
Selective estrogen receptor modulators regulate reactive microglia after penetrating brain injury
Upload
independentCategory
view
0download
0
of July 3, 2014.This information is current as
to Transient Global Cerebral IschemiaResponseUp-Regulated in Brain Microglia in
Complement C1q Is Dramatically
Petry, Michael Loos and Eberhard WeihePatricia Salvati, Marcello Calabresi, Robert B. Sim, Franz Martin K.-H. Schäfer, Wilhelm J. Schwaeble, Claes Post,
http://www.jimmunol.org/content/164/10/5446doi: 10.4049/jimmunol.164.10.5446
2000; 164:5446-5452; ;J Immunol
Referenceshttp://www.jimmunol.org/content/164/10/5446.full#ref-list-1
, 9 of which you can access for free at: cites 43 articlesThis article
Subscriptionshttp://jimmunol.org/subscriptions
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/ji/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/cgi/alerts/etocReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved.Copyright © 2000 by The American Association of9650 Rockville Pike, Bethesda, MD 20814-3994.The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on July 3, 2014http://w
ww
.jimm
unol.org/D
ownloaded from
by guest on July 3, 2014
http://ww
w.jim
munol.org/
Dow
nloaded from

Complement C1q Is Dramatically Up-Regulated in BrainMicroglia in Response to Transient Global CerebralIschemia1,2
Martin K.-H. Schafer,* Wilhelm J. Schwaeble,*† Claes Post,‡§ Patricia Salvati,‡¶
Marcello Calabresi,ঠRobert B. Sim,i Franz Petry,# Michael Loos,# and Eberhard Weihe3*
Recent evidence suggests that the pathophysiology of neurodegenerative and inflammatory neurological diseases has a neuroim-munological component involving complement, an innate humoral immune defense system. The present study demonstrates theeffects of experimentally induced global ischemia on the biosynthesis of C1q, the recognition subcomponent of the classicalcomplement activation pathway, in the CNS. Using semiquantitative in situ hybridization, immunohistochemistry, and confocallaser scanning microscopy, a dramatic and widespread increase of C1q biosynthesis in rat brain microglia (but not in astrocytesor neurons) within 24 h after the ischemic insult was observed. A marked increase of C1q functional activity incerebrospinal fluidtaken 1, 24, and 72 h after the ischemic insult was determined by C1q-dependent hemolytic assay. In the light of the well-establishedrole of complement and complement activation products in the initiation and maintenance of inflammation, the ischemia-inducedincrease of cerebral C1q biosynthesis and of C1q functional activity in the cerebrospinal fluid implies that the proinflammatory activitiesof locally produced complement are likely to contribute to the pathophysiology of cerebral ischemia. Pharmacological modulationof complement activation in the brain may be a therapeutic target in the treatment of stroke. The Journal of Immunology,2000, 164:5446–5452.
T he complement system provides a first line of defense andmediates a large variety of cellular and humoral interac-tions in the immune response, including chemotaxis,
phagocytosis, cell adhesion, B and T cell differentiation (1), andneuronal cell death (2, 3). It consists of;30 plasma proteins withan associated group of cell membrane proteins. Activation of com-plement by the classical, the alternative and the recently discov-ered lectin pathway generates opsonins, inflammatory mediators,and cytolytic protein complexes which play an essential role inclearing microorganisms and tissue damage products (4). It hasrecently been suggested that local activation of complement in theCNS is of pathophysiological significance in both degenerativeand inflammatory neurological diseases including Alzheimer’s (2,5–9) and Parkinson’s dementia (10), supranuclear palsy (11),Pick’s disease (12), multiple sclerosis (13, 14), cerebral malaria
(15), meningoencephalitis (16), scrapie (17, 18), and cerebrovas-cular disorders (19). Increased biosynthesis of various complementfactors in the CNS has also been reported in experimental animalmodels of neurodegeneration or neuroinflammation such as pe-ripheral or central axotomy (20–24), excitotoxic kainic acid le-sions (21, 25), exposure to neurotoxins (26), and experimental al-lergic and virus-induced encephalitis (27) and in culturedmicroglial cells (28–32). By using a combination of techniques tomonitor and localize biosynthesis in vivo, we provide evidencethat experimentally induced cerebral ischemia causes a dramaticincrease of C1q in brain microglia resulting in markedly enhancedlevels of C1q functional activity in cerebrospinal fluid (CSF).
Materials and MethodsMaterials
Restriction enzymes, RNase A, and RNA polymerases were purchasedfrom Boehringer Mannheim (Mannheim, Germany). [35S]CTP was ob-tained from New England Nuclear (Boston, MA). All chemicals, unlessotherwise stated, were purchased from Sigma (Deisenhofen, Germany).X-ray films (Hyperfilm-bmax) were purchased from Amersham-Pharmacia(Uppsala, Sweden). Photo emulsion, developer (full strength), and fixer(rapid fix) were from Kodak (Integra Biosciences, Fernwald, Germany).Wistar rats were from Charles River (Calco, Italy). Animal care and pro-cedures were conducted according to institutional guidelines.
