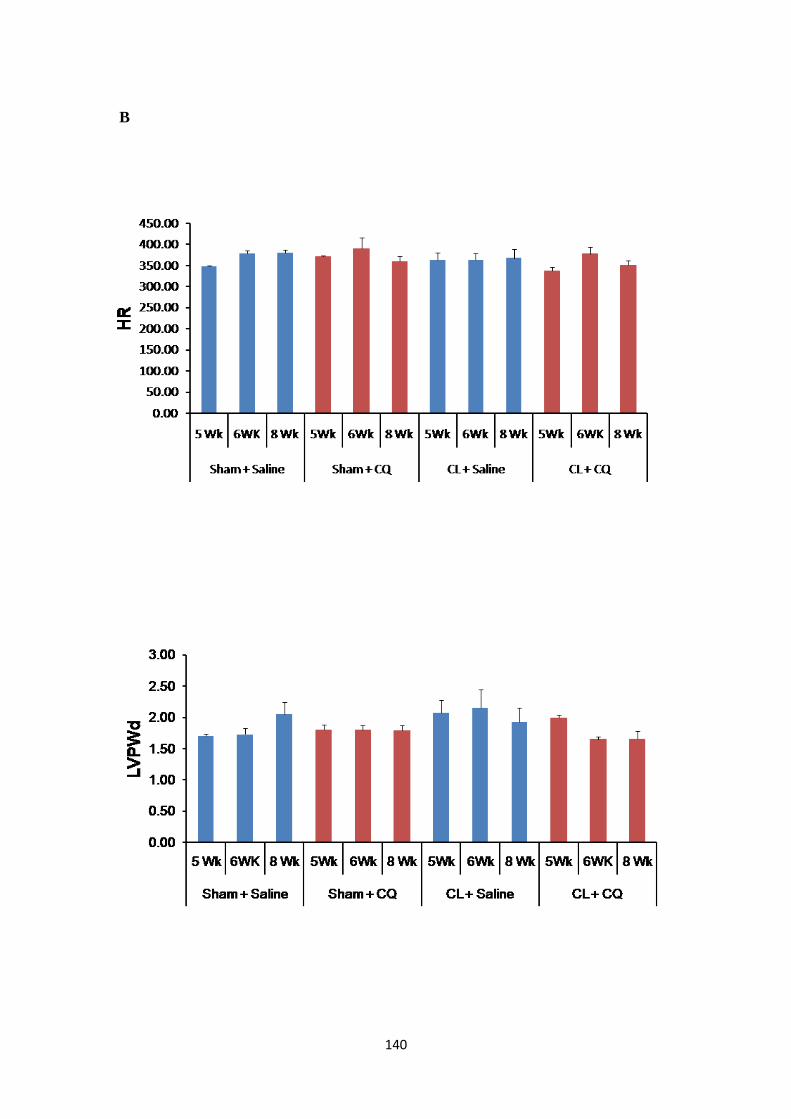
140
B

141
Figure 28: Heart Rate and Left Ventricular Posterior Wall Diameter in Post-MI Rats.
M-Mode transthoracic echocardiography (TTE) was performed at 2, 3 and 4 weeks (for 4
week study groups) or 5, 6, and 8 weeks (for 8 week study groups) after the LAD- ligation
surgery to assess heart rate (HR) and left ventricular posterior wall diameter (LVPWd). In
both 4 week (Panel A) and 8week (Panel B) study groups, HR and LVPWd did not change
following MI, with or without chloroquine treatment (40 mg/kg-1
/day). Data is mean ± SEM
(n=4-6).

142
Discussion
We have investigated the role of autophagy in cardiac fibrosis and its impact on heart
function using an in vivo experimental model of MI in combination with CQ treatment. We
utilized Western blotting and immunofluorescence to assess changes in protein levels for
autophagy and myofibroblast markers in the 4-week post-MI model. We observed significant
increases in LC-3I (in the viable region) and LC-3II (in the scar region) in post-MI animals
treated with CQ (Figure 18, Panels C and D) vs. untreated post-MI hearts, as well as
increased LC-3I in the scar region post-MI compared to sham-operated controls, both of
which were treated with CQ (Figure 18A). Controversy regarding interpretation of LC-3 data
from Western blots exists in the literature [337], and it is suggested that this lies within the
approach used to calculate lipidation ratios [338]. In addition, LC-3II may be degraded by the
autophagolysosome, and needs to be measured against an internal control, such as beta-
tubulin. Increased LC-3II levels are associated with increased autophagosome formation (or
decreased turnover, if trafficking to lysosomes is delayed or there is reduced fusion or
impaired proteolytic activity of lysosomes). However, in the presence of inhibitors such as
Baf-A1 and CQ, increased LC-3II levels are observed, due to inhibition of lysosomal
proteases and impaired degradation of lysosomal contents [155]. Failure of these inhibitors to
enhance LC-3II levels would indicate defects earlier within autophagy, prior to the fusion
events wherein these inhibitors have their effects [338]. In 4 week post-MI experimental
groups, autophagy was significantly elevated in the infarct scar, and was inhibited with CQ
treatment(Figure 18). However this result was not associated with any change in collagen
staining of post-MI hearts, nor in their hydroxyproline content with CQ treatment. Taken
together, these results indicate that CQ treatment diminished α-SMA and inhibited
autophagic flux in treated hearts following MI at 4 weeks, but that these changes were not
linked to any effect on collagen deposition. We suggest that this scenario exists because of

143
the timing of the experiment: that is, with initiation of CQ treatment, fibrosis and collagen
deposition are already well underway, along with reduced EF in experimental animals. Also,
it is known that the biological half-life of collagen is 80-120 days [117], which means that
lag-time of any anti-fibrotic treatment is significant. While CQ may restrict activation of
fibroblasts, the window for the duration of treatment is too narrow to be asscociated with
reduction of collagens. We suggest that an earlier administration of CQ post-MI could have
restricted the formation of collagen and inhibited fibrosis.
The role of autophagy in cardiac pathology remains controversial, as some reports
emphasize a protective role for autophagy in the heart, whereas other reports emphasize its
negative effects on cardiac function. Liu et al. performed in vitro and in vivo autophagy
experiments in cardiac fibroblasts and C5BL/6 mice, respectively, which demonstrated a
protective role for autophagy against lead toxicity [104]. When 3MA was used to inhibit
autophagy, this group observed enhanced lead toxicity in cardiac fibroblasts, resulting in cell
death [339]. Another study by Liu et al. in 2016 used ANGII treatment both in vitro and in
vivo to induce cardiac fibrosis. In this study, the researchers found that enhancing autophagy
(via rapamycin) decreased upregulation of collagen type I and fibronectin expression in rat
cardiac fibroblasts, and inhibition with CQ attenuated these effects. In vivo, rapamycin in
combination with angiotensin II infusion ameliorated both cardiac fibrosis and dysfunction,
while inhibition with CQ worsened these ANGII-mediated effects [340]. Xie et al. found that
chronic metformin treatment of type I diabetic mice enhanced autophagy and improved
cardiac function, indicating a protective role for autophagy in this model [271].
Other reports have provided evidence to underscore autophagy as a maladaptive
response and have demonstrated therapeutic potential for autophagy inhibitors. Xu et al.
found that in a diabetic mouse model, wild type diabetic hearts had increased levels of
oxidative stress, interstitial fibrosis, myocyte apoptosis, and reduced cardiac function

144
compared to heterozygous Beclin-/- and Atg16-/- animals [341]. In that study, Beclin-1
rescue via over-expression restored autophagy and worsened cardiac function, demonstrating
an adverse role for autophagy in the hearts of diabetic mice. Similarly, a report of transverse
aortic constriction (TAC)-induced model of pressure overload, TAC increased cardiac
autophagy to elevated levels for durations reaching 3 weeks. Further enhancement of
autophagy by Beclin-1 overexpression further heightened autophagic activity and increased
adverse remodeling in pressure-overloaded hearts [245]. The variable effects of autophagy in
different models of cardiac pathology indicate a complex role for this process in the heart. It
is also pointed out that multiple cell types exist in the heart and that autophagy may occur in
myocytes, endothelial cells, smooth muscle cells, and fibroblasts, and that some of the
aforementioned studies did not distinguish among them. In our current in vivo work, the
distinction between these cell types was also difficult to resolve. Other limitations of our in
vivo studies may be present in the time of treatment of the post-MI animals with CQ, as the
cardiac collagen levels (4-OH levels) in treated vs. untreated samples was similar between
groups. As mentioned above, we speculate that earlier CQ administration post-MI in our
experimental design may have effected collagen accumulation. Finally we focused the current
study on detection of systolic dysfunction in treated and untreated damaged hearts. This point
could be argued to be a design omission within the experimental design as diastolic left
ventricular (LV) dysfunction is known to be primarily associated with heart failure with
preserved ejection fraction (HFpEF) [342], and while we recognize that this is important
feature of fibrosis, we were not able to fully assess putative subtle changes in cardiac
diastolic function. This issue may have limited our ability to link cardiac filling dysfunction
between treated and untreated groups in damaged hearts, even in the presence of unaltered
global cardiac collagen levels among groups.

145
Other studies using animal models of MI have highlighted the role of autophagy in
post-MI remodeling and cardiac fibrosis, and points to the involvement of temporal changes
in autophagic flux in the injured heart. In 2014,a study by Wu et al. revealed that in mouse
subjected to LAD-induced MI, increased autophagic induction occurred in the acute phase of
MI (0.5-3 days) and was subsequently reduced in the latter phase of MI (5-21 days). One day
post-MI, treatment with rapamycin reduced post-MI remodeling and dysfunction, whereas
inhibition with 3MA worsened these outcomes [290]. Zhang et al. found that berberine-
induced increases in LC3-βII and beclin-1 expression were cardioprotective at 4 weeks post-
MI, reducing interstitial fibrosis and adverse remodeling and improving ejection fraction and
fractional shortening [343]. These studies underscore the importance of autophagy in the
pathogenesis of heart failure as it relates to cardiac fibrosis.
Although the link between autophagy and cardiac fibrosis is still relatively
unexplored, reports in lung, liver and kidney reveal a positive correlation between autophagy
and fibrosis [310, 344] and advocate for anti-autophagic molecules as novel therapeutics.
This is also supported by our earlier work in human atrial fibroblasts demonstrating that
TGF-β-mediated elevation of profibrotic markers collagen type I and fibronectin was
associated with increased autophagic flux [256]. In this study, we also demonstrated
concomitant increases in levels of protein markers of fibrosis, autophagy, and Smad2
phosphorylation (indicating increased TGF-β signaling) in whole scar tissue lysates from the
hearts of post-MI rats at 2 and 4 weeks. Atg5-knockout mouse embryonic fibroblasts also
showed decreased fibronectin synthesis [256]. While our in vitro (Chapter 1) results are
supportive of the involvement of autophagy inhibition in attenuating fibroblast mediated
profibrotic events, our in vivo data (Chapter 2) do not support the argument for the
development of therapeutic agents that influence autophagy in cardiac fibroblasts. We
suggest that addressing the autophagy/cardiac fibrosis hypothesis remains comprehensive in

146
the context of influencing cardiac fibrosis in vivo, but with a refined study using an early time
point for CQ administration or, alternatively, other novel autophagy inhibitory agents with
greater specificity.
Morphological and structural changes are not clinically significant without
corresponding significant changes in cardiac function that leads to improved patient
outcomes. We performed echocardiography in all animal groups, a clinically relevant tool to
assess cardiac function. The procedure is especially useful due to its non-invasiveness and
reflects ventricular dysfunction and hemodynamic alterations resulting from cardiac
pathologies [132]. As expected, MI resulted in decreased functional changes as early as 2
weeks post-MI compared to sham-operated controls, namely ejection fraction and fractional
shortening (Figure 27), which are indicators of pumping efficiency and contractility,
respectively [130, 345]. Notably, these parameters were not altered with CQ treatment of
damaged hearts in either the 4- or 8-week study groups. Similarly, heart rate of post-MI rats
did not change significantly with CQ treatment over the time durations examined in this study
(Figure 28). In the 4week study group, we saw no change in left ventricular posterior wall
diameter (LVPWd), though there was a modest trend towards decreased LVPWd over time
(eg, 5 to 8 weeks) with CQ treatment (Figure 28).The lack of functional changes and lack of
any change in collagen content in treated post-MI hearts vs. controls may be due to
insufficient dosage. Our study used 40 mg/kg/day, but other in vivo studies used from 100 to
500 mg/kg/day[346]. We chose to treat animals with a lower dose with respect to the
possibility of generation of side-effects. In support of this presumption, that serum markers of
tissue damage were not significantly increased with CQ treatment in the current study (Figure
26).Physiologically relevant dosages for CQ will likely differ between species, considering
differences in metabolism and heart size, among a host of other factors affecting the response
to CQ and potential side effects.

