
Characterization of the mycobacterial chromosome segregation protein ParB and identification of its...
11
Characterization of the mycobacterial chromosome segregation protein ParB and identification of its target in Mycobacterium smegmatis Dagmara Jakimowicz, 1,2 Anna Brzostek, 3 Anna Rumijowska-Galewicz, 3 Paulina Z ˙ ydek, 1 Alicja Dolzblasz, 1 Aleksandra Smulczyk-Krawczyszyn, 1 Tomasz Zimniak, 1 Lukasz Wojtasz, 1 Anna Zawilak-Pawlik, 1 Agnieszka Kois, 1 Jaroslaw Dziadek 3 and Jolanta Zakrzewska-Czerwin ´ ska 1,2 Correspondence Jolanta Zakrzewska-Czerwin ´ ska [email protected] 1 Ludwik Hirszfeld Institute of Immunology and Experimental Therapy, Polish Academy of Sciences, ul. Weigla 12, 53-114 Wroclaw, Poland 2 Faculty of Biotechnology, University of Wroclaw, ul. Tamka 2, 50-137 Wroclaw, Poland 3 Medical Biology Center, Polish Academy of Sciences, Lodowa 106, 93-232 Lo ´ dz ´, Poland Received 13 July 2007 Revised 20 August 2007 Accepted 23 August 2007 Bacterial chromosomes (though not Escherichia coli and some other c-proteobacterial chromosomes) contain parS sequences and parAB genes encoding partitioning proteins, i.e. ParA (ATPase) and ParB (DNA-binding proteins) that are components of the segregation machinery. Here, mycobacterial parABS elements were characterized for the first time. parAB genes are not essential in Mycobacterium smegmatis; however, elimination or overexpression of ParB protein causes growth inhibition. Deletion of parB also leads to a rather severe chromosome segregation defect: up to 10 % of the cells were anucleate. Mycobacterial ParB protein uses three oriC-proximal parS sequences as targets to organize the origin region into a compact nucleoprotein complex. Formation of such a complex involves ParB–ParB interactions and is assisted by ParA protein. INTRODUCTION Tuberculosis (TB) is the world’s most common disease caused by an infectious organism, Mycobacterium tuber- culosis (DeAngelis & Flanagin, 2005; and see http:// www.who.int/topics/tuberculosis/en/). Due to the spread of multi-drug-resistant strains of M. tuberculosis and a synergy with HIV, the TB epidemic is growing and becoming more dangerous in both developing and industrialized countries. A unique feature of M. tuberculosis is its ability to maintain a dormant, non-replicating state for extended periods of time under unfavourable condi- tions. The mechanism (or mechanisms) by which this bacterium shifts to a dormant state and reverts to active growth is not well understood (Hampshire et al., 2004; Go ´ mez & Smith, 2000; Wayne & Hayes, 1996: Wayne & Sohaskey, 2001). Knowledge regarding the steps of the mycobacterial cell cycle (replication, chromosome segrega- tion and cell division) seems to be critical for understand- ing the mechanisms that are responsible for the transition from an active to a non-replicative persistent state (and vice versa) of pathogenic mycobacteria, particularly M. tuberculosis. While initiation of chromosome replication (Madiraju et al., 2006; Qin et al., 1999; Rajagopalan et al., 1995; Zawilak et al., 2004) and cell division (FtsZ ring formation) (Chauhan et al., 2006; Dziadek et al., 2002, 2003; Huang et al., 2007; Rajagopalan et al., 2005) are relatively well studied in mycobacteria, nothing is known about the segregation of chromosomes in these bacteria. Bacterial chromosome segregation has been recently found to be an active and complex process closely coupled with replication (see Bartosik & Jagura-Burdzy, 2005; Errington et al., 2005; Hayes & Barilla, 2006; Leonard et al., 2005 for reviews). In bacteria studied to date, the newly synthesized origin (oriC) regions undergo a symmetric or asymmetric segregation process; two copies of the duplicated oriC regions migrate from the cell centre toward opposite cell poles, i.e. to the 1/4 and 3/4 positions (Escherichia coli, Bacillus subtilis, Vibro cholerae chromosome II), or one copy of the newly synthesized origins remains at the pole while the other copy migrates to the opposite pole Abbreviations: EMSA, electrophoretic mobility shift assay; GST, glutathione S-transferase; Ms, M. smegmatis; Mt, M. tuberculosis. Three supplementary figures showing the construction of the M. smegmatis DparB mutant, purified mycobacterial ParAB proteins and protein sequence alignment of homologous ParB proteins, and a supplementary table listing the oligonucleotides used in this study are available with the online version of this paper. Microbiology (2007), 153, 4050–4060 DOI 10.1099/mic.0.2007/011619-0 4050 2007/011619 G 2007 SGM Printed in Great Britain
Transcript of Characterization of the mycobacterial chromosome segregation protein ParB and identification of its...

Characterization of the mycobacterial chromosomesegregation protein ParB and identification of itstarget in Mycobacterium smegmatis
Dagmara Jakimowicz,1,2 Anna Brzostek,3 Anna Rumijowska-Galewicz,3
Paulina Zydek,1 Alicja Dołzbłasz,1 Aleksandra Smulczyk-Krawczyszyn,1
Tomasz Zimniak,1 Łukasz Wojtasz,1 Anna Zawilak-Pawlik,1
Agnieszka Kois,1 Jarosław Dziadek3
and Jolanta Zakrzewska-Czerwinska1,2
Correspondence
Jolanta Zakrzewska-Czerwinska
1Ludwik Hirszfeld Institute of Immunology and Experimental Therapy, Polish Academy of Sciences,ul. Weigla 12, 53-114 Wrocław, Poland
2Faculty of Biotechnology, University of Wroclaw, ul. Tamka 2, 50-137 Wroclaw, Poland
3Medical Biology Center, Polish Academy of Sciences, Lodowa 106, 93-232 Łodz, Poland
Received 13 July 2007
Revised 20 August 2007
Accepted 23 August 2007
Bacterial chromosomes (though not Escherichia coli and some other c-proteobacterial
chromosomes) contain parS sequences and parAB genes encoding partitioning proteins, i.e.
ParA (ATPase) and ParB (DNA-binding proteins) that are components of the segregation
machinery. Here, mycobacterial parABS elements were characterized for the first time. parAB
genes are not essential in Mycobacterium smegmatis; however, elimination or overexpression of
ParB protein causes growth inhibition. Deletion of parB also leads to a rather severe chromosome
segregation defect: up to 10 % of the cells were anucleate. Mycobacterial ParB protein uses
three oriC-proximal parS sequences as targets to organize the origin region into a compact
nucleoprotein complex. Formation of such a complex involves ParB–ParB interactions and is
assisted by ParA protein.
INTRODUCTION
Tuberculosis (TB) is the world’s most common diseasecaused by an infectious organism, Mycobacterium tuber-culosis (DeAngelis & Flanagin, 2005; and see http://www.who.int/topics/tuberculosis/en/). Due to the spreadof multi-drug-resistant strains of M. tuberculosis and asynergy with HIV, the TB epidemic is growing andbecoming more dangerous in both developing andindustrialized countries. A unique feature of M. tuberculosisis its ability to maintain a dormant, non-replicating statefor extended periods of time under unfavourable condi-tions. The mechanism (or mechanisms) by which thisbacterium shifts to a dormant state and reverts to activegrowth is not well understood (Hampshire et al., 2004;Gomez & Smith, 2000; Wayne & Hayes, 1996: Wayne &Sohaskey, 2001). Knowledge regarding the steps of the
mycobacterial cell cycle (replication, chromosome segrega-tion and cell division) seems to be critical for understand-ing the mechanisms that are responsible for the transitionfrom an active to a non-replicative persistent state (andvice versa) of pathogenic mycobacteria, particularly M.tuberculosis. While initiation of chromosome replication(Madiraju et al., 2006; Qin et al., 1999; Rajagopalan et al.,1995; Zawilak et al., 2004) and cell division (FtsZ ringformation) (Chauhan et al., 2006; Dziadek et al., 2002,2003; Huang et al., 2007; Rajagopalan et al., 2005) arerelatively well studied in mycobacteria, nothing is knownabout the segregation of chromosomes in these bacteria.
