
Genetic structure of Plasmodium vivax and ... - eScholarship

Molecular Microbiology (2004)
51
(4), 1039–1049 doi:10.1046/j.1365-2958.2003.03895.x
© 2003 Blackwell Publishing Ltd
Blackwell Science, LtdOxford, UKMMIMolecular Microbiology1365-2958Blackwell Publishing Ltd, 200351
410391049
Original Article
Mapping P. falciparum binding sites on ICAM-1M. T. Tse
et al
.
Accepted 15 October, 2003. *For correspondence. [email protected] and [email protected]; Tel. (+44) 151 705 3161and (+91) 11 2618 7695; Fax (+44) 151 705 3371 and (+91) 11 26162316.
†
Present address: University of Connecticut Health Center,School of Medicine, Farmington, CT 06030, USA.
‡
These authorscontributed equally to this work.
Divergent binding sites on intercellular adhesion molecule-1 (ICAM-1) for variant
Plasmodium falciparum
isolates
Man Tsuey Tse,
1‡
Kausik Chakrabarti,
2†‡
Carolyn Gray,
1
Chetan E. Chitnis
2
* and Alister Craig
1
*
1
Liverpool School of Tropical Medicine, Pembroke Place, Liverpool L3 5QA, UK.
2
International Center for Genetic Engineering and Biotechnology, Aruna Asaf Ali Marg, New Delhi 110067, India.
Summary
Adhesion of human erythrocytes infected with themalaria parasite
Plasmodium falciparum
to hostendothelium has been associated with severe formsof this disease. A number of endothelial receptorshave been identified, and there is evidence that oneof these, intercellular adhesion molecule-1 (ICAM-1),may play an important role in the pathology of cere-bral malaria. Mutagenesis of domain 1 of ICAM-1,which is involved in parasite adhesion, shows that thebinding sites for different parasite variants overlap toa large extent, but that there are subtle differencesbetween them that correlate with their adhesive phe-notypes. This suggests that the ability to bind toICAM-1 has arisen from a common variant, but thatsubsequent changes have led to differences in bind-ing avidity, which may affect pathogenesis. Thedefinition of common binding determinants and theelucidation of links between ICAM-1 binding pheno-type and disease will provide new leads in the designof therapeutic interventions.
Introduction
The human malaria parasite,
Plasmodium falciparum
, isresponsible for around one million deaths every year,mainly in sub-Saharan Africa, and the spread of resis-tance to affordable drugs has resulted in an increase in
malaria-specific mortality over the past 10 years (Snow
et al
., 2001).
P. falciparum
is unique among the humanmalarias in undergoing a process of sequestration,whereby infected erythrocytes are withdrawn from theperipheral circulation through adhesion to post-capillaryvenular endothelium. This process of adhesion has beenassociated with severe disease, for example parasiteaccumulation in the brain and cerebral malaria (CM)(Turner, 1997). The molecular mechanisms underlyingdisease are not understood, but parasite ligands and hostreceptors for the primary adhesive event have been char-acterized. A variant protein on the infected erythrocyte(IE) surface known as
P. falciparum
erythrocyte mem-brane protein 1 (PfEMP1) contains the binding sites for arange of endothelial receptors (Kyes
et al
., 2001). Thisprotein is encoded by a family of genes known as
var
(Baruch
et al
., 1995; Smith
et al
., 1995), and switchingbetween members of this gene family has been associ-ated with changes in antigenicity and adherence pheno-type (Biggs
et al
., 1992; Roberts
et al
., 1992; Smith
et al
.,1995). On the host side, there are a large number ofreceptors (for a review, see Craig and Scherf, 2001), anda recent study has suggested that the ability of parasitesto bind to multiple receptors is correlated with diseaseseverity (Heddini
et al
., 2001). Intercellular adhesion mol-ecule-1 (ICAM-1) has been shown to mediate IE adhesion(Berendt
et al
., 1989) and, with CD36 (Barnwell
et al
.,1989; Ockenhouse
et al
., 1989; Oquendo
et al
., 1989),appears to be responsible for the majority of adhesionseen in isolates from children (Newbold
et al
., 1997). Sev-eral lines of evidence have implicated ICAM-1 as havinga role in severe disease: (i) adhesion to ICAM-1 washigher in isolates from patients with clinical malaria dis-ease and tended to be higher in those with CM (Newbold
et al
., 1997); (ii) ICAM-1 in brain vessels co-localizes withsequestered parasites in autopsy samples from peopledying from CM (Turner
et al
., 1994); (iii) ICAM-1 expres-sion on endothelium is upregulated during malaria infec-tion (Turner
et al
., 1998); and (iv) a genetic polymorphismin the N-terminal immunoglobulin (Ig)-like domain ofICAM-1 is associated with severe disease (Fernandez-Reyes
et al
., 1997). We have also shown recently thatICAM-1 can play a critical role in recruiting IE from flow(Gray
et al
., 2003) and, along with others (McCormick

1040
M. T. Tse
et al.
© 2003 Blackwell Publishing Ltd,
Molecular Microbiology
,
51
, 1039–1049
et al
., 1997; Yipp
et al
., 2000), have shown that ICAM-1and CD36 work synergistically in mediating adhesion toendothelium.
ICAM-1 is an 80–114 kDa variably glycosylated mem-brane glycoprotein consisting of five Ig-like domains thatis expressed on the surface of a wide range of cell types(for a review, see van de Stolpe and van der Saag, 1996).The binding site for IE has been characterized previouslyin two separate studies (Berendt
et al
., 1992; Ockenhouse
et al
., 1992a) and appears to involve the BED ‘side’ of theN-terminal Ig-like domain (Fig. 1). This site is distinct, butshows some overlap with the binding sites for LFA-1 andhuman rhinovirus, and has a high level of overlap with thebinding site for fibrinogen. The two studies used differentICAM-1-binding parasite lines ITO4 and ItG-ICAM, whichhave different avidities for ICAM-1 (Duperray
et al
., 1997),and different panels of ICAM-1 mutants, but both impli-cated regions close to each other on the crystal structureat positions G15, L18, T20 and L43 (Berendt
et al
., 1992;Ockenhouse
et al
., 1992a). Later work showed that a nat-
ural polymorphism at position 29 in the BC loop, replacinga lysine residue with methionine, could also affect IE bind-ing (Adams
et al
., 2000; Craig
et al
., 2000). Therefore, wetargeted residues G14 to L30 and K39 to K50 for alaninereplacement mutagenesis (Cunningham and Wells,1993), covering much of the BED ‘side’ of the ICAM-1molecule, and tested the resulting mutant proteins for theirability to bind erythrocytes infected with three differentICAM-1-binding parasite lines under static and flow con-ditions. Our results show that the binding site for IE islocated on the BED side of the ICAM-1 molecule for allthree parasite lines tested. Importantly, however, the rep-ertoire of ICAM-1 mutants bound by the three parasitelines differed, indicating subtle differences in the contactresidues used by different parasite lines.
