
Cognitive Dysfunction in Mice Infected with Plasmodium berghei Strain ANKA
12
Cognitive Dysfunction in Mice Infected with Plasmodium berghei Strain ANKA Mahalia S. Desruisseaux 1,2,3 , Maria Gulinello 2 , David N. Smith 2 , SunHee C. Lee 1 , Moriya Tsuji 5 , Louis M. Weiss 1,3 , David C. Spray 2,3,4 , and Herbert B. Tanowitz 1,3 1 Department of Pathology, Albert Einstein College of Medicine, Bronx 2 The Dominick P. Purpura Department of Neuroscience, Albert Einstein College of Medicine, Bronx 3 Department of Medicine, Albert Einstein College of Medicine, Bronx 4 Department of Urology, Albert Einstein College of Medicine, Bronx 5 Aaron Diamond AIDS Research Center, The Rockefeller University, New York, New York Abstract Cerebral malaria complicated by cognitive sequelae is a major cause of morbidity in humans infected with Plasmodium falciparum. To model cognitive function after malaria, we created a rodent model of cerebral malaria by infecting C57BL/6 mice with Plasmodium berghei strain ANKA. After 7 days, an object-recognition test of working memory revealed a significant impairment in the visual memory of infected mice. This impairment was observed in the absence of confounding effects of infection. The cognitive dysfunction correlated with hemorrhage and inflammation. Furthermore, microglial activity and morphological changes detected throughout the brains of infected mice were absent from the brains of control mice, and this correlated with the measured cognitive defects. Similar testing methods in human studies could help identify subjects at risk for an adverse cognitive outcome. This murine model should facilitate the study of adjunctive methods to ameliorate adverse neurological outcomes in cerebral malaria. Cerebral malaria due to Plasmodium falciparum infection is a major cause of morbidity and mortality in the developing world. It is estimated that 1 million children die each year of this disease in sub-Saharan Africa [1,2]. Approximately 10%–28% of children who survive an episode of cerebral malaria develop neurological sequelae [1,3,4]. It has become increasingly recognized that cerebral malaria may cause persistent neurological and cognitive deficits that span a wide range of cognitive functions long after the infection has been successfully treated with antimalarial drugs, even in asymptomatic individuals [1–8]. Persistent deficiencies include attentional memory, learning and language impairments, visuospatial and motor deficits, and psychiatric disorders [3,5,6,8–15]. The animal models of cerebral malaria faithfully recapitulate many of the characteristics of the human syndrome and these have been important in investigating pathogenesis of the disease [16]. For example, C57BL/6 mice infected with Plasmodium berghei strain ANKA exhibit neurological and behavioral impairments that are similar to those in humans infected with P. Reprints or correspondence: Dr. Herbert B. Tanowitz, Depts. of Pathology and Medicine, Albert Einstein College of Medicine, 1300 Morris Park Ave., Bronx, NY 10461 (E-mail: [email protected]). Potential conflicts of interest: none reported. Presented in part: VIIIth European Meeting on Glial Cells in Health and Disease, 4 – 8 September 2007, London, United Kingdom; and Burroughs-Wellcome Fund Awardees Meeting, July 2007, Dana Point, California. NIH Public Access Author Manuscript J Infect Dis. Author manuscript; available in PMC 2009 June 8. Published in final edited form as: J Infect Dis. 2008 June 1; 197(11): 1621–1627. doi:10.1086/587908. NIH-PA Author Manuscript NIH-PA Author Manuscript NIH-PA Author Manuscript
Transcript of Cognitive Dysfunction in Mice Infected with Plasmodium berghei Strain ANKA

Cognitive Dysfunction in Mice Infected with Plasmodium bergheiStrain ANKA
Mahalia S. Desruisseaux1,2,3, Maria Gulinello2, David N. Smith2, SunHee C. Lee1, MoriyaTsuji5, Louis M. Weiss1,3, David C. Spray2,3,4, and Herbert B. Tanowitz1,3
1 Department of Pathology, Albert Einstein College of Medicine, Bronx
2 The Dominick P. Purpura Department of Neuroscience, Albert Einstein College of Medicine, Bronx
3 Department of Medicine, Albert Einstein College of Medicine, Bronx
4 Department of Urology, Albert Einstein College of Medicine, Bronx
5 Aaron Diamond AIDS Research Center, The Rockefeller University, New York, New York
AbstractCerebral malaria complicated by cognitive sequelae is a major cause of morbidity in humans infectedwith Plasmodium falciparum. To model cognitive function after malaria, we created a rodent modelof cerebral malaria by infecting C57BL/6 mice with Plasmodium berghei strain ANKA. After 7 days,an object-recognition test of working memory revealed a significant impairment in the visual memoryof infected mice. This impairment was observed in the absence of confounding effects of infection.The cognitive dysfunction correlated with hemorrhage and inflammation. Furthermore, microglialactivity and morphological changes detected throughout the brains of infected mice were absent fromthe brains of control mice, and this correlated with the measured cognitive defects. Similar testingmethods in human studies could help identify subjects at risk for an adverse cognitive outcome. Thismurine model should facilitate the study of adjunctive methods to ameliorate adverse neurologicaloutcomes in cerebral malaria.