Bacterial chromosome segregation has been recently foundto be an active and complex process closely coupled withreplication (see Bartosik & Jagura-Burdzy, 2005; Erringtonet al., 2005; Hayes & Barilla, 2006; Leonard et al., 2005 forreviews). In bacteria studied to date, the newly synthesizedorigin (oriC) regions undergo a symmetric or asymmetricsegregation process; two copies of the duplicated oriCregions migrate from the cell centre toward opposite cellpoles, i.e. to the 1/4 and 3/4 positions (Escherichia coli,Bacillus subtilis, Vibro cholerae chromosome II), or onecopy of the newly synthesized origins remains at the polewhile the other copy migrates to the opposite pole
Abbreviations: EMSA, electrophoretic mobility shift assay; GST,glutathione S-transferase; Ms, M. smegmatis; Mt, M. tuberculosis.
Three supplementary figures showing the construction of the M.smegmatis DparB mutant, purified mycobacterial ParAB proteins andprotein sequence alignment of homologous ParB proteins, and asupplementary table listing the oligonucleotides used in this study areavailable with the online version of this paper.
Microbiology (2007), 153, 4050–4060 DOI 10.1099/mic.0.2007/011619-0
4050 2007/011619 G 2007 SGM Printed in Great Britain

(Caulobacter crescentus, Vibrio cholerae chromosome I)(Fogel & Waldor, 2006; Lewis, 2004; Teleman et al., 1998;Viollier et al., 2004; Webb et al., 1997).
Among the large number of proteins involved inchromosome segregation, ParAB homologues were theearliest to be identified and are the best studied,particularly in B. subtilis (Lewis & Errington, 1997; Linet al., 1997; Webb et al., 1997), C. crescentus (Mohl &Gober, 1997; Mohl et al., 2001), Pseudomonas spp.(Bartosik et al., 2004; Godfrin-Estevenon et al., 2002;Lewis et al., 2002), and Streptomyces coelicolor (Jakimowiczet al., 2002; Kim et al., 2000). Homologues of ParABproteins are encoded in most bacterial chromosomes (theE. coli chromosome is one of the few exceptions) (Bartosik& Jagura-Burdzy, 2005; Bignell & Thomas, 2001). Only inC. crescentus have the parA and parB genes been proved tobe essential (Mohl & Gober, 1997). Recent library screening(transposon mutagenesis) suggested that parAB genes mayalso be indispensable for M. tuberculosis H37Rv (Sassettiet al., 2003). parAB mutants of the other organisms displayperceptible defects in chromosome segregation, includingthe formation of anucleate cells. In all studied bacteria,ParB (Spo0J in B. subtilis) proteins interact with parS sites(14–16-mer palindromic sequences), usually locatedaround the oriC region (Jakimowicz et al., 2002; Lin &Grossman, 1998). Binding of ParB to these sites leads to theformation of large nucleoprotein complexes (segrosomes).ParA (Soj in B. subtilis) homologues have a weak ATPaseactivity and interact with the segrosome (via direct contactwith ParB), but their function in segregation is not clear.Recently, Waldor’s group have demonstrated that ParAIforms a dynamic structure that pulls the ParBI-bound V.cholerae chromosome I to the opposite pole (Fogel &Waldor, 2006).
Besides pathogenic, slow-growing species (M. tuberculosisand Mycobacterium leprae), the genus Mycobacterium alsoincludes saprophytic, ‘fast-growing’ species such asMycobacterium smegmatis, which would be useful forstudying some aspects of mycobacterial biology, includingchromosome segregation. Detailed studies of mycobacterialchromosome segregation and characterization of the keymolecules involved in this process may also be helpful inidentifying novel drug targets.
In this study, we tested whether the parB gene in M.smegmatis is essential and we have demonstrated thatalteration of its expression affects the growth of M.smegmatis. We have also characterized interactions ofmycobacterial ParB with its target, parS sequences.
METHODS
DNA manipulation, bacterial strains and growth conditions.DNA manipulations were carried out by standard protocols(Sambrook et al., 1989). Enzymes were supplied by Fermentas, NewEngland BioLabs or Roche; isotopes were from Amersham–Pharmacia Biotech, and oligonucleotides were from the Institute ofBiochemistry and Biophysics (Warsaw, Poland). E. coli BL21 [B F2
dcm ompT hsdS(r{B m{
B ) gal] was the host for overproduction of the
GST–ParA fusion proteins. E. coli strains were grown in Luria–Bertani
(LB) medium at 37 uC. M. smegmatis mc2155 and its derivatives were
grown in Middlebrook 7H9 medium, on 7H10 agar plates
supplemented with oleic acid albumin/glucose/sodium chloride
(OADC) or in NB broth [8.0 g l21 nutrient broth (Difco), 10.0 g
l21 glucose, supplemented with 0.2 % Tween 80, pH 6.0–6.2]. For
selection, kanamycin (25 mg ml21) was used if required.
Construction of ParB overproduction strains. The M. smegmatis
parB gene was PCR amplified using the primers MsparB_fw and
MsparBrv-2 (see Supplementary Table S1, available with the online
version of this paper) and cloned into the BamHI/XbaI sites of the
pJam2 shuttle vector (Triccas et al., 1998) downstream of the pami
promoter. The resulting construct, carrying parB controlled by pami,
was introduced into M. smegmatis by electroporation. The presence of
plasmid DNA was confirmed by recovery as described previously
(Dziadek et al., 2003).
Targeted gene replacement. To perform unmarked deletions in
the parB (MSMEG6938) gene of M. smegmatis, a suicide recombina-
tion delivery vector was constructed. In the first step, the 59 end of the
parB gene (28 bp) and its upstream region (1319 bp) were amplified
using the primers MsParGR1 and MsParGR2 (see Supplementary
Table S1) and cloned into the HindIII/BamHI sites of p2NIL, creating
pABX. Subsequently, the 39 end of the parB gene (529 bp) and its
downstream region (773 bp) was amplified using the primers
MsParGR3 and MsParGR4 (see Supplementary Table S1) and cloned
into the BamHI/KpnI sites of pABX. The ligated 59 and 39 fragments
of parB gene in the resulting vector (pABY) were out of frame. Finally,
a 6 kb PacI marker cassette from pGOAL17 carrying the lacZ and sacB
genes was cloned into the PacI site of pABY to create pABZ.
The protocol of Parish & Stoker, (2000) was used to disrupt the
investigated genes at their native loci on the chromosome. The
plasmid DNA (pABZ) was treated with NaOH (0.2 mM) and used for
transformation in order to integrate it into the M. smegmatis
chromosome by homologous recombination. The resulting SCO
(single-crossover recombinant) mutant colonies were blue, KanR and
sensitive to sucrose. The SCO strain was further processed to select for
double-crossover (DCO) mutants that were white, KanS and resistant
to sucrose (2 %). PCR and Southern hybridization were used to
distinguish between the wild-type and the DCO mutant (see
Supplementary Fig. S1, available with the online version of this
paper). A probe that hybridizes to the parB gene was generated by
PCR, with MsparB_fw and MsparB_rv as primers (Supplementary
Table S1) and pABY as a template.
Characterization of the DparB mutant strain: growth and
doubling time determination. Culture of the M. smegmatis wild-
type and mutant DparB strains was performed in rich media (NB or
7H9 Middlebrook supplemented with OADC) with an initial OD600
of 0.1. Then the cells were incubated for 24 h and samples were
collected every 3 h for optical density analysis, c.f.u. determination
and microscopic examination.
Microscopy. Mycobacterial cells (48 h cultures) were permeabilized
by exposure to toluene (2 %) for 10 min prior to staining of DNA.
The M. smegmatis cells were washed, resuspended in phosphate-
buffered saline (PBS; 10 mM sodium phosphate, pH 7.4, 150 mM
NaCl, 15 mM KCl), and stained with DAPI (2 mg ml21) for 15 min at
room temperature. The samples were examined under a Zeiss Axio
Imager Z1 equipped with a 6100 objective.
RNA analysis. RNA isolation was performed using a modified Kirby
mix as described previously (Kieser et al., 2000). Briefly, M. smegmatis
was grown in modified Sauton’s broth (Mordarska et al., 1972) at
Mycobacterial chromosome segregation
http://mic.sgmjournals.org 4051

37 uC. After 48 h the cells were centrifuged and the pellet was
resuspended in the modified Kirby mix. The cells were disrupted in
the presence of glass beads by vortexing. Subsequent steps of RNA
isolation were carried out according to Chomczynski & Sacchi (1987).
For RT-PCR, total RNA was treated with RNase-free DNase I (Roche
Molecular Biochemicals), followed by phenol/chloroform extraction
and precipitation. The RT-PCR reactions were carried out using a
RevertAid First Strand cDNA Synthesis kit (MBI Fermentas)
according to the manufacturer’s protocols. RNAs (3.6 mg per
reaction) were reverse transcribed with RevertAid M-MuLV reverse
transcriptase with random hexanucleotides at 42 uC for 90 min A
sample of 2.5 ml of the 20 ml RT solution was subjected to subsequent
PCR. The amplification reaction was carried out in a 50 ml volume
using the pair of appropriate primers (Supplementary Table S1)
for 30 cycles. RT-PCR products were analysed by agarose gel
electrophoresis.
