
Connective tissue growth factor (CTGF/CCN2) is a downstream mediator for TGF-β1-induced...
10
JOURNAL OF CELLULAR PHYSIOLOGY 210:843–852 (2007) Connective Tissue Growth Factor (CTGF/CCN2) Is a Downstream Mediator for TGF-b1-Induced Extracellular Matrix Production in Osteoblasts J.A. ARNOTT, 1 E. NUGLOZEH, 1 M.C. RICO, 2 I. ARANGO-HISIJARA, 1 P.R. ODGREN, 3 F.F. SAFADI, 1 AND S.N. POPOFF 1 * 1 Department of Anatomy and Cell Biology, Temple University School of Medicine, Philadelphia, Pennsylvania 2 Department of Physiology, Temple University School of Medicine, Philadelphia, Pennsylvania 3 Department of Cell Biology, University of Massachusetts Medical School, Worcester, Massachusetts Connective tissue growth factor (CTGF/CCN2) is a cysteine-rich, extracellular matrix (ECM) protein that acts as an anabolic growth factor to regulate osteoblast differentiation and function. Recent studies have identified CTGF as a downstream effector of transforming growth factor-b1 (TGF-b1) for certain functions in specific cell types. In this study, we examined the role of CTGF as a downstream mediator of TGF-b1-induced ECM production and cell growth in osteoblasts. Using primary cultures, we demonstrated that TGF-b1 is a potent inducer of CTGF expression in osteoblasts, and that this induction occurred at all stages of osteoblast differentiation from the proliferative through mineralization stages. TGF-b1 treatment of osteoblasts increased the expression and synthesis of the ECM components, collagen and fibronectin. When CTGF-specific siRNA was used to prevent TGF-b1 induction of CTGF expression, it also inhibited collagen and fibronectin production, thereby demonstrating the requirement of CTGF for their up- regulation. To examine the effects of TGF-b1 on osteoblast cell growth, cultures were treated with TGF-b1 during the proliferative stage. Cell number was significantly reduced and the cells exhibited a decrease in G 1 cyclin expression, consistent with TGF-b1- induced cell-cycle arrest. Cultures transfected with CTGF siRNA prior to TGF-b1 treatment showed an even greater reduction in cell number, suggesting that TGF-b1-induced growth arrest is independent of CTGF in osteoblasts. Collectively, these data demonstrate for the first time that CTGF is an essential downstream mediator for TGF-b1-induced ECM production in osteoblasts, but these two growth factors function independently regarding their opposing effects on osteoblast proliferation. J. Cell. Physiol. 210: 843 – 852, 2007. ß 2006 Wiley-Liss, Inc. The development (modeling) and maintenance (remo- deling) of bone is a complex process that involves the coordinated actions of different cell types and is regu- lated by a multitude of systemic and locally produced growth factors. Osteoblasts, the bone forming cells, originate from mesenchymal (stromal) stem cells and their differentiation is characterized by a temporal pattern of expression of cell growth and osteoblast phenotype-related genes as well as transcriptional factors (Sims and Baron, 2000). The early stages of osteoblast development, from the stromal mesenchymal cell through the pre-osteoblast stages, are characterized by cells that retain the ability to proliferate as they gradually differentiate from a pluripotent cell to a cell that is committed to the osteoblast lineage. As prolifera- tion of pre-osteoblasts ceases, these cells become mature, biosynthetic osteoblasts that produce the organic components of the bone extracellular matrix (ECM), termed osteoid (Sims and Baron, 2000). Osteoid is composed of primarily of type I collagen fibers (90%), with the remainder consisting of an increasing number of non-collagenous proteins (e.g., fibronectin, osteopon- tin) that have been shown to have specific functions. The terminal stage of osteoblast differentiation is character- ized by mineralization of the bone matrix. One secreted growth factor that is known to modulate the differentiation of osteoprogenitor cells into mature osteoblasts, as well as control the synthetic functions of osteoblasts, is transforming growth factor-b1 (TGF-b1) (for review see Bonewald, 2002). Although osteoblasts secrete multiple TGF-b isoforms (TGF-b1, b2, b3) into the bone matrix, the TGF-b1 isoform accounts for 80– 90% of the total expressed in bone (Seyedin et al., 1985; Robey et al., 1987; Sanberg et al., 1988). TGF-b1 promotes recruitment of osteoblast precursors to sites of bone formation (Pfeilschifter et al., 1990), acts as a regulator of osteoblast replication (Bonewald and Mundy, 1990), and stimulates the formation of osteoid by up-regulating the expression of numerous bone ECM proteins such as type I collagen and fibronectin (Centrella and Canalis, 1987; Wrana et al., 1988; Hock et al., 1990; Bonewald and Dallas, 1994). However, the continuous presence of TGF-b1 has been shown to inhibit the terminal differentiation of osteoblasts and the corresponding deposition of mineral (Bonewald and Dallas, 1994). The molecular mechanism(s) by which TGF-b1 functions in osteoblasts are not fully under- stood. Another secreted growth factor with more recently described effects on osteoblast differentiation and function is connective tissue growth factor (CTGF). CTGF is a cysteine-rich, matricellular protein that belongs to the CCN (CTGF, Cyr61, nov, WISP1, WISP2, and WISP3) protein family (Moussad and Brigstock, ß 2006 WILEY-LISS, INC. Contract grant sponsor: National Institutes of Health; Contract grant number: DE 07444. *Correspondence to: S.N. Popoff, Department of Anatomy and Cell Biology, Temple University School of Medicine, 3400 North Broad St., Philadelphia, PA 19040. E-mail: [email protected] Received 7 March 2006; Accepted 11 September 2006 DOI: 10.1002/jcp.20917
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Connective tissue growth factor (CTGF/CCN2) is a downstream mediator for TGF-β1-induced...

JOURNAL OF CELLULAR PHYSIOLOGY 210:843–852 (2007)
Connective Tissue Growth Factor (CTGF/CCN2)Is a Downstream Mediator for TGF-b1-InducedExtracellular Matrix Production in Osteoblasts
J.A. ARNOTT,1 E. NUGLOZEH,1 M.C. RICO,2 I. ARANGO-HISIJARA,1
P.R. ODGREN,3 F.F. SAFADI,1 AND S.N. POPOFF1*1Department of Anatomy and Cell Biology,
Temple University School of Medicine, Philadelphia, Pennsylvania2Department of Physiology, Temple University School of Medicine,
Philadelphia, Pennsylvania3Department of Cell Biology, University of Massachusetts Medical School,
Worcester, Massachusetts
Connective tissue growth factor (CTGF/CCN2) is a cysteine-rich, extracellular matrix (ECM) protein that acts as an anabolic growthfactor to regulate osteoblast differentiation and function. Recent studies have identified CTGF as a downstream effector oftransforming growth factor-b1 (TGF-b1) for certain functions in specific cell types. In this study, we examined the role of CTGF as adownstreammediator of TGF-b1-inducedECMproduction andcell growth in osteoblasts.Using primary cultures,wedemonstratedthat TGF-b1 is a potent inducer of CTGF expression in osteoblasts, and that this induction occurred at all stages of osteoblastdifferentiation from the proliferative through mineralization stages. TGF-b1 treatment of osteoblasts increased the expression andsynthesis of the ECM components, collagen and fibronectin.When CTGF-specific siRNAwas used to prevent TGF-b1 induction ofCTGFexpression, it also inhibited collagenandfibronectin production, therebydemonstrating the requirement ofCTGF for their up-regulation. To examine the effects of TGF-b1 on osteoblast cell growth, cultures were treated with TGF-b1 during the proliferativestage. Cell number was significantly reduced and the cells exhibited a decrease in G1 cyclin expression, consistent with TGF-b1-induced cell-cycle arrest. Cultures transfectedwithCTGF siRNAprior to TGF-b1 treatment showed an even greater reduction in cellnumber, suggesting that TGF-b1-induced growth arrest is independent of CTGF in osteoblasts. Collectively, these data demonstratefor the first time that CTGF is an essential downstreammediator for TGF-b1-induced ECM production in osteoblasts, but these twogrowth factors function independently regarding their opposing effects on osteoblast proliferation. J. Cell. Physiol. 210: 843–852,2007. � 2006 Wiley-Liss, Inc.
The development (modeling) and maintenance (remo-deling) of bone is a complex process that involves thecoordinated actions of different cell types and is regu-lated by a multitude of systemic and locally producedgrowth factors. Osteoblasts, the bone forming cells,originate from mesenchymal (stromal) stem cells andtheir differentiation is characterized by a temporalpattern of expression of cell growth and osteoblastphenotype-related genes as well as transcriptionalfactors (Sims and Baron, 2000). The early stages ofosteoblast development, from the stromal mesenchymalcell through the pre-osteoblast stages, are characterizedby cells that retain the ability to proliferate as theygradually differentiate from a pluripotent cell to a cellthat is committed to the osteoblast lineage. As prolifera-tion of pre-osteoblasts ceases, these cells becomemature, biosynthetic osteoblasts that produce theorganic components of the bone extracellular matrix(ECM), termed osteoid (Sims and Baron, 2000). Osteoidis composed of primarily of type I collagen fibers (90%),with the remainder consisting of an increasing numberof non-collagenous proteins (e.g., fibronectin, osteopon-tin) that have been shown to have specific functions. Theterminal stage of osteoblast differentiation is character-ized by mineralization of the bone matrix.
One secreted growth factor that is known to modulatethe differentiation of osteoprogenitor cells into matureosteoblasts, as well as control the synthetic functions ofosteoblasts, is transforming growth factor-b1 (TGF-b1)(for review see Bonewald, 2002). Although osteoblastssecrete multiple TGF-b isoforms (TGF-b1, b2, b3) intothe bone matrix, the TGF-b1 isoform accounts for 80–90% of the total expressed in bone (Seyedin et al., 1985;
Robey et al., 1987; Sanberg et al., 1988). TGF-b1promotes recruitment of osteoblast precursors to sitesof bone formation (Pfeilschifter et al., 1990), acts asa regulator of osteoblast replication (Bonewald andMundy, 1990), and stimulates the formation of osteoidby up-regulating the expression of numerous boneECM proteins such as type I collagen and fibronectin(Centrella and Canalis, 1987; Wrana et al., 1988; Hocket al., 1990; Bonewald and Dallas, 1994). However, thecontinuous presence of TGF-b1 has been shown toinhibit the terminal differentiation of osteoblasts andthe corresponding deposition of mineral (Bonewald andDallas, 1994). The molecular mechanism(s) by whichTGF-b1 functions in osteoblasts are not fully under-stood.