Induction of global ischemia and experimental groups
Transient forebrain ischemia was induced in 39 male Wistar rats (200–250g) using the four vessel occlusion model according to the design ofPulsinelli and Brierley (33). Briefly, animals were anesthetized with chloralhydrate (350 mg/kg i.p.), and the vertebral arteries were electrocauterizedin the alar foramina of the first cervical vertebra. The carotid arteries wereisolated and transiently occluded with atraumatic clips for 15 min. Animalsnot showing isoelectric cortical electroencephalographic activity duringischemia and animals that did not fully lose their righting reflex after ca-rotid artery occlusion were excluded from the study. Core body tempera-ture was maintained at 376 0.5°C during ischemia and for 2 h post isch-emia with a feedback-regulated heating pad (LSI termist, Milan, Italy). Inthe sham animals, only the vertebral arteries were occluded. In addition, a
*Department of Anatomy and Cell Biology, University of Marburg, Marburg, Ger-many;†Department of Microbiology and Immunology, University of Leicester, Le-icester, United Kingdom;‡Pharmacia & Upjohn SpA, Nerviano Mi, Italy;§MelacureTherapeutics AB, Uppsala, Sweden;¶Newron Pharmaceuticals Spa, Milan, Italy;iMedical Research Council, Immunochemistry Unit, Department of Biochemistry,University of Oxford, Oxford, United Kingdom; and#Institute for Medical Microbi-ology and Hygiene, Johannes Gutenberg-University of Mainz, Mainz, Germany
Received for publication September 22, 1999. Accepted for publication March6, 2000.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby markedadvertisementin accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 This work was supported by the German Research Foundation (ForschergruppeNeuroprotektion and Sonderforschungsbereich 297), the Deutscher AkademischerAustauschdienst, the Kempkes Foundation, and the British Council.2 Data were presented in part at the 26th Annual Meeting of the Society for Neuro-science, Washington, DC, 1996 (45), and the Fourth International Workshop on C1and Collectins, Mainz, Germany, October 3–5, 1997 (46).3 Address correspondence and reprint requests to Dr. Eberhard Weihe, Department ofMolecular Neuroimmunology, Institute of Anatomy and Cell Biology, Philipps-Uni-versity of Marburg, Robert Koch Strasse 6, 35033 Marburg, Germany. E-mail ad-dress: [email protected]
Copyright © 2000 by The American Association of Immunologists 0022-1767/00/$02.00
by guest on July 3, 2014http://w
ww
.jimm
unol.org/D
ownloaded from

group of untreated rats served as controls. Animals were sacrificed at 1, 4,24, and 72 h after the ischemic insult. Their brains were randomly dividedinto two groups and either frozen in isopentane (240°C) for in situ hy-bridization (n5 2–8) or processed for immunocytochemistry (n 5 3–4).
In situ hybridization
For cellular localization of C1q mRNA,35S-labeled sense and antisenseRNA probes were generated by in vitro transcription from an internal425-bpBamHI/PstI subfragment (cloned in pBluescript II KS2) of clonepRB-15, a cDNA transcript of mRNA encoding the B-chain of rat C1q(34). Labeling of the cRNA using [35S]CTP was performed according tothe method of Melten et al. (35). With the use of T7 RNA polymerase, invitro transcription yielded a cRNA in antisense orientation labeled with a
specific activity of 430,000 Ci/mmol. Transcription with T3 RNA poly-merase generated a sense strand probe labeled with a specific activity of450,000 Ci/mmol. In situ hybridization histochemistry was performed ac-cording to the previously reported protocol (34, 36). Briefly, cryostat sec-tions were fixed in 4% formaldehyde, 0.1 M PBS for 1 h at room temper-ature, after three washes in 0.05 M PBS, pH 7.4, for 10 min each. Slideswere transferred to 0.1 M triethanolamine (TEA),4 pH 8.0, and incubatedin TEA supplemented with acetic anhydride (0.25% v/v) for 10 min atroom temperature, stirring rapidly. Sections were then rinsed in 23SSC
4 Abbreviations used in this paper: TEA, triethanolamine; CSF, cerebrospinal fluid;NeuN, antineuronal nuclei; p.i., postischemia.
FIGURE 1. Time course of C1q mRNA expres-sion after ischemia. X-ray autoradiograms of fore-brain sections hybridized with35S-labeled anti-sense cRNA corresponding to the mRNA encodingthe rat C1q B chain. In brains of rats at time point0 and of the sham control (72 h p.i.) low level C1qhybridization signals are seen throughout white andgray matter without preferences to the neuronallayers (Aand F ). Note moderate and widespreadincrease of C1q mRNA levels at 1 h (B) and 4 h p.i.(C) and pronounced increases of C1q mRNA ex-pression throughout the forebrain at 24 h (D) and72 h (E) as compared with nontreated control (A)or to 72 h postsham (F). At 72 h p.i. (E), foci withdramatically enhanced C1q expression are seen inthe hippocampus and in the thalamic region andalso in the neocortex.
FIGURE 2. Localization of C1q expression in thehippocampal region.A, Low power brightfield micro-graph showing nonradioactive in situ hybridizationwith a digoxigenin-labeled C1q antisense riboprobe24 h after ischemia. Note presence of hybridizationsignals in numerous nonneuronal cells throughout thehippocampus.B, Low power brightfield micrographshowing presence of C1q immunoreactivity in numer-ous nonneuronal cells throughout the hippocampalformation 24 h after ischemia.C, High power bright-field micrograph showing presence of moderately in-tense immunostaining for C1q in a microglial cell ofthe ramified type in a sham control animal.D, Highpower brightfield micrograph showing heavilystained microglial cells of less ramified activatedtype. Note increased density of C1q-immunoreactivemicroglial cells inD compared withC. Note also ab-sence of C1q mRNA or immunostaining from neuro-nal cell bodies inA–D. Bar inB (A,) 200mm; bar inD (C), 12.5mm.