Results
Characterization of mutant proteins
An important aspect of functional studies using mutagen-esis is a screen for major disruption of conformation of themutated protein. We used a panel of seven, conformation-ally sensitive, monoclonal antibodies (mAbs) to the N-terminal domain of human ICAM-1, which had beenmapped to different regions of this domain, as probes forcorrect folding (Table 1). Of the 25 mutant proteins usedin this study, only two (C25/A and P28/A) had lost morethan two mAb epitopes. For the C25/A mutant, thisresulted in a complete abrogation of binding of all parasitelines under all conditions but, unusually, the P28/A mutantwas still able to bind IE, indicating conservation of at leastpart of the structure. It is interesting to note that, althoughmutation of the cysteine at position 25 resulted in a lossof all mAb epitopes, changing the cysteine at position 21had no effect on conformation. Mutant proteins not listedhad no significant epitope loss.
Static adhesion assays
Binding of the three parasites lines to different ICAM-1mutant proteins under static conditions is shown as a
Fig. 1.
The crystal structure of the N-terminal domain of human ICAM-1, showing the binding sites for LFA-1, fibrinogen and
P. falci-parum
-infected erythrocytes. Binding to human rhinovirus involves residues distributed over the whole Ig domain, as this ‘docks’ into the canyon formed by the viral proteins VP1, 2 and 3. The strands of the
b
-barrel are labelled A to G. Some residues have been numbered for reference.
Table 1.
Loss of anti-ICAM-1 mAb epitopes in ICAM-1 mutantproteins.
Mutant protein mAb epitopes lost
a
V17/A 15.2C25/A RR1/1, My13, 15.2, 170/6, BBA4, 7.5C2P28/A RR1/1, My13, 15.2, 170/6K29/M BBA4, 7.5C2L42/A RR1/1, 15.2G46/A My13
a.
Epitope loss is defined as
<
20% reactivity as measured by ELISAcompared with reactivity with ICAM-1
ref
; isotype-matched irrelevantmAbs were used as negative controls.
Mapping
P. falciparum
binding sites on ICAM-1
1041
© 2003 Blackwell Publishing Ltd,
Molecular Microbiology
,
51
, 1039–1049
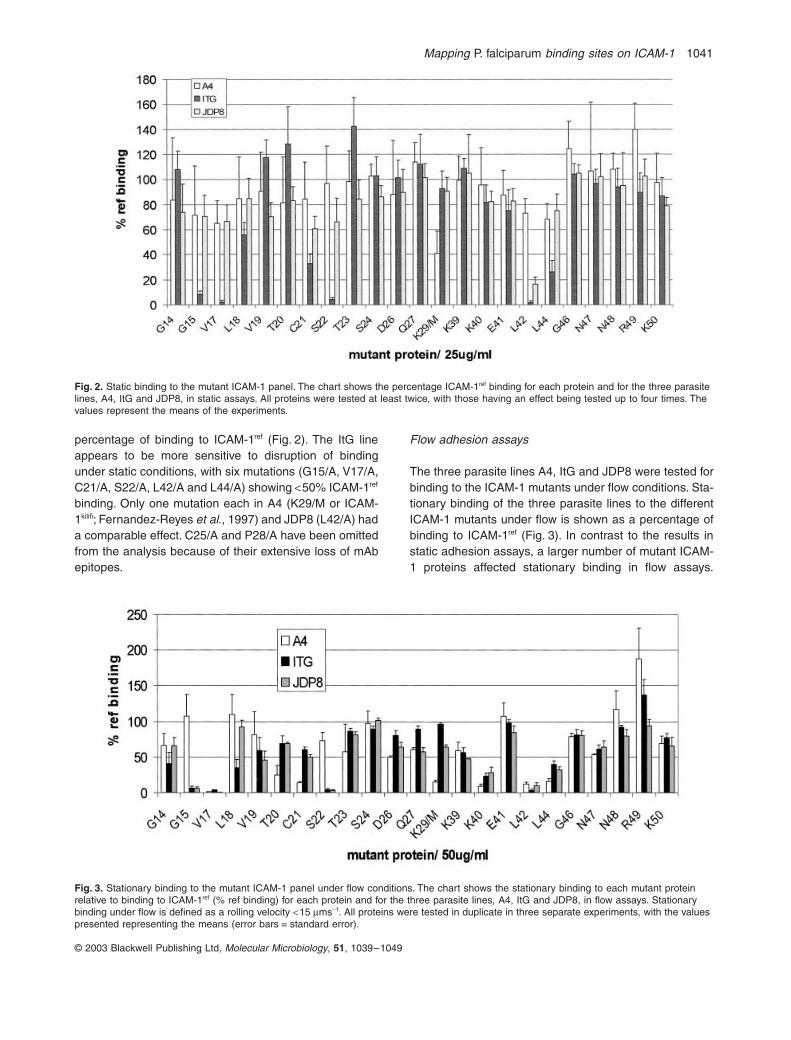
percentage of binding to ICAM-1
ref
(Fig. 2). The ItG lineappears to be more sensitive to disruption of bindingunder static conditions, with six mutations (G15/A, V17/A,C21/A, S22/A, L42/A and L44/A) showing
<
50% ICAM-1
ref
binding. Only one mutation each in A4 (K29/M or ICAM-1
kilifi
; Fernandez-Reyes
et al
., 1997) and JDP8 (L42/A) hada comparable effect. C25/A and P28/A have been omittedfrom the analysis because of their extensive loss of mAbepitopes.