Cerebral malaria due to Plasmodium falciparum infection is a major cause of morbidity andmortality in the developing world. It is estimated that 1 million children die each year of thisdisease in sub-Saharan Africa [1,2]. Approximately 10%–28% of children who survive anepisode of cerebral malaria develop neurological sequelae [1,3,4]. It has become increasinglyrecognized that cerebral malaria may cause persistent neurological and cognitive deficits thatspan a wide range of cognitive functions long after the infection has been successfully treatedwith antimalarial drugs, even in asymptomatic individuals [1–8]. Persistent deficienciesinclude attentional memory, learning and language impairments, visuospatial and motordeficits, and psychiatric disorders [3,5,6,8–15].
The animal models of cerebral malaria faithfully recapitulate many of the characteristics of thehuman syndrome and these have been important in investigating pathogenesis of the disease[16]. For example, C57BL/6 mice infected with Plasmodium berghei strain ANKA exhibitneurological and behavioral impairments that are similar to those in humans infected with P.
Reprints or correspondence: Dr. Herbert B. Tanowitz, Depts. of Pathology and Medicine, Albert Einstein College of Medicine, 1300Morris Park Ave., Bronx, NY 10461 (E-mail: [email protected]).Potential conflicts of interest: none reported.Presented in part: VIIIth European Meeting on Glial Cells in Health and Disease, 4 – 8 September 2007, London, United Kingdom; andBurroughs-Wellcome Fund Awardees Meeting, July 2007, Dana Point, California.
NIH Public AccessAuthor ManuscriptJ Infect Dis. Author manuscript; available in PMC 2009 June 8.
Published in final edited form as:J Infect Dis. 2008 June 1; 197(11): 1621–1627. doi:10.1086/587908.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

falciparum, including ataxia, hemiparaplegias, and seizures [16–18]. These neurologicalsequelae correlate with pathological findings in the brains of humans with cerebral malaria andof experimental animals [2,5,16,17]. There have been few systematic studies of cognitiveoutcomes of cerebral malaria in animal models. However, damage to the brain in cerebralmalaria occurs in regions known to be important in memory, such as the fornix, cortex, andhippocampus [19]. Astrogliosis and microglial activation in particular are found in theseregions [20,21] and precede neurological alterations [20]. In addition, several reports havedocumented increased levels of inflammatory mediators, such as cytokines and chemokines,in the brains of P. berghei ANKA–infected mice [21,22]. In the present study, we demonstratethat mice infected with P. berghei ANKA display demonstrable cognitive impairment that isnot observed in uninfected mice and that this impairment is associated with pathologicalfindings in the brain.
MATERIALS AND METHODSMouse infection
Six-week-old C57BL/6 female mice (Jackson Laboratories) were infected with 5 × 105 redblood cells parasitized with P. berghei ANKA diluted in 500 μL of PBS or were injected withuninfected blood diluted in PBS. The mice were then randomly separated into 2 groups (N=8).Parasitemia was monitored by examining Giemsa-stained blood smears on days 6 and 8 afterinfection. Experiments were performed in accordance with the Institutional Animal Care andUse Committee of the Albert Einstein College of Medicine.
Behavioral assaysThe object-recognition test of working memory [23], which is based on the natural and robusttendency of rodents to preferentially explore novel objects, was performed as describedelsewhere [23]. Briefly, in trial 1, mice were placed in an opaque acrylic arena (area, 97 cm2)and allowed to freely explore 2 identical, nontoxic objects (e.g., plastic, glass, or ceramic items)for 3 min. The time spent exploring the objects was recorded manually with timers, andlocomotor activity was assessed simultaneously with Viewer tracking software (Biobserve),after which the mouse was returned to the home cage. Following a retention interval of 60 min,the mouse was returned to the arena, where one of the familiar objects had been replaced witha novel object (trial 2). The mouse was again allowed to explore for 3 min. Exploration of boththe novel and familiar objects was given a preference score, defined as the time spent exploringthe novel object divided by the total time spent exploring both objects, expressed as apercentage. A preference score of 50% indicated chance exploration (i.e., both novel andfamiliar objects are explored equally). Exploration of the objects was defined as any physicalexploration (whisking, sniffing, rearing on, or touching the object) and approach and obviousorientation within 3 cm of the object.