5447The Journal of Immunology
by guest on July 3, 2014http://w
ww
.jimm
unol.org/D
ownloaded from

5448 MICROGLIAL C1q BIOSYNTHESIS AND CEREBRAL ISCHEMIA
by guest on July 3, 2014http://w
ww
.jimm
unol.org/D
ownloaded from
and dehydrated in ethanol (50–100%). Radioactive cRNA probes werediluted in hybridization buffer (50% formamide, 10% dextran sulfate, 33SSC, 50 mM sodium phosphate (pH 7.4), 13Denhardt’s (0.02% w/v Fi-coll 400, polyvinylpyrrolidone 0.02% w/v, BSA 0.02% w/v), 0.1 mg/mlyeast RNA) to a final concentration of 53 104 dpm/ml. DTT was added toa final concentration of 10 mM. Hybridization mix (40ml) was applied; thecryostat sections were then coverslipped and incubated in a humid chamberat 55°C for 16 h. The next day the coverslips were removed in 23SSC.Sections were treated with RNase A (20mg/ml) at 37°C for 60 min toremove single-stranded RNA molecules. Successive washes were done atroom temperature in 23, 13, 0.53, and 0.23SSC for 10 min and in 0.23SSC at 60°C for 1 h. The tissue was then dehydrated by dipping sequen-tially in 70%, 80%, and absolute ethanol. Hybridized sections were ex-posed to x-ray films (Hyperfilm-bmax) for 96 h.
For microscopic analysis, slides were dipped in Kodak NTB2 nuclearemulsion and stored at 4°C. After 2–3 weeks of exposure, autoradiogramswere developed for 2 min (Kodak D19 full strength) and fixed for 4 min.As controls, sections were incubated in parallel with C1q cRNA probes insense strand orientation, or sections were pretreated with RNase beforehybridization with the antisense probe.
For nonradioactive detection, the C1q riboprobe was labeled withdigoxigenin-UTP according to the manufacturer’s protocol (BoehringerMannheim). The digoxigenin-labeled C1q riboprobe was diluted in hybrid-ization buffer to a final concentration of 0.5 ng/ml, and hybridization wasperformed with the same incubation protocol as for radioactive in situhybridization. Hybridization signals were visualized with sheep anti-digoxigenin Fab fragments coupled to alkaline phosphatase according tothe manufacturer’s protocol (Boehringer Mannheim).
Hybridized sections were analyzed in dark and bright field illuminationand were photographed with the Olympus AX70 microscope (OlympusOptical, Hamburg, Germany).
Semiquantitative image analysis of hybridized sections
Analysis of autoradiograms was conducted using the MCID M4 imageanalysis system (Imaging Research, St. Catharine’s, ON, Canada). Fromeach animal in each group, x-ray film autoradiograms of six 20-mm-thickinterval sections were digitized under constant light and camera conditions.Calibrated densitometry of areas of the parietal cortex from similar Bregmalevels was performed yielding measurements of integrated optical density(IOD; area3 average optical density) as described previously (37). Theoptical density values were calculated after subtraction of the film back-ground, which was defined by hybridization with the sense strand probes.A radioactive standard (ARC 146B, American Radiolabeled Chemicals, St.Louis, MO) was used for calibration. Data were expressed as IOD innanocuries per gram.
Statistics
Intergroup differences in the C1q mRNA levels were evaluated using theKruskal-Wallis one-way ANOVA followed by the Mann-WhitneyU test.In all tests,p , 0.05 was considered significant.
Single enzymatic immunostaining of C1q
Deparaffinized serial sections (5–7mm thick) of Bouin-Hollande (fixativesolution containing 6% (w/v) picric acid, 2.5% cupric acetate, 3.7% form-aldehyde, and 1% glacial acetic acid) fixed brains were incubated overnightwith primary Abs at room temperature. Rat C1q was detected by using apolyclonal goat antiserum directed against mouse C1q (IgG fraction, di-luted 1:400) as described previously (34). After washing in 50 mM PBS(pH 7.4), containing 1% w/v BSA, an anti-goat IgG biotinylated secondaryantiserum (dilution, 1:200) (Dianova, Hamburg, Germany) was applied (45min at 37°C). After repeated rinsing in PBS/BSA, sections were incubatedwith a streptavidin-HRP complex (Amersham, 1:200) for 2 h at 37°C.Staining was performed using H2O2 and 39,3-diaminobenzidine (Sigma)(12.5 mg/100 ml PBS) as a substrate and nickel enhancement by addingammonium nickel sulfate (70 mg/100 ml, Fluka, Buchs, Switzerland). Im-munostained sections were analyzed and photographed with the OlympusAX70 microscope. Specificity of the C1q antiserum was determined bypreabsorption with purified rat and mouse C1q (50mM) as well as byWestern blotting and C1q ELISA (data not shown).