Another secreted growth factor with more recentlydescribed effects on osteoblast differentiation andfunction is connective tissue growth factor (CTGF).CTGF is a cysteine-rich, matricellular protein thatbelongs to the CCN (CTGF, Cyr61, nov, WISP1, WISP2,and WISP3) protein family (Moussad and Brigstock,
� 2006 WILEY-LISS, INC.
Contract grant sponsor: National Institutes of Health; Contractgrant number: DE 07444.
*Correspondence to: S.N. Popoff, Department of Anatomy andCell Biology, Temple University School of Medicine, 3400 NorthBroad St., Philadelphia, PA 19040. E-mail: [email protected]
Received 7 March 2006; Accepted 11 September 2006
DOI: 10.1002/jcp.20917

2000). CTGF has been implicated as a key regulatoryfactor in complex biological and pathological processesincluding wound healing (Igarashi et al., 1993, 1996;Grotendorst, 1997), angiogenesis (Babic et al., 1999; Lauand Lam, 1999), fibrotic disorders (Igarashi et al., 1996;Dammeier et al., 1998; Lasky et al., 1998; di Mola et al.,1999; Paradis et al., 1999), carcinogenesis (Frazier andGrotendorst, 1997; Kubo et al., 1998), endochondralossification (Ivkovic et al., 2003; Takigawa et al., 2003),and bone fracture repair (Nakata et al., 2002; Safadiet al., 2003). Depending on the cell type, CTGF has beenshown to promote cell growth, migration, adhesion,survival, differentiation, and the biosynthesis of ECMproteins (Igarashi et al., 1993; Frazier et al., 1996;Kireeva et al., 1996; Brigstock, 1999; Moussad andBrigstock, 2000).
Our laboratory recently demonstrated that CTGFis produced and secreted by osteoblasts where it actsin an autocrine fashion as an anabolic growth factorto regulate osteoblast differentiation and function (Xuet al., 2000; Safadi et al., 2003). Using recombinantCTGF, we demonstrated that CTGF acts as an osteoin-ductive agent when injected into the marrow cavity invivo and has the ability to promote proliferation, matrixformation (nodule formation), and mineralization indifferentiating primary osteoblast cultures, suggestingthat CTGF plays distinct roles during both early and lateevents of osteoblast differentiation (Safadi et al., 2003).These results have been confirmed by similar findingsusing osteoblast cell lines, where CTGF was shownto promote proliferation, collagen synthesis, alkalinephosphatase (ALPase) activity, and mineralization(Nishida et al., 2000).
Studies in other connective tissue cell types haveshown that TGF-b1 is a potent inducer of CTGFexpression (Blom et al., 2002). Numerous other studieshave shown that CTGF acts as a downstream mediatorof some of the effects of TGF-b1 on cell proliferation,migration, adhesion, and matrix production (Groten-dorst, 1997; Duncan et al., 1999; Takehara, 2000; Blomet al., 2001; Gore-Hyer et al., 2002; Yokoi et al., 2002;Weston et al., 2003; Suzuki et al., 2006). More recently, astudy using CTGF �/� mice (Shi-wen et al., 2006) hascharacterized the requirement of CTGF for TGF-b1-induced responses in embryonic fibroblasts includinginduction of subsets of genes associated with matrixproduction including type I collagen. These studiesdemonstrate the requirement of CTGF expression forTGF-b1-induced responses in these cells. However, it isimportant to note that this TGF-b1/CTGF interaction ishighly cell type specific. Although TGF-b1 and CTGFhave similar effects on osteoblast differentiation bymodulating proliferation and inducing ECM synthesis,the potential interaction between these two factors inosteoblasts has not been examined. Therefore, thepurpose of this study was to: (1) characterize thetemporal and kinetic aspects of CTGF up-regulation byTGF-b1 in primary osteoblast cultures and (2) deter-mine whether CTGF is a downstream mediator of TGB-b1 effects on synthesis of ECM proteins and cell growthin these cultures.
MATERIALS AND METHODSAnimals
Primary osteoblasts were derived from bone (calvaria) ofNorway-Hooded rats that are maintained at Temple Univer-sity School of Medicine (Philadelphia, PA). All animals werehandled according to the principles in the NIH Guide for theCare and Use of Laboratory Animals (U.S. Department of
Health and Human Services, Publ. No. 86-23, 1985) andguidelines established by the IACUC of Temple University.
Primary cell culture
Primary osteoblast cultures were obtained using neonatalrats as previously described (Xu et al., 2000). Briefly, parietalcalvaria from neonatal rat pups were isolated and subjected tosequential digestions of 5, 15,15, 25, and 25 min at 378C in ashaking water-bath with 2% collagenase-P (Roche, Nutley,NJ)/0.25% trypsin (Mediatec, Herndon, VA). Cells from thefirst step of digestion were discarded and those released fromthe 2nd to 5th digestions were plated in 100 mm dishes (Falcon,Bedford, MA) at 5� 105 cells/plate in Earle’s MinimalEssential Medium (EMEM; Mediatec) supplemented with10% fetal calf serum (FCS; Mediatec). The cells were incubatedat 378C with 5% CO2 with change of media every 3 days untilthey reached 80% confluency. Cells were maintained in thisstate or frozen in EMEM with 10% FCS and 10% DMSO forsubsequent utilization. To initiate osteoblast differentiation,the plating medium was replaced with EMEM containing 10%FBS and 50 mg/ml ascorbic acid (Sigma, St. Louis, MO) on the3rd day of culture. Then on the 7th day and every 3 daysthereafter the medium was replaced with EMEM containing10% FCS, 50 mg/ml ascorbic acid, and 10 mM b-glyceropho-sphate (Sigma).
TGF-b1 stimulation
Recombinant TGF-b1 (Calbiochem, La Jolla, CA) wasdiluted to a concentration of 1 mg/ml in sterile 4 mM HClcontaining 0.1% BSA and stored at �208C. Osteoblasts wereserum deprived for 24 h prior to treatment. Cells were treatedwith a standard concentration of 5 ng/ml of TGF-b1 for a 24 hperiod unless otherwise stated.
RNA isolation and Northern blotting
Total cellular RNA was isolated from cell cultures usingTrizol reagent (Invitrogen, Carlsbad, CA) according to themanufacturer’s directions. For Northern blot analysis, 10 mg ofRNA was separated on an agarose gel and transferred to aHybond (Amersham Biosciences, Piscataway, NJ) membranein the presence of 10�SSC. DNA probes used for hybridizationwere obtained from the following sequences: rat CTGF (Xuet al., 2000), 18S (kindly provided by C. Demers, University ofMontreal, Canada); rat type I collagen and rat fibronectin wereamplified from a rat cDNA pool. All DNA probes were labeledwith 32P-dCTP using Rediprime II Random Prime LabelingSystem (Amersham Biosciences) according to the manufac-turer’s instructions. Membranes were hybridized at 658C inChurch buffer consisting of 1% BSA, 1 mM EDTA, 0.5 MNaHPO4 (pH 7.2), and 7% SDS. After hybridization, the blotswere washed twice in 0.1% SDS/2�SSC and twice in 0.1% SDS/1�SSC at room temperature and exposed to Kodak film at�808C. Changes in mRNA levels were determined followingnormalization of each sample to the 18S RNA signal in eachlane. All data are expressed as a percentage of the control foreach experiment.
Protein isolation and Western blotting
Western blot analysis was performed as previouslydescribed (Safadi et al., 2003). Briefly, cell monolayers werehomogenized for 24 h at 48C in protein extraction buffer (RIPAbuffer) consisting of 50 mM Tris-HCl (pH 7.5), 135 mM NaCl,1% Triton X-100, 0.1% sodium deoxycholate, 2 mM EDTA,50 mM NaF, 2 mM sodium orthovanadate, 10 mg/ml aprotinin,10mg/ml leupeptin, and 1 mM PMSF. Cell lysates were clarifiedusing centrifugation. Total protein concentration from theresulting supernatant was measured using the BCA ProteinAssay Reagent Kit (Pierce, Rockford, IL) according to themanufacturer’s instructions. Samples were subjected tosodium dodecyl sulfate–polyacrylamide gel electrophoresis(SDS–PAGE) on a 7.5% acrylimide gel and transferred toPVDF filters by electroblotting. Blots were incubated witheither anti-CTGF, cyclin D1, cyclin A, cyclin E, CDK4 (Santa Cruz,Santa Cruz, CA), anti-fibronectin (Cell Signalling, Danvers,MA) or anti-actin (Sigma) primary antibody, and then withHRP-conjugated secondary antibody (Sigma). Antigens weredetected using the Pierce chemiluminescent substrate system.
844 ARNOTT ET AL.
Journal of Cellular Physiology DOI 10.1002/jcp
Osteoblast collagen synthesis
Collagen synthesis was measured by the incorporation of[3H]-proline into collagen as previously described (Yu et al.,2003). Primary rat osteoblasts were pulsed with 1 mCi/ml L-[2,3,4,5-3H]-proline (Amersham Biosciences) and 50 mg/mlascorbic acid in the last 24 h of the culture. Collagen from thecell layer was solubilized using 0.5 N NaOH and proteins wereprecipitated with 20% trichloroacetic acid (TCA) by centrifu-gation at 3,000g. Samples were split into duplicates, neutra-lized with HCl and one sample from each pair was treated withcollagenase (600 U/ml). Following a 2 h digestion at 378C, acid-insoluble material was precipitated with 10% TCA/0.5% tannicacid. Samples were counted for b-emission. Counts weresubtracted from duplicate samples without collagenase andnormalized to cell number.
Cell proliferation
Cell proliferation was measured using the CyQuant CellProliferation Kit (C-7026) (Molecular Probes, Eugene, OR)according to the manufacturer’s procedure. Briefly, cells wereplated in a 24-well microplate at the indicated density andincubated for 24 h in EMEM supplemented with 10% FCS.Prior to stimulation with TGF-b1 (5 ng/ml) the cells wereserum starved for 24 h. The samples were measured using aWallac 1420 fluorometer. Cell number was calculated based oncomparison to a standard curve generated for primaryosteoblasts.