Novelty-induced exploration in trial 1 was defined as the total time spent exploring both of theidentical objects. This is a reliable indicator of exploratory behavior. Although this measure isnot generally correlated with performance in the testing trials, it is nevertheless an importantassessment of general sickness behavior.
The subjects were tested in a longitudinal (i.e., within-group) design at 3 time points (5, 7, and11 days) after P. berghei ANKA infection or control injection. These time points were chosenon that basis of our previous observations of the time course of the worsening pathologicalstatus after infection with the chosen inoculum.
Desruisseaux et al. Page 2
J Infect Dis. Author manuscript; available in PMC 2009 June 8.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

The functional observation battery (i.e., the primary screen) was performed essentiallyaccording to the SHIRPA protocol [24,25] and assessed general arousal, reflexes, andautonomic, motor, and neurological deficits.
Statistical analysis was performed using 1-way repeated-measured analysis of variance(ANOVA), followed by the Fisher protected least significant difference post hoc t test. Posthoc test results are only shown for data in which findings of ANOVA were statisticallysignificant.
Histological analysis and immunostainingOne day after the second and third behavioral tests, infected mice (4 on day 8 and 2 on day 12)were euthanized using CO2 gas, and their brains were harvested and fixed in 10% bufferedformalin. Four control mice were sacrificed on day 12 after injection. Brain specimens of thefollowing widths relative to bregma were obtained: 0.98 to −0.22 mm, −1.82 to −1.94 mm, and−5.88 to −7.2 mm. Specimens were later embedded in paraffin, sectioned into 5-μm slices, andstained with hematoxylineosin.
For quantification of microglial activity, immunohistochemistry analysis was performed usingrabbit antibody to ionized calcium-binding adaptor molecule 1 (Iba1) (Wako Chemicals USA),a peptide that is selectively expressed in macrophages/microglia. The sections weredeparaffinized and boiled at 95°C for 20 min in sodium citrate solution (DAKO) for antigenretrieval. The sections were then incubated overnight at 4°C with Iba-1 at a dilution of 1:300.A standard avidin-biotin complex method (Vector Immunolab) was used for the secondaryantibody (anti-rabbit), using a 1:200 dilution and a 1-h incubation period. Slides werecounterstained with hematoxylin after immunolabeling.
To quantify the intensity of Iba1 staining, photographs were taken of the cortex and the tissuessurrounding the neuronal cell layers of the hippocampus, using a Nikon Microphot-FXAmicroscope system and a Nikon digital sight DS-5M camera. The photographs were thenanalyzed using ImageJ 1.37v (National Institutes of Health) to quantify the staining intensityas a percentage of the region of interest. Five control mice were compared with 7 infected mice,and a t test was used for statistical analysis.
RESULTSParasitemia
The average parasitemia (defined as the percentage of parasitized RBCs) in infected mice was4.12% on day 6 after infection and 9.63% on day 8 after infection.
Visual memory and object recognitionInfected mice began to perform significantly worse than control mice in the object-recognitiontest on day 7 (figure 1A), based on a repeated-measures ANOVA (F score, 9.9; P < .01). Posthoc tests demonstrated that neither group exhibited significant cognitive impairments on day5; however, on day 7, there were significant treatment-group effects (P < .01). Visual memoryin the infected group was significantly worse than that in the control group (figure 1A).
Most infected mice died by day 11. The remaining 2 infected mice performed worse thancontrol mice on day 12; however, because of the low number of remaining mice, statisticalanalysis could not be performed. Data from testing on day 11 are included in figure 1A onlyfor the purpose of visual comparison.
Desruisseaux et al. Page 3
J Infect Dis. Author manuscript; available in PMC 2009 June 8.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Locomotor activity, novel-object exploration, and functional observation batteryTo verify the absence of potential confounding effects of infection on nonspecific sicknessbehavior, several tests were conducted to assess memory performance. The first of these testsassessed locomotor activity in the open field and found no significant difference betweeninfected mice and control mice over time (figure 1C). Second, the total duration of novel-objectexploration was assessed during trial 1 of the object-recognition test. Infected and control micehad similar durations of novel-object exploration (figure 1B). Third, the functional observationbattery revealed no significant differences in arousal, reflexes, motor coordination, orneurological characteristics (data not shown). These data indicate that the cognitive deficitsexhibited by the infected mice were not purely due to nonspecific behavioral effects of sicknesscaused by the infection.
Histological analysisHematoxylin-eosin staining of brain sections obtained from infected mice demonstratedmargination of inflammatory cells in the vessels and extravasation of red blood cells in severalregions of the brain, including the thalamus, midbrain, and cerebellum (figure 2). Evidence offornical hemorrhage, which is consistent with damage to the major input/output pathway tothe hippocampus, was found in 3 infected mice (75%) sacrificed on day 8 and 2 infected mice(100%) sacrificed on day 12 but was absent in 4 control mice (figure 3).