Confocal laser scanning double-immunofluorescence microscopyfor C1q and markers for astrocytes, neurons, and microglialcells
Double-immunofluorescence detection of anti-C1q reactivity and reactivityfor the different established markers (anti-glial fibrillary acidic protein(GFAP) for astrocytes, antineuronal nuclei (NeuN) for neurons, and isolec-tin B4 for microglia) was performed as follows. Sections were incubatedovernight at room temperature with a mixture of the polyclonal goat anti-mouse C1q Ab (IgG fraction, diluted 1:40) and a polyclonal rabbit anti-GFAP antiserum (1:400, Dianova, Hamburg, Germany) or the mouse
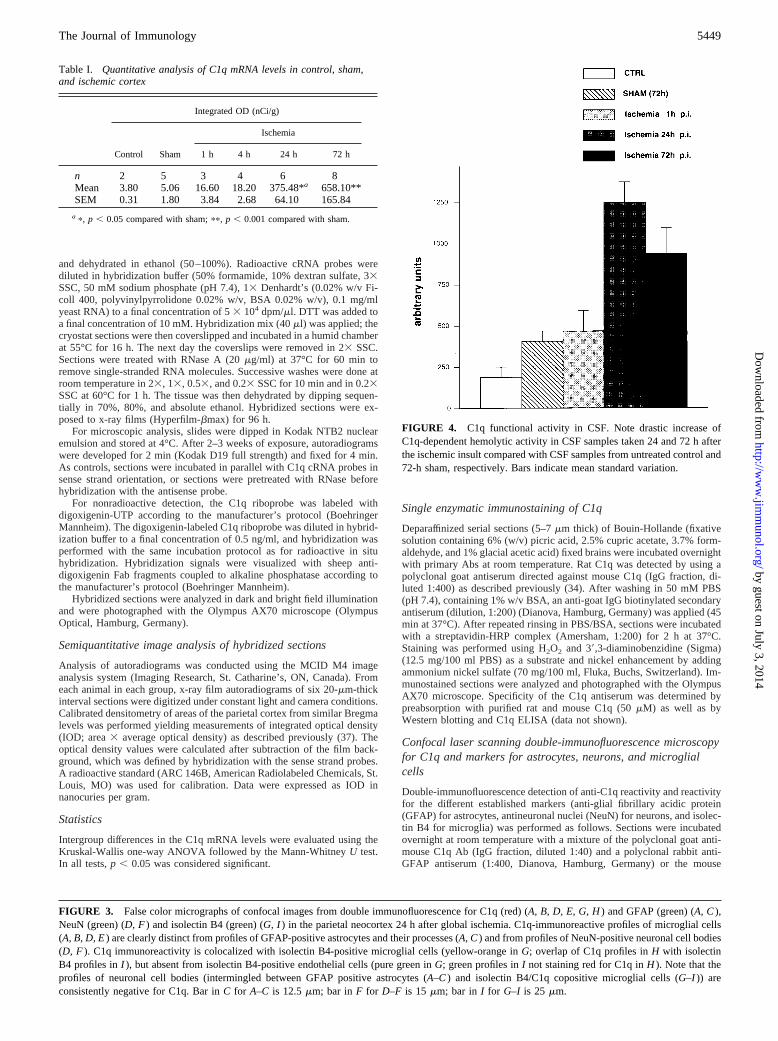
FIGURE 4. C1q functional activity in CSF. Note drastic increase ofC1q-dependent hemolytic activity in CSF samples taken 24 and 72 h afterthe ischemic insult compared with CSF samples from untreated control and72-h sham, respectively. Bars indicate mean standard variation.
Table I. Quantitative analysis of C1q mRNA levels in control, sham,and ischemic cortex
Integrated OD (nCi/g)
Control Sham
Ischemia
1 h 4 h 24 h 72 h
n 2 5 3 4 6 8Mean 3.80 5.06 16.60 18.20 375.48*a 658.10**SEM 0.31 1.80 3.84 2.68 64.10 165.84
a p, p , 0.05 compared with sham;pp, p , 0.001 compared with sham.
FIGURE 3. False color micrographs of confocal images from double immunofluorescence for C1q (red) (A, B, D, E, G, H) and GFAP (green) (A, C),NeuN (green) (D, F) and isolectin B4 (green) (G, I) in the parietal neocortex 24 h after global ischemia. C1q-immunoreactive profiles of microglial cells(A, B, D, E) are clearly distinct from profiles of GFAP-positive astrocytes and their processes (A, C) and from profiles of NeuN-positive neuronal cell bodies(D, F). C1q immunoreactivity is colocalized with isolectin B4-positive microglial cells (yellow-orange inG; overlap of C1q profiles inH with isolectinB4 profiles inI ), but absent from isolectin B4-positive endothelial cells (pure green inG; green profiles inI not staining red for C1q inH ). Note that theprofiles of neuronal cell bodies (intermingled between GFAP positive astrocytes (A–C) and isolectin B4/C1q copositive microglial cells (G–I)) areconsistently negative for C1q. Bar inC for A–C is 12.5mm; bar inF for D–F is 15 mm; bar in I for G–I is 25 mm.
5449The Journal of Immunology
by guest on July 3, 2014http://w
ww
.jimm
unol.org/D
ownloaded from

monoclonal anti-NeuN Ab MAB377 (dilution 1:20, Chemicon Interna-tional, Temecula, CA), or biotin-labeled isolectin B4 (dilution 1:30; Sigma,St. Louis, MO). C1q immunoreactivity was visualized with indocarbocya-nine-conjugated anti-goat IgG (Dianova), diluted 1:200, and applied for 45min at 37°C, resulting in a red-orange fluorescence labeling. GFAP im-munoreactivity (as a specific marker for astrocytes) and NeuN immunore-activity (as a specific marker for neuronal cell bodies and nuclei) werevisualized with biotinylated anti-rabbit IgG (Dianova) and biotinylated anti-mouse IgG (Dianova), respectively, both diluted 1:200. The secondary Abswere applied for 45 min at 37°C, followed by incubation with Alexis 488-conjugated streptavidin (MoBiTec, Gottingen, Germany) for 2 h at 37°C,resulting in a green fluorescence. Biotinylated isolectin B4 was visualizedwith Alexis 488-conjugated streptavidin for 2 h at 37°C, resulting in agreen fluorescence. Sections were analyzed with the Olympus Fluoviewconfocal laser scanning microscope (Olympus Optical) and false color con-focal images were printed with a digital color printer (Sony, Tokyo, Japan).