Caspase 3/7 activity
Caspase 3/7 activity was measured using the Caspase-Glo 3/7 assay (Promega, Madison, WI) according to the manufacturer’sdirections. Briefly, cells were plated in a 96-well microplate(2.4� 104 cells/well) and incubated in EMEM supplementedwith 10% FCS for 24 h. Cells were then serum starved for 24 hand treated with TGF-b1 at the indicated doses for 24 h. Cellsand buffers were equilibrated to RT for 1 h and subsequentlylysed in Caspase 3/7 reagent for 30 min with mild agitation.The samples were then measured using a Wallac 1420fluorometer, normalized to a blank reaction, and graphed asrelative caspase activity.
Osteoblasts tranfection with CTGF siRNA
Sub-confluent primary osteoblast cultures were transfectedwith siRNA 48 h prior to treatment with TGF-b1. Transfectionswere carried out using Lipofectamine 2000 (Invitrogen) andOpti-MEM (Invitrogen) according to the manufacturer’sprotocol. An siRNA SMARTpool consisting of four target-specific 19-nucleotide siRNA duplexes was manufactured fromthe open reading frame of rat CTGF (accession number inGenBank: NM_022266) by Dharmacon Research (Lafayette,CO). Control siRNA was siCONTROL non-targeting siRNA #1(D-001210-0105) (Dharmacon, Lafayette, CO). siGLO RISC-Free siRNA (D-001600-01-05) (Dharmacon) was used to assesstransfection efficiency.
Data analysis
For all quantitative data, analysis of variance (ANOVA) wasemployed to evaluate the effect of one variable on two or moreindependent groups. In the event of a significant group effect,individual pairs of means were compared using the Bonferronipost hoc test. Data are calculated as mean�SEM, and in somecases, converted to percent of control. A P-value <0.05 wasused to determine whether differences were statisticallysignificant.
RESULTSTGF-b1 up-regulation of CTGF expression in
primary osteoblast cultures
In the first series of experiments we assessed theability of TGF-b1 to up-regulate CTGF expression inprimary osteoblast cultures. Northern blot analysisrevealed that treatment of osteoblasts with TGF-b1induced a robust increase in steady-state CTGF tran-script levels compared to mock-treated (diluent only),control cultures (Fig. 1A). This increase in CTGF mRNAoccurred with a similar potency regardless of the dose ofTGF-b1 used (Fig. 1A). Similarly, CTGF protein levelsincreased with TGF-b1 stimulation reaching maximum
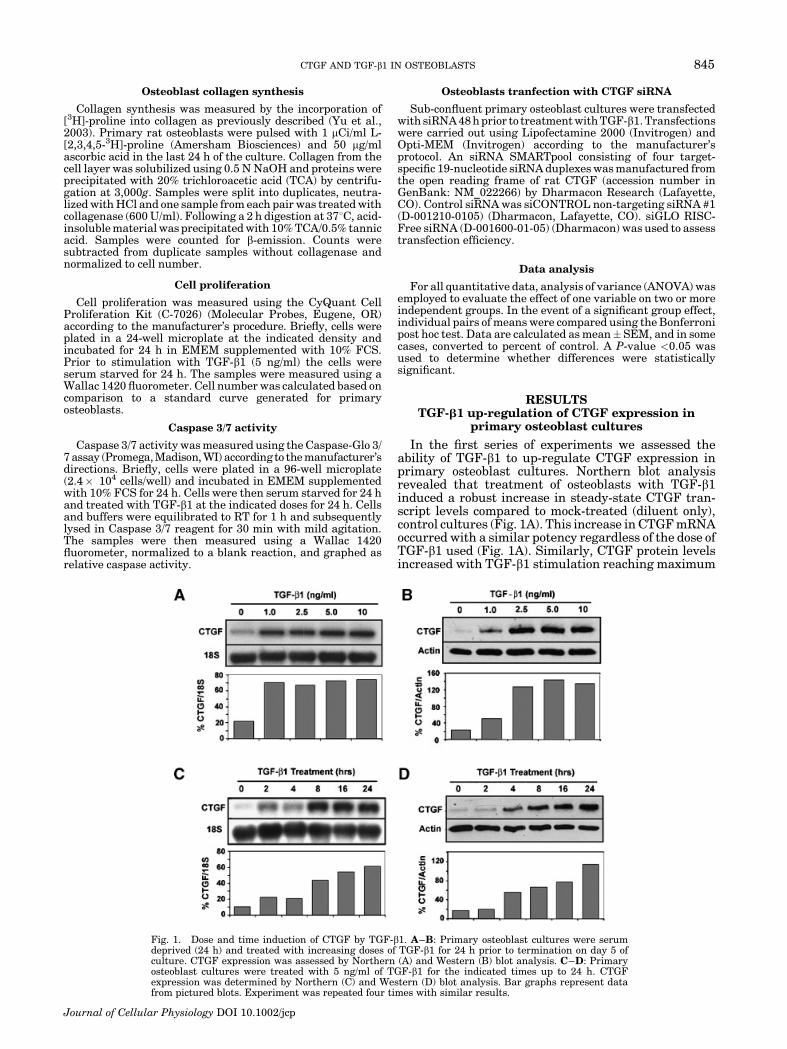
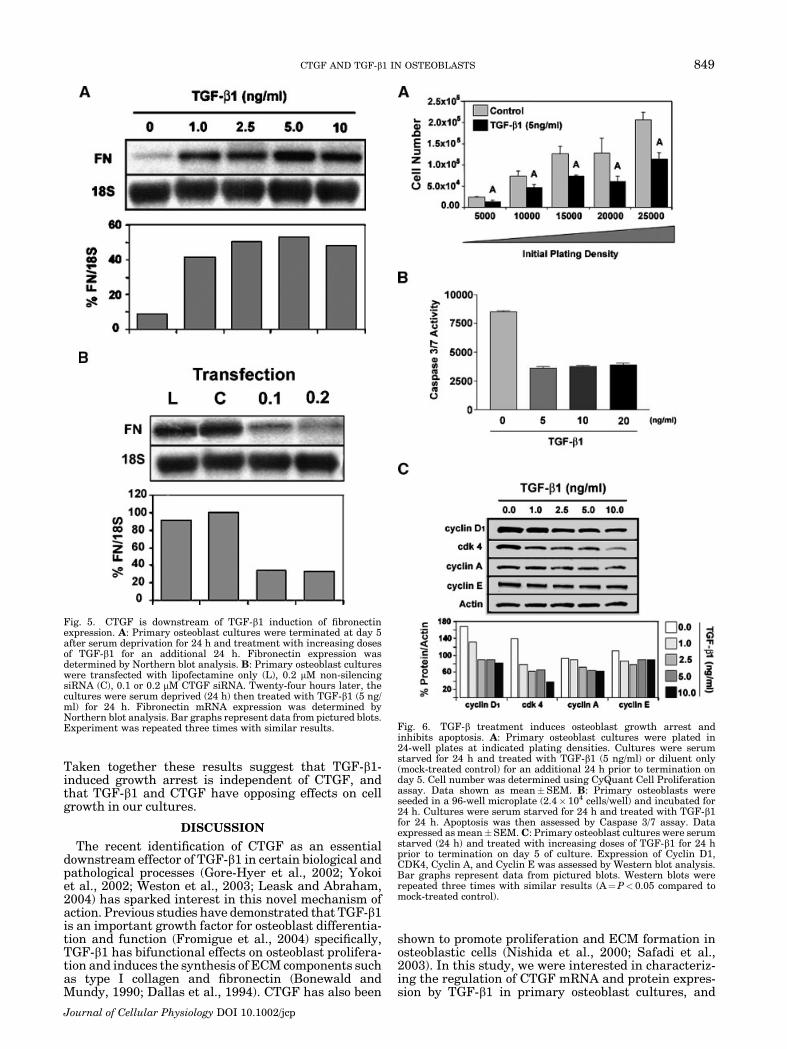
Fig. 1. Dose and time induction of CTGF by TGF-b1. A–B: Primary osteoblast cultures were serumdeprived (24 h) and treated with increasing doses of TGF-b1 for 24 h prior to termination on day 5 ofculture. CTGF expression was assessed by Northern (A) and Western (B) blot analysis. C–D: Primaryosteoblast cultures were treated with 5 ng/ml of TGF-b1 for the indicated times up to 24 h. CTGFexpression was determined by Northern (C) and Western (D) blot analysis. Bar graphs represent datafrom pictured blots. Experiment was repeated four times with similar results.
CTGF AND TGF-b1 IN OSTEOBLASTS 845
Journal of Cellular Physiology DOI 10.1002/jcp
levels at doses of 2.5 ng/ml and above (Fig. 1B). It wasalso determined that none of the TGF-b1 doses used hada toxic effect on the osteoblasts (data not shown). Sincethe standard dose of TGF-b1 previously used in manyother cell/tissue culture systems is 5 ng/ml and this doseresulted in maximal induction of CTGF mRNA andprotein in osteoblast cultures, the dose of TGF-b1 used inall subsequent experiments was 5 ng/ml unless other-wise stated.
We next examined the temporal pattern of CTGFexpression in TGF-b1-treated primary osteoblast cul-tures. CTGF transcripts increased in a time-dependentmanner following TGF-b1 exposure, with maximalinduction occurring between 8 and 24 h post-treatment(Fig. 1C). CTGF mRNA and protein levels were stillelevated but falling at 48 h post-treatment (data notshown). Similar results were obtained by Western blotanalysis with CTGF protein levels increasing in a time-dependent manner and reaching maximum expressionlevels at 24 h post-treatment. Based on these findings,we used a minimum 24 h treatment period for allsubsequent experiments.