Immunostaining revealed robust Iba-1 reactivity in specimens from infected mice,demonstrating that microglia cell activation was prominent throughout the brain, including thecortex (figure 4B) and the hippocampus (figure 4D). There was extensive microglial activationwith retraction and thickening of the microglial processes. In addition, there were Iba-1–positive intravascular inflammatory cells in the cerebellum of an infected mouse, which wasconsistent with sequestration of monocytes (figure 4). In contrast, Iba-1–positive microglialcells in brains obtained from 5 uninfected mice were highly ramified and uniformly maintaineda delicate morphological processes. The percentages (±SD) of the regions of interest in thecortex that stained positive for Iba-1 were 0.8% ± 0.56% in infected mice and 0.23% ± 0.12%in control mice (P < .05); the percentages (± SD) in the hippocampus were 1.07% ± 0.53% ininfected mice and 0.21% ± 0.063% in control mice (P < .05).
DISCUSSIONNeurological and cognitive impairments are common features of human cerebral malaria,primarily when infection is due to P. falciparum. These deficits may include hemiparesis,ataxia, deep and prolonged coma, intracranial hypertension, extrapyramidal tremor andepilepsy, hearing and visual impairments, and behavioral difficulties and cognitive deficits[2]. Some of the cognitive alterations that persist after the resolution of the infection includeimpairment in working memory, learning and language deficits, visuospatial and motordeficits, and psychiatric disorders [3,5,6,8–15]. Here, we used the novel object–recognitiontask, which evaluates working memory, to observe cognitive deficits after infection in anestablished mouse model of cerebral malaria. In the brains of the infected mice, there werefocal areas of inflammation and hemorrhagic damage. This damage, which included inflamedvessels with leukocyte margination and areas of microhemorrhages, was evident in the fornix,thalamus, midbrain, and cerebellum, consistent with the wide-ranging patterns of reportedneurocognitive sequelae that ensue in children after malaria [1–6,8–10,13].
In addition to the sequestration of inflammatory cells in the brain vasculature of infected mice,activation of microglia in all areas of their brains demonstrated extensive intrinsic brainparenchymal inflammation. Microglial activation, as well as increased inflammatory
Desruisseaux et al. Page 4
J Infect Dis. Author manuscript; available in PMC 2009 June 8.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

mediators, can cause neurodegeneration and apoptosis of neuronal cells in the hippocampus,which adversely affects cognitive function [26–28].
Microglia are cells of macrophage/monocyte origin and are important components in thebrain’s response to infection. When microglia are activated by different pathogenic stimuli,such as those associated with malaria, they respond in various ways, which includemorphological alterations and release of inflammatory mediators [20,29–31]. This activationcorresponds with insults to the brain and can contribute to the encephalopathy of cerebralmalaria.
It is difficult to dissect the specific effects of malaria-induced inflammation and damage fromthe effects of general sickness behavior or peripheral inflammation. It is generally recognizedthat inflammation resulting from several etiologies can have an adverse effect on cognitivefunction [32,33]. In this regard, no currently existing behavior test is unaffected by the effectsof systemic inflammatory mediators that result from acute infection or even stress, becausecytokines have been shown to interfere with exploratory behavior, locomotor activity, andsome hippocampal functions, as well as to induce microglial activation and reactive gliosis[32,33]. For this reason, our batteries of tests are designed with inherent controls in an effortto minimize the confounding effects of inflammatory mediators and general sickness. Eachanimal served as its own control for exploratory behavior, locomotor activity, and novel-objectpreference during the training phase of the trial, when there were no significant differencesbetween infected mice and control mice.
Several brain regions are known to play a role in visual working memory, including thehippocampus [34–37], the posterior parahippocampal region [38], and the perirhinal cortex[39,40]. In this regard, the hippocampus is an integral part of the brain because it is requiredfor efficient acquisition of visuospatial tasks, as well as for memory encoding and recall [41,42]. Both functions of the hippocampus are supported by cholinergic and noncholinergicafferents and efferents in the fornix [43,44]. Indeed, this study detected extensive hemorrhageof bilateral fornices in the infected mice that was absent in the control mice. Lesions of thefornix have been shown to cause marked lasting impairment in both memory and spatiallearning; however, fornix lesions are less likely to interfere with short-term recognitionmemory [45–47].
Although P. berghei ANKA infection is generally considered to be a model of cerebral malaria,there is systemic involvement; thus, it is important to distinguish between nonspecific sicknessbehavior and specific cognitive deficits. A previous report demonstrated a decrease inlocomotor activity in infected mice during the immediate ante mortem period [17], which likelyindicated moribund behavior in those mice. However, our observation of the cognitive functionof mice at earlier time points after infection did not detect decreased activity in the open fieldor decreased exploration of novel objects in the training trial. This is consistent with otherreports that suggested that locomotor activity is not altered at earlier time points after infection(even at 1.5 days before death) [17]. Furthermore, we demonstrated normal visual placementin the functional observation battery and normal levels of novel-object exploration up to day11, indicating that novel-object preference, exploratory activity, and visual acuity were intactdespite evident cognitive deficits.