Purification of mycobacterial ParAB proteins. The vectors
pGEX-6P-2 and pET-28a(+) were chosen to overexpress the
recombinant fusion proteins GST–ParA and 6His–ParB of M.
tuberculosis (Mt) and M. smegmatis (Ms). The parA and parB genes
of M. smegmatis or M. tuberculosis were PCR amplified with the
appropriate pairs of primers (Supplementary Table S1). The PCR
products were cloned into the vector pGEM-T Easy (Promega) and
later recloned as BamHI–EcoRI fragments into pGEX-6P-2 (parA
genes) or into pET28a(+) (parB genes). All the PCR-derived clones
were analysed by DNA sequencing to check their fidelity. The fusion
proteins, glutathione S-transferase (GST)–ParA or 6His–ParB were
purified on a glutathione Sepharose 4B or on a Ni2+-NTA-agarose
column (Qiagen), respectively, as described previously (Majka et al.,
1999; Zawilak et al., 2004). For removal of the GST part, the bound
fusion proteins were treated with the PreScission protease
(Amersham–Pharmacia Biotech) and the ParA proteins were released
from the beads. The purified ParA proteins were more than 95 %
homogeneous, as judged by SDS-PAGE analysis (see Supplementary
Fig. S2, available with the online version of this paper).
Electrophoretic mobility shift assay (EMSA). For the binding
assays, 59-end-32P-labelled DNA fragments (5 fmol) were incubated
with ParB protein in the presence of the non-specific competitor
poly[(dA-dC)(dT-dG)] (100 ng) at 20 uC for 20 min in binding
Marians’ buffer (16 Marians’ buffer: 20 mM HEPES/KOH, pH 7.6,
5 mM magnesium acetate, 1 mM EDTA, 4 mM DTT, 0.2 % Triton
X-100, 1 mM ATP, 0.5 mg BSA ml21) (Parada & Marinas, 1991). The
bound complexes were separated by electrophoresis in 4 % poly-
acrylamide gels [0.256TBE (Sambrook et al., 1989), at 4 V cm21,
4 uC]. The gels were dried and analysed with a Typhoon 8600 Variable
Mode Imager.
Electron microscopy. Electron microscopic analysis of ParB–DNA
interactions was performed according to a previously described
method (Szalewska-Palasz et al., 1998). Briefly, the DNA fragments
harbouring parS sequences were PCR-amplified (Supplementary
Table S1). The M. smegmatis ParB protein was incubated at room
temperature with the DNA fragment in Marians’ binding buffer (as
described above, but without BSA). After fixation in 0.2 %
glutaraldehyde (30 min, room temperature), samples were prepared
for electron microscopy by adsorption to mica (Spiess & Lurz, 1988).
They were analysed with a transmission electron microscope (Philips
CM100) at 100 kV.
Preparation of antisera. Antisera were obtained by immunization
of rabbits with purified M. smegmatis ParB protein mixed with
monophosphoryl lipid A (MPL) adjuvant (a derivative of lipid A
from Hafnia alvei PCM 1200) (Chodaczek et al., 2006). The first
booster injection was given at 3 weeks and the second booster after a
further 2 weeks. Serum samples were taken 10 days after the second
booster injection. Cellular particles were removed by centrifugation
(500 g), and the serum was stored at 220 uC. IgG was purified by
ammonium sulfate precipitation (to 40 % saturation) and then
dialysed against 5 mM Tris/HCl. Anti-ParB antibodies were purified
by affinity chromatography. The affinity column was prepared by
immobilization of 5 mg purified recombinant ParB protein on CNBr-
activated Sepharose 4B, according to the manufacturer’s recommen-
dations. Antibodies in PBS (pH 7.3), were applied to the affinity
column, washed with PBS, eluted with 1 mM propionic acid, and
dialysed against PBS. The recombinant MtParB was also recognized
by antibodies (72 % identity between MtParB and MsParB proteins;
data not shown).
SDS-PAGE and Western blotting. SDS-PAGE was performed
according to the method established by Laemmli (1970). Proteins
were separated by SDS-PAGE (10 % or 12 % acrylamide) and
transferred to a nitrocellulose membrane. The membrane was blocked
for 1 h at room temperature with TBST (10 mM Tris/HCl, pH 8.0,
100 mM NaCl, 0.05 % Tween 20) containing 3 % BSA and
subsequently incubated with polyclonal anti-ParB antibody.
Afterwards the membrane was incubated with a goat anti-rabbit
secondary antibody conjugated with alkaline phosphatase. The
membrane was stained with 5-bromo-4-chloro-3-indolyl phosphate
and nitro blue tetrazolium. The amount of cellular ParB protein was
quantified (ImageQuant software, Molecular Dynamics) by scanning
an immunoblot containing different amounts of the 6His–MsParB
fusion protein.
GST pull-down. Affinity chromatography was performed as
described previously (Majka et al., 1997). Briefly, 25 mg ParA protein
fused to the C terminus of GST was bound to glutathione–Sepharose
beads and then used as an affinity reagent to evaluate its interaction
with ParB. Beads bound to GST–ParA were incubated with the
purified ParB protein in buffer containing 20 mM Tris, pH 7.5,
150 mM NaCl, 2.5 mM MgCl2, 0.1 % Tween and 1 mM ATP and
then were extensively washed with buffer containing 150 mM NaCl
and 0.1 % Tween. The bound protein was eluted with increasing salt
concentrations [20 mM Tris, (pH 7.5) 300, 500, 700 or 1000 mM
NaCl] and analysed by Western blotting with anti-MsParB antibodies.
Immunoprecipitation assay. M. smegmatis and its parB deletion
mutant cultivated for 48 h in modified Sauton’s broth (Mordarska
et al., 1972) were cross-linked by addition of formaldehyde (final
concentration: 1 %) for 5 min as described previously (Solomon &
Varshavsky, 1985). Cells not subjected to cross-linking and mutant
strains served as negative controls and were treated subsequently in the
same way as the experimental samples. Subsequent steps of the
immunoprecipitation assay were carried out according to Jakimowicz
et al. (2002). Briefly, cell extracts were prepared by resuspension of
~50 mg cells in 200 ml PBS buffer and sonicated (2620 s). After
centrifugation, 500 ml IMP buffer (50 mM Tris/HCl, pH 7.5, 300 mM
NaCl, 1 mM EDTA, 1 mg BSA ml21, 2 % Nonidet P40, protease
inhibitors) and 20 ml immunopurified anti-ParB antibody were added
to the cell extracts. Immunoprecipitation was carried out overnight
with gentle agitation at 4 uC followed by binding of immunocomplexes
to proteinA–Sepharose (Sigma; 4 h at 4 uC). The Sepharose slurry was
washed five times with IMP-wash buffer (50 mM Tris/HCl, pH 7.5,
300 mM NaCl, 0.5 % Nonidet P40, 0.1 % SDS), and then cross-links
were reversed by overnight incubation of the slurry with proteinase K
(0.2 mg ml21), 1 % SDS at 55 uC.
Immunoprecipitated DNA released from the protein complexes was
treated with phenol, ethanol precipitated and resuspended in TE
buffer (Sambrook et al., 1989). Semiquantitative PCR reactions (25
cycles) on precipitated DNA were set up with six primer sets
(Supplementary Table S1) amplifying DNA fragments of ~400 bp.
D. Jakimowicz and others
4052 Microbiology 153

The input DNA (not immunoprecipitated), subjected to the same
cross-link reversal and purification procedures as the experimentalsamples, served as a positive control for PCR. Following electrophor-
esis, the gel was analysed using a PhosphorImager and ImageQuant
software (Molecular Dynamics).
RESULTS
M. tuberculosis and M. smegmatis chromosomescontain parA and parB genes encodinghomologues of ParA and ParB segregationproteins and putative parS sequences
Sequence analysis identified parA and parB genes in all themycobacterial chromosomes (including M. tuberculosis andM. smegmatis) sequenced so far. As in other bacteria,mycobacterial parAB genes are situated near the chro-mosomal replication origin (5 kbp from oriC) in thevicinity of genes that encode proteins required for basiccellular functions, including a protein subunit of RNase P(RnpA), ribosomal protein L34 (RpmH) and GidB, whichhas been proposed to play a role in cell division (Fig. 1a).The identified parAB homologues are candidates formycobacterial chromosome segregation genes. In addition,we found a third element crucial to chromosomesegregation, a set of parS sequences (see below, Fig. 4a).