CTGF induction by TGF-b1 during differentstages of osteoblast differentiation
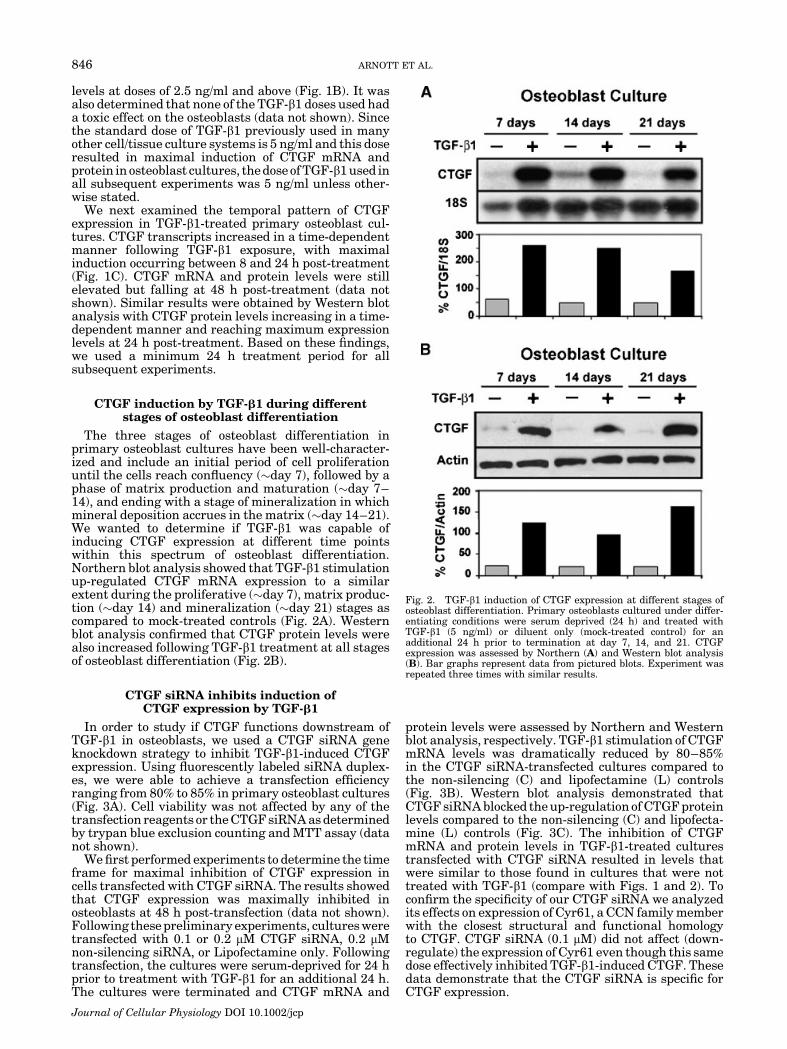
The three stages of osteoblast differentiation inprimary osteoblast cultures have been well-character-ized and include an initial period of cell proliferationuntil the cells reach confluency (�day 7), followed by aphase of matrix production and maturation (�day 7–14), and ending with a stage of mineralization in whichmineral deposition accrues in the matrix (�day 14–21).We wanted to determine if TGF-b1 was capable ofinducing CTGF expression at different time pointswithin this spectrum of osteoblast differentiation.Northern blot analysis showed that TGF-b1 stimulationup-regulated CTGF mRNA expression to a similarextent during the proliferative (�day 7), matrix produc-tion (�day 14) and mineralization (�day 21) stages ascompared to mock-treated controls (Fig. 2A). Westernblot analysis confirmed that CTGF protein levels werealso increased following TGF-b1 treatment at all stagesof osteoblast differentiation (Fig. 2B).
CTGF siRNA inhibits induction ofCTGF expression by TGF-b1
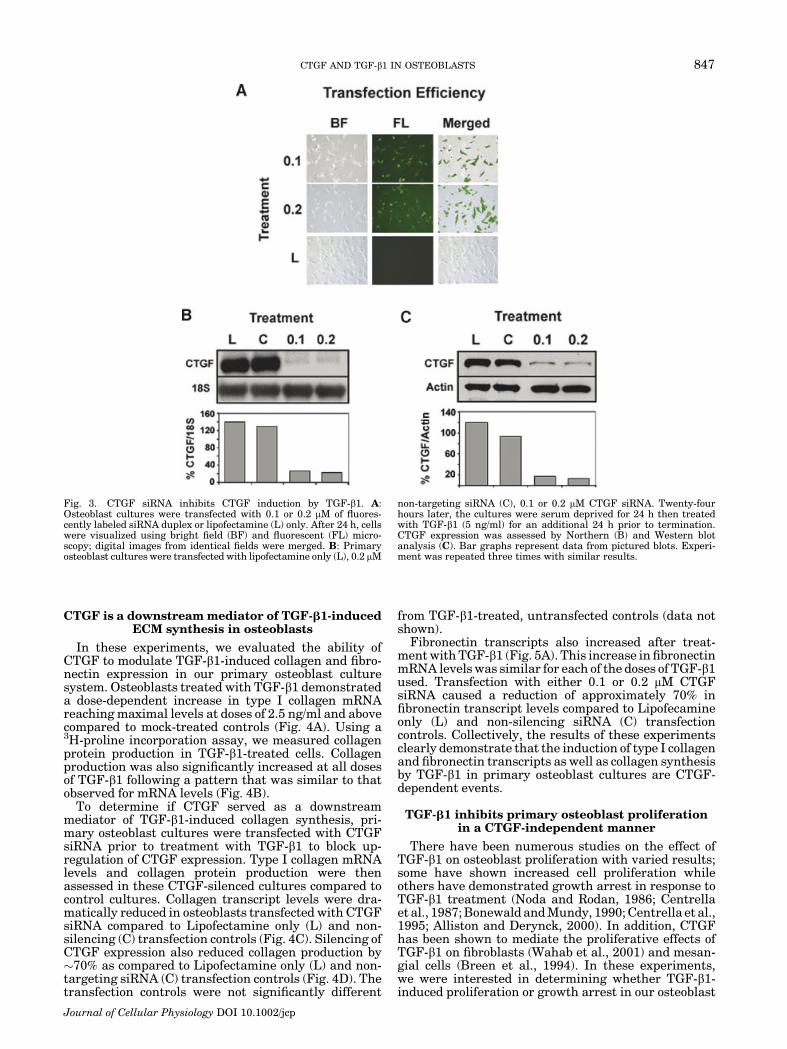
In order to study if CTGF functions downstream ofTGF-b1 in osteoblasts, we used a CTGF siRNA geneknockdown strategy to inhibit TGF-b1-induced CTGFexpression. Using fluorescently labeled siRNA duplex-es, we were able to achieve a transfection efficiencyranging from 80% to 85% in primary osteoblast cultures(Fig. 3A). Cell viability was not affected by any of thetransfection reagents or the CTGF siRNA as determinedby trypan blue exclusion counting and MTT assay (datanot shown).
We first performed experiments to determine the timeframe for maximal inhibition of CTGF expression incells transfected with CTGF siRNA. The results showedthat CTGF expression was maximally inhibited inosteoblasts at 48 h post-transfection (data not shown).Following these preliminary experiments, cultures weretransfected with 0.1 or 0.2 mM CTGF siRNA, 0.2 mMnon-silencing siRNA, or Lipofectamine only. Followingtransfection, the cultures were serum-deprived for 24 hprior to treatment with TGF-b1 for an additional 24 h.The cultures were terminated and CTGF mRNA and
protein levels were assessed by Northern and Westernblot analysis, respectively. TGF-b1 stimulation of CTGFmRNA levels was dramatically reduced by 80–85%in the CTGF siRNA-transfected cultures compared tothe non-silencing (C) and lipofectamine (L) controls(Fig. 3B). Western blot analysis demonstrated thatCTGF siRNA blocked the up-regulation of CTGF proteinlevels compared to the non-silencing (C) and lipofecta-mine (L) controls (Fig. 3C). The inhibition of CTGFmRNA and protein levels in TGF-b1-treated culturestransfected with CTGF siRNA resulted in levels thatwere similar to those found in cultures that were nottreated with TGF-b1 (compare with Figs. 1 and 2). Toconfirm the specificity of our CTGF siRNA we analyzedits effects on expression of Cyr61, a CCN family memberwith the closest structural and functional homologyto CTGF. CTGF siRNA (0.1 mM) did not affect (down-regulate) the expression of Cyr61 even though this samedose effectively inhibited TGF-b1-induced CTGF. Thesedata demonstrate that the CTGF siRNA is specific forCTGF expression.
Fig. 2. TGF-b1 induction of CTGF expression at different stages ofosteoblast differentiation. Primary osteoblasts cultured under differ-entiating conditions were serum deprived (24 h) and treated withTGF-b1 (5 ng/ml) or diluent only (mock-treated control) for anadditional 24 h prior to termination at day 7, 14, and 21. CTGFexpression was assessed by Northern (A) and Western blot analysis(B). Bar graphs represent data from pictured blots. Experiment wasrepeated three times with similar results.
846 ARNOTT ET AL.
Journal of Cellular Physiology DOI 10.1002/jcp
CTGF is a downstream mediator of TGF-b1-inducedECM synthesis in osteoblasts
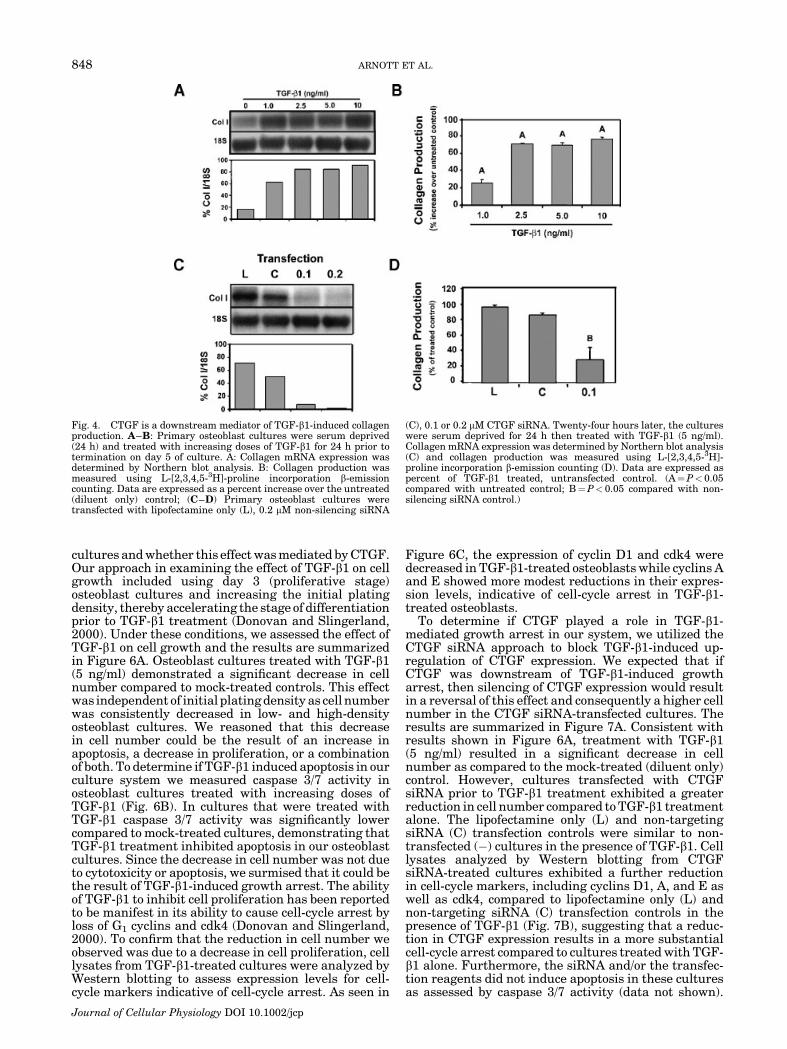
In these experiments, we evaluated the ability ofCTGF to modulate TGF-b1-induced collagen and fibro-nectin expression in our primary osteoblast culturesystem. Osteoblasts treated with TGF-b1 demonstrateda dose-dependent increase in type I collagen mRNAreaching maximal levels at doses of 2.5 ng/ml and abovecompared to mock-treated controls (Fig. 4A). Using a3H-proline incorporation assay, we measured collagenprotein production in TGF-b1-treated cells. Collagenproduction was also significantly increased at all dosesof TGF-b1 following a pattern that was similar to thatobserved for mRNA levels (Fig. 4B).
