
ChemInform Abstract: The Biosynthesis of Polyketide-Derived Polycyclic Ethers
Upload
independentCategory
view
0download
0
Photoinduced Ultrafast Heterogeneous Electron Transfer atMolecule−Semiconductor InterfacesJesus Nieto-Pescador, Baxter Abraham, and Lars Gundlach*
Department of Chemistry & Biochemistry and Department of Physics & Astronomy, University of Delaware, 109 Lammot DuPont,Newark, Delaware 19711, United States
ABSTRACT: This Perspective discusses recent developments inultrafast electron transfer dynamics at interfaces between organic andinorganic materials. Heterogeneous electron transfer (HET) is a keyprocess in important fields like catalysis and solar energy conversion.Furthermore, the solid state nature of the systems gives control overrelevant parameters and allows for investigating excited state dynamicsand electron transfer processes in unprecedented detail. Progress insynthesis, sample preparation, and instrumentation makes it possible toprovide experimental proof of recent prediction from theory concerningthe adiabaticity of the reaction and the influence of coherence. A shortrecapitulation of the field is followed by a discussion of recentexperimental efforts that allowed for studying HET, particularlyfocusing on the influence of energetics and vibrational dynamics.
In heterogeneous electron transfer (HET) an electron istransferred between a molecular species and a solid state
electrode. This process is important for many applications inelectrochemistry, chemical sensing, and energy conversion. Inparticular, photoinduced HET where the electron is donatedfrom an electronic excited state is of great interest because of itsimportance for solar energy conversion. Earlier applicationsincluded xerography and companies like Xerox, Kodak, andPolaroid focused especially on ET between molecules andpolymers for application in photocopying machines. ThisPerspective starts with a short recapitulation of the field thatdoes not claim to be exhaustive. However, many excellentreviews of HET dynamics have been published and are citedhere. In the early days, research was focused on revealing ifphotosensitization results in energy or electron transfer.Photocurrent measurements on merocyanine sensitized CdSsuggested that this sensitization is in fact electron transfer andnot energy transfer.1 The net result of a photodriven redoxreaction gives relative little information about the primaryelectron transfer reaction and its quantum yield because reverseelectron transfer competes with the reaction. However, undercertain circumstances the reverse reaction can be minimized inheterogeneous systems. Such systems have been used toinvestigate photoinduced ET by measuring photo currentdensities.2 Early research focused mostly on the energetics andthe alignment of molecular states and semiconducting bands.3,4
First, time-resolved measurements were performed in the 1970sand 80s using flash photolysis on the μs time scale to measurehomogeneous ET between quinones.5 A short time later,electron transfer was reported between an organic moleculeand semiconducting TiO2 nanoparticles on the nanosecond timescale.6 Picosecond time-resolved energy transfer betweenorganic molecules and organic molecular and inorganic crystals
were measured in the early 1990s using pulsed dye lasers.7,8 HETis, in general, faster than intra- or intermolecular ET.Consequently, time-resolved investigations of HET requiredthe development of subpicosecond techniques. The rapiddevelopment of ultrafast optics in the late 80s made HETreactions accessible to time-resolved spectroscopic techniquesand gave “ultrafast HET” its name. The number of ultrafast time-resolved studies increased shortly after Gratzel’s group publishedthe first papers on solar energy conversion employingRuthenium sensitized TiO2 nanoparticles9 and again afterpublication of the first 12% cell in 1991.10 Gratzel’s discoverywas made in the same year the first commercial Ti:sapphire lasersbecame available and femtosecond measurements could beperformed on a regular basis more easily compared to pumpeddye lasers that were used until then to study interfacial chargetransfer in the few picosecond regime. In the late 1990s, a wealthof femtosecond time-resolved measurements of interfacialelectron transfer were performed, most of them using TiO2 asthe semiconducting electrode.11−15 The present Perspectivefocuses on fundamental experimental studies of the kinetics ofHET.Systems for photoinduced heterogeneous electron transfer
consist in general of a light absorber that is photoexcited and anelectrode that acts as an acceptor for the excited electron. Thephotoexcited electron is transferred to the acceptor leaving theabsorber in an oxidized state and the electron residing in theelectrode. Conventionally, the absorber is an organic or metal−organic molecule, also called dye, that is chemically bound to theelectrode with some sort of anchor group. However, other light
Received: July 22, 2014Accepted: September 25, 2014Published: September 25, 2014
Perspective
pubs.acs.org/JPCL
© 2014 American Chemical Society 3498 dx.doi.org/10.1021/jz501541a | J. Phys. Chem. Lett. 2014, 5, 3498−3507

absorbing materials like nanoparticles have been employedrecently.16 The electrode is usually made from metals orsemiconductors. For generating a long-lived charge separatedstate, a semiconducting electrode is preferable. Usually, asemiconductor with a band gap larger than the absorption ofthe dye is used for preventing direct photoexcitation of thesemiconductor. Themost important requirement for HET is thatthe excited dye state aligns energetically with empty states in theelectrode. This situation results in themost basic picture for HETshown in Figure 1a. The gray areas on the left constitute theconduction and valence bands of the semiconducting electrode.On the right-hand side, ground and excited state of the dyemolecule are shown.Almost all early time-resolved studies of HET were performed
on colloidal semiconductor films for two reasons. First, the mostprominent application, that is, the Gratzel cell, employs suchfilms. Second, the optical methods that were used demandedhigh optical density that can only be achieved by high surface areabecause the covalent nature of the dye/electrode bond restrictsthe dye coverage to less than one monolayer. These studies werevery successful in explaining the first steps of charge separationthat happens in the Gratzel cell, for example, the dynamics of themetal-to-ligand charge transfer state in ruthenium dyes. HET ofruthenium dyes or similar metal−organic sensitizers wasintensively studied because these dyes resulted in the highestconversion efficiencies.11,17,18 However, the colloidal films thatwere used differed greatly in composition and morphology. Forexample, a commonly used commercial TiO2 nanocrystal(Degussa, P25) is a mixture of anatase and rutile. Also, theresults of the most commonly used preparation procedure10 candiffer greatly. For example, it has been found that the sodiumcontent of the glass substrate used for depositing the colloidalfilm influenced electron transfer dynamics considerably.19 Inaddition to problems related to the colloidal film, the Ru dyes areproblematic for fundamental HET studies because they have verybroad absorption bands. This makes them well suited forapplication in solar cells; however, it complicates dynamicmeasurements tremendously because distinguishing contribu-tions from different species, that is, excited state, triplet state, andcation, is very involved due to the spectral overlap.17
Consequently, measurements of electron transfer kinetics arestrongly dependent on specific material properties andexperimental conditions and transfer rates often represent time
scales for a process rather than the exact rate constants. Thesemeasurements showed that already in the N3/TiO2 system thefastest processes happen on the sub-100-fs time scale. Despitetheir long history, Ru dyes are still actively studied and theircomplex intramolecular energy and electron dynamics and HETbehavior still reveal new features like sub-10-fs injection fromnonthermalized dye states.20
With the availability of shorter laser pulses HET systems withstronger electronic coupling and shorter ET times could bestudied. Because of the stronger interaction between the donorand acceptor state, the influence of inhomogeneities in thesystems like nearby surface defects or changes in the compositionof the semiconductor are less pronounced.21 In addition,sensitizers like perylene,13 alizarin,22 and bi-isonicotinic acid23
have simple electronic structures compared to Ru dyes with itspronounced metal to ligand charge transfer and highly efficientintersystem crossing. These systems have the additionaladvantage that they are accessible to ab initio calculationsbecause of the relatively small number of atoms.24,25 Chargetransfer complexes formed by enediol/TiO2 systems give rise tovery strong donor/acceptor coupling to the point were theexcited state is predominantly localized on the Ti 3d states, cf.Figure 1b. Resolving these time scales is still challenging.26
However, such systems are very useful for understanding detailsof the HET process. An intensively studied model system for thiscase is catechol bound to TiO2. TiO2 sensitized with catecholshows the same charge transfer band in ground state absorptionthat has been observed for a catechol/Ti complex in solution,27
indicating the formation of a surface charge-transfer complex.28
In this limiting case, the distinction between excited state andcharge separated state is not possible anymore and the reactioncan be characterized as photoexcitation in the catechol/Ticomplex rather than as adiabatic HET between catechol andTiO2. Enediol sensitizers with slightly weaker coupling to Ti canin principle show a combination of both pathways and can beused for studying electronic coherence in ET by measuringinterference between both pathways (Figure 1b). Because theHET pathway is absent for the catechol/TiO2 system, it allowsfor studying short-lived intermediate states at the surface of thesemiconductor without the influence of molecular states.29
Recently, a similar reaction pathway has been proposed for HETfrom a metal nanoparticle to TiO2.