ParA and ParB proteins from M. tuberculosis and M.smegmatis have extensive identity with chromosomallyencoded orthologues from other bacteria, being mostsimilar to those of S. coelicolor, e.g. 65 % identity ofMsParA to ParA of S. coelicolor and 57 % identity ofMtParB to S. coelicolor ParB. The homology betweenMsParB and MtParB is very high (72 %), especially in themid-part of the protein, which contains a putative DNA-binding motif (helix-turn-helix), and in the C- terminus(Supplementary Fig. S3). Therefore, in this study we use M.smegmatis as the model for in vivo experiments andpurified MsParB and MtParB proteins for in vitro bindingexperiments.
Since mycobacterial segregation genes were identified asputative ORFs we first tested whether these genes wereexpressed in M. smegmatis. Expression of parA and parBgenes was proved by RT-PCR (Fig. 1b) and additionally thepresence of ParB protein was confirmed by Westernblotting. A rabbit polyclonal antibody was raised againstpurified MtParB protein. The affinity-purified anti-6HisMtParB rabbit antibodies specifically detected an ~40 kDaprotein (corresponding in size to the deduced parB geneproduct) in an M. smegmatis strain (Fig. 2). The antibodieswere also used to determine the number of ParB moleculesby Western blotting of cells of M. smegmatis. By usingvarious defined amounts (10–100 ng) of purified 6His–MsParB protein as standards, the amount of ParB protein(at the end of the exponential phase) was found to beapproximately 4000 molecules per single cell (0.1 % of totalsoluble cellular protein) (the intensities of bands werecompared with the standards and the amount of ParB
protein was determined by quantitative densitometry; datanot shown).
Sequence analysis of M. smegmatis and M. tuberculosischromosomes (http://www.tigr.org/) indicates that theparA and parB genes are localized within an eight-genecluster in which the distances between adjacent genes donot exceed 60 bp. This suggests that the M. smegmatisparAB belong to a large operon spanning over 5 kbp, fromrpmH to parB. It has been found previously that rpmH istranscribed from a single promoter (Salazar et al., 2003).To investigate whether transcription of the parAB genes isunder the control of this promoter, a comprehensive RT-PCR analysis was performed (Fig. 1b). Total RNA extractedfrom M. smegmatis was used in RT-PCR experiments withseven sets of primers designed to amplify the regionsspanning the gene junctions (Supplementary Table S1,Fig. 1b). A reaction without reverse transcriptase wascarried out as a negative control in all cases to ensure thatthe resulting DNA fragment was not due to amplificationof contaminant DNA. RT-PCR products were observed foreach pair of primers, while the corresponding products inthe negative controls were absent (Fig. 1b). Thus, thetranscriptional analysis results suggest that parAB as well asother genes from this cluster may be co-transcribed.However, the mRNA used for transcription analysis wasisolated from cells propagated under laboratory conditions;thus the existence of the other parAB promoter(s) thatwere not detected in this particular experimental set cannotbe ruled out.
Lack or excess of ParB protein causes growthinhibition of M. smegmatis
Library screening has suggested previously that ParB proteinis essential in M. tuberculosis H37Rv (Sassetti et al., 2003). Toverify that parB is also essential for the viability of M.smegmatis, we attempted to construct a strain defective inthe production of this protein. A two-step recombinationprotocol (Parish & Stoker, 2000) was used to generate anunmarked deletion within the M. smegmatis chromosome.Surprisingly, the parB gene could be easily deleted from theM. smegmatis chromosome. The resultant strain, M.smegmatis DparB, was verified by PCR, Southern hybridiza-tion (see Methods and Supplementary Fig. S1), and Westernblotting (Fig. 2, lane 3). The viability of this strain shows thatparB is not essential in M. smegmatis (Fig. 3a). However, inrich medium (7H9/OADC), growth of the parB deletionstrain was delayed and it exhibited a longer lag phase thanwas seen with the wild-type (Fig. 3b).
In order to screen for defective chromosome segregation,the M. smegmatis DparB strain was subjected to analysis byfluorescence microscopy. Staining with DAPI to visualizeDNA revealed that 10.3 % of the cells were anucleatecompared with 0.8 % of the wild-type (222 of 2165 DparBcells and 15 of 1915 wt cells counted) (Fig. 3c). Thus, thelack of ParB affects segregation of chromosomes and thismay influence growth of M. smegmatis.
Mycobacterial chromosome segregation
http://mic.sgmjournals.org 4053
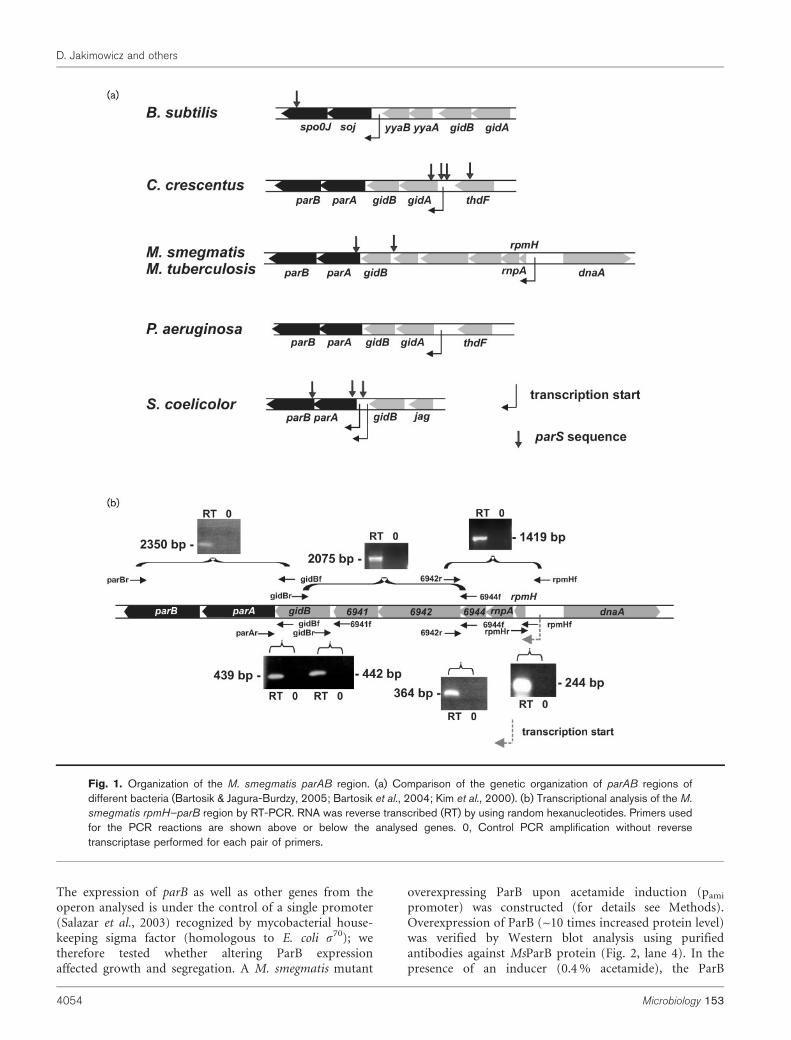
The expression of parB as well as other genes from theoperon analysed is under the control of a single promoter(Salazar et al., 2003) recognized by mycobacterial house-keeping sigma factor (homologous to E. coli s70); wetherefore tested whether altering ParB expressionaffected growth and segregation. A M. smegmatis mutant
overexpressing ParB upon acetamide induction (pami
promoter) was constructed (for details see Methods).Overexpression of ParB (~10 times increased protein level)was verified by Western blot analysis using purifiedantibodies against MsParB protein (Fig. 2, lane 4). In thepresence of an inducer (0.4 % acetamide), the ParB
Fig. 1. Organization of the M. smegmatis parAB region. (a) Comparison of the genetic organization of parAB regions ofdifferent bacteria (Bartosik & Jagura-Burdzy, 2005; Bartosik et al., 2004; Kim et al., 2000). (b) Transcriptional analysis of the M.
smegmatis rpmH–parB region by RT-PCR. RNA was reverse transcribed (RT) by using random hexanucleotides. Primers usedfor the PCR reactions are shown above or below the analysed genes. 0, Control PCR amplification without reversetranscriptase performed for each pair of primers.