To determine if CTGF served as a downstreammediator of TGF-b1-induced collagen synthesis, pri-mary osteoblast cultures were transfected with CTGFsiRNA prior to treatment with TGF-b1 to block up-regulation of CTGF expression. Type I collagen mRNAlevels and collagen protein production were thenassessed in these CTGF-silenced cultures compared tocontrol cultures. Collagen transcript levels were dra-matically reduced in osteoblasts transfected with CTGFsiRNA compared to Lipofectamine only (L) and non-silencing (C) transfection controls (Fig. 4C). Silencing ofCTGF expression also reduced collagen production by�70% as compared to Lipofectamine only (L) and non-targeting siRNA (C) transfection controls (Fig. 4D). Thetransfection controls were not significantly different
from TGF-b1-treated, untransfected controls (data notshown).
Fibronectin transcripts also increased after treat-ment with TGF-b1 (Fig. 5A). This increase in fibronectinmRNA levels was similar for each of the doses of TGF-b1used. Transfection with either 0.1 or 0.2 mM CTGFsiRNA caused a reduction of approximately 70% infibronectin transcript levels compared to Lipofecamineonly (L) and non-silencing siRNA (C) transfectioncontrols. Collectively, the results of these experimentsclearly demonstrate that the induction of type I collagenand fibronectin transcripts as well as collagen synthesisby TGF-b1 in primary osteoblast cultures are CTGF-dependent events.
TGF-b1 inhibits primary osteoblast proliferationin a CTGF-independent manner
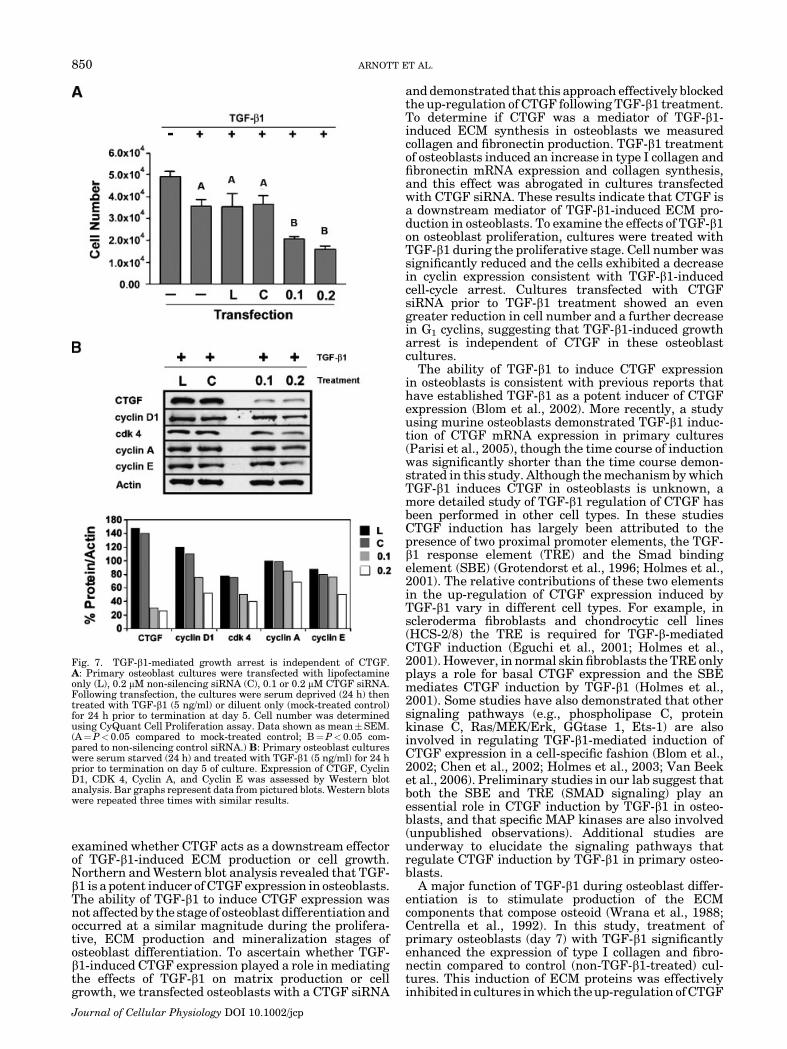
There have been numerous studies on the effect ofTGF-b1 on osteoblast proliferation with varied results;some have shown increased cell proliferation whileothers have demonstrated growth arrest in response toTGF-b1 treatment (Noda and Rodan, 1986; Centrellaet al., 1987; Bonewald and Mundy, 1990; Centrella et al.,1995; Alliston and Derynck, 2000). In addition, CTGFhas been shown to mediate the proliferative effects ofTGF-b1 on fibroblasts (Wahab et al., 2001) and mesan-gial cells (Breen et al., 1994). In these experiments,we were interested in determining whether TGF-b1-induced proliferation or growth arrest in our osteoblast
Fig. 3. CTGF siRNA inhibits CTGF induction by TGF-b1. A:Osteoblast cultures were transfected with 0.1 or 0.2 mM of fluores-cently labeled siRNA duplex or lipofectamine (L) only. After 24 h, cellswere visualized using bright field (BF) and fluorescent (FL) micro-scopy; digital images from identical fields were merged. B: Primaryosteoblast cultures were transfected with lipofectamine only (L), 0.2 mM
non-targeting siRNA (C), 0.1 or 0.2 mM CTGF siRNA. Twenty-fourhours later, the cultures were serum deprived for 24 h then treatedwith TGF-b1 (5 ng/ml) for an additional 24 h prior to termination.CTGF expression was assessed by Northern (B) and Western blotanalysis (C). Bar graphs represent data from pictured blots. Experi-ment was repeated three times with similar results.
CTGF AND TGF-b1 IN OSTEOBLASTS 847
Journal of Cellular Physiology DOI 10.1002/jcp
cultures and whether this effect was mediated by CTGF.Our approach in examining the effect of TGF-b1 on cellgrowth included using day 3 (proliferative stage)osteoblast cultures and increasing the initial platingdensity, thereby accelerating the stage of differentiationprior to TGF-b1 treatment (Donovan and Slingerland,2000). Under these conditions, we assessed the effect ofTGF-b1 on cell growth and the results are summarizedin Figure 6A. Osteoblast cultures treated with TGF-b1(5 ng/ml) demonstrated a significant decrease in cellnumber compared to mock-treated controls. This effectwas independent of initial plating density as cell numberwas consistently decreased in low- and high-densityosteoblast cultures. We reasoned that this decreasein cell number could be the result of an increase inapoptosis, a decrease in proliferation, or a combinationof both. To determine if TGF-b1 induced apoptosis in ourculture system we measured caspase 3/7 activity inosteoblast cultures treated with increasing doses ofTGF-b1 (Fig. 6B). In cultures that were treated withTGF-b1 caspase 3/7 activity was significantly lowercompared to mock-treated cultures, demonstrating thatTGF-b1 treatment inhibited apoptosis in our osteoblastcultures. Since the decrease in cell number was not dueto cytotoxicity or apoptosis, we surmised that it could bethe result of TGF-b1-induced growth arrest. The abilityof TGF-b1 to inhibit cell proliferation has been reportedto be manifest in its ability to cause cell-cycle arrest byloss of G1 cyclins and cdk4 (Donovan and Slingerland,2000). To confirm that the reduction in cell number weobserved was due to a decrease in cell proliferation, celllysates from TGF-b1-treated cultures were analyzed byWestern blotting to assess expression levels for cell-cycle markers indicative of cell-cycle arrest. As seen in
Figure 6C, the expression of cyclin D1 and cdk4 weredecreased in TGF-b1-treated osteoblasts while cyclins Aand E showed more modest reductions in their expres-sion levels, indicative of cell-cycle arrest in TGF-b1-treated osteoblasts.
To determine if CTGF played a role in TGF-b1-mediated growth arrest in our system, we utilized theCTGF siRNA approach to block TGF-b1-induced up-regulation of CTGF expression. We expected that ifCTGF was downstream of TGF-b1-induced growtharrest, then silencing of CTGF expression would resultin a reversal of this effect and consequently a higher cellnumber in the CTGF siRNA-transfected cultures. Theresults are summarized in Figure 7A. Consistent withresults shown in Figure 6A, treatment with TGF-b1(5 ng/ml) resulted in a significant decrease in cellnumber as compared to the mock-treated (diluent only)control. However, cultures transfected with CTGFsiRNA prior to TGF-b1 treatment exhibited a greaterreduction in cell number compared to TGF-b1 treatmentalone. The lipofectamine only (L) and non-targetingsiRNA (C) transfection controls were similar to non-transfected (�) cultures in the presence of TGF-b1. Celllysates analyzed by Western blotting from CTGFsiRNA-treated cultures exhibited a further reductionin cell-cycle markers, including cyclins D1, A, and E aswell as cdk4, compared to lipofectamine only (L) andnon-targeting siRNA (C) transfection controls in thepresence of TGF-b1 (Fig. 7B), suggesting that a reduc-tion in CTGF expression results in a more substantialcell-cycle arrest compared to cultures treated with TGF-b1 alone. Furthermore, the siRNA and/or the transfec-tion reagents did not induce apoptosis in these culturesas assessed by caspase 3/7 activity (data not shown).