30
Figure 1. Schematic alignment of the electronic levels with respect to the semiconductor band structure and reaction pathway for two cases discussed inthe text. (a) Representation of the most basic ET reaction for a weakly coupled molecule. In the absence of electronic/vibrational coupling, HETproceeds iso-energetic. (b) In the case of strong coupling between donor and acceptor state, direct ET is possible and in the intermediate regime bothpathways can interfere.
The Journal of Physical Chemistry Letters Perspective
dx.doi.org/10.1021/jz501541a | J. Phys. Chem. Lett. 2014, 5, 3498−35073499

Apart from its importance for technological applications, HETconstitutes a unique test platform for the intrinsic factors thatgovern chemical reactions. The influence of conformationalchanges, vibrational degrees of freedom, and coherence effectscan be studied by following ultrafast photoinduced reactions.The solid-state nature of the system allows for a high degree oforder and for exclusion of solvation effects. The knowledgegained from these studies is not restricted to photochemistry butgives insight into all processes that involve bond breaking andformation in general chemical reactions.A system that proved to be very suitable for fundamental HET
studies using femtosecond time-resolved spectroscopy is theperylene/TiO2 system. First, because absorption of the differentintermediate species, that is, excited state and cation, are wellseparated from the ground state absorption and they are locatedin the visible region.31 Second, the fluorescence lifetime is on theorder of several ns and contributions from triplet states can beneglected. Third, the HOMO−LUMO gap is several 100 meVsmaller than the band gap of TiO2 allowing for selectiveexcitation of the dye molecule. And last, the electronic couplingbetween perylene and TiO2 can be adjusted by employingdifferent bridge/anchor group combinations. Using perylenederivatives as sensitizers for TiO2, a large number of studies havebeen performed. Among others, HET temperature dependencewas measured,13 vibrational wave packet motion has beenstudied,31 the influence of different bridge groups on HET hasbeen investigated,32 and the influence of the anchor group wasstudied.33 However, the above investigations were all performedusing colloidal films as substrates. As mentioned above, HET inthese films has the disadvantage that dynamics is influenced byinhomogeneities, in particular on longer time scales. This hasbeen shown by comparing measurements on colloidal films withthose employing single-crystal surfaces of TiO2 as substrates forlarge weakly coupled perylene derivatives. The electron transferdynamics was strongly influenced by the presence of adjacentcolloids on time scales above 70 fs, and it was concluded thatmeasurements of slow dynamics in these films have to be takenwith care.21 To circumvent the sensitivity problem that occurswhen studying submonolayer coverage of the dye sensitizer onTiO2 single crystal surfaces, two-photon photoelectron spec-troscopy (2PPE) has been employed for these measurements.This approach turned out to be very successful and allowedamong others for measuring the electronic coupling dependentHET times,34 the orientation of the molecules on the surface,35
and the ET spectrum of the injected electrons.36 Both systems,the colloidal as well as the single crystal system, were also used asmodel systems for several theoretical studies on differentcomputational levels, employing parametrized fully quantummechanical models,37,38 combinations of first principle electronicand time-dependent Hartree calculations,39,40 and DFT/TDDFT calculations.41−43 After giving an overview of thegeneral properties of HET, we will summarize the experimentalwork that involved the unique perylene/TiO2 system and discussthem in light of results gained from different theoretical modelsand proceed with an overview of recent published and
unpublished experiments that mainly address energy depend-ence of HET. Strong focus will also be placed on the vibrational-electronic coupling. This Perspective gives an outlook ofexperimental and theoretical challenges that are faced whenstudying fundamental aspects of HET.The most crucial points that are not well understood in
heterogeneous ET to date are
1. the distinction between adiabatic and nonadiabatic HET,including possible electronic coherence between theexcited donor and the acceptor state.
2. the influence of vibrational degrees of freedom, includingtheir coherence.
3. the influence of the available density of states for reactantand product that includes short-lived intermediates nearthe surface.
Recent results from our group and collaborators that areaddressing points 2 and 3 will be discussed here. Experimentalwork that addresses the first item is rare. However, there areseveral theoretical approaches that focus on the transitionbetween the nonadiabatic and the adiabatic regime in ET.44
General properties of HET.When studying the basic properties ofHET the parameters for the process have to be defined as well aspossible. In general, the observables can be expressed by a simplereaction scheme
ω ω* + ⇒ + ++ −D A k D A k( ) ( ) ( ) ( ( ) e ) (1)
The reactants are the vibrational distribution of occupied exciteddonor statesD*(ω) and the distribution of empty acceptor statesin the semiconductor A(k), the products are the vibrationaldistribution of the oxidized donor D+(ω), and the distribution ofthe electron among the available acceptor states, includingphonon modes A(k) + e−. It is well established that the strengthof the diabatic coupling, that is, the splitting between adiabaticpotentials, dictates the applicable model. For weak adiabaticcoupling, a nonadiabatic picture is appropriate and perturbativetreatment of the adiabatic coupling leads to a Fermi’s golden ruleresult. For strong adiabatic coupling the non-Born−Oppen-heimer coupling is small and can be treated perturbatively.45
Although, a Fermi golden rule expression is only valid in thenonadiabatic limit, the expression helps to define the relevantparameters. Following Nitzan’s notation, the rate can beexpressed as46
π=ℏ
k V2
et D A,
2
(2)
Where VD,A is the strength of the coupling between donor andacceptor potential energy surface (PES) and is the Franck−Condon weighted density of states, which includes thereorganization of the nuclear configuration. In case of strongcoupling donor and acceptor, PES form a single adiabatic state.The height of the barrier E* (the free energy of activation)between donor and acceptor configuration and the relaxationtime from the donor configuration to the acceptor configurationTr are the factors that are governing the transition rate. AdiabaticHET in general is assumed to show Arrhenius type temperaturedependence
∝ − *k
TE
k T1
expETr B (3)
For nonadiabatic (NA) coupling the kinetics of the reactionare dominated by the electronic coupling between the donor and
The influence of conformationalchanges, vibrational degrees offreedom, and coherence effectscan be studied by following
ultrafast photoinduced reactions.