D. Jakimowicz and others
4054 Microbiology 153
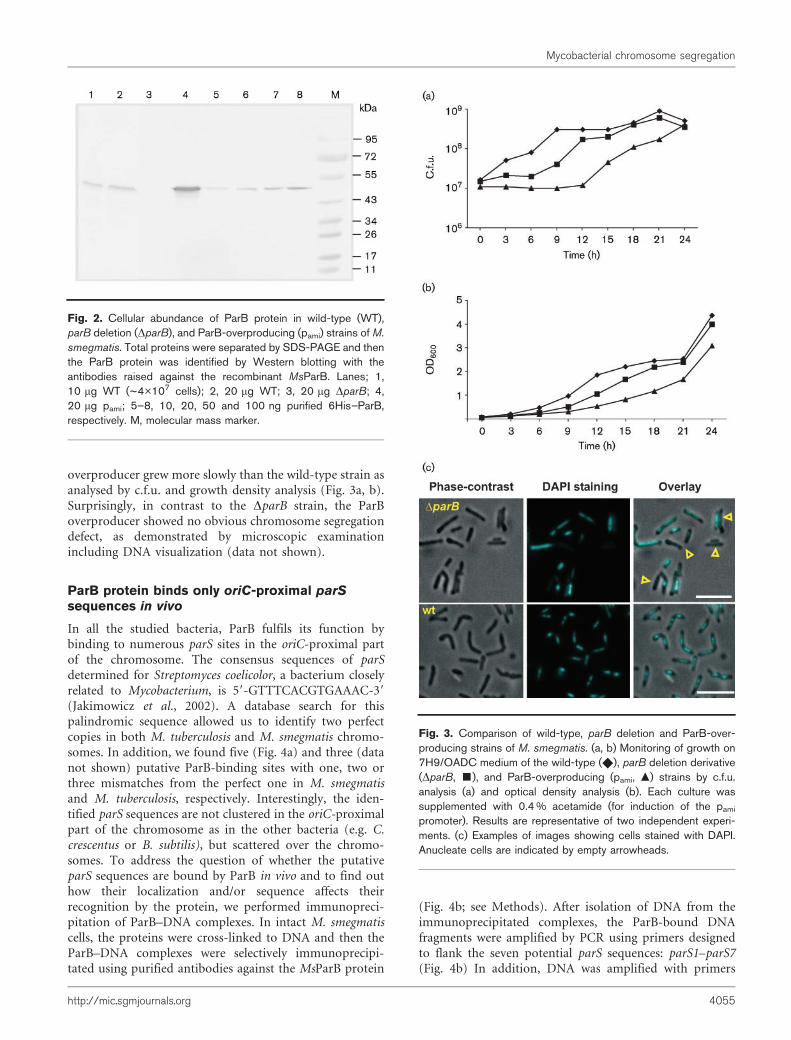
overproducer grew more slowly than the wild-type strain asanalysed by c.f.u. and growth density analysis (Fig. 3a, b).Surprisingly, in contrast to the DparB strain, the ParBoverproducer showed no obvious chromosome segregationdefect, as demonstrated by microscopic examinationincluding DNA visualization (data not shown).
ParB protein binds only oriC-proximal parSsequences in vivo
In all the studied bacteria, ParB fulfils its function bybinding to numerous parS sites in the oriC-proximal partof the chromosome. The consensus sequences of parSdetermined for Streptomyces coelicolor, a bacterium closelyrelated to Mycobacterium, is 59-GTTTCACGTGAAAC-39
(Jakimowicz et al., 2002). A database search for thispalindromic sequence allowed us to identify two perfectcopies in both M. tuberculosis and M. smegmatis chromo-somes. In addition, we found five (Fig. 4a) and three (datanot shown) putative ParB-binding sites with one, two orthree mismatches from the perfect one in M. smegmatisand M. tuberculosis, respectively. Interestingly, the iden-tified parS sequences are not clustered in the oriC-proximalpart of the chromosome as in the other bacteria (e.g. C.crescentus or B. subtilis), but scattered over the chromo-somes. To address the question of whether the putativeparS sequences are bound by ParB in vivo and to find outhow their localization and/or sequence affects theirrecognition by the protein, we performed immunopreci-pitation of ParB–DNA complexes. In intact M. smegmatiscells, the proteins were cross-linked to DNA and then theParB–DNA complexes were selectively immunoprecipi-tated using purified antibodies against the MsParB protein
(Fig. 4b; see Methods). After isolation of DNA from theimmunoprecipitated complexes, the ParB-bound DNAfragments were amplified by PCR using primers designedto flank the seven potential parS sequences: parS1–parS7(Fig. 4b) In addition, DNA was amplified with primers
Fig. 2. Cellular abundance of ParB protein in wild-type (WT),parB deletion (DparB), and ParB-overproducing (pami) strains of M.
smegmatis. Total proteins were separated by SDS-PAGE and thenthe ParB protein was identified by Western blotting with theantibodies raised against the recombinant MsParB. Lanes; 1,10 mg WT (~4�107 cells); 2, 20 mg WT; 3, 20 mg DparB; 4,20 mg pami; 5–8, 10, 20, 50 and 100 ng purified 6His–ParB,respectively. M, molecular mass marker.
Fig. 3. Comparison of wild-type, parB deletion and ParB-over-producing strains of M. smegmatis. (a, b) Monitoring of growth on7H9/OADC medium of the wild-type (X), parB deletion derivative(DparB, &), and ParB-overproducing (pami, m) strains by c.f.u.analysis (a) and optical density analysis (b). Each culture wassupplemented with 0.4 % acetamide (for induction of the pami
promoter). Results are representative of two independent experi-ments. (c) Examples of images showing cells stained with DAPI.Anucleate cells are indicated by empty arrowheads.
Mycobacterial chromosome segregation
http://mic.sgmjournals.org 4055

flanking a DNA fragment that does not contain a putativeparS sequence and is distant from the other parS sequences(Fig. 4a, b, pnull). A parB deletion mutant of M. smegmatisserved as a negative control. As shown in Fig. 4(b), twostrong parS sequences (parS1 and parS2) were clearlydetected in the ParB immunoprecipitate from the wild-type strain and none of them was detected in the equivalentsamples from the deletion parB mutant. Only onesequence, parS3 (located in the vicinity of perfect parSsequences), of the remaining five putative parS sequencescould be amplified, although more weakly than the perfectparS sites. Thus, our results suggest that only the oriC-proximal parS sites are engaged in the formation of thesegregation complex.
Binding of ParB to parS sequences in vitroinvolves ParB–ParB interaction and is assisted byParA
ParB homologues are known to be able to assemble intolarge complexes (Breier & Grossman, 2007; Jakimowicz et al.,2002), which encompass a large fragment of DNA. Thismay result from the formation of high-molecular-mass
complexes on DNA. To study the properties of the ParBcomplexes formed in vitro, electron microscopy (EM) andEMSA were performed. For both types of experiments weused a PCR-amplified DNA fragment (Supplementary TableS1) harbouring two parS sequences separated by 78 bp(Fig. 5). Analysis of electron micrographs showed that at aratio of 0.5 : 1 MsParB protein to parS, one or twonucleoprotein complexes per single DNA fragment wereobserved, while at a ratio of 1 : 1, two complexes were usuallypresent (Fig. 5a). The relative positions of the analysednucleoprotein complexes corresponded to the positions ofthe parS sequences (data not shown).
For gel retardation, a 59-end-32P-labelled DNA fragmentwas incubated with increasing amounts of MsParB orMtParB protein (Fig. 5b). At lower protein concentrations,two nucleoprotein complexes were observed for both ParBproteins. An elevated concentration, particularly in the caseof MtParB, caused the formation of higher-molecular-masscomplexes, suggesting ParB–ParB interaction(s).
To investigate if ParA–ParB interactions contribute to theformation of ParB complexes, EMSA was performed in thepresence of ParA. For this assay we used ‘unfavourable’
Fig. 4. In vivo detection of MsParB–DNA complexes. (a) Localization of parS sequences on the M. smegmatis chromosome.(b) Immunoprecipitation of DNA fragments bound to ParB. Purified antibodies raised against MsParB were used toimmunoprecipitate ParB–DNA complexes after formaldehyde cross-linking. PCR reactions were carried out with primersflanking parS sequences from different regions of the chromosome. Lanes: 1, immunoprecipitated cross-linked DNA; 2,immunoprecipitated DNA, not cross-linked (negative control); 3, input (not immunoprecipitated) cross-linked DNA (positivecontrol); 4, input (not immunoprecipitated) not cross-linked DNA (positive control). The wild-type (WT) and parB deletionmutant (DparB, negative control) were used for immunoprecipitation.