The use of the novel object–recognition task has many benefits in this infection model. It doesnot rely on food or water deprivation to motivate the animal’s behavior, which could be asubstantial confounding factor in infected mice. Furthermore, skilled motor coordination,efficient reaction time, and motor stamina are not required, because the tests are rapid andrequire only normal exploratory behavior. This test is also similar to those conducted toevaluate a range of human disorders [48–50]. Although we did not observe decreased
Desruisseaux et al. Page 5
J Infect Dis. Author manuscript; available in PMC 2009 June 8.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

locomotor activity in infected mice, we did not formally test for cerebellar ataxia, a knowncomplication of malaria [1,4]. Our results clearly demonstrate that the cerebellum is damagedduring cerebral malaria. Therefore, more-focused experiments are currently being performedto validate and quantify the associated functional impairment.
Neurocognitive impairment as a result of infection with certain malarial species represents asubstantial health, social, and economic burden in countries that are already resource poor, andthousands of children are at risk every year [1–3,5,8,9,11]. The mechanisms by which theinfection causes these deficits are not well understood, although inflammation and vascularimpairment have been suggested. Furthermore, there are no uniform standardized methods ofidentifying children at risk of developing neurocognitive sequelae. Here, we suggest a modelof objective cognitive deficit in infected mice, as demonstrated by the decrease in workingmemory that is associated with brain parenchymal hemorrhage and inflammation, as well asmicroglial activation and neuronal degeneration. The use of this model will permit thedevelopment of strategies to modify neurological outcome that can be transferred to humanpopulation-based studies helping to prevent adverse neurocognitive outcomes in those at risk.
AcknowledgmentsIDSA ERF/NFID Colin L. Powell Minority Postdoctoral Fellowship in Tropical Disease Research, sponsored byGlaxoSmithKline; NIH Training Grant in Mechanisms of Cardiovascular Diseases (grant T32 HL-07675 to M.S.D.);Dominick P. Purpura Department of Neuroscience (neuroscience fellowship to M.S.D.) and Department of Psychiatryand Behavioral Sciences (support to M.S.D.), Albert Einstein College of Medicine; Burroughs-Wellcome Funds CareerAwards for Medical Scientists (to M.S.D.); Dana Foundation Program in Brain and Immunoimaging (to H.B.T.); andAlbert Einstein College of Medicine (grant CFAR P30 AI051519 to S.C.L.).
We thank Dr. Kami Kim for review and helpful discussions of the manuscript, Dr. Meng-Liang Zhao for assistancewith immunohistochemistry analyses, and Mr. Dazhi Zhao for assistance with the animal-associated research.
References1. Newton CRJC, Krishna S. Severe falciparum malaria in children: current understanding of
pathophysiology and supportive treatment. Pharmacol Ther 1998;79:1–53. [PubMed: 9719344]2. Idro R, Jenkins NE, Newton CRJC. Pathogenesis, clinical features, and neurological outcomes of
cerebral malaria. Lancet Neurol 2005;4:827–40. [PubMed: 16297841]3. Boivin MJ, Bangirana P, Byarugaba J, et al. Cognitive impairment after cerebral malaria in children:
a prospective study. Pediatrics 2007;119:e360–6. [PubMed: 17224457]4. Taylor, TE.; Molyneux, ME. Clinical features of malaria in children. In: Warrell, DA.; Gilles, HM.,
editors. Essential malariology. Vol. 4. Oxford, United Kingdom: A Hodder Arnold Publication; 2002.p. 206-18.