Flow adhesion assays
The three parasite lines A4, ItG and JDP8 were tested forbinding to the ICAM-1 mutants under flow conditions. Sta-tionary binding of the three parasite lines to the differentICAM-1 mutants under flow is shown as a percentage ofbinding to ICAM-1
ref
(Fig. 3). In contrast to the results instatic adhesion assays, a larger number of mutant ICAM-1 proteins affected stationary binding in flow assays.
Fig. 2.
Static binding to the mutant ICAM-1 panel. The chart shows the percentage ICAM-1
ref
binding for each protein and for the three parasite lines, A4, ItG and JDP8, in static assays. All proteins were tested at least twice, with those having an effect being tested up to four times. The values represent the means of the experiments.
Fig. 3.
Stationary binding to the mutant ICAM-1 panel under flow conditions. The chart shows the stationary binding to each mutant protein relative to binding to ICAM-1
ref
(% ref binding) for each protein and for the three parasite lines, A4, ItG and JDP8, in flow assays. Stationary binding under flow is defined as a rolling velocity
<
15
m
ms
-
1
. All proteins were tested in duplicate in three separate experiments, with the values presented representing the means (error bars = standard error).

1042
M. T. Tse
et al.
© 2003 Blackwell Publishing Ltd,
Molecular Microbiology
,
51
, 1039–1049
Seven ICAM-1 mutations caused a reduction of
>
50% inA4 (V17/A, T20/A, C21/A, K29/M, K40/A, L42/A and L44/A) and ItG adhesion (G15/A, V17/A, L18/A, S22/A, K40/A, L42/A and L44/A), whereas eight mutants reducedJDP8 adhesion by
>
50% (G15/A, V17/A, V19/A, S22/A,K39/A, K40/A, L42/A and L44/A). This higher number ofmutants affecting binding under flow conditions reflectsthe higher sensitivity of the flow-based assay and may bemore relevant in physiological terms, as there is someevidence to suggest that one of the major roles of ICAM-1 in IE adhesion is the initiation of tethers to host endot-helium under flow (Gray
et al
., 2003). The only ICAM-1mutant to increase binding was R49/A, particularly withA4 and, to a lesser extent, with ItG. Increases in mAb
binding as a result of mutations in ICAM-1 have beenreported previously (Berendt
et al
., 1992) and, althoughrarer than reductions in biological interactions, it is notinconceivable that a mutation near the binding site mightact to increase access of the IE to its receptor.
The distribution of velocities for IE rolling on mutantICAM-1 proteins is shown in Fig. 4. Although many ofthese show similar trends to the results seen in stationarycells (Fig. 3), a number of them indicate more subtleeffects of the mutation. For example, the ItG/L44 resultshows decreased stationary adhesion and a matchingincrease in the rolling velocities, indicating a generalreduction in binding in both populations of IE, whereasJDP8/L18 has no effect on the stationary binding but has
Fig. 4.
Velocity distributions of infected erythrocytes rolling on mutant ICAM-1 proteins. Cells with rolling velocities of at least 15
m
ms
-
1
are plotted for the mutant proteins. Each dot represents a single parasitized red blood cell for which the rolling velocity was calculated from the videotape of the flow assay. The points for each protein represent the velocities measured over 1 min in six different areas of the microslide. The mean velocity is shown as a horizontal line.

Mapping
P. falciparum
binding sites on ICAM-1
1043
© 2003 Blackwell Publishing Ltd,
Molecular Microbiology
,
51
, 1039–1049
clearly shifted the distribution of rolling velocities upwards,suggesting a decrease in binding that is only seen in thispopulation of cells. Other situations are harder to interpret,such as ItG/S22, where there is a complete loss of sta-tionary adhesion but rolling adhesion is hardly affected.Uncoupling of the effects on stationary and rolling adhe-sion in malaria have been reported previously in a studyexamining the effect of src family kinase inhibitors onadhesion under flow (Yipp
et al
., 2003), although not viaICAM-1.
Variant parasite lines show differences in their adhesion to ICAM-1
In order to simplify the binding data for analysis, we havecoded them into ‘no difference’, ‘minor decrease’, ‘moder-ate decrease’ and ‘major decrease’ (Table 2). A compari-son of the effects of different mutations on the adhesionof the three variant parasite lines shows that, althoughthey share common binding regions (e.g. K40/A, L42/Aand L44/A), they also have differences in their contactresidues. The pattern produced for strain ItG agrees withthe results of Ockenhouse
et al
. (1992a), which showedthat the region G14 to L18 was important for adhesion,but the use of more sensitive flow-based assays alsoshows that the DE loop is implicated in binding of this
parasite line to ICAM-1. The similar pattern of mutationsaffecting the binding of ItG and the Indian isolate JDP8(Okoyeh
et al
., 1999) may be linked to their shared high-avidity type binding to ICAM-1 (Gray
et al
., 2003; Chatto-padhyay
et al
., 2004). Although A4 had some residues incommon with ItG and JDP8, several differences wereseen (T20/A and K29/M), and A4 also differs from ItG andJDP8 in having a low-avidity ICAM-1-binding phenotype.Although there are differences between the ways in whichthe parasite lines interact with the mutant proteins, onlytwo ICAM-1 mutants showed a major effect in one parasiteline and not in at least one of the other lines, namely A4/K29 and A4/R49. This suggests that the ability of IE tobind to ICAM-1 has a common origin, with subsequentchanges in PfEMP1 altering the quantitative nature of theinteraction. This is highlighted by the fact that all threeparasite strains bound residues on
b
sheets B and D aswell as DE loop of ICAM-1 (Fig. 5). However, A4 alonebound residues on the BC loop of ICAM-1 (Fig. 5).
Discussion
Mutagenesis using alanine replacements for residues inthe binding region on ICAM-1 for
P. falciparum
-infectederythrocytes has allowed a detailed analysis of adhesionto this host receptor with three antigenically distinct,
Table 2.
Summary of binding results.
Results for flow assays are for stationary adhesion.

1044
M. T. Tse
et al.
© 2003 Blackwell Publishing Ltd,
Molecular Microbiology
,
51
, 1039–1049
ICAM-1-binding parasite lines. Using a panel of mutantICAM-1 proteins, we have been able to segregate thesethree isolates on the basis of disruption of adhesion bydifferent alanine replacements. These results show thatvariant parasite lines have differences in their contactresidues on the BED side of the molecule, as defined bythe effect of mutations on their adhesive phenotype.