Measurement of C1q functional activity in the CSF byC1q-dependent hemolysis assay
In a separate set of experiments, rats (n5 3) for each of the different groupswere implanted with an intrathecal catheter 1 week before the experimentto collect CSF samples. At different time points after global ischemia (1 h,24 h and 72 h), 50-ml samples were drawn. C1q levels in these sampleswere determined by a sensitive C1q-dependent hemolysis assay. The assaywas adjusted with serial dilutions of four samples to determine the appro-priate sensitivity range. Thereafter, an aliquot (5ml) of each CSF samplewas assayed in duplicate, by adding the sample to 50ml sheep erythrocytes(Tissue Culture Services, Botolph Claydon, U.K.) sensitized with rabbitanti-sheep erythrocyte Abs at a concentration of 23 108 cells/ml. Both thesample and the EA cells were incubated at 37°C for 30 min, then 50ml ofa 1:50 dilution of C1q-deficient human serum (Sigma, Deisenhofen, Ger-many) was added, and the incubation was continued for 1 h at 37°C. Theremaining unlysed cells were then spun down, and the optical density(OD412) of the supernatant was read and expressed as percentage lysis. Theassay buffer was dextrose-gelatin-Veronal buffer with 0.15 mM CaCl2 and0.5 mM MgCl2. The titer of C1q activity was expressed in arbitrary unitsrelative to a human C1q standard. It was not considered appropriate toexpress C1q in absolute units (e.g., micrograms per milliliter), because therelative activities of rat and human C1q in the assay were not determined.
ResultsQualitative and semiquantitative in situ hybridization using35S-labeled cRNA probes
Biosynthesis of C1q was examined at the mRNA level in rat brainsections at different time points after global transient ischemia andcompared with sham-operated and control rats, respectively, whichis shown on low magnification x-ray micrographs in Fig. 1. In situhybridization demonstrated a slight increase of C1q mRNA levelsthroughout the brain within the first hour (Fig. 1,B andC) afterischemia, with a more pronounced elevation after 24 h (Fig. 1D).A dramatic increase of C1q mRNA levels was observed 72 h post-ischemia (p.i.), especially in the hippocampus, in ventroposteriorthalamic nuclei, and in the parietal neocortex (Fig. 1E). At lowmagnification, it was already obvious that C1q mRNA expressionwas absent from neuronal layers of cerebral cortex and hippocam-pus. High power bright field analysis of emulsion-dipped slides(data not shown) consolidated the observation that C1q mRNAwas absent from neurons in both control and all experimentalgroups and exclusively present in small cells distributed through-out gray and white matter which were identified as microglial cells(see below).
The relative quantity of C1q mRNA was monitored in a definedarea of the lateral parietal neocortex by quantitative image analysisof x-ray autoradiograms (Table I). Control and sham-operated an-imals displayed only low levels of C1q mRNA (3.80 and 5.06nCi/g tissue, respectively). At 1 and 4 h p.i., C1q mRNA expres-sion was significantly increased.3-fold over control values(16.60 and 18.20 nCi/g tissue, respectively). After 24 h, a 70- to100-fold increase of C1q mRNA levels (375.48 nCi/g) was ob-served as compared with control or sham. At 72 h p.i., levels of
C1q mRNA (658.10 nCi/g) were in excess of 100- and 200-fold ascompared with control and sham, respectively. The intensities ofhybridization signals in the sham control at 24 and at 72 h wereidentical (data not shown).
Immunocytochemistry and in situ hybridization withdigoxigenin-labeled cRNA probes
Both low and high magnification immunocytochemistry and in situhybridization demonstrate that microglial cells, but no other resi-dent cells in the brain, are the source of constitutive biosynthesisand of ischemia-induced increase in C1q biosynthesis throughoutthe rat brain. This is shown, for example, in the hippocampal re-gion 24 h after ischemia (Fig. 2).
To identify the C1q-expressing cell types at the protein level,dual color immunofluorescence and confocal laser scanning mi-croscopy was applied using established markers for astrocytes,neurons, and microglial cells. C1q immunostaining was exclu-sively detected in microglial cells throughout the brain identifiedby costaining for isolectin B4, a well-established marker for mi-croglial cells (Fig. 3). Isolectin B4, in addition, also stains endo-thelial cells (Fig. 3I). The endothelial cells stained for isolectin B4are negative for C1q (Fig. 3H). C1q immunoreactivity was com-pletely absent in neuronal cell bodies (Fig. 3,D–F) identified byusing mouse anti neuronal nuclei Ab NeuN, an established markerfor neurons (Fig. 3F). Furthermore, no expression of C1q inGFAP-positive astrocytes was observed (Fig. 3,A–C). The datashown in Fig. 3 are from sections through the parietal cortex 24 hafter ischemia. These patterns of expression were observedthroughout all areas of the brain at any of the time points after theischemic insult.
The spatiotemporal pattern of increased C1q immunoreactivityin microglial cells during the course of ischemia was found to besimilar to that of ischemia-induced C1q mRNA as demonstratedabove, indicating that the increase in C1q mRNA expression con-sequently led to increased C1q protein biosynthesis.
Analysis of C1q functional activity in CSF
To quantify secretion of functionally active C1q into CSF, sampleswere collected at different time points after the ischemic insult, andfunctional activity of C1q was determined in a C1q-dependent he-molytic assay. Samples were assessed for serum leakage by mea-suring OD412 as an indicator of hemoglobin contamination, andCSF samples with serum contamination were excluded. At 24 hp.i., the level of hemolytically active C1q was markedly elevated(.5-fold compared with control and.3-fold compared withsham; see Fig. 4). At 72 h p.i., the levels of C1q functional activityin CSF were still more than twice as high as in sham animals. Therelative lower level of functionally active C1q at 72 h comparedwith 24 h may be explained by C1q consumption (i.e., removalfrom the fluid phase due to classical pathway activation or bindingof C1q to its receptors) (1).