147
In light of the above observations, our study does support the central hypothesis of
attenuating the activation of fibroblast, however it does not support that low-dose CQ
treatment in a rat model of MI could reverse or attenuate adverse cardiac remodeling. This
notwithstanding, our in vitro work provides supportive evidence for future research to
address autophagy inhibition in treatment of cardiac fibrosis in vivo.

148
General Discussion
Interest in cardiac fibrosis as a primary cause amongst the pathogenesis of heart
failure is burgeoning., following MI [11]. Cardiac fibrosis is characterized by the chronic
accumulation of collagen via the persistence of activated myofibroblasts in the infarcted and
distal regions of the heart, long after wound healing associated with scar formation is
complete [347-349]. In recent years, autophagy has been suggested to regulate cardiac
remodeling, and dysregulated levels of autophagy are implicated in various cardiac
pathologies including pressure overload-induced hypertrophy and fibrosis [258, 340, 343].
Gao et al. reported elevated autophagy with in a diabetes-induced cardiac fibrosis rat model
[350]. A role for autophagy in fibrosis of other organs, including the lungs and kidney has
already elucidated [167, 351-353]. However, the relationship of cardiac fibroblast activation
and autophagy in the context of myocardial infarction and heart failure is still unexplored. In
the current study, we have focused on understanding the relationship between autophagy and
fibroblast activation using pharmacological inhibition of autophagy in unpassaged (P0)
primary cardiac fibroblasts (in vitro) and in an in vivo experimental model of MI 4 and 8
weeks post-MI.
In our in vitro study, when unpassaged primary cardiac fibroblasts were exposed to
mechanical stress, we found enhanced activation of fibroblasts with concomitant conversion
to myofibroblasts. This was demonstrated by increased α-SMA and ED-A FN protein levels
as assessed by Western blotting and immunofluorescence (Figures 7 and 11, respectively).
Concomitant to this, we found elevated expression of LC-3βII, which is an important
molecular marker of autophagy (Figure 8). Several reports are available where autophagy
inhibitors have been used to attenuate fibrosis, including well-known inhibitors 3MA and CQ
[343, 354], though one report indicated worsening of ANGII-induced fibrosis following CQ
treatment [340], and there are no studies examining the effects of Baf-A1 on cardiac fibrosis.

149
In our experiments, these inhibitors successfully inhibited autophagy in the in vitro model,
confirmed by both Western blotting and TEM (Figures 8 and 9). These inhibitors, in a dose-
and time-dependent manner, caused reduced activation of P0 rat cardiac fibroblasts to the
myofibroblast phenotype, indicated by significant reductions in α-SMA and ED-A FN
proteins levels compared to controls (Figure 10). Immunofluorescence staining of unpassaged
cardiac fibroblasts treated with Baf-A1, 3MA, or CQ showed concomitant decreases in LC-
3II staining and α-SMA stress fiber formation (Figure 11). Functional myofibroblast
functions, including motility and contractility, were also reduced with autophagy inhibition
(Figures 12 and 13). These lines of evidence provide strong support for the role of autophagy
in the fibroblast activation and the efficacy of autophagy inhibitors in preventing the
myofibroblast phenotype.
Among the three inhibitory drugs, CQ appeared to be the most effective inhibitor of
fibroblast activation, thus we utilized CQ to further understand the mechanism underlying the
relationship between autophagy inhibition and reduced activation of cardiac fibroblasts to
myofibroblasts. The MAPK signaling pathway has been previously implicated in autophagy
in the contexts of cardiac hypertrophy and fibrosis [287]. Our preliminary study on MAPK
signaling proteins such as p38 and ERK showed significant changes with p38 MAPK
phosphorylation, however we did not see any change with ERK (p44/42) with time. Thus,
we monitored p38 MAPK activation by examining the ratio of phosphorylated p38 MAPK
protein to total p38 MAPK protein (via Western blotting) in the presence and absence of
autophagy inhibitors. In the absence of autophagy inhibitors, we found a time-dependent
decrease in p38 MAPK activation with plating on a stiff substrate (Figure 14). This result is
as expected, as decreased p38 MAPK signaling has shown to be associated with the
conversion of fibroblasts to myofibroblasts [101]. The finding that treatment with CQ
increased p38 MAPK activation agrees with our findings that autophagy inhibition prevents

150
fibroblast activation (Figure 14). Co-inhibition of MAPK signaling and autophagy did not
demonstrate any cooperative effects, though this strategy remained effective in attenuating
activation of P0 cardiac fibroblasts (Figure 15). Nonetheless, these findings demonstrate a
putative link between MAPK signaling and autophagy inhibition.
After successfully establishing a link between autophagy and cardiac fibroblast
activation, as well as attenuation of these events with CQ treatment of primary fibroblasts, we
tested the efficacy of CQ in an in vivo experimental model of MI. We confirmed an increase
in myocardial collagen deposition (Figure 21 and 22) as well as α-SMA expression (Figure
19) in untreated experimental animals, indicating increased myofibroblast presence,
concomitant with increased autophagic flux (Figure 18) at both 4 post-MI. These results
agree with previous findings linking increased autophagy and the development of cardiac
fibrosis and subsequent heart failure [255, 290, 355]. In 4 week groups treated with low-dose
CQ for 3 weeks prior to tissue harvesting, α-SMA expression and was attenuated with
inhibition of autophagy in agreement with our findings in vitro. However, when we
compared collagen content and carried out functional assessment of heart was performed by
echocardiography in MI and CQ + MI groups we observed no effect with low-dose CQ
treatment of experimental hearts vs. non-treated controls. Indeed, ejection fraction, heart rate,
and fractional shortening were compromised in MI groups (compared to sham-operated
controls) and were unaltered with CQ treatment (Figure 27 and 28). However, serum markers
for liver, kidney, and cardiac damage were significantly reduced in the CQ + MI group
(compared to MI group), though not to the same level as the control group (Figure 26).As
changes in these markers precede functional alterations in the heart, the discrepancy between
serum marker levels and echocardiography results may be due to the timing of low-dose CQ
treatment. Thus, further optimization of the study timeline, such as commencing CQ

151
treatment earlier, or extending treatment to 8 weeks, may prove fruitful in observing similar
effects between marker levels and functional assessment.

152
Study Conclusions
Overall, the findings of this study lead us to three major conclusions, which are:
1. Plating of primary, unpassaged P0 cardiac fibroblasts on stiff substrates induces the
activation of fibroblasts, and in parallel their conversion to the myofibroblast phenotype,
as indicated by elevated expression of myofibroblast markers.
2. Activation of cardiac fibroblasts is associated with decreased p38 MAPK activation and
increased autophagic flux in myofibroblasts, indicated by the markers LC-3B and p62, as
well as autophagosome formation (assessed via TEM), both in vitro and in vivo.
3. Inhibition of autophagy prevents activation of fibroblasts in vitro and ameliorates -SMA
elevation post-MI in vivo. However the application of relatively low-dose CQ to post-MI
animals in vivo was not associated with any change in systolic cardiac function nor in
cardiac hydroxyproline (collagen) content. Future experiments with different antifibrotic
agents and/or timing of CQ treatment to earlier points after MI are likely warranted.
Thus, the in vitro segment (Chapter 1) of our study provides supportive evidence for
further examination of autophagy inhibition as a therapeutic strategy for attenuating
activation of cardiac fibroblasts and ensuing profibrotic events mediated by cardiac
myofibroblasts. The results of our in vivo study does restrict the phenotypic shift in the 4
week model, but not support the use of relatively low-dose CQ as an inhibitor of post-MI
cardiac fibrosis. Limitations in our in vivo experimental design may have impacted on the de
facto efficacy of our anti-autophagic treatment.

153
Future Directions
The in vitro findings above provide support for the induction of autophagy in driving the
activation of cardiac fibroblasts. However, questions still remain regarding the efficacy of in
vivo autophagy inhibition in preventing or reversing cardiac fibrosis and the specific cellular
mechanisms involved.
1. Pan-specific or inducible genetic knock-out mouse models to exploit the autophagic
pathway is a possible avenue to be explored, in combination with MI induction or
TAC-induced pressure overload. Specifically the knock-out of key autophagic genes
such as ATG7 might be used to assess the specificity of autophagy suppression (both
in vitro and in vivo). This approach could be applied to bypass the usual issues
associated with drug delivery and efficacy, particularly in lieu of the lack of a highly
effective small-molecule inhibitor of autophagy.
2. Additionally, the role of autophagy in cardiac fibrosis can be further understood by
targeting other important signaling pathways such as mTOR and PI3K. To elucidate
the role of mTOR and PI3K pathways, one could use mTOR specific and mTOR and
PI3K dual specificity inhibitors, either alone or in combination [356].
3. Fibroblasts are mechanically responsive cells. In the context of our work, there may
be relevant mechanical signaling pathways involved in triggering autophagy and
activation of fibroblasts. The use of various mechanical stimuli such as culture of
fibroblasts subjected to repeated and controlled mechanical stretch, and co-plating
with other cell types like cardiomyocytes and/or on various substrates would allow
cell-cell and cell-matrix interactions, which could then be studied in terms of
autophagy and fibroblast activation.
4. The question of the precise mechanism(s) of autophagy in fibroblast activation is far
from clear. We speculate that autophagy affects MAPK (p38) signaling, but we know

154
that other molecules may influence autophagy and also have anti-fibrotic properties.
Our lab has recently shown that Ski effectively inhibits autophagy in cardiac
myofibroblasts [357], it will be important to understand which, if any, autophagy-
related genes are affected. To this end, microarray data in myofibroblasts treated with
autophagy inhibitors may provide a starting point to determine the specific genes
involved in the autophagy-fibroblast activation relationship.
5. The effects of enhanced autophagy using rapamycin with such models can also be
studied to examine the integrity of the relationship between autophagy and cardiac
fibrosis. Although other studies have examined cardiomyocyte autophagy in cardiac
fibrosis, it will be important to understand the effects of this agent in myofibroblasts
and to assess for effects on profibrotic events.
6. Altered timing of administration of inhibitors of fibroblast autophagy: If the lack of
efficacy of CQ – mediated autophagic inhibition in vivo are due to alterations in the
activation of fibroblasts, a time course study to address earlier phases (eg, earlier than
4 weeks of post-MI wound healing) will need to be considered. For example,
administering CQ within 2-5 days post-MI could not only limit the activation of
fibroblast but it might also restrict the collagen production and fibrosis. This will also
help to better understand the initial phases of post-MI wound healing and scar
formation in the context of autophagy activation. In addition to a comprehensive time
course study, the use of different doses of CQ will also allow us to determine a dose-
response. In addition to CQ, the use of newer, more potent autophagy inhibitors such
as Lys01 and Lys05 may provide a clearer understanding of the potential for these
drugs as anti-fibrotics in real-world applications. If efficacy can be demonstrated with
optimized timing and drug usage/dosage in experimental MI, this will help to
determine the putative effectiveness in other models of MI and pressure overload. It