D. Jakimowicz and others
4056 Microbiology 153
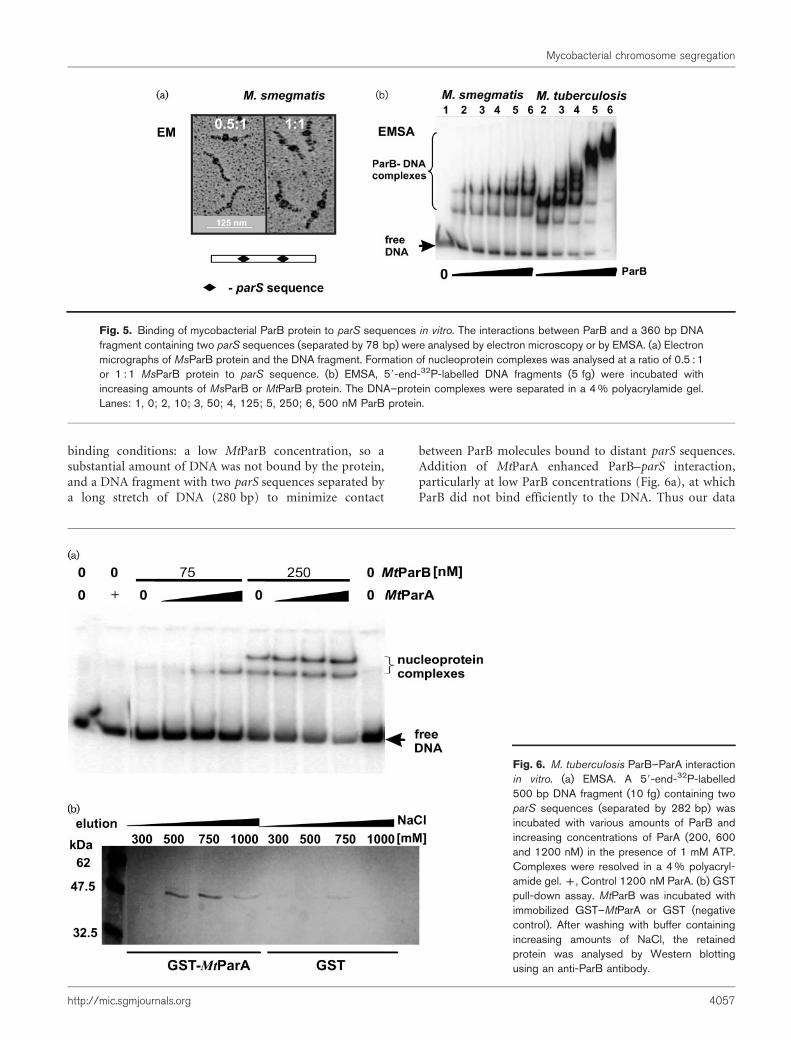
binding conditions: a low MtParB concentration, so asubstantial amount of DNA was not bound by the protein,and a DNA fragment with two parS sequences separated bya long stretch of DNA (280 bp) to minimize contact
between ParB molecules bound to distant parS sequences.Addition of MtParA enhanced ParB–parS interaction,particularly at low ParB concentrations (Fig. 6a), at whichParB did not bind efficiently to the DNA. Thus our data
Fig. 5. Binding of mycobacterial ParB protein to parS sequences in vitro. The interactions between ParB and a 360 bp DNAfragment containing two parS sequences (separated by 78 bp) were analysed by electron microscopy or by EMSA. (a) Electronmicrographs of MsParB protein and the DNA fragment. Formation of nucleoprotein complexes was analysed at a ratio of 0.5 : 1or 1 : 1 MsParB protein to parS sequence. (b) EMSA, 59-end-32P-labelled DNA fragments (5 fg) were incubated withincreasing amounts of MsParB or MtParB protein. The DNA–protein complexes were separated in a 4 % polyacrylamide gel.Lanes: 1, 0; 2, 10; 3, 50; 4, 125; 5, 250; 6, 500 nM ParB protein.
Fig. 6. M. tuberculosis ParB–ParA interactionin vitro. (a) EMSA. A 59-end-32P-labelled500 bp DNA fragment (10 fg) containing twoparS sequences (separated by 282 bp) wasincubated with various amounts of ParB andincreasing concentrations of ParA (200, 600and 1200 nM) in the presence of 1 mM ATP.Complexes were resolved in a 4 % polyacryl-amide gel. +, Control 1200 nM ParA. (b) GSTpull-down assay. MtParB was incubated withimmobilized GST–MtParA or GST (negativecontrol). After washing with buffer containingincreasing amounts of NaCl, the retainedprotein was analysed by Western blottingusing an anti-ParB antibody.
Mycobacterial chromosome segregation
http://mic.sgmjournals.org 4057

suggest that ParA contributes to nucleoprotein complexformation and presumably increases the affinity of ParB forits target (Fig. 6a). In contrast, incubation of ParA with theDNA fragment did not result in formation of anynucleoprotein complexes, indicating that ParA itself didnot bind the parS sequence(s) (Fig. 6a). In order to testwhether a direct ParA–ParB interaction occurs, an affinitychromatography assay was performed. In this assay, theMtParB protein was incubated with the GST–MtParAfusion protein bound to glutathione–Sepharose beads.After extensive washing, the bound protein was analysed byimmunoblotting with anti-MsParB antibodies (Fig. 6b).MtParB was detected in the GST–MtParA sample, whileonly a tiny amount of the protein was observed with GSTalone (negative control). Thus our in vitro data suggest thatParA interacts with ParB protein, enhancing its affinity forDNA.
DISCUSSION
Although M. tuberculosis cell multiplication is critical forestablishing infections, little is known about the cell cycleregulation of this pathogen. Since chromosome segregationis one of the major steps in the cell cycle, studies regardingthis process should reveal interesting aspects of M.tuberculosis growth. Here, we characterize, to our know-ledge for the first time, key elements of the segregation ofthe mycobacterial chromosome, ParAB proteins and parSsequences.
Our transcriptional analysis (Fig. 1b) suggests that theparAB genes of M. smegmatis may belong to a large operon,which, in addition to the partitioning genes, contains sixother genes, including those required for basic cellularfunctions, encoding GidB, a subunit of RNaseP, andribosomal protein L34. Interestingly, it has been demon-strated recently that gidB from this operon is a newstreptomycin-resistance locus in M. tuberculosis (GidBfunctions as an rRNA methyltransferase) (Okamoto et al.,2007). This operon is under the control of a singlepromoter as has been recently demonstrated (Salazar et al.,2003) and is recognized by mycobacterial housekeepingsigma factor, homologous to E. coli s70 (Gomez & Smith,2000; Salazar et al., 2003). In contrast, parAB genes of S.coelicolor are arranged in a two-gene operon whosetranscription proceeds from two promoters. Streptomycesand Mycobacterium belong to the same order ofActinomycetales, but they exhibit substantial differences,particularly in their life cycle: Mycobacterium species arestraight or slightly curved bacilli which divide by binaryfission, while Streptomyces species are filamentous bacteriawith a complex life cycle involving mycelial growth andspore formation (Chater, 2001). In S. coelicolor, one of itstwo parAB promoters is strongly upregulated shortly beforesporulation septation at about the time of the synchronoussegregation of several dozen chromosomes into presporecompartments (Kim et al., 2000). As in Streptomyces, theparAB genes of B. subtilis, a spore-forming organism, are
organized as a bicistronic operon, but they are transcribedfrom a single promoter. In the other organisms studied sofar (C. crescentus, P. aeruginosa and P. putida), parAB genesare, as in mycobacteria, not independently expressed, butare part of the gidAB operon (Bartosik et al., 2004). Thus,comparison of parAB gene expression among differentbacteria suggests that their mode of expression seems to beclosely coupled with life cycle.
As in almost all other bacteria studied so far, with theexception of C. crescentus, deletion of the parB gene is notlethal to M. smegmatis. Thus, it would be interesting toverify in vivo if the M. tuberculosis parB gene is essential, ashas been proposed previously (Sassetti et al., 2003).Substantial differences in parB requirements between theseclosely related organisms may be related to their verydistinct life styles. However, the lack of ParB in M.smegmatis does cause a quite severe phenotype: growthinhibition (Fig. 3a, b) combined with missegregation ofchromosomes – a substantial fraction (10 %) of cells wereanucleate. In Pseudomonas putida, deletion of the parBgene had a similar effect on growth and chromosomesegregation (up 10 % anucleate cells) (Lewis et al., 2002).