Fig. 4. CTGF is a downstream mediator of TGF-b1-induced collagenproduction. A–B: Primary osteoblast cultures were serum deprived(24 h) and treated with increasing doses of TGF-b1 for 24 h prior totermination on day 5 of culture. A: Collagen mRNA expression wasdetermined by Northern blot analysis. B: Collagen production wasmeasured using L-[2,3,4,5-3H]-proline incorporation b-emissioncounting. Data are expressed as a percent increase over the untreated(diluent only) control; (C–D) Primary osteoblast cultures weretransfected with lipofectamine only (L), 0.2 mM non-silencing siRNA
(C), 0.1 or 0.2 mM CTGF siRNA. Twenty-four hours later, the cultureswere serum deprived for 24 h then treated with TGF-b1 (5 ng/ml).Collagen mRNA expression was determined by Northern blot analysis(C) and collagen production was measured using L-[2,3,4,5-3H]-proline incorporation b-emission counting (D). Data are expressed aspercent of TGF-b1 treated, untransfected control. (A¼P< 0.05compared with untreated control; B¼P< 0.05 compared with non-silencing siRNA control.)
848 ARNOTT ET AL.
Journal of Cellular Physiology DOI 10.1002/jcp
Taken together these results suggest that TGF-b1-induced growth arrest is independent of CTGF, andthat TGF-b1 and CTGF have opposing effects on cellgrowth in our cultures.
DISCUSSION
The recent identification of CTGF as an essentialdownstream effector of TGF-b1 in certain biological andpathological processes (Gore-Hyer et al., 2002; Yokoiet al., 2002; Weston et al., 2003; Leask and Abraham,2004) has sparked interest in this novel mechanism ofaction. Previous studies have demonstrated that TGF-b1is an important growth factor for osteoblast differentia-tion and function (Fromigue et al., 2004) specifically,TGF-b1 has bifunctional effects on osteoblast prolifera-tion and induces the synthesis of ECM components suchas type I collagen and fibronectin (Bonewald andMundy, 1990; Dallas et al., 1994). CTGF has also been
shown to promote proliferation and ECM formation inosteoblastic cells (Nishida et al., 2000; Safadi et al.,2003). In this study, we were interested in characteriz-ing the regulation of CTGF mRNA and protein expres-sion by TGF-b1 in primary osteoblast cultures, and
Fig. 5. CTGF is downstream of TGF-b1 induction of fibronectinexpression. A: Primary osteoblast cultures were terminated at day 5after serum deprivation for 24 h and treatment with increasing dosesof TGF-b1 for an additional 24 h. Fibronectin expression wasdetermined by Northern blot analysis. B: Primary osteoblast cultureswere transfected with lipofectamine only (L), 0.2 mM non-silencingsiRNA (C), 0.1 or 0.2 mM CTGF siRNA. Twenty-four hours later, thecultures were serum deprived (24 h) then treated with TGF-b1 (5 ng/ml) for 24 h. Fibronectin mRNA expression was determined byNorthern blot analysis. Bar graphs represent data from pictured blots.Experiment was repeated three times with similar results. Fig. 6. TGF-b treatment induces osteoblast growth arrest and
inhibits apoptosis. A: Primary osteoblast cultures were plated in24-well plates at indicated plating densities. Cultures were serumstarved for 24 h and treated with TGF-b1 (5 ng/ml) or diluent only(mock-treated control) for an additional 24 h prior to termination onday 5. Cell number was determined using CyQuant Cell Proliferationassay. Data shown as mean�SEM. B: Primary osteoblasts wereseeded in a 96-well microplate (2.4� 104 cells/well) and incubated for24 h. Cultures were serum starved for 24 h and treated with TGF-b1for 24 h. Apoptosis was then assessed by Caspase 3/7 assay. Dataexpressed as mean�SEM. C: Primary osteoblast cultures were serumstarved (24 h) and treated with increasing doses of TGF-b1 for 24 hprior to termination on day 5 of culture. Expression of Cyclin D1,CDK4, Cyclin A, and Cyclin E was assessed by Western blot analysis.Bar graphs represent data from pictured blots. Western blots wererepeated three times with similar results (A¼P< 0.05 compared tomock-treated control).
CTGF AND TGF-b1 IN OSTEOBLASTS 849
Journal of Cellular Physiology DOI 10.1002/jcp
examined whether CTGF acts as a downstream effectorof TGF-b1-induced ECM production or cell growth.Northern and Western blot analysis revealed that TGF-b1 is a potent inducer of CTGF expression in osteoblasts.The ability of TGF-b1 to induce CTGF expression wasnot affected by the stage of osteoblast differentiation andoccurred at a similar magnitude during the prolifera-tive, ECM production and mineralization stages ofosteoblast differentiation. To ascertain whether TGF-b1-induced CTGF expression played a role in mediatingthe effects of TGF-b1 on matrix production or cellgrowth, we transfected osteoblasts with a CTGF siRNA
and demonstrated that this approach effectively blockedthe up-regulation of CTGF following TGF-b1 treatment.To determine if CTGF was a mediator of TGF-b1-induced ECM synthesis in osteoblasts we measuredcollagen and fibronectin production. TGF-b1 treatmentof osteoblasts induced an increase in type I collagen andfibronectin mRNA expression and collagen synthesis,and this effect was abrogated in cultures transfectedwith CTGF siRNA. These results indicate that CTGF isa downstream mediator of TGF-b1-induced ECM pro-duction in osteoblasts. To examine the effects of TGF-b1on osteoblast proliferation, cultures were treated withTGF-b1 during the proliferative stage. Cell number wassignificantly reduced and the cells exhibited a decreasein cyclin expression consistent with TGF-b1-inducedcell-cycle arrest. Cultures transfected with CTGFsiRNA prior to TGF-b1 treatment showed an evengreater reduction in cell number and a further decreasein G1 cyclins, suggesting that TGF-b1-induced growtharrest is independent of CTGF in these osteoblastcultures.
The ability of TGF-b1 to induce CTGF expressionin osteoblasts is consistent with previous reports thathave established TGF-b1 as a potent inducer of CTGFexpression (Blom et al., 2002). More recently, a studyusing murine osteoblasts demonstrated TGF-b1 induc-tion of CTGF mRNA expression in primary cultures(Parisi et al., 2005), though the time course of inductionwas significantly shorter than the time course demon-strated in this study. Although the mechanism by whichTGF-b1 induces CTGF in osteoblasts is unknown, amore detailed study of TGF-b1 regulation of CTGF hasbeen performed in other cell types. In these studiesCTGF induction has largely been attributed to thepresence of two proximal promoter elements, the TGF-b1 response element (TRE) and the Smad bindingelement (SBE) (Grotendorst et al., 1996; Holmes et al.,2001). The relative contributions of these two elementsin the up-regulation of CTGF expression induced byTGF-b1 vary in different cell types. For example, inscleroderma fibroblasts and chondrocytic cell lines(HCS-2/8) the TRE is required for TGF-b-mediatedCTGF induction (Eguchi et al., 2001; Holmes et al.,2001). However, in normal skin fibroblasts the TRE onlyplays a role for basal CTGF expression and the SBEmediates CTGF induction by TGF-b1 (Holmes et al.,2001). Some studies have also demonstrated that othersignaling pathways (e.g., phospholipase C, proteinkinase C, Ras/MEK/Erk, GGtase 1, Ets-1) are alsoinvolved in regulating TGF-b1-mediated induction ofCTGF expression in a cell-specific fashion (Blom et al.,2002; Chen et al., 2002; Holmes et al., 2003; Van Beeket al., 2006). Preliminary studies in our lab suggest thatboth the SBE and TRE (SMAD signaling) play anessential role in CTGF induction by TGF-b1 in osteo-blasts, and that specific MAP kinases are also involved(unpublished observations). Additional studies areunderway to elucidate the signaling pathways thatregulate CTGF induction by TGF-b1 in primary osteo-blasts.
A major function of TGF-b1 during osteoblast differ-entiation is to stimulate production of the ECMcomponents that compose osteoid (Wrana et al., 1988;Centrella et al., 1992). In this study, treatment ofprimary osteoblasts (day 7) with TGF-b1 significantlyenhanced the expression of type I collagen and fibro-nectin compared to control (non-TGF-b1-treated) cul-tures. This induction of ECM proteins was effectivelyinhibited in cultures in which the up-regulation of CTGF
Fig. 7. TGF-b1-mediated growth arrest is independent of CTGF.A: Primary osteoblast cultures were transfected with lipofectamineonly (L), 0.2 mM non-silencing siRNA (C), 0.1 or 0.2 mM CTGF siRNA.Following transfection, the cultures were serum deprived (24 h) thentreated with TGF-b1 (5 ng/ml) or diluent only (mock-treated control)for 24 h prior to termination at day 5. Cell number was determinedusing CyQuant Cell Proliferation assay. Data shown as mean�SEM.(A¼P< 0.05 compared to mock-treated control; B¼P<0.05 com-pared to non-silencing control siRNA.) B: Primary osteoblast cultureswere serum starved (24 h) and treated with TGF-b1 (5 ng/ml) for 24 hprior to termination on day 5 of culture. Expression of CTGF, CyclinD1, CDK 4, Cyclin A, and Cyclin E was assessed by Western blotanalysis. Bar graphs represent data from pictured blots. Western blotswere repeated three times with similar results.
850 ARNOTT ET AL.
Journal of Cellular Physiology DOI 10.1002/jcp

expression was blocked using CTGF siRNA. This is thefirst study demonstrating CTGF as a major downstreammediator of TGF-b1-induced ECM protein synthesisin osteoblasts, and these results are consistent withprevious reports in other cell systems. In vitro studies innormal rat kidney fibroblasts demonstrated that TGF-b-induced collagen synthesis can be inhibited with anti-sense CTGF or anti-CTGF antibody (Duncan et al.,1999). Similarly, CTGF anti-sense blocked TGF-b1-induced fibronectin expression and collagen productionin rat renal fibroblasts (Yokoi et al., 2002), and TGF-b1-induced fibronectin synthesis in human prostate stro-mal cells (Suzuki et al., 2006). TGF-b1-induced fibro-nectin synthesis was also inhibited in rat mesangialcells treated with neutralizing antibodies to CTGF(Blom et al., 2001). Although our results clearlydemonstrate that CTGF plays an important role as adownstream effecter of TGF-b1-induced ECM synthesis,we cannot rule out the possibility that other CTGF-independent mechanisms may also contribute to TGF-b1-induced ECM synthesis.