155
will also be interesting to examine the effects of autophagy inhibition in fibrosis of
other tissues and organs, particularly those where fibroblast activation is well-studied,
such as dermal models. Ultimately, the use of statistical modelling and regression
analysis in a review of peer-reviewed publications on the subject of autophagy
inhibition in cardiac remodeling and fibrosis in animal models would provide proof-of
concept evidence to bring these methods to the clinical realm.
7. A perfenidone study: Perfenidone is a late generation antifibrotic drug used to treat
idiopathic pulmonary fibrosis (IPF). This drug has been shown to have anti-fibrotic,
anti-inflammatory and antioxidant properties. In vitro studies have shown that it limits
TGF-β stimulated collagen production by downregulating theCol1 mRNA and
fibroblast proliferation. It also reduces production of pro-inflammatory cytokines and
free radicals[358].It would be interesting to determine whether combination therapy
using CQ as autophagy inhibitor and perfenidone as antifibrotic drug could have
beneficial effects on cardiac fibrosis. Our study CQ shows that CQ is an efficient
blocker of myofibroblast phenotype. A combination both drugs could be a new area of
research in treatment of cardiac fibrosis.

156
Limitations of this Study
This study is focused on putative changes to the autophagic state in heart after MI in rat heart
model of post-MI remodeling. The purpose is to definitively study the role of autophagy in
the activation of cardiac fibroblasts in vivo and in the second phase of the study, of cardiac
fibrosis after the induction of MI. While diastolic dysfunction and cardiac hypertrophy are
important features of heart failure, per se, in post-MI cardiovascular disease, our study does
not address the occurrence of heart failure directly. Rather, we have focused on fibroblast
activation and fibrosis and for this purpose we evaluated the protein markers for phenotype
and collagen deposition in our post-MI rat model. Cardiac performance was analyzed using
M-mode echocardiography (rather than by direct intubation of the left ventricle with a force
transducing catheter), which provides data giving information on systolic dysfunction. Thus,
the limitations of our in vivo study are that we did not monitor parameters of diastolic
dysfunction and heart failure such as E/A ratio (ratio of peak velocity flow in early diastole
(the E wave) to peak velocity flow in late diastole caused by atrial contraction the A wave),
LVEDP, and especially liver, abdominal and lung ascites (wet/dry ratio). In retrospect, our
study would have been more comprehensive had we provided data on cardiac hypertrophy by
calculating heart weight/tibia length ratio.

157
References
1. World Health Organization, Cardiovascular Diseases (CVDs). 2016.
2. Statistics Canada, Table 102-0561 - Leading causes of death, total population, by age
group and sex, Canada, annual. 2015.
3. Statistics Canada, Canadian Health Measures Survey: Household and physical
measures data, 2012 to 2013. 2014.
4. Roche, P.L., et al., Intracellular signaling of cardiac fibroblasts. Compr Physiol,
2015. 5(2): p. 721-60.
5. Kardasinski, M. and T. Thum, Cardiac fibroblasts on the fast track--scleraxis: from
Achilles' heel to anti-fibrotic therapy. J Mol Cell Cardiol, 2009. 47(2): p. 174-6.
6. Burwell, C.S. and E.D. Robin, Diagnosis of diffuse myocardial fibrosis. Circulation,
1959. 20: p. 606-14.
7. Gonzalez, A., B. Lopez, and J. Diez, Fibrosis in hypertensive heart disease: role of
the renin-angiotensin-aldosterone system. Med Clin North Am, 2004. 88(1): p. 83-97.
8. Gonzalez-Vilchez, F., et al., Oxidative stress and fibrosis in incipient myocardial
dysfunction in type 2 diabetic patients. Int J Cardiol, 2005. 101(1): p. 53-8.
9. Ihm, S.H., et al., Serum carboxy-terminal propeptide of type I procollagen (PIP) is a
marker of diastolic dysfunction in patients with early type 2 diabetes mellitus. Int J
Cardiol, 2007. 122(3): p. e36-8.
10. Reilly, K., Cardiac Fibrosis: New Treatments in Cardiovascular Medicine. U.S.
Pharmacist, 2015. 40(2): p. 32-35.
11. Travers, J.G., et al., Cardiac Fibrosis: The Fibroblast Awakens. Circ Res, 2016.
118(6): p. 1021-40.
12. Isoyama, S. and Y. Nitta-Komatsubara, Acute and chronic adaptation to
hemodynamic overload and ischemia in the aged heart. Heart Fail Rev, 2002. 7(1): p.
63-9.

158
13. Thygesen, K., et al., Third universal definition of myocardial infarction. Circulation,
2012. 126(16): p. 2020-35.
14. Gheorghiade, M. and G.C. Fonarow, Management of post-myocardial infarction
patients with left ventricular systolic dysfunction. Am J Med, 2007. 120(2): p. 109-20.
15. Chiong, M., et al., Cardiomyocyte death: mechanisms and translational implications.
Cell Death Dis, 2011. 2: p. e244.
16. Cleutjens, J.P., et al., The infarcted myocardium: simply dead tissue, or a lively target
for therapeutic interventions. Cardiovasc Res, 1999. 44(2): p. 232-41.
17. Cleutjens, J.P., et al., Collagen remodeling after myocardial infarction in the rat
heart. Am J Pathol, 1995. 147(2): p. 325-38.
18. Czubryt, M.P., Common threads in cardiac fibrosis, infarct scar formation, and
wound healing. Fibrogenesis Tissue Repair, 2012. 5(1): p. 19.
19. Frangogiannis, N.G., C.W. Smith, and M.L. Entman, The inflammatory response in
myocardial infarction. Cardiovascular research, 2002. 53(1): p. 31-47.
20. Liehn, E.A., et al., Repair after myocardial infarction, between fantasy and reality:
the role of chemokines. Journal of the American College of Cardiology, 2011. 58(23):
p. 2357-2362.
21. Jaeschke, H. and C.W. Smith, Mechanisms of neutrophil-induced parenchymal cell
injury. Journal of leukocyte biology, 1997. 61(6): p. 647-653.
22. Phatharajaree, W., A. Phrommintikul, and N. Chattipakorn, Matrix
metalloproteinases and myocardial infarction. Can J Cardiol, 2007. 23(9): p. 727-33.
23. Zymek, P., et al., Interleukin-10 is not a critical regulator of infarct healing and left
ventricular remodeling. Cardiovascular research, 2007. 74(2): p. 313-322.

159
24. Crawford, J.R., et al., Origin of developmental precursors dictates the
pathophysiologic role of cardiac fibroblasts. Journal of cardiovascular translational
research, 2012. 5(6): p. 749-759.
25. Nelissen-Vrancken, H.J., et al., Time-related normalization of maximal coronary flow
in isolated perfused hearts of rats with myocardial infarction. Circulation, 1996.
93(2): p. 349-55.
26. Zamilpa, R. and M.L. Lindsey, Extracellular matrix turnover and signaling during
cardiac remodeling following MI: causes and consequences. J Mol Cell Cardiol,
2010. 48(3): p. 558-63.
27. Gabbiani, G., G. Ryan, and G. Majno, Presence of modified fibroblasts in granulation
tissue and their possible role in wound contraction. Experientia, 1971. 27(5): p. 549-
550.
28. Desmouliere, A., et al., Apoptosis mediates the decrease in cellularity during the
transition between granulation tissue and scar. The American journal of pathology,
1995. 146(1): p. 56.
29. Mozaffarian, D., et al., Heart disease and stroke statistics--2015 update: a report
from the American Heart Association. Circulation, 2015. 131(4): p. e29-322.
30. Correction. Circulation, 2016. 133(8): p. e417.
31. Rienks, M., et al., Myocardial extracellular matrix: an ever-changing and diverse
entity. Circ Res, 2014. 114(5): p. 872-88.
32. Ma, Y., G.V. Halade, and M.L. Lindsey, Extracellular matrix and fibroblast
communication following myocardial infarction. J Cardiovasc Transl Res, 2012. 5(6):
p. 848-57.
33. Heymans, S., Inflammation and cardiac remodeling during viral myocarditis. Ernst
Schering Res Found Workshop, 2006(55): p. 197-218.

160
34. Ma, Y., et al., Myofibroblasts and the extracellular matrix network in post-myocardial
infarction cardiac remodeling. Pflugers Arch, 2014. 466(6): p. 1113-27.
35. Waldenstrom, A., et al., Accumulation of hyaluronan and tissue edema in
experimental myocardial infarction. J Clin Invest, 1991. 88(5): p. 1622-8.
36. Hellman, U., et al., Parallel up-regulation of FGF-2 and hyaluronan during
development of cardiac hypertrophy in rat. Cell Tissue Res, 2008. 332(1): p. 49-56.
37. Waldenstrom, A., et al., Coxsackie B3 myocarditis induces a decrease in energy
charge and accumulation of hyaluronan in the mouse heart. Eur J Clin Invest, 1993.
23(5): p. 277-82.
38. Bishop, J.E. and G.J. Laurent, Collagen turnover and its regulation in the normal and
hypertrophying heart. Eur Heart J, 1995. 16 Suppl C: p. 38-44.
39. Luther, D.J., et al., Absence of type VI collagen paradoxically improves cardiac
function, structure, and remodeling after myocardial infarction. Circ Res, 2012.
110(6): p. 851-6.
40. Pankov, R. and K.M. Yamada, Fibronectin at a glance. J Cell Sci, 2002. 115(Pt 20):
p. 3861-3.
41. Lenselink, E.A., Role of fibronectin in normal wound healing. Int Wound J, 2015.
12(3): p. 313-6.
42. Sawicka, K.M., et al., Fibronectin Interaction and Enhancement of Growth Factors:
Importance for Wound Healing. Adv Wound Care (New Rochelle), 2015. 4(8): p.
469-478.
43. Hynes, R.O., Fibronectins. Springer Series in Molecular Biology, 1990.
44. Kornblihtt, A.R., et al., The fibronectin gene as a model for splicing and transcription
studies. FASEB J, 1996. 10(2): p. 248-57.