DiscussionThe present study demonstrates early and widespread up-regula-tion of C1q expression in brain microglial cells and secretion offunctionally active C1q into the CSF in response to experimentallyinduced global cerebral ischemia in the rat.
The sustained rise of C1q mRNA levels and biosynthesis inmicroglial cells in response to global ischemia identifies this com-plement component as a sensitive and specific marker of micro-glial activation. This is in line with our previously published ob-servations that the level of C1q biosynthesis monitored by
5450 MICROGLIAL C1q BIOSYNTHESIS AND CEREBRAL ISCHEMIA
by guest on July 3, 2014http://w
ww
.jimm
unol.org/D
ownloaded from

Northern blot, in situ hybridization, and immunocytochemistry 1)is low and is restricted to microglial cells in normal rat and mousebrain and 2) is strongly up-regulated in CNS microglia after viralinfection and experimental allergic encephalitis (27, 34, 38). Wefound that constitutive and lesion-induced C1q biosynthesis wasabsent in any brain-resident cells other than cells of the microglia/macrophage lineage. This conforms to other reports of experimen-tal brain lesions in vivo (22, 39) and to in vitro data (28, 29)providing strong evidence for microglial biosynthesis of C1q.
These findings, however, are in contrast to recently publishedimmunocytochemical data which were interpreted to indicate thatC1q biosynthesis is absent in normal mouse brain and induced denovo and exclusively in neurons during reperfusion after transientfocal cerebral ischemia in the mouse brain (40). Thus far, thisconclusion has not been corroborated by in situ hybridization anal-ysis. Thus, it cannot be ruled out that the immunostaining for C1qin neurons is a result of C1q deposition on neurons in the infarctarea rather than due to neuronal C1q biosynthesis. In fact, there isevidence that in the course of cerebrovascular disease and Alzhei-mer’s disease, serum proteins like Ig, fibrinogen, and complementfactors are deposited in the brain parenchyma (41). Although thereis accumulating evidence for enhanced cerebral biosynthesis ofC1q in Alzheimer’s disease brain, based on Northern blot, RT-PCR, and Western blot results, the issue of neuronal vs nonneu-ronal biosynthesis of C1q in normal human brains and in Alzhei-mer’s disease brains is still controversial (2, 5, 7, 8, 41, 42).Likewise, in experimental studies of different types of inflamma-tory and neurodegenerative brain lesions, the neuronal vs nonneu-ronal biosynthesis of C1q is a matter of controversy (21, 22,24–27).
It has been postulated that cerebral complement and comple-ment receptor activation is causal for neuronal damage, both inencephalitis and in neurodegeneration (3, 9), suggesting that this isalso the case in cerebral ischemia. The mechanisms by which lo-cally produced complement factors may act cytotoxically on acti-vation include the formation of the membrane attack complex,triggering phagocytosis and free radical release from complementreceptor-bearing cells and synergistic interactions with proinflam-matory cytokines (3).
In this context, it remains to be clarified whether and how anincrease in the local production of C1q in CNS tissue may con-tribute to the pathology of CNS disease. It is conceivable thatraised levels of C1q may promote inflammation in two ways: 1)they may lead to increased levels of functionally active C1 com-plexes, thus driving a local activation of the classical complementactivation cascade (1); 2) they might trigger cellular responses bybinding to C1q receptors (1, 43). The potential benefits of usingcomplement inhibitors as a novel therapeutic approach in the treat-ment of inflammatory and degenerative neurological diseases ishighlighted by a recent report of Huang et al. (40) which demon-strates a reduced cerebral infarct volume and neuronal protectionby infusion of a hybrid molecule that simultaneously inhibits com-plement activation and selectin-mediated adhesion in a murine ex-perimental model of focal cerebral ischemia in mice. These results,however, do not permit assessment of the contribution of enhancedC1q biosynthesis, because the complement-inhibitory activity ofthe hybrid molecule used in this study becomes effective furtherdownstream of C1 activation only and may inhibit each of thethree activation routes of complement. The contribution of CNSbiosynthesis of C1q under pathological conditions can be evalu-ated only in either C1q-deficient mouse strains (43) or using spe-cific inhibitors of C1 activation (44).
AcknowledgmentsWe thank Luciano Dho, Marion Hainmuller, Heidi Hlawaty, Elke Roden-berg, Jorg Schmidt, and Tamara Henke for technical assistance and Hei-demarie Schneider for expert photographic reproduction.
References1. Reid, K. B. M., and A. Law. 1995.Complement.IRL Press, Oxford.2. Shen, Y., R. Li, E. McGeer, and P. McGeer. 1997. Neuronal expression of
mRNAs for complement proteins of the classical pathway in Alzheimer brain.Brain Res. 769:391.
3. Morgan, B. P., and P. Gasque. 1996. Expression of complement in the brain: rolein health and disease.Immunol. Today 17:461.
4. Whaley K., and W. Schwaeble. 1997. Complement and complement deficiencies.Semin. Liver Dis. 17:297.
5. Terai, K., D. G. Walker, E. G. McGeer, and P. L. McGeer. 1997. Neurons expressproteins of the classical complement pathway in Alzheimer disease.Brain Res.769:385.
6. Velazquez, P., D. Cribbs, T. Poulos, and A. Tenner. 1997. Aspartate residue 7 inamyloidb-protein is critical for classical complement pathway activation: impli-cations for Alzheimer’s disease pathogenesis.Nat. Med. 3:77.
7. Afagh, A., B. Cummings, D. Cribbs, C. Cotman, and A. Tenner. 1996. Local-ization and cell association of C1q in Alzheimer’s disease brain.Exp. Neurol.138:22.