The Journal of Physical Chemistry Letters Perspective
dx.doi.org/10.1021/jz501541a | J. Phys. Chem. Lett. 2014, 5, 3498−35073500
all acceptor states, that is, the overlap of the respective states, andby the density of acceptor states. During one oscillation of theexcited state wave packet every crossing point (red circles inFigure 2a) between the reactant PES and all product PES is
probed and transitions occur with the respective transmissionprobability. In the case of adiabatic HET, the distribution ofavailable states is reflected in the barrier height (Figure 2a). In thecase the donor is positioned well above the conduction bandedge such that the whole ET spectrum can be accommodated inempty states below the donor level (referred to as the wide bandlimit) for every energy of the donor state, there is in general anacceptor state that upon mixing with the donor state results in anear barrierless transition. Thus, reactant and product state sharethe same adiabatic PES (Figure 2 b) . In this case, the exponentialterm in eq 3 approaches unity and the temperature dependence isexpected to be negligible. Thus, the temperature dependence ofthe reaction can not be used to distinguish between nonadiabaticand adiabatic reactions. Because nonadiabatic HET can beultrafast in the wide band limit because of the large amount ofavailable acceptor states, the rate of ET is also not a sufficientindicator for the adiabaticity of the reaction. One property thatcan be used to distinguish between both regimes is the excitationof high-energy vibrational modes during ET that indicates theinvolvement of Franck−Condon factors in the reaction andcharacterizes a nonadiabatic pathway.To study the influence of the different parameters on the ET
dynamics it is necessary, first, to choose a system that allowsaccess to the different distributions and their dynamics. Second, asystem is needed that allows for changing the relevantparameters, that is, electronic coupling and distribution ofavailable states, individually. Finally, alternative pathways for thereaction like intramolecular charge transfer, internal conversion,or intersystem crossing have to be excluded. Changing theenergetic position of donor and acceptor states, their density, andtheir coupling individually, without simultaneously changing thesurface reconstruction, binding geometry, and coupling betweenelectronic and vibrational (phonon) modes, is very challenging.In addition, the environment of the molecule should be kept assimple as possible. For such measurements, single crystals arepreferred over colloidal films, and vacuum environment oversolvent.Summary of Earlier Results Gained f rom the Perylene/TiO2 System.Recent approaches for measuring HET rates as a function ofelectronic coupling and energetic position are discussed next.The combination of a perylene dye anchored to single crystalTiO2 discussed above fulfills most of the above-mentioned
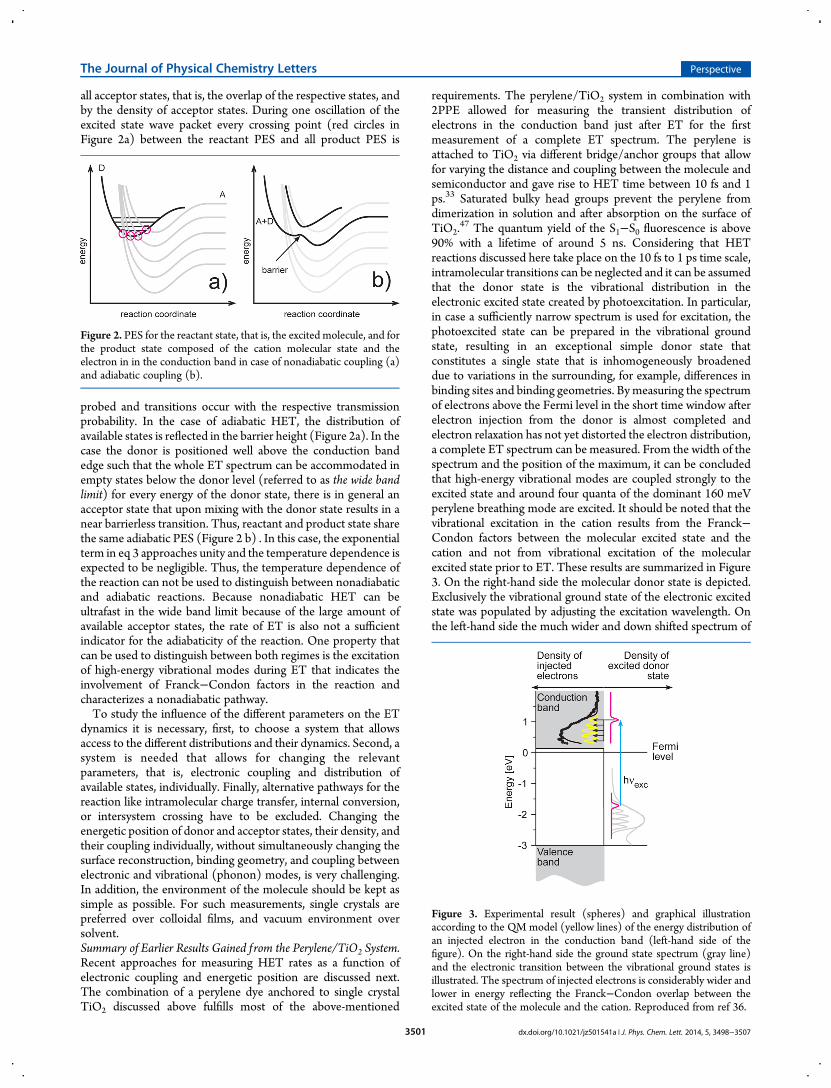
requirements. The perylene/TiO2 system in combination with2PPE allowed for measuring the transient distribution ofelectrons in the conduction band just after ET for the firstmeasurement of a complete ET spectrum. The perylene isattached to TiO2 via different bridge/anchor groups that allowfor varying the distance and coupling between the molecule andsemiconductor and gave rise to HET time between 10 fs and 1ps.33 Saturated bulky head groups prevent the perylene fromdimerization in solution and after absorption on the surface ofTiO2.
47 The quantum yield of the S1−S0 fluorescence is above90% with a lifetime of around 5 ns. Considering that HETreactions discussed here take place on the 10 fs to 1 ps time scale,intramolecular transitions can be neglected and it can be assumedthat the donor state is the vibrational distribution in theelectronic excited state created by photoexcitation. In particular,in case a sufficiently narrow spectrum is used for excitation, thephotoexcited state can be prepared in the vibrational groundstate, resulting in an exceptional simple donor state thatconstitutes a single state that is inhomogeneously broadeneddue to variations in the surrounding, for example, differences inbinding sites and binding geometries. Bymeasuring the spectrumof electrons above the Fermi level in the short time window afterelectron injection from the donor is almost completed andelectron relaxation has not yet distorted the electron distribution,a complete ET spectrum can be measured. From the width of thespectrum and the position of the maximum, it can be concludedthat high-energy vibrational modes are coupled strongly to theexcited state and around four quanta of the dominant 160 meVperylene breathing mode are excited. It should be noted that thevibrational excitation in the cation results from the Franck−Condon factors between the molecular excited state and thecation and not from vibrational excitation of the molecularexcited state prior to ET. These results are summarized in Figure3. On the right-hand side the molecular donor state is depicted.Exclusively the vibrational ground state of the electronic excitedstate was populated by adjusting the excitation wavelength. Onthe left-hand side the much wider and down shifted spectrum of
Figure 2. PES for the reactant state, that is, the excited molecule, and forthe product state composed of the cation molecular state and theelectron in in the conduction band in case of nonadiabatic coupling (a)and adiabatic coupling (b).
Figure 3. Experimental result (spheres) and graphical illustrationaccording to the QM model (yellow lines) of the energy distribution ofan injected electron in the conduction band (left-hand side of thefigure). On the right-hand side the ground state spectrum (gray line)and the electronic transition between the vibrational ground states isillustrated. The spectrum of injected electrons is considerably wider andlower in energy reflecting the Franck−Condon overlap between theexcited state of the molecule and the cation. Reproduced from ref 36.