161
45. Muro, A.F., et al., An essential role for fibronectin extra type III domain A in
pulmonary fibrosis. Am J Respir Crit Care Med, 2008. 177(6): p. 638-45.
46. Liao, Y.F., et al., The EIIIA segment of fibronectin is a ligand for integrins alpha
9beta 1 and alpha 4beta 1 providing a novel mechanism for regulating cell adhesion
by alternative splicing. J Biol Chem, 2002. 277(17): p. 14467-74.
47. Serini, G., et al., The fibronectin domain ED-A is crucial for myofibroblastic
phenotype induction by transforming growth factor-beta1. J Cell Biol, 1998. 142(3):
p. 873-81.
48. Arslan, F., et al., Lack of fibronectin-EDA promotes survival and prevents adverse
remodeling and heart function deterioration after myocardial infarction. Circ Res,
2011. 108(5): p. 582-92.
49. Bagchi, R.A., et al., Regulation of fibronectin gene expression in cardiac fibroblasts
by scleraxis. Cell Tissue Res, 2016. 366(2): p. 381-391.
50. Bagchi, R.A., et al., The transcription factor scleraxis is a critical regulator of
cardiac fibroblast phenotype. BMC Biol, 2016. 14: p. 21.
51. Bagchi, R.A., et al., Regulation of fibronectin gene expression in cardiac fibroblasts
by scleraxis. Cell Tissue Res, 2016.
52. Bornstein, P. and E.H. Sage, Matricellular proteins: extracellular modulators of cell
function. Curr Opin Cell Biol, 2002. 14(5): p. 608-16.
53. Schellings, M.W., Y.M. Pinto, and S. Heymans, Matricellular proteins in the heart:
possible role during stress and remodeling. Cardiovasc Res, 2004. 64(1): p. 24-31.
54. Chiquet-Ehrismann, R., et al., Tenascin-C expression by fibroblasts is elevated in
stressed collagen gels. J Cell Biol, 1994. 127(6 Pt 2): p. 2093-101.
55. Imanaka-Yoshida, K., et al., Serial extracellular matrix changes in neointimal lesions
of human coronary artery after percutaneous transluminal coronary angioplasty:

162
clinical significance of early tenascin-C expression. Virchows Arch, 2001. 439(2): p.
185-90.
56. Trueblood, N.A., et al., Exaggerated left ventricular dilation and reduced collagen
deposition after myocardial infarction in mice lacking osteopontin. Circ Res, 2001.
88(10): p. 1080-7.
57. Ladage, D., et al., Stimulating myocardial regeneration with periostin Peptide in
large mammals improves function post-myocardial infarction but increases
myocardial fibrosis. PLoS One, 2013. 8(5): p. e59656.
58. Ahmed, M.S., et al., Mechanisms of novel cardioprotective functions of CCN2/CTGF
in myocardial ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol, 2011.
300(4): p. H1291-302.
59. Panek, A.N., et al., Connective tissue growth factor overexpression in cardiomyocytes
promotes cardiac hypertrophy and protection against pressure overload. PLoS One,
2009. 4(8): p. e6743.
60. Frangogiannis, N.G., Matricellular proteins in cardiac adaptation and disease.
Physiol Rev, 2012. 92(2): p. 635-88.
61. Baudino, T.A., et al., Cardiac fibroblasts: friend or foe? Am J Physiol Heart Circ
Physiol, 2006. 291(3): p. H1015-26.
62. Camelliti, P., T.K. Borg, and P. Kohl, Structural and functional characterisation of
cardiac fibroblasts. Cardiovasc Res, 2005. 65(1): p. 40-51.
63. Harvey, R.P., Seeking a regulatory roadmap for heart morphogenesis. Semin Cell
Dev Biol, 1999. 10(1): p. 99-107.
64. !!! INVALID CITATION !!!
65. Souders, C.A., S.L. Bowers, and T.A. Baudino, Cardiac fibroblast: the renaissance
cell. Circ Res, 2009. 105(12): p. 1164-76.

163
66. Baxter, S.C., M.O. Morales, and E.C. Goldsmith, Adaptive changes in cardiac
fibroblast morphology and collagen organization as a result of mechanical
environment. Cell biochemistry and biophysics, 2008. 51(1): p. 33-44.
67. Takeda, N. and I. Manabe, Cellular Interplay between Cardiomyocytes and
Nonmyocytes in Cardiac Remodeling. Int J Inflam, 2011. 2011: p. 535241.
68. Souders, C.A., S.L. Bowers, and T.A. Baudino, Cardiac fibroblast the renaissance
cell. Circulation research, 2009. 105(12): p. 1164-1176.
69. Gittenberger-de Groot, A.C., et al., Epicardium-derived cells contribute a novel
population to the myocardial wall and the atrioventricular cushions. Circ Res, 1998.
82(10): p. 1043-52.
70. Munoz-Chapuli, R., et al., The epicardium as a source of mesenchyme for the
developing heart. Ital J Anat Embryol, 2001. 106(2 Suppl 1): p. 187-96.
71. Smith, C.L., et al., Epicardial-derived cell epithelial-to-mesenchymal transition and
fate specification require PDGF receptor signaling. Circ Res, 2011. 108(12): p. e15-
26.
72. Olivey, H.E., et al., Transforming growth factor-beta stimulates epithelial-
mesenchymal transformation in the proepicardium. Dev Dyn, 2006. 235(1): p. 50-9.
73. Acharya, A., et al., The bHLH transcription factor Tcf21 is required for lineage-
specific EMT of cardiac fibroblast progenitors. Development, 2012. 139(12): p. 2139-
49.
74. Vega-Hernandez, M., et al., FGF10/FGFR2b signaling is essential for cardiac
fibroblast development and growth of the myocardium. Development, 2011. 138(15):
p. 3331-40.
75. Lin, F., N. Wang, and T.C. Zhang, The role of endothelial-mesenchymal transition in
development and pathological process. IUBMB Life, 2012. 64(9): p. 717-23.

164
76. Krenning, G., E.M. Zeisberg, and R. Kalluri, The origin of fibroblasts and mechanism
of cardiac fibrosis. J Cell Physiol, 2010. 225(3): p. 631-7.
77. Zeisberg, E.M., et al., Endothelial-to-mesenchymal transition contributes to cardiac
fibrosis. Nat Med, 2007. 13(8): p. 952-61.
78. Lajiness, J.D. and S.J. Conway, Origin, development, and differentiation of cardiac
fibroblasts. J Mol Cell Cardiol, 2014. 70: p. 2-8.
79. Moonen, J.R., et al., Endothelial progenitor cells give rise to pro-angiogenic smooth
muscle-like progeny. Cardiovasc Res, 2010. 86(3): p. 506-15.
80. Timmerman, L.A., et al., Notch promotes epithelial-mesenchymal transition during
cardiac development and oncogenic transformation. Genes Dev, 2004. 18(1): p. 99-
115.
81. Fredj, S., et al., Interactions between cardiac cells enhance cardiomyocyte
hypertrophy and increase fibroblast proliferation. J Cell Physiol, 2005. 202(3): p.
891-9.
82. Lucas, J.A., et al., Inhibition of transforming growth factor-beta signaling induces left
ventricular dilation and dysfunction in the pressure-overloaded heart. Am J Physiol
Heart Circ Physiol, 2010. 298(2): p. H424-32.
83. van Amerongen, M.J., et al., Bone marrow-derived myofibroblasts contribute
functionally to scar formation after myocardial infarction. J Pathol, 2008. 214(3): p.
377-86.
84. Haudek, S.B., et al., Bone marrow-derived fibroblast precursors mediate ischemic
cardiomyopathy in mice. Proc Natl Acad Sci U S A, 2006. 103(48): p. 18284-9.
85. van Amerongen, M.J., et al., Macrophage depletion impairs wound healing and
increases left ventricular remodeling after myocardial injury in mice. Am J Pathol,
2007. 170(3): p. 818-29.

165
86. Abe, R., et al., Peripheral blood fibrocytes: differentiation pathway and migration to
wound sites. J Immunol, 2001. 166(12): p. 7556-62.
87. Deb, A. and E. Ubil, Cardiac fibroblast in development and wound healing. J Mol
Cell Cardiol, 2014. 70: p. 47-55.
88. Sundberg, C., et al., Pericytes as collagen-producing cells in excessive dermal
scarring. Lab Invest, 1996. 74(2): p. 452-66.
89. Humphreys, B.D., et al., Fate tracing reveals the pericyte and not epithelial origin of
myofibroblasts in kidney fibrosis. Am J Pathol, 2010. 176(1): p. 85-97.
90. Gabbiani, G., The cellular derivation and the life span of the myofibroblast. Pathol
Res Pract, 1996. 192(7): p. 708-11.
91. Hinz, B., Myofibroblasts. Exp Eye Res, 2016. 142: p. 56-70.
92. Sun, Y. and K.T. Weber, RAS and connective tissue in the heart. Int J Biochem Cell
Biol, 2003. 35(6): p. 919-31.
93. Talman, V. and H. Ruskoaho, Cardiac fibrosis in myocardial infarction-from repair
and remodeling to regeneration. Cell Tissue Res, 2016. 365(3): p. 563-81.
94. Chauhan, A.K., et al., Alternative splicing of fibronectin: a mouse model
demonstrates the identity of in vitro and in vivo systems and the processing autonomy
of regulated exons in adult mice. Gene, 2004. 324: p. 55-63.
95. Hinz, B. and G. Gabbiani, Mechanisms of force generation and transmission by
myofibroblasts. Curr Opin Biotechnol, 2003. 14(5): p. 538-46.
96. Sandbo, N. and N. Dulin, Actin cytoskeleton in myofibroblast differentiation:
ultrastructure defining form and driving function. Transl Res, 2011. 158(4): p. 181-
96.
97. Shinde, A.V. and N.G. Frangogiannis, Fibroblasts in myocardial infarction: a role in
inflammation and repair. J Mol Cell Cardiol, 2014. 70: p. 74-82.

166
98. Zhang, Y., et al., Receptor-associated Mad homologues synergize as effectors of the
TGF-beta response. Nature, 1996. 383(6596): p. 168-72.
99. Nakao, A., et al., TGF-beta receptor-mediated signalling through Smad2, Smad3 and
Smad4. EMBO J, 1997. 16(17): p. 5353-62.
100. Klingberg, F., et al., Prestress in the extracellular matrix sensitizes latent TGF-beta1
for activation. J Cell Biol, 2014. 207(2): p. 283-97.
101. MacLean, J. and K.B. Pasumarthi, Signaling mechanisms regulating fibroblast
activation, phenoconversion and fibrosis in the heart. Indian J Biochem Biophys,
2014. 51(6): p. 476-82.
102. Wang, J., et al., Mechanical force regulation of myofibroblast differentiation in
cardiac fibroblasts. Am J Physiol Heart Circ Physiol, 2003. 285(5): p. H1871-81.
103. Roche, P.L., et al., Role of scleraxis in mechanical stretch-mediated regulation of
cardiac myofibroblast phenotype. Am J Physiol Cell Physiol, 2016. 311(2): p. C297-
307.
104. Liu, X., et al., cAMP inhibits transforming growth factor-beta-stimulated collagen
synthesis via inhibition of extracellular signal-regulated kinase 1/2 and Smad
signaling in cardiac fibroblasts. Mol Pharmacol, 2006. 70(6): p. 1992-2003.
105. Hinz, B., Formation and function of the myofibroblast during tissue repair. J Invest
Dermatol, 2007. 127(3): p. 526-37.
106. Wipff, P.J., et al., Myofibroblast contraction activates latent TGF-beta1 from the
extracellular matrix. J Cell Biol, 2007. 179(6): p. 1311-23.
107. Hyytiainen, M., C. Penttinen, and J. Keski-Oja, Latent TGF-beta binding proteins:
extracellular matrix association and roles in TGF-beta activation. Crit Rev Clin Lab
Sci, 2004. 41(3): p. 233-64.