5. Boivin MJ. Effects of early cerebral malaria on cognitive ability in Senegalese children. J Dev BehavPediatr 2002;23:353–64. [PubMed: 12394524]
6. Carter JA, Mung’ala-Odera V, Neville BG, et al. Persistent neurocognitive impairments associatedwith severe falciparum malaria in Kenyan children. J Neurol Neurosurg Psychiatry 2005;76:476–81.[PubMed: 15774431]
7. Al Serouri AW, Grantham-McGregor SM, Greenwood B, Costello A. Impact of asymptomatic malariaparasitaemia on cognitive function and school achievement of schoolchildren in the Yemen Republic.Parasitology 2000;121:337–45. [PubMed: 11072896]
8. Kihara M, Carter JA, Newton CRJC. The effect of Plasmodium falciparum on cognition: a systematicreview. Trop Med Int Health 2006;11:386–97. [PubMed: 16553922]
9. Carter JA, Lees JA, Gona JK, et al. Severe falciparum malaria and acquired childhood languagedisorder. Dev Med Child Neurol 2006;48:51–7. [PubMed: 16359594]
10. Carter JA, Ross AJ, Neville BG, et al. Developmental impairments following severe falciparummalaria in children. Trop Med Int Health 2005;10:3–10. [PubMed: 15655008]
Desruisseaux et al. Page 6
J Infect Dis. Author manuscript; available in PMC 2009 June 8.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

11. Fernando D, de Silva D, Wickremasinghe R. Short-term impact of an acute attack of malaria on thecognitive performance of schoolchildren living in a malaria-endemic area of Sri Lanka. Trans R SocTrop Med Hyg 2003;97:633–9. [PubMed: 16117954]
12. Meremikwu MM, Asindi AA, Ezedinachi E. The pattern of neurological sequelae of childhoodcerebral malaria among survivors in Calabar, Nigeria. Cent Afr J Med 1997;43:231–4. [PubMed:9431762]
13. Holding PA, Stevenson J, Peshu N, Marsh K. Cognitive sequelae of severe malaria with impairedconsciousness. Trans R Soc Trop Med Hyg 1999;93:529–34. [PubMed: 10696414]
14. Telgt DS, van der Ven AJ, Schimmer B, Droogleever-Fortuyn HA, Sauerwein RW. Seriouspsychiatric symptoms after chloroquine treatment following experimental malaria infection. AnnPharmacother 2005;39:551–4. [PubMed: 15728331]
15. Lou J, Lucas R, Grau GE. Pathogenesis of cerebral malaria: recent experimental data and possibleapplications for humans. Clin Microbiol Rev 2001;14:810–20. [PubMed: 11585786]
16. Lackner P, Beer R, Heussler V, et al. Behavioural and histopathological alterations in mice withcerebral malaria. Neuropathol Appl Neurobiol 2006;32:177–88. [PubMed: 16599946]
17. Gernaat HB. Malaria presenting as atypical depression. Br J Psychiatry 1990;157:783. [PubMed:2279229]
18. Rest JR. Cerebral malaria in inbred mice. I. A new model and its pathology. Trans R Soc Trop MedHyg 1982;76:410–5. [PubMed: 7051459]
19. Stoltenburg-Didinger G, Neifer S, Bienzle U, Eling WM, Kremsner PG. Selective damage ofhippocampal neurons in murine cerebral malaria prevented by pentoxifylline. J Neurol Sci1993;114:20–4. [PubMed: 8433093]
20. Medana IM, Hunt NH, Chan-Ling T. Early activation of microglia in the pathogenesis of fatal murinecerebral malaria. Glia 1997;19:91–103. [PubMed: 9034826]
21. Medana IM, Hunt NH, Chaudhri G. Tumor necrosis factor-α expression in the brain during fatalmurine cerebral malaria: evidence for production by microglia and astrocytes. Am J Pathol1997;150:1473–86. [PubMed: 9095002]
22. Hunt NH, Grau GE. Cytokines: accelerators and brakes in the pathogenesis of cerebral malaria. TrendsImmunol 2003;24:491–9. [PubMed: 12967673]
23. Ennaceur A, Meliani K. A new one-trial test for neurobiological studies of memory in rats. III. Spatialvs. non-spatial working memory. Behav Brain Res 1992;51:83–92. [PubMed: 1482548]
24. Rogers DC, Fisher EM, Brown SD, Peters J, Hunter AJ, Martin JE. Behavioral and functional analysisof mouse phenotype: SHIRPA, a proposed protocol for comprehensive phenotype assessment.Mamm Genome 1997;8:711–3. [PubMed: 9321461]
25. Rogers DC, Peters J, Martin JE, et al. SHIRPA, a protocol for behavioral assessment: validation forlongitudinal study of neurological dysfunction in mice. Neurosci Lett 2001;306:89–92. [PubMed:11403965]
26. Sparkman NL, Buchanan JB, Heyen JRR, Chen J, Beverly JL, Johnson RW. Interleukin-6 facilitateslipopolysaccharide-induced disruption in working memory and expression of other proinflammatorycytokines in hippocampal neuronal cell layers. J Neurosci 2006;26:10709–16. [PubMed: 17050710]
27. Bessis A, Bechade C, Bernard D, Roumier A. Microglial control of neuronal death and synapticproperties. Glia 2007;55:233–8. [PubMed: 17106878]
28. Griffin R, Nally R, Nolan Y, McCartney Y, Linden J, Lynch MA. The age-related attenuation in long-term potentiation is associated with microglial activation. J Neurochem 2006;99:1263–72. [PubMed:16981890]