The association between antigenic type and cytoadher-ence has been known for some time (Biggs
et al
., 1992;Roberts
et al
., 1992) and is thought to be largely mediatedby the parasite-derived protein PfEMP1 (Smith
et al
.,1995). This protein contains a number of domains thathave been shown to mediate interactions with specifichost receptors, including CD36 (Baruch
et al
., 1997),ICAM-1 (Smith
et al
., 2000) and CR1 (Rowe
et al
., 2000)among several others (for a review, see Craig and Scherf,2001). The exact nature of the parasite binding sites forthese host receptors is not known, as attempts to definea consensus have been impeded by the high degree ofsequence variation seen in these motifs (Smith
et al
.,2001). The N-terminal domain of ICAM-1 on the otherhand is highly conserved, with only one major mutation(ICAM-1
Kilifi
) detected in African populations (Fernandez-Reyes
et al
., 1997). IE have been shown to interact witha region of this molecule that is distinct from, but overlaps,
the binding sites for LFA-1, rhinovirus and fibrinogen (Ber-endt
et al
., 1992; Ockenhouse
et al
., 1992a; Duperray
et al
., 1997). Indeed, the ICAM-1
Kilifi
mutation (K29/M) hasbeen shown to have effects on all these adhesive pheno-types (Craig
et al
., 2000; A. Craig, unpublished observa-tions), placing it at an interesting cross-roads offunctionality. The interaction between ICAM-1 and IE hasbeen studied in some detail. As well as firm adhesion toICAM-1-bearing substrates, IE from some parasite linesalso demonstrate rolling behaviour (Cooke
et al
., 1994).This may be important in terms of capturing IE from thecirculation, and it is assumed that this interaction resultsfrom high kon and koff rates between ICAM-1 and PfEMP1.However the basis of the partition between stationary androlling adhesion on recombinant ICAM-1 protein is notknown, nor is the lack of binding of so-called ‘high-avidity’ICAM-1-binding lines to activated HUVEC (bearing highlevels of ICAM-1 receptor) (Gray et al., 2003). Previousdata have segregated ICAM-1-binding IE into high- andlow-avidity binders (Craig et al., 1997), represented in thiswork by parasite lines ItG and A4 respectively. JDP8 is arecently cultured patient isolate from Madhya Pradesh,India, that binds ICAM-1 with similar avidity to ItG but hasrelatively low binding to CD36 (Gray et al., 2003; Chatto-padhyay et al., 2004). Using a panel of mutant ICAM-1
Fig. 5. Schematic representation of mutations having a major effect on IE binding to ICAM-1 mapped onto the three-dimensional structure of the N-terminal domain of ICAM-1. The positions of mutations having a major effect (<25% reference binding) on IE adhesion under static or flow assay conditions are marked with a circular dark blue zone.

Mapping P. falciparum binding sites on ICAM-1 1045
© 2003 Blackwell Publishing Ltd, Molecular Microbiology, 51, 1039–1049
proteins, we have been able to segregate these threeisolates on the basis of disruption of adhesion by differentalanine replacements. As expected, flow-based adhesionassays are a more sensitive technique in definingresponses to mutations, but even static assays are ableto differentiate between parasite lines (see below).
The similar binding behaviour of ItG and JDP8 wasreflected in the distribution of mutations affecting station-ary binding under flow conditions for both parasite lines(Fig. 5). A4 shared a number of common mutations withItG and JDP8 that reduce binding to ICAM-1, but also hadsome exclusive mutations that only affected its binding.Given the gross differences in binding of A4 and ItG/JDP8to ICAM-1, it is tempting to speculate that the molecularbasis of this differential binding is due to variation in thecontact residues between ICAM-1 and PfEMP1, the par-asite ligand that mediates the interaction. This situation issimilar to that seen in human rhinovirus (HRV), in whichcryoelectron microscopic reconstructions and comparisonwith X-ray crystal structure have shown subtle differencesin the molecular interactions between ICAM-1 and theviral ‘canyon’, defined by proteins VP1, VP2 and VP3 intwo major serotypes, HRV14 and HRV16 (Kolatkar et al.,1999). As in malaria, the pathogenic consequences ofthese differences is not understood, but clear phenotypicdifferences exist in terms of ICAM-1 binding betweenHRV14 and HRV16.
In addition to the major differences between A4 and ItG/JDP8 (Fig. 5), there are a number of mutations thatshowed a differential effect on binding between ItG andJDP8. The clearest demonstration of this comes fromstatic assays (Fig. 5), with ItG showing much greater sen-sitivity to mutations than JDP8. Closer inspection of thedata reveals that these differences are quantitative ratherthan simply ‘binds’ or ‘does not bind’. It is rare for a majoreffect to be observed without some measurable effectbeing detected in another parasite line, the only case herebeing A4/K29. This suggests that the overall interactionbetween ICAM-1 and PfEMP1 is similar between variantsand is indicative of a common origin of ICAM-1 bindingfor P. falciparum, at least within the isolates examined inthis study. Despite this, it is relatively easy to design apanel of mutant proteins that would discriminate betweenA4, ItG and JDP8; for example, a set of assays understatic and flow conditions using only two mutant proteins,S22/A and K29/M, would give definitive results. The useof this type of analysis on a larger scale for typing patientisolates in a field study would allow the definition of differ-ent ICAM-1-binding types and could be important, as aprevious study has identified the ability of patient isolatesto bind to ICAM-1 as being associated with clinical dis-ease (Newbold et al., 1997). Other results (C. Newbold,personal communication) indicate that isolates from CMpatients tended to have higher ICAM-1 binding (‘high avid-
ity’), so it is possible that stratification of the ICAM-1-binding isolates into subtypes might have produced asignificant correlation between adhesive phenotype andCM. This information could also facilitate the elucidationof the mechanisms underlying severe disease, by provid-ing the molecular basis of interactions associated witheither mild disease or CM. The association betweenICAM-1 adhesion and severe disease needs to be treatedwith some caution as, although there is some evidencesupporting this, other data are contradictory. In oneproject looking at patient isolate adhesion profiles anddisease, ICAM-1 binding was lower in isolates from chil-dren with cerebral malaria (Rogerson et al., 1999), andgenetic studies have shown both neutral (Bellamy et al.,1998) and protective (Kun et al., 1999) associations withthe ICAM-1Kilifi polymorphism. Resolution of these issueswill require further examination of the adhesive behaviourof patient isolates from different disease categories.