The Journal of Physical Chemistry Letters Perspective
dx.doi.org/10.1021/jz501541a | J. Phys. Chem. Lett. 2014, 5, 3498−35073501

the injected electron is shown. This spectrum is the ET analogueto an optical absorption or emission spectrum where transitionprobabilities are modulated by Franck−Condon factors andelectronic excitation results in vibrational excitation. Thedominant mode that couples to HET is the same mode that isobserved in gas phase photoionization measurements. This canbe expected because both processes leave the molecule in acationic state.The fully quantummechanical (QM)model mentioned above
has been employed to relate steady-state absorption spectra ofthe adsorbed dye to lifetime broadening that results fromultrafast HET. Although the QM model is complete in the sensethat it includes vibrational modes, it depends on parameters anddoes not allow for first principle calculations. However, thegeneral results and predictions are very useful for understandingthe influence of vibrational modes on HET. The model has beenreviewed recently.48,49 It accounts for coupling betweenelectronic and high-energy vibrational states with energies largerthan kBT and predicts a Franck−Condon-like energy distributionfor the injected electron that is in agreement with theexperimental results shown in Figure 3. Figure 4 shows the
calculated time-dependent probability distribution versus energyfor an electron injected about 0.9 eV above the band edge into aquasicontinuum of acceptor states. A single vibrational modewith 0.1 eV energy was considered. The spacing of the peaks inthe energy dimension accounts for the vibrational mode and theintensity distribution represents the Franck−Condon overlapbetween the electronic excited molecular state and the cation.Along the time-axis vibrational coherence is observed as themodulation of the rising signal. The phase of these oscillations isgiven by the distance on the excited state PES along the reactioncoordinate between the vertically excited population and thecrossing point with the cation PES.The distribution in Figure 3 shows that for the perylene/TiO2
system the wide-band limit is valid. Therefore, this particularsystem gives little information about coherence betweenindividual levels in an energy integrated measurement becauseintegration over the whole spectrum will eliminate theoscillations in the injection yield. However, an energy andtime-resolved measurement can reveal the oscillations shown inFigure 4. The distribution will, in addition, be influenced by anonconstant density of states (DOS) and by an energy-dependent coupling. Measurements of the ET spectrum are
further complicated by the short-lived nature of the distributionthat thermalizes very rapidly into a Boltzmann distribution.Subsequently, cooling shifts the hot distribution toward the bandedge. The perylene/TiO2 system constitutes a good compromiseand allowed for measuring the ET spectrum for the first time.Limited energy resolution obscured details of the Franck−Condon progression in the measurement shown in Figure 3.Although this can be circumvented by employing a higherresolution detector, resolving the quantum beats betweendifferent states will be challenging due to the rapid dephasingin most solid state materials.
These measurements demonstrate how important the levelalignment between donor states and the electrode is. However, areliable method for measuring the level alignment with sufficientprecision to extract the number of vibrational quanta that can beinvolved in the ET reaction is often lacking. At the same time, itremains challenging to change the relative position of the donorstate and the acceptor state without changing other parameterslike the electronic−vibrational coupling in the molecule or thedensity of surface states of the semiconductor. One approach isto take different electronic or vibrational levels of the same dyemolecule as donor states. This approach requires that the ETreaction occurs on a shorter time scale than any intramolecularrelaxation processes, like vibrational relaxation, internal con-version, or intersystem crossing. In the case where differentelectronic levels are chosen as donor states, the tunability of theenergy is limited. However, this approach has the advantage thatthe properties of the electrode and molecule are unaltered.Varying Electron Excess Energy by Employing Dif ferent Sensitizers.Recently, the Piotrowiak group published measurements onazulene attached to TiO2.
50 Here, the vibrational ground state ofthe first excited state is located below the conduction band edgeand is not expected to give rise to electron injection. However,when the quantum yield for ET was measured as a function ofexcess vibrational energy a clear change in quantum yield wasobserved once sufficient vibrational energy was provided toposition the donor state in resonance with the high density ofacceptor states in the conduction band. Figure 5 shows a plot ofthe ET yield as a function of excitation energy. Excitation of thevibrational ground state occurs at 1.8 eV. Higher excitationenergies result in a vibrationally excited S1 state. At 2.1 eV theexcess vibrational energy is sufficient to shift the donor stateabove the conduction band edge. These results show that energy-dependent measurements can be performed by employing hotET and would benefit from dyes with slow IVR. It is remarkablethat even for the case where the donor state is located below theconduction band edge, the quantum yield for ET still reached 0.2.This indicates a high density of near surface states that can act aselectron acceptors. The importance of unoccupied states that areclose to the surface and distinguished from bulk like states bytheir amount of localization, and their energy has attracted moreattention in recent years. Initially, such states were mainlydiscussed in the context of defect states that are located in theband gap and act as trap states for injected electrons. However, itis well known that the surface band structure is distinguishedfrom the bulk band structure by breaking of symmetry and
Figure 4. Time-dependent probability distribution versus the energy ofa quasicontinuum whose width and spacing are similar to Figure 3. Theposition of the injecting molecular level in the energy band of thesubstrate is indicated as “ip”. A single harmonic vibrational mode ofenergy 0.1 eV and an overall transfer time of around 85 fs are consideredin the calculation. Reproduced from ref 49.
The perylene/TiO2 system consti-tutes a good compromise andallowed for measuring the ETspectrum for the first time.
The Journal of Physical Chemistry Letters Perspective
dx.doi.org/10.1021/jz501541a | J. Phys. Chem. Lett. 2014, 5, 3498−35073502

surface reconstruction even in case of defect free single crystalsurfaces. In addition, the covalent bound between the anchor ofthe molecule and the surface can introduce states that act asacceptor states for ET. Including these states into the ET processleads to a three step model, where the electron is first injectedinto states that are localized close to the surface followed by adelocalization into bulk-like states. A qualitative diagram for thelevel alignment in the azulene/TiO2 case is shown in Figure 6a.The black distribution on the right represents the vibrationalstructure of the donor state with its vibrational ground statelocated below the conduction band edge. The dark gray arearepresents the DOS of near surface intermediate states. Thepopulation in the intermediate near-surface states is inherentlydifficult to study with transient absorption (TA) signals becausethe transient change of absorption of the electrons after injectioninto semiconductor states is in general generated by transitionbetween many different states that all show different kinetics anddoes not allow for detailed analysis. One way to study theseprocesses is by employing time-resolved electron spectroscopythat probes the electron in the conduction band directly and
allows for measuring the time scale for the delocalization processdue to its inherent surface sensitivity. Such measurementsshowed that these intermediate near-surface states stronglyinfluence the dynamics. The time scale for the occupancy of thesestates is around 100 fs.29 This number agrees well with TDDFTsimulations for the initial delocalization of injected electrons intoTiO2. The influence of such “reaction intermediates” on HEThas been observed for various interfaces including ZrO2electrodes that show a larger band gap when compared toTiO2 and ZnO.51−53
Altering the energetic position of the donor state can also beachieved by selectively exciting different electronic states of thesame molecule. Recently, our group performed femtosecond−TAmeasurements after selectively exciting the Soret (S2) and Q-bands (S1) of porphyrin derivatives. This situation is illustrated inFigure 6b. Here, we briefly summarize preliminary unpublishedresults. The S2 state is known to be very short-lived due to rapidinternal conversion, which constitutes an effective pathwaycompeting with HET from S2. Consequently, it has beenchallenging to distinguish between the two pathways in thepast.54 In addition, the kinetics of competing intramoleculartransitions have to be known in detail. Porphyrins have beenextensively studied. Their electronic properties are well knownand several time-resolved studies have been published.54,55
Recently, we measured HET from a fluorinated free-basephlorin56 into TiO2 attached via a carboxylic acid group. Bycomparing solution based measurements of the intramolecularrelaxation dynamics and measurements of the molecule attachedto TiO2 we were able to distinguish between IC and HET fromthe S2 state and compare the dynamics with HET from the S1state. Surprisingly, it turned out that the electron transfer timesfrom both states are very similar (Figure 7). They are not affectedby the more than 1 eV difference in energetic position.Unfortunately, a conclusive measurement of the level alignmentin these systems is challenging. The major reason is the wide
Figure 5. Dependence of Φinjection, the quantum yield of electroninjection, on the energy of the incident photons measured on azulenesensitized TiO2. Reproduced from ref 50.