167
108. Yamashita, H., et al., Formation of hetero-oligomeric complexes of type I and type II
receptors for transforming growth factor-beta. J Biol Chem, 1994. 269(31): p. 20172-
8.
109. Massague, J., J. Seoane, and D. Wotton, Smad transcription factors. Genes Dev,
2005. 19(23): p. 2783-810.
110. Gao, Y., et al., SMAD7 antagonizes key TGFbeta superfamily signaling in mouse
granulosa cells in vitro. Reproduction, 2013. 146(1): p. 1-11.
111. Akiyoshi, S., et al., c-Ski acts as a transcriptional co-repressor in transforming
growth factor-beta signaling through interaction with smads. J Biol Chem, 1999.
274(49): p. 35269-77.
112. Xu, W., et al., Ski acts as a co-repressor with Smad2 and Smad3 to regulate the
response to type beta transforming growth factor. Proc Natl Acad Sci U S A, 2000.
97(11): p. 5924-9.
113. Mankoo, B.S., et al., The concerted action of Meox homeobox genes is required
upstream of genetic pathways essential for the formation, patterning and
differentiation of somites. Development, 2003. 130(19): p. 4655-64.
114. Weir, L., et al., Expression of gax, a growth arrest homeobox gene, is rapidly down-
regulated in the rat carotid artery during the proliferative response to balloon injury.
J Biol Chem, 1995. 270(10): p. 5457-61.
115. Valcourt, U., et al., Functional role of Meox2 during the epithelial cytostatic response
to TGF-beta. Mol Oncol, 2007. 1(1): p. 55-71.
116. Cunnington, R.H., et al., The Ski-Zeb2-Meox2 pathway provides a novel mechanism
for regulation of the cardiac myofibroblast phenotype. J Cell Sci, 2014. 127(Pt 1): p.
40-9.

168
117. van den Borne, S.W., et al., Myocardial remodeling after infarction: the role of
myofibroblasts. Nat Rev Cardiol, 2010. 7(1): p. 30-7.
118. Wight, T.N. and S. Potter-Perigo, The extracellular matrix: an active or passive
player in fibrosis? Am J Physiol Gastrointest Liver Physiol, 2011. 301(6): p. G950-5.
119. Daskalopoulos, E.P., B.J. Janssen, and W.M. Blankesteijn, Myofibroblasts in the
infarct area: concepts and challenges. Microsc Microanal, 2012. 18(1): p. 35-49.
120. Selye, H., et al., Simple techniques for the surgical occlusion of coronary vessels in
the rat. Angiology, 1960. 11: p. 398-407.
121. Pfeffer, M.A., et al., Myocardial infarct size and ventricular function in rats. Circ
Res, 1979. 44(4): p. 503-12.
122. Kolk, M.V., et al., LAD-ligation: a murine model of myocardial infarction. J Vis Exp,
2009(32).
123. Wexler, B.C., Myocardial infarction in young vs old male rats: pathophysiologic
changes. Am Heart J, 1978. 96(1): p. 70-80.
124. Lobo Filho, H.G., et al., Experimental model of myocardial infarction induced by
isoproterenol in rats. Rev Bras Cir Cardiovasc, 2011. 26(3): p. 469-76.
125. Adler, N., L.L. Camin, and P. Shulkin, Rat model for acute myocardial infarction:
application to technetium-labeled glucoheptonate, tetracycline, and polyphosphate. J
Nucl Med, 1976. 17(3): p. 203-7.
126. Brooks, W.W., B.A. Garibaldi, and C.H. Conrad, Myocardial injury in the mouse
induced by transthoracic cauterization. Lab Anim Sci, 1998. 48(4): p. 374-8.
127. de Carvalho Frimm, C., M.K. Koike, and M. Curi, Subendocardial fibrosis in remote
myocardium results from reduction of coronary driving pressure during acute
infarction in rats. Arq Bras Cardiol, 2003. 80(5): p. 509-20.

169
128. Clark, J.E. and M.S. Marber, Advancements in pressure-volume catheter technology -
stress remodelling after infarction. Exp Physiol, 2013. 98(3): p. 614-21.
129. Watson, L.E., et al., Baseline echocardiographic values for adult male rats. J Am Soc
Echocardiogr, 2004. 17(2): p. 161-7.
130. Brown, L., et al., Echocardiographic assessment of cardiac structure and function in
rats. Heart Lung Circ, 2002. 11(3): p. 167-73.
131. Popp, R.L., et al., Echocardiography: M-mode and two-dimensional methods. Ann
Intern Med, 1980. 93(6): p. 844-56.
132. Darbandi Azar, A., et al., Echocardiographic evaluation of cardiac function in
ischemic rats: value of m-mode echocardiography. Res Cardiovasc Med, 2014. 3(4):
p. e22941.
133. Csipo, I., et al., Measurement of complement components and alpha 1-antitrypsin
during plasma exchange. Acta Med Hung, 1991. 48(3-4): p. 211-7.
134. Nahrendorf, M., et al., Cardiac magnetic resonance imaging in small animal models
of human heart failure. Med Image Anal, 2003. 7(3): p. 369-75.
135. Thomas, D., et al., Quantitative assessment of regional myocardial function in a rat
model of myocardial infarction using tagged MRI. MAGMA, 2004. 17(3-6): p. 179-
87.
136. Nahrendorf, M., et al., High-resolution imaging of murine myocardial infarction with
delayed-enhancement cine micro-CT. Am J Physiol Heart Circ Physiol, 2007. 292(6):
p. H3172-8.
137. Fuchs, Y. and H. Steller, Programmed cell death in animal development and disease.
Cell, 2011. 147(4): p. 742-58.
138. Sun, X.M., et al., Caspase activation inhibits proteasome function during apoptosis.
Mol Cell, 2004. 14(1): p. 81-93.

170
139. Kerr, J.F., Shrinkage necrosis: a distinct mode of cellular death. J Pathol, 1971.
105(1): p. 13-20.
140. Leist, M., et al., Intracellular adenosine triphosphate (ATP) concentration: a switch
in the decision between apoptosis and necrosis. J Exp Med, 1997. 185(8): p. 1481-6.
141. Glick, D., S. Barth, and K.F. Macleod, Autophagy: cellular and molecular
mechanisms. J Pathol, 2010. 221(1): p. 3-12.
142. Yang, Z. and D.J. Klionsky, Eaten alive: a history of macroautophagy. Nat Cell Biol,
2010. 12(9): p. 814-22.
143. Salvador, N., et al., Import of a cytosolic protein into lysosomes by chaperone-
mediated autophagy depends on its folding state. J Biol Chem, 2000. 275(35): p.
27447-56.
144. Kiffin, R., et al., Activation of chaperone-mediated autophagy during oxidative stress.
Mol Biol Cell, 2004. 15(11): p. 4829-40.
145. Mijaljica, D., M. Prescott, and R.J. Devenish, Microautophagy in mammalian cells:
revisiting a 40-year-old conundrum. Autophagy, 2011. 7(7): p. 673-82.
146. Yoshimori, T., Autophagy as a bulk protein degradation system: it plays various
roles. Tanpakushitsu Kakusan Koso, 2004. 49(7 Suppl): p. 1029-32.
147. Nakatogawa, H., et al., Dynamics and diversity in autophagy mechanisms: lessons
from yeast. Nat Rev Mol Cell Biol, 2009. 10(7): p. 458-67.
148. Kihara, A., et al., Beclin-phosphatidylinositol 3-kinase complex functions at the trans-
Golgi network. EMBO Rep, 2001. 2(4): p. 330-5.
149. Abounit, K., T.M. Scarabelli, and R.B. McCauley, Autophagy in mammalian cells.
World J Biol Chem, 2012. 3(1): p. 1-6.
150. Kang, R., et al., The Beclin 1 network regulates autophagy and apoptosis. Cell Death
Differ, 2011. 18(4): p. 571-80.

171
151. Wu, Y.T., et al., Dual role of 3-methyladenine in modulation of autophagy via
different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase.
J Biol Chem, 2010. 285(14): p. 10850-61.
152. Hanada, T., et al., The Atg12-Atg5 conjugate has a novel E3-like activity for protein
lipidation in autophagy. J Biol Chem, 2007. 282(52): p. 37298-302.
153. Kim, P.K., et al., Ubiquitin signals autophagic degradation of cytosolic proteins and
peroxisomes. Proc Natl Acad Sci U S A, 2008. 105(52): p. 20567-74.
154. Pankiv, S., et al., p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of
ubiquitinated protein aggregates by autophagy. J Biol Chem, 2007. 282(33): p.
24131-45.
155. Rubinsztein, D.C., et al., In search of an "autophagomometer". Autophagy, 2009.
5(5): p. 585-9.
156. Klionsky, D.J., et al., Guidelines for the use and interpretation of assays for
monitoring autophagy. Autophagy, 2012. 8(4): p. 445-544.
157. Jangamreddy, J.R., et al., Salinomycin induces activation of autophagy, mitophagy
and affects mitochondrial polarity: differences between primary and cancer cells.
Biochim Biophys Acta, 2013. 1833(9): p. 2057-69.
158. Ravikumar, B., et al., Regulation of mammalian autophagy in physiology and
pathophysiology. Physiol Rev, 2010. 90(4): p. 1383-435.
159. Tanaka, Y., et al., Accumulation of autophagic vacuoles and cardiomyopathy in
LAMP-2-deficient mice. Nature, 2000. 406(6798): p. 902-6.
160. Jager, S., et al., Role for Rab7 in maturation of late autophagic vacuoles. J Cell Sci,
2004. 117(Pt 20): p. 4837-48.
161. Yang, Z., et al., Atg22 recycles amino acids to link the degradative and recycling
functions of autophagy. Mol Biol Cell, 2006. 17(12): p. 5094-104.

172
162. Ishihara, N., et al., Autophagosome requires specific early Sec proteins for its
formation and NSF/SNARE for vacuolar fusion. Mol Biol Cell, 2001. 12(11): p. 3690-
702.
163. Kochl, R., et al., Microtubules facilitate autophagosome formation and fusion of
autophagosomes with endosomes. Traffic, 2006. 7(2): p. 129-45.
164. Iwata, A., et al., HDAC6 and microtubules are required for autophagic degradation
of aggregated huntingtin. J Biol Chem, 2005. 280(48): p. 40282-92.
165. Ravikumar, B., et al., Dynein mutations impair autophagic clearance of aggregate-
prone proteins. Nat Genet, 2005. 37(7): p. 771-6.
166. Brunet, A., S.R. Datta, and M.E. Greenberg, Transcription-dependent and -
independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr
Opin Neurobiol, 2001. 11(3): p. 297-305.
167. Del Principe, D., et al., Fibroblast autophagy in fibrotic disorders. J Pathol, 2013.
229(2): p. 208-20.
168. Inoki, K., J. Kim, and K.L. Guan, AMPK and mTOR in cellular energy homeostasis
and drug targets. Annu Rev Pharmacol Toxicol, 2012. 52: p. 381-400.
169. Nemchenko, A., et al., Autophagy as a therapeutic target in cardiovascular disease. J
Mol Cell Cardiol, 2011. 51(4): p. 584-93.
170. Jewell, J.L. and K.L. Guan, Nutrient signaling to mTOR and cell growth. Trends
Biochem Sci, 2013. 38(5): p. 233-42.
171. Neufeld, T.P., TOR-dependent control of autophagy: biting the hand that feeds. Curr
Opin Cell Biol, 2010. 22(2): p. 157-68.
172. Inoki, K., et al., TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR
signalling. Nat Cell Biol, 2002. 4(9): p. 648-57.