Recently, Lee et al. (2003) demonstrated that Spo0J is notsufficient to recruit ectopic chromosomal parS sequencesto the cell quarters of B. subtilis. Thus it was suggested thatParB, instead of being involved in origin movements,presumably plays a role in the organization and localizationof this region during the last step of the cell cycle, i.e. celldivision. We demonstrated, using in vivo immunoprecipi-tation of ParB–DNA complexes, that the three origin-proximal parS sequences distributed in an ~60 kb segment(Fig. 4a, b) are bound by ParB protein, while those locatedfar away were not. Thus, as in other organisms, binding ofParB protein only to sites close to oriC presumablyfacilitates proper chromosome orientation during celldivision of M. smegmatis.
In contrast to M. smegmatis, ParB of S. coelicolor binds tonumerous parS sites scattered over a large chromosomalsegment (400 kb) that encompasses the oriC region andassembles into a massive nucleoprotein complex(Jakimowicz et al., 2002). We speculate that organismswhose life cycle includes sporulation (Streptomyces, B.subtilis) possess numerous ParB binding sites (Breier &Grossman, 2007; Jakimowicz et al., 2002; Lee & Grossman,2006; Murray et al., 2006) that are presumably necessaryfor proper compaction of the oriC region and/or itslocalization in tiny prespore compartments.
Although the complex assembled in Mycobacterium seemsto encompass a smaller chromosome fragment, it appar-ently still influences the organization of the nucleoid. Thismay be the result of the higher-order structure subse-quently formed after the binding of parS by ParB. Our invitro EMSA results indicated that ParB binding to parSsequences involves ParB–ParB interactions. Recruitment ofadditional ParB molecules may cause binding to DNAadjacent to parS; spreading of ParB protein along the DNA
D. Jakimowicz and others
4058 Microbiology 153

around its target has been observed in B. subtilis (Breier &Grossman, 2007) and S. coelicolor (Jakimowicz et al., 2002).Moreover, we showed that ParA, interacting directly withParB, promotes ParB–DNA interacton in vitro; particularlybinding to distant parS sequences (Fig. 6a). Thus, all theelements, ParA, ParB and the parS sequences, are believedto be engaged in the formation of the mycobacterialsegregation complex in the oriC-proximal region in vivo.
Overexpression of ParB also caused a delay in growth, evenmore severe than that of the parB disruption mutant.However, neither chromosome segregation nor condensa-tion defects were observed in cells overexpressing ParBprotein. Although we can not exclude that ParB over-expression could be a source of non-specific toxicity in M.smegmatis we assume that excess of ParB presumably led tothe formation of massive nucleoprotein complexes aroundthe oriC region. Such complexes may prevent furtherrounds of replication by reducing origin accessibility forthe initiator protein DnaA and consequently delay growth.A similar effect has been observed in B. subtilis (Lee &Grossman, 2006) and S. coelicolor (D. Jakimowicz,unpublished).
Recent studies showed that M. tuberculosis cells in thedormant state are blocked at the cell division stage. Inmacrophages, M. tuberculosis cells are filamentous anddeficient in FtsZ rings (Chauhan et al., 2006). Sincebacterial cell division is tightly coupled to chromosomesegregation, it would be interesting to examine howchromosome segregation is coordinated with cell divisionin M. tuberculosis.
ACKNOWLEDGEMENTS
We thank Rudi Lurz for help in EM analysis. We are grateful toBarbara Lis for excellent assistance in the laboratory. This work wassupported by the Ministry of Science and Higher Education (grant2P04A 054 29 and partially grant N301 057 32). We are grateful to theFoundation for Polish Science (the Novum Program) for financialsupport for the purchase of a fluorescence microscope. A. K.appreciates a short-term fellowship from the Schering Foundation.
REFERENCES
Bartosik, A. A. & Jagura-Burdzy, G. (2005). Bacterial chromosomesegregation. Acta Biochim Pol 52, 1–34.
Bartosik, A. A., Lasocki, K., Mierzejewska, J., Thomas, C. M. &Jagura-Burdzy, G. (2004). ParB of Pseudomonas aeruginosa: interac-tions with its partner ParA and its target parS and specific effects onbacterial growth. J Bacteriol 186, 6983–6998.
Bignell, C. & Thomas, C. M. (2001). The bacterial ParA-ParBpartitioning proteins. J Biotechnol 91, 1–34.
Breier, A. M. & Grossman, A. D. (2007). Whole-genome analysis ofthe chromosome partitioning and sporulation protein Spo0J (ParB)reveals spreading and origin-distal sites on the Bacillus subtilischromosome. Mol Microbiol 64, 703–718.
Chater, K. F. (2001). Regulation of sporulation in Streptomycescoelicolor A3(2): a checkpoint multiplex? Curr Opin Microbiol 4,667–673.
Chauhan, A., Madiraju, M. V. V. S., Fol, M., Lofton, H., Maloney, E.,Reynolds, R. & Rajagopalan, M. (2006). Mycobacterium tuberculosiscells growing in macrophages are filamentous and deficient in FtsZ
rings. J Bacteriol 188, 1856–1865.
Chodaczek, G., Zimecki, M., Lukasiewicz, J. & Lugowski, C. (2006).A complex of lactoferrin with monophosphoryl lipid A is an efficient
adjuvant of the humoral and cellular immune response in mice. MedMicrobiol Immunol (Berl) 195, 207–216.
Chomczynski, P. & Sacchi, N. (1987). Single-step method of RNA
isolation by acid guanidinium thiocyanate-phenol-chloroform extrac-tion. Anal Biochem 162, 156–159.
DeAngelis, C. D. & Flanagin, A. (2005). Tuberculosis – a globalproblem requiring a global solution. JAMA 293, 2793–2794.
Dziadek, J., Madiraju, M. V., Rutherford, S. A., Atkinson, M. A. &Rajagopalan, M. (2002). Physiological consequences associated withoverproduction of Mycobacterium tuberculosis FtsZ in mycobacterial
hosts. Microbiology 148, 961–971.
Dziadek, J., Rutherford, S. A., Madiraju, M. V., Atkinson, M. A. &Rajagopalan, M. (2003). Conditional expression of Mycobacterium
smegmatis ftsZ, an essential cell division gene. Microbiology 149,
1593–1603.
Errington, J., Murray, H. & Wu, L. J. (2005). Diversity and redundancy
in bacterial chromosome segregation mechanisms. Philos Trans R SocLond B Biol Sci 360, 497–505.
Fogel, M. A. & Waldor, M. K. (2006). A dynamic, mitotic-like
mechanism for bacterial chromosome segregation. Genes Dev 20,3269–3282.
Godfrin-Estevenon, A. M., Pasta, F. & Lane, D. (2002). The parABgene products of Pseudomonas putida exhibit partition activity in both
P. putida and Escherichia coli. Mol Microbiol 43, 39–49.
Gomez, M. & Smith, I. (2000). Determinants of mycobacterial geneexpression. In Molecular Genetics of Mycobacteria pp. 111–129. Edited
by G. F. Hatfull & W. R. Jacobs, Jr. Washington, DC: American
Society for Microbiology.
Hampshire, T., Soneji, S., Bacon, J., James, B. W., Hinds, J., Laing, K.,Stabler, R. A., Marsh, P. D. & Butcher, P. D. (2004). Stationary phasegene expression of Mycobacterium tuberculosis following a progressive
nutrient depletion: a model for persistent organisms? Tuberculosis
(Edinb) 84, 228–238.
Hayes, F. & Barilla, D. (2006). The bacterial segrosome: a dynamic
nucleoprotein machine for DNA trafficking and segregation. Nat Rev
Microbiol 4, 133–143.
Huang, Q., Tonge, P. J., Slayden, R. A., Kirikae, T. & Ojima, I. (2007).FtsZ: a novel target for tuberculosis drug discovery. Curr Top MedChem 7, 527–543.
Jakimowicz, D., Chater, K. & Zakrzewska-Czerwinska, J. (2002). The
ParB protein of Streptomyces coelicolor A3(2) recognizes a cluster ofparS sequences within the origin-proximal region of the linear
chromosome. Mol Microbiol 45, 1365–1377.
Kieser, T., Bibb, M. J., Buttner, M. J., Chater, K. F. & Hopwood, D. A.(2000). Practical Streptomyces Genetics. Norwich: John Innes
Foundation.
Kim, H. J., Calcutt, M. J., Schmidt, F. J. & Chater, K. F. (2000).Partitioning of the linear chromosome during sporulation of
Streptomyces coelicolor A3(2) involves an oriC-linked parAB locus.J Bacteriol 182, 1313–1320.
Laemmli, U. K. (1970). Cleavage of structural proteins during theassembly of the head of bacteriophage T4. Nature 227, 680–685.