Figure 6. Schematic alignment of the electronic and vibrational levels with respect to the semiconductor band structure. (a) Representation of the casewhere the vibrational ground state of the donor is located below the band edge as realized in the azulene/TiO2 system. (b) Representation of the case ofexcitation into higher excited states as realized in the porphyrin/TiO2 system.
These results show that energy-dependent measurements can beperformed by employing hot ETand would benefit from dyes with
slow IVR.
The Journal of Physical Chemistry Letters Perspective
dx.doi.org/10.1021/jz501541a | J. Phys. Chem. Lett. 2014, 5, 3498−35073503

range of values the work function of TiO2 can take, ranging from4.4 to 5.5 eV.57 This introduces significant uncertainty in the levelalignment. However, a rough estimate can be gained from cyclicvoltametric measurements that suggest that the S1 state is locatedabout 1 eV above the band edge. This level alignment wouldresult in a large difference in bulk DOS for the two states that canact as electron acceptors. Calculations of the anatase (101)surface suggest that the density of empty states peaks at around700meV above the conduction band edge and decreases stronglytoward higher energies. This maximum in the conduction bandDOS results mainly fromTi 3d states and can be observed in bulkand surface DOS calculations for rutile and anatase crystals of allorientations. It can be assumed that the wide band limit is alreadyvalid for the S1 state. The similarity in ET rate for states that are inresonance with a significant different DOS can be explained intwo ways. One possible explanation is that injection occurs intointermediate surface states that are different from surface states ofthe clean surface and show a constant DOS over a wide energyrange. This assumption would also require that the electroniccoupling between these states and the excited state is only weaklyenergy dependent. On the other hand, it is possible that theelectronic coupling compensates for the difference in DOSresulting in a stronger coupling for states higher in energy.However, the latter explanation would require a fine balancebetween two unrelated properties and is thus less likely. Thesefindings support the hypothesis that initial HET proceeds intointermediate surface acceptor states that are different from theDOS of the clean crystal. They are pointing in a similar directionas recent experimental and theoretical studies that are focusingon molecular intermediate states on the bridge/anchor groupand go beyond the assumption that bridge/anchor groups act assimple tunneling barrier, instead treating the bridge as a single-molecule conductor.58
Macromolecular HET System. High resolution measurements inthe energy as well as in the time domain on heterogeneouscolloidal systems are complicated due to the strong inhomoge-neity of the electrode. Employing single crystal surfaces insteadof colloidal systems and UHV environment instead of solventhelps reduce these inhomogeneities tremendously. However,even a single crystal surface contains steps and defect states andmay give rise to different binding geometries and local electronicstructure. Such complications are absent in solution-phasespectroscopies of organic molecules because all molecules areidentical and contribute identical signals. Recently it has beenpossible to synthesize HET systems that are based on molecularpolyoxotitanate clusters with well-defined stoichiometry and thatcan be equipped with dye molecules at well-defined binding sites(Figure 8). These clusters showed a mixture of photophysical
properties, including broad band-like absorption featurestraditionally associated with solid state materials and tripletstates traditionally associated with molecules. Measurementssuggested that high frequency modes of the isopropoxidecapping ligands are essential for stabilizing the excited state inthe cluster. Measurements on similar nanosized molecularclusters are still at the very early stage. However, their well-defined structure and limited size make them promisingcandidates for experimental as well as high-level theoretical work.Summary and Outlook. HET becomes considerably morecomplicated to study once the vibrational degrees of freedomon the molecular side and the surface induced states on theelectrode are included. Consequently, it requires much moreeffort from the experimental as well as the theoretical side. Untilnow, no ab initio HET calculations are available that includehigh-energy vibrational modes. Neither has the vibrationalprogression in the molecular cation state yet been measured. Inparticular, the vibrational degrees of freedom are closelyconnected to the question of how the adiabaticity of the systeminfluences its HET dynamics. In general, a nonadiabatic picturehas been adopted for HET systems with the assumption that theultrafast nature of ET is mainly due to the multitude of acceptorlevels and not due to the strength of the donor−acceptor
Figure 7. Injection dynamics is reflected in the rise of the cationabsorption after S2 excitation (blue shaded area) and in the decay of theexcited state after S1 excitation (red shaded area). The long-lived signalpresent in both cases constitutes absorption of the cation. The HETtime is very similar for injection from both states.
These findings support the hy-pothesis that initial HET proceedsinto intermediate surface accept-or states that are different fromthe DOS of the clean crystal.
Figure 8. Ti17 (Ti17(μ4-O)4(μ3-O)16(μ2-O)4(OPri)20) cluster: A
molecular view showing the central tetrahedral and the penta-coordinateTi atoms at the corners of the cluster highlighted in green. Reproducedfrom ref 59.
The Journal of Physical Chemistry Letters Perspective
dx.doi.org/10.1021/jz501541a | J. Phys. Chem. Lett. 2014, 5, 3498−35073504

coupling. However, theoretical and experimental results suggestthat many of the dye molecules that are anchored directly viashort anchor groups, like carboxylic acid, give rise to couplingthat is in the intermediate to strong regime. For example, theparametrized fully quantum mechanical model mentioned abovepredicts a 16 fs ET time with a transfer coupling of about 50 meVthat corresponds to a Landau−Zener transition probability ofaround 0.4. This value is clearly less but not much less than 1, thatis, the limit for nonadiabatic transfer. Such systems are positionedin-between both limiting cases. Depending on the bridge group,the transition probability turns out to be between 0.1 and 0.7 forthe different perylene dyes when attached to TiO2. For the lowestprobabilities; a nonadiabatic treatment is still appropriate,whereas for probabilities around 0.7, strong interaction is alreadyapparent from the strong broadening in the ground stateabsorption spectra upon binding to TiO2.
33 In the intermediaterange of coupling strengths, ET dynamics are expected to show amixture of properties that are characteristic for both regimes. Thedifferences will be most apparent when the influence of high-energy vibrational modes on HET dynamics is measured. In caseof nonadiabatic HET these modes strongly influence thespectrum due to Franck−Condon factors and wave packetdynamics, cf. Figure 4. In the intermediate regime Franck−Condon overlap and vibrational coherence are expected to play amuch smaller role. This intermediate regime was investigatedtheoretically by Prezhdo et al., among others. Their TDDFTapproach allows for distinguishing between adiabatic andnonadiabatic contributions to ET and shows that even in thecase of strongly coupled molecules like isonicotinic acid with ameasured ET time of a few femtoseconds,60 around 15% of ETproceeds via a nonadiabatic mechanism.61 These calculationsonly include vibrational modes with energies around kT. Areview of this work has been published recently.62 Vibrationalenergy distribution and coherent wave packet motion can inprinciple be measured by probing molecular states of the system.Experimental investigations have mostly focused on vibrationalexcitation in the donor state so far.63 First indications of wavepacket dynamics influencing HET have been observed in aperylene/TiO2 system.31 However, these measurements arecomplicated due to the need of very short laser pulses and areonly sensitive to vibrational modes that are coherently excitedand keep coherence in the course of the ET reaction. Suchsystems allow for studying the interplay of nuclear and electrondynamics. They offer a compelling test of our understanding ofthe extent of electron−nuclear coupling in ET reactions, which isneglected in the widely applied Born−Oppenheimer approx-imation. Studying the vibrational energy distribution in theproduct state and its coherence presents one way to study thetransition from adiabatic to nonadiabatic HET experimentallyand test recent theoretical predictions. In addition, vibrationalcoherence opens the way for laser pulse control of HET. Thepossibilities for controlling the rate and the pathway of HET hasbeen explored theoretically, predicting among others things adependence of the controllability of the ET rate as a function ofcoupling strength that agrees with the reduced influence ofvibrational modes near the adiabatic limit.64 Recently, coherenceeffects in biological systems and the prospect of coherent controlof HET has revived the interest in electronic and vibrationalcoherence in excited states and their influence on ET.65−69
Research on electronic coherence between donor and acceptorstates is largely missing and has mostly been computational innature.70,71 In photoinduced HET a short excitation pulse cangenerate a superposition of the photo excited state and the charge
separated state. However, this requires a sufficiently strongcoupling between the two states, that is, a system that is close tothe adiabatic regime. In addition, electronic coherence in generaldephases on the sub 10 fs time scale in solid state materials.Therefore, very short-lived intermediates have to be measuredwith high enough precision for revealing any effect of electroniccoherence in HET.In order to study electronic coherence and adiabatic systems, the
time resolution of the experiments have to be improved.Although the generation of few-femtosecond pulses in theUV−vis range has been demonstrated decades ago, such pulsesare rarely used for studying HET. Research on the coupling ofvibrational degrees of f reedom to HET and its coherence, on theother hand, will benefit from energy-resolved and ultrafast two-dimensional spectroscopic techniques that allow for measuringthe electronic and vibrational dynamics simultaneously. Finally,progress in both fields depends on the synthesis and preparationof reliable well-defined test systems that allow for changing therelevant HET parameters and that gives selective access toproducts and reactants of the HET reaction.