The exact role that TGF-b1 plays in osteoblastproliferation has been controversial. TGF-b1 has beenshown to stimulate proliferation in some osteoblastcultures (Centrella et al., 1987; Alliston and Derynck,2000) and inhibit proliferation in others (Noda andRodan, 1986; Bonewald and Mundy, 1990; Centrellaet al., 1995). The reasons for these discrepancies are notfully understood, but have been attributed to differencesin individual culture conditions and the stage of osteo-blast differentiation at the time of TGF-b1 treatment(Bonewald and Mundy, 1990). Previous studies havealso found that the effect of TGF-b1 on osteoblastproliferation is influenced by the plating density of thecultures at the time of treatment (Canalis et al., 1987;Centrella et al., 1987; Breen et al., 1994). To clarify therole of TGF-b1 on osteoblast proliferation in our culturesystem, we used early (day 3) osteoblast cultures at atime when the cells were actively proliferating (sub-confluent) and plated the cells at increasing densitiesto accelerate their differentiation (Breen et al., 1994).Despite differences in plating density, TGF-b1 treat-ment of our osteoblast cultures consistently inhibitedtheir proliferation.
Osteoblast proliferation is regulated by multiplegrowth checkpoints within its cell cycle at whichcompetency for proliferation is determined and containa G1 restriction checkpoint that can be regulated byTGF-b1 (Stein et al., 2000). In general, TGF-b1 inhibitscell proliferation by arresting cells in late G1 phaseduring their cell cycle (Massague, 1990), and this effectis characterized by a decrease in cyclin D1 and cdk4levels (Donovan and Slingerland, 2000). In this study weshowed that cyclin D1 and cdk4 were reduced therebyproviding evidence that proliferating osteoblasts under-go a late G1 arrest when treated with TGF-b1.
CTGF has been implicated as a mediator of TGF-b1effects on cell growth in some cell types. In fibroblasts,TGF-b1 functions as a potent mitogen through a post-TGF-b1 receptor mechanism that is dependent onCTGF expression (Grotendorst et al., 2004). However,in mesangial cells, TGF-b1 treatment causes a G1 phasegrowth arrest and cell hypertrophy, effects that can bereversed by blocking CTGF expression (Abdel-Wahabet al., 2002). In order to determine if CTGF mediated theG1 growth arrest of osteoblasts by TGF-b1, we blockedthe up-regulation of CTGF expression using CTGFsiRNA. We reasoned that if CTGF acted downstreamof TGF-b1, then blocking its expression would reverse
the growth arrest induced by TGF-b1. However, wefound silencing of CTGF expression resulted in an evengreater reduction in cell number and a further reductionin G1 cyclins compared to cultures treated with TGF-b1alone, suggesting that CTGF itself is mitogenic and thatthe cell-cycle arrest induced by TGF-b1 was indepen-dent of CTGF. Interestingly, although addition ofexogenous CTGF (recombinant protein) itself caninduce osteoblast proliferation as previously demon-strated by our group (Safadi et al., 2003), TGF-b1induction of CTGF does not induce a similar response.This could be due to differences in the signalingpathways and resultant mechanisms activated follow-ing exogenous administration of CTGF (Abreu et al.,2002; Safadi et al., 2003) versus the endogenous up-regulation of CTGF induced by TGF-b1 as demonstratedin this study. Furthermore, the administration of TGF-b1 is likely to activate alternate signaling pathways andmechanisms that are capable of superseding thoseactivated by CTGF alone. Indeed, recent studies inosteoblastic cells have found that TGF-b1 can induceSmad and MAPK (Erk 1/2 and Jnk) signaling pathwaysindependent of each other, and that TGF-b1-inducedErk1/2 and Jnk signaling supersedes that of Smad3,thereby antagonizing ALP activity and mineralizationof osteoblasts induced by Smad3 (Sowa et al., 2002).Perhaps a similar mechanism is at play in mediatingproliferation in osteoblasts. Additional studies to eluci-date the growth control mechanisms through whichTGF-b1 and CTGF function are warranted.
In conclusion, our study demonstrates that CTGF isinduced by TGF-b1 during all phases of osteoblastdifferentiation and that CTGF acts as a downstreammediator of TGF-b1-induced ECM synthesis but notTGF-b1-induced growth arrest in primary osteoblasts.Additional studies are required to elucidate the signal-ing pathways that regulate TGF-b1 induction of CTGFexpression, and to understand the similarities anddifferences in mechanisms of action of these two growthfactors as they relate to osteoblast differentiation andfunction. These studies will not only enhance ourunderstanding of the molecular mechanisms of TGF-b1 and CTGF in osteoblasts, but may also help to identifynovel therapeutic targets to control early stages ofosteoblast differentiation during bone formation.
ACKNOWLEDGMENTS
The authors would like to thank the following peoplefor their constructive criticism and stimulating con-versation: Dr. Judith Litvin, Dr. Archana Sanjay, andDr. Emilia L. Oleszak. This study was supported bygrants AR 47432 (to S.N.P.) and DE 07444 (to P.R.O.)from the National Institutes of Health.
LITERATURE CITED
Abdel-Wahab N, Weston BS, Roberts T, Mason RM. 2002. Connective tissuegrowth factor and regulation of the mesangial cell cycle: Role in cellularhypertrophy. J Am Soc Nephrol 13:2437–2445.
Abreu JG, Ketpura NI, Reversade B, De Robertis EM. 2002. Connective-tissuegrowth factor (CTGF) modulaes cell signaling by BMP and TGF-beta. Nat CellBiol 4:599–604.
Alliston TM, Derynck R. 2000. Transforming growth factor- in skeletaldevelopment and maintenance. In: Canalis E, editor. Skeletal growth factors.Philadelphia, PA: Lippincott, Williams and Wilkins. pp 233–250.
Babic AM, Chen CC, Lau LF. 1999. Fisp12/mouse connective tissue growth factormediates endothelial cell adhesion and migration through integrin alphav-beta3, promotes endothelial cell survival, and induces angiogenesis in vivo. MolCell Biol 19:2958–2966.
Blom IE, van Dijk AJ, Wieten L, Duran K, Ito Y, Kleij L, deNichilo M, RabelinkTJ, Weening JJ, Aten J, Goldschmeding R. 2001. In vitro evidence fordifferential involvement of CTGF, TGFbeta, and PDGF-BB in mesangialresponse to injury. Nephrol Dial Transplant 16:1139–1148.
Blom IE, Goldschmeding R, Leask A. 2002. Gene regulation of connective tissuegrowth factor: New targets for antifibrotic therapy? Matrix Biol 21:473–482.
CTGF AND TGF-b1 IN OSTEOBLASTS 851
Journal of Cellular Physiology DOI 10.1002/jcp

Bonewald LF. 2002. Transforming growth factor-B. In: Bilezikian JP, Raisz LG,Rodan GA, editors. Principles of bone biology, vol. 2. New York: AcademicPress. pp 903–918.
Bonewald LF, Dallas SL. 1994. Role of active and latent transforming growthfactor beta in bone formation. J Cell Biochem 55:350–357.
Bonewald LF, Mundy GR. 1990. Role of transforming growth factor-beta in boneremodeling. Clin Orthop Relat Res 250:261–276.
Breen EC, Ignotz RA, McCabe L, Stein JL, Stein GS, Lian JB. 1994. TGF betaalters growth and differentiation related gene expression in proliferatingosteoblasts in vitro, preventing development of the mature bone phenotype.J Cell Physiol 160:323–325.
Brigstock DR. 1999. The connective tissue growth factor/cysteine-rich61/nephroblastoma overexpressed (CCN) family. Endocr Rev 20:189–206.
Canalis E, McCarthy T, Centrella M. 1987. A bone-derived growth factor isolatedfrom rat calvariae is beta 2 microglobulin. Endocrinology 121:1198–1200.
Centrella M, Canalis E. 1987. Isolation of EGF-dependent transforming growthfactor (TGF beta-like) activity from culture medium conditioned by fetal ratcalvariae. J Bone Miner Res 2:29–36.
Centrella M, McCarthy TL, Canalis E. 1987. Transforming growth factor beta is abifunctional regulator of replication and collagen synthesis in osteoblast-enriched cell cultures from fetal rat bone. J Biol Chem 262:2869–2874.
Centrella M, Casinghino S, Ignotz R, McCarthy TL. 1992. Multiple regulatoryeffects by transforming growth factor-beta on type I collagen levels in osteo-blast-enriched cultures from fetal rat bone. Endocrinology 131:2863–2872.
Centrella M, Casinghino S, Kim J, Pham T, Rosen V, Wozney J, McCarthy TL.1995. Independent changes in type I and type II receptors for transforminggrowth factor beta induced by bone morphogenetic protein 2 parallel expressionof the osteoblast phenotype. Mol Cell Biol 15:3273–3281.
Chen Y, Blom IE, Sa S, Goldschmeding R, Abraham DJ, Leask A. 2002. CTGFexpression in mesangial cells: Involvement of SMADs, MAP kinase, and PKC.Kidney Int 62:1149–1159.
Dallas SL, Park-Snyder S, Miyazono K, Twardzik D, Mundy GR, Bonewald LF.1994. Characterization and autoregulation of latent transforming growthfactor beta (TGF beta) complexes in osteoblast-like cell lines. Production of alatent complex lacking the latent TGF beta-binding protein. J Biol Chem269:6815–6821.
Dammeier J, Brauchle M, Falk W, Grotendorst GR, Werner S. 1998. Connectivetissue growth factor: A novel regulator of mucosal repair and fibrosis ininflammatory bowel disease? Int J Biochem Cell Biol 30:909–922.
di Mola FF, Friess H, Martignoni ME, Di Sebastiano P, Zimmermann A,Innocenti P, Graber H, Gold LI, Korc M, Buchler MW. 1999. Connective tissuegrowth factor is a regulator for fibrosis in human chronic pancreatitis. AnnSurg 230:63–71.
Donovan J, Slingerland J. 2000. Transforming growth factor-beta and breastcancer: Cell cycle arrest by transforming growth factor-beta and its disruptionin cancer. Breast Cancer Res 2:116–124.
Duncan MR, Frazier KS, Abramson S, Williams S, Klapper H, Huang X,Grotendorst GR. 1999. Connective tissue growth factor mediates transforminggrowth factor beta-induced collagen synthesis: Down-regulation by cAMP.FASEB J 13:1774–1786.
Eguchi T, Kubota S, Kondo S, Shimo T, Hattori T, Nakanishi T, Kuboki T, YataniH, Takigawa M. 2001. Regulatory mechanism of human connective tissuegrowth factor (CTGF/Hcs24) gene expression in a human chondrocytic cell line,HCS-2/8. J Biochem (Tokyo) 130:79–87.