8. Fischer, B., H. Schmoll, P. Riederer, J. Bauer, D. Platt, and A. Popa-Wagner.1995. Complement C1q and C3 mRNA expression in the frontal cortex of Alz-heimer’s patients.J. Mol. Med. 73:465.
9. McGeer, P. L., and E. G. McGeer. 1995. The inflammatory response system ofbrain: implications for therapy of Alzheimer and other neurodegenerative dis-eases.Brain Res. Brain Res. Rev. 21:195.
10. Schwab, C., J. Steele, and P. McGeer. 1996. Neurofibrillary tangles of GuamParkinson-dementia are associated with reactive microglia and complement pro-teins.Brain Res. 707:196.
11. Yamada, T., I. Moroo, Y. Koguchi, M. Asahina, and K. Hirayama. 1994. In-creased concentration of C4d complement protein in the cerebrospinal fluids inprogressive supranuclear palsy.Acta Neurol. Scand. J. 89:42.
12. Singhrao, S., J. Neal, P. Gasque, B. Morgan, and G. Newman. 1996. Role ofcomplement in the aetiology of Pick’s disease?J. Neuropathol. Exp. Neurol.55:578.
13. Walsh, M. J., and J. M. Murray. 1998. Dual implication of 29,39-cyclic nucleotide39 phosphodiesterase as major autoantigen and C3 complement-binding protein inthe pathogenesis of multiple sclerosis.J. Clin. Invest. 101:1923.
14. Gasque, P., S. K. Singhrao, J. W. Neal, P. Wang, S. Sayah, M. Fontaine, andB. P. Morgan. 1998. The receptor for complement anaphylatoxin C3a is ex-pressed by myeloid cells and nonmyeloid cells in inflamed human central nervoussystem: analysis in multiple sclerosis and bacterial meningitis.J. Immunol. 160:3543.
15. Adam, C., M. Geniteau, M. Gougerot-Pocidalo, P. Verroust, J. Lebras, C. Gibert,and L. Morel-Maroger. 1981. Cryoglobulins, circulating immune complexes, andcomplement activation in cerebral malaria.Infect. Immun. 31:530.
16. Stahel, P. F., K. Frei, H. P. Eugster, A. Fontana, K. M. Hummel, R. A. Wetsel,R. S. Ames, and S. R. Barnum. 1997. TNF-alpha-mediated expression of thereceptor for anaphylatoxin C5a on neurons in experimentalListeria meningoen-cephalitis. J. Immunol. 159:861.
17. Williams, A. E., L. J. Lawson, V. H. Perry, and H. Fraser. 1994. Characterizationof the microglial response in murine scrapie.Neuropathol. Appl. Neurobiol. 20:47.
18. Dandoy-Dron, F., F. Guillo, L. Benboudjema, J. P. Deslys, C. Lasmezas,D. Dormont, M. G. Tovey, and M. Dron. 1998. Gene expression in scrapie:cloning of a new scrapie-responsive gene and the identification of increased lev-els of seven other mRNA transcripts.J. Biol. Chem. 273:7691.
19. Lindsberg, P. J., J. Ohman, T. Lehto, M. L. Karjalainen-Lindsberg, A. Paetau,T. Wuorimaa, O. Carpen, M. Kaste, and S. Meri. 1996. Complement activationin the central nervous system following blood-brain barrier damage in man.Ann.Neurol. 40:587.
20. Svensson, M., L. Liu, P. Mattsson, B. Morgan, and H. Aldskogius. 1995. Evi-dence for activation of the terminal pathway of complement and upregulation ofsulfated glycoprotein (SGP)-2 in the hypoglossal nucleus following peripheralnerve injury.Mol. Chem. Neuropathol. 24:53.
21. Goldsmith, S. K., P. Wals, I. Rozovsky, T. E. Morgan, and C. E. Finch. 1997.Kainic acid and decorticating lesions stimulate the synthesis of C1q protein inadult rat brain.J. Neurochem. 68:2046.
22. Pasinetti, G. M., S. A. Johnson, I. Rozovsky, M. Lampert-Etchells, D. G. Morgan,M. N. Gordon, T. E. Morgan, D. Willoughby, and C. E. Finch. 1992. Comple-ment C1qB and C4 mRNAs responses to lesioning in rat brain.Exp. Neurol.118:117.
23. Johnson, S., C. Young-Chan, N. Laping, and C. Finch. 1996. Perforant pathtransection induces complement C9 deposition in hippocampus.Exp. Neurol.138:198.
24. Jensen, M., B. Finsen, and J. Zimmer. 1997. Morphological and immunopheno-typic microglial changes in the denervated fascia dentata of adult rats: correlationwith blood-brain barrier damage and astroglial reactions.Exp. Neurol. 143:103.
25. Akiyama, H., I. Tooyama, H. Kondo, K. Ikeda, H. Kimura, E. McGeer, andP. McGeer. 1994. Early response of brain resident microglia to kainic acid-in-duced hippocampal lesions.Brain Res. 635:257.
5451The Journal of Immunology
by guest on July 3, 2014http://w
ww
.jimm
unol.org/D
ownloaded from

26. Rozovsky, I., T. Morgan, D. Willoughby, M. Dugichi-Djordjevich, G. Pasinetti,S. Johnson, and C. Finch. 1994. Selective expression of clusterin (SGP-2) andcomplement C1qB and C4 during responses to neurotoxins in vivo and in vitro.Neuroscience 62:741.
27. Dietzschold, B., W. Schwaeble, M. K.-H. Schafer, F. Petry, Y. Zehng, H. Zheng,T. Fink, M. Loos, and E. Weihe. 1995. The expression of C1q, a subcomponentof the rat complement system, is dramatically enhanced in brains of rats witheither Borna disease or experimental allergic encephalomyelitis.J. Neurol. Sci.130:11.