173
173. Yan, Y. and J.M. Backer, Regulation of class III (Vps34) PI3Ks. Biochem Soc Trans,
2007. 35(Pt 2): p. 239-41.
174. Nobukuni, T., et al., Amino acids mediate mTOR/raptor signaling through activation
of class 3 phosphatidylinositol 3OH-kinase. Proc Natl Acad Sci U S A, 2005.
102(40): p. 14238-43.
175. Findlay, G.M., et al., A MAP4 kinase related to Ste20 is a nutrient-sensitive regulator
of mTOR signalling. Biochem J, 2007. 403(1): p. 13-20.
176. Kim, E., et al., Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell
Biol, 2008. 10(8): p. 935-45.
177. Sancak, Y., et al., The Rag GTPases bind raptor and mediate amino acid signaling to
mTORC1. Science, 2008. 320(5882): p. 1496-501.
178. Chang, Y.Y. and T.P. Neufeld, An Atg1/Atg13 complex with multiple roles in TOR-
mediated autophagy regulation. Mol Biol Cell, 2009. 20(7): p. 2004-14.
179. Guertin, D.A., et al., Ablation in mice of the mTORC components raptor, rictor, or
mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha,
but not S6K1. Dev Cell, 2006. 11(6): p. 859-71.
180. Sarbassov, D.D., et al., Rictor, a novel binding partner of mTOR, defines a
rapamycin-insensitive and raptor-independent pathway that regulates the
cytoskeleton. Curr Biol, 2004. 14(14): p. 1296-302.
181. Mammucari, C., et al., FoxO3 controls autophagy in skeletal muscle in vivo. Cell
Metab, 2007. 6(6): p. 458-71.
182. Corradetti, M.N., et al., Regulation of the TSC pathway by LKB1: evidence of a
molecular link between tuberous sclerosis complex and Peutz-Jeghers syndrome.
Genes & development, 2004. 18(13): p. 1533-1538.

174
183. Woods, A., et al., Ca2+/calmodulin-dependent protein kinase kinase-beta acts
upstream of AMP-activated protein kinase in mammalian cells. Cell Metab, 2005.
2(1): p. 21-33.
184. Hoyer-Hansen, M. and M. Jaattela, AMP-activated protein kinase: a universal
regulator of autophagy? Autophagy, 2007. 3(4): p. 381-3.
185. Browne, G.J., S.G. Finn, and C.G. Proud, Stimulation of the AMP-activated protein
kinase leads to activation of eukaryotic elongation factor 2 kinase and to its
phosphorylation at a novel site, serine 398. J Biol Chem, 2004. 279(13): p. 12220-31.
186. Mizushima, N., The role of the Atg1/ULK1 complex in autophagy regulation. Curr
Opin Cell Biol, 2010. 22(2): p. 132-9.
187. Chan, E.Y., et al., Kinase-inactivated ULK proteins inhibit autophagy via their
conserved C-terminal domains using an Atg13-independent mechanism. Mol Cell
Biol, 2009. 29(1): p. 157-71.
188. Huang, J., G.Y. Lam, and J.H. Brumell, Autophagy signaling through reactive oxygen
species. Antioxid Redox Signal, 2011. 14(11): p. 2215-31.
189. Zhang, H., et al., Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic
response to hypoxia. J Biol Chem, 2008. 283(16): p. 10892-903.
190. Bellot, G., et al., Hypoxia-induced autophagy is mediated through hypoxia-inducible
factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol, 2009.
29(10): p. 2570-81.
191. Li, Y., et al., Bnip3 mediates the hypoxia-induced inhibition on mammalian target of
rapamycin by interacting with Rheb. J Biol Chem, 2007. 282(49): p. 35803-13.
192. Brugarolas, J., et al., Regulation of mTOR function in response to hypoxia by REDD1
and the TSC1/TSC2 tumor suppressor complex. Genes Dev, 2004. 18(23): p. 2893-
904.

175
193. Wang, R.C., et al., Akt-mediated regulation of autophagy and tumorigenesis through
Beclin 1 phosphorylation. Science, 2012. 338(6109): p. 956-9.
194. Zhang, J. and P.A. Ney, Role of BNIP3 and NIX in cell death, autophagy, and
mitophagy. Cell Death Differ, 2009. 16(7): p. 939-46.
195. Okkenhaug, K., Signaling by the phosphoinositide 3-kinase family in immune cells.
Annu Rev Immunol, 2013. 31: p. 675-704.
196. Groenewoud, M.J. and F.J. Zwartkruis, Rheb and Rags come together at the lysosome
to activate mTORC1. Biochem Soc Trans, 2013. 41(4): p. 951-5.
197. Zoncu, R., A. Efeyan, and D.M. Sabatini, mTOR: from growth signal integration to
cancer, diabetes and ageing. Nat Rev Mol Cell Biol, 2011. 12(1): p. 21-35.
198. Huang, J. and B.D. Manning, The TSC1-TSC2 complex: a molecular switchboard
controlling cell growth. Biochem J, 2008. 412(2): p. 179-90.
199. Mizushima, N., Noboru Mizushima: All about autophagy. Interview by Caitlin
Sedwick. J Cell Biol, 2010. 190(6): p. 946-7.
200. Tsukamoto, S., et al., Autophagy is essential for preimplantation development of
mouse embryos. Science, 2008. 321(5885): p. 117-20.
201. Merz, E.A., et al., Protein degradation during preimplantation development of the
mouse. J Reprod Fertil, 1981. 61(2): p. 415-8.
202. Kuma, A., et al., The role of autophagy during the early neonatal starvation period.
Nature, 2004. 432(7020): p. 1032-6.
203. Sandoval, H., et al., Essential role for Nix in autophagic maturation of erythroid cells.
Nature, 2008. 454(7201): p. 232-5.
204. Miller, B.C., et al., The autophagy gene ATG5 plays an essential role in B lymphocyte
development. Autophagy, 2008. 4(3): p. 309-14.

176
205. Singh, R., et al., Autophagy regulates adipose mass and differentiation in mice. J Clin
Invest, 2009. 119(11): p. 3329-39.
206. Lavandero, S., et al., Autophagy in cardiovascular biology. J Clin Invest, 2015.
125(1): p. 55-64.
207. Lee, E., et al., Autophagy is essential for cardiac morphogenesis during vertebrate
development. Autophagy, 2014. 10(4): p. 572-87.
208. Lavine, K.J., et al., Endocardial and epicardial derived FGF signals regulate
myocardial proliferation and differentiation in vivo. Dev Cell, 2005. 8(1): p. 85-95.
209. Zhang, J., et al., FRS2alpha-mediated FGF signals suppress premature differentiation
of cardiac stem cells through regulating autophagy activity. Circ Res, 2012. 110(4):
p. e29-39.
210. Aranguiz-Urroz, P., et al., Beta(2)-adrenergic receptor regulates cardiac fibroblast
autophagy and collagen degradation. Biochim Biophys Acta, 2011. 1812(1): p. 23-
31.
211. Ishida, Y., et al., Autophagic elimination of misfolded procollagen aggregates in the
endoplasmic reticulum as a means of cell protection. Mol Biol Cell, 2009. 20(11): p.
2744-54.
212. Lum, J.J., et al., Growth factor regulation of autophagy and cell survival in the
absence of apoptosis. Cell, 2005. 120(2): p. 237-48.
213. Levine, B. and D.J. Klionsky, Development by self-digestion: molecular mechanisms
and biological functions of autophagy. Dev Cell, 2004. 6(4): p. 463-77.
214. Mizushima, N., The pleiotropic role of autophagy: from protein metabolism to
bactericide. Cell Death Differ, 2005. 12 Suppl 2: p. 1535-41.
215. Kim, I., S. Rodriguez-Enriquez, and J.J. Lemasters, Selective degradation of
mitochondria by mitophagy. Arch Biochem Biophys, 2007. 462(2): p. 245-53.

177
216. Klionsky, D.J., Autophagy: from phenomenology to molecular understanding in less
than a decade. Nat Rev Mol Cell Biol, 2007. 8(11): p. 931-7.
217. Maiuri, M.C., et al., Self-eating and self-killing: crosstalk between autophagy and
apoptosis. Nat Rev Mol Cell Biol, 2007. 8(9): p. 741-52.
218. Komatsu, M., et al., Loss of autophagy in the central nervous system causes
neurodegeneration in mice. Nature, 2006. 441(7095): p. 880-4.
219. Mathew, R., V. Karantza-Wadsworth, and E. White, Role of autophagy in cancer. Nat
Rev Cancer, 2007. 7(12): p. 961-7.
220. Qu, X., et al., Promotion of tumorigenesis by heterozygous disruption of the beclin 1
autophagy gene. J Clin Invest, 2003. 112(12): p. 1809-20.
221. Gutierrez, M.G., et al., Autophagy is a defense mechanism inhibiting BCG and
Mycobacterium tuberculosis survival in infected macrophages. Cell, 2004. 119(6): p.
753-66.
222. Levine, B. and G. Kroemer, Autophagy in the pathogenesis of disease. Cell, 2008.
132(1): p. 27-42.
223. Martinez-Vicente, M. and A.M. Cuervo, Autophagy and neurodegeneration: when the
cleaning crew goes on strike. Lancet Neurol, 2007. 6(4): p. 352-61.
224. Mizushima, N. and D.J. Klionsky, Protein turnover via autophagy: implications for
metabolism. Annu Rev Nutr, 2007. 27: p. 19-40.
225. Hara, T., et al., Suppression of basal autophagy in neural cells causes
neurodegenerative disease in mice. Nature, 2006. 441(7095): p. 885-9.
226. White, E., Deconvoluting the context-dependent role for autophagy in cancer. Nat
Rev Cancer, 2012. 12(6): p. 401-10.
227. Masiero, E., et al., Autophagy is required to maintain muscle mass. Cell Metab, 2009.
10(6): p. 507-15.

178
228. Raben, N., et al., Suppression of autophagy in skeletal muscle uncovers the
accumulation of ubiquitinated proteins and their potential role in muscle damage in
Pompe disease. Hum Mol Genet, 2008. 17(24): p. 3897-908.
229. Ebato, C., et al., Autophagy is important in islet homeostasis and compensatory
increase of beta cell mass in response to high-fat diet. Cell Metab, 2008. 8(4): p. 325-
32.
230. Jung, H.S., et al., Loss of autophagy diminishes pancreatic beta cell mass and
function with resultant hyperglycemia. Cell Metab, 2008. 8(4): p. 318-24.
231. Shibata, M., et al., Regulation of intracellular accumulation of mutant Huntingtin by
Beclin 1. J Biol Chem, 2006. 281(20): p. 14474-85.
232. Cadwell, K., et al., A key role for autophagy and the autophagy gene Atg16l1 in
mouse and human intestinal Paneth cells. Nature, 2008. 456(7219): p. 259-63.
233. Cadwell, K., et al., A common role for Atg16L1, Atg5 and Atg7 in small intestinal
Paneth cells and Crohn disease. Autophagy, 2009. 5(2): p. 250-2.
234. De Meyer, G.R. and W. Martinet, Autophagy in the cardiovascular system. Biochim
Biophys Acta, 2009. 1793(9): p. 1485-95.
235. Nakai, A., et al., The role of autophagy in cardiomyocytes in the basal state and in
response to hemodynamic stress. Nat Med, 2007. 13(5): p. 619-24.
236. Nishida, K. and K. Otsu, Autophagy during cardiac remodeling. J Mol Cell Cardiol,
2016. 95: p. 11-8.
237. Kanamori, H., et al., Autophagy limits acute myocardial infarction induced by
permanent coronary artery occlusion. Am J Physiol Heart Circ Physiol, 2011. 300(6):
p. H2261-71.
238. Minatoguchi, T.W.G.T.H.K.A.T.G.K.A.O.T.T.T.K.K.M.H.F.T.F.S., Caloric
Restriction Augments Cardiomyocyte Autophagy to Mitigate Postinfarction Cardiac