Lee, P. S. & Grossman, A. D. (2006). The chromosome partitioning
proteins Soj (ParA) and Spo0J (ParB) contribute to accuratechromosome partitioning, separation of replicated sister origins,
Mycobacterial chromosome segregation
http://mic.sgmjournals.org 4059

and regulation of replication initiation in Bacillus subtilis. MolMicrobiol 60, 853–869.
Lee, P. S., Lin, D. C., Moriya, S. & Grossman, A. D. (2003). Effects ofthe chromosome partitioning protein Spo0J (ParB) on oriCpositioning and replication initiation in Bacillus subtilis. J Bacteriol185, 1326–1337.
Leonard, T. A., Butler, P. J. & Lowe, J. (2004). Structural analysis ofthe chromosome segregation protein Spo0J from Thermus thermo-philus. Mol Microbiol 53, 419–432.
Leonard, T. A., Moller-Jensen, J. & Lowe, J. (2005). Towardsunderstanding the molecular basis of bacterial DNA segregation.Philos Trans R Soc Lond B Biol Sci 360, 523–535.
Lewis, P. J. (2004). Bacterial subcellular architecture: recent advancesand future prospects. Mol Microbiol 54, 1135–1150.
Lewis, P. J. & Errington, J. (1997). Direct evidence for activesegregation of oriC regions of the Bacillus subtilis chromosome andco-localization with the Spo0J partitioning protein. Mol Microbiol 25,945–954.
Lewis, R. A., Bignell, C. R., Zeng, W., Jones, A. C. & Thomas, C. M.(2002). Chromosome loss from par mutants of Pseudomonas putidadepends on growth medium and phase of growth. Microbiology 148,537–548.
Lin, D. C. H. & Grossman, A. D. (1998). Identification andcharacterization of a bacterial chromosome partitioning site. Cell92, 675–685.
Lin, D. C., Levin, P. A. & Grossman, A. D. (1997). Bipolar localizationof a chromosome partition protein in Bacillus subtilis. Proc Natl AcadSci U S A 94, 4721–4726.
Madiraju, M. V., Moomey, M., Neuenschwander, P. F., Muniruz-zaman, S., Yamamoto, K., Grimwade, J. E. & Rajagopalan, M. (2006).The intrinsic ATPase activity of Mycobacterium tuberculosis DnaApromotes rapid oligomerization of DnaA on oriC. Mol Microbiol 59,1876–1890.
Majka, J., Jakimowicz, D., Zarko-Postawka, M. & Zakrzewska-Czerwinska, J. (1997). Glutathione S-transferase fusion proteins as anaffinity reagent for rapid isolation of specific sequence directly fromgenomic DNA. Nucleic Acids Res 25, 2537–2538.
Majka, J., Jakimowicz, D., Messer, W., Schrempf, H., Lisowski, M. &Zakrzewska-Czerwinska, J. (1999). Interactions of the Streptomyceslividans initiator protein DnaA with its target. Eur J Biochem 260,325–335.
Mohl, D. A. & Gober, J. W. (1997). Cell cycle-dependent polarlocalization of chromosome partitioning proteins in Caulobactercrescentus. Cell 88, 675–684.
Mohl, D. A., Easter, J., Jr & Gober, J. W. (2001). The chromosomepartitioning protein, ParB, is required for cytokinesis in Caulobactercrescentus. Mol Microbiol 42, 741–755.
Mordarska, H., Mordarski, M. & Goodfellow, M. (1972). Chemo-taxonomic characters and classification of some nocardioformbacteria. J Gen Microbiol 71, 77–86.
Murray, H., Ferreira, H. & Errington, J. (2006). The bacterialchromosome segregation protein Spo0J spreads along DNA fromparS nucleation sites. Mol Microbiol 61, 1352–1361.
Okamoto, S., Tamaru, A., Nakajima, C., Nishimura, K., Tanaka, Y.,Tokuyama, S., Suzuki, Y. & Ochi, K. (2007). Loss of a conserved 7-methylguanosine modification in 16S rRNA confers low-levelstreptomycin resistance in bacteria. Mol Microbiol 63, 1096–1106.
Parada, C. A. & Marinas, K. J. (1991). Mechanism of DnaA protein-dependent pBR322 DNA replication. DnaA protein-mediated trans-strand loading of the DNA B protein at the origin of pBR322 DNA.J Biol Chem 266, 18895–18906.
Parish, T. & Stoker, N. G. (2000). Use of a flexible cassette method togenerate a double unmarked Mycobacterium tuberculosis tlyA plcABCmutant by gene replacement. Microbiology 146, 1969–1975.
Qin, M. H., Madiraju, M. V. & Rajagopalan, M. (1999). Charac-terization of the functional replication origin of Mycobacteriumtuberculosis. Gene 233, 121–130.
Rajagopalan, M., Qin, M.-H., Nash, D. R. & Madiraju, M. V. V. S.(1995). Mycobacterium smegmatis dnaA region and autonomousreplication activity. J Bacteriol 177, 6527–6535.
Rajagopalan, M., Maloney, E., Dziadek, J., Poplawska, M., Lofton, H.,Chauhan, A. & Madiraju, M. V. (2005). Genetic evidence thatmycobacterial FtsZ and FtsW proteins interact, and colocalize tothe division site in Mycobacterium smegmatis. FEMS Microbiol Lett250, 9–17.
Salazar, L., Guerrero, E., Casart, Y., Turcios, L. & Bartoli, F. (2003).Transcription analysis of the dnaA gene and oriC region of thechromosome of Mycobacterium smegmatis and Mycobacterium bovisBCG, and its regulation by the DnaA protein. Microbiology 149,773–784.
Sambrook, J., Ftritsch, E. F. & Maniatis, T. (1989). Molecular Cloning:a Laboratory Manual, 2nd edn. Cold Spring Harbor NY: Cold SpringHarbor Laboratory.
Sassetti, C. M., Boyd, D. H. & Rubin, E. J. (2003). Genes required formycobacterial growth defined by high density mutagenesis. MolMicrobiol 48, 77–84.
Solomon, M. J. & Varshavsky, A. (1985). Formaldehyde-mediatedDNA-protein crosslinking: a probe for in vivo chromatin structures.Proc Natl Acad Sci U S A 82, 6470–6474.
Spiess, E. & Lurz, R. (1988). Electron microscopic analysis of nucleicacids and nucleic acid–protein complexes. Methods Microbiol 20,293–323.
Szalewska-Palasz, A., Weigel, C., Speck, C., Srutkowska, S.,Konopa, G., Lurz, R., Marszalek, J., Taylor, K., Messer, W. &Wegrzyn, G. (1998). Interaction of the Escherichia coli DnaA proteinwith bacteriophage l DNA. Mol Gen Genet 259, 679–688.
Teleman, A. A., Graumann, P. L., Lin, D. C., Grossman, A. D. & Losick, R.(1998). Chromosome arrangement within a bacterium. Curr Biol 8,1102–1109.
Triccas, J. A., Parish, T., Britton, W. J. & Gicquel, B. (1998). Aninducible expression system permitting the efficient purification of arecombinant antigen from Mycobacterium smegmatis. FEMS MicrobiolLett 167, 151–156.
Viollier, P. H., Thanbichler, M., McGrath, P. T., West, L., Meewan, M.,McAdams, H. H. & Shapiro, L. (2004). Rapid and sequentialmovement of individual chromosomal loci to specific subcellularlocations during bacterial DNA replication. Proc Natl Acad Sci U S A101, 9257–9262.
Wayne, L. G. & Hayes, L. G. (1996). An in vitro model for sequentialstudy of shiftdown of Mycobacterium tuberculosis through two stagesof nonreplicating persistence. Infect Immun 64, 2062–2069.
Wayne, L. G. & Sohaskey, C. D. (2001). Nonreplicating persistence ofMycobacterium tuberculosis. Annu Rev Microbiol 55, 139–163.
Webb, C. D., Teleman, A., Gordon, S., Straight, A., Belmont, A., Lin,D. C., Grossman, A. D., Wright, A. & Losick, R. (1997). Bipolarlocalization of the replication origin regions of chromosomes invegetative and sporulating cells of B. subtilis. Cell 88, 667–674.
Zawilak, A., Kois, A., Konopa, G., Smulczyk-Krawczyszyn, A. &Zakrzewska-Czerwinska, J. (2004). Mycobacterium tuberculosisDnaA initiator protein – purification and DNA binding requirements.Biochem J 382, 247–252.
Edited by: C. W. Chen.
D. Jakimowicz and others
4060 Microbiology 153




