Part of the rationale for mapping the sites of interactionbetween parasite ligands and host receptors is for thedevelopment of inhibitors of adhesion that may have ther-apeutic value as the ability to reverse adhesion could playa significant role in reducing severe malaria. The mosthighly developed work in antiadhesive therapies is in pla-cental malaria (Gysin et al., 1999) and cytoadherence viaCD36 (Yipp et al., 2002), in which the binding sites havebeen mapped, and soluble proteins based on these struc-tures have been shown to inhibit adhesion. For ICAM-1,it would clearly be desirable to have an inhibitor that wouldeither be specific for highly pathogenic ICAM-1-bindinglines or a pan-blocking molecule, able to interfere with alladhesion via ICAM-1. Our study has shown that the latterroute might be possible using inhibitors based on the L42loop (DE loop), as the L42/A mutation has a major effectunder flow conditions on all three parasite lines. Indeed,this information has been used to screen a library ofcompounds as mimeotopes for the L42 loop structure insilico, producing a lead compound able to block adhesionof A4 and ItG to ICAM-1 protein (A. Craig, manuscript inpreparation). In addition to small-molecule inhibitors, thedefinition of the molecular interaction between ICAM-1and PfEMP1 and the possible subcategorization of bind-ing and progression to severe disease should also allowthe development of a vaccine able to reduce disease (byblocking cytoadherence) while still permitting parasite car-riage and the development of naturally acquired immunity.
ICAM-1 is thought to exist as a dimer on the cell surface(Reilly et al., 1995) with the interface formed by majorcontacts in domain 2, plus the BED face of the N-terminaldomain (domain 1). Our previous work has also shownthat the ICAM-1-Fc protein also exists as a dimer (Craiget al., 1997). Thus, the binding surface for LFA-1 is com-pletely accessible on the outside of the dimer, whereasthe P. falciparum binding site is located in the centre. It is

1046 M. T. Tse et al.
© 2003 Blackwell Publishing Ltd, Molecular Microbiology, 51, 1039–1049
possible that the parasite uses the physical proximity ofthe two binding faces at the core of the dimer tostrengthen the interaction with PfEMP1, such that theparasite ligand sits within the ‘groove’ formed by the twoICAM-1 molecules. The broad nature of the P. falciparumbinding footprint would also facilitate a multistep bindingprocess, also seen in HRV (Kolatkar et al., 1999), whichcould potentially separate the processes involved in adhe-sion or even those of adhesion and signalling.
One of the characteristics of immunoglobulin super-family members that bind integrins (such as ICAM-1/LFA-1) is the presence of two disulphide bonds in the N-terminal domain (Wang and Springer, 1998), in contrastto the ‘normal’ arrangement of only one. The ‘extra’ disul-phide bond in ICAM-1 is located at the top of the domain,joining the BC loop to the F strand and includes thecysteine residue at position 25 (Fig. 1). The other disul-phide bond is located within the b-barrel and joinsstrands B and F, including the cysteine at position 21.Both these cysteines (C21 and C25) were replaced indi-vidually with alanine during this study, but only one ofthem, C25/A, resulted in a complete loss of conformationin the N-terminal domain, as shown by the loss of confor-mationally sensitive epitopes for a range of monoclonalantibodies (Table 1), with no mAb deficit recorded forC21/A. This result indicates that, although other interac-tions within the b-barrel are able to compensate for theloss of the C21 disulphide bridge, removal of the C25disulphide bond causes a major perturbation of thedomain structure. This is rather counterintuitive as itmight be expected that removal of this bond from a loopstructure (loop BC) would have little effect, but it is possi-ble that the correct orientation of this loop with respect tothe rest of the domain is important or that removal of C25may cause an incorrect pairing of the cysteine at the topof the F strand with C21. The involvement of the BC loopin the conformation of the N-terminal domain is strength-ened by the observation that the P28/A mutant alsoshows the loss of multiple mAb epitopes.
Using a panel of ICAM-1 mutant proteins, we haveshown that three different ICAM-1-binding parasite linesbind to a similar region of the ICAM-1 molecule, but thatdifferences exist in the contact residues used. These dif-ferences appear to correlate with the type of binding toICAM-1, with the low-avidity A4 having a markedly differ-ent ‘footprint’ compared with ItG and JDP8. Even the twohigh-avidity binders show subtle differences, with ItGbeing much more sensitive to the effect of mutations understatic assay conditions. The ability to subdivide ICAM-1adhesion of IE into different categories may contribute tothe definition of the mechanisms underlying progressionto severe disease, and thereby facilitate the design oftherapeutic interventions, either through specific inhibitorsdesigned to localized regions of the ICAM-1 molecule or
the transfer of contiguous, non-linear epitopes to a small-molecule framework (Gadek et al., 2002).
Experimental procedures
Parasite culture
Plasmodium falciparum parasites were grown in group O+
human erythrocytes using previously described conditions(Trager and Jensen, 1976). To minimize the effect of antigenicswitching in culture, a batch of stabilates was prepared fromICAM-1-selected cultures, which were serially thawed andmaintained in culture for not more than 3 weeks. Lines A4(Roberts et al., 1992) and ItG (Ockenhouse et al., 1992b) areboth from the IT lineage and are of Brazilian origin. JDP8(Okoyeh et al., 1999; Chattopadhyay et al., 2003) is arecently identified patient isolate from Jagdalpur, MadhyaPradesh, India.
ICAM-1 mutagenesis and protein expression
Single amino acid substitutions were introduced into thehuman ICAM-1 cDNA, cloned as an IgG1-Fc chimera(Craig et al., 1997), using the incorporation of mismatchedmutagenic oligonucleotide primers (QuikChange, Strat-agene). A list of the primers used in this study is shown inTable 3.