179
Remodeling and Heart Failure. American Heart Association: Circulation, 2011.
124(A9424).
239. Gottlieb, R.A. and R.M. Mentzer, Jr., Autophagy: an affair of the heart. Heart Fail
Rev, 2013. 18(5): p. 575-84.
240. Terman, A. and U.T. Brunk, Autophagy in cardiac myocyte homeostasis, aging, and
pathology. Cardiovasc Res, 2005. 68(3): p. 355-65.
241. Kubli, D.A. and A.B. Gustafsson, Mitochondria and mitophagy: the yin and yang of
cell death control. Circ Res, 2012. 111(9): p. 1208-21.
242. Ghavami, S., et al., Autophagy regulates trans fatty acid-mediated apoptosis in
primary cardiac myofibroblasts. Biochim Biophys Acta, 2012. 1823(12): p. 2274-86.
243. Jin, S.M. and R.J. Youle, PINK1- and Parkin-mediated mitophagy at a glance. J Cell
Sci, 2012. 125(Pt 4): p. 795-9.
244. Bjorkoy, G., et al., Monitoring autophagic degradation of p62/SQSTM1. Methods
Enzymol, 2009. 452: p. 181-97.
245. Zhu, H., et al., Cardiac autophagy is a maladaptive response to hemodynamic stress.
J Clin Invest, 2007. 117(7): p. 1782-93.
246. Cao, D.J., et al., Histone deacetylase (HDAC) inhibitors attenuate cardiac
hypertrophy by suppressing autophagy. Proc Natl Acad Sci U S A, 2011. 108(10): p.
4123-8.
247. Valentim, L., et al., Urocortin inhibits Beclin1-mediated autophagic cell death in
cardiac myocytes exposed to ischaemia/reperfusion injury. J Mol Cell Cardiol, 2006.
40(6): p. 846-52.
248. Lu, L., et al., Adriamycin-induced autophagic cardiomyocyte death plays a
pathogenic role in a rat model of heart failure. Int J Cardiol, 2009. 134(1): p. 82-90.

180
249. Kobayashi, S., et al., Transcription factor GATA4 inhibits doxorubicin-induced
autophagy and cardiomyocyte death. J Biol Chem, 2010. 285(1): p. 793-804.
250. Matsui, Y., et al., Distinct roles of autophagy in the heart during ischemia and
reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating
autophagy. Circ Res, 2007. 100(6): p. 914-22.
251. Sciarretta, S., et al., Rheb is a critical regulator of autophagy during myocardial
ischemia: pathophysiological implications in obesity and metabolic syndrome.
Circulation, 2012. 125(9): p. 1134-46.
252. Zhai, P., et al., Differential roles of GSK-3beta during myocardial ischemia and
ischemia/reperfusion. Circ Res, 2011. 109(5): p. 502-11.
253. Xie, M., et al., Tuning flux: autophagy as a target of heart disease therapy. Curr Opin
Cardiol, 2011. 26(3): p. 216-22.
254. Shimomura, H., et al., Autophagic degeneration as a possible mechanism of
myocardial cell death in dilated cardiomyopathy. Jpn Circ J, 2001. 65(11): p. 965-8.
255. Kostin, S., et al., Myocytes die by multiple mechanisms in failing human hearts. Circ
Res, 2003. 92(7): p. 715-24.
256. Ghavami, S., et al., Autophagy is a regulator of TGF-beta1-induced fibrogenesis in
primary human atrial myofibroblasts. Cell Death Dis, 2015. 6: p. e1696.
257. Bernard, M., et al., Autophagy fosters myofibroblast differentiation through MTORC2
activation and downstream upregulation of CTGF. Autophagy, 2014. 10(12): p.
2193-207.
258. Georgescu, S.P., et al., Decreased metalloprotease 9 induction, cardiac fibrosis, and
higher autophagy after pressure overload in mice lacking the transcriptional
regulator p8. Am J Physiol Cell Physiol, 2011. 301(5): p. C1046-56.

181
259. Salabei, J.K. and B.G. Hill, Implications of autophagy for vascular smooth muscle
cell function and plasticity. Free Radic Biol Med, 2013. 65: p. 693-703.
260. Bharath, L.P., et al., Impairment of autophagy in endothelial cells prevents shear-
stress-induced increases in nitric oxide bioavailability. Can J Physiol Pharmacol,
2014. 92(7): p. 605-12.
261. Lavandero, S., et al., Cardiovascular autophagy: concepts, controversies, and
perspectives. Autophagy, 2013. 9(10): p. 1455-66.
262. Esclatine, A., M. Chaumorcel, and P. Codogno, Macroautophagy signaling and
regulation. Curr Top Microbiol Immunol, 2009. 335: p. 33-70.
263. Meijer, A.J. and P. Codogno, Signalling and autophagy regulation in health, aging
and disease. Mol Aspects Med, 2006. 27(5-6): p. 411-25.
264. Floto, R.A., et al., Small molecule enhancers of rapamycin-induced TOR inhibition
promote autophagy, reduce toxicity in Huntington's disease models and enhance
killing of mycobacteria by macrophages. Autophagy, 2007. 3(6): p. 620-2.
265. Meley, D., et al., AMP-activated protein kinase and the regulation of autophagic
proteolysis. J Biol Chem, 2006. 281(46): p. 34870-9.
266. Petrovski, G., et al., Cardioprotection by endoplasmic reticulum stress-induced
autophagy. Antioxid Redox Signal, 2011. 14(11): p. 2191-200.
267. Sarkar, S., et al., Lithium induces autophagy by inhibiting inositol monophosphatase.
J Cell Biol, 2005. 170(7): p. 1101-11.
268. Criollo, A., et al., Regulation of autophagy by the inositol trisphosphate receptor. Cell
Death Differ, 2007. 14(5): p. 1029-39.
269. Zhang, L., et al., Small molecule regulators of autophagy identified by an image-
based high-throughput screen. Proc Natl Acad Sci U S A, 2007. 104(48): p. 19023-8.

182
270. Chan, A.Y., et al., Resveratrol inhibits cardiac hypertrophy via AMP-activated
protein kinase and Akt. J Biol Chem, 2008. 283(35): p. 24194-201.
271. Xie, Z., et al., Improvement of cardiac functions by chronic metformin treatment is
associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes,
2011. 60(6): p. 1770-8.
272. Andres, A.M., et al., Mitophagy is required for acute cardioprotection by simvastatin.
Antioxid Redox Signal, 2014. 21(14): p. 1960-73.
273. Blommaart, E.F., et al., The phosphatidylinositol 3-kinase inhibitors wortmannin and
LY294002 inhibit autophagy in isolated rat hepatocytes. Eur J Biochem, 1997. 243(1-
2): p. 240-6.
274. Tolkovsky, A.M., et al., Mitochondrial disappearance from cells: a clue to the role of
autophagy in programmed cell death and disease? Biochimie, 2002. 84(2-3): p. 233-
40.
275. Xie, M., et al., Histone deacetylase inhibition blunts ischemia/reperfusion injury by
inducing cardiomyocyte autophagy. Circulation, 2014. 129(10): p. 1139-51.
276. Miyata, S., et al., Autophagic cardiomyocyte death in cardiomyopathic hamsters and
its prevention by granulocyte colony-stimulating factor. Am J Pathol, 2006. 168(2): p.
386-97.
277. Yamamoto, A., et al., Bafilomycin A1 prevents maturation of autophagic vacuoles by
inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line,
H-4-II-E cells. Cell Struct Funct, 1998. 23(1): p. 33-42.
278. Amaravadi, R.K. and C.B. Thompson, The roles of therapy-induced autophagy and
necrosis in cancer treatment. Clin Cancer Res, 2007. 13(24): p. 7271-9.
279. Harhaji-Trajkovic, L., et al., Chloroquine-mediated lysosomal dysfunction enhances
the anticancer effect of nutrient deprivation. Pharm Res, 2012. 29(8): p. 2249-63.

183
280. Amaravadi, R.K. and J.D. Winkler, Lys05: a new lysosomal autophagy inhibitor.
Autophagy, 2012. 8(9): p. 1383-4.
281. McAfee, Q., et al., Autophagy inhibitor Lys05 has single-agent antitumor activity and
reproduces the phenotype of a genetic autophagy deficiency. Proc Natl Acad Sci U S
A, 2012. 109(21): p. 8253-8.
282. Sciarretta, S., et al., Pharmacological modulation of autophagy during cardiac stress.
J Cardiovasc Pharmacol, 2012. 60(3): p. 235-41.
283. Piek, A., R.A. de Boer, and H.H. Sillje, The fibrosis-cell death axis in heart failure.
Heart Fail Rev, 2016. 21(2): p. 199-211.
284. Hernandez-Gea, V. and S.L. Friedman, Autophagy fuels tissue fibrogenesis.
Autophagy, 2012. 8(5): p. 849-50.
285. Hernandez-Gea, V., et al., Autophagy releases lipid that promotes fibrogenesis by
activated hepatic stellate cells in mice and in human tissues. Gastroenterology, 2012.
142(4): p. 938-46.
286. van Putten, S., Y. Shafieyan, and B. Hinz, Mechanical control of cardiac
myofibroblasts. J Mol Cell Cardiol, 2016. 93: p. 133-42.
287. Lin, L., et al., Mechanical stress triggers cardiomyocyte autophagy through
angiotensin II type 1 receptor-mediated p38MAP kinase independently of angiotensin
II. PLoS One, 2014. 9(2): p. e89629.
288. Kanamori, H., et al., The role of autophagy emerging in postinfarction cardiac
remodelling. Cardiovasc Res, 2011. 91(2): p. 330-9.
289. Zhang, S., et al., PDCD5 protects against cardiac remodeling by regulating
autophagy and apoptosis. Biochem Biophys Res Commun, 2015. 461(2): p. 321-8.
290. Wu, X., et al., Impaired autophagy contributes to adverse cardiac remodeling in
acute myocardial infarction. PLoS One, 2014. 9(11): p. e112891.

184
291. Hao, J., et al., Interaction between angiotensin II and Smad proteins in fibroblasts in
failing heart and in vitro. Am J Physiol Heart Circ Physiol, 2000. 279(6): p. H3020-
30.
292. Smith, P.K., et al., Measurement of protein using bicinchoninic acid. Anal.Biochem.,
1985. 150(1): p. 76-85.
293. Dixon, I.M., et al., Alterations in cardiac membrane Ca2+ transport during oxidative
stress. Mol Cell Biochem, 1990. 99(2): p. 125-33.
294. Kliment, C.R., et al., A novel method for accurate collagen and biochemical
assessment of pulmonary tissue utilizing one animal. Int J Clin Exp Pathol, 2011.
4(4): p. 349-55.
295. Neuman, R.E. and M.A. Logan, The determination of hydroxyproline. J Biol Chem,
1950. 184(1): p. 299-306.
296. Santiago, J.J., et al., Cardiac fibroblast to myofibroblast differentiation in vivo and in
vitro: expression of focal adhesion components in neonatal and adult rat ventricular
myofibroblasts. Dev Dyn, 2010. 239(6): p. 1573-84.
297. Hinz, B., Masters and servants of the force: the role of matrix adhesions in
myofibroblast force perception and transmission. Eur J Cell Biol, 2006. 85(3-4): p.
175-81.
298. Hinz, B., Tissue stiffness, latent TGF-beta1 activation, and mechanical signal
transduction: implications for the pathogenesis and treatment of fibrosis. Curr
Rheumatol Rep, 2009. 11(2): p. 120-6.
299. Dugina, V., et al., Focal adhesion features during myofibroblastic differentiation are
controlled by intracellular and extracellular factors. J Cell Sci, 2001. 114(Pt 18): p.
3285-96.