29. Kreutzberg GW. Microglia: a sensor of pathological events in the CNS. Trends Neurosci1996;19:312–8. [PubMed: 8843599]
30. Medana IM, Chan-Ling T, Hunt NH. Reactive changes of retinal microglia during fatal murinecerebral malaria: effects of dexamethasone and experimental permeabilization of the blood-brainbarrier. Am J Pathol 2000;156:1055–65. [PubMed: 10702421]
31. Kennan RP, Machado F, Lee SC, et al. Reduced cerebral blood flow and N-acetyl aspartate in a murinemodel of cerebral malaria. Parasitol Res 2005;96:302–7. [PubMed: 15918069]
32. Dantzer R. Cytokine, sickness behavior, and depression. Neurol Clin 2006;24:441–60. [PubMed:16877117]
Desruisseaux et al. Page 7
J Infect Dis. Author manuscript; available in PMC 2009 June 8.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

33. Wilson CJ, Finch CE, Cohen HJ. Cytokines and cognition—the case for a head-to-toe inflammatoryparadigm. J Am Geriatr Soc 2002;50:2041–56. [PubMed: 12473019]
34. Clark RE, Zola SM, Squire LR. Impaired recognition memory in rats after damage to the hippocampus.J Neurosci 2000;20:8853–60. [PubMed: 11102494]
35. Wood ER, Mumby DG, Pinel JP, Phillips AG. Impaired object recognition memory in rats followingischemia-induced damage to the hippocampus. Behav Neurosci 1993;107:51–62. [PubMed:8447957]
36. Zola SM, Squire LR, Teng E, Stefanacci L, Buffalo EA, Clark RE. Impaired recognition memory inmonkeys after damage limited to the hippocampal region. J Neurosci 2000;20:451–63. [PubMed:10627621]
37. Hammond RS, Tull LE, Stackman RW. On the delay-dependent involvement of the hippocampus inobject recognition memory. Neurobiol Learn Mem 2004;82:26–34. [PubMed: 15183168]
38. Malkova L, Mishkin M. One-trial memory for object-place associations after separate lesions ofhippocampus and posterior parahippocampal region in the monkey. J Neurosci 2003;23:1956–65.[PubMed: 12629201]
39. Wan H, Aggleton JP, Brown MW. Different contributions of the hippocampus and perirhinal cortexto recognition memory. J Neurosci 1999;19:1142–8. [PubMed: 9920675]
40. Abe H, Ishida Y, Iwasaki T. Perirhinal N-methyl-D-aspartate and muscarinic systems participate inobject recognition in rats. Neurosci Lett 2004;356:191–4. [PubMed: 15036627]
41. Aggleton JP, Brown MW. Episodic memory, amnesia, and the hippocampal-anterior thalamic axis.Behav Brain Sci 1999;22:425–44. [PubMed: 11301518]
42. Eacott MJ, Gaffan EA. The roles of perirhinal cortex, postrhinal cortex and the fornix in memory forobjects, contexts, and events in the rat. Q J Exp Psychol B 2005;58:202–17. [PubMed: 16194965]
43. Vnek N, Rothblat L. The hippocampus and long-term object memory in the rat. J Neurosci1996;16:2780–7. [PubMed: 8786453]
44. Puma C, Deschaux O, Molimard R, Bizot JC. Nicotine improves memory in an object recognitiontask in rats. Eur Neuropsychopharmacol 1999;9:323–7. [PubMed: 10422893]
45. Ennaceur A, Aggleton JP. Spontaneous recognition of object configurations in rats: effects of fornixlesions. Exp Brain Res 1994;100:85–92. [PubMed: 7813656]
46. Bussey TJ, Duck J, Muir JL, Aggleton JP. Distinct patterns of behavioural impairments resultingfrom fornix transection or neurotoxic lesions of the perirhinal and postrhinal cortices in the rat. BehavBrain Res 2000;111:187–202. [PubMed: 10840144]
47. Warburton EC, Baird AL, Morgan A, Muir JL, Aggleton JP. Disconnecting hippocampal projectionsto the anterior thalamus produces deficits on tests of spatial memory in rats. Eur J Neurosci2000;12:1714–26. [PubMed: 10792449]
48. Caterini F, Della Sala S, Spinnler H, Stangalino C, Tumbull OH. Object recognition and objectorientation in Alzheimer’s disease. Neuropsychology 2002;16:146–55. [PubMed: 11949706]
49. Doniger GM, Foxe JJ, Murray MM, Higgins BA, Javitt DC. Impaired visual object recognition anddorsal/ventral stream interaction in schizophrenia. Arch Gen Psychiatry 2002;59:1011–20. [PubMed:12418934]
50. Laatu S, Revonsuo A, Jaykka H, Portin R, Rinne JO. Visual object recognition in early Alzheimer’sdisease: deficits in semantic processing. Acta Neurol Scand 2003;108:82–9. [PubMed: 12859283]
Desruisseaux et al. Page 8
J Infect Dis. Author manuscript; available in PMC 2009 June 8.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Figure 1.A, Infected mice performed worse in the object recognition test of working memory by 7 daysafter infection (P < .01). Infected animals had significantly worse visual memory than thenoninfected control. Infected mice and control mice demonstrated similar levels of novel objectexploration (B) and locomotor activity (C).