Mutagenesis was attempted on all residues from G14 toL30 and K39 to K50 but, despite repeated attempts, it wasnot possible to produce some mutants. This included K29/A,and so the natural mutation K29/M, ICAM-1Kilifi, was used forthe study. DNA minipreps (Qiagen) were sequenced toscreen for the correct mutations [and lack of other, poly-merase chain reaction (PCR)-induced, non-specific muta-tions], and successful constructs were resequenced aftermaxipreps of plasmid DNA for transfection had beenproduced.
Expression of the ICAM-1-Fc constructs was carried outessentially as described previously (Craig et al., 1997) bytransfection of COS7 cells using Fugene-6 (Roche) accord-ing to the manufacturer’s instructions. Each protein wastested for structural integrity by enzyme-linked immunosor-bent assay (ELISA) using a panel of six mAbs that hadpreviously been mapped to the N-terminal domain [RR1/1,My13, 15.2 (Serotec), 170/6 (G. Turner), BBA4 (R and DSystems) and 7.5C2 (D. Haskard)] and two mAbs from theC-terminal domain [GP89-14 and GP89-23 (J. Johnson)],used as reporters of protein concentration.
Parasite adhesion assays
Static protein-binding assays were performed as describedpreviously (Berendt et al., 1992) using an ICAM-1-Fc spottingconcentration of 25 mg ml-1, which had previously been shownto be within the dynamic range for detecting differences inadhesion. All mutant proteins were screened twice, in dupli-cate dishes, with those showing an effect being included intwo further rounds of experiments. Data shown are the meanof all experiments. Flow assays on protein-coated microslideswere also performed using standard conditions (Cooke et al.,

Mapping P. falciparum binding sites on ICAM-1 1047
© 2003 Blackwell Publishing Ltd, Molecular Microbiology, 51, 1039–1049
1994), with a coating concentration of ICAM-1-Fc of 50 mgml-1. All proteins were tested in duplicate in three separateexperiments. Small variations in the concentrations of mutantproteins were corrected for using the ELISA results for GP89-14 and GP89-23. All results are expressed as percentagebinding of the relevant parasite isolate to that seen on anequivalent concentration of ICAM-1ref. Statistical significancewas determined using the Tamhane T-2 test, with differencesregarded as significant if P < 0.05.
Results have been placed into three main groups (plus oneexample of an increase in binding) (Table 2) to aid interpre-tation of the results. Owing to the inherent variability of bind-ing assays, some results that would normally have beencoded as a moderate decrease were not statistically signifi-cant and have been left as ‘no difference’. All ‘major decrease’results were highly significant.
Acknowledgements
We would like to thank Gareth Turner (170/6), Judith Johnson(GP89-14 and -23) and Dorian Haskard (7.5C2) for their kindgifts of monoclonal antibodies, C. R. Pillai for P. falciparumfield isolate JDP8, Ian Hastings for advice on statistical anal-ysis, and Jordi Bella and R. Craig for help with Figs 1 and 5.This work was supported by grants from the Wellcome Trust(A.C. and C.C.) and Framework 5 of the European Union(QLK2/CT/2000/00109). C.C. is an International SeniorResearch Fellow of The Wellcome Trust, UK, and Interna-tional Research Scholar of the Howard Hughes Medical Insti-tute, USA.
References
Adams, S., Turner, G.D., Nash, G.B., Micklem, K., Newbold,C.I., and Craig, A.G. (2000) Differential binding of clonal
variants of Plasmodium falciparum to allelic forms of intra-cellular adhesion molecule 1 determined by flow adhesionassay. Infect Immun 68: 264–269.
Barnwell, J.W., Asch, A.S., Nachman, R.L., Yamaya, M.,Aikawa, M., and Ingravallo, P. (1989) A human 88-kDmembrane glycoprotein (CD36) functions in vitro as areceptor for a cytoadherence ligand on Plasmodium falci-parum-infected erythrocytes. J Clin Invest 84: 765–772.
Baruch, D.I., Pasloske, B.L., Singh, H.B., Bi, X., Ma, X.C.,Feldman, M., et al. (1995) Cloning the P. falciparum geneencoding PfEMP1, a malarial variant antigen and adher-ence receptor on the surface of parasitized human eryth-rocytes. Cell 82: 77–87.
Baruch, D.I., Ma, X.C., Singh, H.B., Bi, X., Pasloske, B.L.,and Howard, R.J. (1997) Identification of a region ofPfEMP1 that mediates adherence of Plasmodium falci-parum infected erythrocytes to CD36: conserved functionwith variant sequence. Blood 90: 3766–3775.
Bellamy, R., Kwiatkowski, D., and Hill. A.V.S. (1998)Absence of an association between intercellular adhesionmolecule 1, complement receptor 1 and interleukin 1receptor antagonist gene polymorphisms and severemalaria in a West African population. Trans R Soc Trop MedHyg 92: 312–316.
Berendt, A.R., Simmons, D.L., Tansey, J., Newbold, C.I., andMarsh, K. (1989) Intercellular adhesion molecule-1 is anendothelial cell adhesion receptor for Plasmodium falci-parum. Nature 341: 57–59.
Berendt, A.R., McDowall, A., Craig, A.G., Bates, P.A., Stern-berg, M.J., Marsh, K., et al. (1992) The binding site onICAM-1 for Plasmodium falciparum-infected erythrocytesoverlaps, but is distinct from, the LFA-1-binding site. Cell68: 71–81.