185
300. Tanida, I., T. Ueno, and E. Kominami, LC3 and Autophagy. Methods Mol Biol, 2008.
445: p. 77-88.
301. Tuloup-Minguez, V., et al., Autophagy modulates cell migration and beta1 integrin
membrane recycling. Cell Cycle, 2013. 12(20): p. 3317-28.
302. Newton, P.T., et al., Pharmacological inhibition of lysosomes activates the MTORC1
signaling pathway in chondrocytes in an autophagy-independent manner. Autophagy,
2015. 11(9): p. 1594-607.
303. Yang, Y.P., et al., Application and interpretation of current autophagy inhibitors and
activators. Acta Pharmacol Sin, 2013. 34(5): p. 625-35.
304. Jin, Y., et al., Role of autophagy in myocardial reperfusion injury. Front Biosci (Elite
Ed), 2010. 2: p. 1147-53.
305. Cunnington, R.H., et al., Antifibrotic properties of c-Ski and its regulation of cardiac
myofibroblast phenotype and contractility. Am J Physiol Cell Physiol, 2011. 300(1):
p. C176-86.
306. Drobic, V., et al., Differential and combined effects of cardiotrophin-1 and TGF-
beta1 on cardiac myofibroblast proliferation and contraction. Am J Physiol Heart
Circ Physiol, 2007. 293(2): p. H1053-64.
307. Furukawa, F., et al., p38 MAPK mediates fibrogenic signal through Smad3
phosphorylation in rat myofibroblasts. Hepatology, 2003. 38(4): p. 879-89.
308. Madeo, F., et al., Essential role for autophagy in life span extension. J Clin Invest,
2015. 125(1): p. 85-93.
309. Pyo, J.O., et al., Overexpression of Atg5 in mice activates autophagy and extends
lifespan. Nat Commun, 2013. 4: p. 2300.
310. Patel, A.S., et al., Autophagy in idiopathic pulmonary fibrosis. PLoS One, 2012. 7(7):
p. e41394.

186
311. Mallat, A., et al., Autophagy: a multifaceted partner in liver fibrosis. Biomed Res Int,
2014. 2014: p. 869390.
312. Hariharan, N., P. Zhai, and J. Sadoshima, Oxidative stress stimulates autophagic flux
during ischemia/reperfusion. Antioxid Redox Signal, 2011. 14(11): p. 2179-90.
313. Dorn, G.W., 2nd, Apoptotic and non-apoptotic programmed cardiomyocyte death in
ventricular remodelling. Cardiovasc Res, 2009. 81(3): p. 465-73.
314. Shaker, M.E., et al., Comparison of imatinib, nilotinib and silymarin in the treatment
of carbon tetrachloride-induced hepatic oxidative stress, injury and fibrosis. Toxicol
Appl Pharmacol, 2011. 252(2): p. 165-75.
315. Olsen, A.L., et al., Fibronectin extra domain-A promotes hepatic stellate cell motility
but not differentiation into myofibroblasts. Gastroenterology, 2012. 142(4): p. 928-
937.e3.
316. Junker, J.P., et al., Mechanical tension stimulates the transdifferentiation of
fibroblasts into myofibroblasts in human burn scars. Burns, 2008. 34(7): p. 942-6.
317. Masur, S.K., et al., Myofibroblasts differentiate from fibroblasts when plated at low
density. Proc Natl Acad Sci U S A, 1996. 93(9): p. 4219-23.
318. Porter, K.E. and N.A. Turner, Cardiac fibroblasts: at the heart of myocardial
remodeling. Pharmacol Ther, 2009. 123(2): p. 255-78.
319. Grassi, G., et al., Autophagy regulates hepatocyte identity and epithelial-to-
mesenchymal and mesenchymal-to-epithelial transitions promoting Snail
degradation. Cell Death Dis, 2015. 6: p. e1880.
320. He, W., et al., Chloroquine improved carbon tetrachloride-induced liver fibrosis
through its inhibition of the activation of hepatic stellate cells: role of autophagy. Biol
Pharm Bull, 2014. 37(9): p. 1505-9.

187
321. Masiero, E. and M. Sandri, Autophagy inhibition induces atrophy and myopathy in
adult skeletal muscles. Autophagy, 2010. 6(2): p. 307-9.
322. Taneike, M., et al., Inhibition of autophagy in the heart induces age-related
cardiomyopathy. Autophagy, 2010. 6(5): p. 600-6.
323. Indelicato, M., et al., Role of hypoxia and autophagy in MDA-MB-231 invasiveness. J
Cell Physiol, 2010. 223(2): p. 359-68.
324. Zhang, Y., et al., Chloroquine inhibits MGC803 gastric cancer cell migration via the
Toll-like receptor 9/nuclear factor kappa B signaling pathway. Mol Med Rep, 2015.
11(2): p. 1366-71.
325. Klingberg, F., B. Hinz, and E.S. White, The myofibroblast matrix: implications for
tissue repair and fibrosis. J Pathol, 2013. 229(2): p. 298-309.
326. Yong, K.W., et al., Mechanoregulation of cardiac myofibroblast differentiation:
implications for cardiac fibrosis and therapy. Am J Physiol Heart Circ Physiol, 2015.
309(4): p. H532-42.
327. Hinz, B., The extracellular matrix and transforming growth factor-beta1: Tale of a
strained relationship. Matrix Biol, 2015. 47: p. 54-65.
328. Steinhauser, M.L. and R.T. Lee, Regeneration of the heart. EMBO Mol Med, 2011.
3(12): p. 701-12.
329. King, J.H., C.L. Huang, and J.A. Fraser, Determinants of myocardial conduction
velocity: implications for arrhythmogenesis. Front Physiol, 2013. 4: p. 154.
330. Biernacka, A. and N.G. Frangogiannis, Aging and Cardiac Fibrosis. Aging Dis, 2011.
2(2): p. 158-173.
331. Al-Bari, M.A., Chloroquine analogues in drug discovery: new directions of uses,
mechanisms of actions and toxic manifestations from malaria to multifarious
diseases. J Antimicrob Chemother, 2015. 70(6): p. 1608-21.

188
332. Ohm, I.K., et al., Toll-like receptor 9-activation during onset of myocardial ischemia
does not influence infarct extension. PLoS One, 2014. 9(8): p. e104407.
333. Lott, J.A., D.W. Tholen, and C.G. Massion, Determination of reference ranges for
serum enzymes via a large interlaboratory survey. Arch Pathol Lab Med, 1987.
111(1): p. 9-15.
334. Glick, J.H., Jr., Serum lactate dehydrogenase isoenzyme and total lactate
dehydrogenase values in health and disease, and clinical evaluation of these tests by
means of discriminant analysis. Am J Clin Pathol, 1969. 52(3): p. 320-8.
335. Gowda, S., et al., Markers of renal function tests. N Am J Med Sci, 2010. 2(4): p.
170-3.
336. Gowda, S., et al., A review on laboratory liver function tests. Pan Afr Med J, 2009. 3:
p. 17.
337. Mizushima, N. and T. Yoshimori, How to interpret LC3 immunoblotting. Autophagy,
2007. 3(6): p. 542-5.
338. Barth, S., D. Glick, and K.F. Macleod, Autophagy: assays and artifacts. J Pathol,
2010. 221(2): p. 117-24.
339. Sui, L., et al., Lead toxicity induces autophagy to protect against cell death through
mTORC1 pathway in cardiofibroblasts. Biosci Rep, 2015. 35(2).
340. Liu, S., et al., Autophagy activation attenuates angiotensin II-induced cardiac
fibrosis. Arch Biochem Biophys, 2016. 590: p. 37-47.
341. Xu, X., et al., Diminished autophagy limits cardiac injury in mouse models of type 1
diabetes. J Biol Chem, 2013. 288(25): p. 18077-92.
342. Borlaug, B.A. and W.J. Paulus, Heart failure with preserved ejection fraction:
pathophysiology, diagnosis, and treatment. Eur Heart J, 2011. 32(6): p. 670-9.

189
343. Weng, L.Q., et al., Aliskiren ameliorates pressure overload-induced heart
hypertrophy and fibrosis in mice. Acta Pharmacol Sin, 2014. 35(8): p. 1005-14.
344. Song, Y., et al., Autophagy in hepatic fibrosis. Biomed Res Int, 2014. 2014: p.
436242.
345. Miranda, A., et al., Time course of echocardiographic and electrocardiographic
parameters in myocardial infarct in rats. An Acad Bras Cienc, 2007. 79(4): p. 639-
48.
346. Kimura, T., et al., Chloroquine in cancer therapy: a double-edged sword of
autophagy. Cancer Res, 2013. 73(1): p. 3-7.
347. Freed, D.H., et al., Cardiotrophin-1: expression in experimental myocardial
infarction and potential role in post-MI wound healing. Mol Cell Biochem, 2003.
254(1-2): p. 247-56.
348. Ju, H., et al., Effect of AT1 receptor blockade on cardiac collagen remodeling after
myocardial infarction. Cardiovasc Res, 1997. 35(2): p. 223-32.
349. Hao, J., et al., Elevation of expression of Smads 2, 3, and 4, decorin and TGF-beta in
the chronic phase of myocardial infarct scar healing. J Mol Cell Cardiol, 1999. 31(3):
p. 667-78.
350. Gao, H., et al., Sequential changes in autophagy in diabetic cardiac fibrosis. Mol
Med Rep, 2016. 13(1): p. 327-32.
351. Ding, Y. and M.E. Choi, Regulation of autophagy by TGF-beta: emerging role in
kidney fibrosis. Semin Nephrol, 2014. 34(1): p. 62-71.
352. Margaritopoulos, G.A., et al., Self-eating: friend or foe? The emerging role of
autophagy in idiopathic pulmonary fibrosis. Biomed Res Int, 2013. 2013: p. 420497.
353. Mao, Y.Q. and X.M. Fan, Autophagy: A new therapeutic target for liver fibrosis.
World J Hepatol, 2015. 7(16): p. 1982-6.

190
354. Yuan, X., et al., Chloroquine improves left ventricle diastolic function in
streptozotocin-induced diabetic mice. Drug Des Devel Ther, 2016. 10: p. 2729-37.
355. Hein, S., et al., Progression from compensated hypertrophy to failure in the pressure-
overloaded human heart: structural deterioration and compensatory mechanisms.
Circulation, 2003. 107(7): p. 984-91.
356. Zhou, H., Y. Luo, and S. Huang, Updates of mTOR inhibitors. Anticancer Agents
Med Chem, 2010. 10(7): p. 571-81.
357. Zeglinski, M.R., et al., Chronic expression of Ski induces apoptosis and represses
autophagy in cardiac myofibroblasts. Biochim Biophys Acta, 2016. 1863(6 Pt A): p.
1261-8.
358. Margaritopoulos, G.A., E. Vasarmidi, and K.M. Antoniou, Pirfenidone in the
treatment of idiopathic pulmonary fibrosis: an evidence-based review of its place in
therapy. Core Evid, 2016. 11: p. 11-22.




