■ AUTHOR INFORMATION
Corresponding Author*E-mail: [email protected].
NotesThe authors declare no competing financial interest.
Biographies
Jesus Nieto-Pescador is a Ph.D. student at the University of Delawarestudying heterogeneous electron transfer under the supervision of Prof.Lars Gundlach. Born in Durango, Mexico, he obtained a bachelor’s inengineering at Durango Insitute of Technology and a M.Sc. in physicsfrom CINVESTAV-IPN (Mexico City).
Baxter Abraham received his B.S. in Chemistry from the University ofCentral Florida in 2012. Currently a Ph.D. student at the University ofDelaware, his research focuses on the use of third-order spectroscopy tostudy excited state dynamics.
Professor Lars Gundlach received his Master in Physics from the FreeUniversity of Bremen, Germany, in 2000, and his Ph.D. from the FreeUniversity of Berlin, Germany, in 2005. He is currently an AssistantProfessor at the University of Delaware (http://www.udel.edu/chem/gundlach/). His research includes ultrafast studies of charge carrierdynamic in heterogeneous system and in nanoparticle.
■ ACKNOWLEDGMENTS
We thank Dr. Piotr Piotrowiak for discussion and constructivecomments on the manuscript.
■ REFERENCES(1) Meier, H. Sensitization of Electrical Effects in Solids. J. Phys. Chem.1965, 69, 719−729.(2) Willig, F.; Gerischer, H. Reaction of Excited Dye Molecules atElectrodes. In Physical and Chemical Applications of Dyestuffs; Boschke,F., Ed.; Topics in Current Chemistry, Vol. 61, Springer: Berlin, 1976; pp31−84.(3) Gerischer, H.; Tributsch, H. Elektrochemische Untersuchungenzur spektralen Sensibilisierung von “ZnO” Einkristallen. Ber. Bunsenges.Phys. Chem. 1967, 72, 437−445.(4) Tributsch, H.; Gerischer, H. Elektrochemische Untersuchungenuber denMechanismus der Sensibilisierung und Ubersensibilisierung anZnO Einkristallen. Ber. Bunsen-Ges. 1973, 73, 251−260.
The Journal of Physical Chemistry Letters Perspective
dx.doi.org/10.1021/jz501541a | J. Phys. Chem. Lett. 2014, 5, 3498−35073505

(5) Hore, P. J.; McLauchlan, K. A. CIDEP in Radicals UndergoingElectron Transfer. A Time-resolved Flash Photolysis ESR Study. Chem.Phys. Lett. 1980, 75, 582−586.(6) Rossetti, R.; Brus, L. E. Time-Resolved Raman Scattering Study ofAdsorbed, Semioxidized Eosin Y Formed by Excited-State ElectronTransfer into Colloidal TiO2 Particles. J. Am. Chem. Soc. 1984, 106,4336−4340.(7) Eichberger, R.; Willig, F. Ultrafast Electron Injection from ExcitedDye Molecules into Semiconductor Electrodes. Chem. Phys. 1990, 141,159.(8) Lanzafame, J. M.; Miller, R. J. D.; Muenter, A. A.; Parkinson, B. A.Ultrafast Charge-Transfer Dynamics at Tin Disulfide Surfaces. J. Phys.Chem. 1992, 96, 2820.(9) Duonghong, D.; Serpone, N.; Gratzel, M. Integrated Systems forWater Cleavage by Visible Light; Sensitization of TiO2 Particles bySurface Derivatization with Ruthenium Complexes. Helv. Chim. Acta1984, 67, 1012−1018.(10) O’Regan, B.; Gratzel, M. A Low Cost, High Efficiency Solar CellBased On Dye-Sensitized Colloidal TiO2 Films. Nature 1991, 353, 737.(11) Asbury, J.; Ellingson, R.; Ghosh, H.; Ferrere, S.; Nozik, A.; Lian, T.Femtosecond IR Study of Excited-State Relaxation and Electron-Injection Dynamics of Ru(dcbpy)2(NCS)2 in Solution and onNanocrystalline TiO2 and Al2O3 Thin Films. J. Phys. Chem. B 1999,103, 3110−3119.(12) Rehm, J. M.; McLendon, G. L.; Nagasawa, Y.; Moser, J.; Gratzel,M. Femtosecond Electron-Transfer Dynamics at a Sensitizing Dye-Semiconductor (TiO2) Interface. J. Phys. Chem. 1996, 100, 9577−9578.(13) Burfeindt, B.; Hannappel, T.; Storck, W.; Willig, F. MeasurementOf Temperature-Independent Femtosecond Interfacial Electron Trans-fer from an Anchored Molecular Electron Donor to a Semiconductor asAcceptor. J. Phys. Chem. 1996, 100, 16463.(14) Hilgendorff, M.; Sundstrom, V. Dynamics of Electron Injectionand Recombination of Dye-Sensitized TiO2 Particles. J. Phys. Chem. B1998, 102, 10505−10514.(15) Ghosh, H. N.; Asbury, J. B.; Lian, T. Direct Observation ofUltrafast Electron Injection from Coumarin 343 to TiO2 Nanoparticlesby Femtosecond Infrared Spectroscopy. J. Phys. Chem. B 1998, 102,6482.(16) Yang, Y.; Rodríguez-Cordoba, W.; Xiang, X.; Lian, T. StrongElectronic Coupling and Ultrafast Electron Transfer between PbSQuantum Dots and TiO2 Nanocrystalline Films. Nano Lett. 2012, 12,303−309.(17) Benko, G.; Kallioinen, J.; Korppi-Tommola, J. E. I.; Yartsev, A. P.;Sundstrom, V. Photoinduced Ultrafast Dye-to-Semiconductor ElectronInjection from Nonthermalized and Thermalized Donor States. J. Am.Chem. Soc. 2001, 124, 489−493.(18) Tachibana, Y.; Haque, S.; Mercer, I.; Durrant, J.; Klug, D. ElectronInjection and Recombination in Dye Sensitized NanocrystallineTitanium Dioxide Films: A Comparison of Ruthenium Bipyridyl andPorphyrin Sensitizer Dyes. J. Phys. Chem. B 2000, 104, 1198−1205.(19) Ernstorfer, R. Spectroscopic Investigation of PhotoinducedHeterogeneous Electron Transfer. Ph.D. Thesis, Freie Universitat Berlin,2004.(20) Bram, O.; Cannizzo, A.; Chergui, M. Ultrafast FluorescenceStudies of Dye Sensitized Solar Cells. Phys. Chem. Chem. Phys. 2012, 14,7934−7937.(21) Gundlach, L.; Ernstorfer, R.; Willig, F. Pathway DependentElectron Transfer for Rod-Shaped Perylene-Derived MoleculesAdsorbed in Nanometer-Size TiO2 Cavities. J. Phys. Chem. C 2007,111, 13586−1359.(22) Huber, R.; Moser, J.-E.; Gratzel, M.; Wachtveitl, J. Real-TimeObservation of Photoinduced Adiabatic Electron Transfer in StronglyCoupled Dye/Semiconductor Colloidal Systems with a 6 fs TimeConstant. J. Phys. Chem. B 2002, 106, 6494−6499.(23) Schnadt, J.; Bruhwiler, B.; Patthey, L.; O’Shea, J.; Sodergren, S.;Odelius, M.; Ahuja, R.; Karis, O.; Baessler, M.; Persson, P.; et al.Experimental Evidence for Sub-3-fs Charge Transfer from an AromaticAdsorbate to a Semiconductor. Nature 2002, 418, 620.