Biggs, B.A., Anders, R.F., Dillon, H.E., Davern, K.M., Martin,M., Petersen, C., and Brown, G.V. (1992) Adherence ofinfected erythrocytes to venular endothelium selects for
Mutation Primer 5¢-3¢ (forward primer)
G14/A GTC ATC CTG CCC CGG GCA GGC TCC GTG CTG GTGG15/A ATC CTG CCC CGG GGA GCC TCC GTG CTG GTG ACAV17/A CCC CGG GGA GGC TCC GCG CTG GTG ACA TGC AGC ACL18/A CGG GGA GGC TCC GTG GCA GTG ACA TGC AGC ACCV19/A GGA GGC TCC GTG CTG GCG ACA TGC AGC ACC TCCT20/A GGC TCC GTG CTG GTG GCA TGC AGC ACC TCC TGTC21/A TCC GTG CTG GTG ACA GCC AGC ACC TCC TGT GACS22/A GTG CTG GTG ACA TGC GCC ACC TCC TGT GAC CAGT23/A CTG GTG ACA TGC AGC GCC TCC TGT GAC CAG CCCS24/A GTG ACA TGC AGC ACC GCC TGT GAC CAG CCC AAGC25/A ACA TGC AGC ACC TCC GCT GAC CAG CCC AAG TTGD26/A TGC AGC ACC TCC TGT GCA CAG CCC AAG TTG TTGQ27/A AGC ACC TCC TGT GAC GCG CCC AAG TTG TTG GGC AP28/A ACC TCC TGT GAC CAG GCC AAG TTG TTG GGC ATA GK39/A GAG ACC CCG TTG CCT GCA AAG GAG TTG CTC CTGK40/A ACC CCG TTG CCT AAA GCG GAG TTG CTC CTG CCTE41/A CCG TTG CCT AAA AAG GCG TTG CTC CTG CCT GGGL42/A TTG CCT AAA AAG GAG GCG CTC CTG CCT GGG AACL44/A AAA AAG GAG TTG CTC GCG CCT GGG AAC AAC CGGG46/A GAG TTG CTC CTG CCT GCG AAC AAC CGG AAG GTGN47/A TTG CTC CTG CCT GGG GCC AAC CGG AAG GTG TAT GN48/A CTC CTG CCT GGG AAC GCC CGG AAG GTG TAT GAAR49/A CTG CCT GGG AAC AAC GCG AAG GTG TAT GAA CTGK50/A CCT GGG AAC AAC CGG GCG GTG TAT GAA CTG AGC A
Table 3. Oligonucleotides used formutagenesis.

1048 M. T. Tse et al.
© 2003 Blackwell Publishing Ltd, Molecular Microbiology, 51, 1039–1049
antigenic variants of Plasmodium falciparum. J Immunol149: 2047–2054.
Chattopadhyay, R., Taneja, T., Chakrabarti, K., Pillai, C.R.,and Chitnis, C.E. (2004) Molecular analysis of the cytoad-herence phenotype of a Plasmodium falciparum fieldisolate that binds intercellular adhesion molecule-1. MolBiochem Parasitol (in press).
Cooke, B.M., Berendt, A.R., Craig, A.G., MacGregor, J.,Newbold, C.I., and Nash, G.B. (1994) Rolling and station-ary cytoadhesion of red blood cells parasitized by Plasmo-dium falciparum: separate roles for ICAM-1, CD36 andthrombospondin. Br J Haematol 87: 162–170.
Craig, A., and Scherf, A. (2001) Molecules on the surface ofthe Plasmodium falciparum infected erythrocyte and theirrole in malaria pathogenesis and immune evasion. MolBiochem Parasitol 115: 129–143.
Craig, A.G., Pinches, R., Khan, S., Roberts, D.J., Turner,G.D., Newbold, C.I., and Berendt, A.R. (1997) Failure toblock adhesion of Plasmodium falciparum-infected eryth-rocytes to ICAM-1 with soluble ICAM-1. Infect Immun 65:4580–4585.
Craig, A., Fernandez-Reyes, D., Mesri, M., McDowall, A.,Altieri, D.C., Hogg, N., and Newbold, C. (2000) A functionalanalysis of a natural variant of intercellular adhesionmolecule-1 (ICAM-1Kilifi). Hum Mol Genet 9: 525–530.
Cunningham, B.C., and Wells, J.A. (1993) Comparison of astructural and a functional epitope. J Mol Biol 234: 554–563.
Duperray, A., Languino, L.R., Plescia, J., McDowall, A.,Hogg, N., Craig, A.G., et al. (1997) Molecular identificationof a novel fibrinogen binding site on the first domain ofICAM-1 regulating leukocyte-endothelium bridging. J BiolChem 272: 435–441.
Fernandez-Reyes, D., Craig, A.G., Kyes, S.A., Peshu, N.,Snow, R.W., Berendt, A.R., et al. (1997) A high frequencyAfrican coding polymorphism in the N-terminal domain ofICAM-1 predisposing to cerebral malaria in Kenya. HumMol Genet 6: 1357–1360.
Gadek, T.R., Burdick, D.J., McDowell, R.S., Stanley, M.S.,Marsters, J.C., Jr, Paris, K.J., et al. (2002) Generation ofan LFA-1 antagonist by the transfer of the ICAM-1 immu-noregulatory epitope to a small molecule. Science 295:1086–1089.
Gray, C., McCormick, C., Turner, G., and Craig, A. (2003)ICAM-1 can play a major role in mediating P. falciparumadhesion to endothelium under flow. Mol Biochem Parasi-tol 128: 187–193.
Gysin, J., Pouvelle, B., Fievet, N., Scherf, A., and Lepolard,C. (1999) Ex vivo desequestration of Plasmodiumfalciparum-infected erythrocytes from human placenta bychondroitin sulfate A. Infect Immun 67: 6596–6602.
Heddini, A., Pettersson, F., Kai, O., Shafi, J., Obiero, J.,Chen, Q., et al. (2001) Fresh isolates from children withsevere Plasmodium falciparum malaria bind to multiplereceptors. Infect Immun 69: 5849–5856.
Kolatkar, P.R., Bella, J., Olson, N.H., Bator, C.M., Baker,T.S., and Rossmann, M.G. (1999) Structural studies of tworhinovirus serotypes complexed with fragments of their cel-lular receptor. EMBO J 18: 6249–6259.
Kun, J.F., Klabunde, J., Lell, B., Luckner, D., Alpers, M., May,J., et al. (1999) Association of the ICAM-1Kilifi mutation
with protection against severe malaria in Lambarene,Gabon. Am J Trop Med Hyg 61: 776–779.
Kyes, S., Horrocks, P., and Newbold, C. (2001) Antigenicvariation at the infected red cell surface in malaria. AnnuRev Microbiol 55: 673–707.
McCormick, C.J., Craig, A., Roberts, D., Newbold, C.I., andBerendt, A.R. (1997) Intercellular adhesion molecule-1 andCD36 synergize to mediate adherence of Plasmodiumfalciparum-infected erythrocytes to cultured humanmicrovascular endothelial cells. J Clin Invest 100: 2521–2529.
Newbold, C., Warn, P., Black, G., Berendt, A., Craig, A.,Snow, B., et al. (1997) Receptor-specific adhesion andclinical disease in Plasmodium falciparum. Am J Trop MedHyg 57: 389–398.