Frazier KS, Grotendorst GR. 1997. Expression of connective tissue growth factormRNA in the fibrous stroma of mammary tumors. Int J Biochem Cell Biol29:153–161.
Frazier K, Williams S, Kothapalli D, Klapper H, Grotendorst GR. 1996.Stimulation of fibroblast cell growth, matrix production, and granulationtissue formation by connective tissue growth factor. J Invest Dermatol 107:404–411.
Fromigue O, Modrowski D, Marie PJ. 2004. Growth factors and bone formation inosteoporosis: Roles for fibroblast growth factor and transforming growth factorbeta. Curr Pharm Des 10:2593–2603.
Gore-Hyer E, Shegogue D, Markiewicz M, Lo S, Hazen-Martin D, Greene EL,Grotendorst G, Trojanowska M. 2002. TGF-beta and CTGF have overlappingand distinct fibrogenic effects on human renal cells. Am J Physiol Renal Physiol283:F707–F716.
Grotendorst GR. 1997. Connective tissue growth factor: A mediator of TGF-betaaction on fibroblasts. Cytokine Growth Factor Rev 8:171–179.
Grotendorst GR, Okochi H, Hayashi N. 1996. A novel transforming growth factorbeta response element controls the expression of the connective tissue growthfactor gene. Cell Growth Differ 7:469–480.
Grotendorst GR, Rahmanie H, Duncan MR. 2004. Combinatorial signalingpathways determine fibroblast proliferation and myofibroblast differentiation.FASEB J 18:469–479.
Hock JM, Canalis E, Centrella M. 1990. Transforming growth factor-betastimulates bone matrix apposition and bone cell replication in cultured fetal ratcalvariae. Endocrinology 126:421–426.
Holmes A, Abraham DJ, Sa S, Shiwen X, Black CM, Leask A. 2001. CTGF andSMADs, maintenance of scleroderma phenotype is independent of SMADsignaling. J Biol Chem 276:10594–10601.
Holmes A, Abraham DJ, Chen Y, Denton C, Shi-wen X, Black CM, Leask A. 2003.Constitutive connective tissue growth factor expression in sclerodermafibroblasts is dependent on Sp1. J Biol Chem 278:41728–41733.
Igarashi A, Okochi H, Bradham DM, Grotendorst GR. 1993. Regulation ofconnective tissue growth factor gene expression in human skin fibroblasts andduring wound repair. Mol Biol Cell 4:637–645.
Igarashi A, Nashiro K, Kikuchi K, Sato S, Ihn H, Fujimoto M, Grotendorst GR,Takehara K. 1996. Connective tissue growth factor gene expression in tissuesections from localized scleroderma, keloid, and other fibrotic skin disorders.J Invest Dermatol 106:729–733.
Ivkovic S, Yoon BS, Popoff SN, Safadi FF, Libuda DE, Stephenson RC, DaluiskiA, Lyons KM. 2003. Connective tissue growth factor coordinates chondrogene-sis and angiogenesis during skeletal development. Development 130:2779–2791.
Kireeva ML, Mo FE, Yang GP, Lau LF. 1996. Cyr61, a product of a growth factor-inducible immediate-early gene, promotes cell proliferation, migration, andadhesion. Mol Cell Biol 16:1326–1334.
Kubo M, Kikuchi K, Nashiro K, Kakinuma T, Hayashi N, Nanko H, Tamaki K.1998. Expression of fibrogenic cytokines in desmoplastic malignant melanoma.Br J Dermatol 139:192–197.
Lasky JA, Ortiz LA, Tonthat B, Hoyle GW, Corti M, Athas G, Lungarella G,Brody A, Friedman M. 1998. Connective tissue growth factor mRNA expressionis upregulated in bleomycin-induced lung fibrosis. Am J Physiol 275:L365–L371.
Lau LF, Lam SCT. 1999. The CCN family of angiogenic regulators: The integrinconnection. Exp Cell Res 248:44–57.
Leask A, Abraham DJ. 2004. TGF-beta signaling and the fibrotic response.FASEB J 18:816–827.
Massague J. 1990. The Transforming Growth Factor Beta family. Annu Rev CellBiol 6:597–641.
Moussad EE, Brigstock DR. 2000. Connective tissue growth factor: What’s in aname? Mol Genet Metab 71:276–292.
Nakata E, Nakanishi T, Kawai A, Asaumi K, Yamaai T, Asano M, Nishida T,Mitani S, Inoue H, Takigawa M. 2002. Expression of connective tissue growthfactor/hypertrophic chondrocyte-specific gene product 24 (CTGF/Hcs24) duringfracture healing. Bone 31:441–447.
Nishida T, Nakanishi T, Asano M, Shimo T, Takigawa M. 2000. Effects of CTGF/Hcs24, a hypertrophic chondrocyte-specific gene product, on the proliferationand differentiation of osteoblastic cells in vitro. J Cell Physiol 184:197–206.
Noda M, Rodan GA. 1986. Type-beta transforming growth factor inhibitsproliferation and expression of alkaline phosphatase in murine osteoblast-likecells. Biochem Biophys Res Commun 140:56–65.
Paradis V, Dargere D, Vidaud M, De Gouville AC, Huet S, Martinez V, GauthierJM, Ba N, Sobesky R, Ratziu V, Bedossa P. 1999. Expression of connectivetissue growth factor in experimental rat and human liver fibrosis. Hepatology30:968–976.
Parisi MS, Gazzerro E, Rydziel S, Canalis E. 2005. Expression and regulation ofCCN genes in murine osteoblasts. Bone 38:671–677.
Pfeilschifter J, Bonewald L, Mundy GR. 1990. Characterization of the latenttransforming growth factor beta complex in bone. J Bone Miner Res 5:49–58.
Robey PG, Young MF, Flanders KC, Roche NS, Kondaiah P, Reddi AH,Termine JD, Sporn MB, Roberts AB. 1987. Osteoblasts synthesize andrespond to transforming growth factor-type b (TGF-b) in vitro. J Cell Biol105:457–463.
Safadi FF, Xu J, Smock SL, Kanaan RA, Selim AH, Odgren PR, Marks SC, Jr.,Owen TA, Popoff SN. 2003. Expression of connective tissue growth factor inbone: Its role in osteoblast proliferation and differentiation in vitro and boneformation in vivo. J Cell Physiol 196:51–62.
Sanberg M, Vurio T, Hirvonen H, Alitalo K, Vuorio E. 1988. Enhanced expressionof TGF-b and c-fos mRNAs in the growth plates of developing human longbones. Development 102:461–470.
Seyedin SM, Thomas TC, Thompson AY, Rosen DM, Piez K. 1985. Purificationand characterization of two cartilage-inducing factors from bovine deminer-alized bone. Proc Natl Acad Sci USA 82:2267–2271.
Shi-wen X, Stanton LA, Kennedy L, Pala D, Chen Y, Howat SL, Renzoni EA,Carter DE, Bou-Gharios G, Stratton RJ, Pearson JD, Beier F, Lyons KM, BlackCM, Abraham DJ, Leask A. 2006. CCN2 is necessary for adhesive responses totransforming growth factor-beta1 in embryonic fibroblasts. J Biol Chem 281:10715–10726.
Sims N, Baron R. 2000. Bone cells and their function. In: Canalis E, editor.Skeletal growth factors. Philadelphia, PA: Lippincott Williams and Wilkins.pp 1–17.
Sowa H, Kaji H, Yamaguchi T, Sugimoto T, Chihara K. 2002. Smad3 promotesalkaline phosphatase activity and mineralization of osteoblastic MC3T3-E1cells. J Bone Miner Res 17:1190–1199.
Stein GS, Owen TA, van Wijnen AJ, Cho B, Stein JL, Lian JB. 2000. Mechanismsof action of skeletal growth factors in osteoblasts. In: Canalis E, editor. Skeletalgrowth factors. Philadelphia, PA: Lippincott, Williams and Wilkins. pp 51–65.
Suzuki K, Obara K, Kobayashi K, Yamana K, Bilim V, Itoi T, Takahashi K. 2006.Role of connective tissue growth factor in fibronectin synthesis in culturedhuman prostate stromal cells. Urology 67:647–653.
Takehara K. 2000. Growth regulation of skin fibroblasts. J Dermatol Sci 24:S70–S77.
Takigawa M, Nakanishi T, Kubota S, Nishida T. 2003. Role of CTGF/HCS24/ecogenin in skeletal growth control. J Cell Physiol 194:256–266.
Van Beek JP, Kennedy L, Rockel JS, Bernier SM, Leask A. 2006. The induction ofCCN2 by TGFbeta1 involves Ets-1. Arthritis Res Ther 8:R36.
Wahab NA, Yevdokimova N, Weston BS, Roberts T, Li XJ, Brinkman H, MasonRM. 2001. Role of connective tissue growth factor in the pathogenesis ofdiabetic nephropathy. Biochem J 359:77–87.
Weston BS, Wahab NA, Mason RM. 2003. CTGF mediates TGF-beta-inducedfibronectin matrix deposition by upregulating active alpha5beta1 integrin inhuman mesangial cells. J Am Soc Nephrol 14:601–610.
Wrana JL, Maeno M, Hawrylyshyn B, Yao KL, Domenicucci C, Sodek J. 1988.Differential effects of transforming growth factor-beta on the synthesis ofextracellular matrix proteins by normal fetal rat calvarial bone cell popula-tions. J Cell Biol 106:915–924.
Xu J, Smock SL, Safadi FF, Rosenzweig AB, Odgren PR, Marks SC, Jr., Owen TA,Popoff SN. 2000. Cloning the full-length cDNA for rat connective tissue growthfactor: Implications for skeletal development. J Cell Biochem 77:103–115.
Yokoi H, Mukoyama M, Sugawara A, Mori K, Nagae T, Makino H, Suganami T,Yahata K, Fujinaga Y, Tanaka I, Nakao K. 2002. Role of connective tissuegrowth factor in fibronectin expression and tubulointerstitial fibrosis. Am JPhysiol Renal Physiol 282:F933–F942.
Yu C, Le AT, Yeger H, Perbal B, Alman BA. 2003. NOV (CCN3) regulation in thegrowth plate and CCN family member expression in cartilage neoplasia.J Pathol 201:609–615.
Yu L, Border WA, Huang Y, Noble NA. 2003. TGF-beta isoforms in renalfibrogenesis. Kid Int 64:844–856.
852 ARNOTT ET AL.
Journal of Cellular Physiology DOI 10.1002/jcp




