(24) Duncan, W. R.; Stier, W. M.; Prezhdo, O. V. Ab InitioNonadiabatic Molecular Dynamics of the Ultrafast Electron InjectionAcross the Alizarin-TiO2 Interface. J. Am. Chem. Soc. 2005, 127, 7941−7951.(25) Li, Z.; Zhang, X.; Lu, G. Electron Dynamics in Dye-SensitizedSolar Cells: Effects of Surface Terminations andDefects. J. Phys. Chem. B2010, 114, 17077−17083.(26) Kaniyankandy, S.; Rawalekar, S.; Sen, A.; Ganguly, B.; Ghosh, H.N. Does Bridging Geometry Influence Interfacial Electron TransferDynamics? Case of the Enediol−TiO2 System. J. Phys. Chem. C 2012,116, 98−103.(27) Borgias, B. A.; Cooper, S. R.; Koh, Y. B.; Raymond, K. N.Synthetic, Structural, and Physical Studies of Titanium Complexes ofCatechol and 3,5-Di-tert-butylcatechol. Inorg. Chem. 1984, 23, 1009−1016.(28) Moser, J.; Punchihewa, S.; Infelta, P. P.; Gratzel, M. SurfaceComplexation of Colloidal Semiconductors Strongly EnhancesInterfacial Electron-Transfer Rates. Langmuir 1991, 7, 3012−3018.(29) Gundlach, L.; Ernstorfer, R.; Willig, F. Escape Dynamics ofPhotoexcited Electrons at Catechol: TiO2(110). Phys. Rev. B 2006, 74,035324.(30) Long, R.; Prezhdo, O. V. Instantaneous Generation of Charge-Separated State on TiO2 Surface Sensitized with Plasmonic Nano-particles. J. Am. Chem. Soc. 2014, 136, 4343−4354.(31) Zimmermann, C.; Willig, F.; Ramakrishna, S.; Burfeindt, B.;Pettinger, B.; Eichberger, R.; Storck, W. Experimental Fingerprints ofVibrational Wave-packet Motion During Ultrafast HeterogeneousElectron Transfer. J. Phys. Chem. B 2001, 105, 9245.(32) Ernstorfer, R.; Felber, S.; Storck, W.; Galoppini, E.; Wei, Q.;Willig, F. Distance Dependence of Heterogeneous Electron TransferProbed in Ultra-high Vacuum with Femtosecond Transient Absorption.Res. Chem. Intermed. 2005, 31, 643−647.(33) Ernstorfer, R.; Gundlach, L.; Felber, S.; Storck,W.; Eichberger, R.;Willig, F. The Role of Molecular Anchor Groups in Molecule-to-Semiconductor Electron Transfer. J. Phys. Chem. B 2006, 110, 25383−25391.(34) Gundlach, L.; Ernstorfer, R.; Willig, F. Ultrafast InterfacialElectron Transfer from the Excited State of Anchored Molecules into aSemiconductor. Prog. Surf. Sci. 2007, 82, 355−377.(35) Gundlach, L.; Szarko, J.; Socaciu-Siebert, L. D.; Neubauer, A.;Ernstorfer, R.; Willig, F. Different Orientations of Large Rigid OrganicChromophores at The Rutile TiO2 Surface Controlled by DifferentBinding Geometries Of Specific Anchor Groups. Phys. Rev. B 2007, 75,125320.(36) Gundlach, L.; Letzig, T.; Willig, F. Test of Theoretical Models forUltrafast Heterogeneous Electron Transfer with Femtosecond Two-Photon Photoemission Data. J. Chem. Sci. 2009, 121, 561−574.(37) Ramakrishna, S.; Willig, F.; May, V. Photoinduced UltrafastElectron Injection From a Surface Attached Molecule: Control ofElectronic and Vibronic Distributions via Vibrational Wave Packets.Phys. Rev. B 2000, 62, R16330.(38) Wang, L.; Willig, F.; May, V. Ultrafast Heterogeneous ElectronTransfer Reactions: Comparative Theoretical Studies on Time- andFrequency-Domain Data. J. Chem. Phys. 2006, 124, 014712−11.(39) Li, J.; Wang, H.; Persson, P.; Thoss, M. Photoinduced ElectronTransfer Processes in Dye-Semiconductor Systems with DifferentSpacer Groups. J. Chem. Phys. 2012, 137, 22A529.(40) Li, J.; Nilsing, M.; Kondov, I.; Wang, H.; Persson, P.; Lunell, S.;Thoss, M. Dynamical Simulation of Photoinduced Electron TransferReactions in Dye-Semiconductor Systems with Different AnchorGroups. J. Phys. Chem. C 2008, 112, 12326−12333.(41) Persson, P.; Lundqvist, M. J.; Ernstorfer, R.; Goddard, W. A., III;Willig, F. Quantum Chemical Calculations of the Influence of Anchor-Cum-Spacer Groups on Femtosecond Electron Transfer Times in Dye-Sensitized Semiconductor Nanocrystals. J. Chem. Theory Comp. 2006, 2,441.(42) Angelis, F. D. Direct vs. Indirect Injection Mechanisms inPerylene Dye-Sensitized Solar Cells: a DFT/TDDFT Investigation.Chem. Phys. Lett. 2010, 493, 323−327.