Ockenhouse, C.F., Tandon, N.N., Magowan, C., Jamieson,G.A., and Chulay, J.D. (1989) Identification of a plateletmembrane glycoprotein as a falciparum malaria seques-tration receptor. Science 243: 1469–1471.
Ockenhouse, C.F., Betageri, R., Springer, T.A., and Staun-ton, D.E. (1992a) Plasmodium falciparum-infected erythro-cytes bind ICAM-1 at a site distinct from LFA-1, Mac-1, andhuman rhinovirus. Cell 68: 63–69.
Ockenhouse, C.F., Tegoshi, T., Maeno, Y., Benjamin, C., Ho,M., Kan, K.E., et al. (1992b) Human vascular endothelialcell adhesion receptors for Plasmodium falciparum-infected erythrocytes: roles for endothelial leukocyte adhe-sion molecule 1 and vascular cell adhesion molecule 1. JExp Med 176: 1183–1189.
Okoyeh, J.N., Pillai, C.R., and Chitnis, C.E. (1999) Plasmo-dium falciparum field isolates commonly use erythrocyteinvasion pathways that are independent of sialic acid res-idues of glycophorin A. Infect Immun 67: 5784–5791.
Oquendo, P., Hundt, E., Lawler, J., and Seed, B. (1989)CD36 directly mediates cytoadherence of Plasmodium fal-ciparum parasitized erythrocytes. Cell 58: 95–101.
Reilly, P.L., Woska, J.R., Jr, Jeanfavre, D.D., McNally, E.,Rothlein, R., and Bormann, B.J. (1995) The native struc-ture of intercellular adhesion molecule-1 (ICAM-1) is adimer. Correlation with binding to LFA-1. J Immunol 155:529–532.
Roberts, D.J., Craig, A.G., Berendt, A.R., Pinches, R., Nash,G., Marsh, K., and Newbold, C.I. (1992) Rapid switchingto multiple antigenic and adhesive phenotypes in malaria.Nature 357: 689–692.
Rogerson, S.J., Tembenu, R., Dobano, C., Plitt, S., Taylor,T.E., and Molyneux, M.E. (1999) Cytoadherence charac-teristics of Plasmodium falciparum-infected erythrocytesfrom Malawian children with severe and uncomplicatedmalaria. Am J Trop Med Hyg 61: 467–472.
Rowe, J.A., Rogerson, S.J., Raza, A., Moulds, J.M., Kaza-tchkine, M.D., Marsh, K., et al. (2000) Mapping of theregion of complement receptor (CR) 1 required for Plas-modium falciparum rosetting and demonstration of theimportance of CR1 in rosetting in field isolates. J Immunol165: 6341–6346.
Smith, J.D., Chitnis, C.E., Craig, A.G., Roberts, D.J.,Hudson-Taylor, D.E., Peterson, D.S., et al. (1995) Switchesin expression of Plasmodium falciparum var genes corre-late with changes in antigenic and cytoadherent pheno-types of infected erythrocytes. Cell 82: 101–110.

Mapping P. falciparum binding sites on ICAM-1 1049
© 2003 Blackwell Publishing Ltd, Molecular Microbiology, 51, 1039–1049
Smith, J.D., Craig, A.G., Kriek, N., Hudson-Taylor, D., Kyes,S., Fagen, T., et al. (2000) Identification of a Plasmodiumfalciparum intercellular adhesion molecule-1 bindingdomain: a parasite adhesion trait implicated in cerebralmalaria. Proc Natl Acad Sci USA 97: 1766–1771.
Smith, J.D., Gamain, B., Baruch, D.I., and Kyes, S. (2001)Decoding the language of var genes and Plasmodium fal-ciparum sequestration. Trends Parasitol 17: 538–545.
Snow, R.W., Trape, J.F., and Marsh, K. (2001) The past,present and future of childhood malaria mortality in Africa.Trends Parasitol 17: 593–597.
van de Stolpe, A., and van der Saag, P.T. (1996) Intercellularadhesion molecule-1. J Mol Med 74: 13–33.
Trager, W., and Jensen, J.B. (1976) Human malaria parasitesin continuous culture. Science 193: 673–675.
Turner, G. (1997) Cerebral malaria. Brain Pathol 7: 569–582.Turner, G.D., Morrison, H., Jones, M., Davis, T.M., Looaree-
suwan, S., Buley, I.D., et al. (1994) An immunohistochem-ical study of the pathology of fatal malaria. Evidence forwidespread endothelial activation and a potential role forintercellular adhesion molecule-1 in cerebral sequestra-tion. Am J Pathol 145: 1057–1069.
Turner, G.D., Ly, V.C., Nguyen, T.H., Tran, T.H., Nguyen,
H.P., Bethell, D., et al. (1998) Systemic endothelial activa-tion occurs in both mild and severe malaria. Correlatingdermal microvascular endothelial cell phenotype and solu-ble cell adhesion molecules with disease severity. Am JPathol 152: 1477–1487.
Wang, J., and Springer, T.A. (1998) Structural specializationsof immunoglobulin superfamily members for adhesion tointegrins and viruses. Immunol Rev 163: 197–215.
Yipp, B.G., Anand, S., Schollaardt, T., Patel, K.D., Looaree-suwan, S., and Ho, M. (2000) Synergism of multiple adhe-sion molecules in mediating cytoadherence of Plasmodiumfalciparum-infected erythrocytes to microvascular endothe-lial cells under flow. Blood 96: 2292–2298.
Yipp, B.G., Baruch, D.I., Brady, C., Murray, A.G., Looaree-suwan, S., Kubes, P., and Ho, M. (2002) RecombinantPfEMP1 peptide inhibits and reverses cytoadherence ofclinical Plasmodium falciparum isolates in vivo. Blood 101:331–337.
Yipp, B.G., Robbins, S.M., Resek, M.E., Baruch, D.I., Looa-reesuwan, S., and Ho, M. (2003) Src-family kinase signal-ing modulates the adhesion of Plasmodium falciparum onhuman microvascular endothelium under flow. Blood 101:2850–2857.
Copyright © 2022 FDOKUMEN




