Desruisseaux et al. Page 9
J Infect Dis. Author manuscript; available in PMC 2009 June 8.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Figure 2.A, Hematoxylin-eosin staining of cerebellum of an infected mouse demonstrating dilatedvessels with leukocyte margination (arrowheads) and an area of microhemorrhage (arrow).B, Ionized calcium-binding adaptor molecule 1 (Iba-1) immunostaining of the same cerebellarregion, demonstrating Iba-1 positive leukocytes (arrows) and activated microglial cells(arrowheads).
Desruisseaux et al. Page 10
J Infect Dis. Author manuscript; available in PMC 2009 June 8.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
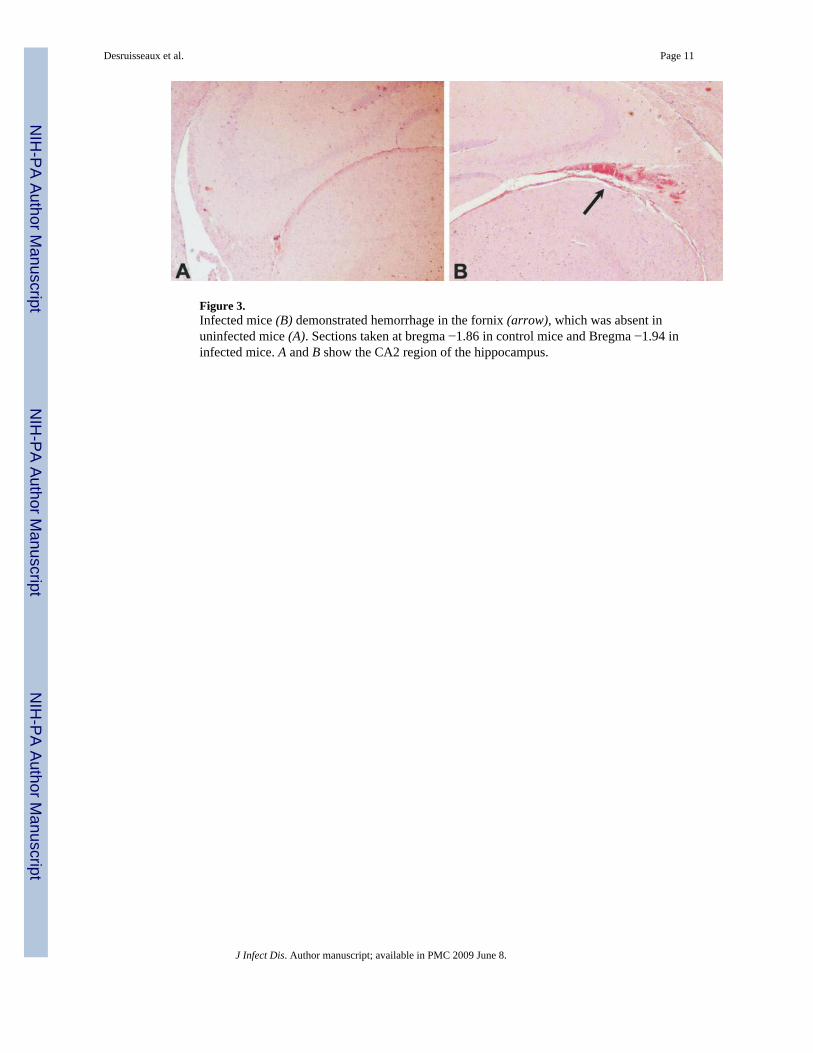
Figure 3.Infected mice (B) demonstrated hemorrhage in the fornix (arrow), which was absent inuninfected mice (A). Sections taken at bregma −1.86 in control mice and Bregma −1.94 ininfected mice. A and B show the CA2 region of the hippocampus.
Desruisseaux et al. Page 11
J Infect Dis. Author manuscript; available in PMC 2009 June 8.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Figure 4.Ionized calcium-binding adaptor molecule 1 (Iba-1) staining of the cortex and hippocampusof uninfected control mice and infected mice. In the cortex (A) and hippocampus (C) of controlmice, Iba-1–positive microglial cells are small with delicate fine processes (arrows). In thecortex (B) and hippocampus (D) of infected mice, microglial cells undergo morphologicalchanges that reflect activation, such as thickening of the processes and strong Iba-1 reactivity(arrows).
Desruisseaux et al. Page 12
J Infect Dis. Author manuscript; available in PMC 2009 June 8.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript




