The Journal of Physical Chemistry Letters Perspective
dx.doi.org/10.1021/jz501541a | J. Phys. Chem. Lett. 2014, 5, 3498−35073506

(43) Pastore, M.; De Angelis, F. Intermolecular Interactions in Dye-Sensitized Solar Cells: A Computational Modeling Perspective. J. Phys.Chem. Lett. 2013, 4, 956−974.(44) Feldberg, S. W.; Sutin, N. Distance Dependence of Heteroge-neous Electron Transfer Through the Nonadiabatic and AdiabaticRegimes. Chem. Phys. 2006, 324, 216−225 TheMolecules and Methodsof Chemical, Biochemical and Nanoscale Electron Transfer, Part 1. Inhonour of Noel S. Hush on his 80th birthday..(45) Wynne, K.; Hochstrasser, R. M. Coherence and Adiabaticity inUltrafast Electron Transfer. In Electron Transfer: From Isolated Moleculesto Biomolecules, Part Two; Jortner, J., Bixon, M., Eds.; JohnWiley & Sons,Inc., 1999; pp 263−309.(46) Nitzan, A. Electron Transmission Through Molecules andMolecular Interfaces. Annu. Rev. Phys. Chem. 2001, 52, 681−750.(47) Mahrt, J. Angeregte Dimere und Excimere des Perylens. Ph.D. thesis,Technical University of Berlin, 1995.(48) Wang, L.; Willig, F.; May, V. Theory of Ultrafast PhotoinducedHeterogeneous Electron Transfer. Mol. Simul. 2006, 32, 765−781.(49) Ramakrishna, S.; Seideman, T.; Willig, F.; May, V. Theory ofCoherent Molecule to Surface Electron Injection: an Analytical Model.J. Chem. Sci. 2009, 121, 589−594.(50) Myahkostupov, M.; Pagba, C. V.; Gundlach, L.; Piotrowiak, P.Vibrational State Dependence of Interfacial Electron Transfer: HotElectron Injection from the S1 State of Azulene into TiO2 Nanoparticles.J. Phys. Chem. C 2013, 117, 20485−20493.(51) Maity, P.; Debnath, T.; Akbar, A.; Verma, S.; Ghosh, H. N.Ultrafast Electron-Transfer and -Trapping Dynamics in the Inter-Band-Gap States of ZrO2 Nanoparticles Sensitized by Baicalein. J. Phys. Chem.C 2013, 117, 17531−17539.(52) Knauf, R. R.; Brennaman, M. K.; Alibabaei, L.; Norris, M. R.;Dempsey, J. L. Revealing the Relationship between SemiconductorElectronic Structure and Electron Transfer Dynamics at MetalOxideChromophore Interfaces. J. Phys. Chem. C 2013, 117, 25259−25268.(53) Strothkamper, C.; Bartelt, A.; Sippel, P.; Hannappel, T.; Schutz,R.; Eichberger, R. Delayed Electron Transfer through Interface States inHybrid ZnO/Organic-Dye Nanostructures. J. Phys. Chem. C 2013, 117,17901−17908.(54) Chang, C.-W.; Luo, L.; Chou, C.-K.; Lo, C.-F.; Lin, C.-Y.; Hung,C.-S.; Lee, Y.-P.; Diau, E. W.-G. Femtosecond Transient Absorption ofZinc Porphyrins with Oligo(phenylethylnyl) Linkers in Solution and onTiO2 Films. J. Phys. Chem. C 2009, 113, 11524−11531.(55) Baskin, J. S.; Yu, H.-Z.; Zewail, A. H. Ultrafast Dynamics ofPorphyrins in the Condensed Phase: I. Free Base Tetraphenylporphyr-in. J. Phys. Chem. A 2002, 106, 9837−9844.(56) Pistner, A. J.; Lutterman, D. A.; Ghidiu, M. J.; Ma, Y.-Z.;Rosenthal, J. Synthesis, Electrochemistry, and Photophysics of a Familyof Phlorin Macrocycles that Display Cooperative Fluoride Binding. J.Am. Chem. Soc. 2013, 135, 6601−6607.(57) Heinrich, V. E.; Cox, P. A. The Surface Science of Metal Oxides;Cambridge University Press: New York, 1994.(58) Negre, C. F. A.; Milot, R. L.; Martini, L. A.; Ding, W.; Crabtree, R.H.; Schmuttenmaer, C. A.; Batista, V. S. Efficiency of Interfacial ElectronTransfer from Zn-Porphyrin Dyes into TiO2 Correlated to the LinkerSingle Molecule Conductance. J. Phys. Chem. C 2013, 117, 24462−24470.(59) Bao, J.; Yu, Z.; Gundlach, L.; Benedict, J. B.; Coppens, P.; Chen,H. C.; Miller, J. R.; Piotrowiak, P. Excitons and Excess Electrons inNanometer SizeMolecular Polyoxotitanate Clusters: Electronic Spectra,Exciton Dynamics, and Surface States. J. Phys. Chem. B 2013, 117,4422−4430.(60) Schnadt, J.; Henningsson, A.; Andersson, M. P.; Karlsson, P. G.;Uvdal, P.; Siegbahn, H.; Bruhwiler, P. A.; Sandell, A. Adsorption andCharge-Transfer Study of Bi-isonicotinic Acid on In Situ-GrownAnatase TiO2 Nanoparticles. J. Phys. Chem. B 2004, 108, 3114−3122.(61) Prezhdo, O. V.; Duncan, W. R.; Prezhdo, V. V. PhotoinducedElectron Dynamics at the Chromophoresemiconductor Interface: aTime-Domain Ab Initio Perspective. Prog. Surf. Sci. 2009, 84, 30−68.
(62) Akimov, A. V.; Neukirch, A. J.; Prezhdo, O. V. Theoretical Insightsinto Photoinduced Charge Transfer and Catalysis at Oxide Interfaces.Chem. Rev. 2013, 113, 4496−4565.(63) Ricks, A. M.; Anfuso, C. L.; Rodrguez-Crdoba, W.; Lian, T.Vibrational Relaxation Dynamics of Catalysts on TiO2 Rutile (1,1,0)Single Crystal Surfaces and Anatase Nanoporous Thin Films. Chem.Phys. 2013, 422, 264−271 A tribute to Dr. Robin Hochstasser..(64) Wang, L.; May, V. Laser Pulse Control of Bridge MediatedHeterogeneous Electron Transfer. Chem. Phys. 2009, 361, 1−8.(65) Engel, G. S.; Calhoun, T. R.; Read, E. L.; Ahn, T.-K.; Mancal, T.;Cheng, Y.-C.; Blankenship, R. E.; Fleming, G. R. Evidence for WavelikeEnergy Transfer through Quantum Coherence in PhotosyntheticSystems. Nature 2007, 446, 782−786.(66) Rego, L. G.; Santos, L. F.; Batista, V. S. Coherent Control ofQuantum Dynamics with Sequences of Unitary Phase-Kick Pulses.Annu. Rev. Phys. Chem. 2009, 60, 293−320.(67) Turner, D. B.; Dinshaw, R.; Lee, K.-K.; Belsley, M. S.; Wilk, K. E.;Curmi, P. M. G.; Scholes, G. D. Quantitative Investigations of QuantumCoherence for a Light-Harvesting Protein at Conditions SimulatingPhotosynthesis. Phys. Chem. Chem. Phys. 2012, 14, 4857−4874.(68) Plenio, M.; Almeida, J.; Huelga, S. Origin of Long-LivedOscillations in 2D-Spectra of a Quantum Vibronic Model: Electronicversus Vibrational Coherence. J. Chem. Phys. 2013, 139, 235102−10.(69) Andrea Rozzi, C.; Maria Falke, S.; Spallanzani, N.; Rubio, A.;Molinari, E.; Brida, D.; Maiuri, M.; Cerullo, G.; Schramm, H.;Christoffers, J.; et al. Quantum Coherence Controls the ChargeSeparation in a Prototypical Artificial Light-Harvesting System. Nat.Commun. 2013, 4, 1602−7.(70) Kondov, I.; ek, M.; Benesch, C.; Wang, H.; Thoss, M. QuantumDynamics of Photoinduced Electron-Transfer Reactions in Dye-Semiconductor Systems: First-Principles Description and Applicationto Coumarin 343-TiO2. J. Phys. Chem. C 2007, 111, 11970−11981.(71) Rasmussen, A. M.; Ramakrishna, S.; Weiss, E. A.; Seideman, T.Theory of Ultrafast Photoinduced Electron Transfer from a BulkSemiconductor to a Quantum Dot. J. Chem. Phys. 2014, 140, 144102−13.
The Journal of Physical Chemistry Letters Perspective
dx.doi.org/10.1021/jz501541a | J. Phys. Chem. Lett. 2014, 5, 3498−35073507
Copyright © 2022 FDOKUMEN




