28. Haga, S., T. Aizawa, T. Ishii, and K. Ikeda. 1996. Complement gene expressionin mouse microglia and astrocytes in culture: comparisons with mouse peritonealmacrophages.Neurosci. Lett. 216:191.
29. Korotzer, A., J. Watt, D. Cribbs, A. Tenner, D. Burdick, C. Glabe, andC. Cotman. 1995. Cultured rat microglia express C1q and receptor for C1q:implications for amyloid effects on microglia.Exp. Neurol. 134:214.
30. Gasque, P., M. Fontaine, and B. Morgan. 1995. Complement expression in hu-man brain: biosynthesis of terminal pathway components and regulators in humanglial cells and cell lines.J. Immunol. 154:4726.
31. Kreutzberg, G. W. 1995. Microglia, the first line of defence in brain pathologies.Arzneim.-Forsch.45:357.
32. Nolte, C., T. Moller, T. Walter, and H. Kettenmann. 1996. Complement 5a con-trols motility of murine microglial cells in vitro via activation of an inhibitoryG-protein and the rearrangement of the actin cytoskeleton.Neuroscience 73:1091.
33. Pulsinelli, W. A., and J. B. Brierley. 1979. A new model of bilateral hemisphericischemia in the unanesthetized rat.Stroke 10:267.
34. Schwaeble, W., M. K.-H. Schafer, F. Petry, T. Fink, D. Knebel, E. Weihe, andM. Loos. 1995. Follicular dendritic cells, interdigitating cells, and cells of themonocyte-macrophage lineage are the C1q-producing sources in the spleen: iden-tification of specific cell types by in situ hybridization and immunohistochemicalanalysis.J. Immunol. 155:4971.
35. Melten, D. A., P. A. Krieg, M. R. Rebagliati, T. Maniatis, K. Zinn, andM. R. Green. 1984. Efficient in vitro synthesis of biologically active RNA andRNA hybridization probes from plasmids containing a bacteriophage SP6 pro-moter.Nucleic Acids Res. 12:7035.
36. Schafer, M. K.-H., J. P. Herman, and S. J. Watson. 1992. In situ hybridizationimmunohistochemistry. InImaging Drug Action in the Brain.E. D. London, ed.CRC Press, Boca Raton, p. 337.
37. Persson, S., M. K.-H. Schafer, D. Nohr, G. Ekstrom, C. Post, F. Nyberg, andE. Weihe. 1994. Spinal prodynorphin gene expression in collagen-induced ar-thritis: influence of the glucocorticosteroid budesonide.Neuroscience 63: 313.
38. Schwaeble, W. J., C. M. Stover, T. J. Schall, D. J. Dairaghi, P. K. E. Trinder,C. Linington, A. Iglesias, A. Schubart, N. J. Lynch, E. Weihe, and M. K.-H.Schafer. 1998. Neuronal expression of fractalkine in the presence and absence ofinflammation.FEBS Lett. 439:203.
39. Johnson, S. A., G. M. Pasinetti, and C. E. Finch. 1994. Expression of complementC1qB and C4 mRNAs during rat brain development.Brain Res. Dev. Brain Res.80:163.
40. Huang, J., L. Kim, R. Mealey, H. Marsh, Jr., Y. Zhang, A. Tenner, E. Connolly,Jr., and D. Pinsky. 1999. Neuronal protection in stroke by an sLe(x)-glycosylatedcomplement inhibitory protein.Science 285:595.
41. Tomimoto, H., I. Akiguchi, H. Wakita, T. Suenaga, S. Nakamura, and J. Kimura.1997. Regressive changes of astroglia in white matter lesions in cerebrovasculardisease and Alzheimer’s disease patients.Acta Neuropathol. 94:146.
42. Yasojima, K., C. Schwab, E. G. McGeer, and P. L. McGeer. 1999. Up-regulatedproduction and activation of the complement system in Alzheimer’s diseasebrain.Am. J. Pathol. 154:927.
43. Botto, M., C. Dell’Agnola, A. E. Bygrave, E. M. Thompson, H. T. Cook, F. Petry,M. Loos, P. P. Pandolfi, and M. J. Walport. 1998. Homozygous C1q deficiencycauses glomerulonephritis associated with multiple apoptotic bodies.Nat. Genet.19:56.
44. Stuart, G. R., N. J. Lynch, J. Lu, A. Geick, B. E. Moffatt, R. B. Sim, andW. J. Schwaeble. 1996. Localisation of the C1q binding site within C1q receptor/calreticulin.FEBS Lett. 397:245.
45. Post, C., P. Salvati, M. K.-H. Schafer, W. Schwaeble, M. Cini, M. Calabresi,F. Vaghi, E. H. F. Wong, and E. Weihe. 1996. Early up-regulation of complementfactors and astrocyte dysfunction in animal models of global ischemia and stroke.Soc. Neurosci. Abstracts 22:2143.
46. Weihe, E., M. K.-H. Schafer, P. Salvati, L. Dho, M. Calabresi, C. Post, R. B. Sim,and W. Schwaeble. Implications of cerebral biosynthesis of classical pathwaycomponents of the complement system in CNS ischemia. InFourth InternationalWorkshop on C1 and Collectins,Mainz, Germany, October 3–5, 1997.
5452 MICROGLIAL C1q BIOSYNTHESIS AND CEREBRAL ISCHEMIA
by guest on July 3, 2014http://w
ww
.jimm
unol.org/D
ownloaded from
Copyright © 2022 FDOKUMEN




















